WO1994005696A1 - Novel peptide, and antithrombotic agent, anticoagulant for extracorporeal circulation, cell fusion inhibitor, cancer metastasis inhibitor, protective for platelet preparation for transfusion and pack of platelet preparation for transfusion - Google Patents
Novel peptide, and antithrombotic agent, anticoagulant for extracorporeal circulation, cell fusion inhibitor, cancer metastasis inhibitor, protective for platelet preparation for transfusion and pack of platelet preparation for transfusion Download PDFInfo
- Publication number
- WO1994005696A1 WO1994005696A1 PCT/JP1993/001262 JP9301262W WO9405696A1 WO 1994005696 A1 WO1994005696 A1 WO 1994005696A1 JP 9301262 W JP9301262 W JP 9301262W WO 9405696 A1 WO9405696 A1 WO 9405696A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- peptide
- derivative
- acid
- blood
- platelet
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 200
- 230000004087 circulation Effects 0.000 title claims abstract description 41
- 238000002360 preparation method Methods 0.000 title claims abstract description 32
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 30
- 201000011510 cancer Diseases 0.000 title claims abstract description 29
- 239000002257 antimetastatic agent Substances 0.000 title claims abstract description 13
- 239000003146 anticoagulant agent Substances 0.000 title abstract description 14
- 229940127219 anticoagulant drug Drugs 0.000 title abstract description 10
- 230000001681 protective effect Effects 0.000 title abstract description 8
- 230000007910 cell fusion Effects 0.000 title abstract 3
- 229960004676 antithrombotic agent Drugs 0.000 title abstract 2
- 229940125777 fusion inhibitor Drugs 0.000 title abstract 2
- 239000004480 active ingredient Substances 0.000 claims abstract description 30
- 210000004369 blood Anatomy 0.000 claims description 89
- 239000008280 blood Substances 0.000 claims description 89
- 150000001875 compounds Chemical class 0.000 claims description 72
- 235000001014 amino acid Nutrition 0.000 claims description 51
- 230000023555 blood coagulation Effects 0.000 claims description 51
- 150000001413 amino acids Chemical class 0.000 claims description 49
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 27
- 239000002253 acid Substances 0.000 claims description 24
- 150000003839 salts Chemical class 0.000 claims description 23
- 229940127218 antiplatelet drug Drugs 0.000 claims description 22
- 239000000106 platelet aggregation inhibitor Substances 0.000 claims description 22
- 230000009471 action Effects 0.000 claims description 20
- 101000783577 Dendroaspis angusticeps Thrombostatin Proteins 0.000 claims description 19
- 101000783578 Dendroaspis jamesoni kaimosae Dendroaspin Proteins 0.000 claims description 19
- 230000021164 cell adhesion Effects 0.000 claims description 19
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 19
- 239000000126 substance Substances 0.000 claims description 19
- 125000001841 imino group Chemical group [H]N=* 0.000 claims description 16
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 claims description 15
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 claims description 15
- 239000003130 blood coagulation factor inhibitor Substances 0.000 claims description 15
- 125000000217 alkyl group Chemical group 0.000 claims description 14
- 125000003277 amino group Chemical group 0.000 claims description 14
- 150000003722 vitamin derivatives Chemical class 0.000 claims description 13
- 239000003223 protective agent Substances 0.000 claims description 12
- 229940088594 vitamin Drugs 0.000 claims description 12
- 239000011782 vitamin Substances 0.000 claims description 12
- 235000013343 vitamin Nutrition 0.000 claims description 12
- 229930003231 vitamin Natural products 0.000 claims description 12
- 125000000524 functional group Chemical group 0.000 claims description 10
- 150000007523 nucleic acids Chemical class 0.000 claims description 10
- 102000039446 nucleic acids Human genes 0.000 claims description 10
- 108020004707 nucleic acids Proteins 0.000 claims description 10
- 239000003112 inhibitor Substances 0.000 claims description 9
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 8
- 150000003862 amino acid derivatives Chemical class 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 7
- PXQPEWDEAKTCGB-UHFFFAOYSA-N orotic acid Chemical group OC(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-N 0.000 claims description 6
- 239000004471 Glycine Substances 0.000 claims description 5
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 5
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 5
- 230000002209 hydrophobic effect Effects 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical group C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 4
- LIXQSGRMEVDRBL-UHFFFAOYSA-N acetic acid;imidazolidine-2,4-dione Chemical compound CC(O)=O.O=C1CNC(=O)N1 LIXQSGRMEVDRBL-UHFFFAOYSA-N 0.000 claims description 4
- 235000004279 alanine Nutrition 0.000 claims description 4
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 4
- DZLNHFMRPBPULJ-VKHMYHEASA-N L-thioproline Chemical compound OC(=O)[C@@H]1CSCN1 DZLNHFMRPBPULJ-VKHMYHEASA-N 0.000 claims description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical group C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 claims description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Chemical group CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 3
- 239000004473 Threonine Chemical group 0.000 claims description 3
- 229960005010 orotic acid Drugs 0.000 claims description 3
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 claims description 3
- OMGHIGVFLOPEHJ-UHFFFAOYSA-N 2,5-dihydro-1h-pyrrol-1-ium-2-carboxylate Chemical compound OC(=O)C1NCC=C1 OMGHIGVFLOPEHJ-UHFFFAOYSA-N 0.000 claims description 2
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 claims description 2
- 125000000266 alpha-aminoacyl group Chemical group 0.000 claims description 2
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 claims description 2
- 229960002591 hydroxyproline Drugs 0.000 claims description 2
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 claims description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical group N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims 2
- SLGBZMMZGDRARJ-UHFFFAOYSA-N Triphenylene Natural products C1=CC=C2C3=CC=CC=C3C3=CC=CC=C3C2=C1 SLGBZMMZGDRARJ-UHFFFAOYSA-N 0.000 claims 1
- 239000011814 protection agent Substances 0.000 claims 1
- ODHXBMXNKOYIBV-UHFFFAOYSA-N triphenylamine Chemical compound C1=CC=CC=C1N(C=1C=CC=CC=1)C1=CC=CC=C1 ODHXBMXNKOYIBV-UHFFFAOYSA-N 0.000 claims 1
- 125000005580 triphenylene group Chemical group 0.000 claims 1
- 230000002401 inhibitory effect Effects 0.000 abstract description 44
- 208000007536 Thrombosis Diseases 0.000 abstract description 8
- 201000010099 disease Diseases 0.000 abstract description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 3
- 208000010125 myocardial infarction Diseases 0.000 abstract description 3
- 230000002785 anti-thrombosis Effects 0.000 abstract description 2
- 230000002537 thrombolytic effect Effects 0.000 abstract description 2
- 206010053567 Coagulopathies Diseases 0.000 abstract 1
- 230000035602 clotting Effects 0.000 abstract 1
- 238000002560 therapeutic procedure Methods 0.000 abstract 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 69
- 238000004458 analytical method Methods 0.000 description 60
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 53
- 229940024606 amino acid Drugs 0.000 description 49
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 46
- 230000000694 effects Effects 0.000 description 42
- 238000000034 method Methods 0.000 description 36
- 238000004128 high performance liquid chromatography Methods 0.000 description 34
- 230000015572 biosynthetic process Effects 0.000 description 32
- 239000011347 resin Substances 0.000 description 31
- 229920005989 resin Polymers 0.000 description 31
- 238000003786 synthesis reaction Methods 0.000 description 30
- 210000004027 cell Anatomy 0.000 description 28
- 230000014759 maintenance of location Effects 0.000 description 28
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 27
- 238000010828 elution Methods 0.000 description 27
- 210000002381 plasma Anatomy 0.000 description 26
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 24
- 239000000243 solution Substances 0.000 description 23
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 22
- -1 diaminopropyl group Chemical group 0.000 description 22
- 238000002474 experimental method Methods 0.000 description 21
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 21
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 21
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 20
- 229960002897 heparin Drugs 0.000 description 20
- 229920000669 heparin Polymers 0.000 description 20
- 238000003860 storage Methods 0.000 description 20
- 239000003814 drug Substances 0.000 description 19
- 238000000502 dialysis Methods 0.000 description 18
- IYMAXBFPHPZYIK-BQBZGAKWSA-N Arg-Gly-Asp Chemical compound NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O IYMAXBFPHPZYIK-BQBZGAKWSA-N 0.000 description 17
- 230000002776 aggregation Effects 0.000 description 16
- 238000004220 aggregation Methods 0.000 description 16
- 229940079593 drug Drugs 0.000 description 16
- 238000002347 injection Methods 0.000 description 15
- 239000007924 injection Substances 0.000 description 15
- 108010035532 Collagen Proteins 0.000 description 12
- 102000008186 Collagen Human genes 0.000 description 12
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 12
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 12
- 229920001436 collagen Polymers 0.000 description 12
- 238000001727 in vivo Methods 0.000 description 12
- 241000282472 Canis lupus familiaris Species 0.000 description 11
- 210000004623 platelet-rich plasma Anatomy 0.000 description 11
- 238000012360 testing method Methods 0.000 description 11
- 239000002504 physiological saline solution Substances 0.000 description 10
- 238000005259 measurement Methods 0.000 description 9
- 230000010412 perfusion Effects 0.000 description 9
- 125000006239 protecting group Chemical group 0.000 description 9
- 238000001179 sorption measurement Methods 0.000 description 9
- 238000011282 treatment Methods 0.000 description 9
- 108010049003 Fibrinogen Proteins 0.000 description 8
- 102000008946 Fibrinogen Human genes 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 229940012952 fibrinogen Drugs 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- 239000012528 membrane Substances 0.000 description 8
- 239000000203 mixture Substances 0.000 description 8
- 239000006228 supernatant Substances 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- 206010027476 Metastases Diseases 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 239000000306 component Substances 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 230000009401 metastasis Effects 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 210000003462 vein Anatomy 0.000 description 7
- NNRFRJQMBSBXGO-CIUDSAMLSA-N (3s)-3-[[2-[[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]acetyl]amino]-4-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-oxobutanoic acid Chemical compound NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(O)=O NNRFRJQMBSBXGO-CIUDSAMLSA-N 0.000 description 6
- 101000829980 Homo sapiens Ral guanine nucleotide dissociation stimulator Proteins 0.000 description 6
- 102000007547 Laminin Human genes 0.000 description 6
- 108010085895 Laminin Proteins 0.000 description 6
- 102100023320 Ral guanine nucleotide dissociation stimulator Human genes 0.000 description 6
- 230000017531 blood circulation Effects 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 238000006482 condensation reaction Methods 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- 239000004033 plastic Substances 0.000 description 6
- 229920003023 plastic Polymers 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 5
- 102000012422 Collagen Type I Human genes 0.000 description 5
- 108010022452 Collagen Type I Proteins 0.000 description 5
- 206010062713 Haemorrhagic diathesis Diseases 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 108010094028 Prothrombin Proteins 0.000 description 5
- 102100027378 Prothrombin Human genes 0.000 description 5
- 125000003275 alpha amino acid group Chemical group 0.000 description 5
- 108010072041 arginyl-glycyl-aspartic acid Proteins 0.000 description 5
- 230000015556 catabolic process Effects 0.000 description 5
- 238000006731 degradation reaction Methods 0.000 description 5
- 239000008187 granular material Substances 0.000 description 5
- 208000031169 hemorrhagic disease Diseases 0.000 description 5
- 238000001802 infusion Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 229940039716 prothrombin Drugs 0.000 description 5
- 210000002784 stomach Anatomy 0.000 description 5
- 238000001356 surgical procedure Methods 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- IADUEWIQBXOCDZ-VKHMYHEASA-N (S)-azetidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCN1 IADUEWIQBXOCDZ-VKHMYHEASA-N 0.000 description 4
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 4
- 210000002469 basement membrane Anatomy 0.000 description 4
- 210000004204 blood vessel Anatomy 0.000 description 4
- 230000015271 coagulation Effects 0.000 description 4
- 238000005345 coagulation Methods 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 201000001441 melanoma Diseases 0.000 description 4
- 238000004321 preservation Methods 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 4
- 239000001509 sodium citrate Substances 0.000 description 4
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 4
- 238000010532 solid phase synthesis reaction Methods 0.000 description 4
- 238000002834 transmittance Methods 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- PGOHTUIFYSHAQG-LJSDBVFPSA-N (2S)-6-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-4-methylsulfanylbutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-hydroxypropanoyl]amino]-4-methylpentanoyl]amino]-3-sulfanylpropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-oxopentanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-oxopentanoyl]amino]-3-phenylpropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-5-oxopentanoyl]amino]hexanoic acid Chemical compound CSCC[C@H](N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1cnc[nH]1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(O)=O PGOHTUIFYSHAQG-LJSDBVFPSA-N 0.000 description 3
- DHBXNPKRAUYBTH-UHFFFAOYSA-N 1,1-ethanedithiol Chemical compound CC(S)S DHBXNPKRAUYBTH-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 239000006145 Eagle's minimal essential medium Substances 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 108010000499 Thromboplastin Proteins 0.000 description 3
- 102000002262 Thromboplastin Human genes 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- LCPVBXOHXMBLFW-JSGCOSHPSA-N Trp-Arg Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)=CNC2=C1 LCPVBXOHXMBLFW-JSGCOSHPSA-N 0.000 description 3
- 108010031318 Vitronectin Proteins 0.000 description 3
- 102100035140 Vitronectin Human genes 0.000 description 3
- 150000008065 acid anhydrides Chemical class 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 230000000593 degrading effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- LMBMDLOSPKIWAP-UHFFFAOYSA-N embutramide Chemical compound OCCCC(=O)NCC(CC)(CC)C1=CC=CC(OC)=C1 LMBMDLOSPKIWAP-UHFFFAOYSA-N 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 208000023589 ischemic disease Diseases 0.000 description 3
- 231100000053 low toxicity Toxicity 0.000 description 3
- 150000003147 proline derivatives Chemical class 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- 230000002792 vascular Effects 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- QABAUCFGPWONOG-UHFFFAOYSA-N 2-Amino-4-hydroxy-6-pteridinecarboxylic acid Chemical compound OC(=O)C1=CN=C2NC(N)=NC(=O)C2=N1 QABAUCFGPWONOG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 2
- FUFZNHHSSMCXCZ-UHFFFAOYSA-N 5-piperidin-4-yl-3-[3-(trifluoromethyl)phenyl]-1,2,4-oxadiazole Chemical compound FC(F)(F)C1=CC=CC(C=2N=C(ON=2)C2CCNCC2)=C1 FUFZNHHSSMCXCZ-UHFFFAOYSA-N 0.000 description 2
- 101710186708 Agglutinin Proteins 0.000 description 2
- 206010002091 Anaesthesia Diseases 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 108010012088 Fibrinogen Receptors Proteins 0.000 description 2
- 102100037362 Fibronectin Human genes 0.000 description 2
- 108010067306 Fibronectins Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 208000032843 Hemorrhage Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101710146024 Horcolin Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 150000008575 L-amino acids Chemical class 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 101710189395 Lectin Proteins 0.000 description 2
- 101710179758 Mannose-specific lectin Proteins 0.000 description 2
- 101710150763 Mannose-specific lectin 1 Proteins 0.000 description 2
- 101710150745 Mannose-specific lectin 2 Proteins 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- YPIGGYHFMKJNKV-UHFFFAOYSA-N N-ethylglycine Chemical compound CC[NH2+]CC([O-])=O YPIGGYHFMKJNKV-UHFFFAOYSA-N 0.000 description 2
- 108010065338 N-ethylglycine Proteins 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- ODHCTXKNWHHXJC-GSVOUGTGSA-N Pyroglutamic acid Natural products OC(=O)[C@H]1CCC(=O)N1 ODHCTXKNWHHXJC-GSVOUGTGSA-N 0.000 description 2
- 108010077895 Sarcosine Proteins 0.000 description 2
- 229920005654 Sephadex Polymers 0.000 description 2
- 239000012507 Sephadex™ Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- UCTWMZQNUQWSLP-UHFFFAOYSA-N adrenaline Chemical compound CNCC(O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-UHFFFAOYSA-N 0.000 description 2
- 239000000910 agglutinin Substances 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 2
- 210000001367 artery Anatomy 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000000740 bleeding effect Effects 0.000 description 2
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001718 carbodiimides Chemical class 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 2
- 230000002612 cardiopulmonary effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 2
- 210000001105 femoral artery Anatomy 0.000 description 2
- 239000003527 fibrinolytic agent Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000001641 gel filtration chromatography Methods 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- HXEACLLIILLPRG-RXMQYKEDSA-N l-pipecolic acid Natural products OC(=O)[C@H]1CCCCN1 HXEACLLIILLPRG-RXMQYKEDSA-N 0.000 description 2
- 150000003951 lactams Chemical class 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 108020004084 membrane receptors Proteins 0.000 description 2
- 102000006240 membrane receptors Human genes 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- CXKWCBBOMKCUKX-UHFFFAOYSA-M methylene blue Chemical compound [Cl-].C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 CXKWCBBOMKCUKX-UHFFFAOYSA-M 0.000 description 2
- 229960000907 methylthioninium chloride Drugs 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 230000010118 platelet activation Effects 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920000098 polyolefin Polymers 0.000 description 2
- 229920000915 polyvinyl chloride Polymers 0.000 description 2
- 239000004800 polyvinyl chloride Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 150000003180 prostaglandins Chemical class 0.000 description 2
- 230000002633 protecting effect Effects 0.000 description 2
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- HNKJADCVZUBCPG-UHFFFAOYSA-N thioanisole Chemical compound CSC1=CC=CC=C1 HNKJADCVZUBCPG-UHFFFAOYSA-N 0.000 description 2
- 229960000103 thrombolytic agent Drugs 0.000 description 2
- RZWIIPASKMUIAC-VQTJNVASSA-N thromboxane Chemical compound CCCCCCCC[C@H]1OCCC[C@@H]1CCCCCCC RZWIIPASKMUIAC-VQTJNVASSA-N 0.000 description 2
- 230000036962 time dependent Effects 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 1
- GMVPRGQOIOIIMI-UHFFFAOYSA-N (8R,11R,12R,13E,15S)-11,15-Dihydroxy-9-oxo-13-prostenoic acid Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CCCCCCC(O)=O GMVPRGQOIOIIMI-UHFFFAOYSA-N 0.000 description 1
- IZFHEQBZOYJLPK-SSDOTTSWSA-N (R)-dihydrolipoic acid Chemical compound OC(=O)CCCC[C@@H](S)CCS IZFHEQBZOYJLPK-SSDOTTSWSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 1
- UFBJCMHMOXMLKC-UHFFFAOYSA-N 2,4-dinitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O UFBJCMHMOXMLKC-UHFFFAOYSA-N 0.000 description 1
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 1
- HEPOIJKOXBKKNJ-UHFFFAOYSA-N 2-(propan-2-ylazaniumyl)acetate Chemical compound CC(C)NCC(O)=O HEPOIJKOXBKKNJ-UHFFFAOYSA-N 0.000 description 1
- BHUGZIJOVAVBOQ-UHFFFAOYSA-N 2-(propylazaniumyl)acetate Chemical compound CCCNCC(O)=O BHUGZIJOVAVBOQ-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- WFIYPADYPQQLNN-UHFFFAOYSA-N 2-[2-(4-bromopyrazol-1-yl)ethyl]isoindole-1,3-dione Chemical compound C1=C(Br)C=NN1CCN1C(=O)C2=CC=CC=C2C1=O WFIYPADYPQQLNN-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 1
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- RCJIVJMTTMAMME-UHFFFAOYSA-N 4-[(2-amino-4-oxo-1h-pteridin-6-yl)methyl-formylamino]benzoic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CN(C=O)C1=CC=C(C(O)=O)C=C1 RCJIVJMTTMAMME-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 1
- VJZTVPVXKYQRJZ-UHFFFAOYSA-N 5-pyridoxic acid Chemical compound CC1=NC=C(C(O)=O)C(CO)=C1O VJZTVPVXKYQRJZ-UHFFFAOYSA-N 0.000 description 1
- LSBDFXRDZJMBSC-UHFFFAOYSA-N Amide-Phenylacetic acid Natural products NC(=O)CC1=CC=CC=C1 LSBDFXRDZJMBSC-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 1
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241000270281 Coluber constrictor Species 0.000 description 1
- CKLJMWTZIZZHCS-UWTATZPHSA-N D-aspartic acid Chemical compound OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000032371 Glanzmann thrombasthenia 1 Diseases 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- HXEACLLIILLPRG-YFKPBYRVSA-N L-pipecolic acid Chemical compound [O-]C(=O)[C@@H]1CCCC[NH2+]1 HXEACLLIILLPRG-YFKPBYRVSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 101100084404 Mus musculus Prodh gene Proteins 0.000 description 1
- 125000003047 N-acetyl group Chemical group 0.000 description 1
- GNMSLDIYJOSUSW-LURJTMIESA-N N-acetyl-L-proline Chemical compound CC(=O)N1CCC[C@H]1C(O)=O GNMSLDIYJOSUSW-LURJTMIESA-N 0.000 description 1
- BAQMYDQNMFBZNA-UHFFFAOYSA-N N-biotinyl-L-lysine Natural products N1C(=O)NC2C(CCCCC(=O)NCCCCC(N)C(O)=O)SCC21 BAQMYDQNMFBZNA-UHFFFAOYSA-N 0.000 description 1
- CWLQUGTUXBXTLF-UHFFFAOYSA-N N-methyl-L-proline monohydrate Natural products CN1CCCC1C(O)=O CWLQUGTUXBXTLF-UHFFFAOYSA-N 0.000 description 1
- CWLQUGTUXBXTLF-YFKPBYRVSA-N N-methylproline Chemical compound CN1CCC[C@H]1C(O)=O CWLQUGTUXBXTLF-YFKPBYRVSA-N 0.000 description 1
- 102000002356 Nectin Human genes 0.000 description 1
- 108060005251 Nectin Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- AFWBWPCXSWUCLB-WDSKDSINSA-N Pro-Ser Chemical compound OC[C@@H](C([O-])=O)NC(=O)[C@@H]1CCC[NH2+]1 AFWBWPCXSWUCLB-WDSKDSINSA-N 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 206010038563 Reocclusion Diseases 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 206010039509 Scab Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 208000000392 Thrombasthenia Diseases 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 231100000215 acute (single dose) toxicity testing Toxicity 0.000 description 1
- 238000011047 acute toxicity test Methods 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229960000711 alprostadil Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 239000010775 animal oil Substances 0.000 description 1
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000002429 anti-coagulating effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 229940127217 antithrombotic drug Drugs 0.000 description 1
- 238000002617 apheresis Methods 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000003305 autocrine Effects 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- BAQMYDQNMFBZNA-MNXVOIDGSA-N biocytin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)NCCCC[C@H](N)C(O)=O)SC[C@@H]21 BAQMYDQNMFBZNA-MNXVOIDGSA-N 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- QPFQYMONYBAUCY-ZKWXMUAHSA-N biotin sulfone Chemical compound N1C(=O)N[C@H]2CS(=O)(=O)[C@@H](CCCCC(=O)O)[C@H]21 QPFQYMONYBAUCY-ZKWXMUAHSA-N 0.000 description 1
- 239000010836 blood and blood product Substances 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 239000012503 blood component Substances 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 238000009530 blood pressure measurement Methods 0.000 description 1
- 229940125691 blood product Drugs 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- LYVYUAHSSSHKHC-UHFFFAOYSA-N butyl(trimethyl)silane Chemical compound CCCC[Si](C)(C)C LYVYUAHSSSHKHC-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- BRPQOXSCLDDYGP-UHFFFAOYSA-N calcium oxide Chemical compound [O-2].[Ca+2] BRPQOXSCLDDYGP-UHFFFAOYSA-N 0.000 description 1
- 239000000292 calcium oxide Substances 0.000 description 1
- ODINCKMPIJJUCX-UHFFFAOYSA-N calcium oxide Inorganic materials [Ca]=O ODINCKMPIJJUCX-UHFFFAOYSA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- FOCAUTSVDIKZOP-UHFFFAOYSA-N chloroacetic acid Chemical compound OC(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-N 0.000 description 1
- 229940106681 chloroacetic acid Drugs 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229930003836 cresol Natural products 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- OZRNSSUDZOLUSN-LBPRGKRZSA-N dihydrofolic acid Chemical compound N=1C=2C(=O)NC(N)=NC=2NCC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OZRNSSUDZOLUSN-LBPRGKRZSA-N 0.000 description 1
- ZJIPHXXDPROMEF-UHFFFAOYSA-N dihydroxyphosphanyl dihydrogen phosphite Chemical compound OP(O)OP(O)O ZJIPHXXDPROMEF-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 210000003191 femoral vein Anatomy 0.000 description 1
- OQZCSNDVOWYALR-UHFFFAOYSA-N flurochloridone Chemical compound FC(F)(F)C1=CC=CC(N2C(C(Cl)C(CCl)C2)=O)=C1 OQZCSNDVOWYALR-UHFFFAOYSA-N 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- 108010053299 glycyl-arginyl-glycyl-aspartyl-seryl-proline Proteins 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 125000004970 halomethyl group Chemical group 0.000 description 1
- 230000023597 hemostasis Effects 0.000 description 1
- 239000012510 hollow fiber Substances 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- SRJOCJYGOFTFLH-UHFFFAOYSA-N isonipecotic acid Chemical class OC(=O)C1CCNCC1 SRJOCJYGOFTFLH-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- AGBQKNBQESQNJD-UHFFFAOYSA-N lipoic acid Chemical compound OC(=O)CCCCC1CCSS1 AGBQKNBQESQNJD-UHFFFAOYSA-N 0.000 description 1
- 235000019136 lipoic acid Nutrition 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000004880 lymph fluid Anatomy 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical class [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 229940055726 pantothenic acid Drugs 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-N phosphoramidic acid Chemical compound NP(O)(O)=O PTMHPRAIXMAOOB-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- HXEACLLIILLPRG-UHFFFAOYSA-N pipecolic acid Chemical compound OC(=O)C1CCCCN1 HXEACLLIILLPRG-UHFFFAOYSA-N 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000003761 preservation solution Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 108010031719 prolyl-serine Proteins 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- GMVPRGQOIOIIMI-DWKJAMRDSA-N prostaglandin E1 Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O GMVPRGQOIOIIMI-DWKJAMRDSA-N 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 230000001012 protector Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- UQDJGEHQDNVPGU-UHFFFAOYSA-N serine phosphoethanolamine Chemical compound [NH3+]CCOP([O-])(=O)OCC([NH3+])C([O-])=O UQDJGEHQDNVPGU-UHFFFAOYSA-N 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229960002663 thioctic acid Drugs 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 206010043554 thrombocytopenia Diseases 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000003966 vascular damage Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 108010047303 von Willebrand Factor Proteins 0.000 description 1
- 102100036537 von Willebrand factor Human genes 0.000 description 1
- 229960001134 von willebrand factor Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/78—Connective tissue peptides, e.g. collagen, elastin, laminin, fibronectin, vitronectin or cold insoluble globulin [CIG]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/745—Blood coagulation or fibrinolysis factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- New peptides and platelet aggregation inhibitors using them blood coagulation inhibitors for extracorporeal circulation, cell adhesion inhibitors, cancer metastasis inhibitors, platelets for blood transfusions, and platelets for blood transfusions.
- the present invention relates to a novel peptide having a platelet aggregation inhibitory activity, a platelet aggregation inhibitor containing the peptide as an active ingredient, a blood coagulation inhibitor for extracorporeal circulation, a cell adhesion inhibitor, and a cancer metastasis inhibitor.
- the present invention relates to a platelet preparation for blood transfusion, which is characterized in that the peptide or the like is contained in a platelet preparation for blood transfusion in a package and a platelet preparation for blood transfusion.
- platelets play a major role in preventing bleeding by adsorbing to the surface of damaged blood vessels.
- thrombus a major cause of thrombus formation, and it is known that this thrombus causes blood vessel obstruction. This obstruction prevents adequate supply of oxygen and nutrients to tissues and organs, which is a significant cause of cardiovascular ischemic diseases such as myocardial infarction and stroke. And today, such ischemic diseases represent the second highest mortality rate after cancer, and represent a major social problem.
- platelets are activated by binding to connective tissue proteins such as collagen present in subendothelial tissues exposed due to vascular damage and the like, and to platelet membrane receptors such as thrombin present in plasma. It is also activated by the release of adenosine diphosphite (ADP), adrenaline, serotonin, and thromboxane (TX) A2, which are present in platelets, by autocrine binding to membrane receptors. It is Then, two types of glycoprotein units constituting the fibrinogen receptor are displayed on the cell surface, associate with each other, and form a receptor complex (gpllbIlia) to form a fibrinogen receptor. Aggregation is induced via brinogen crosslinking.
- ADP adenosine diphosphite
- adrenaline adrenaline
- TX thromboxane
- the monoclonal antibody certainly has potential as a therapeutic agent for suppressing platelet aggregation.
- the monoclonal antibody since it is a high molecular weight protein itself, there is a concern that an antibody acting on the monoclonal antibody itself will be produced if the drug is administered repeatedly.
- a platelet aggregation inhibitor that has properties as an antagonist to the gpllbllla complex and contains a low molecular weight compound having no immunogenicity as an active ingredient is expected.
- a synthetic peptide containing the RGD sequence will inhibit platelet aggregation by inhibiting the binding of the gpHbIIIa complex to fibrinogen. It has been reported that the peptide GRGDSP completely inhibited the aggregation of ADP-activated platelets (Plow et al., Proc. Natl. Acad. Sci. USA., 82, 8057-8061 (1985). ). RGDS has been shown to inhibit platelet aggregation by 80-90% in a concentration-dependent manner at a concentration of 46-50 ⁇ M, and the peptide RGDF is 4-5 times more potent than RGDS. It has been shown to show strong platelet aggregation inhibitory activity (Plow et al., Blood, 70, 110-115 (1987) or Harfinest et al., 71, 132-136 (1988)).
- Tetrapeptide derivatives having an RGD peptide are described in JP-A-190699, JP-A-2-62892, EP-A-422937A1, and US Pat. No. 4,952,562. Derivatives composed of peptides are further described in JP-A-63-215696. The cyclic structure derivatives of the RGD peptide are described in JP-A-3-118331, JP-A-2-62892 or WO 91/01331.
- Platelet products for blood transfusion used for such component blood transfusions may be prepared by treating whole blood obtained by blood donation or by component blood donation using the apheresis method. It is one of the blood products prepared by any of the methods. Immediately after the preparation, these platelet preparations are usually placed in a special storage bag made of polyolefin or polyvinyl chloride and stored with shaking at room temperature.
- cancer metastasis can be divided into two very important steps: detachment of cancer cells from the primary focus into blood and lymphocytes and migration of cancer cells from blood and lymph fluid into tissues. Can be. At both stages, it is thought that the extracellular matrix proteins constituting the basement membrane of blood vessels and the like provide a scaffold for cell migration.
- the vascular basement membrane contains extracellular basal proteins having various RGD sequences such as fibronectin, collagen, vitronectin, and laminin.
- RGD sequence plays a very important role in adhesion to quality. Therefore, compounds that suppress the adhesion between cancer cells and the vascular basement membrane, especially RGD-like compounds, dissociate cancer cells into blood and inhibit the migration of blood from blood to outside blood vessels. The possibility of suppressing cancer metastasis has been pointed out.
- the improvement of the in vivo stability of the compound leads to the maintenance of the platelet aggregation inhibitory action and blood coagulation inhibitory action, leading to a bleeding tendency, etc., thereby stopping the essential physiological action inherent in platelets for a long time. There is a possibility.
- platelet aggregation When a platelet aggregation inhibitor is used for the purpose of temporarily suppressing the ability, it is needless to say that the agent should have excellent platelet aggregation inhibitory ability, but at the same time, it should be suitable for the intended use. It is necessary that the compound has a long duration of drug action and has excellent safety properties that can be rapidly metabolized to a compound without side effects after inactivation of the drug. As mentioned earlier, the platelet aggregation inhibitory activity and blood coagulation inhibitory activity of the RGD peptide itself are not high enough for practical use. However, this RGD peptide has an excellent feature that it is degraded to amino acids that are safe and useful for living organisms by proteolytic enzymes originally present in living organisms.
- the present inventor has made great use of this feature, has a variety of retention times in the living body depending on the purpose of use, has an excellent ability to inhibit platelet aggregation or blood coagulation, and has a structure as close as possible to a natural peptide. It is intended to produce an induced peptide having high activity and to provide a platelet aggregation inhibitor containing this as an active ingredient. In addition, the present invention intends to provide a blood coagulation inhibitor having few side effects, which is suitable for extracorporeal circulation or surgery.
- the function of platelets is significantly reduced with storage at present, which is one of the factors that hinders effective transfusion.
- the present invention intends to provide a platelet preparation protective agent for blood circulation containing the above-mentioned peptide or the like as an active ingredient.
- the decrease in platelet function due to storage is as follows: 1 Blood collection, preparation processing, storage Platelet activation and aggregation during storage caused by various physical stimuli during storage
- the main cause is considered to be a decrease in the pH of the preservative solution.
- preservation solutions and preservation systems for controlling pH have been actively improved in recent years, but at present they are not sufficiently effective.
- the platelet aggregation ability and the number of platelets decrease during storage. Thought that it could be prevented. That is, it is an object of the present invention to develop a compound having an action of protecting platelets in a pharmaceutical pack during storage.
- the present inventor when obtaining a highly active compound that can withstand practical use, adds to the RGD-like sequence a compound useful for living organisms with few side effects, such as aminoaminoacid bisubimin. This led to the creation of peptides with high activity and appropriate action time. When this peptide or the like is decomposed, the added portion is accepted by the living body as an originally useful substance.
- the present application has the following matters as its gist.
- A-B-A rg -Gly -A sp -C -D (where A is an amino acid, an amino acid derivative, a vitamin, a vitamin derivative, a vitamin-like substance)
- B is an amino acid;
- C is a compound having a hydrophobic functional group.
- Amino acid; D represents a hydroxyl group or an amino group
- amino acid or amino acid derivative in A is proline, thioproline, hydroxyproline, dehydroproline, 2-oxo-14-thiazolidincarboxylic acid, N- Alkyl glycine or the following general formula
- R> is a hydrogen atom, a general formula (CH 2 ) P CH] or a general formula
- R 2 is a hydrogen atom or an alkyl group
- R 3 is a hydrogen atom or a monoalkyl group
- R ⁇ is a hydrogen atom or an alkyl group
- R 5 is a hydrogen atom, an amino group or an amino acyl group
- q is an integer of 0 to 3
- the peptide according to the above (1) which is a triptophan derivative, pyroglutamic acid or 2-azetidinone-14 monocarboxylic acid; Peptide derivatives or salts thereof.
- the vitamin-like substance or the derivative of the vitamin-like substance is The peptide, peptide derivative or salt thereof according to the above (1), which is rotinoic acid or hydrorotinic acid.
- a platelet aggregation inhibitor comprising, as an active ingredient, the peptide, the peptide derivative or the salt thereof according to any one of (1) to (5).
- a blood coagulation inhibitor for extracorporeal circulation comprising as an active ingredient the peptide, the peptide derivative or a salt thereof described in any one of the above (1) to (5).
- a cell adhesion inhibitor comprising, as an active ingredient, the peptide, the peptide derivative or the salt thereof described in any one of the above (1) to (5).
- a cancer metastasis inhibitor comprising, as an active ingredient, the peptide, the peptide derivative or the salt thereof described in any one of the above (1) to (5).
- a protective agent for a blood transfusion platelet preparation comprising as an active ingredient the peptide, the peptide derivative or a salt thereof described in any one of the above (1) to (5).
- the platelet protective agent for transfusion described in any one of the above (1) to (5) is contained in a platelet preparation for transfusion in a pack. Platelets for transfusion.
- the present invention will be described in detail.
- A is amino acid, amino acid derivative, vitamin, vitamin derivative, vitamin-like substance, derivative of vitamin-like substance, base of nucleic acid And a compound selected from the group consisting of nucleic acid base derivatives and hydantoinacetic acid.
- amino acid refers to a molecule having an amino group and a carboxy group in the molecule.
- amino acid or the amino acid derivative A preferred in the present invention are imino acid containing proline, and a peptide bond (in the molecule). Lactam), tributophan, and derivatives of these amino acids.
- the peptide of the present invention or the like When the peptide of the present invention or the like is used as an active ingredient of a platelet aggregation inhibitor or a blood coagulation inhibitor for extracorporeal circulation, the presence of proline at the site A is determined by the formula (I).
- the peptide and the like (hereinafter referred to as the peptide of the present invention, etc.) are preferable because they can remarkably amplify the platelet aggregation inhibitory activity and the blood coagulation inhibitory activity.
- proline derivative examples include those having another hetero atom in the ring structure and those having a functional group added to the ring. Specific examples thereof include thioproline, quinproline in the mouth, hydrin in the mouth, oxothiazolidincarboxylic acid, N_methylproline, and N-acetylproline.
- the imino acids preferably include N-alkylglycines and cyclic imino acids represented by the general formula (II) having different ring sizes (R and are hydrogen atoms).
- the alkyl moiety of N-alkylglycine is preferably a lower alkyl chain. Specific examples include N-methylglycine (sarcosine), N-ethylglycine, N-ethylglycine, N-propylglycine, N-isopropylglycine and the like.
- R 1 and R 2 are an alkyl group or a C 0 -alkyl group, and each is preferably a lower alkyl group or a lower C 0 -alkyl group.
- p is an integer of 0 to 5 are preferable in that they have appropriate basicity and have low steric hindrance.
- the size of the ring structure of the cyclic imino acid can be selected from m in the range of 2 to 8.However, it is necessary to reduce the steric hindrance of the ring itself. Is preferably 2-5.
- the position of the carboxylic acid in the general formula (II) does not directly affect the platelet aggregation inhibitory activity of the present invention but the orientation of the basic amino group, imino group. With consideration, the position 2 adjacent to the imino group can be raised, preferably. Also, an alkyl chain may be introduced between the ring and the carbonyl.
- N which represents the number of the alkyl chains, can be selected from the range of 0 to 6, but is preferably 0 to 2 in consideration of the necessity of keeping an appropriate distance from the carbonyl to the imino group.
- preferred specific examples of the cyclic imino acids include, for example, L-2-azetidine carboxylic acid, 0-, m-, and p-piperidine carboxylic acids, and pyrrolidine-13. —Carboxylic acid, pyrrolidine-12-acetic acid and the like.
- the platelet aggregation inhibitor and blood coagulation of the present invention containing the peptide of the present invention as an active ingredient, depending on whether A is a compound which is susceptible to degradation by enzymes in blood or a compound which is less susceptible to the degradation.
- the duration of action of the inhibitor can be ensured according to the purpose of use.
- A a compound which is easily decomposed by enzymes in blood.
- the compound include azetidine carboxylic acid of a four-membered ring compound and proline of a five-membered ring compound.
- the action time in vivo can be extended by making A a compound that is hardly decomposed.
- the compound include a 6-membered ring compound such as L-pipecolic acid.
- the imino group may have a structure (lactam) in which the imino group is changed to an imido group.
- Such a structure also provides a peptide compound having a moderate stability to a degrading enzyme and a high activity.
- Preferable examples include pyroglutamic acid 2- 2-azetidinone 14-carboxylic acid.
- the platelet aggregation inhibitory activity of the peptide of the present invention is also improved. This is thought to be due to the hydrophobic action of the indole ring in the tributane.
- R 2 is a hydrogen atom or an alkyl group, preferably a hydrogen atom or a branched alkyl group
- R 3 is a hydrogen atom or a 0-alkyl group; preferred and rather is hydrogen atom or a lower O-alkyl group
- R ⁇ is a hydrogen atom or an alkyl group, good or to rather a hydrogen atom or a lower alkyl group
- R 5 is a hydrogen atom, ⁇ Mi Roh group or Huh Mi Bruno acyl Group.
- the amino group include, for example, an amino acetyl group and a diaminopropyl group.
- A is hydantoinacetic acid
- the activity is also improved.
- the type of vitamin used as A is not particularly limited.
- nicotinic acid, P-pantothenic acid, piotin, pterylglutamic acid and the like can be mentioned.
- a vitamin-like substance is a substance whose physiological action is close to that of vitamin, but in general, especially in humans and mammals, it is not necessary to take it as a nutrient from outside.
- Self in vivo A group of compounds that can be synthesized. Specific examples include orotic acid and lipoic acid.
- Both vitamins and vitamin-like agents are characterized in that they are covalently bonded to the peptide skeleton, but they need to have a corresponding functional group in order to bind to the peptide skeleton.
- the functional group include a carboxyl group.
- a vitamin or the like which does not have a corresponding functional group may be any as long as the corresponding functional group can be derived by a simple treatment.
- An example thereof is a case in which a carboxylic acid is induced by hydrolysis of an amide in a nicotinic acid amide molecule.
- bonds to the peptide skeleton are easily degraded by enzymes present in the living body, and as a result, useful vitamins or vitamin-like substances or intermediate metabolites or toxic substances useful for living organisms.
- Any compound that can be replaced by a compound having almost no compound may be used. Examples thereof include an amide bond and an ester bond.
- nucleic acid base or the derivative thereof according to the present invention may be In general, it refers to the base components constituting nucleotides and their derivatives, but they have the same structural characteristics as those described for the vitamin-bitamine-like substance or derivatives thereof. is there.
- Preferable examples include pyrimidine derivatives and the like, for example, 5-carboxymethylperacyl, 5-carboxythioperacyl and the like.
- the acid is B, for example, serine, glycine, parin, alanine, threonine or 3-alanine. Particularly preferred is serine because of its high activity.
- C refers to an amino acid having a hydrophobic functional group as a hydrophobic domain that binds to the receptor. Preference is given to tryptophan or phenylalanine.
- D is a hydroxyl group or an amino group.
- D is a hydroxyl group
- the platelet aggregation inhibitory activity, blood coagulation inhibitory activity, and cell adhesion inhibitory activity tend to be higher than when the amino group is D.
- the action time is longer for amino groups, and can be selected according to the purpose.
- the peptides of the present invention are used as an active ingredient of a blood coagulation inhibitor for extracorporeal circulation, it is advantageous in that the peptides of the present invention have different half-lives. Become.
- the half-life of the peptide of the present invention in plasma ranges from about 10 minutes to longer than 4 hours. However, many of them have the property of being rapidly degraded in the body, and most of the peptides of the present invention and the like. In vivo, the half-life of the body is too short to be identifiable (within about 2 minutes) to within 10 minutes. Therefore, when these peptides and the like are applied to extracorporeal circulation, they are stable in the blood circulating in the device, but when they enter the body, they quickly turn into compounds that are beneficial to the living body by various degrading enzymes. It has the advantage of being degraded and losing blood clotting activity.
- the serious side effect of increased bleeding tendency due to anti-thrombotic drugs during extracorporeal circulation which has been a problem in the past, has been overcome by replacing heparin with the peptide of the present invention. It is possible to That is, the peptides of the present invention are very useful as an active ingredient of a blood coagulation inhibitor for extracorporeal circulation.
- the preferred action time of the blood coagulation inhibitor varies depending on the circulatory apparatus and the purpose of use. It can be used properly depending on the length of circulation time and the purpose of use.
- Trp or W Tribute fan
- Fmoc 9-fluorenylmethoxycarbonyl
- the peptide of the present invention and its analogous compound can be easily synthesized by a simple operation using a commercially available amino acid. That is, methods commonly used in peptide chemistry, for example, “The Peptides”, Vol. 1 [Schroder and Luhke, Acad emic Press, New York, USA (1966)], “Peptides” Basics and Experiments on Synthesis ”[Nobuo Izumiya et al., Maruzen (1985)], etc., and can be manufactured by either the liquid phase method or the solid phase method. it can. Furthermore, any of the column method and the batch method can be used.
- Condensation methods for forming peptide bonds include the azide method, the acid chloride method, the acid anhydride method, the sulfoimide method, and the carbodiimide method. Additive method, Activated ester method, Carbonyl midazo method
- Examples thereof include a redox method, an enzymatic method, and a method using the adword reagent K.
- the acid anhydride method, the carbodiimide method, and the active ester method are the main methods.
- the peptide chain is bound to a support such as a resin insoluble in an organic solvent using a C-terminal amino acid.
- a resin in which a functional group is introduced for the purpose of binding amino acid to the resin a spacer in which a spacer is inserted between the resin and the functional group, and a resin that can be cut at various locations depending on conditions.
- Resins into which chains called dollars (handles) are introduced may be used according to the purpose.
- Such resins include, for example, halomethyl resins such as chloromethyl resin, oxymethyl resin, 4- (oxymethyl) phenylacetamide amide methyl resin, and 4— (oxymethyl) phenyloxymethyl resin. Examples include chill resin and resin for C-terminal amide.
- Protecting groups used in the protective means include protecting groups commonly used in the field of organic chemistry, for example, “Protective Groups in Organic Synthesis” [Greened, John Wiley & Sons, Inc. (1981)] and the like.
- Examples of a hydroxyl-protecting group in an amino acid residue containing a hydroxyl group such as a serine residue include tributyl, benzyl, trimethylsilyl, and tetrahydrovinylyl groups. And so on.
- carboxyl-protecting group examples include commonly known protective groups such as various methyl esters, ethyl esters, benzene esters, p-nitrobenzene esters, t-butyl esters, and cyclohexyl esters. Groups can be mentioned.
- amino-protecting group examples include a benzyloxycarbonyl group, a t-butoxycarbonyl group, an isobornyloxycarbonyl group, and a 9-fluoroenylmethoxycarbonyl group. be able to.
- Examples of the protecting group for the guanidino group in the arginine residue include a nitro group, a tosyl group, a mesitylenesulfonyl group, a 4-methoxy-1,2,3,6—trimethylbenzenesulfonyl group , 2,2,5,7,8-Pentamethylchroman-6-sulfonyl group.
- activated carboxyl groups include, for example, an acid anhydride corresponding to the carboxylic group; azide; pentafluorophenol, 2,4-dinitrophenol, and cyanomethyl.
- Alcohol p-Nitrophenol, N-Hydroxysuccinic acid imid, N-Hydroxy-1-5-Nonolebornene-2,3-dicarboxymide, N-Hydroxyphthalimid, 1-Hydroxybenzotriazole, etc. And the like.
- Examples of the activated amino group include a phosphoric acid amide or the like corresponding to the amino group.
- the condensation reaction at the time of peptide synthesis is usually performed in a solvent.
- the solvent include c-form form, dichloromethane, ethyl acetate, N, N-dimethylformamide, dimethylsulfoxide, pyridine, dioxane, tetrahydrofuran, and N_metholane. Tilpyrrolidone, water, methanol, etc., or a mixture thereof can be mentioned.
- the reaction temperature of the condensation reaction can be in the range of ⁇ 30 ° C. to 50 ° C. as in a normal case.
- the type of the elimination reaction of the protective group in the peptide production process of the present invention depends on the type of the protective group to be used as long as the protective group can be eliminated without affecting the peptide bond. You can select one according to your needs. For example, acid treatment with hydrogen chloride, hydrogen bromide, hydrogen fluoride, methanesulfonic acid, trifluoromethansulfonic acid, trifluoracetic acid, or a mixture thereof, sodium hydroxide, water Alkali treatment with calcium oxide, hydrazine, getylamine, pyridin, etc .; sodium treatment in liquid ammonia or reduction with palladium carbon; and trimethylsilyl chloride Examples include silylation treatment such as reflux and trimethylsilyl bromide.
- a cation scavenger such as anisol, phenol, cresol, thioanisole or ethanedithiol is a deprotection group. Preferred in that the reaction is performed efficiently.
- the method for cleaving the peptide of the present invention synthesized by the solid phase method from the solid phase generally follows a known method.
- the above-mentioned treatment with an acid or a silylating agent can be mentioned as the cutting method.
- the peptide of the present invention thus produced, After the end of the series of reactions, known separation and purification means can be used.
- the peptide of the present invention can be obtained in a more pure form by extraction, distribution, reprecipitation, recrystallization, column chromatography, and the like.
- the peptide of the present invention can be obtained in the form of a salt depending on the reaction conditions in the production process.
- the salts include inorganic acid salts such as hydrochloric acid, sulfuric acid, nitric acid, and phosphoric acid; formic acid, acetic acid, propionic acid, glycolic acid, succinic acid, lingoic acid, tartaric acid, citric acid, and triflic acid.
- Organic acids such as chloroacetic acid; alkali metal salts such as sodium and potassium; alkaline earth metal salts such as calcium salts; ammonium, ethanolamine, triethylamine, diamine
- Organic amines such as cyclohexylamine can be exemplified.
- the peptide of the present invention obtained above is used as an active ingredient of a platelet aggregation inhibitor, a cell adhesion inhibitor, a cancer metastasis inhibitor, or a platelet preparation protector for blood transfusion (hereinafter referred to as a platelet aggregation inhibitor).
- a platelet aggregation inhibitor a platelet aggregation inhibitor, a cell adhesion inhibitor, a cancer metastasis inhibitor, or a platelet preparation protector for blood transfusion (hereinafter referred to as a platelet aggregation inhibitor).
- a platelet aggregation inhibitor a platelet preparation protector for blood transfusion
- the ratio of the active ingredient to the carrier ingredient can be varied between 1 and 90% by weight.
- the dosage form and administration form of the preparation can be used in the form of granules, fine granules, powders, tablets, capsules, pills or liquids. In addition, it can be administered orally as it is, and it can be administered as an injection intravenously, intramuscularly, or subcutaneously.
- the peptide of the present invention or the like can be prepared as a powder for injection before use.
- an organic or inorganic carrier or diluent suitable for oral, enteral or parenteral administration, and further used for solid or liquid medicine is used. Can be used.
- a stabilizer, a wetting agent, or an emulsifier can be added, and a salt can be used as an osmotic pressure adjusting agent or a pH adjusting agent, as an adjuvant.
- the platelet aggregation inhibitor of the present invention may be used in the treatment of various diseases.
- An inhibitory component and the like can be contained.
- the active ingredient When it is in the form of granules, fine granules, powders, tablets or capsules, it is preferable to contain 5-80% by weight of the active ingredient. In the case of a liquid, it is preferable to contain the active ingredient in a ratio of 1 to 30% by weight. Further, among parenteral administration preparations, when used as an injection, it is preferable to contain the active ingredient in a ratio of 1 to 10% by weight.
- an adult In the case of oral administration, it is preferable for an adult to take 500 to 1000 mg of the above active ingredient daily for an adult. However, the dosage can be adjusted as appropriate depending on the age, symptoms, etc. of the patient.
- the platelet aggregation inhibitor of the present invention described above can be administered once a day, but it can also be administered at appropriate intervals in two or three divided doses. Wear.
- the peptides of the present invention and the like include those characterized by high degradability in the body, and it is necessary to maintain high blood concentrations of these compounds for several hours to several days continuously. May be continuously infused by drip or other means. In such a case, the injection amount is about 50 to 500 mg / Kg per hour, but it is possible to further reduce the amount when combined with other drugs.
- the peptide of the present invention or the like when used for extracorporeal circulation, it can be used in the form of the above-mentioned injection or infusion.
- the place of administration and the dose vary depending on the extracorporeal circulation system and the duration of the system.For example, it is possible to continuously inject 1 to 100 mg / kg per hour from the entrance to the extracorporeal circulation system. it can.
- the dosage, whether administered alone or in combination with other drugs, is less effective in the extracorporeal circulatory system than in the body, where there are large amounts of degrading enzymes.
- heparin which has been conventionally used as an anticoagulant for extracorporeal circulation, in combination with the peptide of the present invention, etc.
- platelet aggregation and coagulation related to blood coagulation It is thought that the pathway can be suppressed and blood coagulation can be more completely suppressed.
- a synergistic effect of the two can be expected, so that the amount of heparin used can be reduced because of the above-mentioned side effects.
- the platelet protective agent for blood transfusion of the present invention is contained in the platelet preparation for blood transfusion in the package.
- the form is not particularly limited. In other words, all forms that can be used for the platelet product pack for blood transfusion usually used in clinical practice can be adopted. Specifically, it is possible to take the form of a bag, a bottle, or the like. Moreover, those materials are not particularly limited.
- the platelet protective agent for transfusion of the present invention when converted to the amount of the peptide of the present invention, is 1 M to 1 mM as a final concentration, preferably 10 M to 50 M with respect to the platelet component amount. Can be added.
- other components which are usually added to the platelet preparation package for blood transfusion can also be added together with the platelet protective agent for blood transfusion of the present invention.
- FIG. 1 shows the stability of synthetic peptides in human plasma (1).
- Figure 2 shows the stability of synthetic peptides in human plasma. 2)
- Fig. 3 shows the stability of the synthetic peptide in the mouse body
- Fig. 4 shows the platelet aggregation inhibitory activity
- Fig. 5 is a schematic diagram of an artificial dialysis model dialysis circuit using a beagle dog.
- Fig. 6 shows the effect of blood coagulation in an artificial dialysis model using beagle dogs.
- Fig. 7 shows the change in platelet count with storage, and
- Fig. 8 shows the platelet aggregation.
- FIG. 9 is a graph showing the time-dependent changes associated with the preservation of the ability, and FIG.
- FIG. 9 is a graph showing the effect of the peptide and the like of Example 9 on the adhesion of various extracellular matrix proteins to HeLa cells.
- FIG. 0 is a diagram in which the effect of the peptide and the like of Example 9 on the adhesion of various extracellular matrix proteins to B16F10 melanoma cells was examined.
- Trp-Arg (Mtr) -Gly-Asp (0Bu 1 ) -Trp-resin Formula (3) was obtained.
- the resulting protected peptide resin was treated with 1M trimethylsilylbutane and 1M thioanisole for 1 hour in trifluoroacetic acid at 0 ° C in the presence of m-cresol and ethanedithiol. I got it. After distilling off the trimethylsilyl bromide in a nitrogen stream, the resin was removed by filtration, and getyl ether was added to the filtrate under ice-cooling to obtain a peptide cut from the resin as a powder. Then, the powder was washed with getyl ether.
- the washed material was desalted by gel filtration chromatography using Sephadex G-10 (manufactured by Pharmacia) as a support, and the salt was freeze-dried to obtain a crude peptide.
- This crude peptide was subjected to high performance liquid chromatography (HPLC) [column: 0DS 5C, 8 ( (PL bondasphere, ⁇ 20 x 150mm).
- Trp-Ser-Arg-Gly-Asp-Trp-OH 100 mg of the formula (1) was obtained.
- Tributane cannot be detected by this detection method because it is degraded during acid hydrolysis.
- the amino acid used as an external standard for quantification is a standard amino acid, it is not possible to detect even amino acids that are not contained in the standard amino acid.
- Trp- Ser (tBu) -Arg (Mtr) -Gly-Asp (OBu ') - to give Trp-C Suea Mi de resin equation (8).
- the obtained protective peptide resin was obtained in the same manner as in Example 1 to obtain 20 mg of the title peptide.
- Trp- ⁇ Ala-Arg-Gly-Asp-Trp-OH Synthesis of Formula (17): In the same manner as in Example 1, 100 mg of the peptide shown in the title was synthesized.
- the obtained protected peptide resin was treated for 1 hour in trifluoracetic acid at 0 ° C. in the presence of m-cresol, ethanedithiol and thiodisole. After distilling off trifluoroacetic acid with an evaporator, the resin was filtered off, and getyl ether was added to the filtrate under ice-cooling to obtain a peptide cut from the resin as a powder. Then, the powder was washed with getyl ether. The washed product was desalted by gel chromatography using SEF ADEX G-10 (manufactured by Pharmacia) as a support, and lyophilized to obtain a crude peptide.
- SEF ADEX G-10 manufactured by Pharmacia
- HPLC high performance liquid chromatography
- Subjects were healthy men who had not taken any medication for at least two weeks. Blood was collected using a No. 19 injection needle and a plastic syringe pre-filled with 1/10 volume of 3.8% sodium citrate solution, and blood was collected from the vein of the lower stomach on an empty stomach. Was. Immediately after blood collection, the syringe was gently stirred to mix both solutions. The blood was centrifuged at room temperature for 15 minutes (llOOrpm, 250g), the rotation was stopped without applying a brake, and the supernatant was collected with a Komagome pipet and stored at room temperature as platelet-rich plasma (PRP). .
- PRP platelet-rich plasma
- the remaining blood after centrifugation was further centrifuged at room temperature for 15 minutes (3500 rpm, 1500 g), and the supernatant after stopping without applying a brake was taken as platelet-poor plasma (PPP). After PRP adjustment, the number of platelets was counted, and the following experiments were performed only on platelets having a platelet count of 2 ⁇ 10 8 Zml or more.
- Platelet aggregation was measured from the change in the light transmittance of PRP using an 8-channel platelet aggregometer (Hemat racer, Nikoh Bioscience, Tokyo, Japan).
- 200 / zl of PPP and PRP were placed in a glass scab, and after incubating at 37 ° C, the transmittance was measured.
- the transmittance of PPP was set to 100%, and the transmittance of PRP was set to 0%.
- the experiment first confirmed that aggregation occurred using collagen and ADP, and the maximum aggregation rate of collagen was 70%. Only those with more than% were used in the experiments.
- Samples were dissolved in physiological saline so that 2.2 X 10- 2 M, was used to adjust the 2-fold dilution series based on Re this experiment. Samples that were insoluble in saline were dissolved in saline containing 10% DMS ⁇ (Dimethyl sulfoxide).
- Aggregation inhibition rate (1 ——] X inn ⁇ ⁇ ? ⁇ ( ⁇ , maximum aggregation rate when only raw food is added J ⁇ tool ⁇ u) Plot a graph that plots the aggregation inhibition rate against the sample concentration, From this figure, the concentration that inhibits aggregation by 50% (IC 5 ) was calculated.Table 2 shows the IC 5 of each sample.
- the RGD sequence compared to RGDS, which is the amino acid sequence present in the fibrinogen molecule (Table 2, Comparative Example 1), the RGD sequence has a hydrophobic group such as Trp at both ends of the RGD sequence. It was found that introduction of the molecule significantly increased the activity of inhibiting platelet aggregation.
- RGDS-0H purchased from Peptide Research Institute (Minoh City)
- the peptide of the present invention was used. It has been found that the platelet aggregation inhibiting ability of the is significantly improved.
- the samples were analyzed on reversed phase HPLC.
- the peak area of each synthetic peptide was calculated, and the stability in blood or plasma was evaluated using the change in the peak area as an index.
- FIG. 1 to 3 show the general formula
- FIG. 3 is a diagram comparing the stability of a vitamin-like substance or a derivative thereof, a nucleic acid base or a derivative thereof) in plasma. Table 3 summarizes their half-lives.o
- Example 9 240 minutes or more
- Example 18 1 2 0 minutes or more
- Example 11 240 minutes or more In the case where A is a natural amino acid and is a triptophan having a primary amino group, the degradation was very rapid, and its half-life was 11.5 minutes. In contrast, A is a natural amino acid, but proline with a secondary amino group (imino group) is more stable in plasma than proline. The half-life was 74.5 minutes. On the other hand, even in the case of a proline derivative having the same secondary amino group (imino group), the stability in plasma greatly differs depending on the number of carbon atoms in the heterocycle or the presence of the side chain.
- a compound in which azetidine carboxylic acid having a hetero 4-membered ring structure is introduced into proline having a hetero 5-membered ring structure has a half-life of 49 minutes and is more degradable in plasma than proline. High.
- the compound introduced with pipecolic acid having a hetero 6-membered ring structure had a plasma half-life of 120 minutes or more and was very stable.
- Compounds containing amino acids, nucleic acids, and vitamin derivatives having a carboxy group in the heterocyclic structure, such as phosphoric acid were all extremely stable in plasma and had a half-life of 240 minutes or more.
- various compounds having different stability in plasma can be obtained by changing the compounds introduced at both ends of the synthetic peptide into various ones.
- Compounds having such different stability in blood may cause differences in extracorporeal circulation systems such as artificial dialysis and cardiopulmonary bypass, for example, when considering application to blood coagulation inhibitors for extracorporeal circulation.
- mice of the MCH (ICR) strain (oss, body weight 28 to 32 g) were used.
- a solution prepared by dissolving each synthetic peptide in sterile physiological saline at a concentration of 10 mM to 20 mM was used as a sample solution, and intravenously administered at 0.15 ml per mouse via the tail vein (1-2 mg). / mouse).
- blood was collected from the abdominal vein using a syringe that had been parinized. After blood collection, the blood was immediately centrifuged at 4 ° C (1500g [3 minutes]), and the supernatant was stored frozen at -20 ° C. Each supernatant was analyzed by reversed-phase HPLC as in the case of the stability evaluation in plasma described above.
- A-Ser-Arg-G1y—Asp—Trp—OH peptide and its analogous compounds (where A is amino acid or amino acid derivative, vitamin 'Shows the results of examining the in vivo stability of vitamin-like substances or their derivatives, nucleic acid bases or their derivatives).
- the peptide compound represented by the above general formula has a characteristic of being highly degradable in a living body as a whole. This is an extremely significant point when considering the application of the peptides of the present invention to blood coagulation inhibitors for extracorporeal circulation. That is, these compounds maintain a half-life of several minutes to several hours during circulation outside the body and inhibit platelet aggregation, whereas they are degraded within a few minutes when blood returns to the body. In other words, despite its antithrombotic effect in the extracorporeal circulation system, it is a compound having extremely excellent characteristics of not having an effect of extending the hemostasis time in the body.
- the peptide of the present invention and the like have an action of inhibiting platelet aggregation
- blood is collected over time after administering the compound intravenously to the body, and the platelet aggregation inhibitory activity of blood at each time point is measured.
- the platelet aggregation inhibitory activity of blood at each time point is measured.
- Example 9 Male beagle dogs weighing about l OKg were used.
- the peptide of the present invention etc. obtained in Example 9 was dissolved in physiological saline, The drug was administered using a syringe equipped with a 21G needle.
- spontaneous blood flow was performed at 5, 15, 30, 60, 12, 180, and 360 minutes from the beagle dog's caudal vein under anesthesia with an extension tube attached to a 20-G injection needle under anesthesia. Blood was collected at a ratio of 1% of 3.8% sodium citrate and 9 volumes of whole blood.
- the collected blood was centrifuged at 100 rpm for 10 minutes to collect platelet-rich plasma (PRP), and then centrifuged at 3000 rpm for 15 minutes to collect platelet-poor plasma (PPP).
- PRP platelet-rich plasma
- PPPP platelet-poor plasma
- the platelet aggregation was measured using the above PRP, and the platelet aggregation by collagen and ADP was measured with an aggregometer.
- the final concentrations of ADP and collagen were adjusted to 80 to 10,000 / M at a final concentration of 80, and to 7.5 to 10.0 ⁇ g / ml at a final concentration of collagen.
- a of FIG. 4 shows the time course of the recovery of platelet aggregation activity when the peptide of the present invention of Example 9 was administered at a dose of 5 mg / Kg body weight of a beagle dog.
- Agglutinin is 10 / z g / mi of collagen.
- Platelet aggregation was completely inhibited 5 minutes after administration, but platelet aggregation activity was recovered over time, and completely recovered to the level before administration 45 to 60 minutes later.
- Exactly the same results were obtained when the collagen concentration was changed or when ADP was used as agglutinin. This indicates that the peptide or the like was decomposed in the body or excreted from the kidney outside the body within 60 minutes at most, and the concentration in the blood decreased.
- Heparin which is currently used as an anticoagulant, was used as a comparative example for the peptide of the present invention.
- heparin has no effect on platelets, and the method described in (1) above must be used to measure stability in the body. Can not. Therefore, the stability in the body was estimated by measuring changes in the anticoagulant action, which is the main action of heparin, over time.
- heparin dissolved in physiological saline was intravenously administered in the same manner as in (1) above, blood was collected over time, and PPP was prepared.
- the measurement was performed by measuring prothrombin time and activated part thromboplastin time, which are indicators of blood coagulation activity in a test tube. Each measurement was performed at 37 ° C.
- FIG. 4B shows the time course of the prolongation of prothrombin time when beagle dogs were administered 200 U of heparin per kg of body weight intravenously.
- the prothrombin time was increased to 1.5 to 2.0 times that before administration, but as time passed, the prothrombin time gradually returned to the level before administration. It is. However, it took 2-3 hours to completely return to pre-dose levels. Similar results were obtained for the activated part thromboplastin time. This indicates that heparin is slowly decomposed or eliminated from the body, and its effects remain for several hours.
- Example 9 the present invention prepared in Example 9 was compared with heparin conventionally used as an anticoagulant during extracorporeal circulation. Cides etc. lost their effects very quickly. This compensates for the disadvantage of the current anticoagulant heparin, which has a problem that the bleeding tendency continues for several hours even after the use of the extracorporeal circulation system such as dialysis, etc. This indicates that the peptide and the like can be sufficiently expected as a new blood coagulation inhibitor.
- the measurement parameters during the experiment are: 1) pressure at the upstream of the dialysis membrane (perfusion pressure), 2) platelet adhesion, and 3) coagulation time of whole blood.
- perfusion pressure pressure at the upstream of the dialysis membrane
- platelet adhesion a pressure gauge incorporated in the dialysis circuit upstream of the dialysis membrane.
- the dialysis membrane site has a lot of contact with foreign matter and the blood flow passes through a narrow space, so the blood is the most blood in the dialysis circuit This is the area where coagulation is likely to occur. If blood coagulation occurs here, the permeable membrane will be clogged and the blood pressure in the upstream will rise. That is, the change in the perfusion pressure in this part is an indicator of the degree of blood coagulation in the dialysis circuit.
- FIG. 6 shows the results of an experiment in which the inhibitory effect of the compound of Example 9 on the perfusion pressure was measured.
- the horizontal axis shows the time after the start of drug administration, and the vertical axis shows the perfusion pressure.
- the perfusion pressure is 0 mm immediately after mounting the circuit, and this value increases when blood coagulation occurs.
- a sharp increase in perfusion pressure was observed after 10 minutes, and the measured value exceeded 500 minHg in 25 minutes, making measurement impossible.
- blood coagulation occurs rapidly in the dialysis circuit, especially at the dialysis membrane site.
- the platelet aggregation ability and the whole blood clotting time also showed changes almost parallel to the pattern of rising perfusion pressure (not shown). That is, in the control group, the platelet aggregation ability increased and the whole blood clotting time decreased with time. This indicates that platelets were activated and that the blood coagulation system was also activated, and blood coagulation was very likely to occur.
- the peptide of the present invention and the like in Example 9 it was found that during continuous infusion, blood adhesive ability was reduced to almost 0%, and the platelet activity was completely suppressed. . Also, the whole blood coagulation time was significantly prolonged during this period.
- the peptides of the present invention and the like almost completely suppressed blood coagulation in the extracorporeal circulation system.
- the peptide of the present invention can be sufficiently used as an alternative to the currently used heparin.
- heparin completely suppresses blood coagulation in the extracorporeal circulation system, but also has a slow rate of excretion from the body, and suppresses blood coagulation and promotes bleeding tendency for several hours after the system is removed.
- the peptide of the present invention as already shown in FIG. 4, is very good in degradability in the body unlike heparin, and relatively promptly stops blood coagulation before drug administration, when injection is stopped. It has the advantage of restoring normal levels.
- the peptide of the present invention since the peptide of the present invention has extremely low toxicity, it is extremely useful as a completely new blood coagulation inhibitor that compensates for the above-mentioned disadvantage of heparin. And promising.
- the peptide of the present invention is dissolved in physiological saline or citric acid solution and continuously infused at about 3 mg per hour by drip or the like from near the entrance of the extracorporeal circulation system.
- a blood coagulation effect can be expected. In actual application to humans, it may be possible to further reduce the dose.
- the platelet fraction is placed in a polypropylene test tube, covered with a membrane filter to ensure air permeability, placed on a shaker, and shaken under the conditions of 20 cm amplitude and 2 Hz shaking. Store at room temperature Was. All of the above operations were performed aseptically to prevent bacterial growth during storage.
- FIG. 7 is a diagram showing a change in platelet count with storage.
- the platelet count decreased almost in proportion to the storage time.
- the compound of Example 9 formula (14)
- This protective effect was dependent on the concentration of the added peptide, and when 400 fi was added, there was almost no decrease in the number of platelets during storage for 72 hours.
- FIG. 8 shows the time-dependent change of the platelet aggregation ability with storage.
- the aggregation activity of platelets was reduced to 25% after storage for 72 hours, but in the group to which the compound of Example 9 (formula (14)) was added, the decrease in aggregation activity was significantly suppressed. .
- the inhibitory effect on the reduction of the aggregation activity depends on the concentration of the added peptide compound.When 400 is added, 60-70% of the platelet aggregation activity remains even after storage for 72 hours. Was.
- peptide compounds such as RGDS and RGDF, which are conventionally known, are degraded within several hours by the action of enzymes in plasma even when added to the platelet fraction. It is clear that these compounds cannot be used for prolonged platelet storage. Conversely, compounds that are very stable and are not easily degraded when they enter the body will inhibit the function of all platelets in the body after blood transfusion, reducing the effectiveness of blood transfusion.
- the peptide of the present invention is considered to be a very excellent platelet protective agent in that it has high stability in platelet fractionation, high degradability in the body, and low toxicity.
- an effect of adding a buffering effect and suppressing a change in pH accompanying storage of the platelet fraction can be expected.
- an effect can be expected not only by administering the peptide of the present invention alone but also by using it in combination with a platelet aggregation inhibitor having a different action mechanism, such as aspirin.
- Collagen is diluted with saline adjusted to pH 3.0 with hydrochloric acid, The diluted solution adjusted to 100 ⁇ g / ml was used for adsorption. On the other hand, the remaining three proteins were diluted with PBS at pH 7.4, adjusted to 20 g / ml, and used. -:
- a type 1 collagen adsorption plate when preparing a type 1 collagen adsorption plate, add 0.4 ml of a diluted solution of type 1 collagen to a 24-well plastic plate, incubate at 37 ° C for 10 minutes, and plate the type 1 collagen adsorption plate. Was adsorbed. Furthermore, PBS containing 3% bovine serum albumin (manufactured by Sigma) was added to each well to prevent nonspecific cell adsorption, and the cells were treated at 37 ° C for 1 to 2 hours. Finally, the plate was washed three times with PBS to obtain a type 1 collagen adsorption plate. For other extracellular matrix proteins, adsorption plates were prepared in exactly the same manner.
- the peptides of the present invention were diluted in a serum-free EMEM medium (manufactured by Nissi) to prepare a dilution series of 5 mM, 0.5 mM, 50 ⁇ . And 5 (iM.
- the adsorption plate was previously filled with 300 serum-free EMEM media, and then 100 n1 of each concentration of the peptide solution was added to each well. However, as a control group to which no peptide was added, only serum-free EMEM medium was added at 100 a 1.
- HeLa cells or B16F10 melanoma cells were added. suspension (5 X 10 6 cells / ml) were use mind was added in 0.1ml into each well. plastic Kupure preparative lightly rocking the horizontal, after stirring, C0 2 Lee Nkyubeta 1 hour Lee Nki Yupe preparative one among did.
- the concentration of methylene blue released from the cells was measured using a spectrophotometer by the absorbance of light at 600 nm.
- the number of cells attached to the extracellular matrix protein is proportional to the absorbance at 600 ⁇ , so that accurate measurement of the number of cells is possible.
- the peptides of the present invention suppress cell adhesion of various cancer cells to extracellular matrix proteins. This indicates that these peptides and the like can suppress the adhesion of cancer cells to the vascular basement membrane, and strongly suggests their potential as cancer metastasis inhibitors. .
- the compounds of the present invention which have extremely low toxicity, can withstand long-term administration, and have been used in the field of cancer metastasis inhibitors, which have never existed before and are new drugs. It can be expected enough.
- a novel peptide having a platelet aggregation inhibitory action and a blood coagulation inhibitory action, and platelet thrombosis and thrombosis containing the peptide as an active ingredient during and after thrombolysis treatment Provide platelet aggregation inhibitors that are effective against obstruction and can also prevent reocclusion and myocardial infarction, and blood coagulation inhibitors that can suppress blood clotting, which is the main cause of thrombus formation during extracorporeal circulation Is done.
- the present invention provides a platelet preparation protective agent for blood transfusion, a cell adhesion inhibitor, and a cancer metastasis inhibitor.
- a platelet preparation for blood transfusion characterized in that the above-mentioned protective agent for platelet preparation for blood transfusion is contained in the platelet preparation for blood transfusion.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Toxicology (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description




Claims
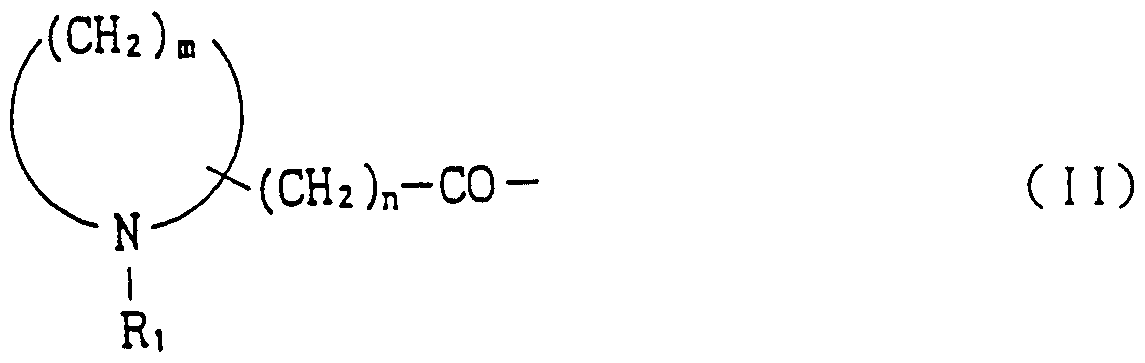

Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP93919619A EP0628571A4 (en) | 1992-09-07 | 1993-09-07 | NOVEL PEPTIDE AND ANTITHROMBOTIC AGENT, ANTICOAGULANT FOR EXTRACORPOREAL CIRCULATION, CELL FUSION INHIBITOR, CANCER METASTASIS INHIBITOR, PLAQUETATIVE PROTECTION AGENT FOR TRANSFUSION, AND PACKAGING CONTAINER. |
| KR1019940701533A KR950701645A (ko) | 1992-09-07 | 1993-09-07 | 신규펩티드와 그것을 사용한 혈소판응집억제제, 체외순환용혈액 응고억제제, 세포접착저지제, 암전이저지제, 수혈용 혈소판제제보호제 및 수혈용혈소판제제팩 |
| US08/232,261 US5498601A (en) | 1992-07-09 | 1993-09-07 | Platelet aggregation-inhibiting peptides |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP23862492 | 1992-09-07 | ||
| JP4/238624 | 1992-09-07 | ||
| JP5203962A JPH06192291A (ja) | 1992-09-07 | 1993-08-18 | 新規ペプチドとそれを用いた血小板凝集抑制剤及び血液凝固抑制剤 |
| JP5/203962 | 1993-08-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994005696A1 true WO1994005696A1 (en) | 1994-03-17 |
Family
ID=26514200
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1993/001262 WO1994005696A1 (en) | 1992-07-09 | 1993-09-07 | Novel peptide, and antithrombotic agent, anticoagulant for extracorporeal circulation, cell fusion inhibitor, cancer metastasis inhibitor, protective for platelet preparation for transfusion and pack of platelet preparation for transfusion |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US5498601A (ja) |
| EP (1) | EP0628571A4 (ja) |
| JP (1) | JPH06192291A (ja) |
| KR (1) | KR950701645A (ja) |
| CA (1) | CA2122912A1 (ja) |
| WO (1) | WO1994005696A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995009185A1 (en) * | 1993-09-30 | 1995-04-06 | Nippon Steel Corporation | Novel peptide, active as inhibitors of platelet aggregation |
| JP2014506319A (ja) * | 2010-12-02 | 2014-03-13 | ベクトン・ディキンソン・アンド・カンパニー | 血液安定剤を含む採血用デバイス |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6231834B1 (en) | 1995-06-07 | 2001-05-15 | Imarx Pharmaceutical Corp. | Methods for ultrasound imaging involving the use of a contrast agent and multiple images and processing of same |
| US6139819A (en) | 1995-06-07 | 2000-10-31 | Imarx Pharmaceutical Corp. | Targeted contrast agents for diagnostic and therapeutic use |
| US6521211B1 (en) | 1995-06-07 | 2003-02-18 | Bristol-Myers Squibb Medical Imaging, Inc. | Methods of imaging and treatment with targeted compositions |
| US5839443A (en) * | 1996-05-16 | 1998-11-24 | The Trustees Of Columbia University In The City Of New York | Method for inhibiting thrombosis in a patient whose blood is subjected to extracorporeal circulation |
| WO1998013527A2 (en) * | 1996-09-24 | 1998-04-02 | Rapigene, Inc. | Compositions and methods for enhancing hybridization specificity |
| US6361940B1 (en) | 1996-09-24 | 2002-03-26 | Qiagen Genomics, Inc. | Compositions and methods for enhancing hybridization and priming specificity |
| EP0952228A3 (en) * | 1996-09-24 | 1999-12-15 | Rapigene, Inc. | Compositions and methods for enhancing hybridization specificity |
| US6120751A (en) | 1997-03-21 | 2000-09-19 | Imarx Pharmaceutical Corp. | Charged lipids and uses for the same |
| US6548047B1 (en) | 1997-09-15 | 2003-04-15 | Bristol-Myers Squibb Medical Imaging, Inc. | Thermal preactivation of gaseous precursor filled compositions |
| US6123923A (en) | 1997-12-18 | 2000-09-26 | Imarx Pharmaceutical Corp. | Optoacoustic contrast agents and methods for their use |
| US20050250688A1 (en) * | 1999-04-01 | 2005-11-10 | The Trustees Of Columbia University In The City Of New York | Methods for treating an ischemic disorder and improving stroke outcome |
| US20050180945A1 (en) * | 2002-05-21 | 2005-08-18 | Chaikof Elliot L. | Multivalent polymers with chan-terminating binding groups |
| US7517954B2 (en) | 2003-03-28 | 2009-04-14 | Fuji Film Manufacturing Europe B.V. | RGD-enriched gelatine-like proteins with enhanced cell binding |
| EP2845601B1 (en) * | 2010-11-01 | 2018-04-25 | Industry-Academic Cooperation Foundation, Yonsei University | Composition for use for dissolving thrombi |
| CN103113456B (zh) * | 2013-03-05 | 2014-07-16 | 中国药科大学 | 具有抗血小板聚集活性的僵蚕多肽及其制备方法和应用 |
| WO2016201374A1 (en) * | 2015-06-11 | 2016-12-15 | University Of Florida Research Foundation, Incorporated | Sulfanide adenosine derivatives and uses thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63215696A (ja) * | 1986-12-15 | 1988-09-08 | アンスティテュ ナシオナル ドウ ラサントゥ エ ドゥ ラ ルシェルシェメディカル(イーエヌエスエーエールエム) | 新規ペプチドおよびこのペプチドを含む血栓治療剤 |
| JPH03118398A (ja) * | 1989-07-28 | 1991-05-20 | Merck & Co Inc | フイブリノーゲンレセプター拮抗剤 |
-
1993
- 1993-08-18 JP JP5203962A patent/JPH06192291A/ja active Pending
- 1993-09-07 EP EP93919619A patent/EP0628571A4/en not_active Withdrawn
- 1993-09-07 WO PCT/JP1993/001262 patent/WO1994005696A1/ja not_active Application Discontinuation
- 1993-09-07 US US08/232,261 patent/US5498601A/en not_active Expired - Fee Related
- 1993-09-07 CA CA002122912A patent/CA2122912A1/en not_active Abandoned
- 1993-09-07 KR KR1019940701533A patent/KR950701645A/ko not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63215696A (ja) * | 1986-12-15 | 1988-09-08 | アンスティテュ ナシオナル ドウ ラサントゥ エ ドゥ ラ ルシェルシェメディカル(イーエヌエスエーエールエム) | 新規ペプチドおよびこのペプチドを含む血栓治療剤 |
| JPH03118398A (ja) * | 1989-07-28 | 1991-05-20 | Merck & Co Inc | フイブリノーゲンレセプター拮抗剤 |
Non-Patent Citations (2)
| Title |
|---|
| FEBS Lett., Vol. 291, No. 1 (1991), MONIQUE AUMAILLEY et al.: "Arg-Gly-Asp Constrained within Cyclic Pentapeptides. Strong and Selective Inhibitors of Cell Adhesion to Vitronectin and Laminin Fragment P1", pp. 50-54. * |
| See also references of EP0628571A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995009185A1 (en) * | 1993-09-30 | 1995-04-06 | Nippon Steel Corporation | Novel peptide, active as inhibitors of platelet aggregation |
| JP2014506319A (ja) * | 2010-12-02 | 2014-03-13 | ベクトン・ディキンソン・アンド・カンパニー | 血液安定剤を含む採血用デバイス |
| US10299714B2 (en) | 2010-12-02 | 2019-05-28 | Becton, Dickinson And Company | Blood collection devices containing blood stabilization agent including variegin or analog thereof and/or a polysulfated disaccharide |
Also Published As
| Publication number | Publication date |
|---|---|
| JPH06192291A (ja) | 1994-07-12 |
| EP0628571A1 (en) | 1994-12-14 |
| KR950701645A (ko) | 1995-04-28 |
| CA2122912A1 (en) | 1994-03-17 |
| US5498601A (en) | 1996-03-12 |
| EP0628571A4 (en) | 1998-03-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1994005696A1 (en) | Novel peptide, and antithrombotic agent, anticoagulant for extracorporeal circulation, cell fusion inhibitor, cancer metastasis inhibitor, protective for platelet preparation for transfusion and pack of platelet preparation for transfusion | |
| KR100438244B1 (ko) | 사이클릭결합저해제 | |
| JPH01190699A (ja) | 血小板凝集インヒビターペプチド誘導体 | |
| RU2151776C1 (ru) | Циклопептиды, способ их получения | |
| MXPA96004100A (en) | Cyclic compounds, adhes inhibitors | |
| EP0665853B1 (en) | Multimeric polyvalent antithrombotic agents | |
| KR100438378B1 (ko) | 고리형부착억제제 | |
| JP2011523634A (ja) | ペプチド、ペプチド模倣物およびそれらの誘導体、それらの製造、ならびに治療および/または予防活性のある医薬組成物を調製するためのそれらの使用 | |
| EP1987063A1 (en) | Peptides and peptide derivatives, the production thereof as well as their use for preparing a therapeutically and/or preventively active pharmaceutical composition | |
| KR100186656B1 (ko) | 신규한 펩타이드, 및 전기 펩타이드를 이용하는 혈소판 응집저해제, 체외순환하는 혈액의 혈액응고 저해제, 세포점착저해제, 종양전이 저해제, 수혈용 혈소판 제제의 보호제, 수혈용 혈소판 제제 및 혈소판 제제 팩 | |
| US5338725A (en) | Anti-aggregatory agents for platelets | |
| WO2010034041A1 (en) | Peptides and peptidomimetic compounds, the manufacturing thereof as well as their use for preparing a therapeutically and/or preventively active pharmaceutical composition | |
| JP3581174B2 (ja) | 新規ペプチド並びにそれを用いた血小板凝集抑制剤、体外循環用血液凝固抑制剤及び輸血用血小板製剤保護剤 | |
| JP2011519957A (ja) | ペプチドおよびそれらの誘導体、それらの製造、ならびに治療および/または予防活性のある医薬組成物を調製するためのそれらの使用 | |
| US11643439B2 (en) | Multi-target compound with anticoagulation and antiplatelet activity, preparation method therefor, and use thereof | |
| WO1995001371A1 (fr) | Nouveau peptdie et agent anti-agregation plaquettaire le renfermant | |
| JP2011519958A (ja) | ペプチドおよびそれらの誘導体、それらの製造、ならびに治療および/または予防活性のある医薬組成物を調製するためのそれらの使用 | |
| JPH0797397A (ja) | 新規ペプチド及びそれを用いた血小板凝集抑制剤 | |
| RU2200166C2 (ru) | Производные циклопептидов в качестве ингибиторов адгезии | |
| JPH069687A (ja) | 新規ペプチドおよびそれを用いた血小板凝集阻害剤 | |
| JPH07242552A (ja) | 輸血用血小板保護剤 | |
| JPH07242563A (ja) | ガン転移抑制剤 | |
| JPH08217692A (ja) | 血管閉塞防止剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CA KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BE CH DE ES FR GB IT NL SE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2122912 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08232261 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993919619 Country of ref document: EP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWP | Wipo information: published in national office |
Ref document number: 1993919619 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993919619 Country of ref document: EP |