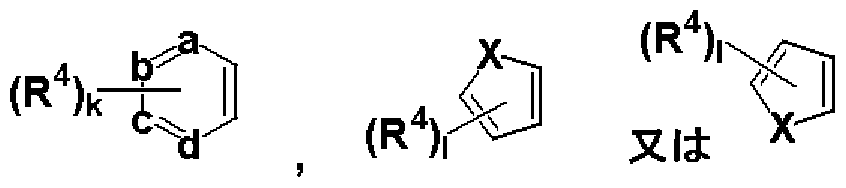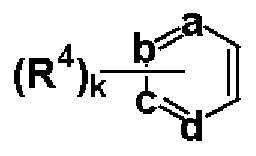明 細 書
縮合ピリダジン誘導体
技術分野
[0001] 本発明は新規な縮合ピリダジン誘導体に関する。より詳しくは、縮合ピリダジン誘導 体及びその塩、並びにそれらを有効成分とする NAD(P)H oxidase抑制薬に関する。 背景技術
[0002] 活性酸素種は生体内に存在するが、過剰産生により様々な血管障害を起こすこと が知られている。この中でもスーパーォキシドア二オン (以下、 .0—と記すことも有る。 )
2
は、 NAD(P)H oxidaseにより NAD(P)Hと酸素から生成され、全ての活性酸素種 (以下 、 ROSと記すことも有る。)の生成源として知られている。病態との関連では、糖尿病、 高脂血症、高血圧、心筋梗塞、狭心症、動脈硬化症等の病態により NAD(P)H oxidaseが 進することが失ロられてレヽる。
心筋梗塞との関係では、心筋梗塞発症後、特異的に梗塞部分において NAD(P)H oxidaseが亢進することが報告されている。 (非特許文献 1、 2)。狭心症との関係では、 狭心症患者の病変において ROSの過剰産生されることが報告されており、 NAD(P)H oxidaseの亢進と狭心症の病態の関連性が示唆される。 (非特許文献 3)、動脈硬化症 との関係では、ヒトのァテローム性動脈硬化症において、スーパーォキシドア二オン の生成と NAD(P)H oxidaseの亢進が報告されている。 (非特許文献 4、 5)。また、狭心 症や心筋梗塞に代表されるように、何らかの原因で心臓の血管内腔が狭窄または閉 塞し、心臓に必要な酸素や栄養が十分に供給されなくなった状態の疾患は、虚血性 心疾患、又は急性冠症候群と称されるが、これらの疾患と NAD(P)H oxidaseの亢進と の関係が明らかとなっている(非特許文献 6、 7)。従って、亢進した NAD(P)H oxidase を抑制することで上記のような障害を予防、治療することが出来ると考えられる。
[0003] NAD(P)H oxidase inhibitorとしては、 DPI(Diphenylene iodonium)やアポシニン( Apocynin)の例が知られている。これらの化合物は正常状態、又は病態に関わらず 非選択的に NAD(P)H oxidaseを阻害する。更に、これらはその薬効用量も高い。従つ て、これらの化合物は薬剤として十分ではないと考えられる。
[0004] 一方で、ピリダジン誘導体としてはいくつかの薬理作用例が報告されている。
[0005] アルコキシ置換フタラジンが PDE4阻害作用を有すること (特許文献 1、 2参照)、イミ タゾーピリグシンィ匕合物が Corticotropin releasing factor antagonistであること (特許 文献 3参照)、ピリダジン化合物力 SAngiogenesis inhibitorであること (特許文献 4参照)、 さらにピリダジンィ匕合物が銅の錯体へ配位する配位子であること(非特許文献 8参照) 等が報告されている。
[0006] しかしながら、これらの報告中に、 NAD(P)H oxidase抑制の作用は見られていない。
その他に、降圧作用を有するヒドララジンは、薬効量以上の用量で NAD(P)H oxidase の抑制作用を示すことが知られているが、その作用は弱い (非特許文献 9参照)。また 、 S_17834 (非特許文献 10参照)という化合物が NAD(P)H oxidase inhibitorとして報 告されている。しかし in vivoの実験における薬効用量は、 130mg/kgと高レ、。従って 、 in vivoにおいても強力な NAD(P)H oxidase抑制能を有する化合物は未だ報告例は ない。
特許文献 1: PCT国際公開 WO00/05219号公報
特許文献 2: PCT国際公開 W099/324565号公報
特許文献 3: PCT国際公開 WO00/01697号公報
特許文献 4: PCT国際公開 W098/58929号公報
^^特 3午文献 1: Biocnemical and Biophysical Research
Communicatons,2001,vol.281, 1200-1206
非特許文献 2 : Journal of Clinical Pathology,2003, 56, 194-199
非特許文献 3 Arteriosclerosis Thrombosis and Vascular Biology,2002, 22, 1838-44 非特許文献 4: Circuration, 2002, 105, 1429-35
非特許文献 5 : Annals New York Academy of Sciences, 2002, 902, 241-7
非特許文献 6: Circuration, 1999, 100, 1494-98
非特許乂 ': Arteriosclerosis thrombosis and Vascular Biology,
2003,23, 1398-1404
非特許文献 8 : Canadian Jounral of Chemistry, 1984, vol. 62, 2755
非特許文献 9 : Circulation, Res. 1997, 80, 45
非特許文献 10 : Arteriosclerosis Thrombosis and Vascular Biology, 2001, 21 : 1577 発明の開示
発明が解決しょうとする課題
[0007] 前述のように、現在、医薬品として十分な抑制能又は選択性を有する NAD(P)H oxidase抑制薬は知られていない。本発明が解決しょうとする課題は、医薬品の候補 と成り得る NAD(P)H oxidase抑制薬を提供することにある。
課題を解決するための手段
[0008] 本発明者らは上記の上記課題を解決すべく鋭意研究した結果、特定の縮合ピリダ ジン誘導体が所望の目的を達成し得ることを見出し、本発明を完成するに至った。 発明の効果
[0009] 本発明の縮合ピリダジン誘導体は NAD(P)H oxidase抑制作用を有し、当該酵素の 関与する疾患の予防又は治療に有効な医薬となりうる。
発明を実施するための最良の形態
[0010] 本発明は、下記の縮合ピリダジン誘導体若しくはその薬理学的に許容できる塩、又 はそれらの水和物若しくは溶媒和物に関する。
[0011] (1) 下記一般式 (I)
[0012] [化 1]
[0013] {式中、 Hetは、少なくとも 1個の窒素原子を環内に含み、ピリダジン環と炭素で結合し た飽和又は不飽和の 5員複素環 [該複素環は、アルキル、ヒドロキシアルキル、アルコ キシアルキル、アミノアルキル、トリフルォロメチル、ヒドロキシ、ァリール、ヘテロァリー ノレ、ハロゲン、シァノ、 -CO R1 (式中、 R1は水素原子、アルキル、又はァラルキルを示 す。)、 -CONR 3 (式中、 R2及び R3は、それぞれ同一または異なっていても良ぐ水素
原子、アルキル、又は置換されていてもよいァリールを示し、もしくは R2及び R3がー緒 になって 4から 7員環を形成してもよい。)、 -CONHSO R2a (式中、 R2aは水素原子、アル
2
キル、又は置換されていてもよいァリールを示す。)、 -NR 3 (式中、 R2及び R3は前記と 同義である。)、 -NHCO R2a (式中、 R2aは前記と同義である。)、 -NHCO R2a (式中、 R2a
2
は前記と同義である。)、 -NHSO R2a (式中、 R2aは前記と同義である。)、又は
2
-P(=〇)(〇R2a)(OR3a) (式中、 R2aは前記と同義であり、 R3aは水素原子、アルキル、又は 置換されていてもよいァリールを示す。)から選ばれる 1以上の置換基で置換されてい てもよレヽ。 ]を示し、
[0015]
[0017] (式中、 a、 b、 c、及び dは、それぞれ置換されていてもよい炭素原子又は窒素原子を 示し、かつ、 a、 b、 c、及び dのうち少なくとも 2つは置換されていてもよい炭素原子を示 し、 Xは酸素原子又は硫黄原子を示し、 R4は、同一又は異なっていても良ぐ水素原 子、ハロゲン、ァノレコキシ、ニトロ、ァミノ、ァノレキノレアミノ、ジァノレキノレアミノ、ァノレキノレ 、又は、ヒドロキシを示し、 kは 0から 4の整数を示し、 1は 0から 2までの整数を示す。た だし、 R4がメトキシを示し、かつ、 Hetが 2-チアゾリルを示す時、 kは 1を示す。)から選 ばれる何れかの環を示し、 Bは、水素原子、ハロゲン原子、アルキル、トリフルォロメチ ル、シァノ、 -CO R5 (式中、 R5は水素原子、アルキル又はァラルキルを示す。)、 -OR5'(
2
式中、 R"はアルキル、又は、置換されていてもよいフエニルを示す。)、 -NR 3 (式中、 R2及び R3は前記と同義である。)、置換されていても良いヒドラジ人置換されていても 良いグァニジノ、置換されていても良いアミジ入置換されていてもよいフエニル、置 換されていてもよいピリジル、置換されていてもよいチェニル、置換されていてもよい フリル、置換されていてもよいピペリジル、置換されていてもよいピロリジル、置換され
ていてもよいモルフォリル、置換されていてもよい 1, 4-ピペラジル、又は置換されてい てもよい 1-ピペリジル、置換されていてよいチアゾリジル、置換されていてよいアジリ ジル又は置換されてレ、てよレ、ァゼチジルを示す。
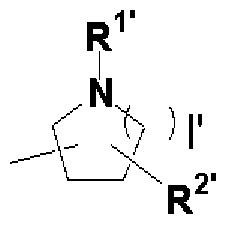
[0018] 但し、 Hetが、 2位でピリダジンと結合する 1, 2, 4-チアジアゾールを示す時、 Bは水 素原子、アルキル、トリフルォロメチル、ヒドロキシ、シァノ、カルボキシル、エステル、 アルコキシ、ジァノレキノレアミノ、置換されていてもよいフエニル、置換されていてもよい ピリジル、置換されていてもよいチェニル、置換されていてもよいフリル、置換されて いてもよいモルフォリル、下記式 (1)
[0020] [式中、 1,は 1又は 2を示し、 R1'は水素原子、アルキル、 -CO R1 (式中、 R1は前記と同 義である。)、 -SO -alkyU -SO - arvl、又は- CONH- alkvlを示し、 R2'はアルキル、ヒドロ キシ、カルボキシル、又は -CO R1 (式中、 R1は前記と同義である。)を示す。 ]、又は下 記式 (2)
[0022] (式中、 Rlb及び i 5'は同一でも異なっていてもよぐそれぞれ水素原子、アルキル、 ァリール、又は置換スルホニルを示す。)を示す。 }で表わされる縮合ピリダジン誘導 体若しくはその薬理学的に許容できる塩、又はそれらの水和物若しくは溶媒和物。 ( 2) B力 水素原子、ハロゲン原子、アルキル、トリフルォロメチル、シァ入 -CO R5 (式 中、 R5は水素原子、アルキル又はァラルキルを示す。)、 -〇R5' (式中、 R5'はアルキル、 又は、置換されていてもよいフエニルを示す。)、 -NR 3 (式中、 R2及び R3は前記と同 義である。)、ァリーノレァノレキノレ、アミノアノレキノレ、ヒドロキシアルキル、アルコキシァノレ キル、置換されていても良いヒドラジ入置換されていても良いグァニジ人置換されて いても良いアミジノ、置換されていてもよいフエニル、置換されていてもよいピリジル、 置換されていてもよいチェニル、置換されていてもよいフリル、置換されていてもよい
ピペリジル、置換されていてもよいピロリジル、置換されていてもよいモルフォリル、置 換されていてもよい 1, 4-ピペラジル、置換されていてもよい 1-ピペリジルを示す請求 項 1に記載の縮合ピリダジン誘導体 {但し、 Hetが、 2位でピリダジンと結合する 1, 2, 4-チアジアゾールを示す時、 Bは水素原子、アルキル、トリフルォロメチル、ヒドロキシ 、シァノ、カルボキシル、エステル、アルコキシ、ジァノレキノレアミノ、置換されていても よいフエニル、置換されていてもよいピリジル、置換されていてもよいチェニル、置換 されていてもよいフリル、置換されていてもよいモルフォリル、下記式 (1)
[0024] [式中、 は 1又は 2を示し、 R1は水素原子、アルキル、 -CO R1 (式中、 R1は前記と同義 である。)、 -SO -alkyl, -SO -aryl、又は -CONH- alkylを示し、 R2'はアルキル、ヒドロキ シ、カルボキシル、又は- CO R1 (式中、 R1は前記と同義である。)を示す。 ]、又は下記 式 (2)
[0026] (式中、 Rlb及び R2b'は同一でも異なっていてもよぐそれぞれ水素原子、アルキル、ァ リール、又は置換スルホニルを示す。)を示す。 }若しくはその薬理学的に許容できる 塩、又はそれらの水和物若しくは溶媒和物。 (3) Hetが少なくとも 1個の窒素原子を 環内に含み、ピリダジン環と炭素で結合した不飽和の 5員複素環である前記(1)又は (2)に記載の縮合ピリダジン誘導体若しくはその薬理学的に許容できる塩、又はそれ らの水和物若しくは溶媒和物。 (4)
[0028] が下式であり、
[0029] [化 9]
[0030] a、 b、 c、及び dのうち少なくとも 3つが置換されていてもよい炭素原子を示す前記( 1)から(3)のいずれかに記載の縮合ピリダジン誘導体若しくはその薬理学的に許容 できる塩、又はそれらの水和物若しくは溶媒和物。 (5) Bが水素原子、アルキル、トリ フルォロメチル、ヒドロキシ、シァノ、カルボキシル、エステル、 -OR5 (式中、 R5はァノレキ ル基又は置換されていてもよいフエ二ル基を示す。)、 _NR 3 (式中、 R2及び R3は前記 と同義である。)、又は下式
[0031] [化 10]
[0032] [式中、 Yは酸素原子または硫黄原子を示し、 はそれぞれ同一もしくは異なって、 水素原子、アルキル、ハロゲン、アルコキシ、ヒドロキシ、シァノ、カルボキシル、 -C〇
R1 (式中、 R1は前記と同義である。)、二卜口、 ミノ、 ノレキノレアミノ、ジ ノレキノレアミノ、 トリフルォロメチル、トリフルォロメトキシを示し、 R7は、水素原子、アルキル、 -CO R'( 式中、 R1は前記と同義である。)、 -SO _alkvl、 -SO _aryl、 _CONH_alkyl、又は置換さ れていてもよいフエニルを示し、 R8はそれぞれ同一もしくは異なって、水素原子、ヒド 口キシ、ヒドロキシメチル、カルボキシル、 -CO R1 (式中、 R1は前記と同義である。)、又 はアルキルを示し、 R9は水素原子、アルキル、ァシル、 -SO -alky 又は- SO -arylを 示し、 。は水素原子、ヒドロキシ、アルキル、カルボキシル、 -CO R1 (式中、 R1は前記 と同義である。)、 _CONR 3 (式中、 R2及び R3は前記と同義である。)を示し、 mは 0から 5の整数を示し、 nは 0から 4の整数を示し、 pは 0から 3の整数を示し、 qは 0から 8の整数 を示す。 ]から選ばれる何れかの複素環である前記(1)から (4)のレ、ずれかに記載の 縮合ピリダジン誘導体若しくはその薬理学的に許容できる塩、又はそれらの水和物
若しくは溶媒和物。 (6)
[0034] が下式であり、
[0036] kが 0、 1又は 2を示す前記(1)から(5)のレ、ずれかに記載の縮合ピリダジン誘導体若 しくはその薬理学的に許容できる塩、又はそれらの水和物若しくは溶媒和物。 (7) 飽和又は不飽和の 5員複素環が、置換基としてアルキル、ヒドロキシァノレキノレ、アルコ キシアルキル、トリフルォロメチル、ヒドロキシ、ァリーノレ、ヘテロァリール、ハロゲン、シ ァノ、カルボキシル、 -CO R11 (式中、 R11はァノレキノレ、又はァラルキルを示す。)、又は
2
-C〇NR12R13 (式中、 R12及び R13は、それぞれ同一または異なっていても良ぐ水素原 子、アルキル、又は置換されていてもよいァリールを示し、もしくは R12及び R13がー緒 になって 4から 7員環を形成してもよい。)のいずれ力 4以上を有していてもよい下記複 素環から選ばれる何れかの複素環である前記(1)から (6)のいずれかに記載の縮合 ピリダジン誘導体若しくはその薬理学的に許容できる塩、又はそれらの水和物若しく は溶媒和物。イミダゾリル、ォキサゾリル、チアゾリル、 1, 2, 4-ォキサジァゾリル、 1, 3,
4-ォキサジァゾリル、 1, 2, 4-トリァゾリル、ピラゾリル、テトラゾリル、ォキソピラゾリル、 2, 5-ジヒドロ- 5-ォキソ -4H-1, 2, 4-チアジアゾリル、 2, 5_ジヒドロ- 5_チォキソ -4H-1,
2, 4-チアジアゾリル、 2, 5-ジヒドロ- 5-ォキソ -4H-1, 2, 4_ォキサジァゾリル、 2, 5_ジ ヒドロ- 5-チォキソ -4H-1, 2, 4-ォキサジァゾリル、 4, 5-ジヒドロイミダゾリル、 4, 5_ジヒ ドロォキサゾリル、及び 4, 5-ジヒドロチアゾリル。 (8) mが 0、 1又は 2であり、 nが 0、 1又 は 2であり、 pが 0、 1又は 2であり、 qが 0、 1又は 2である前記(1)から(7)のいずれかに 記載の縮合ピリダジン誘導体若しくはその薬理学的に許容できる塩、又はそれらの 水和物若しくは溶媒和物。 (9) 以下の化合物から選ばれるいずれかの縮合ピリダジ ン誘導体若しくはその薬理学的に許容できる塩、又はそれらの水和物若しくは溶媒 和物。
[0037]
1-(1-メチル -1H-イミダゾール -2-ィル) -4-フエニルフタラジン、 1-(4-メトキシフエ二 ノレ) -4-(1-メチル -1H-イミダゾール -2-ィル)フタラジン、 1-(1-メチル -1H-イミダゾール -2-ィル) -4-(4-トリフルォロメトキシフエニル)フタラジン、 1-(1_メチル -1H -イミダゾー ル -2-ィル)フタラジン、 1-(2-フリル) -4-(1-メチル -1H-イミダゾール -2-ィル)フタラジン 、 1-[4_(1-メチル -1H-イミダゾール- 2_ィル)フタラジン _1_ィル] -ピペリジン- 4_オール 、 1-(1_メチル -1H -イミダゾール -2-ィル) -4-(4-ピリジル)フタラジン、 3-メチル _2-(4- フエニルフタラジン- 1 -ィル )_3H-イミダゾール- 4_カルボ二トリル、 3-メチル _2_(4-フエ ニルフタラジン- 1 -ィル )_3H -イミダゾール -4-カルボン酸、 1-[5_(4, 4 -ジメチル -4, 5_ ジヒドロ-ォキサゾール -2-ィル) -1-メチル -1H-イミダゾール- 2_ィル] -4-フエニルフタ ラジン、 1 -フエニル _4_(1H -テトラゾル -5-ィル)フタラジン、 1_(2_メチル -2H -テトラゾ ル -5-ィル) -4-フエニルフタラジン、及び 1-ピリジン _4_ィル- 4_(2, 5-ジヒドロ -5-ォキソ -4H-1, 2, 4-チアジアゾル -3-ィル)フタラジン。
[0038] また本発明は以下の治療薬又は抑制薬に関する。 (10)前記(1)から(9)のいずれ 力に記載の縮合ピリダジン誘導体またはその薬理学的に許容出来る塩を有効成分と する、虚血性心疾患の予防または治療薬。 (11)前記(1)から(9)のいずれかに記載 の縮合ピリダジン誘導体またはその薬理学的に許容出来る塩を有効成分とする、急 性冠症候群の予防または治療薬。 (12)前記(1)から(9)のいずれかに記載の縮合 ピリダジン誘導体またはその薬理学的に許容出来る塩を有効成分とする、心筋梗塞 の予防または治療薬。 (13)前記(1)から(9)のレ、ずれかに記載の縮合ピリダジン誘 導体またはその薬理学的に許容出来る塩を有効成分とする、狭心症の予防または 治療薬。 (14)前記(1)から (9)のいずれかに記載の縮合ピリダジン誘導体またはそ の薬理学的に許容出来る塩を有効成分とする、動脈硬化症の予防または治療薬。 ( 15)前記(1)から (9)のレ、ずれかに記載の縮合ピリダジン誘導体またはその薬理学 的に許容出来る塩を有効成分とする、 NAD(P)H oxidase抑制薬。
[0039] 本明細書にぉレ、て、「置換されてレ、てもよレ、フヱニル」の置換基としては、アルキル 、ハロゲン、アルコキシ、ヒドロキシ、シァ人カルボキシル、 -CO R1 (式中、 R1は前記と
2
同義である。)、ニトロ、ァミノ、ァノレキノレアミノ、ジァノレキノレアミノ、トリフルォロメチル、ト
リフルォロメトキシ、スルフイド、スルホキシド、スルホン、 -S-alkyU _SO_alkyl、 -SO
-alkyl等が挙げられ、該フエニルは同一もしくは異なる上記置換基で 1から 5個置換さ れていてもよぐ 2個の置換基が環を形成した結果、「置換されていてもよいフエニル」 力 ¾3 -ジヒドロ- 1-ベンゾフラン等の縮合環であってもよい。
更に「置換されていてもよいフエニル」のより好ましい置換基としては、アルキル、ハロ ゲン、アルコキシ、ヒドロキシ、シァ入カルボキシル、 -C〇 R1 (式中、 R1は前記と同義 である。)、ニトロ、ァミノ、ァノレキノレアミノ、ジァノレキノレアミノ、トリフルォロメチル、トリフ ルォロメトキシ等が挙げられ、該フヱニルは同一もしくは異なる上記置換基で 1から 5 個置換されていてもよい。
[0040] 本明細書において、「置換されていてもよいピリジル」の置換基としては、「置換され ていてもよいフエニル」の置換基と同様の置換基が挙げられ、該ピリジノレは同一もしく は異なる上記置換基で 1から 4個置換されていても良レ、。本明細書において、「置換さ れていてもよいチェニル」の置換基としては、本明細書において、「置換されていても よいフエニル」の置換基と同様の置換基が挙げられ、該チェニルは同一もしくは異な る上記置換基で、 1から 3個置換されていてもよい。
[0041] 本明細書において、「置換されていてもよいフリル」の置換基としては、「置換されて いてもよいフエニル」の置換基と同様の置換基が挙げられ、該フリルは同一もしくは異 なる上記置換基で 1から 3個置換されてレ、てもよレ、。
[0042] 本明細書において、「置換されていてもよいピペリジル」の窒素上の置換基としては
、アルキル、 -CO R1, -SO -alkyl、 -SO _aryl、又は- CONH-alkyl等が挙げられ、環状 炭素上の置換基としてはアルキル、ヒドロキシ、カルボキシル、又は- CO R1等が挙げ られる。
[0043] 本明細書において、「置換されていてもよいピロリジル」窒素上の置換基としては、 アルキル、 -CO
-SO - alkyl、 -SO _aryl、 _CONH_alkyl、環状炭素上の置換基とし ては、アルキル、ヒドロキシ、カルボキシル、又は- CO R
1 (式中、 R
1は前記と同義であ る。 )、ヒドロキシアルキル等が挙げられる。
[0044] さらに「置換されていてもよいピロリジル」窒素上のより好ましい置換基としては、ァ ノレキル、 -CO
-SO _alkyl、 -SO - aryl、 -CONH-alkyl,環状炭素上の置換基とし
ては、アルキル、ヒドロキシ、カルボキシル、又は- CO R
1 (式中、 R
1は前記と同義であ る。)等が挙げられる。
[0045] 本明細書において、「置換されていてもよい 1, 4-ピペラジル」の置換基としては、窒 素上にアルキル、ァシル、 -SO _alkyl、 -SO _arvl、等を有していてもよぐ又これらの 置換基がピペラジノレ環の炭素上に置換してレ、てもよレ、。
[0046] 本明細書において、「置換されていてもよい 1-ピペリジル」の置換基としては、ヒドロ キシ、アルキル、カルボキシル、 -C〇 R1 (式中、 R1は前記と同義である。)、 -CONR2R3( 式中、 R2及び R3は前記と同義である。)、アルコキシ、シァノ等が挙げられる。
[0047] さらに「置換されていてもよい 1-ピペリジル」のより好ましい置換基としては、ヒドロキ シ、アルキル、カルボキシル、 -CO R1 (式中、 R1は前記と同義である。 ), -CONR¾3 (式 中、 R2及び R3は前記と同義である。)等が挙げられる。
[0048] 本明細書にぉレ、て、「置換されてレ、てもよレ、モルフオリル」の置換基としては、ヒドロ キシ、アルキル、カルボキシル、 -CO R1 (式中、 R1は前記と同義である。)、 -CONR¾3( 式中、 R2及び R3は前記と同義である。)が挙げられる。
本明細書において、「置換されていてよいチアゾリジル」の置換基としては、「置換さ れていてもよいフエニル」の置換基と同様の置換基が挙げられ、該チアゾリジルは同 一もしくは異なる上記置換基で 1から 3個置換されてレ、ても良レ、。
本明細書において、「置換されていてよいアジリジル」の置換基、「置換されていてよ レ、ァゼチジル」の置換基としては、「置換されてレ、てもよレ、フエニル」の置換基と同様 の置換基が挙げられ、該アジリジルゃ該ァゼチジノレは同一もしくは異なる上記置換 基で 1から 3個置換されていても良い。
[0049] 本明細書にぉレ、て、「飽和又は不飽和の 5員複素環」としては、イミダゾリル、ォキサ ゾリル、チアゾリル、 1, 2, 4-ォキサジァゾリル、 1, 3, 4-ォキサジァゾリル、 1, 2, 4_トリ ァゾリル、ピラゾリル、テトラゾリル、ォキソピラゾリル、 2, 5-ジヒドロ- 5_ォキソ -4H-1, 2, 4-チアジアゾリル、 2, 5-ジヒドロ _5 -チォキソ -4H-1, 2, 4 -チアジアゾリル、 2, 5_ジヒド 口- 5-ォキソ -4H-1, 2, 4-ォキサジァゾリル、 2, 5 -ジヒドロ- 5-チォキソ -4H-1, 2, 4-ォ キサジァゾリル、 4, 5-ジヒドロイミダゾリル、 4, 5 -ジヒドロォキサゾリル、又は 4, 5-ジヒド 口チアゾリル等が挙げられる。
[0050] また、複素環の置換基として、アルキル、ヒドロキシアルキル、アルコキシアルキル、 トリフルォロメチル、ヒドロキシ、ァリール、ヘテロァリール、ハロゲン、シァノ、カルボキ シル、又は- CO RU(RUは、アルキル又はァラルキルを示す。)、 - CONR12R13(R12及び R
13は同一または異なっていても良ぐ水素原子、アルキル、又は置換されていてもよい ァリールを示し、若しくは R12及び R13が一緒になつて 4から 7員環を形成してもよい。)等 、より好ましくはアルキル、アルコキシアルキル、トリフルォロメチル、ヒドロキシ、ァリー ノレ、ヘテロァリール、ハロゲン、シァノ、又はカルボキシル等から選ばれる置換基を 1 つ以上有していてもよい。
[0051] 本明細書において、「アルキル」とは、好ましくは炭素数 1一 8で直鎖状でも分岐鎖 状でもよぐ例えばメチル、ェチル、ノルマル (以下、 n -と記す。)プロピル、イソプロピ ノレ、ブチル、イソブチル、第 3級 (以下、 t-と記す。)ブチル、 n-ペンチル、 n -へキシル、 又は n -ォクチル等が挙げられる。
[0052] 本明細書において、「アルコキシ」とは、好ましくは炭素数 1一 8で直鎖状でも分岐鎖 状でもよぐ例えばメトキシ、エトキシ、プロポキシ、ブトキシ、ペンチルォキシ、へキシ ノレォキシ、ォクチルォキシ等が挙げられる。アルキルチオは、好ましくは炭素数 1一 8 で直鎖状でも分岐鎖状でもよぐ例えばメチルチオ、ェチルチオ、プロピルチオ、ブ チルチオ、ペンチルチオ、へキシルチオ、ォクチルチオ等が挙げられる。ァシルォキ シは、好ましくは炭素数 1一 8で直鎖状でも分岐鎖状でもよぐ例えばホルミルォキシ、 ァセチルォキシ、プロピオニルォキシ、ブチリルォキシ、バレリルォキシ、ビバロイルォ キシ、又はへキサノィルォキシ等が挙げられる。
[0053] 本明細書にぉレ、て、「ハロゲン」とは、フッ素、塩素、臭素又はヨウ素が挙げられる。
[0054] 本明細書において、「ヒドロキシアルキル」とは、好ましくは炭素数 1一 3のヒドロキシ アルキルであり、ヒドロキシメチル、 2-ヒドロキシェチル、又は 3 -ヒドロキシプロピル等 が挙げられる。
[0055] 本明細書において、「ァリールアルキル」とは、そのァリール部は上記と同等であり、 そのアルキル部は、好ましくは炭素数 1一 3で直鎖状でも分岐鎖状でもよぐ例えばべ ンジル、フヱネチル、 3-フエニルプロピル、 1-ナフチルメチル、 2-(1-ナフチノレ)ェチル 、 2-(2_ナフチル)ェチル、又は 3_(2_ナフチル)プロピル等が挙げられる。
[0056] 本明細書において、アルコキシアルキルとは、そのアルコキシ部は上記と同等であ り、そのアルキル部は、好ましくは炭素数 1一 3で直鎖状でも分岐鎖状でもよぐ例え ばエトキシメチル、メトキシェチル、 3-メトキシプロピル、 3-エトキシプロピル、 2-メトキ シプロピル等が挙げられる。
[0057] 本明細書において、「アミノアノレキノレ」とは、そのアミノ部は任意にアルキル、ァリー ノレ、ヘテロァリール、又はへテロサイクル等で置換されていても良ぐ置換基が窒素 原子を含む環を形成していても良ぐそのアルキル部は、好ましくは炭素数 1一 3で直 鎖状でも分岐鎖状でもよぐ例えばアミノメチル、アミノエチル、ジメチノレアミノエチノレ、 N-ピロリジルェチル、 N-フエニルアミノエチル、又は 3-ァミノプロピル等が挙げられる
[0058] 本明細書において、「ヘテロサイクル」とは、炭素および少なくとも 1個の窒素を有し さらに他のへテロ原子 (酸素または硫黄)を有していてもよい 4一 6員環基であり、具体 的には、ァゼチジニル、ピロリジニル、ピペリジノ、ピペラジニル、モルホリノ、チオモル ホリノ、ォキソチオモルホリノ、又はジォキソチオモルホリノ等が挙げられる。
[0059] 本明細書において、「ァリール」とは、好ましくはフエニル、ナフチル、又はオルト融 合した二環式の基で 8— 10個の環原子を有し少なくとも一つの環が芳香環であるもの (インデニル等)等が挙げられる。
[0060] 本明細書において、「ヘテロァリール」とは、好ましくは炭素及び 1一 4個のへテロ原 子 (酸素、硫黄または窒素)を有する 5— 6員環基、またはそれ力 誘導される 8— 10個 の環原子を有するオルト融合した二環式へテロアリール、特にべンズ誘導体、もしく はプロぺニレン、トリメチレンもしくはテトラメチレン基をそれに融合して導かれるもの、 ならびにその安定な N -ォキシド等が挙げられる。例えば、プロリル、フリル、チェニル 、ォキサゾリル、イソキサゾリル、イミダゾリル、チアゾリル、イソチアゾリル、ピラゾリル、 トリアゾリノレ、テトラゾリル、 1, 3, 5_ォキサジァゾリル、 1, 2, 4_ォキサジァゾリル、 1, 2, 4-チアジアゾリル、ピリジノレ、ピラジュル、ピリミジェノレ、ピリダジニル、 1, 2, 4-トリアジ ニル、 1, 2, 3_トリアジ二ノレ、 1, 3, 5_トリアジ二ノレ、ベンゾキサゾリル、ベンゾチアゾリノレ 、ベンゾイミダゾリル、チアナフテュル、イソチアナフテュル、ベンゾフラニル、イソベン ゾフラニル、クロメニノレ、イソインドリル、インドリル、インダゾリル、イソキノリル、キノリノレ
、フタラジュル、キノキサリニル、キナゾリニル、シンノリニル、又はベンゾキサジニル等 が挙げられる。
[0061] 本明細書において、「R2及び R3が一緒になつて 4から 7員環を形成してもよレ、」の「4 力 7員環」の例としてはァゼチジン、ピロリジン、チアゾリジン、ピぺラジン、ピぺリジン
、モノレフオリン、ァザシクロヘプタン等が挙げられる。
[0062] また、上記「4から 7員環」は環上に置換基を有していても良レ、。その置換基としては
、アルキル、アルコキシ、ヒドロキシ、カルボキシル、ァシル、スルフイド、スルホキシド 等が挙げられる。
[0063] 本明細書にぉレ、て「NAD(P)H oxidase]と表記した酵素は、 NADH (Nicotinamide adenine dinucleotide 以下、 NADHと記す。) oxidase, NADH/NADPH (Nicotinamide adenine dinucleotide phosphate 以下、 NADPHと記す。) oxidase,又は、 NADPH oxidaseと表記することもある力 S、 NADH及び NADPHを基質として · 0 -産生する酵素
2
である限りすベて本発明の NAD(P)H oxidaseに含まれる。当該 NAD(P)H oxidaseの発 現部位としては、血管細胞系、心臓、腎臓、網膜、ミクログリア、又は、腫瘍細胞等の 組織が挙げられる力 特にこれらに限定されるものではない。
[0064] 本明細書において、「NAD(P)H oxidase抑制」とは、 NAD(P)H oxidaseによって産 生される · 0—産生量を低下させるあらゆる抑制を含む。例えば、 NAD(P)H oxidase活
2
性の抑制、活性化の過程の抑制、又はコンポーネントの発現の抑制等を意味する。
[0065] 本明細書において「虚血性心疾患」とは、何らかの原因で心臓の血管内腔が狭窄 または閉塞し、心臓に必要な酸素や栄養が十分に供給されなくなった状態の疾患を 意味する。
[0066] 本明細書にぉレ、て「急性冠症候群」とは、冠動脈の粥腫が破綻し、血栓形成、内腔 閉塞が起こった結果、心臓に必要な酸素や栄養が十分に供給されなくなった状態の 疾患群を意味する。その疾患の例として、急性心筋梗塞、不安定狭心症等があげら れる。
[0067] 本明細書において「心筋梗塞」とは、冠動脈の循環障害に起因する高度の虚血に より、心筋組織が壊死をきたした状態の疾患を意味する。その病態の例として急性心 筋梗塞や陳旧性心筋梗塞、貫壁性心筋梗塞や非貫壁性心筋梗塞等が挙げられる。
[0068] 本明細書にぉレ、て「狭心症」とは、心筋の酸素需要と供給のバランスの破綻により 生じる一過性の心筋虚血を意味する。その疾患の例として、安定狭心症、不安定狭 心症、労作性狭心症、安静狭心症、異型狭心症等が挙げられる。
[0069] 本明細書において「動脈硬化症」とは、動脈の弾力性が失われたり、動脈内で狭窄 、閉鎖、又は動脈瘤等の発生により、組織や臓器全体に血行障害を起こす疾患を意 味する。その疾患の例として動脈血栓症ゃァテローム性動脈硬化等が挙げられる。 本明細書にぉレ、て「予防」とは疾病に罹患してレ、なレ、者が疾患することを防止するこ とや、一度疾患に罹患し、それが治癒あるいは陳旧化してから、この疾患に再度かか ることを防止することを意味する。特に、高脂血症患者、糖尿病患者、高血圧患者、 喫煙者等のように動脈硬化症や虚血性心疾患の発症の危険が高いとされる者に対 して、又は、一度、動脈硬化症や虚血性心疾患を発症した患者に対して、その後に 虚血性心疾患すなわち心筋梗塞や狭心症発作を起こさないようにすることが含まれ る。
[0070] 次に本発明の化合物の製造方法について説明する。
[0071] 本発明の化合物は、 目的とする化合物に適した反応の組合せにより製造することが できる。以下に代表的な反応スキームを例示するが、以下に記載の方法のみに限定 されるものではない。
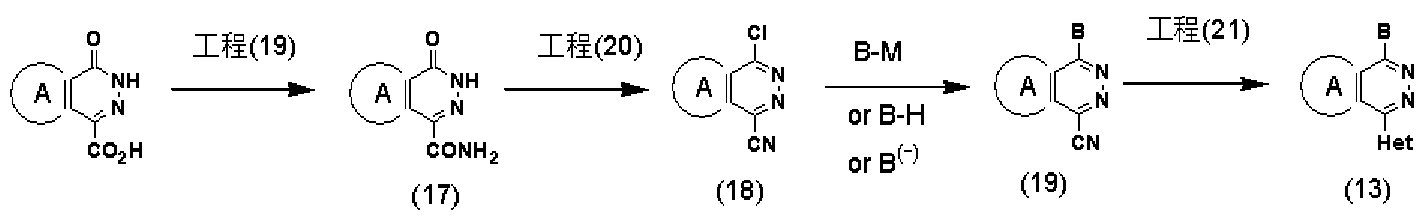
(製造法 1)
一般式 (I)の
の部分が、置換されていても良いベンゼン環、ピリジン、フラン又はチォフェンの場合 の製造方法について、以下に説明する。
[0074] [化 14]
[0075] 工程 (1)は、無水フタル酸から 2-ケト安息香酸 (1)を製造する工程である。(a)有機金 属試薬の酸無水物への付加反応、又は (b)ァリールィヒ合物と酸無水物のフリーデルク ラフッ反応を用レ、ることが出来る。
(a)の場合の有機金属試薬としてはァリール金属又はアルキル金属を用いることが出 来る。金属としては、リチウム、又はマグネシウム等が挙げられる。使用する溶媒として は、ジェチルエーテル、又は t-ブチルメチルエーテル等のエーテル類、テトラヒドロフ ラン (以下、 THFと記す。)、ジォキサン、へキサン、シクロへキサン、ベンゼン、トルェ ン、塩化メチレン、ジクロロェタン等が挙げられ、これらを単独又は混合して用いること が出来る。反応温度は- 78°C 100°C、好ましくは- 78°C 30°Cであり、反応時間は、 5分一 24時間である。
[0076] (b)の場合には、塩化アルミニウム、四塩化チタン、塩化スズ、又は三フッ化ホウ素ジ ェチルエーテラート等を触媒として用レ、、塩化メチレン、ジクロロェタン、ニトロべンゼ ン、又は二硫化炭素等の溶媒を使用する事が出来る。反応温度は- 78°C— 200°C、 好ましくは- 50°C— 100°Cであり、反応時間は、 5分一 24時間である。
[0077] 工程 (2)は縮合ピリダジノン (2)を製造する工程である。反応は (1)とヒドラジン又はヒド ラジン水和物とを反応させることにより実施出来る。溶媒を使用する場合は、水、メタ
ノーノレ、エタノール、エチレングリコール、ベンゼン、又はトルエン等を用いる。反応温 度は 20°C— 200°C、好ましくは 20°C— 100°Cであり、反応時間は、 5分一 48時間であり
、好ましくは、 1時間一 10時間である。
[0078] 工程 (3)はジカルボン酸ハーフエステルと芳香族化合物とのフリーデルクラフツ反応 により 2 -ケト安息香酸エステル (3)を製造する工程である。化合物 (3)における Bは、 2- チェニル、又は 2-フリルを示す。
[0079] この反応には、塩ィ匕ァノレミニゥム、四塩化チタン、塩化スズ、又は三フッ化ホウ素ジ ェチルエーテラート等を触媒として用レ、、塩化メチレン、ジクロロェタン、ニトロべンゼ ン、又は二硫化炭素等の溶媒を使用する事が出来る。反応温度は- 78°C— 200°C、 好ましくは _50°C 100°Cであり、反応時間は、 5分一 24時間である。
[0080] 工程 (4)は縮合ピリダジノン (4)を製造する工程である。反応は (3)とヒドラジン又はヒド ラジン水和物とを反応させることにより実施出来る。
[0081] 溶媒を使用する場合は、水、メタノーノレ、エタノール、エチレングリコール、ベンゼン
、又はトノレエン等を用いる。反応温度は 20°C— 200°C、好ましくは 20°C— 100°Cであり
、反応時間は、 5分一 48時間であり、好ましくは、 1時間一 10時間である。
[0082] 工程 (5)は、アミド化合物にブチルリチウム等の塩基を反応させてォノレトメタレーショ ンした有機金属中間体とアシノレ化試薬とを反応させ、オルト位にアシノレ基を導入し 2- ケトカルボン酸アミド (5)を得る工程である。化合物 (5)において、 Bは、置換された、ベ ンゼン環、芳香族複素環やアルキル等であっても良レ、。
[0083] アミド化合物としては、ァニリド化合物、 N, N-ジェチルアミド化合物、又はトフェニ ノレ- 1-メチルェチルアミド化合物等を用いることが出来る。
[0084] 塩基としては、ブチルリチウム、第 2級ブチルリチウム、又は t_ブチルリチウム等をァ ミド化合物に対して 1一 3等量用いる事が出来る。
[0085] また、本工程において補助的にテトラメチルエチレンジァミン等を添加することが出 来る。
[0086] 溶媒としては、ジェチルエーテル、又は t_ブチルメチルエーテル等のエーテル類、 THF、ジォキサン、へキサン、シクロへキサン、ベンゼン、トルエン、塩化メチレン、又 はジクロロェタン等が挙げられ、これらは単独又は混合して用いても良い。
[0087] ァシル化剤としては、 N, N-ジメチルアミド誘導体、 N-メチル -N-メトキシアミド誘導 体、又は酸クロリド等を用いることが出来る。
反応温度は- 78°C— 100°C、好ましくは- 78°C— 30°Cである。
[0088] 工程 (6)は 2-ケトカルボン酸アミド (5)から、 2-ケトカルボン酸 (6)を得る工程である。反 応は水、硫酸を用いた加水分解、酸化剤を用いた反応が使用出来る。反応温度は
20°C— 200°Cであり、反応時間は 10分一 24時間である。
[0089] 工程 (7)は縮合ピリダジノン (7)を製造する工程である。反応は、(6)とヒドラジン又はヒ ドラジン水和物とを反応させることにより、もしくは (5)とヒドラジン又はヒドラジン水和物 とを反応させることにより実施出来る。溶媒を使用する場合は、水、メタノール、ェタノ ール、エチレングリコール、ベンゼン、又はトノレエン等を用いる。
反応温度は 20°C 200°C、好ましくは 20°C— 100°Cであり、反応時間は、 5分一 48時 間であり、好ましくは、 1時間一 10時間である。
[0090] 工程 (8)は、 2-ョード安息香酸エステルから生成した有機金属中間体とアシノレ化試 薬とを反応させ、オルト位にアシノレ基を導入し 2-ケトカルボン酸エステル (8)を製造す る工程である。
[0091] イソプロピルグリニヤー試薬等を反応させることにより生じた有機金属中間体と無機 亜鉛化合物 (ZnCl、 ZnBr、又は Znl等)とを反応させることで有機亜鉛化合物へと変 換し、さらに触媒存在下、ハロゲン化アシノレ化合物と反応させることにより目的化合 物を製造することが出来る。
[0092] 触媒としては、 0価又は 2価の Pd触媒 (Pd(PPh )、又は Pd(PPh ) C1等)や 0価又は 2価 の M触媒等を用いることが出来る。
[0093] 使用する溶媒としては、ジェチルエーテル、又は t-ブチルメチルエーテル等のエー テノレ類、 THF、ジォキサン、へキサン、シクロへキサン、ベンゼン、トルエン、塩化メチ レン、ジクロロェタン等が挙げられ、これらを単独又は混合して用いることが出来る。
[0094] 反応温度は- 78°C 100°C、好ましくは- 78°C 30°Cである。
[0095] 工程 (9)は縮合ピリダジノン (11)を製造する工程である。
[0096] 反応は、(8)とヒドラジン又はヒドラジン水和物とを反応させることにより実施出来る。
溶媒を使用する場合は、水、メタノーノレ、エタノール、エチレングリコール、ベンゼン、
又はトルエン等を用いる。
[0097] 反応温度は 20°C— 200°C、好ましくは 20°C— 100°Cであり、反応時間は、 5分一 48時 間であり、好ましくは、 1時間一 10時間である。
[0098] 工程 (10)はジメチルォキサゾリン化合物にブチルリチウム等の塩基を反応させてォ ルトメタレーシヨンした有機金属中間体とアシノレ化試薬とを反応させ、オルト位にァシ ル基を導入し 2 -ケトカルボン酸アミド (9)を得る工程である。
[0099] アミド化合物としては、ァニリド化合物、 N, N-ジェチルアミド化合物、又は 1-フエ二 ノレ- 1-メチルェチルアミド化合物等を用いることが出来、塩基としては、 n -ブチルリチ ゥム、第 2級ブチルリチウム、 t-ブチルリチウム等をアミド化合物に対して 1等量一 3等 量用いる事が出来る。
[0100] 反応補助試薬として、テトラメチルエチレンジァミン等を添加することが出来る。溶媒 としては、ジェチルエーテル、又は t-ブチルメチルエーテル等のエーテル類、 THF、 ジォキサン、へキサン、シクロへキサン、ベンゼン、トルエン、塩化メチレン、ジクロロェ タン等が挙げられ、これらは単独又は混合して用いても良レ、。
[0101] ァシル化剤としては、 N, N-ジメチルアミド誘導体、 N-メチル -N-メトキシアミド誘導 体、又は酸クロリド等を用いることが出来る。
[0102] 酸クロリドをアシノレ化剤として用いる時には、触媒として 0価又は 2価の Pd触媒
(Pd(PPh )、 Pd(PPh ) CI等)や 0価又は 2価の Ni触媒等を用いることが出来る。
3 4 3 2 2
[0103] 反応温度は- 78°C— 100°C、好ましくは- 78°C— 30°Cである。
[0104] 工程 (11)は 2-ケトォキサゾリン化合物 (9)から、 2-ケトカルボン酸 (10)を得る工程であ る。
[0105] 反応は塩酸、硫酸、メシノレ酸、又はトシノレ酸存在下で行うことが出来る。溶媒は水、 ジォキサン、 THF、エタノール、又はメタノール等を単独又は混合して用いる。
[0106] 工程 (12)は、縮合ピリダジノン (11)を製造する工程である。反応は、化合物 (10)とヒド ラジン又はヒドラジン水和物とを反応させることにより、実施出来る。溶媒を使用する 場合は、水、メタノーノレ、エタノール、エチレングリコール、ベンゼン、又はトルエン等 を用いる。
[0107] 反応温度は 20°C 200°C、好ましくは 20°C 100°Cであり、反応時間は、 5分一 48時
間であり、好ましくは、 1時間一 10時間である。
[0108] 工程 (13)は化合物 (2)、(4)、(7)、及び (11)の塩素化反応により化合物 (12)を製造する 工程である。
[0109] 塩素化剤としては、塩化チォニル、ォキシ塩化リン、三塩化リン、又は五塩化リン等 を用いることが出来る。反応は、無溶媒あるいはベンゼン、トルエン、クロ口ホルム、又 はジクロ口ェタン等の溶媒中で行う。
[0110] 工程 (14)は化合物 (12)より化合物 (13)を製造する工程である。工程 (14)において、化 合物 (13)の
[0112] の部分は、置換されていても良いベンゼン環を示す。
[0113] 複素環有機ホウ素化合物、複素環有機亜鉛化合物、又は複素環有機スズ化合物 を触媒下で反応させることにより実施出来る。
[0114] 複素環有機スズィ匕合物は、有機リチウム又は有機マグネシウム化合物とトリアルキ ノレ塩化スズ力 調整することが出来、単離する事なく工程 (14)の反応に使用すること あでさる。
[0115] 複素環有機ホウ素化合物、及び複素環有機亜鉛化合物もホウ酸エステル、ハロゲ ン化亜鉛を用いて上記と同様に調整出来、単離する事なく工程 (14)の反応に使用す ることも出来る。また、前記有機リチウム化合物はブチルリチウム等の塩基によるリチ エーシヨン (lithiation)、あるいはハロゲン化合物とのハロゲン-リチウム交換により調整 出来る。
[0116] 触媒としては、 0価又は 2価の Pd触媒 (Pd(PPh )、又は Pd(PPh ) C1等)や 0価又は 2価
3 4 3 2 2
の Ni触媒等を用いることが出来る。
[0117] 反応温度は _78°C 200°C、好ましくは 0°C 120°Cである。
[0118] 使用する溶媒としては、ジェチルエーテル、 t_ブチルメチルエーテル等のエーテル 類、 THF、ジォキサン、へキサン、シクロへキサン、ベンゼン、トルエン、キシレン、塩 化メチレン、ジクロロェタン、等が挙げられ、これらを単独又は混合して用いることが
出来る。また、本工程は化合物 (12)をスルホン体へと変換した後に、有機リチウム化 合物と反応させることによつても実施出来る。
[0120] の部分がピリジン、フラン又はチォフェンである化合物 (13)については、工程 (3)、(5)、 (8)、(10)における出発原料のベンゼン環部分力 ピリジン、フラン、又はチォフェンで ある化合物を選択し、上述と同様の方法で製造することが可能である。
[0121] さらに、本発明化合物は以下の方法によっても合成できる。
(製造法 2)
[0123] 工程 (15)はカルボン酸力 複素環置換のケトン (14)を製造する工程である。反応は カルボン酸を塩ィ匕チォニル、又はオギザリルクロライド等により酸クロリドへと変換した 後に、 N -メチルイミダゾール等の複素環とトリェチルァミン等の塩基で処理することに より実施できる。
[0124] 溶媒を使用する場合は、塩化メチレン、ァセトニトリル、ベンゼン、又はトルエン等を 用いる。
[0125] 反応温度は 0°C 100°C、好ましくは 0°C 30°Cである。反応時間は、 1時間一 48時 間であり、好ましくは、 1時間一 10時間である。
[0126] 工程 (16)は縮合ピリダジノン (15)を製造する工程である。
[0127] 反応は (14)とヒドラジン又はヒドラジン水和物を作用させることにより実施出来る。溶
媒を使用する場合は、水、メタノーノレ、エタノール、エチレングリコール、ベンゼン、又 はトルエン等を用いる。反応温度は 20°C— 200°C、好ましくは 20°C— 100°Cであり、反 応時間は、 5分一 48時間であり、好ましくは、 1時間一 10時間である。
[0128] 工程 (17)は化合物 (15)の塩素化反応により化合物 (16)を製造する工程である。塩素 化剤としては、塩ィ匕チォニル、ォキシ塩化リン、三塩化リン、又は五塩化リン等を用い 、無溶媒あるいはベンゼン、トルエン、クロ口ホルム、又はジクロロエタン等の溶媒中 で反応を行う。
[0129] 工程 (18)は化合物 (16)より本発明化合物 (13)を製造する工程である。 B-Mは、有機 ホウ素化合物、有機亜鉛化合物又は有機スズィ匕合物等の有機金属化合物を表し、 これらを触媒下で化合物 (16)と反応させることにより工程 (18)を実施出来る。有機ホウ 素化合物、有機亜鉛化合物又は有機スズ化合物は、市販の化合物を使用することも 出来るが、自ら調整することも出来る。
[0130] 有機スズィ匕合物は有機リチウム又は有機マグネシウム化合物とトリアルキル塩ィ匕ス ズカ 調整することが出来、単離することなく用いることもできる。有機ホウ素化合物、 有機亜鉛化合物はホウ酸エステル、ハロゲンィヒ亜鉛を用いて同様に調整出来、単離 する事なく用いることも出来る。また前記有機リチウム化合物はブチルリチウム等の塩 基によるリチェーシヨンあるいはハロゲンィ匕合物とのハロゲン-リチウム交換により調整 することが出来る。
[0131] 触媒としては、 0価又は 2価の Pd触媒 (Pd(PPh )、 Pd(PPh ) CI等)や 0価又は 2価の Ni
3 4 3 2 2
触媒等を用いることが出来る。反応温度は- 78°C— 200°C、好ましくは 0°C— 120°Cで ある。使用する溶媒としては、ジェチルエーテル、 t-ブチルメチルエーテル等のエー テノレ類、 THF、ジォキサン、へキサン、シクロへキサン、ベンゼン、トルエン、キシレン 、塩化メチレン、ジクロロェタン、 DMF、 N -メチノレピロリドン、が挙げられ、単独又は混 合して用いることが出来る。
[0132] また、本工程は化合物 (16)と B-Hとの反応によっても実施できる。ここで、 B-H、 B(—〕と は求核剤 (nucleophileXァミン、チオール、又はアルコキシド等)を示す。反応温度は -78°C— 200°C、好ましくは 0°C 160°Cである。使用する溶媒としては、ジェチルエー テル、又は t-ブチルメチルエーテル等のエーテル類、 THF、ジォキサン、メタノーノレ、
エタノール、へキサン、シクロへキサン、ベンゼン、トルエン、キシレン、塩化メチレン、 ジクロロェタン、 DMF、又は N-メチルピロリドンが挙げられ、単独又は混合して用いる ことが出来る。
[0133] 工程 (19)は、カルボン酸をアミド (17)に変換する反応であり、既知の方法を用いるこ とが出来る。
[0134] 工程 (20)は化合物 (17)の塩素化反応により化合物 (18)を製造する工程である。塩素 化剤としては、塩ィ匕チォニル、ォキシ塩化リン、三塩化リン、又は五塩化リン等を単独 又は混合して用い、無溶媒あるいはベンゼン、トルエン、クロ口ホルム、又はジクロロ ェタン等の溶媒中で反応を行う。
[0135] 工程 (21)は、二トリル (19)から本発明化合物 (13)を製造する工程であり、既存の方法 を用いて閉環を行うことが出来る。
[0136] 例えば、ヒドロキシァミンと二トリルを反応させて得られる N -ヒドロキシアミジンを、酸 クロリド又は酸無水物で処理することによりォキサジァゾールが得られる。また、種々 の縮合剤 (CDI、又は DCC(Dicyclohexylcarbodiimide)等を用いてカルボン酸と N-ヒド ロキシアミジンとを縮合し、さらに加熱閉環させることも出来る。また、 N-ヒドロキシアミ ジンにクロロギ酸ェチルゃチォカルボニルジイミダゾールを反応させることにより、そ れぞれォキソォキサジァゾールゃチォキソォキサジァゾールへと変換出来る。
[0137] また、 N-ヒドロキシアミジンにチォカルボ二ルイミダゾールを加え、さらにボロントリフ ルオリド ェチルエーテル錯体で処理して、ォキソチアジアゾールを得ることもできる 。その他の例として、アジ化ナトリウム等のアジド化合物と二トリルを反応させてテトラ ゾールへと変換することが挙げられる。
[0138] また、エチレンジァミン誘導体と二トリルを反応させてジヒドロイミダゾールを得ること も出来る。
[0139] さらに本発明化合物は以下の方法によっても製造出来る。
[0141] 工程 (22)は、前記手法にて製造したフタラジノン化合物に、複素環リチウム化合物 を反応させ、本発明化合物を製造する工程である。
[0142] 複素環リチウム化合物としては、 N-置換 _2_リチウム化合物が挙げられ、フタラジノ ンに対して 200 500モル%量用いることで実施できる。
[0143] 反応温度は _78°C 65°C、好ましくは _50°C 20°Cである。
[0144] 使用する溶媒としては、ジェチルエーテル、又は第 3級ブチルメチルエーテル等の エーテル類、 THF、ジォキサン、メタノーノレ、エタノール、へキサン、シクロへキサン、 ベンゼン、トルエン、キシレン、塩化メチレン、ジクロロェタン等が挙げられ、単独又は 混合して用いることが出来る。
[0145] また、本発明化合物は以下の方法によっても製造出来る。
(製造法 4)
[0147] 工程 (23)は、二トリル体 (出発物質右化合物)からの変換の場合には、工程 (21)の 方法に準じて実施することが出来る。さらに二トリルの加水分解等によって得られる力 ルボン酸化合物 (出発物質左化合物)を既知の方法に従って、種々の複素環に変換 することが出来る。
[0148] 例えば、出発物質左化合物をァミンと縮合剤にてアミドに変換した後に五塩化リン、 トリメチルシリルアジドと反応させるとテトラゾールを得ることが出来る。また、ァセトヒド ラジドとの反応で得られるアミド化合物ポリリン酸中で加熱することにより 1, 3, 4-ォキ サジァゾールを得ることが出来る。さらにこの化合物とパラメトキシベンジルァミンとを 反応させると 1, 3, 4-トリァゾール誘導体へと変換される。さらにトリフルォロ酢酸をカロ えて加熱すると脱べンジノレ化したトリァゾール化合物が得られる。また、 1, 3, 4 -ォキ
サジァゾール化合物をローソン試薬 (Lawesson' s Reagent)にて処理すると 1, 3, 4_チ アジアゾールイ匕合物が得られる。
[0149] また、出発物質左化合物をカルボエルジイミダゾール、マロン酸ェチルカリウム、塩 化マグネシウムで 3-ォキソフタラジン- 1 -ィル -プロピオン酸エステルと変換し、このェ ステルとヒドラジンとを反応させるとピラゾロン誘導体へと変換出来る。
[0150] また、得られたフタラジンィ匕合物の芳香環上 (複素 5員環含む)に置換基を導入する ことも出来る。例えば、ハロゲン化は、塩素、臭素、ヨウ素、 NCS、 NBS、又は MS等を 用いて実施出来る。また導入されたハロゲン原子は種々の金属触媒を用いてさらに 他の誘導体へと導くことが出来る。例えば、 CuCNを用いてのシァノ化が可能である。 また、種々の有機金属化合物 (有機スズ化合物、有機亜鉛化合物、又は有機ホウ素 化合物等)も Pd化合物等を触媒とした反応にてカップリングさせることが出来る。
[0151] また、得られた化合物は保護、脱保護の手法、あるいはアルキル化、加水分解等の 反応によって官能基を変換させることも出来る。
[0152] 以上のようにして得られる各化合物は抽出、結晶化、再結晶、各種クロマトグラフィ 一などの通常の化学操作により単離精製される。
[0153] 本発明の化合物は、通常用いられる適当な希釈剤や他の添加剤とともに適当な投 与形態 (粉末剤、注射剤、錠剤、カプセル剤又は局所外用剤など)に調整した後、そ の投与形態応じた適当な投与方法 (例えば静脈内投与、経口投与、経皮投与又は 局所投与など)によって、ヒト又は動物に投与する事ができる。
[0154] 前記、式 (I)の化合物の塩としては、酸付加塩又は塩基付加塩を用いることができる 力 生理的に許容されるものであれば塩の種類は特に限定されることはない。
一般式 (I)で表わされる縮合ピリダジン誘導体の塩、又はそれらの水和物若しくは溶 媒和物は、縮合ピリダジン誘導体力 公知の方法により製造すればよい。
[0155] 本発明の式 (I)の化合物又はその塩の 1種又は 2種以上をそのまま患者に投与して もよいが、好ましくは、有効成分と薬理学的及び製剤学的に許容しうる添加物を加え 、当業者に周知な形態の製剤として提供されるべきである。
[0156] 薬理学的及び製剤学的に許容しうる添加物としては、賦形剤、崩壊剤、結合剤、滑 沢剤、コーティング剤、色素、希釈剤、基剤、及び等張化剤等を用いることが出来る。
[0157] 経口投与に適する製剤の例としては、錠剤、カプセル剤、散剤、細粒剤、顆粒剤、 液剤、又はシロップ剤等を挙げることができ、非経口投与に適する製剤としては、注 射剤、点滴剤、又は坐剤等を挙げることが出来る。
[0158] 経口投与に適する製剤には、添加物として、賦形剤、崩壊剤、結合剤、滑沢剤、コ 一ティング剤又は基剤等を用いることが出来る。また、本発明の化合物を治療の対象 となる患者に対して投与する場合、対象疾患の治療のために適切な他剤と本発明の 化合物とを併用してもよい。
[0159] 本発明の医薬の投与経路は特に限定されず、経口的又は非経口的に投与するこ とができる。本発明の医薬の投与量は、疾患の予防及び/又は治療の目的、患者の 年齢や状態などの条件に応じて適宜選択可能であるが、一般的には、成人に対して 0.001— 10mg/kg程度、好ましくは 0.0001— lmg/kg程度を注射又は点滴により投与す るか、 0.001— 100mg/kg程度、好ましくは 0.001— 30mg/kg程度を経口的に投与する ことが好ましい。
実施例
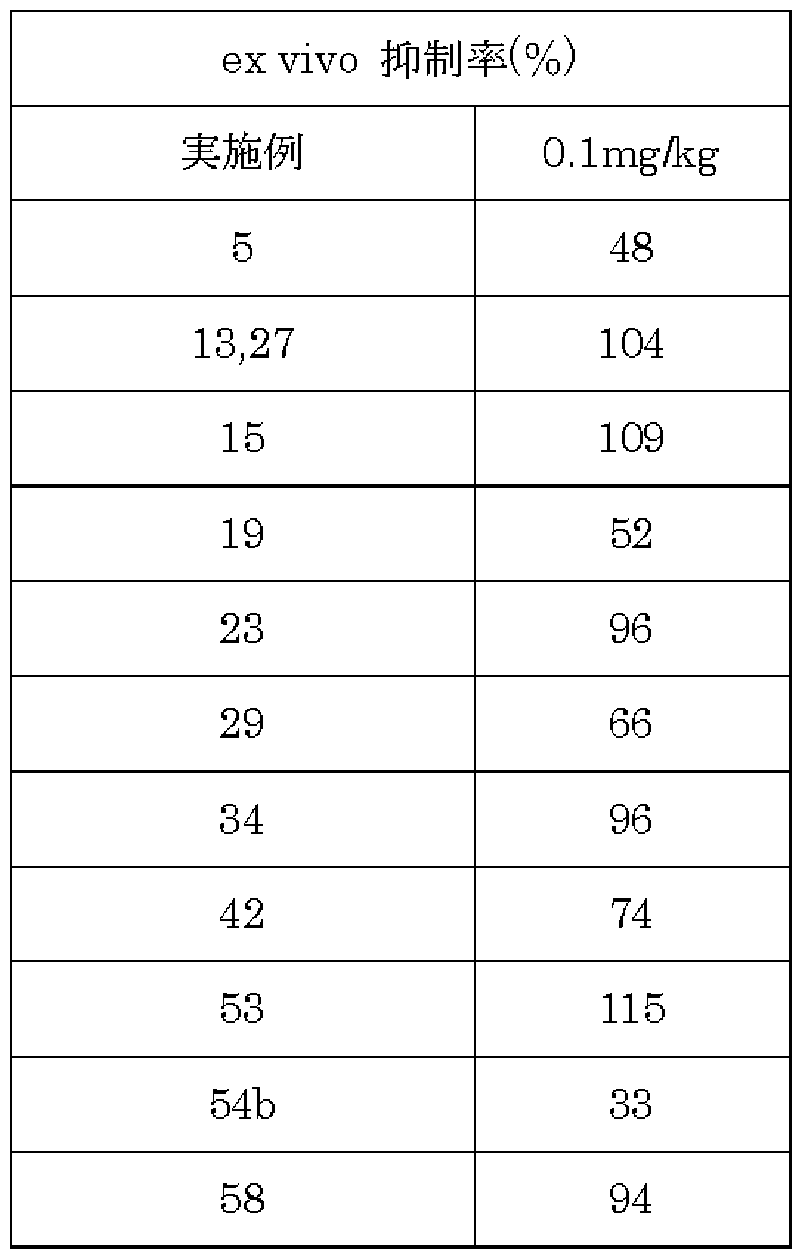
[0160] 以下、本発明を実施例によりさらに具体的に説明するが、本発明の範囲は下記の 実施例に限定されることはない。なお、以下の実施例においては次に示すような慣 用略号を用いるものとする。
[0161] 1¾ =テトラヒドロフラン、 DMF = N, N-ジメチルホルムアミド、 DMS〇 =ジメチルス ルホキシド、 CDI =カルボニルジイミダゾール、 DPPA =ジフヱニルホスホリルアジド 、 Z =ベンジルォキシカルボニル、 Boc =第 3級ブチルォキシカルボニル DMF =ジ メチルフオルムアミド、 1¾ =テトラヒドロフラン NMRは核磁気共鳴スぺクトノレを意味 する。数字は通常化学シフトを表示するのに用いられる δ (デルタ)値であり単位は ppmである。内部標準としては TMS (テトラメチルシラン)あるいは重水素化溶媒中の残 留プロトン (DMSO-d6: 2.5ppm)を用いた。なお、 δ値の次に表示したカツコ内の数字 は水素原子の数であり、それに続く表示は sが単一線、 dが二重線、 tが三重線、 qが 四重線、 mが多重線、 brが巾広い吸収ピークを意味する。
[0162] MSはマススペクトルを意味する。括弧内に測定方法 (イオン化方法)を示した。すな わち、 EIは電子イオン化法、 FritFABはフリット高速原子衝撃法を示す。数字は観測
された分子イオン (M+、 M + H + )、フラグメントイオンを示し、単位は m/zである。
[0163] (実施例 1) 1-(4-エトキシフエニル) -4-(1-メチル -1H-イミダゾール -2-ィル)フタラジ ン 塩酸塩
a) フタル酸 モノメチルエステル
無水フタル酸 30.0g(202.5 mmol)をメタノールに溶解し、 2時間加熱還流する。反応液 を減圧留去し、得られた固体を減圧下乾燥させ表記化合物 36.9g (収率 100%)を無色 固体として得た。 1H- NMR(CDC13): 3·93(3Η, s), 7.57- 7.64(2H, m), 7.69- 7.73(1H, m), 7.94-7.96(lH, m)。
b) 2_(1-メチル -1H-イミダゾール- 2_ィル)カルボニル安息香酸 メチルエステル 実施例 laで得られた化合物 67.94g(377.1 mmol)を塩化チォニル (160ml)に溶解し、 3 時間過熱還流する。反応液を減圧留去し、ァセトニトリル (50ml)に溶解する。
[0164] 別に N -メチルイミダゾール 31.95g(389.2 mmol)をァセトニトリル (300ml)に溶解し、トリ ェチルァミン 57.0ml(410 mmol)を加え、氷冷下攪拌する。この氷冷攪拌中の溶液に 前記ァセトニトリル溶液を滴下し、室温下数時間攪拌する。
[0165] 反応液に水を加え、酢酸ェチルにて抽出し、飽和食塩水にて洗浄後、硫酸ナトリウ ムにて乾燥する。不溶物をろ過した後、溶液を減圧留去する。得られた油状物をシリ 力ゲルカラムクロマトグラフィー (酢酸ェチル -へキサン)にて精製した後、へキサン-ジ ェチルエーテルで洗浄して、表記化合物 49.88g (収率 54%)を固体として得た。
1H-NMR(CDC13): 3.69(3H, s), 4.15(3H, s), 7.06(1H, s), 7.08(1H, s), 7.51— 7·64(3Η, m), 7.96-8.00(lH, m)。
c) 4-(l-メチル -1H-イミダゾール -2-ィル) -2H-フタラジン- 1-オン
実施例 lbで得られた化合物 40.13g(164 mmol)をエタノール 160mlに溶解し、ヒドラジン 1水和物 11.5ml(237 mmol)をカ卩ぇ 6時間加熱還流した。室温に冷却した後、ジェチル エーテル 100mlをカ卩え、 0°Cで 30分撹拌した。析出した固体をろ取し、ジェチルエーテ ルで洗浄し、表記化合物 35.47g (収率 95%)を得た。 1H_NMR(CDC13): 3.87(3H, s), 7·07(1Η, s), 7·25(1Η, s), 7.75— 8.00(2Η, m), 8.45— 8.55(1Η, m), 8·65_8·75(1Η, m), 11.24(1Η, s)。
d) l_クロ口- 4_(1_メチル _1Η -イミダゾール -2-ィル)フタラジン
実施例 lcで得られた化合物 5.40g(23.87 mmol)にォキシ塩化リン (8.0ml)を加え、 3時 間加熱還流した。反応液を冷却後、ォキシ塩化リンを減圧留去し、塩化メチレンに懸 濁させた。飽和重曹水、水酸化ナトリウムをカ卩えて pH > 8とし、分液し、水層を塩化メ チレンで抽出し、有機層を合わせて硫酸ナトリウムで乾燥した。不溶物をろ過した後、 シリカゲルカラムクロマトグラフィー (酢酸ェチル-ジクロロメタン)にて精製後、へキサン で洗浄して表記化合物 5.19g (収率 89%)を無色固体として得た。
[0166] さらに、ろ液を減圧濃縮して、 0.21g (収率 4%)の固体を得た。 1H_NMR(CDC13):
4·08(3Η, s), 7· 16(1Η, s), 7.33(1Η, s), 7.95— 8.05(2Η, m), 8.30— 8.40(1Η, m), 9· 15-9·25(1Η, m)0
e) l-(4-エトキシフエニル) -4-(1_メチル -1Η -イミダゾール -2-ィル)フタラジン 実施例 Idで得られた化合物 296mg(1.210 mmol)に 4-エトキシフヱニルボロン酸 306mg(1.843 mmol),炭酸カリウム 524mg(3.719 mmol),トルエン 5ml、エタノール 2mlを 加え、窒素ガスを懸濁液中に吹き込みながら室温で 1時間撹拌した。テトラキス (トリフ ェニルホスフィン)パラジウム (0)86mg(0.074 mmol)を加え、窒素気流下、 110°Cにて 6時 間撹拌した。反応溶液にクロ口ホルムをカ卩え、セライト、シリカゲル上でろ過した。 ろ 液を減圧濃縮した後、残渣を t_ブチルメチルエーテル -酢酸ェチルに加熱溶解した。 この溶液を冷却し、析出した固体をろ取し、表題化合物 201mg (収率 55%)を得た。 1 H— NMR(CDCl ): 1.49(3H, t), 4.10— 4·20(5Η, m), 7.09— 7.13(2Η, m), 7.16(1Η, s),
3
7.34(1Η, s), 7.76-7.80(2Η, m), 7.80— 8.00(2Η, m), 8.16- 8.20(1Η, m), 9.15- 9·20(1Η, m)。
f) l-(4-エトキシフエニル) -4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン 塩酸塩 実施例 leで得られた化合物 201mg(0.662 mmol)をアセトン 30mlに溶解し、 4モル塩酸 -酢酸ェチル溶液 (0.57ml)をカ卩ぇ室温で撹拌した。エーテルを加えた後、析出物をろ 取して表題化合物 252mgを得た。 1H-NMR(DMS0- d6: 1.41(3H, t), 3.96(3H, t), 4.19(2H, q), 7.21-7.25(2H, m), 7.79— 7.83(2H, m), 7.97(1H, s), 8.07(1H, s), 7.80-8.00(2H, m), 8.16_8.30(4H, m)0
[0167] (実施例 2) 1_(1-メチル -1H-イミダゾール- 2_ィル) _4_(4_トリフルォロメトキシフヱ二 ノレ)フタラジン 塩酸塩
a) 1-(1-メチル -1H -イミダゾール _2-ィル) -4-(4-トリフルォロメトキシフエ二ル)フタラ ジン
実施例 Idで得られた化合物、及び 4-トリフルォロメトキシフヱニルボロン酸を用いて実 施例 leと同様の操作を行うことにより、表記化合物 506mg (収率 68%)を固体として得 た。 1H-NMR(CDC13): 4.14(3H, s), 7· 18(1Η, s), 7.35(1Η, s), 7.43-7.470(2Η, m), 7.80-8.10(5Η, m), 9.23-9.26(1Η, m)MS(EI): 370(Μ+), 342, 285, 241, 209, 183, 156, 129。
b) 1-(1-メチル -1Η-イミダゾール _2 -ィル )_4_(4-トリフルォロメトキシフヱニル)フタラ ジン 塩酸塩
実施例 2aで得られた化合物を用いて実施例 Ifと同様の操作を行うことにより、表記化 合物 320mg (収率 58%)を固体として得た。 lH_NMR(DMSO_d6: 3.96(3H, t),
7.67-7.75(2H, m), 7.98_8·05(3Η, m), 8.10(1H, s), 8.15-8.25(4H, m)0
[0168] (実施例 3) l-(3, 4-ジメトキシフエニル) -4-(l-メチル -1H-イミダゾール -2-ィル)フタ ラジン 塩酸塩
a) 1-(3, 4-ジメトキシフエニル) -4-(1-メチル -1H-イミダゾール -2-ィル)フタラジン 実施例 Idで得られた化合物及び 3, 4-ジメトキシフヱニルボロン酸を用いて実施例 le と同様の操作を行うことにより、表記化合物 531mg (収率 77%)を固体として得た。
1H-NMR(CDC13): 3·99(3Η, s), 4.01(3H, s), 4.14(3H, s), 7.05- 7·50(5Η, m),
7.85— 8.05(2H, m), 8.20— 8.25(1H, m), 9.10— 9·20(1Η, m)。
b) l-(3, 4-ジメトキシフヱニル) -4-(l_メチル -1H -イミダゾール -2-ィル)フタラジン 塩 酸塩
実施例 3aで得られた化合物を用いて実施例 Ifと同様の操作を行うことにより、表記化 合物 404mg (収率 58%)を黄色固体として得た。 lH_NMR(DMSO_d6: 3.87(3H, s), 3.91(3H, s), 3·96(1Η, s), 7.24— 7.28(1H, m), 7.39— 7.45(2H, m), 7.97(1H, s),
8.07(1H, s), 8.15—8.35(4H, m)0
[0169] (実施例 4) 1_(2, 4 -ジメトキシフヱニル) -4-(l-メチル -1H-イミダゾール- 2_ィル)フタ ラジン 塩酸塩
a) 1_(2, 4-ジメトキシフエニル) -4-(1-メチル -1H-イミダゾール- 2_ィル)フタラジン
実施例 Idで得られた化合物及び 2, 4-ジメトキシフヱニルボロン酸を用いて、実施例 leと同様の操作を行うことにより、表記化合物 326mg (収率 78%)を固体として得た。 1H-NMR(CDC13): 3·69(3Η, s), 3·92(3Η, s), 4.13(3Η, s), 6.60-6.75(2Η, m), 7· 15(1Η, s), 7·33(1Η, s), 7.46-7.50(1Η, m), 7.75— 8.00(3Η, m), 9· 10_9· 15(1Η, m)0 b) 1_(2, 4-ジメトキシフヱニル) -4-(l-メチル -1H-イミダゾール- 2_ィル)フタラジン 塩 酸塩
実施例 4aで得られた化合物を用いて実施例 Ifと同様の操作を行うことにより、表記化 合物 316mg (収率 88%)を黄色固体として得た。 lH_NMR(DMSO_d6: 3.71(3H, s), 3.9K3H, s), 3·98(1Η, s), 6.75— 6.90(2H, m), 7.45— 7.48(1H, m), 7·80_8·25(6Η, m)0
[0170] (実施例 5) 1_(1-メチル -1H-イミダゾール- 2_ィル) _4_(4_ピリジル)フタラジン
実施例 Idで得られた化合物 2.80g(11.44 mmol)、及びピリジン- 4_ボロン酸
3.25g(26.44 mmol)を用いて実施例 leと同様の操作を行うことにより、表記化合物 2.21g (収率 67%)を固体として得た。 1H-NMR(CDC13): 4.15(3H, s), 7.20(1H, s), 7.37(1H, s), 7, 75(2H, d), 7.90— 8.10(3H, m), 8.88(2H, d), 9.29(1H, d)MS(EI): 287, 259, 209, 182, 156, 129, 78。
[0171] (実施例 6) 1-メトキシ -4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン 塩酸塩 実施例 Idで得られた化合物 342mg(1.415 mmol)をメタノールに懸濁させ、 _70°Cにて 水素化ナトリウム (60%)65mg(1.63 mmol)を加えた。さらに室温で撹拌した後、 50°Cに て 3時間撹拌した。反応液に水を加え室温にて撹拌した後、析出した固体をろ取し、 得られた粗生成物をへキサン-エタノールで洗浄し、フリー体 180mg (収率 53%)を得 た。 1H-NMR(CDC13): 4.04(3H, s), 4.58(3H, s), 7.10(1H, s), 7.28(1H, s),
7.85-7.95(2H, m), 8.20_8.30(1H, m), 8.90- 9.00(1H, m) この化合物をアセトン 7mlに 溶解し、 4モル塩酸-酢酸ェチル溶液 (0.175ml)を加え室温で撹拌した。析出した固体 をろ取し、表題化合物 109mg (収率 53%)を得た。 lH_NMR(DMSO_d6: 3.88(3H, s), 4.32(3H, s), 7·88(1Η, s), 7.99(1H, s), 8.00— 8.10(3H, m), 8.30— 8.40(1H, m)。
[0172] (実施例 7) 1_(4-シァノフエノキシ )_4_(1_メチル _1H -イミダゾール -2-ィル)フタラジ ン 塩酸塩
4-シァノフヱノール 220mg(0.899 mmol)を DMFに溶解させ、水素化ナトリウム (60%
)126mg(1.06 mmol)を加え室温で撹拌した。これに Idで得られた化合物を加え、 150 °Cで 6時間撹拌した。反応液に水をカ卩ぇ室温にて撹拌した後析出した固体をろ取し、 エタノールに加熱溶解し、冷却後、析出物をろ取してフリー体 132mg (収率 45%)を固 体として得た。ろ液に同様の操作を行レ、、フリー体 42mg (収率 14%)を得た。
1H-NMR(CDC13): 4.01(3H, s), 7.13(1H, s), 7·31(1Η, s), 7.48— 7.52(2H, m), 7.75-7.85(2H, m), 7.90_8.05(2H, m), 8.40- 8.45(1H, m), 9.10- 9.15(1H, m) この化 合物 132mgをアセトン-ジェチルエーテルに溶解し、 4モル塩酸-酢酸ェチル溶液 (0.27ml)をカ卩ぇ室温で撹拌した。析出した固体をろ取して表題化合物 134mg (収率 92 %)を得た。 lH-NMR(DMSO-d6: 3·88(3Η, s), 7.65— 7.70(2H, m), 7.80— 7.82(1H, m), 7.92-7.94(lH, m), 8.00-8.10(2H, m), 8.20— 8.30(3H, m), 8.50— 8.60(1H, m)。
[0173] (実施例 8) 6 -メトキシ メチル _1H -イミダゾール -2-ィル) -4-フエニルフタラジ ン
a) 2-ベンゾィル -4-メトキシ -N-フエニルベンズアミド
4-メトキシベンゾィル クロリド 10.83g(63.48 mmol)のジクロロメタン 20ml溶液を、ァニリ ン 5.99ml(64.32 mmol),トリェチルァミン 9.5mlのジクロロメタン 120ml溶液に氷冷下で 滴下した後、 1時間室温にて撹拌した。反応液を濃縮し、水を加えて酢酸ェチルで抽 出した。 1M水酸化ナトリウム、飽和食塩水で洗浄し、無水硫酸マグネシウムにて乾燥 し、ろ過、濃縮した。へキサン、酢酸ェチルを加え析出した固体をろ取し、へキサンで 洗浄して、 4-メトキシ -N-フエニル-ベンズアミド 5.50g(24.20 mmol、収率 38%)を得た。 同じ方法で得たものと合わせた 4-メトキシ -N-フエニル-ベンズアミド 6.57g(28.91 mmol)に、 THF100ml、 TMEDA (テトラメチルエチレンジァミン) 9.6ml(63.6 mmol)をカロ え、 -78°Cに冷却した。この混合物に窒素気流下にて n-ブチルリチウムへキサン溶液 (1.5M)42ml(63mmol)を 20分かけて滴下した。
[0174] 反応液を 0°Cにて 15分撹拌した後、 _78°Cに冷却し、ジメチルベンズアミド
4.80g(32.2 mmol)の THF溶液 (40ml)を 15分かけて滴下した。反応液を徐々に室温ま で戻した後、メタノールを加えた。溶媒を減圧濃縮し、 THF-酢酸ェチルに溶解し、飽 和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥した。ろ過、減圧濃縮後、へキサ ン-酢酸ェチル = 1 : 1 約 200mlをカ卩え、加熱還流下で洗浄した。室温に冷却し、析
出物をろ取して、表題化合物 5.49g (収率 57%)を得た。さらに、ろ液から 3.4gの粗生成 物を得た。
b) 6-メトキシ- 4-フエニル -2H -フタラジン- 1-オン
実施例 8aにて得られた化合物 8.84g(26.68 mmol)にヒドラジン 1水和物 40ml、エチレン グリコール 20mlをカ卩え、 2時間加熱還流した。反応液を冷却し、水 60ml を加え、析出 した固体をろ取した。これにエタノール 50mlを加え、加熱還流下で洗浄して、表題 化合物 5.92g (収率 88%)を得た。 lH_NMR(DMSO-d6): 3.81(3H, s), 6.95_7.05(1H, m), 7.40-7.65(6H, m), 8.20— 8.30(1H, m), 12.70(1H, s)。
c) 6 -メトキシ -1-(1-メチル -1H -イミダゾール -2-ィル) -4-フエニルフタラジン 実施例 8bにて得られた化合物を THF13mlに懸濁させ- 30°Cに冷却した。別に調整し た 1-メチルイミダゾリル- 2-リチウムの THF溶液 (1 -メチルイミダゾール 1.31g(15.96 mmol), THF 10ml, n_BuLi(1.56M) 10ml 力も調整)を滴下し、徐々に室温に戻し 10 時間撹拌した。氷冷下、反応液に 1モル塩酸 50mlを加え、酢酸ェチルで洗浄した。水 層を、酢酸ェチル -THFで洗浄した後、炭酸カリウムを加えて塩基性とした。これを酢 酸ェチルにて抽出し、飽和食塩水で洗浄し、無水硫酸ナトリウムにて乾燥した。シリ 力ゲル上でろ過、濃縮し、アセトン 5ml、ジェチルエーテル 15mlを加えて 0°Cで洗浄す ることにより、表題化合物 771mg (収率 60%)を淡黄色固体として得た。
1H-NMR(CDC13): 3·87(3Η, s), 4.13(3H.s), 7.14(1H, s), 7.32(1H, s), 7.35— 7·38(1Η, m), 7.50-7.65(4H, m), 7.80_7.85(2H, m), 9.13(1H, d)。
(実施例 9) 6-クロ口- 1-(1-メチル -1H-イミダゾール -2-ィル) -4-フエニルフタラジン a) 2-ベンゾィル -4-クロ口 -N -フエニル -ベンズアミド
4-クロ口安息香酸 15.72g(100.4 mmol)にジクロロメタン 100ml、 DMF0.3mlを加え、塩化 オギザリル 9.2ml(105 mmol)を室温で 30分かけて滴下した後、 30°Cにて約 1時間撹拌 した。得られた反応液を、ァニリン 10.0ml(109.4 mmol),トリェチルァミン 15.3ml(110 mmol)のジクロロメタン 100ml溶液に氷冷下で滴下した後、終夜室温にて撹拌した。反 応液に水 100mlを加え、ジクロロメタンを留去し、水酸化ナトリウム 2g、エタノール 50ml をカロえた。析出した固体をろ取し、水洗した。得られた固体を酢酸ェチル、次いでへ キサン、酢酸ェチルの混合溶媒にて洗浄することにより、 4_クロ口- N-フエニル -ベン
ズアミド 21.19g(91.46 mmol、収率 91%)を得た。この 4_クロ口- N-フエニル-ベンズアミ ド 21.19g(91.46 mmol)を用レ、、実施例 8aと同様の操作を行うことにより、表題化合物 16.04g (収率 52%)が得られた。
b) 6_クロ口 - 4_フエニル -2H-フタラジン- 1-オン
実施例 9aにて得られた化合物 16.04g(47.77 mmol)用レ、、実施例 8bと同様の操作を行 うことにより、表題化合物 4.77g (収率 39%)を得た。 lH-NMR(DMSO_d6):
7·50-7·65(6Η, m), 7.90_7·95(1Η, m), 8.30— 8.40(1Η, m), 12.96(1Η, s)。
c) 6_クロ口- 1-(1_メチル _1Η -イミダゾール -2-ィル) -4-フエニルフタラジン
実施例 9bにて得られた化合物 536mg(2.088 mmol)用レ、、実施例 8cと同様の操作を行 うことにより、表題化合物 210mg (収率 31%)を得た。 1H-NMR(CDC ): 4.15(3H.s), 7· 18(1Η, s), 7.34(1H, s), 7.55— 7.65(3H, m), 7.75— 7.82(2H, m), 7·85_8·00(1Η, m), 8.00-8.10(1H, m), 9.29(1H, d)。
(実施例 10) 8-クロ口- 1-(1-メチル -1H-イミダゾール -2-ィル) -4-フエニルフタラジン a) 6-ベンゾィル -2-クロ口 -N -フエエルベンズアミド
実施例 8aと同様にして合成した 2-クロ口- N-フエエル-ベンズアミド 10.06g(43.
42 mmol)を用い、実施例 8aと同様の操作を行うことにより、表題化合物 10.99g (収率 75
%)を得た。
b) 8-クロ口- 4-フエニル -2H-フタラジン- 1-オン
実施例 10aにて得られた化合物 10.99g(32.73 mmol)に 50%硫酸水溶液 (濃硫酸 14ml 、水 14mはり調整)、 n-ブタノール 10mlをカ卩ぇ 2時間加熱還流させた。反応液に水 200mlを加え、酢酸ェチルで抽出し、飽和食塩水で洗浄した後、減圧濃縮した。これ にエタノール 80ml、ヒドラジン 1水和物 5mlを加え、 6時間加熱還流させた。室温まで冷 却した後、析出した固体をろ取することにより、表題化合物 6.48g (収率 77%)を得た。 lH-NMR(DMSO-d6): 7·50_7·60(6Η, m), 7·75_7·95(2Η, m), 12.82(1Η, s)。
c) 8_クロ口- 1-(1_メチル _1Η -イミダゾール -2-ィル) -4-フエニルフタラジン
実施例 10bにて得られた化合物 51 lmg(l.990 mmol)用レ、、実施例 8cと同様の操作を 行うことにより、表題化合物 554mg (収率 87%)を得た。 1H_NMR(CDC ): 3.77(3H.s), 7· 11(1Η, s), 7·25(1Η, s), 7.50— 8.10(8Η, m)
(実施例 11) 6-フルォ口- 1-(1-メチル -1H-イミダゾール -2-ィル) -4-フエニルフタラジ ン
a) 2-ベンゾィル -4-フルォ口- N-フエニルベンズアミド
実施例 8aと同様にして合成した 4_フルォ口- N-フエニルベンズアミド 4.37g(20.30 mmol)を用レ、、実施例 9aと同様の操作を行うことにより、表題化合物 3.65g (収率 56%) を得た。
b) 6_フルォ口- 4-フエニル -2H-フタラジン- 1-オン
実施例 11aにて得られた化合物 2.55g(7.99 mmol)に 50%硫酸水溶液 (濃硫酸 4ml、水 4mはり調整)を加え 2時間加熱還流させた。反応液を冷却し、水 40mlを加え、ろ取す ることにより、 2-ベンゾィル -4-フルォロ安息香酸 1.40g (収率 72%)を得た。
lH-NMR(DMS-d6): 7.35_7.45(m, 1H), 7.45-7.55(3H, m), 7.60- 7.70(3H, m), 8.00-8.10(1H, m) この化合物 0.73gにエタノール 6ml、ヒドラジン 1水和物 0.16gを加え 、 2時間加熱還流させた。 0°Cまで冷却した後、析出した固体をろ取することにより、表 題化合物 465mg (収率 65%)を得た。 lH-NMR(DMSO_d6): 7.25-7.35(1Η, m), 7.50-7.60(5H, m), 7.70- 7.80(1H, m), 8.35- 8·45(1Η, m)。
c) 6-フルォロ -1-(1-メチル -1H-イミダゾール -2-ィル) -4-フエニルフタラジン 実施例 l ibにて得られた化合物 139mg(0.579 mmol)用い、実施例 8cと同様の操作を 行うことにより、表題化合物 71.0mg (収率 40%)を得た。 1H_NMR(CDC13): 4.15(3H.s), 7.17(1H, s), 7.33(1H, s), 7.55- 7·85(7Η, m), 9.30- 9·40(1Η, m)。
(実施例 12) 1-(1-メチル -1H-イミダゾール -2-ィル) -4-トリフルォロメチルフタラジン a) 2-トリフルォロアセチル安息香酸
4, 4 -ジメチル- 2_フエニル -1, 3 -ォキサゾリン 8.70g(49.65 mmol)を THF35mlに溶解 し、窒素下、 -70°Cにてノルマルブチルリチウム (1.56Mへキサン溶液) 35mlを滴下した 。 0°Cで 40分撹拌した混合物を、 -70°Cにて 1等量のトリフルォロ酢酸ェチルエステル の THF50ml溶液へ 1時間かけて滴下した。反応混合物に氷と 4モル塩酸水溶液をカロ え、酢酸ェチルで 3回抽出した。塩酸一食塩水で洗浄、無水硫酸ナトリウムで乾燥さ せ、シリカゲル上でろ過し減圧濃縮して、 13.95gの油状物質を得た。これに 1, 4 -ジォ キサン 40ml、濃塩酸 50ml、水 30mlを加え、 100°Cにて 6時間撹拌した。酢酸ェチルと
水を反応液に加え、分液し濃縮した。さらに 6モル塩酸水溶液 100mlをカ卩え、 100°Cで 1時間加熱した後、酢酸ェチルと水を反応液に加え、分液し濃縮した。シリカゲルクロ マトグラフィ一にて精製し、表題化合物 8.34g (収率 77%)を褐色油状物質として得た b) 4_トリフルォロメチル -2H-フタラジン- 1-オン
実施例 12aにて得られた化合物 8.34gをエタノールに溶解し、ヒドラジン 1水和物 4.4ml をカロえて 12時間加熱還流した。反応液を 0°Cに冷却した後、析出した固体をろ取し、 表題化合物 5.88g (収率 72%)を得た。 lH_NMR(DMSO-d6): 7.90-8.10(3H, m), 8.30-8.40(1Η, m), 13.13(1H, s)。
c) l_(l_メチル _1H -イミダゾール -2-ィル) -4-トリフルォロメチルフタラジン
実施例 12bで得られた化合物 365mg(1.704 mmol)を THF6mlに懸濁させ- 70°Cに冷却 した。別に調整したトメチルイミダゾリル- 2-リチウムの THF溶液 (1-メチルイミダゾー ノレ 561mg(6.833 mmol), THF60ml, n-BuLi(1.56M)4.4ml 力ら調整)を滴下し、 -70。C で 3時間撹拌した。さらに- 50°Cに昇温し、 1時間撹拌した。
[0178] 反応液に水 20mlを加え、酢酸ェチルで抽出した。これを、 1モル塩酸水溶液で 2回 抽出した水層に、炭酸ナトリウムを加えアルカリ性にした後、酢酸ェチルにて抽出した 。飽和食塩水で洗浄し、無水硫酸ナトリウムにて乾燥した。ろ過、濃縮し、ジェチルェ 一テルをカ卩えて得られた溶液にへキサンをカ卩えた。溶媒を減圧留去すると途中で固 体が得られた。これを- 70°Cにて 10分間へキサンで洗浄して、表題化合物 54.1mg (収 率 11%)を得た。 1H_NMR(CDC 3): 4.15(3H, s), 7.21(1H, s), 7.37(1H, s),
8.00-8.10(2H, m), 8.25— 8.35(1H, m), 9·35— 9·45(1Η, m)。
[0179] (実施例 13) 1_(1-メチル -1H-イミダゾール _2 -ィル )_4 -フエニルフタラジン
4-フエニル -2H-フタラジン _1_オン 5.60g(25.20 mmol)用レ、、実施例 8cと同様の操作を 行うことにより、表題化合物 5.87g (収率 81%)を得た。 1H-NMR(CDC13): 4.13(3H, s), 7· 17(1Η, s), 7.34(1Η, s), 7.55— 7.60(3Η, m), 7.75— 7.95(4Η, m), 8· 10_8· 14(1Η, m), 9· 19-9·25(1Η, m)MS(EI): 286(Μ+), 258, 209, 183, 156, 143, 129, 102, 77。
[0180] (実施例 14) 1_(1Η-イミダゾール _2 -ィル )_4 -フヱニルフタラジン
a) エトキシメチル -1H -イミダゾール -2-ィル) -4-フエニルフタラジン
イミダゾール 6.85g(100.6 mmol)を THF30mlに溶解し、クロロメチルェチルエーテル 4.95ml(51.22 mmol)の THF溶液を滴下した。さらに室温で 2.5時間撹拌した後、トルェ ン 80mlを加えた。塩をろ去し、溶液を濃縮して 1-エトキシメチル -1H -イミダゾール 4.43gの粗生成物を得た (収率 69%)。これを THF30mlに溶解し、 _70°Cにて n_ブチル リチウム (1.56M)35ml(54.6 mmol)を滴下し 20分撹拌し、黄色懸濁液を得た。これを、 4-フエニル -2H-フタラジン _1_オンの THF 15ml懸濁液に、室温で加え、さらに 8時間 撹拌した。水、濃塩酸をカ卩えて pH8 9に調整し、酢酸ェチルで抽出した。 2モル塩酸 にて水層へと抽出した後、炭酸カリウムをカ卩えてアルカリ性とした。これを酢酸ェチル で抽出、飽和食塩水洗浄、無水硫酸ナトリウム乾燥、ろ過、濃縮し、シリカゲルカラム クロマトグラフィー (へキサン:酢酸ェチル = 1:4→酢酸ェチル)にて精製し表題化合物 0.45g (収率 13%)を得た。 1H-NMR(CDC13): 1· 11(3Η, t), 3·52(2Η, q), 5.97(2Η, s), 7·39(1Η, d), 7.44(1Η, d), 7·55_7·65(3Η, m), 7.75-8.15(5Η, m), 9.15(1Η, d)。
b) l-(lH-イミダゾール -2-ィル) -4-フエニルフタラジン
実施例 14aで得られた化合物 0.45g(1.362 mmol)にエタノール 6ml、水 3ml、濃塩酸 3ml を加え、 9時間加熱還流させた。反応液を濃縮しアセトン 10mlを加え 30分撹拌し固体 をろ取して表題化合物 399mg (収率 95%)を得た。 lH-NMR(DMSO_d6):
7.65— 7.75(3H, m), 7.80— 7.85(2H, m), 7.98(2H, s), 8.15— 8.25(3H, m), 8.69(1H, d)。
(実施例 15) 1-(1-メチル -1H-イミダゾール -2-ィル)フタラジン
1-メチルイミダゾール 4.96g(60.41 mmol)を THF50mlに溶解し、窒素下、 -70。Cで、ノ ルマルブチルリチウム (1.57M)38ml(60 mmol)を滴下し 30分撹拌した。塩化トリ- n_ブ チルスズ 20.53gの THF20ml溶液を滴下し、室温まで昇温した。これに塩化リチウム 2.58g(60.86 mmol),既知の方法 (J.Chem.,So , 1956, pl081)に従って合成した 1-クロ 口フタラジン 4.92g(29.89 mmol),テトラキス (トリフエニルホスフィン)パラジウム
(0)4.65g(4.02 mmol),キシレン 80mlを加えた後、窒素ガスを吹き込み 30分加熱撹拌し た。さらに 130°Cにて 3.5時間撹拌した。反応液に酢酸ェチルをカ卩え、シリカゲルカラ ムクロマトグラフィー後、へキサン、酢酸ェチルの混合溶媒で洗浄して、表題化合物 を合わせて 2.46g (収率 39%)得た。 1H-NMR(CDC13): 4.10(3H, s), 7· 16(1Η, s), 7·33(1Η, s), 7.90— 8.05(3Η, m), 9.05-9.15(1Η, m), 9.49(1Η, s)MS(EI), 210輔, 182,
155, 129, 103, 82, 76, 42。
実施例 1から実施例 15に記載の化合物を以下に示す。
[0182] [化 20] 実施例 実施例 実施例
[0183] (実施例 16) 1-(1, 3-チアゾール -2-ィル) -4-フヱニルフタラジン
1,3 -チアゾール 710mg(8.34 mmol)を THF6mlに溶解し、窒素下、 - 70°Cで、ノノレマノレ
ブチルリチウム (1.56M)5.5ml(8.58 mmol)を滴下し 30分撹拌した。塩化トリーノルマル ブチルスズ 2.87gを加え、室温まで昇温した。これに塩化リチウム 0.41g(9.67 mmol), 1-クロロフ- 4-フエニルタラジン 1.30g(5.40 mmol),テトラキス (トリフエニルホスフィン)パ ラジウム (0)0.3g(0.26 mmol),トルエン 13mlを加えた後、窒素下 40時間加熱還流した。 反応液に酢酸ェチルと水を加え、ろ過、分液、飽和食塩水洗浄後、無水硫酸ナトリウ ム乾燥しろ過した。シリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル = 2: 1) 後、シクロへキサン、エタノールの混合溶媒で洗浄して、表題化合物 1.128g (収率 72 %)を得た。 1H-NMR(CDC13): 7·55_7·65(4Η, m), 7·80_7·85(2Η, m), 7.85_7·95(1Η, m), 7.95-8.051H, m), 8.10— 8.20(1H, m), 9.87(1H, d)。
[0184] (実施例 17) 1_(4, 5 -ジヒドロ- 1H-イミダゾール _2 -ィル )_4 -フエニルフタラジン
4-フエニルフタラジン _1_カルボ二トリル 0.71g(3.07 mmol)にエチレンジァミン 8ml,硫 黄 55mgをカ卩えて約 10時間加熱還流した。水 100mlを加えて析出した固体をろ取しェ 一テルで洗浄して、表題化合物 192mg (収率 29%)を得た。 1H_NMR(CDC13):
3.65(2H, t), 4.29(2H, t), 6.45(1H, s), 7.55- 7·65(3Η, m), 7.70- 8.00(4H, m),
8.10(1H, d), 9.73(1H, d)。
[0185] (実施例 18) 1-(1-メチル -4, 5-ジヒドロ- 1H-イミダゾール -2-ィル) -4-フエニルフタラ ジン 塩酸塩
4-フエニルフタラジン- 1-カルボ二トリル 347mg(1.50 mmol)に N_メチルエチレンジアミ ン 0.4ml,硫黄 20mgをカ卩えて約 8時間加熱還流した。反応液を濃縮後、アセトンを加え てろ過した後、 4モル塩ィヒ水素酢酸溶液 0.4mlをカ卩えた。さらに塩ィヒメチレンを加え撹 拌し固体をろ取し、表題化合物 335mg (収率 69%)を得た。 _NMR(DMSO-d6): 3·06(3Η, s), 4.20-4.40(4H, m), 7·70_7·90(5Η, m), 8.15_8·30(3Η, m), 8.30— 8.45(1H, m), 11, 20(1H, s)。
[0186] (実施例 19) 1_(5-メチル -[1, 2, 4]-ォキサジァゾール -2-ィル) -4-フヱニルフタラジ ン
4-フエニルフタラジン - 1_カルボ二トリル 500mg(2.162 mmol)に水 5.5ml,エタノール 14ml,塩酸ヒドロキシァミン 0.54g、 炭酸カリウム 0.67gをカ卩ぇ 4時間加熱還流した。反 応液を室温まで冷却し水 40mlをカ卩えて析出した固体をろ取し、 N-ヒドロキシ- 4_フエ
ニルフタラジン- 1-カルボキシアミジン 524mg (収率 92%)を得た。 1H_NMR(CDC13): 5.93(2H, s), 7.21(1H, s), 7.55- 7·65(3Η, m), 7.7.75-7.80(2H, m), 7.85_8.00(2H, m), 8.11(1H, d), 9.31(1H, d) この化合物 57mg(0.216 mmol)にピリジン lmlを加え 0°Cに冷 却し、塩化ァセチル 0.3mlをカ卩えた。室温で 1時間撹拌した後、 1モル水酸化ナトリウム 水溶液 40mlを加えた。生成した固体をろ取しアセトンに溶解しシリカゲル上でろ過し た。ろ液にへキサンを加えて固体を析出させることにより表題化合物 30mg (収率 49%) を得た。 1H-NMR(CDC13): 2.82(3H, s), 7·55_7·65(3Η, m), 7·75_7·85(2Η, m), 7·90-8·05(2Η, m), 8.19(1Η, d), 8·91(1Η, d)。
[0187] (実施例 20) 1_(5-トリフルォロメチル -[1, 2, 4]-ォキサジァゾール _2 -ィル )_4 -フエ ニルフタラジン
実施例 19と同様の方法を用い、 N -ヒドロキシ -4-フエニルフタラジン- 1-カルボキシァ ミジン 100mg(0.378 mmol),ピリジン lml、トリフルォロ酢酸無水物 0.2πから、表題化合 物 55mg (収率 42%)を得た。 1H-NMR(CDC13): 7.60_7.65(3H, m), 7.80-7.85(2H, m), 7.90-8.15(2H, m), 8.26(1H, d), 8.83(1H, d)。
[0188] (実施例 21) l-(4, 5-ジブロモ -1-メチル -1H-イミダゾール -2-ィル) -4-フエニルフタ ラジン
1-(1-メチル -1H -イミダゾール -2-ィル) -4-フエニルフタラジン 550mg(1.921 mmol)をク ロロホルム 10mlに溶解し、 -20°Cにて臭素 0.22ml(4.4 mmol)のクロロホノレム溶液 (3ml) を滴下した。 0°Cまで昇温し 30分撹拌した。 1モル水酸化ナトリウム水溶液 30mlをカロえ て分液し、飽和食塩水で洗浄、無水硫酸ナトリウム乾燥して、ろ過した。シリカゲル力 ラムクロマトグラフィーにて精製し、エーテルで洗浄し表題化合物 308mg (収率 36%)を 得た。 1H-NMR(CDC13): 4· 11(3Η, s), 7.55- 7.65(3H, m), 7·75_7.85(2H, m),
7-85-8.05(2H, m), 8.16(1H, d), 9.03(1H, d)。
[0189] (実施例 22) 1_(5-ブロモ _1_メチル _1H -イミダゾール -2-ィル) -4-フエニルフタラジ ン
1-(1-メチル -1H-イミダゾール - 2_ィル) _4_フエニルフタラジン 2.54g(8.87 mmol)をクロ 口ホルム 40mlに溶解し、 N-ブロモコハク酸イミド (NBS)をカ卩え、 1時間加熱還流した。減 圧濃縮後、シリカゲルカラムクロマトグラフィーにて精製し、へキサン-酢酸ェチルで
洗浄し表題化合物 2.02g (収率 62%)を得た。 1H-NMR(CDC13): 4.08(3H, s), 7.36(1H, s), 7.55-7.65(3H, m), 7.75— 8·00(4Η, m), 8.14(1H, d), 9.04(1H, d)。
[0190] (実施例 23) 3-メチル -2-(4-フエニルフタラジン- 1-ィル) -3H-イミダゾール -4-カル ボニトリル
実施例 22で得られた化合物 212mg(0.580 mmol)を NMP3mlに溶解し、塩化第一銅 64mg(0.715 mmol)を加えて、 200°C (オイルバス温度)で約 2時間加熱した。室温まで 冷却し、水 40ml、エチレンジァミン四酢酸 2水和物 0.42g、炭酸カリウム、クロ口ホルム を加え、セライトろ過した。分液、飽和食塩水洗浄、無水硫酸ナトリウム乾燥し、ろ過、 濃縮した。シリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル =2: 1)にて精製 し、へキサン-酢酸ェチルで洗浄し表題化合物 55.7mg (収率 31%)を得た。
1H-NMR(CDC13): 4.23(3H, s), 7.55_7·65(3Η, m), 7.80_7·85(2Η, m), 7.85— 8.00(3Η, m), 8.20(1Η, d), 8.98(1Η, d)MS(EI): 311輔, 310, 283, 267, 234, 208, 205, 129, 77。
[0191] (実施例 24) 1-[5-(4, 4_ジメチル _4, 5_ジヒドロォキサゾール _2_ィル) _1_メチル -1H-イミダゾール -2-ィル] -4-フエニルフタラジン
4, 4-ジメチル -2-ォキサゾリン 505mg(5.09 mmol)をジェチルエーテル 10mlに溶解し、 窒素下、 -70°Cにてノルマルブチルリチウム (1.56M)3.6ml(5.62 mmol)を滴下した。さら に 30分撹拌した後、塩化トリーノルマルブチルスズ 1.85g(5.68 mmol)とジェチルエー テル 3mlの溶液をカ卩えた。室温まで徐々に昇温し、テトラキス (トリフエニルホスフィン) パラジウム (0)0.30g(0.26 mmol),実施例 25で得られた化合物 0.88g(2.41 mmol),トル ェン 12mlを加えた。窒素ガスを 20分吹き込んだ後、 14時間加熱還流した。シリカゲル カラムクロマトグラフィー (酢酸ェチル)にて精製し、へキサン-酢酸ェチルで洗浄し表 題化合物 689mg (収率 75%)を得た。 1H_NMR(CDC13): 1.41(6H, s), 4.07(2H, s), 4.34(3H.s), 7.55-7.65(3H, m), 7.75_8.00(5H, m), 8.14(1H, d), 8.85(1H, d)。
[0192] (実施例 25) 3_メチル _2_(4-フエニルフタラジン- 1 -ィル )_3H -イミダゾール -4-カル ボン酸
実施例 24で得られた化合物 689mg(1.796 mmol)に、 1, 4 -ジォキサン 4.5ml、水 3ml、濃 塩酸 3mlを加えて 9時間加熱還流した。反応液を減圧濃縮し、トルエンをカ卩えて減圧
濃縮する操作を 2回繰り返した。得られた油状物質にエタノールをカ卩ぇ減圧濃縮した 。アセトンを加えて析出した固体をろ取した。これに水、エタノールを加えて加熱還流 し溶解させ、室温に冷却した。析出した固体をろ取することにより表題化合物 370mg( 収率 62%)を得た。 1H-NMR(CDC13): 4·07(3Η, s), 7.60- 7.70(3H, m),
7.75-7.85(2H.m), 7.94(1H, s), 8.00-8.10(3H, m), 8.50— 8.60(1H, m), 13.2(1H, broads)MS(FritFAB): 331輔, 287, 232, 207, 149, 77。
(実施例 26) 8_(1-メチル -1H-イミダゾール _2 -ィル )_5 -フエニルピリド [2, 3_d]ピリダ ジン
a) 5 -フエニル- 7H -ピリド [2, 3- d]ピリダジン- 8-オン
既知の方法 (Monatsh.Chem.1990, 121(11)、 909)に従って合成した 3_ベンゾィル-ピリ ジン- 2-カルボン酸 2.26gに、ヒドラジン 1水和物 1.6ml,エタノール 20mlを加えて加熱 還流した。室温まで冷却し析出した固体をろ取することにより、表題化合物を 1.48g( 収率 72%)で得た。
b) 8-クロ口- 5-フエエル-ピリド [2, 3-d]ピリダジン
実施例 26aにて得られた化合物 1.48gにォキシ塩化リン約 6mlを加えて約 30分加熱還 流させた。反応液を氷、水酸化ナトリウム水溶液の混合物へ注ぎ、塩化メチレンで抽 出した。飽和食塩水洗浄、無水硫酸ナトリウム乾燥、ろ過、濃縮の後、シリカゲルカラ ムクロマトグラフィー、へキサンで洗浄して精製し表題化合物 1.05g (収率 65%)を得た c) 8-(1-メチル -1H-イミダゾール -2-ィル) -5-フエニルピリド [2, 3-d]ピリダジン
1-メチルイミダゾール 183mg(2.23 mmol)を THF2.5mlに溶解し、窒素下、 _70°Cで、ノ ルマルブチルリチウム (1.8M)1.5ml(2.81 mmol)を滴下し 15分撹拌した。塩化トリ _n -ブ チルスズ 915mg(2.81 mmol)の THF 2.5ml溶液を加え、室温まで昇温した。これに塩化 リチウム 112mg(2.64 mmol),実施例 26bにて得られた化合物 254mg(1.138 mmol),テト ラキス (トリフエニルホスフィン)パラジウム (0)200mg(0.173 mmol),トルエンを加えた後、 窒素下 90°Cにて 6.5時間撹拌した。反応液にクロ口ホルムをカ卩え、シリカゲルカラムク 口マトグラフィー (クロ口ホルム:酢酸ェチル:トリエチノレアミン)、へキサン-酢酸ェチノレ で洗浄した。エタノール-へキサンに加熱溶解、冷却後、析出した固体をろ取して、表
題化合物 130mg (収率 40%)を得た。 1H-NMR(CDC13): 4.06(3H, s), 7.20(1H, s), 7.43(1H, s), 7.60-7.70(3H, m), 7.75- 7.85(3H, m), 8.49(1H, dd), 9.44(1H, dd)。
[0194] (実施例 27) 1-(1-メチル -1H-イミダゾール -2-ィル) -4-フエニルフタラジン
N-メチルイミダゾール 126mg(1.53 mmol)を THF(lOml)に溶解させ、 _78°Cに冷却した 。窒素雰囲気下にて n_ブチルリチウム-へキサン溶液 (1.56M)1.0mlを滴下し、 _78°C にて 1時間攪拌した。反応溶液に既知の方法 (Heterocycles 1994, 39(1)、 345)にて 合成した 1-フヱニル -4-(p-トルイルスルホニル)フタラジン 0.5g(1.39 mmol)を加え、同 温度で 1時間攪拌した後、室温に昇温しさらに 2.5時間攪拌した。反応溶液に水を加 え、酢酸ェチルにて抽出し、無水硫酸ナトリウムにて乾燥後ろ過、減圧濃縮した。得 られた残さをシリカゲルカラムクロマトグラフィー (ジクロロメタン-酢酸ェチル)にて精製 し、表記化合物 205mg (収率 52%)を得た。 1H_NMR(CDC13): 4.14(3H, s), 7.18(1H, d), 7.26(1H, d), 7.58-7.6K3H, m), 7.80_7.87(3H, m), 7.96(1H, t), 8.14(1H, d), 9.20(1H, d)。
[0195] (実施例 28) 1-(1-メチル -1H-イミダゾール -2-ィル) -4-(p-トルィル)フタラジン
a) 4-(p-トルィル)フタラジン- 1-オン
p-トルィル -0-安息香酸 5.0g(20.8 mmol),ヒドラジン 1水和物 1.6ml(31.2 mmol),ェタノ ール (15ml)の混合物を 6時間加熱還流した。室温に冷却した後、溶媒を減圧濃縮し て得られた固体を水-エタノールで洗浄し、表記化合物 4.86g (収率 99%)を得た。 1H-NMR(CDC13): 2.46(3H, s), 7·33(2Η, d), 7.48(2Η, d), 7.78- 7.83(3Η, m), 8.51-8.55(1Η, m), 10·37(1Η, brs)。
b) 1-クロ口- 4-(p-トルィル)フタラジン
実施例 28aで得られた化合物 2.85g(12.1 mmol)の 1, 2-ジクロロェタン (12ml)溶液に、 ォキシ塩化リン 1.4ml(14.5 mmol)をカ卩ぇ 3時間加熱還流した。室温に冷却した後反応 溶液を氷水中に注ぎ、ジクロロメタンにて抽出した。有機層を無水硫酸ナトリウムにて 乾燥後ろ過、減圧濃縮し、得られた固体をシリカゲルカラムクロマトグラフィー (ジクロ ロメタン-酢酸ェチル)にて精製後、へキサンで洗浄し、表記化合物 2.77g (収率 90%) を得た。 1H-NMR(CDC13): 2.48(3H, s), 7·38(2Η, d), 7.63(2Η, d), 7·88_7·94(1Η, m), 7.97— 8.02(1Η, m), 8· 10(1Η, d), 8.38(1Η, d)。
c) l_(p-トノレィル) _4-(p-トノレイルスルホニノレ)フエニルフタラジン
実施例 28bで得られた化合物 1.76g(6.89 mmol)の DMF(20ml)溶液に、 p-トルエンスル フィン酸ナトリウム 2.46g(13.8 mmol)を加え 90°Cにて 3時間加熱した。室温に冷却した 後反応溶液に水を加え、析出した固体をろ取し水で洗浄した。得られた固体をシリカ ゲルカラムクロマトグラフィー (ジクロロメタン-酢酸ェチル)にて精製後、表記化合物 2.26g (収率 88%)を得た。 1H-NMR(CDC13): 2.48(3H, s), 7.42(2H, d), 7.55_7.57(3H, m), 7.69-7.72(2H, m), 7.97_8.09(4H, m), 8.18(1H, d), 9.21(1H, d)。
d) l_(l-メチル _1H-イミダゾール _2 -ィル )_4_(p -トルィル)フタラジン
実施例 28cで得られた化合物 1.0g(2.67 mmol)を用いて実施例 27と同様の操作を行う ことにより、表記化合物 404mg (収率 50%)を得た。 1H-NMR(CDC13): 7.16(1H, s), 7.34(1H, s), 7.40(2H, d), 7.71(2H, d), 7·83_7·96(2Η, m), 8.15(1H, d), 9.19(1H, d)。
(実施例 29) 1_(4-メトキシフヱニル) _4_(1_メチル _1H -イミダゾール -2-ィル)フタラジ ン 塩酸塩
a) 4-メトキシフエニル -0-安息香酸
無水コハク酸 3.0g(20.3 mmol)を THF(50ml)に溶解させ、 _78°Cに冷却した。窒素雰囲 気下にて 4-メトキシフエニルマグネシウムブロミド THF溶液 (0.5M)44.6mlを滴下し、 室温に昇温し 2.5時間攪拌した。反応溶液に水を加え、酢酸ェチルにて抽出し、無水 硫酸ナトリウムにて乾燥後ろ過した。溶媒を減圧濃縮して得られた固体をヘプタン- 酢酸ェチル-エタノールで洗浄し、表記化合物 3.80g (収率 73%)を得た。
lH-NMR(DMSO-d6): 3.18(3H, s), 7.01(2H, d), 7.36(1H, d), 7.56— 7·72(4Η, m), 7.96(1H, d)。
b) 4_(4-メトキシフエ二ル)- 2H -フタラジン _1_オン
実施例 29aで得られた化合物 3.81g(14.9 mmol)を用いて実施例 28aと同様の操作を行 うことにより、表記化合物 3.87g (収率 100%)を得た。 1H-NMR(CDC13): 3.90(3H, s), 7·05(2Η, d), 7.53(2Η, d), 7·79_7·82(3Η, m), 8.50_8·54(1Η, m), 10.13(1Η, brs)0 c) 1_クロ口- 4-(4 -メトキシフエニル)フタラジン
実施例 29bで得られた化合物 3.87g(15.3 mmol)を用いて実施例 28bと同様の操作を行 うことにより、表記化合物 4.08g (収率 98%)を得た。 1H-NMR(CDC13): 3.92(3H, s),
7.10(2H, d), 7.70(2H, d), 7.92— 8.00(2H, m), 8.12(1H, d), 8.38(1H, d)。
d) l-(4-メトキシフエニル) -4-(p-トノレイルスルホニノレ)フエニルフタラジン
実施例 29cで得られた化合物 1.0g(3.69 mmol)を用いて実施例 31cと同様の操作を行 うことにより、表記化合物 1.05g (収率 79%)を得た。 1H-NMR(CDC13): 2.47(3H, s), 3·90(3Η, s), 7.07(2Η, d), 7.41(2Η, d), 7·69(2Η, d), 7.96— 8.07(4Η, m), 8.23(1Η, d), 9· 19(1Η, d)。
e) l-(4-メトキシフエニル) -4-(1-メチル -1Η-イミダゾール- 2_ィル)フタラジン 実施例 29dで得られた化合物 1.05g(2.69 mmol)を用いて実施例 27と同様の操作を行 うことにより、表記化合物 352mg (収率 41%)を得た。 1H_NMR(CDC13): 3.93(3H, s), 4· 13(3Η, s), 7.11-7.16(3Η, m), 7.34(1Η, d), 7.78— 7.80(2Η, m), 7.86— 7.94(2Η, m), 8· 17(1Η, d), 9.18(1Η, d)MS(EI): 316輔, 300, 288, 272, 244, 209, 183, 156, 129。 f) 1-(4 -メトキシフヱニル) -4-(1-メチル -1H-イミダゾール -2-ィル)フタラジン 塩酸塩 実施例 29eで得られた化合物 100mg(0.32 mmol)のメタノール溶液に、 4モル塩酸-酢 酸ェチル溶液 0.09mlをカ卩え、室温にて 4時間攪拌した。析出した固体をろ取し、表記 化合物 112mg (収率 100%)を得た。 lH-NMR(DMSO-d6): 3·91(3Η, s), 3·96(3Η, s), 7.26(2Η, d), 7.84(2Η, d), 7.99(1Η, s), 8.10(1Η, s), 8.18— 8·29(4Η, m)。
(実施例 30) 4-(3-メトキシフエ二ル) -1-(1-メチル -1H-イミダゾール -2-ィル)フタラジ ン塩酸塩
a) 3-メトキシフエニル -0-安息香酸
無水コハク酸 3.0g(20.3 mmol)及び 3-メトキシフエ二ルマグネシウムブロミド THF溶液 (1.0M)22.3ml(22.3 mmol)を用いて実施例 29aと同様の操作を行うことにより、表記化 合物 4.95g (収率 96%)を得た。 1H_NMR(CDC13): 3.84(3H, s), 7.10- 7.16(2H, m), 7.28(1H, t), 7.37-7.39(2H, m), 7.56_7.59(1H, m), 7.63_7.66(1H, m), 8.08(1H, d)。 b) 4_(3-メトキシフエ二ル)- 2H -フタラジン _1_オン
実施例 30aで得られた化合物 4.95g(19.3 mmol)を用いて実施例 28aと同様の操作を行 うことにより、表記化合物 4.64g (収率 95%)を得た。 1H-NMR(CDC13): 3.87(3H, s), 7·05-7·08(1Η, m), 7.08-7.17(2Η, m), 7.44(1Η, t), 7.78_7·81(3Η, m), 8.51— 8.54(1Η, m), 10·37(1Η, brs)。
c) 1-クロ口- 4-(3-メトキシフエ二ル)フタラジン
実施例 30bで得られた化合物 4.64g(18.4 mmol)を用いて実施例 31bと同様の操作を行 うことにより、表記化合物 4.71g (収率 95%)を得た。 1H-NMR(CDC13): 3·89(3Η, s), 7.11-7.14(1H, m), 7.27-7.29(2H, m), 7.46-7.51(1H, m), 7.93— 7.96(1H, m),
7.99-8.04(lH, m), 8.12(1H, d), 8.40(1H, d)。
d) l- (3-メトキシフエ二ル)- 4- (p-トノレイルスルホニノレ)フエニルフタラジン
実施例 30cで得られた化合物 1.0g(3.69 mmol)を用いて実施例 28cと同様の操作を行 うことにより、表記化合物 725mg (収率 54%)を得た。 1H_NMR(CDC13): 2.48(3H, s), 3·86(3Η, s), 7.10— 7.12(1Η, m), 7·23_7·25(2Η, m), 7.41-7.48(3Η, m), 7.97— 7.99(1Η, m), 8.03-8.1 K3H, m), 8.21(1H, d), 9.21(1H, d)。
e) l-(3_メトキシフエニル) -4-(l-メチル -1H-イミダゾール- 2_ィル)フタラジン 実施例 30dで得られた化合物 725mg(1.86 mmol)を用いて実施例 27と同様の操作を行 うことにより、表記化合物 382mg (収率 65%)を得た。 1H-NMR(CDC13): 3·91(3Η, s), 4.14(3H, s), 7.13— 7·14(1Η, m), 7.18(1H, s), 7.35— 7·38(3Η, m), 7.47- 7.50(1H, m), 7.87-7.90(lH, m), 7.93—7.96(1H, m), 8.16(1H, d), 9, 20(1H, d)。
f) l-(3-メトキシフエニル) -4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン 塩酸塩 実施例 30eで得られた化合物 382mg(1.21 mmol)を用いて実施例 29fと同様の操作を 行うことにより、表記化合物 417mg (収率 64%)を得た。 _NMR(DMSO-d6): 3.87(3H, s), 3·96(3Η, d), 7.22— 7.26(1Η, m), 7.34- 7.37(2Η, m), 7.59(1Η, t), 7.85(1Η, s), 7.98(1Η, s), 8.14-8·18(3Η, m), 8.33_8.36(1Η, m)。
実施例 16から実施例 30に記載の化合物を以下に示す。
[化 21]
(実施例 31) 1-(4-クロ口フエニル) -4-(1-メチル -1H-イミダノール- 2-ィル)フタラジン a) 4-(4-クロ口フエニル) -2H-フタラジン- 1-オン
無水コハク酸 3.0g(20.3 mmol)及び 4-クロ口フエニルマグネシウムブロミド ジェチルェ 一テル溶液 (1.0M)22.3ml(22.3 mmol)を用いて実施例 29aと同様の操作を行うことによ
り、 4-クロ口フエニル -o-安息香酸を得た。 4-クロ口フエニル -0-安息香酸を用いて実 施例 102aと同様の操作を行うことにより、表記化合物 3.28g (収率 63%)を得た。
1H-NMR(CDC13): 7·52_7.56(4Η, m), 7.70- 7·73(1Η, m), 7.80_7·83(2Η, m), 8.52-8.55(1Η, m), 10.19(1Η, brs)。
b) 1_クロ口 _4_(4 -クロロフヱニル)フタラジン
実施例 31aで得られた化合物 3.28g(12.8 mmol)を用いて実施例 28bと同様の操作を行 うことにより、表記化合物 3.30g (収率 94%)を得た。 1H-NMR(CDC13): 7.55_7.58(2H, m), 7.67-7.70(2H, m), 7.94— 7.97(1H, m), 8.00— 8.06(2H, m), 8.39— 8.42(1H, m)0 c) l- (4-クロロフヱ二ル)- 4- (p-トノレイルスルホニノレ)フタラジン
実施例 31bで得られた化合物 1.0g(3.64 mmol)を用いて実施例 28cと同様の操作を行 うことにより、表記化合物 928mg (収率 65%)を得た。 1H-NMR(CDC13): 2.49(3H, s), 7.42-7.44(2H, m), 7.55(2H, d), 7.67(2H, d), 8.00-8.16(5H, m), 9.22(1H, d)。
d) l-(4-クロ口フエニル) -4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン 実施例 31cで得られた化合物 927mg(2.35 mmol)を用いて実施例 27と同様の操作を行 うことにより、表記化合物 369mg (収率 49%)を得た。 1H_NMR(CDC13): 4·14(3Η, s), 7.17(1H, d), 7.35(1H, d), 7.57— 7.59(2H, m), 7.75— 7.78(2H, m), 7·88— 7·91(1Η, m), 7.96- 7.97(1H, m), 8.08(1H, d), 9.24(1H, d)。
(実施例 32) l-(4-フルオロフェニル )-4-(l-メチル -1H-イミダゾール -2-ィル)フタラ ジン
a) 4-(4-フルオロフェニル )-2H-フタラジン- 1-オン
無水コハク酸 3.0g(20.3 mmol)及び 4-フルオロフェニルマグネシウムブロミド THF溶 液 (1.0M)22.3ml(22.3 mmol)を用いて実施例 29aと同様の操作を行うことにより、 4-フル オロフェニル -0-安息香酸を得た。 4_フルオロフヱニル _o_安息香酸を用いて実施例 28aと同様の操作を行うことにより、表記化合物 3.37g (収率 69%)を得た。
1H-NMR(CDC13): 7.23-7.24(1Η, m), 7.59— 7.61(2H, m), 7.74(1H, m),
7.81-7.84(3H, m), 8.58(1H, m), 10.35(1H, brs)。
b) 1_クロ口- 4_(4_フルオロフヱニル)フタラジン
実施例 32aで得られた化合物 3.37g(14.0 mmol)を用いて実施例 28bと同様の操作を行
うことにより、表記化合物 2.78g (収率 77%)を得た。 lH-NMR(DMSO-d6): 7.42(2H, t), 7.78— 7.83(2H, m), 8·03(1Η, d), 8.14— 8·22(2Η, m), 8.41(1H, d)。
c) l-(4-フルオロフヱニル) -4-(p-トノレイルスルホニノレ)フエニルフタラジン
実施例 32bで得られた化合物 1.0g(3.87 mmol)を用いて実施例 28cと同様の操作を行 うことにより、表記化合物 909mg (収率 62%)を得た。 1H-NMR(CDC13): 2.49(3H, s), 7.23-7.29(2H, m), 7.43(2H, d), 7.70— 7.74(2H, m), 8.00-8.18(5H, m), 9.22(1H, d)。 d) l_(4-フルオロフェニル )_4_(1_メチル _1H -イミダゾール -2-ィル)フタラジン 実施例 32cで得られた化合物 900mg(2.38 mmol)を用いて実施例 27と同様の操作を行 うことにより、表記化合物 501mg (収率 69%)を得た。 1H-NMR(CDC13): 4.14(3H, s), 7· 18(1Η, s), 7.29— 7.35(3Η, m), 7·80_7·84(2Η, m), 7.89_7·92(1Η, m), 7.95— 7.97(1Η, m), 8· 10(1Η, d), 9.22(1Η, d)。
[0201] (実施例 33) 1_(2-メチル -2H-ピラゾール -3-ィル) -4-フエニルフタラジン
N-メチルビラゾール 342mg(4.17 mmol)を用いて実施例 27と同様の操作を行うことによ り、表記化合物 254mg (収率 32%)を得た。 1H_NMR(CDC13): 4.13(3H, s), 6.72(1H, d), 7.59-7.62(3H, m), 7.71(1H, d), 7.81_7.84(2H, m), 7.90—7.94(2H, m),
8.18- 8.26(2H, m)。
[0202] (実施例 34) l-(2-フリル) -4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン
a) l-(2-フリル) -4-(p-トノレイルスルホニル)フエニルフタラジン
既知の方法 (特開平 6-135938)に準じて合成した 1-クロ口- 4-(2-フリル)フタラジン 816mg(3.54 mmol)を用いて実施例 28cと同様の操作を行うことにより、表記化合物 1.06g (収率 86%)を得た。 1H-NMR(CDC13): 2.48(3H, s), 6.67(1H, dd), 7.41(2H, d), 7.55(1H, d), 7.79(1H, d), 8·00_8·08(4Η, m), 9.01-9.02(1H, m), 9.18— 9.19(1H, m)。 b) l_(2-フリル)- (1-メチル _1H-イミダゾール- 2_ィル)フタラジン
実施例 34aで得られた化合物 1.06g(3.03 mmol)を用いて実施例 27と同様の操作を行 うことにより、表記化合物 420mg (収率 50%)を得た。 1H-NMR(CDC13): 4.11(3H, s), 6·71(1Η, dd), 7.16(1Η, d), 7·33(1Η, d), 7.51(1Η, d), 7·79_7·80(1Η, m),
7·94-7·98(2Η, m), 8.86_8·90(1Η, m), 9.15— 9.19(1Η, m)MS(EI): 276, 248, 220, 193, 183, 167, 156, 149, 129。
[0203] (実施例 35) 7-(l-メチル -1H-イミダゾール -2-ィル) -4-フエニルチエノ [2, 3_d]ピリ ダジン
a) 4-フエニル -6H-チエノ [2, 3-d]ピリダジン- 7-オン
2- (2-チェニル) -4, 4 -ジメチルォキサゾリン 6.5g(35.9 mmol)を用いて実施例 34aと同 様の操作を行うことにより、表記化合物 4.91g (収率 59%)を得た。
lH-NMR(DMSO-d6): 7·51_7·55(4Η, m), 7·69_7·72(2Η, m), 8.27(1Η, d), 13· 13(1Η, brs)。
b) 7_クロ口- 4_フエニルチエノ [2, 3_d]ピリダジン
実施例 35aで得られた化合物 4.91g(21.3 mmol)を用いて実施例 28bと同様の操作を行 うことにより、表記化合物 5.17g (収率 97%)を得た。 1H-NMR(CDC13): 7.57-7.62(3H, m), 7.73(1H, d), 7.89— 7.92(2H, m), 7.96(1H, d)。
c) 7_(1_メチル _1H -イミダゾール -2-ィル) -4-フエニルチエノ [2, 3_d]ピリダジン 実施例 35bで得られた化合物 500mg(2.03 mmol)を用いて実施例 15と同様の操作を行 うことにより、表記化合物 527mg (収率 89%)を得た。 1H_NMR(CDC13): 4·39(3Η, s), 7.14(1H, s), 7.37(1H, s), 7.55- 7·62(3Η, m), 7.71(1H, d), 7·95— 7·99(3Η, m)。
[0204] (実施例 36) 4-(l-メチル -1H-イミダゾール -2-ィル) -7-フエ二ルフロ [2, 3_d]ピリダ ジン
a) 2- (ヒドロキシフエニルメチル) -3-フランカルボン酸メチルエステル
3-フランカルボン酸 5.0g(44.6 mmol)を THF(lOOml)に溶解させ、 _78°Cに冷却した。窒 素雰囲気下にて別に調製した LDA溶液 (98.1 mmol)を滴下し、- 78°Cにて 30分間攪拌 した。反応溶液にベンズアルデヒド 5.22g(49.1 mmol)の THF(lOml)溶液を加え、同温 度で 1時間攪拌した後、室温に昇温した。反応溶液に希塩酸水溶液を加え、酢酸ェ チルにて抽出し、無水硫酸ナトリウムにて乾燥後ろ過、減圧濃縮し、 2- (ヒドロキシフヱ ニルメチル) _3_フランカルボン酸を得た。 2 -(ヒドロキシフエニルメチル) _3_フランカル ボン酸 1.5g(6.87 mmol)の DMF(13ml)溶液に炭酸カリウム 1.14g(8.25 mmol),ヨウィ匕メ チノレ 1.46g(10.3 mmol)を加え、室温にて 7時間攪拌した。反応溶液に水を加え、酢酸 ェチルにて抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムにて乾燥後 ろ過、減圧濃縮した。残さをシリカゲルカラムクロマトグラフィー (へキサン-酢酸ェチル
)にて精製し、表記化合物 1.33g (収率 84%)を得た。 1H-NMR(CDC13): 3·86(3Η, s), 5.20(1H, d), 6·13(1Η, d), 6.68(1H, d), 7.29-7.43(6H, m)。
b) 7-フエニル -5H-フロ [2, 3-d]ピリダジン- 4-オン
実施例 36aで得られた化合物 1.51g(6.50 mmol)のジクロロメタン (12ml)溶液を
Dess-Martinperiodinane 3.0g(7.15 mmol)のジクロロメタン溶液 (20ml)に加え、室温に て 1時間攪拌した。反応液をチォ硫酸ナトリウム水溶液中に注ぎ、ジクロロメタンにて 抽出後、有機層を飽和重曹水、飽和食塩水で洗浄し、無水硫酸ナトリウムにて乾燥 後ろ過、減圧濃縮した。残さをシリカゲルカラムクロマトグラフィー (へキサン-酢酸ェチ ル)にて精製し、 2_ベンゾィル _3_フランカルボン酸 メチルエステルを得た。 2_ベンゾ ィル- 3_フランカルボン酸 メチルエステル 1.08g(4.69 mmol)を用いて実施例 28aと同 様の操作を行うことにより、表記化合物 787mg (収率 57%)を得た。 1H-NMR(CDC13): 7· 18(1Η, d), 7.49-7.57(3H, m), 7·82(1Η, d), 8.10_8· 13(2Η, m), 10.57(1H, brs)0 c) 4-クロ口- 7-フエ二ルフロ [2, 3-d]ピリダジン
実施例 36bで得られた化合物 787mg(3.71 mmol)を用いて実施例 28bと同様の操作を 行うことにより、表記化合物 704mg (収率 82%)を得た。 1H_NMR(CDC13): 7.04(1H, d), 7.58— 7·60(3Η, m), 7.98(1Η, d), 8.42-8.44(2Η, m)。
d) 4-(l-メチル -1H-イミダゾール -2-ィル) -7-フエエルフ口 [2, 3_d]ピリダジン 実施例 36cで得られた化合物 300mg(1.30 mmol)を用いて実施例 15と同様の操作を行 うことにより、表記化合物 292mg (収率 81%)を得た。 1H_NMR(CDC13): 4·34(3Η, s), 7.12(1H, d), 7.31(1H, d), 7.55_7.63(3H, m), 7.84(1H, d), 7.96(1H, d),
8.51-8.54(2H, m)。
(実施例 37) 4_(4-メトキシフヱニル) _7_(1_メチル _1H -イミダゾール -2-ィル)チエノ [2, 3-d]ピリダジン塩酸塩
a) 7_(1_メチル _1H -イミダゾール -2-ィル) -5H -チェノ [2, 3_d]ピリダジン _4_オン 3-チオフヱンカルボン酸 3.0g(23.4 mmol)を THF(40ml)に溶解させ、 0°Cに冷却した。 窒素雰囲気下にて LDA ヘプタン- THF—ェチルベンゼン溶液 (2.0M)25.8mlを滴下し 、 0°Cにて 30分間攪拌した。反応溶液に N -メチルイミダゾール -2-カルボキシアルデヒ ド 2.89g(25.8 mmol)の THF(lOml)溶液をカ卩え、室温に昇温し 15時間攪拌した。反応溶
液に氷水を加え、酢酸ェチルにて洗浄した。水層に過マンガン酸カリウム 7.40g(46.8 mmol)を加え、室温にて 4時間攪拌した。反応液をセライトろ過後、ろ液を濃塩酸で中 和し、減圧濃縮して 2-(1-メチル -1H-イミダゾール -2-カルボニル) -3-チォフェンカル ボン酸を得た。 2_(1_メチル -1H -イミダゾール -2-カルボ二ル)- 3 -チォフェンカルボン 酸を用いて実施例 28aと同様の操作を行うことにより、表記化合物 3.50g (収率 64%)を 得た。 1H-NMR(CDC13): 4·06(3Η, s), 7.04(1H, d), 7.24(1H, d), 7.81(2H, s),
10.72(1H, brs)。
b) 4_クロ口- 7_(1_メチル _1H -イミダゾール -2-ィル)チエノ [2, 3_d]ピリダジン 実施例 37aで得られた化合物 310mg(1.34 mmol)にォキシ塩化リン lmlを加え、 110°C にて 2時間攪拌した。反応溶液を氷水中に注ぎ、水酸化ナトリウム水溶液にて中和後 、ジクロロメタンにて抽出した。有機層を無水硫酸ナトリウムにて乾燥後ろ過、減圧濃 縮し、得られた固体をシリカゲルカラムクロマトグラフィー (ジクロロメタン-酢酸ェチル) にて精製後、表記化合物 275mg (収率 82%)を得た。 1H-NMR(CDC13): 4.31(3H, s), 7.13(1H, d), 7.34(1H, d), 7.63(1H, d), 8.01(1H, d)。
c) 4-(4-メトキシフエ二ル) -7-(l-メチル -1H-イミダゾール -2-ィル)チエノ [2, 3_d]ピリダ ジン塩酸塩
実施例 37bで得られた化合物 275mg(1.10 mmol)及び 4_メトキシフエニルボロン酸を用 いて実施例 leと同様の操作を行うことにより、 4-(4_メトキシフエ二ル) -7-(1_メチル -1H-イミダゾール -2-ィル)チエノ [2, 3-d]ピリダジンを得た。 4-(4-メトキシフヱニル )-7-(1-メチル -1H-イミダゾール -2-ィル)チエノ [2, 3-d]ピリダジンを用いて実施例 30f と同様の操作を行うことにより、表記化合物 348mg (収率 88%)を得た。
lH-NMR(DMSO-d6): 3·09(3Η, s), 4.25(3H, s), 7.24(2H, d), 7.39(1H, s), 7.65(1H, s), 7.85(2H, d), 7.96(2H, d), 8.52(2H, d)。
(実施例 38) 7_(4-メトキシフヱニル) _4_(1_メチル _1H -イミダゾール -2-ィル)チエノ
[2, 3-d]ピリダジン塩酸塩
a) 4- (1-メチル - 1H -イミダゾール -2-ィル) -5H -チェノ [2, 3- d]ピリダジン- 7-オン
2-チォフェンカルボン酸 0.5g(3.90 mmol)を用いて実施例 37aと同様の操作を行うこと により、表記化合物 168mg (収率 19%)を得た。 1H-NMR(CDC13): 3·98(3Η, s),
7.04(1H, s), 7.21(1H, s), 7.87(1H, d), 8.42(1H, d), 10.11(1H, brs)。
b) 7-クロ口- 4
-(1-メチル -1H-イミダゾール -2-ィル)チエノ [2, 3-d]ピリダジン
実施例 38aで得られた化合物 125mg(0.54 mmol)を用いて実施例 37bと同様の操作を 行うことにより、表記化合物 75mg (収率 56%)を得た。 1H-NMR(CDC13): 4.22(3H, s), 7· 13(1Η, s), 7·30(1Η, s), 7.93(1Η, d), 8.65(1Η, d)。
c) 7_(4 -メトキシフヱニル) _4_(1_メチル _1Η -イミダゾール -2-ィル)チエノ [2, 3_d]ピリダ ジン塩酸塩
実施例 38bで得られた化合物 74mg(0.30 mmol)を用いて実施例 37cと同様の操作を行 うことにより、表記化合物 58mg (収率 54%)を得た。 lH-NMR(DMSO-d6): 3.91(3H, s), 4· 10(3Η, s), 7·26(2Η, d), 7.64(1Η, s), 7.81(1Η, s), 8· 15_8·21(3Η, m), 8.52_8·54(1Η, m)0
[0207] (実施例 39) 1-(1-メチル -1H-イミダゾール -2-ィル) -4-(3-チェニル)フタラジン
実施例 Idで得られた化合物 540mg(2.21 mmol)及び 3_チォフェンボロン酸 566mg(4.42 mmol)を用レ、て実施例 1 eと同様の操作を行うことにより、表記化合物 500mg (収率 77 %)を得た。 1H_NMR(CDC13): 4.13(3H, s), 7.16(1H, d), 7.34(1H, d), 7.55-7.57(lH, m), 7.66-7.68(1H, m), 7.90-7.96(3H, m), 8.30(1H, m), 9.22(1H, m)。
[0208] (実施例 40) 1-(1-メチル -1H-イミダゾール -2-ィル) -4-(2-チェニル)フタラジン
実施例 Idで得られた化合物 540mg(2.21 mmol)及び 2_チォフェンボロン酸 566mg(4.42 mmol)を用レ、て実施例 1 eと同様の操作を行うことにより、表記化合物 390mg (収率 60 %)を得た。 1H_NMR(CDC 3): 4.13(3H, s), 7.17(1H, s), 7·28- 7·31(1Η, m), 7.35(1H, s), 7.64(1H, d), 7.76(1H, d), 7.94—7.98(2H, m), 8·53_8·56(1Η, m), 9.20-9.23(lH, m)。
[0209] (実施例 41) 1_(1-メチル -1H-イミダゾール- 2_ィル) _4_(4_モルホリノ)フタラジン塩 酸塩
実施例 Idで得られた化合物 300mg(1.23 mmol)及びモルホリン 32 lmg(3.68 mmol)の N-メチルピロリドン (0.45ml)溶液を 140°Cにて 8時間攪拌した。室温に冷却後反応溶液 に水を加え、ジクロロメタンにて抽出し、無水硫酸ナトリウムにて乾燥後ろ過、減圧濃
縮した。残さをシリカゲルカラムクロマトグラフィー (へキサン-酢酸ェチル)にて精製し、
1-(1-メチル -1H-イミダゾール -2-ィル) -4-(4-モルホリノ)フタラジンを得た。 1-(1-メチ ル -1H-イミダゾール -2-ィル) -4-モルホリン- 4-ィル-フタラジンを用いて実施例 29f 同様の操作を行うことにより、表記化合物 340mg (収率 84%)を得た。
lH-NMR(DMSO-d6): 3.64-3.67)4H, m), 3·88(3Η, s), 3·90_3·92(4Η, m), 7·89(1Η, s), 7.99— 8.08(4Η, m), 8·28_8·31(1Η, m)0
[0210] (実施例 42) 1_[4_(1_メチル _1H -イミダゾール -2-ィル)フタラジン- 1-ィル]ピベリジ ン -4-ォーノレ
実施例 Idで得られた化合物 250mg(1.02 mmol)及び 4 -ヒドロキシピペリジン
155mg(1.53 mmol), N_メチルモルホリン 155mg(1.53 mmol)を用いて実施例 41と同様 の操作を行うことにより、表記化合物 279mg (収率 88%)を得た。 1H-NMR(CDC13): 1·76(1Η, brs), 1.85— 1.94(2H, m), 2.13— 2.18(2H, m), 3.28—3.36(2H, m),
3.87-3.93(2H, m), 4·00_4.01(1Η, m), 4.04(3H, s), 7.10(1H, s), 7.28(1H, d), 7.82-7.85(2H, m), 8.04-8.07(1H, m), 8·90_8·94(1Η, m)MS(EI): 309(M+), 292, 280, 264, 252, 238, 209, 182, 100, 82, 56。
[0211] (実施例 43) 1-(1-メチル -1H-イミダゾール -2-ィル) -4-(4-メチル-ピペラジン- 1-ィ ノレ)フタラジン 塩酸塩
実施例 Idで得られた化合物 250mg(1.02 mmol)及び N_メチルビペラジン 307mg(3.06 mmol)を用いて実施例 41と同様の操作を行うことにより、表記化合物 330mg (収率 94% )を得た。 lH-NMR(DMSO-d6): 2.89(3H, s), 3.34- 3.56(6H, m), 3·90(3Η, s), 3.97-4.12(2H, m), 7.26(1H, s), 7.52(1H, s), 7.98— 8·05(2Η, m), 8.20— 8.22(1H, m), 8.74-8.77(lH, m)0
[0212] (実施例 44) 1_(1-メチル -1H-イミダゾール- 2_ィル) _4_(2_ピリジル)フタラジン
a) 1_クロ口- 4-ピリジン- 2-ィル-フタラジン
既知の方法 (Synth.Commun.1999, 29(3), 457)に従って合成した 4_(2_ピリジル) -2H- フタラジン- 1-オン 5.0g(22.4 mmol)を用いて実施例 37bと同様の操作を行うことにより、 表記化合物 4.74g (収率 88%)を得た。 1H_NMR(CDC13): 7.48-7.51(1H, m),
7·96-8·02(3Η, m), 8.22(1Η, d), 8.38— 8.41(1Η, m), 8·81_8·85(2Η, m)0
b) l-(l-メチル -1H-イミダゾール -2-ィル) -4-(2-ピリジル)フタラジン
実施例 44aで得られた化合物 300mg(1.24 mmol)を用いて実施例 38dと同様の操作を 行うことにより、表記化合物 169mg (収率 47%)を得た。 1H_NMR(CDC ): 4.12(3H, s), 7· 19(1Η, s), 7·36(1Η, s), 7.50(1H, m), 7·92_7·99(3Η, m), 8.25(1H, m),
8.74-8.75(lH, m), 8.78_8.84(1H, m), 9.17— 9.20(1H, m)。
(実施例 45) 1_(2-ピリジル) -4-(1Η-テトラゾル _5_ィル)フタラジン ナトリウム塩 a) 4- (2-ピリジル)フタラジン- 1-カルボ二トリノレ
実施例 44aで得られた化合物 600mg(2.48 mmol)の DMF(8ml)溶液に、 p -トルエンスル フィン酸ナトリウム 221mg(1.24 mmol),シアン化カリウム 243mg(3.73 mmol)を加え 90度 にて 5.5時間加熱した。室温に冷却した後反応溶液に水を加え、析出した固体をろ取 し水で洗浄した。得られた固体をシリカゲルカラムクロマトグラフィー (ジクロロメタン-酢 酸ェチル)にて精製後、表記化合物 306mg (収率 54%)を得た。 1H_NMR(CDC13): 7.52— 7.54(1H, m), 8.01-8.12(3H, m), 8.34- 8·39(2Η, m), 8.85(1H, m),
9.04-9.08(lH, m)。
b) l-(2-ピリジル) -4-(lH-テトラゾル -5-ィル)フタラジン
実施例 45aで得られた化合物 308mg(1.33 mmol)のトルエン (5ml)溶液に、トリメチルシリ ルアジド 458mg(3.98 mmol),ジ _n_ブチルスズォキシド 33mg(0.13 mmol)を加え、 60時 間加熱還流した。反応液を減圧濃縮後、固体をメタノールにて加熱洗浄し表記化合 物 267mg (収率 73%)を得た。 lH-NMR(DMSO_d6): 7.68-7.72(1Η, m), 8.14_8·29(4Η, m), 8.74(1Η, d), 8·89(1Η, d), 9.42(1Η, d)。
c) l-(2-ピリジル) -4-(1Η-テトラゾル -5-ィル)フタラジン ナトリウム塩
実施例 45bで得られた化合物 135mg(0.49 mmol)のメタノール (5ml)溶液にナトリウムメト キシド メタノール溶液 (28%)l. lmlを加え、室温にて 2時間攪拌した。反応液を減圧 濃縮した後、得られた固体をアセトン-メタノールで洗浄し表記化合物 124mg (収率 85 %)を得た。 lH-NMR(DMSO-d6): 7.63— 7.65(1H, m), 7.98— 8.03(2H, m),
8· 11-8· 15(2Η, m), 8.59_8·60(1Η, m), 8.85(1H, d), 9·04_9·08(1Η, m)0
実施例 31から実施例 45に記載の化合物を以下に示す。
[0214] [化 22] 実施例
[0215] (実施例 46) 1-(3-ピリジル) -4-( -テトラゾル -5-ィル)フタラジン ナトリウム塩
a) 4-(3-ピリジル) -フタラジン- 1-カルボ二トリル
既知の方法 (Synth.Commun.1999, 29(3), 457)に従って合成した 4_(3_ピリジル )-2H-フタラジン- 1-オン 2.33g(10.4 mmol)を用いて実施例 37bと同様の操作を行うこと
により、 1-クロ口- 4-ピリジン- 3-ィル-フタラジン 2.30g (収率 91%)を得た。
1H-NMR(CDC13): 7·54_7.58(1Η, m), 8.01-8.13(4H, m), 8.45(1H, d), 8.82-8.84(1H m), 8.99(1H, d) この化合物 700mg(2.90 mmol)を用いて実施例 45aと同様の操作を 行うことにより、表記化合物 522mg (収率 78%)を得た。 1H_NMR(CDC13):
7·58-7·62(1Η, m), 8.10-8.22(4H, m), 8.43(1H, m), 8.87— 8.89(1H, m), 9.04(1H, d)。 b) l- (3-ピリジル) 4_(1H-テトラゾル- 5-ィル)フタラジン
実施例 46aで得られた化合物 520mg(2.24 mmol)を用いて実施例 45bと同様の操作を 行うことにより、表記化合物 533mg (収率 86%)を得た。 lH_NMR(DMSO_d6):
7.70-7.74(1Η, m), 8.13_8·22(2Η, m), 8.26— 8.31(2Η, m), 8.86— 8.88(1Η, m), 9·02(1Η, d), 9.43(1Η, d)。
c) 1_(3_ピリジル )_4_(1H-テトラゾル _5_ィル)フタラジン ナトリウム塩
塩実施例 46bで得られた化合物 164mg(0.59 mmol)を用いて実施例 45cと同様の操作 を行うことにより、表記化合物 175mg (収率 99%)を得た。 m_NMR(DMSO-d6):
7.66-7.7K1H, m), 7.97- 8.91(3H, d), 8.25(1H, d), 8.82(1H, d), 8.98(1H, s),
9.10(1H, d)。
(実施例 47) l-(4-ピリジル) -4-( -テトラゾル -5-ィル)フタラジン ナトリウム塩 a) 1-クロ口- 4-(4-ピリジル) -フタラジン
実施例 44中記載方法と同様にして得られた 4-(4-ピリジル) -2H-フタラジン- 1-オン 4.13g(18.5 mmol)を用いて実施例 37bと同様の操作を行うことにより、表記化合物 4.43g (収率 99%)を得た。 1H-NMR(CDC13): 7.66_7.68(2H, m), 7.99_8.01(2H, m), 8.03_8.09(1H, m), 8.45(1H, d), 8.86_8·89(2Η, m)。
b) 4_(4-ピリジル) -フタラジン _1_カルボ二トリノレ
実施例 47aで得られた化合物 1.2g(4.97 mmol)を用いて実施例 45aと同様の操作を行 うことにより、表記化合物 987mg (収率 86%)を得た。 1H-NMR(CDC13): 7.70-7.72(2H, m), 8.10-8· 19(3Η, m), 8.44-8.48(1Η, m), 8.91— 8.93(2Η, m)。
c) l_(4_ピリジル )_4_(1Η-テトラゾル _5_ィル)フタラジン
実施例 47bで得られた化合物 510mg(2.20 mmol)を用いて実施例 45bと同様の操作を 行うことにより、表記化合物 487mg (収率 81%)を得た。 lH_NMR(DMSO_d6):
7.84-7.86(2H, m), 8.11- 8.19(2H, m), 8.27- 8·31(1Η, m), 8.89_8.91(2H, m), 9.45(1H, d)。
d) l-(4-ピリジル) -4-(lH -テトラゾル -5-ィル)フタラジン ナトリウム塩
実施例 47cで得られた化合物 487mg(1.77 mmol)を用いて実施例 45cと同様の操作を 行うことにより、表記化合物 489mg (収率 93%)を得た。 lH_NMR(DMSO_d6):
7·80-7·82(2Η, m), 7.98_8·07(3Η, m), 8.84— 8.86(2Η, m), 9.14(1Η, d)。
[0217] (実施例 48) 1_(4-ピリジル) -4-(2, 5 -ジヒドロ- 5_ォキソ _4H_1, 2, 4_ォキサジァゾ一 ノレ- 3-ィル)フタラジン 塩酸塩
a) N -ヒドロキシ- 4_(4_ピリジル)フタラジン _1_カルボキシアミジン
実施例 47bで得られた化合物 450mg(1.94 mmol)のエタノール (15ml)溶液に、塩酸ヒド ロキシルァミン 153mg(2.13 mmol),水酸化カリウム 120mg(2.13 mmol)を加え 4時間加 熱還流した。室温に冷却し、溶媒を減圧濃縮後、析出した固体を含水エタノールで 洗浄し、表記化合物 428mg (収率 83%)を得た。 1H_NMR(CDC13): 5.93(2H, brs), 6.91(1H, s), 7.69-7.71(2H, m), 7.88- 8.04(3H, m), 8.87—8.89(2H, m), 9.40(1H, d)。 b) l-(4-ピリジル) -4-(2, 5-ジヒドロ- 5-ォキソ -4H-1, 2, 4_ォキサジァゾール _3_ィル) フタラジン 塩酸塩
実施例 48aで得られた化合物 200mg(0.75 mmol)の THF(3ml)溶液を 0°Cに冷却し、トリ ェチルァミン 0.15ml(1.06 mmol),クロロギ酸ェチル 117mg(1.01 mmol)を加え、室温に て一晩攪拌した。反応溶液に水をカ卩え、析出した固体をろ取し水で洗浄、乾燥後、ピ リジン (5ml)に溶解させ、 12時間加熱還流した。室温に冷却し、溶媒を減圧濃縮後、 析出した固体をアセトン-メタノールで洗浄し 1-(4-ピリジル) -4-(2, 5-ジヒドロ- 5 -ォキ ソ- 4H-1, 2, 4 -ォキサジァゾール -3-ィル)フタラジンを得た。 1-(4-ピリジル) -4-(2, 5- ジヒドロ- 5_ォキソ -4H-1, 2, 4_ォキサジァゾール- 3_ィル)フタラジンを用いて実施例 29fと同様の操作を行うことにより、表記化合物 193mg (収率 78%)を得た。
lH-NMR(DMSO-d6): 8·04(2Η, m), 8.1ト 8·22(2Η, m), 8.26— 8.31(1H, m), 9.01(1H, d), 9.07(1H, d)。
[0218] (実施例 49) 1_(4-ピリジル) -4-(2, 5 -ジヒドロ- 5_ォキソ _4H_1, 2, 4 -チアジアゾル -3-ィル)フタラジン ナトリウム塩
実施例 48aで得られた化合物 255mg(0.96 mmol)の THF(lOml)溶液にチォカルボニル ジイミダゾール 286mg(1.44 mmol)を加え、室温にて 4時間攪拌した。反応溶液に水を 加え、析出した固体をろ取し水で洗浄、乾燥後、 THF(20ml)に溶解させ、ボロントリフ ルオリド ェチルエーテル錯体 660mg(4.69 mmol)を加えて 31時間加熱還流した。室 温に冷却し溶媒を減圧濃縮後、水を加えて析出した固体をアセトン -メタノールで洗 浄し 1_(4-ピリジル) -4-(2, 5 -ジヒドロ- 5-ォキソ -4H-1, 2, 4-チアジアゾル _3_ィル)フタ ラジンを得た。
[0219] 得られた 1_(4-ピリジル) -4-(2, 5 -ジヒドロ- 5-ォキソ -4H-1, 2, 4-チアジアゾル _3_ィ ノレ)フタラジンのメタノール (10ml)溶液にナトリウムメトキシド メタノール溶液
(0.5M)l. lmlをカ卩え、室温にて一晩攪拌した。反応液を減圧濃縮した後、得られた固 体をアセトン-メタノールで洗浄し表記化合物 94mg (収率 30%)を得た。
lH-NMR(DMSO-d6): 7·79_7·83(2Η, m), 7.97—8.14(3H, m), 8.59_8·70(1Η, m), 8.84- 8.85(2H, m)。
[0220] (実施例 50) l-(2-ピリジル) 4-(2, 5_ジヒドロ- 5_ォキソ _4H_1, 2, 4_ォキサジァゾ一 ノレ- 3-ィル)フタラジン ナトリウム塩
a) N-ヒドロキシ -4-(2-ピリジル) -フタラジン- 1-カルボキシアミジン
実施例 47aで得られた化合物 500mg(2.15 mmol)を用いて実施例 48aと同様の操作を 行うことにより、表記化合物 417mg (収率 73%)を得た。 1H_NMR(CDC13): 5·91(2Η, brs), 6·83(1Η, s), 7.48- 7.51(1Η, m), 7·90— 7·99(3Η, m), 8.22(1Η, d), 8.71_8.75(1Η, m), 8.84(1Η, d), 9.29— 9·32(1Η, m)。
b) 1-(2-ピリジル) 4-(2, 5-ジヒドロ- 5-ォキソ -4H-1, 2, 4_ォキサジァゾール _3_ィル)フ タラジン ナトリウム塩
実施例 50aで得られた化合物 170mg(0.64 mmol)を用いて実施例 48bと同様の操作を 行うことにより、 1_(2_ピリジル) 4_(2, 5-ジヒドロ- 5_ォキソ -4H-1, 2, 4_ォキサジァゾ一 ノレ- 3-ィル)フタラジンを得た。得られた 1_(2-ピリジル) 4-(2, 5 -ジヒドロ- 5-ォキソ -4H-1, 2, 4-ォキサジァゾール -3-ィル)フタラジンのメタノール (3ml)溶液にナトリウム メトキシド メタノール溶液 (0.5M)1.2mlをカ卩え、室温にて一晩攪拌した。反応液を減 圧濃縮した後、得られた固体をアセトン-メタノールで洗浄し表記化合物 140mg (収率
82%)を得た。 lH-NMR(DMSO-d6): 7.63-7.67(1Η, m), 8.02- 8· 16(4Η, m),
8.58— 8.61(1H, m), 8.78— 8.86(2H, m)。
[0221] (実施例 51) l-(2-ピリジル) -4-(2, 5_ジヒドロ- 5_ォキソ _4H_1, 2, 4_チアジアゾル -3-ィル)フタラジン
実施例 50aで得られた化合物 500mg(2.15 mmol)を用いて実施例 49と同様の操作を行 うことにより、表記化合物 98mg (収率 34%)を得た。 lH-NMR(DMSO-d6):
7.68-7.7K1H, m), 8.13-8.22(4H, m), 8.71— 8.74(1H, m), 8.88— 8.89(2H, m)。
[0222] (実施例 52) 5_フエニル _8_(1H-テトラゾル _5_ィル)ピリド [2, 3_d]ピリダジン 塩酸塩 a) 5 -フエニル- 7H -ピリド [2, 3- d]ピリダジン- 8-オン
既知の方法 (Monatsh.Chem.1990, 121(11)、 909)により得られた 6, 7 -ジヒドロ- 5_ヒドロ キシ -5, 6 -ジフエニル -5H-ピロ [3, 4-b]ピリジン - 7_オン 8.14g(26.9 mmol)にヒドラジン 1 水和物 12mlを加えて 5時間加熱還流した。 0°Cに冷却後水をカ卩え、酢酸にて中和した 。析出した固体をろ過後、水で洗浄し表記化合物 3.85g (収率 64%)を得た。
1H-NMR(CDC13): 7.57(5H, m), 7.74(1H, dd), 8.14- 8.17(1H, m), 9.15- 9· 17(1Η, m), 10.26(1H, brs)。
b) 8-クロ口- 5-フエニルピリド [2, 3-d]ピリダジン
実施例 52aで得られた化合物 3.84g(17.2 mmol)を用いて実施例 37bと同様の操作を行 うことにより、表記化合物 2.49g (収率 60%)を得た。 -NMR(CDC13): 7.60-7.62(3H, m), 7.72-7.75(2H, m), 7·86— 7·89(1Η, m), 8.44(1H, dd), 9.36— 9.37(1H, m)。
c) 8-(2-ベンジルォキシメチル -2H-テトラゾル -5-ィル) -5-フエニルピリド [2, 3_d]ピリ ダジン
実施例 52bで得られた化合物 300mg(1.24 mmol)及び既知の方法 (Tetrahedron Lett.2000, 41(16)、 2805)に従って得られた 2-ベンジルォキシメチル _5_ (トリブチルス ズ)テトラゾール 714mg(1.49 mmol)を用いて実施例 15と同様の操作を行うことにより、 表記化合物 165mg (収率 34%)を得た。 1H_NMR(CDC13): 4.84(2H, s), 6· 16(2Η, s), 7.36-7.42(5Η, m), 7.64_7·66(3Η, m), 7.83— 7.89(3Η, m), 8.55— 8.57(1Η, m),
9.40-9.41(1Η, m)0
d) 5_フエニル _8_(1H -テトラゾル- 5_ィル)ピリド [2, 3-d]ピリダジン 塩酸塩
実施例 52cで得られた化合物 160mg(405 mmol)のメタノール (4ml)溶液に、 4モル塩酸 -酢酸ェチル溶液 0.61mlを加え 3時間加熱還流した。室温に冷却し、溶媒を減圧濃 縮後、析出した固体をアセトン-メタノールで洗浄し、表記化合物 113mg (収率 90%)を 得た。 lH-NMR(DMSO-d6): 7.69— 7.71(3H, m), 7.86— 7.89(2H, m), 8.1ト 8.16(1H, m), 8.56-8.59(lH, m), 9.44-9.46(1H, m)。
[0223] (実施例 53) 1 -フエニル _4_(1H-テトラゾル _5_ィル)フタラジン
既知の方法 (Heterocycles 1994, 39(1)、 345)により得られた 4 -フエニルフタラジン- 1- カルボ二トリル 400mg(l .73 mmol)を用レ、て実施例 45bと同様の操作を行うことにより、 表記化合物 225mg (収率 48%)を得た。 1H_NMR(CDC13): 7.67_7.69(3H, m), 7.84-7.87(2H, m), 8.04_8·06(1Η, m), 8.14- 8.17(1H, m), 8.25(1H, d), 9.78(1H, d)MS(FritFAB): 275(M+H+), 247, 232, 205, 189, 77。
[0224] (実施例 54)
a) 1-(1-メチル -1H-テトラゾル -5-ィル) -4-フエニルフタラジン
b) l_(2-メチル -2H-テトラゾル -5-ィル) -4-フエニルフタラジン
実施例 53で得られた化合物 280mg(1.02 mmol)を DMF(3.5ml)に溶解させ、 0°Cに冷却 した。窒素雰囲気下にて水素化ナトリウム (60%)65mg(1.63 mmol)を加え、 0°Cにて 30 分攪拌した。反応溶液にヨウ化メチル 274mg(1.63 mmol)をカ卩え、室温でー晚攪拌し た。反応溶液に水をカ卩え、析出した固体をろ取し、シリカゲルカラムクロマトグラフィー (ジクロロメタン-酢酸ェチル)にて精製後、表記化合物を得た。
a) 1-(1-メチル -1H -テトラゾル -5-ィル) -4-フエニルフタラジン: 97mg (収率 33%)。 RM).525(ジクロロメタン/酢酸ェチル =5/l)lH_NMR(CDC ): 4.59(3H, s),
7.62-7.64(3H, m), 7.81_7·85(2Η, m), 7.99_8.02(1H, m), 8.05_8.08(1H, m), 8.24(1H, d), 9.24(1H, d)
b) 1_(2-メチル _2H-テトラゾル- 5 -ィル )_4 -フヱニルフタラジン: 123mg (収率 42%)。 Ri 3.175(ジクロロメタン/酢酸ェチル =5/l)lH_NMR(CDC13): 4.59(3H, s),
7.59-7.6K3H, m), 7.80_7·83(2Η, m), 7.93— 7.96(1H, m), 7.98— 8.01(1H, m), 8.17-8.21(1H, m), 9.06(1H, d)MS(EI): 288(M+), 232, 217, 203, 189, 176, 163, 77。
[0225] (実施例 55) 5-(4-フエニルフタラジン- 1-ィル) -2, 4_ジヒドロピラゾール _3_オン a) 4-フエニルフタラジン- 1-カルボン酸
4-フエニルフタラジン- 1-カルボ二トリル 2.2g(9.51 mmol)に 50%硫酸 (30ml)を加え、 7 時間加熱還流した。反応液を 0°Cに冷却後水酸化ナトリウム水溶液にて中和し、析出 した固体をろ過後、氷水で洗浄し表記化合物 1.77g (収率 75%)を得た。
lH-NMR(DMSO-d6): 7·63_7·74(5Η, m), 8·01_8·08(3Η, m), 8.48(1Η, m)。
b) 5_(4_フエニルフタラジン- 1-ィル) -2, 4 -ジヒドロピラゾール _3_オン
実施例 55aで得られた化合物 975mg(3.90 mmol)の THF(lOml)溶液にカルボニルジイミ ダゾール 695mg(4.29 mmol)を加え、 50°Cにて 2時間攪拌した。反応溶液を 0°Cに冷却 後、マロン酸ェチルカリウム 729mg(4.29 mmol),塩化マグネシウム 408mg(4.29 mmol) を加え、 5時間加熱還流した。反応液を冷却後、希塩酸水溶液を加え、酢酸ェチル にて抽出し、無水硫酸ナトリウムにて乾燥後、ろ過、減圧濃縮した。得られた残さをシ リカゲルカラムクロマトグラフィー (へキサン-酢酸ェチル)にて精製し、 3-ォキソ -3-(4- フエニルフタラジン- 1-ィル)プロピオン酸メチルエステル 571mg (収率 46%)を得た。 得られた 3-ォキソ -3-(4-フエニルフタラジン- 1-ィル)プロピオン酸メチルエステル 236mg(0.74 mmol)のエタノール (3ml)溶液にヒドラジン 1水和物 0.7mlを加え 6時間加熱 還流した。室温に冷却した後、溶媒を減圧濃縮して得られた固体をェチルエーテル- エタノールにて洗浄し、表記化合物 189mg (収率 89%)を得た。 1H-NMR(CD30D): 7.63— 7.65(3H, m), 7.72— 7.76(2H, m), 7.98-8.12(4H, m), 8.85(1H, brs)。
[0226] (実施例 56) l-(5-メチル -[1, 3, 4]ォキサジァゾール _2_ィル) _4_フエニルフタラジン a) 2-ァセチル -1-(4-フエニルフタラジン- 1-カルボニル)ヒドラジン
実施例 55aで得られた化合物 600mg(2.40 mmol)の THF(12ml)溶液にカルボニルジイミ ダゾール 505mg(3.12. mmol)をカ卩え、 50°Cにて 2時間攪拌した。反応溶液を 0°Cに冷 却後ァセチルヒドラジン 195mg(2.64 mmol)をカ卩え、室温にて 3時間攪拌した。反応液 に水を加え、ジクロロメタンにて抽出し、無水硫酸ナトリウムにて乾燥後ろ過、減圧濃 縮した。得られた固体をへキサン-酢酸ェチルにて洗浄し、表記化合物 597mg (収率 70%)を得た。 1H-NMR(CDC13): 2.20(3H, s), 7.60- 7.62(3H, m), 7·75_7·78(2Η, m), 7.94(1H, t), 7.98-8.02(lH, m), 8.15(1H, d), 8.21(1H, brs), 9.43(1H, d)。
b) l-(5-メチル -[1, 3, 4]ォキサジァゾール -2-ィル) -4-フエニルフタラジン 実施例 56aで得られた化合物 250mg(0.82 mmol)及びポリリン酸 5.0gの混合物を 120°C にて 2時間攪拌した。反応溶液を室温に冷却し氷水を加え、炭酸ナトリウムにて中和 後、ジクロロメタンにて抽出し、無水硫酸ナトリウムにて乾燥後ろ過、減圧濃縮した。 得られた固体をへキサン-酢酸ェチルにて洗浄し、表記化合物 203mg (収率 86%)を 得た。 1H-NMR(CDC13): 2.78(3H, s), 7.61- 7.63(3H, m), 7.81- 7.84(2H, m), 7.97(1H, t), 8.08(1H, t), 8.22(1H, d), 9.54(1H, d)。
[0227] (実施例 57) 1_(5-メチル -[1, 3, 4]チアジアゾル -2-ィル) -4-フエニルフタラジン 実施例 56aで得られた化合物 336mg(1.10 mmol)のトルエン (11ml)溶液にローソン試薬 577mg(1.43. mmol)を加え、 1時間加熱還流した。反応溶液を室温に冷却後飽和重曹 水をカ卩え、酢酸ェチルにて抽出し、無水硫酸ナトリウムにて乾燥後ろ過、減圧濃縮し た。得られた残さをシリカゲルカラムクロマトグラフィー (ジクロロメタン-酢酸ェチル)に て精製し、表記化合物 208mg (収率 62%)を得た。 m_NMR(CDC13): 2.92(3H, s), 7.60-7.63(3H, m), 7.80—7.84(2H, m), 7.95_7.98(1H, m), 8.05- 8·10(1Η, m), 8.20(1H, d), 9.84(1H, d)。
[0228] (実施例 58) l-(5-メチル -4H-[1, 2, 4]トリァゾル _3_ィル) _4_フエニルフタラジン 実施例 56bで得られた化合物 95mg(0.33 mmol)及びベンジルァミン 0.7mlの混合物を 150°Cにて 2日間攪拌した。反応溶液を室温に冷却後水を加え、酢酸ェチルにて抽 出し、無水硫酸ナトリウムにて乾燥後、ろ過、減圧濃縮した。得られた残さにトリフル ォロ酢酸 (1.3ml)を加え、 10時間加熱還流した。反応溶液を減圧濃縮後ジクロロメタン にて希釈し、希塩酸水溶液にて洗浄した。水層を 0°Cに冷却して水酸化ナトリウム水 溶液にて中和して得られた固体をろ過し、表記化合物 67mg (収率 71%)を得た。 1H-NMR(CDC13): 2.63(3H, s), 7.59_7·62(3Η, m), 7.78-7.82(2Η, m), 7.94(1Η, t), 8·06(1Η, t), 8· 16(1Η, d), 9.81(1Η, brs)0
[0229] (実施例 59) 1_(1-メチル -1H-イミダゾール- 2_ィル) _4_フエニルピリド [3, 4_d]ピリダ ジン 塩酸塩
a) 4-フエニル -2H-ピリド [3, 4-d]ピリダジン _1_オン
既知の方法 (Monatsh.Chem.1990, 121(11)、 909)により得られた 5, 6 -ジヒドロ- 7_ヒドロ
キシ -6, 7-ジフエニル -7H-ピロ [4, 3-c]ピリジン- 5-オン 8.63g(28.6 mmol)を用いて実 施例 54aと同様の操作を行うことにより、表記化合物 6.13g (収率 96%)を得た。
1H-NMR(CDC13): 7.58- 7.62(5H, m), 8.29(1H, d), 9.03(1H, d), 9.21(1H, s), 10.1 5(1H, brs)。
b) 1_クロ口- 4-フエニルピリド [3, 4-d]ピリダジン
実施例 59aで得られた化合物 6.13g(27.5 mmol)を用いて実施例 37bと同様の操作を行 うことにより、表記化合物 3.28g (収率 49%)を得た。 1H-NMR(CDC13): 7.63_7.65(3H, m), 7.79-7.82(2H, m), 8.13(1H, d), 9.16(1H, d), 9.56(1H, s)。
c) l_(l_メチル _1H -イミダゾール -2-ィル) -4-フエニルピリド [3, 4-d]ピリダジン 塩酸塩 実施例 59bで得られた化合物 300mg(1.24 mmol)を用いて実施例 15と同様の操作を行 うことにより、 メチル -1H -イミダゾール -2-ィル) -4-フエニルピリド [3, 4-d]ピリダジ ン 291mg (収率 82%)を得た。 メチル -1H -イミダゾール -2-ィル) -4-フエニルピリド [3, 4-d]ピリダジンを用いて実施例 30fと同様の操作を行うことにより、表記化合物 351mg (収率 96%)を得た。 lH-NMR(DMSO_d6): 4·04(3Η, s), 7.71-7.73(3H, m), 7.78(1H, m), 7·94— 7·96(3Η, m), 8.47(1H, m), 9.20(1H, d), 9.55(1H, s)。
実施例 46から実施例 59の化合物を以下に示す。
[化 23] 実施例 実施例 実施例
(実施例 60) 4-(1-メチル -1H-イミダノール- 2-ィル) -7-(2-ピリジル)チエノ [2, 3d]ピ リダジン 2塩酸塩
a) 7-(2-ピリジル) -5H-チエノ [2, 3d]ピリダジン- 4_オン
3-チォフェンカルボン酸 8.90g(69.4 mmol)及び 2-ピリジンカルボアルデヒド 8.18g(76.3
mmol)を用いて実施例 37aと同様の操作を行うことにより、表記化合物 7.62g (収率 48 %)を得た。 lH-NMR(DMSO-d6): 7.72-7.79(1Η, m), 7.88(1H, d), 8.02-8.09(1H, m), 8.71(1H, d), 9.04—9.06(2H, m)。
b) 4_クロ口- 7_(2_ピリジル) -チエノ [2, 3d]ピリダジン
実施例 60aで得られた化合物 7.62g(33.2 mmol)を用いて実施例 37bと同様の操作を行 うことにより、表記化合物 7.92g (収率 97%)を得た。 1H-NMR(CDC13): 7.43-7.48(1H, m), 7.75(1H, d), 7.91— 7.98(1H, m), 8.04(1H, d), 8.80— 8.88(2H, m)。
c) 4_(1_メチル _1H -イミダゾール -2-ィル) -7-(2_ピリジル)チエノ [2, 3d]ピリダジン 2 塩酸塩
実施例 60bで得られた化合物 300mg(1.21 mmol)を用いて実施例 15と同様の操作を行 うことにより、 4- (1-メチル - 1H -イミダゾール -2-ィル) -7-(2-ピリジル)チエノ [2, 3d]ピリ ダジンを得た。 4-(1-メチル -1H-イミダゾール _2 -ィル )_7_(2-ピリジル)チエノ [2, 3d]ピ リダジンを用いて実施例 29fと同様の操作を行うことにより、表記化合物 166mg (収率 37 %)を得た。 lH-NMR(DMSO-d6): 4.12(3H, s), 7·65_7.71(2Η, m), 7.82(1Η, s), 8.14— 8.20(2Η, m), 8·55(1Η, d), 8.89(1Η, d), 8·95(1Η, d)。
(実施例 61) 4-(1-メチル -1H-イミダゾール -2-ィル) -7-(4-ピリジル)チエノ [2, 3d]ピ リダジン
a) 7-ピリジン- (4-ピリジル) -5H-チエノ [2, 3d]ピリダジン- 4_オン
3-チォフェンカルボン酸 11.7g(90.9 mmol)及び 4-ピリジンカルボアルデヒド 10.7g(100 mmol)を用いて実施例 37aと同様の操作を行うことにより、表記化合物 5.90g (収率 28% )を得た。 lH-NMR(DMSO-d6): 7.90(1H, d), 8.40- 8.44(2H, m), 8.66(1H, d), 9.06-9.11(2H, m)0
b) 4_クロ口- 7_(4_ピリジル)チエノ [2, 3d]ピリダジン
実施例 61aで得られた化合物 5.90g(25.7 mmol)を用いて実施例 37bと同様の操作を行 うことにより、表記化合物 1.98g (収率 31%)を得た。 1H-NMR(CDC13): 7.75(1H, d), 8.02-8.04(3H, m), 8.88_8·90(2Η, m)0
c) 4_(l_メチル _1H -イミダゾール -2-ィル) -7-(4_ピリジル)チエノ [2, 3d]ピリダジン 実施例 61ヒで得られた化合物30011¾(1.21 mmol)を用いて実施例 15と同様の操作を行
うことにより、表記化合物 43mg (収率 12%)を得た。 1H-NMR(CDC13): 4·30(3Η, s), 7.17(1H, s), 7.34(1H, s), 7.96(1H, d), 8.11— 8.13(2H, m), 8.77(1H, d),
8.88-8.90(2H, m)。
[0234] (実施例 62) 7_(1-メチル -1H-イミダゾール- 2_ィル) _4_(2_ピリジル)チエノ [2, 3d]ピ リダジン 塩酸塩
実施例 37bで得られた化合物 300mg(1.20 mmol)及びトリ- n_ブチル (2-ピリジル)スズ 529mg(1.44 mmol)を用いて実施例 15と同様の操作を行うことにより、 7_(1_メチル -1H-イミダゾール _2 -ィル )_4_(2-ピリジル)チエノ [2, 3d]ピリダジンを得た。 7-(1_メチ ノレ- 1H-イミダゾール -2-ィル) -4- (2-ピリジル)チエノ [2, 3d]ピリダジンを用いて実施例 29fと同様の操作を行うことにより、表記化合物 103mg (収率 26%)を得た。
lH-NMR(DMSO-d6): 4.30(3H, s), 7·35(1Η, s), 7.61— 7.64(2Η, m), 8.08—8.13(1Η, m), 8.43(1Η, d), 8.57— 8.60(2Η, m), 8·87(1Η, d)。
[0235] (実施例 63) 7-(1-メチル -1H-イミダゾール -2-ィル) -4-(4-ピリジル)チエノ [2, 3d]ピ リダジン
実施例 37bで得られた化合物 300mg(1.20 mmol)及びトリ- n_ブチル (4-ピリジル)スズ 556mg(1.44 mmol)を用いて実施例 15と同様の操作を行うことにより、表記化合物 121mg (収率 34%)を得た。 1H-NMR(CDC13): 4.40(3H, s), 7.17(1H, s), 7.39(1H, d), 7.71(1H, d), 7.89-7.9K2H, m), 8.05(1H, d), 8.86— 8.88(2H, m)。
[0236] (実施例 64) l-(5-ォキソ -4, 5_ジヒドロ- [1, 2, 4]ォキサジァゾール _3_ィル) _4_フエ ニルフタラジン
a) N-ヒドロキシ -4-フエニルフタラジン- 1-カルボキシアミジン
4-シァノフタラジン _1_カルボ二トリル 1.057g(4.569 mmol),塩酸ヒドロキシルァミン 442mg(6.36 mmol)及び酢酸ナトリウム 447mg(5.45 mmol)をエタノーノレ:水 =5: 1混合 溶液 (60ml)中に加え、 80分加熱還流した。反応液を室温まで冷却し、エタノールを減 圧留去した後、反応液を水で希釈した。析出した固体をろ取し、水にて洗浄後、減圧 下乾燥して表記化合物 1.066g (収率 88%)を淡黄色固体として得た。
lH-NMR(DMSO-d6): 6.24(2H, s, NH2), 7·62_7·66(3Η, m), 7.73_7·77(2Η, m), 7.99-8.10(3Η, m), 9.03_9·07(1Η, m), 10.36(1Η, s, ΟΗ)。
b) l-(5-ォキソ -4, 5-ジヒドロ- [1, 2, 4]ォキサジァゾール -3-ィル) -4-フエエルフタラジ ン
実施例 64aで得られた化合物 302mg(1.143 mmol)をピリジン (6ml)に溶解し、室温にて 攪拌しながらクロロギ酸ェチル 0.117ml(1.23 mmol)をカ卩え、昇温し 4時間加熱還流し た。反応液を冷却後、クェン酸水溶液をカ卩ぇ pHく 4とし、析出した固体をろ取し、水に て洗浄後、減圧下乾燥して表記化合物 298mg (収率 90%)を淡黄色固体として得た。 lH-NMR(DMSO-d6): 7·36_7·42(1Η, m), 7·66_7·69(2Η, m), 7.76-7.82(2Η, m), 8· 13-8· 15(1Η, m), 8.19_8·24(1Η, m), 8.57— 8.59(1Η, m), 9.02(1Η, d, J=8.4), 13·67(1Η, brs)。
[0237] (実施例 65) 1 -フヱニル _4_(5 -チォキソ -4, 5 -ジヒドロ- [1, 2, 4]ォキサジァゾ一ノレ -3-ィル)フタラジン
実施例 64aで得られた化合物 249mg(0.942 mmol)をァセトニトリル (7ml)に溶解し、室温 にて攪拌しながらチォカルボニルジイミダゾール (90%)368mg(1.86 mmol)を加え、引 き続いて DBU 0.56ml(3.75 mmol)を加え、同温にて 6時間半攪拌した。反応液中にク ェン酸水溶液を加え pHく 4とし、析出した固体をろ取し、水にて洗浄後、減圧下乾燥 して表記化合物 293mg (収率 100%)を淡黄色固体として得た。 m_NMR((DMSO-d6): 7.67-7.70(3H, m), 7·79_7.82(2Η, m), 8.15— 8·27(3Η, m), 8·93(1Η, d, J=8.2)。
[0238] (実施例 66) 1-{4-(1Η-テトラゾール -5-ィル)フタラジン- 1-ィル }ピペリジン- 4-ォー ノレ
a) 4-ォキソ一 3, 4-ジヒドロ- 3H-フタラジン- 1-カルボン酸
石 J 11らの方法(文献 Chem.Pharm.Bull.28(9)2770-2778(1980))を参考にして表題化合 物を合成した。水酸化カリウム (85%grade)8.85g(134.1 mmol)を水 (250ml)に溶解し、 そこに 2-ァセチル安息香酸 8.28g(50.44 mmol)を加え、室温下攪拌した。そこに水 (350ml)に溶解した過マンガン酸カリウム 15.94g(100.9 mmol)を 20分かけて滴下し、同 温にて、さらに 90分攪拌した。不溶物をセライトろ過し、水 (500ml)で洗浄した。母液中 にドライアイスをゆっくり加え、二酸化炭素を飽和させた後、ヒドラジン水和物 (40ml)を 加え 80°Cで 4時間加熱攪拌した。反応液を氷冷し 6モル塩酸水溶液を加え pH 1とし た。析出した固体をろ取し、減圧下乾燥して表記化合物 7.30g (収率 76%)を無色固体
として得た。 lH-NMR(DMSO-d6): 7.90(1H, t, J=7.5), 7·96_8·02(1Η, m), 8.29(1H, d, J=7.5), 8.58(1H, d, J=8.1), 13.11(1H, s), 13.2-14.0(1H, broad)0
b) 4-ォキソ一 3, 4-ジヒドロ- 3H-フタラジン- 1-カルボン酸アミド
実施例 66aで得られた化合物 4.19g(22.03 mmol)をジクロロメタン (150ml)中に加え、氷 冷下攪拌した。そこにトリェチルァミン 3.40ml(24.4 mmol),およびクロロギ酸イソブチ ノレ 3.10ml(23.6 mmol)を順次加え、同温にて 30分攪拌後、 28%アンモニア水溶液 (30ml)を一気にカ卩え、室温下 45分攪拌した。反応液にジェチルエーテルを加え、析 出した固体をろ取し、水、希塩酸、水で順次洗浄し、減圧下乾燥して表記化合物 3.49g (収率 84%)を無色固体として得た。 lH_NMR(DMSO-d6): 7.70(1H, s),
7·85-7·98(3Η, m), 8.28(1Η, d, J=7.6), 8·59(1Η, d, J=8.0), 12.8— 13.2(1H, broad)0 c) 4_クロ口フタラジン- 1-カルボ二トリノレ
実施例 66bで得られた化合物 3.48g(18.40 mmol)を塩化チォニル (30ml)_ォキシ塩化リ ン (30ml)混合溶液中に加え、 110°Cで 3時間半攪拌した。反応液を減圧留去し、得ら れた油状物をジクロロメタンに溶解し、飽和重曹水、ぼうしょう水で洗浄後、硫酸マグ ネシゥムにて乾燥後、ろ過し、溶媒を減圧留去後、得られた油状物をシリカゲルカラ ムクロマトグラフィー (酢酸ェチル -ジクロロメタン)にて精製し、表記化合物 1.99g (収率 57%)を淡黄色固体として得た。 1H-NMR(CDC 3): 8.17_8·23(2Η, m), 8.34_8.38(1H, m), 8.43- 8.47(1H, m)。
d) 4-(4-ヒドロキシピペリジン _1-ィル)フタラジン- 1-カルボ二トリル
実施例 66cで得られた化合物 488mg(2.574 mmol)および 4_ヒドロキシピペリジン 830mg(8.21 mmol)を N-メチルビペリドンに溶解し、 110°Cで 90分過熱攪拌した。反応 液を室温まで冷却し、水をカ卩えると固体が析出した。これをろ取、水にて洗浄後、減 圧下乾燥させることにより表記化合物 519mg (収率 79%)を淡黄色固体として得た。 lH-NMR(DMSO-d6): 1·5_1·7(2Η, m), 1·8_2·0(2Η, m), 3.41_3·50(2Η, m),
3.80-3.87(1Η, m), 3.97_4·05(2Η, m), 4.86(1Η, d, J=4.2, OH), 8.00-8.16(4H, m)。 e) 4-{4-(lH -テトラゾール -5-ィル)フタラジン- 1 -ィル }ピペリジン- 4_オール 実施例 66dで得られた化合物 294mg(1.156 mmol),トリメチルシリルアジド (7ml)、及び ジブチルスズォキシド 34.5mg(0.139 mmol)をトルエン (10ml)に溶解し、 110。Cにて 30時
間過熱攪拌する。溶媒を減圧留去し、得られた油状物をシリカゲルカラムクロマトダラ フィー (メタノール-ジクロロメタン)にて精製し、表記化合物 114mg (収率 33%)を淡黄色 固体として得た。 lH_NMR(DMSO-d6): 1·64_1·76(2Η, m), 1·96_2·02(2Η, m), 3.2-3.5(2Η, m), 3.81_3·95(3Η, m), 4.85(1Η, brs, OH), 8.02-8.13(2H, m), 8.18(1H, d, J=8.5), 9.23(1H, d, J=8.0)。
(実施例 67) 1_{4- (5-ォキソ -4, 5-ジヒドロ- [1, 2, 4]ォキサジァゾール _3_ィル)フタ ラジン- 1 -ィル }ピペリジン- 4-オール
a) 4_[4-(t -ブチルジメチルシリルォキシ)ピぺリジン- 1-ィル]フタラジン- 1-カルボ二トリ ノレ
実施例 66dで得られた化合物 522mg(2.053 mmol)及びイミダゾール 310mg(4.553 mmol)を DMF(5ml)に溶解し、氷冷下攪拌した。そこにクロ口- 1 -ブチルジメチルシラン 371mg(2.462 mmol)をカ卩え、同温にて 3時間攪拌した。反応液を酢酸ェチルで希釈し 、飽和食塩水にて洗浄後、硫酸マグネシウムにて乾燥後、ろ過、溶媒を減圧留去し て得られた油状物をシリカゲルカラムクロマトグラフィー (ジクロロメタン)にて精製する ことにより、表記化合物 635mg (収率 84%)を淡黄色固体として得た。
1H-NMR(CDC13): 0· 11(6Η, s), 0.93(9H, s), 1.77- 1.84(2H, m), 1.99—2.06(2H, m), 3.62-3.71(2H, m), 3.92-4.00(2H, m), 4.07- 4· 10(1Η, m), 7.84-7.97(2H, m),
8.02(1H, d, J=8.4), 8.15(1H, d, J=7.8)。
b) 4-[4-(t-ブチルジメチルシリルォキシ)ピぺリジン- 1-ィル]フタラジン- 1-カルボキシ アミジン
実施例 67aで得られた化合物を用いて、実施例 64aと同様の操作を行うことにより表記 化合物 649mg (収率 97%)を淡黄色固体として得た。 1H_NMR(CDC13): 0.11(6H, s), 0.92(9H, s), 1.80— 1.87(2H, m), 1·99_2·08(2Η, m), 3.37_3.57(2H, m), 3.78— 3.83(2H, m), 3.97-4.05(lH, m), 5.79(2H, s, NH2), 7.76— 7.84(2H, m), 8.00— 8.04(1H, m), 9.08-9.12(1H, m)0
c) l- {4- (5-ォキソ _4, 5-ジヒドロ- [1, 2, 4]ォキサジァゾール -3-ィル)フタラジン- 1-ィ ノレ }ピペリジン -4-オール
実施例 67bで得られた化合物を用いて、実施例 64bと同様の操作を行うことにより
246mgの淡黄色無定形固体を得た。これを THFに溶解し、 1モル テトラプチルアンモ ニゥムフルオリド (2.7ml)を加え、一昼夜放置する。反応液を減圧濃縮した後、残查を シリカゲルカラムクロマトグラフィー (メタノール-ジクロロメタン)にて精製し、ェチルエー テル-へキサン混合溶媒で洗浄し表記化合物 146mg (収率 53%)を淡黄色固体として 得た。 lH-NMR(DMSO-d6): 1.64— 1.75(2H, m), 1.96—2.01(2H, m), 3.34— 3.42(2H, m), 3.81-3.96(3H, m), 4.85(1H, s), 7.99_8.08(2H, m), 8.14— 8.18(1H, m),
8.79-8.83(lH, m), 13.30(1H, s, NH)。
[0240] (実施例 68) 1_{4- (5-チォキソ -4, 5-ジヒドロ- [1, 2, 4]ォキサジァゾール _3_ィル)フ タラジン _1 -ィル }ピペリジン- 4_オール
実施例 67aで得られた化合物を用いて実施例 65と同様の操作を行うことにより 269mg の淡黄色固体を得た。この化合物を実施例 67cと同様の操作を行い脱保護し、表記 化合物 142mg (収率 67%)を淡黄色固体として得た。 lH_NMR(DMSO_d6):
1.64-1.75(2H, m), 1.95-2.02(2H, m), 3.36- 3·43(2Η, m), 3·82_3·96(3Η, m), 4.6-5.0(1H, broad), 8.00—8.09(2H, m), 8· 15_8· 19(1Η, m), 8.68_8·72(1Η, m)。
[0241] (実施例 69) 4-{(l-メチル -1H-イミダゾール -2-ィル) -フタラジン- 1-ィル }-フエノー ノレ
a) l-(l-メチル -1H-イミダゾール -2-ィル) -4-(4-tert-ブチルジメチルシリルォキシフ ェニル)フタラジン
実施例 Idで得られた化合物 411mg及び 4-4-t-ブチルジメチルシリルォキシフエニル ボロン酸 0.66gを用いて、実施例 leと同様の操作を行うことにより表記化合物 478mg( 収率 68%)を固体として得た。 1H-NMR(CDC13): 0.28(6H, s), 1·04(9Η, s), 4.12(3Η, s), 7·05(2Η, d), 7.16(1Η, s), 7.34(1Η, s), 7.72(2Η, d), 7.80— 8.00(2Η, m), 8.17(1Η, d), 9.18(1Η, d)。
b) 4_{(l-メチル _1Η-イミダゾール- 2_ィル) -フタラジン- 1 -ィル }-フエノール 実施例 69aで得られた化合物 307mgを THF6mlに溶解し、テトラプチルアンモニゥムフ ルオリド (1M THF溶液) 0.8mlを加え室温で終夜撹拌した。反応液を濃縮し、残渣にァ セトンを加えた後、 4モル塩酸酢酸ェチル溶液 0.2mlを加えた。これにイソプロピルァ ルコールを加え加熱還流し、室温まで冷却した。析出した固体をろ取して表記化合
物 150mg (収率 67%)を固体として得た。 lH-NMR(DMSO_d6: 3.97(3H, t), 7.01(2H, d), 7.26(1H, s), 7.53(1H, s), 7.65(2H, d), 8.00-8.15(3H, m), 8.92(1H, d), 9.98(1H, brs)MS(EI): 302(M+), 274, 209, 183, 156, 151, 129, 65。 (実施例 70) 1_(4_メトキシ -ピペリジン- 1_ィル) -4-(1-メチル -1H-イミダゾール- 2_ィル)フタラジン 塩酸塩 実施例 42で得られた化合物 200mg(0.65mmol)及び水素化ナトリウム
28mg(0.71mmol)の DMF(1.5 ml)溶液を 0°Cにて 30分間攪拌した。反応溶液にヨウ化メ チル 0.05 ml(0.78mmol)を加え、室温にて一昼夜攪拌した。反応溶液に水を加え、酢 酸ェチルにて抽出し、無水硫酸ナトリウムにて乾燥後ろ過、減圧濃縮した。残さをシリ 力ゲルカラムクロマトグラフィー (ジクロロメタン-メタノール)にて精製し、 1-(4-メトキシ- ピぺリジン _1_ィル) -4-(1-メチル -1H-イミダゾール- 2_ィル)フタラジンを得た。 1-(4- メトキシ-ピペリジン- 1_ィル) _4_(1-メチル -1H-イミダゾール- 2_ィル)フタラジンの酢 酸ェチル溶液に、 4モル塩酸-酢酸ェチル溶液 0.14mlをカ卩え、室温にてー晚攪拌し た。析出した固体をろ取し、表記化合物 183mg (収率 79%)を得た。
1H-NMR(CD30D): 1.94(2H, m), 2.20- 2.23(2H, m), 3.45(3H, s), 3.63— 3.70(3H, m), 4.05(2H, m), 7.74(1H, d), 7.80(1H, d), 8.05—8.12(3H, m), 8.34- 8·37(1Η, m)。
(実施例 71) l-[4-(l-メチル -1H-イミダゾール -2-ィル) -フタラジン- 1-ィル] -ピペリ ジン- 4-カルボン酸 ェチルエステル
実施例 Idで得られた化合物 400mg(1.64 mmol)及び 4_ピぺリジンカルボン酸ェチル エステル 307mg(3.06 mmol), N-メチルモルホリン 0.27ml(2.45mmol)の tert-ブタノール (0.65ml)溶液を 4.5時間加熱還流した。室温に冷却後溶媒を減圧濃縮して得られた 固体をへキサン-酢酸ェチルにて懸洗し、表記化合物 500mg (収率 84%)を得た。
1H-NMR(CDC13): 1.31(3H, t), 2.07-2.15(4H, m), 2.59— 2.62(1H, m), 3.17— 3.25(2H, m), 3.96-4.00(2H, m), 4.05(3H, s), 4.17-4.24(2H, q), 7.10(1H, d), 7.28(1H, d), 7.82-7.85(2H, m), 8.05_8.08(1H, m), 8.93— 8.97(1H, m)。
(実施例 72) {(2R)_1_[4-(1_メチル _1H -イミダゾール -2-ィル)フタラジン- 1-ィル]ピロ リジン- 2 -ィル }メタノール
実施例 Idで得られた化合物 300mg(1.23 mmol)及び D -プロリノール 0.15ml(1.47 mmol), N-メチルモルホリン 0.16ml(1.47 mmol)を用いて実施例 71と同様の操作を行う
ことにより、表記化合物 102mg (収率 27%)を得た。 1H-NMR(CDC13): 1.83_1.90(2H, m), 2.08-2.09(1H, m), 2.28- 2·30(1Η, m), 3·80_3·86(1Η, m), 3.90_4·03(5Η, m), 4.15— 4.18(1H, m),4.89-4.91(lH, m), 7.08(1H, d), 7.26(1H, d), 7.76-7.84(2H, m), 8.18-8.21(1H, m), 8.74_8.77(1H, m)0
(実施例 73) 1-[4_(1-メチル -1H-イミダゾール- 2_ィル)フタラジン _1_ィル]ァゼチジ ン -3-ォーノレ
実施例 Idで得られた化合物 200mg(0.82 mmol)及び 3_ァゼチジノール 171mg (0.98 mmol), N-メチルモルホリン 0.22ml(0.98 mmol)を用いて実施例 71と同様の操作を行う ことにより、表記化合物 167mg (収率 73%)を得た。 1H-NMR(CDC13): 3.71_3.75(1H, m), 3.93(3H, s), 4·00_4·05(1Η, m), 4.51(2H, m), 4.67— 4.69(1H, m), 7.17(1H, d), 7.29(1H, d), 8.05— 8.11(2H, m), 8.45—8.88(1H, m), 9·05_9·08(1Η, m)0
(実施例 74) {(2S)-1-[4_(1_メチル _1H -イミダゾール -2-ィル)フタラジン- 1-ィル]ピ 口リジン- 2-ィル }メタノール
実施例 Idで得られた化合物 300mg(1.23 mmol)及び L_プロリノール 0.15ml(1.47 mmol), N-メチルモルホリン 0.16ml(1.47 mmol)を用いて実施例 71と同様の操作を行う ことにより、表記化合物 186mg (収率 49%)を得た。 1H_NMR(CDC13): 1.7(2H, m), 2.09(1H, m), 2.28 (1H, m), 3·80_3·86(1Η, m), 3.93_4·03(5Η, m), 4.16_4.18(1H, m),4.89 (1H, m), 7.08(1H, d), 7.25(1H, d), 7.73-7.84(2H, m), 8.18— 8·21(1Η, m), 8.73-8.76(lH, m)。
実施例 60から実施例 74に記載の化合物を以下に示す。
[0243] [化 24] 実施例 実施例 実施例
[0244] (実施例 75) { 3-メチル -2-(4-フエニルフタラジン- 1-ィル) -3H-イミダゾール -4-ィル } 一メタノーノレ
実施例 25の方法で得られたカルボン酸 303 mg(0.916 mmol)に、 THF12ml、トリェチ ルァミン 0.15 ml (0.997 mmol)を加えて室温で 5分間撹拌した。 _25°Cに冷却し、クロ口
ギ酸イソブチノレ 0.16 ml(1.22 mmol)をカ卩えて 20分間撹拌した。析出した固体をろ過 して除き THF 6mlで洗浄した。ろ液を _45°Cに冷却し、テトラヒドロホウ酸ナトリウム 34mg (0.899 mmol)を加えて 40分撹拌しながら _25°Cまで昇温した。反応液を -45 °Cに冷却し飽和食塩水を加えた後、 THFで抽出した。飽和食塩水で洗浄し無水硫 酸ナトリウムで乾燥させろ過し減圧濃縮した。これに水 10ml、濃塩酸 3mlを加え、 50 °Cで 3時間撹拌した。酢酸ェチルで洗浄し、 0°Cで炭酸カリウムを加えアルカリ性にし てから酢酸ェチルで抽出した。飽和食塩水洗浄、無水硫酸ナトリウム乾燥、ろ過、減 圧濃縮を順次行い、酢酸ェチルを溶出液としてカラムクロマトグラフィーを行った。得 られた物質にへキサン:酢酸ェチル = 20 : 1をカ卩えて撹拌し析出した固体をろ取する ことにより表題化合物 47.6 mg (収率 16%)を得た。 1H_NMR(CDC13): 4.09(3H, s), 4.78(2H, s), 7·26(1Η, s), 7.50— 7.70(3H, m), 7.70— 8.00(4H.m), 8.14(2H, d), 9.00(2H, d)
(実施例 76) 4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン- 1-カルボ二トリノレ
1—メチルイミダゾール 496 mg(6.04 mmol)を THF 8mlに溶解し _70°Cにて、 n_ブ チルリチウム 4.1 ml(1.58Mへキサン溶液、 6.47mmol)を力 Pえた。 20分間撹拌後、塩 化トリブチルスズ 2.13g (6.54 mmol)をカロえ、約 1 · 5時間かけて室温まで温度を上昇 させた。塩化リチウム 0.15g (3.5 mmol),実施例 66cで得られた 4_クロ口フタラジン- 1_ カノレボニトリノレ 567 mg(2.990 mmol),テトラキス(トリフエニルホスフィン)パラジウム(0) 0.35g (0.30mmol)、トルエン 20mlを加えて 60_70°Cで 10—20分加熱した後、約 3 時間加熱還流させた。反応液をカラムクロマトグラフィーを用いて精製すると(へキサ ン:酢酸ェチル:塩化メチレン)、褐色固体が得られた。これを酢酸ェチル、ジェチノレ エーテルから再結晶し、 188.7 mg(0.802 mmol, 27%)の目的物を得た。さらにろ液から 、 25.6mg(0.109 mmol, 3.6%)の目的物を得た。 1H-NMR(CDC13): 4.17(3H, s),
7.23(1H, s), 7.39(1H, s), 8.00— 8.20(2H, m), 8.30— 8.40(lH.m), 9.40_9.50(1H, m) (実施例 77) 1-(1_メチル -1H -イミダゾール -2-ィル) -4-(l,3_チアゾリジン- 3_ィル)フ タラジン 塩酸塩
実施例 Idで得られた化合物 200mg(0.82 mmol)及びチアゾリジン 0.08ml(0.98 mmol) 、 N_メチルモルホリン 0.11ml(0.98 mmol)を用いて実施例 71と同様の操作を行うことに
より、 1-(1-メチル -1H-イミダゾール -2-ィル) -4-(l,3-チアゾリジン- 3-ィル)フタラジン を得た。 1-(1-メチル -1H-イミダゾール -2-ィル) -4-(1,3-チアゾリジン- 3-ィル)フタラジ ンの酢酸ェチル溶液に、 4モル塩酸-酢酸ェチル溶液 0.1 lmlを加え、室温にてー晚 攪拌した。析出した固体をろ取し、表記化合物 112mg (収率 81%)を得た。
lH-NMR(DMSO-d6): 3.22 (2H, t), 3.87(3H, s), 4.23(2H, t), 5.07(2H, s),
7.92-8.08(5H, m), 8.46-8.49(lH, m)0
(実施例 78) 1_[4-(1_メチル -1H -イミダゾール -2-ィル)フタラジン- 1-ィル]ピぺリジン -4-カルボ二トリノレ
a) 4 -シァノピペリジン 塩酸塩
N- tert-ブトキシカルボニル -4-シァノビペリジンの THF (9.0ml)溶液に、 4モル塩酸- 酢酸ェチル溶液 3.6mlをカ卩え、室温にて一晩攪拌した。析出した固体をエーテル-酢 酸ェチルにて懸洗し、表記化合物 667mg (収率 96%)を得た。 lH_NMR(DMSO_d6): 1.87- 1.94(2H, m), 2.06-2.09(2H, m), 2·95_3·03(2Η, m), 3.12- 3·20(3Η, m), 9.05(1H, brs)。
b) l-[4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン- 1-ィル]ピぺリジン- 4-カルボ 二トリル
実施例 Idで得られた化合物 200mg(0.82 mmol)及び実施例 78aで得られた化合物 144mg (0.98 mmol), N_メチルモルホリン 0.22ml(0.98 mmol)を用いて実施例 71と同様 の操作を行うことにより、表記化合物 192mg (収率 74%)を得た。 1H-NMR(CDC13): 2.16-2.26(4H, m), 2·93_2.94(1Η, m), 3.43-3.50(2Η, m), 3·82_3·88(2Η, m), 4.05(3Η, s), 7.12(1Η, d), 7.29(1Η, d), 7.83_7.89(2Η, m), 8.00_8.04(1Η, m), 8·97-9·00(1Η, m)0
(実施例 79) 4_[4-(1-メチル -1H-イミダゾール- 2_ィル)フタラジン _1_ィル]ベンゾニ トリル
実施例 Idで得られた化合物 200mg(0.82 mmol),及び 4-シァノフヱニルボロン酸 180mg (1.23mmol)を用いて実施例 leと同様の操作を行うことにより、表記化合物 194mg (収率 76%)を得た。 1H— NMR(CDC13): 4· 15(3Η, s), 7·20(1Η, d), 7.36(1Η, d), 7.89-8.03(7Η, m), 9.27_9·30(1Η, m)0
(実施例 80) 4-[4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン- 1-ィル]ベンゾィ ル酸
実施例 Idで得られた化合物 200mg(0.82 mmol)、及び 4_カルボキシフヱニルボロン 酸 136mg (0.82mmol)を用いて実施例 leと同様の操作を行うことにより、表記化合物 137mg (収率 76%)を得た。 1H_NMR(DMS0- d6): 4.00(3H, s), 7.29(1H, d), 7.57(1H, d), 7.92(2H, d), 8.04-8.13(3H, m), 8.19(2H, d), 9.00— 9.03(1H, m)。
(実施例 81) 4_(1_メチル _1H -イミダゾール -2-ィル) -1-フエニルピリド [3, 4_d]ピリダ ジン
a) 4_(l_メチル -1H -イミダゾール -2-ィル)ピリド [3,4-d]ピリダジン- 1(2H) -オン
4-ピリジンカルボン酸 5.0g (40.6mmol)、ァニリン3.91111 (42.611111101)の DMF(50ml)溶 液に、氷冷下にてシァノホスホン酸ジェチル 7.5ml (44.7mmol)及びトリェチルァミン 6.2ml (44.7mmol)をカ卩え、 1時間攪拌した。室温にてー晚攪拌後、水をカ卩えて析出し た固体をへキサン-酢酸ェチルにて懸洗し、 N-フエニルイソニコチンアミド 5.6gを得た 。 N-フエニルイソニコチンアミド 3.0g(15.1 mmol)、テトラメチルエチレンジァミン 5.0ml ( 33.3mmol)を THF(40ml)に溶解させ、 _78°Cに冷却した。窒素雰囲気下にて n_ブチル リチウム-へキサン溶液 (1.58M)21.1mlを滴下し、 _78°Cにて 1時間攪拌した。反応溶液 に既知の方法 (J.Chem.Soc. Perkin Transl 1994, 3, 239)にて合成した 1_メチル _1H_ イミダゾール -2-カルボン酸ェチルエステル 2.8g(18.2 mmol)を加え、同温度で 2時間 攪拌した後、室温に昇温しさらに 2.5時間攪拌した。反応溶液にメタノールをカ卩え、溶 媒を減圧濃縮した。得られた残さに水を加え、塩酸にて中和後析出した固体をへキ サン-酢酸ェチルにて懸洗し、 3-ヒドロキシ -3-(1-メチル -1H-イミダゾル -2-ィル) -2- フエニル- 2,3-ジヒドロ_lH-ピロロ[3,4-c]ピリジン-l_ォン3· lgを得た。 3_ヒドロキシ -3-(1_メチル -1H-イミダゾル -2-ィル) - 2 -フエニル _2,3-ジヒドロ-1^^_ピロロ[3,4-0]ピリ ジン- 1-オン、及びヒドラジン 1水和物 15mlを用いて実施例 26aと同様の操作を行うこと により、表記化合物 1.67g (収率 49%)を得た。 lH_NMR(DMSO-d6): 3.85(3H, s), 7· 19(1Η, s), 7.45(1Η, s), 8.14(1Η, d), 9.01(1Η, d), 9·96(1Η, d), 13.25(1Η, brs)。 b) l_クロ口- 4_(1_メチル _1Η -イミダゾール -2-ィル)ピリド [3,4-d]ピリダジン
実施例 8 laにて得られた化合物 1.56g、及びォキシ塩化リン約 5mlを用いて実施例
26bと同様の操作を行うことにより、表記化合物 607mg (収率 36%)を得た。
1H-NMR(CDC13): 4· 15(3Η, s), 7·21(1Η, d), 7.38(1Η, d), 8.06—8.08(1Η, m), 9.15(1Η, d), 10.70(1Η, s)。
c) 4_(1_メチル _1H -イミダゾール -2-ィル) -1-フエニルピリド [3, 4-d]ピリダジン
実施例 81bで得られた化合物 300mg(1.22 mmol)、及びフヱニルボロン酸 184mg ( 1.46mmol)を用レ、て実施例 1 eと同様の操作を行うことにより、表記化合物 298mg (収率 85%)を得た。 1H-NMR(CDC13): 4.21(3H, s), 7·22(1Η, s), 7.40(1Η, s),
7.61-7.64(3Η, m), 7.82-7.85(2Η, m), 7.90— 7.92(1Η, m), 9.02(1Η, d), 10·70(1Η, d)。 (実施例 82) 1-(5_メチル -2-フリル) -4-(1-メチル -1H-イミダゾール -2-ィル)フタラジ ン 塩酸塩
a) 4_(5_メチル _2_フリル)- 2H -フタラジン _1_オン
フタル酸モノメチルエステル 2.0g (ll.lmmol)、及び 2 -メチルフラン
1.5ml(16.7mmol)を用いて既知の方法 (特開平 6-135938)に準じて合成し、表記化合 物 348mg (収率 14%)を得た。 1H-NMR(CDC13): 2.47(3H, s), 6.21(1H, m), 6.83(1H, d), 7.83-7.92(2H, m), 8.37(1H, d), 8.49- 8.52(1H, m), 10.02(1H, brs)。
b) l-クロ口- 4-(5-メチル -2-フリル) -2H-フタラジン
実施例 82aで得られた化合物 343mg(1.52 mmol)及びォキシ塩化リン 0.17mlを用いて 実施例 28bと同様の操作を行うことにより、表記化合物 266mg (収率 72%)を得た。 1H-NMR(CDC13): 2.53(3H, s), 6.28— 6.29(1H, m), 7.35(1H, d), 7.98— 8·03(2Η, m), 8.33-8.36(lH, m), 8.86—8.89(1H, m)。
c) l-(5-メチル -2-フリル) -4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン 塩酸塩 実施例 82bで得られた化合物 263mg(1.08 mmol)及び N-メチルイミダゾール
0.13ml(1.61mmol)を用いて実施例 26cと同様の操作を行うことにより、 1_(5_メチル _2_ フリル)- 4_(1-メチル -1H-イミダゾール- 2_ィル)フタラジンを得た。 1_(5-メチル _2 -フリ ル) -4-(1_メチル -1H -イミダゾール -2-ィル)フタラジンの酢酸ェチル溶液に、 4モル塩 酸-酢酸ェチル溶液 0.18mlをカ卩え、室温にて一晩攪拌した。析出した固体をろ取し、 表記化合物 189mg (収率 54%)を得た。 lH_NMR(DMSO_d6): 2.55(3H, s), 3.94(3H, s), 6.54(1H, d), 7.55(1H, d), 7.77(1H, m), 7.91(1H, m), 8.12— 8.24(2H, m), 8.34(1H,
m), 8.96(1H, d)。
(実施例 83) 3-メチル -2- (フエニルフタラジン- 1-ィル) -3H-イミダゾール -4-カルボン 酸
実施例 25と同様にして表題化合物 75mgを得た。 lH-NMR(DMSO_d6): 4.04(3H, s), 7·92(1Η, s), 8.00-8.15(2H.m), 8·25_8·35(1Η, m), 8.48(1H, d), 9.80(1H, s) (実施例 84) l-(4_トリフルォロメチルフヱ二ル)- 4_(1-メチル _1H-イミダゾール _2 -ィ ノレ)フタラジン
実施例 Idで得られた化合物 0.63g及び 4-トリフルォロメチルフヱニルボロン酸 0.53g を用いて、実施例 leと同様の操作を行うことにより、表記化合物 518mg (収率 57%)を 固体として得た。
1H-NMR(CDC13): 4.15(3H, s), 7.19(1H, s), 7·36(1Η, s), 7.80—8.10(7H, m),
9.25(1H, d)。
(実施例 85) ジメチル -4-{4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン- 1-ィル } フエニノレアミン
実施例 Idで得られた化合物 0.58g及び 4-ジメチルァミノフエニルボロン酸 0.47gを 用いて、実施例 leと同様の操作を行うことにより、表記化合物 161mg (収率 21%)を固 体として得た。 1H-NMR(CDC13): 3.09(6H, s), 4.12(3H, s), 6.90(2H, d), 7.15(1H, d),
7.33(1H, d), 7.78(2H, d), 7.80— 8.00(2H, m), 8.27(1H, d), 9.12(1H, d)。
(実施例 86) l-(2-メトキシフエニル) -4-(l-メチル -1H-イミダゾール -2-ィル)フタラジ ン 塩酸塩
実施例 Idで得られた化合物 0.50g及び 2-メトキシフヱニルボロン酸 0.35gを用いて、 実施例 leと同様の操作を行うことにより、ト (2 -メトキシフエニル) -4- (トメチル -1H -イミ ダゾール -2-ィル)フタラジンを油状物質として得た。これを酢酸ェチル、ヘプタンの混 合溶媒に溶解し 4モル塩酸-酢酸ェチル溶液をカ卩え析出した固体をろ取することに より表題化合物 277 mg (収率 39%)を得た。 lH_NMR(DMSO_d6): 3.71(3H, s), 3·99(3Η, s), 7·23(1Η, t), 7·33(1Η, d), 7.51(1Η, dd), 7.70—7.85(1Η, m), 7.92(1Η, d), 8.04(1Η, d), 8.05— 8.20(2Η, m), 8.20— 8.30(1Η, m)。
(実施例 87) 1-(4 -メチルチオフエニル) _4_(1_メチル _1H -イミダゾール -2-ィル)フタ
ラジン
実施例 Idで得られた化合物 1.45g及び 4-メチルチオフエニルボロン酸 1.02gを用い て、実施例 leと同様の操作を行うことにより、表記化合物 886mg (収率 45%)を固体とし て得た。 1H-NMR(CDC13): 2.59(3H, s), 4.13(3H, s), 7.17(1H, s), 7.34(1H, s), 7.46(2H, d), 7.76(2H, d), 7.80-8.10(2H, m), 8.15(1H, d), 9.19(1H, d)。
(実施例 88) 1_(2,3 -ジヒドロ- 1-ベンゾフラン- 5 -ィル )_4_(1-メチル -1H-イミダゾー ル -2-ィル)フタラジン
5-ブロモ -2, 3-ジヒドロ -1-ベンゾフラン 620mg(3.11mmol)の THF 10ml溶液に、 -70。Cにて n_ブチルリチウムをカ卩えて 30分撹拌した。この反応液に 4-(1-メチル -1H-イミダゾール _2 -ィル )_2H -フタラジン _1_オン(実施例 lc参照)を加え、徐々に室 温に昇温させ終夜放置した。反応液に水を加え、酢酸ェチルで抽出した。これを 1モ ル塩酸で抽出した後、 1モル水酸化ナトリウム水溶液をカ卩えてアルカリ性にし、酢酸 ェチルで抽出した。飽和食塩水で洗浄、無水硫酸マグネシウムで乾燥させて、ろ過、 減圧濃縮した。得られた固体にジェチルエーテルをカ卩えて撹拌しながら洗浄し、ろ取 すると表題化合物が 130.9 mg (収率 37% )で得られた。 1H_NMR(CDC13):
3.35(2H, t), 4.12(3H, s), 4.70(2H, t), 6.98(1H, d), 7.16(1H, s), 7.33(1H, s), 7.57(1H, d), 7.71(1H, s), 7.80— 8.00(2H, m), 8.19(1H, d), 9.17(1H, d)。
(実施例 89) l-{4- (ピリジン- 2-ィル) -3-メチル -1H-イミダゾール -2-ィル }フタラジン 実施例 22と同様の方法にてト (1-メチル -1H -イミダゾール -2-ィル)フタラジンから 合成した 1- {4-ブロモ -3-メチル -1H-イミダゾール -2-ィル }フタラジン
268mg(0.927mmol)、 2_ピリジルトリブチルスズ 378 mg(1.027mmol)、塩化リチウム 0.06g(1.4mmol)、テトラキス(トリフエニルホスフィン)パラジウム(0) 160mg(0.138mmol) 、トルエン 7.5mlの混合物を約 6時間加熱還流した。カラムクロマトグラフィー(酢酸ェ チル:クロ口ホルム一酢酸ェチル)にて精製して得られた固体を酢酸ェチルで洗浄す ると、 157mg (収率 59%)の表題化合物が得られた。 1H_NMR(CDC13): 4.27(3H, s), 4.70(2H, t), 7· 10-7· 13(1Η, M), 7.60_7·70(1Η, M), 7.33(1H, s), 7.70— 7.90(1H, m), 7.90-8.10(3H, m), 8.71(1H, d), 8.86(1H, d), 9.54(1H, s)。
実施例 75から実施例 89に記載の化合物を以下に示す。
[0246] [化 25]
[0247] (実施例 90) 1_イソプチル -4-(1_メチル _1H -イミダゾール -2-ィル)フタラジン
実施例 Idの方法で得られた 1_クロ口- 4_(1_メチル -1H -イミダゾール -2-ィル)フタラ ジン 481mg(1.97mmol)、イソブチルボロン酸 408mg(4.00mmol)、ジクロロ(1, 1,-ビスジ フエニルホスフイノフエ口セン)パラジウム 塩化メチレン錯体 112mg(0.137mmol)、炭
酸カリウム 562mg(4.07mmol)、 1,4-ジォキサン 8mlの混合物を 100°Cにて約 20時間 加熱撹拌した。反応液に水をカ卩えて塩ィ匕メチレンで抽出し、これを 1モル塩酸で抽出 した。不溶固体をろ過して除いた後に水酸化ナトリウムを加えてアルカリ性とし、塩化 メチレンで抽出した。飽和食塩水洗浄、無水硫酸ナトリウム乾燥、ろ過、減圧濃縮の 後、カラムクロマトグラフィー(酢酸ェチル:メタノール グラジェント)にて精製して得ら れた固体をへキサンで洗浄すると、 117mg (収率 22%)の表題化合物が得られた。
1H-NMR(CDC13): 1.06(6H, d), 2.37(1H, hep), 3·28(2Η, d), 4.09(3H, s), 7.14(1H, s), 7.30(1H, s), 7.80-8.00(2H, m), 8.ト 8.25(1H, m), 9.00-9.15(1H, m)0
(実施例 91) 1_メチル -4-(l_メチル _1H -イミダゾール -2-ィル)フタラジン
1-クロ口- 4-(1-メチル -1H-イミダゾール - 2_ィル)フタラジン 506mg(2.07mmol)、メチ ルボロン酸 408mg(4.83mmol)、ジクロロ(1, 1, -ビスジフエニルホスフイノフエ口セン)パ ラジウム 塩化メチレン錯体 170mg(0.208mmol)、炭酸カリウム 852mg(6.16mmol)、 1,4-ジォキサン 15mlの混合物を約 1日加熱還流した。反応液に水を加えて塩化メ チレンで抽出し、これを 1モル塩酸で抽出した。不溶固体をろ過して除いた後に水酸 化ナトリウムをカ卩えてアルカリ性とし、塩ィ匕メチレンで抽出した。飽和食塩水洗浄、無 水硫酸ナトリウム乾燥、ろ過、減圧濃縮の後、シリカゲルショートカラム(酢酸ェチル: メタノーノレ グラジェント)にて精製して得られた固体をさらに HPLCにて分取精製し、 173mg (収率 37%)の表題化合物が得られた。 1H_NMR(CDC13): 3.07(3H, s), 4.07(3H, s), 7.14(1H, s), 7.31(1H, s), 7.80— 8·00(2Η, m), 8.05— 8· 15(1Η, m), 9.00-9.15(1H, m)。
(実施例 92)メチル 4- {4-(l-メチル -1H-イミダゾール -2-ィル)フタラジン- 1-ィル }フ ェニルスルホン
実施例 87で得られた化合物 618mg (1.859mmol)を塩化メチレン 16mlに溶解し、 氷冷下で mCPBA (m -クロ口過安息香酸、純度〉 65%) 0.74gをカ卩えて約 1時間撹拌し た。反応液に亜硫酸ナトリウム水溶液、水酸化ナトリウム水溶液をカ卩えて分液した。有 機層を水酸化ナトリウム水溶液、飽和食塩水で洗浄無水硫酸ナトリウムで乾燥した後 ろ過し、減圧濃縮した。シリカゲルカラムクロマトグラフィー(酢酸ェチル→酢酸ェチル :メタノール =4 : 1)により先に流出したフラクションを濃縮し、 t_ブチルメチルエーテ
ルで洗浄することにより表題化合物 316.7 mg (収率 47%)を固体として得た。
1H-NMR(CDC13): 3· 17(3Η, s), 4.15(3H, s), 7.20(1H, s), 7.37(1H, s), 7.80-8.10(5H, m), 8.15- 8.25(2H, m), 9.20- 9.30(1H, m)。
MS (LC-MS): 365(M+1)
(実施例 93)メチル 4-{4-(l_メチル _1H -イミダゾール -2-ィル)フタラジン- 1-ィル }フ ヱニルスルホキシド
実施例 92で行ったカラムクロマトグラフィーにより後から流出したフラクションを濃 縮し、アセトン、 t-ブチルメチルエーテルで洗浄することにより表題化合物 174.7 mg ( 収率 27%)を固体として得た。 1H-NMR(CDC13): 2.84(3H, s), 4.14(3H, s), 7.19(1H, s), 7.36(1H, s), 7.80-8.05(6H, m), 8.07(1H, d), 9.26 (1H, d)。 MS (LC-MS):
349(M+1)
(実施例 94) l-[4_(2-メチル -2H-ピラゾール -3-ィル)フタラジン- 1-ィル]ピぺリジン -4-オール 2塩酸塩
a) 2-(2-メチル -2H-ピラゾール -3-ィル)カルボニル安息香酸 メチルエステル
フタル酸モノメチルエステル 27.51g (153mmol)、塩化メチレン 50ml、塩化チォニル 12.5ml(171 mmol)、 DMF 0.5mlを室温で約 4· 5時間撹拌した。減圧濃縮した後 THF 100mlをカ卩えて酸クロリド溶液を得た。別の容器にて 1-メチルビラゾール 12.52g (152mmol)、 t_ブチルメチルエーテノレ 200mlの混合物を _70°Cに冷却し、 1.54M n_ブ チルリチウムへキサン溶液を滴下し約 1時間撹拌した。これに臭化亜鉛を加え約 30 分撹拌した。テトラキス (トリフエニルホスフィン)パラジウム (0)8.2 g(7.1 mmol)を _60°C にて加え、先に調整した酸クロリド溶液を加え徐々に室温まで温度上昇させ終夜放 置した。反応液に水、酢酸ェチルを加え分液し水層は酢酸ェチルで再抽出した。合 わせた有機層を飽和食塩水で洗浄後、無水硫酸ナトリウム乾燥しろ過そてから減圧 濃縮した。これをカラムクロマトグラフィー(へキサン:酢酸ェチル = 100 : 1→ 3 : 1) にて精製して得られた生成物を酢酸ェチル 5ml、へキサン 100mlの混合溶媒に懸 濁させ撹拌した。 0°Cに冷却後ろ取することにより、 2_(2-メチル -2H-ピラゾール -3-ィ ノレ)カルボニル安息香酸 メチルエステル 12.46g (収率 33%)を得た。
1H-NMR(CDC13): 3.71(3H, s), 4.32(3H, s), 6· 19(1Η, d), 7.38(1H, d), 7.48(1H, dd),
7.50-7.70(2H, m), 8.00 (1H, dd)。
b) 4-(2-メチル -2H-ピラゾール -3-ィル) -2H-フタラジン- 1-オン
実施例 94aにて得られたケトカルボン酸にヒドラジン 1水和物 2.7ml、エタノール 100mlを加え 7. 5時間加熱還流させた。析出した固体を冷却後にろ取しることによつ て 4-(2-メチル -2H-ピラゾール- 3 -ィル )_2H -フタラジン _1_オン 9.89g (収率 86%)を 固体として得た。 lH-NMR(DMSO-d6): 3.81(3H, s), 6.64(1H, d), 7.60— 7.70(2H, m), 7.80-8.00(2H, m), 8.34(1H, dd), 13.06 (1H, s)。
c) l_クロ口- 4-(2_メチル _2H -ピラゾール- 3_ィル)フタラジン
実施例 Idと同様にして実施例 lbで得られた化合物 5.05g (22.32mmol)とォキシ塩 化リン 5.2mlを用い、表題化合物 4.95g (収率 91%)を固体として得た。
1H-NMR(CDC13): 4.07(3H, s), 6.68(1H, d), 7.69(1H, d), 7.95-8.20(2H, m), 8.19(1H, d), 8.42(1H, d)。
d) l-[4-(2-メチル -2H-ピラゾール -3-ィル)フタラジン- 1-ィル]ピぺリジン- 4-オール 2塩酸塩
実施例 94cで得られた化合物 339mg(1.385mmol)、 4_ヒドロキシピペリジン
320mg(3.164 mmol)、 DMF 2mlを 150°Cにて約 8時間加熱撹拌した。溶媒を全て留 去し、アセトンに溶解させた後、 4M塩酸一酢酸ェチル溶液をカ卩えて析出した固体を ろ取すると、表題化合物 395mg (収率 75%)が得られた。 lH-NMR(DMSO_d6):
1.70-1.85 (2H, m), 1.95- 2.15(2H, m), 3.60- 3.75(2H, m), 3.88(3H, s),3.90_4.00(lH, m), 4.00-4.15(2H, m), 6.78(1H, d), 7.72(1H, d), 7.90— 8.05(1H, m), 8.05— 8·25(2Η, m), 8.35-8.45 (1H, m)。
(実施例 95) l-(2_メチル -2H -ピラゾール- 3_ィル)フタラジン
実施例 94cで得られた化合物 689mg (2.816 mmol)、トリェチルァミン
0.6ml(4.3mmol)、イソプロピルアルコール 12ml、 THF 6ml、 50%含水パラジウム炭素 (10%)約 60mgを水素雰囲気下、 40_50°Cにて約 8時間撹拌した。反応液にクロロホ ノレムをカロえ、セライト上でろ過し、減圧濃縮した。これをクロ口ホルムに溶解し、 1M水 酸化ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過、減圧濃縮す ると、 1_(2-メチル -2H -ピラゾール- 3 -ィル )_3,4 -ジヒドロ- 3H-フタラジン力 820mgの
粗生成物として得られた。 1H-NMR(CDC13): 3.94(3H, s), 4.30(2H, s), 6.22(1H, s), 6.40(1H, d), 7.10-7.20(2H, m), 7.20-7.50(2H, m), 7.54(1H, d)。
このうちの 393 mgにトルエン 5ml、活性化二酸化マンガン 0.35gを加え約 2時間 加熱還流した。反応液にクロ口ホルムをカ卩ぇセライト上でろ過し減圧濃縮した後に、 t- ブチルメチルエーテル 2ml、へキサン 10mlを加えて撹拌した。沈殿している固体を ろ取すると表題化合物が 279.2 mg (収率 72%)で得られた。 1H_NMR(CDC13): 4·08(3Η, s), 6·69(1Η, d), 7.69(1Η, d), 7.90-8.30(4Η, m), 9·56(1Η, s)。
(実施例 96) 1_ヒドラジノ _4_(1-メチル -1H-イミダゾール- 2_ィル)フタラジン 実施例 Idで得られた化合物 420 mg(1.717mmol)、イソプロピルアルコール 8ml、ヒ ドラジン 1水和物 1mlの混合物を室温にて約 4時間撹拌した。減圧濃縮した後、イソ プロピルアルコール 4mlをカ卩えて 0°Cにて 1時間撹拌した。固体をろ取することにより 表題化合物 297.3mg (収率 72%)を得た。 1H_NMR(CDC13): 3.91(3H, s), 7.07(1H, s), 7.27(1H, s), 7.60-7.75(2H, m), 7.90(1H, d), 8.60(1H, d)。
(実施例 97) N-メチル -N' -{3-メチル -2-(4-フエニルフタラジン- 1-ィル) -3H-イミダ ゾール -4-カルボキサミド}ピペラジン
実施例 25にて得られるカルボン酸 253mg (0.766mmol)に塩化メチレン 15ml、トリエ チルァミン 0.2ml (1.43mmol)を加え氷冷した。ここにクロ口蟻酸イソブチル 0.13 ml (1.00mmol)、を加え約 1時間撹拌した。さらに N—メチルビペラジン 0.1ml
(0.90mmol)を加えて室温にて撹拌した。反応液に水酸化ナトリウム水溶液を加えて分 液し、有機層を水酸化ナトリウム水溶液、飽和食塩水にて順次洗浄して無水硫酸ナト リウムにて乾燥した後、ろ過した。これを減圧濃縮して得られた黄色固体をエーテル で洗浄することによる表題化合物 153 mg (収率 49%)が得られた。
1H-NMR(CDC13): 2.38(3H, s), 2.40-2.60(m, 4H), 3.70_3·90(4Η, m), 4.11(3H, s), 7.44(1H, s), 7.50— 7.7— (3H, m), 7·70_8·00(4Η, m), 8.10_8.20(1H, m), 8.70— 8.90(1H, m)0
実施例 90から実施例 97に記載の化合物を以下に示す。
[化 26] 実施例 実施例 実施例
[0250] 実施例の化合物についての薬理試験結果を以下に記す。
[0251] (実験例 1) NAD(P)H oxidase抑制作用(vitro) (方法) ヒト臍帯静脈内皮細胞 (以下 、 HUVECと記す。 BioWhittaker社より購入)に 44mM Glucoseを添加し、高糖下約 1週 間培養し、界面活性剤を含む Lysis bufferに細胞を溶解させた。高糖下過剰発現又 は活性化した NAD(P)H oxidaseにより産生増加した Interleukin-8(以下、 IL-8と記す 。)を固相酵素免疫検定法 (ELISA= enzyme-linked immuno solvent assay)で測定し た。産生増加した IL-8量を指標に化合物の評価をおこなった。
[0252] 被験化合物を添加し高糖培養した細胞の IL-8産生を A、溶媒を添加し高糖培養し た細胞の IL-8産生を B、正常糖濃度で培養した細胞の IL-8産生を Cとし、以下のよう に抑制率を算出した。
[0253] 抑制率(%)= 100- ((A-C)/(B_C)) X 100
結果を下記表に記す。
[0254] [表 1]
[0255] (実験例 2) NAD(P)H oxidase抑制作用(vivo) (方法) Wistar系ラット (雄性、 日本 SLCより購入)の尾静脈より 0.05Mクェン酸バッファー (pH4.5)に溶解したストレプトゾト シン (以下、 STZと記す )40mg/kgを静脈内投与し、糖尿病ラットを作成した。 STZ静注 1 週間後以降に、尾静脈よりへパリン処理したキヤピラリーを用いて採血し、直ちに氷 冷後、 3000rpm, 15min, 4°Cで遠心分離して血清を得た。血糖値は、グルコース測定 用キッド GLUネオ シノテスド' (シノテスト社)を用いて酵素法で測定した。測定はマイ クロプレートリーダー (SPCTRAMAX250, MolecularDevices社)で行った。
[0256] 糖尿病ラットに 3日間化合物を 1日 1回経口投薬した。大動脈の · 0 -産生測定日に
2
は化合物は投薬しなかった。
[0257] NAD(P)H oxidase活性は大動脈の · 0 -産生を指標とし、 David.G.Harrisonらの方
2
法 (J.Clin.Invest.91, 2546-2551, 1993)を改変して行った。詳しい方法は以下の通り である。
[0258] 正常ラットおよび糖尿病ラットの腹部大動脈より放血後、胸部大動脈を摘出した。摘 出した大動脈を Krebs_H印 esbuffer中にいれ、周辺組織を除き、約 5mmのリング標本 を作製した。 Krebs-H印 esbuffer中にリング標本をいれ 37°C、 10分間プレインキュべ ートした後、リング標本を 0.25mM lucigeninを含む Krebs-H印 esbuffer中に移した。
500 μ Μの NADHを添加し、 chemiluminescence (ィヒ学発光数:単位 RLU: relativ elightunits)を、ルミノメーター (MULTト BIOLUMATLB9505C又は AutoLumatPlus,ヮ
ラック'ベルトールド社)を用いて 37°Cで 10分間測定した。測定時間 (横軸)に対し、 chemiluminescence (縦軸)をプロットした曲線の曲線下面積 (AUC)により評価した。 chemiluminescence測定後、血管リング標本の湿重量を測定し、 - 0 -産生量を標準
2
化した。
[0259] 投薬した糖尿病ラットの標準化した chemiluminescenceを A、投薬してレ、なレ、糖尿病 ラットの標準化した chemiluminescenceを B、正常ラットの標準化した
chemiluminescenceを Cとし、下記式により抑制率 (。/。)を算出した。
[0260] 抑制率 (%)= 100- ((A-C)/(B_C)) X 100
結果を下表に記す。
[0261] [表 2]
[0262] 本発明の縮合ピリダジン誘導体は NAD(P)H oxidase抑制作用を有し、当該酵素の 関与する疾患の予防又は治療に有効な医薬となりうる。
[0263] なお本出願は、 2004年 2月 24日付で出願された日本特許出願(特願 2004—047
129号)に基づいており、その全体が引用により援用される。