WO2000063171A1 - Composes tricycliques - Google Patents
Composes tricycliques Download PDFInfo
- Publication number
- WO2000063171A1 WO2000063171A1 PCT/JP2000/002573 JP0002573W WO0063171A1 WO 2000063171 A1 WO2000063171 A1 WO 2000063171A1 JP 0002573 W JP0002573 W JP 0002573W WO 0063171 A1 WO0063171 A1 WO 0063171A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- substituents
- alkyl group
- ring
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 558
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 189
- 125000002252 acyl group Chemical group 0.000 claims abstract description 76
- 150000003839 salts Chemical class 0.000 claims abstract description 69
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 55
- 101710151321 Melanostatin Proteins 0.000 claims abstract description 48
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 42
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 41
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 claims abstract description 40
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 38
- 125000003118 aryl group Chemical group 0.000 claims abstract description 30
- 125000002947 alkylene group Chemical group 0.000 claims abstract description 25
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims abstract description 23
- 201000010099 disease Diseases 0.000 claims abstract description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 20
- 239000004480 active ingredient Substances 0.000 claims abstract description 18
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 17
- 125000000304 alkynyl group Chemical group 0.000 claims abstract description 16
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims abstract description 15
- 230000001105 regulatory effect Effects 0.000 claims abstract description 13
- 239000001257 hydrogen Substances 0.000 claims abstract description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 8
- 125000001183 hydrocarbyl group Chemical group 0.000 claims abstract 7
- 102400000064 Neuropeptide Y Human genes 0.000 claims abstract 3
- 125000001424 substituent group Chemical group 0.000 claims description 192
- -1 phosphoryl group Chemical group 0.000 claims description 120
- 238000000034 method Methods 0.000 claims description 85
- 125000003545 alkoxy group Chemical group 0.000 claims description 66
- 125000005843 halogen group Chemical group 0.000 claims description 41
- 239000000126 substance Substances 0.000 claims description 39
- 125000005647 linker group Chemical group 0.000 claims description 36
- 125000000623 heterocyclic group Chemical group 0.000 claims description 33
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 33
- 239000003814 drug Substances 0.000 claims description 32
- 125000003282 alkyl amino group Chemical group 0.000 claims description 30
- 238000004519 manufacturing process Methods 0.000 claims description 30
- 125000003277 amino group Chemical group 0.000 claims description 29
- 239000012453 solvate Substances 0.000 claims description 23
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 17
- 125000005842 heteroatom Chemical group 0.000 claims description 16
- 125000004442 acylamino group Chemical group 0.000 claims description 15
- 229910052799 carbon Inorganic materials 0.000 claims description 14
- 125000003368 amide group Chemical group 0.000 claims description 13
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 13
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 12
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 11
- 125000004414 alkyl thio group Chemical group 0.000 claims description 11
- 125000004429 atom Chemical group 0.000 claims description 11
- 230000002265 prevention Effects 0.000 claims description 11
- 125000002993 cycloalkylene group Chemical group 0.000 claims description 10
- 206010012601 diabetes mellitus Diseases 0.000 claims description 10
- 235000012631 food intake Nutrition 0.000 claims description 9
- 241000124008 Mammalia Species 0.000 claims description 8
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 8
- 150000004677 hydrates Chemical class 0.000 claims description 8
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 7
- 208000035150 Hypercholesterolemia Diseases 0.000 claims description 7
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 7
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 7
- 230000037406 food intake Effects 0.000 claims description 7
- 239000003446 ligand Substances 0.000 claims description 7
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 6
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 claims description 6
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 6
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims description 6
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 6
- 150000001721 carbon Chemical group 0.000 claims description 6
- 125000002883 imidazolyl group Chemical group 0.000 claims description 6
- 125000004076 pyridyl group Chemical group 0.000 claims description 6
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 6
- 125000001425 triazolyl group Chemical group 0.000 claims description 6
- 125000005741 alkyl alkenyl group Chemical group 0.000 claims description 5
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 5
- 229910052717 sulfur Inorganic materials 0.000 claims description 5
- 125000004434 sulfur atom Chemical group 0.000 claims description 5
- 125000005120 alkyl cycloalkyl alkyl group Chemical group 0.000 claims description 4
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 claims description 4
- 125000002757 morpholinyl group Chemical group 0.000 claims description 4
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical group [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 3
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims description 3
- 235000005911 diet Nutrition 0.000 claims description 3
- 230000000378 dietary effect Effects 0.000 claims description 3
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 3
- 229910052726 zirconium Inorganic materials 0.000 claims description 3
- 241000282412 Homo Species 0.000 claims description 2
- 125000005083 alkoxyalkoxy group Chemical group 0.000 claims description 2
- 125000005157 alkyl carboxy group Chemical group 0.000 claims description 2
- 108050002826 Neuropeptide Y Receptor Proteins 0.000 claims 3
- 102000012301 Neuropeptide Y receptor Human genes 0.000 claims 3
- 229940126062 Compound A Drugs 0.000 claims 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 claims 1
- YZCKVEUIGOORGS-UHFFFAOYSA-N Hydrogen atom Chemical compound [H] YZCKVEUIGOORGS-UHFFFAOYSA-N 0.000 claims 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M acrylate group Chemical group C(C=C)(=O)[O-] NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 claims 1
- 125000006413 ring segment Chemical group 0.000 claims 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 abstract description 14
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 abstract description 8
- 239000003795 chemical substances by application Substances 0.000 abstract description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 abstract description 2
- 239000001301 oxygen Substances 0.000 abstract description 2
- 229910052760 oxygen Inorganic materials 0.000 abstract description 2
- 206010020710 Hyperphagia Diseases 0.000 abstract 1
- 230000036528 appetite Effects 0.000 abstract 1
- 235000019789 appetite Nutrition 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 218
- 238000003786 synthesis reaction Methods 0.000 description 156
- 230000015572 biosynthetic process Effects 0.000 description 154
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 151
- 238000005481 NMR spectroscopy Methods 0.000 description 129
- 238000006243 chemical reaction Methods 0.000 description 103
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 98
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 97
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 96
- 239000000243 solution Substances 0.000 description 80
- 239000000203 mixture Substances 0.000 description 74
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 67
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 63
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 57
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 57
- 239000002904 solvent Substances 0.000 description 52
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 51
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 49
- 239000012044 organic layer Substances 0.000 description 48
- 102100028427 Pro-neuropeptide Y Human genes 0.000 description 45
- 238000010898 silica gel chromatography Methods 0.000 description 44
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 41
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 37
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 33
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 32
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 31
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 30
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 28
- 239000007864 aqueous solution Substances 0.000 description 28
- 108020003175 receptors Proteins 0.000 description 28
- 102000005962 receptors Human genes 0.000 description 27
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 26
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- ABRVLXLNVJHDRQ-UHFFFAOYSA-N [2-pyridin-3-yl-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound FC(C1=CC(=CC(=N1)C=1C=NC=CC=1)CN)(F)F ABRVLXLNVJHDRQ-UHFFFAOYSA-N 0.000 description 24
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 24
- 239000003960 organic solvent Substances 0.000 description 22
- 239000002585 base Substances 0.000 description 21
- 238000003756 stirring Methods 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 19
- 239000003480 eluent Substances 0.000 description 18
- 150000002430 hydrocarbons Chemical group 0.000 description 18
- 229920006395 saturated elastomer Polymers 0.000 description 18
- 238000012360 testing method Methods 0.000 description 18
- 101710198055 Neuropeptide Y receptor type 5 Proteins 0.000 description 17
- 102100029549 Neuropeptide Y receptor type 5 Human genes 0.000 description 17
- 125000006239 protecting group Chemical group 0.000 description 17
- 229940125904 compound 1 Drugs 0.000 description 16
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 16
- 239000000706 filtrate Substances 0.000 description 15
- 230000035484 reaction time Effects 0.000 description 15
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- 238000001914 filtration Methods 0.000 description 14
- 238000009833 condensation Methods 0.000 description 13
- 230000005494 condensation Effects 0.000 description 13
- 235000017557 sodium bicarbonate Nutrition 0.000 description 13
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 12
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 12
- 229910000027 potassium carbonate Inorganic materials 0.000 description 12
- 239000002994 raw material Substances 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 11
- ZEEBGORNQSEQBE-UHFFFAOYSA-N [2-(3-phenylphenoxy)-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound C1(=CC(=CC=C1)OC1=NC(=CC(=C1)CN)C(F)(F)F)C1=CC=CC=C1 ZEEBGORNQSEQBE-UHFFFAOYSA-N 0.000 description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 11
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 10
- MZSAMHOCTRNOIZ-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-phenylaniline Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(NC2=CC=CC=C2)C=CC=1 MZSAMHOCTRNOIZ-UHFFFAOYSA-N 0.000 description 10
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 10
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- SAHIZENKTPRYSN-UHFFFAOYSA-N [2-[3-(phenoxymethyl)phenoxy]-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound O(C1=CC=CC=C1)CC=1C=C(OC2=NC(=CC(=C2)CN)C(F)(F)F)C=CC=1 SAHIZENKTPRYSN-UHFFFAOYSA-N 0.000 description 9
- 238000001704 evaporation Methods 0.000 description 9
- 239000002244 precipitate Substances 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- 208000008589 Obesity Diseases 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 238000006482 condensation reaction Methods 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 235000020824 obesity Nutrition 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 7
- 239000012153 distilled water Substances 0.000 description 7
- 239000011259 mixed solution Substances 0.000 description 7
- 125000002950 monocyclic group Chemical group 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- 238000002360 preparation method Methods 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- 208000032841 Bulimia Diseases 0.000 description 6
- 206010012289 Dementia Diseases 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 206010028980 Neoplasm Diseases 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 230000005856 abnormality Effects 0.000 description 6
- 150000001413 amino acids Chemical class 0.000 description 6
- 208000022531 anorexia Diseases 0.000 description 6
- VHRGRCVQAFMJIZ-UHFFFAOYSA-N cadaverine Chemical compound NCCCCCN VHRGRCVQAFMJIZ-UHFFFAOYSA-N 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 6
- 206010061428 decreased appetite Diseases 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 206010015037 epilepsy Diseases 0.000 description 6
- 230000003054 hormonal effect Effects 0.000 description 6
- 208000030159 metabolic disease Diseases 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- GIAFURWZWWWBQT-UHFFFAOYSA-N 2-(2-aminoethoxy)ethanol Chemical compound NCCOCCO GIAFURWZWWWBQT-UHFFFAOYSA-N 0.000 description 5
- LCZAYEFIEDNEOV-UHFFFAOYSA-N 9-ethyl-4-methoxycarbazole Chemical compound C1=CC=C2N(CC)C3=CC=CC=C3C2=C1OC LCZAYEFIEDNEOV-UHFFFAOYSA-N 0.000 description 5
- SAIKVEAWIXLGIN-UHFFFAOYSA-N 9-propan-2-yl-5,6,7,8-tetrahydrocarbazol-3-amine Chemical compound C12=CC(N)=CC=C2N(C(C)C)C2=C1CCCC2 SAIKVEAWIXLGIN-UHFFFAOYSA-N 0.000 description 5
- 206010006550 Bulimia nervosa Diseases 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 229940024606 amino acid Drugs 0.000 description 5
- 235000001014 amino acid Nutrition 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 5
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 5
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 5
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 5
- 239000008194 pharmaceutical composition Substances 0.000 description 5
- 108090000765 processed proteins & peptides Proteins 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 5
- AFBBKYQYNPNMAT-UHFFFAOYSA-N 1h-1,2,4-triazol-1-ium-3-thiolate Chemical compound SC=1N=CNN=1 AFBBKYQYNPNMAT-UHFFFAOYSA-N 0.000 description 4
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 150000003863 ammonium salts Chemical class 0.000 description 4
- 150000001540 azides Chemical class 0.000 description 4
- 230000027455 binding Effects 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 229940126214 compound 3 Drugs 0.000 description 4
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical compound C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 description 4
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 4
- 239000012280 lithium aluminium hydride Substances 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 4
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 4
- AHWALFGBDFAJAI-UHFFFAOYSA-N phenyl carbonochloridate Chemical compound ClC(=O)OC1=CC=CC=C1 AHWALFGBDFAJAI-UHFFFAOYSA-N 0.000 description 4
- 229910052700 potassium Inorganic materials 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- 102000004196 processed proteins & peptides Human genes 0.000 description 4
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- 229940124530 sulfonamide Drugs 0.000 description 4
- 150000003456 sulfonamides Chemical class 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- INQJZLMMPOYKCF-UHFFFAOYSA-N 6-nitro-9-propan-2-yl-1,2,3,4-tetrahydrocarbazole Chemical compound C12=CC([N+]([O-])=O)=CC=C2N(C(C)C)C2=C1CCCC2 INQJZLMMPOYKCF-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 101100515517 Arabidopsis thaliana XI-I gene Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical group C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 3
- 101000633401 Homo sapiens Neuropeptide Y receptor type 5 Proteins 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- REAYFGLASQTHKB-UHFFFAOYSA-N [2-[3-(1H-pyrazol-4-yl)phenoxy]-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound N1N=CC(=C1)C=1C=C(OC2=NC(=CC(=C2)CN)C(F)(F)F)C=CC=1 REAYFGLASQTHKB-UHFFFAOYSA-N 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 210000004727 amygdala Anatomy 0.000 description 3
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 210000003169 central nervous system Anatomy 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- WBKFWQBXFREOFH-UHFFFAOYSA-N dichloromethane;ethyl acetate Chemical compound ClCCl.CCOC(C)=O WBKFWQBXFREOFH-UHFFFAOYSA-N 0.000 description 3
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 3
- 230000020595 eating behavior Effects 0.000 description 3
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 3
- 125000004372 methylthioethyl group Chemical group [H]C([H])([H])SC([H])([H])C([H])([H])* 0.000 description 3
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 3
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 125000002971 oxazolyl group Chemical group 0.000 description 3
- FRTSLTNCECJJDA-UHFFFAOYSA-N phenyl n-(9-propan-2-yl-5,6,7,8-tetrahydrocarbazol-3-yl)carbamate Chemical compound C=1C=C2N(C(C)C)C=3CCCCC=3C2=CC=1NC(=O)OC1=CC=CC=C1 FRTSLTNCECJJDA-UHFFFAOYSA-N 0.000 description 3
- 125000004193 piperazinyl group Chemical group 0.000 description 3
- 238000012746 preparative thin layer chromatography Methods 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 125000000335 thiazolyl group Chemical group 0.000 description 3
- 125000004149 thio group Chemical group *S* 0.000 description 3
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- ZOBPZXTWZATXDG-UHFFFAOYSA-N 1,3-thiazolidine-2,4-dione Chemical group O=C1CSC(=O)N1 ZOBPZXTWZATXDG-UHFFFAOYSA-N 0.000 description 2
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 2
- FYELSNVLZVIGTI-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-5-ethylpyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1CC)CC(=O)N1CC2=C(CC1)NN=N2 FYELSNVLZVIGTI-UHFFFAOYSA-N 0.000 description 2
- UVHFABZTBXPFPC-UHFFFAOYSA-N 2-ethoxy-6,7,8,9-tetrahydro-5h-carbazole-3-carboxylic acid Chemical compound C1CCCC2=C1NC1=C2C=C(C(O)=O)C(OCC)=C1 UVHFABZTBXPFPC-UHFFFAOYSA-N 0.000 description 2
- RJPVXQXPKDRSIA-UHFFFAOYSA-N 2-nitro-1,2,3,4,4a,5-hexahydrocyclohepta[b]indole Chemical compound N1C2=CC=CC=CC2=C2C1CCC([N+](=O)[O-])C2 RJPVXQXPKDRSIA-UHFFFAOYSA-N 0.000 description 2
- WDGCBNTXZHJTHJ-UHFFFAOYSA-N 2h-1,3-oxazol-2-id-4-one Chemical group O=C1CO[C-]=N1 WDGCBNTXZHJTHJ-UHFFFAOYSA-N 0.000 description 2
- CJYDQTAWSHWBIT-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-(2-hydroxy-2-methylpropyl)benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCC(C)(C)O)C=CC=1 CJYDQTAWSHWBIT-UHFFFAOYSA-N 0.000 description 2
- GRFNBEZIAWKNCO-UHFFFAOYSA-N 3-pyridinol Chemical compound OC1=CC=CN=C1 GRFNBEZIAWKNCO-UHFFFAOYSA-N 0.000 description 2
- BLFRQYKZFKYQLO-UHFFFAOYSA-N 4-aminobutan-1-ol Chemical compound NCCCCO BLFRQYKZFKYQLO-UHFFFAOYSA-N 0.000 description 2
- VGVHNLRUAMRIEW-UHFFFAOYSA-N 4-methylcyclohexan-1-one Chemical compound CC1CCC(=O)CC1 VGVHNLRUAMRIEW-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- JJMDCOVWQOJGCB-UHFFFAOYSA-N 5-aminopentanoic acid Chemical compound [NH3+]CCCCC([O-])=O JJMDCOVWQOJGCB-UHFFFAOYSA-N 0.000 description 2
- OXEUETBFKVCRNP-UHFFFAOYSA-N 9-ethyl-3-carbazolamine Chemical compound NC1=CC=C2N(CC)C3=CC=CC=C3C2=C1 OXEUETBFKVCRNP-UHFFFAOYSA-N 0.000 description 2
- VJFOQHMJEZRHHJ-UHFFFAOYSA-N 9-ethyl-5,6,7,8-tetrahydrocarbazole-3-carboxylic acid Chemical compound C12=CC(C(O)=O)=CC=C2N(CC)C2=C1CCCC2 VJFOQHMJEZRHHJ-UHFFFAOYSA-N 0.000 description 2
- VAQTVYMSLBECQB-UHFFFAOYSA-N 9-ethylcarbazol-4-ol Chemical compound C(C)N1C2=CC=CC=C2C=2C(=CC=CC1=2)O VAQTVYMSLBECQB-UHFFFAOYSA-N 0.000 description 2
- QZQNANUJNRAGPZ-UHFFFAOYSA-N 9-ethylcarbazole-3-carboxylic acid Chemical compound OC(=O)C1=CC=C2N(CC)C3=CC=CC=C3C2=C1 QZQNANUJNRAGPZ-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- YNXLOPYTAAFMTN-SBUIBGKBSA-N C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 Chemical compound C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 YNXLOPYTAAFMTN-SBUIBGKBSA-N 0.000 description 2
- 235000008733 Citrus aurantifolia Nutrition 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 208000010412 Glaucoma Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 206010020772 Hypertension Diseases 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 2
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 2
- 108700020479 Pancreatic hormone Proteins 0.000 description 2
- 102100029909 Peptide YY Human genes 0.000 description 2
- 108010088847 Peptide YY Proteins 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 235000011941 Tilia x europaea Nutrition 0.000 description 2
- 206010047163 Vasospasm Diseases 0.000 description 2
- 239000006096 absorbing agent Substances 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 230000006229 amino acid addition Effects 0.000 description 2
- 125000002431 aminoalkoxy group Chemical group 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O ammonium group Chemical group [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 230000003042 antagnostic effect Effects 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 125000005129 aryl carbonyl group Chemical group 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 210000004204 blood vessel Anatomy 0.000 description 2
- YAGCIXJCAUGCGI-UHFFFAOYSA-N butoxycarbonyl butyl carbonate Chemical compound CCCCOC(=O)OC(=O)OCCCC YAGCIXJCAUGCGI-UHFFFAOYSA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 210000003710 cerebral cortex Anatomy 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 210000003890 endocrine cell Anatomy 0.000 description 2
- AULAUQBCOBPZTQ-UHFFFAOYSA-N ethyl 2-ethoxy-6,7,8,9-tetrahydro-5h-carbazole-3-carboxylate Chemical compound C1CCCC2=C1C(C=C(C(=C1)OCC)C(=O)OCC)=C1N2 AULAUQBCOBPZTQ-UHFFFAOYSA-N 0.000 description 2
- ZVPZZSPBUHJPKI-UHFFFAOYSA-N ethyl 4-ethoxy-6,7,8,9-tetrahydro-5h-carbazole-3-carboxylate Chemical compound C1CCCC2=C1NC1=CC=C(C(=O)OCC)C(OCC)=C12 ZVPZZSPBUHJPKI-UHFFFAOYSA-N 0.000 description 2
- LBKPGNUOUPTQKA-UHFFFAOYSA-N ethyl n-phenylcarbamate Chemical compound CCOC(=O)NC1=CC=CC=C1 LBKPGNUOUPTQKA-UHFFFAOYSA-N 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000003205 fragrance Substances 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- WJRBRSLFGCUECM-UHFFFAOYSA-N hydantoin Chemical group O=C1CNC(=O)N1 WJRBRSLFGCUECM-UHFFFAOYSA-N 0.000 description 2
- 150000007857 hydrazones Chemical class 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000002198 insoluble material Substances 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- 210000003127 knee Anatomy 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 239000004571 lime Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 229960002748 norepinephrine Drugs 0.000 description 2
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- 210000001428 peripheral nervous system Anatomy 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 2
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 239000012286 potassium permanganate Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- SSOLNOMRVKKSON-UHFFFAOYSA-N proguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C=C1 SSOLNOMRVKKSON-UHFFFAOYSA-N 0.000 description 2
- 125000002294 quinazolinyl group Chemical class N1=C(N=CC2=CC=CC=C12)* 0.000 description 2
- 125000005493 quinolyl group Chemical group 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- VWDWKYIASSYTQR-UHFFFAOYSA-N sodium nitrate Chemical compound [Na+].[O-][N+]([O-])=O VWDWKYIASSYTQR-UHFFFAOYSA-N 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- IBFKCAFDQSYGFJ-UHFFFAOYSA-N tert-butyl n-(1-hydroxybutyl)-n-methylcarbamate Chemical compound CCCC(O)N(C)C(=O)OC(C)(C)C IBFKCAFDQSYGFJ-UHFFFAOYSA-N 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 125000005505 thiomorpholino group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- BZVJOYBTLHNRDW-UHFFFAOYSA-N triphenylmethanamine Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(N)C1=CC=CC=C1 BZVJOYBTLHNRDW-UHFFFAOYSA-N 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 125000001834 xanthenyl group Chemical class C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- ZWHOTPNCEFWATE-CQSZACIVSA-N (3R)-3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-phenylpyrrolidine-1-carboxamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)O[C@H]1CN(CC1)C(=O)NC1=CC=CC=C1 ZWHOTPNCEFWATE-CQSZACIVSA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- UOAJUPONZOXFNX-UHFFFAOYSA-N (4-methylphenyl)sulfonyl acetate Chemical compound CC(=O)OS(=O)(=O)C1=CC=C(C)C=C1 UOAJUPONZOXFNX-UHFFFAOYSA-N 0.000 description 1
- UOCLXMDMGBRAIB-UHFFFAOYSA-N 1,1,1-trichloroethane Chemical compound CC(Cl)(Cl)Cl UOCLXMDMGBRAIB-UHFFFAOYSA-N 0.000 description 1
- OXFSTTJBVAAALW-UHFFFAOYSA-N 1,3-dihydroimidazole-2-thione Chemical compound SC1=NC=CN1 OXFSTTJBVAAALW-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- JPIGSMKDJQPHJC-UHFFFAOYSA-N 1-(2-aminoethoxy)ethanol Chemical compound CC(O)OCCN JPIGSMKDJQPHJC-UHFFFAOYSA-N 0.000 description 1
- KHKQQQLOLBEJES-UHFFFAOYSA-N 1-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]pyridin-2-one Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)N1C(C=CC=C1)=O KHKQQQLOLBEJES-UHFFFAOYSA-N 0.000 description 1
- HHCLNZBCCQDVOQ-UHFFFAOYSA-N 1-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]pyrazol-3-yl]methyl]piperazin-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C(=NN(C=1)CC(N1CC2=C(CC1)NN=N2)=O)CN1C(CNCC1)=O HHCLNZBCCQDVOQ-UHFFFAOYSA-N 0.000 description 1
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 1
- BTUGGGLMQBJCBN-UHFFFAOYSA-N 1-iodo-2-methylpropane Chemical compound CC(C)CI BTUGGGLMQBJCBN-UHFFFAOYSA-N 0.000 description 1
- 125000005955 1H-indazolyl group Chemical group 0.000 description 1
- CRZDNISJUXVSKX-UHFFFAOYSA-N 1h-imidazol-2-ylmethanamine Chemical compound NCC1=NC=CN1 CRZDNISJUXVSKX-UHFFFAOYSA-N 0.000 description 1
- HNVIQLPOGUDBSU-UHFFFAOYSA-N 2,6-dimethylmorpholine Chemical compound CC1CNCC(C)O1 HNVIQLPOGUDBSU-UHFFFAOYSA-N 0.000 description 1
- JONTXEXBTWSUKE-UHFFFAOYSA-N 2-(2-aminoethylsulfanyl)ethanamine Chemical compound NCCSCCN JONTXEXBTWSUKE-UHFFFAOYSA-N 0.000 description 1
- KKFDCBRMNNSAAW-UHFFFAOYSA-N 2-(morpholin-4-yl)ethanol Chemical compound OCCN1CCOCC1 KKFDCBRMNNSAAW-UHFFFAOYSA-N 0.000 description 1
- WOXFMYVTSLAQMO-UHFFFAOYSA-N 2-Pyridinemethanamine Chemical compound NCC1=CC=CC=N1 WOXFMYVTSLAQMO-UHFFFAOYSA-N 0.000 description 1
- APDYPEOKIUKUQV-UHFFFAOYSA-N 2-[1-(2-oxo-2-piperidin-1-ylethyl)indol-4-yl]oxy-6-(trifluoromethyl)pyridine-4-carbonitrile Chemical compound O=C(CN1C=CC2=C(C=CC=C12)OC=1C=C(C#N)C=C(N=1)C(F)(F)F)N1CCCCC1 APDYPEOKIUKUQV-UHFFFAOYSA-N 0.000 description 1
- UJORPFQWUKFXIE-UHFFFAOYSA-N 2-[1-[(4-ethoxy-2,6-difluorophenyl)methyl]-5,6-dihydro-4h-cyclopenta[c]pyrazol-3-yl]-5-methoxy-n-pyridin-4-ylpyrimidin-4-amine Chemical compound FC1=CC(OCC)=CC(F)=C1CN1C(CCC2)=C2C(C=2N=C(NC=3C=CN=CC=3)C(OC)=CN=2)=N1 UJORPFQWUKFXIE-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- ZRPAUEVGEGEPFQ-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2 ZRPAUEVGEGEPFQ-UHFFFAOYSA-N 0.000 description 1
- JVKRKMWZYMKVTQ-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-N-(2-oxo-3H-1,3-benzoxazol-6-yl)acetamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1)CC(=O)NC1=CC2=C(NC(O2)=O)C=C1 JVKRKMWZYMKVTQ-UHFFFAOYSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- IBZKBSXREAQDTO-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)ethanamine Chemical compound COCCNCCOC IBZKBSXREAQDTO-UHFFFAOYSA-N 0.000 description 1
- HDECRAPHCDXMIJ-UHFFFAOYSA-N 2-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC=C1S(Cl)(=O)=O HDECRAPHCDXMIJ-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- WKHXVTIIUHSRJX-UHFFFAOYSA-N 2-nitrocyclohex-2-en-1-ol Chemical compound OC1CCCC=C1[N+]([O-])=O WKHXVTIIUHSRJX-UHFFFAOYSA-N 0.000 description 1
- PTHDBHDZSMGHKF-UHFFFAOYSA-N 2-piperidin-2-ylethanol Chemical compound OCCC1CCCCN1 PTHDBHDZSMGHKF-UHFFFAOYSA-N 0.000 description 1
- LDSQQXKSEFZAPE-UHFFFAOYSA-N 2-piperidin-4-ylethanol Chemical compound OCCC1CCNCC1 LDSQQXKSEFZAPE-UHFFFAOYSA-N 0.000 description 1
- ZSLUVFAKFWKJRC-IGMARMGPSA-N 232Th Chemical compound [232Th] ZSLUVFAKFWKJRC-IGMARMGPSA-N 0.000 description 1
- ULRPISSMEBPJLN-UHFFFAOYSA-N 2h-tetrazol-5-amine Chemical compound NC1=NN=NN1 ULRPISSMEBPJLN-UHFFFAOYSA-N 0.000 description 1
- HQNOODJDSFSURF-UHFFFAOYSA-N 3-(1h-imidazol-2-yl)propan-1-amine Chemical compound NCCCC1=NC=CN1 HQNOODJDSFSURF-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- MROVZCRMXJZHCN-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-(2-hydroxyethyl)benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCCO)C=CC=1 MROVZCRMXJZHCN-UHFFFAOYSA-N 0.000 description 1
- SHBHYINHXNTBRP-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-(2-methylsulfonylethyl)benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCCS(=O)(=O)C)C=CC=1 SHBHYINHXNTBRP-UHFFFAOYSA-N 0.000 description 1
- GDSLUYKCPYECNN-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-[(4-fluorophenyl)methyl]benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCC2=CC=C(C=C2)F)C=CC=1 GDSLUYKCPYECNN-UHFFFAOYSA-N 0.000 description 1
- FJPUKTJEFOXMJX-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-[1-(hydroxymethyl)cyclopropyl]benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NC2(CC2)CO)C=CC=1 FJPUKTJEFOXMJX-UHFFFAOYSA-N 0.000 description 1
- ZUNFPBNHELLPPP-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-[2-(dimethylamino)ethyl]benzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NCCN(C)C)C=CC=1 ZUNFPBNHELLPPP-UHFFFAOYSA-N 0.000 description 1
- HAEQAUJYNHQVHV-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-phenylbenzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NC2=CC=CC=C2)C=CC=1 HAEQAUJYNHQVHV-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- PVNNOLUAMRODAC-UHFFFAOYSA-N 4-(ethylamino)butan-1-ol Chemical compound CCNCCCCO PVNNOLUAMRODAC-UHFFFAOYSA-N 0.000 description 1
- DBKSSENEKWOVKL-UHFFFAOYSA-N 4-(methylamino)butan-1-ol Chemical compound CNCCCCO DBKSSENEKWOVKL-UHFFFAOYSA-N 0.000 description 1
- XGYKKVTZDQDYJQ-UHFFFAOYSA-N 4-aminobutanenitrile Chemical compound NCCCC#N XGYKKVTZDQDYJQ-UHFFFAOYSA-N 0.000 description 1
- IMLXLGZJLAOKJN-UHFFFAOYSA-N 4-aminocyclohexan-1-ol Chemical compound NC1CCC(O)CC1 IMLXLGZJLAOKJN-UHFFFAOYSA-N 0.000 description 1
- GSDQYSSLIKJJOG-UHFFFAOYSA-N 4-chloro-2-(3-chloroanilino)benzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1NC1=CC=CC(Cl)=C1 GSDQYSSLIKJJOG-UHFFFAOYSA-N 0.000 description 1
- 125000000242 4-chlorobenzoyl group Chemical group ClC1=CC=C(C(=O)*)C=C1 0.000 description 1
- WUXBUDFYYIHRLC-UHFFFAOYSA-N 4-ethoxy-6,7,8,9-tetrahydro-5h-carbazole-3-carboxylic acid Chemical compound C1CCCC2=C1NC1=C2C(OCC)=C(C(O)=O)C=C1 WUXBUDFYYIHRLC-UHFFFAOYSA-N 0.000 description 1
- SIEMSXQGGVJXOK-UHFFFAOYSA-N 4-ethoxy-9-ethylcarbazole-3-carboxylic acid Chemical compound CCN1C2=CC=CC=C2C2=C1C=CC(C(O)=O)=C2OCC SIEMSXQGGVJXOK-UHFFFAOYSA-N 0.000 description 1
- JRTLLYUWSKIWBR-UHFFFAOYSA-N 4-hydrazinyl-2-hydroxybenzoic acid Chemical compound NNC1=CC=C(C(O)=O)C(O)=C1 JRTLLYUWSKIWBR-UHFFFAOYSA-N 0.000 description 1
- GCNTZFIIOFTKIY-UHFFFAOYSA-N 4-hydroxypyridine Chemical compound OC1=CC=NC=C1 GCNTZFIIOFTKIY-UHFFFAOYSA-N 0.000 description 1
- XADCKKKOYZJNAR-UHFFFAOYSA-N 4-methoxycyclohexan-1-one Chemical compound COC1CCC(=O)CC1 XADCKKKOYZJNAR-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- FUFZNHHSSMCXCZ-UHFFFAOYSA-N 5-piperidin-4-yl-3-[3-(trifluoromethyl)phenyl]-1,2,4-oxadiazole Chemical compound FC(F)(F)C1=CC=CC(C=2N=C(ON=2)C2CCNCC2)=C1 FUFZNHHSSMCXCZ-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- RUFDYIJGNPVTAY-UHFFFAOYSA-N 6-[(2-methylpropan-2-yl)oxycarbonylamino]hexanoic acid Chemical compound CC(C)(C)OC(=O)NCCCCCC(O)=O RUFDYIJGNPVTAY-UHFFFAOYSA-N 0.000 description 1
- HCHXCJKIVMIBAD-UHFFFAOYSA-N 6-amino-n-(9-ethylcarbazol-1-yl)hexanamide Chemical compound C1=CC(NC(=O)CCCCCN)=C2N(CC)C3=CC=CC=C3C2=C1 HCHXCJKIVMIBAD-UHFFFAOYSA-N 0.000 description 1
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- NRLQBVLOUUPAMI-UHFFFAOYSA-N 8-[3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxybenzoyl]-1-oxa-3,8-diazaspiro[4.5]decan-2-one Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)N2CCC3(CNC(O3)=O)CC2)C=CC=1 NRLQBVLOUUPAMI-UHFFFAOYSA-N 0.000 description 1
- PLAZXGNBGZYJSA-UHFFFAOYSA-N 9-ethylcarbazole Chemical compound C1=CC=C2N(CC)C3=CC=CC=C3C2=C1 PLAZXGNBGZYJSA-UHFFFAOYSA-N 0.000 description 1
- QGJXVBICNCIWEL-UHFFFAOYSA-N 9-ethylcarbazole-3-carbaldehyde Chemical compound O=CC1=CC=C2N(CC)C3=CC=CC=C3C2=C1 QGJXVBICNCIWEL-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- IYHHRZBKXXKDDY-UHFFFAOYSA-N BI-605906 Chemical compound N=1C=2SC(C(N)=O)=C(N)C=2C(C(F)(F)CC)=CC=1N1CCC(S(C)(=O)=O)CC1 IYHHRZBKXXKDDY-UHFFFAOYSA-N 0.000 description 1
- 108010001478 Bacitracin Proteins 0.000 description 1
- 206010004716 Binge eating Diseases 0.000 description 1
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- RENMDAKOXSCIGH-UHFFFAOYSA-N Chloroacetonitrile Chemical compound ClCC#N RENMDAKOXSCIGH-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 1
- 101800000414 Corticotropin Proteins 0.000 description 1
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 1
- 241001440267 Cyclodes Species 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- YIIMEMSDCNDGTB-UHFFFAOYSA-N Dimethylcarbamoyl chloride Chemical compound CN(C)C(Cl)=O YIIMEMSDCNDGTB-UHFFFAOYSA-N 0.000 description 1
- 241000255925 Diptera Species 0.000 description 1
- TVZNFYXAPXOARC-UHFFFAOYSA-N Dl-norleucine methyl ester Chemical compound CCCCC(N)C(=O)OC TVZNFYXAPXOARC-UHFFFAOYSA-N 0.000 description 1
- UNBMQQNYLCPCHS-UHFFFAOYSA-N Ebelactone B Natural products CCC(C)C(O)C(C)C(=O)C(C)C=C(C)CC(C)C1OC(=O)C1CC UNBMQQNYLCPCHS-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101000652582 Homo sapiens Antigen peptide transporter 2 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102000016267 Leptin Human genes 0.000 description 1
- 108010092277 Leptin Proteins 0.000 description 1
- 102000009151 Luteinizing Hormone Human genes 0.000 description 1
- 108010073521 Luteinizing Hormone Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- MKYBYDHXWVHEJW-UHFFFAOYSA-N N-[1-oxo-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propan-2-yl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(C(C)NC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 MKYBYDHXWVHEJW-UHFFFAOYSA-N 0.000 description 1
- QCOGKXLOEWLIDC-UHFFFAOYSA-N N-methylbutylamine Chemical compound CCCCNC QCOGKXLOEWLIDC-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 1
- 102100038991 Neuropeptide Y receptor type 2 Human genes 0.000 description 1
- 101710197945 Neuropeptide Y receptor type 2 Proteins 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229930184840 Phorbazole Natural products 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical group OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 1
- 102000003946 Prolactin Human genes 0.000 description 1
- 108010057464 Prolactin Proteins 0.000 description 1
- WUGQZFFCHPXWKQ-UHFFFAOYSA-N Propanolamine Chemical compound NCCCO WUGQZFFCHPXWKQ-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 101500025021 Rattus norvegicus Neuropeptide Y Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229910052776 Thorium Inorganic materials 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- GYDJEQRTZSCIOI-UHFFFAOYSA-N Tranexamic acid Chemical compound NCC1CCC(C(O)=O)CC1 GYDJEQRTZSCIOI-UHFFFAOYSA-N 0.000 description 1
- 206010047139 Vasoconstriction Diseases 0.000 description 1
- GXBMIBRIOWHPDT-UHFFFAOYSA-N Vasopressin Natural products N1C(=O)C(CC=2C=C(O)C=CC=2)NC(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CCCN=C(N)N)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C1CC1=CC=CC=C1 GXBMIBRIOWHPDT-UHFFFAOYSA-N 0.000 description 1
- 102000002852 Vasopressins Human genes 0.000 description 1
- 108010004977 Vasopressins Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- HVVNJUAVDAZWCB-YFKPBYRVSA-N [(2s)-pyrrolidin-2-yl]methanol Chemical compound OC[C@@H]1CCCN1 HVVNJUAVDAZWCB-YFKPBYRVSA-N 0.000 description 1
- RWQJLIWMOBYOTI-AWEZNQCLSA-N [(3S)-3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxypiperidin-1-yl]-pyridin-3-ylmethanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)O[C@@H]1CN(CCC1)C(=O)C=1C=NC=CC=1 RWQJLIWMOBYOTI-AWEZNQCLSA-N 0.000 description 1
- QDPOOGQUCJJZAO-FCXRPNKRSA-N [(E)-3-(3,4-dihydroxyphenyl)prop-2-enoyl] (E)-3-(3,4-dihydroxyphenyl)prop-2-enoate Chemical compound C1=C(O)C(O)=CC=C1\C=C\C(=O)OC(=O)\C=C\C1=CC=C(O)C(O)=C1 QDPOOGQUCJJZAO-FCXRPNKRSA-N 0.000 description 1
- JOSCNYCOYXTLTN-GFCCVEGCSA-N [3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxyphenyl]-[(3R)-3-(hydroxymethyl)pyrrolidin-1-yl]methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=CC=1)C(=O)N1C[C@@H](CC1)CO JOSCNYCOYXTLTN-GFCCVEGCSA-N 0.000 description 1
- LJHFUFVRZNYVMK-CYBMUJFWSA-N [3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxyphenyl]-[(3R)-3-hydroxypyrrolidin-1-yl]methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=CC=1)C(=O)N1C[C@@H](CC1)O LJHFUFVRZNYVMK-CYBMUJFWSA-N 0.000 description 1
- KDSYNTPPPISIJB-UHFFFAOYSA-N [3-[[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxymethyl]phenyl]-(3-fluoro-4-hydroxypyrrolidin-1-yl)methanone Chemical compound NCc1cc(OCc2cccc(c2)C(=O)N2CC(O)C(F)C2)nc(c1)C(F)(F)F KDSYNTPPPISIJB-UHFFFAOYSA-N 0.000 description 1
- UGKIKJFPXNOHHA-UHFFFAOYSA-N [5-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxypyridin-3-yl]-(3-fluoro-4-hydroxypyrrolidin-1-yl)methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=NC=1)C(=O)N1CC(C(C1)O)F UGKIKJFPXNOHHA-UHFFFAOYSA-N 0.000 description 1
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- NGCGMRBZPXEPOZ-HBBGHHHDSA-N acetic acid;(2s)-n-[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2s)-1-[[2-[[(2s)-1-[[(2s)-1-[(2s)-2-[(2-amino-2-oxoethyl)carbamoyl]pyrrolidin-1-yl]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-2-oxoethyl]amino]-3-(4-hydroxyphenyl)- Chemical compound CC(O)=O.C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 NGCGMRBZPXEPOZ-HBBGHHHDSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 238000005904 alkaline hydrolysis reaction Methods 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004471 alkyl aminosulfonyl group Chemical group 0.000 description 1
- 125000005094 alkyl carbonyl amino alkyl group Chemical group 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- 150000003927 aminopyridines Chemical class 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 210000003403 autonomic nervous system Anatomy 0.000 description 1
- ZSIQJIWKELUFRJ-UHFFFAOYSA-N azepane Chemical compound C1CCCNCC1 ZSIQJIWKELUFRJ-UHFFFAOYSA-N 0.000 description 1
- 229960003071 bacitracin Drugs 0.000 description 1
- 229930184125 bacitracin Natural products 0.000 description 1
- CLKOFPXJLQSYAH-ABRJDSQDSA-N bacitracin A Chemical compound C1SC([C@@H](N)[C@@H](C)CC)=N[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]1C(=O)N[C@H](CCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2N=CNC=2)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)NCCCC1 CLKOFPXJLQSYAH-ABRJDSQDSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- UUQMNUMQCIQDMZ-UHFFFAOYSA-N betahistine Chemical compound CNCCC1=CC=CC=N1 UUQMNUMQCIQDMZ-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 208000014679 binge eating disease Diseases 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- KMGBZBJJOKUPIA-UHFFFAOYSA-N butyl iodide Chemical compound CCCCI KMGBZBJJOKUPIA-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 210000001638 cerebellum Anatomy 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 230000001055 chewing effect Effects 0.000 description 1
- ITVPBBDAZKBMRP-UHFFFAOYSA-N chloro-dioxido-oxo-$l^{5}-phosphane;hydron Chemical compound OP(O)(Cl)=O ITVPBBDAZKBMRP-UHFFFAOYSA-N 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 1
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 1
- 229960000258 corticotropin Drugs 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 description 1
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- USLKCMBGQFYUFI-UHFFFAOYSA-N dichloromethane;tribromoborane Chemical compound ClCCl.BrB(Br)Br USLKCMBGQFYUFI-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical group O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- UNBMQQNYLCPCHS-VYNDPHDASA-N ebelactone b Chemical compound CC[C@@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)\C=C(/C)C[C@H](C)[C@@H]1OC(=O)[C@H]1CC UNBMQQNYLCPCHS-VYNDPHDASA-N 0.000 description 1
- 230000002996 emotional effect Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 230000004634 feeding behavior Effects 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000004088 foaming agent Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical compound C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 102000054264 human TAP2 Human genes 0.000 description 1
- 125000000717 hydrazino group Chemical group [H]N([*])N([H])[H] 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- YSLDOTFAFZJPOC-UHFFFAOYSA-N hydron;methyl 6-aminohexanoate;chloride Chemical compound Cl.COC(=O)CCCCCN YSLDOTFAFZJPOC-UHFFFAOYSA-N 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 230000002267 hypothalamic effect Effects 0.000 description 1
- 210000003016 hypothalamus Anatomy 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- IENZCGNHSIMFJE-UHFFFAOYSA-N indole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2NC=CC2=C1 IENZCGNHSIMFJE-UHFFFAOYSA-N 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 230000003871 intestinal function Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 210000003140 lateral ventricle Anatomy 0.000 description 1
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 1
- 229940039781 leptin Drugs 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 229940040129 luteinizing hormone Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- CEAJFNBWKBTRQE-UHFFFAOYSA-N methanamine;methanol Chemical compound NC.OC CEAJFNBWKBTRQE-UHFFFAOYSA-N 0.000 description 1
- DGRLUQTYWVBYCB-UHFFFAOYSA-N methyl 9-ethyl-5,6,7,8-tetrahydrocarbazole-3-carboxylate Chemical compound C12=CC(C(=O)OC)=CC=C2N(CC)C2=C1CCCC2 DGRLUQTYWVBYCB-UHFFFAOYSA-N 0.000 description 1
- MQWCXKGKQLNYQG-UHFFFAOYSA-N methyl cyclohexan-4-ol Natural products CC1CCC(O)CC1 MQWCXKGKQLNYQG-UHFFFAOYSA-N 0.000 description 1
- 239000004200 microcrystalline wax Substances 0.000 description 1
- 235000019808 microcrystalline wax Nutrition 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- XMWFMEYDRNJSOO-UHFFFAOYSA-N morpholine-4-carbonyl chloride Chemical compound ClC(=O)N1CCOCC1 XMWFMEYDRNJSOO-UHFFFAOYSA-N 0.000 description 1
- FZEKDGOINZEYAW-UHFFFAOYSA-N n-(6,7,8,9-tetrahydro-5h-carbazol-3-yl)morpholine-4-carboxamide Chemical compound C=1C=C2NC=3CCCCC=3C2=CC=1NC(=O)N1CCOCC1 FZEKDGOINZEYAW-UHFFFAOYSA-N 0.000 description 1
- SFMJNHNUOVADRW-UHFFFAOYSA-N n-[5-[9-[4-(methanesulfonamido)phenyl]-2-oxobenzo[h][1,6]naphthyridin-1-yl]-2-methylphenyl]prop-2-enamide Chemical compound C1=C(NC(=O)C=C)C(C)=CC=C1N1C(=O)C=CC2=C1C1=CC(C=3C=CC(NS(C)(=O)=O)=CC=3)=CC=C1N=C2 SFMJNHNUOVADRW-UHFFFAOYSA-N 0.000 description 1
- WSTNFGAKGUERTC-UHFFFAOYSA-N n-ethylhexan-1-amine Chemical compound CCCCCCNCC WSTNFGAKGUERTC-UHFFFAOYSA-N 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- VJUDVVHGQMPPEI-UHFFFAOYSA-N n-methyl-1-(oxolan-2-yl)methanamine Chemical compound CNCC1CCCO1 VJUDVVHGQMPPEI-UHFFFAOYSA-N 0.000 description 1
- WWXPJKGWWPWBTB-UHFFFAOYSA-N n-methyl-2-morpholin-4-ylethanamine Chemical compound CNCCN1CCOCC1 WWXPJKGWWPWBTB-UHFFFAOYSA-N 0.000 description 1
- WKRUEUNOCVALAM-UHFFFAOYSA-N n-methyl-2-pyridin-3-ylethanamine Chemical compound CNCCC1=CC=CN=C1 WKRUEUNOCVALAM-UHFFFAOYSA-N 0.000 description 1
- QZBUGNURVLWJPK-UHFFFAOYSA-N n-methyl-2-pyridin-4-ylethanamine Chemical compound CNCCC1=CC=NC=C1 QZBUGNURVLWJPK-UHFFFAOYSA-N 0.000 description 1
- XHFGWHUWQXTGAT-UHFFFAOYSA-N n-methylpropan-2-amine Chemical compound CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 125000005327 perimidinyl group Chemical group N1C(=NC2=CC=CC3=CC=CC1=C23)* 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 125000005543 phthalimide group Chemical group 0.000 description 1
- PRAYXGYYVXRDDW-UHFFFAOYSA-N piperidin-2-ylmethanol Chemical compound OCC1CCCCN1 PRAYXGYYVXRDDW-UHFFFAOYSA-N 0.000 description 1
- VUNPWIPIOOMCPT-UHFFFAOYSA-N piperidin-3-ylmethanol Chemical compound OCC1CCCNC1 VUNPWIPIOOMCPT-UHFFFAOYSA-N 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- FYRHIOVKTDQVFC-UHFFFAOYSA-M potassium phthalimide Chemical compound [K+].C1=CC=C2C(=O)[N-]C(=O)C2=C1 FYRHIOVKTDQVFC-UHFFFAOYSA-M 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940097325 prolactin Drugs 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- DRINJBFRTLBHNF-UHFFFAOYSA-N propane-2-sulfonyl chloride Chemical compound CC(C)S(Cl)(=O)=O DRINJBFRTLBHNF-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- HDOUGSFASVGDCS-UHFFFAOYSA-N pyridin-3-ylmethanamine Chemical compound NCC1=CC=CN=C1 HDOUGSFASVGDCS-UHFFFAOYSA-N 0.000 description 1
- TXQWFIVRZNOPCK-UHFFFAOYSA-N pyridin-4-ylmethanamine Chemical compound NCC1=CC=NC=C1 TXQWFIVRZNOPCK-UHFFFAOYSA-N 0.000 description 1
- ATBIAJXSKNPHEI-UHFFFAOYSA-N pyridine-3-carbonyl chloride Chemical compound ClC(=O)C1=CC=CN=C1 ATBIAJXSKNPHEI-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- JHHZLHWJQPUNKB-UHFFFAOYSA-N pyrrolidin-3-ol Chemical compound OC1CCNC1 JHHZLHWJQPUNKB-UHFFFAOYSA-N 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 230000029865 regulation of blood pressure Effects 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- JWHOQZUREKYPBY-UHFFFAOYSA-N rubonic acid Natural products CC1(C)CCC2(CCC3(C)C(=CCC4C5(C)CCC(=O)C(C)(C)C5CC(=O)C34C)C2C1)C(=O)O JWHOQZUREKYPBY-UHFFFAOYSA-N 0.000 description 1
- HBEFYGYBMKPNSZ-UHFFFAOYSA-N s-phenyl chloromethanethioate Chemical compound ClC(=O)SC1=CC=CC=C1 HBEFYGYBMKPNSZ-UHFFFAOYSA-N 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- 239000004317 sodium nitrate Substances 0.000 description 1
- 235000010344 sodium nitrate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- ZRRGOUHITGRLBA-UHFFFAOYSA-N stattic Chemical compound [O-][N+](=O)C1=CC=C2C=CS(=O)(=O)C2=C1 ZRRGOUHITGRLBA-UHFFFAOYSA-N 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- NVBFHJWHLNUMCV-UHFFFAOYSA-N sulfamide Chemical class NS(N)(=O)=O NVBFHJWHLNUMCV-UHFFFAOYSA-N 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002889 sympathetic effect Effects 0.000 description 1
- 210000002820 sympathetic nervous system Anatomy 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- FKHIFSZMMVMEQY-UHFFFAOYSA-N talc Chemical compound [Mg+2].[O-][Si]([O-])=O FKHIFSZMMVMEQY-UHFFFAOYSA-N 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- AXMNAUFSNXCVOE-UHFFFAOYSA-N tert-butyl N-(4-methoxybutyl)-N-methylcarbamate Chemical compound COCCCCN(C)C(=O)OC(C)(C)C AXMNAUFSNXCVOE-UHFFFAOYSA-N 0.000 description 1
- BHLOFKXQIGGRMD-UHFFFAOYSA-N tert-butyl n-[6-[(9-ethylcarbazol-3-yl)amino]-6-oxohexyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCCCC(=O)NC1=CC=C2N(CC)C3=CC=CC=C3C2=C1 BHLOFKXQIGGRMD-UHFFFAOYSA-N 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- UGNWTBMOAKPKBL-UHFFFAOYSA-N tetrachloro-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(Cl)=C(Cl)C1=O UGNWTBMOAKPKBL-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 210000001103 thalamus Anatomy 0.000 description 1
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 238000007070 tosylation reaction Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- HRXKRNGNAMMEHJ-UHFFFAOYSA-K trisodium citrate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O HRXKRNGNAMMEHJ-UHFFFAOYSA-K 0.000 description 1
- 229940038773 trisodium citrate Drugs 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 230000025033 vasoconstriction Effects 0.000 description 1
- 239000005526 vasoconstrictor agent Substances 0.000 description 1
- 229960003726 vasopressin Drugs 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/4035—Isoindoles, e.g. phthalimide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/572—Five-membered rings
- C07F9/5728—Five-membered rings condensed with carbocyclic rings or carbocyclic ring systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B57/00—Other synthetic dyes of known constitution
Definitions
- the present invention relates to a tricyclic compound useful in the field of medicine and a drug containing the compound as an active ingredient.
- Neuropeptide Y (hereinafter sometimes abbreviated as “NPY” in the present specification) is a peptide consisting of 36 amino acids, and was first isolated from Tsubmoto et al. ), 296, 659 (1982)]. NPY was found to belong to the pancreatic polypeptide (PP) family from its amino acid primary sequence homology. As polypeptides belonging to this family, pancreatic polypeptide (PP) produced in endocrine cells of the knee and peptide YY (PYY) produced in endocrine cells of the digestive tract are known.
- ⁇ family peptides all consist of 36 amino acids, but the amino acid sequence at the carboxy terminus (C-terminus) is well conserved, especially at the C-terminus (36th amino acid: ⁇ 36). Tyrosine.
- receptors for the ⁇ family of peptides are called type ⁇ receptors.
- the type I receptor has also been found to be a seven-transmembrane receptor coupled to a G protein.
- ⁇ is widely distributed in the central nervous system and peripheral nervous system, and as one of the most abundant peptides in the nervous system, plays various functions in living organisms. For example, regulation of blood pressure, regulation of eating behavior, regulation of intestinal function, inhibition of regulation of insulin rhythm and insulin secretion, prolactin, luteinizing hormone, ACTH, gonadotropin-releasing hormone, vasopressin, etc. It is involved in the suppression of hormone secretion.
- ⁇ is continuously administered intraventricularly, It is known to induce obesity and insulin resistance. It is also involved in emotional control and the function of the central autonomic nervous system.
- NPY coexists with norepinephrine at sympathetic endings and has been linked to the tone of the sympathetic nervous system.
- Peripheral administration of NPY is known to cause vasoconstriction and enhance the effects of other vasoconstrictors, including norepinephrine [Inna Yuni Nationale 'journale' off, 'Obecity 1 (Internatin 1) ional j ournal of obes ity), 19, 517 (1995); Endocrinology, 133, 1753 (1993); Pretty journal / ob pharmacology-(Brit ish Journal of Pharmacology) 95, 419 (1988)].
- NPY The function of NPY is expressed by binding to the NPY Y-type receptor present in the central or peripheral nervous system.
- NPY receptors At least six subtypes have been identified as NPY receptors, and their genes have been isolated except for Y3.
- Y1 is the first cloned receptor [FEBS Lett., Vol. 271, p. 81 (1990)], which is mainly distributed in blood vessels at the periphery and constricts blood vessels (blood pressure). Rise). In the center, it is mainly distributed in the cerebral cortex, thalamus, and amygdala, and it is said that anxiety in the amygdala is expressed via the Y1 receptor.
- the history of the Y2 receptor which has been classified pharmacologically as a receptor different from the Y1 receptor, has been isolated and its existence has been clarified [Journal-of-biological- Chemistry (J. Biol. Chem.), 270, 22661 (1995)].
- This receptor is expressed mainly in the brain, especially in the cerebral cortex, hippocampus, and amygdala, but has not been detected in the cerebellum or spinal cord.
- the ⁇ 3 receptor has been classified pharmacologically, but no gene has yet been isolated.
- the ⁇ 4 receptor has been found using the human Y1 receptor cDNA as a probe and the gene has been isolated [Journal of Biological Chemistry (J. Biol. Chem.), 270, 26762 (1995 Year) ].
- Expression sites are specific to the prostate, colon, knee, and small intestine, and have not been detected in the brain, kidney, lung, heart, spleen, and the like.
- NPY neuropeptide Y5 receptor
- the Y5 receptor has low homology to other NPY receptors of 35% or less, and its expression site is restricted to the hypothalamus of the brain, and is most involved in the control of feeding.
- the Y6 receptor is found only in mice and does not function as a pseudogene in humans.
- Substances that have an affinity for these Y-type receptors and act as agonists or antagonists for these receptors can regulate the expression of NPY.
- Substances having such properties are used for various diseases involving NPY, such as hypertension, kidney disease, heart disease, and circulatory diseases such as vasospasm, such as bulimia, depression, epilepsy, and dementia. It is expected to be useful in the prevention or treatment of diseases such as obesity, diabetes, hyperlipidemia, metabolic diseases such as hormonal abnormalities, and anorexia and glaucoma in cancer patients. [Trend-in 'Pharmacology' Science ( Trends in Pharmacological Sciences), 15, 153 (1994)].
- NPY / Y5 receptor substances that have a selective affinity for the Y5 receptor of the NPY receptor
- NPY / Y5 receptor substances that have a selective affinity for the Y5 receptor of the NPY receptor. It is expected to be useful for prevention and / or treatment and to be used without side effects such as enhancing or antagonizing the function of other Y-type receptors.
- Y5 receptor Since Y5 receptor is most involved in the control of eating, it can be used as a food intake regulator such as anorexia in bulimia and cancer patients, as well as central nervous system such as depression, epilepsy and dementia. It can be used for the prevention or treatment of metabolic diseases such as diseases, obesity, diabetes, hyperlipidemia, and hormonal abnormalities.
- International Publication W097 / 19682 states that Sulfonamide and sulfamide derivatives, International Publication W097 / 20820, International Publication W097 / 20821, International Publication W097 / 20822, and International Publication W097 / 20823, quinazoline derivatives, International Publication W098 / 35944, and International Publication W098 / 35957 describes amide derivatives, International Publication W098 / 40356 describes aminopyridine derivatives, International Publication W098 / 24768, International Publication W098 / 25907, International Publication W098 / 25908, and International Publication W098 / 27063.
- An object of the present invention is to provide a substance having an affinity for an NPY receptor, particularly a substance having a selective affinity for an NPY / Y5 receptor.
- Another object of the present invention is to provide a medicament having an action of controlling eating, which is useful as a drug for regulating eating, such as anorexia in bulimia and cancer patients.
- Still another object of the present invention is to provide a medicament useful for the prevention or treatment of central diseases such as depression, epilepsy, and dementia, and metabolic diseases such as obesity, diabetes, hyperlipidemia, and hormonal abnormalities. It is to provide.
- the present inventors have conducted intensive studies to solve the above-mentioned problems, and as a result, a novel compound represented by the following general formula (I) has affinity for the NPY receptor, and the action of NPY They have been found to have the effect of regulating expression. Further, they have found that a compound represented by the following general formula (IV) also has a similar effect. Furthermore, these substances have a selective affinity especially for the NPY / Y5 receptor, and these substances are useful as medicaments for regulating food intake or preventing or treating the above diseases. It was found that The present invention has been completed based on these findings.
- A is a 5- to 7-membered hydrocarbon ring group (one or two or more selected from the group consisting of a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, and a halogen atom on the ring)
- L may have a substituent
- the lower alkyl group, the lower acryl group, or the lower alkoxy group may have one or more substituents).
- R 3 represents a hydrogen atom, a lower alkyl group, or a lower acyl group, and the lower alkyl group or the lower acyl group may have one or more substituents.
- a linking group that is
- M is an alkylene linking group having 2 to 10 carbon atoms
- the alkylene linking group may have one or more substituents, and a carbon atom constituting the carbon chain of the alkylene linking group (at least 1 May be substituted with a nitrogen atom, an oxygen atom, a zirconium atom, or a 3- to 8-membered cycloalkylene group, and the nitrogen atom is substituted with a lower alkyl group or a lower acyl group.
- the cycloalkylene group may have one or more substituents], provided that when L represents —NR 3 —CO—, M may be a single bond. ;
- X is - S -, -0-, - NR 4 -, -NR 5 - CO -, -NR 5 - CS -, and - NR 5 - S0 2 -
- R 4 is a hydrogen atom, an alkyl group, Or the alkyl group or the lower acyl group may have one or more substituents, the alkyl group may have a ring structure, and R 5 is hydrogen.
- R 4 is a hydrogen atom, an alkyl group, Or the alkyl group or the lower acyl group may have one or more substituents, the alkyl group may have a ring structure, and R 5 is hydrogen.
- Y is an alkyl group having 1 to 20 carbon atoms (the alkyl group may have a ring structure), an aryl group having 6 to 12 carbon atoms, an amino group, a monoalkylamino group having 1 to 8 carbon atoms A dialkylamino group having 2 to 16 carbon atoms, an azacycloalkyl group having 4 to 8 carbon atoms, a phosphoryl group, a monoalkylphosphoryl group having 1 to 8 carbon atoms, and a dialkylphosphoryl having 2 to 16 carbon atoms A substituent selected from the group consisting of a group, an aromatic heterocyclic group, and a 5- to 7-membered non-aromatic heterocyclic group (the above group further has one or more substituents; May be combined with R 5 to form a ring).
- Y is an aromatic heterocyclic group or a 5- to 7-membered non-aromatic
- R 4 and Y may combine with each other to form a ring together with the nitrogen atom to which they are attached (wherein R 4 and Y One or more hetero atoms may be contained as ring-constituting atoms other than the nitrogen atom to be bonded, and one or more substituents may be present on the ring):
- R 1 is a substituent selected from the group consisting of a lower alkyl group, a lower alkenyl group, a lower alkynyl group, and a lower acyl group (the above group may have a ring structure, and may have one or more substituents; May be present);
- R 21 , R 22 , and R 23 each independently represent a hydrogen atom, a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, a halogen atom, an amino group, a mono-lower alkylamino group, a di-lower alkylamino group, a lower A substituent selected from the group consisting of an acylamino group and an amide group (the substituent may have one or more substituents) or a salt thereof. It is.
- A is a 5- to 7-membered hydrocarbon ring group (having one or more substituents selected from the group consisting of a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, and a halogen atom on the ring;
- the lower alkyl group, the lower acyl group, or the lower alkoxy group may have one or more substituents);
- L is, - NR 3 - C0-, - CO- NR 3 -, - NR 3 - CS -, - CS- NR 3 -, - NR 3 - S0 2 -, and - S0 2 - NR 3 - (in the formula And R 3 represents a hydrogen atom, a lower alkyl group or a lower acyl group, and the lower alkyl group or the lower acyl group may have one or more substituents. Indicates a linking group to be selected;
- M is an alkylene linking group having 2 to 10 carbon atoms
- the alkylene linking group may have one or more substituents, and a carbon atom constituting the carbon chain of the alkylene linking group (at least 1 May be substituted with a nitrogen atom, an oxygen atom, a thio atom, or a 3- to 8-membered cycloalkylene group, and the nitrogen atom is a lower alkyl group.
- the cycloalkylene group may have one or more substituents.
- X is -S -, - 0-, - NR 4 -, -NR 5 Hiroshi, - NR 5 _CS -, and - NR 5 - S0 2 - (wherein, the R 4 and R 5 Waso respectively independently A hydrogen atom, a lower alkyl group, or a lower acyl group; the lower alkyl group or the lower acyl group may have one or more substituents, and R 4 is linked to M to form a ring;
- R 1 is a substituent selected from the group consisting of a lower alkyl group, a lower alkenyl group, a lower alkynyl group, and a lower acyl group (the above group may have a ring structure, and may have one or more substituents; May be present);
- R 21 , R 22 , and R 23 each independently represent a hydrogen atom, a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, a halogen atom, an amino group, a mono-lower alkylamino group, a di-lower alkylamino group, a lower And a substituent selected from the group consisting of an acylamino group and an amide group (the substituent may have one or more substituents).
- A represents the following formula (la), (lb), or (Ic):
- the above ring may have one or more substituents selected from the group consisting of a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, and a halogen atom, and the lower alkyl group, the lower
- L is - NR 3 - CO - in and, X is - NR 5 -C0- or - NR 5 - S0 2 - the compound represented by the above general formula (I) is or salts thereof;
- Oyobi Shi is - CO- NR 3 - a and, X is - NR 5 - CO- or - NR 5 - S0 2 - above general formula or a salt thereof (I) is provided which is Is done.
- A is a 5- to 7-membered hydrocarbon ring group (having one or more substituents selected from the group consisting of a lower alkyl group, a lower alkoxy group, and a halogen atom on the ring.
- the lower alkyl group or the lower alkoxy group may have one or more substituents).
- R 101 represents a lower alkyl group or a lower acryl group (the lower alkyl group or the lower acryl group may have a ring structure and may have one or more substituents);
- R lfl2 represents a hydrogen atom or an alkyl group having 1 to 20 carbon atoms (the alkyl group may have a ring structure and may have one or more substituents);
- R 1Q3 represents the total carbon number of 1 to 20 alkyl group (said alkyl group rather it may also contain a ring structure, may have one or more substituents), R IQ2 and R 1113 may be bonded to each other to form a ring together with the nitrogen atom to which they are bonded (the ring has one or more heteroatoms in addition to the nitrogen atom to which R lfl2 and R 1Q3 are bonded).
- R 1Q4 , R lfl5 , and R lfl6 each independently represent a hydrogen atom, a hydroxyl group, a lower alkyl group, or may have one or more substituents on the ring.
- A represents the following general formula (la), (lb), or (Ic):
- the ring may have one or two or more substituents selected from the group consisting of a lower alkyl group, a lower alkoxy group, and a halogen atom, and the lower alkyl group or the lower alkoxy group may have one or more substituents.
- R 1 () 1 is a lower alkyl group (the alkyl group may have a ring structure, and 1 or 2 or more A compound represented by the above general formula (XXI) or a salt thereof, wherein R 1Q3 is a heteroatom selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom
- R 1Q3 is a heteroatom selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom
- R 1Q3 is a heteroatom selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom
- R 1Q3 is a heteroatom selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom
- R 1Q3 is a heteroatom selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom
- R 1Q3 is a heteroatom selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur
- R ID2 and R IQ3 are bonded to each other to form a ring together with the nitrogen atom to which they are bonded, represented by the following general formula (XXI I)
- X is - CH 2 -, - 0-, - S -, - NH -, or - NR lG8 -
- R LQ8 is a lower alkyl group, a lower Ashiru group, heterocyclic phenyl group, or to Group (the lower alkyl group, the lower acyl group
- a group, the phenyl group, or the heterocyclic group may have one or more substituents).
- n an integer of 1 to 4.
- R 1 ( "is a hydroxyl group, an amino group, a cyano group, a lower alkyl group, a lower alkoxy group, a lower alkylthio group, a lower alkyl group, a propyl group (the lower alkyl group, the lower alkoxy group, the lower alkylthio group, or the lower alkyl group)
- the alkylcarbonyl group may have a ring structure and may have one or more substituents, an aryl group (the aryl group has one or more substituents). Or a heterocyclic group;
- n represents an integer of 0 to 4, and when there are a plurality of R 1Q7 , each R 1 [) 7 is independent and may be the same or different.] or a salt thereof XXI); and X is - CH 2 -, - 0-, or - or a salt thereof and S- is the general formula (XXI) is provided.
- the compound represented by the above general formula (I) or a salt thereof has an affinity for the NPY receptor, and in particular, acts as a ligand for the NPY / Y5 receptor and regulates the expression of NPY action. Can be. Therefore, the compound represented by the above general formula (I) or a salt thereof is useful for preventing and treating or treating NPY-related diseases, particularly NPY / Y5 receptor-related diseases.
- a compound selected from the group consisting of the compound represented by the general formula (I) and a physiologically acceptable salt thereof, and a hydrate and a solvate thereof is used as an active ingredient.
- the above-mentioned medicament is useful, for example, as a feed-regulating drug or a medicament for preventing and / or treating diabetes, or a medicament for preventing and / or treating hypercholesterolemia, hyperlipidemia, or arteriosclerosis. is there.
- a method for regulating use and eating comprising a compound represented by the above general formula (I), a physiologically acceptable salt thereof, a hydrate thereof, and a solvent thereof.
- a method comprising administering an effective amount of a substance selected from the group consisting of a human to a mammal including a human, and a method for treating and / or preventing a disease involving NPY, the method comprising: A step of administering an effective amount of a compound selected from the group consisting of the compound represented by I), a physiologically acceptable salt thereof, and a hydrate and a solvate thereof to a mammal including a human.
- the present invention provides a compound represented by the following general formula (IV):
- a ' is a 5- to 7-membered hydrocarbon ring group (a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, a halogen atom, an amino group, a mono-lower alkylamino group, a di-lower alkyl It may have one or two or more substituents selected from the group consisting of an amino group, a lower acylamino group, and an amide group, and the lower alkyl group, the lower acyl group, or the lower alkoxy group has one or more substituents. Which may have two or more substituents);
- L is, - NR 63 -C0-, - CO- NR 63 -, -NR 63 -CS-, _CS-NR 63 -, - NR 63 - S0 2 -, and - S0 2 - NR 63 - (in the formula, R 63 represents a hydrogen atom, a lower alkyl group, or a lower acyl group, and the lower alkyl group or the lower acyl group may have one or more substituents.
- Q represents an alkyl group, an alkenyl group, an alkynyl group, an alkylalkenyl group, a cycloalkyl group, an alkylcycloalkylalkyl group, an aryl group, a heterocyclic group, an alkylcycloalkyl group, a cycloalkylalkyl group, and an alkylazacycloalkyl group.
- R 41 is a substituent selected from the group consisting of a lower alkyl group, a lower alkenyl group, a lower alkynyl group, and a lower acyl group (the substituent may have a ring structure, and may have one or more substituents; May be present);
- R 52, and R 53 are each independently a hydrogen atom, a hydroxyl group, a lower alkyl group, a lower ⁇ sill group, a lower alkoxy group, a halogen atom, an amino group, a mono-lower alkylamino group, a di-lower alkylamino group, a lower A substituent selected from the group consisting of an acylamino group and an amide group (the substituent may have one or more substituents); or a physiologically acceptable compound
- An NPY receptor ligand containing the salt as an active ingredient is provided. According to a preferred embodiment of the present invention, there is provided the above NPY receptor ligand, wherein L ′ is —CONR 63 —.
- the present invention is selected from the group consisting of the compound represented by the above general formula (IV) and a physiologically acceptable salt thereof, and a hydrate and a solvate thereof according to the present invention.
- a drug for regulating food intake containing a substance as an active ingredient, and a drug for preventing and / or treating diabetes containing the substance as an active ingredient, or for hypercholesterolemia, hyperlipidemia, or arteriosclerosis A medicament for prevention and / or treatment is provided.
- a method for regulating dietary savings wherein the compound represented by the general formula (IV) and a physiologically acceptable salt thereof, and a hydrate and a solvate thereof are effectively used.
- a method comprising administering an amount to a mammal including a human, and a method for treating and / or preventing a disease associated with NPY, wherein the compound represented by the above general formula (IV) and a physiologically acceptable compound
- a method comprising the step of administering to a mammal, including a human, an effective amount of a substance selected from the group consisting of salts thereof, and hydrates and solvates thereof.
- the alkyl moiety in the “alkyl group” or a substituent containing an alkyl moiety may be straight-chain, branched, cyclic, Or any combination thereof.
- the cyclic alkyl group may be a polycyclic alkyl group.
- C 2 is an alkyl group.
- Alkyl group can be preferably used C c 12 alkyl groups, more preferably c r c 8 alkyl group, more preferably a c r c 6 alkyl group, particularly preferably c r c 4 alkyl group.
- lower alkyl groups include methyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-butyl, sec-butyl, tert-butyl, cyclobutyl, cyclobutylmethyl, Examples include, but are not limited to, n-pentyl, neopentyl, n-hexyl, cyclohexyl, n-heptyl and the like.
- halogen atom may be any of a fluorine atom, a chlorine atom, a bromine atom and an iodine atom.
- a monocyclic or condensed polycyclic aromatic group can be used, for example, a monocyclic to tetracyclic aromatic group, preferably a monocyclic to tricyclic aromatic group
- An aromatic group, more preferably a monocyclic or bicyclic aromatic group can be used.
- the aryl group has 6 to 20, preferably 6 to 16, more preferably 6 to 12, and even more preferably 6 to 10, carbon atoms.
- aryl group examples include a phenyl group, a naphthyl group, an anthryl group, a phenanthryl group, a biphenyl group and the like, and preferably a phenyl group, a naphthyl group, a 2-naphthyl group and the like.
- An aryl group can be attached at any position on the ring.
- heterocyclic group refers to a monocyclic to tetracyclic heterocyclic group containing one or more heteroatoms such as a nitrogen atom, an oxygen atom, and a zeolite atom.
- a monocyclic to tricyclic heterocyclic group more preferably, a monocyclic or bicyclic heterocyclic group can be used.
- heteroatom Means an atom other than a carbon atom, such as a nitrogen atom, an oxygen atom, and a zeo atom, unless otherwise specified. If they contain two or more heteroatoms, they may be the same or different.
- the hetero ring may be a saturated, partially saturated, or aromatic ring.
- “Aromatic heterocyclic group” means a heterocyclic group in which the heterocyclic part is an aromatic ring
- non-aromatic heterocyclic group means a heterocyclic part in which the heterocyclic part is saturated or partially saturated.
- Te means a ring group.
- Heterocyclic groups can be attached at any position on the ring.
- heterocyclic group examples include, for example, an isochromanyl group, a chromanyl group, a pyrrolidinyl group, a pyrrolinyl group, an imidazolidinyl group, an imidazolinyl group, a bilazolidinyl group, a bilazolinyl group, a piperidyl group, a piperidino group, a morpholinyl group, a morpholino group, and thiomorpholino group.
- a functional group when referred to as "optionally having a substituent", the functional group may have one or more optional substituents unless otherwise specified. It means that it may have. When it has two or more substituents, they may be the same or different. Position of substituents is limited It is not defined and can be present at any substitutable site.
- substituent is not particularly limited, for example, (: C 2 "alkyl group, (: ⁇ C 7 Luque alkenyl group, C 2 - C 2Q alkynyl group, C 6 -C 2 () ⁇ re Ichiru group, Heterocyclic group, halogen atom (in the present specification, a halogen atom may be any of a fluorine atom, a chlorine atom, a bromine atom or an iodine atom), a hydroxy group, an oxo group, an amino group, an ammonium group, an imino group group, a mercapto group, Chiokiso group, Shiano group, a nitro group, carboxyl group, phosphoric acid group, a sulfo group, a hydrazino group, C, - C 15 ureido group, C r C 15 imide groups, Isochioshiana one Bok group, Isoshiana one C, C 2
- Ararukiruo alkoxy group a heterocyclic Arukiruokishi group, C r C 2Q alkoxycarbonyl group, C 6 -C 2 ⁇ rie Ruo carboxymethyl local Poni group, a heterocyclic Okishikaruponiru group, c 2 - C 1G alkylcarboxy alkylsulfonyl group, c 6 -.
- substituents exemplified above may be further substituted with one or more other substituents.
- substituents for example, hydroxy C r C 2 () alkyl group, a halogenated c r c 2.
- ( ⁇ alkylsulfonylamino alkylthio group, c 3 etc. -c 2Q alkylcarbonylaminoalkyl thio group can be exemplified also In any case, the substituents described above are for the purpose of illustration, and the present invention is not limited thereto.
- an arylcarbonyl group such as a benzoyl group or an alkylcarbonyl group such as an acetyl group can be used, and these may have a substituent.
- the arylcarbonyl group having a substituent include a P-methoxybenzoyl group and a p-chlorobenzoyl group.
- the alkylcarbonyl group having a substituent include a chloroacetyl group, Examples thereof include a trifluoroacetyl group and a benzylcarbonyl group.
- alkoxy group examples include methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, neopentoxy, n -Hexoxy groups and the like.
- the number of double bonds present in the “alkenyl group” is not particularly limited, but is preferably 1 to 3, more preferably 1 to 2, and still more preferably 1. When it contains two or more double bonds, they may be either conjugated or non-conjugated.
- the number of triple bonds present in the “alkynyl group” is not particularly limited, but is preferably! The number is preferably from 3 to 3, more preferably from 1 to 2, and still more preferably 1.
- the alkynyl group may contain one or more double bonds.
- the two alkyl groups present in the “dialkylamino group” or “dialkyl phosphoryl group” may be the same or different.
- the number of nitrogen atoms contained as ring-constituting atoms of the “azacycloalkyl group” is not particularly limited, but is preferably 1 to 3, more preferably 1 to 2, and still more preferably 1.
- A represents a 5- to 7-membered hydrocarbon ring group.
- the hydrocarbon ring group may contain one or two double bonds.
- A for example, Alternatively, a hydrocarbon ring group represented by the above (Ia), (lb), or (Ic) can be used.
- A is a 6-membered hydrocarbon ring group, and particularly preferably a benzene ring or a hydrocarbon group represented by (lb).
- the ring of A may have one or more substituents selected from the group consisting of a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, and a halogen atom, and the lower alkyl group,
- the lower acyl group or the lower alkoxy group may have one or more substituents.
- a lower alkyl group and a lower alkoxy group are preferable.
- L represents a hydrogen atom, a lower alkyl group, or a lower acyl group, and preferably, a hydrogen atom, a methyl group, an ethyl group, or the like can be used.
- the lower alkyl group or the lower acyl group may have one or more substituents. Examples of such a substituent include a halogen atom.
- L is - NR 3 -C0- or - C0-NR 3 - a, and still more preferably - CO- NR 3 -, and particularly preferably - a CO- NH-.
- M represents an alkylene linking group having 2 to 10 carbon atoms
- the alkylene linking group may have one or more substituents
- the carbon chain of the alkylene linking group has 1 or 2 or more It may have a branched chain.
- carbon atoms constituting the carbon chain of the alkylene linking group carbon atoms excluding at least one carbon atom are substituted with a nitrogen atom, an oxygen atom, an iodine atom, or a 3- to 8-membered cycloalkylene group.
- the nitrogen atom may be substituted with a lower alkyl group or a lower acyl group
- the cycloalkylene group may have one or more substituents.
- L is - NR 3 - to indicate CO-
- M is other alkylene linking groups described above may be a single binding.
- R 3 is preferably a hydrogen atom.
- Examples of the alkylene linking group represented by M include an alkylene group, an alkyleneoxyalkylene group, an alkylenethioalkylene group, a cycloalkylenealkylene group, an alkylenecycloalkylene group, Group, or - ZLZ 2 - Z 3 -, a group represented by [Z 1 and Z 3 are each independently Al Killen group having 2 to 7 carbon atoms, alkylene O alkoxy alkylene group, alkylene wrench O alkylene group, consequent opening alkylene alkylene Z 2 represents an oxygen atom, an iodine atom, or NR 6 (R 6 represents a hydrogen atom, a lower alkyl group, a lower acyl group, and the lower alkyl group or the lower acyl group represents 1 Or a group which has two or more substituents and is represented by the following formula:]
- Examples of M include, for example,-(CH 2 ) 4 -,-((3 ⁇ 4) 5- ,-(CH 2
- substituents present M for example, A hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, a mono-lower alkylamino group, a di-lower alkylamino group, or a lower acylamino group; and the like.
- the lower alkyl group, the lower alkoxy group, The lower acylamino group may have a substituent.
- X is - S -, - 0-, - NR and, - NR 5 - C0_, -NR 5 - CS -, and - NR 5 - S0 2 - represents a linkage group or a single bond selected from the group consisting of.
- R 4 represents a hydrogen atom, an alkyl group, or a lower acyl group; the alkyl group or the lower acyl group may have one or more substituents; and the alkyl group has a ring structure.
- R 5 represents a hydrogen atom, lower alkyl group, or a lower Ashiru group, lower alkyl group or said lower Ashiru group may have one or more substituents
- R 4 May combine with M to form a ring.
- R 4 and R 5 preferably include a hydrogen atom, a methyl group, or an ethyl group.
- the alkyl group or lower acyl group represented by R 4 or the lower alkyl group or lower acyl group represented by R 5 may have a substituent.
- As X preferably -NR 5 _C0- or - NR 5 - S0 2 - may be used, - NR 5 - S0 2 - is particularly preferred.
- X is - NR 4 - represents a group represented by, in this case R represents a hydrogen atom or an alkyl group, the alkyl group may contain a ring structure It may have one or more substituents.
- R represents a hydrogen atom or an alkyl group
- the alkyl group may contain a ring structure It may have one or more substituents.
- X is the - NR 5 - C0-, - NR 5 - CS -, and - NR 5 - S0 2 - (wherein, R 5 is as defined above der Represents a linking group selected from the group consisting of
- Examples of the substituent of the alkyl group or lower acryl group represented by R 4 include a hydroxyl group, an alkoxy group, an alkylthio group, a carbamoyl group, a cyano group, and a halogen atom.
- R 4 a hydrogen atom, a hydroxymethyl group, a hydroxyethyl group, a methoxymethyl group, a methoxyethyl group, a methylthiomethyl group, a methylthioethyl group, a cyanomethyl group, a cyanoethyl group, a hydroxymethyl group, a hydroxyethyl group And a carbamoylmethyl group.
- R 4 may be linked to M to form a ring.
- R 4 can be linked to Z 1 or Z 2 to form a ring, and preferably forms a 5- to 7-membered ring.
- a piperazine ring, a piperidine ring, a pyrrolidine ring and the like can be formed.
- L is - NR 3 -C0-, - NR 3 - CS -, or - NR 3 -S0 2 - is preferably a linking group selected from, in particular, L is - NR 3 - Preferably it is CO-.
- the substituent of the lower alkyl group or lower acyl group represented by R 5 include a halogen atom.
- R 5 is preferably a hydrogen atom or a methyl group.
- Y is an alkyl group having 1 to 20 carbon atoms (the alkyl group may have a ring structure), an aryl group having 6 to 12 carbon atoms, an amino group, a monoalkylamino group having 1 to 8 carbon atoms A dialkylamino group having 2 to 16 carbon atoms, an azacycloalkyl group having 4 to 8 carbon atoms, a phosphoryl group, a monoalkylphosphoryl group having 1 to 8 carbon atoms, and a dialkylphosphoryl having 2 to 16 carbon atoms A substituent selected from the group consisting of a group, an aromatic heterocyclic group, and a 5- to 7-membered non-aromatic heterocyclic group.
- alkyl group having 1 to 20 carbon atoms represented by Y a linear or branched alkyl group having 1 to 12 carbon atoms or a cycloalkyl group having 3 to 12 carbon atoms is preferably used.
- the cycloalkyl group includes, for example, a bicyclic or tricyclic cycloalkyl group such as an adamantyl group.
- the above substituents represented by Y may further have one or more substituents.
- substituents include a hydroxyl group, a halogen atom, a dimethylamino group and the like.
- the substituents indicated Y is in the form a ring with R 5 May be implemented.
- R 5 May be implemented as an example of the case where Y and R 5 are bonded to form a ring, there is a case where a phthalimide ring is formed.
- Y represents an aromatic heterocyclic group or a 5- to 7-membered non-aromatic heterocyclic group.
- R 4 and Y may form a ring together with the nitrogen atom to which they are attached, (said ring in addition to the nitrogen atom to which R 4 and Y are bonded It may contain one or more hetero atoms as ring-constituting atoms, and may have one or more substituents on the ring).
- X is - NR 5 - C0-, - NRS -CS -, or - NR 5 - S0 2 - to indicate a linking group selected from as Y, preferably a straight chain having 1 to 6 carbon atoms Alternatively, a branched-chain alkyl group, aryl group, heterocyclic group, monoalkylamino group, dialkylamino group, or azacycloalkyl group having 5 to 7 carbon atoms can be used.
- X is - S -, - 0-, or - NR 4 - in the case illustrated a linking group represented as Y
- good Mashiku is Ariru group, dialkyl phosphoryl group, heterocyclic group or a non-aromatic Aromatic heterocyclic groups are preferred.
- preferable examples include a tetrazolyl group, a triazolyl group, an imidazolyl group, an oxazolyl group, a thiazolyl group, a getyl phosphoryl group, a hydantoin ring, a thiazolidinedione ring, an oxazolidone ring, and a pyrrolodion ring.
- Y represents an aromatic heterocyclic group or a 5- to 7-membered non-aromatic heterocyclic group, specifically, for example, a tetrazolyl group, a triazolyl group, an imidazolyl group
- Preferred examples include a group, an oxazolyl group, a thiazolyl group, a hydantoin ring, a thiazolidinedione ring, an oxazolidone ring and a pyrrolodion ring.
- R 1 represents a substituent selected from the group consisting of a lower alkyl group, a lower alkenyl group, a lower alkynyl group, and a lower acyl group, and these groups may have a ring structure.
- a lower alkyl group or a lower acyl group can be preferably used.
- the above group represented by R 1 may have one or more substituents. Examples of the substituent of the above group represented by include a hydroxyl group, an alkoxy group, an alkylthio group, a carbamoyl group, a cyano group, and a halogen atom.
- R 1 examples include, for example, methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, sec-butyl group, tert-butyl group, cyclopropyl group, cyclopropylmethyl group, Methoxymethyl group, methoxyethyl group, methylthiomethyl group, methylthioethyl group, cyanomethyl group, cyanoethyl group, propargylmethyl group, hydroxymethyl group, hydroxyethyl group, acetyl group, carbamoylmethyl group, and the like.
- R 21 , R 22 , and R 23 each independently represent a hydrogen atom, a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, an halogen atom, an amino group, a mono-lower alkylamino group, a di-lower alkylamino group, a lower And a substituent selected from the group consisting of an acylamino group and an amide group. It is preferred that R 21 , R 22 and R 23 are all hydrogen atoms.
- R 21 , R 22 , or R 23 is a substituent other than a hydrogen atom, preferably a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, or a halogen atom
- a substituent other than a hydrogen atom preferably a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, or a halogen atom
- An atom and a di-lower alkylamino group can be used, and more preferably, a hydroxyl group, a methyl group, a methoxy group, a halogen atom, a carbamoyl group, an amino group, a dimethylamino group and the like can be used.
- the above groups represented by R 21 , R 22 and R 23 may have one or more substituents, for example, may have a halogen atom or the like.
- R 41 , R 51 , R 52 , R 53 , R 63 , and L the groups described in the aforementioned R 1 R 21 , R 22 , R 23 , R 3 and L are used be able to.
- a 'in general formula (IV) In this case, a 5- to 7-membered hydrocarbon ring group specifically described for A can be used.
- substituents selected from the group consisting of: a lower alkyl group, a lower acyl group, and a lower alkoxy group having one or more substituents. Is also good.
- a hydroxyl group, an amino group, a mono-lower alkylamino group, a di-lower alkylamino group, and a lower acylamino group are preferable.
- Q is an alkyl group, an alkenyl group, an alkynyl group, an alkylalkenyl group, a cycloalkyl group, an alkylcycloalkylalkyl group, an aryl group, a heterocyclic group, an alkylcycloalkyl group, a cycloalkylalkyl group, and an alkylazaalkyl group.
- an ethyl group, a propyl group, an isopropyl group, a butyl group, a sec-butyl group, a tert-butyl group, a cyclopropyl group, a cyclopropylmethyl group and the like can be used.
- the above group represented by Q may have one or more substituents.
- Examples of such a substituent include a hydroxyl group, a carbamoyl group, a sulfamoyl group, a sulfamoylalkoxy group, a sulfamoylalkylthio group, a sulfamoylalkoxy group, a sulfamoylalkylthio group, a dialkylphosphoryl group, Examples include a monoalkyl phosphoryl group and a phosphoryl group.
- the hydrocarbon ring group may contain one or two double bonds.
- the 5- to 7-membered hydrocarbon ring groups specifically described can be used.
- a hydrocarbon ring group represented by the above (Ia), (lb), or (Ic) is particularly preferred.
- One or more substituents selected from the group consisting of a lower alkyl group, a lower alkoxy group, and a halogen atom may be present on the ring of A, and the lower alkyl group or the lower alkoxy group Alternatively, it may have two or more substituents.
- a substituent present on the ring of A Lower alkyl groups are preferred.
- Preferred examples of the lower alkyl group or lower acyl group represented by R lfll include, for example, methyl group, ethyl group, n-propyl group, isopropyl group, cyclopropyl group, n-butyl group, isobutyl group, sec-butyl group.
- R 102 represents, preferably alkyl group containing a total number of 1-10 carbons, more preferably a lower alkyl group. Particularly preferred is a methyl group.
- the alkyl group having 1 to 20 carbon atoms represented by R 1D3 it preferably has one or more substituents having one or more hetero atoms consisting of a nitrogen atom, an oxygen atom, and a sulfur atom
- An alkyl group having 1 to 20 carbon atoms in total preferably, the alkyl moiety is a linear or cyclic lower alkyl group having 1 to 4 carbon atoms, and the total carbon number is the number of carbon atoms of the substituent; including).
- Preferred examples of the substituent having one or more hetero atoms selected from the group consisting of a nitrogen atom, an oxygen atom, and a sulfur atom present on the alkyl group having 1 to 20 carbon atoms represented by Rlfl3 include: For example, a hydroxyl group, an amino group, a cyano group, a sulfamoyl group, a sulfamoyl group, a lower alkoxy group, a lower alkylthio group, a lower alkylsulfonylamino group, a lower alkylcarbonylamino group, a hydroxyalkyl group, a hydroxyalkyloxy group, Alkoxyalkyloxy group, monoalkylamino group, dialkylamino group, lower alkylsulfonylaminoalkoxy group, lower alkylcarbonylcarbonylaminoalkoxy group, lower alkylsulfonylaminoalkylthio group, lower alky
- tetrazolyl, triazolyl, imidazolyl, pyridyl, morpholinyl Preferred examples include a heterocyclic group such as a group, a morpholino group, a thiomorpholino group, a piperazino group, a piperazinyl group, a piperidino group, a piperidinyl group, and a pyrrolidinyl group; and a heterocyclic thio group such as a triazolylthio group or an imidazolylthio group.
- No. More preferred are a lower alkoxy group and a pyridyl group, and particularly preferred are a methoxy group, a 3-pyridyl group and a 4-pyridyl group.
- R IQ2 and R 1113 may combine with each other to form a ring together with the nitrogen atom to which they are attached.
- the ring has one or more hetero atoms other than the nitrogen atom to which R 1D3 is bonded, preferably one or more hetero atoms selected from the group consisting of a nitrogen atom, an oxygen atom, and a sulfur atom as ring members. Is also good.
- one or more substituents may be present on the ring, and when a plurality of substituents are present, they may be the same or different.
- the ring to be formed is preferably a 5- to 8-membered ring, particularly preferably a group represented by the above general formula (XXII).
- R 1Q4 , R 1D5 , and R 1 () 6 each independently represent a hydrogen atom, a hydroxyl group, a lower alkyl group, a lower acyl group, a lower alkoxy group, a halogen atom, an amino group, a mono-lower alkylamino group, a di-lower alkylamino A substituent selected from the group consisting of a lower acylamino group, and an amido group (the substituent may have one or more substituents, and the above R 21 , R 22 And the groups specifically described for R 23. It is preferred that R 1M , 105 , and ⁇ 6 are all hydrogen atoms, in which case either R lfl5 or R 1Q6 is halogen. Also preferred is an atom, preferably a fluorine atom.
- the compound represented by the general formula (I) or the general formula (IV) may have one or more asymmetric carbons depending on the type of the substituent, and may have one or more asymmetric carbons.
- Stereoisomers such as optically active isomers based on carbon and diastereoisomers based on two or more asymmetric carbons, may exist.
- the configuration may be either Z or E.
- Salts include acid addition salts such as inorganic acid salts and organic acid salts; metal salts and ammonium salts Base addition salts such as salts and organic ammonium salts; and amino acid addition salts can be used.
- Acid addition salts include inorganic acid salts such as hydrochloride, nitrate, hydrobromide, sulfate, hydrogen sulfate, monohydrogen phosphate and dihydrogen phosphate, as well as aliphatic monocarboxylic acids, Organic acids such as dicarboxylic acids, hydroxyalkanoic acids, hydroxyalkane diacids, amino acids, aromatic carboxylic acids, or aliphatic or aromatic sulfonic acids can be used.
- inorganic acid salts such as hydrochloride, nitrate, hydrobromide, sulfate, hydrogen sulfate, monohydrogen phosphate and dihydrogen phosphate, as well as aliphatic monocarboxylic acids
- Organic acids such as dicarboxylic acids, hydroxyalkanoic acids, hydroxyalkane diacids, amino acids, aromatic carboxylic acids, or aliphatic or aromatic sulfonic acids can be used.
- Organic acids include formate, acetate, propionate, benzoate, maleate, malonate, fumarate, phthalate, cooctarate, tartrate, citrate, mandelic acid
- Organic salts such as salt, oxalate, methanesulfonate, P-toluenesulfonate, benzenesulfonate, lactate, malate, glycolate, aspartate, or glutamate
- the metal salt include alkali metal salts such as lithium salt, sodium salt and potassium salt, alkaline earth metal salts such as magnesium salt and calcium salt, aluminum salt, zinc salt and the like.
- ammonium salt examples include ammonium and tetramethylammonium salts
- organic ammonium salt examples include addition salts such as morpholine and piperidine.
- amino acid addition salt examples include addition salts such as glycine, phenylalanine, glutamic acid, and lysine.
- the compound represented by the general formula (I) or the general formula (IV) or a salt thereof may exist as a hydrate or a solvate.
- the type of the solvent that forms the solvate is not particularly limited, and examples thereof include alcohols such as methanol, ethanol, and isopropanol, and ethers such as tetrahydrafuran.
- the compounds of general formula (I) in free form or any salts thereof, or hydrates or solvates thereof, are all included in the scope of the present invention.
- the isomers, pure mixtures of the above-mentioned isomers, arbitrary mixtures, racemic isomers and the like of the present invention represented by the general formula (I) are all included in the scope of the present invention.
- the active ingredient of the medicament of the present invention includes a compound in free form represented by the general formula (I) or a physiologically acceptable salt thereof, Alternatively, hydrates or solvates thereof can be used.
- the active ingredient of the medicament of the present invention the above-mentioned isomer in a pure form, an arbitrary mixture of the above-mentioned isomers, a racemate and the like may be used. Further, a biological equivalent or a chemical equivalent of the compound represented by the general formula (I) or (IV) may be used as an active ingredient of the medicament of the present invention. For example, dimers or prodrugs of these compounds may be used as the active ingredient of the medicament of the present invention.
- Compound 12-3 The compound represented by the general formula (I) or (IV) can be produced, for example, as follows, but the method for producing the compound is not limited to the following method.
- L is - NR3-C0-, - NR 3 - CS -, or - NR 3 - S0 2 - and is, X is - NR 5 - C0-, - NR 5 - CS -, or -NR 5 -S0 2 -
- X is - NR 5 - C0-, - NR 5 - CS -, or -NR 5 -S0 2 -
- R′X 1 (wherein, R 1 is as defined above, and X 1 is Is reacted with an organic solvent in the presence of a base in an organic solvent to give a compound of the general formula (VI):
- the leaving group X 1 of R′X 1 used in the above reaction is preferably a halogen atom, a tosyl group, or a mesyl group.
- the type of organic solvent used in the reaction is not particularly limited as long as it is inert in the reaction.For example, a common organic solvent such as acetonitrile, tetrahydrofuran, dimethylformamide, dimethylacetamide, dimethylsulfoxide, and acetone is used. be able to.
- As the base used for example, Common bases such as sodium hydride, sodium hydroxide, potassium carbonate, sodium carbonate, sodium hydrogen carbonate, and triethylamine can be exemplified.
- the reaction temperature is usually from 20 ° C to 100 ° C, preferably from 0 ° C to room temperature.
- the reaction time is generally 1 minute to 3 days, preferably 1 hour to 1 day.
- A, RR 21 , R 22 , and R 23 have the same meanings as described above).
- Various general methods can be used as the reduction method, and a typical method is reduction using iron.
- Acetic acid can be used as a preferred reaction solvent.
- the reaction temperature is usually 0 to 100 ⁇ , preferably room temperature to 70 ° C.
- the reaction time is generally 1 minute to 3 days, preferably 1 hour to 1 day.
- X 2 is -C00H
- a general condensation method such as DCC condensation, DCC / HOBt method, WSC method, mixed acid anhydride method, CDI method, or DPPA method can be employed for the condensation reaction.
- DCC condensation, DCC / HOBt method, or WSC method is preferred.
- a general base such as potassium carbonate or tritylamine is present in a common organic solvent such as tetrahydrofuran, dimethylformamide, dimethylacetamide, or dichloromethane.
- the reaction temperature of the condensation reaction is usually 1 ( ⁇ ⁇ ⁇ ( ⁇ , preferably (re ⁇ room temperature.
- the reaction time is usually 10 minutes ⁇ 3 days, preferably 1 hour to 1 day.
- amino protecting group for X 3 various protecting groups can be employed (see, for example, Protective Groups in Organic Synthes is, TW Greene, John Wiley & Sons, Inc., 1981). And the like.
- an appropriate method can be adopted depending on the protecting group used.
- a method using a dioxane solution of hydrochloric acid or trifluoroacetic acid is preferable.
- the reaction temperature is usually from 120 ° C to 50 ° C, preferably from 120 ° C to room temperature.
- the reaction time is generally 10 minutes to 3 days, preferably 30 minutes to 3 hours.
- R 3 is other than a hydrogen atom
- a compound represented by the general formula (VI I) and a compound represented by the general formula: (R 3 has the same meaning as described above and X 1 represents a leaving group) Is reacted in an organic solvent in the presence of NaOH, and then the amino protecting group is deprotected.
- a fluorinated group is preferred as the amino protecting group represented by X 3 , but this protecting group can be deprotected using hydrazine.
- Q is an alkyl group, an alkenyl group, an alkynyl group, an alkyl alkenyl group, a cycloalkyl group, alkylcycloalkyl Al
- a compound represented by the general formula (VI I) is condensed with a compound represented by the general formula: X 2 -Q (wherein X 2 has the same meaning as described above) by the same method as in production method 1, and The compound of IV) can be produced.
- L is - C0-NR 3 -
- X is - NR 5 I, -NR 5 - CS -, or - NR 5 - S0 2 - at a production method of general formula (I) compounds represented by
- the leaving group X 1 used is preferably a halogen, a tosyl group, or a mesyl group.
- the type of the organic solvent is not particularly limited as long as it is inert in the reaction, and a common organic solvent such as acetonitrile, tetrahydrofuran, dimethylformamide, dimethylacetamide, dimethylsulfoxide, and acetone can be used.
- a common organic solvent such as acetonitrile, tetrahydrofuran, dimethylformamide, dimethylacetamide, dimethylsulfoxide, and acetone can be used.
- common bases such as potassium carbonate, sodium carbonate, sodium bicarbonate and triethylamine.
- reaction temperature is usually from 20 to 100 ° C, preferably from 0 ° C to room temperature.
- reaction time is usually 10 minutes to 3 days, preferably 1 hour to 1 day.
- a compound represented by the general formula (X) and a compound represented by the general formula: HR 3 N-M-NR 5 X 3 (wherein X 3 , M, R 3 , and R 5 are as defined above)
- the reaction temperature of the condensation reaction is usually ⁇ 2 (T to 10 (rC, preferably 0 to room temperature.)
- the reaction time is usually 10 minutes to 3 days, preferably 1 hour to 1 day.
- various protecting groups can be employed, for example, a Boc group is preferable, etc.
- reaction temperature is usually -20T: ⁇ 50 ° C, preferably from 1-20 ° C to room temperature, and the reaction time is usually from 10 minutes to 3 days. Preferably, it is 30 minutes to 3 hours.
- the compound of the general formula: R3 ⁇ 4N-MX-Y (wherein R 3 , M, X, and Y are as defined above) is condensed to form a compound of the general formula (I) (where L is- CO- NR 3 - a and, X is - NR 3 - CO-, - NR 3 -CS -, or - NR 3 -S0 2 - and is) can be obtained.
- ordinary DCC condensation, DCC / HOBt method, or WSC method can be employed.
- the reaction temperature is usually from 120 to 100 ° C., preferably (TC to room temperature.
- the reaction time is usually from 10 minutes to 3 days, preferably from 1 hour to 1 day.
- the reaction sequence may be changed according to the suitability of the target compound.
- R 1 R 21 , R 22 and R 23 have the same meanings as defined above, to produce the carboxylic acid represented by the general formula (X).
- oxidation method a method using a permanganic acid-containing realm in acetone is preferable.
- L ′ is —CO—NR 63 —
- Q is an alkyl group, an alkenyl group, an alkynyl group, an alkylalkenyl group, a cycloalkyl group, an alkylcycloalkylalkyl group, an aryl group, a heterocyclic group, an alkylcycloalkyl group,
- a compound of the general formula (XI) is condensed with a compound of the general formula (XI) and a compound of the general formula: R3 ⁇ 4N-Q (wherein R 3 and Q are as defined above) in the same manner as in Production method 3 to give a compound of the general formula (IV) Can be manufactured.
- L is _NH- CO- or - CO- NR 3 - a and, X is - S -, - 0 -, -N -, or preparation of a compound represented by a single bond and is the general formula (I)
- the compound represented by the general formula (VI I) obtained in the above-mentioned production method 1 is condensed with a compound represented by HOOC-M-0H (wherein M is as defined above), or
- the compound represented by the general formula (X) obtained by the method 3 is condensed with a compound represented by HR 3 N-M-0H (wherein M is as defined above), and the compound represented by the general formula (XI I):
- U represents a leaving group
- the leaving group represented by U is preferably a tosyl group, a mesyl group, or a halogen atom.
- U is a tosyl group
- the usual reaction conditions for tosylation can be employed, but a method of reacting with tosyl chloride in pyridine is preferred.
- the reaction temperature is usually from 20 ° C (: to 100 ° C, preferably from 0 ° C to room temperature.
- the reaction time is usually from 10 minutes to 3 days, preferably from 1 hour to 1 day.
- U is a halogen atom
- ordinary halogenation conditions can be employed. For example, when U is a bromine atom, a method using carbon tetrabromide and phosphine in dichloromethane at room temperature is preferable.
- the compound represented by the general formula (XI II) By reacting the compound represented by the general formula (XI II) with the compound represented by HXY in an organic solvent in the presence of a base, the compound represented by the general formula (I) (L-NH-C0- or- CO- NR 3 - a and, X is - S -, - 0-, -NR 4 -, or a single bond) can be obtained.
- the reaction temperature is usually ⁇ 2 (T to 100, preferably 0 ° C. to room temperature.
- the reaction time is usually 10 minutes to 3 days, preferably 1 hour to 1 day.
- Acetonitrile can be used as a preferable organic solvent, and triethylamine or carbonated lime can be used as a preferable base.
- ⁇ - ⁇ (Y represents a dialkylphosphoryl group) by reacting the compound represented by the general formula (I) wherein X is —NR 4 — and Y is a dialkylphosphoryl group. Can be produced.
- the reaction can be performed in an organic solvent in the presence of a base.
- the reaction temperature is usually
- the temperature is from 20 ° C to 100 ° C, preferably from 0 ⁇ to room temperature.
- the reaction time is usually 10 minutes to 3 days, preferably 1 hour to 1 day.
- Acetonitrile can be used as a preferred organic solvent, and triethylamine or potassium carbonate can be given as a preferred base.
- the compound represented by the general formula (XIV) can also be synthesized by tosylating the compound represented by the general formula (XI I), converting the compound to an azide, and subjecting the compound to hydrogenolysis. .
- the compound represented by the general formula (XXI) included in the general formula (I) can be produced, for example, as follows, but the method for producing the above compound is not limited to the following method. Absent.
- the leaving group X 1 of RHX 1 used in the above reaction is preferably a logene atom, a tosyl group, or a mesyl group.
- the type of organic solvent used in the reaction is not particularly limited as long as it is inert in the reaction.For example, a common organic solvent such as acetonitrile, tetrahydrofuran, dimethylformamide, dimethylacetamide, dimethylsulfoxide, and acetone can be used.
- Examples of the base used include common bases such as sodium hydride, sodium hydroxide, potassium carbonate, sodium carbonate, sodium hydrogen carbonate, and triethylamine.
- the reaction temperature is usually from 20 to 100 ° C, preferably (T to room temperature.
- the reaction time is usually from 1 minute to 3 days, preferably from 1 hour to 1 day.
- reaction temperature is It is always 0 ° C to 100 ° C, preferably room temperature to 70 ° C.
- the reaction time is generally 1 minute to 3 days, preferably 1 hour to 1 day.
- a "and R lfll have the same meanings as described above.)
- organic solvents such as tetrahydrofuran, dimethylformamide, dimethylacetamide, dichloromethane, and acetonitrile.
- the condensation can be carried out in the presence of a general base such as potassium carbonate, tritylamine, etc.
- the reaction temperature of the condensation reaction is usually from 120 to 100, preferably from 120 to room temperature.
- the time is usually from 10 minutes to 3 days, preferably from 1 hour to 1 day.
- a compound represented by the general formula (XXVI) obtained above and a compound represented by the general formula: HN (R 1 () 2 ) (R 103 ) (wherein, R lfl2 and R 103 are as defined above) are converted to an organic compound.
- the compound of the present invention represented by the general formula (XXI) can be obtained.
- the organic solvent a general organic solvent such as tetrahydrofuran, dimethylformamide, dimethylacetamide, dichloromethane, and acetonitrile, or a mixed solvent thereof can be used.
- the base a general solvent such as potassium carbonate or triethylamine can be used. Bases can be used.
- the reaction temperature of the condensation reaction is usually 0 ° (: to 20 (T, preferably room temperature to 120.
- the reaction time is usually 10 minutes to 3 days, preferably 1 hour to 1 day.
- the compound of the general formula (XXI) can also be obtained.
- R 1Q2 , R lfl3 , and X 1 have the same meanings as described above), and in the final step, a general formula: R ⁇ -X 1 (where R I (M is as defined above) Wherein X 1 represents a leaving group) to produce a compound represented by the general formula (XXI) in an organic solvent in the presence of a base.
- the preparation of representative compounds is described specifically and in detail in the Examples herein. Therefore, based on the description of the general production method and the examples described above, by appropriately selecting the starting compounds, the reaction reagents, the reaction conditions, and the like, and as necessary, the method disclosed in the examples.
- a person skilled in the art can produce any of the compounds encompassed by the general formulas (I) and (IV) by making appropriate modifications or alterations to the compounds.
- the reaction yield may be increased in some cases by appropriately protecting the reactive functional group.
- the protecting group can be appropriately selected according to the type of the reactive functional group.For example, the protecting group can be selected by referring to Protective Groups in Organic Synthes is, TW Greene, John Wiley & Sons, Inc., 1981. It will be easier.
- the compound of the present invention represented by the general formula (I) and the compound represented by the general formula (IV) have an affinity for the NPY Y-type receptor, and are particularly selective for the Y5 receptor. It has the characteristic of having affinity. Therefore, the compound of the present invention represented by the general formula (I) and the compound represented by the general formula (IV) have an effect of regulating the expression of the action of NPY, and are used for various diseases associated with NPY. Cardiovascular diseases such as hypertension, kidney disease, heart disease, and vasospasm; central diseases such as binge eating, depression, epilepsy, and dementia; obesity, It is useful for preventing or treating metabolic diseases such as diabetes, hyperlipidemia, and hormonal abnormalities, or anorexia and glaucoma in cancer patients.
- metabolic diseases such as diabetes, hyperlipidemia, and hormonal abnormalities, or anorexia and glaucoma in cancer patients.
- the above-mentioned compounds have an effect of regulating eating, such as anorexia in bulimia and cancer patients, as well as depression, epilepsy, dementia, etc. It is useful for prevention and treatment of metabolic diseases such as central diseases, obesity, diabetes, hypercholesterolemia, hyperlipidemia, arteriosclerosis, and hormonal abnormalities.
- the medicament provided by the present invention comprises a compound selected from the group consisting of a compound represented by the general formula (I) and a physiologically acceptable salt thereof, and a hydrate or a solvate thereof. Or a compound selected from the group consisting of a compound represented by the general formula (IV) and a physiologically acceptable salt thereof, and a hydrate or a solvate thereof as an active ingredient. I have.
- the medicament of the present invention can be administered orally or parenterally. Parenteral administration can include routes of administration such as intratracheal, intrarectal, subcutaneous, intramuscular, and intravenous.
- the above-mentioned substance as an active ingredient may be administered as it is, but generally, a pharmaceutical composition is prepared and administered using one or more pharmaceutical additives.
- a pharmaceutical composition is prepared and administered using one or more pharmaceutical additives.
- suitable formulations include, for example, tablets, granules, fine granules, powders, syrups, solutions, capsules, chewables, and suspensions.
- suitable formulations include: injections, drops, inhalants, sprays, suppositories, transdermal absorbers, transmucosal absorbers, eye drops, ear drops, nasal drops, or patches. And so on.
- Liquid preparations such as injections and infusions are provided, for example, as a powdered pharmaceutical composition in lyophilized form, and dissolved in water or other appropriate medium (eg, saline, glucose infusion, buffer, etc.) at the time of use Alternatively, they may be used after being suspended.
- water or other appropriate medium eg, saline, glucose infusion, buffer, etc.
- Pharmaceutical additives can be appropriately selected according to the form of the pharmaceutical composition, and the type thereof is not particularly limited. Examples thereof include a stabilizer, a surfactant, a plasticizer, a lubricant, a solubilizer, and a buffer. Agents, sweeteners, bases, adsorbents, flavoring agents, binders, suspending agents, brighteners, coating agents, flavoring agents, fragrances, wetting agents, wetting regulators, fillers, defoamers, Chewing agents, fresheners, coloring agents, sugar coatings, tonicity agents, pH regulators, softeners, emulsifiers, adhesives, increased adhesion Strongeners, thickeners, thickeners, foaming agents, excipients, dispersants, propellants, disintegrants, disintegration aids, fragrances, moisture inhibitors, preservatives, preservatives, soothing agents, solvents, Examples thereof include solubilizers, solubilizers, and fluidizers, and these may be used in combination
- a pharmaceutical composition in a desired form can be produced according to a method commonly used in the art.
- the above-mentioned pharmaceutical composition is prepared so that the above-mentioned substance as an active ingredient is 1.0 to 100% (W / W), preferably 1.0 to 60% (W / W). be able to.
- gelatin lactose, sucrose, titanium oxide, starch, crystalline cellulose, hydroxypropylmethylcellulose, carboxymethylcellulose, corn starch, microcrystalline wax, white petrolatum, magnesium metasilicate, magnesium aluminate, anhydrous calcium phosphate , Cunic acid, trisodium citrate, hydroxypropylcellulose, sorbitol, sorbitan fatty acid ester, polyisopate, sucrose fatty acid ester, polyoxyethylene hydrogenated castor oil, polyvinylpyrrolidone, magnesium stearate, light caffeic anhydride , Talc, vegetable oil, benzyl alcohol, gum arabic, propylene glycol, polyalkylene glycol, cyclodextrin or hydroxypropyl cyclode It can be used pharmaceutical additives such as stringent, but it has a name that is not limited thereto.
- the dose and frequency of administration of the medicament of the present invention are not particularly limited, but may be appropriately determined according to various conditions such as the purpose of treatment or prevention, the type of disease, the age, weight, and symptoms of the patient.
- the number of times can be determined. In the case of oral administration, it can be administered once or several times a day so that the active ingredient content is 0.1 to 100 mg / kg per day for adults. It is preferable to administer 0.001 to 10 mg / kg once or several times a day.
- Example 2 Compound 1-2
- N-Boc-6-aminocaproic acid prepared according to the method described in Journal of Medicinal 'Chemistry (J. Med. Cheni. Vol. 35, p. 272 (1993)) (7. 58 g) and 7.58 g of 3-amino-9-ethylcarbazole were dissolved in 75 mL of dimethylformamide, 10.4 g of WSC hydrochloride was added, and the mixture was stirred at room temperature for 3.5 hours. The organic layer was washed with a 1N aqueous sodium hydroxide solution, a 10% aqueous citric acid solution and a saturated saline solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- 6-aminocaproylamino-9-ethylcarbazole (530 mg) was dissolved in 18 mL of acetonitrile, and 494 mg of sodium hydrogen carbonate and 255 mg of nicotinic acid chloride were added. Stirred. Water was added to the reaction solution, neutralized with a 10% aqueous solution of citric acid, and extracted with ethyl acetate. The organic layer is dried over anhydrous magnesium sulfate, After evaporating the solvent under reduced pressure, the residue is purified by silica gel chromatography.
- 6-Nitro-1,, 3,4-tetrahydrophenol (5.04 g) prepared by the method of Journal of The Chemical Society, page 833 (1924) It was dissolved in 50 mL of acetone, 2.25 g of potassium hydroxide and 8.45 g of isopropyl iodide were added, and the mixture was heated to 50 and stirred for 3 hours. Water was added to the reaction solution, and the precipitated precipitate was collected to obtain 2.60 g of N-isopropyl-6-nitro-1,2,3,4-tetrahydrocarbazol.
- N-isopropyl-6-nitro-1,2,3,4-tetrahydrocarbazole (2.60 g) was dissolved in 100 mL of acetic acid, and 2.75 g of iron powder was added. Warmed and stirred for 3 hours. The reaction solution was filtered, and the filtrate was diluted with water. The reaction solution was made basic with 1N-sodium hydroxide solution and extracted with dichloromethane. After the organic layer was dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure.
- N-isopropyl-6-amino-1,2,3,4-tetrahydrocarbazole (228 mg) obtained above, 6- (tonafurensurensulfonyl) aminocaproic acid (321 mg) obtained above , DCC (226 mg) and HOBt (153 mg) were dissolved in 3 mL of DMF and stirred at room temperature for 12 hours.
- the reaction solution was filtered, a 10% aqueous solution of citric acid was added to the filtrate, and the mixture was extracted with ethyl acetate.
- N-ethylcarbazole-3-carboxylic acid (4.78 g) and the journal 'OB' ⁇ - ⁇ -Boc-aminopentylamine (4.04 g), DCC (4.32 g), and HOBt (3.06 g) prepared by the method described in Medicinal Chemistry, Vol. 40, p. 2643 (1997) Dissolved in DMF (mL) and stirred at room temperature for 6 hours.
- the reaction solution was filtered, a 10% aqueous solution of citric acid was added to the filtrate, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with a saturated aqueous solution of hydrogen carbonate and dried over anhydrous sodium sulfate.
- N-ethyl-3- ( ⁇ -aminopentylaminopotassium rubonyl) -potassium (162 mg), indole-5-carboxylic acid (95 mg), DCC ( 103 mg) and HOBt (77 mg) were dissolved in 3 mL of DMF and stirred at room temperature for 24 hours.
- the reaction solution was filtered, a 10% aqueous solution of citric acid was added to the filtrate, and the mixture was extracted with dichloromethane.
- the organic layer was washed with a saturated aqueous solution of hydrogen carbonate and dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure.
- Example 26 Compound 1-26
- Example 27 FAB-MS (m / e) 532 (M + H) + The materials used in Example 27 were replaced with the materials corresponding to the desired compounds, respectively. -The compound of Example 34 was synthesized.
- the trans_4-aminomethylcyclohexanecarboxylic acid (6.04) was dissolved in a 1N aqueous solution of sodium hydroxide, and 8.71 sodium trisulfonyl chloride was added, followed by stirring at room temperature for 3 hours.
- the reaction mixture was acidified with 4N hydrochloric acid, diluted with water, and the precipitated solid was washed with water. The solid was collected by filtration to give 10.1 g of sulfonamide.
- the obtained sulfonamide (2.57 g) was dissolved in 20 mL of toluene, and 1.6 mL of diphenylphosphoryl azide and i. OniL triethylamine were added, followed by stirring at 70 ° C. for 2 hours.
- silica gel column chromatography (Methanol) Purification was carried out using the same method as in Example 1 (1.5%) to obtain 1.09 g of an isocyanate.
- the obtained isocyanate was dissolved in 40 mL of toluene, 1.5 mL of concentrated hydrochloric acid was added dropwise, and the mixture was heated and refluxed at 120-130 for 2 hours.
- the precipitated white solid was washed with water, dried, dissolved in 15 mL of dimethylformamide, and 0.76 g of (9-ethylcarbazole) -3-carboxylic acid and 0.61 g of WSC hydrochloride were added. And stirred at room temperature for 30 minutes. Water was added to the reaction solution, and the mixture was extracted with ethyl acetate.
- ⁇ -NR 300MHz, CDC 1 3) ⁇ 0. 8- 1. 0 (m, 2H), 1. 0-1. 3 (m, 3H), 1. 35 (t, 3H), 1. 6- 2.0 (m, 4H), 2.70 (m, 2H), 3.83 (m, 1H), 4.27 (Q, 2H), 5.48 (bs, 1H), 5.63 (bs , 1H), 7.1-7.7 (m, 8H), 7.8-8.3 (m, 5H), 8.69 (m, 1H)
- Example 3 8 Compound 1 3 8 7-Ethoxytetrahydrocarbazole-6-carboxylic acid obtained in Example 37 was dissolved in 8 mL of toluene, 2.14 g of chloranil was added, and the mixture was refluxed for 2 hours and 30 minutes. The reaction mixture was filtered, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate) to obtain 336 mg of 9-ethyl-5-ethoxycarbazole-6-carboxylic acid. Was.
- Example 41 FAB-MS (m / e) 530 (M + H) + The raw materials used in Example 41 were changed to the raw materials corresponding to the desired compounds, respectively. The compound of Example 44 was synthesized.
- 1,2,3,4-tetrahydrohydrorubazol-6-carboxylate methyl ester (460 mg) prepared by the method of Journal of Chemical Society, page 809 (1992).
- Example 46 Compound 1-1 46
- N-ethyl-3- ( ⁇ -aminopentylaminocarbonyl) -caproluvyl (162 mg) and phthalic anhydride (74 mg) obtained by the method of Example 10 in 3 mL of chloroform After dissolving in mouth form and stirring for 5 hours under reflux conditions, the solvent was distilled off under reduced pressure, and the mixture was allowed to stand at 100 under reduced pressure for 5 hours. The obtained residue was purified by silica gel chromatography (eluent: dichloromethane / ethyl acetate 82) to give 20 mg of compound 1-56.
- Example 58 Compound 1-157
- Negation R (300MHz, CDC1 3) ⁇ 1. 8-2. 0 (m, 4H), 2. 72 (m, 4H), 3. 28 (s, 3H), 3. 5-3. 8 (m, , 4.21 (t, 2H), 6.52 (m, 1H), 7.18 (m, 1H), 7.29 (d, 1H), 7.57 (dd, 1H), 7.96 (d, 1H)
- Example 65 Compound 2-2
- i-band R 300MHz, CDC I 3) ⁇ 1.2-1.4 (m, 9H), 1.8-2.0 (m, 4H), 2.70 (m, 4 ⁇ ), 3.0-3.2 (m, 3H), 3.55 (t, 2H), 3.68 (b, 4H), 4.0-4.2 (m, 6H), 6.65 (m, 1H), 7.25 (d, 1H), 7.58 (dd, 1H), 7.98 (d, 1H)
- 6-Nitro-1,2,3,4-tetrahydrophenol (5.04 g) prepared by the method of Journal of Chemical Society, page 833 (1924) Dissolved in 50 mL of acetone, added 2.25 g of potassium hydroxide and 8.45 g of isopropyl iodide, and heated to 5 (TC and stirred for 3 hours. Water was added to the reaction solution to precipitate it. The precipitate was collected to give 2.60 N-Isopropyl-6-nitro-1,1,2,3,4-tetrahydrobenzene.
- Example 71 N-isopropyl-6-amino-1,5 Synthesis of 2,3,4-tetrahydrocarbazole
- N-Isopropyl-6-phenoxycarbonylamino-1,2,3,4-tetrahydropyrazole (88 mg) was dissolved in 3 mL of a mixture of dichloromethane and acetonitrile (11), and 40% methyl Amine methanol solution (96 mg) was added, and the mixture was stirred under reflux conditions for 8 hours. The reaction solution was allowed to stand at room temperature, and the precipitated crystals were collected and washed with acetonitrile to obtain 63 mg of compound 4-1.
- N-Isopropyl-6-phenoxycarbonylamino-1,2,3,4-tetrahydrophenol 88 mg was dissolved in dichloromethane, and 31 mg of hydroxyethylamine was added. The mixture was stirred at for 8 hours. The reaction solution was allowed to stand at room temperature, and the precipitated crystals were collected and washed with acetonitrile to obtain 35 mg of compound 412.
- Example 7 5 Synthesis of compound 413 In the description of Example 74, hydroxybutylamine was used in place of hydroxyethylamine, and thereafter, compound 4-3 was synthesized in the same manner as in Example 74.
- Example 74 4-aminobutyronitrile was used in place of hydroxyethylamine, and then compound 4_4 was synthesized in the same manner as in Example 74.
- the compound 4-5 was synthesized in the same manner as in Example 74, except that hydroxyethoxyethylamine was used in place of hydroxyethylamine in the description in Example 74.
- ⁇ -NMR 300MHz, CDC1 3 ) ⁇ 1. 57 (d, 6H), 1. 8-2. 0 (m, 4H), 2. 6-2. 8 (m, 4H), 3. 42 (t , 2H), 3.54 (m, 4H), 3.66 (m, 2H), 4.59 (m, 1H), 6.95 (dd, 1H), 7.33 (d, 1H), 7 . 38 (d, 1H)
- Example 74 2-picolylamine was used in place of hydroxyethylamine, and then compound 417 was synthesized in the same manner as in Example 74.
- ⁇ -Marauder R (300MHz, CDC1 3 ) (5 1.57 (d, 6H), 1.8-2.0 (m, 4H), 2.64-2.78 (m, 4H), 4.50 -4.62 (in, 3H), 5.93 (br, 1H), 6.61 (br, 1H), 6.98 (dd, 1H), 7.17 (ddd, 1H), 7. 32- 7.40 (m, 3H), 7.67 (ddd, 1H), 8.46 (dd, 1H)
- Example 74 3-picolylamine was used in place of hydroxyethylamine, and thereafter, Compound 4-8 was synthesized in the same manner as in Example 74.
- Example 74 4-picolylamine was used in place of hydroxyethylamine, and then the compound 419 was synthesized in the same manner as in Example 74.
- Example 74 In the description in Example 74, imidazolylmethylamine was used in place of hydroxyethylamine, and then compound 4 10 was synthesized in the same manner as in Example 74.
- Example 71 N-isopropyl-6-amino-1,2,3,4-tetrahydrocarbazole (228 mg) obtained in 1 (1), 161 mg of dimethylaminocarbonyl chloride, and 152 mg of triethylamine were added in 2 mL. It was dissolved in dichloromethane and stirred at room temperature. The reaction solution was concentrated under reduced pressure, and then ethyl acetate was added. The organic layer was washed with a 10% aqueous solution of citric acid, then with a saturated aqueous solution of sodium hydrogen carbonate, and dried over anhydrous sodium sulfate.
- Example 84 Synthesis of compound 5-2 In the description in Example 74, N-methylisopropylamine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 N-methylbutylamine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 N-methylhydroxystylamine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 In the description in Example 74, ⁇ , ⁇ , ⁇ ′-trimethylethylenediamine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 4-methylaminobutanol was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 8 Compound 5-6 (195 mg) synthesized in 8 was dissolved in 5 mL of pyridine, 50.6 methanesulfonyl chloride was added, and the mixture was stirred at room temperature for 20 minutes. The reaction was quenched with water, extracted with ethyl acetate, and the organic layer was washed twice with 1N hydrochloric acid, once with water and saturated saline, and then concentrated under reduced pressure.
- the residue was dissolved without purification in 5 mL of dimethylformamide, added 106 mg of sodium azide and heated in an oil bath at 90 for 30 minutes. After terminating the reaction with water and extracting with ethyl acetate, the organic layer was washed three times with water and once with a saturated saline solution, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (form: chloroform) to give an azide (205 mg). The obtained azide (205 nig) was added to 6 mL of ethanol. After dissolving, 40 nig of 10% palladium on carbon was added and replaced with hydrogen (normal pressure).
- Example 74 3-methylaminopentionitrile was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- benzyloxycarbonylamino compound (0.96 g) was taken in 200 mL of trilofurasco and replaced with nitrogen, and dissolved in 4 mL of anhydrous tetrahydrofuran. After ice cooling, slowly add 310 mg of lithium aluminum hydride, heat to 60 ° C and stir for 30 minutes did. The reaction was stopped with methanol, the solid was removed by filtration, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was dissolved in 10 mL of chloroform without purification, and the N-isopropyl-6-phenoxycarbonylamino-1,2,3,4-tetrahydrocarbazole obtained in Example 3 (150 mg) and heated under reflux for 12 hours.
- Example 92 cyclohexylamine was used in place of 2- (2-aminoethoxy) ethanol, and the synthesis was performed in the same manner as in Example 92.
- Example 92 trans-4-aminocyclohexanol was used in place of 2- (2-aminoethoxy) ethanol, and the synthesis was performed in the same manner as in Example 92.
- Example 74 N-methyltetrahydrofurfurylamine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 instead of hydroxyethylamine, it is obtained by the method described in Tetrahedron, 48, 199, (1992) (2-piberidinoethyl). The synthesis was performed in the same manner as in Example 74, using -N-methylamine.
- Example 74 instead of hydroxyethylamine, it can be obtained by the method described in Tetrahedron, 48, 199, (1992) (2- (4 Using -N-methylbiperazino) ethyl) -N-methylamine, the subsequent synthesis was carried out in the same manner as in Example 74.
- Example 74 instead of hydroxyethylamine, (2-morpholinoethyl) -N-methylamine obtained by the method described in Tetrahedron, 48, 1999 (1992) is used. The compound was synthesized in the same manner as in Example 74.
- Example 92 2-aminomethylviridine was used in place of 1- (2-aminoethoxy) ethanol, and the synthesis was performed in the same manner as in Example 92.
- Example 74 2- (2-methylaminoethyl) -pyridine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 instead of hydroxyethylamine, it was obtained by the method described in Journal "OB” Heterocycle Chemistry, vol. 27, p. 147, (1990). Using the obtained 3- (2-methylaminoethyl) -pyridine, the subsequent synthesis was carried out in the same manner as in Example 74.
- Example 74 4- (2-methylaminoethyl) -pyridine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 88 Compound 5-6 (195 mg) obtained by the method described in 8 was dissolved in 5 mL of pyridine, methanesulfonyl chloride (50.) was added, and the mixture was stirred at room temperature for 2.5 hours. The reaction was quenched with water, extracted with ethyl acetate, and the organic layer was washed twice with 1N hydrochloric acid, once with water and saturated saline, and then concentrated under reduced pressure. The residue was dissolved in 5 mL of dimethylformamide without purification, and 111 mg of potassium phthalimide was added. The mixture was stirred at room temperature for 1.5 hours.
- Toluene (3-aminopropyl) imidazole (5.00 g) was dissolved in 40 mL of acetonitrile, 5.03 g of sodium hydrogen carbonate was added, and the mixture was cooled with ice. 6. 55 mL of benzyloxycarbonyl chloride was slowly added, and the mixture was stirred at room temperature for 1 hour. Then, the solvent was distilled off under reduced pressure and diluted with water. After dilution with water and extraction with ethyl acetate, the organic layer was washed with water and saturated saline and concentrated under reduced pressure. The obtained residue is subjected to silica gel column chromatography.
- benzyloxycarponylamino compound (1.31 g) was placed in 200 mL of triloflasco and replaced with nitrogen.
- Anhydrous tetrahydrofuran (15 mL) was added, and after ice cooling, 336 mg of lithium aluminum hydride was slowly added, and the mixture was heated to 60 and stirred for 30 minutes.
- the reaction was stopped with methanol, insolubles were removed by filtration, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the residue was dissolved without purification in 30 mL of chloroform and heated for 1.5 h with 230 mg of N-isopropyl-6-phenoxycarbonylamino-1,2,3,4-tetrahydrocarbazole did.
- the reaction solution was concentrated under reduced pressure, and purified by silica gel column chromatography (methanol Z-form: 1-20) to obtain 220 mg of compound 5-222.
- benzyloxycarbonylamino compound (5.40 g) was dissolved in 30 mL of pyridine, and after cooling with ice, 6.33 g of tosyl sulfide was added and stirred for 1.5 hours. The reaction was stopped with water, extracted with ethyl acetate, and the organic layer was washed twice with 1N hydrochloric acid, once with water and saturated brine, and then concentrated under reduced pressure. The precipitated solid was purified by washing with ethyl acetate to obtain a tosyl acetate (5.40 g).
- the obtained triazole-substituted product (552 mg) was placed in a 200-mL three-necked flask, purged with nitrogen, cooled with ice, 15 mL of anhydrous tetrahydrofuran, and 151 mg of lithium aluminum hydride were added. Stirred for hours. After cooling, ethyl acetate was poured, a small amount of water was added, and the mixture was filtered. The filtrate was dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
- Example 74 instead of hydroxyethylamine, 3-methylamino Using -1,2-propanediol, synthesis was carried out in the same manner as in Example 74.
- Example 74 4-ethylaminobutanol was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 diethanolamine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 di (2-methoxyethyl) amine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 the synthesis was carried out in the same manner as in Example 74 after using pyrrolidine instead of hydroxyethylamine.
- Example 74 In the description of Example 74, piperidine was used in place of hydroxyethylamine, and the synthesis was carried out in the same manner as in Example 74.
- Example 74 hexamethyleneimine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 3-pyrrolidinol was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 In the description in Example 74, (s)-(+)-2-pyrrolidinemethanol was used instead of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 In the description in Example 74, (R) -H-2-pyrrolidinemethanol was used instead of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 4-hydroxypyridine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- ⁇ -NMR 300MHz, CDC1 3 ) ⁇ 1.50-1.80 (m + d, 8H), 1.80-2.00 (m, 6H), 2.60-2.80
- Example 74 3-hydroxypyridine was used in place of hydroxyethylamine, and the subsequent synthesis was performed in the same manner as in Example 74.
- Example 74 2-piperidine methanol was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 3-piperidine methanol was used in place of hydroxyethylamine, and the subsequent synthesis was performed in the same manner as in Example 74.
- Example 74 2-piperidineethanol was used in place of hydroxyethylamine, and the subsequent synthesis was performed in the same manner as in Example 74.
- Example 74 4-piperidineethanol was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 4-piperidine noviperidine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 125 Synthesis of compound 7-14 In the description in Example 74, thiazolidine was used in place of hydroxyethylamine, and then synthesized in the same manner as in Example 74.
- Example 74 thiomorpholine was used in place of hydroxyethylamine, and the subsequent synthesis was performed in the same manner as in Example 74.
- Example 74 2,6-dimethylmorpholine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 2-hydroxyethylmorpholine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- ⁇ -NMR 300MHz, dmso-d 6 ) ⁇ 5 1.50-1.75 (i + d, 8H), 1.75-2.00 (m, 4H), 2.33 (br, 1H), 2.60-2.75 (m, 5H), 2.80-3.04 (m, 1H), 3.45-3.60 (m, 2H), 3.65-3.90 (m, 5H ), 4.55 (sep, 1H), 6.64 (br, 1H), 6.99 (dd, 1H), 7.31 (d, 1H), 7.39 (d, 1H)
- Example 74 In the description of Example 74, piperazine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 N-methylbiperazine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 tri (2-pyridyl) piperazine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- Example 74 tri (2-pyrimidyl) piperazine was used in place of hydroxyethylamine, and the synthesis was performed in the same manner as in Example 74.
- NZ- (N-Boc-piperazino) ethylamine (1.33 g) was dissolved in 5 mL of dioxane, 5 mL of 4N dioxane hydrochloride solution was added, and the mixture was stirred at room temperature for 2 hours.
- the reaction solution was concentrated under reduced pressure, and the residue was dissolved in 1N hydrochloric acid.
- the aqueous layer was washed with ethyl acetate, adjusted to pH 9 with a 40% aqueous sodium hydroxide solution, and extracted with ethyl acetate. The extract was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 610 mg of NZ-ethylamine.
- NZ-ethylethylamine (610 mg) and N-isopropyl-6-phenoxycarbonylamino-1,2,3,4-tetrahydropotassium (810 mg) obtained by the method of Example 3 were combined.
- Dissolve in 3 ml dichloromethane, 232 mg triethylamine, 3 ml acetate Nitril was added, and the mixture was stirred at 80 ° C for 20 hours.
- the reaction solution was concentrated under reduced pressure, a saturated aqueous sodium hydrogen carbonate solution was added to the residue, and the mixture was extracted with dichloromethane.
- the phorbazole derivative (258 mg) obtained above was dissolved in 20 mL of methanol, 50 mg of 10% palladium on carbon was added, and the mixture was stirred under a hydrogen atmosphere for 16 hours.
- -6- (2-Aminoethyl) piperazinocarbonylamino-1,2,3,4-tetrahydrol-rubazole was obtained.
- Ammonium chloride (1.27 g) was dissolved in 24 ml of water, 240 ml of isopropyl alcohol and 13.1 g of iron powder were added, and the mixture was refluxed for 15 minutes with stirring. Then, jar Add 6-dito-1,2,3,4-tetrahydropyrazole (10.0 g) prepared according to the method described in p. 833 (1 924) of Naruov Chemical Society. For 5 hours, and ammonium chloride (0.53 g) and iron powder (5.19 g) were further added and stirred for 4 hours. After allowing the reaction solution to cool to room temperature, insolubles were removed by filtration, and the filtrate was concentrated.
- the 6-morpholinocarbonylamino-1,2,3,4-tetrahydrophenol (503 mg) obtained above was dissolved in 8 ml of dimethylformamide, and 414 mg of potassium hydroxide was added. A mixed solution of 237 mg of methyl iodide and 8 ml of dimethylformamide was added dropwise. After stirring at room temperature for 15 minutes, water was added, and 2N hydrochloric acid was further added, and the resulting precipitate was collected by filtration. The obtained precipitate was recrystallized from a mixed solvent of methanol and ethyl acetate to give 89 mg of compound 7-1.
- Example 135 propyl iodide was used in place of thiol iodide, and the synthesis was performed in the same manner as in Example 135.
- Example 71 N-Isopropyl-6-amino-1,2,3,4-tetrahydrocarbazole (2.28 g) obtained in 1, 1.80 g of 4-morpholinocarbonyl chloride, 1.20 g Triethylamine was dissolved in 20 mL of tetrahydrofuran, followed by stirring at room temperature. After the reaction solution was concentrated under reduced pressure, ethyl acetate was added, and the organic layer was washed with a 10% aqueous solution of citric acid, subsequently with a saturated aqueous solution of sodium hydrogen carbonate, and dried over anhydrous sodium sulfate.
- Example 70 butyl iodide was used in place of isopropyl iodide, and the subsequent synthesis was carried out in the same manner as in Examples 71 and 137.
- Example 135 isobutyl iodide was used in place of ethyl iodide, and the synthesis was performed in the same manner as in Example 135.
- Example 135 bromomethylcyclopropane was used in place of thiol iodide, and the synthesis was performed in the same manner as in Example 135.
- Example 135 In the description in Example 35, bromoethylmethyl ether was used in place of thiol iodide, and the synthesis was performed in the same manner as in Example 135.
- Example 135 bromoethanol was used in place of thiol iodide, and the synthesis was performed in the same manner as in Example 135.
- ⁇ -NR 300MHz, CDC1 3 ) ⁇ 1.80-2.00 (m, 4H), 2.60-2.80 (m, 4H), 3.50 (t, 4H), 3.77 (t, 4H), 3.91 (t, 2H), 4.19 (t, 2H), 6.42 (brs, 1H), 7.05 (dd, 1H), 7.14 (d, 1H), 7.48 (d, 1H)
- Example 70 chloroacetonitrile was used in place of isopropyl iodide, and the subsequent synthesis was performed in the same manner as in Examples 71 and 137.
- Compound 912 was synthesized in the same manner as in Example 144, except that 4-methoxycyclohexanone was used as a starting material.
- Windole was prepared from cyclopentene according to the method of Journal of Chemical Society, pp. 833 (1924), followed by Example 70, Compound 10-1 was synthesized in the same manner as in Example 71, Example 72, and Example 88.
- Example 149 Synthesis of compound 10-3 2-Nitro-hexahydrocyclopentz [b] indole was prepared from cyclopene according to the method of Journal of Chemical Society, pp. 833 (1924), followed by the implementation. Compound 10-3 was synthesized in the same manner as in Example 70, Example 71, Example 72, and Example 101.
- 2-Nitro-hexahydrocyclopentz [b] indole was prepared from cyclopentene according to the method of Journal of Chemical Society, p. 833 (1924), followed by Examples.
- Compound 10-4 was synthesized in the same manner as in Example 70, Example 71, Example 72, and Example 137.
- Example 70 Preparation of 2-nitro-hexahydrocycloheptyl [b] indole from cyclohexanone according to the method of Journal of Chemical Society, p. 833 (1 924) Then, Example 70, Example 71, Example 72, and Example 1 Compound 111 was synthesized in the same manner as 37.
- the obtained amino compound (500 mg) was dissolved in chloroform (10 ml), and phenyl chloroformate (190 mg) was added dropwise thereto, followed by stirring for 2 hours. Water was added to the reaction solution for extraction. The extract was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by column chromatography (carrier: silica gel, developing solvent: ethyl acetate: hexane), and phenyl urethane (263 mg, 61%).
- phenyl urethane (900 mg) and (4-pyridino) ethylamine (400 mg) were dissolved in acetonitrile (15 ml) and refluxed for 2 hours. Water was added to the reaction solution in the solvent for extraction. The extract was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The residue obtained was purified by column chromatography (carrier: silica gel, developing solvent: ethyl acetate: hexane) to give compound 12-1 ( 814 mg, 81%).
- Example 70 N-isopropyl-6-nitro-one was prepared by using 2-fluoro-4-12-trophenylenylhydrazine according to the method described in Journal of Chemical Society, page 833 (1924). 8—Fluoro 1,
- Compound 12-3 was synthesized in the same manner as in Example 102, except that phenylthiochloroformate was used in place of phenyl chloroformate in Example 72.
- the human ⁇ ⁇ 5 receptor gene was isolated by amplifying a gene fragment by PCR based on its cDNA sequence [Nature, 382, 168 (1996)] and incorporating it into the expression vector pcDNA3. went. The sequence of the obtained human Y5 gene was analyzed using ABI PRISM Dye Terminator Kit (manufactured by Perkin Elmer), and it was confirmed that the sequence was correct. Human ⁇ ⁇ 5 receptor was expressed using a baculovirus expression system. Using a baculovirus expression system kit (Li fe Teclmokogies), a recombinant virus containing the human Y5 gene was prepared and infected into High Five insect cells to express a large amount of the human Y5 receptor.
- baculovirus expression system kit Li fe Teclmokogies
- a membrane preparation prepared from an insect cell expressing the human Y5 receptor was combined with a test compound (10 M) and 3 H-NPY (manufactured by Amersham Pharmacia Biotech) in an Atssey buffer (1 mM magnesium chloride, 0 mM). Incubated for 2 hours at 41: in 50 mM HEPES buffer, pH 7.4) containing 25 mg / ml bacitracin, 10 g / ml leptin, lg / ml ebelactone B, 1% fetal bovine serum albumin. . The radioactivity bound to the membrane was recovered by a filtration method using a 96-well Dunifilter.
- Test Example 2 Animal test for eating behavior induced by NPY
- mice Male ddY mice (5-week-old, 25-35 g) were kept under anesthesia, and neuropeptide Y (human) was injected into the lateral ventricle (1.0 band to the right of bregma) using a two-stage needle (2.5 mm). / rat NPY, 300 pmol / mouse). The test compound was dissolved in distilled water and orally administered 1 hour before NPY administration. Food consumption was measured over 4 hours after NPY administration. The compound of the present invention significantly reduced the amount of food intake induced by NPY as compared with the control group to which only distilled water was orally administered.
- Test Example 3 Animal test for eating behavior induced by fasting
- mice Male ddY mice (5 weeks old, 25-35 g) were fasted from noon on the day before the experiment and feeding was resumed 24 hours later.
- the test compound was dissolved in distilled water and orally administered 1 hour before the start of refeeding. Food consumption was measured over 4 hours after refeeding.
- the compounds of the present invention significantly reduced the food intake induced by fasting as compared to the control group to which only distilled water was orally administered.
- Test example 4 Continuous injection test for obese diseased animals
- mice Male ob / ob mice (8 weeks old, 41 g-53 g) were treated daily with test compound dissolved in distilled water. Oral administration was performed twice a week for 2 weeks, and the amount of food consumed was measured. The compound of the present invention significantly reduced the amount of food consumed as compared with the control group to which only distilled water was orally administered. In addition, as a result of measuring blood parameters at the end of continuous administration, the compound of the present invention reduced glucose, insulin, lipid and corticosterone concentrations as compared with the control group to which only distilled water was orally administered. Industrial applicability
- the compounds represented by the general formula (I) or (IV) of the present invention include, for example, diseases involving NPY, particularly various diseases involving NPY / Y5 receptor, such as bulimia and cancer patients.
- Eating regulators such as anorexia, central illnesses such as depression, epilepsy and dementia, and metabolic diseases such as obesity, diabetes, hypercholesterolemia, hyperlipidemia, arteriosclerosis, and hormonal abnormalities It is useful as an active ingredient of a medicament for treatment and / or prevention.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Ophthalmology & Optometry (AREA)
- Endocrinology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Emergency Medicine (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Child & Adolescent Psychology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description







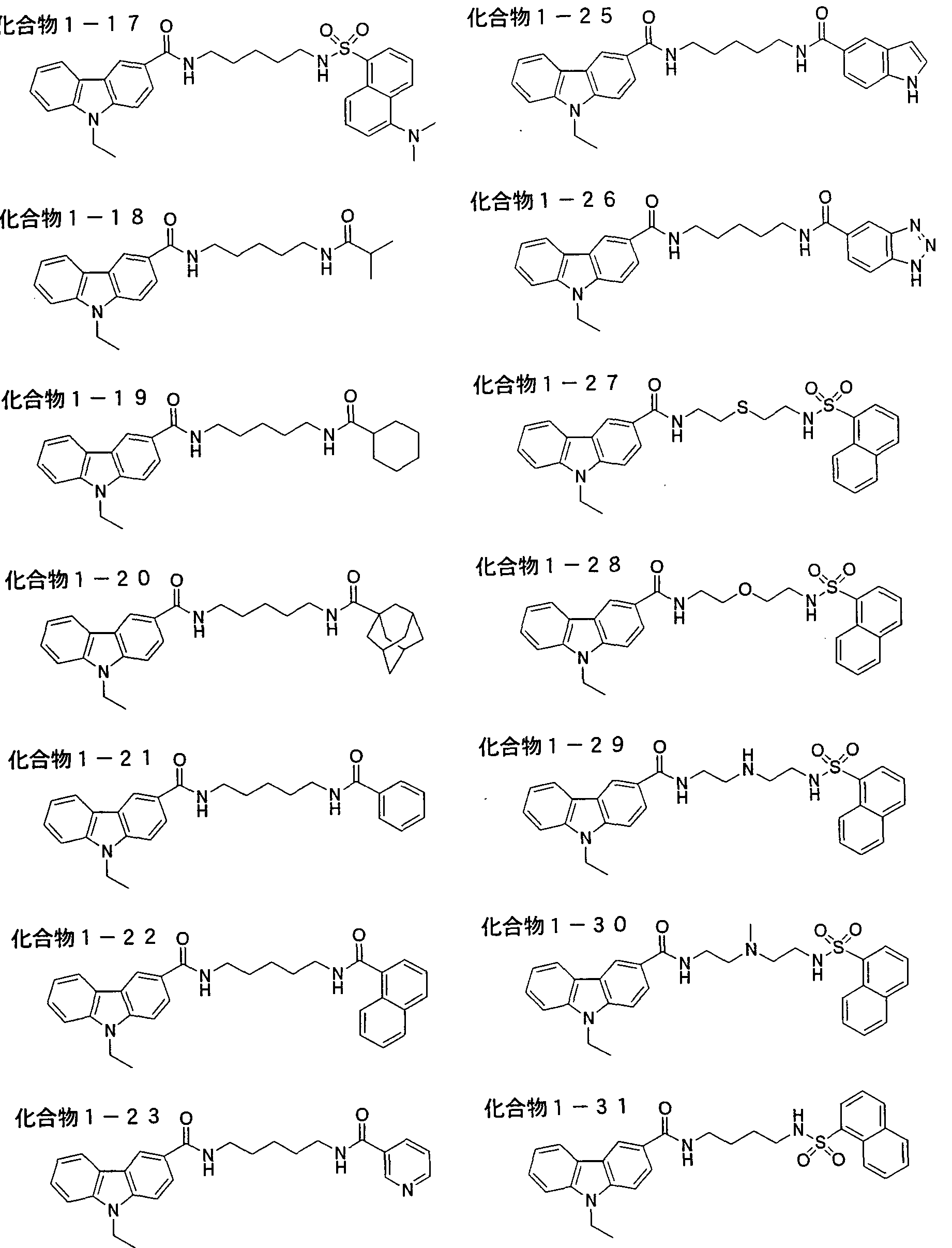











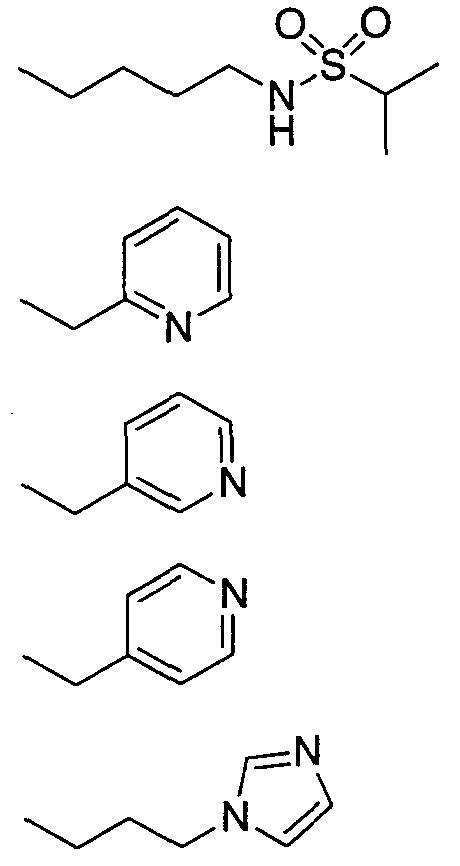



























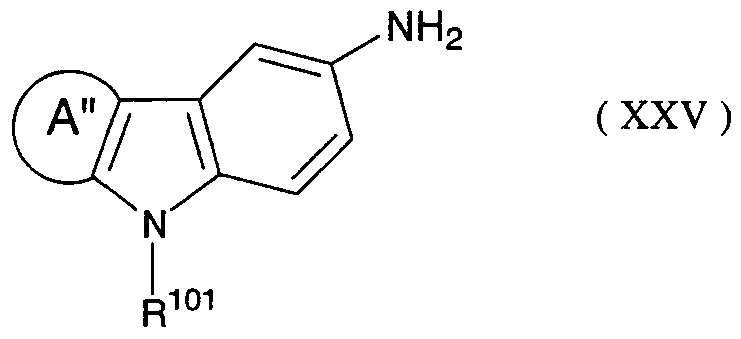


Claims






Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU38400/00A AU3840000A (en) | 1999-04-20 | 2000-04-20 | Tricyclic compounds |
| US09/926,355 US6713473B1 (en) | 1999-04-20 | 2000-04-20 | Tricyclic compounds |
| EP00917373A EP1184373A4 (en) | 1999-04-20 | 2000-04-20 | TRICYCLIC CONNECTIONS |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP11169899 | 1999-04-20 | ||
| JP11/111698 | 1999-04-20 | ||
| JP20022899 | 1999-07-14 | ||
| JP11/200228 | 1999-07-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000063171A1 true WO2000063171A1 (fr) | 2000-10-26 |
Family
ID=26451032
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2000/002573 WO2000063171A1 (fr) | 1999-04-20 | 2000-04-20 | Composes tricycliques |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US6713473B1 (ja) |
| EP (1) | EP1184373A4 (ja) |
| AU (1) | AU3840000A (ja) |
| WO (1) | WO2000063171A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001037826A1 (fr) * | 1999-11-26 | 2001-05-31 | Shionogi & Co., Ltd. | Antagonistes npyy5 |
| US6399631B1 (en) | 1999-07-23 | 2002-06-04 | Pfizer Inc. | Carbazole neuropeptide Y5 antagonists |
| WO2002051806A1 (en) * | 2000-12-22 | 2002-07-04 | Astrazeneca Ab | Carbazole derivatives and their use as neuropeptide y5 receptor ligands |
| WO2002096902A1 (en) * | 2001-05-29 | 2002-12-05 | Boehringer Ingelheim International Gmbh | Carbazole derivatives and their use for the preparation of pharmaceutical compositions for the treatment of npy related diseases |
| WO2003022849A1 (en) * | 2001-09-11 | 2003-03-20 | Astrazeneca Ab | Carbazole derivatives and their use as npy5 receptor antagonists |
| EP1343503A1 (en) * | 2000-12-21 | 2003-09-17 | Schering Corporation | Heteroaryl urea neuropeptide y y5 receptor antagonists |
| US6723730B2 (en) | 2000-07-20 | 2004-04-20 | Neurogen Corporation | Capsaicin receptor ligands |
| US6967216B2 (en) | 2000-05-05 | 2005-11-22 | Astrazeneca Ab | Amino substituted dibenzothiophene derivatives for the treatment of disorders mediated by NP Y5 receptor |
| US7309704B2 (en) | 2000-12-21 | 2007-12-18 | Schering Corporation | Heteroaryl urea neuropeptide Y Y5 receptor antagonists |
| JP2012500197A (ja) * | 2008-08-15 | 2012-01-05 | ジョージタウン ユニバーシティー | Rassf1a発現およびヒト癌細胞増殖の蛍光調節剤 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6958347B2 (en) | 2002-12-18 | 2005-10-25 | Pfizer Inc. | Aminophenanthridinone and aminophenanthridine as NPY-5 antagonists |
| GB2422828A (en) * | 2005-02-03 | 2006-08-09 | Hunter Fleming Ltd | Tricyclic cytoprotective compounds comprising an indole residue |
| CN1807413B (zh) * | 2005-09-28 | 2010-05-05 | 中国医学科学院医药生物技术研究所 | 咔唑磺酰胺衍生物及其制备方法 |
| TW200808807A (en) * | 2006-03-02 | 2008-02-16 | Incyte Corp | Modulators of 11-β hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| WO2007130898A1 (en) * | 2006-05-01 | 2007-11-15 | Incyte Corporation | TETRASUBSTITUTED UREAS AS MODULATORS OF 11-β HYDROXYL STEROID DEHYDROGENASE TYPE 1 |
| JP5220013B2 (ja) * | 2006-08-07 | 2013-06-26 | アクテリオン ファーマシューティカルズ リミテッド | (3−アミノ−1,2,3,4−テトラヒドロ−9h−カルバゾール−9−イル)−酢酸誘導体 |
| WO2008119017A1 (en) * | 2007-03-28 | 2008-10-02 | High Point Pharmaceuticals, Llc | 11beta-hsd1 active compounds |
| WO2010057101A2 (en) * | 2008-11-17 | 2010-05-20 | Schering Corporation | Compounds useful as hiv blockers |
| EP2887804B1 (en) | 2012-05-22 | 2021-01-27 | Trustees of Dartmouth College | Cycloalkanyl[b]indoles useful as glp-1 modulators |
| US11053255B2 (en) | 2015-06-22 | 2021-07-06 | Georgetown University | Synthesis of mahanine and related compounds |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0555824A1 (de) * | 1992-02-13 | 1993-08-18 | Dr. Karl Thomae GmbH | N-alpha-arylsulfonylierte Benzimidazolyl-alaninamid Derivate, diese enthaltende Arzneimittel sowie Verfahren zu ihrer Herstellung |
| WO1997009308A1 (en) * | 1995-09-01 | 1997-03-13 | Eli Lilly And Company | Indolyl neuropeptide y receptor antagonists |
| JPH09157253A (ja) * | 1995-12-12 | 1997-06-17 | Yamanouchi Pharmaceut Co Ltd | 新規アミノ酸誘導体 |
| WO1998006717A1 (en) * | 1996-08-12 | 1998-02-19 | Neurogen Corporation | Fused indolecarboxamides: dopamine receptor subtype specific ligands |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3932456A (en) | 1969-06-16 | 1976-01-13 | Richardson-Merrell Inc. | Bis-basic esters and amides of carbazole |
| JPS4854061A (ja) | 1971-11-15 | 1973-07-30 | ||
| DE2731175C3 (de) | 1977-07-09 | 1980-12-11 | Hoechst Ag, 6000 Frankfurt | Pigmentzubereitungen |
| JPH05507942A (ja) | 1991-03-07 | 1993-11-11 | ジー.ディー.サール アンド カンパニー | 新規セロトニン作動性剤としての新規なmeso―アザ環式芳香族酸アミド化合物およびエステル化合物 |
| GB9226566D0 (en) | 1992-12-21 | 1993-02-17 | Smithkline Beecham Plc | Compounds |
| CA2167154A1 (en) | 1993-08-10 | 1995-02-16 | Sarkis Barret Kalindjian | Gastrin and cck receptor ligands |
| US5602024A (en) | 1994-12-02 | 1997-02-11 | Synaptic Pharmaceutical Corporation | DNA encoding a hypothalamic atypical neuropeptide Y/peptide YY receptor (Y5) and uses thereof |
| JPH08301846A (ja) | 1995-05-01 | 1996-11-19 | Nippon Steel Chem Co Ltd | カルバゾール誘導体、その製造方法及びこれを有効成分とする農薬用殺菌剤 |
| EP0749962B1 (en) | 1995-06-23 | 2000-11-02 | Eli Lilly And Company | 6-substituted-1,2,3,4-tetrahydro-9H-carbazoles and 7-substituted-10H-cyclohepta(7,6-B)indoles |
| WO1997019682A1 (en) | 1995-12-01 | 1997-06-05 | Synaptic Pharmaceutical Corporation | Aryl sulfonamide and sulfamide derivatives and uses thereof |
| WO1997020821A1 (en) | 1995-12-01 | 1997-06-12 | Novartis Ag | Heteroaryl derivatives |
| AU7626496A (en) | 1995-12-01 | 1997-06-27 | Ciba-Geigy Ag | Heteroaryl compounds |
| WO1997020823A2 (en) | 1995-12-01 | 1997-06-12 | Novartis Ag | 2-amino quinazoline derivatives as npy receptor antagonists |
| AU7692896A (en) | 1995-12-01 | 1997-06-27 | Novartis Ag | Quinazolin-2,4-diazirines as NPY receptor antagonist |
| AU2381397A (en) | 1996-04-19 | 1997-11-12 | Novo Nordisk A/S | Modulators of molecules with phosphotyrosine recognition units |
| AU3295297A (en) | 1996-06-04 | 1998-01-05 | Synaptic Pharmaceutical Corporation | Methods of modifying feeding behavior, compounds useful in such methods, and dna encoding a hypothalamic atypical neuropeptide y/peptide yy receptor (y5) |
| US5708187A (en) | 1996-06-27 | 1998-01-13 | Eli Lilly And Company | 6-substituted-1,2,3,4-tetrahydro-9H-carbazoles and 7-substituted-10H-cyclohepta 7,6-B!indoles: New 5-HT1F agonists |
| KR100771092B1 (ko) | 1996-07-08 | 2007-10-30 | 기린 파마 가부시끼가이샤 | 칼슘 수용체 활성 화합물 |
| US5962473A (en) | 1996-08-16 | 1999-10-05 | Eli Lilly And Company | Methods of treating or ameliorating the symptoms of common cold or allergic rhinitis with serotonin 5-HT1F |
| AU4074897A (en) | 1996-09-18 | 1998-04-14 | Eli Lilly And Company | A method for the prevention of migraine |
| US6043246A (en) | 1996-12-03 | 2000-03-28 | Banyu Pharmaceutical Co., Ltd. | Urea derivatives |
| DE69719544T2 (de) | 1996-12-12 | 2003-11-06 | Banyu Pharmaceutical Co., Ltd. | Pyrazolderivate |
| CA2274593A1 (en) | 1996-12-13 | 1998-06-18 | Banyu Pharmaceutical Co., Ltd. | Novel aminopyrazole derivatives |
| AU7738198A (en) | 1996-12-15 | 1998-07-15 | Banyu Pharmaceutical Co., Ltd. | Aminopyrazole derivatives |
| AU6267198A (en) | 1997-02-14 | 1998-09-08 | Bayer Corporation | Amides as npy5 receptor antagonists |
| JP2000510164A (ja) | 1997-02-14 | 2000-08-08 | バイエル・コーポレーシヨン | 選択的神経ペプチドy受容体アンタゴニストとしてのアミド誘導体 |
| WO1998040356A1 (fr) | 1997-03-12 | 1998-09-17 | Banyu Pharmaceutical Co., Ltd. | Medicaments contenant des derives d'aminopyridine comme ingredient actif |
| JP4228398B2 (ja) | 1997-04-23 | 2009-02-25 | 萬有製薬株式会社 | 神経ペプチドy受容体拮抗剤 |
| ID23053A (id) | 1997-06-04 | 2000-01-20 | Lilly Co Eli | Karboksamida yang digunakan sebagai agonis 5-ht <if> |
| AU1262199A (en) | 1997-11-28 | 1999-06-16 | Banyu Pharmaceutical Co., Ltd. | Antihyperlipemic agents |
-
2000
- 2000-04-20 AU AU38400/00A patent/AU3840000A/en not_active Abandoned
- 2000-04-20 WO PCT/JP2000/002573 patent/WO2000063171A1/ja active Application Filing
- 2000-04-20 EP EP00917373A patent/EP1184373A4/en not_active Withdrawn
- 2000-04-20 US US09/926,355 patent/US6713473B1/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0555824A1 (de) * | 1992-02-13 | 1993-08-18 | Dr. Karl Thomae GmbH | N-alpha-arylsulfonylierte Benzimidazolyl-alaninamid Derivate, diese enthaltende Arzneimittel sowie Verfahren zu ihrer Herstellung |
| WO1997009308A1 (en) * | 1995-09-01 | 1997-03-13 | Eli Lilly And Company | Indolyl neuropeptide y receptor antagonists |
| JPH09157253A (ja) * | 1995-12-12 | 1997-06-17 | Yamanouchi Pharmaceut Co Ltd | 新規アミノ酸誘導体 |
| WO1998006717A1 (en) * | 1996-08-12 | 1998-02-19 | Neurogen Corporation | Fused indolecarboxamides: dopamine receptor subtype specific ligands |
Non-Patent Citations (3)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 55, Columbus, Ohio, US; abstract no. 18702E, RN=102659-65-4, PANOS GRAMMATICAKIS: "Preparation and absorption in the visible and ultraviolet region of various amino and nitro N-substituted-1,2,3,4-tetrahydrocarbazoles" XP002932485 * |
| COMPT. REND., vol. 251, 1960, pages 2728 - 2730 * |
| See also references of EP1184373A4 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6399631B1 (en) | 1999-07-23 | 2002-06-04 | Pfizer Inc. | Carbazole neuropeptide Y5 antagonists |
| US8115027B2 (en) | 1999-11-26 | 2012-02-14 | Shionogi & Co., Ltd. | NPY Y5 antagonist |
| US7781461B2 (en) | 1999-11-26 | 2010-08-24 | Yasuyuki Kawanishi | NPY Y5 antagonist |
| EP2014285A1 (en) * | 1999-11-26 | 2009-01-14 | Shionogi&Co., Ltd. | NPYY5 antagonists |
| US6699891B1 (en) | 1999-11-26 | 2004-03-02 | Shionogi & Co., Ltd. | Npyy5 antagonists |
| US7265130B2 (en) | 1999-11-26 | 2007-09-04 | Shionogi & Co., Ltd. | NPY Y5 antagonist |
| WO2001037826A1 (fr) * | 1999-11-26 | 2001-05-31 | Shionogi & Co., Ltd. | Antagonistes npyy5 |
| US6967216B2 (en) | 2000-05-05 | 2005-11-22 | Astrazeneca Ab | Amino substituted dibenzothiophene derivatives for the treatment of disorders mediated by NP Y5 receptor |
| US7332492B2 (en) | 2000-05-05 | 2008-02-19 | Astrazeneca Ab | Amino substituted dibenzothiophene derivatives for the treatment of disorders mediated by NP Y5 receptor |
| US6723730B2 (en) | 2000-07-20 | 2004-04-20 | Neurogen Corporation | Capsaicin receptor ligands |
| EP1343503A4 (en) * | 2000-12-21 | 2004-03-17 | Schering Corp | HETEROARYL-urea neuropeptide Y Y5 receptor antagonists |
| US7309704B2 (en) | 2000-12-21 | 2007-12-18 | Schering Corporation | Heteroaryl urea neuropeptide Y Y5 receptor antagonists |
| EP1343503A1 (en) * | 2000-12-21 | 2003-09-17 | Schering Corporation | Heteroaryl urea neuropeptide y y5 receptor antagonists |
| WO2002051806A1 (en) * | 2000-12-22 | 2002-07-04 | Astrazeneca Ab | Carbazole derivatives and their use as neuropeptide y5 receptor ligands |
| WO2002096902A1 (en) * | 2001-05-29 | 2002-12-05 | Boehringer Ingelheim International Gmbh | Carbazole derivatives and their use for the preparation of pharmaceutical compositions for the treatment of npy related diseases |
| US7408064B2 (en) | 2001-09-11 | 2008-08-05 | Astrazeneca Ab | Carbazole derivatives and their use as NPY5 receptor antagonists |
| WO2003022849A1 (en) * | 2001-09-11 | 2003-03-20 | Astrazeneca Ab | Carbazole derivatives and their use as npy5 receptor antagonists |
| JP2012500197A (ja) * | 2008-08-15 | 2012-01-05 | ジョージタウン ユニバーシティー | Rassf1a発現およびヒト癌細胞増殖の蛍光調節剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1184373A1 (en) | 2002-03-06 |
| US6713473B1 (en) | 2004-03-30 |
| AU3840000A (en) | 2000-11-02 |
| EP1184373A4 (en) | 2004-10-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2000063171A1 (fr) | Composes tricycliques | |
| US11639353B2 (en) | Cyclobutanes- and azetidine-containing mono and spirocyclic compounds as αV integrin inhibitors | |
| AU761719B2 (en) | Antiviral indoleoxoacetyl piperazine derivatives | |
| RU2195455C2 (ru) | Замещенные 1-фенил-3-карбоксамиды и их фармацевтически приемлемые соли, способ их получения, промежуточные соединения и фармацевтическая композиция на их основе, обладающая сродством к человеческим рецепторам нейротензина | |
| US7442693B2 (en) | Diazepine compounds as ligands of the melanocortin 1 and/or 4 receptors | |
| WO2007055418A1 (ja) | アザ置換スピロ誘導体 | |
| TW200404796A (en) | Nitrogen-containing compound | |
| MXPA01011884A (es) | Nuevos compuestos farmaceuticamente activos.. | |
| EP1255751A2 (fr) | Derives de 1,3-dihydro-2h-indol-2-one et leur utilisation en tant que ligands des recepteurs v1b ou v1b et v1a de l'arginine-vasopressine | |
| FR2827604A1 (fr) | Nouveaux derives de 1-phenylsulfonyl-1,3-dihydro-2h-indol-2- one, un procede pour leur preparation et les compositions pharmaceutiques en contenant | |
| US6057335A (en) | Pyrazole derivatives | |
| JPH049396A (ja) | 新規化合物、その製法及びそれを含む医薬組成物 | |
| HU225157B1 (en) | 1,3-dihydro-2h-indol-2-one derivatives, process for preparation of the compounds and pharmaceutical compositions containing the same | |
| FR2805536A1 (fr) | Nouveaux derives de 1,3-dihydro-2h-indol-2-one, un procede pour leur preparation et les compositions pharmaceutiques en contenant | |
| FR2722190A1 (fr) | Derives de 1-benzyl-1,3-dihydro-2h-benzimidazol-2-one, leur preparation, les compositions pharmaceutique en contenant | |
| TW396155B (en) | Novel triazole derivatives, process for their preparation and pharmaceutical compositions containing them | |
| WO1998008867A1 (fr) | Nouveaux derives de peptides presentant un residu de thiazolyle-alanine | |
| FR2927625A1 (fr) | Nouveaux derives de 3-aminoalkyl-1,3-dihydro-2h-indol-2-one, leur preparation et leur application en therapeutique | |
| JPH09510720A (ja) | エンドセリンレセプターのアンタゴニスト | |
| CZ294233B6 (cs) | Acylaminoalkenylenamidové deriváty, způsob jejich přípravy a farmaceutické prostředky, které je obsahují | |
| KR20080065674A (ko) | 신규한 인돌 함유 베타 효능제, 이의 제조 방법 및약제로서의 이의 용도 | |
| TWI258469B (en) | Aryl-substituted alicyclic compounds and pharmaceutical composition containing the same | |
| TW593337B (en) | Compounds with growth hormone releasing properties | |
| CN101087757B (zh) | 二肽基肽酶ⅳ抑制剂、含有它们的药用组合物及其制备方法 | |
| JP4679778B2 (ja) | アミドスピロピペリジン類による成長ホルモン放出の促進 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 612266 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000917373 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09926355 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000917373 Country of ref document: EP |