WO1999032634A2 - Compositions derived from mycobacterium vaccae and methods for their use - Google Patents
Compositions derived from mycobacterium vaccae and methods for their use Download PDFInfo
- Publication number
- WO1999032634A2 WO1999032634A2 PCT/NZ1998/000189 NZ9800189W WO9932634A2 WO 1999032634 A2 WO1999032634 A2 WO 1999032634A2 NZ 9800189 W NZ9800189 W NZ 9800189W WO 9932634 A2 WO9932634 A2 WO 9932634A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- vaccae
- cells
- polypeptide
- seq
- antigen
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 95
- 241000187644 Mycobacterium vaccae Species 0.000 title claims abstract description 81
- 239000000203 mixture Substances 0.000 title claims abstract description 44
- 239000000427 antigen Substances 0.000 claims abstract description 147
- 108091007433 antigens Proteins 0.000 claims abstract description 147
- 102000036639 antigens Human genes 0.000 claims abstract description 147
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 68
- 239000000706 filtrate Substances 0.000 claims abstract description 58
- 238000011282 treatment Methods 0.000 claims abstract description 57
- 230000028993 immune response Effects 0.000 claims abstract description 37
- 238000001514 detection method Methods 0.000 claims abstract description 24
- 239000002253 acid Substances 0.000 claims abstract description 20
- 150000007513 acids Chemical class 0.000 claims abstract description 14
- 208000035473 Communicable disease Diseases 0.000 claims abstract description 13
- 229920000189 Arabinogalactan Polymers 0.000 claims abstract description 12
- 235000019312 arabinogalactan Nutrition 0.000 claims abstract description 12
- SATHPVQTSSUFFW-UHFFFAOYSA-N 4-[6-[(3,5-dihydroxy-4-methoxyoxan-2-yl)oxymethyl]-3,5-dihydroxy-4-methoxyoxan-2-yl]oxy-2-(hydroxymethyl)-6-methyloxane-3,5-diol Chemical compound OC1C(OC)C(O)COC1OCC1C(O)C(OC)C(O)C(OC2C(C(CO)OC(C)C2O)O)O1 SATHPVQTSSUFFW-UHFFFAOYSA-N 0.000 claims abstract description 11
- 239000001904 Arabinogalactan Substances 0.000 claims abstract description 11
- 230000002708 enhancing effect Effects 0.000 claims abstract description 10
- 208000026278 immune system disease Diseases 0.000 claims abstract description 9
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 211
- 108090000623 proteins and genes Proteins 0.000 claims description 208
- 229920001184 polypeptide Polymers 0.000 claims description 204
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 202
- 102000004169 proteins and genes Human genes 0.000 claims description 157
- 210000004027 cell Anatomy 0.000 claims description 137
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 66
- 239000002671 adjuvant Substances 0.000 claims description 63
- 201000010099 disease Diseases 0.000 claims description 47
- 239000000523 sample Substances 0.000 claims description 42
- 208000006673 asthma Diseases 0.000 claims description 40
- 229960005486 vaccine Drugs 0.000 claims description 35
- 201000004681 Psoriasis Diseases 0.000 claims description 32
- 230000002163 immunogen Effects 0.000 claims description 30
- 108091033319 polynucleotide Proteins 0.000 claims description 30
- 102000040430 polynucleotide Human genes 0.000 claims description 30
- 239000002157 polynucleotide Substances 0.000 claims description 30
- 239000002773 nucleotide Substances 0.000 claims description 28
- 125000003729 nucleotide group Chemical group 0.000 claims description 28
- 210000003491 skin Anatomy 0.000 claims description 26
- 230000004044 response Effects 0.000 claims description 24
- 239000003153 chemical reaction reagent Substances 0.000 claims description 22
- 208000035475 disorder Diseases 0.000 claims description 20
- 208000027531 mycobacterial infectious disease Diseases 0.000 claims description 20
- 239000008194 pharmaceutical composition Substances 0.000 claims description 20
- 206010062207 Mycobacterial infection Diseases 0.000 claims description 18
- 239000013604 expression vector Substances 0.000 claims description 16
- 239000007787 solid Substances 0.000 claims description 16
- 239000012472 biological sample Substances 0.000 claims description 15
- 102000037865 fusion proteins Human genes 0.000 claims description 15
- 108020001507 fusion proteins Proteins 0.000 claims description 15
- 125000006853 reporter group Chemical group 0.000 claims description 15
- 210000002966 serum Anatomy 0.000 claims description 14
- 230000015788 innate immune response Effects 0.000 claims description 13
- 210000002345 respiratory system Anatomy 0.000 claims description 11
- 241000588724 Escherichia coli Species 0.000 claims description 10
- 239000011230 binding agent Substances 0.000 claims description 9
- 210000004369 blood Anatomy 0.000 claims description 8
- 239000008280 blood Substances 0.000 claims description 8
- 102000004190 Enzymes Human genes 0.000 claims description 7
- 108090000790 Enzymes Proteins 0.000 claims description 7
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 claims description 6
- 238000009007 Diagnostic Kit Methods 0.000 claims description 5
- 239000002245 particle Substances 0.000 claims description 5
- 208000022535 Infectious Skin disease Diseases 0.000 claims description 4
- 240000004808 Saccharomyces cerevisiae Species 0.000 claims description 4
- 210000001175 cerebrospinal fluid Anatomy 0.000 claims description 4
- 210000002381 plasma Anatomy 0.000 claims description 4
- 210000003296 saliva Anatomy 0.000 claims description 4
- 210000004927 skin cell Anatomy 0.000 claims description 4
- 210000002700 urine Anatomy 0.000 claims description 4
- 229960002685 biotin Drugs 0.000 claims description 3
- 235000020958 biotin Nutrition 0.000 claims description 3
- 239000011616 biotin Substances 0.000 claims description 3
- 241000238631 Hexapoda Species 0.000 claims description 2
- 108090001090 Lectins Proteins 0.000 claims description 2
- 102000004856 Lectins Human genes 0.000 claims description 2
- 101710120037 Toxin CcdB Proteins 0.000 claims description 2
- 239000002523 lectin Substances 0.000 claims description 2
- 210000004962 mammalian cell Anatomy 0.000 claims description 2
- 229940072221 immunoglobulins Drugs 0.000 claims 1
- 210000005253 yeast cell Anatomy 0.000 claims 1
- 206010028980 Neoplasm Diseases 0.000 abstract description 13
- 201000011510 cancer Diseases 0.000 abstract description 6
- 230000002265 prevention Effects 0.000 abstract description 4
- 235000018102 proteins Nutrition 0.000 description 150
- 201000008827 tuberculosis Diseases 0.000 description 111
- 101000865179 Phyllomedusa distincta Dermaseptin-DI3 Proteins 0.000 description 98
- 241000699670 Mus sp. Species 0.000 description 94
- 210000001744 T-lymphocyte Anatomy 0.000 description 78
- 230000000694 effects Effects 0.000 description 67
- 230000014509 gene expression Effects 0.000 description 51
- 108020004414 DNA Proteins 0.000 description 45
- 108091028043 Nucleic acid sequence Proteins 0.000 description 41
- 208000015181 infectious disease Diseases 0.000 description 39
- 239000007924 injection Substances 0.000 description 38
- 238000002347 injection Methods 0.000 description 38
- 229940092253 ovalbumin Drugs 0.000 description 37
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 36
- 238000004519 manufacturing process Methods 0.000 description 36
- 239000002953 phosphate buffered saline Substances 0.000 description 36
- 108010074328 Interferon-gamma Proteins 0.000 description 35
- 108010058846 Ovalbumin Proteins 0.000 description 35
- 210000002540 macrophage Anatomy 0.000 description 34
- 239000012528 membrane Substances 0.000 description 33
- 238000002649 immunization Methods 0.000 description 30
- 210000004379 membrane Anatomy 0.000 description 29
- 210000004072 lung Anatomy 0.000 description 27
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 26
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 26
- 239000002609 medium Substances 0.000 description 25
- 238000002360 preparation method Methods 0.000 description 25
- 241000282693 Cercopithecidae Species 0.000 description 23
- 102100037850 Interferon gamma Human genes 0.000 description 23
- 241001467552 Mycobacterium bovis BCG Species 0.000 description 22
- 210000004443 dendritic cell Anatomy 0.000 description 22
- 239000012634 fragment Substances 0.000 description 22
- 108010065805 Interleukin-12 Proteins 0.000 description 21
- 102000013462 Interleukin-12 Human genes 0.000 description 21
- 229940117681 interleukin-12 Drugs 0.000 description 21
- 238000012360 testing method Methods 0.000 description 21
- 238000003556 assay Methods 0.000 description 20
- 229960000190 bacillus calmette–guérin vaccine Drugs 0.000 description 20
- 150000001875 compounds Chemical class 0.000 description 19
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- 229940024606 amino acid Drugs 0.000 description 18
- 235000001014 amino acid Nutrition 0.000 description 18
- 150000001413 amino acids Chemical class 0.000 description 18
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 18
- 230000000638 stimulation Effects 0.000 description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 16
- 229930186217 Glycolipid Natural products 0.000 description 16
- 108091034117 Oligonucleotide Proteins 0.000 description 16
- 238000005119 centrifugation Methods 0.000 description 16
- 230000003053 immunization Effects 0.000 description 16
- 210000001165 lymph node Anatomy 0.000 description 16
- 238000000746 purification Methods 0.000 description 16
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- 238000002965 ELISA Methods 0.000 description 15
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 15
- 241001465754 Metazoa Species 0.000 description 15
- 210000003979 eosinophil Anatomy 0.000 description 15
- 210000000822 natural killer cell Anatomy 0.000 description 15
- 230000035755 proliferation Effects 0.000 description 15
- 102000004127 Cytokines Human genes 0.000 description 14
- 108090000695 Cytokines Proteins 0.000 description 14
- 239000000499 gel Substances 0.000 description 14
- 230000003902 lesion Effects 0.000 description 14
- 208000020816 lung neoplasm Diseases 0.000 description 14
- 208000004631 alopecia areata Diseases 0.000 description 13
- 239000008103 glucose Substances 0.000 description 13
- 230000001965 increasing effect Effects 0.000 description 13
- 230000001939 inductive effect Effects 0.000 description 13
- 238000002955 isolation Methods 0.000 description 13
- 238000003752 polymerase chain reaction Methods 0.000 description 13
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 13
- 201000000306 sarcoidosis Diseases 0.000 description 13
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 12
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 12
- 102000008070 Interferon-gamma Human genes 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 12
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 12
- 210000003719 b-lymphocyte Anatomy 0.000 description 12
- 239000000872 buffer Substances 0.000 description 12
- 238000002474 experimental method Methods 0.000 description 12
- 238000001155 isoelectric focusing Methods 0.000 description 12
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 11
- 206010061218 Inflammation Diseases 0.000 description 11
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 11
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 11
- 230000000890 antigenic effect Effects 0.000 description 11
- 238000006243 chemical reaction Methods 0.000 description 11
- 231100000433 cytotoxic Toxicity 0.000 description 11
- 230000001472 cytotoxic effect Effects 0.000 description 11
- 238000000338 in vitro Methods 0.000 description 11
- 230000004054 inflammatory process Effects 0.000 description 11
- 229960003130 interferon gamma Drugs 0.000 description 11
- 230000002829 reductive effect Effects 0.000 description 11
- 241000894007 species Species 0.000 description 11
- 206010003645 Atopy Diseases 0.000 description 10
- 101150013553 CD40 gene Proteins 0.000 description 10
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- 201000004624 Dermatitis Diseases 0.000 description 10
- 206010039085 Rhinitis allergic Diseases 0.000 description 10
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 10
- 201000010105 allergic rhinitis Diseases 0.000 description 10
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 10
- 206010012442 Dermatitis contact Diseases 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 229910021502 aluminium hydroxide Inorganic materials 0.000 description 9
- 230000003321 amplification Effects 0.000 description 9
- 238000010367 cloning Methods 0.000 description 9
- 239000003246 corticosteroid Substances 0.000 description 9
- 229960001334 corticosteroids Drugs 0.000 description 9
- 238000011161 development Methods 0.000 description 9
- 230000018109 developmental process Effects 0.000 description 9
- 238000010790 dilution Methods 0.000 description 9
- 239000012895 dilution Substances 0.000 description 9
- 210000004698 lymphocyte Anatomy 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 238000003199 nucleic acid amplification method Methods 0.000 description 9
- 230000036961 partial effect Effects 0.000 description 9
- 210000004989 spleen cell Anatomy 0.000 description 9
- 230000000699 topical effect Effects 0.000 description 9
- 239000013598 vector Substances 0.000 description 9
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 8
- 241000894006 Bacteria Species 0.000 description 8
- 206010004146 Basal cell carcinoma Diseases 0.000 description 8
- 102000009016 Cholera Toxin Human genes 0.000 description 8
- 108010049048 Cholera Toxin Proteins 0.000 description 8
- 229940046168 CpG oligodeoxynucleotide Drugs 0.000 description 8
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 230000014828 interferon-gamma production Effects 0.000 description 8
- 230000019734 interleukin-12 production Effects 0.000 description 8
- 150000002632 lipids Chemical class 0.000 description 8
- 239000008188 pellet Substances 0.000 description 8
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 8
- 230000001681 protective effect Effects 0.000 description 8
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 8
- 230000009257 reactivity Effects 0.000 description 8
- 238000012216 screening Methods 0.000 description 8
- 238000012163 sequencing technique Methods 0.000 description 8
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 8
- 238000010561 standard procedure Methods 0.000 description 8
- 230000009885 systemic effect Effects 0.000 description 8
- 238000011740 C57BL/6 mouse Methods 0.000 description 7
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 7
- 108010036949 Cyclosporine Proteins 0.000 description 7
- 108090000978 Interleukin-4 Proteins 0.000 description 7
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 7
- 208000002029 allergic contact dermatitis Diseases 0.000 description 7
- 230000004663 cell proliferation Effects 0.000 description 7
- 238000002512 chemotherapy Methods 0.000 description 7
- 229960001265 ciclosporin Drugs 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 238000009169 immunotherapy Methods 0.000 description 7
- 238000011534 incubation Methods 0.000 description 7
- 201000005202 lung cancer Diseases 0.000 description 7
- 201000001441 melanoma Diseases 0.000 description 7
- 230000001185 psoriatic effect Effects 0.000 description 7
- 210000000952 spleen Anatomy 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- 238000002255 vaccination Methods 0.000 description 7
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 6
- 102000005416 ATP-Binding Cassette Transporters Human genes 0.000 description 6
- 108010006533 ATP-Binding Cassette Transporters Proteins 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 229930105110 Cyclosporin A Natural products 0.000 description 6
- 206010014950 Eosinophilia Diseases 0.000 description 6
- 102000014150 Interferons Human genes 0.000 description 6
- 108010050904 Interferons Proteins 0.000 description 6
- 208000019693 Lung disease Diseases 0.000 description 6
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 6
- 230000001580 bacterial effect Effects 0.000 description 6
- 239000000470 constituent Substances 0.000 description 6
- 229930182912 cyclosporin Natural products 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 229940088598 enzyme Drugs 0.000 description 6
- 230000006872 improvement Effects 0.000 description 6
- 210000000265 leukocyte Anatomy 0.000 description 6
- 210000001616 monocyte Anatomy 0.000 description 6
- 229920002401 polyacrylamide Polymers 0.000 description 6
- 206010041823 squamous cell carcinoma Diseases 0.000 description 6
- 238000010186 staining Methods 0.000 description 6
- 230000004936 stimulating effect Effects 0.000 description 6
- 238000007920 subcutaneous administration Methods 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 208000027930 type IV hypersensitivity disease Diseases 0.000 description 6
- 102100038222 60 kDa heat shock protein, mitochondrial Human genes 0.000 description 5
- 108010058432 Chaperonin 60 Proteins 0.000 description 5
- 206010012438 Dermatitis atopic Diseases 0.000 description 5
- 108700026244 Open Reading Frames Proteins 0.000 description 5
- 208000000453 Skin Neoplasms Diseases 0.000 description 5
- 206010041067 Small cell lung cancer Diseases 0.000 description 5
- 206010053613 Type IV hypersensitivity reaction Diseases 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 239000013566 allergen Substances 0.000 description 5
- 210000000612 antigen-presenting cell Anatomy 0.000 description 5
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 5
- 201000008937 atopic dermatitis Diseases 0.000 description 5
- 208000010668 atopic eczema Diseases 0.000 description 5
- 230000006399 behavior Effects 0.000 description 5
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000012228 culture supernatant Substances 0.000 description 5
- 230000009089 cytolysis Effects 0.000 description 5
- 238000003745 diagnosis Methods 0.000 description 5
- 208000024711 extrinsic asthma Diseases 0.000 description 5
- 238000009396 hybridization Methods 0.000 description 5
- 230000002519 immonomodulatory effect Effects 0.000 description 5
- 230000036039 immunity Effects 0.000 description 5
- 230000004957 immunoregulator effect Effects 0.000 description 5
- 239000002198 insoluble material Substances 0.000 description 5
- 210000002510 keratinocyte Anatomy 0.000 description 5
- 238000005374 membrane filtration Methods 0.000 description 5
- 102000039446 nucleic acids Human genes 0.000 description 5
- 108020004707 nucleic acids Proteins 0.000 description 5
- 150000007523 nucleic acids Chemical class 0.000 description 5
- 210000003024 peritoneal macrophage Anatomy 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 230000021986 positive regulation of interferon-gamma production Effects 0.000 description 5
- 230000009465 prokaryotic expression Effects 0.000 description 5
- 230000009696 proliferative response Effects 0.000 description 5
- 238000000926 separation method Methods 0.000 description 5
- 208000000587 small cell lung carcinoma Diseases 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- 230000005951 type IV hypersensitivity Effects 0.000 description 5
- QFVHZQCOUORWEI-UHFFFAOYSA-N 4-[(4-anilino-5-sulfonaphthalen-1-yl)diazenyl]-5-hydroxynaphthalene-2,7-disulfonic acid Chemical compound C=12C(O)=CC(S(O)(=O)=O)=CC2=CC(S(O)(=O)=O)=CC=1N=NC(C1=CC=CC(=C11)S(O)(=O)=O)=CC=C1NC1=CC=CC=C1 QFVHZQCOUORWEI-UHFFFAOYSA-N 0.000 description 4
- 208000023275 Autoimmune disease Diseases 0.000 description 4
- 238000011725 BALB/c mouse Methods 0.000 description 4
- 239000008001 CAPS buffer Substances 0.000 description 4
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 4
- 102000053602 DNA Human genes 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 4
- 108060003951 Immunoglobulin Proteins 0.000 description 4
- 108010002616 Interleukin-5 Proteins 0.000 description 4
- 102000018697 Membrane Proteins Human genes 0.000 description 4
- 108010052285 Membrane Proteins Proteins 0.000 description 4
- 229920001213 Polysorbate 20 Polymers 0.000 description 4
- 239000005700 Putrescine Substances 0.000 description 4
- 206010040914 Skin reaction Diseases 0.000 description 4
- 230000006052 T cell proliferation Effects 0.000 description 4
- 230000005867 T cell response Effects 0.000 description 4
- 241000700605 Viruses Species 0.000 description 4
- 230000002159 abnormal effect Effects 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 208000026935 allergic disease Diseases 0.000 description 4
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 4
- 210000002421 cell wall Anatomy 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 230000001332 colony forming effect Effects 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 239000002299 complementary DNA Substances 0.000 description 4
- 230000016396 cytokine production Effects 0.000 description 4
- 230000007402 cytotoxic response Effects 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 231100000517 death Toxicity 0.000 description 4
- 238000001400 expression cloning Methods 0.000 description 4
- 229930182830 galactose Natural products 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 230000002209 hydrophobic effect Effects 0.000 description 4
- 230000001900 immune effect Effects 0.000 description 4
- 102000018358 immunoglobulin Human genes 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 229960000367 inositol Drugs 0.000 description 4
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 4
- 230000002147 killing effect Effects 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 210000000207 lymphocyte subset Anatomy 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 108020004999 messenger RNA Proteins 0.000 description 4
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 4
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 4
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 4
- 229920000136 polysorbate Polymers 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 210000004761 scalp Anatomy 0.000 description 4
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 4
- 230000001932 seasonal effect Effects 0.000 description 4
- 230000028327 secretion Effects 0.000 description 4
- 208000017520 skin disease Diseases 0.000 description 4
- 230000035483 skin reaction Effects 0.000 description 4
- 231100000430 skin reaction Toxicity 0.000 description 4
- 229940063673 spermidine Drugs 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 238000001356 surgical procedure Methods 0.000 description 4
- 108091005703 transmembrane proteins Proteins 0.000 description 4
- 102000035160 transmembrane proteins Human genes 0.000 description 4
- 102000003390 tumor necrosis factor Human genes 0.000 description 4
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 4
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 3
- IQFYYKKMVGJFEH-OFKYTIFKSA-N 1-[(2r,4s,5r)-4-hydroxy-5-(tritiooxymethyl)oxolan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound C1[C@H](O)[C@@H](CO[3H])O[C@H]1N1C(=O)NC(=O)C(C)=C1 IQFYYKKMVGJFEH-OFKYTIFKSA-N 0.000 description 3
- HVCOBJNICQPDBP-UHFFFAOYSA-N 3-[3-[3,5-dihydroxy-6-methyl-4-(3,4,5-trihydroxy-6-methyloxan-2-yl)oxyoxan-2-yl]oxydecanoyloxy]decanoic acid;hydrate Chemical compound O.OC1C(OC(CC(=O)OC(CCCCCCC)CC(O)=O)CCCCCCC)OC(C)C(O)C1OC1C(O)C(O)C(O)C(C)O1 HVCOBJNICQPDBP-UHFFFAOYSA-N 0.000 description 3
- 102000021527 ATP binding proteins Human genes 0.000 description 3
- 108091011108 ATP binding proteins Proteins 0.000 description 3
- 201000004384 Alopecia Diseases 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- 108090001008 Avidin Proteins 0.000 description 3
- 241000304886 Bacilli Species 0.000 description 3
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 3
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 3
- 206010020751 Hypersensitivity Diseases 0.000 description 3
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 3
- 108010047761 Interferon-alpha Proteins 0.000 description 3
- 102000006992 Interferon-alpha Human genes 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- 206010024229 Leprosy Diseases 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 241000186359 Mycobacterium Species 0.000 description 3
- 241000186366 Mycobacterium bovis Species 0.000 description 3
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 3
- 239000000020 Nitrocellulose Substances 0.000 description 3
- 239000004677 Nylon Substances 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 206010036790 Productive cough Diseases 0.000 description 3
- 108010059712 Pronase Proteins 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 230000005856 abnormality Effects 0.000 description 3
- 201000009961 allergic asthma Diseases 0.000 description 3
- 230000007815 allergy Effects 0.000 description 3
- 231100000360 alopecia Toxicity 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 230000000692 anti-sense effect Effects 0.000 description 3
- 208000003373 basosquamous carcinoma Diseases 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000004202 carbamide Substances 0.000 description 3
- 230000003915 cell function Effects 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 239000011651 chromium Substances 0.000 description 3
- 229910001873 dinitrogen Inorganic materials 0.000 description 3
- 239000002158 endotoxin Substances 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 238000000684 flow cytometry Methods 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 210000003780 hair follicle Anatomy 0.000 description 3
- 230000008004 immune attack Effects 0.000 description 3
- 239000002955 immunomodulating agent Substances 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- 229940079322 interferon Drugs 0.000 description 3
- 229940047124 interferons Drugs 0.000 description 3
- 229920006008 lipopolysaccharide Polymers 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 238000013160 medical therapy Methods 0.000 description 3
- 206010061289 metastatic neoplasm Diseases 0.000 description 3
- 239000004005 microsphere Substances 0.000 description 3
- 150000002772 monosaccharides Chemical class 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 101150056832 mpt70 gene Proteins 0.000 description 3
- 229920001220 nitrocellulos Polymers 0.000 description 3
- 229920001778 nylon Polymers 0.000 description 3
- 244000052769 pathogen Species 0.000 description 3
- 230000001717 pathogenic effect Effects 0.000 description 3
- 210000005259 peripheral blood Anatomy 0.000 description 3
- 239000011886 peripheral blood Substances 0.000 description 3
- 229920003023 plastic Polymers 0.000 description 3
- 239000004033 plastic Substances 0.000 description 3
- 239000011148 porous material Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 230000000770 proinflammatory effect Effects 0.000 description 3
- 238000001959 radiotherapy Methods 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 108020004418 ribosomal RNA Proteins 0.000 description 3
- 239000012723 sample buffer Substances 0.000 description 3
- 210000003802 sputum Anatomy 0.000 description 3
- 208000024794 sputum Diseases 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 229940071127 thioglycolate Drugs 0.000 description 3
- CWERGRDVMFNCDR-UHFFFAOYSA-M thioglycolate(1-) Chemical compound [O-]C(=O)CS CWERGRDVMFNCDR-UHFFFAOYSA-M 0.000 description 3
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 3
- 230000029069 type 2 immune response Effects 0.000 description 3
- 230000003827 upregulation Effects 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 2
- 108020004465 16S ribosomal RNA Proteins 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 2
- YLDCUKJMEKGGFI-QCSRICIXSA-N 4-acetamidobenzoic acid;9-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purin-6-one;1-(dimethylamino)propan-2-ol Chemical compound CC(O)CN(C)C.CC(O)CN(C)C.CC(O)CN(C)C.CC(=O)NC1=CC=C(C(O)=O)C=C1.CC(=O)NC1=CC=C(C(O)=O)C=C1.CC(=O)NC1=CC=C(C(O)=O)C=C1.O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(NC=NC2=O)=C2N=C1 YLDCUKJMEKGGFI-QCSRICIXSA-N 0.000 description 2
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 108010088751 Albumins Proteins 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- 241000588832 Bordetella pertussis Species 0.000 description 2
- 238000009010 Bradford assay Methods 0.000 description 2
- 238000011746 C57BL/6J (JAX™ mouse strain) Methods 0.000 description 2
- 102100035793 CD83 antigen Human genes 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- 102000016938 Catalase Human genes 0.000 description 2
- 108010053835 Catalase Proteins 0.000 description 2
- 101710098119 Chaperonin GroEL 2 Proteins 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 241000991587 Enterovirus C Species 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 2
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 2
- 108090000174 Interleukin-10 Proteins 0.000 description 2
- 108090001005 Interleukin-6 Proteins 0.000 description 2
- 108091092195 Intron Proteins 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- ZFMITUMMTDLWHR-UHFFFAOYSA-N Minoxidil Chemical compound NC1=[N+]([O-])C(N)=CC(N2CCCCC2)=N1 ZFMITUMMTDLWHR-UHFFFAOYSA-N 0.000 description 2
- 241000187480 Mycobacterium smegmatis Species 0.000 description 2
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 2
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 description 2
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 2
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 description 2
- 239000005642 Oleic acid Substances 0.000 description 2
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 239000012614 Q-Sepharose Substances 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- 230000024932 T cell mediated immunity Effects 0.000 description 2
- 210000004241 Th2 cell Anatomy 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 239000011149 active material Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 230000000172 allergic effect Effects 0.000 description 2
- 229940037003 alum Drugs 0.000 description 2
- 150000003862 amino acid derivatives Chemical class 0.000 description 2
- NUZWLKWWNNJHPT-UHFFFAOYSA-N anthralin Chemical compound C1C2=CC=CC(O)=C2C(=O)C2=C1C=CC=C2O NUZWLKWWNNJHPT-UHFFFAOYSA-N 0.000 description 2
- 230000001355 anti-mycobacterial effect Effects 0.000 description 2
- 239000003926 antimycobacterial agent Substances 0.000 description 2
- 229940030999 antipsoriatics Drugs 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 125000006267 biphenyl group Chemical group 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 229940041514 candida albicans extract Drugs 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 210000000038 chest Anatomy 0.000 description 2
- 208000037976 chronic inflammation Diseases 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000012875 competitive assay Methods 0.000 description 2
- 208000010247 contact dermatitis Diseases 0.000 description 2
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 2
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 230000004041 dendritic cell maturation Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 229960002311 dithranol Drugs 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 238000002481 ethanol extraction Methods 0.000 description 2
- 239000000469 ethanolic extract Substances 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 210000004209 hair Anatomy 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 210000004408 hybridoma Anatomy 0.000 description 2
- 230000009390 immune abnormality Effects 0.000 description 2
- 230000005965 immune activity Effects 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000009851 immunogenic response Effects 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 210000003000 inclusion body Anatomy 0.000 description 2
- 239000000411 inducer Substances 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 206010022000 influenza Diseases 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 239000007928 intraperitoneal injection Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- 238000004255 ion exchange chromatography Methods 0.000 description 2
- 239000002085 irritant Substances 0.000 description 2
- 231100000021 irritant Toxicity 0.000 description 2
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 2
- 210000001821 langerhans cell Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 230000015654 memory Effects 0.000 description 2
- 230000001394 metastastic effect Effects 0.000 description 2
- 238000003808 methanol extraction Methods 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 229960003632 minoxidil Drugs 0.000 description 2
- 101150023079 mpt83 gene Proteins 0.000 description 2
- 229950006780 n-acetylglucosamine Drugs 0.000 description 2
- OHDXDNUPVVYWOV-UHFFFAOYSA-N n-methyl-1-(2-naphthalen-1-ylsulfanylphenyl)methanamine Chemical compound CNCC1=CC=CC=C1SC1=CC=CC2=CC=CC=C12 OHDXDNUPVVYWOV-UHFFFAOYSA-N 0.000 description 2
- 239000007922 nasal spray Substances 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 231100000957 no side effect Toxicity 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- 210000003200 peritoneal cavity Anatomy 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 239000004800 polyvinyl chloride Substances 0.000 description 2
- 229920000915 polyvinyl chloride Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 208000023504 respiratory system disease Diseases 0.000 description 2
- 230000037390 scarring Effects 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- 238000010254 subcutaneous injection Methods 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 210000003437 trachea Anatomy 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 239000012137 tryptone Substances 0.000 description 2
- 229960001005 tuberculin Drugs 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 238000000108 ultra-filtration Methods 0.000 description 2
- 108700026220 vif Genes Proteins 0.000 description 2
- 239000012138 yeast extract Substances 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- GMKMEZVLHJARHF-UHFFFAOYSA-N (2R,6R)-form-2.6-Diaminoheptanedioic acid Natural products OC(=O)C(N)CCCC(N)C(O)=O GMKMEZVLHJARHF-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- 108020005097 23S Ribosomal RNA Proteins 0.000 description 1
- 102000041092 ABC transporter family Human genes 0.000 description 1
- 108091060858 ABC transporter family Proteins 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 102000005345 Acetyl-CoA C-acetyltransferase Human genes 0.000 description 1
- 108010006229 Acetyl-CoA C-acetyltransferase Proteins 0.000 description 1
- 108010042708 Acetylmuramyl-Alanyl-Isoglutamine Proteins 0.000 description 1
- 208000000884 Airway Obstruction Diseases 0.000 description 1
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 description 1
- 206010001766 Alopecia totalis Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- 241000024188 Andala Species 0.000 description 1
- 101000787278 Arabidopsis thaliana Valine-tRNA ligase, chloroplastic/mitochondrial 2 Proteins 0.000 description 1
- 101000787296 Arabidopsis thaliana Valine-tRNA ligase, mitochondrial 1 Proteins 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 108020004652 Aspartate-Semialdehyde Dehydrogenase Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 230000003844 B-cell-activation Effects 0.000 description 1
- 238000011731 BALB/cByJ (JAX™ mouse strain) Methods 0.000 description 1
- 208000005440 Basal Cell Neoplasms Diseases 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- FRPHFZCDPYBUAU-UHFFFAOYSA-N Bromocresolgreen Chemical compound CC1=C(Br)C(O)=C(Br)C=C1C1(C=2C(=C(Br)C(O)=C(Br)C=2)C)C2=CC=CC=C2S(=O)(=O)O1 FRPHFZCDPYBUAU-UHFFFAOYSA-N 0.000 description 1
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 101710104159 Chaperonin GroEL Proteins 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- PJWWRFATQTVXHA-UHFFFAOYSA-N Cyclohexylaminopropanesulfonic acid Chemical compound OS(=O)(=O)CCCNC1CCCCC1 PJWWRFATQTVXHA-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 108010041986 DNA Vaccines Proteins 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 239000003298 DNA probe Substances 0.000 description 1
- 229940021995 DNA vaccine Drugs 0.000 description 1
- 206010061619 Deformity Diseases 0.000 description 1
- 101710088334 Diacylglycerol acyltransferase/mycolyltransferase Ag85B Proteins 0.000 description 1
- 101000787280 Dictyostelium discoideum Probable valine-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 201000010374 Down Syndrome Diseases 0.000 description 1
- 206010013786 Dry skin Diseases 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 108010093223 Folylpolyglutamate synthetase Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 102000006354 HLA-DR Antigens Human genes 0.000 description 1
- 108010058597 HLA-DR Antigens Proteins 0.000 description 1
- 241000606768 Haemophilus influenzae Species 0.000 description 1
- 208000025309 Hair disease Diseases 0.000 description 1
- 241001622557 Hesperia Species 0.000 description 1
- 101000599940 Homo sapiens Interferon gamma Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010020633 Hyperglobulinaemia Diseases 0.000 description 1
- 102000015271 Intercellular Adhesion Molecule-1 Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 241000274177 Juniperus sabina Species 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229920006063 Lamide® Polymers 0.000 description 1
- 101001018085 Lysobacter enzymogenes Lysyl endopeptidase Proteins 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 208000005647 Mumps Diseases 0.000 description 1
- MSFSPUZXLOGKHJ-UHFFFAOYSA-N Muraminsaeure Natural products OC(=O)C(C)OC1C(N)C(O)OC(CO)C1O MSFSPUZXLOGKHJ-UHFFFAOYSA-N 0.000 description 1
- 101001044384 Mus musculus Interferon gamma Proteins 0.000 description 1
- 101000928686 Mycobacterium avium Alanine and proline-rich secreted protein Apa Proteins 0.000 description 1
- 101100131080 Mycobacterium bovis (strain ATCC BAA-935 / AF2122/97) mpb70 gene Proteins 0.000 description 1
- 241000186362 Mycobacterium leprae Species 0.000 description 1
- 101000944608 Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Chaperonin GroEL 2 Proteins 0.000 description 1
- 241000204051 Mycoplasma genitalium Species 0.000 description 1
- CLMZMILVSHKNLI-HONWWXKESA-N N-Glycolyl-Muramic Acid Chemical compound OC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)OC(O)[C@@H]1NC(=O)CO CLMZMILVSHKNLI-HONWWXKESA-N 0.000 description 1
- 108091061960 Naked DNA Proteins 0.000 description 1
- 206010028735 Nasal congestion Diseases 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- 101710116435 Outer membrane protein Proteins 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 102000010562 Peptide Elongation Factor G Human genes 0.000 description 1
- 108010077742 Peptide Elongation Factor G Proteins 0.000 description 1
- 102000007079 Peptide Fragments Human genes 0.000 description 1
- 108010033276 Peptide Fragments Proteins 0.000 description 1
- 108010013639 Peptidoglycan Proteins 0.000 description 1
- 108010030544 Peptidyl-Lys metalloendopeptidase Proteins 0.000 description 1
- 108010090127 Periplasmic Proteins Proteins 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- PGFBYAIGHPJFFJ-PWIZWCRZSA-N Plicatic acid Chemical compound C1([C@@H]2[C@](O)([C@](O)(CO)CC=3C=C(C(=CC=32)O)OC)C(O)=O)=CC(O)=C(O)C(OC)=C1 PGFBYAIGHPJFFJ-PWIZWCRZSA-N 0.000 description 1
- PGFBYAIGHPJFFJ-UHFFFAOYSA-N Plicatic acid Natural products C1=2C=C(O)C(OC)=CC=2CC(O)(CO)C(O)(C(O)=O)C1C1=CC(O)=C(O)C(OC)=C1 PGFBYAIGHPJFFJ-UHFFFAOYSA-N 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 101710132632 Protein C4 Proteins 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 208000003251 Pruritus Diseases 0.000 description 1
- 108010007131 Pulmonary Surfactant-Associated Protein B Proteins 0.000 description 1
- 102100032617 Pulmonary surfactant-associated protein B Human genes 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 101710086015 RNA ligase Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 241001068263 Replication competent viruses Species 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- 206010039094 Rhinitis perennial Diseases 0.000 description 1
- 206010039101 Rhinorrhoea Diseases 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 206010039580 Scar Diseases 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 208000035286 Spontaneous Remission Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 241000218638 Thuja plicata Species 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 206010044688 Trisomy 21 Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 206010046306 Upper respiratory tract infection Diseases 0.000 description 1
- 206010046851 Uveitis Diseases 0.000 description 1
- 206010046865 Vaccinia virus infection Diseases 0.000 description 1
- 102000013625 Valine-tRNA Ligase Human genes 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 208000000260 Warts Diseases 0.000 description 1
- 208000021017 Weight Gain Diseases 0.000 description 1
- UZQJVUCHXGYFLQ-AYDHOLPZSA-N [(2s,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-4-[(2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-6-(hydroxymethyl)-4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-6-(hy Chemical compound O([C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1CC[C@]2(C)[C@H]3CC=C4[C@@]([C@@]3(CC[C@H]2[C@@]1(C=O)C)C)(C)CC(O)[C@]1(CCC(CC14)(C)C)C(=O)O[C@H]1[C@@H]([C@@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O[C@H]4[C@@H]([C@@H](O[C@H]5[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O5)O)[C@H](O)[C@@H](CO)O4)O)[C@H](O)[C@@H](CO)O3)O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO)O1)O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O UZQJVUCHXGYFLQ-AYDHOLPZSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- PMZXXNPJQYDFJX-UHFFFAOYSA-N acetonitrile;2,2,2-trifluoroacetic acid Chemical compound CC#N.OC(=O)C(F)(F)F PMZXXNPJQYDFJX-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 229960005339 acitretin Drugs 0.000 description 1
- 230000000240 adjuvant effect Effects 0.000 description 1
- 239000013567 aeroallergen Substances 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 208000037883 airway inflammation Diseases 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- IHUNBGSDBOWDMA-AQFIFDHZSA-N all-trans-acitretin Chemical compound COC1=CC(C)=C(\C=C\C(\C)=C\C=C\C(\C)=C\C(O)=O)C(C)=C1C IHUNBGSDBOWDMA-AQFIFDHZSA-N 0.000 description 1
- 150000004347 all-trans-retinol derivatives Chemical class 0.000 description 1
- 230000000735 allogeneic effect Effects 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 230000006023 anti-tumor response Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 238000011319 anticancer therapy Methods 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 230000002917 arthritic effect Effects 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 238000002820 assay format Methods 0.000 description 1
- 238000000376 autoradiography Methods 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 239000002981 blocking agent Substances 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 229960002882 calcipotriol Drugs 0.000 description 1
- LWQQLNNNIPYSNX-UROSTWAQSA-N calcipotriol Chemical compound C1([C@H](O)/C=C/[C@@H](C)[C@@H]2[C@]3(CCCC(/[C@@H]3CC2)=C\C=C\2C([C@@H](O)C[C@H](O)C/2)=C)C)CC1 LWQQLNNNIPYSNX-UROSTWAQSA-N 0.000 description 1
- 229960005084 calcitriol Drugs 0.000 description 1
- 235000020964 calcitriol Nutrition 0.000 description 1
- 239000011612 calcitriol Substances 0.000 description 1
- GMRQFYUYWCNGIN-NKMMMXOESA-N calcitriol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C GMRQFYUYWCNGIN-NKMMMXOESA-N 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000007910 cell fusion Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000007969 cellular immunity Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 1
- 208000023819 chronic asthma Diseases 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000003501 co-culture Methods 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- -1 cofactors Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 229960000265 cromoglicic acid Drugs 0.000 description 1
- IMZMKUWMOSJXDT-UHFFFAOYSA-N cromoglycic acid Chemical compound O1C(C(O)=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C(O)=O)O2 IMZMKUWMOSJXDT-UHFFFAOYSA-N 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 238000000315 cryotherapy Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 108010005400 cutinase Proteins 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 102000003675 cytokine receptors Human genes 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 1
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 1
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical group O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000015961 delipidation Effects 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- HCIBTBXNLVOFER-UHFFFAOYSA-N diphenylcyclopropenone Chemical compound O=C1C(C=2C=CC=CC=2)=C1C1=CC=CC=C1 HCIBTBXNLVOFER-UHFFFAOYSA-N 0.000 description 1
- 230000009266 disease activity Effects 0.000 description 1
- 208000034653 disorder of pilosebaceous unit Diseases 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 230000037336 dry skin Effects 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 210000000883 ear external Anatomy 0.000 description 1
- 238000013399 early diagnosis Methods 0.000 description 1
- 230000002996 emotional effect Effects 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 239000000147 enterotoxin Substances 0.000 description 1
- 231100000655 enterotoxin Toxicity 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 210000005175 epidermal keratinocyte Anatomy 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 210000003237 epithelioid cell Anatomy 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 210000004709 eyebrow Anatomy 0.000 description 1
- 230000001815 facial effect Effects 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000011152 fibreglass Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229940044627 gamma-interferon Drugs 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 102000006602 glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 1
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 1
- 208000010758 granulomatous inflammation Diseases 0.000 description 1
- 230000003779 hair growth Effects 0.000 description 1
- 230000003659 hair regrowth Effects 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 230000004727 humoral immunity Effects 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000006054 immunological memory Effects 0.000 description 1
- 238000013394 immunophenotyping Methods 0.000 description 1
- 230000001759 immunoprophylactic effect Effects 0.000 description 1
- 229960001438 immunostimulant agent Drugs 0.000 description 1
- 239000003022 immunostimulating agent Substances 0.000 description 1
- 230000003308 immunostimulating effect Effects 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 230000001861 immunosuppressant effect Effects 0.000 description 1
- 229940124589 immunosuppressive drug Drugs 0.000 description 1
- 230000001024 immunotherapeutic effect Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 208000033065 inborn errors of immunity Diseases 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 229960003971 influenza vaccine Drugs 0.000 description 1
- 230000004941 influx Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 230000007803 itching Effects 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- GZQKNULLWNGMCW-PWQABINMSA-N lipid A (E. coli) Chemical compound O1[C@H](CO)[C@@H](OP(O)(O)=O)[C@H](OC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)[C@@H](NC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H]1OC[C@@H]1[C@@H](O)[C@H](OC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](OP(O)(O)=O)O1 GZQKNULLWNGMCW-PWQABINMSA-N 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 229960001226 live attenuated influenza Drugs 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 230000000527 lymphocytic effect Effects 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 210000005015 mediastinal lymph node Anatomy 0.000 description 1
- GMKMEZVLHJARHF-SYDPRGILSA-N meso-2,6-diaminopimelic acid Chemical compound [O-]C(=O)[C@@H]([NH3+])CCC[C@@H]([NH3+])C([O-])=O GMKMEZVLHJARHF-SYDPRGILSA-N 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000007799 mixed lymphocyte reaction assay Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 210000002864 mononuclear phagocyte Anatomy 0.000 description 1
- 229940035032 monophosphoryl lipid a Drugs 0.000 description 1
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 1
- 101150006328 mpb83 gene Proteins 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 208000010805 mumps infectious disease Diseases 0.000 description 1
- BSOQXXWZTUDTEL-ZUYCGGNHSA-N muramyl dipeptide Chemical compound OC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)O[C@@H](O)[C@@H]1NC(C)=O BSOQXXWZTUDTEL-ZUYCGGNHSA-N 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 231100000243 mutagenic effect Toxicity 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 239000000133 nasal decongestant Substances 0.000 description 1
- 208000010753 nasal discharge Diseases 0.000 description 1
- 210000002850 nasal mucosa Anatomy 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 230000037311 normal skin Effects 0.000 description 1
- 239000002853 nucleic acid probe Substances 0.000 description 1
- 208000007892 occupational asthma Diseases 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 230000004768 organ dysfunction Effects 0.000 description 1
- 210000003254 palate Anatomy 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 210000004976 peripheral blood cell Anatomy 0.000 description 1
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 208000015768 polyposis Diseases 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920000131 polyvinylidene Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000016412 positive regulation of cytokine production Effects 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 208000028529 primary immunodeficiency disease Diseases 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- 230000001823 pruritic effect Effects 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 201000003651 pulmonary sarcoidosis Diseases 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 201000004193 respiratory failure Diseases 0.000 description 1
- 208000020029 respiratory tract infectious disease Diseases 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 108010037379 ribosome releasing factor Proteins 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 238000007480 sanger sequencing Methods 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 231100000489 sensitizer Toxicity 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 201000009890 sinusitis Diseases 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 238000007390 skin biopsy Methods 0.000 description 1
- 201000008261 skin carcinoma Diseases 0.000 description 1
- 201000010153 skin papilloma Diseases 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 206010041232 sneezing Diseases 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 238000011895 specific detection Methods 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 208000017572 squamous cell neoplasm Diseases 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 238000009121 systemic therapy Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- DVKJHBMWWAPEIU-UHFFFAOYSA-N toluene 2,4-diisocyanate Chemical compound CC1=CC=C(N=C=O)C=C1N=C=O DVKJHBMWWAPEIU-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000006433 tumor necrosis factor production Effects 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 108010010296 urea carboxylase (hydrolyzing) Proteins 0.000 description 1
- 208000007089 vaccinia Diseases 0.000 description 1
- 230000008728 vascular permeability Effects 0.000 description 1
- QYSXJUFSXHHAJI-YRZJJWOYSA-N vitamin D3 Chemical class C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)CCCC(C)C)=C\C=C1\C[C@@H](O)CCC1=C QYSXJUFSXHHAJI-YRZJJWOYSA-N 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/35—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Mycobacteriaceae (F)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/04—Mycobacterium, e.g. Mycobacterium tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
Definitions
- the present invention relates generally to compositions which are present in or may be derived from Mycobacterium vaccae and their use in the treatment, prevention and detection of disorders including infectious diseases, immune disorders and cancer.
- the invention is related to compounds and methods for the treatment of diseases of the respiratory system, such as mycobacterial infections, asthma, sarcoidosis and lung cancers, and disorders of the skin, such as psoriasis, atopic dermatis, allergic contact dermatitis, alopecia areata, and the skin cancers basal cell carcinoma, squamous cell carcinoma and melanoma.
- the invention is further related to compounds that function as non-specific immune response amplifiers, and the use of such non-specific immune response amplifiers as adjuvants in vaccination or immunotherapy against infectious disease, and in certain treatments for immune disorders and cancer.
- Tuberculosis is a chronic, infectious disease, that is caused by infection with Mycobacterium tuberculosis (M. tuberculosis). It is a major disease in developing countries, as well as an increasing problem in developed areas of the world, with about 8 million new cases and 3 million deaths each year. Although the infection may be asymptomatic for a considerable period of time, the disease is most commonly manifested as a chronic inflammation of the lungs, resulting in fever and respiratory symptoms. If left untreated, significant morbidity and death may result.
- tuberculosis can generally be controlled using extended antibiotic therapy, such treatment is not sufficient to prevent the spread of the disease. Infected individuals may be asymptomatic, but contagious, for some time. In addition, although compliance with the treatment regimen is critical, patient behaviour is difficult to monitor. Some patients do not complete the course of treatment, which can lead to ineffective treatment and the development of drug resistant mycobacteria.
- Inhibiting the spread of tuberculosis requires effective vaccination and accurate, early diagnosis of the disease.
- vaccination by subcutaneous or intradermal injection with live bacteria is the most efficient method for inducing protective immunity.
- the most common mycobacterium employed for this purpose is Bacillus Calmette-Guerin (BCG), an avirulent strain of Mycobacterium bovis (M. bovis).
- BCG Bacillus Calmette-Guerin
- M. bovis Mycobacterium bovis
- the safety and efficacy of BCG is a source of controversy and some countries, such as the United States, do not vaccinate the general public.
- Diagnosis of M. tuberculosis infection is commonly achieved using a skin test, which involves intradermal exposure to tuberculin PPD (protein-purified derivative).
- Antigen-specific T cell responses result in measurable induration at the injection site by 48-72 hours after injection, thereby indicating exposure to mycobacterial antigens. Sensitivity and specificity have, however, been a problem with this test, and individuals vaccinated with BCG cannot be distinguished from infected individuals.
- M. vaccae Mycobacterium vaccae
- U.S. Patent 4,716,038 discloses diagnosis of, vaccination against and treatment of autoimmune diseases of various types, including arthritic diseases, by administering mycobacteria, including M. vaccae.
- U.S. Patent 4,724,144 discloses an immunotherapeutic agent comprising antigenic material derived from M. vaccae for treatment of mycobacterial diseases, especially tuberculosis and leprosy, and as an adjuvant to chemotherapy.
- International Patent Publication WO 91/01751 discloses the use of antigenic and or immunoregulatory material from M.
- U.S. Patent 5,599,545 discloses the use of mycobacteria, especially whole, inactivated M. vaccae, as an adjuvant for administration with antigens which are not endogenous to M. vaccae. This publication theorises that the beneficial effect as an adjuvant may be due to heat shock protein 65 (hsp 65).
- International Patent Publication WO 92/08484 discloses the use of antigenic and/or immunoregulatory material derived from M. vaccae for the treatment of uveitis.
- International Patent Publication WO 93/16727 discloses the use of antigenic and/or immunoregulatory material derived from M. vaccae for the treatment of mental diseases associated with an autoimmune reaction initiated by an infection.
- International Patent Publication WO 95/26742 discloses the use of antigenic and/or immunoregulatory material derived from M. vaccae for delaying or preventing the growth or spread of tumors.
- International Patent Publication WO 91/02542 discloses the use of autoclaved M. vaccae in the treatment of chronic inflammatory disorders in which a patient demonstrates an abnormally high release of IL-6 and/or TNF or in which the patient's IgG shows an abnormally high proportion of agalactosyl IgG.
- psoriasis rheumatoid arthritis
- mycobacterial disease Crohn's disease
- primary biliary cirrhosis sarcoidosis
- ulcerative colitis systemic lupus erythematosus
- multiple sclerosis Guillain-Barre syndrome
- primary diabetes mellitus and some aspects of graft rejection.
- M. vaccae is apparently unique among known mycobacterial species in that heat- killed preparations retain vaccine and immunotherapeutic properties.
- M. tuberculosis BCG vaccines used for vaccination against tuberculosis, employ live strains. Heat-killed M. bovis BCG and M. tuberculosis have no protective properties when employed in vaccines.
- a number of compounds have been isolated from a range of mycobacterial species which have adjuvant properties. The effect of such adjuvants is essentially to stimulate a particular immune response mechanism against an antigen from another species.
- DD-M vaccae contains highly polymerised cell wall.
- DD-M vaccae contains 50% w/w protein, comprising a number of distinct protein species.
- Sarcoidosis is a disease of unknown cause characterised by granulomatous inflammation affecting many organs of the body and especially the lungs, lymph nodes and liver.
- Sarcoid granulomata are composed of mononuclear phagocytes, with epithelioid and giant cells in their centre, and T lymphocytes.
- CD4 T lymphocytes are closely associated with the epithelioid cells while both CD4 and CD8 T lymphocytes accumulate at the periphery.
- the characteristic immunological abnormalities in sarcoidosis include peripheral blood and bronchoalveolar lavage hyper-globulinaemia and depression of 'delayed type' hypersensitivity reactions in the skin to tuberculin and other similar antigens, such as Candida and mumps.
- Peripheral blood lymphocyte numbers are reduced and CD4: CD8 ratios in peripheral blood are depressed to approximately 1-1.5:1. These are not manifestations of a generalised immune defect, but rather the consequence of heightened immunological activity which is 'compartmentalised' to sites of disease activity.
- the total number of cells recovered by bronchoalveolar lavage is increased five- to ten-fold and the proportion of lymphocytes increased from the normal of less than 10-14% to between 15% and 50%. More than 90% of the lymphocytes recovered are T lymphocytes and the CD4-.CD8 ratio has been reported to be increased from the value of 1.8:1 in normal controls to 10.5:1.
- the T lymphocytes are predominantly of the Thl class, producing IFN- ⁇ and IL-2 cytokines, rather than of the Th2 class. Following treatment, the increase in Thl lymphocytes in sarcoid lungs is corrected.
- sarcoidosis involves the lungs in nearly all cases. Even when lesions are predominantly seen in other organs, subclinical lung involvement is usually present. While some cases of sarcoidosis resolve spontaneously, approximately 50% of patients have at least a mild degree of permanent organ dysfunction. In severe cases, lung fibrosis develops and progresses to pulmonary failure requiring lung transplantation.
- the mainstay of treatment for sarcoidosis is corticosteroids. Patients initially responding to corticosteroids often relapse and require treatment with other immunosuppressive drugs such as methotrexate or cyclosporine.
- Asthma is a common disease, with a high prevalence in the developed world. Asthma is characterised by increased responsiveness of the tracheobronchial tree to a variety of stimuli, the primary physiological disturbance being reversible airflow limitation, which may be spontaneous or drug-related, and the pathological hallmark being inflammation of the airways. Clinically, asthma can be subdivided into extrinsic and intrinsic variants.
- Extrinsic asthma has an identifiable precipitant, and can be thought of as being atopic, occupational and drug-induced.
- Atopic asthma is associated with the enhancement of a Th2- type of immune response with the production of specific immunoglobulin E (IgE), positive skin tests to common aeroallergens and/or atopic symptoms. It can be divided further into seasonal and perennial forms according to the seasonal timing of symptoms.
- IgE immunoglobulin E
- the airflow obstruction in extrinsic asthma is due to nonspecific bronchial hyperesponsiveness caused by inflammation of the airways. This inflammation is mediated by chemicals released by a variety of inflammatory cells including mast cells, eosinophils and lymphocytes. The actions of these mediators result in vascular permeability, mucus secretion and bronchial smooth muscle constriction.
- atopic asthma the immune response producing airway inflammation is brought about by the Th2 class of T cells which secrete IL-4, IL-5 and IL-10. It has been shown that lymphocytes from the lungs of atopic asthmatics produce IL-4 and IL-5 when activated. Both IL-4 and IL-5 are cytokines of the Th2 class and are required for the production of IgE and involvement of eosinophils in asthma.
- Occupational asthma may be related to the development of IgE to a protein hapten, such as acid anhydrides in plastic workers and plicatic acid in some western red cedar-induced asthma, or to non-IgE related mechanisms, such as that seen in toluene diisocyanate-induced asthma.
- Drug-induced asthma can be seen after the administration of aspirin or other non-steroidal anti-inflammatory drugs, most often in a certain subset of patients who may display other features such as nasal polyposis and sinusitis.
- Intrinsic or cryptogenic asthma is reported to develop after upper respiratory tract infections, but can arise de novo in middle-aged or older people, in whom it is more difficult to treat than extrinsic asthma.
- Asthma is ideally prevented by the avoidance of triggering allergens but this is not always possible nor are triggering allergens always easily identified.
- the medical therapy of asthma is based on the use of corticosteroids and bronchodilator drugs to reduce inflammation and reverse airway obstruction. In chronic asthma, treatment with corticosteroids leads to unacceptable adverse side effects.
- Allergic rhinitis is a common disorder and is estimated to affect at least 10% of the population. Allergic rhinitis may be seasonal (hay fever) caused by allergy to pollen. Non- seasonal or perennial rhinitis is caused by allergy to antigens such as those from house dust mite or animal dander.
- the abnormal immune response in allergic rhinitis is characterised by the excess production of IgE antibodies specific against the allergen.
- the inflammatory response occurs in the nasal mucosa rather than further down the airways as in asthma.
- local eosinophilia in the affected tissues is a major feature of allergic rhinitis.
- patients develop sneezing, nasal discharge and congestion.
- the inflammation extends to the eyes (conjunctivitis), palate and the external ear. While it is not life threatening, allergic rhinitis may be very disabling, prevent normal activities, and interfere with a person's ability to work.
- Current treatment involves the use of antihistamines, nasal decongestants and, as for asthma, sodium cromoglycate and corticosteroids.
- Lung cancer is the leading cause of death from cancer.
- the incidence of lung cancer continues to rise and the World Health Organisation estimates that by 2000AD there will be 2 million new cases annually.
- Lung cancers may be broadly classified into two categories: small cell lung cancer (SCLC) which represents 20-25% of all lung cancers, and non-small cell lung cancer (NSCLC) which accounts for the remaining 75%.
- SCLC small cell lung cancer
- NSCLC non-small cell lung cancer
- the majority of SCLC is caused by tobacco smoke.
- SCLC tends to spread early and 90% of patients present at diagnosis with involvement of the mediastinal lymph nodes in the chest.
- SCLC is treated by chemotherapy, or a combination of chemotherapy and radiotherapy. Complete response rates vary from 10% to 50%. For the rare patient without lymph node involvement, surgery followed by chemotherapy may result in cure rates exceeding 60%.
- the prognosis for NSCLC is more dismal, as most patients have advanced disease by the time of diagnosis. Surgical removal of the tumor is possible in a very small number of patients and the five
- this invention deals with treatment of disorders of skin which appear to be associated with factors that influence the balance of thymus-derived (T) immune cells known as Thl and Th2. These T cells are identified by their cytokine secretion phenotype.
- T thymus-derived
- a common feature of treatment is the use of compounds prepared from M vaccae which have immunomodulating properties that alter the balance of activities of these T cells as well as other immune cells.
- Psoriasis is a common, chronic inflammatory skin disease which can be associated with various forms of arthritis in a minority of patients.
- the defect in psoriasis appears to be overly rapid growth of keratinocytes and shedding of scales from the skin surface.
- Drug therapy is directed at slowing down this process.
- the disease may become manifest at any age. Spontaneous remission is relatively rare, and life-long treatment is usually necessary.
- Psoriasis produces chronic, scaling red patches on the skin surface.
- Psoriasis is a very visible disease, it frequently affects the face, scalp, trunk and limbs. The disease is emotionally and physically debilitating for the patient, detracting significantly from the quality of life. Between one and three million individuals in the United States have psoriasis with nearly a quarter million new cases occurring each year. Conservative estimates place the costs of psoriasis care in the United States currently at $248 million a year.
- the first is that genetic factors determine abnormal proliferation of epidermal keratinocytes. The cells no longer respond normally to external stimuli such as those involved in maintaining epidermal homeostasis. Abnormal expression of cell membrane cytokine receptors or abnormal transmembrane signal transduction might underlie cell hyperproliferation. Inflammation associated with psoriasis is secondary to the release of pro-inflammatory molecules from hyperproliferative keratinocytes.
- T cells interacting with antigen-presenting cells in skin release pro-inflammatory and keratinocyte-stimulating cytokines Hancock, G.E. et al, J. Exp. Med. 765:1395-1402, 1988.
- the keratinocytes themselves may be the antigen-presenting cell.
- the cellular infiltrate in psoriatic lesions show an influx of CD4+ T cells and, more prominently, CD8+ T cells (Bos, J.D. et al., Arch. Dermatol. Res. 281:23-3, 1989; Baker, B.S., Br. J. Dermatol. 110:555-564, 1984).
- Atopic dermatitis is a chronic pruritic inflammatory skin disease which usually occurs in families with an hereditary predisposition for various allergic disorders such as allergic rhinitis and asthma.
- Atopic dermatitis occurs in approximately 10% of the general population.
- the main symptoms are dry skin, dermatitis (eczema) localised mainly in the face, neck and on the flexor sides and folds of the extremities accompanied by severe itching. It typically starts within the first two years of life. In about 90% of the patients this skin disease disappears during childhood but the symptoms can continue into adult life. It is one of the commonest forms of dermatitis world-wide. It is generally accepted that in atopy and in atopic dermatitis, a T cell abnormality is primary and that the dysfunction of T cells which normally regulate the production of IgE is responsible for the excessive production of this immunoglobulin.
- Allergic contact dermatitis is a common non-infectious inflammatory disorder of the skin.
- immunological reactions cannot develop until the body has become sensitised to a particular antigen.
- Subsequent exposure of the skin to the antigen and the recognition of these antigens by T cells result in the release of various cytokines, proliferation and recruitment of T cells, and finally in dermatitis (eczema).
- T cells CD4 + cells
- MHC class II antigens CD4 + cells
- Keratinocytes can produce interleukin- 1 which can facilitate the antigen presentation to T cells.
- IFN- ⁇ interferon-gamma
- TNF tumor necrosis factor
- IFN- ⁇ interferon-gamma
- An inhibitory effect of cyclosporin has been observed in delayed-type hypersensitivity on the pro-inflammatory function(s) of primed T cells in vitro (Shidani, B. et al, Eur. J. Immunol. 74:314-318, 1984). The inhibitory effect of cyclosporin on the early phase of T cell activation in mice has also been reported (Milon, G. et al., Ann. Immunol. (Inst. Pasteur) 135d: 237-245, 1984).
- Alopecia areata is a common hair disease, which accounts for about 2% of the consultations at dermatological outpatient clinics in the United States.
- the hallmark of this disease is the formation of well-circumscribed round or oval patches of non-scarring alopecia which may be located in any hairy area of the body.
- the disease may develop at any age. The onset is usually sudden and the clinical course is varied.
- alopecia areata At present, it is not possible to attribute all or indeed any case of alopecia areata to a single cause (Rook, A. and Dawber, R, Diseases of the Hair and Scalp; Blackwell Scientific Publications 1982: 272-30). There are many factors that appear to be involved. These include genetic factors, atopy, association with disorders of supposed autoimmune etiology, Down's syndrome and emotional stress. The prevalence of atopy in patients with alopecia areata is increased. There is evidence that alopecia areata is an autoimmune disease.
- Immunophenotyping studies on scalp biopsy specimens shows expression of HLA-DR on epithelial cells in the presumptive cortex and hair follicles of active lesions of alopecia areata, as well as a T cell infiltration with a high proportion of helper/inducer T cells in and around the hair follicles, increased numbers of Langerhans cells and the expression of ICAM- 1 (Messenger, A.G. et al., J. Invest. Dermatol. 85:569-516, 1985; Gupta, A.K. et al., J. Am. Acad. Dermatol. 22:242-250, 1990).
- alopecia The large variety of therapeutic modalities in alopecia areata can be divided into four categories: (i) non-specific topical irritants; (ii) 'immune modulators' such as systemic corticosteroids and PUVA; (iii) 'immune enhancers' such as contact dermatitis inducers, cyclosporin and inosiplex; and (iv) drugs of unknown action such as minoxidil (Dawber, R.P.R. et al., Textbook of Dermatology, Blackwell Scientific Publications, 5 th Ed, 1982:2533- 2638).
- Non-specific topical irritants such as dithranol may work through as yet unidentified mechanisms rather than local irritation in eliciting regrowth of hair.
- Topical corticosteroids may be effective but prolonged therapy is often necessary.
- Intralesional steroids have proved to be more effective but their use is limited to circumscribed patches of less active disease or to maintain regrowth of the eyebrows in alopecia totalis.
- Photochemotherapy has proved to be effective, possibly by changing functional subpopulations of T cells.
- Topical immunotherapy by means of induction and maintenance of allergic contact dermatitis on the scalp may result in hair regrowth in as many as 70% of the patients with alopecia areata. Diphencyprone is a potent sensitiser free from mutagenic activity.
- Oral cyclosporin can be effective in the short term (Gupta, A.K. et al., J. Am. Acad. Dermatol. 22:242-250, 1990).
- Inosiplex an immunostimulant, has been used with apparent effectiveness in an open trial.
- Topical 5% minoxidil solution has been reported to be able to induce some hair growth in patients with alopecia areata. The mechanism of action is unclear.
- Carcinomas of the skin are a major public health problem because of their frequency and the disability and disfigurement that they cause. Carcinoma of the skin is principally seen in individuals in their prime of life, especially in fair skinned individuals exposed to large amounts of sunlight. The annual cost of treatment and time loss from work exceeds $250 million dollars a year in the United States alone. The three major types - basal cell cancer, squamous cell cancer, and melanoma - are clearly related to sunlight exposure.
- Basal cell carcinomas are epithelial tumours of the skin. They appear predominantly on exposed areas of the skin. In a recent Australian study, the incidence of basal cell carcinomas was 652 new cases per year per 100,000 of the population. This compares with 160 cases of squamous cell carcinoma or 19 of malignant melanoma (Giles, G. et al., Br. Med. J. 296:13- 1, 1988). Basal cell carcinomas are the most common of all cancers. Lesions are usually surgically excised. Alternate treatments include retinoids, 5-fluorouracil, cryotherapy and radiotherapy.
- Alpha or gamma interferon have also been shown to be effective in the treatment of basal cell carcinomas, providing a valuable alternative to patients unsuitable for surgery or seeking to avoid surgical scars (Cornell et al., J Am. Acad. Dermatol. 23:694-700, 1990; Edwards, L. et al., J. Am. Acad. Dermatol. 22:496-500, 1990).
- SCC Squamous cell carcinoma
- interferons have also been used in the treatment of melanoma (Kirkwood, J.M. et al., J. Invest. Dermatol. °5:180S-4S, 1990).
- Response rates achieved with systemic IFN- were in the range 5-30%.
- Recently, encouraging results (30%) response were obtained with a combination of IFN- ⁇ and DTIC.
- Preliminary observations indicate a beneficial effect of IFN- ⁇ in an adjuvant setting in patients with high risk melanoma.
- interferon Of all the available therapies for treating cutaneous viral lesions, only interferon possesses a specific antiviral mode of action, by reproducing the body's immune response to infection. Interferon treatment cannot eradicate the viruses however, although it may help with some manifestations of the infection. Interferon treatment is also associated with systemic adverse effects, requires multiple injections into each single wart and has a significant economic cost (Kraus, SJ. et al., Review of Infectious Diseases 2(6):S620-S632, 1990; Frazer, I.H., Current Opinion in Immunology 5(4):484-491, 1996).
- the present invention provides compositions present in or derived from M vaccae and methods for their use in the prevention, treatment and diagnosis of diseases, including mycobacterial infection, immune disorders of the respiratory system, and skin disorders.
- the inventive methods comprise administering a composition having antigenic and/or adjuvant properties.
- Diseases of the respiratory system which may be treated using the inventive compositions include mycobacterial infections (such as infection with M tuberculosis and/or M avium), asthma, sarcoidosis and lung cancers.
- disorders of the skin which may be treated using the inventive compositions include psoriasis, atopic dermatis, allergic contact dermatitis, alopecia areata, and the skin cancers basal cell carcinoma, squamous cell carcinoma and melanoma.
- Adjuvants for use in vaccines or immunotherapy of infectious diseases and cancers are also provided.
- isolated polypeptides derived from Mycobacterium vaccae comprising an immunogenic portion of an antigen, or a variant of such an antigen.
- the antigen includes an amino acid sequence selected from the group consisting of: (a) sequences recited in SEQ ID NO: 143, 145, 147, 152, 154 156, 158, 160, 162, 165, 166, 170, 172, 174, 177, 178, 181, 182, 184, 186, 187, 192, 194, 196, 197, 199, 201, 203, 205 and 207; (b) sequences having at least about 50% identical residues to a sequence recited in SEQ ID NO: 143, 145, 147, 152, 154 156, 158, 160, 162, 165, 166, 170, 172, 174, 177, 178, 181, 182, 184, 186, 187, 192, 194, 196,
- DNA sequences encoding the inventive polypeptides, expression vectors comprising these DNA sequences, and host cells transformed or transfected with such expression vectors are also provided.
- the present invention provides fusion proteins comprising at least one polypeptide of the present invention.
- the present invention provides pharmaceutical compositions that comprise at least one of the inventive polypeptides, or a DNA molecule encoding such a polypeptide, and a physiologically acceptable carrier.
- the invention also provides vaccines comprising at least one of the above polypeptides, or at least one DNA sequence encoding such polypeptides, and a non-specific immune response amplifier.
- the non-specific immune response enhancer is selected from the group consisting of: delipidated and deglycolipidated M. vaccae cells; inactivated M.
- vaccae cells delipidated and deglycolipidated M.vaccae cells depleted of mycolic acids; delipidated and deglycolipidated M.vaccae cells depleted of mycolic acids and arabinogalactan; and M vaccae culture filtrate.
- methods for enhancing an immune response in a patient, comprising administering to a patient an effective amount of one or more of the above pharmaceutical compositions and/or vaccines.
- the immune response is a Thl response.
- methods are provided for the treatment of a disorder in a patient, comprising administering to the patient a pharmaceutical composition or vaccine of the present invention.
- the disorder is selected from the group consisting of immune disorders, infectious diseases, skin diseases and diseases of the respiratory system. Examples of such diseases include mycobacterial infections, asthma and psoriasis.
- the invention provides methods for the treatment of immune disorders, infectious diseases, skin diseases or diseases of the respiratory system, comprising administering a composition comprising inactivated M vaccae cells, delipidated and deglycolipidated M vaccae cells or M vaccae culture filtrate.
- Methods for enhancing an immune response to an antigen are also provided.
- such methods comprising administering a polypeptide that comprises an immunogenic portion of a M vaccae antigen which includes a sequence of SEQ ID NO: 89 or 201, or a variant thereof.
- such methods comprise administering a composition comprising a component selected from the group consisting of: delipidated and deglycolipidated M.vaccae cells depleted of mycolic acids, and delipidated and deglycolipidated M.vaccae cells depleted of mycolic acids and arabinogalactan.
- the method comprises contacting dermal cells of a patient with one or more of the above polypeptides and detecting an immune response on the patient's skin.
- the method comprises contacting a biological sample with at least one of the above polypeptides; and detecting in the sample the presence of antibodies that bind to the polypeptide or polypeptides, thereby detecting M tuberculosis infection in the biological sample.
- suitable biological samples include whole blood, sputum, serum, plasma, saliva, cerebrospinal fluid and urine.
- Diagnostic kits comprising one or more of the above polypeptides in combination with an apparatus sufficient to contact the polypeptide with the dermal cells of a patient are provided.
- the present invention also provides diagnostic kits comprising one or more df the inventive polypeptides in combination with a detection reagent.
- the present invention provides antibodies, both polyclonal and monoclonal, that bind to the polypeptides described above, as well as methods for their use in the detection of mycobacterial infection.
- Figs. 1A and IB illustrate the protective effects of immunizing mice with autoclaved M vaccae or unfractionated M vaccae culture filtrates, respectively, prior to infection with live M tuberculosis H37Rv.
- Figs. 2A and B show the percentage of eosinophils in mice immunized intranasally with either 10 or 1000 ⁇ g of heat-killed M vaccae or 200-100 ⁇ g of DD-M vaccae, respectively, 4 weeks prior to challenge with ovalbumin, as compared to control mice.
- Figs. 2C and D show the percentage of eosinophils in mice immunized intranasally with either 100 ⁇ g of heat-killed M vaccae or 200 ⁇ g of DD-M vaccae, respectively, as late as one week prior to challenge with ovalbumin.
- Fig. 2A and B show the percentage of eosinophils in mice immunized intranasally with either 10 or 1000 ⁇ g of heat-killed M vaccae or 200-100 ⁇ g of DD-M vaccae, respectively, 4 weeks prior to challenge with ovalbumin, as compared to control mice.
- Figs. 2C and D
- 2E shows the percentage of eosinophils in mice immunized either intranasally (i.n.) or subcutaneously (s.c.) with either BCG of the Pasteur strain (BCG-P), BCG of the Connought strain (BCG-C), 1 mg of heat-killed M. vaccae, or 200 ⁇ g of DD-M vaccae prior to challenge with ovalbumin.
- BCG-P BCG of the Pasteur strain
- BCG-C BCG of the Connought strain
- 1 mg of heat-killed M. vaccae or 200 ⁇ g of DD-M vaccae prior to challenge with ovalbumin.
- Fig. 3A illustrates the effect of immunizing mice with heat-killed M vaccae or delipidated and deglycolipidated M vaccae (DD-M vaccae) prior to infection with tuberculosis.
- Fig. 3B illustrates the effect of immunizing mice with heat-killed M vaccae, recombinant M vaccae proteins, or a combination of heat-killed M vaccae and M vaccae recombinant proteins prior to infection with tuberculosis.
- Fig. 4 illustrates the induction of IL-12 by autoclaved M vaccae, lyophilized M vaccae, delipidated and deglycolipidated M vaccae and M vaccae glycolipids.
- Fig. 5 compares the in vitro stimulation of interferon-gamma production in spleen cells from Severe Combined ImmunoDeficient (SCID) mice by different concentrations of heat-killed (autoclaved) M vaccae, delipidated and deglycolipidated M vaccae, and M vaccae glycolipids.
- Figs. 6A, B and C illustrate the stimulation of interferon-gamma production by different concentrations of M vaccae recombinant proteins, heat-killed M vaccae, delipidated and deglycolipidated M vaccae (referred to in the figure as "delipidated M vaccae' ' '), M. vaccae glycolipids and lipopolysaccharide, in peritoneal macrophages from C57BL/6 mice (Fig. 6A), BALB/C mice (Fig. 6B) or C3H/HeJ mice (Fig. 6C).
- Fig. 6A C57BL/6 mice
- Fig. 6B BALB/C mice
- Fig. 6C C3H/HeJ mice
- FIG. 7A(i) - (iv) illustrate the non-specific immune amplifying effects of 10 ⁇ g, 100 ⁇ g and lmg autoclaved M vaccae and 75 ⁇ g unfractionated culture filtrates of M vaccae, respectively.
- Fig. 7B(i) and (ii) illustrate the non-specific immune amplifying effects of autoclaved M vaccae, and delipidated and deglycolipidated M vaccae, respectively.
- Fig. 7C(i) illustrates the non-specific immune amplifying effects of whole autoclaved M vaccae.
- Fig. 7C(ii) illustrates the non-specific immune amplifying effects of soluble M vaccae proteins extracted with SDS from delipidated and deglycolipidated M vaccae.
- Fig. 7C(iii) illustrates that the non-specific amplifying effects of the preparation of Fig. 7C(ii) are destroyed by treatment with the proteolytic enzyme Pronase.
- Fig. 7D illustrates the nonspecific immune amplifying effects of heat-killed M vaccae (Fig. 7D(i)), whereas a nonspecific immune amplifying effect was not seen with heat-killed preparations of M tuberculosis (Fig. 7D(ii)), M bovis BCG (Fig. 7D(iii)), M phlei (Fig. 7D(iv)) and M. smegmatis (Fig. 7D(v)).
- Figs. 8A and B illustrate the stimulation of CD69 expression on ⁇ T cells, ⁇ T cells and NK cells, respectively, by the M vaccae protein GV23, the Thl -inducing adjuvants MPL/TDM/CWS and CpG ODN, and the Th2 -inducing adjuvants aluminium hydroxide and cholera toxin.
- Figs. 9A-D illustrate the effect of heat-killed M vaccae, DD-M vaccae and M vaccae recombinant proteins on the production of IL-l ⁇ , TNF- ⁇ , IL-12 and IFN- ⁇ , respectively, by human PBMC.
- Figs. 10A-C illustrate the effects of varying concentrations of the recombinant M vaccae proteins GV-23 and GV-45 on the production of IL-l ⁇ , TNF- ⁇ and IL-12, respectively, by human PBMC.
- Figs. 11A-D illustrate the stimulation of IL-l ⁇ , TNF- ⁇ , IL-12 and IFN- ⁇ production, respectively, in human PBMC by the M vaccae protein GV23, the Thl -inducing adjuvants MPL/TDM/CWS and CpG ODN, and the Th2-inducing adjuvants aluminium hydroxide and cholera toxin.
- Figs. 12A-C illustrate the effects of varying concentrations of the recombinant M vaccae proteins GV-23 and GV-45 on the expression of CD40, CD80 and CD86, respectively, by dendritic cells.
- Fig. 13 illustrates the enhancement of dendritic cell mixed leukocyte reaction by the recombinant M vaccae protein GV-23.
- the present invention is generally directed to compositions and methods for preventing, treating and diagnosing infectious diseases and immune disorders.
- Disorders which may be effectively treated using the inventive compositions include diseases of the respiratory system, such as mycobacterial infections, asthma, sarcoidosis and lung cancers, and disorders of the skin, such as psoriasis, atopic dermatis, allergic contact dermatitis, alopecia areata, and the skin cancers basal cell carcinoma, squamous cell carcinoma and melanoma.
- Effective vaccines that provide protection against infectious microorganisms contain at least two functionally different components.
- the first is an antigen, which may be polypeptide or carbohydrate in nature, and which is processed by macrophages and other antigen-presenting cells and displayed for CD4 + T cells or for CD8 + T cells. This antigen forms the "specific" target of an immune response.
- the second component of a vaccine is a non-specific immune response amplifier, termed an adjuvant, with which the antigen is mixed or is inco ⁇ orated into.
- An adjuvant amplifies either cell-mediated or antibody immune responses to a structurally unrelated compound or polypeptide.
- Several known adjuvants are prepared from microbes such as Bordetella pertussis, M.
- Adjuvants may also contain components designed to protect polypeptide antigens from degradation, such as aluminum hydroxide or mineral oil. While the antigenic component of a vaccine contains polypeptides that direct the immune attack against a specific pathogen, such as M tuberculosis, the adjuvant is often capable of broad use in many different vaccine formulations. Certain known proteins, such as bacterial enterotoxins, can function both as an antigen to elicit a specific immune response and as an adjuvant to enhance immune responses to unrelated proteins.
- pathogens such as M tuberculosis, as well as certain cancers, are effectively contained by an immune attack directed by CD4 + and CD8 + T cells, known as cell-mediated immunity.
- Other pathogens such as poliovirus, also require antibodies, produced by B cells, for containment.
- T cell or B cell are controlled by different subpopulations of CD4 + T cells, commonly referred to as Thl and Th2 cells.
- Thl and Th2 cells are controlled by different subpopulations of CD4 + T cells, commonly referred to as Thl and Th2 cells.
- Thl and Th2 cells A desirable property of an adjuvant is the ability to selectively amplify the function of either Thl or Th2 populations of CD4 + T cells.
- Many skin disorders including psoriasis, atopic dermatitis, alopecia, and skin cancers appear to be influenced by differences in the activity of these Th cell subsets.
- Th cell subsets have been well characterized in a murine model and are defined by the cytokines they release upon activation.
- the Thl subset secretes IL-2, IFN- ⁇ and tumor necrosis factor, and mediates macrophage activation and delayed-type hypersensitivity response.
- the Th2 subset releases IL-4, IL-5, IL-6 and IL-10, which stimulate B cell activation.
- the Thl and Th2 subsets are mutually inhibiting, so that IL-4 inhibits Thl -type responses, and IFN- ⁇ inhibits Th2-type responses. Similar Thl and Th2 subsets have been found in humans, with release of the identical cytokines observed in the murine model.
- Th2 subset a subset of T-cell clones from atopic human lymphocytes resemble the murine Th2 cell that produces IL-4, whereas very few clones produce IFN- ⁇ . Therefore, the selective expression of the Th2 subset with subsequent production of IL-4 and decreased levels of IFN- ⁇ -producing cells could lead to preferential enhancement of IgE production.
- Amplification of Thl -type immune responses is central to a reversal of disease state in many disorders, including disorders of the respiratory system such as tuberculosis, sarcoidosis, asthma, allergic rhinitis and lung cancers.
- Inactivated M vaccae and many compounds derived from M vaccae have both antigen and adjuvant properties which function to enhance Thl -type immune responses.
- the methods of the present invention employ one or more of these antigen and adjuvant compounds from M vaccae and/or its culture filtrates to redirect immune activities of T cells in patients. Mixtures of such compounds are particularly effective in the methods disclosed herein. While it is well known that all mycobacteria contain many cross-reacting antigens, it is not known whether they contain adjuvant compounds in common.
- inactivated M vaccae and a modified (delipidated and deglycolipidated) form of inactivated M vaccae have been found to have adjuvant properties of the Thl -type which are not shared by a number of other mycobacterial species. Furthermore, it has been found that M vaccae produces compounds in its own culture filtrate which amplify the immune response to M vaccae antigens also found in culture filtrate, as well as to antigens from other sources.
- the present invention provides methods for the immunotherapy of respiratory and/or lung disorders, including tuberculosis, sarcoidosis, asthma, allergic rhinitis and lung cancers, in a patient to enhance Thl -type immune responses.
- the compositions are delivered directly to the mucosal surfaces of airways leading to and/or within the lungs.
- the compositions may also be administered via intradermal or subcutaneous routes.
- Compositions which may be usefully employed in such methods comprise at least one of the following components: inactivated M vaccae cells; M vaccae culture filtrate; delipidated and deglycolipidated M vaccae cells (DD-M vaccae); and compounds present in or derived from M vaccae and/or its culture filtrate.
- compositions results in specific T cell immune responses and enhanced protection against M tuberculosis infection, and is also effective in the treatment of asthma. While the precise mode of action of these compositions in the treatment of diseases such as asthma is unknown, they are believed to suppress an asthma-inducing Th2 immune response.
- the term "respiratory system” refers to the lungs, nasal passageways, trachea and bronchial passageways.
- airways leading to or located in the lung includes the nasal passageways, mouth, tonsil tissue, trachea and bronchial passageways.
- a "patient” refers to any warm-blooded animal, preferably a human. Such a patient may be afflicted with disease or may be free of detectable disease. In other words, the inventive methods may be employed to induce protective immunity for the prevention or treatment of disease.
- the present invention provides methods for the immunotherapy of skin disorders, including psoriasis, atopic dermatitis, alopecia, and skin cancers in patients, in which immunotherapeutic agents are employed to alter or redirect an existing state of immune activity by altering the function of T cells to a Thl -type of immune response.
- Compositions which may be usefully employed in the inventive methods comprise at least one of the following components: inactivated M vaccae cells; M vaccae culture filtrate; modified M vaccae cells; and constituents and compounds present in or derived from M vaccae and/or its culture filtrate.
- multiple administrations of such compositions preferably by intradermal injection, have been shown to be highly effective in the treatment of psoriasis.
- the term “inactivated M vaccae” refers to M vaccae that have either been killed by means of heat, as detailed below in Example 7, or subjected to radiation, such as 60 Cobalt at a dose of 2.5 megarads.
- the term “modified M vaccae” includes delipidated M vaccae cells, deglycolipidated M vaccae cells and M vaccae cells that have been both delipidated and deglycolipidated (DD-M vaccae).
- Example 7 The preparation of DD-M vaccae and its chemical composition are described below in Example 7. As detailed below, the inventors have shown that removal of the glycolipid constituents from M vaccae results in the removal of molecular components that stimulate interferon-gamma production in natural killer (NK) cells, thereby significantly reducing the non-specific production of a cytokine that has numerous harmful side-effects.
- NK natural killer
- the present invention provides isolated polypeptides that comprise at least one immunogenic portion of a M vaccae antigen, or a variant thereof, or at least one adjuvant porition of an M. vaccae protein.
- such polypeptides comprise an immunogenic portion of an antigen, or a variant thereof, wherein the antigen includes a sequence selected from the group consisting of SEQ ID NO: 1-4, 9-16, 18-21, 23, 25, 26, 28, 29, 44, 45, 47, 52-55, 63, 64, 70, 75, 89, 94, 98, 100-105, 109, 110, 112, 121, 124, 125, 134, 135, 140, 141, 143, 145, 147, 152, 154, 156, 158, 160, 165, 166, 170, 172, 174, 177, 178, 181, 182, 184, 186, 187, 192, 194, 201, 203, 205 and 207.
- polypeptide encompasses amino acid chains of any length, including full length proteins (i.e., antigens), wherein the amino acid residues are linked by covalent peptide bonds.
- a polypeptide comprising an immunogenic portion of one of the above antigens may consist entirely of the immunogenic portion, or may contain additional sequences.
- the additional sequences may be derived from the native M vaccae antigen or may be heterologous, and such sequences may (but need not) be immunogenic.
- polypeptides of the present invention may be isolated from M vaccae cells or culture filtrate, or may be prepared by synthetic or recombinant means.
- Immunogenic refers to the ability to elicit an immune response in a patient, such as a human, or in a biological sample.
- immunogenic antigens are capable of stimulating cell proliferation, interleukin- 12 production or interferon- ⁇ production in biological samples comprising one or more cells selected from the group of T cells, NK cells, B cells and macrophages, where the cells are derived from an M tuberculosis-immune individual. Exposure to an immunogenic antigen generally results in the generation of immune memory such that upon re-exposure to that antigen, an enhanced and more rapid response occurs.
- Immunogenic portions of the antigens described herein may be prepared and identified using well known techniques, such as those summarised in Paul, Fundamental Immunology, 3d ed., Raven Press, 1993, pp. 243-247. Such techniques include screening polypeptide portions of the native antigen or protein for immunogenic properties. The representative proliferation and cytokine production assays described herein may be employed in these screens.
- An immunogenic portion of an antigen is a portion that, within such representative assays, generates an immune response (e.g., cell proliferation, interferon- ⁇ production or interleukin- 12 production) that is substantially similar to that generated by the full-length antigen.
- an immunogenic portion of an antigen may generate at least about 20%, preferably about 65%, and most preferably about 100% of the proliferation induced by the full-length antigen in the model proliferation assay described herein.
- An immunogenic portion may also, or alternatively, stimulate the production of at least about 20%, preferably about 65% and most preferably about 100%, of the interferon- ⁇ and/or interleukin- 12 induced by the full length antigen in the model assay described herein.
- a M vaccae adjuvant is a compound found in M vaccae cells or M vaccae culture filtrates which non-specifically stimulates immune responses.
- Adjuvants enhance the immune response to immunogenic antigens and the process of memory formation. In the case of M vaccae proteins, these memory responses favour Thl -type immunity.
- Adjuvants are also capable of stimulating interleukin- 12 production or interferon- ⁇ production in biological samples comprising one or more cells selected from the group of T cells, NK cells, B cells and macrophages, where the cells are derived from healthy individuals. Adjuvants may or may not stimulate cell proliferation.
- Such M vaccae adjuvants include, for example, polypeptides comprising a sequence recited in SEQ ID NO: 89, 117, 160, 162 or 201.
- polynucleotide(s), means a single or double-stranded polymer of deoxyribonucleotide or ribonucleotide bases and includes DNA and corresponding RNA molecules, including HnRNA and mRNA molecules, both sense and anti-sense strands, and comprehends cDNA, genomic DNA and recombinant DNA, as well as wholly or partially synthesized polynucleotides.
- An HnRNA molecule contains introns and corresponds to a DNA molecule in a generally one-to-one manner.
- An mRNA molecule corresponds to an HnRNA and DNA molecule from which the introns have been excised.
- a polynucleotide may consist of an entire gene, or any portion thereof.
- Operable anti-sense polynucleotides may comprise a fragment of the corresponding polynucleotide, and the definition of "polynucleotide” therefore includes all such operable anti-sense fragments.
- compositions and methods of this invention also encompass variants of the above polypeptides and polynucleotides.
- variant covers any sequence which has at least about 40%, more preferably at least about 60%, more preferably yet at least about 75% and most preferably at least about 90% identical residues (either nucleotides or amino acids) to a sequence of the present invention.
- the percentage of identical residues is determined by aligning the two sequences to be compared, determining the number of identical residues in the aligned portion, dividing that number by the total length of the inventive, or queried, sequence and multiplying the result by 100.
- Polynucleotide or polypeptide sequences may be aligned, and percentage of identical nucleotides in a specified region may be determined against another polynucleotide, using computer algorithms that are publicly available.
- Two exemplary algorithms for aligning and identifying the similarity of polynucleotide sequences are the BLASTN and FASTA algorithms.
- the similarity of polypeptide sequences may be examined using the BLASTP algorithm. Both the BLASTN and BLASTP software are available on the NCBI anonymous FTP server (ftp://ncbi.nlm.nih.gov) under /blast executables/.
- the computer algorithm FASTA is available on the Internet at the ftp site ftp://ftp.virginia.edu/pub/fasta/. Version 2.0u4, February 1996, set to the default parameters described in the documentation and distributed with the algorithm, is preferred for use in the determination of variants according to the present invention.
- the use of the FASTA algorithm is described in W.R. Pearson and D.J. Lipman, "Improved Tools for Biological Sequence Analysis,” Proc. Natl. Acad. Sci. USA ⁇ 5:2444-2448 (1988) and W.R. Pearson, “Rapid and Sensitive Sequence Comparison with FASTP and FASTA," Methods in Enzymology 183:63-98 (1990).
- the "hits" to one or more database sequences by a queried sequence produced by BLASTN, BLASTP, FASTA, or a similar algorithm align and identify similar portions of sequences.
- the hits are arranged in order of the degree of similarity and the length of sequence overlap. Hits to a database sequence generally represent an overlap over only a fraction of the sequence length of the queried sequence.
- the BLASTN and FASTA algorithms also produce "Expect" values for alignments.
- the Expect value (E) indicates the number of hits one can "expect” to see over a certain number of contiguous sequences by chance when searching a database of a certain size.
- the Expect value is used as a significance threshold for determining whether the hit to a database, such as the preferred EMBL database, mdicates true similarity. For example, an E value of 0.1 assigned to a hit is inte ⁇ reted as meaning that in a database of the size of the EMBL database, one might expect to see 0.1 matches over the aligned portion of the sequence with a similar score simply by chance.
- the aligned and matched portions of the sequences then have a probability of 90% of being the same.
- the probability of finding a match by chance in the EMBL database is 1% or less using the BLASTN or FASTA algorithm.
- variant polynucleotides with reference to each of the polynucleotides of the present invention, preferably comprise sequences having the same number or fewer nucleic acids than each of the polynucleotides of the present invention and producing an E value of 0.01 or less when compared to the polynucleotide of the present invention. That is, a variant polynucleotide is any sequence that has at least a 99% probability of being the same as the polynucleotide of the present invention, measured as having an E value of 0.01 or less using the BLASTN or FASTA algorithms set at the default parameters.
- a variant polynucleotide is a sequence having the same number or fewer nucleic acids than a polynucleotide of the present invention that has at least a 99%o probability of being the same as the polynucleotide of the present invention, measured as having an E value of 0.01 or less using the BLASTN or FASTA algorithms set at the default parameters.
- Variant polynucleotide sequences will generally hybridize to the recited polynucleotide sequence under stringent conditions.
- stringent conditions refers to prewashing in a solution of 6X SSC, 0.2% SDS; hybridizing at 65 °C, 6X SSC, 0.2% SDS overnight; followed by two washes of 30 minutes each in IX SSC, 0.1% SDS at 65 °C and two washes of 30 minutes each in 0.2X SSC, 0.1% SDS at 65 °C.
- M vaccae polypeptides may be generated by synthetic or recombinant means.
- Synthetic polypeptides having fewer than about 100 amino acids, and generally fewer than about 50 amino acids may be generated using techniques well known to those of ordinary skill in the art.
- such polypeptides may be synthesized using any of the commercially available solid-phase techniques, such as the Merrifield solid-phase synthesis method, where amino acids are sequentially added to a growing amino acid chain. See Merrifield, J Am. Chem. Soc. 85:2149-2146, 1963. Equipment for automated synthesis of polypeptides is commercially available from suppliers such as Perkin Elmer/Applied BioSystems, Inc.
- Variants of a native antigen or adjuvant may be prepared using standard mutagenesis techniques, such as oligonucleotide-directed site specific mutagenesis. Sections of the DNA sequence may also be removed using standard techniques to permit preparation of truncated polypeptides.
- a polypeptide of the present invention may be conjugated to a signal (or leader) sequence at the N-terminal end of the protein which co-translationally or post-translationally directs transfer of the protein.
- the polypeptide may also be conjugated to a linker or other sequence for ease of synthesis, purification or identification of the polypeptide (e.g., poly- His), or to enhance binding of the polypeptide to a solid support.
- a polypeptide may be conjugated to an immunoglobulin Fc region.
- M vaccae antigens and DNA sequences encoding such antigens, may be prepared using any of a variety of procedures.
- soluble antigens may be isolated from M vaccae culture filtrate as described below.
- Antigens may also be produced recombinantly by inserting a DNA sequence that encodes the antigen into an expression vector and expressing the antigen in an appropriate host. Any of a variety of expression vectors known to those of ordinary skill in the art may be employed. Expression may be achieved in any appropriate host cell that has been transformed or transfected with an expression vector containing a DNA molecule that encodes a recombinant polypeptide. Suitable host cells include prokaryotes, yeast and higher eukaryotic cells.
- the host cells employed are E. coli, mycobacteria, insect, yeast or a mammalian cell line such as COS or CHO.
- the DNA sequences expressed in this manner may encode naturally occurring antigens, portions of naturally occurring antigens, or other variants thereof.
- DNA sequences encoding M vaccae antigens may be obtained by screening an appropriate M vaccae cDNA or genomic DNA library for DNA sequences that hybridize to degenerate oligonucleotides derived from partial amino acid sequences of isolated soluble antigens. Suitable degenerate oligonucleotides may be designed and synthesized, and the screen may be performed as described, for example in Sambrook et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY, 1989. As described below, polymerase chain reaction (PCR) may be employed to isolate a nucleic acid probe from genomic DNA, or a cDNA or genomic DNA library. The library screen may then be performed using the isolated probe. DNA molecules encoding M vaccae antigens may also be isolated by screening an appropriate M vaccae expression library with anti-sera (e.g., rabbit or monkey) raised specifically against M vaccae antigens.
- anti-sera e.g., rabbit or monkey
- the antigens described herein have the ability to induce an immunogenic response. More specifically, the antigens have the ability to induce cell proliferation and/or cytokine production (for example, interferon- ⁇ and/or interleukin- 12 production) in T cells, NK cells, B cells or macrophages derived from an M tuberculosis- immune individual.
- cytokine production for example, interferon- ⁇ and/or interleukin- 12 production
- An M tuberculosis-immune individual is one who is considered to be resistant to the development of tuberculosis by virtue of having mounted an effective T cell response to M tuberculosis.
- Such individuals may be identified based on a strongly positive (i.e., greater than about 10 mm diameter induration) intradermal skin test response to tuberculosis proteins (PPD), and an absence of any symptoms of tuberculosis infection.
- Assays for cell proliferation or cytokine production in T cells, NK cells, B cells or macrophages may be performed, for example, using the procedures described below.
- the selection of cell type for use in evaluating an immunogenic response to an antigen will depend on the desired response. For example, interleukin- 12 production is most readily evaluated using preparations containing T cells, NK cells, B cells and macrophages derived from M tuberculosis-immune individuals may be prepared using methods well known in the art.
- PBMCs peripheral blood mononuclear cells
- PBMCs may be prepared, for example, using density centrifugation through FicollTM (Winthrop Laboratories, NY).
- T cells for use in the assays described herein may be purified directly from PBMCs.
- an enriched T cell line reactive against mycobacterial proteins, or T cell clones reactive to individual mycobacterial proteins may be employed.
- Such T cell clones may be generated by, for example, culturing PBMCs from M tuberculosis-immune individuals with mycobacterial proteins for a period of 2-4 weeks. This allows expansion of only the mycobacterial protein- specific T cells, resulting in a line composed solely of such cells. These cells may then be cloned and tested with individual proteins, using methods well known in the art, to more accurately define individual T cell specificity.
- the polypeptides disclosed herein are prepared in an isolated, substantially pure, form.
- the polypeptides are at least about 80%) pure, more preferably at least about 90% pure and most preferably at least about 99% pure.
- the substantially pure polypeptides are inco ⁇ orated into pharmaceutical compositions or vaccines for use in one or more of the methods disclosed herein.
- the present invention also provides fusion proteins comprising a first and a second inventive polypeptide or, alternatively, a polypeptide of the present invention and a known M tuberculosis antigen, such as the 38 kDa antigen described in Andersen and Hansen, Infect. Immun. 57:2481-2488, 1989, together with variants of such fusion proteins.
- the fusion proteins of the present invention may also include a linker peptide between the first and second polypeptides.
- a DNA sequence encoding a fusion protein of the present invention is constructed using known recombinant DNA techniques to assemble separate DNA sequences encoding the first and second polypeptides into an appropriate expression vector.
- the 3' end of a DNA sequence encoding the first polypeptide is ligated, with or without a peptide linker, to the 5' end of a DNA sequence encoding the second polypeptide so that the reading frames of the sequences are in phase to permit mRNA translation of the two DNA sequences into a single fusion protein that retains the biological activity of both the first and the second polypeptides.
- a peptide linker sequence may be employed to separate the first and the second polypeptides by a distance sufficient to ensure that each polypeptide folds into its secondary and tertiary structures.
- Such a peptide linker sequence is inco ⁇ orated into the fusion protein using standard techniques well known in the art.
- Suitable peptide linker sequences may be chosen based on the following factors: (1) their ability to adopt a flexible extended conformation; (2) their inability to adopt a secondary structure that could interact with functional epitopes on the first and second polypeptides; and (3) the lack of hydrophobic or charged residues that might react with the polypeptide functional epitopes.
- Preferred peptide linker sequences contain Gly, Asn and Ser residues.
- linker sequences which may be usefully employed as linkers include those disclosed in Maratea et al., Gene 40:39-46, 1985; Mu ⁇ hy et al., Proc. Natl. Acad. Sci. USA 53:8258-8262, 1986; U.S. Patent No. 4,935,233 and U.S. Patent No. 4,751,180.
- the linker sequence may be from 1 to about 50 amino acids in length.
- Peptide linker sequences are not required when the first and second polypeptides have non-essential N-terminal amino acid regions that can be used to separate the functional domains and prevent steric interference.
- the ligated DNA sequences encoding the fusion proteins are cloned into suitable expression systems using techniques known to those of ordinary skill in the art.
- M vaccae, DD-M. vaccae and recombinant M vaccae proteins of the present invention may be employed to activate T cells and NK cells; to stimulate the production of cytokines (in particular Thl class of cytokines) in human PBMC; to enhance the expression of co-stimulatory molecules on dendritic cells and monocytes (thereby enhancing activation); and to enhance dendritic cell maturation and function.
- cytokines in particular Thl class of cytokines
- GV-23 may thus be employed in the treatment of diseases that involve enhancing a Thl immune response.
- GV-23 may be employed as a dendritic cell or NK cell enhancer in the treatment of immune deficiency disorders, such as HIV, and to enhance immune responses and cytotoxic responses to, for example, malignant cells in cancer and following immunosuppressive anti-cancer therapies, such as chemotherapy.
- the inactivated M vaccae, M. vaccae culture filtrate, modified M vaccae cells, M vaccae polypeptide, fusion protein (or polynucleotides encoding such polypeptides or fusion proteins) is generally present within a pharmaceutical composition or a vaccine.
- Pharmaceutical compositions may comprise one or more components selected from the group consisting of inactivated M vaccae cells, M vaccae culture filtrate, modified M vaccae cells, and compounds present in or derived from M vaccae and/or its culture filtrate, together with a physiologically acceptable carrier.
- Vaccines may comprise one or more components selected from the group consisting of inactivated M vaccae cells, M vaccae culture filtrate, modified M vaccae cells, and compounds present in or derived from M vaccae and/or its culture filtrate, together with a non-specific immune response amplifier.
- Such pharmaceutical compositions and vaccines may also contain other mycobacterial antigens, either, as discussed above, inco ⁇ orated into a fusion protein or present within a separate polypeptide.
- a vaccine of the present invention may contain DNA encoding one or more polypeptides as described above, such that the polypeptide is generated in situ.
- the DNA may be present within any of a variety of delivery systems known to those of ordinary skill in the art, including nucleic acid expression systems, bacterial and viral expression systems. Appropriate nucleic acid expression systems contain the necessary DNA sequences for expression in the patient (such as a suitable promoter and terminator signal).
- Bacterial delivery systems involve the administration of a bacterium (such as Bacillus- Calmette-Guerin) that expresses an immunogenic portion of the polypeptide on its cell surface.
- the DNA may be introduced using a viral expression system (e.g., vaccinia or other poxvirus, retrovirus, or adenovirus), which may involve the use of a non-pathogenic, or defective, replication competent virus.
- a viral expression system e.g., vaccinia or other poxvirus, retrovirus, or adenovirus
- Techniques for inco ⁇ orating DNA into such expression systems are well known in the art.
- the DNA may also be "naked,” as described, for example, in Ulmer et al., Science 259: 1745- 1749, 1993 and reviewed by Cohen, Science 259: 1691 -1692, 1993.
- the uptake of naked DNA may be increased by coating the DNA onto biodegradable beads, which are efficiently transported into the cells.
- a DNA vaccine as described above may be administered simultaneously with or sequentially to either a polypeptide of the present invention or a known mycobacterial antigen, such as the 38 kDa antigen described above.
- administration of DNA encoding a polypeptide of the present invention may be followed by administration of an antigen in order to enhance the protective immune effect of the vaccine.
- Routes and frequency of administration, as well as dosage will vary from individual to individual and may parallel those currently being used in immunization using BCG.
- the pharmaceutical compositions and vaccines may be administered by injection (e.g., intradermal, intramuscular, intravenous or subcutaneous), intranasally (e.g., by aspiration) or orally.
- a suitable dose is an amount of polypeptide or DNA that, when administered as described above, is capable of raising an immune response in a patient sufficient to protect the patient from mycobacterial infection for at least 1-2 years.
- the amount of polypeptide present in a dose ranges from about 1 pg to about 100 mg per kg of host, typically from about 10 pg to about 1 mg, and preferably from about 100 pg to about 1 ⁇ g.
- Suitable dose sizes will vary with the size of the patient, but will typically range from about 0.1 ml to about 5 ml.
- the pharmaceutical composition or vaccine is in a form suitable for delivery to the mucosal surfaces of the airways leading to or within the lungs.
- the pharmaceutical composition or vaccine may be suspended in a liquid formulation for delivery to a patient in an aerosol form or by means of a nebulizer device similar to those currently employed in the treatment of asthma.
- the pharmaceutical composition or vaccine is in a form suitable for administration by injection (intracutaneous, intramuscular, intravenous or subcutaneous) or orally. While any suitable carrier known to those of ordinary skill in the art may be employed in the pharmaceutical compositions of this invention, the type of carrier will depend on the suitability for the chosen route of administration.
- the carrier preferably comprises water, saline, alcohol, a lipid, a wax or a buffer.
- a solid carrier such as mannitol, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, glucose, sucrose, and magnesium carbonate.
- Biodegradable microspheres e.g., polylactic galactide
- Suitable biodegradable microspheres are disclosed, for example, in U.S. Patent Nos. 4,897,268 and 5,075,109.
- adjuvants may be employed in the vaccines of this invention to non-specifically enhance the immune response.
- Most adjuvants contain a substance designed to protect the antigen from rapid catabolism, such as aluminum hydroxide or mineral oil, and a non-specific stimulator of immune responses, such as lipid A, Bordetella pertussis, M. tuberculosis, or, as discussed below, M vaccae.
- Suitable adjuvants are commercially available as, for example, Freund's Incomplete Adjuvant and Freund's Complete Adjuvant (Difco Laboratories, Detroit, MI), and Merck Adjuvant 65 (Merck and Company, Inc., Rahway, NJ).
- Other suitable adjuvants include alum, biodegradable microspheres, monophosphoryl lipid A and Quil A.
- this invention provides methods for using one or more of the inventive polypeptides to diagnose tuberculosis using a skin test.
- a skin test is any assay performed directly on a patient in which a delayed-type hypersensitivity (DTH) reaction (such as swelling, reddening or dermatitis) is measured following intradermal injection of one or more polypeptides as described above.
- DTH delayed-type hypersensitivity
- the reaction is measured at least 48 hours after injection, more preferably 48-72 hours.
- the DTH reaction is a cell-mediated immune response, which is greater in patients that have been exposed previously to the test antigen (i.e., the immunogenic portion of the polypeptide employed, or a variant thereof).
- the response may be measured visually, using a ruler.
- a response that is greater than about 0.5 cm in diameter, preferably greater than about 1.0 cm in diameter, is a positive response, indicative of tuberculosis infection.
- the polypeptides of the present invention are preferably formulated, as pharmaceutical compositions containing a polypeptide and a physiologically acceptable carrier, as described above.
- Such compositions typically contain one or more of the above polypeptides in an amount ranging from about 1 ⁇ g to about 100 ⁇ g, preferably from about 10 ⁇ g to about 50 ⁇ g in a volume of 0.1 ml.
- the carrier employed in such pharmaceutical compositions is a saline solution with appropriate preservatives, such as phenol and/or Tween 80TM.
- a polypeptide employed in a skin test is of sufficient size such that it remains at the site of injection for the duration of the reaction period.
- polypeptide that is at least 9 amino acids in length is sufficient.
- the polypeptide is also preferably broken down by macrophages or dendritic cells within hours of injection to allow presentation to T-cells.
- Such polypeptides may contain repeats of one or more of the above sequences or other immunogenic or nonimmunogenic sequences.
- a biological sample is any antibody-containing sample obtained from a patient.
- the sample is whole blood, sputum, serum, plasma, saliva, cerebrospinal fluid or urine. More preferably, the sample is a blood, serum or plasma sample obtained from a patient or a blood supply.
- the polypeptide(s) are used in an assay, as described below, to determine the presence or absence of antibodies to the polypeptide(s) in the sample, relative to a predetermined cut-off value. The presence of such antibodies indicates the presence of mycobacterial infection.
- the polypeptides used are preferably complementary (i.e., one component polypeptide will tend to detect infection in samples where the infection would not be detected by another component polypeptide).
- Complementary polypeptides may generally be identified by using each polypeptide individually to evaluate serum samples obtained from a series of patients known to be infected with a Mycobacterium. After determining which samples test positive (as described below) with each polypeptide, combinations of two or more polypeptides may be formulated that are capable of detecting infection in most, or all, of the samples tested. For example, approximately 25-30% of sera from tuberculosis-infected individuals are negative for antibodies to any single protein, such as the 38 kDa antigen mentioned above.
- Complementary polypeptides may, therefore, be used in combination with the 38 kDa antigen to improve sensitivity of a diagnostic test.
- a variety of assay formats employing one or more polypeptides to detect antibodies in a sample are well known in the art. See, e.g., Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988.
- the assay involves the use of polypeptide immobilized on a solid support to bind to and remove the antibody from the sample. The bound antibody may then be detected using a detection reagent that contains a reporter group.
- Suitable detection reagents include antibodies that bind to the antibody /polypeptide complex and free polypeptide labelled with a reporter group (e.g., in a semi-competitive assay).
- a competitive assay may be utilized, in which an antibody that binds to the polypeptide is labelled with a reporter group and allowed to bind to the immobilized antigen after incubation of the antigen with the sample. The extent to which components of the sample inhibit the binding of the labelled antibody to the polypeptide is indicative of the reactivity of the sample with the immobilized polypeptide.
- the solid support may be any solid material to which the antigen may be attached. Suitable materials are well known in the art.
- the solid support may be a test well in a microtiter plate or a nitrocellulose or other suitable membrane.
- the support may be a bead or disc, such as glass, fiberglass, latex or a plastic material such as polystyrene or polyvinylchloride.
- the support may also be a magnetic particle or a fiber optic sensor, such as those disclosed, for example, in U.S. Patent No. 5,359,681.
- the polypeptides may be bound to the solid support using a variety of techniques well known in the art.
- the term "bound” refers to both noncovalent association, such as adso ⁇ tion, and covalent attachment, which may be a direct linkage between the antigen and functional groups on the support or a linkage by way of a cross-linking agent. Binding by adso ⁇ tion to a well in a microtiter plate or to a membrane is preferred. In such cases, adso ⁇ tion may be achieved by contacting the polypeptide, in a suitable buffer, with the solid support for a suitable amount of time. The contact time varies with temperature, but is typically between about 1 hour and 1 day.
- contacting a well of a plastic microtiter plate such as polystyrene or polyvinylchloride
- an amount of polypeptide ranging from about 10 ng to about 1 ⁇ g, and preferably about 100 ng
- Covalent attachment of polypeptide to a solid support may generally be achieved by first reacting the support with a bifiinctional reagent that will react with both the support and a functional group, such as a hydroxyl or amino group, on the polypeptide.
- polypeptide may be bound to supports having an appropriate polymer coating using benzoquinone or by condensation of an aldehyde group on the support with an amine and an active hydrogen on the polypeptide (see, e.g., Pierce Immunotechnology Catalog and Handbook, 1991, at A12-A13).
- the assay is an enzyme-linked immunosorbent assay (ELISA).
- ELISA enzyme-linked immunosorbent assay
- This assay may be performed by first contacting a polypeptide antigen that has been immobilized on a solid support, with the sample, such that antibodies to the polypeptide within the sample are allowed to bind to the immobilized polypeptide. Unbound sample is then removed from the immobilized polypeptide and a detection reagent capable of binding to the immobilized antibody-polypeptide complex is added. The amount of detection reagent that remains bound to the solid support is then determined using a method appropriate for the specific detection reagent.
- the remaining protein binding sites on the support are typically blocked. Any suitable blocking agent known to those of ordinary skill in the art, such as bovine serum albumin or Tween 20TM (Sigma Chemical Co., St. Louis, MO) may be employed.
- the immobilized polypeptide is then incubated with the sample, and antibody is allowed to bind to the antigen.
- the sample may be diluted with a suitable diluent, such as phosphate-buffered saline (PBS) prior to incubation.
- PBS phosphate-buffered saline
- an appropriate contact time, or incubation time is that period of time that is sufficient to detect the presence of antibody within a M tuberculosis-infected sample.
- the contact time is sufficient to achieve a level of binding that is at least 95%o of that achieved at equilibrium between bound and unbound antibody.
- the time necessary to achieve equilibrium may be readily determined by assaying the level of binding that occurs over a period of time. At room temperature, an incubation time of about 30 minutes is generally sufficient. Unbound sample may be removed by washing the solid support with an appropriate buffer, such as PBS containing 0.1% Tween 20TM. Detection reagent may then be added to the solid support.
- An appropriate detection reagent is any compound that binds to the immobilized antibody-polypeptide complex and that can be detected by any of a variety of means known in the art.
- the detection reagent contains a binding agent (such as, for example, Protein A, Protein G, immunoglobulin, lectin or free antigen) conjugated to a reporter group.
- a binding agent such as, for example, Protein A, Protein G, immunoglobulin, lectin or free antigen
- reporter groups include enzymes (such as horseradish peroxidase), substrates, cofactors, inhibitors, dyes, radionuclides, luminescent groups, fluorescent groups and biotin.
- the conjugation of binding agent to reporter group may be achieved using standard methods known in the art. Common binding agents may also be purchased conjugated to a variety of reporter groups from many commercial sources (e.g., Zymed Laboratories, San Francisco, CA, and Pierce, Rockford, IL).
- the detection reagent is incubated with the immobilized antibody-polypeptide complex for an amount of time sufficient to detect the bound antibody.
- An appropriate amount of time may generally be determined from the manufacturer's instructions or by assaying the level of binding that occurs over a period of time.
- Unbound detection reagent is then removed and bound detection reagent is detected using the reporter group.
- the method employed for detecting the reporter group depends upon the nature of the reporter group. For radioactive groups, scintillation counting or autoradiographic methods are generally appropriate. Spectroscopic methods may be used to detect dyes, luminescent groups and fluorescent groups. Biotin may be detected using avidin, coupled to a different reporter group (commonly a radioactive or fluorescent group or an enzyme). Enzyme reporter groups may be detected by the addition of substrate (generally for a specific period of time), followed by spectroscopic or other analysis of the reaction products.
- the signal detected from the reporter group that remains bound to the solid support is generally compared to a signal that corresponds to a predetermined cut-off value.
- the cut-off value is the average mean signal obtained when the immobilized antigen is incubated with samples from an uninfected patient.
- the cut-off value is determined using a Receiver Operator Curve, according to the method of Sackett et al., Clinical Epidemiology: A Basic Science for Clinical Medicine, Little Brown and Co., 1985, pp. 106-107. In general, signals higher than the predetermined cut-off value are considered to be positive for mycobacterial infection.
- the assay may also be performed in a rapid flow-through or strip test format, wherein the antigen is immobilized on a membrane, such as nitrocellulose.
- a membrane such as nitrocellulose.
- a detection reagent e.g., protein A-colloidal gold
- a detection reagent then binds to the antibody-polypeptide complex as the solution containing the detection reagent flows through the membrane.
- the detection of bound detection reagent may then be performed as described above.
- the strip test format one end of the membrane to which polypeptide is bound is immersed in a solution containing the sample. The sample migrates along the membrane through a region containing detection reagent and to the area of immobilized polypeptide.
- Concentration of detection reagent at the polypeptide indicates the presence of anti- mycobacterial antibodies in the sample.
- concentration of detection reagent at that site generates a pattern, such as a line, that can be read visually. The absence of such a pattern indicates a negative result.
- the amount of polypeptide immobilized on the membrane is selected to generate a visually discernible pattern when the biological sample contains a level of antibodies that would be sufficient to generate a positive signal in an ELISA, as discussed above.
- the amount of polypeptide immobilized on the membrane ranges from about 25 ng to about 1 ⁇ g, and more preferably from about 50 ng to about 500 ng.
- Such tests can typically be performed with a very small amount (e.g. , one drop) of patient serum or blood.
- the present invention also provides antibodies to the inventive polypeptides.
- Antibodies may be prepared by any of a variety of techniques known to those of ordinary skill in the art. See, e.g., Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988.
- an immunogen comprising the antigenic polypeptide is initially injected into any of a wide variety of mammals (e.g., mice, rats, rabbits, sheep and goats).
- the immunogen is injected into the animal host, preferably according to a predetermined schedule inco ⁇ orating one or more booster immunizations, and the animals are bled periodically.
- Polyclonal antibodies specific for the polypeptide may then be purified from such antisera by, for example, affinity chromatography using the polypeptide coupled to a suitable solid support.
- Monoclonal antibodies specific for the antigenic polypeptide of interest may be prepared, for example, using the technique of Kohler and Milstein, Eur. J. Immunol. (5:511-519, 1976, and improvements thereto. Briefly, these methods involve the preparation of immortal cell lines capable of producing antibodies having the desired specificity (i.e., reactivity with the polypeptide of interest). Such cell lines may be produced, for example, from spleen cells obtained from an animal immunized as described above. The spleen cells may then be immortalized by fusion with a myeloma cell fusion partner, preferably one that is syngeneic with the immunized animal, using one of a variety of techniques well known in the art.
- Monoclonal antibodies may be isolated from the supernatants of the resulting hybridoma colonies.
- various techniques may be employed to enhance the yield, such as injection of the hybridoma cell line into the peritoneal cavity of a suitable vertebrate host, such as a mouse.
- Monoclonal antibodies may then be harvested from the ascites fluid or the blood.
- Antibodies may be used in diagnostic tests to detect the presence of mycobacterial antigens using assays similar to those detailed above and other techniques well known to those of skill in the art, thereby providing a method for detecting mycobacterial infection, such as M tuberculosis infection, in a patient.
- Diagnostic reagents of the present invention may also comprise polynucleotides encoding one or more of the above polypeptides, or one or more portions thereof.
- primers comprising at least 10 contiguous oligonucleotides of an inventive polynucleotide may be used in polymerase chain reaction (PCR) based tests.
- probes comprising at least 18 contiguous oligonucleotides of an inventive polynucleotide may be used for hybridizing to specific sequences. Techniques for both PCR based tests and hybridization tests are well known in the art.
- Primers or probes may thus be used to detect M tuberculosis and other mycobacterial infections in biological samples, preferably sputum, blood, serum, saliva, cerebrospinal fluid or urine.
- DNA probes or primers comprising oligonucleotide sequences described above may be used alone, in combination with each other, or with previously identified sequences, such as the 38 kDa antigen discussed above.
- This example illustrates the effect of immunization with heat-killed M vaccae or M vaccae culture filtrate in mice prior to challenge with live M tuberculosis.
- M. vaccae (ATCC Number 15483) was cultured in sterile Medium 90 (yeast extract, 2.5 g/1; tryptone, 5 g/1; glucose, 1 g/1) at 37 °C. The cells were harvested by centrifugation, and transferred into sterile Middlebrook 7H9 medium (Difco Laboratories, Detroit, MI, USA) with glucose at 37 °C for one day. The medium was then centrifuged to pellet the bacteria, and the culture filtrate removed. The bacterial pellet was resuspended in phosphate buffered saline at a concentration of 10 mg/ml, equivalent to 10 10 M vaccae organisms per ml. The cell suspension was then autoclaved for 15 min at 120 °C. The culture filtrate was passaged through a 0.45 ⁇ m filter into sterile bottles.
- sterile Medium 90 yeast extract, 2.5 g/1; tryptone, 5 g/1; glucose, 1 g/1
- the cells were harvested by centrifugation, and
- Fig.l A when mice were immunized with 1 mg, 100 ⁇ g or 10 ⁇ g of M vaccae and infected three weeks later with 5x10 5 colony forming units (CFU) of live M tuberculosis H37Rv, significant protection from infection was seen.
- CFU colony forming units
- spleen, liver and lung tissue was harvested from mice three weeks after infection, and live bacilli determined (expressed as CFU).
- the reduction in bacilli numbers when compared to tissue from non-immunized control mice, exceeded 2 logs in liver and lung tissue, and 1 log in spleen tissue.
- Fig. IB shows that when mice were immunized with 100 ⁇ g of M vaccae culture filtrate, and infected three weeks later with 5x10 5 CFU of M tuberculosis H37Rv, significant protection was also seen.
- CFU live bacilli numbers
- This example illustrates the effect of immunisation with heat-killed M vaccae or M vaccae culture filtrate through intradermal and intralung routes in cynomolgous monkeys prior to challenge with live M tuberculosis.
- Heat-killed M vaccae and M vaccae culture filtrate were prepared as described above in Example 1.
- Five groups of cynomolgous monkeys were used, with each group containing 2 monkeys. Two groups of monkeys were immunised with whole heat-killed M vaccae either intradermally or intralung; two groups of monkeys were immunised with M vaccae culture filtrate either intradermally or intralung; and a control group received no immunisations. All immunogens were dissolved in phosphate buffered saline. The composition employed for immunisation, amount of immunogen, and route of administration for each group of monkeys are provided in Table 1.
- ESR mm/hr erythrocyte sedimentation rate
- LPA lymphocyte proliferation
- body weight, temperature, ESR and LPA to PPD were measured, then all monkeys were infected with 10 3 colony forming units of the Erdman strain of Mycobacterium tuberculosis by inserting the organisms directly in the right lungs of immunised animals. Twenty eight days following infection, body weight, temperature, ESR and LPA to PPD were measured in all monkeys, and the lungs were x-rayed to determine whether infection with live M tuberculosis had resulted in the onset of pneumonia.
- Table 2A Twenty-eight days after infection with M tuberculosis Erdman, chest x-rays of control (non-immunised) monkeys revealed haziness over the right suprahilar regions of both animals, indicating the onset of pneumonia. This progressed and by day 56 post-infection x- rays indicated disease in both lungs. As expected, as disease progressed both control animals lost weight and showed significant LPA responses to PPD, indicating strong T cell reactivity to M tuberculosis. The ESR measurements were variable but consistent with strong immune reactivity.
- Table 2C The two monkeys immunised twice with 500 ⁇ g M vaccae intralung showed almost identical results to those animals of Table 2B. There was no sign of lung disease 84 days post infection with M tuberculosis, with consistent weight gains. Both animals showed LPA response to PPD in the immunisation phase (day 0-62) and post-infection, indicating strong T cell reactivity had developed as a result of using the lung as the route of immunisation and subsequent infection.
- mice were given 2 ⁇ g ovalbumin in 100 ⁇ l alum adjuvant by the intraperitoneal route at time 0 and 14 days, and subsequently given 100 ⁇ g ovalbumin in 50 ⁇ l phosphate buffered saline (PBS) by the intranasal route on day 28.
- PBS phosphate buffered saline
- the mice accumulated eosinophils in their lungs as detected by washing the airways of the anaesthetised mice with saline, collecting the washings (broncheolar lavage or BAL), and counting the numbers of eosinophils.
- mice groups of seven mice administered either 10 or 1000 ⁇ g of heat-killed M vaccae (Fig. 2A), or 10 , 100 or 200 ⁇ g of DD-M vaccae, prepared as described below (Fig. 2B) intranasally 4 weeks before intranasal challenge with ovalbumin, had reduced percentages of eosinophils in the BAL cells collected 5 days after challenge with ovalbumin compared to control mice.
- Control mice were given intranasal PBS.
- Live M bovis BCG at a dose of 2 x 10 5 colony forming units also reduced lung eosinophilia.
- the data in Figs. 2A and B show the mean and SEM per group of mice.
- Figs. 2C and D show that mice given either 1000 ⁇ g of heat-killed M vaccae (Fig. 2C) or 200 ⁇ g of DD-M vaccae (Fig. 2D) intranasally as late as one week before challenge with ovalbumin had reduced percentages of eosinophils compared to control mice. In contrast, treatment with live BCG one week before challenge with ovalbumin did not inhibit the development of lung eosinophilia when compared with control mice.
- immunization with BCG of the Pasteur (BCG-P) and Connought (BCG-C) strains prior to challenge with ovalbumin also reduced the percentage of eosinophils in the BAL of mice.
- Eosinophils are blood cells that are prominent in the airways in allergic asthma.
- the secreted products of eosinophils contribute to the swelling and inflammation of the mucosal linings of the airways in allergic asthma.
- the data shown in Figs. 2A-E indicate that treatment with heat-killed M vaccae or DD-M vaccae reduces the accumulation of lung eosinophils, and may be useful in reducing inflammation associated with eosinophilia in the airways, nasal mucosal and upper respiratory tract.
- Mycolic acids were depleted from DD-Mv ⁇ cc ⁇ e by treatment with potassium hydroxide (0.5% KOH) in ethanol for 48 hours at 37°C.
- Mycolic acid depleted OO-M.vaccae cells were then washed with ethanol and ether and dried.
- Arabinogalactans were depleted from the KOH treated OO-M.vaccae by further treatment with 1% periodic acid in 3% acetic acid for 1 hr at room temperature followed by treatment with sodium borohydride 0.1M for 1 hour at room temperature. After arabinogalactan depletion, samples were washed with water and lyophilized.
- mice infected with BCG had higher levels of ovalbumin specific IgGl than sera from PBS controls.
- mice immunized with M vaccae or DD-M vaccae had similar or lower levels of ovalbumin- specific IgGl.
- IgGl antibodies are characteristic of a Th2 immune response, these results are consistent with the suppressive effects of heat-killed M vaccae and DD-M. vaccae on the asthma-inducing Th2 immune responses.
- mice per group At least 7 mice per group.
- Ovalbumin-specific IgGl was detected using anti-mouse IgGl (Serotec). Group means are expressed as the reciprocal of the EU50 end point titre.
- This example illustrates the effect of immunization with heat-killed M.vaccae, OO- M.vaccae or recombinant M vaccae proteins without additional adjuvants, or a combination of heat-killed M.vaccae with a pool of recombinant proteins derived from M.vaccae.
- mice were injected intraperitoneally with one of the following preparations on two occasions three weeks apart: a) Phosphate buffered saline (PBS, control); b) Heat-killed M vaccae (500 ug); c) OO-M.vaccae (50 ug); d) A pool of recombinant proteins containing 15 ug of each of GV4P, GN7, GN9, GN27B, GN33 protein (prepared as described below); and e) Heat-killed M vaccae plus the pool of recombinant proteins
- mice were infected with 5 X 10 5 live H37Rv M. tuberculosis organisms. After a further three weeks, the mice were sacrificed, and their spleens homogenized and assayed for colony forming units (CFU) of M.tuberculosis as an indicator of severity of infection.
- CFU colony forming units
- Figs. 3A and 3B show data in which each point represents individual mice.
- the numbers of CFU recovered from control mice immunised with PBS alone were taken as the baseline. All data from experimental mice were expressed as number of logarithms of CFUs below the baseline for control mice (or log protection).
- mice immunized with heat-killed M.vaccae or OO-M.vaccae showed a mean reduction of >1 or 0.5 logs CFU, respectively.
- the spleens of mice immunized with the pool of recombinant proteins containing GV4P, GV7, GV9, GV27B and GV33 had CFUs slightly less than control mice.
- GV4P, GV7, GN9, GN27B and GV33 were given in combination with heat-killed M.vaccae, the reduction in CFUs exceeded a mean of > 1.5 logs.
- the data demonstrates the effectiveness of immunization with M.vaccae, DD- M.vaccae or recombinant proteins derived from M.vaccae against subsequent infection with tuberculosis, and further indicates that M.vaccae, DD-M.vaccae and recombinant proteins may be developed as vaccines against tuberculosis.
- This example illustrates the effect of two intradermal injections of heat-killed Mycobacterium vaccae on psoriasis in human patients.
- M vaccae (ATCC Number 15483) was cultured in sterile Medium 90 (yeast extract, 2.5g/l; tryptone, 5 g/1; glucose, 1 g/1) at 37 °C. The cells were harvested by centrifugation, and transferred into sterile Middlebrook 7H9 medium (Difco Laboratories, Detroit, MI, USA) with glucose at 37 °C for one day. The medium was then centrifuged to pellet the bacteria, and the culture filtrate removed. The bacterial pellet was resuspended in phosphate buffered saline at a concentration of 10 mg/ml, equivalent to 10 10 M vaccae organisms per ml.
- sterile Medium 90 yeast extract, 2.5g/l; tryptone, 5 g/1; glucose, 1 g/1
- the cells were harvested by centrifugation, and transferred into sterile Middlebrook 7H9 medium (Difco Laboratories, Detroit, MI, USA) with glucose at 37 °C for one day. The medium was then centr
- the cell suspension was then autoclaved for 15 min at 120 °C and stored frozen at -20 °C. Prior to use the M vaccae suspension was thawed, diluted to a concentration of 5 mg/ml in phosphate buffered saline, autoclaved for 15 min at 120 °C and 0.2 ml aliquoted under sterile conditions into vials for use in patients.
- PASI scores are a measure of the location, size and degree of skin scaling in psoriatic lesions on the body.
- a PASI score of above 12 reflects widespread disease lesions on the body. The study commenced with a washout period of four weeks where the patients did not have systemic anti-psoriasis treatment or effective topical therapy.
- the PASI scores were determined at -4, 0, 3, 6 and 12 weeks;
- PASI score 0
- the remission or improvement of PASI score may be long lasting.
- Patient PS-003 achieved remission by week 20 and was still in remission at week 80.
- Overall 13 of 24 patients showed a greater than 50% improvement in PASI scores.
- Patient PS-001 achieved remission at week 16, relapsed at week 48 (PASI 2.7), was re- vaccinated with injections of M.vaccae and subsequently improved with PASI falling from 17.8 (Week 60) to 0.8 (week 84). Thus patients may benefit from repeated treatment.
- This example illustrates the effect of two intradermal injections of DD-M. vaccae on psoriasis.
- PASI scores are a measure of the location, size and degree of skin scaling in psoriatic lesions on the body.
- a PASI score of above 12 reflects widespread disease lesions on the body.
- the study commenced with a washout period of four weeks where the four patients did not have systemic antipsoriasis treatment or effective topical therapy.
- the seven patients were then injected intradermally with 0.1 ml DD-M vaccae (equivalent to 100 ⁇ g). This was followed three weeks later with a second intradermal injection with the same dose of DD-M vaccae (100 ⁇ g).
- Psoriasis was evaluated from four weeks before the first injection of M vaccae to six weeks after the first injection as follows:
- Table 9 The data shown in Table 9 are the PASI scores of the seven patients at the time of the first injection of DD-M. vaccae (Day 0), 3, 6, 12 and 24 weeks later.
- the PASI score of patient PS-025 was reduced to less than 1 for more than 12 weeks.
- PASI 15.8
- PASI 15.8
- treatment of psoriasis with DD-Mv ⁇ cc ⁇ e may lead to disease remission or provide prolonged benefit. Patients may also benefit with repeated treatment.
- Heat-killed M vaccae was prepared as described as above in Example 1. To prepare delipidated M.vaccae, the autoclaved M.vaccae was pelleted by centrifugation, the pellet washed with water, collected again by centrifugation and then freeze-dried. An aliquot of this freeze-dried M.vaccae was set aside and referred to as lyophilised M.vaccae. When used in experiments it was resuspended in PBS to the desired concentration. Freeze-dried M vaccae was treated with chloroform methanol (2:1) for 60 mins at room temperature to extract lipids, and the extraction was repeated once.
- the delipidated residue from chloroform/methanol extraction was further treated with 50% ethanol to remove glycolipids by refluxing for two hours.
- the 50% ethanol extraction was repeated two times.
- the pooled 50% ethanol extracts were used as a source of M vaccae glycolipids (see below).
- the residue from the 50% ethanol extraction was freeze-dried and weighed.
- the amount of delipidated and deglycolipidated M.vaccae prepared was equivalent to 11.1% of the starting wet weight of M.vaccae used.
- the delipidated and deglycolipidated M vaccae (DD-M. vaccae) was resuspended in phosphate-buffered saline by sonication, and sterilised by autoclaving.
- compositional analyses of heat-killed M vaccae and DD-M. vaccae are presented in Table 9. Major changes are seen in the fatty acid composition and amino acid composition of DD-M vaccae as compared to the insoluble fraction of heat-killed M vaccae.
- the data presented in Table 9 show that the insoluble fraction of heat-killed M.vaccae contains 10% w/w of lipid, and the total amino acid content is 2750 nmoles/mg, or approximately 33% w/w.
- DD-M. vaccae contains 1.3% w/w of lipid and 4250 nmoles/mg amino acids, which is approximately 51% w/w.
- the insoluble fraction of heat-killed M vaccae contains 10% w/w of lipid, and DD-M. vaccae contains 1.3% w/w of lipid.
- the total amino acid content of the insoluble fraction of heat-killed M vaccae is 2750 nmoles/mg, or approximately 33% w/w.
- the total amino acid content of DD-M vaccae is 4250 nmoles/mg, or approximately 51% w/w.
- Delipidated and deglycolipidated M tuberculosis and M smegmatis were prepared using the procedure described above for delipidated and deglycolipidated M vaccae. As indicated in Table 10, the profiles of the percentage composition of amino acids in DD-M. vaccae, DD-M. tuberculosis and DD-M smegmatis showed no significant differences. However, the total amount of protein varied - the two batches of DD-M vaccae contained 34% and 55% protein, whereas DD-M tuberculosis and DD- M smegmatis contained 79% and 72% protein, respectively.
- DD-M vaccae Analysis of the monosaccharide composition shows significant differences between DD-M vaccae, and DD-M tuberculosis and DD-M. smegmatis.
- the monosaccharide composition of two batches of DD-M. vaccae was the same and differed from that of DD-M tuberculosis and M smegmatis. Specifically, DD-M. vaccae was found to contain free glucose while both DD-M tuberculosis and M smegmatis contain glycerol, as shown in Table 11.
- the pooled 50% ethanol extracts described above were dried by rotary evaporation, redissolved in water, and freeze-dried.
- the amount of glycolipid recovered was 1.2% of the starting wet weight of M vaccae used.
- the glycolipids were dissolved in phosphate-buffered saline.
- a group of C57BL/6J mice were injected intraperitoneally with DIFCO thioglycolate and after three days, peritoneal macrophages were collected and placed in cell culture with interferon-gamma for three hours.
- the culture medium was replaced and various concentrations of whole heat-killed (autoclaved) M v ⁇ cc ⁇ e, lyophilized M. v ⁇ cc ⁇ e, DD-M v ⁇ cc ⁇ e and M v ⁇ cc ⁇ e glycolipids, prepared as described above, were added.
- the culture supernatants were assayed for the presence of IL-12 produced by macrophages.
- the M v ⁇ cc ⁇ e preparations stimulated the production of IL-12 from macrophages.
- NK cells Natural Killer cells
- Spleen cells were prepared from Severe Combined Immunodeficient (SCID) mice. These populations contain 75-80% NK cells. The spleen cells were incubated at 37 °C in culture with different concentrations of heat-killed M v ⁇ cc ⁇ e, DD-M. v ⁇ cc ⁇ e, or M v ⁇ cc ⁇ e glycolipids. The data shown in Fig. 5 demonstrates that, while heat-killed M vaccae and M.
- v ⁇ cc ⁇ e glycolipids stimulate production of interferon-gamma, DD-M v ⁇ cc ⁇ e stimulated relatively less interferon-gamma.
- the combined data from Figs. 4 and 5 indicate that, compared with whole heat-killed M v ⁇ cc ⁇ e, DD-M.
- v ⁇ cc ⁇ e is a better stimulator of IL-12 than interferon gamma.
- Figs. 6 A, B, and C show data from separate experiments in which groups of C57BL/6 mice (Fig. 6A), BALB/c mice (Fig. 6B) or C3H/HeJ mice (Fig. 6C) were given DIFCO thioglycolate intraperitoneally. After three days, peritoneal macrophages were collected and placed in culture with interferon- gamma for three hours.
- the culture supernatants were assayed for the presence of IL-12 produced by macrophages.
- the recombinant proteins and M v ⁇ cc ⁇ e preparations stimulated the production of IL-12 from macrophages.
- IF ⁇ -primed peritoneal macrophages from BALB/c mice were stimulated with 40 ug/ml of M v ⁇ cc ⁇ e recombinant proteins in culture for 3 days and the presence of IL-12 produced by macrophages was assayed.
- IF ⁇ -primed macrophages produced IL-12 when cultured with a control protein, ovalbumin (ova).
- the recombinant proteins GV 24B, 38BP, 38AP, 27, 5, 27B, 3, 23 and 22B stimulated more than twice the amount of IL-12 detected in control macrophage cultures.
- M. vaccae culture supernatant S/N
- killed M vaccae delipidated M vaccae and delipidated and deglycolipidated M vaccae (DD-M vaccae)
- DD-M vaccae deglycolipidated M vaccae
- vaccae delipidated M vaccae; delipidated M vaccae with glycolipids also extracted (DD-M vaccae) and proteins extracted with SDS; the SDS protein extract treated with Pronase (an enzyme which degrades protein); whole M vaccae culture filtrate; and heat- killed M tuberculosis or heat-killed M bovis BCG, M phlei or M smegmatis or M vaccae culture filtrate.
- E.G7 cells which are EL4 cells (a C57BL/6-derived T cell lymphoma) transfected with the ovalbumin gene and thus express ovalbumin.
- the spleen cells were then assayed for their ability to kill non-specifically EL4 target cells or to kill specifically the E.G7 ovalbumin expressing cells. Killing activity was detected by the release of 51 Chromium with which the EL4 and E.G7 cells have been labelled (100 ⁇ Ci per 2x10 6 ), prior to the killing assay. Killing or cytolytic activity is expressed as % specific lysis using the formula:
- cytotoxic cells are generated only in mice immunized with ovalbumin with an adjuvant but not in mice immunized with ovalbumin alone.
- the diagrams that make up Fig. 7 show the effect of various M vaccae derived adjuvant preparations on the generation of cytotoxic T cells to ovalbumin in C57BL/6 mice.
- cytotoxic cells were generated in mice immunized with (i) 10 ⁇ g, (ii) 100 ⁇ g or (iii) 1 mg of autoclaved M vaccae or (iv) 75 ⁇ g of M vaccae culture filtrate.
- Fig. 7 A cytotoxic cells were generated in mice immunized with (i) 10 ⁇ g, (ii) 100 ⁇ g or (iii) 1 mg of autoclaved M vaccae or (iv) 75 ⁇ g of M vaccae culture filtrate.
- FIG. 7B shows that cytotoxic cells were generated in mice immunized with (i) 1 mg whole autoclaved M vaccae or (ii) 1 mg delipidated and deglycolipidated (DD-) M vaccae.
- cytotoxic cells were generated in mice immunized with 1 mg whole autoclaved M vaccae;
- Fig. 7C(ii) shows the active material in M vaccae soluble proteins extracted with SDS from DD-M vaccae.
- Fig. 7C(iii) shows that active material in the adjuvant preparation of Fig. 7C(ii) was destroyed by treatment with the proteolytic enzyme Pronase.
- 100 ⁇ g of the SDS-extracted proteins had significantly stronger immune-enhancing ability (Fig. 7C(ii)) than did 1 mg whole autoclaved M vaccae (Fig. 7C(i)).
- mice immunized with 1 mg heat-killed M vaccae generated cytotoxic cells to ovalbumin, but mice immunized separately with 1 mg heat-killed M tuberculosis (Fig. 7D(ii)), 1 mg M bovis BCG (Fig. 7D(iii)), 1 mg M phlei (Fig. 7D(iv)), or 1 mg M smegmatis (Fig. 7D(v)) failed to generate cytotoxic cells.
- mice immunised with ovalbumin plus 200 ug of DD- M.vaccae depleted of mycolic acids and arabinogalactan were also able to generate cytotoxic cells (28% to 46% maximum specific lysis compared with ⁇ 8% specific lysis for control mice immunised with ovalbumin alone).
- the M vaccae culture filtrate described above was fractionated by iso-electric focusing and the fractions assayed for adjuvant activity in the anti-ovalbumin-specific cytotoxic response assay in C57BL/6 mice as described above. Peak adjuvant activities were demonstrated in fractions corresponding to pl of 4.2-4.32 (fraction nos. 7-9), 4.49-4.57 (fraction nos. 13-17) and 4.81-5.98 (fraction nos. 23-27).
- mice were immunised intra-peritoneally with 50 ug of DD-M. vaccae once a week for 5 weeks. At the 6 th week mice were sacrificed and their serum collected. The sera were tested for antibodies to recombinant M v ⁇ cc ⁇ e-derived proteins, prepared as described below, in standard enzyme-linked immunoassays.
- the antisera did not react with several M vaccae recombinant proteins nor with ovalbumin, which served as an irrelevant negative control protein in the enzyme-linked assays (data not shown).
- Antisera from mice immunised with DD-M vaccae reacted with 12 M. v ⁇ cc ⁇ e-derived GN antigens.
- the results are shown in Table 12 below.
- the antisera thus identified GV3, 5P, 5, 7, 9, 22B, 24, 27, 27A, 27B, 33 and 45 as being present in DD-M. vaccae.
- mice were injected in each footpad with 100 ug DD-M v ⁇ cc ⁇ e in combination with incomplete Freund's adjuvant and 10 days later were sacrificed to obtain popliteal lymph node cells.
- the cells from immunized and non-immunized control mice were stimulated in vitro with recombinant M v ⁇ cc ⁇ e-derived GV proteins. After 3 days, cell proliferation and IF ⁇ production were assessed. T cell proliferative responses of lymph node cells from OO-M.vaccae immunized mice to GV proteins.
- Lymph node cells from DD-M. v ⁇ cc ⁇ e-immunized mice did not proliferate in response to an irrelevant protein, ovalbumin, (data not shown). As shown in Table 13, lymph node cells from immunized mice showed proliferative responses to GV 3, 7, 9, 23, 27, 27B, and 33. The corresponding cells from non-immunized mice did not proliferate in response to these GV proteins suggesting that mice immunized with DD-M vaccae have been immunized with these proteins. Thus, the M.vaccae derived proteins GV 3, 7, 9, 23, 27, 27B and 33 are likely to be present in DD-M v ⁇ cc ⁇ e.
- Stimulation index cpm from tritiated Thymidine uptake in presence of GV protein/cpm in absence of GV protein
- lymph node cells from DD-M vaccae immunized mice did not produce IFN ⁇ upon stimulation with GV proteins.
- lymph node cells from DD-M v ⁇ cc ⁇ e immunized mice secrete IFN ⁇ in a dose dependent manner when stimulated with GV 3, 5, 23, 27A, 27B, 33, 45 or 46, suggesting that the mice have been immunized with these proteins.
- No IFN ⁇ production was detectable when cells from immunized mice were stimulated with the irrelevant protein, ovalbumin (data not shown).
- the proteins GV 3, 5, 23, 27A, 27B, 33, 45 and 46 are thus likely to be present in DD-M vaccae.
- the five proteins GV27, 27A, 27B, 23 and 45 were used as non-specific immune amplifiers with ovalbumin antigen to immunize mice as described above in Example 6.
- 50 ug of any one of the recombinant proteins GV27, 27A, 27B, 23 and 45 when injected with 50-100 ug of ovalbumin, demonstrated adjuvant properties in being able to generate cytotoxic cells to ovalbumin.
- This example illustrates the ability of killed M vaccae to stimulate cytotoxic CD8 T cells which preferentially kill macrophages that have been infected with M tuberculosis.
- mice were immunized by the intraperitoneal injection of 500 ⁇ g of killed M vaccae which was prepared as described in Example 1.
- the spleen cells of immunized mice were passed through a CD8 T cell enrichment column (R&D Systems, St. Paul, MN, USA).
- the spleen cells recovered from the column have been shown to be enriched up to 90% CD8 T cells.
- These T cells, as well as CD8 T cells from spleens of non-immunized mice were tested for their ability to kill uninfected macrophages or macrophages which have been infected with M tuberculosis.
- Macrophages were obtained from the peritoneal cavity of mice five days after they have been given 1 ml of 3% thioglycolate intraperitoneally.
- the macrophages were infected overnight with M tuberculosis at the ratio of 2 mycobacteria per macrophage. All macrophage preparations were labelled with 51 Chromium at 2 ⁇ Ci per 10 4 macrophages.
- the macrophages were then cultured with CD8 T cells overnight (16 hours) at killer to target ratios of 30:1. Specific killing was detected by the release of 51 Chromium and expressed as % specific lysis, calculated as in Example 5.
- ELISA enzyme-linked immunosorbent assay
- CD 8 T cells from spleens of mice immunized with M vaccae were cytotoxic for macrophages infected with M tuberculosis and did not lyse uninfected macrophages.
- the CD8 T cells from non-immunized mice did not lyse macrophages.
- CD8 T cells from naive or non-immunized mice do produce IFN- ⁇ when cocultured with infected macrophages. The amount of IFN- ⁇ produced in coculture was greater with CD8 T cells derived from M vaccae immunized mice.
- M vaccae (ATCC Number 15483) was cultured in sterile Medium 90 at 37 °C. The cells were harvested by centrifugation, and transferred into sterile Middlebrook 7H9 medium with glucose at 37 °C for one day. The medium was then centrifuged (leaving the bulk of the cells) and filtered through a 0.45 ⁇ m filter into sterile bottles.
- the culture filtrate was concentrated by lyophilization, and redissolved in MilliQ water. A small amount of insoluble material was removed by filtration through a 0.45 ⁇ m membrane.
- the culture filtrate was desalted by membrane filtration in a 400 ml Amicon stirred cell which contained a 3kDa molecular weight cut-off (MWCO) membrane. The pressure was maintained at 50 psi using nitrogen gas.
- the culture filtrate was repeatedly concentrated by membrane filtration and diluted with water until the conductivity of the sample was less than 1.0 mS. This procedure reduced the 20 1 volume to approximately 50 ml. Protein concentrations were determined by the Bradford protein assay (Bio-Rad, Hercules, CA, USA).
- the desalted culture filtrate was fractionated by ion exchange chromatography on a column of Q-Sepharose (Pharmacia Biotech, Uppsala, Sweden) (16 X 100 mm) equilibrated with lOmM Tris HC1 buffer pH 8.0. Polypeptides were eluted with a linear gradient of NaCI from 0 to 1.0 M in the above buffer system. The column eluent was monitored at a wavelength of 280 nm.
- the pool of polypeptides eluting from the ion exchange column was concentrated in a 400 ml Amicon stirred cell which contained a 3 kDa MWCO membrane. The pressure was maintained at 50 psi using nitrogen gas. The polypeptides were repeatedly concentrated by membrane filtration and diluted with 1% glycine until the conductivity of the sample was less than 0.1 mS.
- the purified polypeptides were then fractionated by preparative isoelectric focusing in a Rotofor device (Bio-Rad, Hercules, CA, USA).
- the pH gradient was established with a mixture of Ampholytes (Pharmacia Biotech) comprising 1.6% pH 3.5-5.0 Ampholytes and 0.4% pH 5.0 - 7.0 Ampholytes.
- Acetic acid 0.5 M was used as the anolyte, and 0.5 M ethanolamine as the catholyte.
- Isoelectric focusing was carried out at 12W constant power for 6 hours, following the manufacturer's instructions. Twenty fractions were obtained.
- the polypeptide fractions which were shown to contain significant contamination were further purified using a Mono Q column (Pharmacia Biotech) 10 micron particle size (5 x 50 mm) or a Vydac Diphenyl column (Separations Group) 300 Angstrom pore size, 5 micron particle size (4.6 x 250 mm).
- Mono Q column polypeptides were eluted with a linear gradient from 0-0.5 M NaCI in 10 mM Tris HC1 pH 8.0.
- polypeptides were eluted with a linear gradient of acetonitrile (20-60% v/v) in 0.1% TFA.
- the flow-rate was 1.0 ml/min and the column eluent was monitored at 220 nm for both columns.
- the polypeptide peak fractions were collected and analysed for purity on a 15% polyacrylamide gel as described above.
- polypeptides were individually dried onto BiobreneTM (Perkin Elmer/ Applied BioSystems Division, Foster City, CA)-treated glass fiber filters.
- the filters with polypeptide were loaded onto a Perkin Elmer/Applied BioSystems Procise 492 protein sequencer and the polypeptides were sequenced from the amino terminal end using traditional Edman chemistry.
- the amino acid sequence was determined for each polypeptide by comparing the retention time of the PTH amino acid derivative to the appropriate PTH derivative standards.
- GVc-1 six soluble M vaccae antigens, designated GVc-1, GVc-2, GVc-7, GVc-13, GVc-20 and GVc-22, were isolated.
- N-terminal and internal sequences for GVc-1 are shown in SEQ ID NOS: 1, 2 and 3, respectively; the N- terminal sequence for GVc-2 is shown in SEQ ID NO: 4; internal sequences for GVc-7 are shown in SEQ ID NOS: 5-8; internal sequences for GVc-13 are shown in SEQ ID NOS: 9-11; internal sequence for GVc-20 is shown in SEQ ID NO: 12; and N-terminal and internal sequences for GVc-22 are shown in SEQ ID NO: 56-59, respectively.
- Each of the internal peptide sequences provided herein begins with an amino acid residue which is assumed to exist in this position in the polypeptide, based on the known cleavage specificity of cyanogen bromide (Met) or Lys-C (Lys).
- GVc-16, GVc-18 and GVc-21 Three additional polypeptides, designated GVc-16, GVc-18 and GVc-21, were isolated employing a preparative sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS- PAGE) purification step in addition to the preparative isoelectric focusing procedure described above. Specifically, fractions comprising mixtures of polypeptides from the preparative isoelectric focusing purification step previously described were purified by preparative SDS-PAGE on a 15% polyacrylamide gel. The samples were dissolved in reducing sample buffer and applied to the gel.
- SDS- PAGE preparative sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- the separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane by electroblotting in 10 mM 3- (cyclohexylamino)-l-propanesulfonic acid (CAPS) buffer pH 11 containing 10% (v/v) methanol.
- PVDF polyvinylidene difluoride
- CAPS cyclohexylamino-l-propanesulfonic acid
- the transferred protein bands were identified by staining the PVDF membrane with Coomassie blue. Regions of the PVDF membrane containing the most abundant polypeptide species were cut out and directly introduced into the sample cartridge of the Perkin Elmer/ Applied BioSystems Procise 492 protein sequencer. Protein sequences were determined as described above.
- the N-terminal sequences for GVc-16, GVc-18 and GVc-21 are provided in SEQ ID NOS: 13, 14 and 15, respectively.
- Additional antigens designated GVc-12, GVc-14, GVc-15, GVc-17 and GVc-19, were isolated employing a preparative SDS-PAGE purification step in addition to the chromatographic procedures described above. Specifically, fractions comprising a mixture of antigens from the Vydac C4 HPLC purification step previously described were fractionated by preparative SDS-PAGE on a polyacrylamide gel. The samples were dissolved in non- reducing sample buffer and applied to the gel. The separated proteins were transferred to a PVDF membrane by electroblotting in 10 mM CAPS buffer, pH 11 containing 10% (v/v) methanol. The transferred protein bands were identified by staining the PVDF membrane with Coomassie blue.
- Regions of the PVDF membrane containing the most abundant polypeptide species were cut out and directly introduced into the sample cartridge of the Perkin Elmer/Applied BioSystems Procise 492 protein sequencer. Protein sequences were determined as described above. The determined N-terminal sequences for GVc-12, GVc-14, GVc-15, GVc-17 and GVc-19 are provided in SEQ ID NOS: 16-20, respectively.
- GVc-22B The determined nucleotide sequence of the gene encoding GV-22B and the predicted amino acid sequence are provided in SEQ ID NOS: 144 and 145 respectively.
- Amplifications primers AD86 and AD112 were designed from the amino acid sequence of GVc-1 (SEQ ID NO: 1) and the M tuberculosis MPT70 gene sequence. Using these primers, a 310 bp fragment was amplified from M vaccae genomic DNA and cloned into EcoRV-digested vector pBluescript II SK + (Stratagene). The sequence of the cloned insert is provided in S ⁇ Q ID NO: 62. The insert of this clone was used to screen a M vaccae genomic DNA library constructed in lambda ZAP- ⁇ xpress (Stratagene, La Jolla, CA).
- the clone isolated contained an open reading frame with homology to the M. tuberculosis antigen MPT83 and was re-named GV-1/83. This gene also had homology to the M bovis antigen MPB83.
- the determined nucleotide sequence and predicted amino acid sequences are provided in S ⁇ Q ID NOS: 146 and 147 respectively.
- degenerate oligonucleotides ⁇ V59 and EV61 (SEQ ID NOS: 148 and 149 respectively) were designed.
- a 100 bp fragment was amplified, cloned into plasmid pBluescript II SK + and sequenced (SEQ ID NO: 150) following standard procedures (Sambrook et al. Ibid).
- the cloned insert was used to screen a M vaccae genomic DNA library constructed in lambda ZAP-Express.
- the clone isolated had homology to M tuberculosis antigen MPT70 and M bovis antigen MPB70, and was named GV-1/70.
- the determined nucleotide sequence and predicted amino acid sequence for GV-1/70 are provided in SEQ ID NOS: 151 and 152 respectively.
- the genes encoding GV1/83, GV1/70, GVc-13, GVc- 14 and GV-22B were sub-cloned into the expression vector pET16 (Novagen, Madison, WI). Expression and purification were performed according to the manufacturer's protocol.
- the purified polypeptides were screened for the ability to induce T-cell proliferation and IFN- ⁇ in peripheral blood cells from immune human donors. These donors were known to be PPD (purified protein derivative from M tuberculosis) skin test positive and their T cells were shown to proliferate in response to PPD.
- Donor PBMCs and crude soluble proteins from M vaccae culture filtrate were cultured in medium comprising RPMI 1640 supplemented with 10% (v/v) autologous serum, penicillin (60 ⁇ g/ml), streptomycin (100 ⁇ g/ml), and glutamine (2 mM). After 3 days, 50 ⁇ l of medium was removed from each well for the determination of IFN- ⁇ levels, as described below.
- the plates were cultured for a further 4 days and then pulsed with l ⁇ Ci/well of tritiated thymidine for a further 18 hours, harvested and tritium uptake determined using a scintillation counter. Fractions that stimulated proliferation in both replicates two-fold greater than the proliferation observed in cells cultured in medium alone were considered positive.
- IFN- ⁇ was measured using an enzyme-linked immunosorbent assay (ELISA).
- ELISA plates were coated with a mouse monoclonal antibody directed to human IFN- ⁇ (Endogen, Wobural, MA) 1 ⁇ g/ml phosphate-buffered saline (PBS) for 4 hours at 4 °C.
- Wells were blocked with PBS containing 0.2% Tween 20 for 1 hour at room temperature. The plates were then washed four times in PBS/0.2% Tween 20, and samples diluted 1 :2 in culture medium in the ELISA plates were incubated overnight at room temperature.
- polypeptides containing sequences that stimulate peripheral blood mononuclear cells (PBMC) T cells to proliferate and produce IFN- ⁇ are shown in Table 16, wherein (-) indicates a lack of activity, (+/-) indicates polypeptides having a result less than twice higher than background activity of control media, (+) indicates polypeptides having activity two to four times above background, and (++) indicates polypeptides having activity greater than four times above background.
- M vaccae soluble proteins were isolated from culture filtrate using 2-dimensional polyacrylamide gel electrophoresis as described below. Unless otherwise noted, all percentages in the following example are weight per volume.
- M vaccae (ATCC Number 15483) was cultured in sterile Medium 90 at 37 °C.
- M tuberculosis strain H37Rv (ATCC number 27294) was cultured in sterile Middlebrook 7H9 medium with Tween 80 and oleic acid/albumin/dextrose/catalase additive (Difco Laboratories, Detroit, Michigan). The cells were harvested by centrifugation, and transferred into sterile Middlebrook 7H9 medium with glucose at 37 °C for one day. The medium was then centrifuged (leaving the bulk of the cells) and filtered through a 0.45 ⁇ m filter into sterile bottles. The culture filtrate was concentrated by lyophilisation, and redissolved in MilliQ water.
- a small amount of insoluble material was removed by filtration through a 0.45 ⁇ m membrane filter.
- the culture filtrate was desalted by membrane filtration in a 400 ml Amicon stirred cell which contained a 3 kDa MWCO membrane. The pressure was maintained at 60 psi using nitrogen gas.
- the culture filtrate was repeatedly concentrated by membrane filtration and diluted with water until the conductivity of the sample was less than 1.0 mS. This procedure reduced the 20 1 volume to approximately 50 ml. Protein concentrations were determined by the Bradford protein assay (Bio-Rad, Hercules, CA, USA).
- the desalted culture filtrate was fractionated by ion exchange chromatography on a column of Q-Sepharose (Pharmacia Biotech) (16 x 100 mm) equilibrated with lOmM TrisHCl buffer pH 8.0. Polypeptides were eluted with a linear gradient of NaCI from 0 to 1.0 M in the above buffer system. The column eluent was monitored at a wavelength of 280 nm.
- the pool of polypeptides eluting from the ion exchange column were fractionated by preparative 2D gel electrophoresis.
- Samples containing 200-500 ⁇ g of polypeptide were made 8M in urea and applied to polyacrylamide isoelectric focusing rod gels (diameter 2mm, length 150 mm, pH 5-7).
- the first dimension gels were equilibrated with reducing buffer and applied to second dimension gels (16% poly aery lamide).
- Polypeptides from the second dimension separation were transferred to PVDF membranes by electroblotting in lOmM CAPS buffer pH 11 containing 10% (v/v) methanol.
- the PVDF membranes were stained for protein with Coomassie blue.
- Regions of PVDF containing polypeptides of interest were cut out and directly introduced into the sample cartridge of the Perkin Elmer/ Applied BioSystems Procise 492 protein sequencer.
- the polypeptides were sequenced from the amino terminal end using traditional Edman chemistry.
- the amino acid sequence was determined for each polypeptide by comparing the retention time of the PTH amino acid derivative to the appropriate PTH derivative standards. Using these procedures, eleven polypeptides, designated GVs-1, GVs-3, GVs-4, GVs-5, GVs-6, GVs-8, GVs-9, GVs-10, GVs-11, GV-34 and GV-35 were isolated.
- the determined N- terminal sequences for these polypeptides are shown in SEQ ID NOS: 21-29, 63 and 64, respectively.
- SEQ ID NOS: 21-29, 63 and 64 The determined N- terminal sequences for these polypeptides are shown in SEQ ID NOS: 21-29, 63 and 64, respectively.
- the extended amino acid sequence for GVs-9 is provided in SEQ ID NO: 65.
- Further studies resulted in the isolation of DNA sequences for GVs-9 (SEQ ID NO: 111) and GV-35 (SEQ ID NO: 155).
- the corresponding predicted amino acid sequences are provided in SEQ ID NO: 112 and 156, respectively.
- An extended DNA sequence for GVs-9 is provided in SEQ ID NO: 153, with the corresponding predicted amino acid sequence being provided in SEQ ID NO: 154.
- the predicted amino acid sequence for GVs-9 has been amended in SEQ ID NO: 197.
- GVs-3, GVs-4 and GVs-5 were found to bear some similarity to the antigen 85A and 85B proteins from M leprae (SEQ ID NOS: 30 and 31, respectively), M tuberculosis (SEQ ID NOS: 32 and 33, respectively) and M bovis (SEQ ID NOS: 34 and 35, respectively), and the antigen 85C proteins from M. leprae (SEQ ID NO: 36) and M tuberculosis (SEQ ID NO: 37).
- Probes for antigens 85A, 85B, and 85C were prepared by polymerase chain reaction
- PCR using degenerate oligonucleotides (SEQ ID NOS: 38 and 39) designed to regions of antigen 85 genomic sequence that are conserved between family members in a given mycobacterial species, and between mycobacterial species. These oligonucleotides were used under reduced stringency conditions to amplify target sequences from M vaccae genomic
- An M vaccae genomic library was created in lambda Zap-Express (Stratagene, La Jolla, CA) by cloning BamHl partially-digested M vaccae genomic DNA into similarly- digested ⁇ vector, with 3.4 x 10 5 independent plaque-forming units resulting.
- Twenty-seven thousand plaques from this non-amplified library were plated at low density onto eight 100 cm 2 plates.
- duplicate plaque lifts were taken onto Hybond-N + nylon membrane (Amersham International, United Kingdom), and hybridised under reduced-stringency conditions (55 °C) to the radiolabelled antigen 85C PCR product. Autoradiography demonstrated that seventy-nine plaques consistently hybridised to the antigen 85C probe under these conditions.
- Sequence data from the 5' and 3' ends of inserts from several representatives of each group was obtained using the Perkin Elmer/Applied Biosystems Model 377 automated sequencer and the T3 and T7 primers. Sequence homologies were determined using BLASTN analysis of the EMBL database. Two of these sets of clones were found to be homologous to M bovis and M tuberculosis antigen 85A genes, each containing either the 5' or 3' ends of the M vaccae gene (this gene was cleaved during library construction as it contains an internal BamHI site). The remaining clones were found to contain sequences homologous to antigens 85B and 85C from a number of mycobacterial species.
- the M vaccae antigens GVs-3 and GVs-5 were expressed and purified as follows.
- Amplification primers were designed from the insert sequences of GVs-3 and GVs-5 (SEQ ID NO: 40 and 42, respectively) using sequence data downstream from the putative leader sequence and the 3' end of the clone.
- the sequences of the primers for GVs-3 are provided in SEQ ID NO: 66 and 67
- the sequences of the primers for GVs-5 are provided in SEQ ID NO: 68 and 69.
- a Xhol restriction site was added to the primers for GVs-3, and EcoRI and BamRl restriction sites were added to the primers for GVs-5 for cloning convenience. Following amplification from genomic M.
- vaccae DNA fragments were cloned into the appropriate site of pPro ⁇ X HT prokaryotic expression vector (Gibco BRL, Life Technologies, Gaithersburg, MD) and submitted for sequencing to confirm the correct reading frame and orientation. Expression and purification of the recombinant protein was performed according to the manufacturer's protocol.
- the insert from one of the clones was subcloned into the EcoRI/ATzoI sites of pPro ⁇ X HT prokaryotic expression vector (Gibco BRL), expressed and purified according to the manufacturer's protocol.
- This clone was renamed GV-4P because only a part of the gene was expressed.
- the amino acid and DNA sequences for the partial clone GV-4P are provided in S ⁇ Q ID NO: 70 and 106, respectively.
- the amplification primers AD58 and AD59 were used to amplify a 485 bp fragment from a clone containing GVs-5 (SEQ ID NO:42). This fragment was cloned into the expression vector pET16 and was called GV-5P.
- the determined nucleotide sequence and predicted amino acid sequence of GV-5P are provided in SEQ ID NOS: 157 and 158, respectively.
- GVs-3, GV- 4P and GVs-5 were re-cloned into the alternative vector pET16 (Novagen, Madison, WI).
- An 84 bp probe for the M vaccae antigen GVc-7 was amplified using degenerate oligonucleotides designed to the determined amino acid sequence of GVc-7 (SEQ ID NOS: 5-
- This probe was used to screen a M vaccae genomic DNA library as described in Example 12.
- the determined nucleotide sequence for GVc-7 is shown in SEQ ID NO: 46 and predicted amino acid sequence in SEQ ID NO: 47. Comparison of these sequences with those in the databank revealed homology to a hypothetical 15.8 kDa membrane protein of M tuberculosis.
- SEQ ID NO: 46 The sequence of SEQ ID NO: 46 was used to design amplification primers (provided in SEQ ID NO: 71 and 72) for expression cloning of the GVc-7 gene using sequence data downstream from the putative leader sequence. A Xhol restriction site was added to the primers for cloning convenience. Following amplification from genomic M vaccae DNA, fragments were cloned into the .ATzoI-site of pProEX HT prokaryotic expression vector (Gibco BRL) and submitted for sequencing to confirm the correct reading frame and orientation. Expression and purification of the fusion protein was performed according to the manufacturer's protocol. In subsequent studies, GVc-7 was re-cloned into the vector pET16 (Novagen).
- a redundant oligonucleotide probe (SEQ ID NO 73; referred to as MPG15) was designed to the GVs-8 peptide sequence shown in SEQ ID NO: 26 and used to screen a M vaccae genomic DNA library using standard protocols. Two genomic clones containing genes encoding four different antigens was isolated. The determined DNA sequences for GVs-8A (re-named GV-30), GVs-8B (re-named GV-31), GVs-8C (re-named GV-32) and GVs-8D (re-named GV-33) are shown in SEQ ID NOS: 48-51, respectively, with the corresponding amino acid sequences being shown in SEQ ID NOS: 52-55, respectively.
- GV- 30 contains regions showing some similarity to known prokaryotic valyl-tRNA synthetases; GV-31 shows some similarity to M smegmatis aspartate semialdehyde dehydrogenase; and GV-32 shows some similarity to the H influenza folylpolyglutamate synthase gene. GV-33 contains an open reading frame which shows some similarity to sequences previously identified in M tuberculosis and M leprae, but whose function has not been identified.
- the determined partial DNA sequence for GV-33 is provided in SEQ ID NO: 74 with the corresponding predicted amino acid sequence being provided in SEQ ID NO: 75. Sequence data from the 3' end of the clone showed homology to a previously identified 40.6 kDa outer membrane protein of M tuberculosis. Subsequent studies led to the isolation of a full-length DNA sequence for GV-33 (SEQ ID NO: 193). The corresponding predicted amino acid sequence is provided in SEQ ID NO: 194.
- the gene encoding GV-33 was amplified from M vaccae genomic DNA with primers based on the determined nucleotide sequence. This DNA fragment was cloned into EcoRv- digested pBluescript II SK + (Stratagene), and then transferred to p ⁇ T16 expression vector. Recombinant protein was purified following the manufacturer's protocol.
- M. vaccae bacteria were cultured, pelleted and autoclaved as described in Example 1.
- Culture filtrates of live M vaccae refer to the supernatant from 24 hour cultures of M vaccae in 7H9 medium with glucose.
- the resulting pellet was suspended in 100 ml of chloroform/methanol (2:1), incubated at room temperature for 1 hour, re-centrifuged, and the chloroform/methanol extraction repeated.
- the pellet was obtained by centrifugation, dried in vacuo, weighed and resuspended in PBS at 50 mg (dry weight) per ml as delipidated M vaccae.
- Glycolipids were removed from the delipidated M vaccae preparation by refluxing in 50% v/v ethanol for 2 hours.
- the insoluble material was collected by centrifugation and washed in PBS. Proteins were extracted by resuspending the pellet in 2% SDS in PBS at 56 °C for 2 hours. The insoluble material was collected by centrifugation and the extraction with 2% SDS/PBS at 56 °C was repeated twice more.
- the SDS-extracted proteins derived from DD-M vaccae were analysed by polyacrylamide gel electrophoresis. Three major bands were observed after staining with silver. In subsequent experiments, larger amounts of SDS-extracted proteins from DD- M.vaccae, were analysed by polyacrylamide gel electrophoresis. The proteins, on staining with Coomassie blue, showed several bands.
- a protein represented by a band of approximate molecular weight of 30 kDa was designated GV-45.
- the determined N-terminal sequence for GV-45 is provided in SEQ ID NO: 187.
- a protein of approximate molecular weight of 14 kDa was designated GV-46.
- the determined N-terminal amino acid sequence of GV-46 is provided in SEQ ID NO: 208.
- the sequence of the first ten amino acid residues is provided in SEQ ID NO: 76. Comparison of this sequence with those in the gene bank as described above, revealed homology to the heat shock protein 65 (GroEL) gene from M tuberculosis, indicating that this protein is an M vaccae member of the GroEL family.
- RhoEL heat shock protein 65
- An expression library of M vaccae genomic DNA in 5 ⁇ mHl -lambda ZAP-Express (Stratagene) was screened using sera from cynomolgous monkeys immunised with M vaccae secreted proteins prepared as described above. Positive plaques were identified using a colorimetric system. These plaques were re-screened until plaques were pure following standard procedures.
- pBK-CMV phagemid 2-1 containing an insert was excised from the lambda ZAP Express (Stratagene) vector in the presence of ExAssist helper phage following the manufacturer's protocol.
- the base sequence of the 5' end of the insert of this clone was determined using Sanger sequencing with fluorescent primers on Perkin Elmer/Applied Biosystems Division automatic sequencer.
- the determined nucleotide sequence of the partial M vaccae GroEL-homologue clone GV-27 is provided in SEQ ID NO: 77 and the predicted amino acid sequence in SEQ ID NO: 78.
- This clone was found to have homology to M tuberculosis GroEL.
- a partial sequence of the 65 kDa heat shock protein of M vaccae has been published by Kapur et al. (Arch. Pathol. Lab. Med. 119 :131-138, 1995).
- the nucleotide sequence of the Kapur et al. fragment is shown in SEQ ID NO: 79 and the predicted amino acid sequence in SEQ ID NO: 80.
- GV-27A Two peptide fragments, comprising the N-terminal sequence
- GV-27B the carboxy terminal sequence of GV-27
- SEQ ID NO: 115 and 116 The nucleotide sequences for GV-27A and GV-27B are provided in SEQ ID NO: 115 and 116, respectively, with the corresponding amino acid sequences being provided in SEQ ID NO: 117 and 118.
- SEQ ID NO: 161 is provided in SEQ ID NO: 161, with the corresponding amino acid sequence being provided in SEQ ID NO: 162.
- the sequence of GV-27 A is 95.8% identical to the M tuberculosis GroEL sequence and contains the shorter M vaccae sequence of Kapur et al. discussed above.
- the sequence for GV-27B shows about 92.2% identity to the corresponding region of M tuberculosis HSP65.
- pBK-CMV phagemid 3-1 was isolated.
- the antigen encoded by this DNA was named GV-29.
- the determined nucleotide sequences of the 5' and 3' ends of the gene are provided in SEQ ID NOS: 163 and 164, respectively, with the predicted corresponding amino acid sequences being provided in SEQ ID NOS: 165 and 166 respectively.
- GV-29 showed homology to yeast urea amidolyase.
- the determined DNA sequence for the full-length gene encoding GV-29 is provided in SEQ ID NO: 198, with the corresponding predicted amino acid sequence in SEQ ID NO: 199.
- the DNA encoding GV-29 was sub-cloned into the vector pET16 (Novagen, Madison, WI) for expression and purification according to standard protocols.
- M vaccae (ATCC Number 15483) was grown in sterile Medium 90 at 37 °C for 4 days and harvested by centrifugation. Cells were resuspended in 1 ml Trizol (Gibco BRL, Life Technologies, Gaithersburg, Maryland) and RNA extracted according to the standard manufacturer's protocol. M tuberculosis strain H37Rv (ATCC Number 27294) was grown in sterile Middlebrook 7H9 medium with Tween 80TM and oleic acid/ albumin/dextrose/catalase additive (Difco Laboratories, Detroit, Michigan) at 37 °C and harvested under appropriate laboratory safety conditions. Cells were resuspended in 1 ml Trizol (Gibco BRL) and RNA extracted according to the manufacturer's standard protocol.
- Total M tuberculosis and M vaccae RNA was depleted of 16S and 23 S ribosomal RNA (rRNA) by hybridisation of the total RNA fraction to oligonucleotides AD 10 and AD11 (SEQ ID NO: 81 and 82) complementary to M tuberculosis rRNA.
- oligonucleotides were designed from mycobacterial 16S rRNA sequences published by Bottger (FEMS Microbiol. Lett. 65:11 - 16, 1989) and from sequences deposited in the databanks. Depletion was done by hybridisation of total RNA to oligonucleotides AD 10 and AD11 immobilised on nylon membranes (Hybond N, Amersham International, United Kingdom).
- oligonucleotide, AD 12 (SEQ ID NO: 83), consisting of 20 dATP -residues, was ligated to the 3' ends of the enriched mRNA fraction using RNA ligase.
- First strand cDNA synthesis was performed following standard protocols, using oligonucleotide AD7 (SEQ ID NO: 84) containing a poly(dT) sequence.
- the M tuberculosis and M vaccae cDNA was used as template for single-sided- specific PCR (3S-PCR).
- a degenerate oligonucleotide ADl (SEQ ID NO:85) was designed based on conserved leader sequences and membrane protein sequences. After 30 cycles of amplification using primer ADl as 5'-primer and AD7 as 3'-primer, products were separated on a urea polyacrylamide gel. DNA bands unique to M vaccae were excised and re-amplified using primers ADl and AD7. After gel purification, bands were cloned into pGEM-T (Promega) and the base sequence determined.
- transmembrane genes encode proteins each characteristically having six membrane-spanning regions. Homologues (by similarity) of this ABC transporter have been identified in the genomes of Haemophilus influenza (Fleischmann et al. Science 269 :496-512, 1995) and Mycoplasma genitalium (Fraser, et al. Science, 270:391-403, 1995).
- the nucleotide sequence of the full-length M vaccae homologue of pota (ATP-binding protein) was identified by subcloning of the 4.5 kb fragment and base sequencing.
- the nucleotide and predicted amino acid sequences of the M vaccae pota homologue are provided in SEQ ID NO: 88 and 89, respectively.
- the nucleotide sequence of the M vaccae pota gene was used to design primers EV24 and EV25 (SEQ ID NO: 90 and 91) for expression cloning.
- the amplified DNA fragment was cloned into pProEX HT prokaryotic expression system (Gibco BRL) and expression in an appropriate E.coli host was induced by addition of 0.6 mM isopropylthio- ⁇ -galactoside (IPTG).
- IPTG isopropylthio- ⁇ -galactoside
- the recombinant protein was named GV-23 and purified from inclusion bodies according to the manufacturer's protocol. In subsequent studies, GV-23 (SEQ ID NO: 88) was re-cloned into the alternative vector pET16 (Novagen).
- SEQ ID NO: 89 contains an ATP binding site at residues 34 to 41. At residues 116 to 163 of SEQ ID NO: 89, there is a conserved region that is found in the ATP -transporter family of proteins. These findings suggest that GV-23 is an ATP binding protein.
- a 322 bp Sall-BamRl subclone at the 3'-end of the 4.5 kb insert described above showed homology to the potd gene, (periplasmic protein), of the spermidine/putrescine ABC transporter complex of E. coli.
- the nucleotide sequence of this subclone is shown in SEQ ID NO:92.
- the radiolabelled insert of this subclone was used to probe a M vaccae genomic DNA library constructed in the S ⁇ /1-site of lambda Zap Express (Stratagene) following standard protocols.
- a clone was identified of which 1342 bp showed homology with the potd gene of E. coli.
- the potd homologue of M vaccae was identified by subcloning and base sequencing. The determined nucleotide and predicted amino acid sequences are shown in SEQ ID NO: 93 and 94.
- primers EV-26 and EV-27 were designed from the determined M vaccae potd homologue.
- the amplified fragment was cloned into pProEX HT Prokaryotic expression system (Gibco BRL). Expression in an appropriate E. coli host was induced by addition of 0.6 mM IPTG and the recombinant protein named GV-24.
- the recombinant antigen was purified from inclusion bodies according to the protocol of the supplier.
- GV-24 SEQ ID NO: 93
- GV-24 was re-cloned into the alternative vector pET16 (Novagen).
- the gene encoding GV- 24, but excluding the signal peptide was re-cloned into the expression vector, employing, amplification primers EV101 and EV102 (SEQ ID NOS: 167 and 168).
- the construct was designated GV-24B.
- the nucleotide sequence of GV-24B is provided in SEQ ID NO: 169 and the predicted amino acid sequence in SEQ ID NO: 170. This fragment was cloned into pET16 for expression and purification of GV-24B according to the manufacturer's protocols.
- M vaccae potd gene-homologue Base sequence adjacent to the M vaccae potd gene-homologue was found to show homology to the potb gene of the spermidine/putrescine ABC transporter complex of E.coli, which is one of two transmembrane proteins in the ABC transporter complex.
- the M vaccae potb homologue (referred to as GV-25) was identified through further subcloning and base sequencing. The determined nucleotide and predicted amino acid sequences for GV-25 are shown in SEQ ID NOS: 97 and 98, respectively.
- the 3S- PCR band 12B28 (SEQ ID NO: 119) was used to screen the M vaccae genomic library constructed in the BamHI-site of lambda ZAP Express (Stratagene).
- the clone isolated from the library contained a novel open reading frame and the antigen encoded by this gene was named GV-38A.
- the determined nucleotide sequence and predicted amino acid sequence of GV-38A are shown in SEQ ID NO: 120 and 121, respectively.
- the corresponding amino acid sequence is provided in SEQ ID NO: 172. Comparison of these sequences with those in the gene bank, revealed some homology to an unknown M tuberculosis protein previously identified in cosmid MTCY428.12. (SPTREMBL:P71915).
- GV-38B Upstream of the GV-38A gene, a second novel open reading frame was identified and the antigen encoded by this gene was named GV-38B.
- the determined 5' and 3' nucleotide sequences for GV-38B are provided in SEQ ID NO: 122 and 123, respectively, with the corresponding predicted amino acid sequences being provided in SEQ ID NO: 124 and 125, respectively. Further studies led to the isolation of the full-length DNA sequence for GV- 38B, provided in SEQ ID NO: 173. The corresponding amino acid sequence is provided in SEQ ID NO: 174.
- This protein was found to show homology to an unknown M tuberculosis protein identified in cosmid MTCY428.11 (SPTREMBL: P71914).
- GV-38A and GV-38B antigens were amplified for expression cloning into pET16 (Novagen).
- GV-38A was amplified with primers KR11 and KR12 (SEQ ID NO: 126 and 127) and GV-38B with primers KR13 and KR14 (SEQ ID NO: 128 and 129).
- Protein expression in the host cells BL21(DE3) was induced with 1 mM IPTG, however no protein expression was obtained from these constructs. Hydrophobic regions were identified in the N-termini of antigens GV-38A and GV-38B which may inhibit expression of these constructs.
- the hydrophobic region present in GV-38A was identified as a possible transmembrane motif with six membrane spanning regions.
- primers KR20 for GV-38A, (SEQ ID NO: 130) and KR21 for GV-38B (SEQ ID NO: 131) were designed.
- the truncated GV-38A gene was amplified with primers KR20 and KR12, and the truncated GV-38B gene with KR21 and KR14.
- the determined nucleotide sequences of truncated GV38A and GV-38B are shown in SEQ ID NO: 132 and 133, respectively, with the corresponding predicted amino acid sequences being shown in SEQ ID NO: 134 and 135, respectively.
- Extended DNA sequences for truncated GV-38A and GV- 38B are provided in SEQ ID NO: 175 and 176, respectively, with the corresponding amino acid sequences being provided in SEQ ID NO: 177 and 178, respectively.
- M vaccae soluble proteins were isolated from culture filtrate using preparative isoelectric focusing and preparative polyacrylamide gel electrophoresis as described below. Unless otherwise noted, all percentages in the following example are weight per volume.
- M vaccae (ATCC Number 15483) was cultured in 250 1 sterile Medium 90 which had been fractionated by ultrafiltration to remove all proteins of greater than 10 kDa molecular weight. The medium was centrifuged to remove the bacteria, and sterilised by filtration through a 0.45 ⁇ m filter. The sterile filtrate was concentrated by ultrafiltration over a 10 kDa molecular weight cut-off membrane.
- Proteins were isolated from the concentrated culture filtrate by precipitation with 10% trichloroacetic acid. The precipitated proteins were re-dissolved in 100 mM Tris.HCl pH 8.0. and re-precipitated by the addition of an equal volume of acetone. The acetone precipitate was dissolved in water, and proteins were re-precipitated by the addition of an equal volume of chloroform: methanol 2:1 (v/v). The chloroform:methanol precipitate was dissolved in water, and the solution was freeze-dried.
- the freeze-dried protein was dissolved in iso-electric focusing buffer, containing 8 M deionised urea, 2% Triton X-100, 10 mM dithiothreitol and 2% ampholytes (pH 2.5 - 5.0).
- the sample was fractionated by preparative iso-electric focusing on a horizontal bed of Ultrodex gel at 8 watts constant power for 16 hours. Proteins were eluted from the gel bed fractions with water and concentrated by precipitation with 10% trichloroacetic acid. Pools of fractions containing proteins of interest were identified by analytical polyacrylamide gel electrophoresis and fractionated by preparative polyacrylamide gel electrophoresis.
- Eluted proteins were assayed for their ability to induce proliferation and interferon- ⁇ secretion from the peripheral blood lymphocytejs of immune donors as detailed above. Proteins inducing a strong response in these assays were selected for further study.
- Selected proteins were further purified by reversed-phase chromatography on a Vydac Protein C4 column, using a trifluoroacetic acid-acetonitrile system. Purified proteins were prepared for protein sequence determination by SDS-polyacrylamide gel electrophoresis, and electroblotted onto PVDF membranes. Protein sequences were determined as in Example 3. The proteins were named GV-40, GV-41, GV-42, GV-43 and GV-44. The determined N- terminal sequences for these polypeptides are shown in SEQ ID NOS: 101-105, respectively. Subsequent studies led to the isolation of a 5', middle fragment and 3' DNA sequence for GV- 42 (SEQ ID NO: 136, 137 and 138, respectively). The corresponding predicted amino acid sequences are provided in SEQ ID NO: 139, 140 and 141, respectively.
- GV-41 and GV-42 were cloned.
- the determined nucleotide sequences are provided in SEQ ID NOS: 179 and 180, respectively, and the predicted amino acid sequences in SEQ ID NOS: 181 and 182. Further experiments lead to the cloning of the full-length gene encoding GV-41, which was named GV-41B.
- the determined nucleotide sequence and the predicted amino acid sequence of GV-41B are provided in SEQ ID NOS: 202 and 203, respectively.
- GV-41 had homology to the ribosome recycling factor of M tuberculosis and M leprae, and GV-42 had homology to a M avium fibronectin attachment protein FAP-A.
- the amino acid sequence determined for GV-43 (SEQ ID NO: 104) was identified, indicating that the amino acid sequences for GV-42 and GV-43 were obtained from the same protein.
- Murine polyclonal antisera were prepared against GV-40 and GV-44 following standard procedures. These antisera were used to screen a M vaccae genomic DNA library consisting of randomly sheared DNA fragments. Clones encoding GV-40 and GV-44 were identified and sequenced.
- the determined nucleotide sequence of the partial gene encoding GV-40 is provided in SEQ ID NO: 183 and the predicted amino acid sequence in SEQ ID NO :184.
- the complete gene encoding GV-40 was not cloned, and the antigen encoded by this partial gene was named GV-40P.
- An extended DNA sequence for GV-40P is provided in SEQ ID NO: 206 with the corresponding predicted amino acid sequence being provided in SEQ ID NO 207.
- the determined nucleotide sequence of the gene encoding GV-44 is provided in SEQ ID NO: 185, and the predicted amino acid sequence in SEQ ID NO: 186.
- the determined DNA sequence for the full-length gene encoding GV-44 was obtained and is provided in SEQ ID NO 204, with the corresponding predicted amino acid sequence being provided in SEQ ID NO: 205.
- Homology of GV-40 to M leprae Elongation factor G was found and GV-44 had homology to M leprae glyceraldehyde-3- phosphate dehydrogenase.
- GV-45 a protein represented by a band of approximate molecular weight of 30 kDa, designated GV-45, was isolated.
- the determined N-terminal sequence for GV-45 is provided in SEQ ID NO: 187.
- a protein of approximate molecular weight of 14 kDa, designated GV-46 was obtained.
- the determined N-terminal amino acid sequence of GV-46 is provided in SEQ ID NO: 208.
- GV-46 is homologous to the highly conserved mycobacterial host integration factor of M tuberculosis and M smegmatis.
- degenerate oligonucleotides KR32 and KR33 (SEQ ID NOS: 188 and 189, respectively) were designed.
- a 100 bp fragment was amplified, cloned into plasmid pBluescript II SK + (Stratagene, La Jolla, CA) and sequenced (SEQ ID NO:190) following standard procedures (Sambrook, Ibid).
- the cloned insert was used to screen a M vaccae genomic DNA library constructed in the 5 ⁇ rnHI-site of lambda ZAP -Express (Stratagene).
- the isolated clone showed homology to a 35 kDa M tuberculosis and a 22 kDa M leprae protein containing bacterial histone-like motifs at the N-terminus and a unique C-terminus consisting of a five amino acid basic repeat.
- the determined nucleotide sequence for GV-45 is provided in SEQ ID NO: 191, with the corresponding predicted amino acid sequence being provided in SEQ ID NO: 192.
- the determined DNA sequence for the full-length gene encoding GV-45 was obtained and is provided in SEQ ID NO: 200, with the corresponding predicted amino acid sequence in SEQ ID NO: 201.
- GV recombinant proteins The immunogenicity of Mycobacterium vaccae recombinant proteins (GV recombinant proteins) was tested by injecting female BALB/cByJ mice in each hind foot-pad with 10 ug of recombinant GV proteins emulsified in incomplete Freund's adjuvant (IF A). Control mice received phosphate buffered saline in IF A. The draining popliteal lymph nodes were excised 10 days later and the cells obtained therefrom were stimulated with the immunizing GV protein and assayed for proliferation by measuring the uptake of tritiated thymidine. The amount of interferon gamma (IFN ⁇ ) produced and secreted by these cells into the culture supernatants was assayed by standard enzyme-linked immunoassay.
- IFN ⁇ interferon gamma
- the GV proteins are thus immunogenic in being able to stimulate T cell proliferation and/or IFN ⁇ production when administered by subcutaneous injection.
- the antigen-specific stimulatory effects on T cell proliferation and IFN ⁇ production are two advantageous properties of candidate vaccines for tuberculosis.
- PBMC from normal donors (5 x 10 6 cells/ml) were stimulated with 20 ug/ml of either heat-killed M vaccae cells, DD-M vaccae or recombinant GV-22B (SEQ ID NO: 145), GV- 23 (SEQ ID NO: 89), GV-27 (SEQ ID NO: 160), GV27A (SEQ ID NO: 117), GV-27B (SEQ ID NO: 162) or GV-45 (SEQ ID NO: 201) for 24 hours.
- CD69 expression was determined by staining cultured cells with monoclonal antibody against CD56, ⁇ T cells or ⁇ T cells, in combination with monoclonal antibodies against CD69, followed by flow cytometry analysis
- Table 23 shows the percentage of ⁇ T cells, ⁇ T cells and NK cells expressing CD69 following stimulation with heat-killed M vaccae, DD-M. vaccae or recombinant M vaccae proteins.
- PBMC from normal donors (5 x 10° cells/ml) were stimulated with 20 ug/ml of either heat-killed M vaccae cells, DD-M vaccae, or recombinant GV-22B (SEQ ID NO: 145), GV-23 (SEQ ID NO: 89), GV-27 (SEQ ID NO: 160), GV27A (SEQ ID NO: 117), GV-27B (SEQ ID NO: 162) or GV-45 (SEQ ID NO: 201) for 24 hours.
- Figs. 9A-D show the stimulation of IL-l ⁇ , TNF- ⁇ , IL-12 and IFN- ⁇ production, respectively. Heat-killed M vaccae and DD-M vaccae were found to stimulate the production of all four cytokines examined, while recombinant GV-23 and GV- 45 were found to stimulate the production of IL-l ⁇ , TNF- ⁇ and IL-12.
- Figs. 10A-C show the stimulation of IL-l ⁇ , TNF- ⁇ and IL-12 production, respectively, in human PBMC (determined as described above) by varying concentrations of GV-23 and GV-45.
- Figs. 11A-D show the stimulation of IL-l ⁇ , TNF- ⁇ , IL-12 and IFN- ⁇ production, respectively, in PBMC by GV-23 as compared to that by the adjuvants MPL/TDM/CWS (at a final dilution of 1 :20), CpG ODN (20 ug/ml), aluminium hydroxide (at a final dilution of 1:400) and cholera toxin (20 ug/ml).
- GV-23, MPL/TDM CWS and CpG ODN induced significant levels of the four cytokines examined, with higher levels of IL-l ⁇ production being seen with GV-23 than with any of the known adjuvants. Aluminium hydroxide and cholera toxin induced only negligible amounts of the four cytokines.
- Peripheral blood mononuclear cells depleted of T cells and comprising mainly B cells, monocytes and dendritic cells were stimulated with 20 ug/ml of either heat-killed M vaccae cells, DD-M vaccae, or recombinant GV-22B (SEQ ID NO: 145), GV-23 (SEQ ID NO: 89), GV-27 (SEQ ID NO: 160), GV27A (SEQ ID NO: 117), GV-27B (SEQ ID NO: 162) or GV- 45 (SEQ ID NO: 201) for 48 hours.
- Stimulated cells were harvested and analyzed for upregulation of CD40, CD80 and CD86 using 3 color flow cytometric analysis. Tables 24, 25 and 26 show the fold increase in mean fluorescence intensity from control (non-stimulated cells) for dendritic cells, monocytes, and B cells, respectively.
- Figs. 12A-C show the stimulation of expression of CD40, CD80 and CD86, respectively, in dendritic cells by varying concentrations of GV-23 and GV-45.
- GV-23 The ability of GV-23 to stimulate CD40, CD80 and CD86 expression in dendritic cells was compared to that of the Thl -inducing adjuvants MPL/TDM/CWS (at a final dilution of 1:20) and CpG ODN (20 ug/ml), and the known Th2-inducing adjuvants aluminium hydroxide (at a final dilution of 1 :400) and cholera toxin (20 ug/ml).
- GV23, MPL/TDM/CWS and CpG ODN caused significant up-regulation of CD40, CD80 and CD86, whereas cholera toxin and aluminium hydroxide induced modest or negligible dendritic cell activation, respectively.
- Purified dendritic cells (5 x 10 4 - 10 5 cells/ml) were stimulated with GV-23 (20 ug/ml) or LPS (10 ug/ml) as a positive control. Cells were cultured for 20 hour and then analyzed for CD83 (a maturation marker) and CD80 expression by flow cytometry. Non-stimulated cells were used as a negative control. The results are shown below in Table 27.
- GV-23 The ability of GV-23 to enhance dendritic cell function as antigen presenting cells was determined by mixed lymphocyte reaction (MLR) assay.
- MLR mixed lymphocyte reaction
- Purified dendritic cells were culture in medium alone or with GV-23 (20 ug/ml) for 18-20 hours and then stimulated with allogeneic T cells (2 x 10 5 cells/well). After 3 days of incubation, ( 3 H)-thymidine was added. Cells were harvested 1 day later and the uptake of radioactivity was measured.
- Fig. 13 shows the increase in uptake of ( 3 H)-thymidine with increase in the ratio of dendritic cells to T cells. Significantly higher levels of radioactivity uptake were seen in GV-23 stimulated dendritic cells compared to non-stimulated cells, showing that GV-23 enhances dendritic cell mixed leukocyte reaction.
Abstract
Description















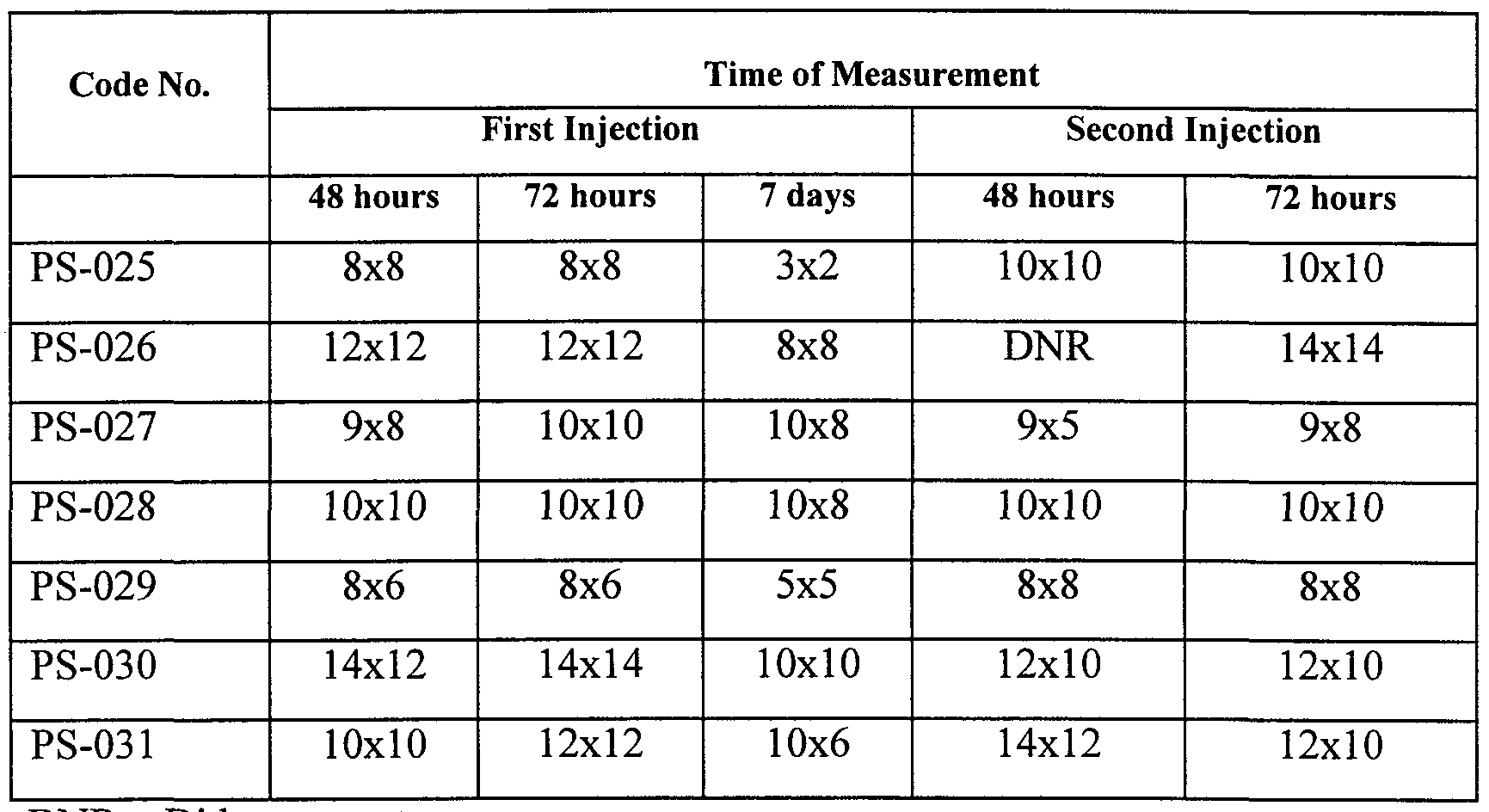








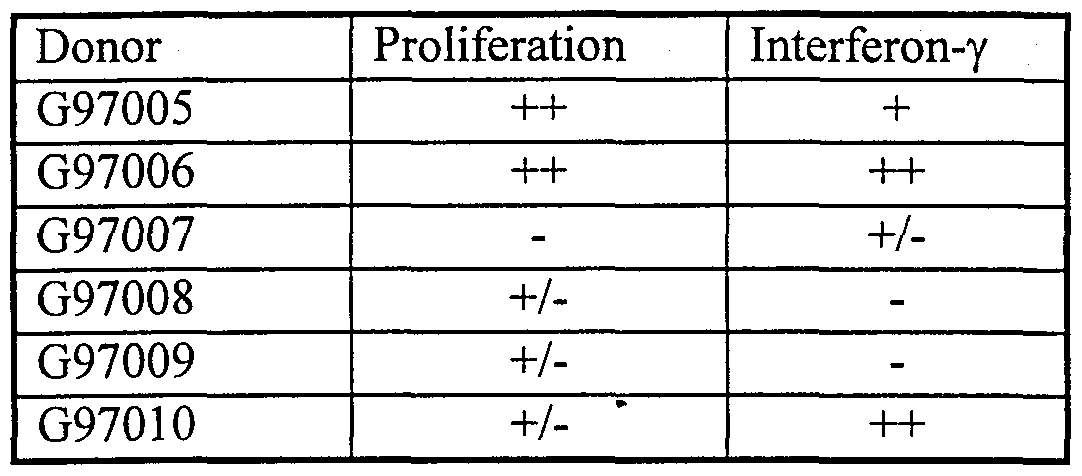







Claims
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020007006505A KR20010033132A (en) | 1997-12-23 | 1998-12-23 | Compositions derived from mycobacterium vaccae and methods for their use |
| JP2000525553A JP2002514385A (en) | 1997-12-23 | 1998-12-23 | COMPOSITIONS DERIVED FROM MYCOBACTERIUM BACKEY AND USE THEREOF |
| CA002315539A CA2315539A1 (en) | 1997-12-23 | 1998-12-23 | Compositions derived from mycobacterium vaccae and methods for their use |
| EP98963665A EP1044273A2 (en) | 1997-12-23 | 1998-12-23 | COMPOSITIONS DERIVED FROM $i(MYCOBACTERIUM VACCAE) AND METHODS FOR THEIR USE |
| MXPA00006168A MXPA00006168A (en) | 1997-12-23 | 1998-12-23 | Compositions derived from mycobacterium vaccae. |
| AU18936/99A AU746311B2 (en) | 1997-12-23 | 1998-12-23 | Compositions derived from (mycobacterium vaccae) and methods for their use |
| HU0100352A HUP0100352A2 (en) | 1997-12-23 | 1998-12-23 | Compositions derived from mycobacterium vaccae and methods for their use |
| IL13682198A IL136821A0 (en) | 1997-12-23 | 1998-12-23 | Compositions derived from mycobacterium vaccae and methods for their use |
| NZ505834A NZ505834A (en) | 1997-12-23 | 1998-12-23 | Compositions containing polypeptides isolated from mycobacterium vaccae and methods for their use |
| BR9814432-4A BR9814432A (en) | 1997-12-23 | 1998-12-23 | Compositions derived from mycobacterium vaccae and process for their use |
| NO20003261A NO20003261L (en) | 1997-12-23 | 2000-06-22 | Mixtures derived from Mycobacterium vaccae and methods for their use |
Applications Claiming Priority (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US99662497A | 1997-12-23 | 1997-12-23 | |
| US08/997,080 | 1997-12-23 | ||
| US08/996,624 | 1997-12-23 | ||
| US08/997,362 | 1997-12-23 | ||
| US08/997,362 US5985287A (en) | 1996-08-29 | 1997-12-23 | Compounds and methods for treatment and diagnosis of mycobacterial infections |
| US08/997,080 US5968524A (en) | 1997-12-23 | 1997-12-23 | Methods and compounds for the treatment of immunologically-mediated psoriasis |
| US09/095,855 US6160093A (en) | 1996-08-29 | 1998-06-11 | Compounds and methods for treatment and diagnosis of mycobacterial infections |
| US09/095,855 | 1998-06-11 | ||
| US15618198A | 1998-09-17 | 1998-09-17 | |
| US09/156,181 | 1998-09-17 | ||
| US09/205,426 | 1998-12-04 | ||
| US09/205,426 US6406704B1 (en) | 1996-08-29 | 1998-12-04 | Compounds and methods for treatment and diagnosis of mycobacterial infections |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1999032634A2 true WO1999032634A2 (en) | 1999-07-01 |
| WO1999032634A3 WO1999032634A3 (en) | 1999-12-02 |
Family
ID=27557521
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/NZ1998/000189 WO1999032634A2 (en) | 1997-12-23 | 1998-12-23 | Compositions derived from mycobacterium vaccae and methods for their use |
Country Status (15)
| Country | Link |
|---|---|
| EP (1) | EP1044273A2 (en) |
| JP (1) | JP2002514385A (en) |
| CN (1) | CN1294632A (en) |
| AU (1) | AU746311B2 (en) |
| BR (1) | BR9814432A (en) |
| CA (1) | CA2315539A1 (en) |
| HU (1) | HUP0100352A2 (en) |
| ID (1) | ID26327A (en) |
| IL (1) | IL136821A0 (en) |
| MX (1) | MXPA00006168A (en) |
| NO (1) | NO20003261L (en) |
| NZ (1) | NZ505834A (en) |
| PL (1) | PL341697A1 (en) |
| TR (1) | TR200001948T2 (en) |
| WO (1) | WO1999032634A2 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000044905A1 (en) * | 1999-01-29 | 2000-08-03 | Otsuka Pharmaceutical Co., Ltd. | Slow growing acid-fast bacterium polypeptide |
| WO2000048615A2 (en) * | 1999-02-16 | 2000-08-24 | Stanford Rook Limited | TREATMENT OF CHRONIC VIRAL INFECTIONS WITH $i(M. VACCAE) |
| WO2000074715A1 (en) * | 1999-06-02 | 2000-12-14 | Genesis Research & Development Corporation Limited | Methods and compounds for the treatment of immunologically-mediated diseases using mycobacterium vaccae |
| WO2001016327A2 (en) * | 1999-08-31 | 2001-03-08 | Michael Niederweis | Method for producing a channel-forming protein |
| WO2001016285A2 (en) * | 1999-08-31 | 2001-03-08 | Novozymes A/S | Novel proteases and variants thereof |
| WO2001040282A1 (en) * | 1999-12-06 | 2001-06-07 | Genesis Research & Development Corporation Limited | Compositions isolated from m. vaccae and their use in the modulation of immune responses |
| WO2001087332A1 (en) * | 2000-05-19 | 2001-11-22 | Pneumobiotics, Pty, Ltd | Compositions and methods for treatment of mucosal infections |
| EP1254953A1 (en) * | 1999-12-28 | 2002-11-06 | Toyoshima, Kumao | Maturation-promoting agent for immature dendritic cells |
| WO2002094184A2 (en) * | 2001-05-11 | 2002-11-28 | Corixa Corporation | Vaccines for the treatment of autoimmune disease |
| WO2003013595A1 (en) * | 2001-07-26 | 2003-02-20 | Genesis Research And Development Corporation Limited | Compounds and methods for the modulation of immune responses |
| US6569436B1 (en) | 1998-10-05 | 2003-05-27 | The Malaghan Institute Of Medical Research | Method of using a vaccine |
| WO2003049751A1 (en) * | 2001-12-10 | 2003-06-19 | Bakulesh Mafatlal Khamar | The process of manufacturing a pharmaceutical composition useful for management of cancer |
| WO2004071387A2 (en) * | 2003-02-14 | 2004-08-26 | Novartis Ag | Hsp60 from arthrobacter |
| US7217554B2 (en) | 1999-08-31 | 2007-05-15 | Novozymes A/S | Proteases and variants thereof |
| CN100379759C (en) * | 1999-07-12 | 2008-04-09 | 吉尼西斯研究及发展有限公司 | Compounds for treatment of infectious and immune system disorders and method for their use |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2936131A1 (en) * | 2014-01-09 | 2015-07-16 | Transgene Sa | Fusion of heterooligomeric mycobacterial antigens |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991002542A1 (en) * | 1989-08-25 | 1991-03-07 | University College London | Treatment of chronic inflammatory conditions |
| EP0763361A2 (en) * | 1992-02-21 | 1997-03-19 | University College London | Mycobacterium vaccae for treatment of long term autoimmune conditions |
| WO1998008542A2 (en) * | 1996-08-29 | 1998-03-05 | Genesis Research & Development Corporation Limited | Compounds and methods for treatment and diagnosis of mycobacterial infections |
-
1998
- 1998-12-23 CN CN98813781A patent/CN1294632A/en active Pending
- 1998-12-23 TR TR2000/01948T patent/TR200001948T2/en unknown
- 1998-12-23 ID IDW20001433A patent/ID26327A/en unknown
- 1998-12-23 CA CA002315539A patent/CA2315539A1/en not_active Abandoned
- 1998-12-23 WO PCT/NZ1998/000189 patent/WO1999032634A2/en not_active Application Discontinuation
- 1998-12-23 NZ NZ505834A patent/NZ505834A/en unknown
- 1998-12-23 BR BR9814432-4A patent/BR9814432A/en not_active IP Right Cessation
- 1998-12-23 HU HU0100352A patent/HUP0100352A2/en unknown
- 1998-12-23 IL IL13682198A patent/IL136821A0/en unknown
- 1998-12-23 JP JP2000525553A patent/JP2002514385A/en active Pending
- 1998-12-23 EP EP98963665A patent/EP1044273A2/en not_active Withdrawn
- 1998-12-23 PL PL98341697A patent/PL341697A1/en unknown
- 1998-12-23 MX MXPA00006168A patent/MXPA00006168A/en unknown
- 1998-12-23 AU AU18936/99A patent/AU746311B2/en not_active Ceased
-
2000
- 2000-06-22 NO NO20003261A patent/NO20003261L/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991002542A1 (en) * | 1989-08-25 | 1991-03-07 | University College London | Treatment of chronic inflammatory conditions |
| EP0763361A2 (en) * | 1992-02-21 | 1997-03-19 | University College London | Mycobacterium vaccae for treatment of long term autoimmune conditions |
| WO1998008542A2 (en) * | 1996-08-29 | 1998-03-05 | Genesis Research & Development Corporation Limited | Compounds and methods for treatment and diagnosis of mycobacterial infections |
Non-Patent Citations (6)
| Title |
|---|
| KAPUR V. ET AL.: "RAPID MYCOBACTERIUM SPECIES ASSIGNMENT AND UNAMBIGUOUS IDENTIFICATION OF MUTATIONS ASSOCIATED WITH ANTIMICROBIALS RESISTANCE IN MYCOBACTERIUM TUBERCULOSIS BY AUTOMATED DNA SEQUENCING" ARCHIVES OF PATHOLOGY & LABORATORY MEDICINE, vol. 119, no. 2, 1 February 1995, pages 131-138, XP000572767 & EMBL database entry MV17958; accession number U17958; 22-Dec-1994; Kapur V. et al.: 'Mycobacterium vaccae 65 kDa heat shock protein gene, partial cds.' * |
| SKINNER M. A. ET AL.: "IMMUNIZATION WITH HEAT-KILLED MYCOBACTERIUM VACCAE STIMULATES CD8+CYTOTOXIC T CELLS SPECIFIC FOR MACROPHAGES INFECTED WITH MYCOBACTERIUM TUBERCULOSIS" INFECTION AND IMMUNITY, vol. 65, no. 11, 1 November 1997, pages 4525-4530, XP002060474 * |
| SOINI H. AND VILJANEN M. K.: "Diversity of the 32-kilodalton protein gene may form a basis for species determination of potentially pathogenic mycobacterial species." JOURNAL OF CLINICAL MICROBIOLOGY, vol. 35, no. 3, March 1997, pages 769-773, XP002094599 * |
| STANFORD J L: "IMPROVING ON BCG" APMIS, vol. 99, no. 2, 1 January 1991, pages 103-113, XP000616012 * |
| STANFORD J. L. ET AL.: "Mycobacterium vaccae in immunoprophylaxis and immunotherapy of leprosy and tuberculosis." VACCINE, vol. 8, December 1990, pages 525-530, XP002106918 * |
| YOUNG R. A. ET AL.: "DISSECTION OF MYCOBACTERIUM TUBERCULOSIS ANTIGENS USING RECOMBINANT DNA" PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF USA, vol. 82, 1 May 1985, pages 2583-2587, XP002034045 * |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6569436B1 (en) | 1998-10-05 | 2003-05-27 | The Malaghan Institute Of Medical Research | Method of using a vaccine |
| WO2000044905A1 (en) * | 1999-01-29 | 2000-08-03 | Otsuka Pharmaceutical Co., Ltd. | Slow growing acid-fast bacterium polypeptide |
| WO2000048615A2 (en) * | 1999-02-16 | 2000-08-24 | Stanford Rook Limited | TREATMENT OF CHRONIC VIRAL INFECTIONS WITH $i(M. VACCAE) |
| WO2000048615A3 (en) * | 1999-02-16 | 2000-12-14 | Stanford Rook Ltd | TREATMENT OF CHRONIC VIRAL INFECTIONS WITH $i(M. VACCAE) |
| US6596282B1 (en) | 1999-02-16 | 2003-07-22 | Stanford Rook Limited | Treatment of chronic viral infections with M. vaccae |
| WO2000074715A1 (en) * | 1999-06-02 | 2000-12-14 | Genesis Research & Development Corporation Limited | Methods and compounds for the treatment of immunologically-mediated diseases using mycobacterium vaccae |
| AU779163B2 (en) * | 1999-06-02 | 2005-01-06 | Genesis Research And Development Corporation Limited | Methods and compounds for the treatment of immunologically-mediated diseases using mycobacterium vaccae |
| CN100379759C (en) * | 1999-07-12 | 2008-04-09 | 吉尼西斯研究及发展有限公司 | Compounds for treatment of infectious and immune system disorders and method for their use |
| WO2001016285A3 (en) * | 1999-08-31 | 2001-09-13 | Novozymes As | Novel proteases and variants thereof |
| US7217554B2 (en) | 1999-08-31 | 2007-05-15 | Novozymes A/S | Proteases and variants thereof |
| WO2001016327A3 (en) * | 1999-08-31 | 2001-05-31 | Michael Niederweis | Method for producing a channel-forming protein |
| US7537922B2 (en) | 1999-08-31 | 2009-05-26 | Novozymes A/S | Proteases and variants thereof |
| WO2001016327A2 (en) * | 1999-08-31 | 2001-03-08 | Michael Niederweis | Method for producing a channel-forming protein |
| EP2336331A1 (en) * | 1999-08-31 | 2011-06-22 | Novozymes A/S | Novel proteases and variants thereof |
| US8119386B2 (en) | 1999-08-31 | 2012-02-21 | Novozymes Als | Proteases and variants thereof |
| EP2206786A1 (en) * | 1999-08-31 | 2010-07-14 | Novozymes A/S | Novel proteases and variants thereof |
| WO2001016285A2 (en) * | 1999-08-31 | 2001-03-08 | Novozymes A/S | Novel proteases and variants thereof |
| US7901918B2 (en) | 1999-08-31 | 2011-03-08 | Novozymes A/S | Proteases and variants thereof |
| WO2001040282A1 (en) * | 1999-12-06 | 2001-06-07 | Genesis Research & Development Corporation Limited | Compositions isolated from m. vaccae and their use in the modulation of immune responses |
| EP1254953A1 (en) * | 1999-12-28 | 2002-11-06 | Toyoshima, Kumao | Maturation-promoting agent for immature dendritic cells |
| EP1254953A4 (en) * | 1999-12-28 | 2004-06-02 | Toyoshima Kumao | Maturation-promoting agent for immature dendritic cells |
| US8637051B2 (en) | 2000-05-19 | 2014-01-28 | Hunter Immunology Limited | Compositions and methods for treatment of mucosal infections |
| WO2001087332A1 (en) * | 2000-05-19 | 2001-11-22 | Pneumobiotics, Pty, Ltd | Compositions and methods for treatment of mucosal infections |
| WO2002094184A3 (en) * | 2001-05-11 | 2003-04-10 | Corixa Corp | Vaccines for the treatment of autoimmune disease |
| WO2002094184A2 (en) * | 2001-05-11 | 2002-11-28 | Corixa Corporation | Vaccines for the treatment of autoimmune disease |
| WO2003013595A1 (en) * | 2001-07-26 | 2003-02-20 | Genesis Research And Development Corporation Limited | Compounds and methods for the modulation of immune responses |
| WO2003049751A1 (en) * | 2001-12-10 | 2003-06-19 | Bakulesh Mafatlal Khamar | The process of manufacturing a pharmaceutical composition useful for management of cancer |
| WO2004071387A3 (en) * | 2003-02-14 | 2005-03-24 | Novartis Ag | Hsp60 from arthrobacter |
| US7803924B2 (en) | 2003-02-14 | 2010-09-28 | Novartis Ag | HSP60 from arthrobacter |
| CN1751064B (en) * | 2003-02-14 | 2010-05-26 | 诺瓦提斯公司 | Hsp60 from arthrobacter |
| AU2004212322B2 (en) * | 2003-02-14 | 2008-01-17 | Novartis Tiergesundheit Ag | Hsp60 from arthrobacter |
| US7297783B2 (en) | 2003-02-14 | 2007-11-20 | Novartis Animal Health Us, Inc. | Hsp60 from arthrobacter |
| WO2004071387A2 (en) * | 2003-02-14 | 2004-08-26 | Novartis Ag | Hsp60 from arthrobacter |
Also Published As
| Publication number | Publication date |
|---|---|
| AU1893699A (en) | 1999-07-12 |
| WO1999032634A3 (en) | 1999-12-02 |
| NO20003261L (en) | 2000-08-22 |
| BR9814432A (en) | 2000-10-10 |
| IL136821A0 (en) | 2001-06-14 |
| AU746311B2 (en) | 2002-04-18 |
| JP2002514385A (en) | 2002-05-21 |
| CA2315539A1 (en) | 1999-07-01 |
| MXPA00006168A (en) | 2005-02-24 |
| NZ505834A (en) | 2002-12-20 |
| ID26327A (en) | 2000-12-14 |
| CN1294632A (en) | 2001-05-09 |
| PL341697A1 (en) | 2001-04-23 |
| EP1044273A2 (en) | 2000-10-18 |
| TR200001948T2 (en) | 2001-02-21 |
| HUP0100352A2 (en) | 2001-06-28 |
| NO20003261D0 (en) | 2000-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6410720B1 (en) | Compounds and methods for treatment and diagnosis of mycobacterial infections | |
| EP1523331B1 (en) | Therapeutic tb vaccine | |
| US10519202B2 (en) | Tuberculosis TB vaccine to prevent reactivation | |
| EP2163255B1 (en) | Tuberculosis vaccines comprising antigens expressed during the latent infection phase | |
| EP1044273A2 (en) | COMPOSITIONS DERIVED FROM $i(MYCOBACTERIUM VACCAE) AND METHODS FOR THEIR USE | |
| JP2001501832A (en) | Compounds and methods for immunotherapy and diagnosis of tuberculosis | |
| US5985287A (en) | Compounds and methods for treatment and diagnosis of mycobacterial infections | |
| US6406704B1 (en) | Compounds and methods for treatment and diagnosis of mycobacterial infections | |
| US20030007976A1 (en) | Methods and compounds for the treatment of immunologically-mediated skin disorders | |
| US6328978B1 (en) | Methods for the treatment of immunologically-mediated skin disorders | |
| US7041295B2 (en) | Compounds for treatment of infectious and immune system disorders and methods for their use | |
| AU741016B2 (en) | Compounds and methods for treatment and diagnosis of mycobacterial infections | |
| CZ20002321A3 (en) | Composition obtained from Mycobacterium vaccae and application methods thereof | |
| US7192590B2 (en) | Compounds for treatment of infectious and immune system disorders and methods for their use | |
| KR20010033132A (en) | Compositions derived from mycobacterium vaccae and methods for their use | |
| US20020197265A1 (en) | Methods and compounds for the treatment of immunologically - mediated diseases of the respiratory system using mycobacterium vaccae | |
| Coler | Identification and characterization of novel antigen genes of Mycobacterium tuberculosis | |
| AU2011203012A1 (en) | Tuberculosis vaccines comprising antigens expressed during the latent infection phase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 136821 Country of ref document: IL Ref document number: 98813781.X Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007006505 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18936/99 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2315539 Country of ref document: CA Ref document number: 2315539 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2000-2321 Country of ref document: CZ Ref document number: PA/a/2000/006168 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000/01948 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998963665 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 505834 Country of ref document: NZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998963665 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-2321 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007006505 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 18936/99 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998963665 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1020007006505 Country of ref document: KR |