KR20190073426A - 저장 또는 전송된 생물정보학 데이터의 선택적 액세스를 위한 방법 및 시스템 - Google Patents
저장 또는 전송된 생물정보학 데이터의 선택적 액세스를 위한 방법 및 시스템 Download PDFInfo
- Publication number
- KR20190073426A KR20190073426A KR1020197013567A KR20197013567A KR20190073426A KR 20190073426 A KR20190073426 A KR 20190073426A KR 1020197013567 A KR1020197013567 A KR 1020197013567A KR 20197013567 A KR20197013567 A KR 20197013567A KR 20190073426 A KR20190073426 A KR 20190073426A
- Authority
- KR
- South Korea
- Prior art keywords
- data
- genomic
- type
- class
- genome
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims description 120
- 238000012163 sequencing technique Methods 0.000 claims description 65
- 229910052757 nitrogen Inorganic materials 0.000 claims description 20
- 229910052698 phosphorus Inorganic materials 0.000 claims description 18
- 238000012217 deletion Methods 0.000 claims description 17
- 230000037430 deletion Effects 0.000 claims description 13
- 238000003780 insertion Methods 0.000 claims description 13
- 230000037431 insertion Effects 0.000 claims description 13
- 230000007246 mechanism Effects 0.000 abstract description 36
- 230000005540 biological transmission Effects 0.000 abstract description 15
- 238000013507 mapping Methods 0.000 description 52
- 230000008569 process Effects 0.000 description 47
- 239000002773 nucleotide Substances 0.000 description 42
- 125000003729 nucleotide group Chemical group 0.000 description 42
- 238000012545 processing Methods 0.000 description 34
- 102100024441 Dihydropyrimidinase-related protein 5 Human genes 0.000 description 29
- 101001053479 Homo sapiens Dihydropyrimidinase-related protein 5 Proteins 0.000 description 29
- 238000007906 compression Methods 0.000 description 28
- 230000006835 compression Effects 0.000 description 28
- 238000004458 analytical method Methods 0.000 description 27
- 230000006870 function Effects 0.000 description 22
- 238000013459 approach Methods 0.000 description 21
- 238000006467 substitution reaction Methods 0.000 description 21
- 230000008901 benefit Effects 0.000 description 20
- 239000013598 vector Substances 0.000 description 19
- 239000012634 fragment Substances 0.000 description 14
- 238000002372 labelling Methods 0.000 description 11
- 230000009466 transformation Effects 0.000 description 10
- 108020004414 DNA Proteins 0.000 description 8
- 108090000623 proteins and genes Proteins 0.000 description 8
- 108091028043 Nucleic acid sequence Proteins 0.000 description 7
- 238000000605 extraction Methods 0.000 description 7
- 108020004707 nucleic acids Proteins 0.000 description 7
- 102000039446 nucleic acids Human genes 0.000 description 7
- 150000007523 nucleic acids Chemical class 0.000 description 7
- 239000012472 biological sample Substances 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 230000000295 complement effect Effects 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- 108010038083 amyloid fibril protein AS-SAM Proteins 0.000 description 5
- 230000008859 change Effects 0.000 description 4
- 238000013144 data compression Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 238000012165 high-throughput sequencing Methods 0.000 description 4
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 4
- 238000000844 transformation Methods 0.000 description 4
- 210000000349 chromosome Anatomy 0.000 description 3
- 238000004891 communication Methods 0.000 description 3
- 230000006837 decompression Effects 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 238000012268 genome sequencing Methods 0.000 description 3
- 230000010365 information processing Effects 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 238000002864 sequence alignment Methods 0.000 description 3
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical group O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 238000004220 aggregation Methods 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 238000007405 data analysis Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 238000010230 functional analysis Methods 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000012916 structural analysis Methods 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 101000911390 Homo sapiens Coagulation factor VIII Proteins 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 238000007792 addition Methods 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000013399 early diagnosis Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000011331 genomic analysis Methods 0.000 description 1
- 102000057593 human F8 Human genes 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 229940047431 recombinate Drugs 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000007723 transport mechanism Effects 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
Images






















































Classifications
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/10—Sequence alignment; Homology search
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F16/00—Information retrieval; Database structures therefor; File system structures therefor
- G06F16/20—Information retrieval; Database structures therefor; File system structures therefor of structured data, e.g. relational data
- G06F16/22—Indexing; Data structures therefor; Storage structures
- G06F16/2282—Tablespace storage structures; Management thereof
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F16/00—Information retrieval; Database structures therefor; File system structures therefor
- G06F16/20—Information retrieval; Database structures therefor; File system structures therefor of structured data, e.g. relational data
- G06F16/23—Updating
- G06F16/2365—Ensuring data consistency and integrity
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F16/00—Information retrieval; Database structures therefor; File system structures therefor
- G06F16/20—Information retrieval; Database structures therefor; File system structures therefor of structured data, e.g. relational data
- G06F16/28—Databases characterised by their database models, e.g. relational or object models
- G06F16/284—Relational databases
- G06F16/285—Clustering or classification
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F21/00—Security arrangements for protecting computers, components thereof, programs or data against unauthorised activity
- G06F21/60—Protecting data
- G06F21/602—Providing cryptographic facilities or services
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F21/00—Security arrangements for protecting computers, components thereof, programs or data against unauthorised activity
- G06F21/60—Protecting data
- G06F21/62—Protecting access to data via a platform, e.g. using keys or access control rules
- G06F21/6218—Protecting access to data via a platform, e.g. using keys or access control rules to a system of files or objects, e.g. local or distributed file system or database
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F21/00—Security arrangements for protecting computers, components thereof, programs or data against unauthorised activity
- G06F21/60—Protecting data
- G06F21/62—Protecting access to data via a platform, e.g. using keys or access control rules
- G06F21/6218—Protecting access to data via a platform, e.g. using keys or access control rules to a system of files or objects, e.g. local or distributed file system or database
- G06F21/6245—Protecting personal data, e.g. for financial or medical purposes
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F3/00—Input arrangements for transferring data to be processed into a form capable of being handled by the computer; Output arrangements for transferring data from processing unit to output unit, e.g. interface arrangements
- G06F3/01—Input arrangements or combined input and output arrangements for interaction between user and computer
- G06F3/048—Interaction techniques based on graphical user interfaces [GUI]
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F7/00—Methods or arrangements for processing data by operating upon the order or content of the data handled
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/10—Ploidy or copy number detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/20—Sequence assembly
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
- G16B40/10—Signal processing, e.g. from mass spectrometry [MS] or from PCR
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B45/00—ICT specially adapted for bioinformatics-related data visualisation, e.g. displaying of maps or networks
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B50/00—ICT programming tools or database systems specially adapted for bioinformatics
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B50/00—ICT programming tools or database systems specially adapted for bioinformatics
- G16B50/10—Ontologies; Annotations
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B50/00—ICT programming tools or database systems specially adapted for bioinformatics
- G16B50/30—Data warehousing; Computing architectures
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B50/00—ICT programming tools or database systems specially adapted for bioinformatics
- G16B50/40—Encryption of genetic data
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B50/00—ICT programming tools or database systems specially adapted for bioinformatics
- G16B50/50—Compression of genetic data
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B99/00—Subject matter not provided for in other groups of this subclass
-
- H—ELECTRICITY
- H03—ELECTRONIC CIRCUITRY
- H03M—CODING; DECODING; CODE CONVERSION IN GENERAL
- H03M7/00—Conversion of a code where information is represented by a given sequence or number of digits to a code where the same, similar or subset of information is represented by a different sequence or number of digits
- H03M7/30—Compression; Expansion; Suppression of unnecessary data, e.g. redundancy reduction
- H03M7/3084—Compression; Expansion; Suppression of unnecessary data, e.g. redundancy reduction using adaptive string matching, e.g. the Lempel-Ziv method
- H03M7/3086—Compression; Expansion; Suppression of unnecessary data, e.g. redundancy reduction using adaptive string matching, e.g. the Lempel-Ziv method employing a sliding window, e.g. LZ77
-
- H—ELECTRICITY
- H03—ELECTRONIC CIRCUITRY
- H03M—CODING; DECODING; CODE CONVERSION IN GENERAL
- H03M7/00—Conversion of a code where information is represented by a given sequence or number of digits to a code where the same, similar or subset of information is represented by a different sequence or number of digits
- H03M7/30—Compression; Expansion; Suppression of unnecessary data, e.g. redundancy reduction
- H03M7/70—Type of the data to be coded, other than image and sound
Landscapes
- Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Theoretical Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medical Informatics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Bioinformatics & Computational Biology (AREA)
- Biotechnology (AREA)
- Evolutionary Biology (AREA)
- Biophysics (AREA)
- Databases & Information Systems (AREA)
- Bioethics (AREA)
- General Engineering & Computer Science (AREA)
- General Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Analytical Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Data Mining & Analysis (AREA)
- Software Systems (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Computer Security & Cryptography (AREA)
- Computer Hardware Design (AREA)
- Artificial Intelligence (AREA)
- Computer Vision & Pattern Recognition (AREA)
- Epidemiology (AREA)
- Evolutionary Computation (AREA)
- Public Health (AREA)
- Human Computer Interaction (AREA)
- Signal Processing (AREA)
- Management, Administration, Business Operations System, And Electronic Commerce (AREA)
- Information Retrieval, Db Structures And Fs Structures Therefor (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Compression, Expansion, Code Conversion, And Decoders (AREA)
- Labeling Devices (AREA)
- Television Signal Processing For Recording (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
Abstract
게놈 데이터의 저장 또는 전송은 파일 또는 게놈 데이터의 스트림에서 구조화된 압축 게놈 데이터 세트를 사용함으로써 실현된다. 특정 게놈 영역에 해당하는 데이터 또는 데이터의 하위 세트에 대한 선택적 액세스는 데이터 분류 및 특정 인덱싱 메커니즘을 기반으로 사용자-정의된 라벨을 사용함으로써 달성된다.
Description
본 출원은 생물정보학 데이터, 특히 게놈 서열화 데이터를 압축된 형태로 효율적으로 저장, 전송 및 멀티플렉싱하는 신규한 방법을 제공하며, 이는 게놈 데이터 세트를 구성하는 상이한 데이터 카테고리들의 효율적인 선택적 액세스 및 선택적 보호를 가능하게 한다.
게놈 서열화 데이터의 적절한 표현은, 서열화 데이터 및 메타데이터를 처리함으로써 다양한 목적으로 수행되는 게놈 변이체 호칭 및 모든 분석과 같은 분석 애플리케이션을 가능하게 하고 용이하게 하는 게놈 데이터의 효율적인 처리, 저장 및 전송을 가능하게 하기 위한 기본이다. 오늘날, 게놈 서열 정보는 정의된 어휘로부터 일련의 문자들로 표현되는 뉴클레오티드(염기라고도 함) 서열의 형태로 고 처리량 서열화(HTS; High Throughput Sequencing) 기계에 의해 생성된다.
이러한 서열화 기계는 전체 게놈이나 유전자를 판독하는 것이 아니라 서열 판독이라고 알려진 뉴클레오티드 서열의 무작위의 짧은 단편들을 생성한다. 품질 점수는 서열 판독에서 각각의 뉴클레오티드와 관련된다. 이러한 숫자는 뉴클레오티드 서열의 특정 위치에서 특정 뉴클레오티드를 판독하기 위해 기계에 의해 주어진 신뢰 수준을 나타낸다.
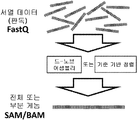
NGS 기계에 의해 생성되는 이러한 원시(raw) 서열 데이터는 일반적으로 FASTQ 파일에 저장된다(도 1 참조).
서열화 과정에 의해 얻은 뉴클레오티드의 서열을 나타내는 가장 작은 어휘는 5 개의 기호 A, C, G, T, N로 구성되며, A, C, G, T, N은 DNA에 존재하는 뉴클레오티드의 네 가지 형태, 즉 아데닌(Adenine), 사이토신(Cytosine), 구아닌(Guanine) 및 티민(Thymine)과, 서열화 기계가 충분한 신뢰 수준으로 염기를 호칭할 수 없어서 이러한 위치에 있는 염기의 형태가 판독 과정에서 결정되지 않은 채로 남아 있음을 가리키는 기호 N을 나타낸다. RNA에서 티민은 우라실(U)로 대체된다. 서열화 기계에 의해 생성된 뉴클레오티드 서열을 "판독"이라고 한다. 쌍으로 된 판독의 경우에는 판독 쌍이 추출된 원래 서열을 지정하기 위해 "템플릿(template)"이라는 용어를 사용한다. 서열 판독은 수십 내지 수천 범위의 뉴클레오티드 수로 구성될 수 있다. 일부 기법은 두 개의 DNA 가닥 중 하나로부터 각각의 판독을 시작할 수 있는 쌍의 서열 판독을 생성한다.
게놈 서열 분야에서, 용어 "커버리지(coverage)"는 기준 게놈에 대한 서열 데이터의 중복 수준을 표현하는데 사용된다. 예를 들어, 인간 게놈 (32 억개의 염기 길이)에서 30 배의 커버리지에 도달하기 위해서는 서열화 기계가 총 약 30×32 억 개의 염기를 생성하여 각각의 기준 위치가 평균 30 배로 "커버"되어야 한다.
(기술 솔루션의 상태)
서열 데이터에 대해 가장 많이 사용되는 게놈 정보 표현은 최초 크기를 줄이기 위해 일반적으로 압축 형태로 제공되는 FASTQ 및 SAM 파일 포맷을 기반으로 한다. 비-정렬 및 정렬된 서열 데이터에 대한 통상적인 파일 포맷인 FASTQ 및 SAM은 각각 일반적인 텍스트 문자로 구성되므로, LZ(Lempel 및 Ziv) 체계 (zip, gzip 등으로 널리 알려짐)와 같은 범용 접근 방식을 사용하여 압축된다. gzip과 같은 범용 압축기가 사용될 때 압축 결과는 보편적으로 이진 데이터의 단일 블롭(blob)이다. 이러한 모놀리식 형태의 정보는 데이터 양이 극도로 방대할 때, 특히 많은 처리량의 서열화의 경우에 파일을 보관하고 전송하고 정교하게 하기가 매우 어렵다.
서열화 후에 게놈 정보 처리 파이프 라인의 각 단계는 실제로 생성된 데이터의 작은 부분만 이전 단계와 관련하여 새로운 것임에도 불구하고 완전히 새로운 데이터 구조 (파일 포맷)로 표시된 데이터를 생성한다.
도 1은 관련 파일 포맷 표현의 지시를 갖는 전형적인 게놈 정보 처리 파이프 라인의 주요 단계를 나타낸다.
보편적으로 사용되는 해결책은 몇 가지 단점을 제시한다: 다수의 데이터 복제를 의미하는 게놈 정보 처리 파이프 라인의 각 단계마다 다른 파일 포맷을 사용하여 결과적으로 필요한 저장 공간이 급격히 증가하기 때문에 데이터 보관이 비효율적이다. 이것은 비효율적이면서도 불필요하며, 또한 HTS 기계에 의해 생성된 데이터 양의 증가로 지속 가능하지 않다. 이것은 실제로 사용 가능한 저장 공간 및 발생되는 비용의 측면에 따른 결과이며, 또한 의료 분야에서 게놈 분석의 잇점을 인구의 대다수에 미치지 못하게 막고 있다. 저장 및 분석되는 서열 데이터의 기하 급수적인 증가로 인해 발생하는 IT 비용의 영향은 현재 과학계와 의료 업계가 당면한 주요 과제 중 하나이다(참조: Scott D. Kahn, "On the future of genomic data" - Science 331, 728(2011) 및 Pavlichin, D. S., Weissman, T. 및 G. Yona. 2013. "The human genome contracts again" Bionformatics 29(17): 2199-2202). 동시에, 수 개의 선택된 개체로부터 대규모의 개체군으로 게놈 서열을 확장하려는 시도가 여러 번 있었다(참조: Josh P. Roberts "Million Veterans Sequenced" - Nature Biotechnology 31, 470(2013).
게놈 데이터의 전송은 현재 사용되는 데이터 포맷이 최대 수백 기가바이트 크기의 모놀리식 파일로 구성되어 있으며 이러한 파일로 수신 측에서 처리하여 완전히 전송해야하기 때문에 느리고 비효율적이다. 이것은 데이터의 작은 부분을 분석할 때 소모된 대역폭과 대기 시간 측면에서 상당한 비용으로 전체 파일을 전송해야 한다는 것을 의미한다. 종종 대용량의 데이터를 온라인으로 전송하는 것은 엄두도 못 낼 정도로 비싸므로, 그러한 데이터 전송은 하드 디스크 드라이브 또는 스토리지 서버와 같은 저장 미디어를 한 위치에서 다른 위치로 물리적으로 이동하여 수행한다.
종래 기술의 방식을 사용할 때 발생하는 이러한 한계들은 본 발명에 의해 극복된다.
일반적으로 사용되는 분석 애플리케이션에서 필요로 하는 데이터 및 메타데이터의 다른 클래스의 부분을 전체 데이터에 액세스하지 않고는 검색할 수 없게 한 방식으로 정보가 구조화되어 있지 않기 때문에 데이터 처리는 느리고 비효율적이다. 이러한 사실은 공통 분석 파이프 라인이 몇 일 또는 몇 주일 동안 가동해야 하므로 귀중하고 값 비싼 처리 재원을 낭비한다는 것을 의미하며, 그 이유는, 특정 분석 목적에 관련된 데이터의 부분이 매우 적을 지라도 액세스하는 각각의 단계에서 많은 양의 데이터를 파싱(parsing)하고 필터링해야 하기 때문이다.
이러한 한계로 인해 의료 전문가가 게놈 분석 보고서를 적시에 수집하지 못하게되고 질병 발병에 신속히 대응할 수 없게 된다. 본 발명은 이러한 필요에 대한 해결책을 제공한다.
본 발명에 의해 극복되는 또 다른 기술적 한계가 있다.
사실상, 본 발명은 게놈 서열 데이터 및 메타데이터의 압축을 최대화하고 선택적인 액세스 및 증분적 업데이트에 대한 지원 및 많은 기타들과 같은 몇몇 기능을 효율적으로 가능하게 하도록 데이터를 조직화 하고 분할함으로써 적절한 게놈 서열 데이터 및 메타데이터 표현을 제공하는 것을 목적으로 한다.
본 발명의 주요 양태는 적절한 소스 모델에 의해 표현되고, 특정 계층으로 구조화됨으로써 개별적으로 코딩(즉, 압축)되는 데이터 및 메타데이터의 클래스를 특정적으로 정의한다. 현존하는 종래 기술의 방법에 대해 본 발명의 가장 중요한 업적은 다음과 같다:
· 데이터 또는 메타데이터의 각 클래스에 대한 효율적인 모델을 제공함으로써 구성되는 정보 소스 엔트로피의 감소로 인한 압축 성능의 증가;
· 압축 도메인에서 직접적으로 임의의 추가 처리 목적을 위해 압축 데이터 및 메타데이터의 일부에 대한 선택적인 액세스를 수행하는 가능성;
· 게놈 파일 헤더(header)에 포함된 "라벨 리스트(label list)"를 파싱함으로써 압축 데이터에 대한 효율적인 선택적 액세스를 가능하게 하는 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합을 식별하는 사용자-지정된 "라벨"을 정의하는 가능성;
· 라벨에 의해 식별되는 상이한 게놈 영역 또는 하위 영역에 대한 액세스 제어 및 보호를 구현하는 가능성;
· 새로운 서열 데이터 및/또는 메타데이터 및/또는 새로운 분석 결과로 인코딩된 데이터 및 메타데이터를 (재 인코딩할 필요없이) 점진적으로 업데이트하고 추가하는 가능성;
· 서열화 또는 정렬 과정의 종료를 기다릴 필요 없이 서열화 기계 또는 정렬 수단(tool)에 의해 데이터가 생성되는 대로 생성된 데이터를 효율적으로 처리하는 가능성.
본 출원은 멀티플렉싱 기술과 결합된 구조화된 액세스 유닛 접근법을 사용함으로써 매우 많은 양의 게놈 서열 데이터의 효율적인 조작, 저장 및 전송 문제를 해결하는 방법 및 시스템을 개시한다.
본 출원은 게놈 데이터 접근성의 기능, 선택적 데이터 보호, 데이터 하위 세트의 효율적인 처리, 효율적인 압축과 결합된 전송 및 스트리밍 (streaming)기능과 관련된 종래 기술 접근법의 모든 한계를 극복한다.
오늘날 게놈 데이터에 가장 많이 사용되는 표현 포맷은 SAM (Sequence Alignment Mapping) 텍스트 포맷과 그의 이진 대응 BAM이다. SAM 파일은 인간이 판독 가능한 ASCII 텍스트 파일인 반면에, BAM은 gzip의 블록 기반 변이체를 채택한다. BAM 파일은 한정된 방식의 랜덤 액세스(random access)를 가능하게 하기 위해 인덱싱될 수 있다. 이것은 별도의 인덱스 파일을 작성함으로써 지원된다.
BAM 포맷은 다음과 같은 이유로 압축 성능이 불량함을 특징으로 한다:
1. SAM 파일에 의해 전달되는 실제 게놈 정보를 추출하고 이를 압축하기 위한 적절한 모델을 사용하는 것 보다는 비효율적이고 중복되는 SAM 파일 포맷을 압축하는데 중점을 두고 있다.
2. 각 데이터 소스의 특정 성격 (게놈 정보 자체)을 이용하는 것이 아니라 gzip과 같은 범용 텍스트 압축 알고리즘을 사용한다.
3. 개념이 없으며, 게놈 데이터의 특정 클래스에 대한 선택적인 액세스를 제공하는 메커니즘을 구현하게 하는 데이터 분류와 관련된 기능을 지원하지 않는다.
BAM보다는 덜 일반적으로 사용되지만 더욱 효율적인 게놈 데이터 압축에 대한 보다 정교한 접근 방식은 CRAM(CRAM 사양: https://samtools.github.io/hts-specs/CRAMv3.pdf)이다. CRAM은 기존의 기준(데이터 소스 중복성을 부분적으로 악용함)과 관련하여 차등 인코딩을 채택하는데 보다 효율적인 압축을 제공하지만, 증분적인 업데이트, 스트리밍을 위한 지원 및 압축된 데이터의 특정 클래스에 대한 선택적 액세스와 같은 기능을 여전히 결여하고 있다 .
CRAM은 CRAM 레코드의 개념에 의존한다. 각각의 CRAM 레코드는 재구성하는데 필요한 모든 요소들을 인코딩함으로써 단일 매핑된 또는 매핑되지 않은 판독을 인코딩 한다.
CRAM은 본 명세서에서 기술된 발명에 의해 해결되고 제거되는 다음과 같은 단점과 한계를 제시한다:
1. CRAM은 특정 특징들을 공유하는 데이터 하위 세트에 대한 데이터 인덱싱 및 랜덤 액세스를 지원하지 않는다. 데이터 인덱싱은 명세서의 범위를 벗어나며(참조: CRAM 규격 v 3.0의 12 절), 별도의 파일로 구현된다. 반대로, 본 명세서에서 기술된 본 발명의 접근법은 인코딩 과정과 통합된 데이터 인덱싱 방법을 사용하며, 인덱스는 인코딩된(즉, 압축된) 비트 스트림(bit stream)에 내장된다.
2. CRAM은 선택적 액세스가 효율적이고 실행 분리(즉, 실제 유기 샘플에서 게놈 정보를 추출하는 과정)가 유지되도록 여러 번의 서열화 실행과 관련된 데이터의 집계를 지원하지 않는다. CRAM은 판독을 다른 그룹에 속한 것으로 라벨링할 수 있는 가능성을 제공하지만, 이는 판독 기반에 의한 판독에 제공된 다음 다른 그룹의 판독이 파일 구조 내에 혼합된다. 본 발명에 따라서, 효과적이면서도 선택적인 액세스가 이용 가능하기 위하여 상이한 서열화 실행들 간에 격리를 유지하도록 데이터를 구조화하는 방법이 기술된다.
3. CRAM은 어떠한 유형의 매핑된 판독(완전히 일치하는 판독, 대체로만 된 판독, 삽입 또는 삭제로된 판독("삽입-결실(indel)"이라고도 함))이라도 포함할 수 있는 코어 데이터 블록에 의해 작성된다. 기준 서열에 대한 매핑 결과에 따라서 클래스에서 판독의 데이터 분류 및 그룹화 개념은 없다. 이는 특정 특징들을 갖는 판독만 검색하는 경우에도 모든 데이터를 검사할 필요가 있음을 의미한다. 이러한 한계는 코딩 전에 클래스 내의 데이터를 분류 및 분할함으로써 본 발명에 의해 해결된다.
4. CRAM은 각각의 판독을 "CRAM 레코드"내로 캡슐화하는 개념에 기반하고 있다. 이는 특정 생물학적 특징에 의해 특성화되는 판독(예를 들어, 대체된 판독이지만 "삽입-결실"이 없는 판독 또는 완전하게 매핑된 판독)이 검색될 때 각각의 완전한 "레코드"를 조사할 필요가 있음을 의미한다.
반대로, 본 발명에서는 별도의 정보 계층에 개별적으로 코딩된 데이터 클래스의 개념은 있지만, 각 판독을 캡슐화하는 레코드의 개념은 없다. 이에 의해, 특정 생물학적 특성을 갖는 판독 세트(예를 들어, 대체된 판독이지만 "삽입-결실"이 없는 판독 또는 완전하게 매핑된 판독)에 대해 보다 효율적으로 액세스할 수 있으며 각각의 판독의 (블록)을 디코딩하지 않아도 해당 특징들을 검사할 수 있다.
5. CRAM 레코드에서 레코드의 각 필드는 특정 플래그(flag)와 연결되며, 각각의 CRAM 레코드에는 다른 유형의 데이터가 포함될 수 있으므로 각각의 플래그는 항상 컨텍스트 개념이 없는 것과 같은 의미를 가져야 한다. 이러한 코딩 메커니즘은 중복 정보를 도입하고 효율적인 컨텍스트 기반 엔트로피 코딩의 사용을 막는다.
반대로, 본 발명에서는 데이터를 나타내는 플래그의 개념이 없으며, 이는 데이터가 속하는 정보 "계층"에 의해 본질적으로 정의되기 때문이다. 이는 사용되는 기호들의 수가 상당히 줄어들고 결과적으로 정보 소스 엔트로피가 감소하여 보다 효율적인 압축을 수행함을 의미한다. 이러한 개선은 서로 다른 "계층들"을 사용함으로써 인코더가 컨텍스트에 따라 각기 다른 의미로 각 계층 전반야에 걸쳐 동일한 기호를 재사용할 수 있기 때문에 가능하다. CRAM에서, 각 플래그는 항상 컨텍스트의 개념이 없는 것과 동일한 의미를 가져야 하며, 각 CRAM 레코드는 모든 유형의 데이터를 포함 할 수 있다.
6. CRAM에서 대체, 삽입 및 삭제는 정보 소스 알파벳의 크기를 늘리고 더 높은 소스 엔트로피를 산출하는 옵션(option)인 다양한 구문 요소를 사용함으로써 표현된다. 반대로, 개시된 발명의 접근법은 대체, 삽입 및 삭제를 위한 단일 알파벳 및 인코딩을 사용한다. 이는 인코딩 및 디코딩 프로세스를 보다 간단하게 만들고 코딩이 높은 압축 성능을 특징으로 하는 비트 스트림을 산출하는 보다 낮은 엔트로피 소스 모델을 생성한다.
7. CRAM은 게놈 데이터의 특정 영역 또는 하위 영역 또는 그의 집합을 고유하게 식별하는 메커니즘을 제공하지 않는다. CRAM 사양에 따라서, 기준 서열의 시작 및 종료 위치의 관점에서 유전자 자리의 정의를 제외하곤 다음과 같은 방법은 없다:
- 영역을 라벨링하고 게놈 시작 및 종료 위치 대신에 정의된 라벨을 사용하여 영역을 액세스 하는 방법. 새로운 기준 서열이 발표되면 동일한 게놈 영역의 시작 및 종료 위치가 변경될 수 있는 반면에, 정의된 라벨은 최종 사용자에게 이러한 변경을 숨길 것이다. 인코딩 및 디코딩 시스템은 라벨에 의해 식별된 실제 영역을 새롭게 발표된 기준 서열로 채택할 것이다.
- 동일한 라벨하에 몇 몇 영역 또는 하위 영역을 합계하여 최종 사용자가 복잡한 중첩 쿼리(query)를 수반하지 않는 단일 쿼리를 통해 필요한 데이터를 선택할 수 있는 방법. 전체 집합 메커니즘은 본 명세서에서 기술된 바와 같이 인코딩 및 디코딩 시스템에 포함된다.
8. CRAM은 게놈 데이터의 특정 영역 또는 하위 영역 또는 그의 집합이 사전에 정의되지 않았을 뿐더러 적절한 "라벨"을 삽입한 사용자에 의해 명시되지 않았을 때 이러한 영역 또는 집합에 대한 선택적 보호 및 액세스 제어를 구현하기 위한 어떠한 메커니즘도 제공하거나 지원하지 않는다.
CRAM 이외에도, 게놈 데이터 압축 및 처리에 대한 다른 접근법은 대부분의 필요한 기능에 대한 확고한 한계를 제시하며, 본 명세서에서 다음과 같이 기술되고 명시된 바와 같이 본 발명에 의해 제공되는 특징들을 지원하지 않는다.
종래의 기술에서 사용되는 게놈 압축 알고리즘은 다음과 같은 카테고리로 분류될 수 있다:
· 변환 기반
· LZ 기반
· 판독 기록
· 어셈블리 기반
· 통계 모델링
첫 번째 두 개의 카테고리는 데이터 소스의 특정 특성(게놈 서열 판독)을 활용하지 않는 단점을 공유하며, 이러한 종류의 정보의 특정 속성(예를 들어, 판독값 중의 중복값, 기존의 샘플에 대한 기준)을 고려하지 않고 게놈 데이터를 압축할 텍스트의 문자열(string)로서 처리한다. 게놈 데이터 압축을 위한 가장 진보된 두 가지 툴킷(toolkit), 즉, CRAM과 Goby("구조화된 높은 처리량 서열 데이터의 압축", F. Campagne, K. C. Dorff, N. Chambwe, J. T. Robinson, J. P. Mesirov, T. D. Wu)은 기하학적 분포에 의해 독립적으로 동일하게 분산된 데이터를 암시적으로 모델링하므로 산술적 코딩의 사용을 불량하게 한다. Goby는 모든 필드를 정수 목록으로 변환하고 각 목록을 컨텍스트의 사용없이 산술적 코딩을 사용하여 독립적으로 인코딩하므로 약간 더 정교하다. 가장 효율적인 작동 모드에서, Goby는 정수 목록에 대한 일부 내부 목록 모델링을 수행하여 압축을 개선할 수 있다. 이러한 종래 기술의 해결책은 불량한 압축율을 산출하고, 일단 압축되면 선택적으로 액세스하고 조작하는 것이 불가능하지는 않더라도 곤란한 데이터 구조를 산출한다. 다운스트림 분석 단계는 간단한 조작을 수행하거나 게놈 데이터 세트의 선택된 영역에 액세스할 때 조차도 크고 단단한 데이터 구조를 처리할 필요가 있기 때문에 비효율적이며 매우 느릴 수 있다.
게놈 프로세싱 파이프 라인에 사용되는 파일 포맷들 간의 관계에 대한 단순화된 비전은 도 1에 도시되어 있다. 이러한 다이어그램에서 파일 내포는 중첩된 파일 구조의 존재를 의미하지 않지만 각 포맷에 대해 인코딩할 수 있는 정보의 유형과 양을 나타낸다(예를 들어, SAM은 FASTQ의 모든 정보를 포함하지만 다른 파일 구조로 구성됨). CRAM은 SAM/BAM과 동일한 게놈 정보를 포함하지만 사용할 수 있는 압축의 유형에 대해 더욱 융통성을 갖기 때문에 SAM/BAM의 상위 집합(superset)으로서 대표된다.
게놈 정보를 저장하기 위해 다수의 파일 포맷을 사용하는 것은 매우 비효율적이며 비용이 많이 든다. 게놈 정보 수명주기의 다른 단계에서 다른 파일 포맷을 갖는 것은 증분적 정보가 최소인 경우에도 활용된 저장 공간의 선형 성장을 내포하고 있다. 종래 기술의 해결책의 추가 단점을 이하에 열거한다.
1. 압축된 FastQ 파일 또는 그의 조합에 저장된 원시 데이터에 대한 주석(메타데이터)을 액세스하거나 분석하거나 또는 추가하려면 계산 자원과 시간의 광범위한 사용으로 전체 파일을 압축 해제하고 다시 압축해야 한다.
2. 판독 매핑 위치, 판독 변이체 위치 및 유형, 삽입-결실 위치 및 유형 또는 BAM 파일에 저장된 정렬 데이터에 포함된 기타 메타데이터 및 주석과 같은 정보의 특정 하위 세트를 검색하려면 각각의 판독과 관련된 전체 데이터 부피에 대한 액세스를 필요로 한다. 선행 기술 해결책에서는 단일 클래스의 메타데이터에 선택적으로 액세스하는 것은 가능하지 않다.
3. 선행 기술의 파일 포맷은 처리가 시작되기 전에 최종 사용자가 전체 파일을 수신할 것을 요구한다. 예를 들어, 서열화 과정이 적절한 데이터 표현에 의존하여 완료되기 전에 판독 정렬이 시작될 수 있다. 서열화, 정렬 및 분석은 병행하여 처리되고 진행될 수 있다.
4. 선행 기술의 해결책은 구조화를 지원하지 않으며, 다른 서열화 과정에 의해 얻어진 게놈 데이터를 그들의 특정 의미 세대(예를 들어, 동일한 개체의 수명의 상이한 시간에서 획득된 서열)에 따라 구별할 수 없다. 동일 개체의 다른 유형의 생물학적 샘플에 의해 얻어진 서열에 대해서도 동일한 한계가 발생한다.
5. 데이터의 전체 또는 선택된 부분의 액세스 제어 메커니즘(예를 들어, 암호화, 워터 마킹, 디지털 서명, 해싱(hasing))에 의한 보호는 선행 기술의 해결책으로 지원되지 않는다. 예를 들면,
a. 선택된 DNA 영역,
b. 변이체를 함유하는 서열들만,
c. 키메라 서열들만,
d. 매핑되지 않은 서열들만,
e. 사용자-정의된 라벨로 식별되는 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합, 및
f. 특정 메타데이터 (예를 들어, 서열화된 샘플의 시초, 서열화된 개체의 식별, 샘플의 유형)의 보호는 종래 기술의 해결책의 파일 및 데이터 포맷에서는 지원되지 않는다.
6. 주어진 기준(즉, SAM/BAM 파일)에 정렬된 서열 데이터를 새로운 기준으로 변환하는 것은 새로운 기준이 이전 기준에서 단일 뉴클레오티드 위치만 다른 경우에도 전체 데이터 부피를 처리해야 할 필요가 있다.
따라서, 효율적인 압축을 가능하게 하고, 국부적 및 원격으로 저장된 데이터의 압축된 도메인에서 선택적 액세스 및 보호 기능을 지원하고, 게놈 데이터 처리의 다른 단계의 모든 수준에서 압축된 도메인내의 이질적인 메타데이터의 점진적 추가를 지원하는 적절한 게놈 정보 스토리지 포맷(게놈 파일 포맷) 및 전송 메커니즘이 명백히 필요하다.
본 발명은 수반되는 특허 청구 범위에 청구된 바와 같은 방법, 장치 및 컴퓨터 프로그램을 사용함으로써 종래 기술의 한계에 대한 해결책을 제공한다.
도 1은 전형적인 게놈 파이프 라인의 주요 단계 및 관련 파일 포맷을 나타낸다.
도 2는 가장 많이 사용되는 게놈 파일 포맷들 간에 상호 관계를 나타낸다.
도 3은 게놈 서열 판독이 드-노브 어셈블리(de-novo assembly) 또는 기준 기반 정렬을 통해 전체 또는 부분 게놈에서 어떻게 조합되는지를 나타낸다.
도 4는 기준 서열상의 판독 맵핑 위치가 어떻게 계산되는지를 나타낸다.
도 5는 판독 페어링(pairing) 거리를 계산하는 방법을 나타낸다.
도 6은 페어링 오류를 계산하는 방법을 나타낸다.
도 7은 판독 메이트(mate) 쌍이 다른 염색체에 매핑될 때 페어링 거리가 인코딩되는 방법을 나타낸다.
도 8은 서열 판독이 게놈의 제 1 또는 제 2 DNA 가닥으로부터 어떻게 생성될 수 있는지를 나타낸다.
도 9는 가닥 2에 매핑된 판독이 가닥1에 상응하는 역 보완 판독을 갖는 방법을 나타낸다.
도 10은 판독 쌍을 구성하는 판독과 rcomp 계층에서 각각의 인코딩의 네 가지 가능한 조합을 나타낸다.
도 11은 "n 유형" 불일치가 nmis 계층에서 인코딩되는 방법을 나타낸다.
도 12는 매핑된 판독 쌍의 대체의 예시를 도시한다.
도 13은 대체 위치를 절대값 또는 차등값으로 계산하는 방법을 나타낸다.
도 14는 IUPAC 코드가 없는 대체 인코딩 기호를 계산하는 방법을 나타낸다.
도 15는 대체 유형이 snpt 계층에서 인코딩되는 방법을 나타낸다.
도 16은 IUPAC 코드를 갖는 대체 인코딩 기호를 계산하는 방법을 나타낸다.
도 17은 위치만 인코딩되지만 대체 유형 당 하나의 계층이 사용되는 대체에 대한 대안적 소스 모델을 나타낸다.
도 18은 IUPAC 코드가 사용되지 않을 때 클래스 I의 판독 쌍에서 대체, 삽입 및 삭제를 인코딩하는 방법을 나타낸다.
도 19는 IUPAC 코드가 사용될 때 클래스 I의 판독 쌍에서 대체, 삽입 및 삭제를 인코딩하는 방법을 나타낸다.
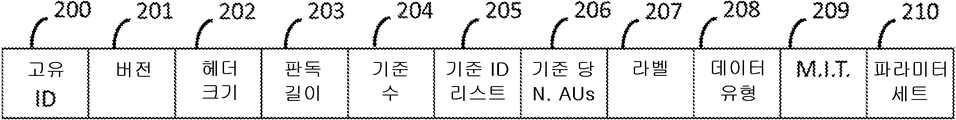
도 20은 본 발명에 의해 개시된 게놈 정보 데이터 구조의 게놈 데이터 세트 헤더의 구조를 도시한다.
도 21은 마스터 인덱스 테이블이 각 액세스 유닛에서 제 1 판독값의 기준 서열 위치를 어떻게 포함하는지를 나타낸다.
도 22는 클래스 P의 각 posAU 내에서 제 1 판독값의 매핑 위치를 도시한 부분적인 MIT의 예시를 나타낸다.
도 23은 계층 헤더의 로컬 인덱스 테이블이 페이로드(payload)의 AU에 대해 어떻게 포인터 벡터(vector of pointer)가 되는 지를 나타낸다.
도 24는 로컬 인덱스 테이블의 예시를 나타낸다.
도 25는 마스터 인덱스 테이블과 로컬 인덱스 테이블 간의 함수 관계를 보여준다.
도 26은 액세스 유닛이 여러 계층에 속한 데이터 블록으로 구성되는 방법을 보여준다. 계층은 패킷으로 세분화된 블록으로 구성된다.
도 27은 제 1 유형의 게놈 액세스 유닛(위치, 쌍, 역 보완 및 판독 길이 정보를 함유함)이 어떻게 게놈 데이터 멀티플렉스에 패킷화되고 캡슐화되는지를 나타낸다.
도 28은 동종 데이터의 하나 이상의 계층에 속한 멀티플렉스화된 블록 및 헤더에 의해 액세스 유닛이 구성되는 방법을 보여준다. 각 블록은 게놈 정보의 실제 디스크립터(descriptor)를 포함하는 하나 이상의 패킷으로 구성될 수 있다.
도 29는 액세스되거나 또는 디코딩 및 액세스되는 다른 액세스 유닛으로부터의 정보를 참조할 필요가 없는 유형 0의 액세스 유닛의 구조를 도시한다.
도 30은 유형 1의 액세스 유닛의 구조를 보여준다.
도 31은 유형 1의 액세스 유닛을 지칭하는 데이터를 포함하는 유형 2의 액세스 유닛의 구조를 도시한다. 이들은 인코딩된 판독에서 N 개의 염기의 위치이다.
도 32은 유형 1의 액세스 유닛을 지칭하는 데이터를 포함하는 유형 3의 액세스 유닛의 구조를 도시한다. 이들은 인코딩된 판독의 불일치의 위치 및 유형이다.
도 33은 유형 1의 액세스 유닛을 지칭하는 데이터를 포함하는 유형 4의 액세스 유닛의 구조를 도시한다. 이들은 인코딩된 판독의 불일치의 위치 및 유형이다.
도 34는 액세스 유닛의 첫 번째 다섯 가지 유형을 보여준다.
도 35는 유형 1의 액세스 유닛이 유형 0의 액세스 유닛을 참조하여 디코딩되는 것을 도시한다.
도 36은 유형 2의 액세스 유닛이 유형 0 및 유형 1의 액세스 유닛을 참조하여 디코딩되는 것을 나타낸다.
도 37은 유형 3의 액세스 유닛이 유형 0 및 유형 1의 액세스 유닛을 참조하여 디코딩되는 것을 나타낸다.
도 38은 유형 4의 액세스 유닛이 유형 0 및 유형 1의 액세스 유닛을 참조하여 디코딩되는 것을 나타낸다.
도 39는 기준 서열의 제 2 단편(AU 0-2)에 매핑된 불일치로 서열 판독을 디코딩하는데 필요한 액세스 유닛을 보여준다.
도 40은 이용 가능한 원시 게놈 서열 데이터를 사전 인코딩된 게놈 데이터에 점진적으로 추가할 수 있는 방법을 보여준다.
도 41은 액세스 유닛 기반의 데이터 구조가 서열화 과정을 완료하기 전에 게놈 데이터 분석을 시작하게 하는 방법을 보여준다.
도 42는 기존 데이터에서 수행된 새로운 분석이 유형 4의 AU에서 유형 3의 Au로 판독값을 이동하는 것을 어떻게 의미할 수 있는 지를 보여준다.
도 43은 새로 생성된 분석 데이터가 유형 8의 새로운 AU에 캡슐화되고 상응하는 인덱스가 MIT에서 어떻게 작성되는 지를 보여준다.
도 44는 새로운 기준 서열(게놈)의 발표로 인해 데이터를 어떻게 트랜스코드(transcord)하는 지를 보여준다.
도 45는 보다 우수한 품질(예를 들어, 삽입-결실이 없는)을 갖는 새로운 게놈 영역에 매핑된 판독이 유형 4의 AU에서 유형 3의 Au로 어떻게 이동되는 지를 보여준다.
도 46은 새로운 매핑 위치가 발견된 경우(예를 들어, 불일치가 적은 경우)에, 관련된 판독을 하나의 AU에서 동일한 유형의 또 다른 Au로 어떻게 이동될 수 있는 지를 보여준다.
도 47은 유형 4의 액세스 유닛이 보호될 민감한 정보를 포함하고 있음에 따라 선택적 암호화를 유형 4의 액세스 유닛에만 어떻게 적용할 수 있는 지를 보여준다.
도 48은 하나 이상의 게놈 데이터 세트(482-483)가 게놈 스트림(484) 및 게놈 데이터 세트 매핑 테이블 리스트의 스트림(481), 게놈 데이터 세트 매핑 테이블의 스트림(485) 및 기준 식별자 매핑 테이블의 스트림(487)을 포함하는 게놈 멀티플렉스 내의 데이터 캡슐화를 도시한다. 각각의 게놈 스트림은 헤더(488) 및 액세스 유닛(486)에 의해 구성된다. 액세스 유닛은 패킷(4810)에 의해 구성된 블록(489)을 캡슐화 한다.
도 49는 원시 게놈 서열 데이터(499) 또는 정렬된 게놈 데이터(요소(491)에 의해 생성됨)가 게놈 멀티플렉스 내에서 캡슐화되도록 처리되는 방법을 도시한다. 인코딩을 위한 데이터를 준비하기 위해 정렬(491) 및 기준 게놈 구성(492) 단계가 필요할 수 있다. 데이터 분류 유닛(494)에 의해 생성된 데이터 클래스(498)는 기준 변환 유닛(4919)에 의해 생성된 하나 이상의 변환된 기준과 관련하여 추가로 분류될 수 있다. 이 때, 변환된 클래스(4918)는 계층 인코더(495-497)로 전송된다. 생성된 계층(4911)은 게놈 멀티플렉서(4916)에 공급되는 액세스 유닛(4915)의 게놈 스트림을 생성하는 엔트로피 코더(4912-4914)에 의해 인코딩된다.
도 50은 게놈 디멀티플렉서(500)가 게놈 멀티플렉스(5010)로부터 게놈 스트림(501)을 추출하고, AU 유형 당 하나의 디코더(502-504)가 게놈 계층을 추출하고 나서, 예를 들어, FASTQ 및 SAM/BAM과 같은 게놈 포맷을 재구성하기 위해 클래스 디코더(509)에 의해 사용되는 다양한 데이터 클래스(5011)내로 디코딩(506-507)되는 방법을 보여준다. 멀티플렉스화된 비트 스트림(5010)내에 존재할 때, 하나 이상의 기준 변환을 포함하는 게놈 스트림이 엔트로피 디코더(504)에 의해 디코딩되어 기준 변환 디스크립터(5012)를 생성한다. 기준 변환 디스크립터는 기준 변환 유닛(5013)에 의해 처리되어 클래스 디코더(509)에 의해 사용될 하나 이상의 변환된 기준(5014)을 생성하도록 하나 이상의 "외부" 기준을 변환한다.
도 51은 6 개의 디스크립터 계층을 사용하여 자체-생성 기준 서열을 사용하여 클래스 U에 속하는 서열 판독을 인코딩하는 과정을 도시한다. 4 개의 계층들은 다른 클래스 P, N, M, I에 대해 사용된 것과 동일한 반면에, 두 개의 계층들은 클래스 U의 판독값에 대해 특이적이다.
도 52는 두 개의 서로 다른 기준에 속하는 게놈 영역을 합계하기 위해 라벨을 어떻게 작성하는 지를 보여준다.
도 53은 새로운 분석 결과가 기존 영역(R1, R2 및 R3)에 추가 영역(R4)을 부가해야 하는 경우 기존 라벨을 어떻게 업데이트할 수 있는 지를 보여준다.
도 54는 특정 게놈 영역 또는 하위 영역에 대한 액세스 제어 및 데이터 보호를 구현하기 위해 라벨링 메커니즘을 어떻게 사용할 수 있는 지를 보여준다. 간단한 경우는 하나의 라벨에 의해 식별된 모든 게놈 영역에 대해 하나의 액세스 제어 규칙(AC)과 하나의 보호 메커니즘(예를 들어, 암호화)을 사용한다.
도 55는 동일한 라벨에 의해 식별되는 다른 게놈 영역이 몇 개의 다른 액세스 제어 규칙(AC) 및 몇 개의 다른 암호화 키로 어떻게 보호될 수 있는 지를 보여준다.
도 56은 서명된 POS 디스크립터가 계산된 기준에서 판독의 매핑 위치를 인코딩하는데 사용되는 클래스 U의 판독을 대안적으로 인코딩하는 방법을 보여준다.
도 57은 절반 매핑된 판독 쌍이 매핑되지 않은 판독값과 보다 긴 컨틱(contig)을 조합함으로써 기준 서열의 미지의 영역을 채우는 것을 도울 수 있는 방법을 도시한다.
도 58은 본 발명에서 기술된 구조에 따라 저장된 게놈 데이터에 대한 헤더의 계층 구조를 나타낸다.
도 59는 본 발명에 의해 기술된 라벨링 메카니즘을 구현하는 장치가 몇 개의 게놈 영역들이 데이터베이스의 다른 기록에 저장될 때 이들 영역과 관련된 데이터에 대한 동시 액세스를 어떻게 가능하게 하는지를 나타낸다. 이는 제어된 액세스가 존재하거나 존재하지 않는 경우에 일어날 수 있다.
도 60은 데이터의 분리된 하위 클래스를 생성하기 위해 클래스 N, M 및 I의 인코더에서 임계 벡터가 어떻게 사용되는지를 나타낸다.
도 61은 불일치의 전부 또는 하위 세트가 제거되었을 때(즉, 기준 변환이 적용된 후 변환이 클래스 P에 할당되기 전에 클래스 M에 속한 판독값)에 속하는 클래스 판독값을 변경할 수 있는 방법의 예시를 제공한다.
도 62는 판독값으로부터 불일치(MM)를 제거하기 위해 기준 변환을 적용하는 방법을 보여준다. 일부 경우에 있어서, 기준 변환은 새로운 불일치를 생성하거나 변환이 적용되기 전에 기준을 참조할 때 발견된 불일치의 유형을 변경할 수 있다.
도 63은 동일한 기준 변환 A0이 데이터의 모든 클래스에 대해 사용될 수 있거나 또는 다른 변환 AN, AM, AI이 각각의 클래스 N, M, I에 대해 사용되는 방법을 도시 한다.
도 2는 가장 많이 사용되는 게놈 파일 포맷들 간에 상호 관계를 나타낸다.
도 3은 게놈 서열 판독이 드-노브 어셈블리(de-novo assembly) 또는 기준 기반 정렬을 통해 전체 또는 부분 게놈에서 어떻게 조합되는지를 나타낸다.
도 4는 기준 서열상의 판독 맵핑 위치가 어떻게 계산되는지를 나타낸다.
도 5는 판독 페어링(pairing) 거리를 계산하는 방법을 나타낸다.
도 6은 페어링 오류를 계산하는 방법을 나타낸다.
도 7은 판독 메이트(mate) 쌍이 다른 염색체에 매핑될 때 페어링 거리가 인코딩되는 방법을 나타낸다.
도 8은 서열 판독이 게놈의 제 1 또는 제 2 DNA 가닥으로부터 어떻게 생성될 수 있는지를 나타낸다.
도 9는 가닥 2에 매핑된 판독이 가닥1에 상응하는 역 보완 판독을 갖는 방법을 나타낸다.
도 10은 판독 쌍을 구성하는 판독과 rcomp 계층에서 각각의 인코딩의 네 가지 가능한 조합을 나타낸다.
도 11은 "n 유형" 불일치가 nmis 계층에서 인코딩되는 방법을 나타낸다.
도 12는 매핑된 판독 쌍의 대체의 예시를 도시한다.
도 13은 대체 위치를 절대값 또는 차등값으로 계산하는 방법을 나타낸다.
도 14는 IUPAC 코드가 없는 대체 인코딩 기호를 계산하는 방법을 나타낸다.
도 15는 대체 유형이 snpt 계층에서 인코딩되는 방법을 나타낸다.
도 16은 IUPAC 코드를 갖는 대체 인코딩 기호를 계산하는 방법을 나타낸다.
도 17은 위치만 인코딩되지만 대체 유형 당 하나의 계층이 사용되는 대체에 대한 대안적 소스 모델을 나타낸다.
도 18은 IUPAC 코드가 사용되지 않을 때 클래스 I의 판독 쌍에서 대체, 삽입 및 삭제를 인코딩하는 방법을 나타낸다.
도 19는 IUPAC 코드가 사용될 때 클래스 I의 판독 쌍에서 대체, 삽입 및 삭제를 인코딩하는 방법을 나타낸다.
도 20은 본 발명에 의해 개시된 게놈 정보 데이터 구조의 게놈 데이터 세트 헤더의 구조를 도시한다.
도 21은 마스터 인덱스 테이블이 각 액세스 유닛에서 제 1 판독값의 기준 서열 위치를 어떻게 포함하는지를 나타낸다.
도 22는 클래스 P의 각 posAU 내에서 제 1 판독값의 매핑 위치를 도시한 부분적인 MIT의 예시를 나타낸다.
도 23은 계층 헤더의 로컬 인덱스 테이블이 페이로드(payload)의 AU에 대해 어떻게 포인터 벡터(vector of pointer)가 되는 지를 나타낸다.
도 24는 로컬 인덱스 테이블의 예시를 나타낸다.
도 25는 마스터 인덱스 테이블과 로컬 인덱스 테이블 간의 함수 관계를 보여준다.
도 26은 액세스 유닛이 여러 계층에 속한 데이터 블록으로 구성되는 방법을 보여준다. 계층은 패킷으로 세분화된 블록으로 구성된다.
도 27은 제 1 유형의 게놈 액세스 유닛(위치, 쌍, 역 보완 및 판독 길이 정보를 함유함)이 어떻게 게놈 데이터 멀티플렉스에 패킷화되고 캡슐화되는지를 나타낸다.
도 28은 동종 데이터의 하나 이상의 계층에 속한 멀티플렉스화된 블록 및 헤더에 의해 액세스 유닛이 구성되는 방법을 보여준다. 각 블록은 게놈 정보의 실제 디스크립터(descriptor)를 포함하는 하나 이상의 패킷으로 구성될 수 있다.
도 29는 액세스되거나 또는 디코딩 및 액세스되는 다른 액세스 유닛으로부터의 정보를 참조할 필요가 없는 유형 0의 액세스 유닛의 구조를 도시한다.
도 30은 유형 1의 액세스 유닛의 구조를 보여준다.
도 31은 유형 1의 액세스 유닛을 지칭하는 데이터를 포함하는 유형 2의 액세스 유닛의 구조를 도시한다. 이들은 인코딩된 판독에서 N 개의 염기의 위치이다.
도 32은 유형 1의 액세스 유닛을 지칭하는 데이터를 포함하는 유형 3의 액세스 유닛의 구조를 도시한다. 이들은 인코딩된 판독의 불일치의 위치 및 유형이다.
도 33은 유형 1의 액세스 유닛을 지칭하는 데이터를 포함하는 유형 4의 액세스 유닛의 구조를 도시한다. 이들은 인코딩된 판독의 불일치의 위치 및 유형이다.
도 34는 액세스 유닛의 첫 번째 다섯 가지 유형을 보여준다.
도 35는 유형 1의 액세스 유닛이 유형 0의 액세스 유닛을 참조하여 디코딩되는 것을 도시한다.
도 36은 유형 2의 액세스 유닛이 유형 0 및 유형 1의 액세스 유닛을 참조하여 디코딩되는 것을 나타낸다.
도 37은 유형 3의 액세스 유닛이 유형 0 및 유형 1의 액세스 유닛을 참조하여 디코딩되는 것을 나타낸다.
도 38은 유형 4의 액세스 유닛이 유형 0 및 유형 1의 액세스 유닛을 참조하여 디코딩되는 것을 나타낸다.
도 39는 기준 서열의 제 2 단편(AU 0-2)에 매핑된 불일치로 서열 판독을 디코딩하는데 필요한 액세스 유닛을 보여준다.
도 40은 이용 가능한 원시 게놈 서열 데이터를 사전 인코딩된 게놈 데이터에 점진적으로 추가할 수 있는 방법을 보여준다.
도 41은 액세스 유닛 기반의 데이터 구조가 서열화 과정을 완료하기 전에 게놈 데이터 분석을 시작하게 하는 방법을 보여준다.
도 42는 기존 데이터에서 수행된 새로운 분석이 유형 4의 AU에서 유형 3의 Au로 판독값을 이동하는 것을 어떻게 의미할 수 있는 지를 보여준다.
도 43은 새로 생성된 분석 데이터가 유형 8의 새로운 AU에 캡슐화되고 상응하는 인덱스가 MIT에서 어떻게 작성되는 지를 보여준다.
도 44는 새로운 기준 서열(게놈)의 발표로 인해 데이터를 어떻게 트랜스코드(transcord)하는 지를 보여준다.
도 45는 보다 우수한 품질(예를 들어, 삽입-결실이 없는)을 갖는 새로운 게놈 영역에 매핑된 판독이 유형 4의 AU에서 유형 3의 Au로 어떻게 이동되는 지를 보여준다.
도 46은 새로운 매핑 위치가 발견된 경우(예를 들어, 불일치가 적은 경우)에, 관련된 판독을 하나의 AU에서 동일한 유형의 또 다른 Au로 어떻게 이동될 수 있는 지를 보여준다.
도 47은 유형 4의 액세스 유닛이 보호될 민감한 정보를 포함하고 있음에 따라 선택적 암호화를 유형 4의 액세스 유닛에만 어떻게 적용할 수 있는 지를 보여준다.
도 48은 하나 이상의 게놈 데이터 세트(482-483)가 게놈 스트림(484) 및 게놈 데이터 세트 매핑 테이블 리스트의 스트림(481), 게놈 데이터 세트 매핑 테이블의 스트림(485) 및 기준 식별자 매핑 테이블의 스트림(487)을 포함하는 게놈 멀티플렉스 내의 데이터 캡슐화를 도시한다. 각각의 게놈 스트림은 헤더(488) 및 액세스 유닛(486)에 의해 구성된다. 액세스 유닛은 패킷(4810)에 의해 구성된 블록(489)을 캡슐화 한다.
도 49는 원시 게놈 서열 데이터(499) 또는 정렬된 게놈 데이터(요소(491)에 의해 생성됨)가 게놈 멀티플렉스 내에서 캡슐화되도록 처리되는 방법을 도시한다. 인코딩을 위한 데이터를 준비하기 위해 정렬(491) 및 기준 게놈 구성(492) 단계가 필요할 수 있다. 데이터 분류 유닛(494)에 의해 생성된 데이터 클래스(498)는 기준 변환 유닛(4919)에 의해 생성된 하나 이상의 변환된 기준과 관련하여 추가로 분류될 수 있다. 이 때, 변환된 클래스(4918)는 계층 인코더(495-497)로 전송된다. 생성된 계층(4911)은 게놈 멀티플렉서(4916)에 공급되는 액세스 유닛(4915)의 게놈 스트림을 생성하는 엔트로피 코더(4912-4914)에 의해 인코딩된다.
도 50은 게놈 디멀티플렉서(500)가 게놈 멀티플렉스(5010)로부터 게놈 스트림(501)을 추출하고, AU 유형 당 하나의 디코더(502-504)가 게놈 계층을 추출하고 나서, 예를 들어, FASTQ 및 SAM/BAM과 같은 게놈 포맷을 재구성하기 위해 클래스 디코더(509)에 의해 사용되는 다양한 데이터 클래스(5011)내로 디코딩(506-507)되는 방법을 보여준다. 멀티플렉스화된 비트 스트림(5010)내에 존재할 때, 하나 이상의 기준 변환을 포함하는 게놈 스트림이 엔트로피 디코더(504)에 의해 디코딩되어 기준 변환 디스크립터(5012)를 생성한다. 기준 변환 디스크립터는 기준 변환 유닛(5013)에 의해 처리되어 클래스 디코더(509)에 의해 사용될 하나 이상의 변환된 기준(5014)을 생성하도록 하나 이상의 "외부" 기준을 변환한다.
도 51은 6 개의 디스크립터 계층을 사용하여 자체-생성 기준 서열을 사용하여 클래스 U에 속하는 서열 판독을 인코딩하는 과정을 도시한다. 4 개의 계층들은 다른 클래스 P, N, M, I에 대해 사용된 것과 동일한 반면에, 두 개의 계층들은 클래스 U의 판독값에 대해 특이적이다.
도 52는 두 개의 서로 다른 기준에 속하는 게놈 영역을 합계하기 위해 라벨을 어떻게 작성하는 지를 보여준다.
도 53은 새로운 분석 결과가 기존 영역(R1, R2 및 R3)에 추가 영역(R4)을 부가해야 하는 경우 기존 라벨을 어떻게 업데이트할 수 있는 지를 보여준다.
도 54는 특정 게놈 영역 또는 하위 영역에 대한 액세스 제어 및 데이터 보호를 구현하기 위해 라벨링 메커니즘을 어떻게 사용할 수 있는 지를 보여준다. 간단한 경우는 하나의 라벨에 의해 식별된 모든 게놈 영역에 대해 하나의 액세스 제어 규칙(AC)과 하나의 보호 메커니즘(예를 들어, 암호화)을 사용한다.
도 55는 동일한 라벨에 의해 식별되는 다른 게놈 영역이 몇 개의 다른 액세스 제어 규칙(AC) 및 몇 개의 다른 암호화 키로 어떻게 보호될 수 있는 지를 보여준다.
도 56은 서명된 POS 디스크립터가 계산된 기준에서 판독의 매핑 위치를 인코딩하는데 사용되는 클래스 U의 판독을 대안적으로 인코딩하는 방법을 보여준다.
도 57은 절반 매핑된 판독 쌍이 매핑되지 않은 판독값과 보다 긴 컨틱(contig)을 조합함으로써 기준 서열의 미지의 영역을 채우는 것을 도울 수 있는 방법을 도시한다.
도 58은 본 발명에서 기술된 구조에 따라 저장된 게놈 데이터에 대한 헤더의 계층 구조를 나타낸다.
도 59는 본 발명에 의해 기술된 라벨링 메카니즘을 구현하는 장치가 몇 개의 게놈 영역들이 데이터베이스의 다른 기록에 저장될 때 이들 영역과 관련된 데이터에 대한 동시 액세스를 어떻게 가능하게 하는지를 나타낸다. 이는 제어된 액세스가 존재하거나 존재하지 않는 경우에 일어날 수 있다.
도 60은 데이터의 분리된 하위 클래스를 생성하기 위해 클래스 N, M 및 I의 인코더에서 임계 벡터가 어떻게 사용되는지를 나타낸다.
도 61은 불일치의 전부 또는 하위 세트가 제거되었을 때(즉, 기준 변환이 적용된 후 변환이 클래스 P에 할당되기 전에 클래스 M에 속한 판독값)에 속하는 클래스 판독값을 변경할 수 있는 방법의 예시를 제공한다.
도 62는 판독값으로부터 불일치(MM)를 제거하기 위해 기준 변환을 적용하는 방법을 보여준다. 일부 경우에 있어서, 기준 변환은 새로운 불일치를 생성하거나 변환이 적용되기 전에 기준을 참조할 때 발견된 불일치의 유형을 변경할 수 있다.
도 63은 동일한 기준 변환 A0이 데이터의 모든 클래스에 대해 사용될 수 있거나 또는 다른 변환 AN, AM, AI이 각각의 클래스 N, M, I에 대해 사용되는 방법을 도시 한다.
하기 청구 범위의 특징은 라벨을 사용하여 게놈 데이터의 영역을 선택적으로 액세스하는 방법을 제공함으로써 기존의 종래 기술의 해결책의 문제점을 해결하며, 여기서, 상기 라벨은 기준 게놈 서열의 식별자(521), 상기 게놈 영역의 식별자(522) 및 상기 게놈 데이터의 데이터 클래스의 식별자(523)를 포함한다.
본 방법의 다른 양태에서, 상기 게놈 데이터는 게놈 판독의 서열이다.
본 방법의 또 다른 양태에서, 데이터 클래스는 다음과 같은 유형 또는 그의 하위 세트로 될 수 있다:
· 기준 서열에 대해 어떠한 불일치도 나타내지 않는 게놈 판독을 포함하는 "클래스 P",
· 서열화 기계가 어떠한 "염기"도 호출할 수 없는 위치의 불일치만을 포함한 게놈 판독을 포함하고 상기 불일치의 수가 주어진 임계값을 초과하지 않는 "클래스 N",
· 서열화 기계가 "n 유형" 불일치라고 명명된 어떠한 염기라도 호출할 수 없는 위치 및/또는 서열화 기계가 "s 유형" 불일치라고 명명된 기준 서열과는 다른 염기를 호출하는 위치에 의해 상기 불일치가 구성되는 게놈 판독을 포함하고, 그리고 불일치의 수가 "n 유형", "s 유형" 및 주어진 함수(f (n, s))로부터 얻은 임계값의 불일치 수에 대해 주어진 임계값을 초과하지 않는 "클래스 M",
· 게놈 판독이, "삽입"("i 유형"), "삭제"("d 유형"), 소프트 클립 ("c 유형") 중 적어도 하나의 불일치 이외에도, "클래스 M"과 동일한 유형의 불일치를 가능하게 가질 수 있을 때의 "클래스 I": 여기서, 각각의 유형에 대한 불일치의 수가 상응하는 주어진 임계값 및 주어진 함수(w(n, s, i, d, c))에 의해 제공된 임계값을 초과하지 않음,
· 클래스 P, N, M, I에서 어떠한 분류도 찾지 못하는 모든 판독을 포함하는 "클래스 U".
본 방법의 또 다른 양태에서, 상기 게놈 데이터는 게놈 판독의 쌍으로 된 서열이다.
본 방법의 또 다른 양태에서, 상기 쌍으로 된 판독의 상기 데이터 클래스는 다음과 같은 유형들 또는 이들의 하위 세트일 수 있다:
· 기준 서열에 대해 어떠한 불일치도 나타내지 않는 게놈 판독 쌍을 포함하는 "클래스 P",
· 서열화 기계가 어떠한 "염기"도 호출할 수 없는 위치의 불일치만을 포함한 게놈 판독을 포함하고 각각의 판독에 대한 상기 불일치의 수가 주어진 임계값을 초과하지 않는 "클래스 N",
· 서열화 기계가 어떠한 염기라도 호출할 수 없는 위치의 불일치만을 포함한 게놈 판독쌍을 포함하며 각각의 판독에 대한 상기 불일치의 수는 "n 유형" 불일치라고 명명된 주어진 임계값을 초과하지 않고, 및/또는 서열화 기계가 "s 유형" 불일치라고 명명된 기준 서열과는 다른 염기를 호출하는 위치의 불일치만을 포함한 게놈 판독쌍을 포함하며 상기 불일치의 수가 "n 유형", "s 유형" 및 주어진 함수(f (n, s))로부터 얻은 임계값의 불일치 수에 대해 주어진 임계값을 초과하지 않는 "클래스 M",
· "삽입"("i 유형"), "삭제"("d 유형"), 소프트 클립 ("c 유형") 중 적어도 하나의 불일치 이외에도, "클래스 M" 쌍과 동일한 유형의 불일치를 가능하게 가질 수 있는 판독 쌍을 포함하는 "클래스 I": 여기서, 각각의 유형에 대한 불일치의 수가 상응하는 주어진 임계값 및 주어진 함수(w (n, s, i, d, c))에 의해 제공된 임계값을 초과하지 않음,
· 하나의 판독 메이트(mate)만이 클래스 P, N, M, I 중 어느 하나로 분류되기 위한 매칭(matching) 규칙을 만족시키지 않는 판독 쌍을 포함하는 "클래스 HM",
· 두 개의 판독값이 클래스 P, N, M, I 로 분류되기 위한 매칭 규칙을 만족하지 않는 모든 판독 쌍을 포함하는 클래스 "U".
본 방법의 또 다른 양태에서, 상기 게놈 영역의 상기 식별자는 마스터 인덱스 테이블에 포함된다.
본 방법의 다른 양태에서, 상기 게놈 데이터 및 상기 라벨은 엔트로피 코딩된다.
본 방법의 다른 양태에서, 상기 마스터 인덱스 테이블(4812)은 게놈 데이터 세트 헤더(4813)에 포함된다.
본 방법의 또 다른 양태에서, 상기 게놈 데이터의 영역은 개별적인 액세스 유닛(524, 486) 사이에 분산되어 있다.
본 방법의 다른 양태에서, 파일 내의 게놈 데이터의 상기 영역의 위치는 로컬 인덱스 테이블(525)에 표시된다.
본 방법의 또 다른 양태에 있어서, 상기 라벨은 사용자가 지정한다.
본 방법의 다른 양태에서, 상기 영역은 전체 게놈 파일을 암호화하지 않고 별도의 방식으로 보호 및/또는 암호화된다.
본 방법의 다른 양태에서, 상기 라벨은 게놈 라벨 리스트(GLL)내에 저장된다.
또 다른 양태에 있어서, 본 방법은 이전에 정의된 게놈 데이터의 영역에 선택적으로 액세스하여 게놈 데이터를 인코딩하는 단계를 추가로 포함한다.
본 방법의 또 다른 양태에서, 상기 게놈 라벨 리스트는 다수의 동기화 포인트를 가능하게 하기 위하여 주기적으로 재전송되거나 또는 업데이트된다.
또 다른 양태에 있어서, 본 방법은 앞서 정의된 게놈 데이터의 영역에 선택적으로 액세스하여 게놈 데이터의 스트림 또는 파일을 디코딩하는 단계를 추가로 포함한다.
본 발명은 앞서 정의된 게놈 데이터를 인코딩하기 위한 장치를 추가로 제공한다.
본 발명은 앞서 정의된 게놈 데이터를 디코딩하기 위한 장치를 추가로 제공한다.
본 발명은 상기 정의된 바와 같은 인코딩된 게놈 데이터를 저장하기 위한 저장 수단을 추가로 제공한다.
본 발명은 실행시 적어도 하나의 프로세서로 하여금 이전에 정의된 인코딩 방법을 수행하게 하는 명령어를 포함하는 컴퓨터 판독 가능한 매체를 추가로 제공한다.
본 발명은 실행시 적어도 하나의 프로세서로 하여금 이전에 정의된 디코딩 방법을 수행하게 하는 명령어를 포함하는 컴퓨터 판독 가능한 매체를 추가로 제공한다.
(상세한 설명)
본 발명은 파일 포맷으로 저장된 압축 게놈 데이터의 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합에 대한 선택적 액세스 및 선택적 액세스 제어를 제공하는 라벨링 메커니즘 및/또는 분자를 표현하는 기호의 서열 형태로 게놈 또는 단백질 유전 정보를 저장, 전송, 액세스 및 처리하는데 사용되는 관련 액세스 유닛을 기술한다.
이들 분자로는, 예를 들어, 뉴클레오티드, 아미노산 및 단백질을 들 수 있다. 기호의 서열로서 표현되는 가장 중요한 정보 중 하나는 높은 처리량의 게놈 서열화 장치에 의해 생성되는 데이터이다.
어떤 생물체의 게놈은 보편적으로 그 생물체를 특성화하는 핵산(염기)의 사슬을 나타내는 기호의 문자열로서 표현된다. 종래의 게놈 서열화 기술의 현재 상태는 메타데이터(식별자, 정확도 수준 등)에 관련된 핵산의 여러(수십억 개까지) 문자열의 형태로 게놈의 단편화된 표현만을 생성할 수 있다. 이러한 문자열은 일반적으로 "서열 판독" 또는 "판독"이라고 한다.
게놈 정보 수명주기의 전형적인 단계는 서열 판독 추출(Sequence reads extraction), 맵핑 및 정렬(Mapping and Alignment), 변이체 검출(Variant Detection), 변이체 주석(Variant Annotation), 및 기능적 및 구조적 분석(Functional and Structural Analysis)을 포함한다(도 1 참조).
서열 판독 추출은 인간 조작자 또는 기계에 의해 수행되는 과정으로서, 생물학적 샘플을 구성하는 분자를 나타내는 기호들의 서열 형태로 유전 정보의 단편을 표현하는 과정이다. 핵산의 경우 이러한 분자를 "뉴클레오티드"라고 한다. 추출에 의해 생성된 기호의 서열을 일반적으로 "판독"이라고 한다. 선행 기술에서 이러한 정보는 보편적으로 텍스트 헤더 및 서열화된 분자를 나타내는 기호의 서열을 포함하는 FASTA 파일로서 인코딩된다.
생물체의 DNA를 추출하기 위하여 생물학적 샘플을 서열화할 때 알파벳은 기호(A, C, G, T, N)로 구성된다.
생물체의 RNA를 추출하기 위하여 생물학적 샘플을 서열화할 때 알파벳은 기호(A, C, G, U, N)로 구성된다.
또한, 소위 "모호성 코드(ambiguity codes)"라고 하는 기호의 IUPAC 확장 세트가 서열화 기계에 의해 생성되는 경우, 판독을 구성하는 기호에 사용되는 알파벳은 (A, C, G, T, U, W, S, M, K, R, Y, B, D, H, V, N 또는 -) 이다.
IUPAC 모호성 코드가 사용되지 않을 때, 서열의 품질 점수는 각각의 서열 판독값과 연관될 수 있다. 이러한 경우, 종래 기술의 해결책은 결과의 정보를 FASTQ 파일로서 인코딩한다. 서열화 장치는 다음과 같은 서열 판독에서 오류를 도입할 수 있다:
1. 서열화된 샘플에 실제로 존재하는 핵산을 나타내는 잘못된 기호(즉, 상이한 핵산을 나타내는)의 식별 - 이를 보편적으로 "대체 오류"(불일치)라고 한다 -;
2. 실제로 존재하는 핵산을 지칭하지 않는 부가적인 기호를 하나의 서열 판독에 삽입 - 이를 보편적으로 "삽입 오류"라고 한다 -;
3. 서열화된 샘플에 실제로 존재하는 핵산을 나타내는 기호를 하나의 서열 판독으로부터 삭제 - 이를 보편적으로 "삭제 오류"라고 한다 -;
4. 하나 이상의 단편을 원래 서열의 실체를 반영하지 않는 단일 단편으로 재조합.
"커버리지(coverage)"라는 용어는 기준 게놈 또는 그의 일부가 이용 가능한 서열 판독에 의해 커버될 수 있는 정도를 정량화하기 위하여 문헌에서 사용된다. 커버리지는 다음과 같다:
· 기준 게놈의 일부분이 임의의 이용 가능한 서열에 의해 매핑되지 않을 때에는 partial(1X 미만),
· 기준 게놈의 모든 뉴클레오티드가 서열 판독에 존재하는 하나의 기호 및 서열 판독에 존재하는 단 하나의 기호에 의해 매핑될 때에는 single(1X),
· 기준 게놈의 각 뉴클레오티드가 여러 번 매핑될 때에는 multiple(2X, 3X, NX).
서열 정렬은 서열 간의 기능적, 구조적 또는 진화적 관계의 결과일 수 있는 유사성의 영역을 발견함으로써 서열 판독을 배열하는 과정을 지칭한다. 정렬이 "기준 게놈"이라고 하는 기존의 뉴클레오티드 서열을 참조하여 수행될 때, 이러한 공정을 "매핑"이라고 한다. 또한, 서열 정렬은 기존의 서열(즉, 기준 게놈)없이 수행될 수 있으며, 이 경우의 과정은 종래 기술에서 "드-노브(de novo)" 정렬로서 알려져 있다. 선행 기술의 해결책은 이러한 정보를 SAM, BAM 또는 CRAM 파일에 저장한다. 부분적인 또는 완전한 게놈을 재구성하기 위하여 서열을 정렬하는 개념은 도 3에 예시되어 있다.
변이체 검출(변이체 호출이라고도 함)은 게놈 서열화 기계의 정렬된 출력(NGS 장치에 의해 생성되어 정렬된 서열 판독)을, 다른 기존의 서열에서는 발견될 수 없거나 또는 극히 일부의 기존의 서열에서만 발견될 수 있는 서열화된 유기체의 고유한 특성의 개요로 번역하는 공정이다. 이러한 특성은 연구 중인 유기체의 게놈과 기준 게놈 사이의 차이로서 표현되기 때문에 "변이체"라고 칭한다. 선행 기술의 해결책은 이러한 정보를 VCF 파일이라고 하는 특정 파일 포맷으로 저장한다.
변이체 주석은 변이체 호출 과정에 의해 식별된 게놈 변이체에 대한 기능적 정보를 할당하는 프로세스이다. 이는 게놈 내의 코딩 서열에 대한 그들의 관계 및 코딩 서열과 유전자 생성물에 대한 그들의 영향에 따른 변이체의 분류를 의미한다. 이는 보편적으로 MAF 파일에 저장되는 선행 기술에 속한다.
유전자(및 단백질)의 기능과 구조의 관계를 정의하기 위해 DNA(변이체, CNV = 복제 수 변이, 메틸화, 등) 가닥을 분석하는 과정을 기능적 또는 구조적 분석이라고 한다. 이러한 데이터를 저장하기 위한 종래의 기술에는 몇 가지 다른 해결책이 존재한다.
(게놈 파일 포맷)
본 명세서에서 개시된 발명은 적어도 다음과 같은 양태에 대해 종래 기술의 해결책과는 다른 게놈 서열화 데이터를 표현하고 처리하고 조작하고 전송하기 위하여 압축된 데이터 구조에 적용되는 선택적으로 제어된 데이터 액세스의 정의로 구성된다:
- 본 발명의 양태는 게놈 정보(즉, FASTQ, SAM)의 선행 기술의 표현 포맷에 의존하지 않는다.
- 본 발명의 양태는 다수의 게놈 데이터 세트로 조직된 다수의 서열화 실행에 의해 생성된 데이터에 대한 효율적인 처리 및 선택적인 랜덤 액세스를 지원한다. 다른 서열화 실행으로부터 나온 데이터를 동일한 데이터 구조내로 분할하면 분석자들이 그들에 대한 쿼리를 동시에 수행할 수 있으므로, 집단 유전학 연구에 대한 커다란 장점을 제공한다.
- 본 발명의 양태는 구체적인 특성에 따라 게놈 데이터와 메타데이터의 새로운 독창적인 분류를 구현한다. 서열 판독은 기준 서열로 매핑되고 정렬 과정의 결과에 따라 별개의 클래스로 그룹화된다. 그 결과, 허프만(Huffman) 코딩, 산술 코딩 (CABAC, CAVLAC), 비대칭 수치 시스템, Lempel-Ziv 및 그의 유도물과 같은 다른 특정 압축 알고리즘을 적용하여 보다 효율적으로 인코딩할 수 있는 정보 엔트로피가 낮은 데이터 클래스를 생성한다.
- 본 발명의 양태는 특정 게놈 영역 또는 하위 영역, 또는 영역과 하위 영역의 집합에 상응하는 상기 압축된 데이터 클래스의 선택적인 액세스 및 보호를 가능하게 하는 사용자-정의된 라벨에 의해, 데이터 클래스 또는 데이터 클래스의 하위 세트를 특정 게놈 영역 또는 하위 영역, 또는 영역과 하위 영역의 집합에 연관시키는 신규 방법을 구현한다.
- 본 발명의 양태는 구문(syntex) 요소들, 및 하류 분석 애플리케이션을 위해 더욱 효율적으로 처리되는 표현으로 서열 판독 및 정렬 정보를 전송하는 관련 인코딩/디코딩 프로세스를 정의한다.
계층들(위치 계층, 메이트 거리 계층, 불일치 유형의 계층, 등)에 저장되는 디스크립터를 사용하여 판독값들을 이들의 매핑 및 코딩 한 결과에 따라 분류하면 다음과 같은 이점을 나타낸다:
· 다른 구문 요소가 더 높은 압축 성능을 산출하는 특정 소스 모델에 의해 모델링될 때 정보 엔트로피의 감소.
· 하류 분석 단계에 대한 특정 의미를 가지며 압축된 영역에서 개별적으로 또는 독립적으로 직접 액세스할 수 있는 그룹/계층으로 이미 조직된 데이터에 대해 보다 효율적인 액세스.
· 전체 데이터 컨텐츠를 디코딩(즉, 압축 해제)할 필요없이 필요한 정보만을 액세스함으로써 점진적으로 업데이트할 수 있는 모듈식 데이터 구조의 존재.
· 서열화 기계에 의해 생성된 게놈 정보는 정보 그 자체의 성격과 서열화 과정에 내재된 오류를 완화할 필요성으로 인해 본질적으로 상당히 중복된다. 이는 식별 및 분석되어야 하는 관련 유전자 정보(기준에 대해 변이)가 생성된 데이터의 일부에 지나지 않음을 의미한다. 선행 기술의 게놈 데이터 표현 포맷은 주어진 분석 단계에서 의미 있는 정보를 나머지 정보로부터 "단리"하여 분석 애플리케이션에 의해 즉시로 사용 가능하게 하고 이해할 수 있게 하도록 고안되지 않았다.
· 개시된 발명에 의해 성취되는 해결책은 데이터의 관련 부분이 데이터 전체를 액세스 및 압축 해제할 필요 없이 분석 애플리케이션에 용이하게 이용 가능하고 데이터의 중복이 효과적인 압축에 의해 효율적으로 감소되어 필요한 저장 공간과 전송 대역폭을 최소화하는 방식으로 게놈 데이터를 표현하는데 있다.
본 발명의 핵심 요소는 다음과 같다:
1. 압축된 형태로 소위 액세스 유닛(Au)이라고 하는 구조화 및 사용자-정의된 선택적 액세스 가능한 데이터 요소를 "함유"하는 파일 포맷의 사양서. 이러한 접근법은, 예를 들면, 데이터가 비압축 형태로 구조화되고 나서 전체 파일이 압축되는 종래 기술의 접근법인 SAM 및 BAM과 정반대인 것으로 보여질 수 있다. 본 접근법의 첫번째 명백한 장점은 종래 기술의 접근법에서 불가능하거나 매우 곤란한 압축 도메인내의 데이터 요소에 대해 다양한 형태의 사용자-정의된 구조화된 선택적 액세스를 효율적이고 자연스럽게 제공할 수 있다는 것이다.
2. 게놈 정보를 동종 데이터 및 메타데이터의 특정 "계층"으로 구조화하면 낮은 엔트로피로 특성화된 정보 소스의 다른 모델을 정의하는 것을 가능하게 하는 상당한 장점이 있다. 이러한 모델은 계층들마다 다를 수 있을뿐만 아니라 계층 내의 압축된 데이터가 액세스 유닛에 포함된 데이터 블록으로 분할될 때 각 계층 내에서 다를 수 있다. 이러한 구조화는 데이터 또는 메타데이터의 각 클래스 및 그들의 일부분에 대해 가장 적절한 압축을 사용하여 종래 기술의 접근법에 비해 코딩 효율을 현저하게 향상시킬 수 있게 한다.
3. 상기 정보는 액세스 유닛(AU)에서 구조화되어 게놈 분석 애플리케이션에 의해 사용되는 관련 데이터의 하위 세트가 적절한 인터페이스의 수단으로 효율적이고 선택적으로 액세스할 수 있도록 한다. 이러한 특징들은 데이터에 대한 보다 빠른 액세스를 가능하게 하고 보다 효율적인 처리를 수행할 수 있다.
4. 마스터 인덱스 테이블 및 로컬 인덱스 테이블의 정의는 압축된 데이터의 전체 부피를 디코딩할 필요없이 인코딩된(즉, 압축된) 데이터의 계층에 의해 전달되는 정보에 선택적으로 액세스할 수 있게 한다.
5. 파일 헤더에 존재하는 "라벨 리스트"를 파싱함으로써 특정 사용자-정의된 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역과 관련 데이터 클래스의 집합에 상응하는 AU만 액세스하는 가능성.
6. 연관된 게놈 영역을 식별하는 사용자-정의된 "라벨"에 따라서 AU에 포함된 데이터의 일부 및 다른 Au에 대해 다른 유형의 액세스 제어를 제공하는 가능성.
7. 압축된 도메인에서 선택된 데이터 부분의 효율적인 트랜스코딩을 수행함으로써 새로 발표된 기준 게놈에 비해 재정렬될 필요가 있을 때 이미 정렬 및 압축된 게놈 데이터 세트의 재정렬을 수행하는 가능성. 새로운 기준 게놈의 빈번한 출현은, 모든 데이터 부피가 처리되어야 할 필요가 있기 때문에 트랜스코딩 과정이 새로 발표된 기준에 대해 이미 압축 및 저장된 게놈 데이터를 재정렬하는데 따른 재원 소비 및 시간을 현재 필요로 한다.
본 명세서에서 개시된 방법은 감소된 엔트로피로 구문 요소의 알파벳을 정의하기 위해 게놈 데이터에 대해 이용 가능한 선험적 지식을 활용하는 것을 목표로 한다. 유전체학에서, 이용 가능한 지식은 보편적으로 처리되는 것과 동일한 종의 기존 게놈 서열에 의해 표현되지만, 반드시 필요한 것은 아니다. 예를 들어, 다른 개체의 인간 게놈은 1 %의 일부분 만 다르다. 그러나, 이러한 소량의 데이터는 조기 진단, 개인화된 의학, 맞춤형 약물 합성, 등을 가능하게 하는데 충분한 정보를 포함한다. 본 발명은 관련 정보가 효율적으로 액세스 가능하고 액세스가 선택적으로 제어되고 데이터가 보호될 수 있는 게놈 정보 표현 포맷을 정의하는 것을 목적으로 하며, 상기 정보는 효율적으로 전송 가능하고, 이러한 모든 처리는 압축된 데이터 구조를 처리하도록 수행된다.
본 발명에 사용된 기술적 특징은 다음과 같다:
1. 하나 이상의 가용 데이터 세트를 쿼리할 때 효율적인 데이터 검색 및 처리를 가능하게 하도록 다른 서열화 실행에 의해 생성된 게놈 정보를 다른 게놈 데이터 세트로 분할;
2. 공통 특징을 공유하는 "클래스"에서 게놈 서열 데이터와 메타데이터를 분할;
3. 가능한 한 정보 엔트로피를 줄이기 위해 게놈 데이터가 디스크립터의 "계층" 세트로 분할되는 각 데이터 클래스에 의해 전달된 게놈 정보의 구조를 정의;
4. 전체 코딩된 게놈 정보를 디코딩할 필요없이 코딩된 정보(즉, 압축)의 원하는 계층만을 액세스함으로써 데이터 클래스 및 관련 정보에 대한 선택적 액세스를 가능하게 하는 마스터 인덱스 테이블 및 로컬 인덱스 테이블의 정의;
5. 포인트 2에서 명시된 바와 같이 정의된 데이터 클래스의 다른 계층에 속하는 구문 요소를 코딩하기 위해 다른 소스 모델 및 엔트로피 코더를 사용;
6. 필요하지 않거나 원하는 경우, 모든 계층을 디코딩할 필요없이 데이터에 대한 선택적 액세스를 가능하게 하기 위하여 종속 계층간에 일치성을 확립하는 특정 메커니즘의 정의;
7. 효율적인 선택적 액세스를 가능하게 하는 "라벨"에 의해 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합에 대응하는 압축된 데이터 및 상응하는 데이터 "클래스" 또는 데이터 클래스의 하위 세트를 라벨링하기 위한 메커니즘의 정의;
8. 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합 및 상응하는 데이터 "클래스" 또는 데이터 클래스의 하위 세트, 및 이들의 임의의 조합의 선택적 보호를 위한 메커니즘의 정의;
9. 서열 데이터 표현의 엔트로피를 줄이기 위해 추가로 변환될 수 있는 하나 이상의 기존 또는 구조화된 기준 서열에 대한 데이터 세트 또는 데이터 "클래스"의 코딩.
효율적인 압축 표현에 의해 효율적인 전송 및 저장을 유지하면서 특정 데이터 "클래스", 특정 게놈 영역 또는 하위 영역 또는 영역 또는 하위 영역의 집합에 대한 효율적인 선택적 액세스 및 선택적 액세스 제어의 관점에서 선행 기술의 상기 언급된 모든 문제를 해결하기 위하여, 본 발명의 애플리케이션은 선행 기술의 게놈 데이터 분석 애플리케이션에 의해 요구되는 끊김없는 액세스 및 처리를 가능하게 하는 동종 및/또는 의미적으로 중요한 데이터의 액세스 가능한 유닛으로 적절한 데이터 기록을 구현하는 특정 데이터 구조 사양을 제공한다.
특히, 본 발명은 액세스 유닛의 개념, "라벨" 및 관련 데이터의 멀티플렉싱을 기반으로 한 데이터 구조를 채택하며, 상기 개념은 종래 기술의 모든 게놈 데이터 포맷에서 빠져있다.
게놈 데이터는 다른 액세스 유닛으로 구조화되고 인코딩된다. 이후부터는 다른 액세스 유닛에 포함되어 있고 게놈 데이터를 기준 게놈 대 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합과 연관시킨 "라벨"에 의해 식별될 수 있는 게놈 데이터를 기술할 것이다.
(매칭 규칙에 따른 게놈 데이터 분류)
서열화 기계에 의해 생성된 서열 판독은 개시된 발명에 의해 하나 이상의 기존의 기준 서열에 대한 정렬의 매칭 결과에 따라서 5 개의 상이한 "클래스"로 분류된다.
기준 서열과 관련하여 뉴클레오티드의 DNA 서열을 정렬하는 경우, 다음과 같은 경우를 확인할 수 있다:
1. 기준 서열의 영역은 오류없이 서열 판독과 일치하는 것으로 나타난다(즉, 완벽한 매핑). 이러한 뉴클레오티드의 서열은 "완벽한 매칭 판독" 또는 "클래스 P"로 표시된다.
2. 기준 서열의 영역은 서열 판독을, 판독을 생성하는 서열화 기계가 임의의 염기(또는 뉴클레오티드)를 호출할 수 없는 위치의 수에 의해서만 결정된 불일치의 유형 및 수와 일치하는 것으로 나타난다. 이러한 유형의 불일치는 정의되지 않은 뉴클레오티드 염기를 나타내기 위해 사용되는 문자 "N"으로 표시된다. 본 명세서에서는 이러한 유형의 불일치를 "n 유형" 불일치라고 한다. 이러한 서열은 "N 불일치 판독" 또는 "클래스 N"이라고 한다. 일단 판독이 "클래스 N"에 속하는 경우, 매칭 부정확성 정도를 주어진 상한으로 제한하고 유효한 매칭으로 간주되는 것과 그렇지 않은 것 사이의 경계를 설정하는 것이 유용하다. 그러므로, 클래스 N에 할당된 판독은 또한 판독에 포함될 수 있는 정의되지 않은 염기(즉, "N"이라고 하는 염기)의 최대 수를 정의하는 임계값(MAXN)을 설정함으로써 제한된다. 이러한 분류는 상응하는 기준 서열을 언급할 때 클래스 N에 속하는 모든 판독이 공유하는 필요한 최소 매칭 정확도(또는 최대 불일치 정도)를 암시적으로 정의하며, 이는 압축된 데이터에 선택적 데이터 검색을 적용하는데 유용한 척도를 구성한다.
3. 기준 서열의 영역은 판독을 생성하는 서열화 기계가, 존재하는 경우, 임의의 뉴클레오티드 염기를 호출할 수 없는 위치의 수에 의해 결정된 불일치의 유형 및 수(즉, "n 유형" 불일치) + 기준에 존재하는 염기보다는 다른 염기가 호출되는 불일치의 수와 서열 판독을 일치시키는 것으로 밝혀졌다. 또한, "대체"로 표시되는 이러한 유형의 불일치는 단일 뉴클레오티드 변이(SNV) 또는 단일 뉴클레오티드 다형성(SNP)이라고도 불린다. 본 명세서에서 이러한 유형의 불일치는 "s 유형" 불일치라고도 한다. 이 때, 서열 판독은 "M 불일치 판독"으로서 언급되고 "클래스 M"에 할당된다. "클래스 N"의 경우와 마찬가지로, "클래스 M"에 속한 모든 판독에 대해서도, 매칭 부정확도를 주어진 상한으로 제한하고 유효한 매칭으로 간주되는 대상과 그렇지 않은 대상 사이의 경계를 설정하는 것이 유용하다. 따라서, 클래스 M에 할당된 판독은, 존재한다면, "n 유형"의 불일치 수 "n"(MAXN)에 대한 임계값과 대체 수 "s"(MAXS)에 대한 또 다른 임계값인 한 세트의 임계값을 정의함으로써 제한된다. 세 번째 제약 조건은 숫자 "n"과 "s"의 함수 f(n,s)에 의해 정의된 임계값이다. 이러한 세 번째 제약은 임의의 의미있는 선택적 액세스 척도에 따라서 매칭 부정확도의 상한을 갖는 클래스를 생성할 수 있게 한다. 예를 들어, f(n,s)는 (n + s)1/2 또는 (n + s) 또는 "클래스 M"에 속한 판독에 대해 인정되는 최대 매칭 부정확도 수준으로 경계를 설정하는 선형 또는 비선형 표현이 될 수 있지만, 이에 한정되어 있지 않다. 이러한 경계는 다양한 목적으로 서열 판독을 분석할 때 원하는 선택적 데이터 검색을 압축된 데이터에 적용하는데 매우 유용한 척도를 구성하며, 그 이유는 하나의 유형 또는 다른 유형에 적용된 단순 임계값을 넘는 "n 유형" 불일치 및 "s 유형" 불일치(대체)의 수의 임의의 가능한 조합으로 추가 경계를 설정할 수 있기 때문이다.
4. 제 4 클래스는 "삽입", "삭제"(삽입-결실이라고도 함) 및 "클립된(clipped)" 중 적어도 하나의 불일치 유형 및, 존재한다면, 클래스 N 또는 M에 속하는 불일치 유형을 나타내는 서열 판독에 의해 구성된다. 이러한 서열은 "I 불일치 판독"으로 언급하고 "클래스 I"로 할당된다. 삽입은 기준에는 존재하지 않지만 판독 서열에는 존재하는 하나 이상의 뉴클레오티드의 부가적인 서열로 구성된다. 본 명세서에서는 이러한 유형의 불일치를 "i 유형" 불일치라고 한다. 문헌에서, 삽입된 서열이 서열의 가장자리에 있을 때 이를 "소프트 클립된(soft clipped)"라고도 부른다(즉, 뉴클레오티드는 기준과 일치하지 않지만, 폐기되는 "하드 클립된 (hard clipped)" 뉴클레오티드와는 반대로 정렬된 판독으로 유지된다). 본 명세서에서는 이러한 유형의 불일치를 "c 유형" 불일치라고 한다. 뉴클레오티드를 유지하거나 또는 폐기하는 것은 서열화 기계에 의해 또는 다음과 같은 정렬 단계에 의해 판독이 결정됨에 따라 판독을 수신하고 처리하는, 본 발명에 개시된 판독 분류기가 아니라 얼라이너 스테이지(aligner stage)에 의해 취해지는 결정이다. 삭제는 기준에 대하여 판독의 "홀(hole)"(누락된 뉴클레오티드)이다. 본 명세서에서는 이러한 유형의 불일치를 "d 유형" 불일치라고 한다. 클래스 "N"및 "M"의 경우에서와 같이, 매칭 부정확도에 대한 제한을 정의하는 것은 가능하고 적절하다. "클래스 I"에 대한 제약 조건 세트의 정의는 "클래스 M"에 사용된 것과 동일한 원리를 기반으로 하며 표 1의 마지막 라인에 보고되어 있다. 클래스 I 데이터에 대해 허용 가능한 각 유형의 불일치의 임계값 이외에도, 불일치 "n", "s", "d", "i"및 "c"의 수의 임의의 함수 w(n, s, d, i, c)에 의해 결정되는 임계값으로 추가적인 제약 조건이 정의된다. 이러한 추가적인 제약 조건은 의미있는 사용자-정의된 선택적 액세스 척도에 따라서 매칭 부정확도의 상한을 갖는 클래스를 생성하는 것을 가능하게 한다. 예를 들어, w(n, s, d, i, c)는 (n + s + d + i + c)1/5 또는 (n + s + d + i + c), 또는 "Class I"에 속하는 판독에 대해 허용되는 최대 매칭 부정확도 수준으로 경계를 설정하는 임의의 선형 또는 비선형 표현일 수 있다. 이러한 경계는 허용 가능한 불일치의 각 유형에 적용된 단순한 임계값을 넘어서 "클래스 Ⅰ"에서 허용 가능한 불일치 수의 임의의 가능한 조합으로 추가적인 경계를 설정하게 할 수 있기 때문에 다양한 목적으로 서열 판독을 분석할 때 원하는 선택적 데이터 검색을 압축된 데이터에 적용하는데 매우 유용한 척도를 구성한다.
5. 제 5 클래스는 기준 서열을 참조할 때 각 데이터 클래스에 대해 유효한 것으로 간주된 임의의 매칭을 찾는 모든 판독을 포함한다(즉, 표 1에 명시된 바와 같이 최대 매칭 부정확도에 대한 상한을 정의하는 매칭 규칙 세트를 충족하지 않음). 이러한 서열은 기준 서열을 언급할 때 "매핑되지 않은"이라고 말하며 "클래스 U"에 속하는 것으로 분류된다.
(매칭 규칙에 따른 판독 쌍의 분류)
이전 단락에서 명시된 분류는 단일 서열 판독에 관한 것이다. 2 개의 판독이 가변 길이의 미지의 서열에 의해 분리되는 것으로 알려진 쌍으로 된 판독을 생성하는 서열화 기술(예를 들어, Illumina Inc.)의 경우에, 전체 쌍을 단일 데이터 클래스로 분류하는 것을 고려하는 것이 적절하다. 다른 판독과 결합된 판독을 "메이트"라고 한다.
쌍으로 된 판독이 모두 동일한 클래스에 속하고 전체 쌍의 클래스로 할당한 것이 분명하다면, 전체 쌍은 임의의 클래스(즉, P, N, M, I, U)에 대해 동일한 클래스로 할당된다. 두 개의 판독들이 다른 클래스에 속하지만 그들 중 어떠한 것도 "클래스 U"에 속하지 않는 경우, 전체 쌍은 다음과 같은 수식에 따라 정의된 가장 높은 우선 순위를 가진 클래스에 할당된다:
P <N <M <I
상기식에서, "클래스 P"는 가장 낮은 우선 순위를 가지며, "클래스 I"는 가장 높은 우선 순위를 갖는다.
판독 중 하나만이 "클래스 U"와 임의의 클래스 P, N, M, I 중 하나와의 메이트에 속하는 경우, 여섯 번째 클래스는 "절반 매핑된(Half Mapped)"을 나타내는 "클래스 HM"으로 정의된다.
이러한 특정 클래스의 판독의 정의는 기준 게놈에 존재하는 갭(gap) 또는 미지 영역(거의 알려지지 않거나 미지의 영역이라고도 함)을 결정하려는 시도로 사용된다는 사실에 의해 동기 부여된다. 이러한 영역은 알려진 영역에 매핑될 수 있는 쌍 판독을 사용하여 가장자리에서 쌍을 매핑함으로써 재구성된다. 이 때, 매핑되지 않은 메이트는 도 57에 나타난 바와 같이 미지의 영역의 소위 "콘틱"을 만드는데 사용된다. 따라서, 오로지 이러한 유형의 판독 쌍에 대한 선택적 액세스를 제공하면 연관된 연산 부담을 크게 줄이므로 종래 기술의 해결책을 사용하여 전체 검사해야 하는 대량의 데이터 세트에 의해 발생하는 데이터를 훨씬 효율적으로 처리할 수 있다.
하기 표에는 각각의 판독이 속하는 데이터 클래스를 정의하기 위해 판독에 적용되는 매칭 규칙이 요약되어 있다. 상기 규칙은 표의 첫 번째 5 컬럼에서 불일치 유형(n, s, d, i 및 c 유형 불일치)의 유무에 따라 정의되어 있다. 여섯 번째 컬럼은 각각의 불일치에 대한 최대 임계값 및 가능한 불일치 유형의 임의의 함수 f (n,s) 및 w (n, s, d, i, c)에 관한 규칙을 제공한다.

(매칭 규칙은 서열 판독 데이터 클래스 N, M 및 I를 다른 매칭 정확도를 갖는 하위 클래스로 분류한다.)
이전 단락에서 정의된 N, M 및 I 유형의 데이터 클래스는 다른 매칭 정확도를 갖는 별개의 하위 클래스의 무작위 수로 추가로 분해될 수 있다. 이러한 선택은 보다 정밀한 세분성을 제공하고 결과적으로 각 데이터 클래스에 대한 훨씬 더 효율적인 선택적 액세스를 제공하는데 있어서 중요한 기술적 장점이다. 이에 제한되어 있지 않지만 예시로서, 클래스 N을 하위 클래스의 수 k(하위 클래스 N1,..., 하위클래스Nk)로 분할하기 위해서는 대응하는 구성요소 MAXN1, MAXN2,..., MAXN(k-1), MAXN(k)을 갖는 벡터를 정의할 필요가 있으며, 이를 위한 조건으로는 MAXN1<MAXN2<...<MAXN(k-1)<MAXN이고 벡터의 각 성분에 대해 평가할 때 표1에 명시된 제약 조건을 만족하는 가장 낮은 등급의 하위 클래스에 각각의 판독값이 할당되어야 한다. 이는 데이터 분류 유닛(601)이 클래스 P, N, M, I, U, HM 인코더 및 주석 및 메타데이터에 대한 인코더를 포함하는 도 60에 나타나 있다. 클래스 N 인코더는 N 데이터(606)의 k 하위 클래스를 생성하는 임계값의 벡터인 MAXN1 내지 MAXNk(606)으로 구성된다.
유형 M과 I의 클래스의 경우, MAXM과 MAXTOT 각각에 대해 동일한 특성을 갖는 벡터를 정의함으로써 동일한 원리를 적용하고 함수 f (n, s) 및 w( n, s, d, i, c)가 제약 조건을 만족시키는 지를 검사하기 위한 임계값으로서 각 벡터 성분을 사용한다. N 유형의 하위 클래스에서와 같이, 제한 조건이 충족되는 최하위 클래스에 할당이 제공된다. 각 클래스 유형에 대한 하위 클래스의 수는 독립적이며 하위 분할의 어떠한 조합도 허용 가능하다. 이는 클래스 M 인코더 및 클래스 I 인코더가 각각 임계값 MAXM1 내지 MAXMj(603) 및 MAXTOT1 내지 MAXTOTh(604)의 벡터로 구성되는 도 60에 나타나 있다. 2개의 인코더는 M데이터(607)의 j하위 클래스 및 I데이터(608)의 h하위 클래스를 각각 생성한다. 한 쌍의 두 개의 판독이 동일한 하위 클래스로 분류되면 그 쌍은 동일한 하위 클래스에 속한다.
한 쌍의 두 개의 판독이 다른 클래스의 하위 클래스로 분류될 때 그 쌍은 다음과 같은 수식에 따라서 보다 높은 우선 순위의 클래스의 하위 클래스에 속한다:
N <M <I
여기서, N은 가장 낮은 우선 순위를 가지며, I는 가장 높은 우선 순위를 가진다.
두 개의 판독이 클래스 N 또는 M 또는 I 중 하나의 다른 하위 클래스에 속하면, 그 쌍은 다음과 같은 수식에 따라서 가장 높은 우선 순위를 갖는 하위 클래스에 속한다:
N1<N2<...<Nk
M1<M2<...<Mj
I1<I2<...<Ih
여기서, 가장 높은 인덱스가 가장 높은 우선 순위를 갖는다.
("외부" 기준 서열의 변환)
클래스 N, M 및 I로 분류된 판독에 대해 밝혀진 불일치는 판독 표현을 보다 효율적으로 압축하기 위해 사용되는 "변환된 기준"을 생성하는데 사용될 수 있다.
클래스 N, M 또는 I에 속하는 것으로 분류된 판독(RS0으로 표시된 기존(즉, "외부")의 기준 서열과 관련 있음)은 변환된 기준과의 실제 불일치의 발생에 따라서 "변환된"기준 서열 RS1에 대해 코딩될 수 있다. 예를 들어, (클래스 M의 i 번째 판독으로서 표시된) 클래스 M에 속하는 readM in 이 기준서열 RSn에 대한 불일치를 포함하면, "변환" 후에 readM in = readP i(n+1) 은 A (Refn) = Refn+1(여기서, A는 기준 서열 RSn+1로부터 RSn으로의 변환이다)로 얻어질 수 있다.
도 61은 기준 서열 1(RS1)에 대해 (클래스 M에 속하는) 불일치를 함유한 판독이 불일치 위치에 대응하는 염기를 변성시킴으로써 수득한 기준 서열 2(RS2)에 대해 완벽한 매칭 판독으로 어떻게 변환될 수 있는지에 대한 예시를 나타낸다. 이들은 분류된 채로 남아 있으며 동일한 데이터 클래스 액세스 유닛에서 다른 판독과 함께 코딩되지만, 코딩은 클래스 P 판독에 필요한 디스크립터 및 디스크립터 값만 사용하여 수행된다. 이러한 변환은 다음과 같이 나타낼 수 있다:
RS2 = A(RS1)
RS1에 적용될 때 RS2를 생성하는 변환 A의 표현과 판독 대 RS2의 표현이 클래스 M 대 RS1의 판독의 표현보다 낮은 엔트로피에 대응하면, 변환 A의 표현 및 판독 대 RS2의 대응하는 표현을 전송하는 것이 유리하며, 그 이유는 데이터 표현의 보다 높은 압축이 달성되기 때문이다.
압축된 비트 스트림에서 전송하기 위한 변환 A의 코딩은 하기 표에 정의된 바와 같이 두 개의 부가적인 구문 요소의 정의를 필요로 한다.

도 62는 맵핑된 판독상에 코딩되는 불일치 수를 줄이기 위해 기준 변환이 어떻게 적용되는 지에 대한 예시를 나타낸다.
몇 몇 경우에, 기준에 적용된 변환은 다음과 같이 관측되어야 한다:
· 변환을 적용하기 전에 기준을 참조할 때 존재하지 않았던 판독의 표현에서 불일치가 생길 수 있다.
· 불일치의 유형을 수정할 수 있다. 판독에는 G 대신 A가 포함될 수 있는 반면에, 다른 모든 판독에서는 G 대신 C가 포함지만 불일치는 동일한 위치에 남아 있다.
· 상이한 데이터 클래스 및 각 데이터 클래스의 데이터의 하위 세트는 동일한 변환된 기준 서열, 또는 동일한 기존의 기준 서열에 상이한 변환을 적용함으로써 얻어진 기준 서열을 지칭할 수 있다.
도 61은 기준 변환이 적용되고 변환된 기준을 사용하여 판독이 판독이 표현된 후, 적절한 세트의 디스크립터(예를 들어, 클래스 M의 판독을 코딩하기 위하여 클래스 P의 디스크립터를 사용)를 사용함으로써 데이터 클래스로부터의 코딩 유형을 다른 코딩 유형으로 어떻게 변경할 수 있는 지의 예시를 나타낸다. 이는, 예를 들어, 변환이 판독에 실제로 존재하는 염기의 판독 불일치에 상응하는 모든 염기를 변경시키고 이에 따라 클래스 M에 속하는 판독(변환되지 않은 원래의 기준 서열을 나타낼 때)을 클래스 P의 가상 판독(변환된 기준을 나타낼 때)으로 가상적으로 변환시킬 때 일어난다. 각 데이터 클래스에 사용되는 디스크립터 세트의 정의는 다음 단락에서 제공된다.
도 63은 다른 클래스의 데이터가 판독을 재 인코딩하기 위해 동일한 "변환된" 기준 R1 = A0(R0)(630)을 어떻게 사용하는지 또는 상이한 형질변환 AN(631), AM(632), AI(633)이 데이터의 각 클래스에 개별적으로 어떻게 적용될 수 있는지를 나타낸다.
(전역 파라미터에 대한 게놈 데이터 헤더)
상기 게놈 데이터의 데이터 구조는 디코딩 엔진에 의해 사용되는 전역 파라미터 및 메타데이터의 저장을 필요로 한다. 이러한 데이터는 다음과 같은 구조로 조직된다:
파일 기반 저장의 경우:
· 데이터 세트 멀티플렉스 헤더
· 데이터 세트 헤더
· 디스크립터 계층 헤더
· 블록 헤더
이들 헤더 간의 계층적 관계는 도 58에 나타나 있다.
스트리밍 시나리오로 전송하는 경우:
· 데이터 세트 매핑 테이블 리스트
· 데이터 세트 매핑 테이블
· 전송 블록 헤더
· 패킷 헤더
데이터 세트는 단일 게놈 서열화 실행 및 모든 후속적인 분석과 관련된 게놈 정보를 재구성하는데 필요한 코딩 요소의 총체로서 정의된다. 동일한 게놈 샘플이 두 개의 별개의 실행에서 두 번 서열화되면, 획득된 데이터는 두 개의 별개의 데이터 세트로 인코딩될 것이다.
(데이터 세트 멀티플렉스 헤더)
이것은 "멀티플렉스"로 합쳐진 하나 이상의 데이터 세트 앞에 추가된 데이터 구조이다.

이것은 인코딩된 데이터 세트 앞에 추가되는 데이터 구조이다.


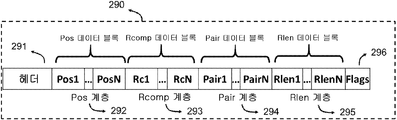
디스크립터(구문 요소라고도 함)는 본 명세서의 다음 단락에서 기술되며 본 발명에 의해 기술된 게놈 정보 표현의 빌딩 블록이다. 이들은 각 디스크립터의 특정 통계적 속성에 따라서 분할된 동종 요소의 계층(디스크립터 스트림이라고도 함)으로 구성된다. 이는 각 계층의 엔트로피를 줄이고 압축 효율을 향상시키는 이점이 있다.
각 계층은 하기 설명된 디스크립터 계층 헤더에 의해 앞에 추가된다.

(블록 헤더)
모든 디스크립터 계층은 하나 또는 다수의 게놈 데이터 블록으로 구성된다. 다른 계층으로부터의 하나 이상의 블록은 데이터 클래스에 따라 액세스 유닛을 구성한다.
액세스 유닛은 전역적으로 사용 가능한 데이터(예를 들어, 디코더 구성)만을 사용하거나 또는 다른 액세스 유닛에 포함된 정보를 사용함으로써 다른 액세스 유닛과 독립적으로 디코딩할 수 있는 게놈 블록의 세트이다.

(서열 판독을 디스크립터 계층으로 표현하는데 필요한 정보의 정의)
일단 클래스의 정의로 판독의 분류가 완료되면, 추가 처리는 주어진 기준 서열에 매핑되는 것으로 표현될 때 DNA 판독 서열의 재구성을 가능하게 하는 나머지 정보를 나타내는 일련의 별개의 구문 요소를 정의하는 것으로 구성된다.
주어진 기준 서열에 대한 서열 판독(예를 들어, DNA 단편)은 다음과 같이 완전히 나타낼 수 있다:
· 기준 서열의 시작 위치 pos(292).
· 판독값이 역 보완 대 기준으로서 간주되어야 하는지를 신호하는 플래그rcomp(293).
· 쌍으로 된 판독 쌍의 경우 메이트 쌍까지의 거리(294).
· 서열화 기술의 경우 판독 길이(295)의 값은 가변성 길이 판독값을 생성한다. 일정한 판독 길이의 경우, 각 판독과 관련된 판독 길이는 명백히 생략될 수 있고 게놈 데이터 세트 헤더에 저장될 수 있다.
· 각 불일치에 대하여:
- 클래스 N에 대한 불일치 위치 nmis(300), 클래스 M에 대해서는 snpp(311) 및 클래스 I에 대해서는 indp(321)
- 불일치 유형(클래스 N에는 존재하지 않음, 클래스 M의 snpt(312), 클래스 I의 indt(322))
· 다음과 같은 서열 판독의 특정 특성을 나타내는 플래그(296):
- 서열 내에 다수의 단편이 있는 템플릿
- 정렬기에 따라서 적당히 정렬된 각 단편
- 매핑되지 않은 단편
- 매핑되지 않은 템플릿내의 다음 단편
- 첫 번째 또는 마지막 단편의 신호화
- 품질 관리 실패
- PCR 또는 광학 복제
- 2 차 정렬
- 보조 정렬
· 클래스 I에 대해 존재할 때 소프트 클립된 뉴클레오티드 문자열(323)
· 적용 가능한 경우 (디스크립터 rtype) 정렬 및 압축에 사용된 기준(예를 들어, 클래스 U의 내부 기준)을 나타내는 플래그.
· 클래스 U에 대해, 디스크립터 indc는 일치하지 않는 판독 부분(일반적으로 가장자리)을, 매칭 정확도 제약 조건의 명시된 세트와 "내부"기준 서열과 함께 식별한다.
· 디스크립터 ureads는 "외부"(즉, 실제 기준 게놈과 같이 이전에 존재하는) 또는 "내부" 기준 서열인 사용 가능한 기준상에 매핑할 수 없는 판독을 축어적으로 인코딩하는데 사용된다.
이러한 분류는 게놈 서열 판독을 명료하게 표현하는데 사용할 수 있는 디스크립터(구문 요소)의 그룹을 생성한다. 하기 표는 "기존"(즉, "외부") 또는 "구조화된"(즉, "내부") 기준과 정렬된 각 판독 클래스에 필요한 구문 요소를 요약하고 있다.

클래스 P에 속한 판독은 특성화되며, 메이트 쌍, 일부 플래그 및 판독 길이를 산출하는 서열화 기술에 의해 수득되는 경우에 위치, 역 보완 정보 및 메이트들 간의 오프셋만으로 완벽하게 재구성될 수 있다.
다음 단락에서는 이들 디스크립터가 클래스 P, N, M 및 I에 대해 어떻게 정의되는 지를 설명하지만, 클래스 U에 대해서는 이후 단락에서 설명한다.
클래스 HM은 판독 쌍에만 적용되며, 그것은 하나의 판독이 클래스 P, N, M 또는 I에 속하고 다른 판독이 클래스 U에 속하는 특별한 경우이다.
(위치 디스크립터 계층)
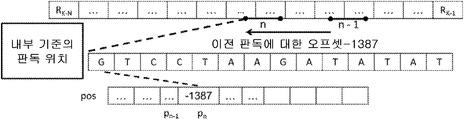
각 액세스 유닛에서, 첫 번째 인코딩된 판독의 매핑 위치만이 기준 게놈의 절대 위치로서 AU 헤더에 저장된다. 다른 모든 위치는 이전 위치와의 차이로 표시되며 특정 계층에 저장된다. 판독 위치의 서열에 의해 정의되는 정보 소스의 이러한 모델링은 일반적으로 높은 커버리지 결과를 산출하는 서열 과정에 대해 감소 된 엔트로피를 특징으로 한다. 첫 번째 정렬의 절대 위치가 저장되면 다른 판독의 모든 위치가 첫 번째 정렬과의 차이(거리)로서 표시된다.
예를 들어, 도 4는 첫 번째 정렬의 시작 위치를 기준 서열의 위치 "10000"으로 인코딩한 후 위치 10180에서 시작하는 두 번째 판독 위치를 "180"으로서 코딩하는 방법을 나타낸다. 높은 커버리지 데이터(> 50x)를 사용하면 위치 벡터의 대부분의 디스크립터는 0과 1 및 기타 작은 정수와 같은 낮은 값이 매우 많이 발생할 것이다. 도 10은 3 개의 판독 쌍의 위치가 pos 계층에서 인코딩되는 방법을 나타낸다.
동일한 소스 모델은 클래스 N, M, P 및 I에 속하는 판독 위치에 사용된다. 데이터에 대한 선택적 액세스 조합을 가능하게 하기 위하여, 4 개의 클래스에 속하는 판독 위치는 표 1에 나타낸 바와 같이 별개의 계층으로 인코딩된다.
(역 보완 디스크립터 계층)
서열화 기술에 의해 생성된 판독 쌍의 각각의 판독은 서열화된 유기 샘플의 게놈 가닥으로부터 유래될 수 있다. 그러나, 두 가닥 중 하나만 기준 서열로 사용된다. 도 8은 판독 쌍에서 하나의 판독(판독 1)이 한 가닥에서 유래되고 다른 하나(판독 2)가 다른 가닥에서 유래될 수 있는 방법을 보여준다.
가닥 1이 기준 서열로서 사용될 때, 판독 2는 가닥 1상의 상응하는 단편의 역 보완으로서 인코딩될 수 있다. 이는 도 9에 나타나 있다.
결합된 판독의 경우, 4 가지가 직접 및 역 보완 메이트 쌍의 가능한 조합이다. 이는 도 10에 나타나 있다. rcomp 계층은 4 가지 가능한 조합을 코딩한다.
클래스 P, N, M, I에 속하는 판독들의 역 보완 정보에 대해서도 동일한 코딩이 사용된다. 데이터에 대한 선택적인 액세스를 향상시키기 위해, 4 개의 클래스에 속하는 판독들의 역 보완 정보는 표 3에 나타낸 바와 같이 상이한 계층으로 코딩된다.
(페어링(Paring) 디스크립터 계층)
페어링 디스크립터는 쌍 계층에 저장된다. 이러한 계층은 사용된 서열화 기술이 쌍으로 판독을 생성할 때 원래 판독 쌍을 재구성하는데 필요한 정보를 인코딩하는 디스크립터를 저장한다. 본 발명의 개시 시점에서, 대다수의 서열 데이터가 쌍으로 된 판독을 생성하는 기술을 사용하여 생성된다 하더라도 이것이 모든 기술의 경우는 아니다. 이는 관련 게놈 데이터의 서열화 기술이 쌍으로 된 판독 정보를 생성하지 않으면 모든 서열 데이터 정보를 재구성하는데 상기 계층의 존재가 필요하지 않기 때문이다.
정의:
· 메이트 쌍: 판독 쌍에서 또 다른 판독과 연관된 판독(예를 들어, 판독 2는 도 4의 예시에서 판독 1의 메이트 쌍임).
· 한 쌍의 거리: 두 번째 판독의 한 위치(예를 들어, 두 번째 판독의 첫 번째 뉴클레오티드)에서부터 제 1 판독의 한 위치(페어링 앵커(pairing anchor), 제 1 판독의 마지막 뉴클레오티드)를 분리하는 기준 서열상의 뉴클레오티드 위치의 번호
· 가장 유망한 페어링 거리(MPPD): 이것은 뉴클레오티드 위치의 번호로 표현되는 가장 유망한 페어링 거리이다.
· 위치 페어링 거리(PPD): PPD는 특정 위치 디스크립터 계층에 존재하는 각각의 메이트로부터 하나의 판독을 분리하는 판독 번호의 관점에서 페어링 거리를 표현하는 방식이다.
· 가장 유망한 위치 페어링 거리(MPPPD): 특정 위치 디스크립터 계층에 존재하는 메이트 쌍으로부터 하나의 판독을 분리하는 가장 유망한 판독 번호이다.
· 위치 페어링 오류(PPE): MPPD 또는 MPPPD와 메이트의 실제 위치 간의 차이로 정의된다.
· 페어링 앵커: 뉴클레오티드 위치 번호 또는 판독 위치 번호의 관점에서 메이트 쌍의 거리를 계산하기 위한 기준으로서 사용된 쌍에서 마지막 뉴클레오티드의 제 1 판독 위치.
도 5는 판독 쌍 간의 페어링 거리를 계산하는 방법을 보여준다.
쌍 디스크립터 계층은 정의된 디코딩 페어링 거리에 대해 쌍의 제 1 판독의 메이트 쌍에 도달하기 위해 건너뛰는 판독 번호로서 계산된 페어링 오류 벡터이다.
도 6은 페어링 오류가 절대값 및 차등 벡터(높은 커버리지에 대한 낮은 엔트로피를 특징으로 함) 둘 다로서 계산되는 방법의 예시를 보여준다.
클래스 N, M, P, I에 속하는 판독의 페어링 정보에 대해서는 동일한 디스크립터가 사용된다. 다른 데이터 클래스에 대한 선택적 액세스를 가능하게 하기 위하여, 4 개의 클래스에 속한 판독의 페어링 정보는 도 6에 나타낸 바와 같이 다른 계층으로 인코딩된다.
(다른 기준에 매핑된 판독의 경우에서 페어링 정보)
기준 서열상의 서열 판독을 매핑하는 과정에서, 하나의 기준(예를 들어, 염색체 1) 및 다른 기준(예를 들어, 염색체 4)에 매핑된 한 쌍의 제 1 판독을 갖는 것은 드문 일이 아니다. 이 경우에, 전술한 페어링 정보는 판독들 중 하나를 매핑하는데 사용된 기준 서열과 관련된 추가 정보에 의해 통합되어야 한다. 이것은 다음의 코딩에 의해 달성된다:
1. 두 개의 다른 서열상에 쌍이 매핑되는 것을 나타내는 예약값(플래그) (다른 값은 판독1 또는 판독2가 현재 인코딩되지 않은 서열상에 매핑되는지의 여부를 나타낸다).
2. 표 2에 설명된 바와 같이 게놈 데이터 세트 헤더 구조로 인코딩된 기준 식별자를 참조하는 고유한 기준 식별자.
3. 위 2번에서 식별된 기준에 대한 매핑 정보를 포함하고 마지막 인코딩된 위치에 대하여 오프셋으로서 표현되는 세 번째 요소.
도 7은 이러한 시나리오의 예시를 제공한다.
도 7에서, 판독 4는 현재 인코딩된 기준 서열상에 매핑되지 않으므로, 게놈 인코더는 쌍 계층에서 부가적인 디스크립터를 만들어 상기 정보를 신호한다. 도 7에 도시된 예시에서, 쌍(2)의 판독(4)은 도면 부호(4)에 맵핑되는 반면에, 현재 인코딩된 기준은 도면 부호(1)이다. 이러한 정보는 다음 3 개의 구성 요소를 사용하여 인코딩된다:
1) 하나의 특정 예약값이 페어링 거리로서 인코딩된다(이 경우, 0xffffff).
2) 두 번째 디스크립터는 게놈 데이터 세트 헤더(이 경우 4에서)에 나열된 바와 같은 기준 ID를 제공한다.
3) 세 번째 요소는 관련 기준(170)에 대한 매핑 정보를 포함한다.
(클래스 N 판독에 대한 불일치 디스크립터)
클래스 N은 A, C, G 또는 T 염기의 위치에서 "n 유형" 불일치만이 존재하는 모든 판독을 포함하며 N은 염기라고 한다. 판독의 다른 모든 염기는 기준 서열과 완벽하게 일치한다.
도 1은 다음과 같은 방법을 보여준다:
판독 1에서 "N"의 위치는
· 판독 1의 절대 위치 또는
· 동일 판독에서 이전의 "N"에 대한 차등 위치로서 코딩된다.
판독 2에서 "N"의 위치는
· 판독 2 + 판독 1 길이에서의 절대 위치 또는
· 이전의 N에 대한 차등 위치로서 코딩된다.
nmis 계층에서, 각 판독 쌍의 코딩은 특정 "세퍼레이터(separator)" 기호에 의해 종료된다.
(인코딩 대체 (불일치 또는 SNP))
대체는 동일한 위치에서 기준 서열에 존재하는 것과 관련하여 상이한 뉴클레오티드의 매핑된 판독에서의 존재로서 정의된다(도 12 참조).
각각의 대체는
· "위치"(snpp 계층) 및 "유형"(snpt 계층) (도 13, 도 14, 도 16 및 도 15 참조), 또는
· 불일치 유형 당 하나의 snpp 계층만 사용하는 "위치" (도 17 참조)로서 인코딩될 수 있다.
(대체 위치)
대체 위치는 nmis 계층의 값에 대해 계산된다. 즉,
판독 1에서의 대체는
· 판독 1의 절대 위치 또는
· 동일한 판독에서 이전 대체에 대한 차등 위치로서 인코딩된다.
판독 2에서의 대체는
· 판독 2 + 판독 1 길이에서 절대 위치 또는
· 이전 대체에 대한 차등 위치로서 인코딩된다. 도 13은 대체 위치가 계층 snpp에서 인코딩되는 방법을 보여준다. 대체 위치는 절대값 또는 차등값으로서 계산될 수 있다.
snpp 계층에서, 각 판독 쌍의 인코딩은 특정 "세퍼레이터" 기호에 의해 종료된다.
(대체 유형 디스크립터)
클래스 M(및 다음 단락에서 기술된 바와 같은 I)에 대하여, 불일치는 판독 {A, C, G, T, N, Z}에 있는 상응하는 대체 기호와 관련하여 존재하는 실제 기호로부터 인덱스(오른쪽에서 왼쪽으로 이동)에 의해 코딩된다. 예를 들어, 정렬된 판독값이 기준 내의 동일한 위치에 존재하는 T 대신에 C를 나타내는 경우, 불일치 인덱스는 "4"로서 표시될 것이다. 디코딩 프로세스는 인코딩된 구문 요소인, 기준에 대한 주어진 위치에서 뉴클레오티드를 판독하고 왼쪽에서 오른쪽으로 이동하여 디코딩된 기호를 검색한다. 예를 들어, G가 기준에 존재하는 위치에 대해 수신된 "2"는 "N"으로서 디코딩될 것이다. 도 14는 IUPAC 모호성 코드가 사용되지 않을 때 모든 가능한 대체 및 각각의 인코딩 기호을 나타내며, 도 15는 snpt 계층에서 대체 유형의 인코딩의 예시를 제공한다.
IUPAC 모호성 코드가 있는 경우 대체 인덱스는 도 16에 나타난 바와 과 같이 변경된다.
상기 기술된 대체 유형의 인코딩이 높은 정보 엔트로피를 제공하는 경우, 대체 인코딩의 택일적 방법은 도 17에 예시된 바와 같이, 불일치 위치만을 뉴클레오티드 당 하나의 분리된 계층에 저장하는데 있다.
(삽입 및 삭제 인코딩)
클래스 I에 대해, 불일치 및 삭제는 기준에 존재하는 실제 기호에서부터 판독값 {A, C, G, T, N, Z}에 존재하는 상응하는 대체 기호까지 인덱스(오른쪽에서 왼쪽으로 이동)에 의해 코딩된다. 예를 들어, 정렬된 판독값이 기준에서 동일한 위치에 있는 T 대신 C를 나타내는 경우 불일치 인덱스는 "4"가 된다. 판독값이 A가 기준에 존재하는 삭제를 나타내는 경우 코딩된 기호는 "5"가 된다. 디코딩 과정은 기준에 대해 주어진 위치에서의 뉴클레오티드인 코딩된 구문 요소를 판독하고 왼쪽에서 오른쪽으로 이동하여 디코딩된 심볼을 검색한다. 예를 들어, G가 기준에 존재하는 위치에 대해 수신된 "3"은 서열 판독에서 삭제의 존재를 나타내는 "Z"로서 디코딩될 것이다.
삽입물은 삽입된 A, C, G, T, N에 대해 각각 6, 7, 8, 9, 10으로서 코딩된다.
IUPAC 모호성 코드의 채택의 경우 대체 메커니즘은 정확히 동일하지만, 대체 벡터는 S = A, C, G, T, N, Z, M, R, W, S, Y, K, V, H, D, B로서 확장되고, 삽입은 다른 코드: 16, 17, 18, 19, 20를 사용한다.
도 18 및 도 19는 클래스 I의 판독 쌍에서 대체, 삽입 및 삭제를 인코딩하는 방법의 예시를 보여준다.
파일 포맷, 액세스 유닛 및 멀티플렉싱의 다음 구조는 전술한 코딩 요소들을 참조하여 설명된다. 그러나, 액세스 유닛, 파일 포멧 및 멀티플렉싱은 소스 모델링 및 게놈 데이터 압축의 다른 알고리즘을 사용하는 경우에도 동일한 기술적 이점을 제공한다.
("클래스 U" 및 "클래스 HM"의 맵핑되지 않은 판독에 대한 "내부" 기준의 구성)
클래스 U에 속하는 판독 또는 "클래스 HM"의 매핑되지 않은 쌍의 경우, 이들은 클래스 P, N, M 또는 I 중 임의의 것에 속하기 위해 매칭 정확성 제약 조건의 특정 세트를 만족시키는 임의의 "외부" 기준 서열에 매핑될 수 없기 때문에, 하나 이상의 "내부" 기준 서열은 이들 데이터 클래스에 속하는 판독의 압축된 표현에 대해 구성되어 사용된다.
예를 들어, 다음과 같은 적절한 "내부" 기준을 구성하기 위한 몇 가지 접근법이 가능하며, 이에 제한되어 있지 않다:
· 매핑되지 않은 판독을 적어도 최소 크기(서명)의 공통 연속 게놈 서열을 공유하는 판독을 포함하는 클러스터(cluster)로 분할하는 것. 각 클러스터는 그의 서명으로 고유하게 식별될 수 있다.
· 유의한 순서(예를 들어, 사전식 순서)로 판독을 분류하고 마지막 N 판독을 N + 1의 인코딩에 대한 "내부" 기준으로서 사용하는 것. 이러한 방법은 도 51에 나타나 있다.
· 특정 매칭 정확성 제약 조건 또는 새로운 제약 조건의 세트들에 따라 상기 클래스에 속하는 판독들 모두 또는 관련된 하위 세트를 정렬 및 인코딩할 수 있도록 클래스 U의 판독들의 하위 세트에 대해 소위 "드-노브 어셈블리"를 수행하는 것.
특정 세트의 매칭 정확성 제약 조건들을 충족시키는 "내부" 기준에 대해 코딩된 판독을 매핑할 수 있는 경우, 압축 후 판독을 재구성하는데 필요한 정보는 다음과 같은 유형의 구문 요소를 사용하여 코딩된다:
1. 내부 기준의 판독 번호의 관점에서 내부 기준상의 매칭 부분의 시작 위치(pos 계층). 이러한 위치는 이전에 인코딩된 판독과 관련하여 절대값 또는 차등값으로 인코딩될 수 있다.
2. 내부 기준에서 상응하는 판독의 초반부터 시작 위치의 오프셋(pair 계층). 예를 들어, 일정한 판독 길이의 경우 실제 위치는 pos *length + pair 이다.
3. 불일치 위치(snpp 계층) 및 유형(snpt 계층)으로서 코딩되는 가능성 있게 존재하는 불일치.
4. 내부 기준과 일치하지 않는(또는 일치하지 않지만 정의된 임계값을 초과하는 많은 불일치가 있는) 판독 부분들(전형적으로 쌍에 의해 식별되는 가장자리)은 indc 계층에 인코딩된다. 패딩 동작은 도 51에 나타난 바와 같이 indc 계층에 인코딩된 불일치의 엔트로피를 줄이기 위하여 사용되는 내부 기준 부분의 가장자리에 수행될 수 있다. 가장 적절한 패딩 전략은 처리되는 게놈 데이터의 통계적 특성에 따라서 인코더에 의해 선택될 수 있다. 가능한 패딩 전략은 다음과 같다:
a. 패딩 없음.
b. 현재 인코딩된 데이터의 빈도에 따라 선택되는 일정한 패딩 패턴.
c. 최근 N 인코딩된 판독의 관점에서 정의된 현재 컨텍스트의 통계적 특성에 따른 가변성 패딩 패턴
특정 유형의 패딩 전략은 indc 계층 헤더의 특정값에 의해 신호받을 것이다.
5. 내부 자체 생성된 기준, 외부 기준 또는 비-기준을 사용하여 판독값이 인코딩되었는지 여부를 나타내는 플래그(rtype 계층).
6. 축어적으로 인코딩된 판독(uread).
도 51은 이러한 인코딩 절차의 예시를 제공한다.
도 56은 pos + pair 구문 요소가 기호화된 pos로 대체되는 내부 기준에서 맵핑되지 않은 판독의 대체 인코딩을 보여준다. 이 경우, pos는 판독 n-1의 가장 왼쪽 뉴클레오티드 위치에 대하여 판독 n의 가장 왼쪽 뉴클레오티드 위치의 거리 - 기준 서열상의 위치 관점에서 - 를 나타낼 것이다.
이러한 코딩 접근법은 판독 당 N 개의 시작 위치를 지원하도록 확장될 수 있으므로 두 개 이상의 기준 위치에서 판독이 분할될 수 있다. 이것은 서열화 방법론에서 루프(loop)에 의해 생성된 반복 패턴을 일반적으로 나타내는 매우 긴 판독 (50K + 염기들)를 생성하는 서열화 기술(예를 들어, Pacific Bioscience)에 의해 생성되는 판독을 인코딩하는데 특히 유용할 수 있다. 동일한 접근법은 거의 겹치지 않거나 전혀 겹치지 않는 게놈의 두 개의 별개 부분에 정렬하는 판독으로서 정의된 키메라 서열 판독을 인코딩하는데 사용될 수 있다.
상기 기술된 접근법은 간단한 클래스 U를 넘어 명확하게 적용할 수 있으며 판독 위치(pos 계층)와 관련된 구문 요소를 포함하는 모든 계층에 적용할 수 있다.
(파일 포맷: 마스터 인덱스 테이블의 사용에 의한 게놈 데이터의 영역에 대한 선택적 액세스)
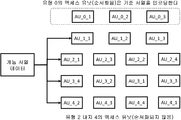
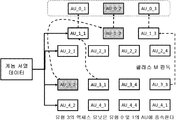
정렬된 데이터의 특정 영역에 대한 선택적 액세스를 지원하기 위하여, 본 문에서 설명하는 데이터 구조는 마스터 인덱스 테이블(MIT)이라고 하는 인덱싱 수단을 구현한다. 이것은 사용된 기순 서열에서 특정 판독값이 매핑되는 위치를 포함하는 다차원 배열이다. MIT에 포함된 값은 각 액세스 유닛에 비- 서열 액세스가 지원되도록 각각의 pos 계층에 대한 제 1 판독의 매핑 위치이다. MIT는 데이터의 각 클래스(P, N, M, I, U 및 HM)와 각 기준 서열 당 하나의 섹션을 포함한다. MIT는 인코딩된 데이터의 게놈 데이터 세트 헤더에 포함되어 있다. 도 20은 게놈 데이터 세트 헤더의 구조를 도시하고, 도 21은 MIT의 일반적인 시각적 표현을 나타내고, 도 22는 인코딩된 판독의 클래스 P에 대한 MIT의 예시를 도시한다.
도 22에 예시된 MIT에 포함된 값은 압축 영역에서 관심 영역(및 상응하는 AU)에 직접 액세스하는데 사용된다.
예를 들어, 도 22를 참조하면, 기준 2에서 위치 150,000과 250,000 사이에 포함된 영역에 액세스해야 하는 경우, 디코딩 애플리케이션은 MIT의 두 번째 기준을 건너 뛰고 두 개의 값 k1과 k2를 검색하여 k1 <150,000 및 k2> 250,000이 되게 할 것이다. 여기서, k1과 k2는 MIT로부터의 2 개의 인덱스 판독값이다. 도 22의 예시에서, 이것은 MIT의 두 번째 벡터의 위치 3과 4가 된다. 이 때, 이들 반환값은 디코딩 애플리케이션에 의해 다음 단락에서 기술되는 바와 같이 pos 계층 로컬 인덱스 테이블로부터 적절한 데이터의 위치를 가져오는데 사용될 것이다.
상기 기술된 게놈 데이터의 4 개의 클래스에 속한 데이터를 포함하는 계층에 대한 포인터와 함께, MIT는 수명주기 동안 게놈 데이터에 추가된 부가적인 메타데이터 및/또는 주석의 인덱스로서 사용할 수 있다.
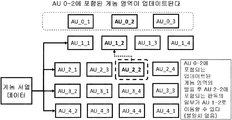
(로컬 인덱스 테이블)
상기 기술된 각 데이터 계층에는 로컬 헤더라고 하는 데이터 구조가 접두어로 붙는다. 로컬 헤더에는 계층의 고유한 식별자, 각 기준 서열 당 액세스 유닛 카운터의 벡터, 로컬 인덱스 테이블(LIT) 및 선택적으로 일부 계층 특정 메타데이터가 포함된다. LIT는 계층 페이로드의 각 AU에 속한 데이터의 물리적 위치에 대한 포인터의 벡터이다. 도 23은 LIT가 비-서열 방식으로 인코딩된 데이터의 특정 영역에 액세스하는데 사용되는 일반적인 계층 헤더 및 페이로드를 예시한다.
상기 예시에서, 기준 서열 번호 2 상에 정렬된 판독의 150,000 내지 250,000 영역에 액세스하기 위하여 디코딩 애플리케이션은 MIT로부터 위치 3 및 4를 검색한다. 이들 값은 디코딩 과정에 의해 LIT의 상응하는 섹션의 3 번째 및 4 번째 요소에 액세스하는데 사용되어야 한다. 도 24에 도시된 예시에서, 계층 헤더에 포함된 총 액세스 유닛 카운터는 기준 1(이 예시에서는 5)과 관련된 AU에 관련되는 LIT 인덱스를 건너뛰는(skip)데 사용된다. 따라서, 인코딩된 스트림에서 요청된 AU의 물리적 위치를 포함하는 인덱스는 다음과 같이 계산된다:
요청된 AU에 속하는 데이터 블록의 위치 = 기준 1의 AU에 속하여 건너 뛰는데이터 블록 + MIT를 사용하여 검색된 위치, 즉,
첫 번째 블록 위치 : 5 + 3 = 8
마지막 블록 위치 : 5 + 4 = 9
로컬 인덱스 테이블이라는 인덱싱 메커니즘을 사용하여 검색된 데이터 블록은 요청된 액세스 유닛의 일부이다.
도 26은 MIT와 LIT를 사용하여 검색된 데이터 블록이 하나 이상의 액세스 유닛을 구성하는 방법을 보여준다.
(액세스 유닛)
데이터 클래스로 분류되고 압축 또는 비 압축 계층으로 구조화된 게놈 데이터는 서로 다른 액세스 유닛으로 구성된다.
게놈 액세스 유닛(AU)은 뉴클레오티드 서열 및/또는 관련 메타데이터 및/또는 DNA/RNA 서열(예를 들어, 가상 기준) 및/또는 게놈 서열화 기계 및/또는 게놈 프로세싱 디바이스 또는 분석 애플리케이션에 의해 생성된 주석 데이터를 재구성하는 (압축되거나 압축되지 않은 형태로) 게놈 데이터의 섹션으로서 정의된다. 액세스 유닛의 예시는 도 26에 제공되어 있다.
액세스 유닛은 전역적으로 사용 가능한 데이터 (예를 들어, 디코더 구성)만을 사용하거나 또는 다른 액세스 유닛에 포함된 정보를 사용함으로써 다른 액세스 유닛과 독립적으로 디코딩할 수 있는 데이터 블록이다.
액세스 유닛은 다음과 같이 차별화된다:
· 게놈 데이터의 성격 및 그들이 지닌 데이터 세트 및 그들이 액세스될 수 있는 방식을 특성화하는 유형,
· 동일한 유형에 속하는 액세스 유닛에 고유한 명령을 제공하는 순서.
모든 유형의 액세스 유닛은 다른 "카테고리"로 추가로 분류할 수 있다.
이후에는 서로 다른 유형의 게놈 액세스 유닛에 대한 정의의 비 한정적인 리스트를 따른다:
1) 유형 0의 액세스 유닛은 액세스하거나 또는 디코딩하여 액세스할 다른 액세스 유닛에서 들어오는 정보를 참조할 필요가 없다. 그들이 포함하는 데이터 또는 데이터 세트에 의해 전송되는 전체 정보는 디코딩 장치 또는 프로세싱 애플리케이션에 의해 독립적으로 판독되고 처리될 수 있다.
2) 유형 1의 액세스 유닛은 유형 0의 액세스 유닛이 전송하는 데이터를 참조하는 데이터를 포함한다. 유형 1의 액세스 유닛에 포함된 데이터를 판독하거나 또는 디코딩하여 처리하는 것은 유형 0의 하나 이상의 액세스 유닛에 액세스해야 함을 필요로 한다. 유형 1의 액세스 유닛은 "클래스 P"의 서열 판독과 관련된 게놈 데이터를 인코딩한다.
3) 유형 2의 액세스 유닛은 유형 0의 액세스 유닛이 전송하는 데이터를 참조하는 데이터를 포함한다. 유형 2의 액세스 유닛에 포함된 데이터를 판독하거나 또는 디코딩하여 처리하는 것은 유형 0의 하나 이상의 액세스 유닛에 액세스해야 함을 필요로 한다. 유형 2의 액세스 유닛은 "클래스 N"의 서열 판독과 관련된 게놈 데이터를 인코딩한다.
4) 유형 3의 액세스 유닛은 유형 0의 액세스 유닛이 전송하는 데이터를 참조하는 데이터를 포함한다. 유형 3의 액세스 유닛에 포함된 데이터를 판독하거나 또는 디코딩하여 처리하는 것은 유형 0의 하나 이상의 액세스 유닛에 액세스해야 함을 필요로 한다. 유형 3의 액세스 유닛은 "클래스 M"의 서열 판독과 관련된 게놈 데이터를 인코딩한다.
5) 유형 4의 액세스 유닛은 유형 0의 액세스 유닛이 전송하는 데이터를 참조하는 데이터를 포함한다. 유형 4의 액세스 유닛에 포함된 데이터를 판독하거나 또는 디코딩하여 처리하는 것은 유형 0의 하나 이상의 액세스 유닛에 액세스해야 함을 필요로 한다. 유형 4의 액세스 유닛은 "클래스 I"의 서열 판독과 관련된 게놈 데이터를 인코딩한다.
6) 유형 5의 액세스 유닛에는 임의의 사용 가능한 기준 서열( "클래스 U")에 매핑할 수 없는 판독을 포함하며 내부적으로 구성된 기준 서열을 사용하여 인코딩된다. 유형 5의 액세스 유닛은 유형 0의 액세스 유닛이 전송하는 데이터를 참조하는 데이터를 포함한다. 유형 5의 액세스 유닛에 포함된 데이터를 판독하거나 또는 디코딩하여 처리하는 것은 유형 0의 하나 이상의 액세스 유닛에 액세스해야 함을 필요로 한다.
7) 유형 6의 액세스 유닛은 하나의 판독이 P, N, M, I의 4 개의 클래스 중 하나에 속할 수 있고 다른 판독이 임의의 사용 가능한 기준 서열( "클래스 HM")에 매핑될 수 없는 판독 쌍을 포함한다. 유형 6의 액세스 유닛은 유형 0의 액세스 유닛이 전송하는 데이터를 참조하는 데이터를 포함한다. 유형 6의 액세스 유닛에 포함된 데이터를 판독하거나 또는 디코딩하여 처리하는 것은 유형 0의 하나 이상의 액세스 유닛에 액세스해야 함을 필요로 한다.
8) 유형 7의 액세스 유닛은 유형 1의 액세스 유닛에 포함된 데이터 또는 데이터 세트와 연관된 메타데이터(예를 들어, 품질 점수) 및/또는 주석 데이터를 포함한다. 유형 7의 액세스 유닛은 다른 계층으로 분류되어 라벨링될 수 있다.
9) 유형 8의 액세스 유닛은 주석 데이터로서 분류된 데이터 또는 데이터 세트를 포함한다. 유형 8의 액세스 유닛은 계층으로 분류되어 라벨링될 수 있다.
10) 추가 유형의 액세스 유닛은 본 문에 기술된 구조와 메커니즘을 확장할 수 있다. 예시로서, 제한적이지 않지만 게놈 변이체 호출, 구조 및 함수 분석의 결과는 새로운 유형의 액세스 유닛으로 인코딩될 수 있다. 본 문에 기술된 액세스 유닛의 데이터 조직은 인코딩된 데이터의 성격과 관련하여 완전히 투명한 메커니즘인 액세스 유닛에 캡슐화 되는 모든 유형의 데이터를 차단하지 않는다.
유형 0의 액세스 유닛은 순서가 매겨지지만(예를 들어, 번호가 매겨짐), 순서대로 저장 및/또는 전송할 필요가 없다(기술적 이점: 병렬 처리/병렬 스트리밍, 멀티플렉싱).
유형 1, 2, 3, 4, 5 및 6의 액세스 유닛은 순서화될 필요가 없으며, 순서대로 저장 및/또는 전송할 필요가 없다(기술적 이점: 병렬 처리/병렬 스트리밍).
도 26은 헤더 및 동종 데이터의 하나 이상의 계층으로 액세스 유닛을 구성하는 방법을 보여준다. 각 계층은 하나 이상의 블록으로 구성될 수 있다. 각 블록은 여러 개의 패킷을 포함하며, 패킷은, 예를 들어, 판독 위치, 페어링 정보, 역 보완 정보, 불일치 위치 및 유형 등을 나타내기 위해 상기 소개된 디스크립터의 구조화된 서열이다.
각각의 액세스 유닛은 각 블록에서 다른 수의 패킷을 가질 수 있지만, 액세스 유닛 내에서 모든 블록은 동일한 수의 패킷을 갖는다.
각 데이터 패킷은 3 개의 식별자 XYZ의 조합으로 식별될 수 있다: 여기서,
· X는 자신이 속한 액세스 유닛을 식별하고,
· Y는 자신이 속한 블록을 식별하고(즉, Y가 캡슐화하는 데이터 유형),
· Z는 동일한 블록 내의 다른 패킷들에 대한 패킷 순서를 나타내는 식별자이다.
도 28은 액세스 유닛 및 패킷 라벨링의 예시를 보여주며, 여기서 AU_T_N 은 액세스 유닛 유형에 따라 순서의 개념을 의미하거나 의미하지 않을 수 있는 식별자 N을 가진 유형 T의 액세스 유닛이다. 식별자는 한 유형의 액세스 유닛을 전송된 게놈 데이터를 완전히 디코딩하는데 필요한 다른 유형의 액세스 유닛과 고유하게 연관시키는데 사용된다.
모든 유형의 액세스 유닛은 다른 서열화 과정에 따라 다른 "카테고리"로 추가로 분류되고 라벨링될 수 있다. 예를 들어, 제한되어 있지 않지만, 분류 및 라벨링은 다음과 같은 경우에 발생할 수 있다:
1. 서로 다른 시간에 동일한 유기체를 서열화하는 경우(액세스 유닛에는 게놈 정보가 "시간적 의미"로 포함되어 있음),
- 동일한 유기체의 서로 다른 성질의 유기 샘플(예를 들어, 인간 샘플에 대한 피부, 혈액, 모발)을 서열화 하는 경우. 이들은 "생물학적" 의미를 가진 액세스 유닛이다.
유형 1, 2, 3, 4, 5 및 6의 액세스 유닛은 그들이 참조하는 유형 0의 액세스 유닛들에서 인코딩된 기준 서열에 대해 게놈 서열 단편들(일명 판독들)에 적용된 매칭 함수의 결과에 따라 구축된다.
예를 들어, 유형 1의 액세스 유닛(AU)(도 30 참조)은 매칭 함수가 유형 0의 AU로 인코딩된 기준 서열의 특정 영역에 적용될 때 완전한 매칭(또는 선택된 매칭 함수에 대응하는 가능한 한 최대 점수)을 유도하는 판독의 위치 및 역 보완 플래그를 포함할 수 있다. 유형 0의 AU에 포함된 데이터와 함께, 이러한 매칭 함수 정보는 유형 1의 액세스 유닛에 의해 운반되는 데이터 세트에 의해 표현된 모든 게놈 서열 판독을 완전히 재구성하기에 충분하다.
본 명세서에서 전술한 게놈 데이터 분류와 관련하여, 상술한 유형 1의 액세스 유닛은 클래스 P의 게놈 서열 판독과 관련된 정보(완벽한 매칭)를 포함한다.
가변성 판독 길이 및 쌍으로 된 판독의 경우, 이전 예시에서 언급된 유형 1의 AU에 포함된 데이터는 판독 쌍의 연관을 비롯하여 게놈 데이터를 완전히 재구성할 수 있도록 판독 페어링 및 판독 길이에 대한 정보를 나타내는 데이터와 통합되어야 한다. 본 명세서에서 이전에 도입된 데이터 분류와 관련하여, pair 와 rlen 계층은 유형 1의 AU로 인코딩된다.
유형 2, 3 및 4에 대한 AU의 콘텐츠를 분류하기 위해 유형 1의 액세스 유닛과 관련하여 적용된 매칭 함수는 다음과 같은 결과를 제공할 수 있다:
1. 유형 1의 AU에 포함된 각 서열은 특이적 위치에 대응하여 유형 0의 AU에 포함된 서열을 완벽하게 매칭한다;
2. 유형 2의 AU에 포함된 각 서열은, 유형 2의 AU 내의 서열에 존재하는 "N" 기호(서열화 디바이스에 의해 호출되지 않은 염기)를 제외하곤, 특이적 위치에 대응하여 유형 0의 AU에 포함된 서열과 완전히 매칭한다;
3. 유형 3의 AU에 포함된 각 서열은 특이적 위치에 대응하여 유형 0의 AU에 포함된 서열에 대한 대체된 기호(변이체)의 형태로 변이체를 포함한다;
4. 유형 4의 AU에 포함된 각 서열은 특이적 위치에 대응하여 유형 0의 AU에 포함된 서열에 대한 대체된 기호(변이체), 삽입 및/또는 삭제의 형태로 변이체를 포함한다;
5. 유형 5의 AU에 포함된 각 서열은 유형 0의 AU에 포함된 서열을 매핑하지 않는다;
6. 유형 6의 AU에 포함된 각각의 서열 쌍은 임의의 클래스 P, N, M 및 I(상기 번호 1 내지 4)에 속할 수 있는 하나의 서열을 나타내는 반면에 다른 서열은 유형 0의 AU에 포함된 서열을 매핑하지 않는다.
유형 0의 액세스 유닛은 순서가 매겨지지만(예를 들어, 번호가 매겨짐), 순서대로 저장 및/또는 전송할 필요가 없다(기술적 이점: 병렬 처리/병렬 스트리밍, 멀티플렉싱).
유형 1, 2, 3, 4, 5 및 6의 액세스 유닛은 순서가 매겨질 필요가 없으며 순서대로 저장 및/또는 전송할 필요가 없다(기술적 이점: 병렬 처리/병렬 스트리밍).
(특정 게놈 영역과 관련된 "라벨"의 사용에 의한 액세스 유닛의 식별)
특정 게놈 영역 또는 하위 영역 또는, 영역 또는 하위 영역의 집합을 나타내는 데이터 클래스에 대한 사용자-정의된 선택적 액세스를 가능하게 하는 추가 메커니즘이 본 발명에 의해 제공된다.
"라벨"은 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합에 할당된 식별자이다. 라벨은 기준 서열 ID("Ref ids"), 기준 서열의 원하는 영역에 대응하는 MIT의 인덱스 및 데이터 클래스를 명시함으로써 게놈 영역을 식별한다. 예시가 도 52에 제공된다.
단일, 하위 세트 또는 모든 데이터 클래스는 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합과 관련된 데이터의 하위 세트에만 선택적으로 액세스를 가능하게 하기 위하여 라벨에 의해 참조될 수 있다.
라벨 목록은 선택적 액세스를 저장 또는 스트리밍된 데이터에 적용하는 분석 애플리케이션에 대해 유용 가능한 라벨을 사용할 수 있게 하기 위하여 스토리지 시나리오 및/또는 스트리밍 시나리오하에 유전체 라벨 발생기(Genomic Labels Generator; 4917 도 49)에 의해 작성되어야 한다.
라벨 리스트에는 다음과 같은 요소가 포함될 수 있다:
· 라벨 번호
· 리스트 내의 각 라벨에 대해:
· 라벨 ID
· 라벨과 관련된 기준 서열 번호
· 각 기준 서열에 대해
· 기준 식별자
· 라벨에 의해 커버된 영역의 수,
· 각 영역에 대해:
· 클래스 ID
· 게놈 영역의 시작 위치
· 게놈 영역의 종료 위치
하기 표는 일반적인 "라벨 리스트"에 대한 의사 구문을 보고한다.

게놈 데이터가 압축되고 스트리밍되는 경우, 블록 헤더 필드(Block Header field) (Label ID")에 의해 특정 "라벨"을 사용하여 하나 이상의 액세스 유닛이 식별될 수 있으며, 상기 블록 헤더 필드는 현재 블록이 속한 "라벨 리스트" 내의 "라벨 "에 대한 식별자로서 역활을 한다. 이러한 필드는 전형적인 스트리밍 시나리오에서 블록을 "라벨"로의 동적 매핑을 가능하게 한다.
게놈 파일 포맷에서, "start_pos" 및 "end_pos" 필드는 다음과 같이 특정 "라벨"에 속한 모든 "블록"을 참조하는 블록 번호에 의해 대체될 수 있다:

"start_pos" 및 "end_pos" 대신에 "블록 번호"의 사용은 관련된 기술적 이점을 제공하는데, 이는 MIT 자체를 직접적으로 어드레스하기 위해 "ref_num", "class_id" 및 "block_num"로 구성된 3차원 벡터가 좌표로 직접 사용될 수 있다는 점을 고려하여 "마스터 인덱스 테이블"(MIT)에 대한 직접적인 액세스를 가능하게 하기 때문이다.
스토리지 시나리오에서, "라벨 리스트"는 게놈 라벨 발생기(4917)에 의해 생성되어 게놈 멀티플렉서로 보내진다(도 49 참조). 디멀티플렉서(Demultiplexer)는 라벨 리스트 구문을 파싱하고 사용 가능한 라벨을 데이터 액세스 애플리케이션에 표시하며, 이는 필요한 특정 데이터 액세스에 따라 "라벨"의 하위 세트에 해당하는 액세스 유닛을 선택한다.
특정 게놈 영역과 관련된 액세스 유닛을 식별하기 위해 "라벨"을 사용할 수 있는 가능성은 MIT 및 LIT와 같은 인덱스 도구를 사용하여 "라벨"없이 랜덤 데이터 액세스 기능을 사용할 수 있게 한다. 일반적인 랜덤 액세스는 관련 MIT 및 LIT 좌표(기준 ID, 위치 영역 및 클래스)를 결정하는 3 차원 벡터를 명시하고 라벨 리스트에 포함된 정보를 무시함으로써 성취될 수 있다.
도 51은 라벨이 MIT에 포함된 인덱스를 사용함으로써 여러 게놈 영역을 집계하고 고유하게 식별하는데 사용되는 방법을 보여준다.
도 59는 본 발명에 의해 개시된 라벨링 메커니즘을 구현하는 장치(592)가 데이터베이스(595)에 저장된 데이터의 여러 레코드(596)에 동시 액세스를 가능하게 하는 방법을 도시한다. 동일한 라벨에 의해 식별된 하나 이상의 영역의 선택적 보호는 쿼리(591)의 파싱을 담당하고 요구된 메타데이터를 액세스 제어의 시행을 담당하는 보안 모듈(594)에 발송하는 전용 모듈(591)에 의해 또한 지원된다. 라벨 디코더(593)는 보호될 수 있거나(따라서, 액세스는 보안 모듈(594)에 의해 제어됨) 또는 보호될 수 없는 객체 식별자로 라벨 구문을 번역하는 역할을 한다.
(기술적 효과)
본 명세서에서 기술된 바와 같이 액세스 유닛 또는 라벨에 의해 식별되는 액세스 유닛에서 게놈 정보를 구조화하는 기술적 효과는 다음과 같은 게놈 데이터의 역할이다:
1. 게놈 데이터는 하기의 액세스를 위하여 선택적으로 쿼리될 수 있다:
- 전체 게놈 데이터 또는 데이터 세트 및/또는 관련 메타데이터의 압축 해제없이 데이터의 특정 "카테고리" (예를 들어, 특정 시간 의미 또는 생물학적 의미),
- 게놈의 다른 영역을 압축 해제할 필요없이 (관련 메타데이터가 있거나 없는) 모든 "카테고리" "카테고리"의 하위 세트, 단일 "카테고리" 에 대한 게놈의 특정 영역,
- "라벨 리스트" 메인 헤더만을 파싱하고 상응하는 액세스 유닛만을 액세싱(즉, 검색 및 압축 해제)함으로써 사용자-정의된 "라벨들"에 의해 식별되는 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합,
2. 게놈 데이터는 다음과 같은 경우에 사용할 수 있는 새로운 데이터로 점진적으로 업데이트될 수 있다:
- 게놈 데이터 또는 데이터 세트에 대해 새로운 분석이 수행되는 경우,
- 새로운 게놈 데이터 또는 데이터 세트가 동일한 유기체 (상이한 생물학적 샘플, 동일한 유형의 다른 생물학적 샘플, 예를 들어, 혈액 샘플이지만 다른 시간에서 획득된 것 등)를 서열화함으로써 생성되는 경우,
3. 게놈 데이터는 다음과 같은 경우에 새로운 데이터 포멧으로 효율적으로 트랜스코딩될 수 있다:
- 새로운 기준으로서 사용될 새로운 게놈 데이터 또는 데이터 세트(예를 들어, 유형 0의 AU에 의해 운반되는 새로운 기준 게놈),
- 인코딩 포멧 사양서의 업데이트.
4. 게놈 데이터는 액세스 제어(예를 들어, 암호화) 및 권한 실행 둘 다와 관련하여 다양한 수준의 세밀성으로 보호될 수 있다. 예를 들어, 다음과 같은 시나리오를 사용할 수 있다:
- 하나의 라벨에 의해 식별된 모든 게놈 영역 또는 하위 영역에 동일한 액세스 제어 규칙 및 암호화 키를 적용할 수 있다(예를 들어, 도 54 참조),
- 서로 다른 액세스 제어 규칙 및 서로 다른 암호화 키를 사용하여 동일한 라벨하에 집계된 각 단일 영역 또는 하위 영역을 보호할 수 있다(예를 들어, 도 55 참조).
SAM/BAM과 같은 종래 기술의 해결책과 관련하여, 설명된 기술적 특징들은 전체 데이터가 인코딩된 포맷으로부터 검색되고 압축 해제될 때 데이터 필터링을 애플리케이션 수준으로 요구하는 문제를 다룬다.
액세스 유닛 구조, 파일 포멧 및 라벨링 메커니즘의 연관성이 기술적 이점을 위한 수단이 되는 애플리케이션 시나리오의 예시들을 이후에 설명할 것이다.
(선택적인 액세스)
특히, 사용자-정의된 "라벨"을 가능하게 포함하는 상이한 유형의 액세스 유닛을 기반으로 하는 개시된 데이터 구조는:
- 연관된 메타데이터 정보도 압축 해제함이 없이 모든 "카테고리" 또는 하위 세트(즉, 하나 이상의 계층) 또는 단일 "카테고리"의 전체 서열화의 판독 정보(데이터 또는 데이터 세트)만 추출하는 것을 가능하게 하고(종래 기술의 한계: 서로 다른 카테고리들 또는 계층들 간의 구별을 지원조차할 수 없는 SAM/BAM);
- 게놈의 다른 영역도 압축 해제할 필요없이 (관련 메타데이터가 있거나 없는) 모든 카테고리, 카테고리의 하위 세트 또는 단일 카테고리에 대해 가정된 기준 서열의 특정 영역상에 정렬된 모든 판독을 추출하는 것을 가능하게 하고 (종래 기술의 한계: SAM/BAM);
- 게놈의 다른 영역과 관련된 다른 데이터도 압축 해제할 필요없이 (관련 메타데이터가 있거나 없는) 모든 카테고리 카테고리의 하위 세트 또는 단일 카테고리에 대해 사용자-지정된 "라벨" 에 의해 식별되는 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합상에 정렬된 단일, 하위 세트 또는 모든 데이터 "클래스"에 속하는 모든 판독을 추출하는 것을 가능하게 한다(종래 기술의 한계: SAM/BAM).
도 39는 기준 서열 (AU 0-2)의 제 2 단편에 불일치하게 매핑된 게놈 정보에 대한 액세스가 AU 0-2, 1-2 및 3-2만을 디코딩하는데 필요한 방법을 도시한다. 이것은 매핑 영역(즉, 기준 서열상의 위치)과 관련된 척도 및 기준 서열에 대해 인코딩된 서열 판독에 적용된 매칭 함수와 관련된 척도에 따른 선택적인 액세스의 예시(예를 들어, 이러한 예시에서는 불일치만)이다.
추가적인 기술적 이점은 데이터 액세스 및 실행 속도면에서 데이터 쿼리가 훨씬 효율적이라는 것이며, 그 이유는 선택된 "카테고리",보다 긴 게놈 서열의 특정 영역 및 적용된 쿼리의 척도와 일치하는 유형 1, 2, 3, 4의 액세스 유닛에 대한 특정 계층, 및 이들의 조합만을 액세스하고 디코딩하는 것을 기반으로 하고 있기 때문이다.
유형 1, 2, 3, 4의 액세스 유닛을 계층내로 조직화하는 것은
- 하나 이상의 기준 게놈과 관련하여 특이적 변이(예를 들어, 불일치, 삽입, 삭제)로 뉴클레오티드 서열의 효율적인 추출을 허락하며;
- 고려된 기준 게놈 중 어떠한 것에도 매핑되지 않은 뉴클레오티드 서열의 효율적인 추출을 허락하며;
- 하나 이상의 기준 게놈을 완벽하게 매핑하는 뉴클레오티드 서열의 효율적인 추출을 허락하며;
- 하나 이상의 정확도 수준으로 매핑하는 뉴클레오티드 서열의 효율적인 추출을 허락한다.
도 52는 사용자-정의된 "라벨"과 연관된 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합에만 관련된 게놈 정보에 대해 액세스하는 방법을 나타낸다. 라벨의 구문은 라벨과 연관된 각 영역 또는 하위 영역을 다음과 같이 고유하게 식별될 수 있는 세 개의 좌표계를 기반으로 한다:
1. 기준 ID,
2. 데이터 유형 (클래스)
3. MIT의 블록 번호 (게놈 영역에 상응함).
이들 세 개의 좌표는 다음을 식별하는데 사용될 수 있다:
· 상응하는 기준상의 영역의 게놈 위치를 포함하는MIT 위치, 및
· 각각의 게놈 영역 또는 하위 영역을 나타내는 데이터의 물리적 위치를 포함하는 LIT 위치.
특정 게놈 영역과 관련된 데이터를 액세스하는 경우와 마찬가지로, 추가의 기술적 이점은 데이터 접근성 및 실행 속도면에서 훨씬 효율적으로 데이터를 쿼리한다는 것이며, 이는 라벨링된 특정 영역의 선택된 데이터 및 적용된 쿼리의 "라벨" 에 해당하는 유형 1, 2, 3, 4의 액세스 유닛에 대한 특정 계층과 그의 조합만을 액세스하고 디코딩하는 것을 기반으로할 수 있기 때문이다.
이러한 라벨링 메커니즘의 또 다른 기술적 이점은 기준 게놈상의 위치, 기준에 대한 불일치 유형(524)과 같은 특성으로 인해 여러 액세스 유닛간에 분산된 인코딩된 게놈 정보를 효율적으로 검색할 수 있다는 것이다.
매핑된 판독의 특성에 따라 게놈 데이터를 필터링(예를 들어, 완벽하게 매칭, 대체만 수행, 등)하는 것은 BAM 및 CRAM과 같은 기존 포멧을 사용할 때 시간이 걸릴 수 있다. 이는 데이터가 압축된 포멧 내에서 부족하고 명령 파이프 라인을 사용하여 압축 해제 및 필터링을 필요로 하기 때문이다. 본 발명은 몇 초 내에 데이터 필터링을 가능하게하는 데이터 구조를 설명한다. 본 발명은 전체 파일의 디코딩 (즉, 메모리 할당)을 필요로 하지 않기 때문에 메모리 사용을 파일 크기(10x 에서 100x 까지)에 비례하는 인자만큼 줄일 수 있다.
("스토리지" 및 "스트리밍" 시나리오에서 사용자-지정된 "라벨"로 식별되는 특정 게놈 영역에 대한 선택적인 액세스)
예를 들어, 서열 데이터가 압축되어 있고 "GeneXY" 및 "GeneWZ"에 대한 선택적인 액세스가 필요한 것으로 가정하자. "GeneXY" 및 "GeneWZ"에 해당하는 압축된 파일 포멧 또는 압축된 스트림의 두 개의 게놈 영역은 라벨링되어야 한다. 압축된 데이터 파일이 스토리지용으로 생성되는지 또는 압축된 데이터 스트리밍이 스트리밍용으로 생성되는지에 따라 두 가지 방법이 사용된다.
압축된 데이터 파일의 경우, 멀티플렉서는 "Label_ID"= GeneXY 및 "Label_ID"= GeneWZ를 갖는 2 개의 라벨을 포함하는 "라벨 리스트"를 생성한다. 라벨 매개 변수 "Label_lenght_in_blocks" 및 각 블록에 대한 매개 변수: "ref_num", "class_ID", "block_num"는 "GeneXY" 및 "GeneWZ" 영역의 기준 위치 및 선택적 액세스가 필요한 데이터 클래스를 기반으로 한 멀티플렉서에 의해 결정된다. 완전한 구문은 표 5에 나와 있다.
압축된 스트림의 경우, 멀티플렉서는 "Label_ID"= GeneXY 및 "Label_ID"= GeneWZ을 갖는 2 개의 라벨을 포함하는 "라벨 리스트"를 생성한다. 라벨 매개 변수 "ref ID" "class_ID" "start_pos" 및 "end_pos" 는 "GeneXY" 및 "GeneWZ"영역의 기준 위치 및 선택적 액세스가 요구되는 데이터 클래스를 기반으로 한 멀티플렉서에 의해 결정된다. 완전한 구문은 표 4에 나와 있다.
압축된 스트림의 경우에 사용되는 방법은 일반적이며 스토리지용 압축 파일의 경우에도 사용될 수 있지만, 상응하는 구현은 압축 파일의 경우에 설명된 바와 같이 블록 번호를 사용하여 "마스터 인덱스 테이블"(MIT)에 직접 액세스할 수 있기 때문에 효율성이 떨어진다.
상기 언급한 두 경우 (스트리밍 및 스토리지)에서 라벨에 의해 식별되는 게놈 데이터 검색 메커니즘은 동일하다.
라벨을 파싱할 때 디코딩 장치는:
1. 라벨의 첫 번째 요소에서 기준 서열을 식별하고,
2. 라벨의 두 번째 요소에서 데이터 클래스를 식별하고,
3. 라벨의 세 번째 요소에서 MIT의 블록(게놈 영역에 상응함)을 식별하고,
4. 상기 1과 2에서 파싱된 2 개의 좌표가 디코더로 하여금 필요한 게놈 스트림(484)을 식별할 수 있게 하고,
5. 각 게놈 스트림이 각 게놈 영역에 매핑된 데이터를 인코딩하는 디스크립터에 대한 포인터를 함유한 LIT(525)를 포함하는 헤더로 시작하고, 상기 3에서 파싱된 세 번째 좌표가 각 게놈 스트림의 LIT에서 올바른 포인터에 액세스하는데 사용되고,
6. 디코더가 비록 상이한 액세스 유닛(524) 사이에 분산되어 있어도 디코딩된 게놈 라벨에 의해 식별된 게놈 데이터를 디코딩하기 위해 모든 디스크립터를 효율적으로 검색할 수 있다.
(증분적 업데이트)
유형 7 및 8의 액세스 유닛은 전체 파일을 역 패킷화/디코딩/압축 해제할 필요없이 주석을 쉽게 삽입할 수 있게하여, 종래 기술 접근법의 한계인 파일의 효율적인 처리를 추가한다. 기존의 압축 해결책은 원하는 게놈 데이터에 액세스하기 전에 다량의 압축 데이터에 액세스하여 처리해야 할 수도 있다. 이는 하드웨어 구현에서 비효율적인 RAM 대역폭 사용과 더 많은 전력 소모를 유발한다. 전력 소모 및 메모리 액세스 문제는 본 원에서 설명된 액세스 유닛에 기반한 접근 방식을 사용하여 완화할 수 있다.
액세스 유닛의 이용과 함께 마스터 인덱스 테이블(도 21 참조)에서 기술된 데이터 인덱싱 메커니즘 및 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합에 관련된 사용자-정의된 "라벨"로 액세스 유닛을 식별할 수 있는 가능성은 아래에 설명되는 바와 같이 인코딩된 컨텐츠의 증분적 업데이트를 가능하게 한다. 이 메커니즘은 도 53에 나와 있다.
(추가적인 데이터 삽입)
새로운 게놈 정보는 여러 이유로 인해 기존 게놈 데이터에 주기적으로 추가될 수 있다. 예를 들면:
· 유기체가 서로 다른 순간에 시간에 맞춰 서열화될 때;
· 동일한 개체의 몇 개의 다른 샘플이 동시에 서열화될 때;
· 서열화 과정(스트리밍)에 의해 새로운 데이터가 생성될 때이다.
위에서 언급한 상황하에, 상기 기술된 액세스 유닛 및 파일 포멧 섹션에서 기술된 데이터 구조를 사용하여 데이터를 구조화하면 기존 데이터를 다시 인코딩할 필요없이 새로 생성된 데이터를 점진적으로 통합할 수 있다. 증분적 업데이트 과정은 다음과 같이 구현될 수 있다:
1. 새로 생성된 AU는 기존 AU로 파일에서 간단하게 연결할 수 있다.
2. 새로 생성된 데이터 또는 데이터 세트의 인덱싱은 본 명세서의 파일 포멧 섹션에서 설명된 마스터 인덱스 테이블에 포함된다. 하나의 인덱스는 새로 생성된 AU를 기존의 기준 서열상에 위치시키고, 다른 인덱스는 물리적 파일에서 새로 생성된 AU의 포인터로 구성되어 직접 및 선택적 액세스를 가능하게 한다.
3. 기존 및/또는 새로 생성된 AU는 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합에 상응하는 사용자-정의된 "라벨"로 식별할 수 있으며, "라벨 리스트"이 포함되거나 업데이트될 수 있다.
이 메커니즘은 도 40에 예시되어 있으며, 여기서 유형 1의 3 AU 및 2에서부터 4까지의 각 유형 당 4 AU로 인코딩된 기존 데이터는 유형 당 3 AU로 업데이트되고 인코딩 데이터는, 예를 들어, 동일한 개체에 대한 새로운 서열 실행으로부터 얻는다.
"라벨" 및 "라벨 리스트"를 생성하거나 업데이트하는 메커니즘은 도 52 및 도 53에 나와 있다.
게놈 데이터 및 데이터 세트를 압축된 형태로 스트리밍하는 특정 용도의 경우, 기존 데이터 세트의 증분적 업데이트는 서열화 기계에 의해 생성되는 즉시로 및 실제 서열화가 완료되기 전에 데이터를 분석할 때 유용할 수 있다. 인코딩 엔진(압축기)은 선택된 기준 서열의 동일한 영역에 매핑되는 서열 판독을 "클러스터링"함으로써 여러 개의 AU를 병렬로 어셈블링할 수 있다. 첫 번째 AU에 미리 구성된 임계값/매개 변수보다 많은 판독 수가 포함되면 AU는 분석 애플리케이션으로 보낼 준비가 된다. 새로 인코딩된 액세스 유닛과 함께 인코딩 엔진(압축기)은 새로운 AU가 의존하는 모든 액세스 유닛이 이미 수신 종료로 전송되었거나 또는 그와 함께 전송되는지를 확인해야 한다. 예를 들어, 유형 3의 AU는 적절하게 디코딩되기 위해 유형 0 및 유형 1의 적절한 AU가 수신 종료에 존재할 것을 요구할 것이다.
기술된 메커니즘에 의해, 수신 변이체 호출 애플리케이션은 서열화 과정이 송신 측에서 완료되기 전에 수신된 AU에 대해 변이체를 호출하기 시작할 수 있다. 이러한 과정의 개략도는 도 41에 나와 있다.
(새로운 결과 분석)
게놈 처리의 수명 주기동안 게놈 분석의 몇 번 반복을 동일한 데이터에 적용할 수 있다(예를 들어, 다른 처리 알고리즘을 사용하여 다른 변이체 호출). 본 명세서에서 정의된 바와 같은 AU 및 본 명세서의 파일 포멧 섹션에서 기술된 데이터 구조를 사용하면 기존 압축 데이터를 새로운 분석 결과로 점진적으로 업데이트할 수 있다.
예를 들어, 기존의 압축된 데이터에 대해 수행된 새로운 분석은 다음과 같은 경우에 새로운 데이터를 생성할 수 있다:
1. 새로운 분석은 이미 인코딩된 데이터와 관련된 기존의 결과를 수정할 수 있다. 이러한 사용 사례는 도 42에 예시되어 있으며 하나의 액세스 유닛의 콘텐츠 전체 또는 일부를 한 유형에서 다른 유형으로 옮김으로써 구현된다. 새로운 AU를 생성해야 하는 경우, (AU 당 사전 정의된 최대 크기로 인해) 마스터 인덱스 테이블 내에서 관련 인덱스가 만들어 져야 하고 필요에 따라 관련 벡터가 분류되어야 한다.
2. 새로운 데이터는 새로운 분석으로부터 생성되며 기존의 인코딩된 데이터와 연관되어야 한다. 이 경우, 유형 7의 새로운 AU가 생성되어 동일한 유형의 AU의 기존 벡터와 연결될 수 있다. 이것과 마스터 인덱스 테이블의 관련 업데이트는 도 43에 예시되어 있다.
상기 기술되고 도 42 및 43에 예시된 사용 사례는 다음과 같은 것들을 가능하게 한다:
1. 매핑 품질이 좋지 않은 데이터에만 직접 액세스할 수 있는 가능성(예를 들어, 유형 4의 AU);
2. 새로운 유형에 속할 가능성이 있는 새로운 액세스 유닛을 간단하게 생성함으로써 판독을 새로운 게놈 영역으로 다시 매핑할 수 있는 가능성(예를 들어, 유형 4 AU에 포함된 판독은 더 적은 (유형 2-3) 불일치를 갖는 새로운 영역으로 다시 매핑되어 새로 생성된 Au에 포함될 수 있음);
3. 새로 생성된 분석 결과 및/또는 관련 주석만 포함하는 유형 8(433)의 AU를 생성할 수 있는 가능성. 이 경우, 새로 생성된 Au는 이들이 참조하는 기존의 AU에 대한 "포인터"만 포함할 필요가 있다;
4. 각 단일 게놈 영역 또는 하위 영역에서 분석을 반복할 필요가 없이 동일한 라벨에 의해 식별된 여러 게놈 영역 및 하위 영역에 대해 새로운 분석을 단일 실행으로 수행할 수 있는 가능성. 본 명세서에서 기술된 바와 같은 라벨은 사용자로 하여금 단일 게놈 서열인 것처럼 연속하지 않은 게놈 단편을 조작할 수 있게 한다;
5. 단일 라벨에 의해 식별된 여러 게놈 영역 또는 하위 영역을 새로운 분석 결과로 업데이트할 수 있는 가능성. 새로운 결과(일반적으로 메타데이터의 형태로 표현됨)는 그 결과로부터 각 게놈 영역 또는 하위 영역으로의 몇 개의 링크를 만들 필요없이 잠재적으로 여러 게놈 영역 및 하위 영역의 집합을 식별하는 라벨에 연결된다.
(트랜스코딩)
압축된 게놈 데이터는, 예를 들어, 다음과 같은 경우에 트랜스코딩을 요구할 수 있다:
· 새로운 기준 서열의 공개;
· 다른 매핑 알고리즘 사용(재 매핑).
게놈 데이터가 기존의 공개된 기준 게놈 상에 맵핑되는 경우, 상기 기준 서열의 새로운 버전의 공개 또는 다른 처리 알고리즘을 사용하여 데이터를 매핑하고자 하는 요구는 오늘날 재 맵핑 과정을 필요로 한다. SAM 또는 CRAM과 같은 종래 기술의 파일 포맷을 사용하여 압축된 데이터를 재 매핑하는 경우, 압축된 데이터 전체는 "원시" 형태로 압축 해제되어 새롭게 이용 가능한 기준 서열 또는 다른 매핑 알고리즘을 사용하여 다시 맵핑되어야 한다. 이는 새로 공개된 기준이 이전의 것과 약간 다르거나 또는 사용된 다른 매핑 알고리즘이 이전 매핑과 매우 유사한 (또는 동일한) 매핑을 생성하는 경우에도 마찬가지이다.
본 원에서 설명된 액세스 유닛을 사용하여 구조화된 게놈 데이터를 트랜스코딩하는 이점은 다음과 같다:
1. 매핑 대 새로운 기준 게놈은 단지 변경된 게놈 영역에 매핑되는 AU의 데이터를 다시 인코딩(압축 해제 및 압축)하는 것을 필요로 한다. 또한, 사용자는 판독이 원래 변경된 영역에 매핑하지 않더라도 어떤 이유로든 다시 매핑해야 할 수도 있는 압축된 판독을 선택할 수 있다(이것은 사용자가 이전 매핑의 품질이 좋지 않다고 생각되는 경우 발생할 수 있음). 이러한 사용 사례는 도 44에 예시되어 있다.
2. 새로 공개된 기준 게놈이 이전의 것과 다른 경우, 다른 게놈 위치( "loci")로 변동된 전체 영역의 관점에서만 볼 때, 트랜스코딩 작업이 특히 간단하고 효율적이다. 사실, "변동된" 영역에 매핑된 모든 판독값을 이동시키기 위해서는 관련된 AU 헤더(의 세트)에 포함된 절대 위치 값만을 변경하는 것만으로 충분하다. 각 AU 헤더는 AU에 포함된 제 1 판독이 기준 서열에서 매핑된 절대 위치를 포함하고, 다른 판독 위치는 모두 첫 번째 것과 관련하여 차등적으로 인코딩된다. 따라서, 제 1 판독의 절대 위치 값을 간단히 업데이트함으로써 AU의 모든 판독이 그에 따라 이동된다. 게놈 데이터 위치를 압축된 페이로드에 인코딩하는 것은 모든 게놈 데이터 세트의 완전한 압축 해제 및 재 압축을 필요로 하기 때문에 이러한 메커니즘은 CRAM 및 BAM과 같은 종래 기술의 접근법으로 구현할 수 없다.
3. 다른 매핑 알고리즘을 사용하는 경우 불량한 품질로 매핑된 압축 판독 부분에만 상기 알고리즘을 적용하는 것이 가능하다. 예를 들어, 기준 게놈에서 완벽하게 일치하지 않는 판독에만 새로운 매핑 알고리즘을 적용하는 것이 적절할 수 있다. 기존 포멧의 경우, 오늘날 매핑 품질(즉, 불일치의 존재 및 불일치 수)에 따라 판독을 추출할 수 없다(또는, 일부 제한으로 부분적으로만 가능함). 새로운 매핑 수단으로 새로운 매핑 결과가 보상되면, 관련 판독은 동일한 유형의 다른 Au로부터의 한 AU(도 46) 또는 한 유형의 한 AU에서 다른 유형의 AU(도 45)로 트랜스코딩될 수 있다.
더우기, 종래 기술의 압축 해결책은 원하는 게놈 데이터가 액세스될 수 있기 전에 다량의 압축 데이터를 액세스하고 처리해야 할 수도 있다. 이로 인해, 비효율적인 RAM 대역폭 사용과 더 많은 전력 소비를 야기하여 하드웨어의 구현 문제가 발생한다. 전력 소모 및 메모리 액세스 문제는 본 원에서 설명된 액세스 유닛에 기반한 접근 방식을 사용하여 완화할 수 있다.
본 원에서 설명된 게놈 액세스 유닛의 채택의 또 다른 이점은 병렬 처리의 용이성과 하드웨어 구현에 대한 적합성이다. SAM/BAM 및 CRAM과 같은 현재의 해결책은 단일 - 스레드(single - threaded) 소프트웨어 구현을 위해 고안되었다.
(선택적 보호)
본 명세서에서 설명된 바와 같이, 여러 유형의 계층으로 조직된 액세스 유닛을 기반으로 한 접근 방식은 종래 기술의 모놀리식 해결책으로는 불가능한 콘텐츠 보호 메커니즘을 구현할 수 있게 한다.
당업자는 유기체의 유전자 프로파일과 관련된 대부분의 게놈 정보가 기지의 서열(예를 들어, 기준 게놈 또는 게놈 집단)에 대한 차이(변이체)에 의존한다는 것을 알고 있다. 그러므로, 허가되지 않은 액세스로부터 보호받을 개별 유전자 프로파일은 본 명세서에서 기술된 바와 같이 유형 3과 4의 액세스 유닛으로 인코딩될 것이다. 따라서, 서열화 및 분석 과정에 의해 생성된 가장 민감한 게놈 정보에 대한 제어된 액세스의 구현은 유형 3 및 4의 AU의 페이로드만 암호화함으로써 구현할 수 있다(예를 들어, 도 47 참조). 이는 암호화 프로세스를 사용하는 리소스가 데이터의 하위 세트에만 적용되어야 하므로 처리 전력과 대역폭 측면에서 상당한 절감 효과를 얻을 수 있다.
("라벨"에 의해 식별된 특정 게놈 영역의 선택적 보호)
라벨링 메커니즘은 데이터 보호 및 액세스 제어의 다른 메커니즘을 가능하게 한다. 예를 들어, 도 54는 동일한 라벨로 식별되는 여러 게놈 영역에 하나의 보호 메커니즘(예를 들어, 암호화)과 하나의 액세스 제어 규칙(AC)을 적용하는 방법을 보여준다. 보다 정교한 시나리오에서, 데이터 보호는 라벨에 의해 식별된 각 영역에 다른 액세스 제어 규칙과 다른 보호 메커니즘(암호화)을 적용함으로써 구현될 수 있다. 이는 도 55에 나타나 있다.
부가적으로, 다른 "라벨" 에 의해 식별되는 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합의 선택적 암호화는 파일 및 스트리밍된 시나리오 모두에 대해 "라벨"에 해당하는 압축된 데이터에만 암호화를 적용함으로써 쉽게 구현할 수 있다. 예를 들어, "스토리지" 및 "스트리밍" 시나리오에서 사용자-지정된 "라벨"에 의해 식별되는 특정 유전체 영역에 대한 선택적 액세스"섹션의 예시에서와 같이, "GeneXY" 및 "GeneWZ"로서 라벨링된 두 개의 게놈 영역은 "GeneXY"에 의해 라벨링된 데이터만을 암호화하고 "GeneWZ"로서 라벨링된 압축된 데이터를 명확하게 유지함으로써 차별화할 수 있다. 암호화 규칙은 (스토리지 및 스트리밍 시나리오 둘다에서) 메타데이터 필드에 의해 전달될 수 있고 "라벨 리스트"의 각 요소와 연관될 수 있다.
(게놈 액세스 유닛의 전송)
(게놈 데이터 멀티플렉스)
게놈 액세스 유닛은 게놈 데이터 멀티플렉스 내의 통신 네트워크를 통해 전송할 수 있다. 게놈 데이터 멀티플렉스는 패킷 손실과 같은 오류가 발생할 수 있는 네트워크 환경에서 전송되는, 본 발명의 일부로서 개시된 데이터 분류에 따라서 표현된 패킷화된 게놈 데이터 및 메타데이터의 서열로서 정의된다.
게놈 데이터 멀티플렉스는 다른 환경(일반적으로 네트워크 환경)에서 게놈 코딩된 데이터의 전송을 보다 효율적으로 용이하게 하기 위해 고안되었으며, 종래 기술의 해결책에는 없는 다음과 같은 장점들을 가진다:
1. 인코딩 수단에 의해 생성된 스트림 또는 일련의 게놈 데이터(하기에서 설명됨) 또는 게놈 파일 포맷을 하나 이상의 게놈 데이터 멀티플렉서로 캡슐화하여, 네트워크 환경을 통해 이를 전송하고 이어서 유효하고 동일한 스트림 또는 파일 포멧을 회복시켜 정보의 전송 및 액세스를 보다 효율적으로 만드는 것을 가능하게 한다.
2. 디코딩 및 프리젠테이션을 위해 캡슐화된 게놈 데이터 스트림으로부터 인코딩된 게놈 데이터를 선택적으로 검색할 수 있게 한다.
3. 여러 게놈 데이터 세트를 전송을 위한 정보의 단일 컨테이너로 멀티플렉싱할 수 있게 하며, 운반된 정보의 하위 세트를 새로운 게놈 데이터 멀티플렉스로 디멀티플렉싱할 수 있게 한다.
4. (결과적으로 개별 액세스를 통해) 다른 소스에서 생성된 데이터 및 메타데이터의 멀티플렉싱 및/또는 서열화/분석 프로세스를 가능하게 하고 네트워크 환경을 통해 결과의 게놈 데이터 멀티플렉스를 전송한다.
5. 패킷 손실과 같은 오류 식별을 지원한다.
6. 네트워크 지연으로 인해 순서가 맞지 않을 수도 있는 적절한 레코더 데이터를 지원하므로, 종래 기술의 해결책과 비교했을 때 게놈 데이터의 전송을 보다 효율적으로 수행한다.
게놈 데이터 멀티플렉싱의 예시는 도 49에 나타나 있다.
(게놈 데이터 세트)
본 발명의 컨텍스트 있어서, 게놈 데이터 세트는, 예를 들어, 살아있는 유기체의 게놈 데이터, 게놈 데이터 과정의 여러 단계에 의해 생성된 하나 이상의 서열 및 메타데이터, 또는 살아있는 유기체의 게놈 서열화를 포함하는 게놈 데이터의 구조화된 세트로서 정의된다. 하나의 게놈 데이터 멀티플렉스는 각 데이터 세트가 다른 유기체를 언급하는 (다중 채널 시나리오에서와 같이) 다수의 게놈 데이터 세트를 포함할 수 있다. 단일 게놈 데이터 멀티플렉스에 대한 여러 데이터 세트의 멀티플렉싱 메커니즘은 소위 게놈 데이터 세트 목록(GDL), 게놈 데이터 세트 매핑 테이블 목록(GDMTL) 및 게놈 데이터 세트 매핑 테이블(GDMT)이라고 하는 데이터 구조에 포함된 정보에 의해 관리된다.
(게놈 데이터 세트 목록)
게놈 데이터 세트 목록(GDL)은 게놈 데이터 멀티플렉스에서 사용 가능한 모든 게놈 데이터 세트를 나열하는 데이터 구조로서 정의된다. 나열된 각 게놈 데이터 세트는 게놈 데이터 세트 ID(GID)라고 하는 고유값으로 식별된다.
GDL에서 나열된 각 게놈 데이터 세트는:
· 하나의 게놈 데이터 세트 매핑 테이블(GDMT)을 가지며 스트림 ID (genomic_dataset_map_SID)의 특정값에 의해 식별되는 하나의 게놈 데이터 스트림;
· 하나의 기준 ID 매핑 테이블(RIDMT)을 가지며 스트림 ID (reference_id_map_SID)의 특정값에 의해 식별되는 하나의 게놈 데이터 스트림.
GDL은 게놈 데이터 스트림 전송의 시작에서 단일 전송 패킷의 페이로드로서 전송되며, 스트림에 대한 임의의 액세스를 가능하게 하기 위해 주기적으로 재전송할 수 있다.
GDL 데이터 구조의 구문은 각 구문 요소와 관련된 데이터 유형의 표시와 함께 하기 표에서 제공된다.

상기 기술된 GDL을 구성하는 구문 요소는 다음과 같은 의미 및 함수를 갖는다.

(게놈 데이터 세트 매핑 테이블)
게놈 데이터 세트 매핑 테이블(GDMT)은 스트리밍 프로세스의 시작에서 생성되고 전송된다(그리고, 스트리밍된 데이터의 대응점 및 관련 종속성을 업데이트 가능하게 하도록 주기적으로 재전송되거나 업데이트되거나 동일해야 함). GDMT는 게놈 데이터 세트 목록 다음에 단일 패킷에 의해 운반되며 하나의 게놈 데이터 세트를 구성하는 게놈 데이터 스트림을 식별하는 SID를 나열하고 있다. GDMT는 게놈 멀티플렉스에 의해 운반되는 하나의 게놈 데이터 세트를 구성하는 게놈 데이터 스트림의 모든 식별자(예를 들어, 게놈 서열, 기준 게놈, 메타데이터 등)의 전체 수집이다. 게놈 데이터 세트 매핑 테이블은 각 게놈 데이터 세트와 관련된 게놈 데이터 스트림의 식별자를 제공함으로써 게놈 서열에 대한 임의 액세스를 가능하게 하는데 유용하다.
GDMT 데이터 구조의 구문은 각 구문 요소와 연관된 데이터 유형의 표시와 함께 하기 표에 제공되어 있다.

상기 기술된 GDMT를 구성하는 구문 요소는 다음과 같은 의미 및 함수를 갖는다.

extension_fields 는 게놈 데이터 세트 또는 하나의 게놈 데이터 세트 구성 요소를 추가로 설명하는데 사용할 수 있는 선택적인 디스크립터이다.data_type 필드는 다음 값을 가질 수 있다.

(게놈 데이터 세트 매핑 테이블 목록)
이 구조는 게놈 데이터 세트 멀티플렉스와 관련된 모든 데이터 세트 매핑 테이블에 대한 정보를 지닌다.

(기준 ID 매핑 테이블)
기준 ID 매핑 테이블(RIDMT)은 스트리밍 과정이 시작될 때 생성되고 전송된다. RIDMT는 게놈 데이터 세트 목록 다음의 단일 패킷에 의해 운반된다. RIDMT는 액세스 유닛의 블록 헤더에 포함된 기준 서열의 숫자 식별자(REFID)와 표 2에 명시된 게놈 데이터 세트 헤더에 포함된 (전형적으로 리터럴(literal)) 기준 식별자 간의 매핑을 지정한다.
RIDMT는:
· 대응점 및 스트리밍된 데이터의 관련 종속성의 업데이트를 가능하게 하고,
· 기존의 기준 서열에 추가된 새로운 기준 서열의 통합(예를 들어, 드-노브 어셈블리 과정에 의해 생성된 합성 기준)을 지원하기 위하여 주기적으로 재전송될 수 있다.
RIDMT 데이터 구조의 구문은 각 구문 요소와 연관된 데이터 유형의 표시와 함께 하기 표에 제공된다.

상기 기술된 RIDMT를 구성하는 구문 요소는 다음과 같은 의미 및 함수를 갖는다.

(게놈 라벨 리스트)
전술한 바와 같이, 라벨은 특정 게놈 영역 또는 하위 영역, 또는 영역 또는 하위 영역의 집합에 할당된 식별자이다.
라벨은 기준 서열 ID, 기준 서열에 대한 위치 영역 및 라벨이 식별하는 데이터 클래스를 지정함으로써 게놈 영역을 식별한다.
이러한 목적을 위해, 게놈 라벨 리스트(GLL)는 멀티플렉서에 의한 패킷화 과정 중에 생성되어 전송된다.
디멀티플렉서의 역 패킷화기는 GLL 구문을 파싱하고, 사용 가능한 "Label"을, 데이터의 원하는 하위 세트를 선택하고 액세스할 수 있는 데이터 액세스 애플리케이션에 제공한다.
GLL은 스트림의 시작에서 (선택적으로) 생성되고 전송되며, 일반적으로 다중 동기화 포인트(4811)를 활성화하기 위해 주기적으로 전송되며, multiplex_id 및 dataset_id 필드로 식별되는 멀티플렉스 및 데이터 세트와 연관된 "Label" 리스트를 제공한다.
GLL 데이터 구조의 구문은 각 구문 요소와 관련된 데이터 유형의 표시와 함께 하기 표에 제공된다.

상기 설명한 GLL을 구성하는 구문 요소는 다음과 같은 의미와 함수를 가진다.

(게놈 데이터 스트림)
게놈 데이터 멀티플렉스는 각 스트림이 다음과 같은 데이터를 전송할 수 있는 하나 또는 여러 개의 게놈 데이터 스트림을 포함한다:
· 전송 정보를 포함하는 데이터 구조(예를 들어, 게놈 데이터 세트 목록, 게놈 데이터 세트 매핑 테이블, 등).
· 본 발명에서 기술된 게놈 데이터 계층 중 하나에 속하는 데이터.
· 게놈 데이터와 관련된 메타데이터.
· 기타 임의의 데이터.
게놈 데이터를 포함하는 게놈 데이터 스트림은 필수적으로 게놈 데이터 계층의 패킷화된 버전이며, 여기서 각 패킷에는 패킷 콘텐츠를 설명하는 헤더가 앞에 추가되며 멀티플렉스의 다른 요소와 어떻게 관련되어 있는지가 추가된다.
본 명세서에 기술된 게놈 데이터 스트림 포멧 및 본 명세서에 기술된 파일 포멧은 상호 변환할 수 있다. 모든 데이터가 수신된 후에만 전체 파일 포멧을 전체적으로 재구성할 수 있지만, 스트리밍의 경우에 디코딩 툴은 언제든지 부분 데이터를 재구성하여 액세스하고 부분 데이터의 처리를 시작할 수 있다.
게놈 데이터 스트림은 각각 하나 이상의 게놈 데이터 패킷을 포함하는 여러 게놈 데이터 블록으로 구성된다. 게놈 데이터 블록(GDB)은 하나의 게놈 AU를 구성하는 게놈 정보의 컨테이너이다. GDB는 통신 채널 요건에 따라 여러 게놈 데이터 패킷으로 나눌 수 있다.
게놈 접근 유닛은 다른 게놈 데이터 스트림에 속한 하나 이상의 게놈 데이터 블록으로 구성된다.
게놈 데이터 패킷(GDP)은 하나의 GDB를 구성하는 전송 단위이다. 패킷 크기는 일반적으로 통신 채널 요건에 따라 설정된다.
도 27은 본 발명에서 정의된 바와 같이 P 클래스에 속하는 데이터를 인코딩할 때 게놈 멀티플렉스, 스트림, 액세스 유닛, 블록 및 패킷 간의 관계를 나타낸다. 이러한 예시로, 3 개의 게놈 스트림은 서열 판독의 위치, 페어링 및 역 보완에 대한 정보를 캡슐화한다.
게놈 데이터 블록은 헤더, 압축된 데이터의 페이로드 및 패딩 정보로 구성된다.
하기 표는 각 필드에 대한 설명과 일반적인 데이터 유형을 가진 GDB 헤더의 구현예를 제공한다.

AUID, POS 및 BS의 사용으로 인해 디코더는 본 발명에서 마스터 인덱스 테이블(MIT) 및 로컬 인덱스 테이블(LIT)로 지칭되는 데이터 인덱싱 메커니즘을 재구성할 수있다. 데이터 스트리밍 시나리오에서, AUID 및 BS의 사용은 수신단으로 하여금 여분의 데이터를 전송할 필요없이 로컬에서 LIT를 동적으로 재생성할 수 있게 한다. AUID, BS 및 POS를 사용하면 추가 데이터를 전송할 필요없이 로컬에서 MIT를 재생성할 수 있다.
이것은 다음과 같은 기술적 이점을 가진다:
· 전체 LIT가 전송되는 경우, 광범위할 수도 있는 인코딩 오버 헤드를 감소시키고;
· 스트리밍 시나리오에서 일반적으로 사용할 수 없는 게놈 위치와 액세스 단위 간의 완전한 매핑을 필요로 하지 않는다.
게놈 데이터 블록은 최대 패킷 크기, 패킷 손실율, 등과 같은 네트워크 계층 제약에 따라 하나 이상의 게놈 데이터 패킷으로 분할될 수 있다. 게놈 데이터 패킷은 헤더, 및 하기 표에 기재된 바와 같이 인코딩 또는 암호화된 게놈 데이터의 페이로드로 구성된다.

게놈 멀리플렉스는 적어도 하나의 게놈 데이터 목록, 하나의 게놈 데이터 세트 매핑 테이블 및 하나의 기준 ID 매핑 테이블이 수신되어 모든 패킷을 특정 게놈 데이터 세트 구성 요소로 매핑할 수 있을 때만 제대로 디코딩될 수 있다.
(게놈 패킷 헤더)
모든 게놈 데이터 블록은 단편으로 분리될 수 있으며, 이는 패킷 손실율, 프로토콜 최대 패킷 크기, 등과 같은 채널 요건에 의존하여 게놈 데이터 패킷의 페이로드에서 전송될 수 있다.
게놈 데이터 패킷은 다음과 같이 정의된다.

(멀티플렉스 인코딩 과정)
도 49는 본 발명에서 제시된 데이터 구조에서 변환되기 전에 원시 유전체 서열 데이터가 연역적으로 알려진 하나 이상의 기준 서열(4920) 상에 어떻게 매핑(491)될 필요가 있는지를 보여준다. 기준 서열이 이용 가능하지 않은 경우, "구조화된" 기준이 원시 서열 데이터(492)로부터 구축될 수 있다. 이미 정렬된 데이터는 정보 엔트로피를 줄이기 위해 다시 정렬될 수 있다. 정렬 후, 게놈 분류기(494)는 표 1에 기술된 매칭 함수에 따라서 데이터 클래스를 생성하고, 게놈 서열로부터 메타데이터(예컨대, 품질값) 및 주석 데이터를 분리한다. 생성된 데이터 클래스(498)의 엔트로피를 추가로 감소시키기 위해, 기준 변환(4919)이 외부 기준(4920)에 적용될 수 있다. 변환된 데이터 클래스(4918)는 계층 엔코더(495-497)에 공급되어 게놈 계층(491)을 생성하고, 이어서 엔트로피 인코더(4912-4914)에 의해 인코딩된다. 그 다음, 엔트로피 인코더에 의해 생성된 게놈 스트림은 게놈 멀티플렉서(4916)로 전송되어 게놈 멀티플렉스를 생성한다. 게놈 라벨 발생기(4917)에 의해 생성된 게놈 라벨은 멀티플렉서(4916)에 의해 게놈 스트림(4915)에 회합될 수 있다.
Claims (21)
- 기준 게놈 서열의 식별자(521), 게놈 영역의 식별자(522), 및 게놈 데이터의 데이터 클래스의 식별자(523)를 포함하는 라벨을 사용해서, 게놈 데이터의 영역을 선택적으로 액세스하는 방법.
- 제 1 항에 있어서,
상기 게놈 데이터는 게놈 판독의 서열인
방법.
- 제 2 항에 있어서,
상기 데이터 클래스는
- 기준 서열에 대해 어떠한 불일치도 나타내지 않는 게놈 판독을 포함하는 "클래스 P"와,
- 서열화 기계가 어떠한 "염기"도 호출할 수 없는 위치의 불일치만을 포함하는 게놈 판독을 포함하고, 상기 불일치의 수는 주어진 임계값을 초과하지 않는 "클래스 N"과,
- 서열화 기계가 "n 유형" 불일치라고 명명된 어떠한 염기도 호출할 수 없고 및/또는 서열화 기계가 "s 유형" 불일치라고 명명된 기준 서열과는 다른 염기를 호출한 위치에 의해 불일치가 구성되는 게놈 판독을 포함하며, 상기 불일치의 수는 "n 유형"의 불일치와 "s 유형"의 불일치의 수에 대한 주어진 임계값 및 주어진 함수 (f(n, s))로부터 얻은 임계값을 초과하지 않는 "클래스 M"과,
- 게놈 판독이 "클래스 M"과 동일한 유형의 불일치 및 나아가 "삽입"("i 유형"), "삭제"("d 유형"), 소프트 클립("c 유형") 중 적어도 하나의 불일치를 가질 수 있고, 각각의 유형에 대한 불일치의 수가 대응하는 주어진 임계값 및 주어진 함수 (w(n, s, i, d, c))에 의해 제공되는 임계값을 초과하지 않는 "클래스 I"와,
- 클래스 P, N, M, I에서 어떠한 분류도 찾지 못하는 모든 판독을 포함하는 "클래스 U"
와 같은 유형 또는 이들의 하위 세트일 수 있는
방법.
- 제 2 항에 있어서,
상기 게놈 데이터는 게놈 판독의 쌍으로 된 서열인
방법.
- 제 4 항에 있어서,
상기 쌍으로 된 판독의 상기 데이터 클래스는
- 기준 서열에 대해 어떠한 불일치도 나타내지 않는 게놈 판독 쌍을 포함하는 "클래스 P"와,
- 서열화 기계가 어떠한 "염기"도 호출할 수 없는 위치의 불일치만을 포함하는 게놈 판독 쌍을 포함하고, 각각의 판독에 대한 상기 불일치의 수는 주어진 임계값을 초과하지 않는 "클래스 N"과,
- 서열화 기계가 어떠한 "염기"도 호출할 수 없으며 각각의 판독에 대한 상기 불일치의 수가 "n 유형" 불일치라고 명명된 주어진 임계값을 초과하지 않고, 및/또는 서열화 기계가 "s 유형" 불일치라고 명명된 기준 서열과는 다른 염기를 호출하고 상기 불일치의 수가 "n 유형"의 불일치와 "s 유형"의 불일치의 수에 대한 주어진 임계값 및 주어진 함수 (f(n, s))로부터 얻은 임계값을 초과하지 않는 위치의 불일치만을 포함한 게놈 판독 쌍을 포함하는 "클래스 M"과,
- "클래스 M" 쌍과 동일한 유형의 불일치 및 "삽입"("i 유형"), "삭제"("d 유형"), 소프트 클립("c 유형") 중 적어도 하나의 불일치를 가질 수 있는 판독 쌍을 포함하며, 각각의 유형에 대한 불일치의 수가 대응하는 주어진 임계값 및 주어진 함수 (w(n, s, i, d, c))에 의해 제공되는 임계값을 초과하지 않는 "클래스 I"와,
- 하나의 판독 메이트(mate)만이 클래스 P, N, M, I 중 어느 하나로 분류되기 위한 매칭(matching) 규칙을 만족시키지 않는 판독 쌍을 포함하는 "클래스 HM"와,
- 두 개의 판독이 클래스 P, N, M, I로 분류되기 위한 매칭 규칙을 만족시키지 않는 모든 판독 쌍을 포함하는 클래스 "U"
와 같은 유형 또는 이들의 하위 세트일 수 있는
방법.
- 제 3 항 또는 제 5 항에 있어서,
상기 게놈 영역의 상기 식별자는 마스터 인덱스 테이블에 포함되는
방법.
- 제 6 항에 있어서,
상기 게놈 데이터 및 상기 라벨은 엔트로피 코딩되는 것인
방법.
- 제 7 항에 있어서,
상기 마스터 인덱스 테이블(4812)은 게놈 데이터 세트 헤더(4813)에 포함되는
방법.
- 제 1 항 내지 제 8 항 중 어느 한 항에 있어서,
상기 게놈 데이터의 상기 영역은 개별 액세스 유닛(524, 486) 간에 분산되는
방법.
- 제 9 항에 있어서,
파일 내의 상기 게놈 데이터 영역의 위치는 로컬 인덱스 테이블(525)에 표시되는
방법.
- 제 1 항 내지 제 10 항 중 어느 한 항에 있어서,
상기 라벨은 사용자 지정되는
방법.
- 제 1 항 내지 제 11 항 중 어느 한 항에 있어서,
상기 영역은 전체 게놈 파일을 암호화하지 않고 별도의 방식으로 보호 및/또는 암호화되는
방법.
- 제 1 항 내지 제 12 항 중 어느 한 항에 있어서,
상기 라벨은 게놈 라벨 리스트(GLL)에 저장되는
방법.
- 제 1 항 내지 제 13 항 중 어느 한 항에 따른 게놈 데이터의 영역에 선택적으로 액세스하여 게놈 데이터를 인코딩하는
방법.
- 제 14 항에 있어서,
상기 게놈 라벨 리스트는 다중 동기화 포인트를 가능하게 하도록 주기적으로 재전송되거나 또는 업데이트되는
방법.
- 제 1 항 내지 제 13 항 및 제 15 항 중 어느 한 항에 따른 게놈 데이터의 영역에 선택적으로 액세스하여 게놈 데이터의 스트림 또는 파일을 디코딩하는
방법.
- 제 14 항 또는 제 15 항에 따른 게놈 데이터를 인코딩하기 위한 장치.
- 제 16 항에 따른 게놈 데이터를 디코딩하기 위한 장치.
- 제 14 항 또는 제 15 항에 따른 인코딩된 게놈 데이터를 저장하기 위한 저장 수단.
- 실행될 때 적어도 하나의 프로세서로 하여금 제 14 항 또는 제 15 항의 인코딩 방법을 수행하게 하는 명령을 포함하는 컴퓨터 판독 가능한 매체.
- 실행될 때 적어도 하나의 프로세서가 제 16 항의 디코딩 방법을 수행하게 하는 명령을 포함하는 컴퓨터 판독 가능한 매체.
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2016/074301 WO2018068828A1 (en) | 2016-10-11 | 2016-10-11 | Method and system for storing and accessing bioinformatics data |
| EPPCT/EP2016/074301 | 2016-10-11 | ||
| EPPCT/EP2016/074307 | 2016-10-11 | ||
| EPPCT/EP2016/074311 | 2016-10-11 | ||
| PCT/EP2016/074307 WO2018068829A1 (en) | 2016-10-11 | 2016-10-11 | Method and apparatus for compact representation of bioinformatics data |
| EPPCT/EP2016/074297 | 2016-10-11 | ||
| PCT/EP2016/074311 WO2018068830A1 (en) | 2016-10-11 | 2016-10-11 | Method and system for the transmission of bioinformatics data |
| PCT/EP2016/074297 WO2018068827A1 (en) | 2016-10-11 | 2016-10-11 | Efficient data structures for bioinformatics information representation |
| PCT/US2017/017841 WO2018071054A1 (en) | 2016-10-11 | 2017-02-14 | Method and system for selective access of stored or transmitted bioinformatics data |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20190073426A true KR20190073426A (ko) | 2019-06-26 |
Family
ID=61905752
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020197013567A KR20190073426A (ko) | 2016-10-11 | 2017-02-14 | 저장 또는 전송된 생물정보학 데이터의 선택적 액세스를 위한 방법 및 시스템 |
| KR1020197013418A KR20190062541A (ko) | 2016-10-11 | 2017-07-11 | 참조 서열을 사용한 생물정보학 데이터의 표현 및 처리를 위한 방법 및 시스템 |
| KR1020197013419A KR20190069469A (ko) | 2016-10-11 | 2017-07-11 | 생물정보학 데이터의 인덱싱을 위한 방법 및 시스템 |
| KR1020197026863A KR20190117652A (ko) | 2016-10-11 | 2017-12-14 | 압축된 게놈 서열 리드로부터 게놈 참조 서열의 복원 방법 및 시스템 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020197013418A KR20190062541A (ko) | 2016-10-11 | 2017-07-11 | 참조 서열을 사용한 생물정보학 데이터의 표현 및 처리를 위한 방법 및 시스템 |
| KR1020197013419A KR20190069469A (ko) | 2016-10-11 | 2017-07-11 | 생물정보학 데이터의 인덱싱을 위한 방법 및 시스템 |
| KR1020197026863A KR20190117652A (ko) | 2016-10-11 | 2017-12-14 | 압축된 게놈 서열 리드로부터 게놈 참조 서열의 복원 방법 및 시스템 |
Country Status (17)
| Country | Link |
|---|---|
| US (6) | US20200042735A1 (ko) |
| EP (3) | EP3526694A4 (ko) |
| JP (4) | JP2020505702A (ko) |
| KR (4) | KR20190073426A (ko) |
| CN (6) | CN110168651A (ko) |
| AU (3) | AU2017342688A1 (ko) |
| BR (7) | BR112019007359A2 (ko) |
| CA (3) | CA3040138A1 (ko) |
| CL (6) | CL2019000968A1 (ko) |
| CO (6) | CO2019003638A2 (ko) |
| EA (2) | EA201990916A1 (ko) |
| IL (3) | IL265879B2 (ko) |
| MX (2) | MX2019004130A (ko) |
| PE (7) | PE20191058A1 (ko) |
| PH (6) | PH12019550060A1 (ko) |
| SG (3) | SG11201903270RA (ko) |
| WO (4) | WO2018071054A1 (ko) |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2526598B (en) * | 2014-05-29 | 2018-11-28 | Imagination Tech Ltd | Allocation of primitives to primitive blocks |
| US11574287B2 (en) | 2017-10-10 | 2023-02-07 | Text IQ, Inc. | Automatic document classification |
| US11030324B2 (en) * | 2017-11-30 | 2021-06-08 | Koninklijke Philips N.V. | Proactive resistance to re-identification of genomic data |
| WO2019191083A1 (en) * | 2018-03-26 | 2019-10-03 | Colorado State University Research Foundation | Apparatuses, systems and methods for generating and tracking molecular digital signatures to ensure authenticity and integrity of synthetic dna molecules |
| MX2020012672A (es) * | 2018-05-31 | 2021-02-09 | Koninklijke Philips Nv | Sistema y metodo para interpretacion de alelos usando un genoma de referencia basado en graficos. |
| CN108753765B (zh) * | 2018-06-08 | 2020-12-08 | 中国科学院遗传与发育生物学研究所 | 一种构建超长连续dna序列的基因组组装方法 |
| US20200058379A1 (en) * | 2018-08-20 | 2020-02-20 | The Board Of Trustees Of The Leland Stanford Junior University | Systems and Methods for Compressing Genetic Sequencing Data and Uses Thereof |
| GB2585816A (en) * | 2018-12-12 | 2021-01-27 | Univ York | Proof-of-work for blockchain applications |
| US20210074381A1 (en) * | 2019-09-11 | 2021-03-11 | Enancio | Method for the compression of genome sequence data |
| CN110797087B (zh) * | 2019-10-17 | 2020-11-03 | 南京医基云医疗数据研究院有限公司 | 测序序列处理方法及装置、存储介质、电子设备 |
| JP2022553199A (ja) | 2019-10-18 | 2022-12-22 | コーニンクレッカ フィリップス エヌ ヴェ | 多様な表形式データの効果的な圧縮、表現、および展開のためのシステムおよび方法 |
| CN111243663B (zh) * | 2020-02-26 | 2022-06-07 | 西安交通大学 | 一种基于模式增长算法的基因变异检测方法 |
| CN111370070B (zh) * | 2020-02-27 | 2023-10-27 | 中国科学院计算技术研究所 | 一种针对大数据基因测序文件的压缩处理方法 |
| US20210295949A1 (en) * | 2020-03-17 | 2021-09-23 | Western Digital Technologies, Inc. | Devices and methods for locating a sample read in a reference genome |
| US11837330B2 (en) | 2020-03-18 | 2023-12-05 | Western Digital Technologies, Inc. | Reference-guided genome sequencing |
| EP3896698A1 (en) * | 2020-04-15 | 2021-10-20 | Genomsys SA | Method and system for the efficient data compression in mpeg-g |
| CN111459208A (zh) * | 2020-04-17 | 2020-07-28 | 南京铁道职业技术学院 | 针对地铁供电系统电能的操纵系统及其方法 |
| AU2021342166A1 (en) * | 2020-09-14 | 2023-01-05 | Illumina, Inc. | Custom data files for personalized medicine |
| CN112836355B (zh) * | 2021-01-14 | 2023-04-18 | 西安科技大学 | 一种预测采煤工作面顶板来压概率的方法 |
| ES2930699A1 (es) * | 2021-06-10 | 2022-12-20 | Veritas Intercontinental S L | Metodo de analisis genomico en una plataforma bioinformatica |
| CN113670643B (zh) * | 2021-08-30 | 2023-05-12 | 四川虹美智能科技有限公司 | 智能空调测试方法及系统 |
| CN113643761B (zh) * | 2021-10-13 | 2022-01-18 | 苏州赛美科基因科技有限公司 | 一种用于解读二代测序结果所需数据的提取方法 |
| US20230187020A1 (en) * | 2021-12-15 | 2023-06-15 | Illumina Software, Inc. | Systems and methods for iterative and scalable population-scale variant analysis |
| CN115391284B (zh) * | 2022-10-31 | 2023-02-03 | 四川大学华西医院 | 基因数据文件快速识别方法、系统和计算机可读存储介质 |
| CN116541348B (zh) * | 2023-03-22 | 2023-09-26 | 河北热点科技股份有限公司 | 数据智能存储方法及终端查询一体机 |
| CN116739646B (zh) * | 2023-08-15 | 2023-11-24 | 南京易联阳光信息技术股份有限公司 | 网络交易大数据分析方法及分析系统 |
| CN117153270B (zh) * | 2023-10-30 | 2024-02-02 | 吉林华瑞基因科技有限公司 | 一种基因二代测序数据处理方法 |
Family Cites Families (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6303297B1 (en) * | 1992-07-17 | 2001-10-16 | Incyte Pharmaceuticals, Inc. | Database for storage and analysis of full-length sequences |
| JP3429674B2 (ja) | 1998-04-28 | 2003-07-22 | 沖電気工業株式会社 | 多重通信システム |
| EP1410301A4 (en) * | 2000-04-12 | 2008-01-23 | Cleveland Clinic Foundation | SYSTEM FOR IDENTIFYING AND ANALYZING GENE EXPRESSION CONTAINING ELEMENTS RICH IN ADENYLATE URIDYLATE (ARE) |
| FR2820563B1 (fr) * | 2001-02-02 | 2003-05-16 | Expway | Procede de compression/decompression d'un document structure |
| US20040153255A1 (en) * | 2003-02-03 | 2004-08-05 | Ahn Tae-Jin | Apparatus and method for encoding DNA sequence, and computer readable medium |
| DE10320711A1 (de) * | 2003-05-08 | 2004-12-16 | Siemens Ag | Verfahren und Anordnung zur Einrichtung und Aktualisierung einer Benutzeroberfläche zum Zugriff auf Informationsseiten in einem Datennetz |
| WO2005024562A2 (en) * | 2003-08-11 | 2005-03-17 | Eloret Corporation | System and method for pattern recognition in sequential data |
| US7805282B2 (en) * | 2004-03-30 | 2010-09-28 | New York University | Process, software arrangement and computer-accessible medium for obtaining information associated with a haplotype |
| US8340914B2 (en) * | 2004-11-08 | 2012-12-25 | Gatewood Joe M | Methods and systems for compressing and comparing genomic data |
| US20130332133A1 (en) * | 2006-05-11 | 2013-12-12 | Ramot At Tel Aviv University Ltd. | Classification of Protein Sequences and Uses of Classified Proteins |
| SE531398C2 (sv) | 2007-02-16 | 2009-03-24 | Scalado Ab | Generering av en dataström och identifiering av positioner inuti en dataström |
| KR101369745B1 (ko) * | 2007-04-11 | 2014-03-07 | 삼성전자주식회사 | 비동기화된 비트스트림들의 다중화 및 역다중화 방법 및장치 |
| US8832112B2 (en) * | 2008-06-17 | 2014-09-09 | International Business Machines Corporation | Encoded matrix index |
| GB2477703A (en) * | 2008-11-14 | 2011-08-10 | Real Time Genomics Inc | A method and system for analysing data sequences |
| US20100217532A1 (en) * | 2009-02-25 | 2010-08-26 | University Of Delaware | Systems and methods for identifying structurally or functionally significant amino acid sequences |
| DK2494060T3 (en) * | 2009-10-30 | 2016-08-01 | Synthetic Genomics Inc | Coding of text for nucleic acid sequences |
| EP2362657B1 (en) * | 2010-02-18 | 2013-04-24 | Research In Motion Limited | Parallel entropy coding and decoding methods and devices |
| US20140228223A1 (en) * | 2010-05-10 | 2014-08-14 | Andreas Gnirke | High throughput paired-end sequencing of large-insert clone libraries |
| CA2797645C (en) * | 2010-05-25 | 2020-09-22 | The Regents Of The University Of California | Bambam: parallel comparative analysis of high-throughput sequencing data |
| JP6420543B2 (ja) * | 2011-01-19 | 2018-11-07 | コーニンクレッカ フィリップス エヌ ヴェKoninklijke Philips N.V. | ゲノムデータ処理方法 |
| US9215162B2 (en) * | 2011-03-09 | 2015-12-15 | Annai Systems Inc. | Biological data networks and methods therefor |
| CN103797486A (zh) * | 2011-06-06 | 2014-05-14 | 皇家飞利浦有限公司 | 用于组装核酸序列数据的方法 |
| PL2721819T3 (pl) * | 2011-06-16 | 2024-02-19 | Ge Video Compression, Llc | Kodowanie entropijne obsługujące przełączanie trybów |
| US8707289B2 (en) * | 2011-07-20 | 2014-04-22 | Google Inc. | Multiple application versions |
| CN104081772B (zh) * | 2011-10-06 | 2018-04-10 | 弗劳恩霍夫应用研究促进协会 | 熵编码缓冲器配置 |
| EP2776962A4 (en) * | 2011-11-07 | 2015-12-02 | Ingenuity Systems Inc | METHODS AND SYSTEMS FOR IDENTIFICATION OF CAUSAL GENOMIC VARIANTS |
| KR101922129B1 (ko) * | 2011-12-05 | 2018-11-26 | 삼성전자주식회사 | 차세대 시퀀싱을 이용하여 획득된 유전 정보를 압축 및 압축해제하는 방법 및 장치 |
| KR20190016149A (ko) * | 2011-12-08 | 2019-02-15 | 파이브3 제노믹스, 엘엘씨 | 게놈 데이터의 동적 인덱싱 및 시각화를 제공하는 분산 시스템 |
| EP2608096B1 (en) * | 2011-12-24 | 2020-08-05 | Tata Consultancy Services Ltd. | Compression of genomic data file |
| US9600625B2 (en) * | 2012-04-23 | 2017-03-21 | Bina Technologies, Inc. | Systems and methods for processing nucleic acid sequence data |
| CN103049680B (zh) * | 2012-12-29 | 2016-09-07 | 深圳先进技术研究院 | 基因测序数据读取方法及系统 |
| US9679104B2 (en) * | 2013-01-17 | 2017-06-13 | Edico Genome, Corp. | Bioinformatics systems, apparatuses, and methods executed on an integrated circuit processing platform |
| WO2014145503A2 (en) * | 2013-03-15 | 2014-09-18 | Lieber Institute For Brain Development | Sequence alignment using divide and conquer maximum oligonucleotide mapping (dcmom), apparatus, system and method related thereto |
| JP6054790B2 (ja) * | 2013-03-28 | 2016-12-27 | 三菱スペース・ソフトウエア株式会社 | 遺伝子情報記憶装置、遺伝子情報検索装置、遺伝子情報記憶プログラム、遺伝子情報検索プログラム、遺伝子情報記憶方法、遺伝子情報検索方法及び遺伝子情報検索システム |
| GB2512829B (en) * | 2013-04-05 | 2015-05-27 | Canon Kk | Method and apparatus for encoding or decoding an image with inter layer motion information prediction according to motion information compression scheme |
| WO2014186604A1 (en) * | 2013-05-15 | 2014-11-20 | Edico Genome Corp. | Bioinformatics systems, apparatuses, and methods executed on an integrated circuit processing platform |
| KR101522087B1 (ko) * | 2013-06-19 | 2015-05-28 | 삼성에스디에스 주식회사 | 미스매치를 고려한 염기 서열 정렬 시스템 및 방법 |
| CN103336916B (zh) * | 2013-07-05 | 2016-04-06 | 中国科学院数学与系统科学研究院 | 一种测序序列映射方法及系统 |
| US20150032711A1 (en) * | 2013-07-06 | 2015-01-29 | Victor Kunin | Methods for identification of organisms, assigning reads to organisms, and identification of genes in metagenomic sequences |
| KR101493982B1 (ko) * | 2013-09-26 | 2015-02-23 | 대한민국 | 품종인식 코드화 시스템 및 이를 이용한 코드화 방법 |
| CN104699998A (zh) * | 2013-12-06 | 2015-06-10 | 国际商业机器公司 | 用于对基因组进行压缩和解压缩的方法和装置 |
| US10902937B2 (en) * | 2014-02-12 | 2021-01-26 | International Business Machines Corporation | Lossless compression of DNA sequences |
| US9916313B2 (en) * | 2014-02-14 | 2018-03-13 | Sap Se | Mapping of extensible datasets to relational database schemas |
| US9886561B2 (en) * | 2014-02-19 | 2018-02-06 | The Regents Of The University Of California | Efficient encoding and storage and retrieval of genomic data |
| US9354922B2 (en) * | 2014-04-02 | 2016-05-31 | International Business Machines Corporation | Metadata-driven workflows and integration with genomic data processing systems and techniques |
| US20150379195A1 (en) * | 2014-06-25 | 2015-12-31 | The Board Of Trustees Of The Leland Stanford Junior University | Software haplotying of hla loci |
| GB2527588B (en) * | 2014-06-27 | 2016-05-18 | Gurulogic Microsystems Oy | Encoder and decoder |
| US20160019339A1 (en) * | 2014-07-06 | 2016-01-21 | Mercator BioLogic Incorporated | Bioinformatics tools, systems and methods for sequence assembly |
| US10230390B2 (en) * | 2014-08-29 | 2019-03-12 | Bonnie Berger Leighton | Compressively-accelerated read mapping framework for next-generation sequencing |
| US10116632B2 (en) * | 2014-09-12 | 2018-10-30 | New York University | System, method and computer-accessible medium for secure and compressed transmission of genomic data |
| US20160125130A1 (en) * | 2014-11-05 | 2016-05-05 | Agilent Technologies, Inc. | Method for assigning target-enriched sequence reads to a genomic location |
| WO2016202918A1 (en) * | 2015-06-16 | 2016-12-22 | Gottfried Wilhelm Leibniz Universität Hannover | Method for compressing genomic data |
| CN105956417A (zh) * | 2016-05-04 | 2016-09-21 | 西安电子科技大学 | 云环境下基于编辑距离的相似碱基序列查询方法 |
| CN105975811B (zh) * | 2016-05-09 | 2019-03-15 | 管仁初 | 一种智能比对的基因序列分析装置 |
-
2017
- 2017-02-14 WO PCT/US2017/017841 patent/WO2018071054A1/en active Search and Examination
- 2017-02-14 CA CA3040138A patent/CA3040138A1/en not_active Abandoned
- 2017-02-14 CN CN201780062919.5A patent/CN110168651A/zh active Pending
- 2017-02-14 JP JP2019540510A patent/JP2020505702A/ja not_active Withdrawn
- 2017-02-14 WO PCT/US2017/017842 patent/WO2018071055A1/en active Application Filing
- 2017-02-14 KR KR1020197013567A patent/KR20190073426A/ko unknown
- 2017-02-14 SG SG11201903270RA patent/SG11201903270RA/en unknown
- 2017-02-14 BR BR112019007359A patent/BR112019007359A2/pt not_active IP Right Cessation
- 2017-02-14 PE PE2019000804A patent/PE20191058A1/es unknown
- 2017-02-14 MX MX2019004130A patent/MX2019004130A/es unknown
- 2017-02-14 AU AU2017342688A patent/AU2017342688A1/en not_active Abandoned
- 2017-02-14 US US16/341,426 patent/US20200042735A1/en not_active Abandoned
- 2017-02-14 EP EP17859972.6A patent/EP3526694A4/en not_active Withdrawn
- 2017-07-11 PE PE2019000805A patent/PE20191227A1/es unknown
- 2017-07-11 SG SG11201903272XA patent/SG11201903272XA/en unknown
- 2017-07-11 EP EP17860980.6A patent/EP3526657A4/en active Pending
- 2017-07-11 BR BR112019007357A patent/BR112019007357A2/pt not_active Application Discontinuation
- 2017-07-11 US US16/337,639 patent/US20190214111A1/en not_active Abandoned
- 2017-07-11 AU AU2017341684A patent/AU2017341684A1/en not_active Abandoned
- 2017-07-11 BR BR112019007363A patent/BR112019007363A2/pt not_active Application Discontinuation
- 2017-07-11 JP JP2019540512A patent/JP2019537172A/ja not_active Withdrawn
- 2017-07-11 BR BR112019007360A patent/BR112019007360A2/pt not_active Application Discontinuation
- 2017-07-11 AU AU2017341685A patent/AU2017341685A1/en not_active Abandoned
- 2017-07-11 US US16/337,642 patent/US11404143B2/en active Active
- 2017-07-11 PE PE2019000802A patent/PE20191056A1/es unknown
- 2017-07-11 WO PCT/US2017/041591 patent/WO2018071080A2/en unknown
- 2017-07-11 SG SG11201903271UA patent/SG11201903271UA/en unknown
- 2017-07-11 IL IL265879A patent/IL265879B2/en unknown
- 2017-07-11 CN CN201780063014.XA patent/CN110121577B/zh active Active
- 2017-07-11 EA EA201990916A patent/EA201990916A1/ru unknown
- 2017-07-11 CA CA3040147A patent/CA3040147A1/en not_active Abandoned
- 2017-07-11 MX MX2019004128A patent/MX2019004128A/es unknown
- 2017-07-11 JP JP2019540513A patent/JP2020500383A/ja not_active Withdrawn
- 2017-07-11 CN CN201780062885.XA patent/CN110114830B/zh active Active
- 2017-07-11 KR KR1020197013418A patent/KR20190062541A/ko active Search and Examination
- 2017-07-11 EA EA201990917A patent/EA201990917A1/ru unknown
- 2017-07-11 EP EP17860868.3A patent/EP3526707A4/en not_active Withdrawn
- 2017-07-11 CA CA3040145A patent/CA3040145A1/en not_active Abandoned
- 2017-07-11 JP JP2019540511A patent/JP7079786B2/ja active Active
- 2017-07-11 PE PE2019000803A patent/PE20191057A1/es unknown
- 2017-07-11 KR KR1020197013419A patent/KR20190069469A/ko not_active Application Discontinuation
- 2017-07-11 WO PCT/US2017/041585 patent/WO2018071079A1/en active Search and Examination
- 2017-07-11 CN CN201780063013.5A patent/CN110506272B/zh active Active
- 2017-12-14 CN CN201780086529.1A patent/CN110603595B/zh active Active
- 2017-12-14 US US16/485,623 patent/US20190385702A1/en active Pending
- 2017-12-14 KR KR1020197026863A patent/KR20190117652A/ko not_active Application Discontinuation
- 2017-12-14 PE PE2019001667A patent/PE20200323A1/es unknown
- 2017-12-14 BR BR112019016230A patent/BR112019016230A2/pt not_active Application Discontinuation
- 2017-12-15 US US16/485,649 patent/US20200051667A1/en active Pending
- 2017-12-15 PE PE2019001669A patent/PE20200226A1/es unknown
- 2017-12-15 BR BR112019016232A patent/BR112019016232A2/pt not_active Application Discontinuation
- 2017-12-15 CN CN201780086770.4A patent/CN110678929B/zh active Active
-
2018
- 2018-02-14 PE PE2019001668A patent/PE20200227A1/es unknown
- 2018-02-14 US US16/485,670 patent/US20200051665A1/en active Pending
- 2018-02-14 BR BR112019016236A patent/BR112019016236A2/pt unknown
-
2019
- 2019-04-08 IL IL265928A patent/IL265928B/en active IP Right Grant
- 2019-04-10 CL CL2019000968A patent/CL2019000968A1/es unknown
- 2019-04-10 CL CL2019000972A patent/CL2019000972A1/es unknown
- 2019-04-10 CL CL2019000973A patent/CL2019000973A1/es unknown
- 2019-04-11 PH PH12019550060A patent/PH12019550060A1/en unknown
- 2019-04-11 CO CONC2019/0003638A patent/CO2019003638A2/es unknown
- 2019-04-11 PH PH12019550059A patent/PH12019550059A1/en unknown
- 2019-04-11 CO CONC2019/0003639A patent/CO2019003639A2/es unknown
- 2019-04-11 PH PH12019550058A patent/PH12019550058A1/en unknown
- 2019-04-11 CO CONC2019/0003595A patent/CO2019003595A2/es unknown
- 2019-04-11 IL IL265972A patent/IL265972A/en unknown
- 2019-04-11 PH PH12019550057A patent/PH12019550057A1/en unknown
- 2019-04-15 CO CONC2019/0003842A patent/CO2019003842A2/es unknown
- 2019-08-12 CL CL2019002277A patent/CL2019002277A1/es unknown
- 2019-08-12 CL CL2019002275A patent/CL2019002275A1/es unknown
- 2019-08-12 CL CL2019002276A patent/CL2019002276A1/es unknown
- 2019-08-13 PH PH12019501881A patent/PH12019501881A1/en unknown
- 2019-08-13 PH PH12019501879A patent/PH12019501879A1/en unknown
- 2019-09-12 CO CONC2019/0009920A patent/CO2019009920A2/es unknown
- 2019-09-12 CO CONC2019/0009922A patent/CO2019009922A2/es unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20190073426A (ko) | 저장 또는 전송된 생물정보학 데이터의 선택적 액세스를 위한 방법 및 시스템 | |
| CA3039688C (en) | Efficient data structures for bioinformatics information representation | |
| US11386979B2 (en) | Method and system for storing and accessing bioinformatics data | |
| KR102421458B1 (ko) | 액세스 유닛으로 구조화된 생물정보학 데이터에 액세스하기 위한 방법 및 장치 | |
| JP6949970B2 (ja) | バイオインフォマティクスデータを送信する方法及びシステム | |
| JP2020509473A (ja) | 複数のゲノム記述子を用いた生体情報データのコンパクト表現方法及び装置 | |
| KR20190113971A (ko) | 다중 게놈 디스크립터를 이용한 생명정보학 데이터의 압축 표현 방법 및 장치 | |
| CN110663022B (zh) | 使用基因组描述符紧凑表示生物信息学数据的方法和设备 | |
| NZ753247B2 (en) | Efficient data structures for bioinformatics information representation |