KR20130100105A - 류코트리엔 생성 억제제인 옥사디아졸 - Google Patents
류코트리엔 생성 억제제인 옥사디아졸 Download PDFInfo
- Publication number
- KR20130100105A KR20130100105A KR1020137003938A KR20137003938A KR20130100105A KR 20130100105 A KR20130100105 A KR 20130100105A KR 1020137003938 A KR1020137003938 A KR 1020137003938A KR 20137003938 A KR20137003938 A KR 20137003938A KR 20130100105 A KR20130100105 A KR 20130100105A
- Authority
- KR
- South Korea
- Prior art keywords
- alkyl
- independently
- optionally substituted
- mmol
- methyl
- Prior art date
Links
- 0 *C(c1ccc(*)cc1*)(C#N)I Chemical compound *C(c1ccc(*)cc1*)(C#N)I 0.000 description 4
- LKRQCIAJVZMFST-VWLOTQADSA-N CC(C)(C[n]1ncc(-c2nc([C@](C)(C3CC3)c(cc3)ccc3-c3ccc(N)nc3)n[o]2)c1)O Chemical compound CC(C)(C[n]1ncc(-c2nc([C@](C)(C3CC3)c(cc3)ccc3-c3ccc(N)nc3)n[o]2)c1)O LKRQCIAJVZMFST-VWLOTQADSA-N 0.000 description 1
- WAZOVRRSGVOESP-DEOSSOPVSA-N CC(C)(C[n]1ncc(-c2nc([C@](C)(C3CC3)c(cc3)ccc3-c3ncc(N)nc3)n[o]2)c1)O Chemical compound CC(C)(C[n]1ncc(-c2nc([C@](C)(C3CC3)c(cc3)ccc3-c3ncc(N)nc3)n[o]2)c1)O WAZOVRRSGVOESP-DEOSSOPVSA-N 0.000 description 1
- RXBLDHGAAFXQJE-UHFFFAOYSA-N CC(C)C(C)(c1n[o]c(-c2c[n](CCN3CCOCC3)nc2)n1)c(cc1)ccc1-c1cnc(N)nc1 Chemical compound CC(C)C(C)(c1n[o]c(-c2c[n](CCN3CCOCC3)nc2)n1)c(cc1)ccc1-c1cnc(N)nc1 RXBLDHGAAFXQJE-UHFFFAOYSA-N 0.000 description 1
- AFIBYTLXVHYHGF-XMMPIXPASA-N CC(C)[C@@](C)(c1n[o]c(-c2c[n](CC(C)(C)O)nc2)n1)c(cc1)ccc1-c1cnc(N)nc1 Chemical compound CC(C)[C@@](C)(c1n[o]c(-c2c[n](CC(C)(C)O)nc2)n1)c(cc1)ccc1-c1cnc(N)nc1 AFIBYTLXVHYHGF-XMMPIXPASA-N 0.000 description 1
- KQCAPJKLNQUTAS-UHFFFAOYSA-N CC(C1CC1)(/C(/N)=N/O)c(cc1)ccc1Br Chemical compound CC(C1CC1)(/C(/N)=N/O)c(cc1)ccc1Br KQCAPJKLNQUTAS-UHFFFAOYSA-N 0.000 description 1
- KFWNLBZFQKXLKN-UHFFFAOYSA-N CC(C1CC1)(C(/N=C(/c1c[n](CC(N(C)C)=O)nc1)\O)=N)c(cc1)ccc1-c(nc1)c(C#N)nc1N Chemical compound CC(C1CC1)(C(/N=C(/c1c[n](CC(N(C)C)=O)nc1)\O)=N)c(cc1)ccc1-c(nc1)c(C#N)nc1N KFWNLBZFQKXLKN-UHFFFAOYSA-N 0.000 description 1
- HWEAEPQUGRBMKZ-UHFFFAOYSA-O CC(C1CC1)(C(/N=C(/c1c[n](CC(N(C)C)=O)nc1)\[OH2+])=N)c1ccc(B2OC(C)(C)C(C)(C)O2)cc1 Chemical compound CC(C1CC1)(C(/N=C(/c1c[n](CC(N(C)C)=O)nc1)\[OH2+])=N)c1ccc(B2OC(C)(C)C(C)(C)O2)cc1 HWEAEPQUGRBMKZ-UHFFFAOYSA-O 0.000 description 1
- NXBUGRSMWYWAOA-UHFFFAOYSA-N CC(C1CC1)(c1n[o]c(-c2c[nH]nc2)n1)c(cc1)ccc1Br Chemical compound CC(C1CC1)(c1n[o]c(-c2c[nH]nc2)n1)c(cc1)ccc1Br NXBUGRSMWYWAOA-UHFFFAOYSA-N 0.000 description 1
- GOWQSVCMKPYCHX-UHFFFAOYSA-N CNc1nccc(F)n1 Chemical compound CNc1nccc(F)n1 GOWQSVCMKPYCHX-UHFFFAOYSA-N 0.000 description 1
- HNPZTDGIKKMDNU-INIZCTEOSA-N C[C@@](C1CC1)(c(cc1)ccc1-c1cnc(N)nc1)C#N Chemical compound C[C@@](C1CC1)(c(cc1)ccc1-c1cnc(N)nc1)C#N HNPZTDGIKKMDNU-INIZCTEOSA-N 0.000 description 1
- VVDSIQPQUUDQHC-FQEVSTJZSA-O C[C@](C1CC1)(C(/N=C(/c1c[nH]nc1)\[OH2+])=N)c(cc1)ccc1-c1cnc(N)nc1 Chemical compound C[C@](C1CC1)(C(/N=C(/c1c[nH]nc1)\[OH2+])=N)c(cc1)ccc1-c1cnc(N)nc1 VVDSIQPQUUDQHC-FQEVSTJZSA-O 0.000 description 1
- FAEDYVRHTVWKBU-VWLOTQADSA-N C[C@](C1CC1)(C(/N=C(/c1c[n](CC(N(C)C)=O)nc1)\O)=N)c(cc1)ccc1-c1nc(C#N)c(N)nc1 Chemical compound C[C@](C1CC1)(C(/N=C(/c1c[n](CC(N(C)C)=O)nc1)\O)=N)c(cc1)ccc1-c1nc(C#N)c(N)nc1 FAEDYVRHTVWKBU-VWLOTQADSA-N 0.000 description 1
- AWKUXQZVCDWEDI-PMERELPUSA-O C[C@](C1CC1)(C(/N=C(/c1c[n](CCN(C(c2c3cccc2)=O)C3=O)nc1)\[OH2+])=N)c(cc1)ccc1-c1cnc(N)nc1 Chemical compound C[C@](C1CC1)(C(/N=C(/c1c[n](CCN(C(c2c3cccc2)=O)C3=O)nc1)\[OH2+])=N)c(cc1)ccc1-c1cnc(N)nc1 AWKUXQZVCDWEDI-PMERELPUSA-O 0.000 description 1
- UCARIQLDASWRIK-VWLOTQADSA-N C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c(c(C)nc(N)n1)c1F Chemical compound C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c(c(C)nc(N)n1)c1F UCARIQLDASWRIK-VWLOTQADSA-N 0.000 description 1
- AKHAUJKTKYSPFH-VWLOTQADSA-N C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c(cc1)cnc1N Chemical compound C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c(cc1)cnc1N AKHAUJKTKYSPFH-VWLOTQADSA-N 0.000 description 1
- KEGOKBSLUSAYSD-HKBQPEDESA-N C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c(cnc(N)n1)c1OCc1ccccc1 Chemical compound C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c(cnc(N)n1)c1OCc1ccccc1 KEGOKBSLUSAYSD-HKBQPEDESA-N 0.000 description 1
- FMPQMNVONTYNSI-VWLOTQADSA-N C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c1cc(F)c(N)nc1 Chemical compound C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c1cc(F)c(N)nc1 FMPQMNVONTYNSI-VWLOTQADSA-N 0.000 description 1
- CKLNNECEUZSQCN-DEOSSOPVSA-N C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c1cnc(N)nc1F Chemical compound C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1-c1cnc(N)nc1F CKLNNECEUZSQCN-DEOSSOPVSA-N 0.000 description 1
- IPIRVHUYPHQCQL-DEOSSOPVSA-N C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1C1=CN=C(N)NC1=O Chemical compound C[C@](C1CC1)(c1n[o]c(-c2c[n](CC(N(C)C)=O)nc2)n1)c(cc1)ccc1C1=CN=C(N)NC1=O IPIRVHUYPHQCQL-DEOSSOPVSA-N 0.000 description 1
- QUYZBNHTYCLZLW-UHFFFAOYSA-N Nc(nc1)c(C(F)(F)F)cc1Br Chemical compound Nc(nc1)c(C(F)(F)F)cc1Br QUYZBNHTYCLZLW-UHFFFAOYSA-N 0.000 description 1
- YWOWJQMFMXHLQD-UHFFFAOYSA-N Nc1c(C(F)(F)F)cccn1 Chemical compound Nc1c(C(F)(F)F)cccn1 YWOWJQMFMXHLQD-UHFFFAOYSA-N 0.000 description 1
- DBGFGNCFYUNXLD-UHFFFAOYSA-N Nc1nccc(Cl)n1 Chemical compound Nc1nccc(Cl)n1 DBGFGNCFYUNXLD-UHFFFAOYSA-N 0.000 description 1
- IMBBXSASDSZJSX-UHFFFAOYSA-N OC(c1c[nH]nc1)=O Chemical compound OC(c1c[nH]nc1)=O IMBBXSASDSZJSX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Abstract
본 발명은 화학식 I의 화합물 또는 약제학적으로 허용되는 이의 염에 관한 것이다.
화학식 I

상기 화학식 I에서,
R1 내지 R5는 본 명세서에서 정의한 바와 같다.
본 발명은 또한 이들 화합물을 포함하는 약제학적 조성물, 다양한 질환 및 장애의 치료시 이들 화합물의 사용 방법, 이들 화합물의 제조 방법 및 이들 방법에 유용한 중간체에 관한 것이다.
화학식 I
상기 화학식 I에서,
R1 내지 R5는 본 명세서에서 정의한 바와 같다.
본 발명은 또한 이들 화합물을 포함하는 약제학적 조성물, 다양한 질환 및 장애의 치료시 이들 화합물의 사용 방법, 이들 화합물의 제조 방법 및 이들 방법에 유용한 중간체에 관한 것이다.
Description
본 발명은 옥사디아졸에 관한 것으로, 상기 옥사디아졸은 5-리폭시게나제 활성 단백질(FLAP; five lipoxygenase activating protein)의 억제제로서 유용하므로, 천식, 알러지, 류마티스 관절염, 다발성 경화증, 염증성 통증, 급성 심장 증후군 및 죽상 동맥경화증, 심근경색과 뇌졸중을 포함한 심혈관 질환을 포함하는 류코트리엔의 활성을 통해 매개되거나 유지되는 다양한 질환 및 장애의 치료에 유용하다. 본 발명은 또한 이들 화합물을 포함하는 약제학적 조성물, 다양한 질환 및 장애의 치료시 이들 화합물의 사용 방법, 이들 화합물 및 이들 방법에 유용한 중간체의 제조 방법에 관한 것이다.
류코트리엔(LT) 및 이들의 생성을 유도하는 아라키돈산으로부터의 생합성 경로는 20년에 걸쳐 약물을 발견하기 위한 대상이 되었다. LT는 호중구, 비만세포, 호산구, 호염기구, 단핵구 및 대식세포를 포함한 몇몇 세포 형태에 의해 생성된다. LT의 세포 내 합성에서 처음 수반되는 단계는 5-리폭시게나제(5-LO)에 의한 아라키돈산의 LTA4로의 산화를 포함하고, 이 과정은 18kD 통합 막단백질 5-리폭시게나제-활성화 단백질(FLAP)의 존재를 필요로 한다(참조: D.K. Miller et al., Nature, 1990, 343, 278-281; R.A.F. Dixon et al., Nature, 1990, 343, 282-284). LTA4의 후속적인 대사작용은 LTB4, 및 시스테이닐 LT- LTC4, LTD4와 LTE4(참조: B. Samuelsson, Science, 1983, 220, 568-575)를 유도한다. 시스테이닐 LT는 효능있는 평활근 수축 및 기관지수축 효과를 가지며, 이들은 점막 분비 및 혈관 누수를 자극한다. LTB4는 백혈구에 대해 효능있는 화학주성 제제이고, 부착, 응집 및 효소 방출을 자극한다.
LT 영역에 있어서 초기 약물을 발견하기 위한 다수의 시도는 알러지, 천식 및 다른 염증성 상태의 치료에 관한 것이었다. 시도된 연구는 5-리폭시게나제(5-LO)의 억제제, LTA4 하이드롤라제 및 5-리폭시게나제-활성화 단백질(FLAP)의 억제제 뿐만 아니라, LTB4의 길항제 및 시스테이닐 류코트리엔 LTC4, LTD4와 LTE4를 포함한 경로에서 수많은 표적에 관한 것이었다(참조: R.W. Friesen and D. Riendeau, Leukotriene Biosynthesis Inhibitors, Ann. Rep. Med. Chem., 2005, 40, 199-214). 상기 영역에서 수년간의 노력으로 5-LO 억제제, 질루톤, 및 LT 길항제, 몬테루카스트, 프란루카스트 및 자피르루카스트를 포함한 천식 치료용 몇몇 시판 제품을 수득하였다.
보다 최근의 작업은 심근경색, 뇌졸중 및 죽상 동맥경화증을 포함한 심혈관 질환에 LT를 연루시켰다(참조: G. Riccioni et al., J. Leukoc. Biol., 2008, 1374-1378). FLAP 및 5-LO는 죽상 동맥경화증 병변에서 발견되는 5-LO 및 LT 캐스케이트의 성분 중에 속하고, 종죽 발생에 있어서 이들의 관여를 제시하는 것이다(참조: R. Spanbroek et al., Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 1238-1243). FLAP의 약물학적 억제는 동물 모델에서 죽상 동맥경화증 병변 크기를 감소시키는 것으로 보고되어 왔다. 한 연구에서, 2 내지 6개월된 고-지방 식이를 공급한 apoE/LDL-R 더블 녹아웃(double knockout) 마우스에 FLAP 억제제 MK-886의 경구 투약은 대동맥에서 플라크 커버리지(plaque coverage)의 56% 감소 및 대동맥 근부에서 43% 감소를 유도한다(참조: J. Jawien et al., Eur. J. Clin. Invest., 2006, 36, 141-146). 이 플라크 효과는 플라크-대식세포 함량의 감소 및 콜라겐과 평활근 함량의 수반되는 증가와 결합되고, 이는 보다 안정한 플라크 표현형으로의 전환을 제시한다. 다른 연구에서, ApoE-/-xCD4dnTβRII 마우스(시스템으로부터 모든 TGF-베타를 효과적으로 제거하는 우세한-네가티브 TGF-베타 수용체를 발현하는 apoE KO 마우스)에 주입을 통한 MK-886의 투여는 대동맥 근부에서 플라크 영역의 약 40% 감소를 일으킨다고 보고되었다(참조: M. Back et al., Circ. Res., 2007, 100, 946-949). 마우스는 플라크 성장이 이미 다소 성숙된 후(12주) 4주 동안 단지 처리됨으로써, 이 메카니즘을 통해 죽상 동맥경화증을 치료학적으로 치료할 가능성이 증가된다. 사람의 죽상 동맥경화증 병변을 검사하는 연구에서, FLAP, 5-LO 및 LTA4 하이드롤라제의 발현은 건강한 대조군에 비하여 상당히 증가되는 것으로 밝혀졌다(참조: H. Qiu et al., Proc. Natl. Acad. Sci. U.S.A., 103, 21, 8161-8166). 유사한 연구는 예를 들면, FLAT의 억제에 의한 LT 경로의 억제가 죽상 동맥경화증의 치료에 유용하다고 제시하고 있다(검토를 위해, 참조: M. Back Curr. Athero. Reports, 2008 10, 244-251 및 Curr. Pharm. Des., 2009, 15, 3116-3132).
상기 인용된 작업 이외에, 많은 다른 연구는 질환에서 LT의 생물학적 작용 및 LT의 역할을 이해하는 것에 관한 것이었다. 이들 연구는 수많은 질환 또는 상태에서 가능할 수 있는 역할을 갖는 것으로서 LT를 연루시켜왔다(검토를 위해, 참조: M. Peters-Golden and W.R. Henderson, Jr., M.D., N. Engl. J. Med., 2007, 357, 1841-1854). 상기 인용된 특정 질환 이외에, LT는 암 뿐만 아니라, 수많은 알러지, 폐, 섬유증성, 염증성 및 심혈관 질환에서 가능할 수 있는 역할을 갖는 것으로 연루되어 왔다. FLAP의 억제는 또한 당뇨병-유도 단백뇨와 같은 신장 질환의 치료에 유용한 것으로 보고되고 있다[참조: J. M. Valdivieso et al., Journal of Nephrology, 2003, 16, 85-94 및 A Montero et al., Journal of Nephrology, 2003, 16, 682-690].
수많은 FLAP 억제제가 과학 문헌[참조: J.F. Evans et al., Trends in Pharmacological Sciences, 2008, 72-78] 및 미국 특허에 보고되어 왔다. 일부는 DG-031로서 또한 공지된 MK-886, MK-591 및 BAY X1005를 포함한 천식을 위한 임상적 시도에서 평가되어 왔다. 보다 최근에는, FLAP 억제제 AM-103(참조: J.H. Hutchinson et al., J. Med. Chem. 52, 5803-5815)이 이의 항-염증 특성을 근거로 하는 임상적 시도에서 평가되었다(참조: D.S. Lorrain et al., J. Pharm. Exp. Ther., 2009, DOI:10.1124/jpet.109.158089). 이어서, 그는 호흡기 질환의 치료를 위한 백-업 화합물 AM-803(GSK-2190915)에 의해 대체되었다. DG-031은 또한 심근경색 위험에 대한 바이오마커(biomarker)로서 이의 효과를 평가하기 위한 임상적 시도가 있었고, 상기 질환에 대한 몇몇 바이오마커의 용량-의존성 억제를 나타내었다(참조: H. Hakonarson et al., JAMA, 2005, 293, 2245-2256). MK-591은 사람의 사구체신염에서 단백뇨를 감소시키기 위한 임상적 시도에서 제시되었다[참조: A. Guash et al., Kidney International, 1999, 56, 291-267].
그러나, 지금까지, FLAP 억제제가 시판 약제로서 승인된 것은 없었다.
발명의 간단한 요약
본 발명은, 5-리폭시게나제 활성화 단백질(FLAP)을 억제함으로써, 알러지성, 폐, 섬유증성, 염증성 및 심혈관 질환과 암을 포함하는, 류코트리엔의 활성을 통해 매개되거나 유지되는 다양한 질환 및 장애의 치료에 유용한 신규 화합물을 제공하는 것이다. 본 발명은 또한 이들 화합물을 포함하는 약제학적 조성물, 다양한 질환 및 장애의 치료시 이들 화합물의 사용 방법, 이들 화합물의 제조 방법 및 이들 방법에 유용한 중간체에 관한 것이다.
본 발명의 첫 번째 가장 광범위한 양태에서, 본 발명은 하기 화학식 I의 화합물 또는 약제학적으로 허용되는 이의 염에 관한 것이다:
화학식 I

상기 화학식 I에서,
R1 및 R2는 각각 독립적으로 수소, C1 -7 알킬 또는 C3 -10 카보사이클이고, 단, R1 및 R2는 모두 수소가 아니고;
R3은 질소, 산소 및 황으로부터 선택되는 1 내지 3개의 헤테로 원자를 함유하는 5원 내지 11원 헤테로아릴 환이고, 여기서, 상기 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1 -5 알킬, C1 -5 알콕시, C1 -3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -6 알킬아미노 및 C1-3 디알킬아미노로부터 선택된 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R4는 수소, C1 -3 알킬, 할로겐 또는 니트릴이고;
R5는 C1 -6 알킬, C3 -10 카보사이클, 3원 내지 11원 헤테로사이클, 아릴, 5원 내지 11원 헤테로아릴, -C(O)-R6, 하이드록시 또는 NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R6은 C3 -8 헤테로사이클 또는 -NH-5원 내지 6원 헤테로사이클이고, 이들 각각은 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8은 각각 독립적으로 수소, C1 -6 알킬에 의해 임의로 치환된 5원 내지 6원 헤테로사이클, 하이드록시에 의해 임의로 치환된 C3 -10 카보사이클, 또는 C1 -6 알킬이고;
R9, R10 및 R11은 독립적으로
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 3원 내지 6원 헤테로사이클, C1 -6 알콕시, C1-6알콕시-O-C1 - 6알킬, -CO2R12, -C(O)N(R12)(R13) 또는 -S(O)nC1- 6알킬에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 6알콕시,
(h) -N(R12)(R13),
(i) -S(O)nC1- 6알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 1 내지 3개의 C1 -6 알킬 그룹에 의해 임의로 치환된 3원 내지 10원 헤테로사이클릭 그룹,
(n') 옥소,
(o) -C(O)-C1 -3 알킬로부터 선택되고;
R12 및 R13은 각각 독립적으로 -H, -C1 - 6알킬, C(O)C1- 6알킬 및 3원 내지 6원 헤테로사이클릭 그룹으로부터 선택되고, 이들 각각은 1 내지 3개의 C1 - 6알킬 그룹, -OH, C1 - 6알콕시, -C(O)N(R14)(R15), -S(O)nC1- 6알킬, CN, 3원 내지 6원 헤테로사이클릭 그룹, -OC1 - 6알킬, CF3에 의해 임의로 독립적으로 치환되거나;
R12 및 R13은, 이들이 부착된 질소 환과 함께, 1 내지 3개의 -OH, CN, -OC1 - 6알킬 또는 옥소에 의해 임의로 치환된 헤테로사이클릴 환을 형성하고;
R14 및 R15는 각각 독립적으로 -H 및 -C1 - 6알킬로부터 선택되고;
n은 0, 1 또는 2이다.
두 번째 양태에서, 본 발명은
R1 및 R2는 각각 독립적으로 수소, 메틸, 에틸, 프로필, 이소프로필, 부틸, 이소부틸, 3급-부틸, 펜틸, 헥실, 사이클로프로필, 사이클로부틸, 사이클로펜틸 또는 사이클로헥실이고, 단, R1 및 R2는 모두 수소가 아니고;
R3은 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 피롤릴, 이미다졸릴, 티에닐, 푸라닐 또는 티아졸릴이고, 여기서, 각각의 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1 -3 알킬, C1 -3 알콕시, C1 -3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -6 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되거나;
R3은 피리도옥사지닐, 디하이드로-피리도옥사지닐, 디하이드로-피롤로피리디닐, 피롤로피리디닐 또는 피롤로피라지닐이고, 여기서, 각각의 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1 -5 알킬, C1 -3 알콕시, C1 -3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -3 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R4는 수소, 메틸 또는 플루오로이고;
R5는 메틸, 에틸, 프로필, 이소프로필, 부틸, 이소부틸, 3급-부틸, 펜틸, 헥실, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 사이클로헵틸, 사이클로옥틸, 페닐, 피페리디닐, 피페라지닐, 모르폴리닐, 티오모르폴리닐, 아제티디닐, 피롤리디닐, 테트라하이드로피라닐, 피롤릴, 티에닐, 푸라닐, 티아졸릴, 옥사졸릴, 이속사졸릴, 피라졸릴, 이미다졸릴, 트리아졸릴, 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 퀴놀리닐, 이소퀴놀리닐, 인돌릴, 피롤로피리디닐, 피롤로피리미디닐, -C(O)-R6, 하이드록시 또는 -NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R6은 피페리디닐, 피페라지닐, 테트라하이드로피라닐, 모르폴리닐, 티오모르폴리닐 또는 -NH-피페라디닐이고, 이들 각각은 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8은 각각 독립적으로 수소, 메틸에 의해 임의로 치환된 5원 내지 6원 헤테로사이클, 하이드록시에 의해 임의로 치환된 C3 -6 카보사이클, 또는 C1 -5 알킬이고;
R9, R10 및 R11은 독립적으로
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 3원 내지 6원 헤테로사이클, C1 -6 알콕시, C1-6알콕시-O-C1 - 6알킬, -CO2R12, -C(O)N(R12)(R13) 또는 -S(O)nC1- 6알킬에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 6알콕시,
(h) -N(R12)(R13),
(i) -S(O)nC1- 6알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 1 내지 3개의 C1 -6 알킬 그룹에 의해 임의로 치환된 3원 내지 8원 헤테로사이클릭 그룹,
(n') 옥소,
(o) -C(O)-C1 -3 알킬로부터 선택되고;
R12 및 R13은 각각 독립적으로 -H, -C1 - 6알킬, C(O)C1- 6알킬 및 3원 내지 6원 헤테로사이클릭 그룹으로부터 선택되고, 이들 각각은 1 내지 3개의 C1 - 6알킬 그룹, -OH, C1 - 6알콕시, -C(O)N(R14)(R15), -S(O)nC1- 6알킬, CN, 3원 내지 6원 헤테로사이클릭 그룹, -OC1 - 6알킬, CF3에 의해 임의로 독립적으로 치환되거나;
R12 및 R13은, 이들이 부착된 질소 환과 함께, 1 내지 3개의 -OH, CN, -OC1 - 6알킬 또는 옥소에 의해 임의로 치환된 헤테로사이클릴 환을 형성할 수 있고;
R14 및 R15는 각각 독립적으로 -H 및 -C1 - 4알킬로부터 선택되고;
n은 1 또는 2인, 상기 가장 광범위한 양태로 기술된 화합물 또는 약제학적으로 허용되는 이의 염에 관한 것이다.
세 번째 양태에서, 본 발명은
R1 및 R2는 각각 독립적으로 수소, 메틸, 에틸, 프로필, 이소프로필, 3급-부틸, 사이클로프로필 또는 사이클로부틸이고, 단, R1 및 R2는 모두 수소가 아닌, 상기 전술한 양태 중 어느 것에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염에 관한 것이다.
네 번째 양태에서,
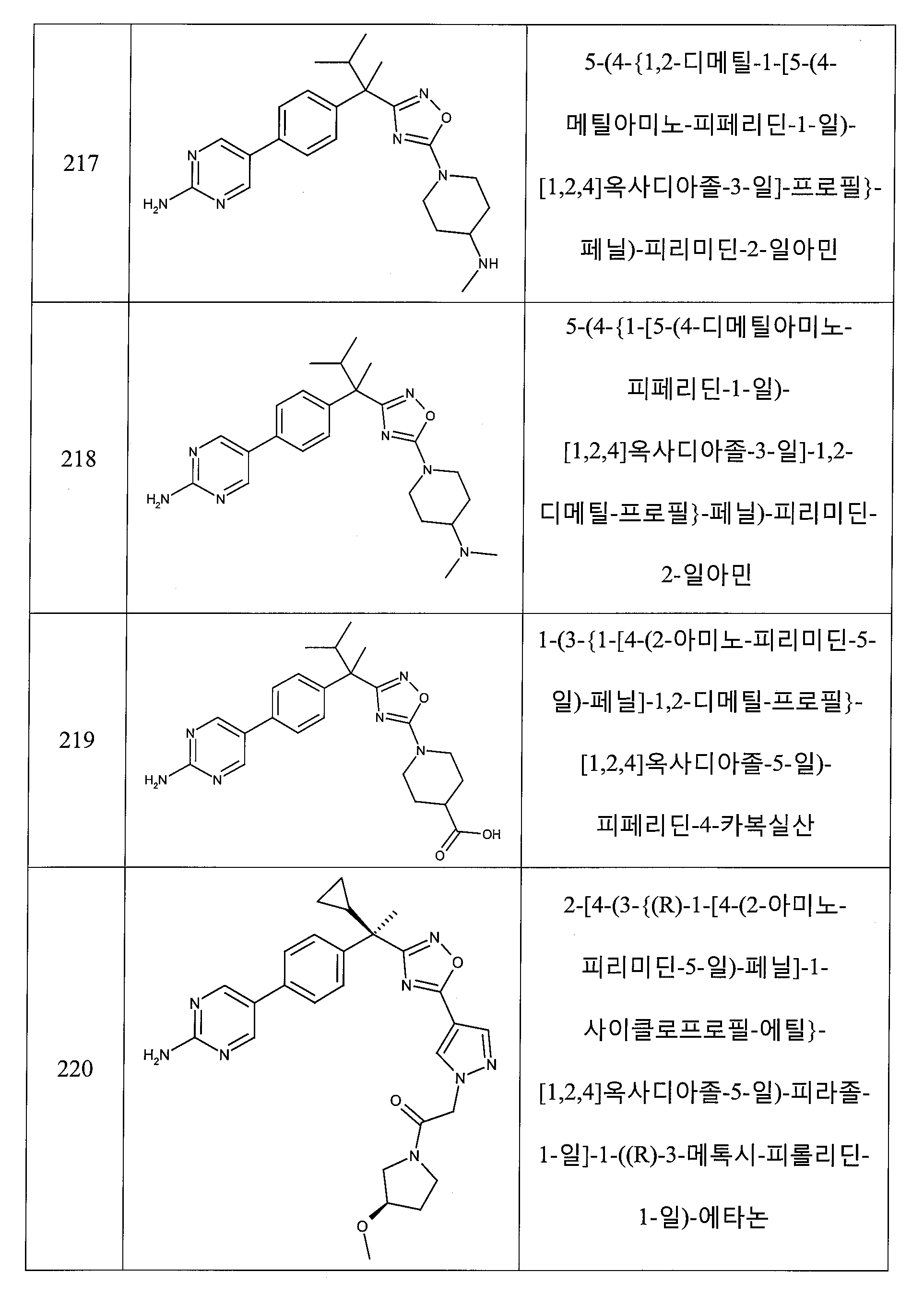
R3은 피리디닐, 피라지닐, 피리다지닐 또는 피리미디닐이고, 여기서, 각각의 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1 -3 알킬, C1 -3 알콕시, C1 -3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3-6 카보사이클, C1 -5 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 2개의 그룹에 의해 임의로 독립적으로 치환되거나;
R3은 피리도옥사지닐, 디하이드로-피리도옥사지닐, 디하이드로-피롤로피리디닐, 피롤로피리디닐 또는 피롤로피라지닐인, 상기 전술한 양태 중 어느 것에서 기술된 화학식 I의 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
다섯 번째 양태에서,
R5는 메틸, 에틸, 프로필, 이소프로필, 부틸, 펜틸, 헥실, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 페닐, 아제티디닐, 피페리디닐, 피페라지닐, 모르폴리닐, 테트라하이드로피라닐, 티아졸릴, 옥사졸릴, 이속사졸릴, 피라졸릴, 이미다졸릴, 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 퀴놀리닐, 이소퀴놀리닐, -C(O)-피페리지닐, -C(O)-피페리디닐, -C(O)-모르폴리닐, -C(O)-NH-피페리디닐, 하이드록시 또는 -NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8은 각각 독립적으로 수소, 메틸에 의해 임의로 치환된 5원 내지 6원 헤테로사이클, 하이드록시에 의해 임의로 치환된 C3 -6 카보사이클, 또는 C1 -C5 알킬이고;
R9, R10 및 R11은 독립적으로
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 모르폴리닐, 피페라지닐, C1 -6 알콕시, C1-3알콕시-O-C1 - 3알킬, -CO2R12 또는 -C(O)N(R12)(R13)에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 6알콕시,
(h) -N(R12)(R13),
(i) -S(O)nC1- 6알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 모르폴리닐, 피페라지닐, 피페리디닐 또는 옥세타닐(이들 각각은 메틸 그룹에 의해 임의로 치환됨),
(n') 옥소,
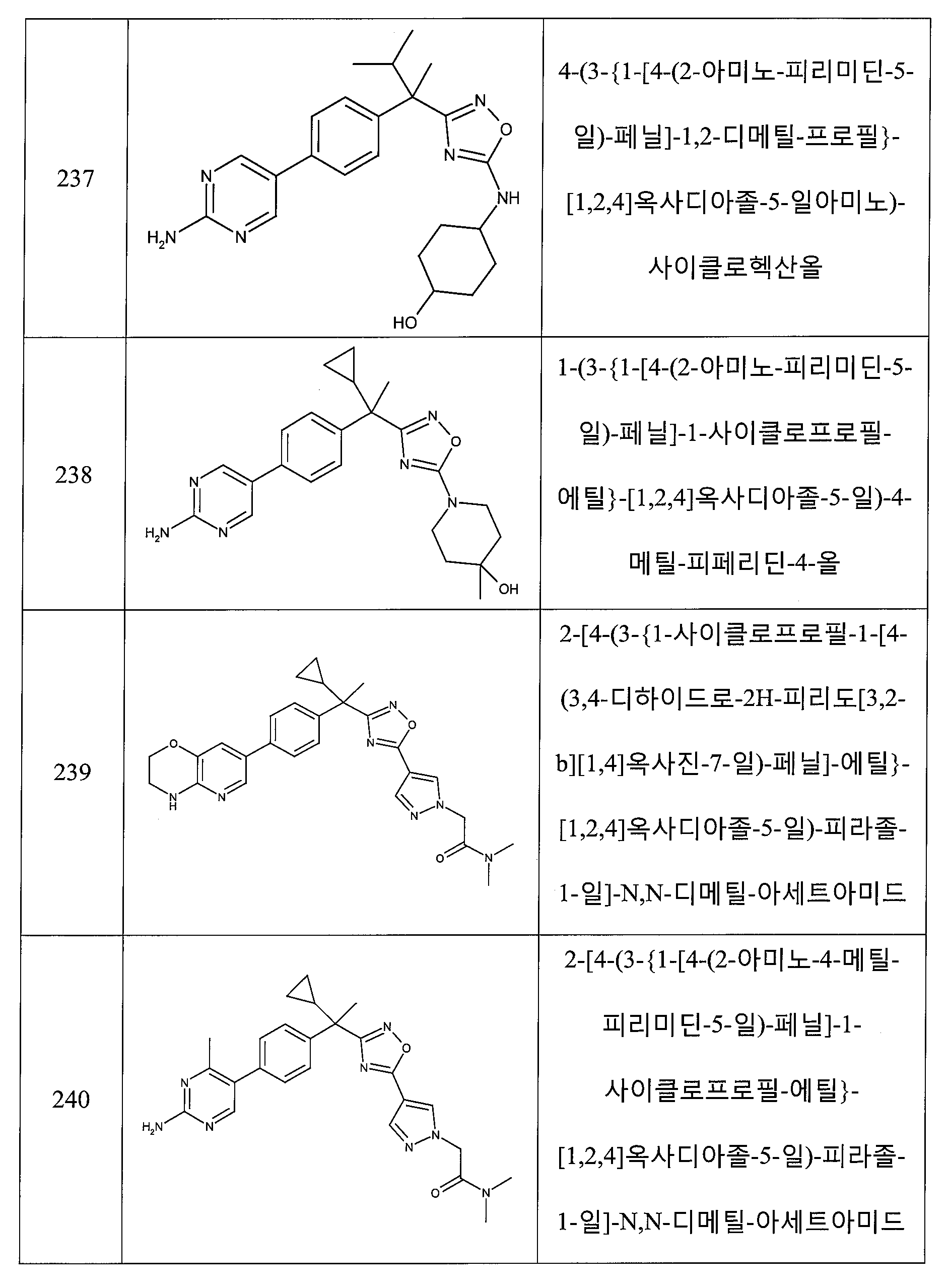
(o) -C(O)-CH3으로부터 선택되고;
R12 및 R13은 각각 독립적으로 -H 및 -C1 - 6알킬로부터 선택되고, 여기서, 알킬 그룹은 1 내지 3개의 -OH, C1 - 6알콕시, -C(O)N(R14)(R15) 또는 -S(O)nC1- 6알킬에 의해 임의로 치환되고;
R14 및 R15는 각각 독립적으로 -H 및 -C1 - 4알킬로부터 선택되고;
n은 2인, 상기 전술한 양태 중 어느 것에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
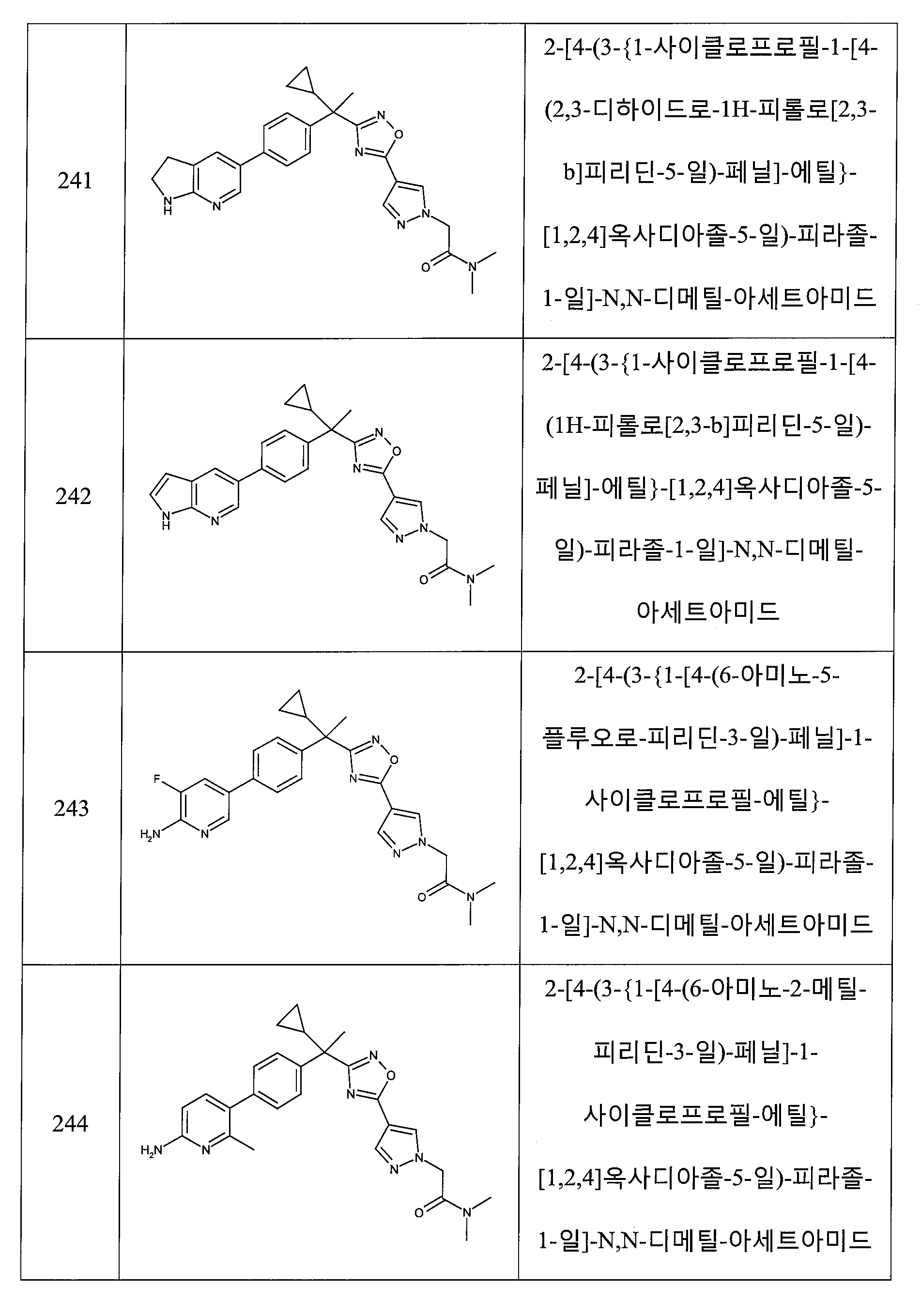
여섯 번째 양태에서,
R1 및 R2는 각각 독립적으로 수소, 메틸, 에틸, 프로필, 이소프로필, 3급-부틸, 사이클로프로필 또는 사이클로부틸이고, 단, R1 및 R2는 모두 수소가 아니고;
R3은 피리디닐, 피라지닐, 피리다지닐 또는 피리미디닐이고, 여기서, 각각의 헤테로아릴 환은 메틸, 메톡시, -CH2OH, 트리플루오로메틸, 브로모, 클로로, 플루오로, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -4 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 2개의 그룹에 의해 임의로 독립적으로 치환되거나;
R3은 피리도옥사지닐, 디하이드로-피리도옥사지닐, 디하이드로-피롤로피리디닐, 피롤로피리디닐 또는 피롤로피라지닐이고;
R4는 수소이고;
R5는 메틸, 에틸, 프로필, 이소프로필, 부틸, 펜틸, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 페닐, 아제티디닐, 피페리디닐, 피페라지닐, 모르폴리닐, 테트라하이드로피라닐, 티아졸릴, 옥사졸릴, 이속사졸릴, 피라졸릴, 이미다졸릴, 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 퀴놀리닐, 이소퀴놀리닐, -C(O)-피페라지닐, -C(O)-모르폴리닐, -C(O)-NH-피페리디닐, 하이드록시 또는 -NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8은 각각 독립적으로 수소, 메틸 그룹에 의해 임의로 치환된 피페리디닐, 하이드록시 그룹에 의해 임의로 치환된 사이클로헥실, 메틸 또는 에틸이고;
R9, R10 및 R11은 독립적으로
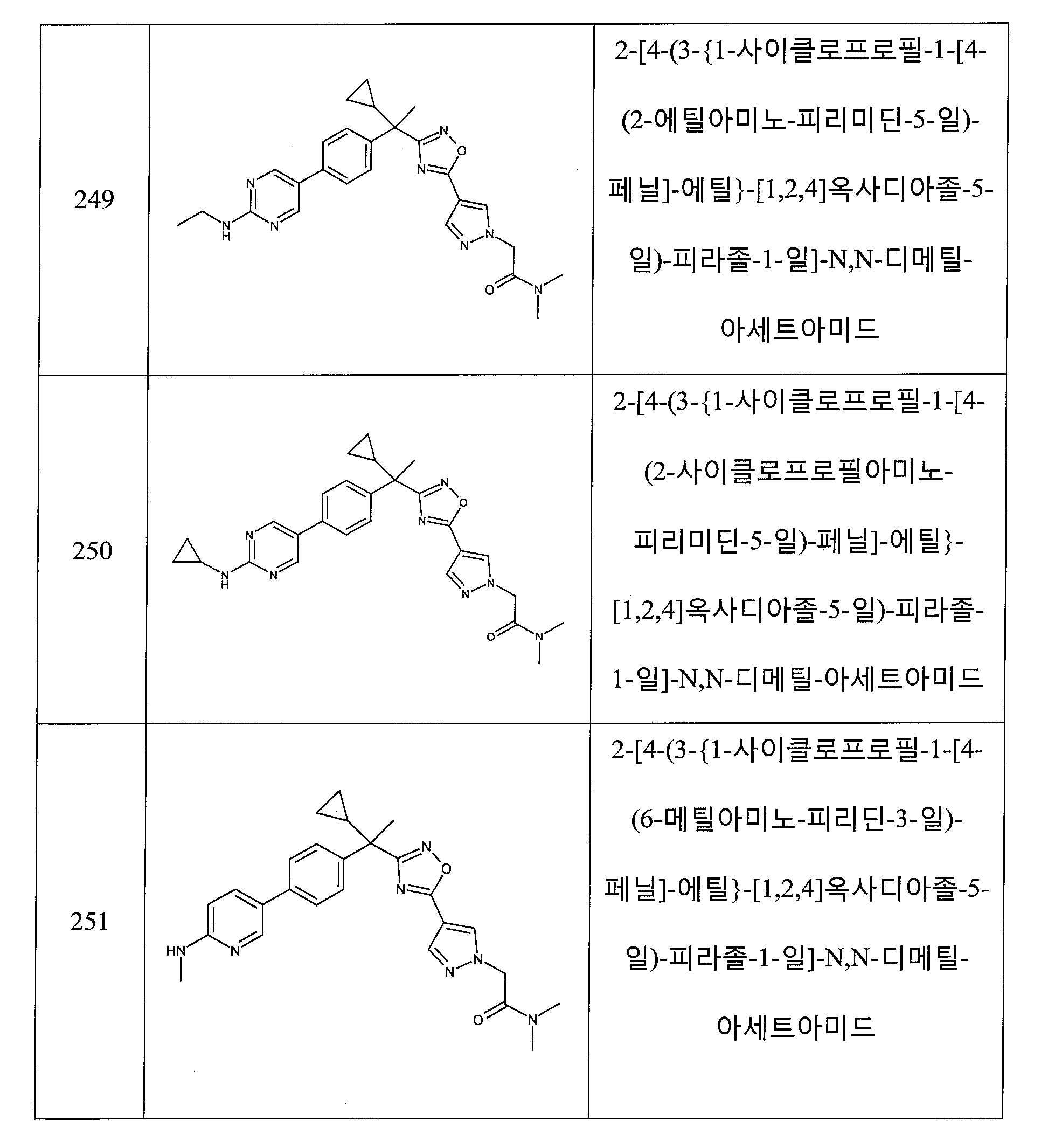
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 모르폴리닐, 피페라지닐, C1 -3 알콕시, C1-3알콕시-O-C1 - 3알킬, -CO2H 또는 -C(O)N(R12)(R13)에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 3알콕시,
(h) -N(R12)(R13),
(i) -S(O)2C1 -2알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 모르폴리닐, 피페라지닐 또는 옥세타닐(이들 각각은 메틸 그룹에 의해 임의로 치환됨),
(n') 옥소,
(o) -C(O)-CH3으로부터 선택되고;
R12 및 R13은 각각 독립적으로 -H 및 -C1 - 6알킬로부터 선택되고, 여기서, 알킬 그룹은 1 내지 3개의 -OH, C1 - 6알콕시, -C(O)N(R14)(R15) 또는 -S(O)2C1 - 6알킬에 의해 임의로 독립적으로 치환되고;
R14 및 R15는 각각 독립적으로 -H 및 -C1 - 4알킬로부터 선택되는, 상기 두 번째 양태로 기술된 화학식 I의 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
일곱 번째 양태에서,
R1은 메틸이고,
R2는 메틸, 에틸, 이소프로필, 3급-부틸, 사이클로프로필 및 사이클로부틸로부터 선택되는, 바로 상기 양태에 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
여덟 번째 양태에서,
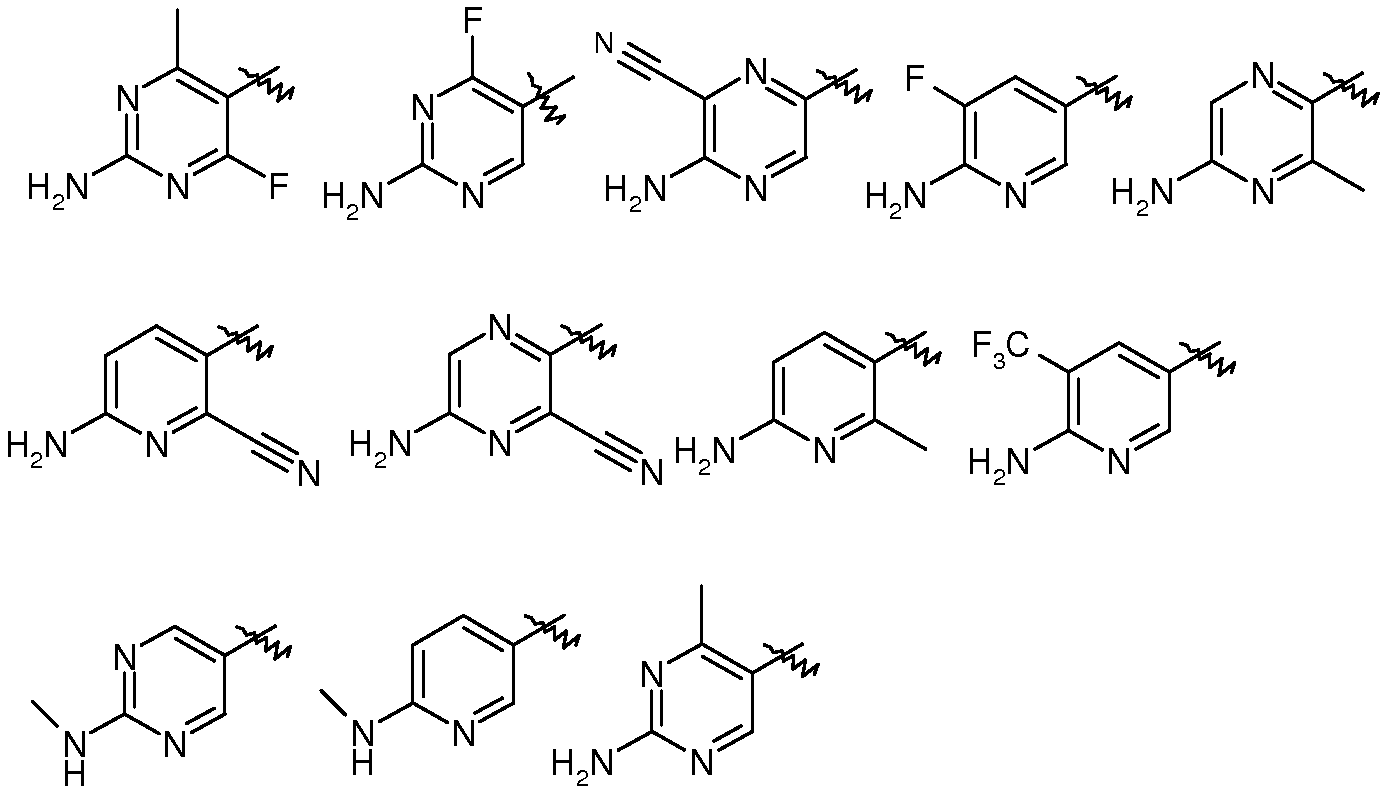
R3은  로부터 선택되는, 상기 여섯 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
로부터 선택되는, 상기 여섯 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
아홉 번째 양태에서,
R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환된 피라졸릴인, 상기 여섯 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
열 번째 양태에서,
R1은 메틸이고,
R2는 메틸, 에틸, 이소프로필, 3급-부틸, 사이클로프로필 및 사이클로부틸로부터 선택되고,
R3은  로부터 선택되고,
로부터 선택되고,
R4는 수소이고,
R5는  로부터 선택되는, 상기 여섯 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
로부터 선택되는, 상기 여섯 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
열한 번째 양태에서,
R2는 사이클로프로필 또는 사이클로부틸인, 상기 열 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
열두 번째 양태에서,
R2는 메틸, 에틸, 이소프로필 및 3급-부틸로부터 선택되는, 상기 열 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
열세 번째 양태에서,
R3은  로부터 선택되는, 상기 여섯 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
로부터 선택되는, 상기 여섯 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
열네 번째 양태에서,
R3은
열다섯 번째 양태에서,
R1은 메틸이고,
R2는 사이클로프로필이고,
R3은  로부터 선택되고,
로부터 선택되고,
R4는 수소이고,
R5는  로부터 선택되는, 상기 열 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
로부터 선택되는, 상기 열 번째 양태에서 기술된 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
열여섯 번째 양태에서,
R1은 메틸이고,
R2는 사이클로프로필이고,
R3은  로부터 선택되고,
로부터 선택되고,
R4는 수소이고,
R5는  로부터 선택되는, 화학식 I의 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
로부터 선택되는, 화학식 I의 화합물 또는 약제학적으로 허용되는 이의 염이 제공된다.
다음은 일반적인 합성 반응식, 실시예 및 당해 분야에 공지된 방법에 의해 제조될 수 있는 본 발명의 대표적인 화합물들이다.


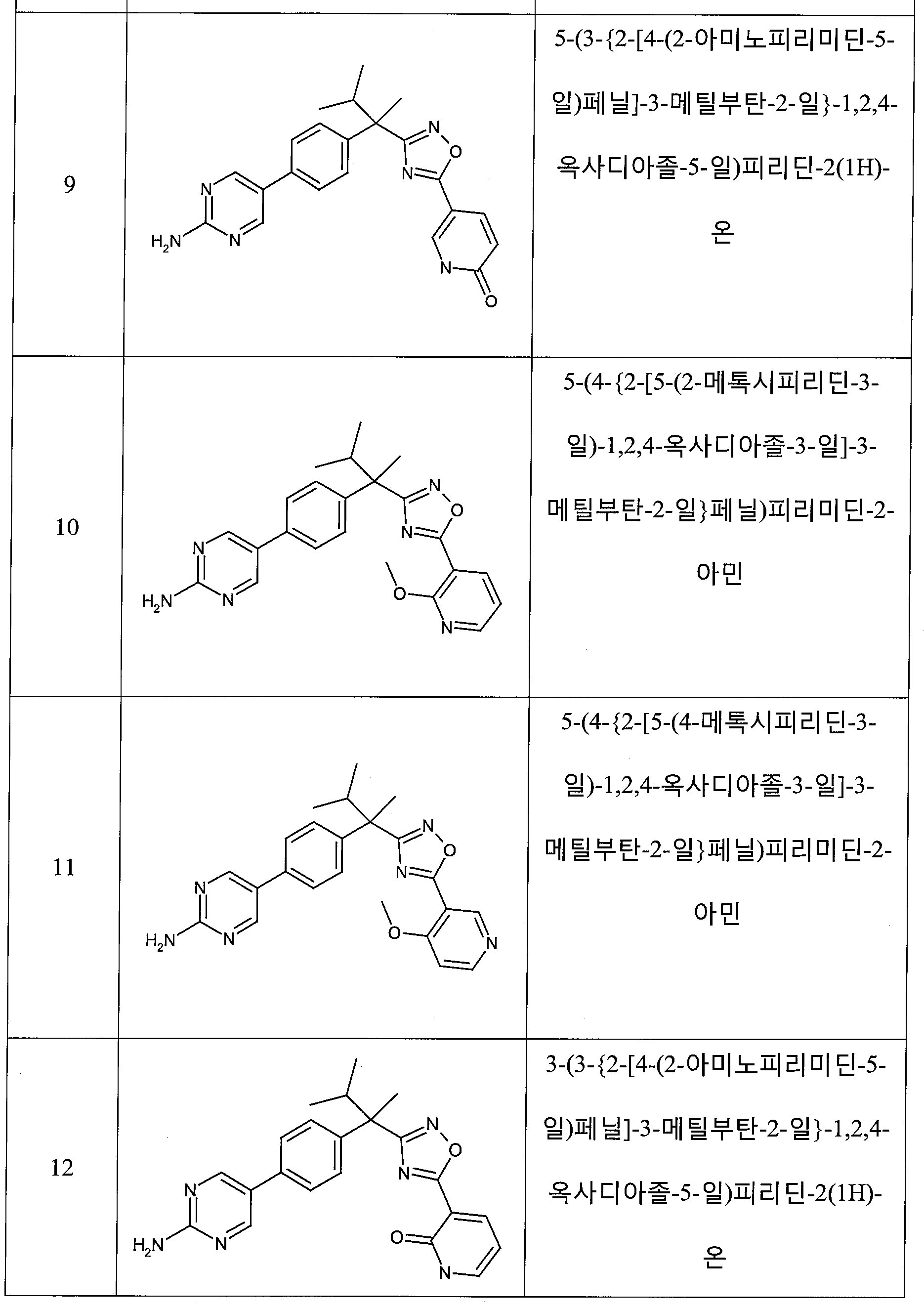



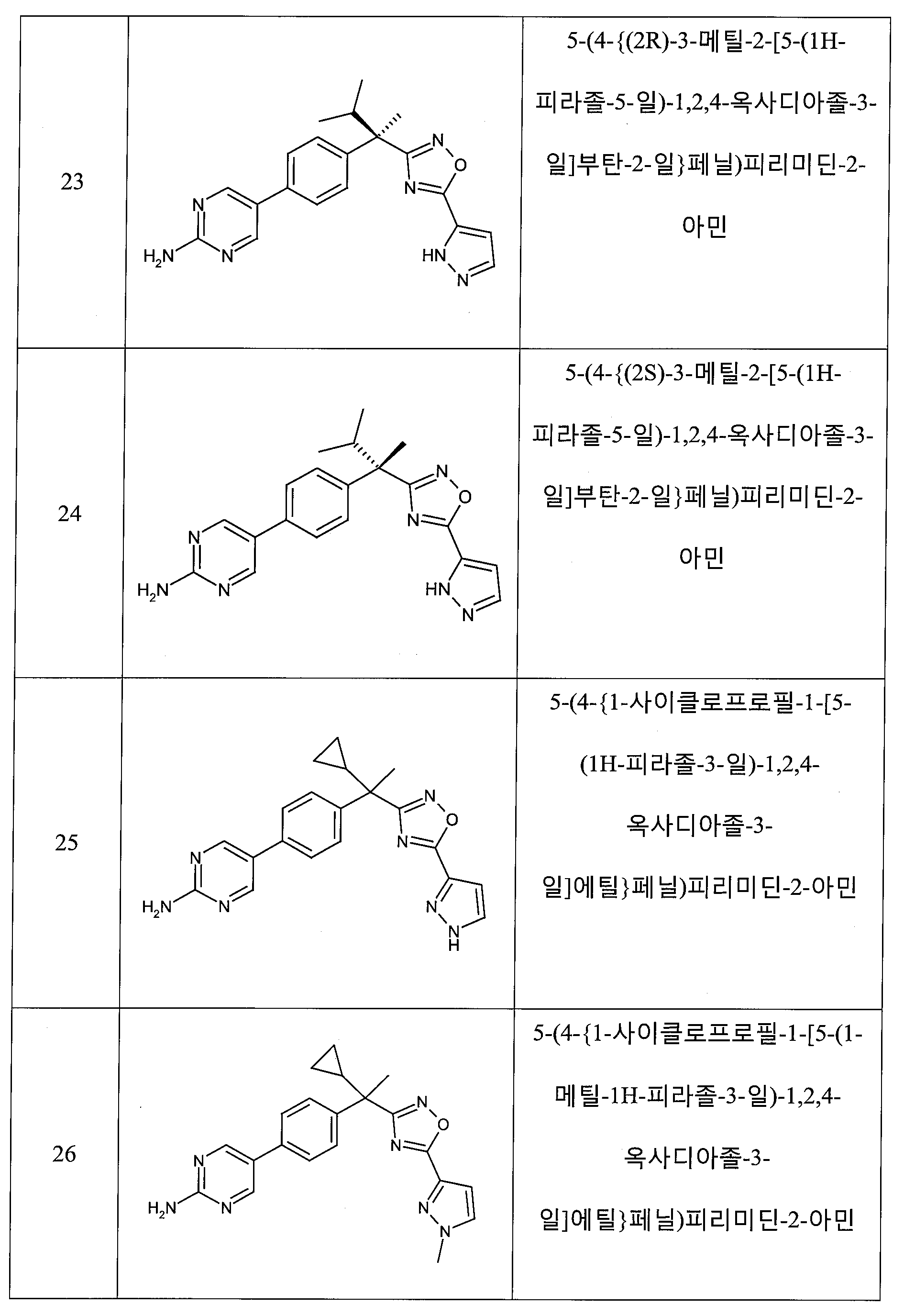

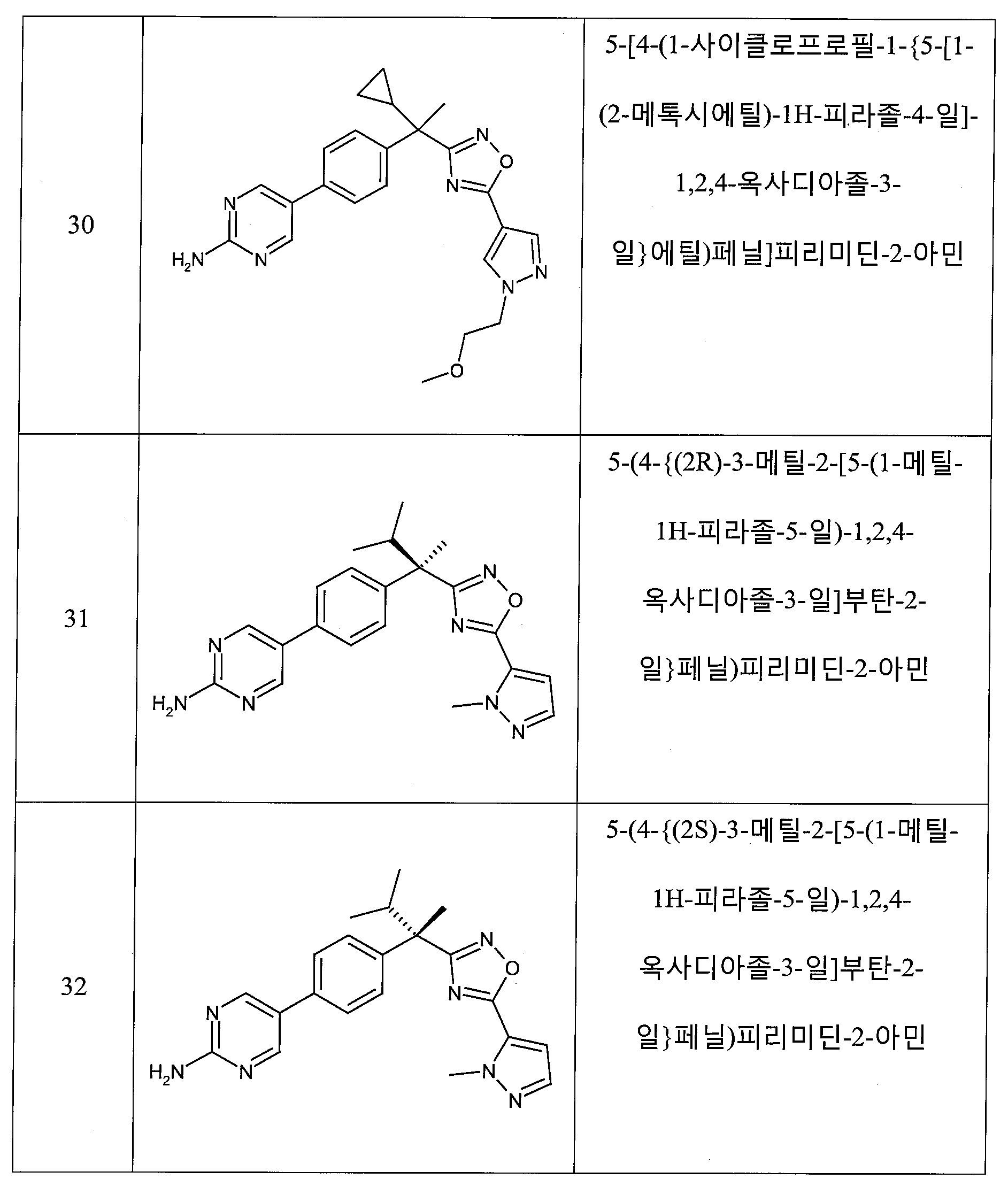






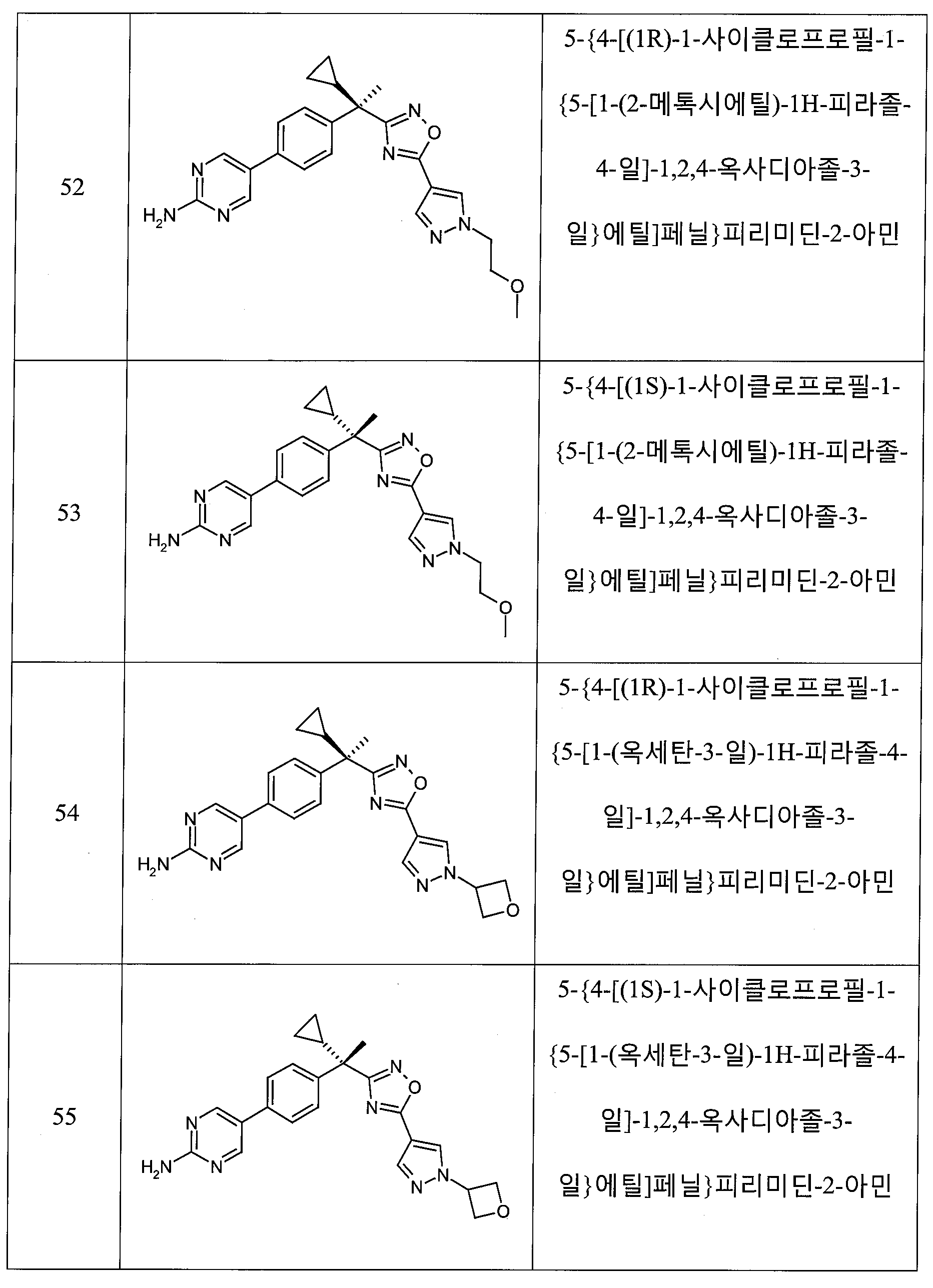

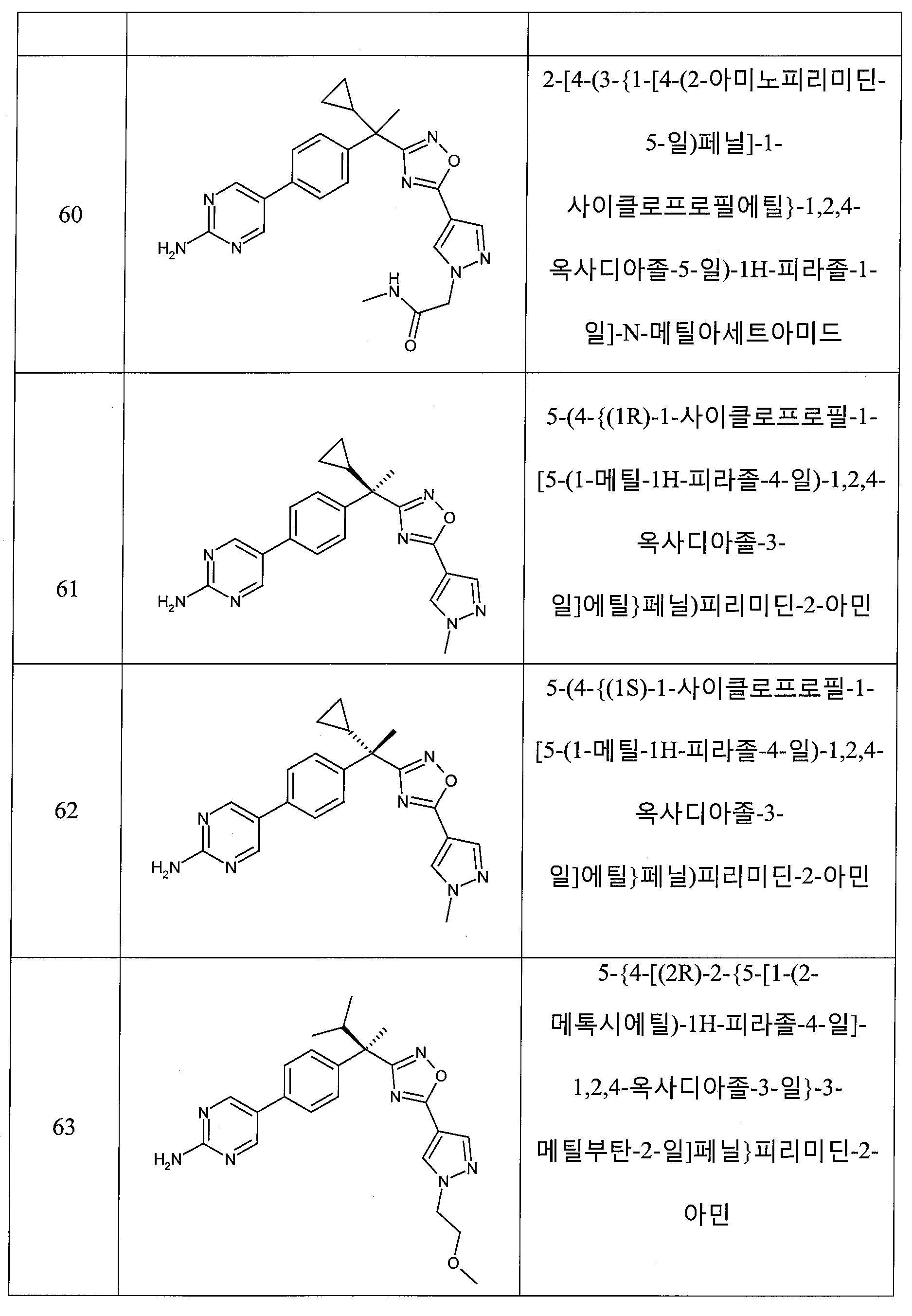











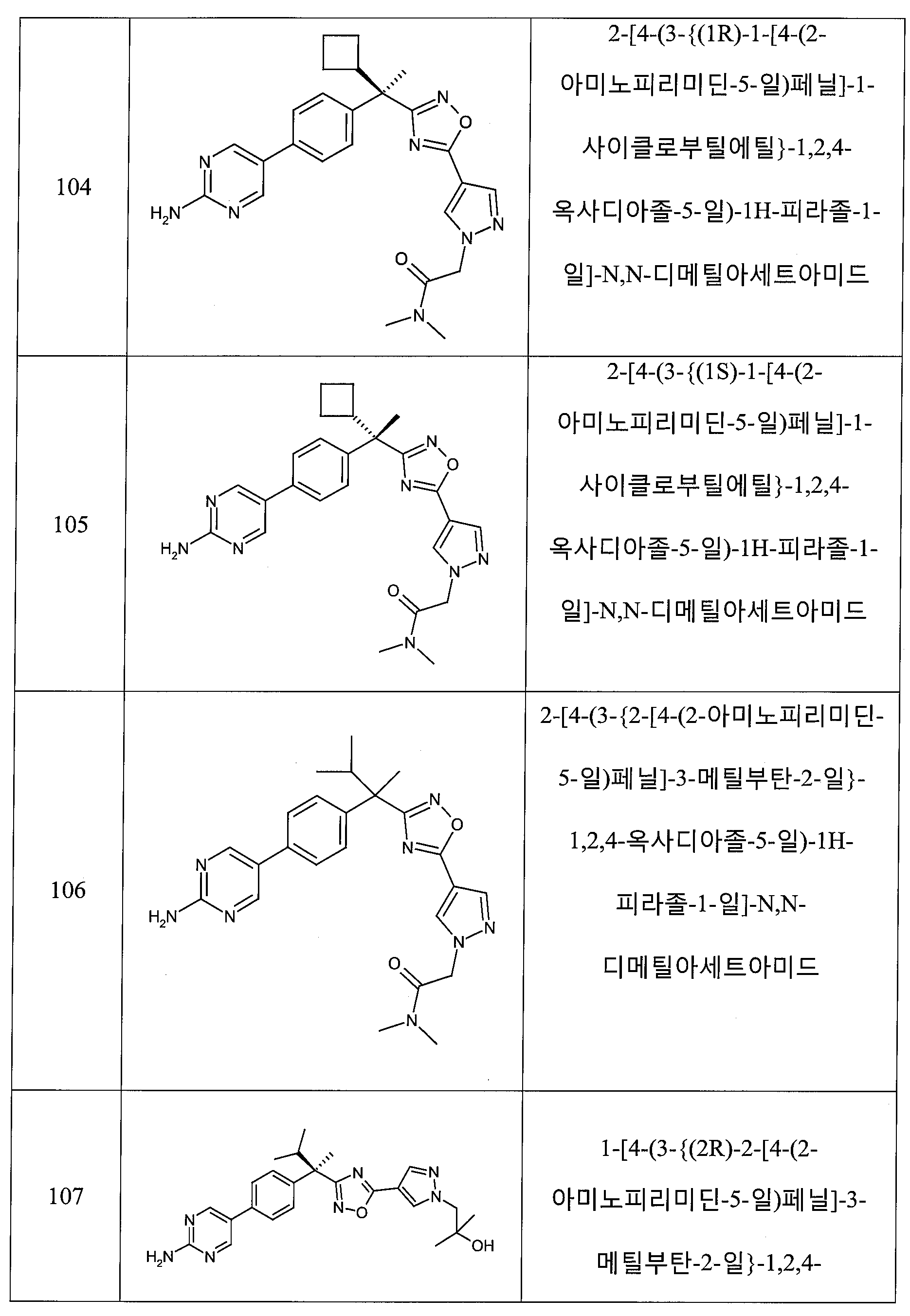



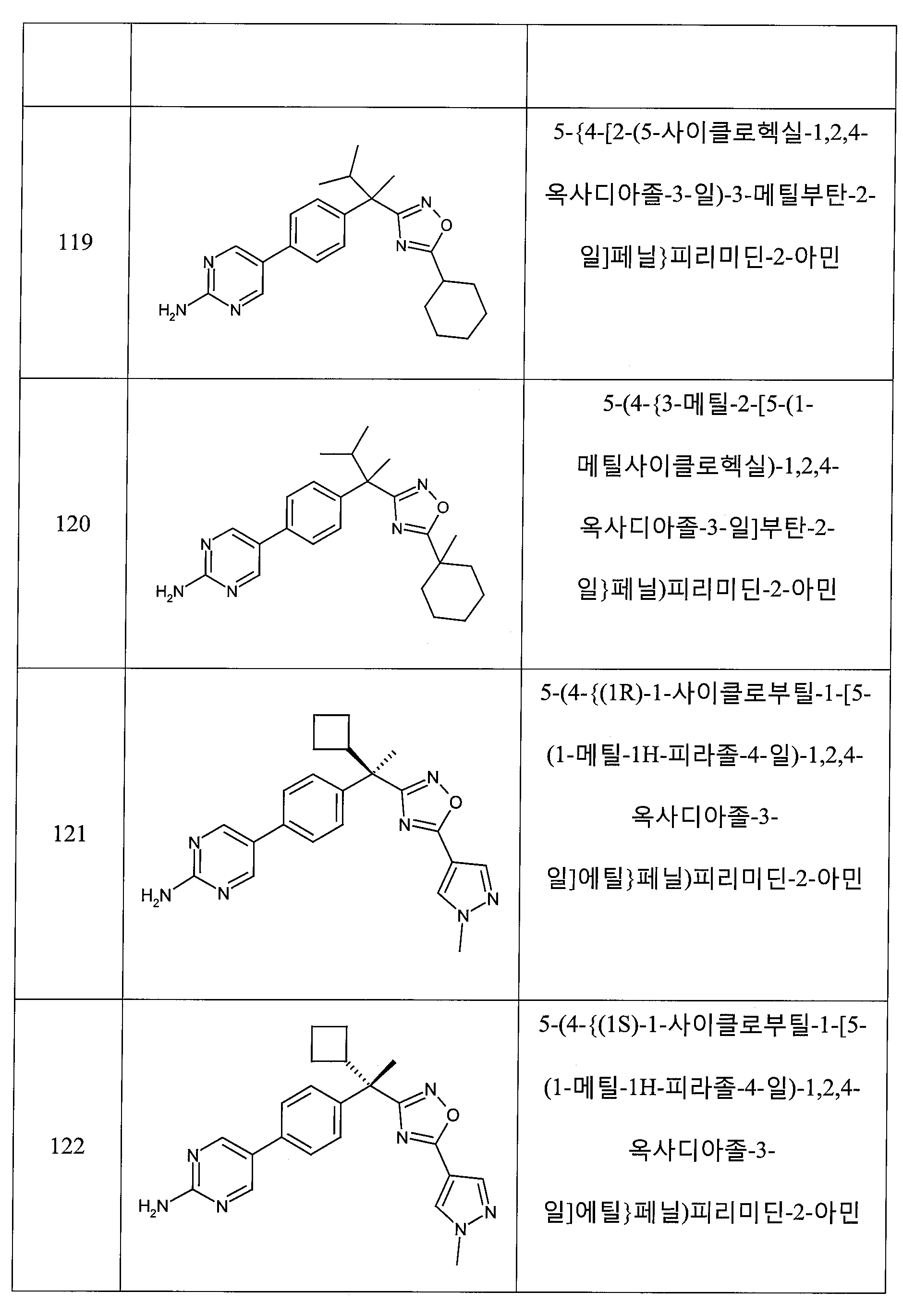











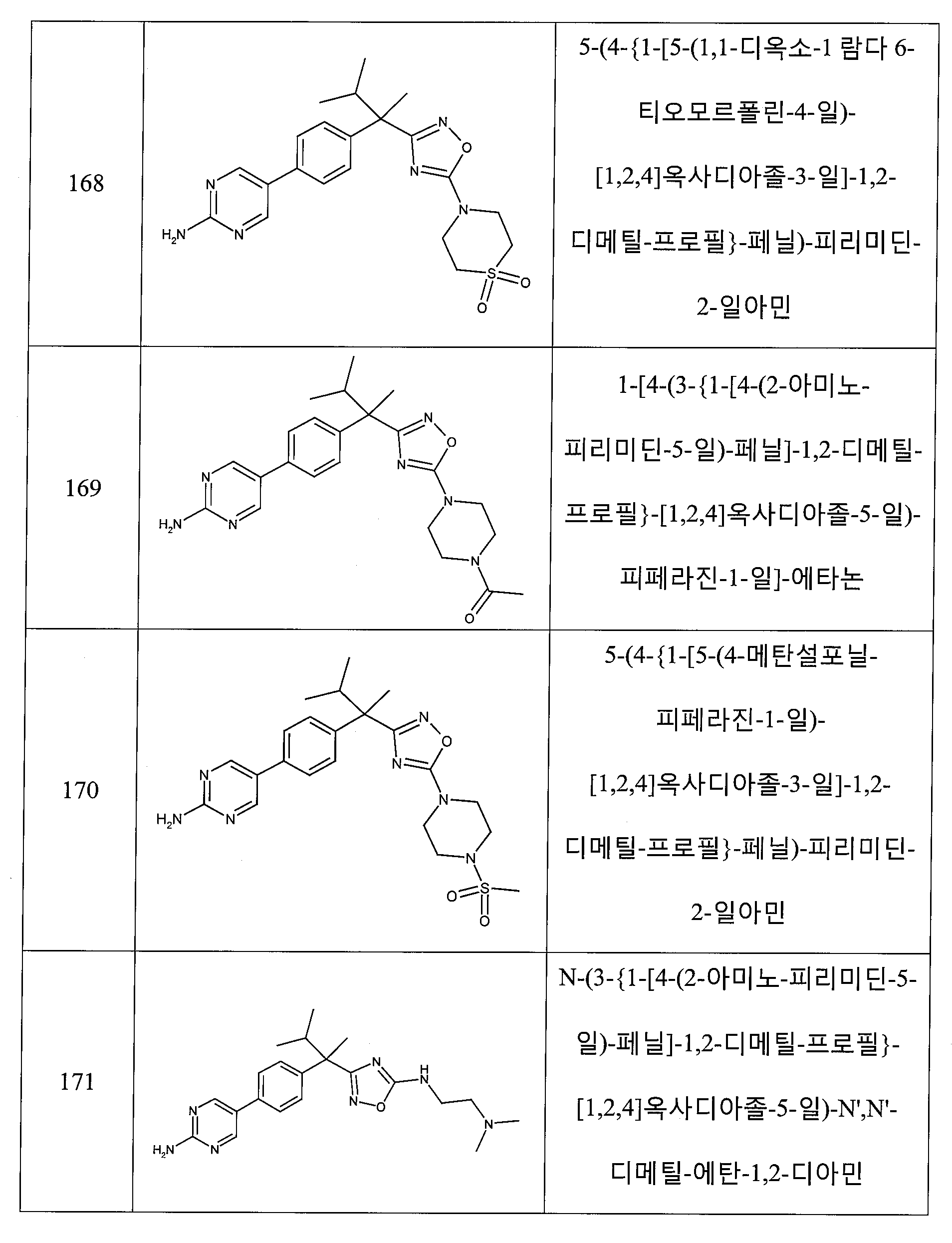

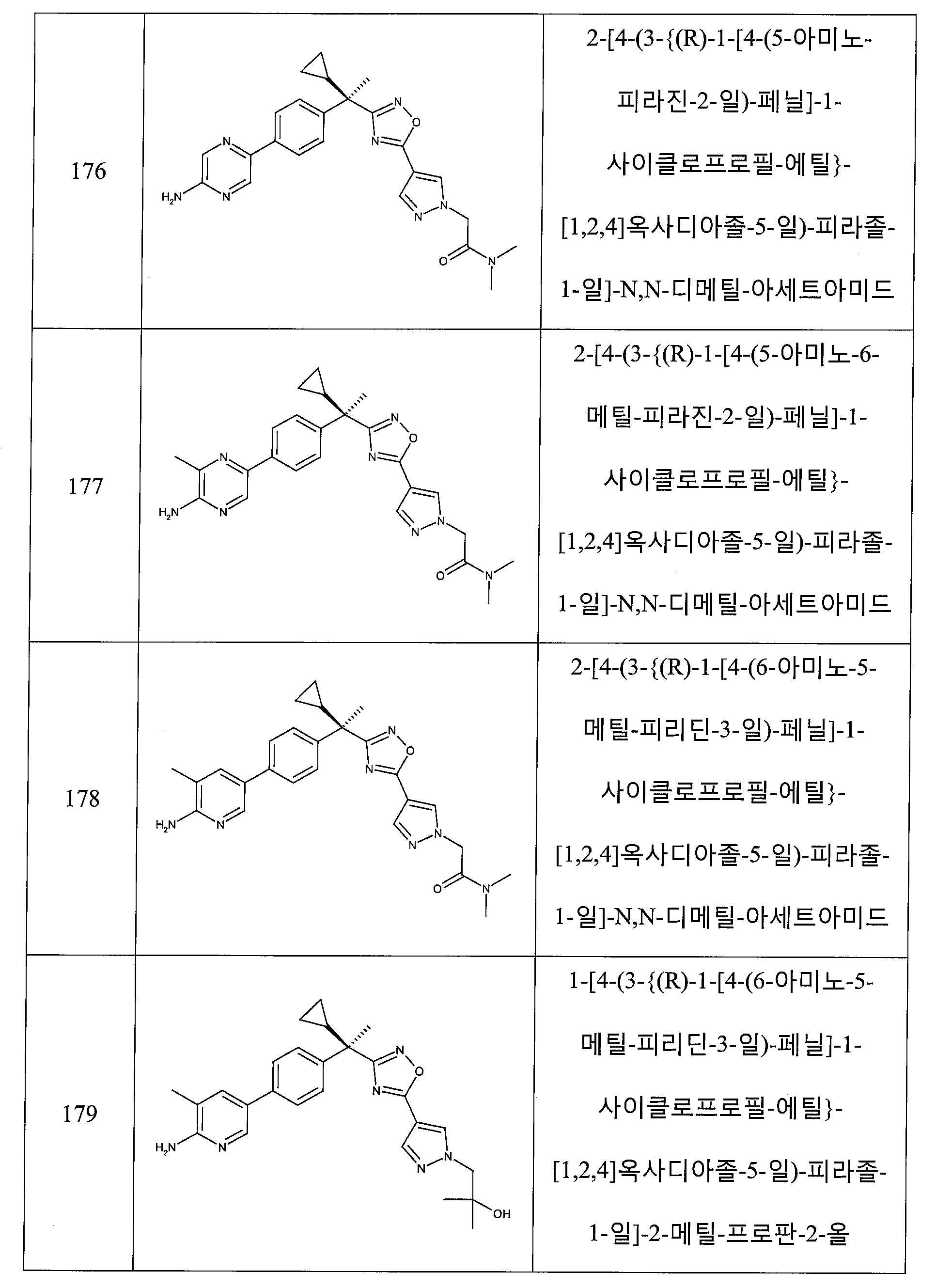
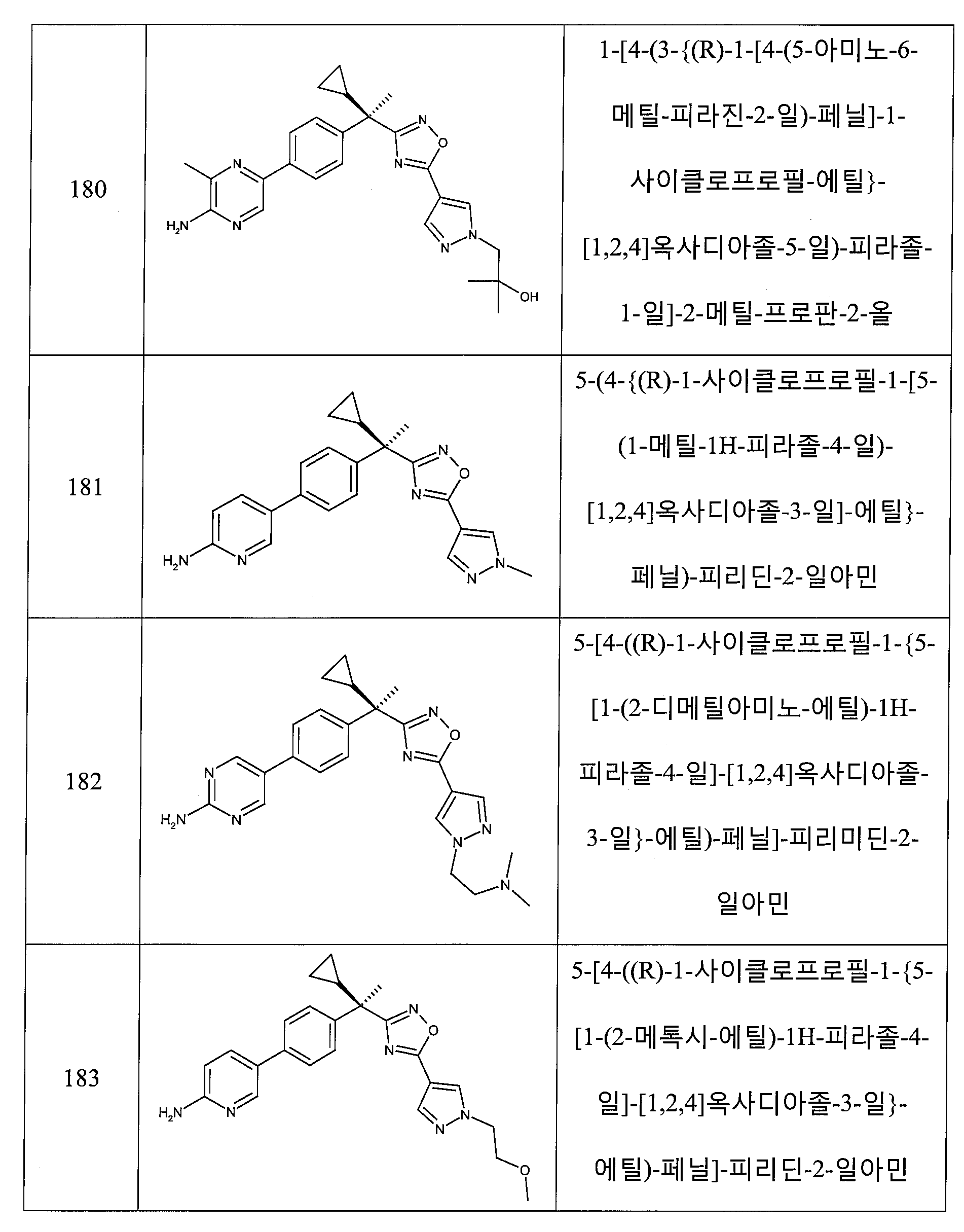






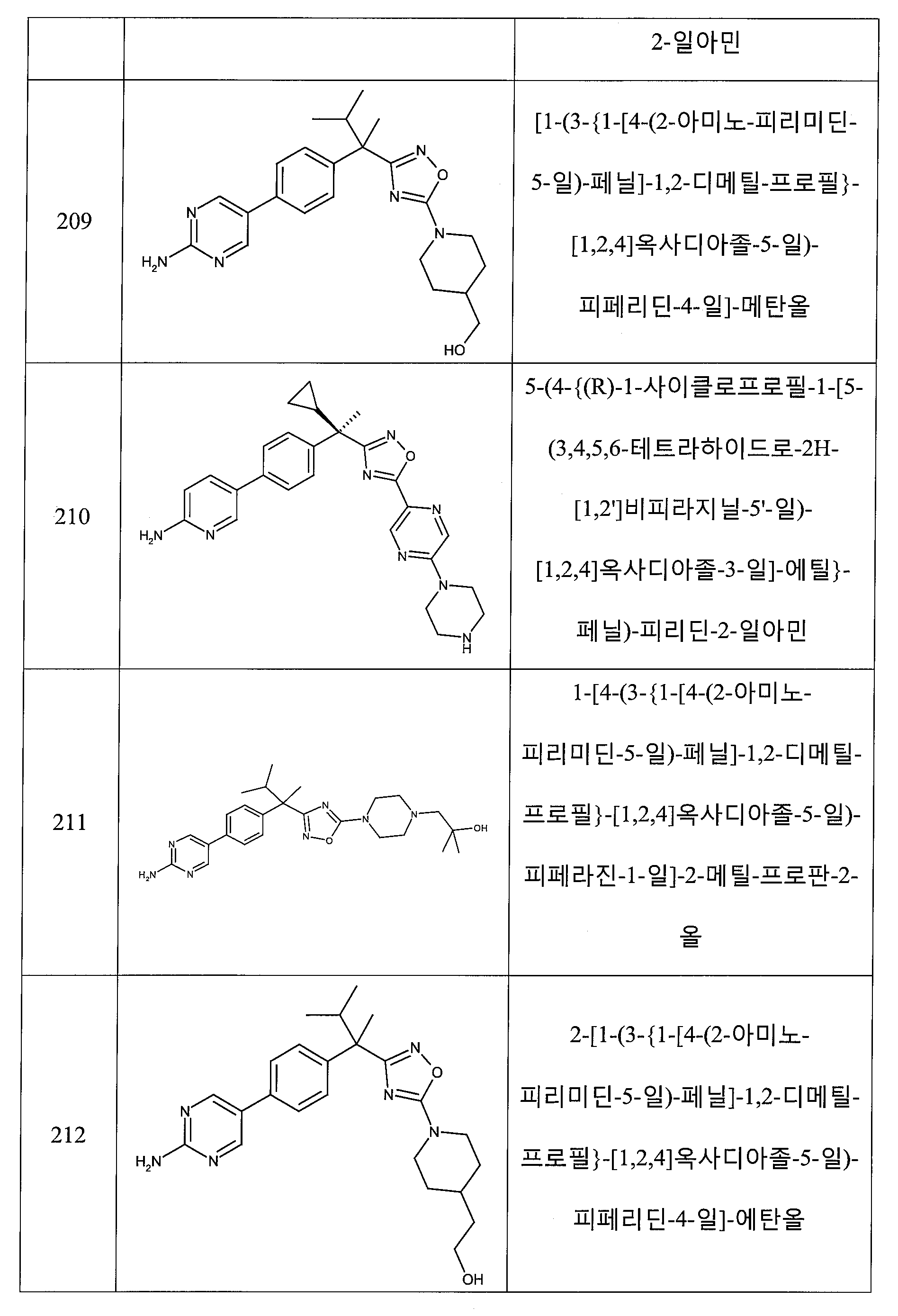











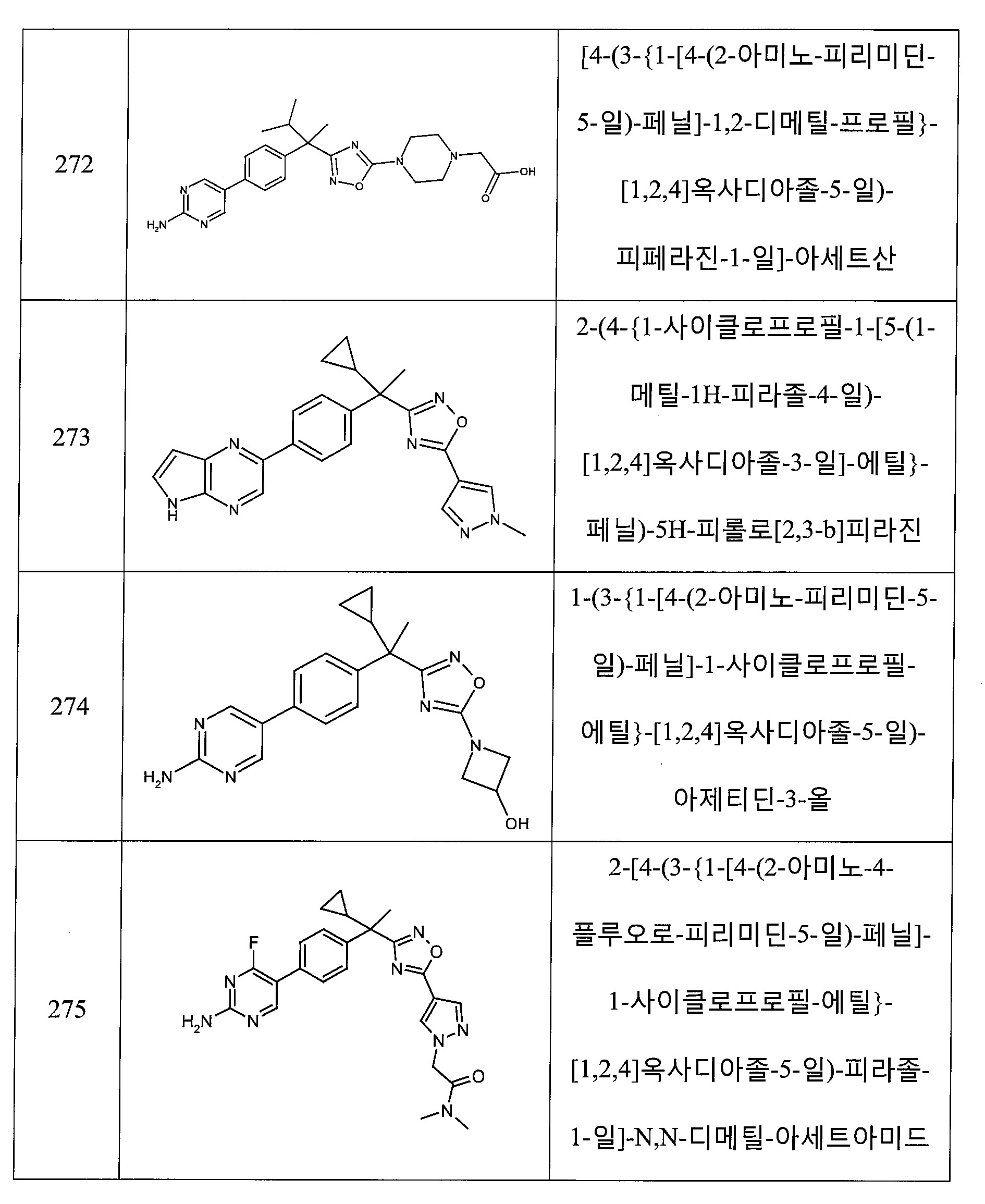






하나의 양태에서, 본 발명은 상기 표 1에 도시된 화합물 중 어느 것 또는 약제학적으로 허용되는 이의 염에 관한 것이다.
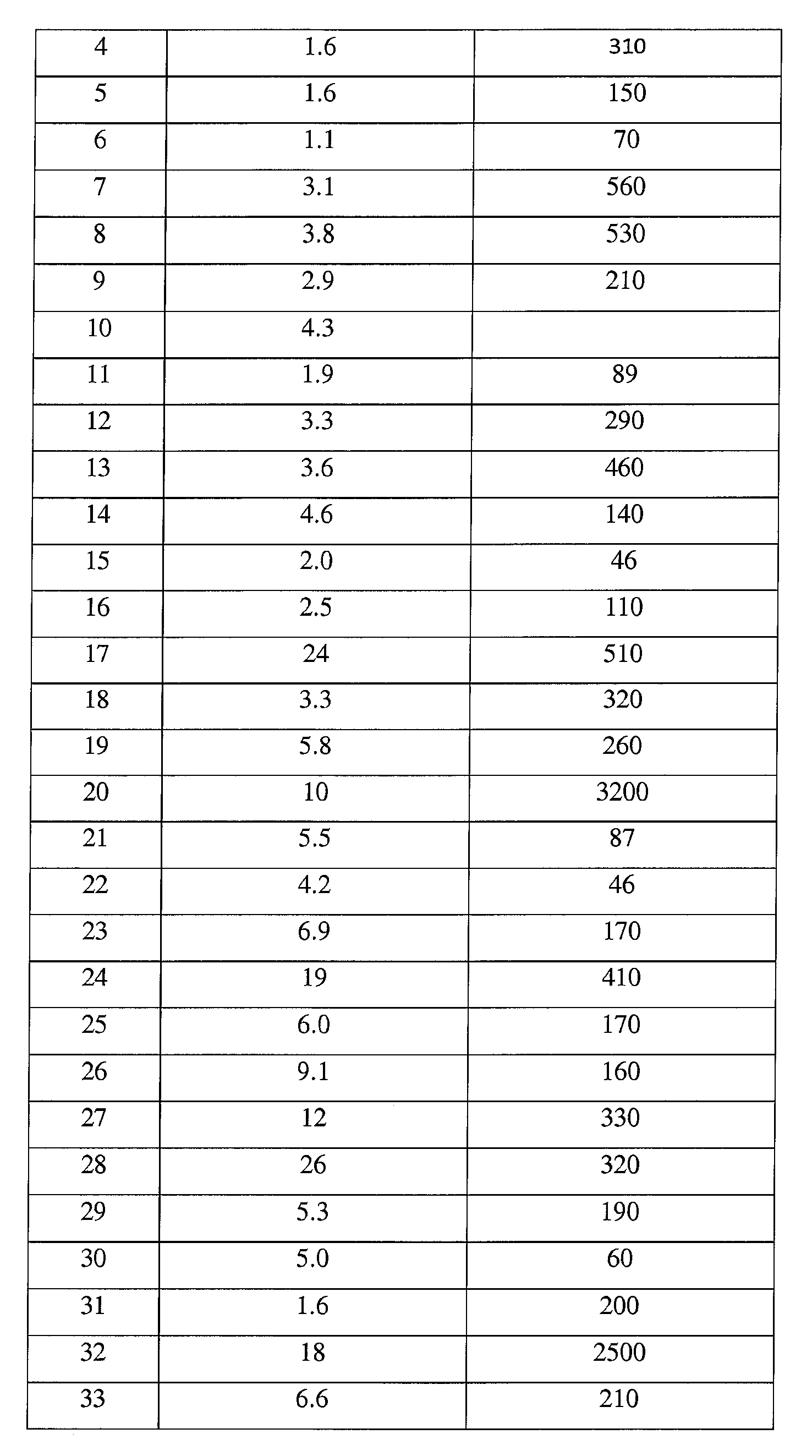
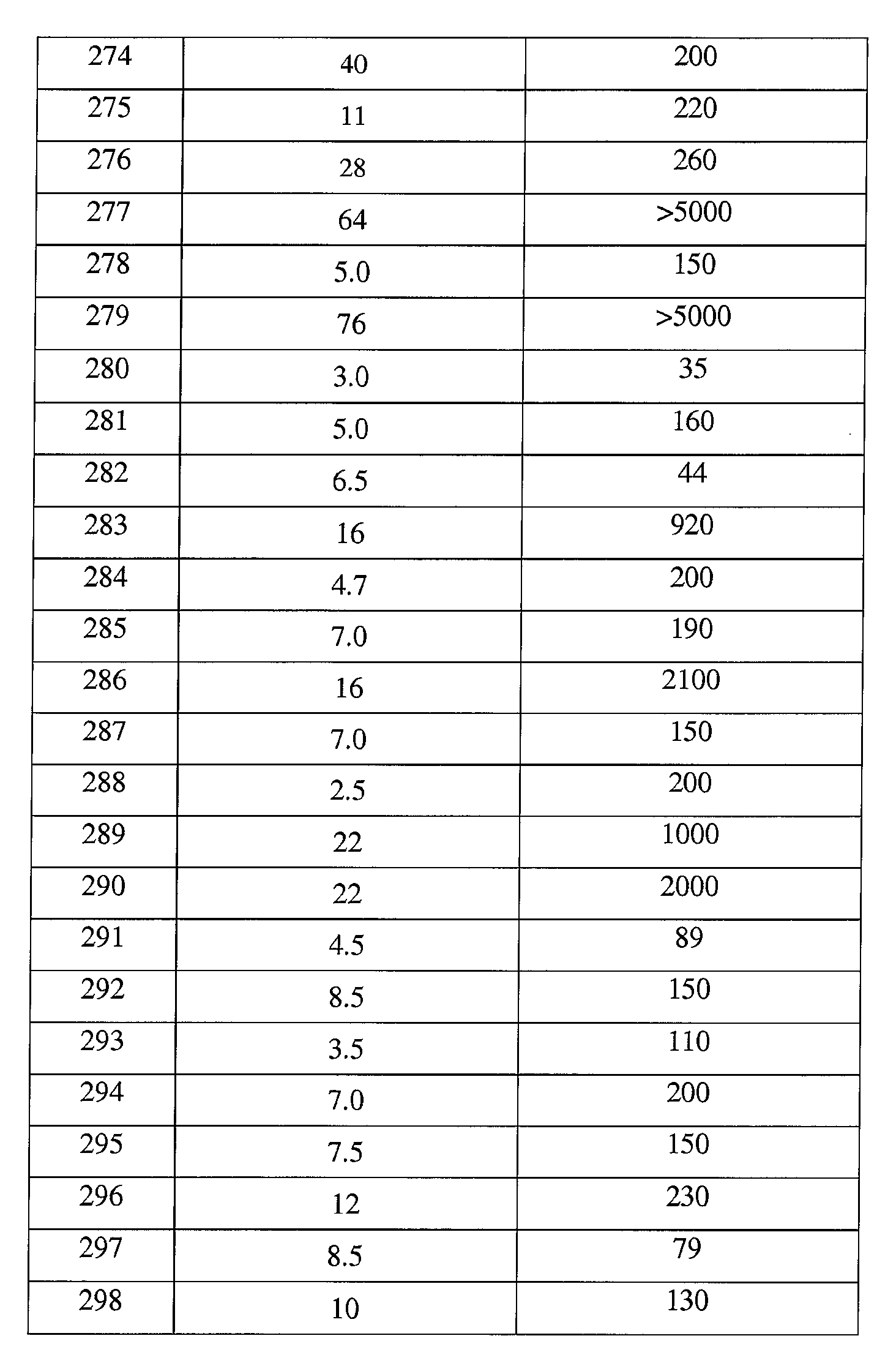
본 발명의 대표적인 화합물은 표 2에 제시된 바와 같이, 생물학적 특성 섹션의 평가에서 기술되는, FLAP 결합 검정에서 활성을 나타낸다.









본 발명은 또한 임의로 통상적인 부형제 및/또는 담체와 함께, 활성 물질로서 하나 이상의 본 발명의 화합물, 또는 약제학적으로 허용되는 이의 유도체를 함유하는 약제학적 제제에 관한 것이다.
본 발명의 화합물은 또한 이들의 동위원소-표지된 형태를 포함한다. 본 발명의 조합물의 활성 제제의 동위원소-표지된 형태는 상기 활성 제제와 동일하지만, 상기 활성 제제 중 하나 이상의 원자가 대개 자연적으로 발견되는 당해 원자의 원자량 또는 원자 번호와 상이한 원자량 또는 원자 번호를 갖는 원자 또는 원자들에 의해 대체되었다는 사실이다. 용이하게 시판되고 있고 잘 확립된 방법에 따라 본 발명의 조합물의 활성 제제로 혼입될 수 있는 동위원소의 예는 수소, 탄소, 질소, 산소, 인, 불소 및 염소의 동위원소, 예를 들면, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F 및 36Cl을 각각 포함한다. 본 발명의 조합물의 활성 제제, 이의 전구약물, 또는 상기 언급한 동위원소 및/또는 다른 원자의 다른 동위원소 중 하나 이상을 함유하는 이것들 중 어느 것의 약제학적으로 허용되는 염이 본 발명의 범위 내에서 시도되었다.
본 발명은 라세미체 및 라세미 혼합물, 단일 에난티오머, 부분입체이성체 혼합물 및 개별 부분입체이성체로서 생성될 수 있는 하나 이상의 비대칭 탄소 원자를 함유하는 상기 기술된 임의의 화합물의 사용을 포함한다. 이성체는 에난티오머 및 부분입체이성체로서 정의될 것이다. 이들 화합물의 상기 모든 이성체 형태는 본 발명에 확실히 포함된다. 각각의 입체 탄소는 R 또는 S 배위나, 배위들의 조합으로 존재할 수 있다.
본 발명의 화합물 중 일부는 하나 이상의 호변이성체 형태로 존재할 수 있다. 본 발명은 상기 모든 호변이성체를 사용하는 방법을 포함한다.
본 명세서에서 사용된 모든 용어는 달리 언급되지 않는 한, 당해 분야에 공지된 이들의 통상적인 의미로 이해될 것이다. 예를 들면, "C1 -6 알콕시"는 말단 산소를 갖는 C1 -6 알킬, 예를 들면, 메톡시, 에톡시, 프로폭시, 부톡시이다. 모든 알킬, 알케닐 및 알키닐 그룹은 달리 명시되지 않는 한 구조적으로 가능한 분지형 또는 비분지형으로서 이해될 것이다. 다른 보다 특정한 정의는 다음과 같다:
용어 "알킬"은 분지형 및 비분지형 알킬 그룹을 모두 의미한다. "알크" 또는 "알킬" 접두어를 사용하는 임의의 조합 용어는 상기 "알킬"의 정의에 따르는 동족체를 의미하는 것으로 이해해야 한다. 예를 들면, "알콕시", "알킬티오"와 같은 용어는 산소 또는 황 원자를 통해 제2 그룹에 결합된 알킬 그룹을 의미한다. "알카노일"은 카보닐 그룹(C=O)에 결합된 알킬 그룹을 의미한다.
모든 알킬 그룹 또는 탄소 쇄에서, 하나 이상의 탄소 원자는 O, S 또는 N와 같은 헤테로 원자에 의해 임의로 대체될 수 있다. N이 치환되지 않는다면, 그것은 NH인 것으로 이해해야 할 것이다. 또한 헤테로 원자가 분지형 또는 비분지형 탄소 쇄에서 말단 탄소 원자 또는 내부 탄소 원자를 대체할 수도 있음을 이해해야 할 것이다. 상기 그룹은 옥소와 같은 그룹에 의해 본 명세서에서 상기 기술된 바와 같이 치환되어 이로써 제한되는 것은 아니지만: 알콕시카보닐, 아실, 아미도 및 티옥소와 같은 정의를 나타낼 수 있다. 본 명세서에 사용된 "질소" 및 "황"은 질소 및 황의 임의의 산화된 형태 및 임의의 염기성 질소의 사급화 형태를 포함한다. 예를 들면, -S-C1 -6 알킬 라디칼의 경우, 달리 명시되지 않는 한, -S(O)-C1 -6 알킬 및 -S(O)2-C1 -6 알킬을 포함하는 것으로 이해해야 할 것이다.
용어 C1 -3 하이드록시는 또한 -C1 - 3알킬-하이드록시 또는 -C1 - 3알킬-OH를 의미한다.
용어 "C3 -10 카보사이클"은 비방향족 3-원 내지 10-원(그러나 바람직하게는 3-원 내지 6-원) 모노사이클릭 카보사이클릭 라디칼 또는 비방향족 6-원 내지 10-원 융합 바이사이클릭, 브릿지 바이사이클릭 또는 스피로사이클릭 카보사이클릭 라디칼을 의미한다. C3 -10 카보사이클은 포화 또는 부분 포화될 수 있고, 카보사이클은 안정한 구조의 생성을 야기하는 사이클의 임의의 원자에 의해 부착될 수 있다. 3-원 내지 10-원 모노사이클릭 카보사이클의 비제한적 예는 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로펜테닐, 사이클로헥실, 사이클로헥세닐, 사이클로헵타닐, 사이클로헵테닐 및 사이클로헥사논을 포함한다. 6-원 내지 10-원 융합 바이사이클릭 카보사이클릭 라디칼의 비제한적 예는 바이사이클로[3.3.0]옥탄, 바이사이클로[4.3.0]노난 및 바이사이클로[4.4.0]데카닐(데카하이드로나프탈레닐)을 포함한다. 6-원 내지 10-원 브릿지 바이사이클릭 카보사이클릭 라디칼의 비제한적 예는 바이사이클로[2.2.2]헵타닐, 바이사이클로[2.2.2]옥타닐 및 바이사이클로[3.2.1]옥타닐을 포함한다. 6-원 내지 10-원 스피로사이클릭 카보사이클릭 라디칼의 비제한적 예는, 이로써 제한되는 것은 아니지만, 스피로[3,3]헵타닐, 스피로[3,4]옥타닐 및 스피로[4,4]헵타닐을 포함한다.
용어 ""C6 -10 아릴" 또는 "아릴"은 6 내지 10개의 탄소 환 원자를 함유하는 방향족 탄화수소 환을 의미한다. 용어 C6 -10 아릴은 모노사이클릭 환 및 바이사이클릭 환을 포함하고, 여기서, 적어도 하나의 환은 방향족이다. C6 -10 아릴의 비제한적 예는 페닐, 인다닐, 인데닐, 벤조사이클로부타닐, 디하이드로나프틸, 테트라하이드로나프틸, 나프틸, 벤조사이클로헵타닐 및 벤조사이클로헵테닐을 포함한다.
용어 "5-원 내지 11-원 헤테로사이클"은 안정한 비방향족 4-원 내지 8-원 모노사이클릭 헤테로사이클릭 라디칼 또는 안정한 비방향족 6-원 내지 11-원 융합 바이사이클릭, 브릿지 바이사이클릭 또는 스피로사이클릭 헤테로사이클릭 라디칼을 의미한다. 5-원 내지 11-원 헤테로사이클은 탄소 원자와, 질소, 산소 및 황으로부터 선택되는 하나 이상, 바람직하게는 1 내지 4개의 헤테로 원자로 이루어진다. 헤테로사이클은 포화 또는 부분 포화될 수 있다. 비방향족 4-원 내지 8-원 모노사이클릭 헤테로사이클릭 라디칼의 비제한적 예는 테트라하이드로푸라닐, 테트라하이드로피라닐, 옥세타닐, 아제티디닐, 피롤리디닐, 피라닐, 테트라하이드로피라닐, 디옥사닐, 티오모르폴리닐, 1,1-디옥소-1λ6-티오모르폴리닐, 모르폴리닐, 피페리디닐, 피페라지닐 및 아제피닐을 포함한다. 비방향족 6-원 내지 11-원 융합 바이사이클릭 라디칼의 비제한적 예는 옥타하이드로인돌릴, 옥타하이드로벤조푸라닐 및 옥타하이드로벤조티오페닐을 포함한다. 비방향족 6-원 내지 11-원 브릿지 바이사이클릭 라디칼의 비제한적 예는 2-아자바이사이클로[2.2.1]헵타닐, 3-아자바이사이클로[3.1.0]헥사닐 및 3-아자바이사이클로[3.2.1]옥타닐을 포함한다. 6-원 내지 11-원 스피로사이클릭 헤테로사이클릭 라디칼의 비제한적 예는 7-아자-스피로[3,3]헵타닐, 7-스피로[3,4]옥타닐 및 7-아자-스피로[3,4]옥타닐을 포함한다.
용어 "5-원 내지 11-원 헤테로아릴"은 방향족 5-원 내지 6-원 모노사이클릭 헤테로아릴 또는 적어도 하나의 환이 방향족인 방향족 7-원 내지 11-원 헤테로아릴 바이사이클릭 환을 의미하고, 여기서, 헤테로아릴 환은 1 내지 4개의 헤테로 원자(예: N, O 및 S)를 함유한다. 5-원 내지 6-원 모노사이클릭 헤테로아릴 환의 비제한적 예는 푸라닐, 옥사졸릴, 이속사졸릴, 옥사디아졸릴, 티아졸릴, 피라졸릴, 피롤릴, 이미다졸릴, 테트라졸릴, 트리아졸릴, 티에닐, 티아디아졸릴, 피리디닐, 피리미디닐, 피리다지닐, 피라지닐, 트리아지닐 및 푸리닐을 포함한다. 7-원 내지 11-원 헤테로아릴 바이사이클릭 헤테로아릴 환의 비제한적 예는 벤즈이미다졸릴, 퀴놀리닐, 디하이드로-2H-퀴놀리닐, 이소퀴놀리닐, 퀴나졸리닐, 인다졸릴, 티에노[2,3-d]피리미디닐, 인돌릴, 이소인돌릴, 벤조푸라닐, 벤조피라닐, 벤조디옥솔릴, 벤조옥사졸릴, 피리도옥사지닐, 디하이드로-피리도옥사지닐, 디하이드로-피롤로피리디닐, 피롤로피리디닐, 피롤로피라지닐 및 벤조티아졸릴을 포함한다.
C3 -10 카보사이클릭 환, 5-원 내지 11-원 헤테로사이클릭 환, 바이사이클릭 아릴 환의 비방향족 부분 및 바이사이클릭 헤테로아릴 환의 비방향족 부분의 각각에서 1 내지 3개의 탄소 환 잔기는 카보닐, 티오카보닐 또는 이미닐 잔기, 즉 -C(=O)-, -C(=S)- 및 -C(=NR8)-(여기서, R8은 상기 정의한 바와 같다)에 의해 각각 독립적으로 치환될 수 있음을 이해할 것이다. 본 명세서에 사용된 용어 "헤테로 원자"는 O, N 및 S와 같은, 탄소가 아닌 다른 원자를 의미하는 것으로 이해해야 할 것이다.
본 명세서에 사용된 용어 "할로겐"은 브롬, 염소, 불소 또는 요오드를 의미하는 것으로 이해해야 할 것이다. 정의 "할로겐화(halogenated)"; "부분 또는 완전 할로겐화"; 부분 또는 완전 플루오르화; "하나 이상의 할로겐 원자에 의해 치환됨"은, 예를 들면, 하나 이상의 탄소 원자 상에 모노, 디 또는 트리 할로 유도체를 포함한다. 알킬의 경우, 비제한적 예는 -CH2CHF2, -CF3 등이다.
본 명세서에 기술된 각각의 알킬, 카보사이클, 헤테로사이클 또는 헤테로아릴이나, 이들의 동족체는 임의로 부분적으로 또는 완전히 할로겐화되는 것으로 이해해야 할 것이다.
본 발명의 화합물은 단지 당해 분야의 숙련가에 의해 이해되는 바와 같이 "화학적으로 안정"한 것으로 고려되는 것들이다. 예를 들면, '단글링 원자가(dangling valency)' 또는 '카르보 음이온(carbanion)'을 갖는 화합물은 본 명세서에 기술된 본 발명의 방법에 의해 고려되는 화합물이 아니다.
본 발명은 화학식 I의 화합물의 약제학적으로 허용되는 유도체를 포함한다. "약제학적으로 허용되는 유도체"는 약제학적으로 허용되는 염이나 에스테르, 또는 환자에 투여시, 본 발명에 유용한 화합물을 (직접 또는 간접적으로) 제공할 수 있는 임의의 다른 화합물, 또는 이의 약물학적으로 활성인 대사물질이나 약물학적으로 활성인 잔사를 의미한다. 약물학적으로 활성인 대사물질은 효소적으로 또는 화학적으로 대사될 수 있는 본 발명의 임의의 화합물을 의미하는 것으로 이해해야 할 것이다. 이는, 예를 들면, 본 발명의 하이드록실화 또는 산화 유도체 화합물을 포함한다.
약제학적으로 허용되는 염은 약제학적으로 허용되는 무기 및 유기산과 염기로부터 유도되는 것들을 포함한다. 적절한 산의 예는 염산, 브롬화수소산, 황산, 질산, 과염소산, 푸마르산, 말레산, 인산, 글리콜산, 락트산, 살리실산, 석신산, 톨루엔-p-황산, 타르타르산, 아세트산, 시트르산, 메탄설폰산, 포름산, 벤조산, 말론산, 나프탈렌-2-황산 및 벤젠설폰산을 포함한다. 다른 산(예: 옥살산)은 이들 자체가 약제학적으로 허용되지 않지만, 화합물 및 이들의 약제학적으로 허용되는 산 부가염을 수득하는데 중간체로서 유용한 염의 제조시 사용될 수 있다. 적절한 염기로부터 유도된 염은 알칼리 금속(예: 나트륨), 알칼리 토금속(예: 마그네슘), 암모늄 및 N-(C1-C4 알킬)4 + 염을 포함한다.
또한, 본 발명의 화합물의 전구약물의 사용이 본 발명의 범위 내에 속한다. 전구약물은 간단한 화학적 변형시, 본 발명의 화합물을 제조하기 위해 개질되는 화합물을 포함한다. 간단한 화학적 변형은 가수분해, 산화 및 환원을 포함한다. 특히, 전구약물을 환자에 투여하는 경우에, 전구약물은 상기 기술된 화합물로 변형됨으로써, 목적하는 약물학적 효과를 부여할 수 있다.
화학식 I의 화합물은 하기 기술되는 일반적인 합성법을 사용하여 제조할 수 있고, 이는 또한 본 발명의 일부를 구성한다.
일반적인 합성법
본 발명은 또한 화학식 I의 화합물의 제조 방법을 제공한다. 모든 반응식에서, 달리 명시되지 않는 한, 하기 화학식에서 R1, R2, R3, R4 및 R5는 상기 본 명세서에 기술된 본 발명의 화학식 I의 R1, R2, R3, R4 및 R5의 의미를 가질 것이다.
최적의 반응 조건 및 반응 시간은 사용된 특별한 반응물에 따라 변할 수 있다. 달리 명시되지 않는 한, 용매, 온도, 압력 및 다른 반응 조건은 당해 분야의 통상의 숙련가에 의해 용이하게 선택될 수 있다. 특정 방법이 합성 실시예 섹션에 제공된다. 통상적으로, 반응 진행은, 경우에 따라, 박층 크로마토그래피(TLC) 또는 LC-MS로 모니터할 수 있고, 중간체 및 생성물은 실리카 겔 상에서 크로마토그래피, 재결정화 및/또는 분취용 HPLC에 의해 정제할 수 있다.
하기의 실시예는 예시이고, 당해 분야의 숙련가에 의해 인지되는 바와 같이, 특별한 시약 또는 조건은, 과도한 실험 없이, 개개 화합물에 대해 필요한 경우 변형될 수 있다. 하기 반응식에서 사용된 출발 물질 및 중간체는 시판중이거나, 당해 분야의 숙련가에 의해 시판중인 물질로부터 용이하게 제조된다.
화학식 I의 화합물은 하기 반응식 1에 따라 합성할 수 있다:
반응식 1

반응식 1에 제시된 바와 같이, 적절한 촉매의 존재하에, 적절한 용매 속에서, 상기 반응식에 제시된 화학식 II의 화합물과 보론산 또는 상응하는 보론산 에스테르의 반응은 화학식 I의 화합물을 제공한다. Ra 및 Rb는 수소이거나, Ra 및 Rb는 이들이 결합된 산소 원자와 함께 2 내지 4개의 메틸 그룹에 의해 임의로 치환된 5원 내지 6원 환을 형성한다.
대안적으로, 표준 반응 조건하에, 화학식 II의 화합물과 디보란의 반응은 화학식 III의 화합물을 제공한다. 적절한 촉매의 존재하에, 적절한 용매 속에서, 화학식 III의 중간체와 할라이드 또는 트리플레이트 R3X의 커플링은 화학식 I의 화합물을 제공한다. X는 클로로, 브로모, 트리플레이트 또는 요오도이다.
화학식 I의 화합물은 반응식 2에 따라 제조할 수 있다:
반응식 2

반응식 2에 제시된 바와 같이, 적절한 염기의 존재하에, 적절한 용매 속에서, 화학식 IV의 화합물과 산 클로라이드 R5COCl의 반응은 화학식 I의 화합물을 제공한다.
대안적으로, 카보닐 디이미다졸 또는 다른 적절한 아미드 커플링제의 존재하에, 적절한 용매 속에서 화학식 IV의 화합물과 산 R5COOH의 반응은 화학식 I의 화합물을 제공한다.
화학식 II의 중간체는 반응식 3에 제시된 바와 같이 합성할 수 있다:
반응식 3

반응식 3에 제시된 바와 같이, 적절한 염기(예: 수소화나트륨 또는 칼륨 3급-부톡사이드)의 존재하에, 적절한 용매 속에서, 화학식 V의 니트릴과 할라이드 R1X의 반응은 화학식 VI의 치환된 니트릴을 제공한다. 적절한 염기의 존재하에, 적절한 용매 속에서, 화학식 VI의 중간체와 할라이드 R2X의 추가 반응은 화학식 VII의 상응하는 이치환된 니트릴을 제공한다. X는 클로로, 브로모 또는 요오도이다. 표준 반응 조건하에, 화학식 VII의 화합물과 하이드록실아민의 반응은 화학식 VIII의 화합물을 제공한다. 적절한 염기의 존재하에, 적절한 용매 속에서, 화학식 VIII의 화합물과 산 클로라이드 R5COCl의 반응은 화학식 II의 화합물을 제공한다. 대안적으로, 카보닐 디이미다졸 또는 다른 적절한 아미드 커플링제의 존재하에, 적절한 용매 속에서 화학식 VIII의 화합물과 산 R5COOH의 반응은 화학식 II의 화합물을 제공한다.
대안적으로, 화학식 VIII의 화합물과 카보닐디이미다졸과 같은 시약과의 반응은 화학식 II의 화합물(여기서,, R5는 -OH임)을 제공한다. 이 -OH의 추가 변형은 당해 분야에 공지된 방법에 의해 수행하여 화학식 II의 부가 화합물을 제공할 수 있다.
화학식 VII의 니트릴 중간체는 또한 당해 분야의 숙련가에게 공지된 분할 기술을 통해 분할시켜 에난티오머 VIIA 및 VIIA'를 제공할 수 있다. 이들 에난티오머는 각각 반응식 3에서 상기 제시된 반응 순서에 의해 화학식 I의 화합물로 추가로 전환시킬 수 있다.
화학식 II의 중간체는 또한 반응식 4에 제시된 바와 같이 합성할 수 있다:
반응식 4

반응식 4에 제시된 바와 같이, 적절한 용매 속에서 화학식 IX의 카보닐 화합물과 그리나드 시약 R2MgX의 반응은 화학식 X의 하이드록시 화합물을 제공한다. 표준 방법을 사용하여 화학식 X의 화합물에서 하이드록실 그룹의 시아노 그룹으로의 전환은 화학식 VII의 화합물을 제공한다. 화학식 VII의 화합물은 반응식 3에 제시된 반응에 의해 화학식 II의 중간체로 전환시킨다. R2MgX에서 X는 클로로, 브로모 또는 요오도이다.
화학식 IV의 중간체는 반응식 5에 따라 합성할 수 있다:
반응식 5

반응식 5에 제시된 바와 같이, 적절한 촉매의 존재하에, 적절한 용매 속에서, 상기 반응식에 제시된 화학식 VII의 니트릴과 보론산 또는 상응하는 보론산 에스테르의 반응은 화학식 XI의 화합물을 제공한다. Ra 및 Rb는 수소이거나, Ra 및 Rb는 이들이 결합된 산소 원자와 함께 2 내지 4개의 메틸 그룹에 의해 임의로 치환된 5원 내지 6원 환을 형성한다. 표준 반응 조건하에 화학식 XI의 화합물과 하이드록실아민의 반응은 화학식 IV의 화합물을 제공한다.
화학식 VII의 니트릴 중간체는 반응식 6에 따라 합성할 수 있다:
반응식 6
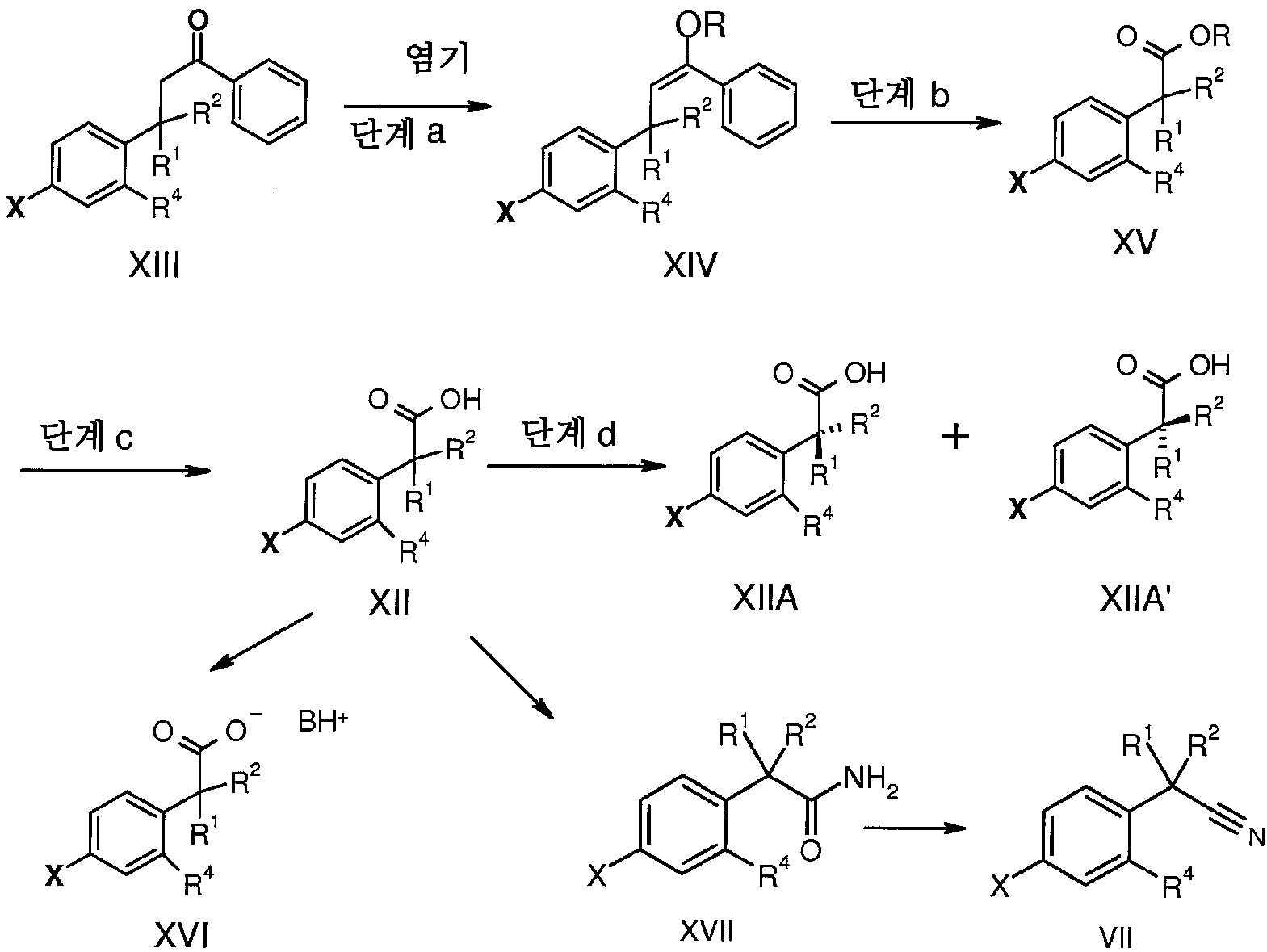
반응식 6에 제시된 바와 같이, 적절한 용매 속에서, 적절한 염기의 존재하에, 화학식 XIII의 케톤과 메틸화제의 반응은 화학식 XIV의 엔올에테르를 제공한다. 적절한 조건하에 엔올에테르 XIV와 산화제(예: 오존)의 반응은 화학식 XV의 에스테르를 제공한다. 적절한 염기의 존재하에, 적절한 용매 속에서 화학식 XV의 에스테르의 가수분해는 화학식 XII의 산을 제공한다. 이 라세미산은 에난티오머 XIIA 및 XIIA'를 제공하기 위하여 분할될 수 있다. 대안적으로, 산 XII는 적절한 용매 속에서 유기 염기(예: 1차 또는 2차 아민)와 반응하여 상응하는 염을 제공할 수 있다.
적절한 용매 속에서, 화학식 XII의 카복실산과 시약(예: 암모니아)의 반응은 화학식 XVII의 아미드를 제공한다. 적절한 용매 속에서, 화학식 XVII의 아미드와 적절한 탈수제의 반응은 화학식 VII의 니트릴을 제공한다. 단계 (a)에 유용한 염기의 비제한적 예는 칼륨 3급-부톡사이드, 나트륨 3급-부톡사이드, 리튬 3급-부톡사이드, 수소화나트륨, 수소화칼륨, 수소화리튬, 나트륨 헥사메틸디실라지드, 칼륨 헥사메틸디실라지드, 리튬 헥사메틸디실라지드, 나트륨 메톡사이드, 칼륨 메톡사이드, 리튬 메톡사이드, 나트륨 에톡사이드, 칼륨 에톡사이드, 리튬 에톡사이드, LDA, n-부틸리튬, 2급-부틸리튬 또는 3급-부틸리튬을 포함한다. 단계 (a)에 유용한 용매의 비제한적 예는 디메틸포름아미드, 디클로로메탄, 에틸 아세테이트, 헥산, 헵탄, 아세토니트릴, 메틸 3급-부틸 에테르, 이소프로필 아세테이트, 톨루엔 및 사이클로프로필메틸 에테르를 포함한다. 단계 (a)에 유용한 알킬화제의 비제한적 예는 디메틸 설페이트, 디메틸 카보네이트, 브로모메탄, 메틸 트리플루오로메탄설포네이트 및 요오도메탄을 포함한다. 단계 (a)에 유용한 실릴화제의 비제한적 예는 트리메틸클로로실란, 3급-부틸디메틸클로로실란, 트리페닐클로로실란 및 트리이소프로필클로로실란, 트리에틸클로로실란을 포함한다.
단계 (b)에 유용한 용매의 비제한적 예는 디메틸포름아미드, 디클로로메탄, 에틸 아세테이트, 헥산, 헵탄, 아세토니트릴, 메틸 3급-부틸 에테르, 이소프로필 아세테이트, 톨루엔 및 사이클로프로필메틸 에테르를 포함한다. 단계 (b)에 유용한 염기의 비제한적 예는 1,8-아자바이사이클로운덱-7-엔(DBU), 트리에틸아민, 피리딘, 4-메틸모르폴린, 디이소프로필에틸아민 및 디메틸아민을 포함한다. 단계 (b)에 유용한 탈수제의 비제한적 예는 아세트산 무수물, 메탄설포닐 클로라이드, 트리플루오로아세트산 무수물, 톨루엔설포닐 클로라이드, 차아염소산나트륨, 차아염소산칼슘 및 3급-부틸 하이포클로라이트를 포함한다.
단계 (c)에 유용한 염기의 비제한적 예는 수산화칼륨, 수산화나트륨, 수산화리튬 및 수산화세슘을 포함한다. 단계 (c)에 유용한 용매의 비제한적 예는 메탄올, 메탄올-물 혼합물, 디메틸포름아미드, 디클로로메탄, 에틸 아세테이트, 헥산, 헵탄, 아세토니트릴, 메틸 3급-부틸 에테르, 이소프로필 아세테이트, 톨루엔 및 사이클로프로필메틸 에테르를 포함한다.
단계 d)에 기술된 화학식 XII의 라세미산의 분할은, 예를 들면, 분별결정화 및 키랄 크로마토그래피를 포함한 당해 분야에 공지된 방법을 사용하여 수행할 수 있다.
하나의 양태에서, 본 발명은 상기 반응식 6에 따르는 중간체 산 XII, XIIA 또는 XIIA'의 제조 방법에 관한 것이다. 다른 양태에서, 본 발명은 화학식 XII, XIIA 또는 XIIA'의 중간체 산에 관한 것이다.
상기 방법에 의해 제조되는 중간체 뿐만 아니라, 화학식 I의 화합물은 당해 분야에 공지되고 하기 합성 실시예 섹션에 예시된 방법에 의해 부가적인 중간체 또는 화학식 I의 화합물로 추가로 전환시킬 수 있다.
합성
실시예
다음은 일반적인 합성 반응식, 실시예 및 당해 분야에 공지된 방법에 의해 제조될 수 있는 본 발명의 대표적인 화합물이다.
하기 화합물에 대한 LCMS 체류시간 및 관찰된 m/z 데이터는 하기 방법 중 하나에 의해 수득된다:






합성 방법
본 발명의 화합물은 하기 기술되는 방법에 의해 제조할 수 있다. 최적의 반응 조건 및 반응 시간은 사용된 특별한 반응물에 따라 변할 수 있다. 달리 명시되지 않는 한, 용매, 온도, 압력 및 다른 반응 조건은 당해 분야의 통상의 숙련가에 의해 용이하게 선택될 수 있다. 특정 방법이 합성 실시예 섹션에 제공된다. 통상적으로, 반응 진행은, 경우에 따라, 박층 크로마토그래피(TLC) 또는 HPLC-MS로 모니터할 수 있다. 중간체 및 생성물은 실리카 겔 상에서 크로마토그래피, 재결정화 및/또는 역상 HPLC에 의해 정제할 수 있다. HPLC 정제 방법은 물 중 0 내지 100% 아세토니트릴이면 어느 경우나 사용되고, 0.1% 포름산, 0.1% TFA 또는 0.2% 수산화암모늄을 함유할 수 있고, 하기 칼럼 중 하나를 사용한다:
a) Waters Sunfire OBD C18 5μM 30x150 mm 칼럼
b) Waters XBridge OBD C18 5μM 30x150 mm 칼럼
c) Waters ODB C8 5μM 19x150 mm 칼럼
d) Waters Atlantis ODB C18 5μM 19x50 mm 칼럼
e) Waters Atlantis T3 OBD 5μM 30x100 mm 칼럼
f) Phenomenex Gemini Axia C18 5μM 30x100 mm 칼럼
g) Waters SunFire C18 Prep OBD 5um 19 x 100 mm
h) Waters XBridge Prep C18 5um 19 x 100 mm
출발 물질 및 시약은 시판중이거나, 화학 문헌에 기술된 방법을 사용하여 당해 분야의 숙련가에 의해 제조될 수 있다.
니트릴 중간체의 합성:
2-(4-
브로모
-
페닐
)-2,3-
디메틸부티로니트릴의
합성

0℃에서 DMF(300mL) 중 R-1(20.0g, 0.102mol)의 용액에 NaH(오일 현탁액 중 60%, 4.28g, 0.107mol)를 서서히 가한다. 이어서, 혼합물을 추가로 15분 동안 교반하고, 2-브로모프로판(9.60mL, 0.107mol)을 가한다. 반응 혼합물을 실온으로 가온하고, 2시간 동안 계속 교반한 다음, 진공하에 농축시킨다. 잔사를 CH2Cl2와 염수 사이에 분배시킨다. 합한 유기층을 Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 헵탄 중 0-15% EtOAc)로 정제하여 I-1(21.3g)을 수득한다; m/z 238.3, 240.2 [M/M+2H].
I-1(21.3g, 89.6mmol)을 DMF(300mL)에 용해시킨다. 혼합물을 0℃로 냉각시키고, NaH(오일 현탁액 중 60%, 3.76g, 94.1mmol)를 서서히 가한다. 이어서, 혼합물을 추가로 15분 동안 교반하고, 메틸 요오다이드(5.9mL, 94.1mmol)를 가한다. 반응 혼합물을 2시간 동안 0℃에서 실온에서 교반한 다음, 진공하에 농축시킨다. 잔사를 메틸 클로라이드와 염수 사이에 분배시킨다. 합한 유기층을 Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 헵탄 중 0-15% EtOAc)로 정제하여 표제 중간체(21.7g)를 수득한다; m/z 252.3, 254.3 [M/M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

2-(4-
브로모
-
페닐
)-2-
사이클로프로필프로피오니트릴의
합성

THF(30mL) 중 R-2(5.00g, 22mmol)의 용액에 MeMgBr(부틸 에테르 중 1.0M, 27.0mL)의 용액을 가한다. 용액을 30분 동안 교반한 다음, 포화 수성 NaHCO3로 처리한다. 혼합물을 CH2Cl2와 염수 사이에 분배시킨다. 유기층을 수집하고, MgSO4로 건조시키고, 여과한 다음 농축시켜 R-3(5.35g)을 수득한다. CH2Cl2(100mL) 중 R-3(5.35g, 22.2mmol)의 용액에 TMSCN(5.9mL, 44mmol) 및 InBr3(790mg, 2.22mmol)를 가한다. 반응물을 밤새 교반한 다음, 20% 수성 Na2CO3로 붓는다. 혼합물을 CH2Cl2로 추출하고, MgSO4로 건조시킨 다음, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 헵탄 중 0-15% EtOAc)로 정제하여 표제 중간체(3.82g)를 수득한다; 1H-NMR, 400 MHz, DMSO-d6 ppm: 7.65 (2H)(d: J=12 ㎐); 7.52 (2H)(d: J=12 Hz); 1.69 (3H) (s); 1.41 (1H) (m); 0.68 (1H) (m); 0.58 (2H)(m); 0.41 (1H) (m).
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

2-(4-
브로모
-
페닐
)-2-
사이클로프로필프로피오니트릴은
또한 하기 방법으로 제조할 수 있다:
-78℃에서 THF(3.0L) 중 R-2(309g, 1.37mol)의 용액에 MeMgBr(Et2O 중 3M, 1.37L, 4.12mol)을 적가한다. 혼합물을 10분 동안 -78℃에서, 그리고 이어서 실온에서 2시간 동안 교반한다. 반응 혼합물을 포화 수성 NH4Cl로 급냉시키고, EtOAc로 추출한다. 합한 유기층을 염수로 세척하고, 무수 Na2SO4로 건조시킨 다음 농축시켜 조 화합물 R-3(330g)을 수득하고, 이를 추가의 정제 없이 다음 단계에서 사용한다.
-78℃에서 CH2Cl2(2.4L) 중 R-3(330g, 1.37mol)의 용액에 BF3.EtO2(198g, 1.37mol)를 적가한다. 혼합물을 동일한 온도에서 30분 동안 교반한다. TMSCN(272g, 2.74mol)를 -78℃에서 적가한다. 부가 후, 혼합물을 2시간 동안 실온에서 교반시킨다. 반응 혼합물을 냉수로 급냉시키고, 유기층을 분리한다. 수성상을 CH2Cl2로 추출한다. 유기층을 염수로 세척하고, 무수 Na2SO4로 건조시킨 다음 농축시킨다. 잔사는 석유 에테르/EtOAc(50:1)를 사용하여 실리카 겔 상에서 크로마토그래피로 정제하여 표제 중간체(160g)를 수득한다.
(R)-2-(4-
브로모
-
페닐
)-2-
사이클로프로필프로피오니트릴
(I-6) 및 (S)-2-(4-브로모-
페닐
)-2-
사이클로프로필프로피오니트릴(I-7)의
제조

에난티오머 I-6 및 I-7은 ChiralPak AY-H 300x20mm SFC 칼럼(용출 85:15 SF CO2:에탄올, 80mL/min 유량) 상에서 I-4(150g)의 분할에 의해 제조한다. 보다 빨리 용출되는 이성체는 I-7로 측정되었다; 1H-NMR, 400 MHz, CDCl3-d6 ppm: 7.54-7.50 (2H)(m); 7.41-7.37 (2H)(m); 1.73 (3H) (s); 1.26-1.19 (1H) (m); 0.74-0.50 (4H) (m); 보다 느리게 용출되는 이성체는 I-6이다; 1H-NMR, 400 MHz, CDCl3-d6 ppm: 7.54-7.50 (2H)(m); 7.41-7.37 (2H)(m); 1.73 (3H) (s); 1.26-1.19 (1H) (m); 0.74-0.50 (4H) (m).
2-(4-
브로모
-
페닐
)-3,3-
디메틸부티로니트릴의
합성

t-BuMgBr(110mL, THF 중 1.0 M)의 용액에 THF(50mL) 중 R-4(10g, 54mmol)의 용액을 가한다. 용액을 10분 동안 교반한 다음, 포화 수성 NaHCO3로 처리한다. 혼합물을 메틸렌 클로라이드와 염수 사이에 분배시키고, 유기층은 수집하고, MgSO4로 건조시키고, 여과하고 농축시킨다. 조 물질을 플래시 크로마토그래피(SiO2, 헵탄 내지 헵탄 중 15% EtOAc)에 의해 정제하여 황색 고체를 수득하고, 이를 헵탄에서 슬러리화하여 추가로 정제함으로써 여과 후 R-5(4.67g)가 수득된다. CH3CN(100mL) 중 R-5(4.63g, 19.0mmol)의 용액에 이미다졸(3.89g, 57.1mmol)에 이어서, Ph3PBr2(24.1g, 57.1mmol)를 가한다. 혼합물을 40℃에서 6시간 동안 가열한 다음, 23℃로 냉각시키고, EtOAc와 포화 수성 NaHCO3 사이에 분배시킨다. 유기층을 수집하고, 물로 세척하고, MgSO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사는 헵탄에서 슬러리화시키고, 생성된 고체는 여과한다. 여액을 수집하고, 휘발성 물질은 진공하에 제거한다. 잔사를 DMSO(100mL)에 용해시키고, NaCN(1.11g, 22.7mmol)으로 처리한다. 혼합물을 140℃에서 3시간 동안 가열한 다음, 23℃로 냉각시킨다. 혼합물을 Et2O와 물 사이에 분배시킨다. 유기층을 물로 세척하고, MgSO4로 건조시키고, 여과하고, 농축시킨다. 조 물질을 플래시 크로마토그래피(SiO2, Hep 내지 Hep 중 15%EtOAc)에 의해 정제하여 표제 중간체(2.37g)가 수득된다; 1H-NMR, 400 MHz, CDCl3 ppm: 7.59 (2H)(d: J=12 Hz); 7.33 (2H)(d: J=12 Hz); 4.26 (1H)(brs); 1.35 (9H)(s).
카복스아미딘
중간체의 합성
2-(4-
브로모
-
페닐
)-N-
하이드록시
-2,3-
디메틸부티르아미딘의
합성

EtOH(50mL) 중 I-2(10.0g, 40mmol)의 용액을 5% 수성 하이드록실아민(50mL)으로 처리한다. 반응물을 밤새 80℃에서 가열한 다음, 진공하에 농축시킨다. 고체를 여과하고, 물에 이어서 헵탄으로 세척한다. 고체를 수집하고, EtOAc로 연마한 다음, 여과하고, 수집하고 건조시켜 표제 중간체(10.4g)를 수득한다; m/z 285.4;287.2 [M/M+2H]
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

2-[4-(2-
아미노피리미딘
-5-일)-
페닐
]-N-
하이드록시
-2,3-
디메틸부탄이미드아미드의
합성
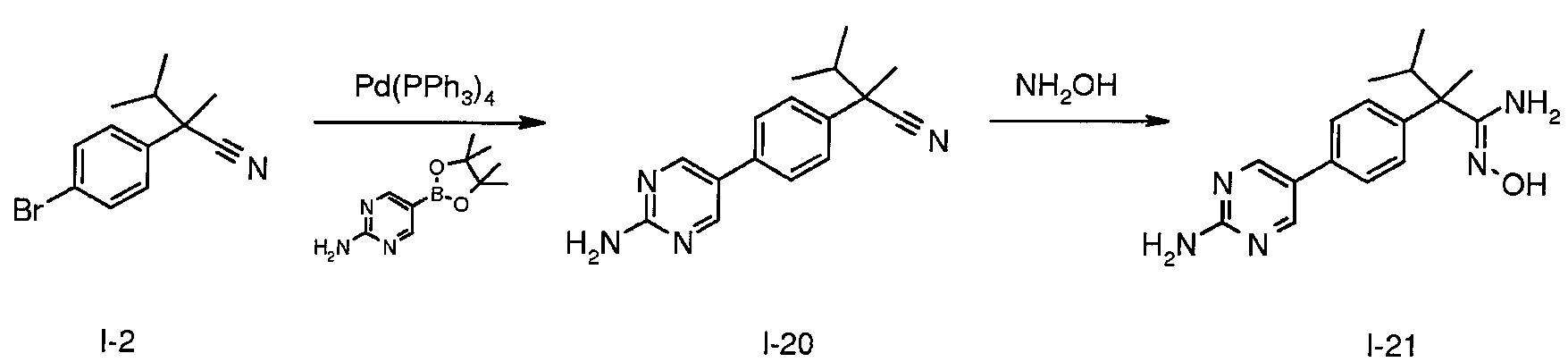
THF(20mL) 및 포화 수성 Na2CO3(10mL) 중 I-2(2.00g, 7.93mmol), 2-아미노피리미딘-5-보론산 피나콜 에스테르(2.63g, 11.0mmol) 및 테트라키스(트리페닐포스핀)팔라듐(0)(459㎎, 0.397mmol)의 용액을 3시간 동안 80℃에서 가열한다. 혼합물을 23℃로 냉각시킨 다음, EtOAc와 염수 사이에 분배시킨다. 유기층을 수집하고, MgSO4로 건조시키고, 여과하고, 농축시켜 잔사를 수득하고, 이를 플래시 크로마토그래피(SiO2, CH2Cl2 내지 CH2Cl2 중 3% MeOH)로 정제하여 I-20(m/z 267.5 [M+H])을 수득한다.
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:
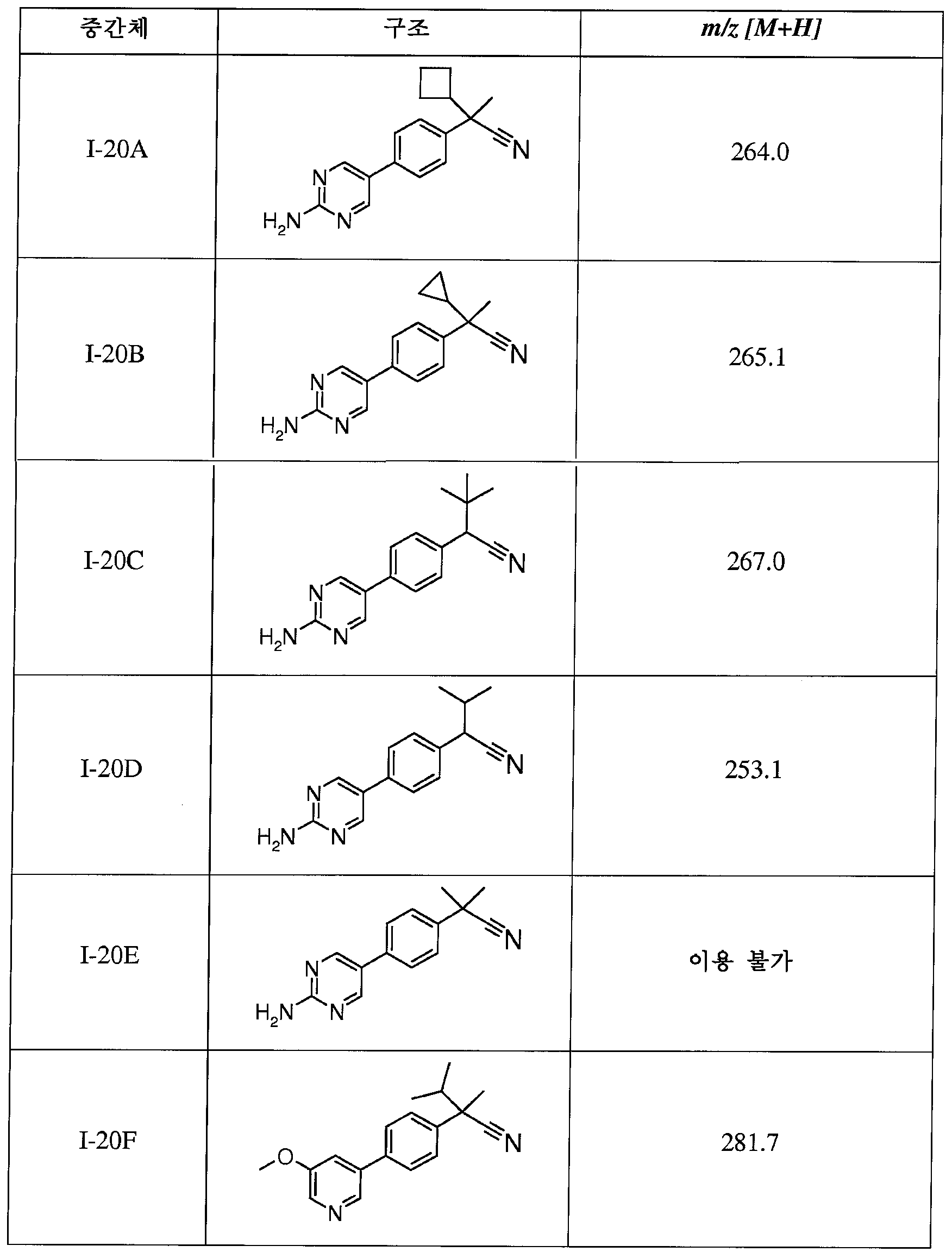
I-20을 EtOH(30mL)에 용해시키고, 50% 수성 하이드록실아민(12mL)으로 처리한다. 반응물을 48시간 동안 80℃에서 가열한 다음, 23℃로 냉각시키고, 셀라이트를 통해 여과한다. 여액을 EtOAc와 물 사이에 분배시킨다. 유기층을 수집하고, MgSO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 조 물질을 플래시 크로마토그래피(SiO2, CH2Cl2 내지 CH2Cl2 중 10% MeOH)에 의해 정제하여 표제 중간체(1.56g)를 제공한다; m/z 300.4 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

(R)-2-[4-(2-아미노-피리미딘-5-일)-
페닐
]-2-
사이클로프로필
-N-
하이드록시
-프로피온아미딘의 합성

THF(300mL) 중 I-6(18.5g, 0.074mol)의 혼합물에 5-(4,4,5,5-테트라메틸-[1,3,2]디옥사보롤란-2-일)-피리미딘-2-일아민(19.6g, 0.089mol), 테트라키스(트리페닐포스핀)팔라듐(0)(8.5g, 0.007mol) 및 2M Na2CO3(74mL, 0.148mol)을 가한다. 혼합물을 24시간 동안 80℃로 가열한다. 용액을 실온으로 냉각시키고, EtOAc 및 물로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 CH2Cl2에 재현탁시킨다. 용액으로부터 침전된 고체는 여과에 의해 수집한다. 고체를 건조시키고, 이는 I-28(14.8g)인 것으로 확인되었다; m/z 265.4 [M+H].
I-28(14.8g, 0.056mol), KOH(15.7g, 0.28mol) 및 H2O(50중량%)(34mL, 0.56mol) 중 하이드록실아민 용액의 현탁액을 48시간 동안 85℃에서 교반한다. 혼합물을 냉각시키고, 고체를 여과하고 건조시켜, 표제 중간체(12.5g)를 수득한다; m/z 298.4 [M+H].
(R)-2-
사이클로프로필
-N-
하이드록시
-2-[4-(2-
메틸아미노
-피리미딘-5-일)-
페닐
]-
프로피온아미딘의
합성
5mL 마이크로파 반응 용기에 톨루엔(5mL) 중 5-브로모-2-(메틸아미노)피리디민(451㎎, 2.39mmol) 및 헥사메틸디스타난(0.456ml, 2.19mmol)을 합한다. 혼합물은 아르곤을 사용하여 탈기시킨 후, 테트라키스(트리페닐포스핀)팔라듐(0)(115mg, 0.10mmol)을 가한다. 반응물을 1회 이상 탈기시키고, 캡핑하여 1시간 동안 115℃로 가온한다. 주위 온도로 냉각되면, I-6(500mg, 1.99mmol)을 테트라키스(트리페닐포스핀)팔라듐(0)(115mg, 0.10mmol)과 함께 도입시킨다. 용기를 캡핑하여 밤새 115℃로 가온한다. 이 시간 후, 반응물을 냉각하고 농축시킨다. 생성된 고체를 플래시 크로마토그래피(실리카 겔, 0-100% EtOAc/헵탄)로 정제하여 I-29bis(134mg)를 수득한다; m/z 279.4 [M+H].
EtOH(3.2ml) 중 I-29bis(134mg, 0.481mmol)의 현탁액에 H2O(50중량%)(1.18mL, 19.24mmol) 중 하이드록실아민 용액을 가하고, 72시간 동안 85℃에서 교반한다. 혼합물을 냉각하고 농축시키고, 물 및 에틸 아세테이트로 희석한다. 백색 고체를 여과하고 건조시키고, 유기층은 플래시 크로마토그래피로 정제하고 고체와 합하여, 표제 중간체(110㎎)를 수득한다; m/z 312.4 [M+H].
아릴 브로마이드 중간체의 합성
3-[2-(4-
브로모페닐
)-3-
메틸부탄
-2-일]-5-
사이클로프로필
-1,2,4-
옥사디아졸
의 합성

피리딘(2mL) 중 I-14(150㎎, 0.53mmol) 및 사이클로프로필카보닐 클로라이드(60㎎, 0.58mmol)의 혼합물을 실온에서 15분 동안 교반한 다음, 110℃에서 18시간 동안 가열한다. 반응 혼합물을 진공하에 농축시킨 다음, CH2Cl2와 포화 수성 NaHCO3 사이에 분배시킨다. 유기층을 Na2SO4로 건조시키고, 여과하고 진공하에 농축시켜 표제 중간체(167㎎)를 수득한다; m/z 336.0 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

5-{3-[2-(4-
브로모페닐
)-3-
메틸부탄
-2-일]-1,2,4-
옥사디아졸
-5-일}피리미딘의 합성
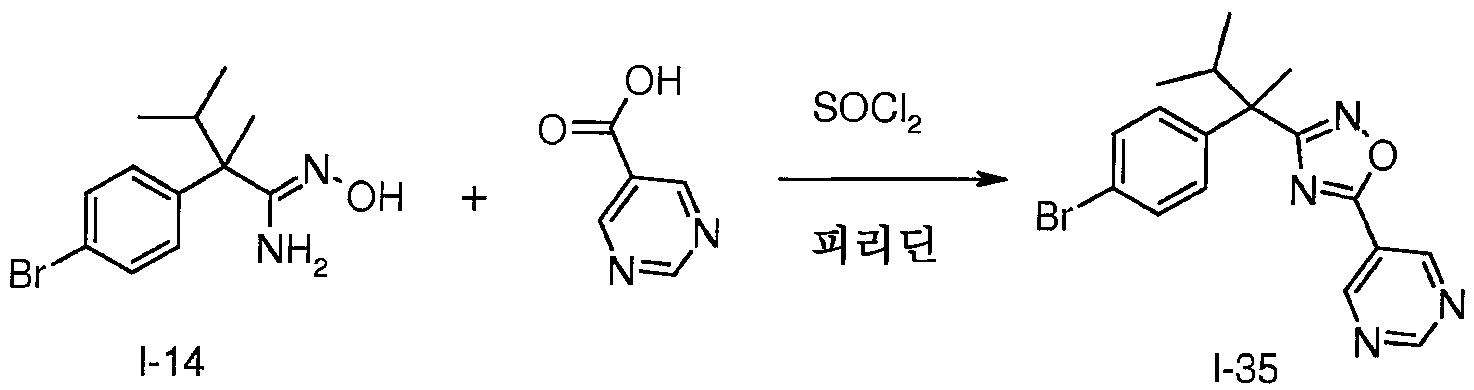
피리딘(1.0mL) 중 피리미딘-5-카복실산(200㎎, 0.70mmol)의 용액에 티오닐 클로라이드(61㎕, 0.84mmol)를 가한다. 혼합물을 15분 동안 실온에서 교반한 다음, I-14(91㎎, 0.74mmol)를 가한다. 생성된 혼합물을 18시간 동안 110℃에서 가열한 다음, 진공하에 농축시킨다. 잔사를 EtOAc와 포화 수성 NaHCO3 사이에 분배시키고, 염수로 세척하고, Na2SO4로 건조시키고, 여과하고 진공하에 농축시켜 표제 화합물(263㎎)을 수득한다; m/z 373.0, 375.0 [M, M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:



3-[1-(4-
브로모
-
페닐
)-1-
사이클로프로필
-에틸]-5-(1H-
피라졸
-4-일)-[1,2,4]옥사디아졸의 합성

1,1'-카보닐디이미다졸(4.9g, 30.7mmol)을 1,4-디옥산(150mL) 중 1H-피라졸-4-카복실산(3.4g, 30.7mmol)의 혼합물에 가한다. 혼합물을 30분 동안 50℃에서 교반하고, I-16을 가하여, 반응 혼합물을 48시간 동안 85℃에서 가열한다. 반응 혼합물을 실온으로 냉각시키고, 포화 NaHCO3 용액으로 부은 다음, EtOAc로 추출한다. 유기층을 MgSO4로 건조시키고, 여과하고 농축시켜 조 생성물을 수득하고, 이를 플래시 크로마토그래피(SiO2, 0-6% MeOH/CH2Cl2)를 통해 정제하여 표제 중간체(6.9g)를 수득한다; m/z 359, 361 [M, M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

3-[(R)-1-(4-
브로모
-
페닐
)-1-
사이클로프로필
-에틸]-5-(1H-
피라졸
-4-일)-[1,2,4]옥
사디
아졸의 합성

밀봉된 관에 1,4-디옥산(8mL) 중 1H-피라졸-4-카복실산(484㎎, 4.2mmol)을 가한 다음, 1,1'-카보닐디이미다졸(679㎎, 4.2mmol)을 가한다. 반응 혼합물을 30분 동안 55℃에서 교반한다. 이어서, 1,4-디옥산(5mL) 중 I-17(1.1g, 4.0mmol)을 상기 혼합물에 가한다. 반응 혼합물을 120℃에서 18시간 동안 교반한다. 반응 혼합물을 진공하에 농축시킨다. 잔사를 EtOAc로 희석하고, 물 및 염수로 세척한 다음, 무수 Na2SO4로 건조시키고, 여과하고 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 0-5% MeOH/CH2Cl2)로 정제하여 표제 중간체(1.3g)를 수득한다; m/z 359.0, 361.0 [M, M+2H].
2-(4-{3-[1-(4-
브로모
-
페닐
)-1-
사이클로프로필
-에틸]-[1,2,4]
옥사디아졸
-5-일}-
피라졸
-1-일)-N,N-디메틸-
아세트아미드의
합성
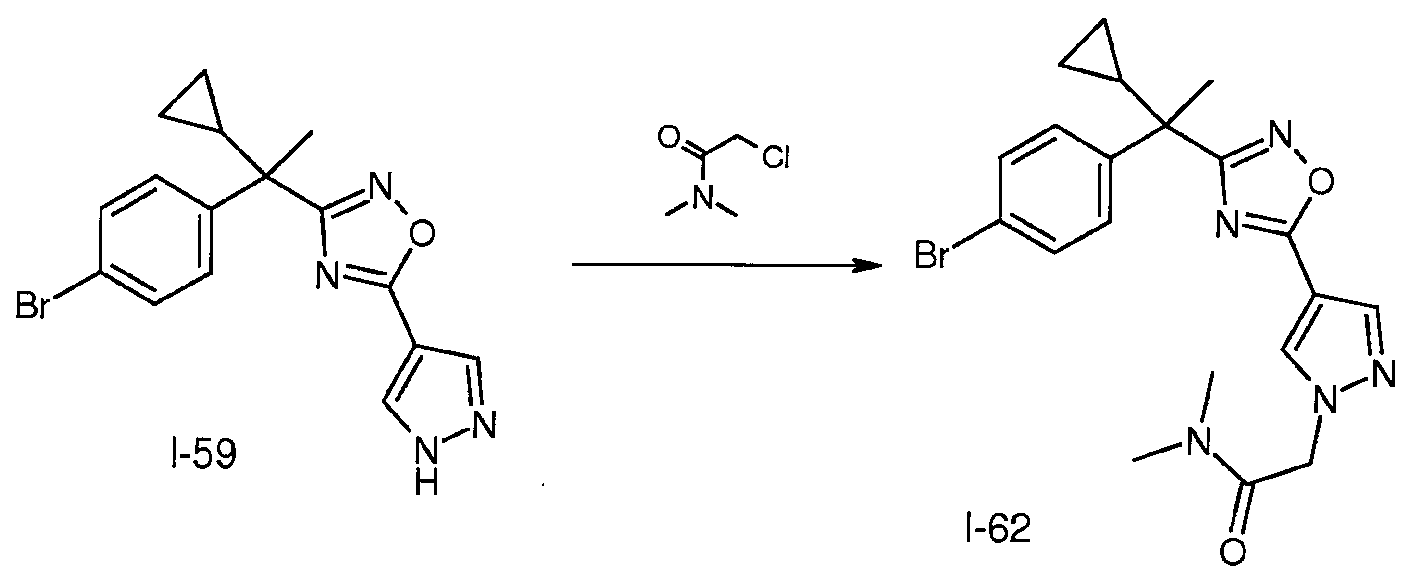
DMF(80mL) 중 I-59(6.9g, 19mmol)의 용액에 실온에서 K2CO3(5.3g, 38mmol) 및 2-클로로-N,N-디메틸아세트아미드(2.9g, 28mmol)를 가한다. 혼합물을 동일한 온도에서 24시간 동안 교반한다. 물(200mL)을 가하고, 혼합물을 EtOAc(300mL)로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 나머지 잔사는 용출제로서 CH2Cl2 중 8% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피를 통해 정제하여 표제 중간체(8.3g)를 수득한다; m/z 444.2, 446.2 [M, M+2].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

a) 반응 혼합물을 80℃에서 48시간 동안 교반한다.
b) 반응은 상응하는 요오다이드로부터 출발하여 수행하고, 혼합물은 80℃에서 밤새 교반한다.
3-[(R)-1-(4-
브로모
-
페닐
)-1-
사이클로프로필
-에틸]-5-(1-
메틸
-1H-
피라졸
-4-일)-[1,2,4]
옥사디아졸의
합성

바이알에 DMF 6mL 중 I-61(550㎎, 1.531mmol), 요오도메탄(0.191mL, 3.062mmol) 및 K2CO3(423㎎, 3.062mmol)를 가한다. 반응 혼합물을 실온에서 2시간 동안 교반한 다음, 물과 염수로 붓고, EtOAc(4x25mL)로 추출한다. 합한 유기 분획을 황산나트륨으로 건조시키고, 여과하고 진공하에 농축시켜 표제 중간체(516㎎)를 수득한다; m/z 374.0, 376.0 [M/M+2].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

2-[4-(3-{1-
사이클로프로필
-1-[4-(4,4,5,5-
테트라메틸
-[1,3,2]
디옥사보롤란
-2-일)-
페닐
]-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드의
합성

가압 바이알에서 1,4-디옥산(20mL) 중 I-62(2.6g, 5.9mmol)의 용액에 비스(피나콜레이토)디보론(2.2g, 8.8mmol), KOAc(2.3g, 23mmol) 및 테트라키스(트리페닐포스핀)팔라듐(0)(481㎎, 0.6mmol)을 가한다. 반응 혼합물을 100℃에서 4시간 동안 Ar하에 교반한다. 혼합물을 냉각시키고, 진공하에 농축시킨다. 잔사를 EtOAc(100mL)로 희석하고, 셀라이트 플러그를 통해 통과시키고, EtOAc(20mL)로 철저히 세정한다. 여액을 황산마그네슘으로 건조시키고 여과하여, 표제 중간체(1.9g)를 수득한다; m/z 492.3 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:
a) 1,1'-비스(디페닐포스피노)페로센디클로로팔라듐(II) 디클로로메탄이 대신에 사용된다.
b) 1,1'-비스(디페닐포스피노)페로센디클로로팔라듐(II) 디클로로메탄이 대신에 사용되고, 반응 혼합물은 밤새 100℃에서 교반한다.
Boc-피페리딘 중간체의 합성
5'-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-2,3,5,6-
테트라하이드로
-[1.2']
비피라지닐
-4-
카복실산
3급-부틸 에스테르의 합성

250mL RB 플라스크에 NMP 100mL 중 R-7(5.4g, 28.99mmol)을 채운다. R-8(5.00g, 28.99mmol)에 이어서, 트리에틸아민(4.85mL, 34.79mmol)을 가한다. 반응물을 질소하에 밤새 60℃로 가열한다. 반응물을 실온으로 냉각시키고, 빙수로 부은 다음, 침전된 I-76(8.60g)을 여과에 의해 분리한다; m/z 323.4 [M+H].
에탄올(250mL) 중 I-76(8.60g, 26.68mmol)의 교반된 현탁액에 실온에서 5M NaOH(26.68mL, 133.39mmol)를 가한다. 혼합물은 균질해진 후 지속되는 침전을 형성하고 이는 고체 덩어리가 된다. 물(200mL)을 가하고, 혼합물을 4시간 동안 교반한 다음, 반응은 완결되는 것으로 나타난다. 밝은 갈색 슬러지를 비이커에 붓고, 물로 처리한다. AcOH를 가하여 산성 pH에 이르도록 하고, 생성물은 DCM(2x)으로 추출한다. 합한 유기층을 무수 MgSO4로 건조시키고, 여과하고 농축시켜 헵탄 중에 현탁되는 고체로서 생성물을 수득한다. 고체는 여과를 통해 수집하고, 헵탄으로 세척하여 I-77(7.90g)을 수득한다; m/z 309.4 [M+H].
THF(40mL) 중 I-77(3.0g, 9.71mmol)의 현탁액에 실온에서 1,1'-카보닐디이미다졸(1.6g, 9.71mmol)을 가한다. 혼합물을 50℃에서 30분 동안 교반한다. 이 시간 후, I-16(2.5g, 8.83mmol)을 가하고, 생성된 혼합물은 80℃에서 3시간 동안 가열한다. 혼합물을 냉각시키고, AcOH(8mL)로 처리한다. 혼합물을 80℃로 가온하고, 밤새 교반한다. 실온으로 냉각되면, 반응물을 농축시키고, 물로 희석한다. 생성물을 DCM(2x)으로 추출한다. 합한 유기층을 염수로 세척하고, 무수 MgSO4로 건조시킨다. 혼합물을 여과하고 농축시킨다. 나머지 조 생성물은 플래시 크로마토그래피(실리카 겔, 0-5% MeOH/DCM)를 통해 정제하여 I-78(2.2g)을 수득한다. 마이크로파 반응 용기에, DMF 15mL 중 I-78(0.50g, 0.90mmol)을 가한 다음, 2-아미노피리미딘-5-보론산 피나콜 에스테르(0.30g, 1.35mmol), 테트라키스(트리페닐포스핀)팔라듐(0)(105㎎, 0.09mmol) 및 수성 Na2CO3(2.0M, 1.8mL)을 가한다. 반응 혼합물을 85℃에서 16시간 동안 교반한다. 이 시간 후, 반응 혼합물을 염수로 붓고, EtOAc(3x)로 추출한다. 합한 유기 분획을 무수 MgSO4로 건조시키고, 여과한 다음, 진공하에 농축시켜 조 물질을 수득한다. 플래시 크로마토그래피(실리카 겔, 0-5% MeOH/DCM)를 통한 정제로 표제 중간체(150㎎)를 수득한다; m/z 570.4 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

5'-(3-{(S)-1-[4-(5-아미노-피라진-2-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-2,3,5,6-
테트라하이드로
-[1.2']
비피라지닐
-4-
카복실산
3급-부틸 에스테르의 합성
THF(20mL) 중 I-77(1.0g, 3.53mmol)의 현탁액에 실온에서 1,1'-카보닐디이미다졸(0.63g, 3.88mmol)을 가한다. 혼합물을 50℃에서 30분 동안 교반한다. 이 시간 후, I-19(1.2g, 3.88mmol)를 THF 용액(15mL)으로서 가하고, 생성된 혼합물은 80℃에서 3시간 동안 가열한다. 혼합물을 냉각시키고, AcOH(8mL)로 처리한 다음, 80℃로 가온하고, 밤새 교반한다. 이 시간 후, 반응물을 실온으로 냉각시키고, 농축시키고, 물로 희석한다. 생성물을 DCM(2x)으로 추출한다. 합한 유기층을 염수로 세척하고, 건조(MgSO4)시킨다. 여과하고 농축시킨다. 나머지 조 생성물은 플래시 크로마토그래피(실리카 겔, 0-5% MeOH/DCM)를 통해 정제하여, I-84(1.2g)를 수득한다. 5mL 마이크로파 반응 용기에, 톨루엔(2mL) 중 5-아미노-2-브로모피라진(60㎎, 0.34mmol) 및 헥사메틸디스타난(120㎎, 0.38mmol)을 합한다. 혼합물은 아르곤을 사용하여 탈기시킨 후, 테트라키스(트리페닐포스핀)팔라듐(0)(40㎎, 0.03mmol)을 가한다. 반응물을 1회 이상 탈기시키고, 캡핑시킨 다음, 1시간 동안 115℃로 가온한다. 주위 온도로 냉각되면, I-84(270㎎, 0.48mmol)를 테트라키스(트리페닐포스핀)팔라듐(0)(30㎎, 0.05mmol)과 함께 도입시킨다. 용기를 캡핑시키고, 밤새 115℃로 가온한다. 이 시간 후, 반응물을 냉각시키고 농축시킨다. 조 생성물을 MeOH/DCM 중에 현탁시키고, 실리카 겔로 처리하고, 농축시킨다. 생성된 고체플 플래시 크로마토그래피(실리카 겔, 0-10% MeOH/DCM)를 통해 정제하여 표제 중간체(100㎎)를 수득한다.
4-
플루오로
-피리미딘-2-
일아민의
합성

CH3CN(10mL) 중 R-9(100mg, 0.77mmol)의 현탁액에 실온에서 Et3N 중 HF(0.26mL, 1.5mmol)를 가한다. 용액을 48시간 동안 80℃로 가열한다. 용액을 냉각시키고, 물(10mL)을 가한다. 용액을 EtOAc(20mL) 및 H2O(5mL)로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시켜 I-86(25mg)을 수득한다; m/z 113.9 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

5-
브로모
-4-
플루오로
-피리미딘-2-
일아민의
합성

CH3CN(20mL) 중 I-86(280mg, 2.5mmol)의 용액에 실온에서 N-브로모석신이미드(881㎎, 4.9mmol)를 가한다. 용액을 12시간 동안 동일한 온도에서 교반한다. 용액으로부터 침전된 고체를 수집하고 건조시켜, 표제 중간체(250mg)를 수득한다; m/z 191.9, 193.9 [M, M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

5-
브로모
-3-
트리플루오로메틸
-피리딘-2-
일아민의
합성

DMF(15mL) 중 R-10(2.70g, 16.66mmol)의 교반된 용액에 DMF 용액(15mL, 적가)으로서 N-브로모석신이미드(3.00g, 16.85mmol)를 가한다. 4시간 후, 반응물을 얼음으로 붓는다. 생성된 침전물은 여과를 통해 수집하여 회백색 고체로서 생성물을 수득한다. DCM에 용해시키고, 염수로 세척한다. 층을 분리하고, 유기상은 건조(MgSO4)시키고, 여과하고 농축시켜, 표제 중간체(3.8g)를 수득한다; m/z 241.2/243.2 [M/M+2H].
5-
브로모
-3-
플루오로
-피리딘-2-
일아민의
합성

환저 플라스크에 0℃에서 CH3CN(120mL) 중 R-11(500㎎, 4.46mmol)을 가한 다음, N-브로모석신이미드(397㎎, 2.23mmol)를 가한다. 반응 혼합물을 15분 동안 강력히 교반(빛으로부터 보호)한 다음, 실온에서 1시간 동안 교반한다. 부가 부분의 N-브로모석신이미드(397㎎, 2.23mmol)를 0℃에서 가한 다음, 반응 혼합물을 실온에서 2시간 동안 교반한다. 반응 혼합물을 진공하에 농축시킨다. 잔사를 EtOAc에 용해시키고, 포화 Na2S2O3(20mL), 염수로 세척한 다음, 무수 Na2SO4 상에서 건조시키고, 여과하고 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 0-20% EtOAc/헵탄)로 정제하여 표제 중간체(772㎎)를 수득한다; m/z 190.89/192.86 [M/M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

(5-
브로모
-피리미딘-2-일)-3급-부틸-
아민의
합성

바이알에 DMF(5mL) 중 R-12(200㎎, 1.13mmol)를 가한 다음, K2CO3(312㎎, 2.26mmol) 및 이소프로필아민(134㎎, 2.27mmol)을 가한다. 반응 혼합물을 3시간 동안 70℃에서 교반한다. 반응 혼합물을 진공하에 농축시킨다. 잔사를 EtOAc에 용해시키고, 물 및 염수로 세척한 다음, 무수 Na2SO4로 건조시키고, 여과하고 농축시켜, 표제 중간체(221㎎)를 수득한다; m/z 216.0/218.0 [M/M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

3-
벤질옥시
-5-
브로모
-피리딘의 합성

바이알에 0℃에서 THF(5mL) 중 3-브로모-5-하이드록시피리딘(200㎎, 1.15mmol), 벤질 알콜(137㎎, 1.27mmol) 및 트리페닐포스핀(332㎎, 1.27mmol)을 가한 다음, 디이소프로필 아조디카복실레이트(256㎎, 1.27mmol)를 가한다. 반응 혼합물을 실온에서 18시간 동안 교반한다. 반응 혼합물을 진공하에 농축시킨다. 잔사를 EtOAc로 희석하고, 포화 NaHCO3, 물 및 염수로 세척한 다음, 무수 Na2SO4로 건조시키고, 여과하고 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 0-5% MeOH/CH2Cl2)로 정제하여 표제 화합물(97㎎)을 수득한다; m/z 264.0, 266.0[M, M+2H].
5-
브로모
-3-
트리플루오로메틸
-피라진-2-
일아민의
합성

Ar하에 교반하는 DMSO(3mL) 중 2-아미노-5-브로모피라진(174㎎, 1.27mmol)의 용액에 페로센(56㎎, 0.3mmol)을 가하고, Ar을 사용하여 5분 동안 탈기시킨다. DMSO 중 1N H2SO4 2mL에 이어서, DMSO(2mL) 중 CF3I(0.276mL, 3mmol)를 가하여 다소 황색의 용액을 수득한다. 30% H2O2 0.2mL를 서서히 가하여, 반응물의 색이 황색으로부터 암녹색으로 변하게 된다. 반응물을 Ar하에 50℃에서 2시간 동안 가열한다. 실온으로 냉각시킨 후, 반응 혼합물을 염수로 붓고, 생성물을 EtOAc(4x20ml)로 추출한다. 합한 유기 분획을 황산마그네슘으로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 조 물질은 플래시 크로마토그래피(SiO2, Biotage SNAP 10g, 0-50% EtOAc/hept)로 정제하여 표제 화합물 70㎎을 수득한다; m/z 242.0, 244.0(M, M+2H).
4-
벤질옥시
-5-
브로모
-피리미딘-2-
일아민의
합성

20ml 마이크로파 반응 용기에 벤질 알콜(7.0ml) 및 나트륨(145㎎, 6.33mmol)을 가한다. 용기를 캡핑시키고, 나트륨이 소모될 때까지 주위 온도에서 교반한다. 이 시간 후, I-90(1.10g, 5.28mmol)을 가하고, 반응물을 2시간 동안 130℃로 가온한다. 실온으로 냉각되면, 반응 혼합물을 저용적으로 농축시킨다. 나머지 잔사는 물로 희석한다. 물을 경사분리하고, 나머지 오일은 메탄올로 처리한다. 침전된 고체는 여과를 통해 수집하고, 메탄올로 세척하여, 표제 중간체(0.86g)를 수득한다; m/z 282.0 [M+H].
(5-
브로모
-피리딘-2-일)-
메틸
-
아민의
합성

20ml 마이크로파 반응 용기에 2,5-디브로모-피리딘(2.00g, 8.44mmol)을 채우고, 메틸아민(에탄올 중 33% 용액 10.45ml, 84.43mmol)으로 처리한 다음, 3일 동안 80℃로 가온한다. 이 시간 후, 반응물을 농축시키고, 나머지 고체는 1M HCl(50ml) 및 DCM으로 처리한다. 층을 분리하고, 수성상은 1N NaOH(pH 약 11까지)를 사용하여 염기성화시킨다. 생성물을 DCM(2x)으로 추출하고, 합한 유기층은 건조(MgSO4)시키고, 여과하고 농축시켜, 목적하는 생성물 I-96bis(1.20g)를 수득한다. 1H-NMR (400MHz, DMSO-d6): 2.75 ppm (d, 3H), 6.44 ppm (d, 1H), 6.72 ppm (bs, 1H), 7.51 ppm (dd, 1H), 8.05 ppm (s, 1H)
2-[4-(3-{(R)-1-[4-(2-아미노-4-
벤질옥시
-피리미딘-5-일)-
페닐
]-1-
사이클로
프로필-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드의
합성

I-97은 방법 26(16시간 동안 85℃에서 팔라듐 테트라키스, 2M Na2CO3 및 DMF를 사용함)에 따라 제조한다; m/z 565.0 [M+H].
2-{2-[4-(3-{(R)-1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-에틸}-
이소인돌
-1,3-
디온의
합성

실시예 48(100㎎, 0.266mmol)을 DMF(2.5mL), 2-(2-브로모-에틸)-이소인돌-1,3-디온(101mg, 0.399mmol) 및 Cs2CO3(83.0mg, 0.599mmol)로 처리하고, 반응물을 밤새 교반한다. 생성된 혼합물을 물 및 에틸 아세테이트로 희석하고, 상을 분리한다. 유기상을 물 및 염수로 세척하고, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 생성된 잔사는 0-10% 메탄올/CH2Cl2로 용출시키면서 실리카 겔 상에서 플래시 크로마토그래피로 정제하여 I-98(120mg)을 수득한다.
최종 화합물의 합성
방법 1
2-(3-{2-[4-(5-
메톡시피리딘
-3-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사디아졸
-5-일)피라진(
실시예
1, 표 1)의 합성

DMF(5mL) 중 I-27(200㎎, 0.64mmol)의 용액에 휘니그 염기(0.3mL, 1.6mmol)에 이어서, 피라진-2-카보닐 클로라이드(110㎎, 0.80mmol)를 가한다. 반응 혼합물을 2시간 동안 120℃에서 가열한 다음, 휘발성 물질을 진공하에 제거한다. 잔사를 플래시 크로마토그래피(SiO2, 헵탄 내지 헵탄 중 60% EtOAc)로 정제하여 표제 화합물(165㎎)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 2 내지 5, 표 1
실시예 7, 표 1
실시예 117 내지 118, 표 1
실시예 120, 표 1
방법 2
[2-아미노-5-(4-{3-
메틸
-2-[5-(피리딘-3-일)-1,2,4-
옥사디아졸
-3-일]부탄-2-일}
페닐
)피리딘-3-일]메탄올(
실시예
6, 표 1)의 합성

I-58(0.450g, 1.21mmol)을 1,4-디옥산(3.0mL)에 현탁시킨다. 비스(피나콜레이토)디보란(0.364g, 1.43mmol), 칼륨 아세테이트(0.500g, 5.09mmol) 및 1,1'-비스(디페닐포스피노)페로센디클로로팔라듐(II) 디클로로메탄 착물(0.100g, 0.122mmol)을 가한다. 이 반응 혼합물을 탈기시키고, 아르곤하에 100℃에서 4시간 동안 가열한다. 혼합물을 실온으로 냉각시킨 다음, EtOAc로 희석하고, 물로 세척한다. 유기층을 수집하고, 진공하에 농축시켜, 잔사를 수득하고, 이를 플래시 크로마토그래피(SiO2, 헥산 내지 헥산 중 30% EtOAc)로 정제하여 I-99(0.362g)를 수득한다; m/z 420.61 [M+1]
I-99(0.100g, 0.238mmol)를 DMF(2.0mL)에 용해시키고, 2-아미노-5-브로모-3(하이드록시메틸)피리딘(0.051g, 0.25mmol), 테트라키스(트리페닐포스핀)팔라듐(0)(0.029g, 0.025mmol) 및 수성 Na2CO3(2.0 M, 1.0mL, 1.0mmol)로 처리한다. 이 반응 혼합물을 탈기시키고, 아르곤 하에 100℃에서 4시간 동안 가열한다. 혼합물을 실온으로 냉각시킨 다음, EtOAc로 희석하고, 물로 세척한다. 유기층을 수집하고, 진공하에 농축시켜, 잔사를 수득하고, 이를 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-10% MeOH)로 정제하여 표제 화합물(0.025g)을 수득한다.
방법 3
5-(4-{2-[5-(6-
메톡시피리딘
-3-일)-1,2,4-
옥사디아졸
-3-일]-3-
메틸부탄
-2-일}
페닐
)피리미딘-2-아민(
실시예
8, 표 1)의 합성
실시예 7(35mg, 0.083mmol)을 MeOH(2.0mL)에 용해시킨다. MeOH(50㎕) 중 25% (w/w) NaOMe 용액을 가한다. 반응 혼합물을 70℃에서 6시간 동안 가열한 다음, 휘발성 물질을 진공하에 제거한다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-10% MeOH)로 정제하여 표제 화합물(26mg)을 수득한다.
하기 화합물들은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 9, 표 1
방법 4
3-(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사디아졸
-5-일)피리딘-2(1H)-온(
실시예
12, 표 1)의 합성

I-21(100mg, 0.255mmol)을 1-메틸-2-피롤리디논(1mL)에 용해시킨다. 에틸-디이소프로필아민(0.3mL, 1.6mmol)에 이어서, 2-클로로-니코티닐 클로라이드(62mg, 0.35mmol)를 가한다. 반응 혼합물을 120℃에서 1시간 동안 가열한 다음, 실온으로 냉각시키고, CH2Cl2와 물 사이에 분배시킨다. 유기층을 수집하고, 휘발성 물질은 진공하에 제거한다. 잔사를 플래시 크로마토그래피(SiO2, 헵탄 중 0-100% 에틸 아세테이트)로 정제하여 I-100(78mg)을 수득한다; m/z 421.48 [M+1].
I-100(35mg, 0.083mmol)을 1,4-디옥산(2.0mL)에 용해시킨다. 10% (w/w) 수성 LiOH 용액(50㎕)을 가한다. 반응 혼합물을 70℃에서 2시간 동안 가열한다. 용매를 진공하에 제거하고, 잔사는 물(2.0mL)에 현탁시킨다. 침전물을 여과에 의해 수집하고, 물로 세척한 다음, 공기-건조시킨다. 고체는 추가로 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-10% MeOH)로 정제하여 표제 화합물(28mg)을 수득한다.
하기 화합물들은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 10, 표 1
방법 5
5-(4-{2-[5-(4-
메톡시피리딘
-3-일)-1,2,4-
옥사디아졸
-3-일]-3-
메틸부탄
-2-일}
페닐
)피리미딘-2-아민(
실시예
11, 표 1)의 합성

4-메톡시-니코틴산(54mg, 0.35mmol)을 1-메틸-2-피롤리디논(1mL)에 용해시키고, 카보닐디이미다졸(57mg, 0.35mmol)을 가한다. 혼합물을 15분 동안 교반한 다음, I-21(100mg, 0.255mmol)을 가한다. 이 반응 혼합물을 120℃에서 1시간 동안 가열한 다음, 실온으로 냉각시키고, 물로 희석한다. 고체는 여과에 의해 수집하고, 플래시 크로마토그래피(SiO2, 헵탄 중 0-100% EtOAc)로 정제하여 표제 화합물(19mg)을 수득한다
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 13 내지 15, 표 1
실시예 18 내지 22, 표 1
실시예 25 내지 29, 표 1
실시예 33 내지 34, 표 1
실시예 37, 표 1
실시예 58, 표 1
실시예 67 내지 69, 표 1
실시예 119, 표 1
5-(4-{(R)-1-
사이클로프로필
-1-[5-(1H-
피라졸
-4-일)-[1,2,4]
옥사디아졸
-3-일]-에틸}-
페닐
)-피리미딘-2-
일아민(실시예 48, 표 1)의
합성

THF(200mL) 중 1H-피라졸-4-카복실산(7.1g, 0.063mol)의 현탁액에 실온에서 1,1'-카보닐디이미다졸(10.2g, 0.063mol)을 가한다. 혼합물을 50℃에서 30분 동안 교반한다. THF(100mL) 중 I-29(12.5g, 0.042mol)의 현탁액을 상기 혼합물에 가하고, 생성된 혼합물은 환류하에 24시간 동안 가열한다. 혼합물을 냉각시키고, 고체는 여과에 의해 수집한다. 이어서, 고체를 실온에서 AcOH(150mL)에 현탁시킨다. 혼합물을 2시간 동안 90℃로 가열한다. 용액을 냉각시키고, 진공하에 농축시킨다. 잔사를 EtOAc(100mL)에 용해시키고, 용액은 H2O(200mL) 및 포화 NaHCO3 용액(200mL)으로 세척한다. 유기층을 농축시켜 표제 화합물(14.7g, 0.040mol)을 수득한다.
방법 6
4-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-1,2,4-
옥사
디아졸-5-일)-피페리딘-1-
카복실산
3급-부틸 에스테르(
실시예
109, 표 1) 및 5-(4-{3-메틸-2-[5-(피페리딘-4-일)-1,2,4-
옥사디아졸
-3-일]부탄-2-일}
페닐
)피리미딘-2-아민(
실시예 110, 표 1)의
합성

THF(1mL) 중 N-Boc-이소니포코트산(115㎎, 0.50mmol)의 현탁액에 카보닐디이미다졸(81㎎, 0.50mmol)을 가한다. 혼합물을 55℃에서 20분 동안 가열한 다음, I-21(100㎎, 0.33mmol)로 처리한다. 반응 혼합물은 55℃에서 17시간 동안 가열한 다음, 150℃인 마이크로파에서 20분 동안 가열한다. 혼합물을 실온으로 냉각시킨 다음, 직접 플래시 크로마토그래피(SiO2, 헵탄 중 15-100% EtOAc)로 정제하여 실시예 109(89mg)를 수득한다
실시예 109(83㎎, 0.17mmol)를 CH2Cl2(1mL)에 용해시키고, 1,4-디옥산 중 HCl의 용액(4.0M, 0.4mL)으로 처리한다. 혼합물을 3.5시간 동안 실온에서 교반한 다음, 생성된 고체를 여과하고, CH2Cl2로 세척하고, 수집하고, 건조시켜 표제 화합물(63㎎)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 111 내지 114, 표 1
방법 7
5-(4-{3-
메틸
-2-[5-(1H-
피라졸
-4-일)-1,2,4-
옥사디아졸
-3-일]부탄-2-일}
페닐
)피리미딘-2-아민(
실시예
146, 표 1)의 합성

DMF(1mL) 중 I-47(70mg, 0.19mmol), 2-아미노피리미딘-5-보론산 피나콜 에스테르(51mg, 0.23mmol) 및 2M 수성 Na2CO3(0.2mL)의 혼합물을 N2하에 5분 동안 탈기시킨다. 이 혼합물에 PdCl2(PPh3)2(14mg, 0.02mmol)를 가한다. 혼합물을 80℃에서 18시간 동안 교반한 다음, EtOAc와 물 사이에 분배시킨다. 유기층을 물로 세척하고, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사는 분취용 HPLC로 정제하여 표제 화합물(13mg)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 125, 표 1 - I-38과 -아미노피리미딘-5-보론산, PdCl2dppf(0.05eq) 및 dppf(0.05eq)의 반응시, 단지 실시예 125가 실시예 128 대신에 형성된다
실시예 126 내지 133, 표 1
실시예 139, 표 1
실시예 143, 표 1
실시예 146 내지 157, 표 1
방법 8
5-[4-(3-
메틸
-2-{5-[6-(
트리플루오로메틸
)피리딘-3-일]-1,2,4-
옥사디아졸
-3-일}부탄-2-일)
페닐
]피리미딘-2-아민(
실시예
136, 표 1)의 합성
압력 관에서 에탄올:톨루엔(4:1, 2ml) 중 I-33(156mg, 0.35mmol), 2-아미노피리미딘-5-보론산(58mg, 0.42mmol) 및 2M 수성 Na2CO3(0.53mL)의 현탁액에 [1,1'-비스(디페닐포스피노)-페로센]디클로로 팔라듐(II)(25mg, 0.030mmol) 및 1,1'-비스(디페닐포스피노)-페로센(15mg, 0.02mmol)을 가한다. 반응 혼합물을 90℃에서 2시간 동안 교반한다. 반응 혼합물은 셀라이트 패드를 통해 여과하고, EtOAc 및 CH2Cl2로 세척한다. 수집한 여액은 진공하에 농축시킨다. 분취용 HPLC로 정제하여 표제 화합물(72mg)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 135 내지 138, 표 1
실시예 140 내지 141, 표 1
방법 9
2-[4-(3-{1-[4-(2-
아미노피리미딘
-5-일)
페닐
]-1-
사이클로프로필에틸
}-1,2,4-옥사디아졸-5-일)-1H-
피라졸
-1-일]-N,N-
디메틸아세트아미드(실시예 59, 표 1)의
합성

DMF(1mL) 중 실시예 21(350mg, 0.94mmol)의 용액에 K2CO3(260mg, 1.9mmol) 및 2-클로로-N,N-디메틸아세트아미드(0.19mL, 1.9mmol)를 가한다. 반응 혼합물을 실온에서 20시간 동안 교반한 다음, 분취용 HPLC(0.1% TFA를 함유하는 물 중 10-60% CH3CN)로 직접 정제하여 표제 화합물(200mg)을 수득한다.
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

a) 반응 혼합물을 50℃에서 가열한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 30, 표 1
실시예 43, 표 1
실시예 60, 표 1
실시예 75 내지 76, 표 1
실시예 79, 표 1
실시예 97 내지 99, 표 1
실시예 106, 표 1
실시예 271, 표 1 - 반응은 상응하는 브로마이드로부터 출발하여 130℃에서 48시간 동안 수행한다
2--[4-(3-{(R)-1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드(실시예 115, 표 1)의
합성
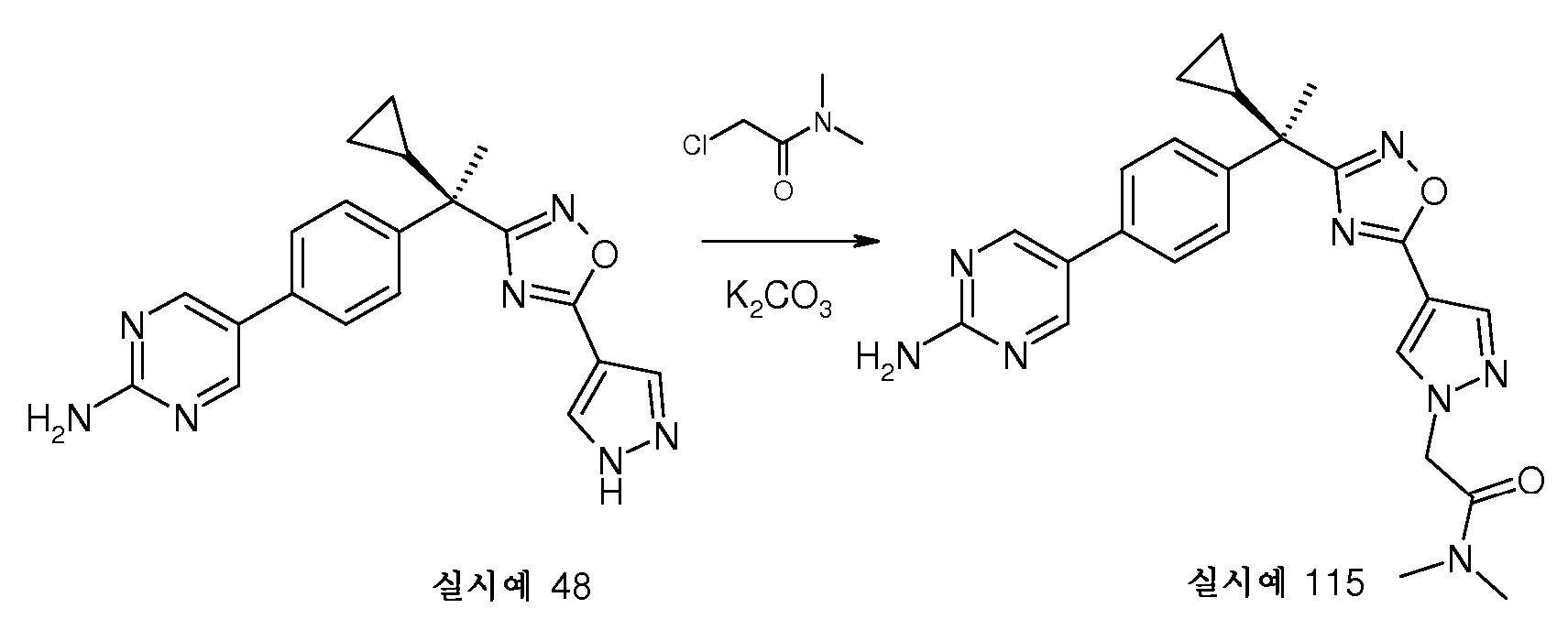
DMF(150mL) 중 실시예 48(14.7g, 0.040mol)의 용액에 실온에서 2-클로로-N,N-디메틸아세트아미드(6.1mL, 0.059mol) 및 K2CO3(10.9g, 0.079mol)를 가한다. 혼합물을 동일한 온도에서 2시간 동안 교반한다. 물(100mL)을 가하고, 혼합물은 EtOAc(200mL)로 추출한다. 합한 유기층을 MgSO4(20g)으로 건조시키고 여과한다. 여액을 농축시키고, 나머지 고체는 소량의 아세토니트릴(30mL)에 10분 동안 재현탁시킨다. 고체는 여과에 의해 수집하고, 냉각된 아세토니트릴로 세척한다. 생성된 고체는 진공하에 건조시키고, 이는 표제 화합물(10g)인 것으로 확인되었다.
표제 화합물은, 경우에 따라, 에탄올, 메탄올 또는 THF로부터 재결정화에 의해 추가로 정제한다. 대안적으로, 표제 화합물은 유기 염기를 에탄올 또는 이소프로필 알콜에 용해시킨 다음, 수성 염산을 용액에 부가함으로써, 표제 화합물을 이의 하이드로클로라이드 염으로 전환시킨다.
방법 10
2-[(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사디
아졸-5-일)아미노]에탄올(
실시예
80, 표 1)의 합성

톨루엔(35mL) 중 I-21(900mg, 3.0mmol)의 용액에 트리클로로아세트산 무수물(0.69mL, 3.6mmol)을 가한다. 반응 혼합물을 환류하에 2.5시간 동안 가열한 다음, 실온으로 냉각시킨다. 혼합물을 EtOAc로 희석하고, 포화 수성 NaHCO3로 세척하고, Na2SO4로 건조시키고, 여과한 다음 농축시켜, I-101(1.25g)을 수득한다; m/z 426.31/428.22 [M/M+2H].
DMSO(1mL) 중 I-101(80mg, 0.19mmol) 및 KOH(19mg, 0.28mmol)의 용액에 에탄올아민(20㎕, 0.28mmol)을 가한다. 반응 혼합물을 실온에서 1시간 동안 교반한 다음, 물로 처리한다. 혼합물을 EtOAc로 추출하고, 염수로 세척한 다음, Na2SO4로 건조시키고, 여과하고, 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-10% MeOH)로 정제하여 표제 화합물(45mg)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 81 내지 83, 표 1
실시예 84, 표 1 - 실시예 83을 형성하기 위하여 수행되는 반응으로부터 분리된 부산물
실시예 86, 표 1 - 의도하는 아민 유도체는 분리되지 않고, 아미드 부산물이 유일하게 분리되는 물질이다
실시예 87, 표 1
실시예 89, 표 1
실시예 90, 표 1 - 실시예 89를 형성하기 위하여 수행되는 반응으로부터 분리된 부산물
실시예 91, 표 1 - 의도하는 아민 유도체는 분리되지 않고, 아미드 부산물이 유일하게 분리되는 물질이다
실시예 134, 표 1
실시예 171, 표 1
방법 11
1-{[5-(3-{1-[4-(2-
아미노피리미딘
-5-일)
페닐
]-1-
사이클로프로필에틸
}-1,2,4-옥
사
디아졸-5-일)피라진-2-일]아미노}-2-
메틸프로판
-2-올(
실시예
44, 표 1)의 합성

NMP(3mL) 중 5-클로로-피라진-2-카복실산(490mg, 3.1mmol)의 용액에 카보닐디이미다졸(500mg, 3.1mmol)을 가한다. 혼합물을 0.5시간 동안 50℃에서 교반한 다음, I-23(830mg, 2.8mmol)으로 처리하고, 2시간 동안 110℃에서 가열한다. 혼합물을 실온으로 냉각시키고, 물로 처리하여, 18시간 동안 교반한다. 생성된 고체를 여과하고, 건조시키고 수집하여, I-102(1.0g)를 수득한다. 1H NMR (DMSO-d6) d ppm 9.40 (1H, s), 9.20 (1H, s), 8.75 (1H, s), 8.55 (2H, s), 8.10 (1H, s), 7.60 (2H, d), 7.40 (2H, d), 7.20 (1H, s), 6.75 (2H, s), 1.65-1.75 (1H, m), 1.55 (3H, s), 0.3-0.75 (4H, m).
I-102(300mg, 0.66mmol)를 1-아미노-2-메틸-프로판-2-올(1.5mL)에 용해시키고, 80℃에서 4시간 동안 가열한다. 혼합물을 실온으로 냉각시키고, 물로 처리한다. 생성된 고체를 여과하고 수집한 다음, CH3CN으로부터 재결정화로 추가로 정제하여 표제 화합물(155㎎)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 42, 표 1
실시예 45, 표 1
실시예 35 내지 36, 표 1
실시예 73 내지 74, 표 1
실시예 162 내지 163, 표 1
방법 12
5-{4-[1-
사이클로프로필
-1-(5-{6-[(2-
메톡시에틸
)아미노]피리딘-3-일}-1,2,4-옥
사
디아졸-3-일)에틸]
페닐
}피리미딘-2-아민(
실시예
38, 표 1)의 합성

NMP(7mL) 중 6-클로로니코틴산(500mg, 3.2mmol)의 용액에 카보닐디이미다졸(520mg, 2.9mmol)을 가한다. 혼합물을 실온에서 1시간 동안 교반한 다음, I-23(860mg, 2.8mmol)으로 처리하고, 130℃에서 2시간 동안 가열한다. 혼합물을 실온으로 냉각시키고, 물로 처리한다. 생성된 고체를 여과하고, 건조시키고 수집하여, I-103(350㎎)을 수득한다; m/z 419.33 [M+H]
I-103(150mg, 0.36mmol)을 2-메톡시에틸아민(0.5mL)에 용해시키고, 80℃에서 2시간 동안 가열한다. 혼합물을 실온으로 냉각시키고, 물로 처리하여 잔사를 수득한다. 물을 경사분리하고, 잔사는 분취용 HPLC(0.1% TFA를 함유하는 물 중 10-60% CH3CN)로 정제하여 표제 화합물(98㎎)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 39 내지 41, 표 1
실시예 70 내지 72, 표 1
방법 13
1-[4-(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사
디아졸-5-일)-1H-
피라졸
-1-일]-2-
메틸프로판
-2-올(
실시예
159, 표 1)의 합성
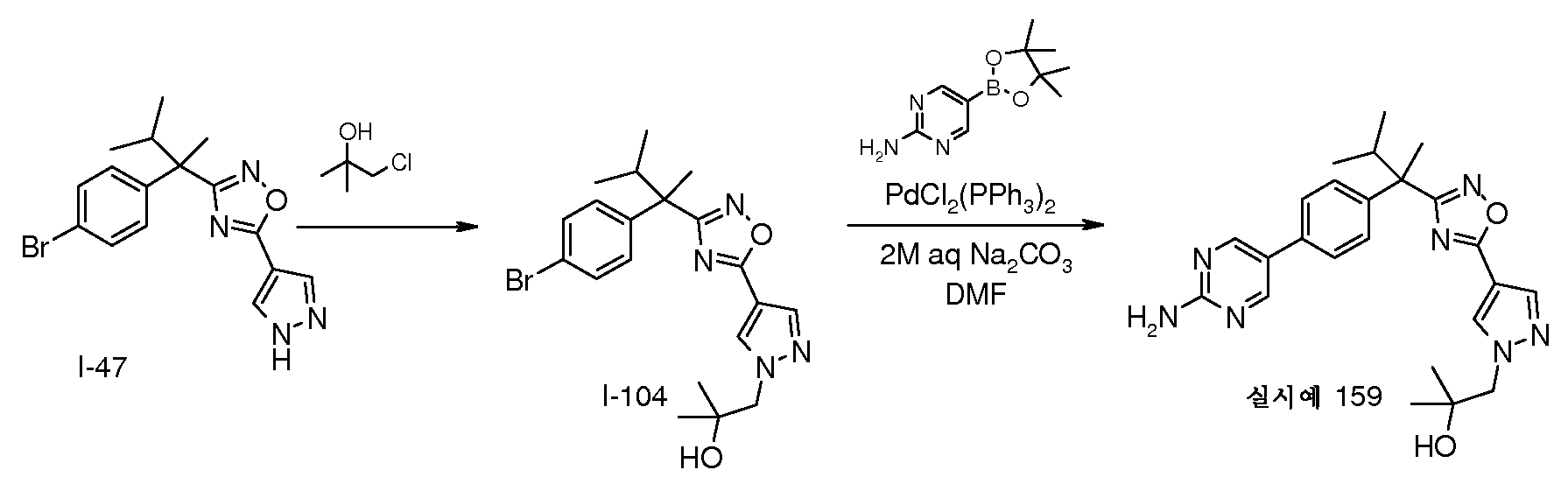
DMF(6mL) 중 I-47(336mg, 0.93mmol) 및 탄산칼륨(154mg, 1.12mmol)의 현탁액에 1-클로로-2-메틸-프로판-2-올(200㎕, 0.98mmol)을 가한다. 반응 혼합물을 80℃에서 16시간 동안 교반한 다음, 진공하에 농축시킨다. 잔사를 CH2Cl2로 추출하고, 포화 수성 NH4Cl로 세척하고, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시켜, I-104(365㎎)를 수득한다; m/z 434 [M+H]
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

DMF(4mL) 중 I-104(365mg, 0.84mmol), 2-아미노피리미딘-5-보론산 피나콜 에스테르(279mg, 1.26mmol) 및 2M 수성 Na2CO3(0.85mL)의 혼합물을 질소하에 5분 동안 탈기시킨다. 이 혼합물에 비스(트리페닐포스핀)팔라듐(II)클로라이드(59mg, 0.08mmol)를 가한다. 혼합물은 80℃에서 18시간 동안 교반한 다음, 진공하에 농축시킨다. 잔사는 CH2Cl2로 추출하고, 포화 수성 NaHCO3로 세척하고, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-5% MeOH)로 정제하여 표제 화합물(268㎎)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 142, 표 1
실시예 144, 표 1
실시예 158, 표 1
실시예 161, 표 1
방법 14
1-(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사디아
졸-5-일)
사이클로프로판카복실산
(
실시예
95, 표 1)의 합성

DMF(1mL) 중 I-21(189mg, 0.63mmol)의 용액에 DMF(1mL) 중 1-클로로카보닐-1-사이클로프로판카복실산 메틸 에스테르(113㎎, 0.69mmol)의 용액을 가한다. 반응 혼합물을 실온에서 15분 동안 교반한 다음, 120℃에서 2시간 동안 가열한다. 혼합물을 냉각시킨 다음, 물로 처리하고, EtOAc로 추출하고, 염수로 세척하고, Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-10% MeOH)로 정제하여 I-105(30㎎)를 수득한다; m/z 408 [M+H].
MeOH(0.5mL) 중 I-105(30mg, 0.074mmol)의 용액에 NaOH(4.0M, 90㎕)의 수용액을 가한다. 반응 혼합물을 실온에서 1시간 동안 교반한 다음, 진공하에 농축시킨다. 잔사를 EtOAc와 물 사이에 분배시킨 다음, 수성층은 1M 수성 HCl을 사용하여 pH 약 4로 산성화시킨다. 수성 혼합물은 EtOAc로 추출하고, 진공하에 농축시켜 잔사를 수득하고, 이를 분취용 HPLC로 정제하여 표제 화합물(20㎎)을 수득한다.
방법 15
1-(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사디아
졸-5-일)
사이클로헥산카복실산
(
실시예
145, 표 1)의 합성

DMF(2.5mL) 중 사이클로헥산-1,1-디카복실산 모노에틸 에스테르(142mg, 0.50mmol), HATU(199mg, 0.52mmol) 및 트리에틸아민(80㎕, 0.55mmol)의 혼합물을 5분 동안 교반한 다음, I-14(100mg, 0.50mmol)를 가한다. 혼합물을 실온에서 2시간에 이어서, 90℃에서 18시간 동안 교반한 다음, 진공하에 농축시킨다. 생성된 잔사는 1M HCl과 EtOAc 사이에 분배시킨다. 유기층을 염수로 세척하고, MgSO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 사이클로헥산 중 15% EtOAc)로 정제하여 I-106(251㎎)을 수득한다; m/z 449/451 [M/M+2H].
DMF(5mL) 중 I-106(224mg, 0.50mmol), 2-아미노피리미딘-5-보론산 피나콜 에스테르(121mg, 0.55mmol) 및 2M 수성 Na2CO3(1.9mL)의 혼합물을 질소하에 5분 동안 탈기시킨다. 이 혼합물에 비스(트리페닐포스핀)팔라듐(II)클로라이드(35mg, 0.05mmol)를 가한다. 혼합물은 80℃에서 1시간 동안 교반한 다음, 진공하에 농축시킨다. 잔사는 EtOAc로 추출하고, 포화 수성 NaHCO3로 세척하고, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 사이클로헥산 중 0-20% EtOAc)로 정제하여 I-107(182㎎)을 수득한다; m/z 464 [M+H].
1:1 THF:물(3mL) 중 I-107(175mg, 0.38mmol)의 용액에 LiOH-H2O(17mg, 0.40mmol)를 가한다. 반응 혼합물을 실온에서 2일 동안 교반한 다음, THF를 진공하에 제거한다. 수성 혼합물을 EtOAc로 세척한 다음, 포화 수성 NH4Cl에 의해 산성화시킨다. 수성 혼합물은 EtOAc로 추출하고, 염수로 세척하고, MgSO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 생성된 고체는 Et2O로 연마한 다음, 여과하고, 수집하고, 건조시켜 표제 화합물(88㎎)을 수득한다.
방법 16
3-[4-(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사
디아졸-5-일)-1H-
피라졸
-1-일]-2,2-
디메틸프로판산(실시예 164, 표 1)의
합성

DMF(8mL) 중 I-47(407mg, 1.13mmol) 및 3-클로로-2,2-디메틸 프로피온산 에틸 에스테르(557mg, 3.38mmol)의 혼합물에 Cs2CO3(734mg, 2.25mmol) 및 테트라부틸암모늄 요오다이드(832mg, 2.25mmol)를 가한다. 혼합물을 80℃에서 36시간 동안 교반한 다음, 진공하에 농축시킨다. 잔사는 CH2Cl2와 포화 수성 NaHCO3 사이에 분배시킨다. 유기층을 Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 사이클로헥산 중 10% EtOAc)로 정제하여 I-108(299㎎)을 수득한다; m/z 489/491 [M/M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

a) 반응은 DMF 중 K2CO3를 사용하여 8시간 동안 80℃에서 수행한다
DMF(6mL) 중 I-108(290mg, 0.46mmol), 2-아미노피리미딘-5-보론산 피나콜 에스테르(307mg, 1.39mmol) 및 2M 수성 Na2CO3(0.46mL)의 혼합물을 N2하에 5분 동안 탈기시킨다. 이 혼합물에 비스(트리페닐포스핀)팔라듐(II)클로라이드(65mg, 0.09mmol)를 가한다. 혼합물은 80℃에서 3시간 동안 교반한 다음, 진공하에 농축시킨다. 잔사는 EtOAc로 추출하고, 포화 수성 NaHCO3로 세척하고, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-1% MeOH)로 정제하여 I-109(171㎎)를 수득한다; m/z 504.90 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:
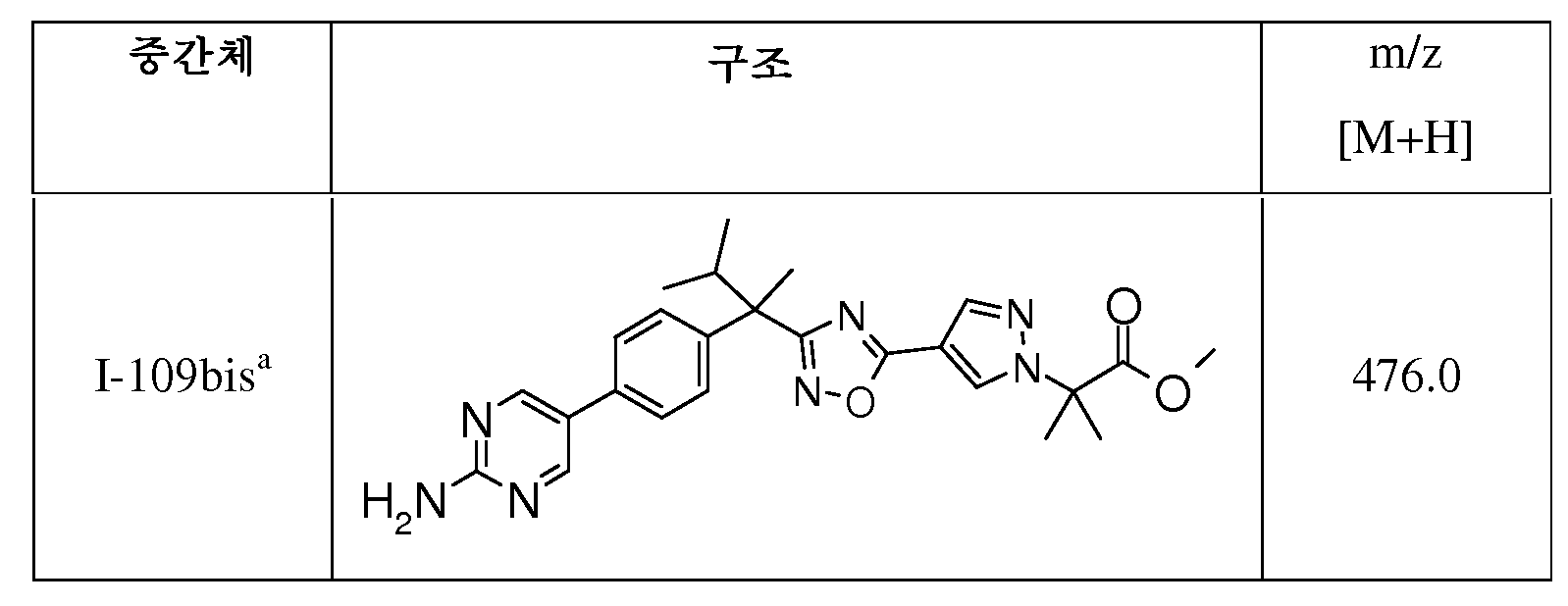
I-109(171mg, 0.282mmol), LiOH.H2O(12mg, 0.286mmol), 메탄올(2.5mL), THF(2.5mL) 및 물(1.3mL)의 혼합물을 40℃에서 8시간 동안 가열한 다음, 진공하에 농축시킨다. 잔사는 분취용 HPLC로 정제하여 표제 화합물(33mg)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 160, 표 1
방법 17
6-(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사디아
졸-5-일)-1H-
메틸피리딘
-2(1H)-온(
실시예
165, 표 1)의 합성

DMF(3mL) 중 2-카복실산 6-옥소-피리딘(161mg, 1.16mmol)의 용액에 카보닐디이미다졸(188mg, 1.16mmol)을 가한다. 혼합물을 50℃에서 20분 동안 교반한 다음, I-14(300mg, 1.05mmol)로 처리하고, 110℃에서 3시간 동안 교반한다. 혼합물을 진공하에 농축시키고, 잔사는 EtOAc로 추출하고, 포화 수성 NaHCO3로 세척하고, MgSO4로 건조시키고, 여과한 다음 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 2% MeOH)로 정제하여 I-110(223㎎)을 수득한다; m/z 389.95 [M+1].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

THF(3mL) 중 I-110(223mg, 0.41mmol)의 용액에 K2CO3(68mg, 0.49mmol) 및 MeI(30㎕, 0.49mmol)를 가한다. 혼합물을 40℃에서 20시간 동안 교반하고, 3시간 및 15시간에서 부가의 MeI(30㎕, 0.49mmol)로 처리한다. 혼합물을 포화 수성 암모니아 및 MeOH로 처리한 다음, 휘발성 물질은 진공하에 제거한다. 잔사는 EtOAc로 추출하고, 포화 수성 NaHCO3, 물 및 염수로 세척하고, MgSO4로 건조시키고, 여과한 다음 농축시킨다. 플래시 크로마토그래피(SiO2, 사이클로헥산 중 0-10% EtOAc)에 의한 조 생성물의 정제로 I-111(80㎎)이 수득된다; m/z 403.85 [M+1].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

DMF(2mL) 중 I-111(80mg, 0.19mmol), 2-아미노피리미딘-5-보론산 피나콜 에스테르(124mg, 0.56mmol) 및 2M 수성 Na2CO3(0.30mL)의 혼합물을 질소하에 5분 동안 탈기시킨다. 이 혼합물에 PdCl2(PPh3)2(26mg, 0.094mmol)를 가한다. 혼합물은 80℃에서 3시간 동안 교반한 다음, 진공하에 농축시킨다. 잔사는 EtOAc로 추출하고, 포화 수성 NaHCO3로 세척하고, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 0-1% MeOH)로 정제하여 표제 화합물(30㎎)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 166 내지 167, 표 1 - 반응은 I-110bis 및 I-111bis의 혼합물로부터 출발하여 수행한다. 실리카 겔 칼럼 크로마토그래피로 실시예 166 및 167이 수득된다.
방법 18
2-[4-(3-{(1R)-1-[4-(2-
아미노피리미딘
-5-일)
페닐
]-1-
사이클로프로필에틸
}-1,2,4-옥
사
디아졸-5-일)-1H-
피라졸
-1-일]-N,N-
디메틸아세트아미드
(
실시예
115) 및 2-[4-(3-(1S)-1-[4-(2-아미노피리미딘-5-일)
페닐
]-1-
사이클로프로필에틸
}-1,2,4-
옥
사디아졸-5-일)-1H-
피라졸
-1-일]-N,N-
디메틸아세트아미드(실시예 116, 표 1)의
합성

에난티오머 115 및 116은 Chiralpak® AD-H(제조원: Chiral Technologies, Inc., Exton, PA) 반분취용(250 x 20 mm) HPLC 칼럼 (0.1% 디에틸아민을 함유하는 헵탄 중 95% EtOH로 용출시킴) 상에서 실시예 59(100mg)의 분할에 의해 제조한다. 체류시간이 약 35분인 보다 빨리 용출되는 에난티오머 115 및 체류시간이 약 73분인 보다 느리게 용출되는 에난티오머 116. 용출물은 농축시켜 실시예 115(32mg) 및 실시예 116(27mg)을 제공한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 23 내지 24, 표 1
실시예 31 내지 32, 표 1 - 0.1% 디에틸아민/헵탄을 포함하는 70% EtOH로 용출시킴
실시예 61 내지 62, 표 1
실시예 65 내지 66, 표 1
실시예 77 내지 78, 표 1
실시예 102 내지 103, 표 1
실시예 104 내지 105, 표 1 - 40℃에서 8ml/min으로 헵탄 중 95% EtOH + 0.05% 디에틸아민
실시예 107 내지 108, 표 1
실시예 115 내지 116, 표 1 - 55ml/min으로 헵탄 중 95% EtOH + 0.05% 디에틸아민
실시예 121 내지 122, 표 1 - 55ml/min으로 헵탄 중 95% EtOH + 0.4% 디에틸아민
실시예 123 내지 124, 표 1
실시예 227 내지 228, 표 1
실시예 230 내지 231, 표 1
실시예 233 내지 234, 표 1
실시예 254 내지 255, 표 1
실시예 257 내지 258, 표 1
실시예 282 내지 283, 표 1
방법 19
5-(4-{(2R)-3-
메틸
-2-[5-(1-
메틸
)-1H-
피라졸
-4-일)-1,2,4-
옥사디아졸
-3-일]부탄-2-일}
페닐
)피리미딘-2-아민(
실시예
46) 및 5-(4-{(2S)-3-
메틸
-2-[5-(1-
메틸
)-1H-피라졸-4-일)-1,2,4-
옥사디아졸
-3-일]부탄-2-일}
페닐
)피리미딘-2-아민(
실시예
47, 표 1)의 제조

에난티오머 46 및 47은 250bar에서 Chiralpak® AD-H(제조원: Chiral Technologies, Inc., Exton, PA) 반분취용(250 x 30 mm) HPLC 칼럼 (CO2 중 70% MeOH로 용출시킴) 상에서 실시예 15(50mg)의 분할에 의해 제조한다. 체류시간이 약 5분인 보다 빨리 용출되는 에난티오머 46(Chiralpak® AD-H 분석 HPLC 칼럼 4.6 x 100 mm) 및 체류시간이 약 13분인 보다 느리게 용출되는 에난티오머 47. 용출물은 농축시켜 실시예 46(12mg) 및 실시예 47(15mg)을 제공한다.
하기 화합물은 유사한 방법으로 분할한다:
실시예 16 내지 17, 표 1
실시예 48 내지 49, 표 1
방법 20
2-{[5-(3-{(1R)-1-[4-(2-
아미노피리미딘
-5-일)
페닐
]-1-
사이클로프로필에틸
}-1,2,4-옥
사
디아졸-5-일)피라진-2-일]아미노}-2-
메틸프로판
-1-올(
실시예
56, 표 1) 및 2-{[5-(3-{(1S)-1-[4-(2-
아미노피리미딘
-5-일)
페닐
]-1-
사이클로프로필에틸
}-1,2,4-옥
사
디아졸-5-일)피라진-2-일]아미노}-2-
메틸프로판
-1-올(
실시예
57, 표 1)의 제조
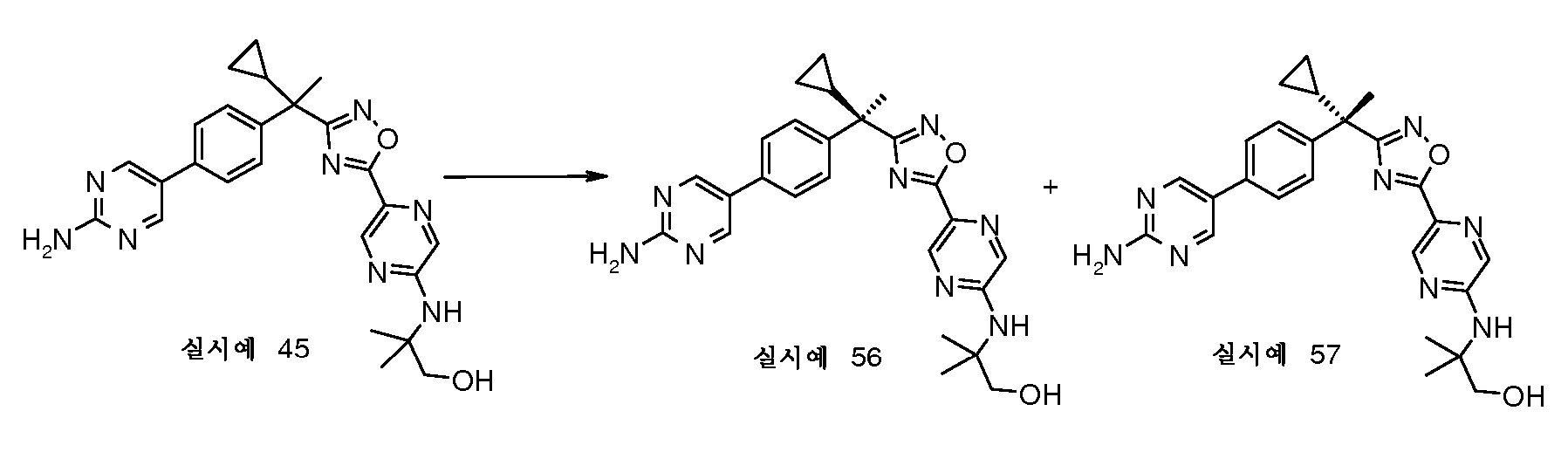
에난티오머 56 및 57은 250bar에서 RegisPak(제조원: Chiral Technologies, Inc., Exton, PA) 반분취용(250 x 30 mm) HPLC 칼럼 (CO2 중 70% MeOH로 용출시킴) 상에서 실시예 45(89mg)의 분할에 의해 제조한다. 체류시간이 약 5분인 보다 빨리 용출되는 에난티오머 56(Chiralpak® AD-H 분석 HPLC 칼럼 4.6 x 100 mm) 및 체류시간이 약 13분인 보다 느리게 용출되는 에난티오머 57. 용출물은 농축시켜 실시예 56(12mg) 및 실시예 57(15mg)을 제공한다.
하기 화합물은 유사한 방법으로 분할한다:
실시예 50 내지 51, 표 1 - CO2 중 55% MeOH로 용출시킴
실시예 52 내지 53, 표 1 - CO2 중 45% 3/1/0.1 MeOH/이소프로판올/이소프로필아민으로 용출시킴
실시예 54 내지 55, 표 1 - CO2 중 55% EtOH로 용출시킴
실시예 56 내지 57, 표 1 - 45% 1/1 메탄올/이소프로판올로 용출시킴
실시예 100 내지 101, 표 1 - 150bar에서 0.5% 이소프로필아민을 포함하는 1:1 메탄올:이소프로판올의 40% 공용매로 용출시킴
실시예 63 내지 64, 표 1
방법 21
5-(4-{3-
메틸
-2-[5-(4-
메틸피페라진
-1-일)-1,2,4-
옥사디아졸
-3-일]부탄-2-일}
페닐
)피리미딘-2-아민(
실시예
96, 표 1)의 합성

I-14(1.029g, 3.61mmol) 및 카보닐디이미다졸(702mg, 7.33mmol)의 혼합물에 아세토니트릴(20mL)을 가한다. 반응 혼합물을 75℃에서 18시간 동안 가열한다. 이 시간 후, 반응 혼합물을 진공하에 농축시키고, 플래시 크로마토그래피(SiO2, 헵탄 중 12-100% EtOAc)로 정제하여 I-112(373mg)를 수득한다; m/z 311.2/313.2 [M/M+2H].
피리딘(0.5mL) 중 I-112(163mg, 0.523mmol)의 용액에 POCl3(0.479mL, 5.23mmol)을 가한다. 반응 혼합물을 90℃에서 18시간 동안 가열한다. 반응 혼합물을 실온으로 냉각시키고, 주의해서 빙수로 부은 다음, EtOAc로 2회 추출한다. 유기층을 합하고, 염수로 세척하고, Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 헵탄 중 12-100% EtOAc)로 정제하여 I-113(59mg)을 수득한다; 1H-NMR (DMSO-d6) δ ppm 7.50 (2H, d), 7.35 (2H, d), 2.62 (1H, m), 1.60 (3H, s), 0.83 (3H, d), 0.6 (3H, d).
DMSO(1mL) 중 I-113(44mg, 0.133mmol)의 용액에 1-메틸피페라진(0.148mL, 1.33mmol)을 가하고, 반응 혼합물을 실온에서 1.5시간 동안 교반한다. 반응 혼합물은 물로 급냉시키고, EtOAc로 2회 추출한다. 유기층을 합하고, 물에 이어서, 염수로 세척한 다음, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 10-100% MeOH)로 정제하여 I-114(45mg)를 수득한다; m/z 393.0/395.0 [M/M+2H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:




a) 아민 1.2당량 및 디이소프로필에틸아민 1.2당량이 사용된다.
b) 아민 1.2당량(유리 염기 또는 하이드로클로라이드 염으로서) 및 디이소프로필에틸아민 2.5당량이 사용된다.
c) 디하이드로클로라이드 염으로서 아민 1.2당량 및 디이소프로필에틸아민 5당량이 사용된다.
바이알에서 I-114(45.000mg, 0.114mmol), 2-아미노피리미딘-5-보론산 피나콜 에스테르(30.286mg, 0.137mmol) 및 테트라키스(트리페닐포스핀)팔라듐(0)(13.173mg, 0.011mmol)의 혼합물을 배출시키고, Ar에 의해 3회 역충전시킨다. 이어서, THF(1mL) 및 포화 Na2CO3 수용액을 가하고, 혼합물을 65℃에서 18시간 동안 가열한다. 이 시간 후, 반응 혼합물을 물로 급냉시키고, EtOAc로 2회 추출한다. 유기층을 합하고, 염수로 세척한 다음, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 헵탄 중 12-100% EtOAc)로 정제하여 실시예 96(12㎎)을 수득한다.
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

a) 반응은 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 168, 표 1
실시예 169, 표 1 - 마지막 단계는 1시간 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 170, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 186, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 192, 표 1
실시예 198 - 202, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 208 내지 209, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 212 내지 213, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 218, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 235, 표 1 - 마지막 단계는 2시간 동안 100℃의 마이크로파 오븐에서 수행한다.
실시예 236 내지 237, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 260, 표 1
실시예 261 내지 262, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 264, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 266, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
실시예 269 내지 270, 표 1 - 마지막 단계는 45분 동안 110℃의 마이크로파 오븐에서 수행한다.
방법 22
4-(3-{2-[4-(2-
아미노피리미딘
-5-일)
페닐
]-3-
메틸부탄
-2-일}-1,2,4-
옥사디아
졸-5-일)-2,2-
디메틸부탄산(실시예 85, 표 1)의
합성

DMF(1mL) 중 I-21(100mg, 0.334mmol)의 용액에 2,2-디메틸글루타르산 무수물(52mg, 0.367mmol)을 가한다. 반응 혼합물을 120℃에서 2.5시간 동안 가열한다. 이 시간 후, 반응 혼합물을 물로 급냉시키고, EtOAc로 2회 추출한다. 유기층을 합하고, 물에 이어서 염수로 세척한 다음, Na2SO4로 건조시키고, 여과하고, 진공하에 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, CH2Cl2 중 10-100% MeOH)로 정제한 다음, 가열된 MeOH로 연마하여 실시예 85(40㎎)를 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 92 내지 94, 표 1
방법 23
5-(4-{(R)-1-
사이클로프로필
-1-[5-(3,4,5,6-
테트라하이드로
-2H-[1.2']
비피라
지닐-5'-일)-[1,2,4]
옥사디아졸
-3-일]-에틸}-
페닐
)-피라진-2-
일아민(실시예 195,
표 1)의 합성
단계 1:
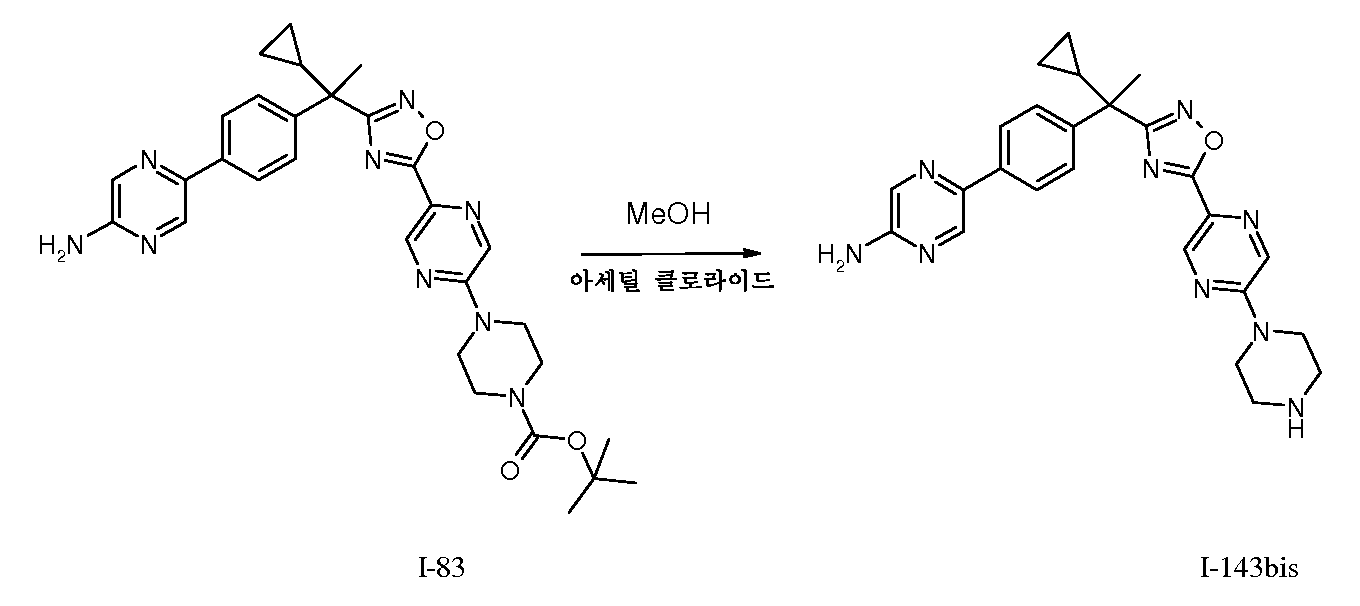
냉각된(0℃) 메탄올(20mL)에 아세틸 클로라이드 1mL를 가한다(적가). 완전히 부가한 후, I-83(350mg, 0.61mmol)을 메탄올 용액(5mL)으로서 가한다. 서서히 RT로 가온하고, 밤새 교반한다. 이 시간 후, 반응물은 7N 암모니아를 사용하여 염기성화시키고, 농축하여 건조시킨다. 나머지 잔사를 플래시 크로마토그래피(SiO2, 0-10% MeOH/DCM)를 통해 정제하여 I-135를 수득한다.
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 214, 표 1 - 조 생성물은 플래시 크로마토그래피(실리카 겔, 0-100% MeOH/DCM)를 통해 정제한다
실시예 267, 표 1
실시예 268, 표 1 - 조 생성물을 농축하여 건조시키고, 조 생성물을 플래시 크로마토그래피(실리카 겔, 0.5% NH4OH를 포함하는 0-10% MeOH/DCM)를 통해 정제한다
단계 2:

I-135의 키랄 분할은 ChiralPak® AD-H 칼럼(3.0 x 25.0 cm, 제조원: Chiral Technologies, West Chester PA)을 사용하여 수행한다. 150bar에서 1% 이소프로필아민을 포함하는 메탄올/IPA(1:3)로 용출시켜 실시예 195(6mg)를 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 196, 표 1 - 키랄 분할은 125psi에서 수행한다
실시예 210, 표 1 - 키랄 분할은 용출제 중 이소프로필아민의 사용없이, 125psi에서 수행한다
방법 24
1-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-[1,2,4]
옥
사디아졸-5-일)-2-
메틸
-프로판-2-올(
실시예
225, 표 1)의 합성

THF(10mL) 중 R-13(118mg, 1.0mmol)의 현탁액에 실온에서 1,1'-카보닐디이미다졸(162mg, 1.0mmol)을 가한다. 혼합물을 50℃에서 30분 동안 교반한다. 이 시간 후, I-21(200mg, 0.67mmol)을 가하고, 생성된 혼합물은 환류하에 3시간 동안 가열한다. 이 시간 후, 반응물을 RT로 냉각시키고, HOAc(1ml)로 처리하여, 80℃로 가온한다. 3일 동안 교반한 후, 혼합물을 RT로 냉각시키고, 농축시킨다. 나머지 잔사를 플래시 크로마토그래피(실리카 겔, 0-8% MeOH/DCM)를 통해 정제하여 표제 화합물(80mg)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 229, 표 1 - 반응 혼합물에 AcOH를 가하지 않음
실시예 232, 표 1 - 반응 혼합물에 AcOH를 가하지 않음
방법 25
5-(4-{1-
사이클로프로필
-1-[5-(3-
옥세탄
-3-일-3H-
이미다졸
-4-일)-[1,2,4]
옥
사디아졸-3-일]-에틸}-
페닐
)-피리미딘-2-
일아민(실시예 206, 표 1)의
합성 및 5-(4-{1-사
이클로
프로필-1-[5-(1-
옥세탄
-3-일-1H-
이미다졸
-4-일)-[1,2,4]
옥사디아졸
-3-일]-에틸}-
페닐
)-피리미딘-2-
일아민(실시예 207, 표 1)의
합성

THF(10mL) 중 1-트리틸-1H-이미다졸-4-카복실산(893mg, 2.5mmol)의 현탁액에 실온에서 1,1'-카보닐디이미다졸(409mg, 2.5mmol)을 가한다. 혼합물을 50℃에서 30분 동안 교반한다. THF(5mL) 중 I-23(500mg, 1.7mmol)의 현탁액을 상기 혼합물에 가하고, 생성된 혼합물은 2시간 동안 마이크로파 반응기에서 130℃에서 가열한다. 혼합물을 냉각시키고, 진공하에 농축시킨다. 잔사를 H2O(10mL) 및 EtOAc(20ml)로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 I-144(150mg)를 수득한다; m/z 374 [M-트리틸 그룹].
CH2Cl2(10ml) 중 I-144(80mg)의 용액에 실온에서 TFA(0.015mL, 0.19mmol)를 가한다. 용액을 동일한 온도에서 24시간 동안 교반한다. 용액을 진공하에 농축시켜 I-145(48mg)를 수득한다; m/z 374 [M+H].
환저 플라스크에 DMF(10mL) 중 I-145(100mg, 0.27mmol), 3-요오도옥세탄(98mg, 0.54mmol) 및 K2CO3(111mg, 0.8mmol)를 가한다. 반응 혼합물을 12시간 동안 80℃에서 교반한다. 반응물을 냉각시키고, 물(10mL)을 가한다. 용액을 EtOAc(20ml) 및 H2O(10mL)로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 용출제로서 TBME 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 표제 화합물(실시예 206: 8mg; 실시예 207: 10mg)을 수득한다.
방법 26
2-[4-(3-{1-[4-(2-아미노-4-
플루오로
-피리미딘-5-일)-
페닐
]-1-
사이클로프로
필에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드(실시예 275, 표 1)의
합성

DMF(10mL) 중 I-69(300mg, 0.6mmol)의 용액에 I-87(140mg, 0.7mmol), 테트라키스(트리페닐포스핀)팔라듐(0)(70mg, 0.06mmol) 및 2M Na2CO3(1.5mL, 3.0mmol)를 가한다. 혼합물을 마이크로파 반응기에서 1시간 동안 100℃로 가열한다. 혼합물을 냉각시키고, H2O(20mL) 및 EtOAc(30ml)로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 표제 화합물(200mg)을 수득한다
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 175, 표 1 - 반응은 120℃에서 수행한다
실시예 176 내지 180, 표 1 - 반응은 120℃에서 수행한다
실시예 184 내지 185, 표 1 - 반응은 120℃에서 수행한다
실시예 187 내지 189, 표 1 - 반응은 120℃에서 수행한다
실시예 191, 표 1 - 반응은 120℃에서 수행한다
실시예 239 내지 243, 표 1 - 반응은 오일욕에서 100℃에서 6시간 동안 수행한다
실시예 244 내지 246, 표 1
실시예 247, 표 1 - 반응은 오일욕에서 100℃에서 6시간 동안 수행한다
실시예 249 내지 250, 표 1 - 반응은 오일욕에서 100℃에서 6시간 동안 수행한다
실시예 251, 표 1 - 반응은 85℃에서 밤새 수행한다
실시예 253, 표 1 - 반응은 오일욕에서 100℃에서 6시간 동안 수행한다
실시예 256, 표 1 - 반응은 오일욕에서 100℃에서 6시간 동안 수행한다
실시예 265, 표 1 - 반응은 오일욕에서 100℃에서 6시간 동안 수행한다
실시예 273, 표 1 - 반응은 오일욕에서 100℃에서 6시간 동안 수행한다
실시예 275, 표 1
실시예 279, 표 1
실시예 280 내지 281, 표 1 - 반응은 80℃에서 48시간 동안 수행한다
실시예 284 내지 286, 표 1
실시예 289 내지 291, 표 1
실시예 292, 표 1 - 반응은 85℃에서 16시간 동안 수행한다
실시예 293 내지 294, 표 1
실시예 298, 표 1
방법 27
2-[4-(3-{(R)-1-[4-(6-아미노-피리딘-3-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드
(
실시예
172, 표 1)
마이크로파 바이알에 DMF(2mL) 중 I-63(100mg, 0.225mmol)을 가한 다음, 2-아미노피리딘-5-보론산 피나콜 에스테르(55mg, 0.25mmol), 테트라키스(트리페닐포스핀)팔라듐(0)(26mg, 0.023mmol) 및 2M 수성 Na2CO3(0.4ml, 0.8mmol)를 가한다. 반응 혼합물을 마이크로파 반응기에서 1시간 동안 120℃에서 교반한다. 잔사를 EtOAc로 희석하고, 물 및 염수로 세척하고, 무수 Na2SO4로 건조시키고 농축시킨다. 잔사를 플래시 크로마토그래피(SiO2, 0-5% MeOH/CH2Cl2)로 정제하여 표제 화합물(39mg)을 수득한다
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 173 내지 174, 표 1
실시예 181, 표 1
실시예 183, 표 1
실시예 263, 표 1 - 반응은 80℃에서 밤새 수행한다
방법 28
5-(4-{1,2-디메틸-1-[5-(4-
메틸아미노
-피페리딘-1-일)-[1,2,4]
옥사디아졸
-3-일]-프로필}-
페닐
)-피리미딘-2-
일아민(실시예 217, 표 1)의
합성
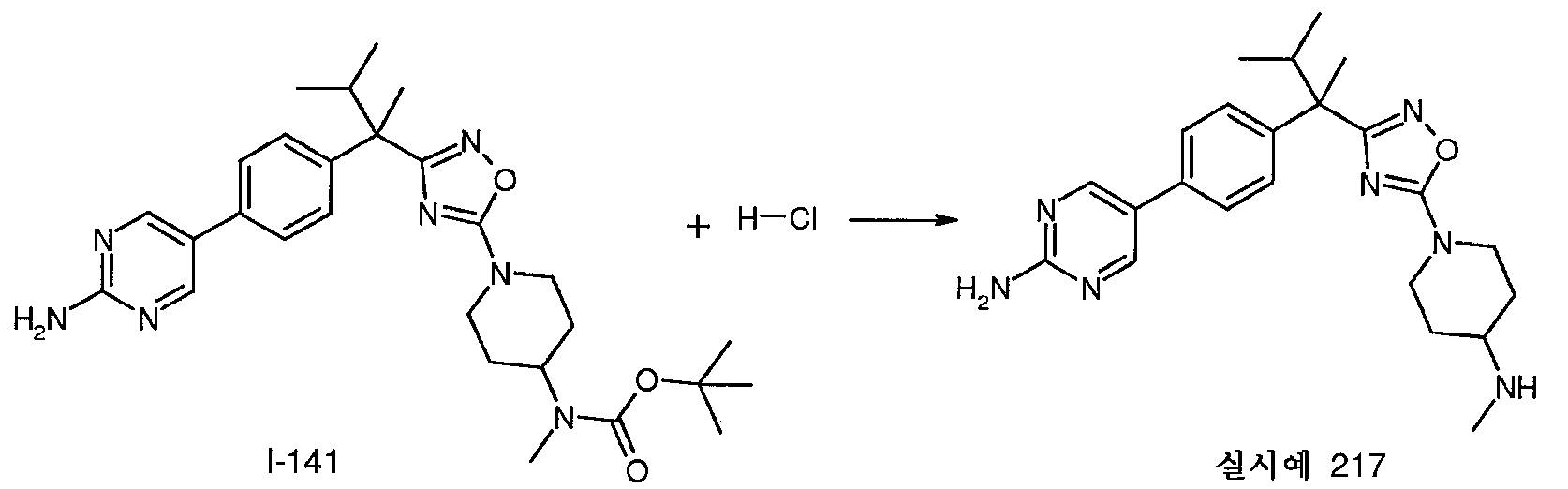
I-141(138mg, 0.265mmol)을 DCM(2mL)에 용해시키고, 1,4-디옥산 중 4N HCl(0.663mL, 2.65mmol)을 가하여, 반응 혼합물을 실온에서 3시간 동안 교반한다. 이 시간 후, 반응 혼합물을 진공하에 농축시키고, 잔사는 MeOH에 용해시키고, PL-HCO3 MP-수지 칼럼을 통해 유리 염기 생성물로 통과시킨다. 여액을 진공하에 농축시키고, 조 생성물은 용매 혼합물로서 10% MeOH/DCM을 사용하여 분취용 TLC로 정제하여 표제 화합물(71mg)을 수득한다; m/z 422.4 [M+1].
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 222, 표 1
방법 29
2-[4-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-[1,2,4]옥사디아졸-5-일)-피페라진-1-일]-N,N-디메틸-
아세트아미드(실시예 193, 표 1)의
합성

2-[4-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-[1,2,4]옥사디아졸-5-일)-피페라진-1-일]-N,N-디메틸-
아세트아미드의
합성
합성은 적절한 시약을 사용하여 방법 9에 사용된 유사한 조건에서 수행한다.
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 211, 표 1
방법 30
1-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-[1,2,4]
옥
사디아졸-5-일)-피페리딘-4-
카복실산(실시예 219, 표 1)의
합성

합성은 적절한 시약을 사용하여 방법 14, 단계 2에 사용된 조건과 유사하다; m/z 437.4 [M+1]. 화합물은 가열된 MeOH로부터 연마에 의해 정제한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 272, 표 1
방법 31
1-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-[1,2,4]
옥사디아졸
-5-일)-3-
메틸
-
아제티딘
-3-올(
실시예
203, 표 1)의 합성

1-{3-[1-(4-
브로모
-
페닐
)-1,2-디메틸-프로필]-[1,2,4]
옥사디아졸
-5-일}-
아제
티딘-3-온의 합성
합성은 적절한 시약을 사용하여 방법 21, 단계 3에 사용된 유사한 조건에서 수행한다; m/z 364 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:
1-{(3-[1-(4-
브로모
-
페닐
)-1,2-디메틸-프로필]-[1,2,4]
옥사디아졸
-5-일}-3-메틸-
아제티딘
-3-올의 합성
-78℃에서 THF(1mL) 중 I-147(132.6mg, 0.364mmol)의 냉각된 용액에 THF 중 3M 메틸 마그네슘 클로라이드(0.485mL, 1.456mmol)를 가한다. 반응 혼합물을 -78℃에서 10분 동안에 이어서, 실온에서 20분 동안 교반한다. 이 시간 후, 반응 혼합물을 물로 급냉시키고, EtOAc로 2회 추출한다. 유기층을 합하고, 염수로 세척하고, Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시켜 표제 화합물(122㎎)을 수득한다; m/z 380 [M+H].
하기 중간체를 적절한 시약으로부터 유사한 방법으로 합성한다:

1-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-[1,2,4]
옥사디아졸
-5-일)-3-
메틸
-
아제티딘
-3-올의 합성
합성은 적절한 시약을 사용하여 방법 21, 단계 4에 사용된 유사한 조건에서 수행한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 216, 표 1
실시예 238, 표 1
실시예 276, 표 1
방법 32
2-[4-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-피페라진-1-일]-에탄올(
실시예
252, 표 1)의 합성
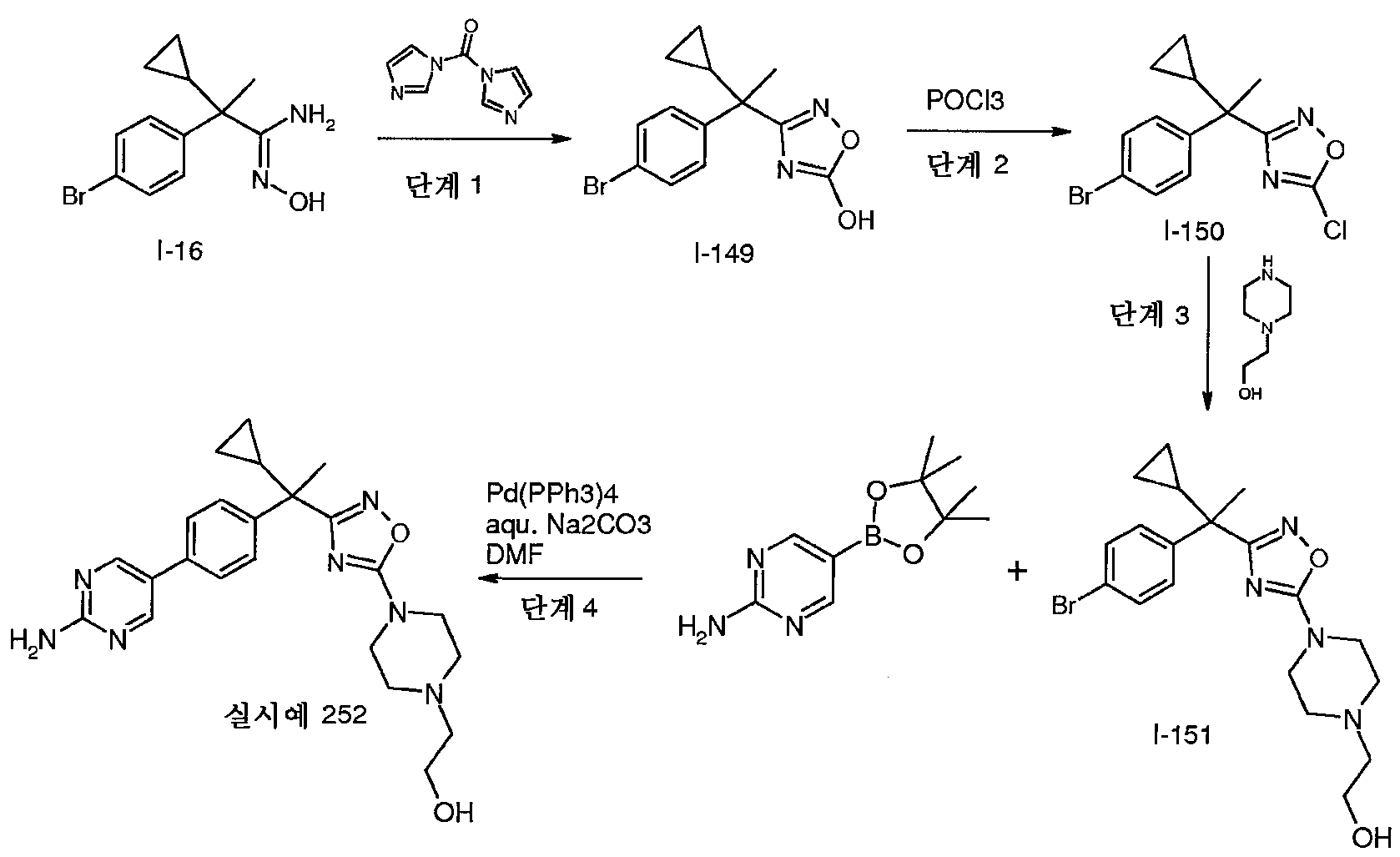
3-[1-(4-
브로모
-
페닐
)-1-
사이클로프로필
-에틸]-[1,2,4]
옥사디아졸
-5-올의 합성
합성은 적절한 시약을 사용하여 방법 21, 단계 1에 사용된 유사한 조건에서 수행한다; m/z 307 [M+H].
3-[1-(4-
브로모
-
페닐
)-1-
사이클로프로필
-에틸]-5-
클로로
-[1,2,4]
옥사디아졸
의 합성
마이크로파 바이알에서 DCM(8mL) 중 I-149(847mg, 2.74mmol)의 용액에 POCl3(0.401mL, 4.384mmol) 및 피리딘(1.107mL, 13.7mmol)을 가한다. 반응 혼합물을 120℃인 마이크로파 오븐에서 1시간 동안 가열한다. 이 시간 후, 반응 혼합물을 물로 급냉시키고, CH2Cl2로 2회 추출한다. 유기층을 합하고, 염수로 세척하고, Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 조 생성물은 플래시 크로마토그래피(SiO2, 6-50% EA/Hep)로 정제하여 표제 중간체(694㎎)를 수득한다;
2-(4-{3-[1-(4-
브로모
-
페닐
)-1-
사이클로프로필
-에틸]-[1,2,4]
옥사디아졸
-5-일}-피페라진-1-일)-에탄올의 합성
합성은 적절한 시약을 사용하여 방법 21, 단계 3에 사용된 조건과 유사하다; m/z 421/423[M/M+2H].
2-[4-(3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-피페라진-1-일]-에탄올의 합성
마이크로파 바이알에서 I-151(193mg, 0.458mmol) 및 Pd(PPh3)4(53mg, 0.046mmol)의 혼합물에 2-아미노피리미딘-5-보론산 피나콜 에스테르(121.6mg, 0.55mmol) 및 2M Na2CO3 수용액(0.92mL)의 DMF(5mL) 용액을 가한다. 반응 혼합물을 Ar로 퍼징시킨 다음, 110℃의 마이크로파 오븐에서 45분 동안 가열한다. 이 시간 후, 반응 혼합물을 물로 급냉시키고, EtOAc로 2회 추출한다. 유기층을 합하고, 염수로 세척하고, Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 조 생성물은 플래시 크로마토그래피(SiO2, 1.2-10% MeOH/DCM)로 정제하여 표제 화합물(83㎎)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 204 내지 205, 표 1
실시예 248, 표 1
실시예 252, 표 1
실시예 259, 표 1
실시예 274, 표 1
실시예 287 내지 288, 표 1
실시예 297, 표 1
방법 33
1-(3-{(R)-1-[4-(2-아미노-피리미딘-5-일)- 페닐 ]-1,2-디메틸-프로필}-[1,2,4]옥사디아졸-5-일)-피페리딘-4-올( 실시예 223, 표 1) 및 1-(3-{(S)-1-[4-(2-아미노-피리미딘-5-일)- 페닐 ]-1,2-디메틸-프로필}-[1,2,4] 옥사디아졸 -5-일)-피페리딘-4-올( 실시예 224, 표 1)의 합성
실시예 223 및 실시예 224는 100bar에서 RegisPak(제조원: Regis Technologies, Morton Grove, IL) 반분취용(30x250 mm) HPLC 칼럼(CO2 중 0.1% 이소프로필아민을 함유하는 이소프로판올 중 35% 1:1 MeOH로 용출시킴) 상에서 실시예 262(90.3mg)의 분할에 의해 제조한다. 체류시간이 2.07분인 보다 빨리 용출되는 에난티오머 실시예 223(RegisPack 4.6x100 mm, 제조원: Regis Technologies, Morton Grove, IL) 및 체류시간이 2.68분인 보다 느리게 용출되는 에난티오머 실시예 224. 용출물은 농축시켜 실시예 223(36mg) 및 실시예 224(34mg)를 제공한다.
방법 34:
5-[4-((R)-1-
사이클로프로필
-1-{5-[1-(2-디메틸아미노-에틸)-1H-
피라졸
-4-일]-[1,2,4]
옥사디아졸
--일}-에틸)-
페닐
]-피리미딘-2-
일아민(실시예 182, 표 1)의
합성

실시예 48(75.0mg, 0.201mmol)을 (2-클로로-에틸)-디메틸-아민 하이드로클로라이드(43.3mg, 0.301mmol), Cs2CO3(147mg, 0.452mmol) 및 DMF(1.5mL)로 처리하고, 생성된 혼합물은 60℃에서 1시간 동안 교반한다. 여기서,, 혼합물은 (2-클로로-에틸)-디메틸-아민 하이드로클로라이드(14.5mg, 0.100mmol), Cs2CO3(32.7mg, 0.100mmol)로 처리하고, 1시간 동안 더 오래 교반한다. 이어서, 반응물은 10-60% 아세토니트릴/물/0.1% 트리플루오로아세트산으로 용출시키면서 C-18 실리카 상에서 플래시 크로마토그래피로 직접 정제한다. 생성된 반-순수한 물질을 CH2Cl2 및 포화 수성 NaHCO3로 처리하고, 상을 분리한다. 생성된 수성상은 CH2Cl2로 5회 더 추출하고, 합한 유기상은 Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시켜 잔사를 수득하고, 이를 8% 메탄올/CH2Cl2로 용출시키면서 분취용 TLC로 추가로 정제하여 표제 화합물(35.0mg)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 215, 표 1
방법 35:
2-[4-(3-{(R)-1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-
피라졸
-1-일]-1-((R)-3-
메톡시
-
피롤리딘
-1-일)-에타논(
실
시예 220, 표 1)의 합성

[4-(3-{(R)-1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-
피라졸
-1-일]-아세트산 에틸 에스테르의 합성
실시예 48은 방법 9에 따라 알킬화시킨다; m/z 460 [M+H].
[4-(3-{(R)-1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]옥사디아졸-5-일)-
피라졸
-1-일]-아세트산의 합성
I-152는 방법 16의 마지막 단계에 따라 가수분해시킨다; m/z 432 [M+H].
I-153(94.0mg, 0.218mmol)은 (R)-3-메톡시-피롤리딘(33.1mg, 0.327mmol), HATU(125mg, 0.327mmol), DIEA(114㎕, 0.654mmol) 및 DMF(1.50mL)로 처리하고, 생성된 혼합물은 1시간 동안 교반한다. 반응물은 30-70% 아세토니트릴/물/0.1% 포름산으로 용출시키면서 역상 분취용 HPLC로 직접 정제하여 표제 화합물(16.0mg)을 수득한다.
하기 화합물은 적절한 중간체로부터 유사한 방법으로 합성한다:
실시예 194, 표 1
실시예 221, 표 1
방법 36
2-[4-(3-{(R)-1-[4-(2-아미노-6-옥소-1,6-
디하이드로
-피리미딘-5-일)-
페닐
]-1-사
이클로프로
필-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-아세트아미드(
실시예
197, 표 1)의 합성
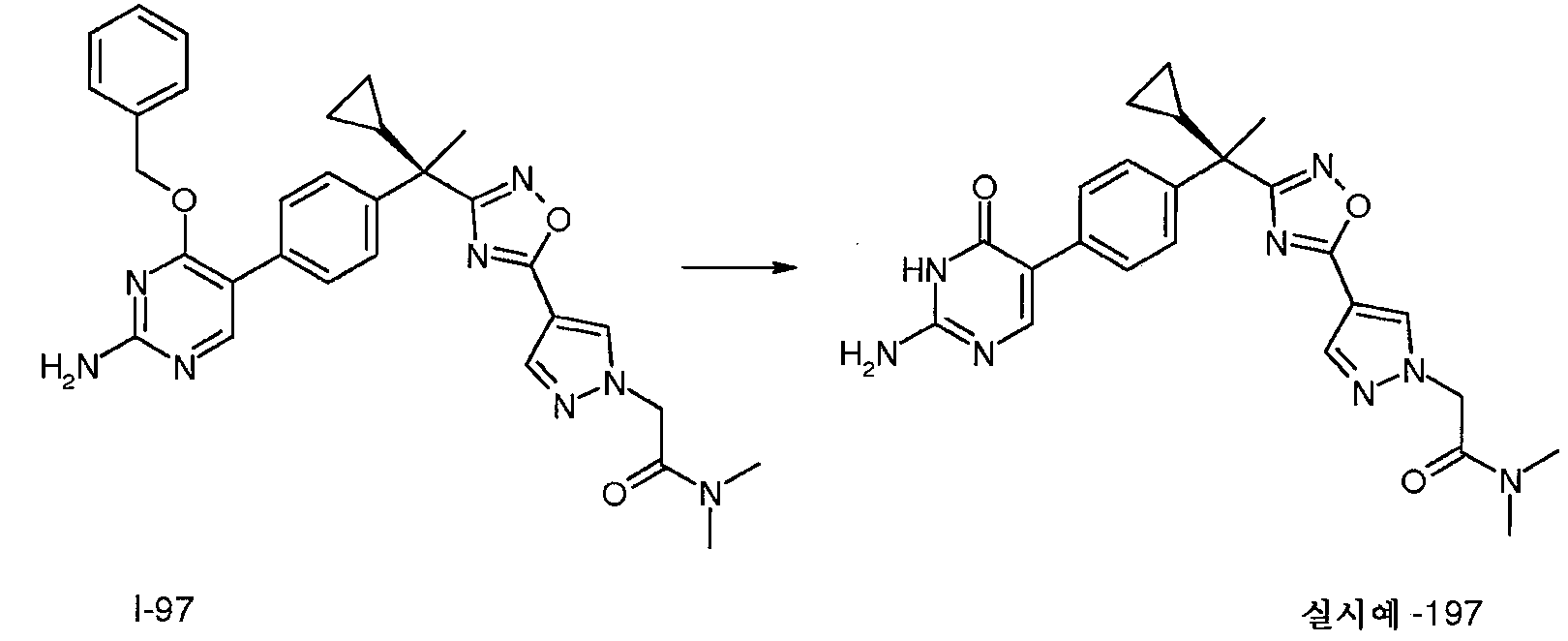
I-97(170mg, 0.30mmol)을 에탄올(10ml)에 용해시키고, 펄맨(Pearlman) 촉매(수산화팔라듐, 35mg, 50% 수분함유, 0.12mmol)로 처리한다. 용기를 탈기시키고, 수소하에 둔다(벌룬). 전환이 완결되면, 반응물은 셀라이트 패드를 통해 여과하고, 고체는 메탄올로 세척한다. 합한 여액을 농축시키고, 나머지 조 생성물은 플래시 칼럼 크로마토그래피(0-10% MeOH/DCM)를 통해 정제하여 표제 화합물(100mg)을 수득한다.
방법 37
5-[4-((R)-1-{5-[1-(2-아미노-에틸)-1H-
피라졸
-4-일]-[1,2,4]
옥사디아졸
-3-일}-1-
사이클로프로필
-에틸)-
페닐
]-피리미딘-2-
일아민(실시예 190, 표 1)의
제조

중간체 I-98(120mg, 0.22mmol)을 에탄올(2.7mL) 및 THF(0.5mL)로 처리한 다음, 히드라진(97mg, 1.93mmol)을 가한다. 생성된 혼합물을 50℃에서 2시간 동안 교반한다. 생성된 혼합물을 여과하고, 에탄올로 세정한 다음, 에틸 아세테이트 및 물로 희석하고, 상을 분리한다. 유기상을 염수로 세척하고, Na2SO4로 건조시키고, 여과한 다음, 진공하에 농축시킨다. 잔사는 10-80% 아세토니트릴/물/트리플루오로아세트산으로 용출시키면서 역상 분취용 HPLC로 정제하여 표제 화합물(33.0mg)을 수득한다.
방법 38
2-[4-(3-{1-[4-(2-아미노-6-
플루오로
-피리딘-3-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드
(
실시예
277, 표 1) 및 2-[4-(3-{1-[4-(6-아미노-2-
플루오로
-피리딘-3-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-아세트아미드(실
시예
278, 표 1)의 합성

1,4-디옥산(10mL) 중 I-69(300mg, 0.68mmol)의 혼합물에 2,6-디플루오로피리딘-3-보론산(128mg, 0.81mmol), PdCl2(PPh3)2(47mg, 0.068mmol) 및 2M Na2CO3 용액(2M 수용액)(1mL, 02mmol)을 가한다. 혼합물을 마이크로파 반응기에서 1시간 동안 100℃로 가열한다. 용액을 냉각시키고, H2O 및 EtOAc로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 I-154(200mg)(m/z: 479.2 [M++H])를 수득한다.
I-154(150mg, 0.31mmol)를 MeOH 중 NH3(2M 용액)(10mL)에 용해시키고, 용액은 72시간 동안 100℃로 가열한다. 용액을 냉각시키고, 농축시킨다. 잔사는 분취용 실리카 겔 TLC로 정제하여 표제 화합물(실시예 277: 10mg; 실시예 278: 14mg)을 수득한다.
방법 39
2-[4-(3-{1-[4-(5-아미노-3-
시아노
-피라진-2-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드
(
실시예
295, 표 1)의 합성

DMF 중 I-69(100mg, 0.2mmol)의 용액에 2-아미노-5-브로모-6-클로로피라진(51mg, 0.24mmol), 테트라키스(트리페닐포스핀)팔라듐(0)(24mg, 0.02mmol) 및 2M Na2CO3 용액(2M 수용액)(0.5mL, 1mmol)을 가한다. 혼합물을 마이크로파 반응기에서 1시간 동안 100℃로 가열한다. 혼합물을 냉각시키고, H2O 및 EtOAc로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 I-155(86mg)(m/z: 493.2 [M++H])를 수득한다.
마이크로파 반응기에 DMF(8mL) 중 I-155(80mg, 0.16mmol)를 용해시킨다. 시안화아연(23mg, 0.19mmol) 및 Pd(PPh3)4(18mg, 0.016mmol)를 가하고, 용액을 마이크로파에서 2시간 동안 120℃로 가열한다. 용액을 냉각시켜 물에 붓고, 생성물을 EtOAc에서 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과하고, 농축시킨다. 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 표제 화합물(38mg)을 수득한다.
방법 40
2-[4-(3-{1-[4-(6-아미노-2-
시아노
-피리딘-3-일)-
페닐
]-1-
사이클로프로필
-에틸}-[1,2,4]
옥사디아졸
-5-일)-
피라졸
-1-일]-N,N-디메틸-
아세트아미드(실시예 296,
표 1)의 합성

DMF(10mL) 중 2-브로모-6-아미노피리딘(200mg, 1.2mmol)의 용액에, 실온에서 Zn(CN)2(163mg, 1.4mmol) 및 Pd(PPh3)4(134mg, 0.12mmol)를 가한다. 용액을 마이크로파 반응기에서 2시간 동안 120℃로 가열한다. 용액을 냉각시키고, 물을 가한다. 용액을 EtOAc에서 추출하고, 합한 유기층은 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 I-156(54mg)(m/z: 119.9 [M+])을 수득한다.
CH3CN(10mL) 중 I-156(54mg, 0.46mmol)의 용액에 실온에서 NBS(162mg, 0.9mmol)를 가한다. 용액을 동일한 온도에서 12시간 동안 교반한다. 용액을 농축시키고, 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 I-157(35mg)(m/z: 197.9 [M+])을 수득한다.
DMF(8mL) 중 I-69(50mg, 0.1mmol)의 용액에 I-157(24mg, 0.12mmol), 테트라키스(트리페닐포스핀)팔라듐(0)(11mg, 0.01mmol) 및 2M Na2CO3 용액(0.25mL, 0.51mmol)을 가한다. 혼합물을 마이크로파 반응기에서 1시간 동안 100℃로 가열한다. 혼합물을 냉각시키고, H2O 및 EtOAc로 추출한다. 합한 유기층을 MgSO4로 건조시키고 여과한다. 여액을 농축시키고, 잔사는 용출제로서 CH2Cl2 중 10% MeOH를 사용하여 실리카 겔 플래시 칼럼 크로마토그래피로 정제하여 표제 화합물(26mg)을 수득한다.
방법 41:
3-{1-[4-(2-아미노-피리미딘-5-일)-
페닐
]-1,2-디메틸-프로필}-[1,2,4]
옥사디
아졸-5-올(
실시예
88, 표 1)의 합성

DMF(1mL) 중 모르폴린-4-카보닐 클로라이드(32.907mg, 0.220mmol)의 현탁액에 I-21(60.000mg, 0.200mmol)을 가한다. 반응 혼합물을 실온에서 20분 동안 교반한 다음, 휘니그 염기(0.038mL, 0.22mmol)를 가하고, 반응 혼합물을 1시간 동안 55℃에서 가열한 다음, 150℃의 마이크로파 오븐에서 30분 동안 가열한다. 이 시간 후, 반응 혼합물을 진공하에 농축시킨다. 조 생성물을 플래시 크로마토그래피(SiO2, DCM 중 0-10% MeOH)에 이어서, 분취용-TLC(DCM 중 10% MeOH)에 의해 정제하여 실시예 88(23mg)을 백색 고체로서 수득한다; m/z 326.0 [M+H].
최종 화합물: 표 3
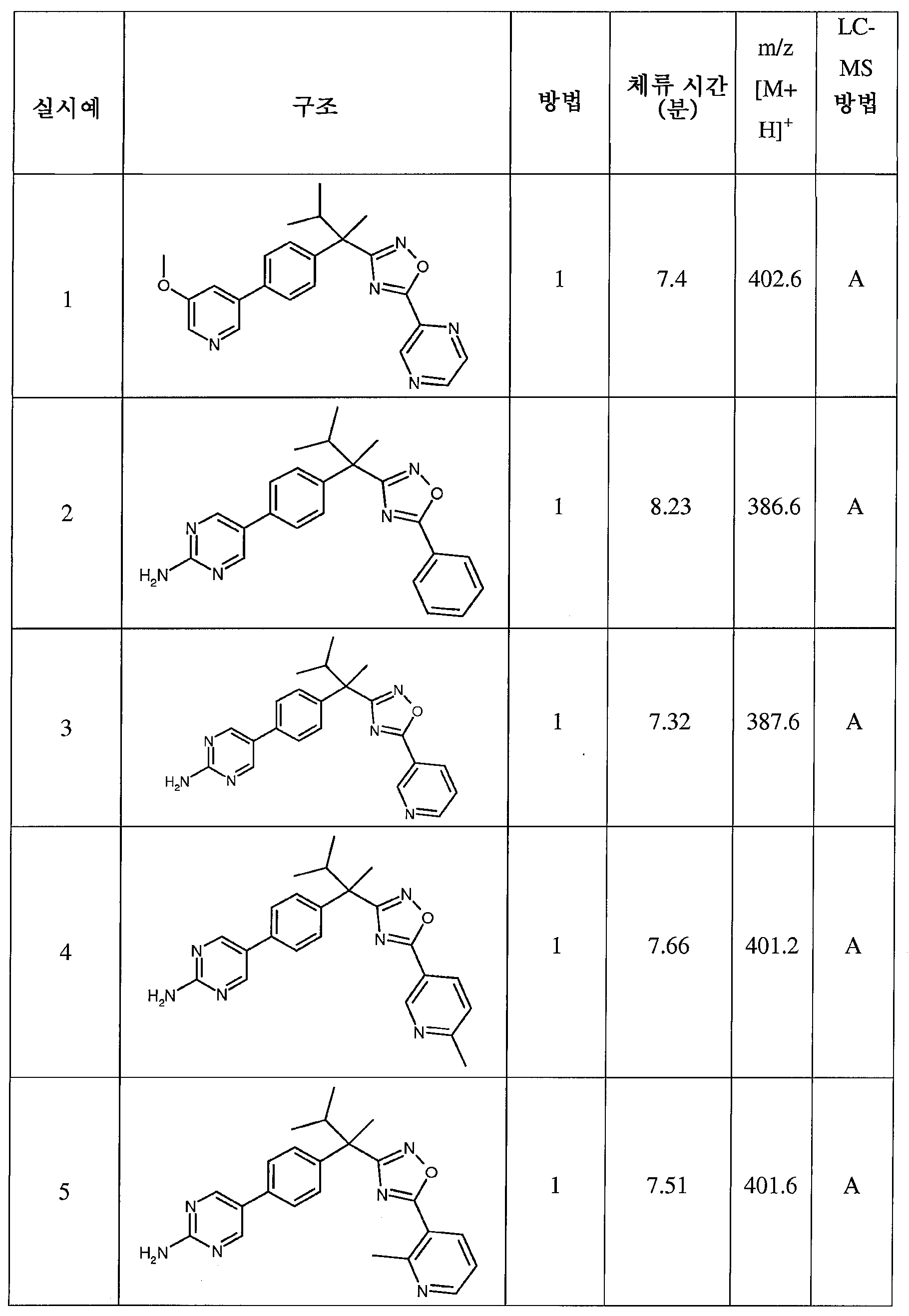


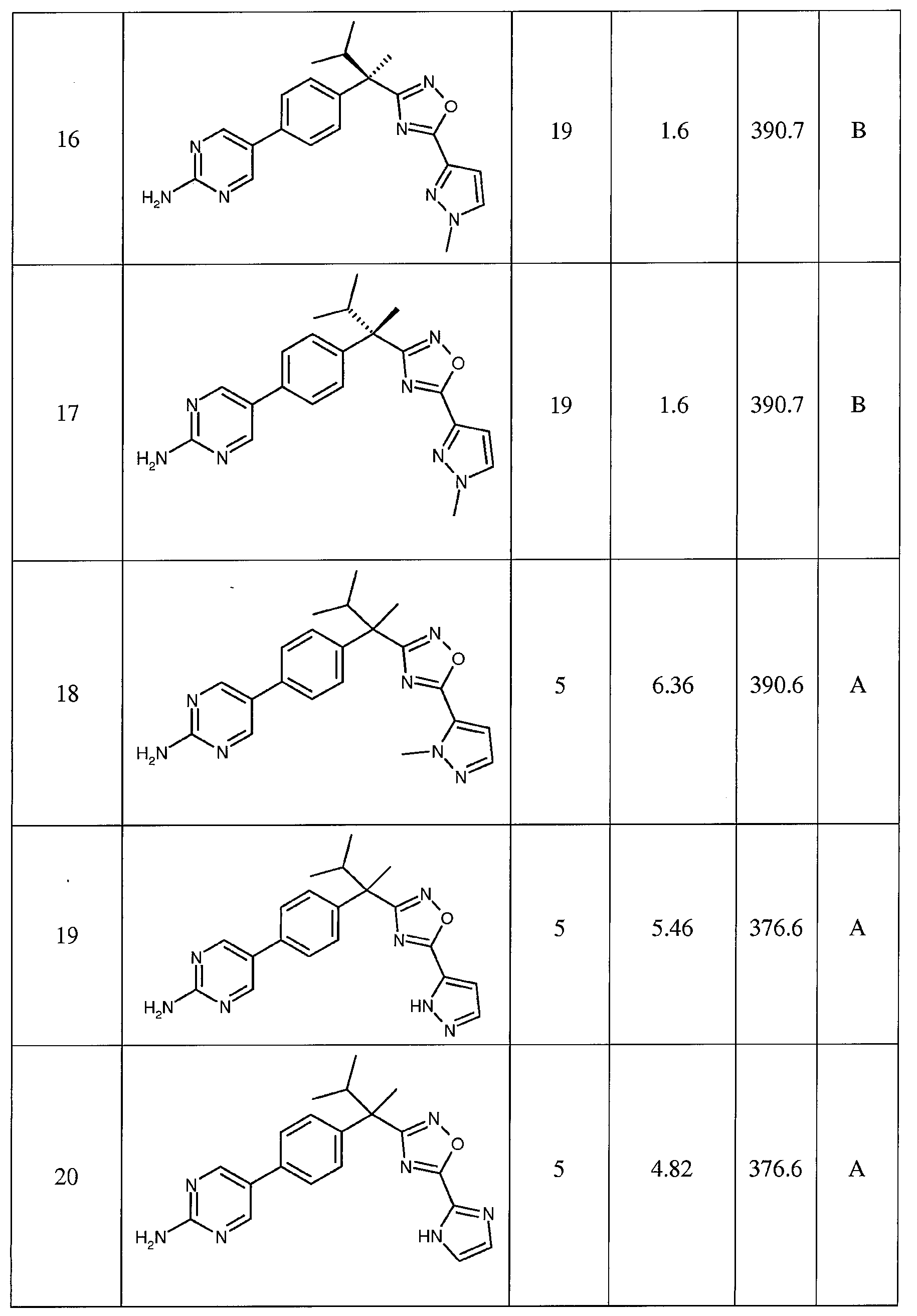

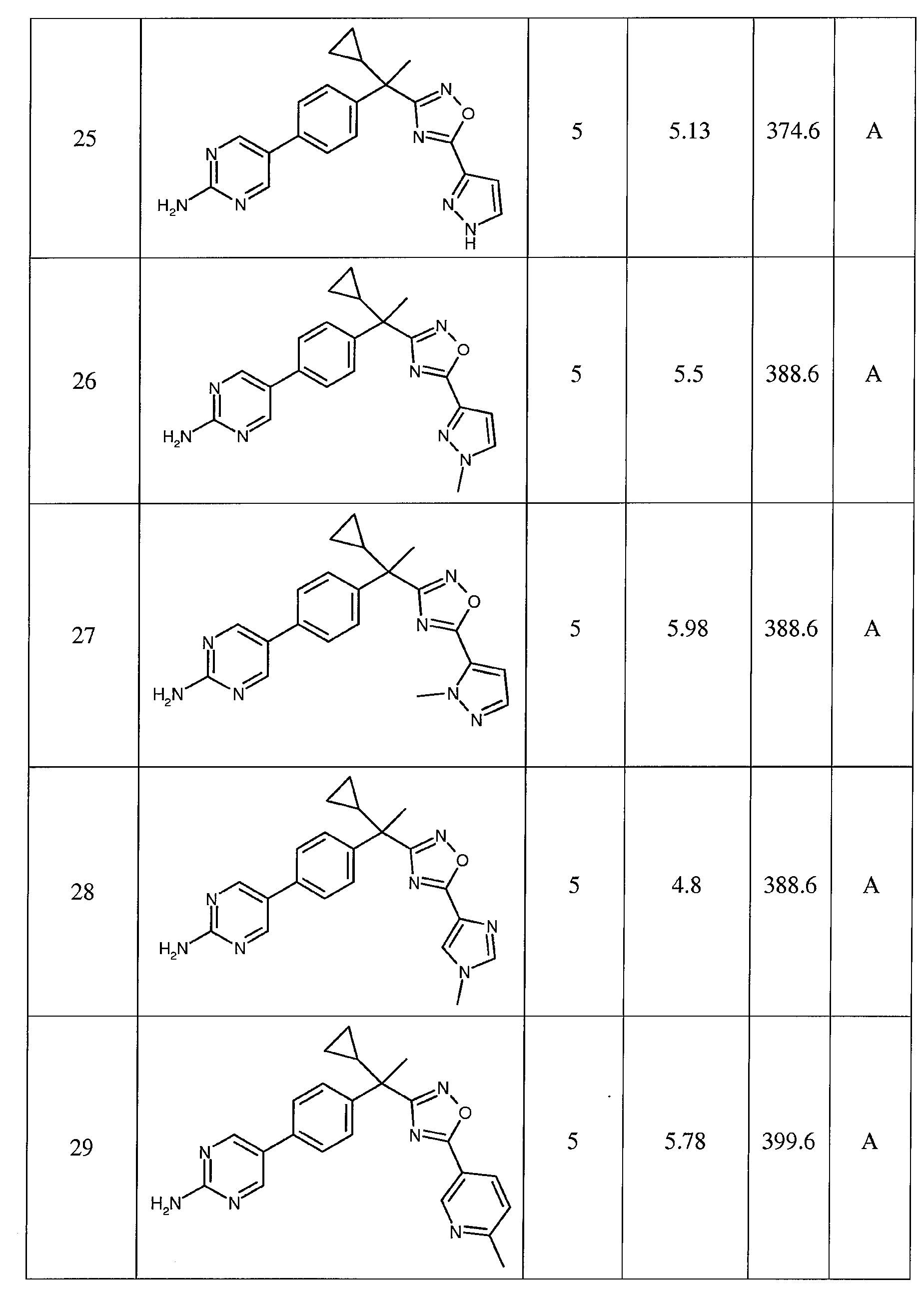
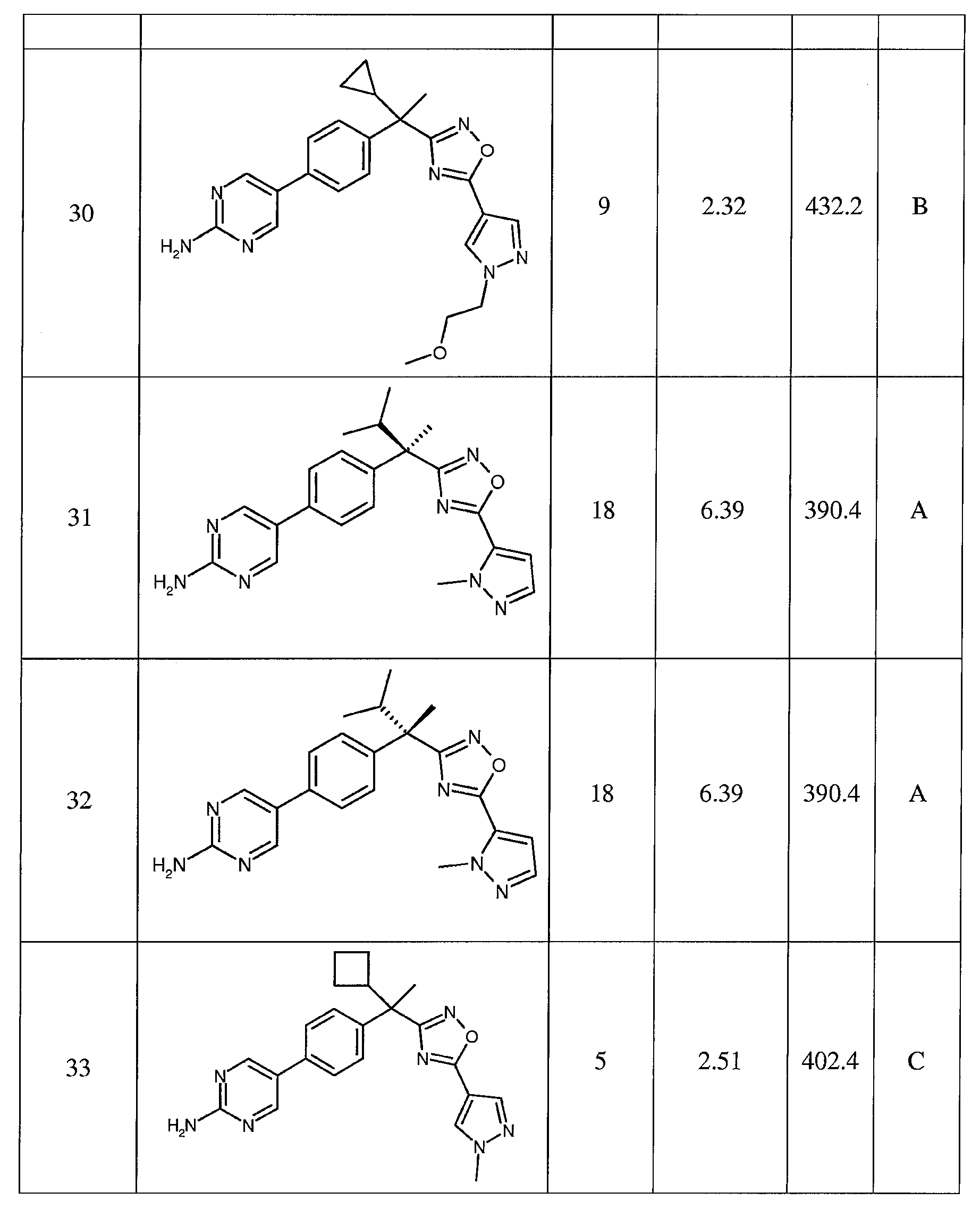


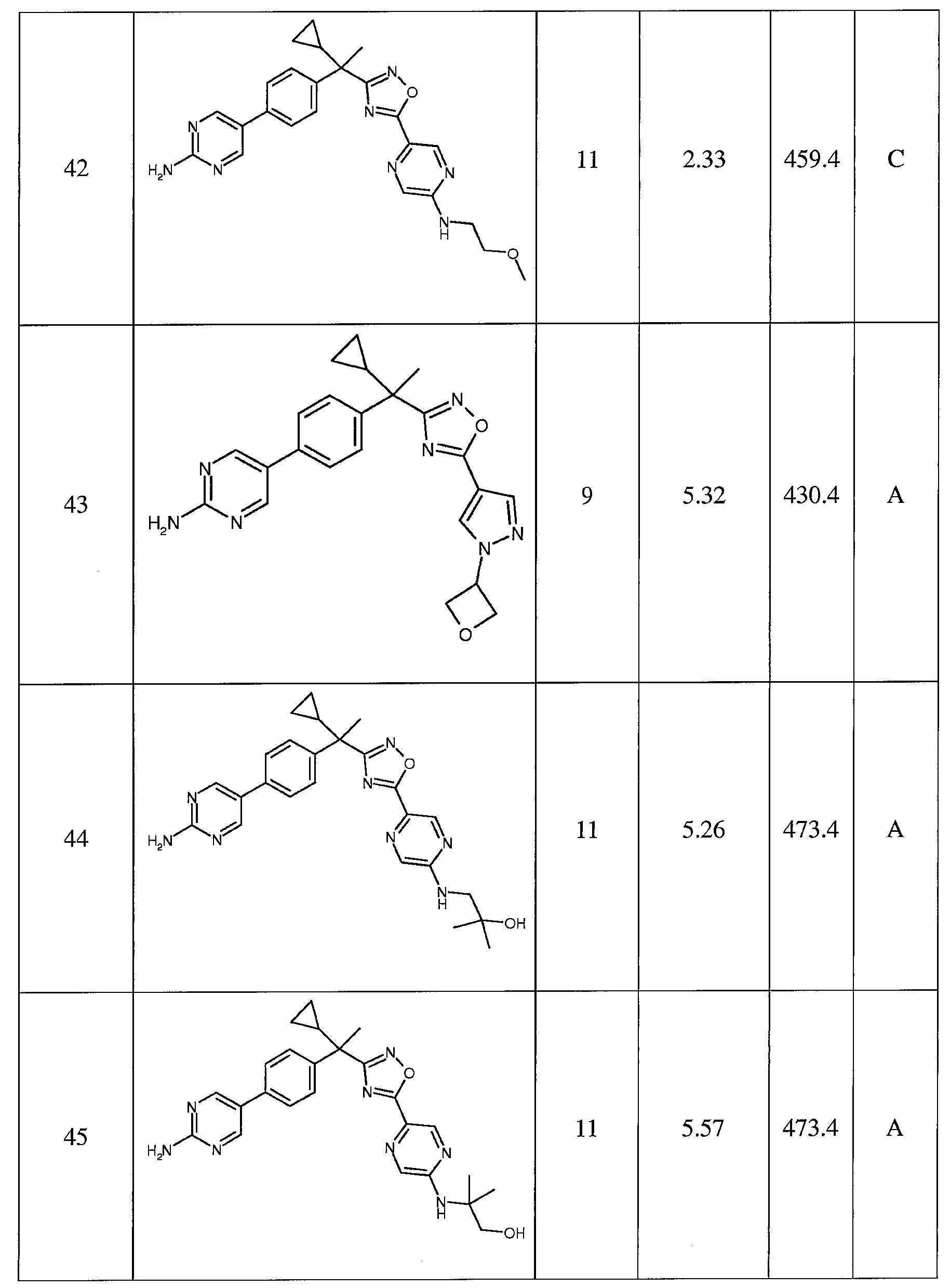


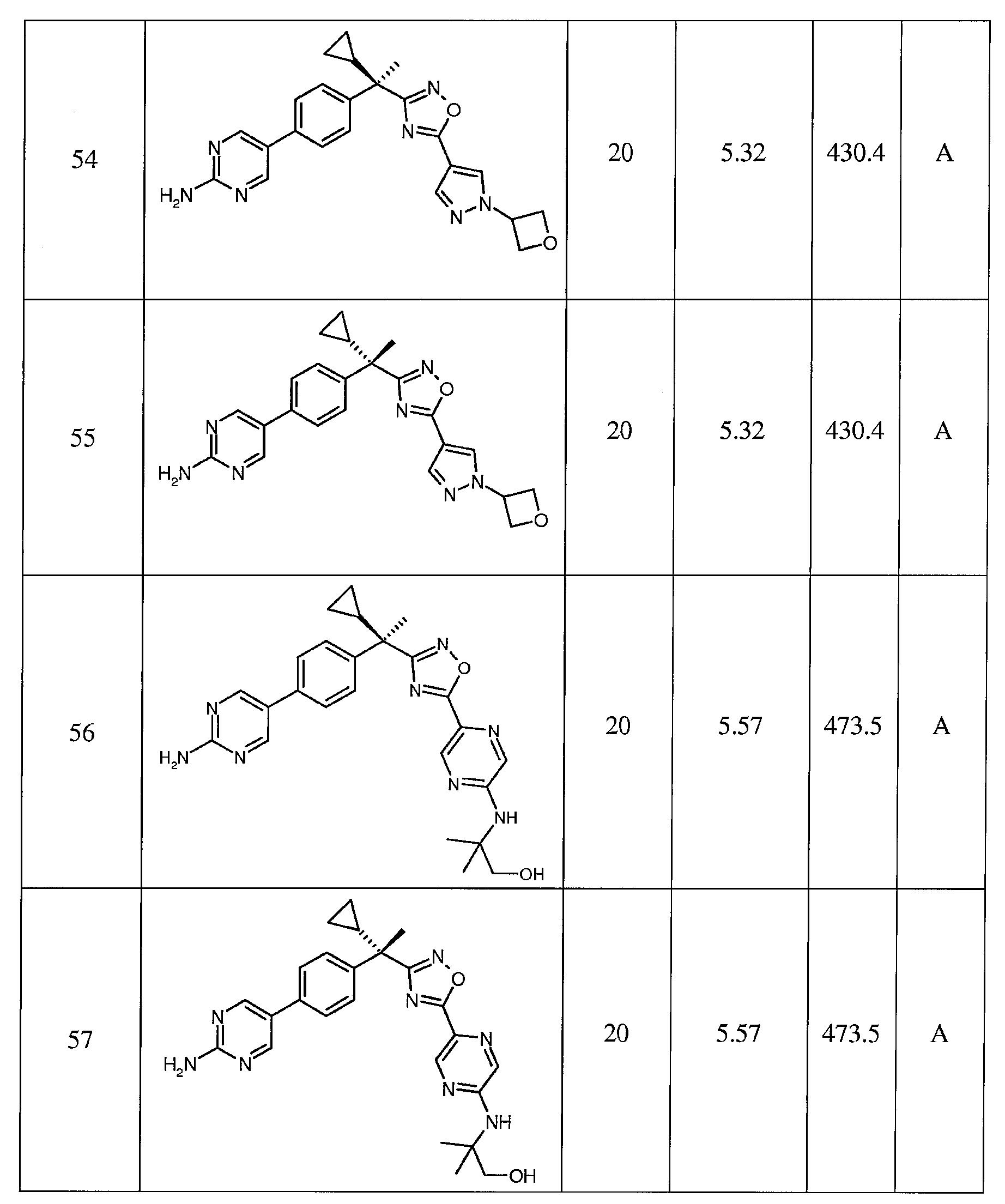









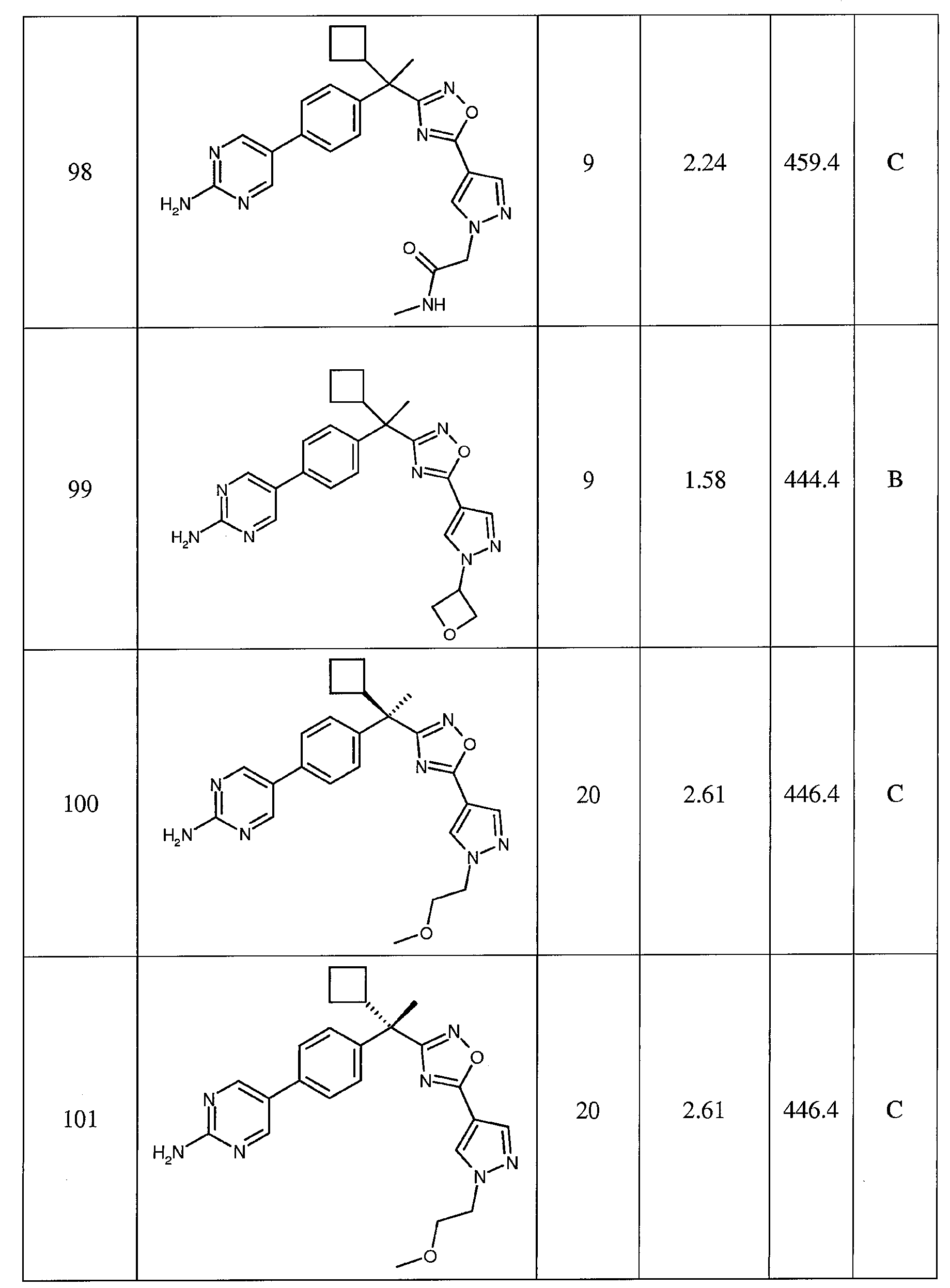





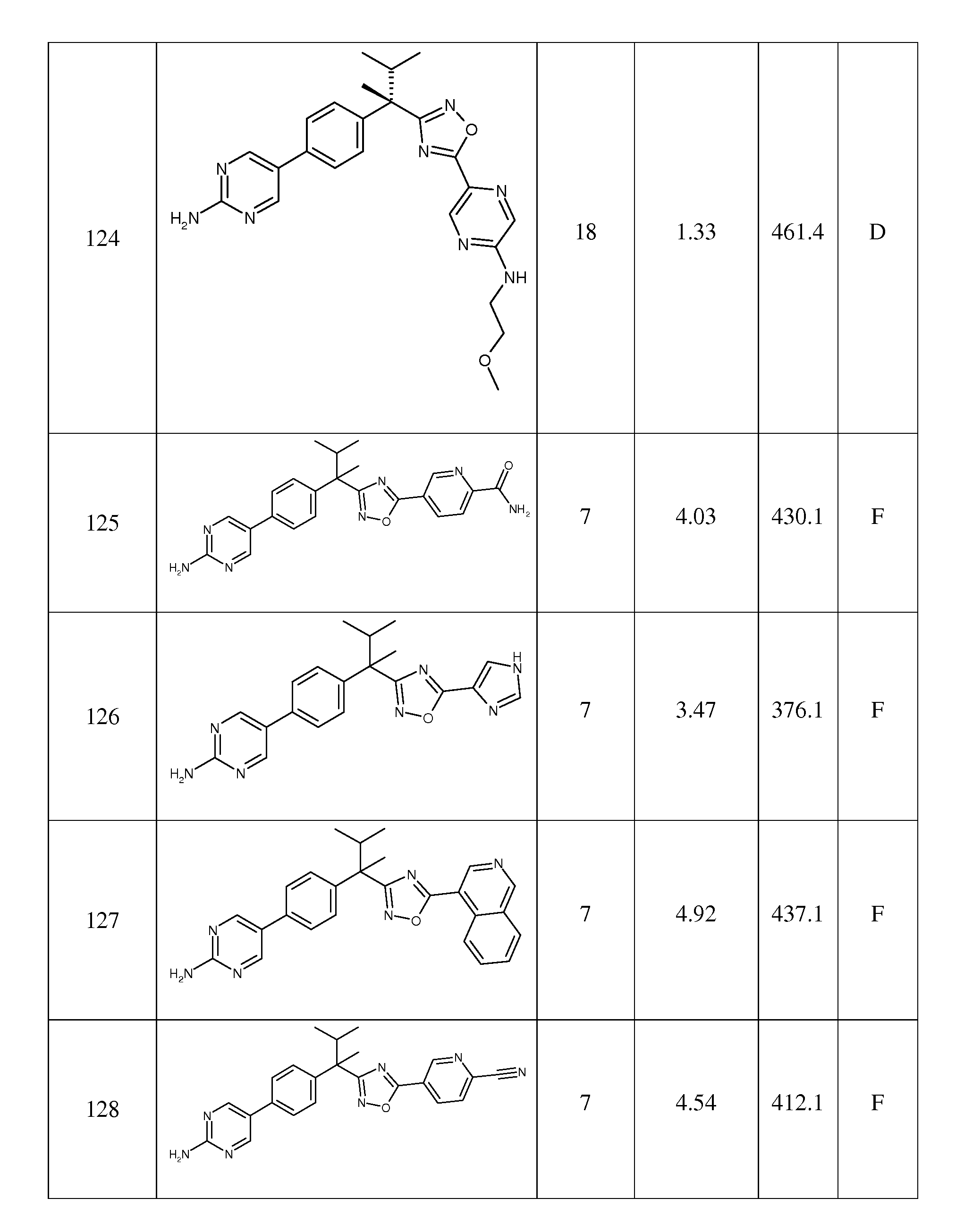



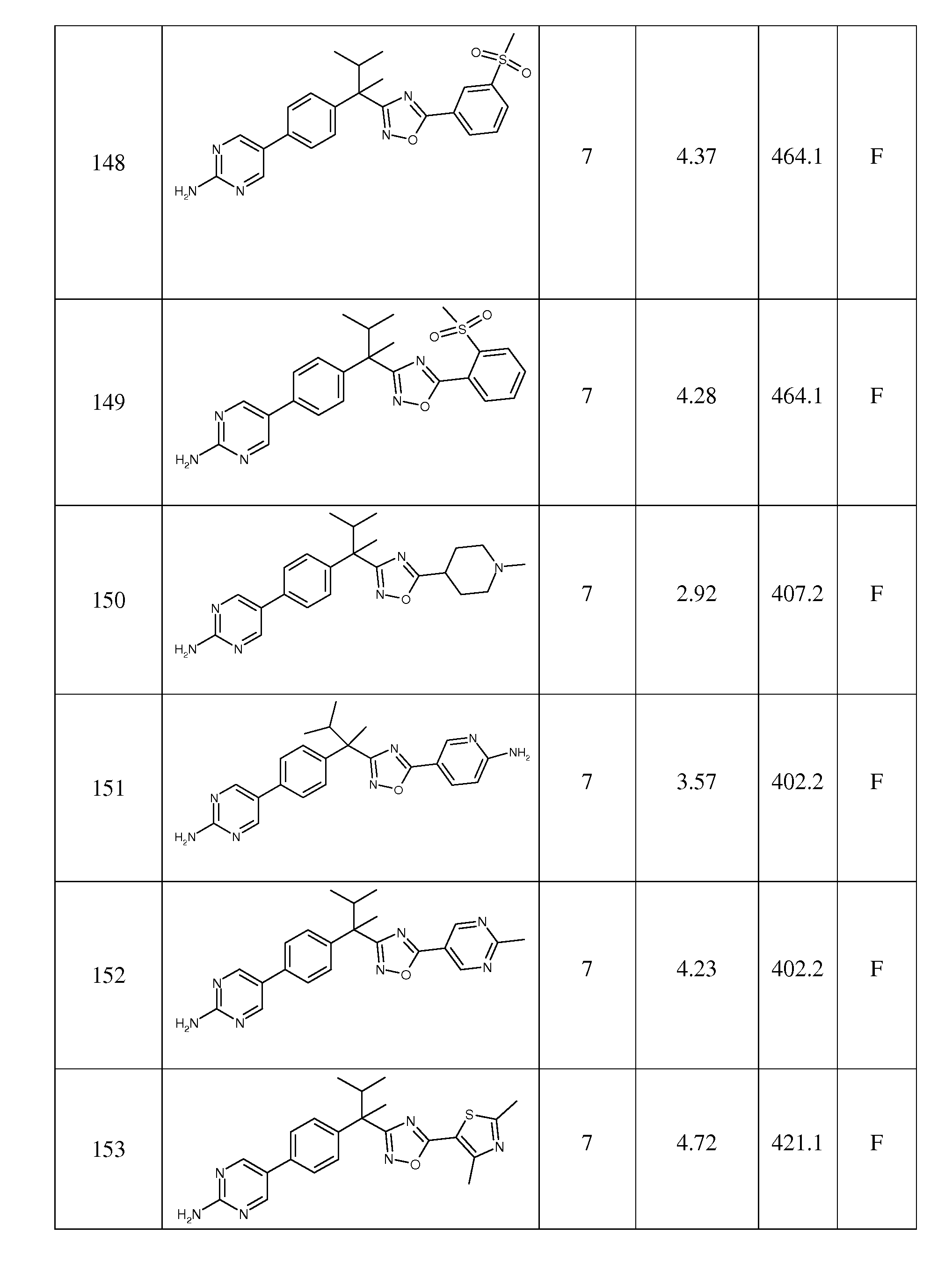




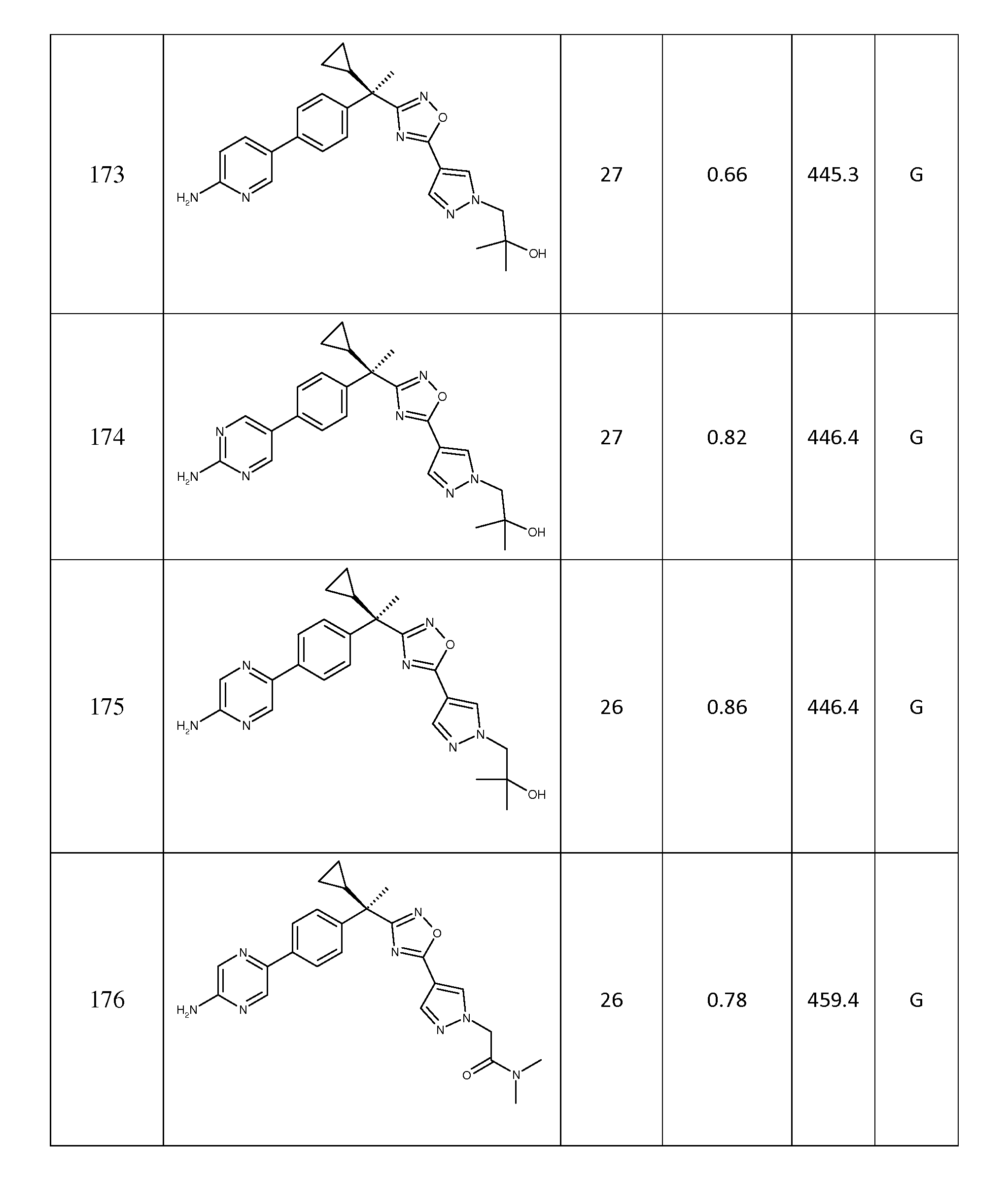

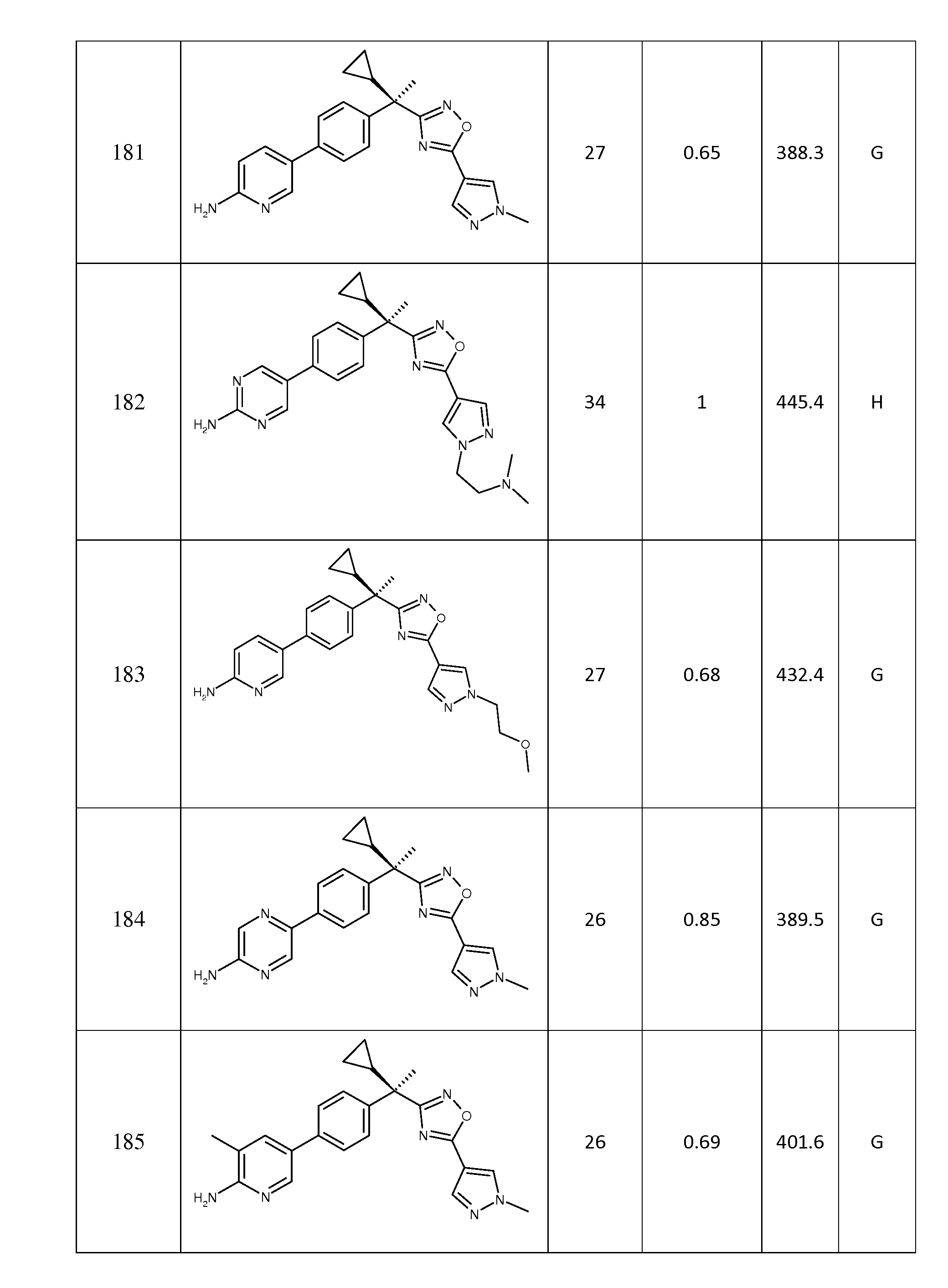

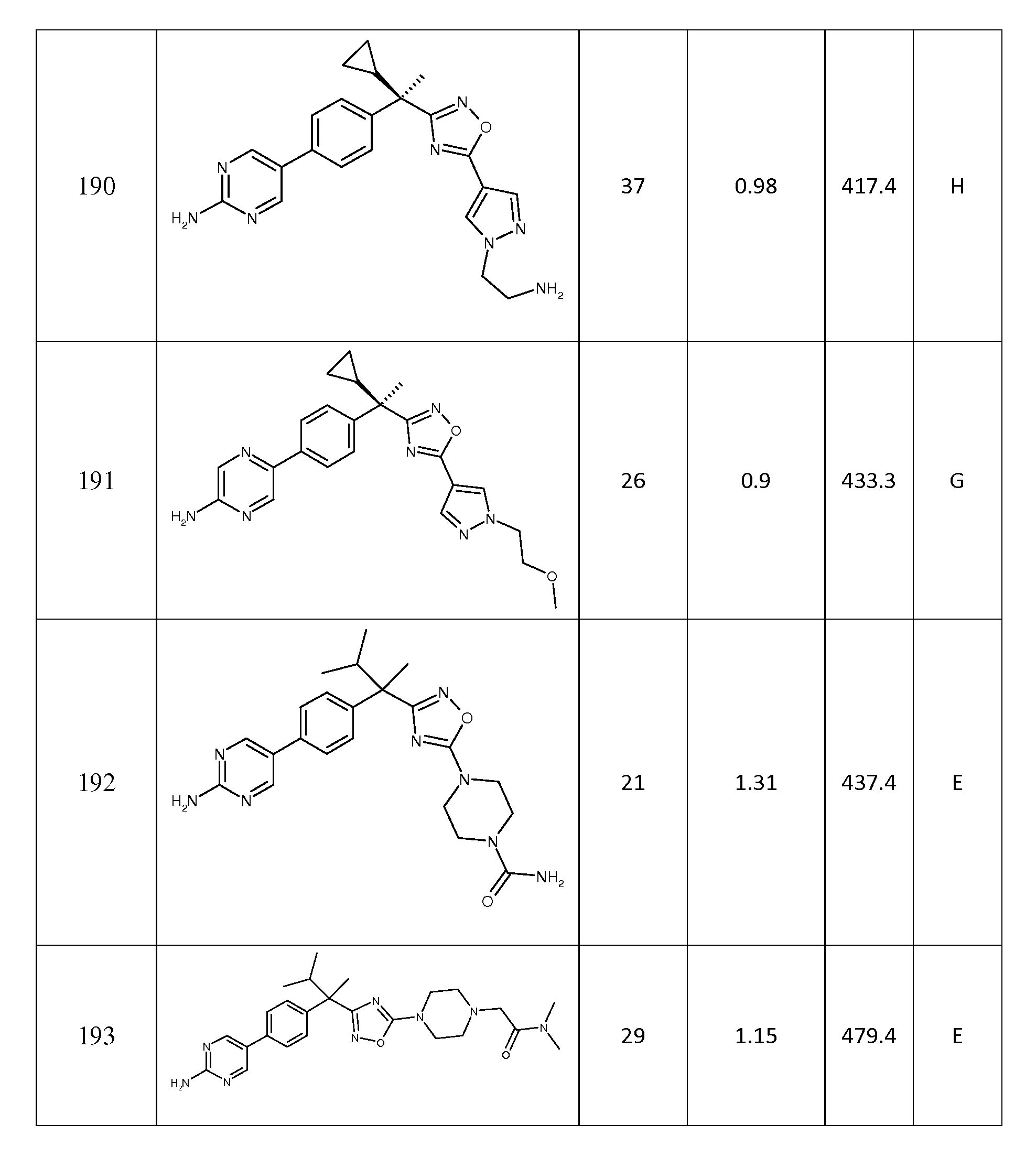
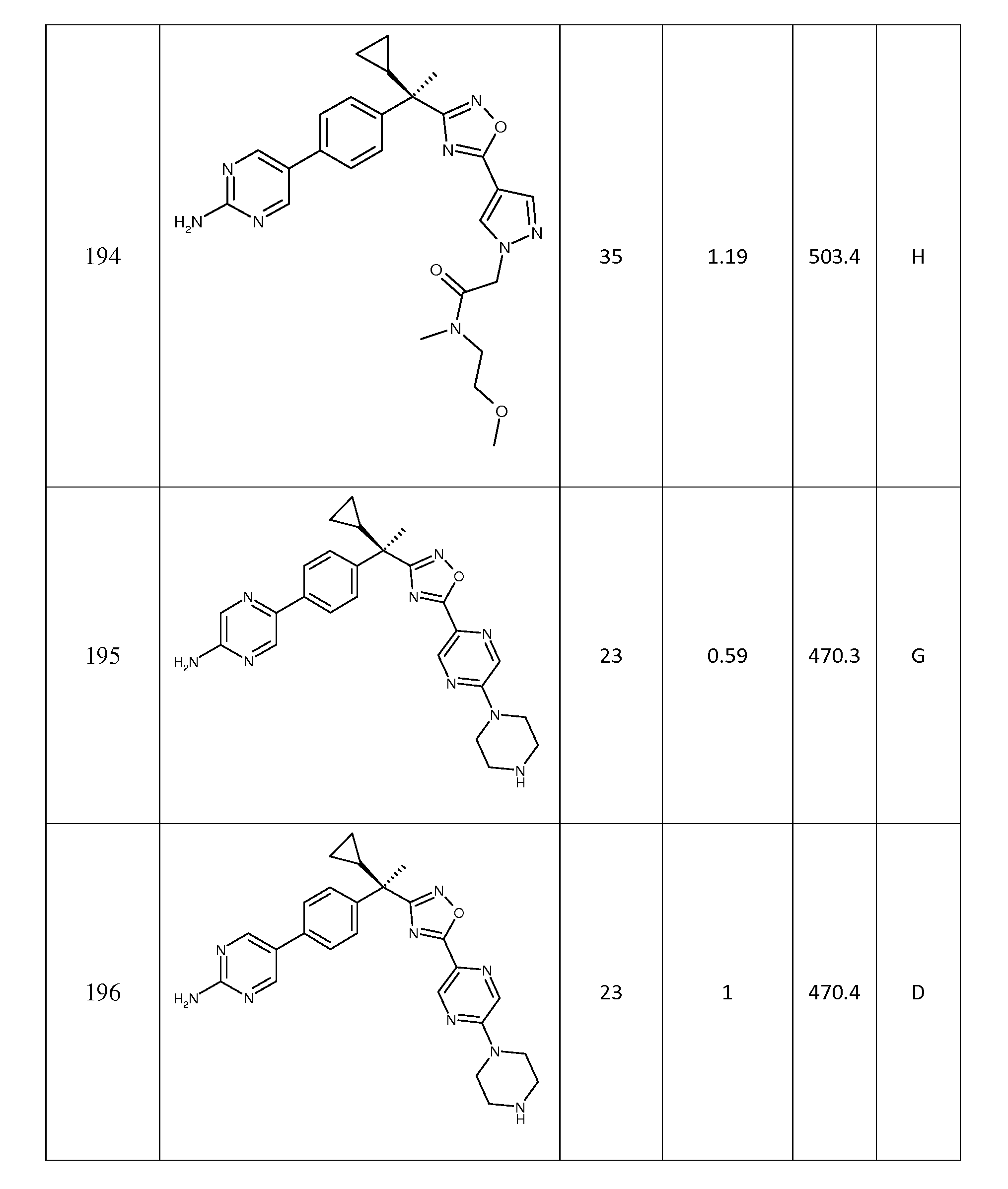




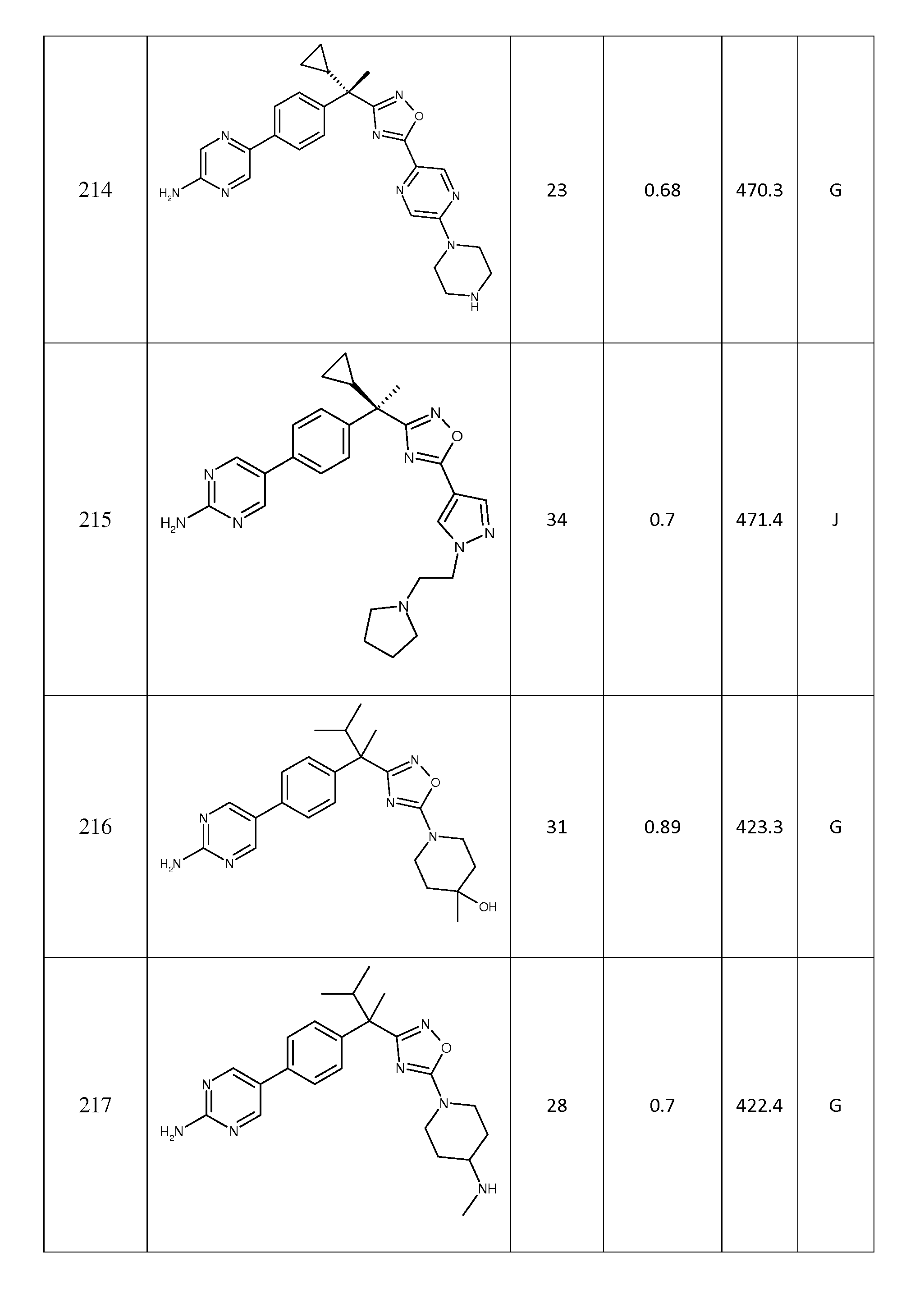



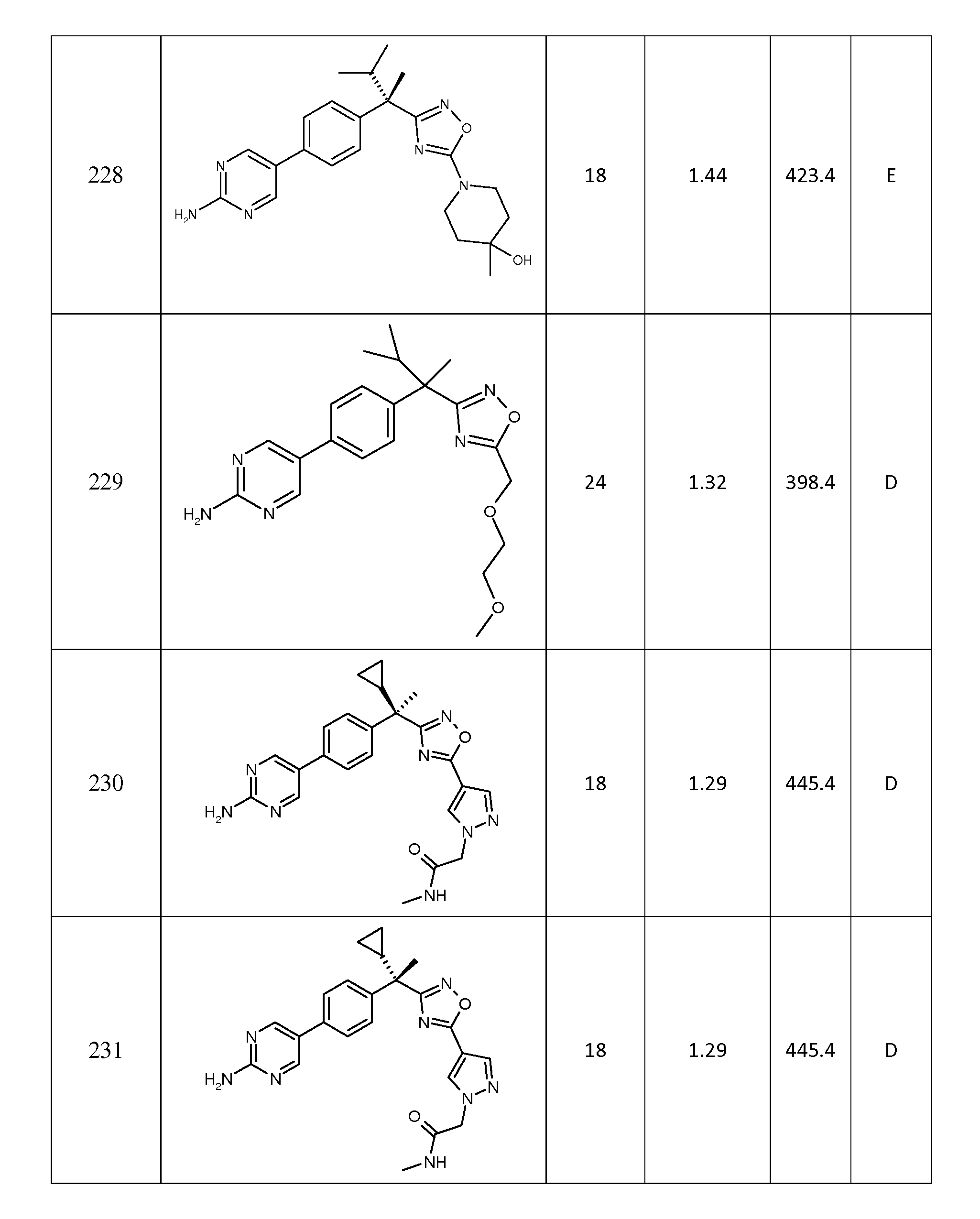


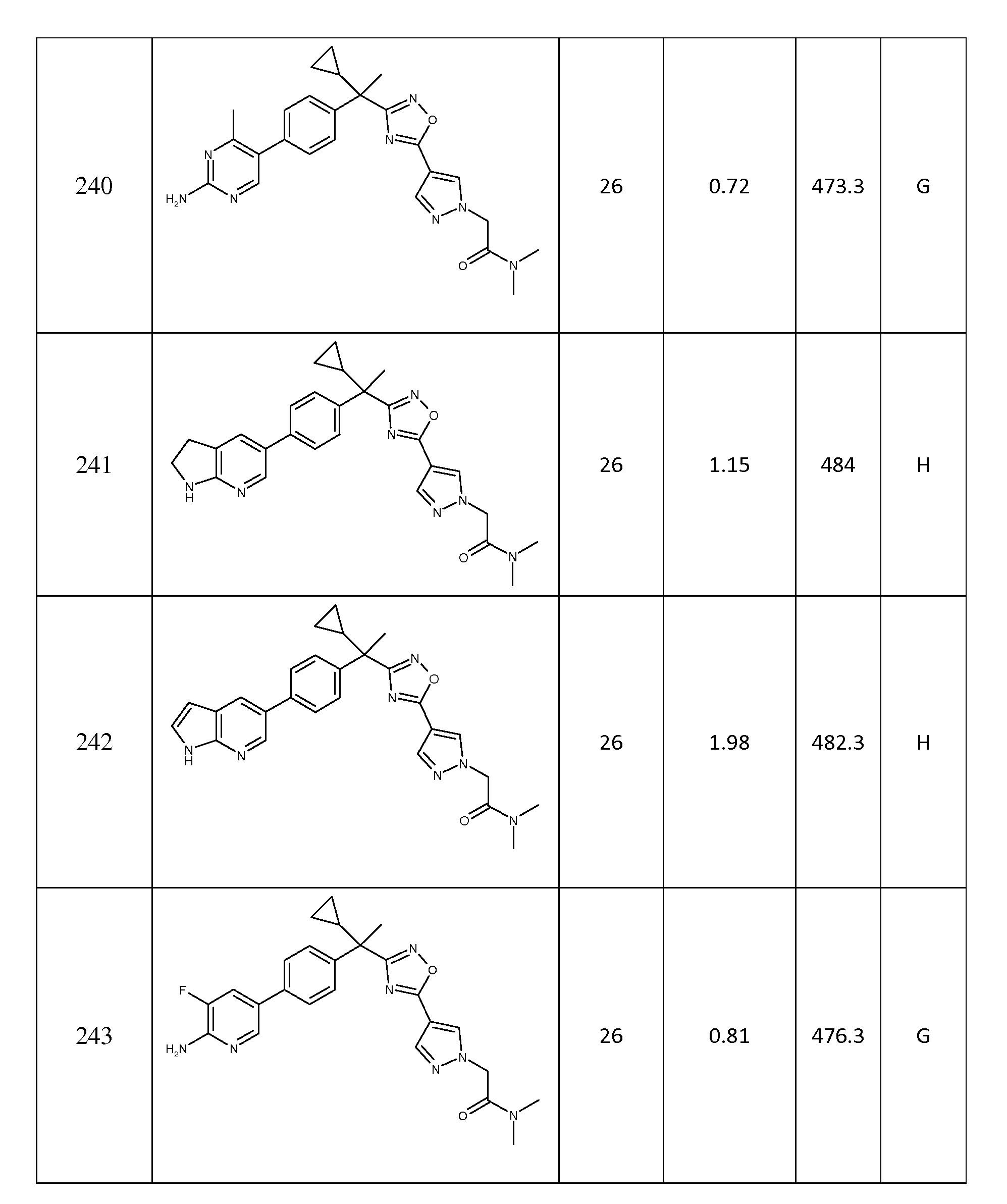







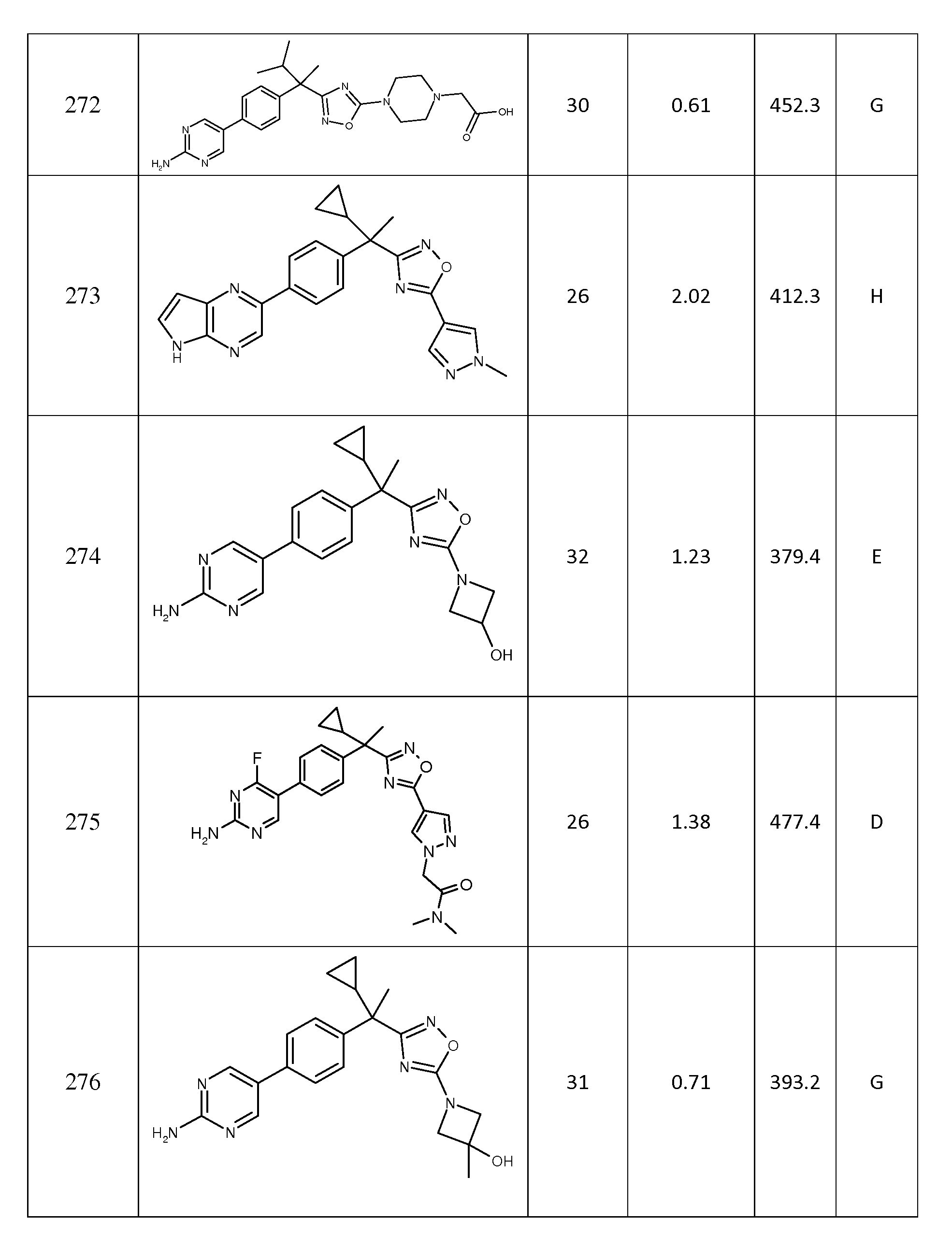





생물학적 특성의 평가
1. 결합 검정
화합물은 섬광 근접 검정 포맷(Scintillation Proximity Assay format)(S. Charleson et al., Mol. Pharmacol., 1992, 41, 873-879로부터 채택됨)을 통해 요오드화(125I) FLAP 억제제의 화합물-특이적 치환을 측정하는 결합 검정에서 FLAP에 결합되는 능력에 대해 평가한다.
재조합 사람 FLAP 단백질을 발현하는 sf9 곤충 세포로부터 생성된 세포 펠릿을 완충액 A[15 mM Tris-HCl (pH 7.5), 2 mM MgCl2, 0.3 mM EDTA, 1 mM PMSF]에 재현탁시킨다. 세포는 Dounce 균질화기에 의해 용해시키고, 물질은 10,000 x g로 10분 동안 원심분리한다. 이어서, 상층액을 수집하고, 100,000 x g로 60분 동안 원심분리한다. 검정을 위한 막단백질을 제조하기 위하여, 100,000 x g 펠릿의 모액을 1ml의 완충액 A에 재현탁시키고, Dounce 균질화시키고, 마지막으로 폴리트론 혼합에 적용시킨다(30초). 막단백질(25㎕, 5㎍)을 WGA SPA 비이드(Amersham)와 혼합하고, 1시간 동안 교반한다. 검정 플레이트(Perkin Elmer FlexiPlate)에 결합 완충액[100 mM Tris (pH 7.5), 140 mM NaCl, 5% 글리세롤, 2 mM EDTA, 0.5 mM TCEP, 0.05% Tween 20]으로 제조된 시험 화합물 25㎕, [125I]L-691,831(MK-591의 요오드화 유사체, Charleson et al . Mol . Pharmacol ., 41, 873-879, 1992) 25㎕ 및 마지막으로 비이드/단백질 혼합물(최종 농도: 비이드, 200 ㎍/웰; 단백질, 5㎕/웰; [125I] 프로브, 0.08 nM/웰(17 nCi/웰) 50㎕를 가한다. 플레이트는 마이크로베타(Microbeta) 플레이트 판독기로 판독하기 전에 2시간 동안 진탕한다. 비-특이적 결합은 냉각된 L-691,831 화합물 10μM의 부가에 의해 측정된다.
일반적으로, 상기 검정에서 화합물의 바람직한 효능 범위(IC50)는 0.1 nM 내지 10μM이고, 보다 바람직한 효능 범위는 0.1 nM 내지 1μM이고, 가장 바람직한 효능 범위는 0.1 nM 내지 100 nM이다.
2. 전혈 검정( Whole Blood Assay ):
화합물은 세포계에서 LTB4의 합성을 억제하는 이들의 능력을 측정하기 위하여 사람 전혈 검정에서 추가로 시험한다. 화합물을 헤파린 처리된 사람 전혈과 합하고, 37℃에서 15분 동안 항온배양시킨다. 이어서, 칼시마이신(20μM 최종, 포스페이트-완충된 염수에서 제조됨, pH 7.4)을 가하고, 혼합물을 37℃에서 추가로 30분 동안 배양시킨다. 샘플을 저속(1500 xg)으로 5분 동안 원심분리하고, 혈장 층은 제거한다. 이어서, 항체-기본 균질한 시-분할 형광법(time-resolved fluorescence method)(CisBio, Bedford, MA)을 사용하여 혈장 LTB4 농도를 측정한다.
사용 방법
본 발명의 화합물은 5-리폭시게나제 활성화 단백질(FLAP)의 효과적인 억제제이므로, 이들이 류코트리엔 생성을 억제한다고 제시하고 있다. 따라서, 본 발명의 하나의 양태에서, 본 발명의 화합물 또는 이들의 약제학적으로 허용되는 염을 사용하여 류코트리엔-매개된 장애를 치료하는 방법이 제공된다. 또다른 양태에서, 본 발명의 화합물 또는 이들의 약제학적으로 허용되는 염을 사용하는 심혈관, 염증성, 알러지, 폐 및 섬유증성 질환, 신장 질환 및 암의 치료 방법이 제공된다.
이론에 결부시키지 않고, FLAP의 활성을 억제함으로써, 본 발명의 화합물은 5-LO에 의한 아라키돈산의 산화 및 후속 대사작용으로부터 야기되는 LT의 생성을 차단한다. 따라서, FLAP 활성의 억제는 LT에 의해 매개되는 다양한 질환의 예방 및 치료를 위해 유리한 방법이다. 이들은 다음을 포함한다:
죽상 동맥경화증, 심근경색, 뇌졸중, 대동맥류, 겸상적혈구발작증(sickle cell crisis), 허혈-재관류 손상, 폐동맥 고혈압 및 패혈증을 포함한 심혈관 질환;
천식, 알러지성 비염, 비부비동염, 아토피성 피부염 및 두드러기를 포함한 알러지 질환;
천식에서 기도 재형성, 특발성 폐섬유증, 공피증, 석면증을 포함한 섬유증성 질환;
성인 호흡곤란 증후군, 바이러스성 모세기관지염, 폐쇄성 수면 무호흡증, 만성 폐쇄성 폐질환, 낭포성 섬유증 및 기관지폐 형성 이상을 포함한 폐 증후군;
류마티스 관절염, 골관절염, 통풍, 사구체신염, 간질성 방광염, 건선, 염증성 장질환, 염증성 통증, 다발성 경화증, 전신성 홍반성 낭창, 이식 거부, 염증성 및 알러지성 안질환을 포함한 염증성 질환;
고형 종양, 백혈병 및 림프종을 포함한 암; 및
사구체신염을 포함한 신장 질환.
상기 기술한 질환 및 상태의 치료를 위해, 치료학적 유효 용량은 일반적으로 본 발명의 화합물의 용량당 체중 1㎏당 약 0.01 내지 약 100㎎; 바람직하게는 용량당 체중 1㎏당 약 0.1 내지 약 20㎎의 범위이다. 예를 들면, 70㎏인 사람에 투여시, 용량 범위는 본 발명의 화합물의 용량당 약 0.7 내지 약 700㎎이고, 바람직하게는 용량당 약 7.0 내지 약 1400㎎이다. 어느 정도의 통상적인 용량 최적화가 최적의 투약 수준 및 패턴을 결정하는데 필요할 수 있다. 활성 성분은 하루에 1 내지 6회로 투여할 수 있다.
일반적인 투여 및 약제학적 조성물
약제로서 사용되는 경우에, 본 발명의 화합물은 통상적으로 약제학적 조성물의 형태로 투여된다. 당해 조성물은 약제 분야에 잘 공지된 방법을 사용하여 제조하고, 적어도 하나의 본 발명의 화합물을 포함한다. 본 발명의 화합물은 또한 단독으로 투여되거나, 본 발명의 화합물의 안정성을 개선하고, 특정 양태로 이들을 함유하는 약제학적 조성물의 투여를 용이하게 하고, 증가된 용해 또는 분산, 증가된 길항제 활성을 제공하고, 보조 치료를 제공하는 등의 보조제와 배합하여 투여될 수 있다. 본 발명에 따르는 화합물은 이들 자체로, 또는 본 발명에 따르는 다른 활성 물질과 함께, 임의로 또한 다른 약물학적 활성 물질과 함께 사용될 수 있다. 일반적으로, 본 발명의 화합물은 치료학적 또는 약제학적 유효량으로 투여될 수 있지만, 진단 또는 다른 목적을 위해 보다 적은 양으로 투여될 수 있다.
순수한 형태로서 또는 적절한 약제학적 조성물 형태로서 본 발명의 화합물의 투여는 약제학적 조성물의 허용되는 투여 형태 중 임의의 것을 사용하여 수행할 수 있다. 따라서, 고체, 반고체, 동결건조 분말 또는 액체 용량형(예: 정제, 좌제, 환제, 연질 탄성 및 경질 젤라틴 캡슐제, 산제, 용제, 현탁제 또는 에어로졸 등)으로, 바람직하게는 정밀한 용량의 간단한 투여에 적합한 단위 용량형으로, 예를 들면, 경구, 구강내(예: 설하), 비내, 비경구, 국소, 경피, 질내 또는 직장내 투여할 수 있다. 약제학적 조성물은 일반적으로 통상적인 약제학적 담체 또는 부형제 및 활성제로서 본 발명의 화합물을 포함하고, 또한 다른 의학 제제, 약제학적 제제, 담체, 보조제, 희석제, 비히클 또는 이들의 혼합물을 포함할 수 있다. 다양한 형태 또는 투여를 위한 약제학적 조성물의 제조 방법 뿐만 아니라, 상기 약제학적으로 허용되는 부형제, 담체 또는 부가제는 당해 분야의 숙련가에게 잘 공지되어 있다. 최신기술은, 예를 들면, 문헌[참조: Remington : The Science and Practice of Pharmacy, 20th Edition, A. Gennaro (ed.), Lippincott Williams & Wilkins, 2000; Handbook of Pharmaceutical Additives, Michael & Irene Ash (eds.), Gower, 1995; Handbook of Pharmaceutical Excipients, A.H. Kibbe (ed.), American Pharmaceutical Ass'n, 2000; H.C. Ansel and N.G. Popovish, Pharmaceutical Dosage Forms and Drug Delivery Systems, 5th ed., Lea and Febiger, 1990; 이들 각각은 최신기술을 보다 잘 기술하기 위하여 이들의 전문이 본 명세서에 참조로 인용된다]에 의해 입증된다.
당해 분야의 숙련가가 예상하는 바와 같이, 제형을 효능있게 하는데 필요한 적절한 물리적 특성(예: 수용해도)을 갖는 특별한 약제학적 제형에 이용되는 본 발명의 화합물의 형태가 선택될 것이다(예: 염).
Claims (25)
- 화학식 I의 화합물 또는 약제학적으로 허용되는 이의 염.
화학식 I

상기 화학식 I에서,
R1 및 R2는 각각 독립적으로 수소, C1 -7 알킬 또는 C3 -10 카보사이클이고, 단, R1 및 R2는 모두 수소가 아니고;
R3은 질소, 산소 및 황으로부터 선택되는 1 내지 3개의 헤테로 원자를 함유하는 5원 내지 11원 헤테로아릴 환이고, 여기서, 상기 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1 -5 알킬, C1 -5 알콕시, C1 -3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -6 알킬아미노 및 C1-3 디알킬아미노로부터 선택된 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R4는 수소, C1 -3 알킬, 할로겐 또는 니트릴이고;
R5는 C1 -6 알킬, C3 -10 카보사이클, 3원 내지 11원 헤테로사이클, 아릴, 5원 내지 11원 헤테로아릴, -C(O)-R6, 하이드록시 또는 NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R6은 C3 -8 헤테로사이클 또는 -NH-5원 내지 6원 헤테로사이클이고, 이들 각각은 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8은 각각 독립적으로 수소, C1 -6 알킬에 의해 임의로 치환된 5원 내지 6원 헤테로사이클, 하이드록시에 의해 임의로 치환된 C3 -10 카보사이클 또는 C1 -6 알킬이고;
R9, R10 및 R11은 독립적으로
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 3원 내지 6원 헤테로사이클, C1 -6 알콕시, C1-6알콕시-O-C1 - 6알킬, -CO2R12, -C(O)N(R12)(R13) 또는 -S(O)nC1- 6알킬에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 6알콕시,
(h) -N(R12)(R13),
(i) -S(O)nC1- 6알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 1 내지 3개의 C1 -6 알킬 그룹에 의해 임의로 치환된 3원 내지 10원 헤테로사이클릭 그룹,
(n') 옥소,
(o) -C(O)-C1 -3 알킬로부터 선택되고;
R12 및 R13은 각각 독립적으로 -H, -C1 - 6알킬, C(O)C1- 6알킬 및 3원 내지 6원 헤테로사이클릭 그룹으로부터 선택되고, 이들 각각은 1 내지 3개의 C1 - 6알킬 그룹, -OH, C1 - 6알콕시, -C(O)N(R14)(R15), -S(O)nC1- 6알킬, CN, 3원 내지 6원 헤테로사이클릭 그룹, -OC1 - 6알킬, CF3에 의해 임의로 독립적으로 치환되거나;
R12 및 R13은, 이들이 부착된 질소 환과 함께, 1 내지 3개의 -OH, CN, -OC1 - 6알킬 또는 옥소에 의해 임의로 치환된 헤테로사이클릴 환을 형성하고;
R14 및 R15는 각각 독립적으로 -H 및 -C1 - 6알킬로부터 선택되고;
n은 0, 1 또는 2이다. - 제1항에 있어서,
R1 및 R2가 각각 독립적으로 수소, 메틸, 에틸, 프로필, 이소프로필, 부틸, 이소부틸, 3급-부틸, 펜틸, 헥실, 사이클로프로필, 사이클로부틸, 사이클로펜틸 또는 사이클로헥실이고, 단, R1 및 R2는 모두 수소가 아니고;
R3이 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 피롤릴, 이미다졸릴, 티에닐, 푸라닐 또는 티아졸릴이고, 여기서, 각각의 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1-3 알킬, C1-3 알콕시, C1-3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -6 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되거나;
R3이 피리도옥사지닐, 디하이드로-피리도옥사지닐, 디하이드로-피롤로피리디닐, 피롤로피리디닐 또는 피롤로피라지닐이고, 여기서, 각각의 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1 -3 알킬, C1 -3 알콕시, C1 -3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -3 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R4가 수소, 메틸 또는 플루오로이고;
R5가 메틸, 에틸, 프로필, 이소프로필, 부틸, 이소부틸, 3급-부틸, 펜틸, 헥실, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 사이클로헵틸, 사이클로옥틸, 페닐, 피페리디닐, 피페라지닐, 모르폴리닐, 티오모르폴리닐, 아제티디닐, 피롤리디닐, 테트라하이드로피라닐, 피롤릴, 티에닐, 푸라닐, 티아졸릴, 옥사졸릴, 이속사졸릴, 피라졸릴, 이미다졸릴, 트리아졸릴, 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 퀴놀리닐, 이소퀴놀리닐, 인돌릴, 피롤로피리디닐, 피롤로피리미디닐, -C(O)-R6, 하이드록시 또는 -NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R6이 피페리디닐, 피페라지닐, 테트라하이드로피라닐, 모르폴리닐, 티오모르폴리닐 또는 -NH-피페라디닐이고, 이들 각각은 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8이 각각 독립적으로 수소, 메틸에 의해 임의로 치환된 5원 내지 6원 헤테로사이클, 하이드록시에 의해 임의로 치환된 C3 -6 카보사이클, 또는 C1 -5 알킬이고;
R9, R10 및 R11이 독립적으로
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 3원 내지 6원 헤테로사이클, C1 -6 알콕시, C1-6알콕시-O-C1 - 6알킬, -CO2R12, -C(O)N(R12)(R13) 또는 -S(O)nC1- 6알킬에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 6알콕시,
(h) -N(R12)(R13),
(i) -S(O)nC1- 6알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 1 내지 3개의 C1 -6 알킬 그룹에 의해 임의로 치환된 3원 내지 8원 헤테로사이클릭 그룹,
(n') 옥소,
(o) -C(O)-C1 -3 알킬로부터 선택되고;
R12 및 R1 3이 각각 독립적으로 -H, -C1 - 6알킬, C(O)C1- 6알킬 및 3원 내지 6원 헤테로사이클릭 그룹으로부터 선택되고, 이들 각각은 1 내지 3개의 C1 - 6알킬 그룹, -OH, C1 - 6알콕시, -C(O)N(R14)(R15), -S(O)nC1- 6알킬, CN, 3원 내지 6원 헤테로사이클릭 그룹, -OC1 - 6알킬, CF3에 의해 임의로 독립적으로 치환되거나;
R12 및 R1 3이, 이들이 부착된 질소 환과 함께, 1 내지 3개의 -OH, CN, -OC1 - 6알킬 또는 옥소에 의해 임의로 치환된 헤테로사이클릴 환을 형성할 수 있고;
R14 및 R1 5가 각각 독립적으로 -H 및 -C1 - 4알킬로부터 선택되고;
n이 1 또는 2인, 화합물 또는 약제학적으로 허용되는 이의 염. - 제1항 또는 제2항에 있어서,
R1 및 R2가 각각 독립적으로 수소, 메틸, 에틸, 프로필, 이소프로필, 3급-부틸, 사이클로프로필 또는 사이클로부틸이고, 단, R1 및 R2는 모두 수소가 아닌, 화합물 또는 약제학적으로 허용되는 이의 염. - 제1항 내지 제3항 중의 어느 한 항에 있어서,
R3이 피리디닐, 피라지닐, 피리다지닐 또는 피리미디닐이고, 여기서, 각각의 헤테로아릴 환은, 1 내지 3개의 할로겐 원자에 의해 임의로 치환된 C1 -3 알킬, C1 -3 알콕시, C1 -3 하이드록시, 할로겐, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3-6 카보사이클, C1 -5 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 2개의 그룹에 의해 임의로 독립적으로 치환되거나;
R3이 피리도옥사지닐, 디하이드로-피리도옥사지닐, 디하이드로-피롤로피리디닐, 피롤로피리디닐 또는 피롤로피라지닐인, 화합물 또는 약제학적으로 허용되는 이의 염. - 제1항 내지 제4항 중의 어느 한 항에 있어서,
R5가 메틸, 에틸, 프로필, 이소프로필, 부틸, 펜틸, 헥실, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 페닐, 아제티디닐, 피페리디닐, 피페라지닐, 모르폴리닐, 테트라하이드로피라닐, 티아졸릴, 옥사졸릴, 이속사졸릴, 피라졸릴, 이미다졸릴, 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 퀴놀리닐, 이소퀴놀리닐, -C(O)-피페리지닐, -C(O)-피페리디닐, -C(O)-모르폴리닐, -C(O)-NH-피페리디닐, 하이드록시 또는 -NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8이 각각 독립적으로 수소, 메틸에 의해 임의로 치환된 5원 내지 6원 헤테로사이클, 하이드록시에 의해 임의로 치환된 C3 -6 카보사이클, 또는 C1 -C5 알킬이고;
R9, R10 및 R11이 독립적으로
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 모르폴리닐, 피페라지닐, C1 -6 알콕시, C1-3알콕시-O-C1 - 3알킬, -CO2R12 또는 -C(O)N(R12)(R13)에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 6알콕시,
(h) -N(R12)(R13),
(i) -S(O)nC1- 6알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 모르폴리닐, 피페라지닐, 피페리디닐 또는 옥세타닐(이들 각각은 메틸 그룹에 의해 임의로 치환됨),
(n') 옥소,
(o) -C(O)-CH3으로부터 선택되고;
R12 및 R1 3이 각각 독립적으로 -H 및 -C1 - 6알킬로부터 선택되고, 여기서, 알킬 그룹은 1 내지 3개의 -OH, C1 - 6알콕시, -C(O)N(R14)(R15) 또는 -S(O)nC1- 6알킬에 의해 임의로 치환되고;
R14 및 R1 5가 각각 독립적으로 -H 및 -C1 - 4알킬로부터 선택되고;
n이 2인, 화합물 또는 약제학적으로 허용되는 이의 염. - 제1항 또는 제2항에 있어서,
R1 및 R2가 각각 독립적으로 수소, 메틸, 에틸, 프로필, 이소프로필, 3급-부틸, 사이클로프로필 또는 사이클로부틸이고, 단, R1 및 R2는 모두 수소가 아니고;
R3이 피리디닐, 피라지닐, 피리다지닐 또는 피리미디닐이고, 여기서, 각각의 헤테로아릴 환은 메틸, 메톡시, -CH2OH, 트리플루오로메틸, 브로모, 클로로, 플루오로, 하이드록시, -O-벤질, 옥소, 시아노, 아미노, -NH-C3 -6 카보사이클, C1 -4 알킬아미노 및 C1 -3 디알킬아미노로부터 선택된 1 내지 2개의 그룹에 의해 임의로 독립적으로 치환되거나;
R3이 피리도옥사지닐, 디하이드로-피리도옥사지닐, 디하이드로-피롤로피리디닐, 피롤로피리디닐 또는 피롤로피라지닐이고;
R4가 수소이고;
R5가 메틸, 에틸, 프로필, 이소프로필, 부틸, 펜틸, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 페닐, 아제티디닐, 피페리디닐, 피페라지닐, 모르폴리닐, 테트라하이드로피라닐, 티아졸릴, 옥사졸릴, 이속사졸릴, 피라졸릴, 이미다졸릴, 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 퀴놀리닐, 이소퀴놀리닐, -C(O)-피페라지닐, -C(O)-모르폴리닐, -C(O)-NH-피페리디닐, 하이드록시 또는 -NR7R8이고, 여기서, 각각의 R5는 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환되고;
R7 및 R8이 각각 독립적으로 수소, 메틸 그룹에 의해 임의로 치환된 피페리디닐, 하이드록시 그룹에 의해 임의로 치환된 사이클로헥실, 메틸 또는 에틸이고;
R9, R10 및 R11이 독립적으로
(a) -H,
(b) -OH,
(c) 할로겐,
(d) -CN,
(e) -CF3,
(f) 1 내지 3개의 -OH, -N(R12)(R13), 모르폴리닐, 피페라지닐, C1 -3 알콕시, C1-3알콕시-O-C1 - 3알킬, -CO2H 또는 -C(O)N(R12)(R13)에 의해 임의로 치환된 C1 - 6알킬,
(g) C1 - 3알콕시,
(h) -N(R12)(R13),
(i) -S(O)2C1 - 2알킬,
(j) -CO2R12,
(k) -C(O)N(R12)(R13),
(l) -S(O)2N(R12)(R13),
(m) 모르폴리닐, 피페라지닐 또는 옥세타닐(이들 각각은 메틸 그룹에 의해 임의로 치환됨),
(n') 옥소,
(o) -C(O)-CH3으로부터 선택되고;
R12 및 R1 3이 각각 독립적으로 -H 및 -C1 - 6알킬로부터 선택되고, 여기서, 알킬 그룹은 1 내지 3개의 -OH, C1 - 6알콕시, -C(O)N(R14)(R15) 또는 -S(O)2C1 - 6알킬에 의해 임의로 독립적으로 치환되고;
R14 및 R1 5가 각각 독립적으로 -H 및 -C1 - 4알킬로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염. - 제6항에 있어서,
R1이 메틸이고,
R2가 메틸, 에틸, 이소프로필, 3급-부틸, 사이클로프로필 및 사이클로부틸로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염. - 제6항에 있어서,
R3이로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염.
- 제6항에 있어서,
R5가 R9, R10 및 R11로부터 선택되는 1 내지 3개의 그룹에 의해 임의로 독립적으로 치환된 피라졸릴인, 화합물 또는 약제학적으로 허용되는 이의 염. - 제6항에 있어서,
R1이 메틸이고,
R2가 메틸, 에틸, 이소프로필, 3급-부틸, 사이클로프로필 및 사이클로부틸로부터 선택되고,
R3이로부터 선택되고,
R4가 수소이고,
R5가로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염.
- 제10항에 있어서,
R2가 사이클로프로필 또는 사이클로부틸인, 화합물 또는 약제학적으로 허용되는 이의 염. - 제10항에 있어서,
R2가 메틸, 에틸, 이소프로필 및 3급-부틸로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염. - 제6항에 있어서,
R3이로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염.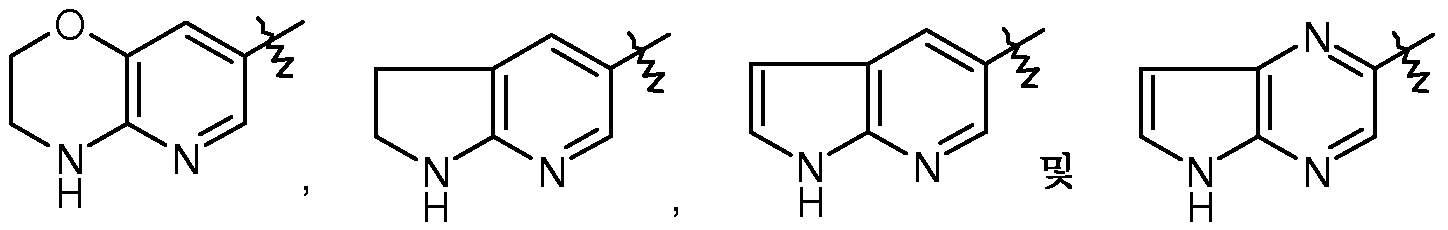
- 제6항에 있어서,
R3이
로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염.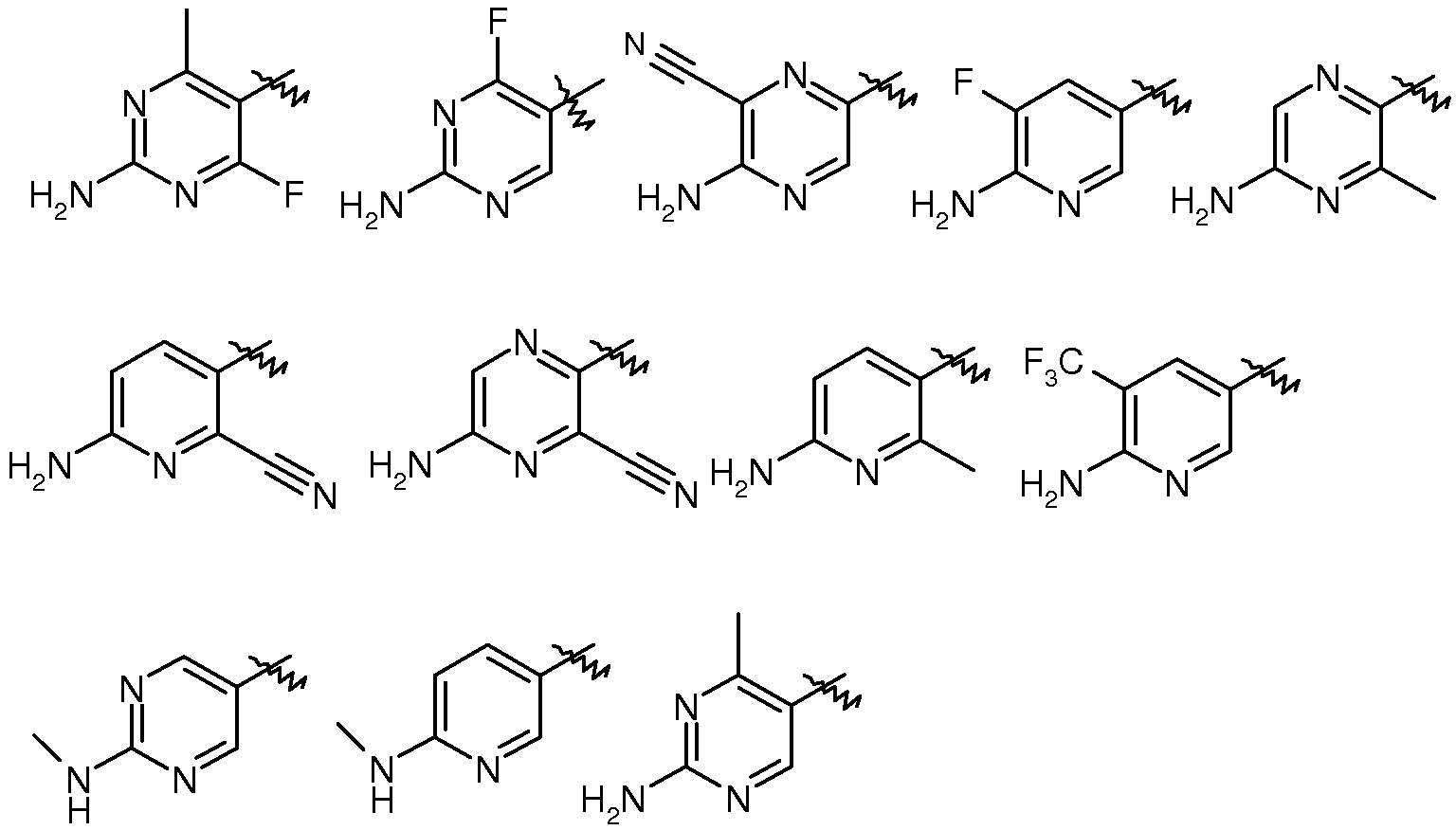
- 제10항에 있어서,
R1이 메틸이고,
R2가 사이클로프로필이고,
R3이로부터 선택되고,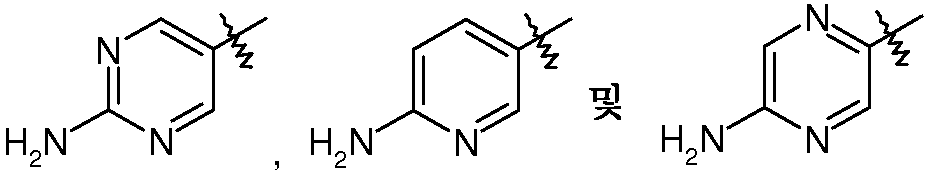
R4가 수소이고,
R5가로부터 선택되는, 화합물 또는 약제학적으로 허용되는 이의 염.
- 다음으로 이루어진 그룹으로부터 선택되는 화합물 또는 약제학적으로 허용되는 이의 염:



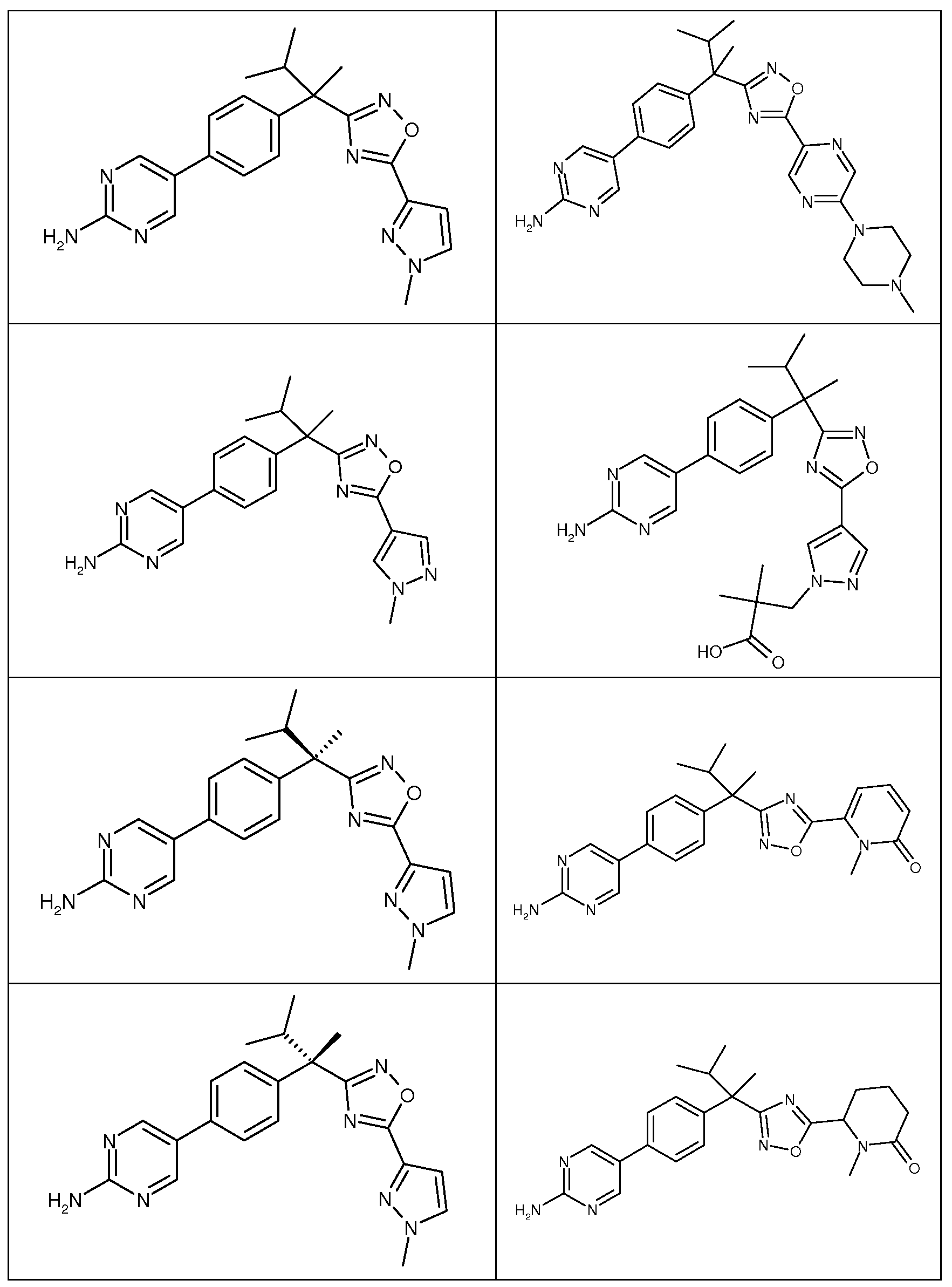




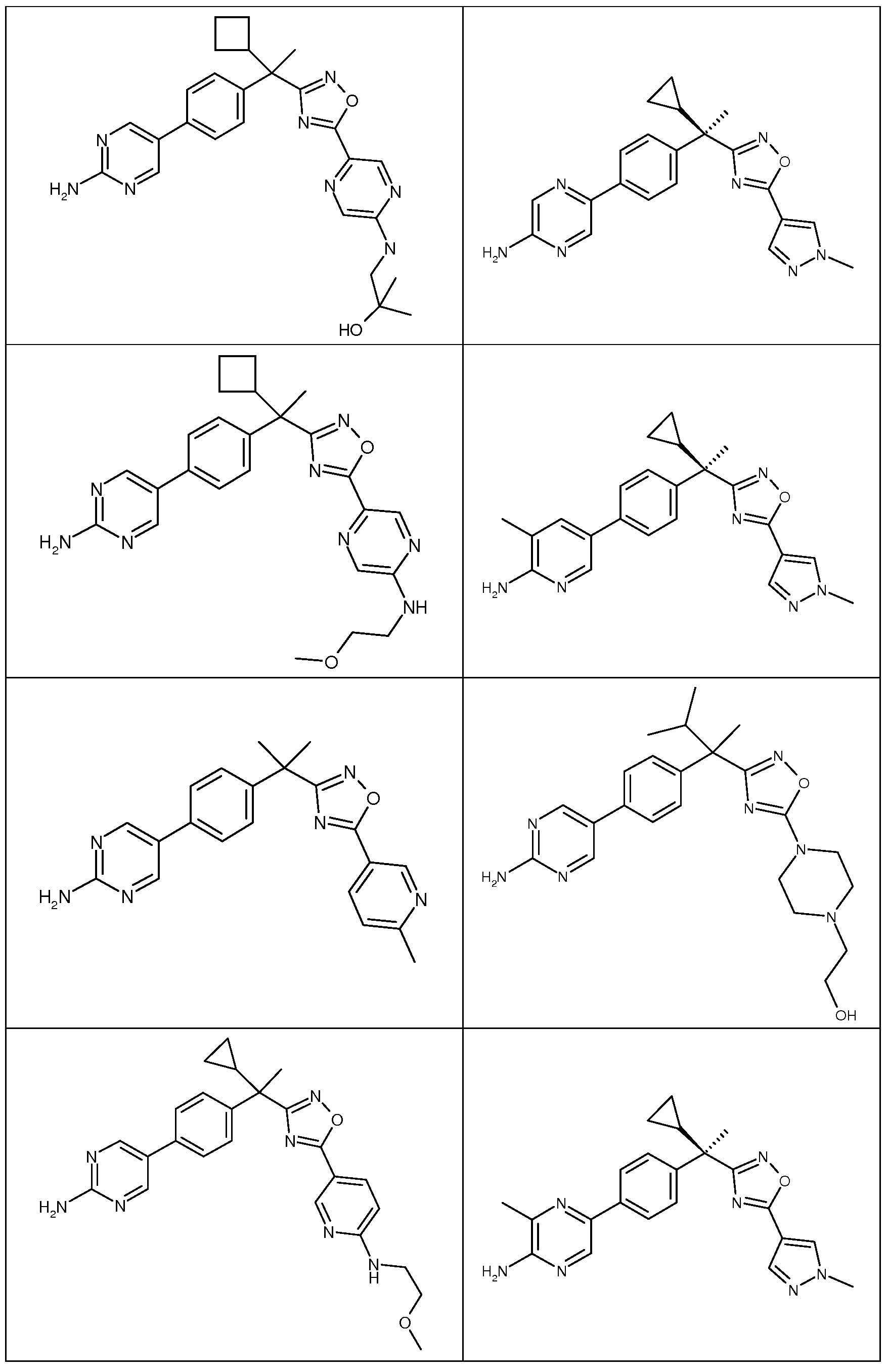
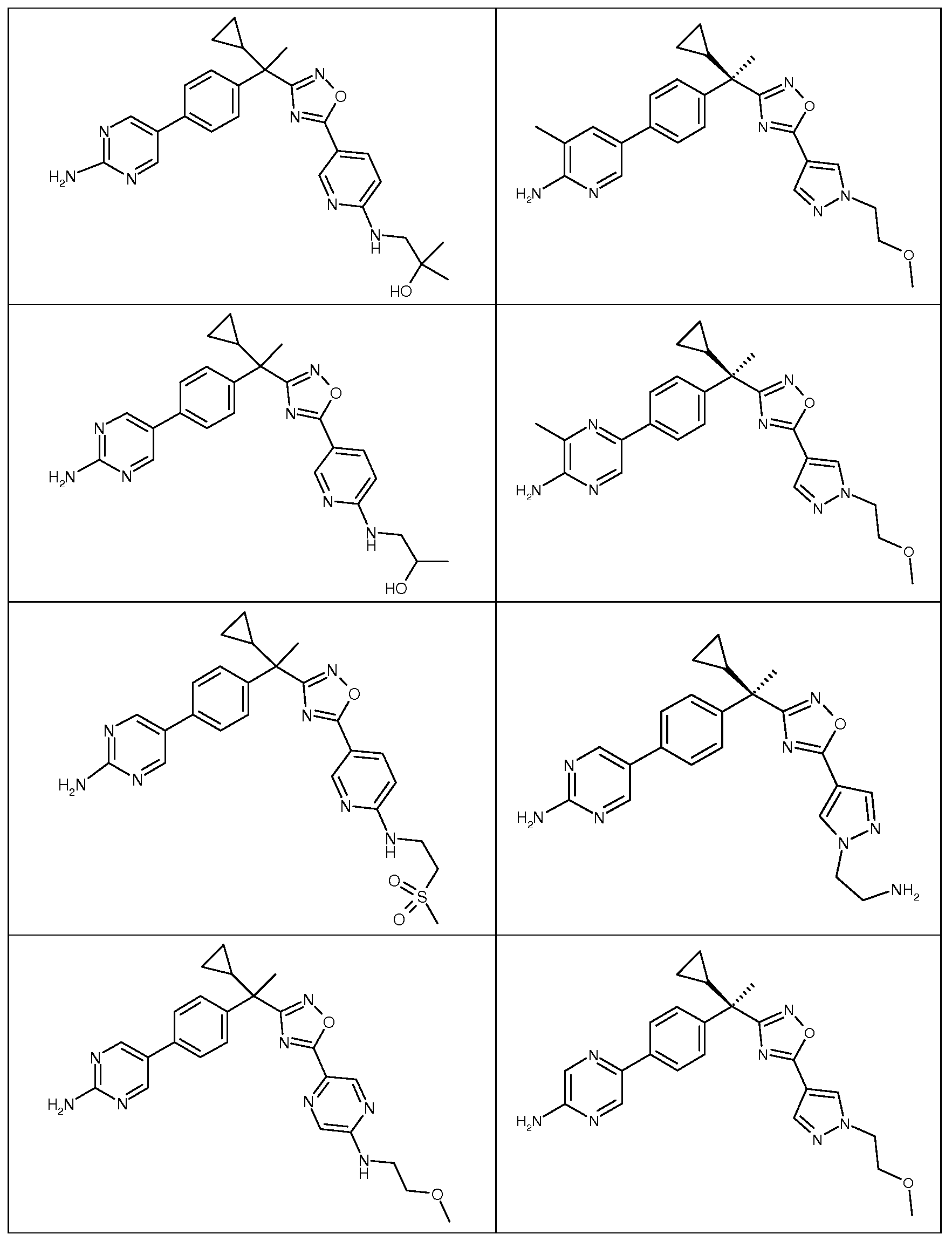


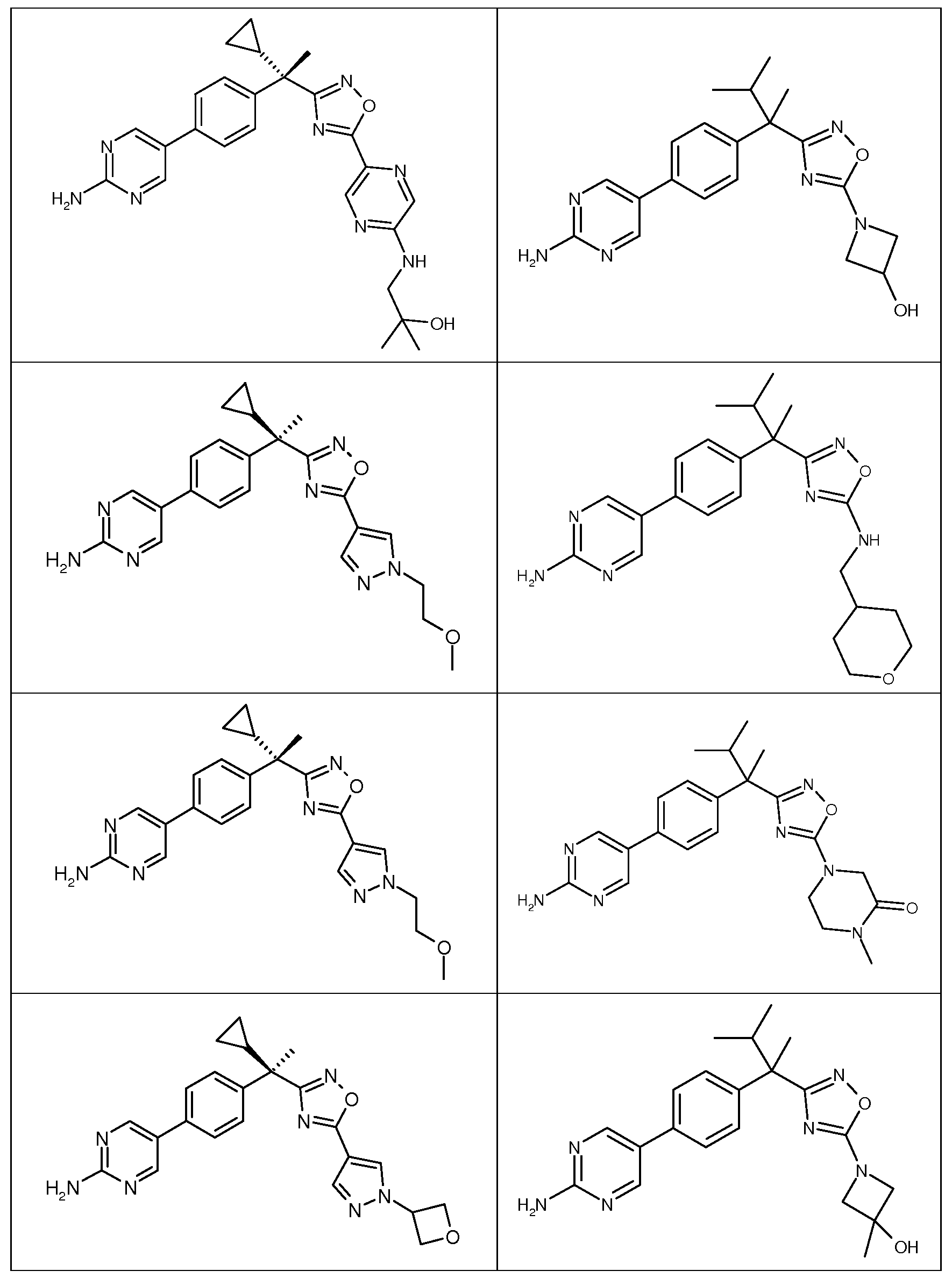
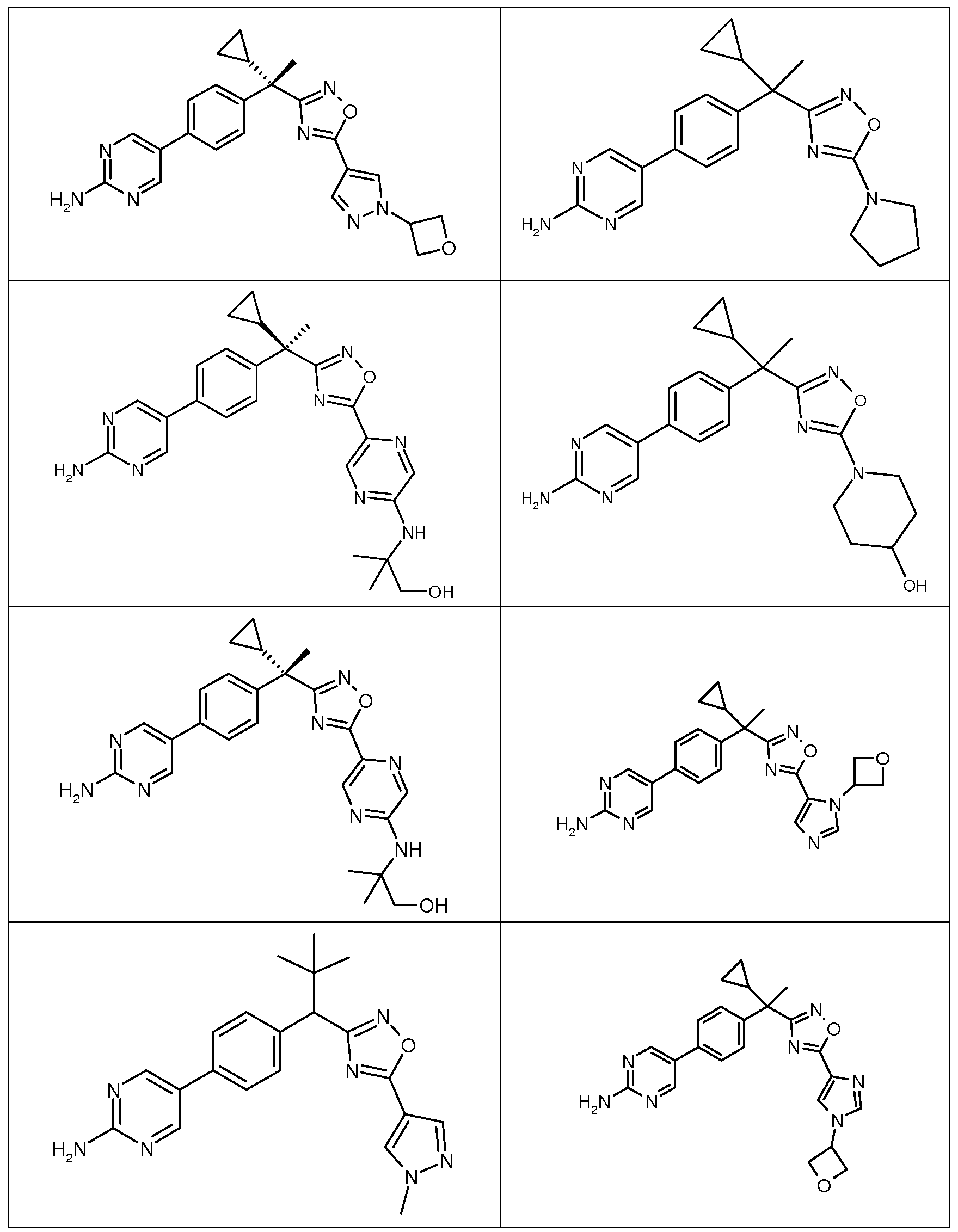

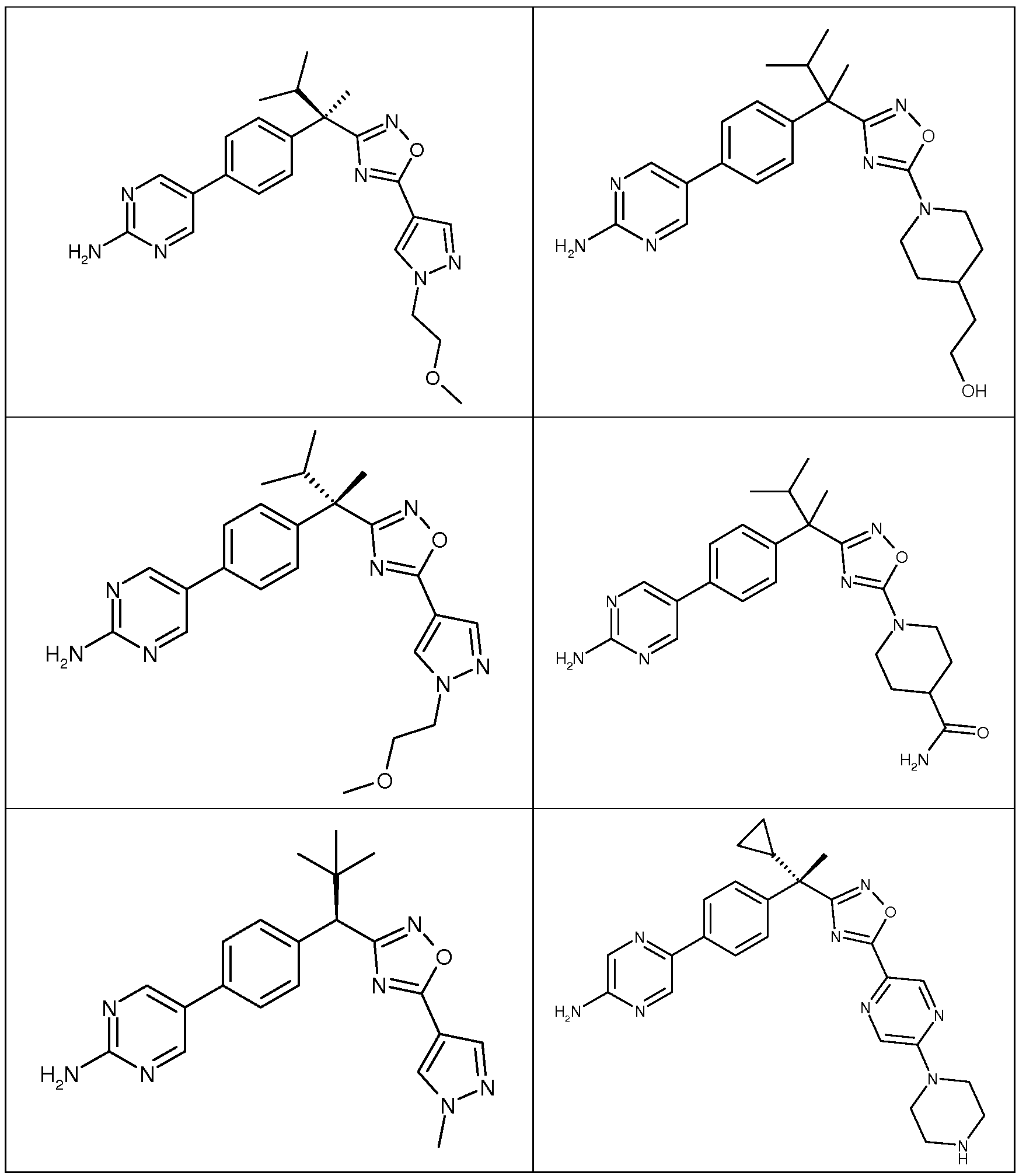




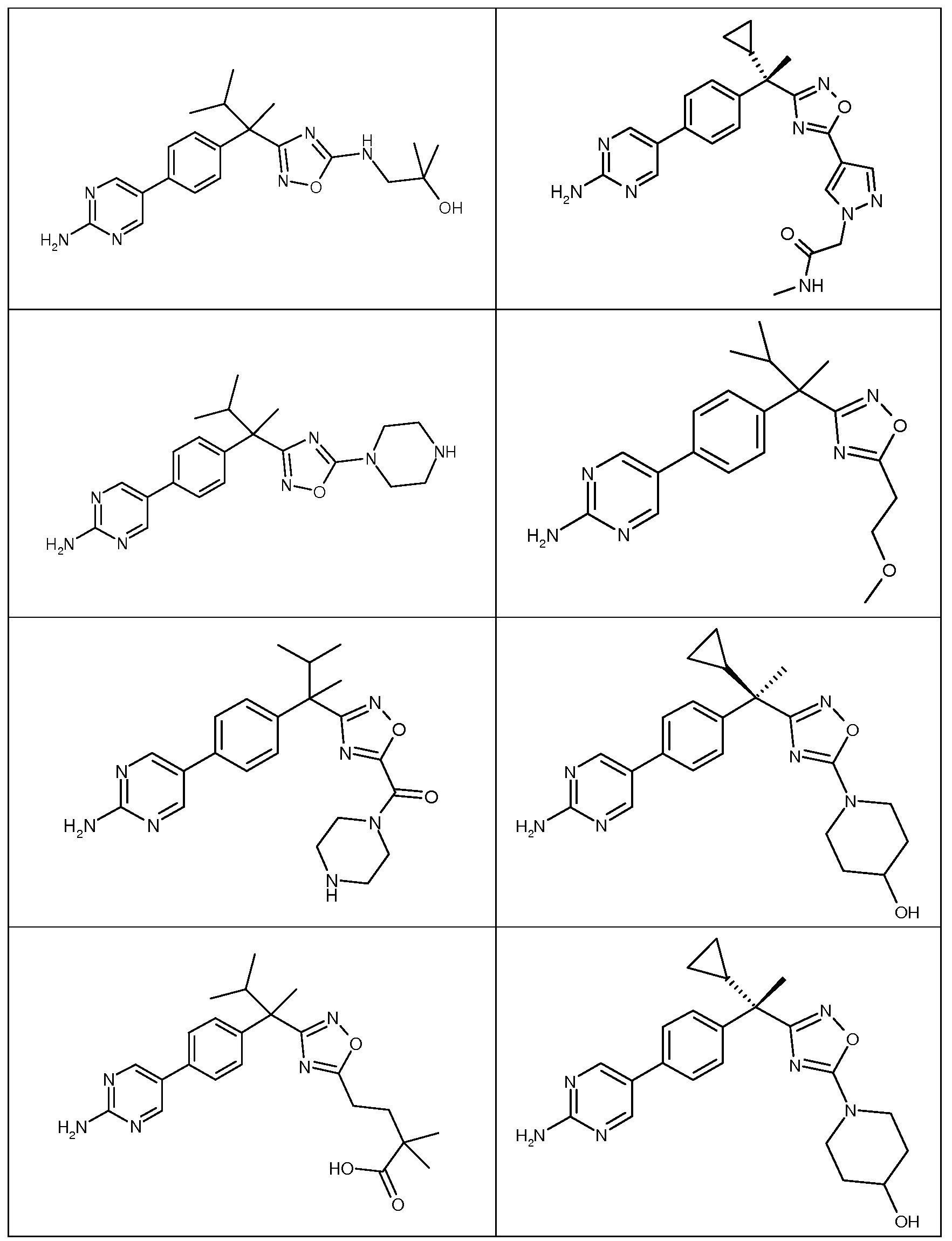




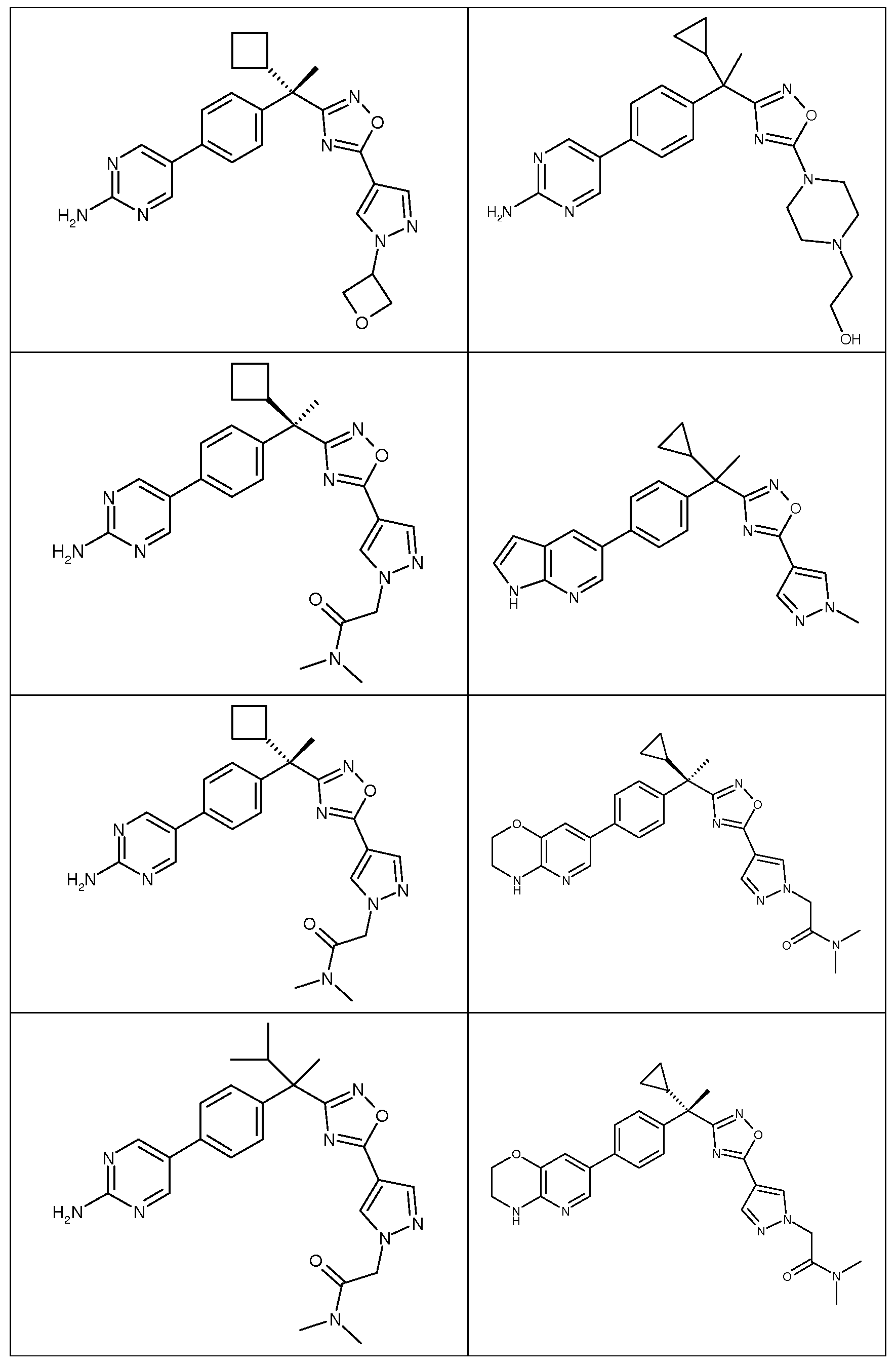










- 제16항에 있어서, 다음으로 이루어진 그룹으로부터 선택되는 화합물 또는 약제학적으로 허용되는 이의 염:
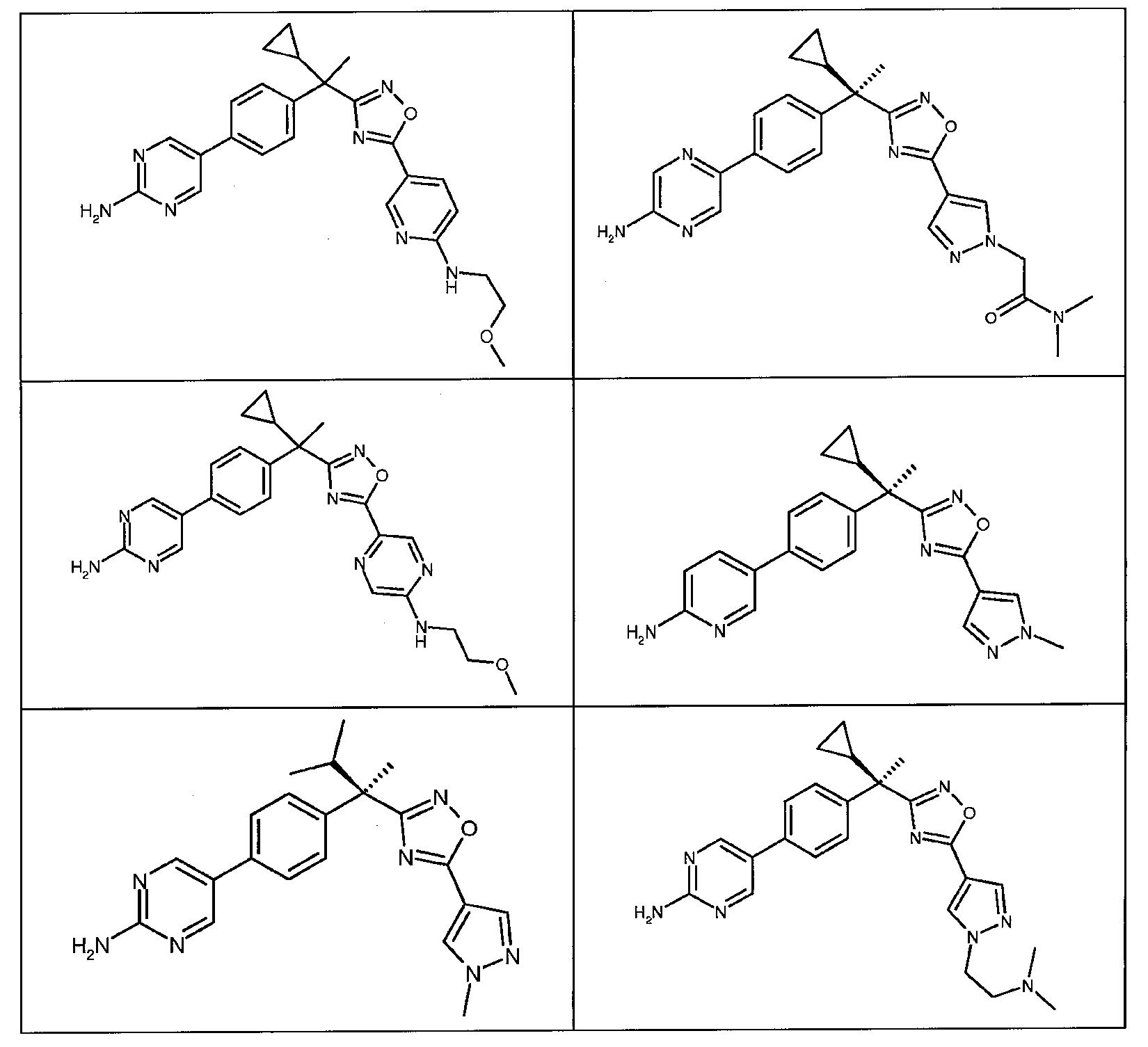
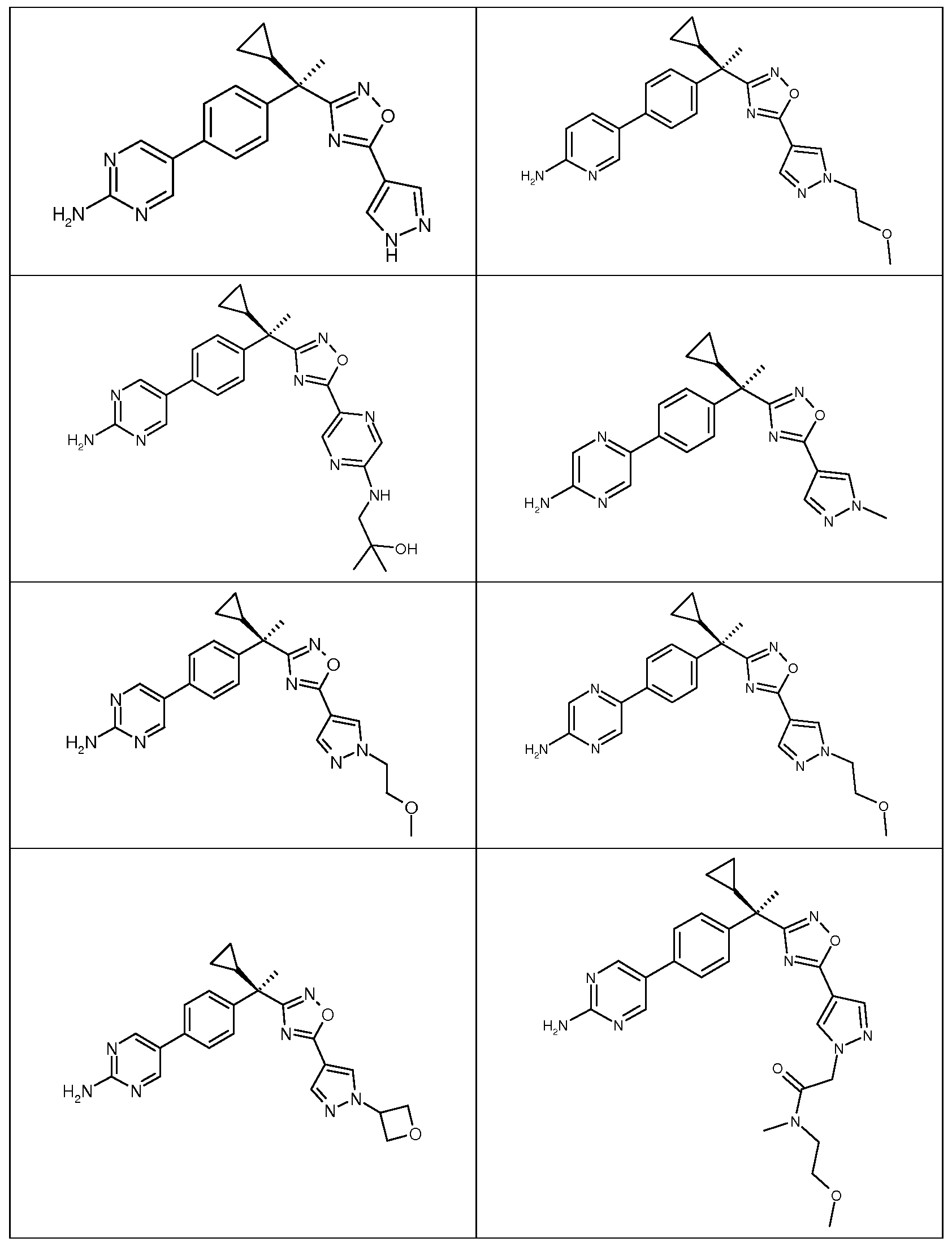
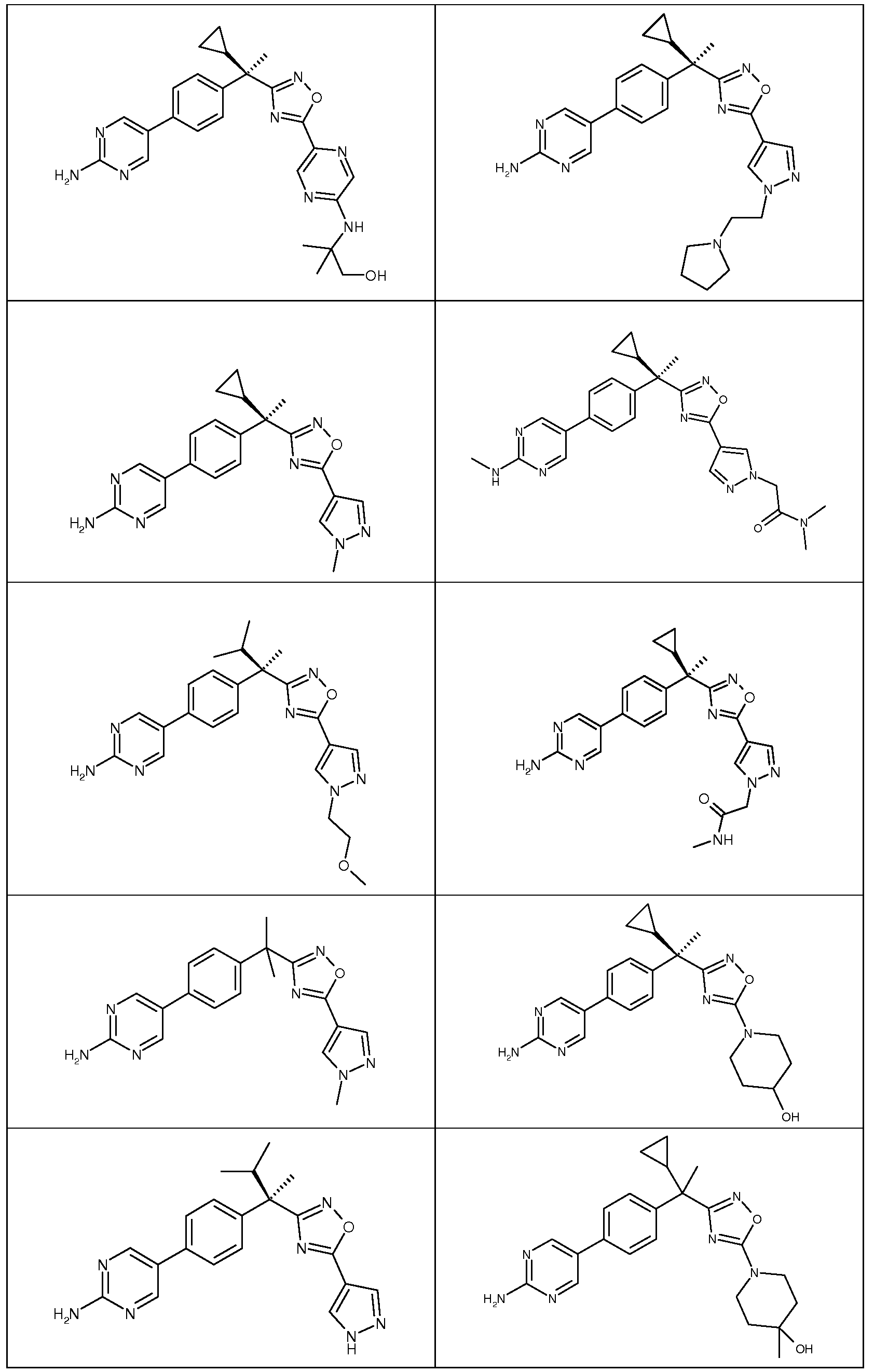



- 제17항에 있어서, 다음으로 이루어진 그룹으로부터 선택되는 화합물 또는 약제학적으로 허용되는 이의 염:


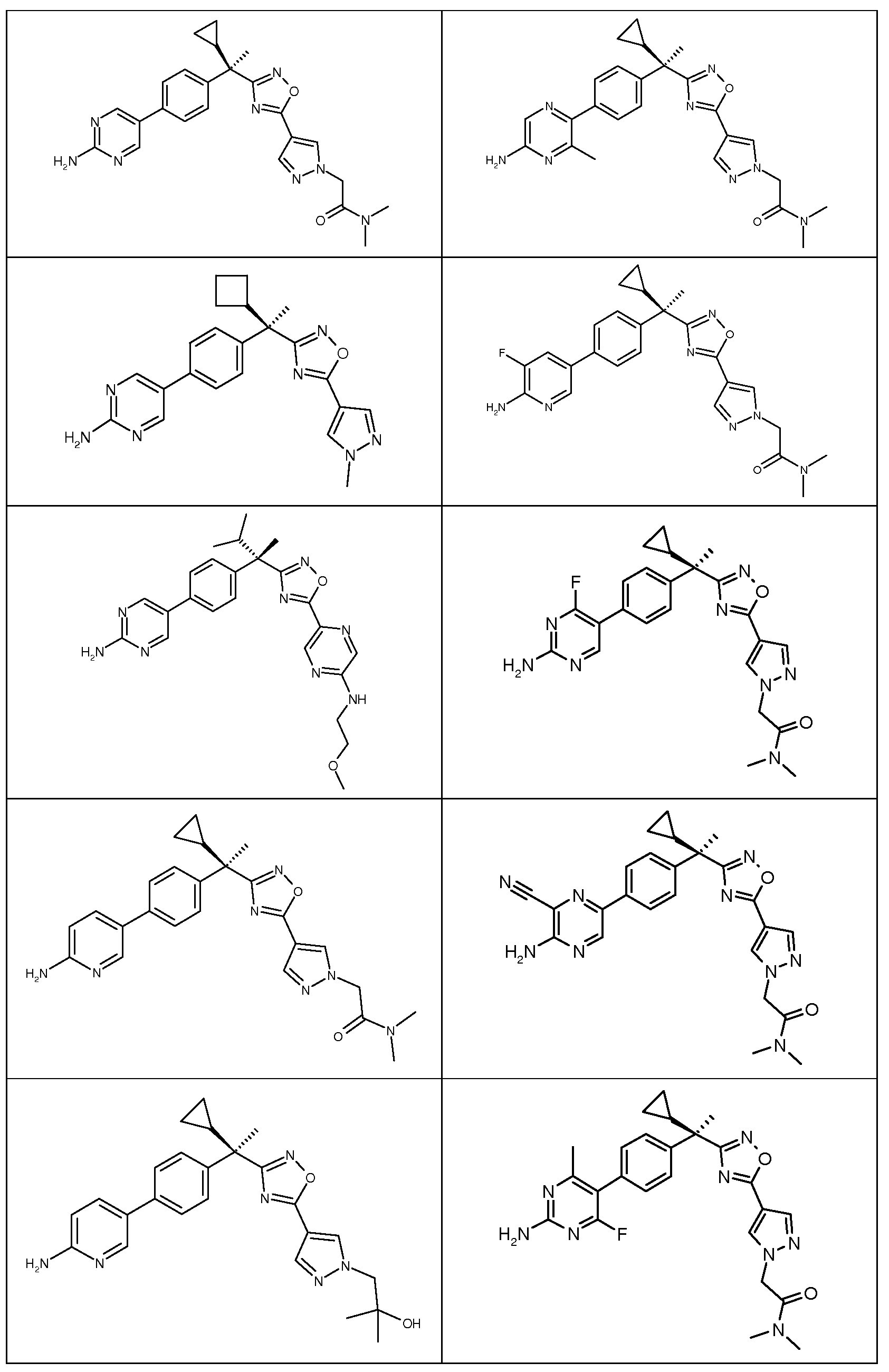
- 제1항 내지 제18항 중의 어느 한 항에 따르는 화합물, 또는 약제학적으로 허용되는 이의 염 및 약제학적으로 허용되는 부형제 및/또는 담체를 포함하는 약제학적 조성물.
- 제1항 내지 제18항 중의 어느 한 항에 따르는 화합물 또는 약제학적으로 허용되는 이의 염의 유효량을 류코트리엔-매개된 장애의 치료를 필요로 하는 환자에게 투여함을 포함하는, 류코트리엔-매개된 장애를 치료하는 방법.
- 제20항에 있어서, 상기 류코트리엔-매개된 장애가 심혈관, 염증성, 알러지, 폐 및 섬유증성 질환, 신장 질환 및 암으로부터 선택되는, 방법.
- 제21항에 있어서, 상기 류코트리엔-매개된 장애가 죽상 동맥경화증인, 방법.
- 약제로서 사용하기 위한 제1항 내지 제18항 중의 어느 한 항에 따르는 화합물 또는 약제학적으로 허용되는 이의 염.
- 류코트리엔-매개된 장애의 치료를 위한 제1항 내지 제18항 중의 어느 한 항에 따르는 화합물 또는 약제학적으로 허용되는 이의 염.
- 심혈관, 염증성, 알러지, 폐 및 섬유증성 질환, 신장 질환 및 암으로부터 선택되는 류코트리엔-매개된 장애의 치료를 위한 제1항 내지 제18항 중의 어느 한 항에 따르는 화합물 또는 약제학적으로 허용되는 이의 염.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37392510P | 2010-08-16 | 2010-08-16 | |
| US61/373,925 | 2010-08-16 | ||
| US201161492176P | 2011-06-01 | 2011-06-01 | |
| US61/492,176 | 2011-06-01 | ||
| PCT/US2011/047356 WO2012024150A1 (en) | 2010-08-16 | 2011-08-11 | Oxadiazole inhibitors of leukotriene production |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20130100105A true KR20130100105A (ko) | 2013-09-09 |
Family
ID=44511591
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020137003938A KR20130100105A (ko) | 2010-08-16 | 2011-08-11 | 류코트리엔 생성 억제제인 옥사디아졸 |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US8658661B2 (ko) |
| EP (1) | EP2606045B1 (ko) |
| JP (1) | JP5619284B2 (ko) |
| KR (1) | KR20130100105A (ko) |
| CN (1) | CN103180314A (ko) |
| AP (1) | AP2013006678A0 (ko) |
| AR (1) | AR082696A1 (ko) |
| AU (1) | AU2011292285A1 (ko) |
| BR (1) | BR112013003439A2 (ko) |
| CA (1) | CA2807364A1 (ko) |
| CL (1) | CL2013000455A1 (ko) |
| CO (1) | CO6680645A2 (ko) |
| EA (1) | EA201300250A1 (ko) |
| EC (1) | ECSP13012494A (ko) |
| MX (1) | MX2013001872A (ko) |
| NZ (1) | NZ605690A (ko) |
| PE (1) | PE20130751A1 (ko) |
| SG (1) | SG187549A1 (ko) |
| TW (1) | TW201238947A (ko) |
| UY (1) | UY33557A (ko) |
| WO (1) | WO2012024150A1 (ko) |
| ZA (1) | ZA201300238B (ko) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2606045B1 (en) | 2010-08-16 | 2016-01-06 | Boehringer Ingelheim International GmbH | Oxadiazole inhibitors of leukotriene production |
| JP5862669B2 (ja) * | 2010-08-26 | 2016-02-16 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | ロイコトリエン産生のオキサジアゾール阻害剤 |
| PE20131398A1 (es) * | 2010-09-23 | 2013-12-19 | Boehringer Ingelheim Int | Inhibidores de oxadiazol de la produccion de leucotrienos |
| US8580825B2 (en) * | 2010-09-23 | 2013-11-12 | Boehringer Ingelheim International Gmbh | Oxadiazole inhibitors of leukotriene production |
| EP2651930B1 (en) * | 2010-12-16 | 2015-10-28 | Boehringer Ingelheim International GmbH | Biarylamide inhibitors of leukotriene production |
| JP5945870B2 (ja) | 2011-03-18 | 2016-07-05 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | カルボン酸の調製方法 |
| WO2013116182A1 (en) * | 2012-01-31 | 2013-08-08 | Boehringer Ingelheim International Gmbh | Heterocyclic compounds as inhibitors of leukotriene production |
| AR089853A1 (es) * | 2012-02-01 | 2014-09-24 | Boehringer Ingelheim Int | Inhibidores de oxadiazol de la produccion de leucotrienos para terapia de combinacion, composicion farmaceutica, uso |
| US9029592B2 (en) * | 2012-02-09 | 2015-05-12 | Boehringer Ingelheim International Gmbh | Process for preparing carboxamidine compounds |
| EP2746256A1 (en) | 2012-12-19 | 2014-06-25 | Basf Se | Fungicidal imidazolyl and triazolyl compounds |
| WO2014095381A1 (en) | 2012-12-19 | 2014-06-26 | Basf Se | Fungicidal imidazolyl and triazolyl compounds |
| WO2014095249A1 (en) | 2012-12-19 | 2014-06-26 | Basf Se | Fungicidal imidazolyl and triazolyl compounds |
| EP2746277A1 (en) | 2012-12-19 | 2014-06-25 | Basf Se | Fungicidal imidazolyl and triazolyl compounds |
| EP2746279A1 (en) | 2012-12-19 | 2014-06-25 | Basf Se | Fungicidal imidazolyl and triazolyl compounds |
| CN103724279B (zh) * | 2014-01-20 | 2016-04-06 | 新发药业有限公司 | 一步成环制备2-甲基-4-氨基-5-氨基甲基嘧啶的便捷合成方法 |
| US10562869B2 (en) | 2014-03-17 | 2020-02-18 | Remynd Nv | Oxadiazole compounds |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3141019A (en) * | 1964-07-14 | Chaohj | ||
| GB1053825A (ko) * | 1963-03-22 | |||
| JP2719742B2 (ja) * | 1991-04-30 | 1998-02-25 | 株式会社大塚製薬工場 | フェニルチアゾール誘導体 |
| EP1000040B1 (en) * | 1997-07-10 | 2007-06-13 | Janssen Pharmaceutica N.V. | Il-5 inhibiting 6-azauracil derivatives |
| EP0987265A1 (en) * | 1998-09-18 | 2000-03-22 | Janssen Pharmaceutica N.V. | Interleukin-5 inhibiting 6-azauracil derivatives |
| BR0013014A (pt) | 1999-08-06 | 2002-04-16 | Janssen Pharmaceutica Nv | Derivados de 6-aza uracila inibindo interleucina-5 |
| US7319108B2 (en) | 2004-01-25 | 2008-01-15 | Sanofi-Aventis Deutschland Gmbh | Aryl-substituted heterocycles, process for their preparation and their use as medicaments |
| DE602005013272D1 (de) * | 2004-10-18 | 2009-04-23 | Merck & Co Inc | Diphenyl-substituierte alkane als flap-inhibitoren |
| GB0516464D0 (en) * | 2005-08-10 | 2005-09-14 | Smithkline Beecham Corp | Novel compounds |
| JP2009504592A (ja) * | 2005-08-10 | 2009-02-05 | スミスクライン・ビーチャム・コーポレイション | 選択的hm74aアゴニストとしてのキサンチン誘導体 |
| US8399666B2 (en) | 2005-11-04 | 2013-03-19 | Panmira Pharmaceuticals, Llc | 5-lipoxygenase-activating protein (FLAP) inhibitors |
| US7977359B2 (en) | 2005-11-04 | 2011-07-12 | Amira Pharmaceuticals, Inc. | 5-lipdxygenase-activating protein (FLAP) inhibitors |
| GB2431927B (en) * | 2005-11-04 | 2010-03-17 | Amira Pharmaceuticals Inc | 5-Lipoxygenase-activating protein (FLAP) inhibitors |
| US20070219206A1 (en) * | 2005-11-04 | 2007-09-20 | Amira Pharmaceuticals, Inc. | 5-lipoxygenase-activating protein (flap) inhibitors |
| WO2007120574A2 (en) | 2006-04-11 | 2007-10-25 | Merck & Co., Inc. | Diaryl substituted alkanes |
| US20070287699A1 (en) * | 2006-05-01 | 2007-12-13 | Virobay, Inc. | Antiviral agents |
| EP2452683A3 (en) * | 2006-06-26 | 2012-08-22 | Amgen Inc. | Methods for treating atherosclerosis |
| AU2007293373A1 (en) * | 2006-09-01 | 2008-03-13 | Merck Sharp & Dohme Corp. | Inhibitors of 5 -lipoxygenase activating protein (FLAP) |
| FR2906866A1 (fr) * | 2006-10-10 | 2008-04-11 | Felix Ifrah | Luminaire comprenant au moins un moyen d'eclairage dont l'activation s'effectue par l'emission d'un rayonnement lumineux |
| WO2008050200A1 (en) * | 2006-10-24 | 2008-05-02 | Pfizer Products Inc. | Oxadiazole compounds as calcium channel antagonists |
| JP2010524861A (ja) | 2007-04-20 | 2010-07-22 | メルク フロスト カナダ リミテツド | ステアロイル−補酵素aデルタ−9デサチュラーゼの阻害剤としての新規な複素環式芳香族化合物 |
| EP2546232A1 (en) | 2007-06-20 | 2013-01-16 | Merck Sharp & Dohme Corp. | Diphenyl Substituted Alkanes |
| WO2009048547A1 (en) | 2007-10-10 | 2009-04-16 | Merck & Co., Inc. | Diphenyl substituted cycloalkanes |
| JP2012516885A (ja) * | 2009-02-04 | 2012-07-26 | ファイザー・インク | 4−アミノ−7,8−ジヒドロピリド[4,3−d]ピリミジン−5(6H)−オン誘導体 |
| MX2012013031A (es) | 2010-05-12 | 2012-12-17 | Univ Vanderbilt | Potenciadores alostericos mglur4 de sulfona heterociclica, composiciones y metodos para tratar disfuncion neurologica. |
| EP2606045B1 (en) | 2010-08-16 | 2016-01-06 | Boehringer Ingelheim International GmbH | Oxadiazole inhibitors of leukotriene production |
| JP5862669B2 (ja) | 2010-08-26 | 2016-02-16 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | ロイコトリエン産生のオキサジアゾール阻害剤 |
| US8580825B2 (en) | 2010-09-23 | 2013-11-12 | Boehringer Ingelheim International Gmbh | Oxadiazole inhibitors of leukotriene production |
-
2011
- 2011-08-11 EP EP11748842.9A patent/EP2606045B1/en active Active
- 2011-08-11 WO PCT/US2011/047356 patent/WO2012024150A1/en active Application Filing
- 2011-08-11 CN CN2011800500399A patent/CN103180314A/zh active Pending
- 2011-08-11 MX MX2013001872A patent/MX2013001872A/es not_active Application Discontinuation
- 2011-08-11 BR BR112013003439A patent/BR112013003439A2/pt not_active IP Right Cessation
- 2011-08-11 SG SG2013003694A patent/SG187549A1/en unknown
- 2011-08-11 EA EA201300250A patent/EA201300250A1/ru unknown
- 2011-08-11 NZ NZ605690A patent/NZ605690A/en not_active IP Right Cessation
- 2011-08-11 AU AU2011292285A patent/AU2011292285A1/en not_active Abandoned
- 2011-08-11 CA CA2807364A patent/CA2807364A1/en not_active Abandoned
- 2011-08-11 AP AP2013006678A patent/AP2013006678A0/xx unknown
- 2011-08-11 KR KR1020137003938A patent/KR20130100105A/ko not_active Application Discontinuation
- 2011-08-11 PE PE2013000279A patent/PE20130751A1/es not_active Application Discontinuation
- 2011-08-11 JP JP2013524874A patent/JP5619284B2/ja active Active
- 2011-08-12 US US13/208,582 patent/US8658661B2/en active Active
- 2011-08-15 TW TW100129127A patent/TW201238947A/zh unknown
- 2011-08-15 UY UY0001033557A patent/UY33557A/es not_active Application Discontinuation
- 2011-08-15 AR ARP110102966A patent/AR082696A1/es unknown
-
2013
- 2013-01-10 ZA ZA2013/00238A patent/ZA201300238B/en unknown
- 2013-02-14 CL CL2013000455A patent/CL2013000455A1/es unknown
- 2013-02-15 CO CO13031460A patent/CO6680645A2/es not_active Application Discontinuation
- 2013-03-15 EC ECSP13012494 patent/ECSP13012494A/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CO6680645A2 (es) | 2013-05-31 |
| US20120220561A1 (en) | 2012-08-30 |
| AR082696A1 (es) | 2012-12-26 |
| AP2013006678A0 (en) | 2013-01-31 |
| US8658661B2 (en) | 2014-02-25 |
| JP2013534245A (ja) | 2013-09-02 |
| UY33557A (es) | 2012-03-30 |
| CN103180314A (zh) | 2013-06-26 |
| NZ605690A (en) | 2014-06-27 |
| MX2013001872A (es) | 2013-09-02 |
| ZA201300238B (en) | 2014-06-25 |
| ECSP13012494A (es) | 2013-04-30 |
| AU2011292285A1 (en) | 2013-01-31 |
| PE20130751A1 (es) | 2013-06-21 |
| SG187549A1 (en) | 2013-03-28 |
| EA201300250A1 (ru) | 2013-08-30 |
| WO2012024150A1 (en) | 2012-02-23 |
| JP5619284B2 (ja) | 2014-11-05 |
| EP2606045A1 (en) | 2013-06-26 |
| BR112013003439A2 (pt) | 2019-09-24 |
| TW201238947A (en) | 2012-10-01 |
| CL2013000455A1 (es) | 2013-05-03 |
| CA2807364A1 (en) | 2012-02-23 |
| EP2606045B1 (en) | 2016-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5619284B2 (ja) | ロイコトリエン産生のオキサジアゾール阻害薬 | |
| US9512122B2 (en) | Spirocyclic compounds as tryptophan hydroxylase inhibitors | |
| CN101511796B (zh) | 咪唑衍生物 | |
| JP5690839B2 (ja) | ロイコトリエン産生のベンゾイミダゾール阻害薬 | |
| JP6500010B2 (ja) | 新規ピリジン誘導体 | |
| KR101615779B1 (ko) | 면역조절제로서 피리딘-2-일 유도체 | |
| JP5828188B2 (ja) | ロイコトリエン産生のオキサジアゾール阻害剤 | |
| KR20200081436A (ko) | 파르네소이드 x 수용체 조정제로서의 알켄 화합물 | |
| KR20200083529A (ko) | 파르네소이드 x 수용체 조정제로서의 알켄 스피로시클릭 화합물 | |
| CN110041316A (zh) | Tam家族激酶/和csf1r激酶抑制剂及其用途 | |
| KR20190026917A (ko) | 아미노피리딘 유도체 및 이의 선택적 alk-2 억제제로서의 용도 | |
| JP6069771B2 (ja) | ロイコトリエン生成の阻害薬としてのフェニル−c−オキサジアゾール誘導体併用療法 | |
| JP5713511B2 (ja) | ロイコトリエン産生のオキサジアゾール阻害剤 | |
| EP2632924A1 (en) | Diaza-spiro[5.5]undecanes useful as orexin receptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |