JP7288904B2 - ビアリールエーテル型キナゾリン誘導体 - Google Patents
ビアリールエーテル型キナゾリン誘導体 Download PDFInfo
- Publication number
- JP7288904B2 JP7288904B2 JP2020529034A JP2020529034A JP7288904B2 JP 7288904 B2 JP7288904 B2 JP 7288904B2 JP 2020529034 A JP2020529034 A JP 2020529034A JP 2020529034 A JP2020529034 A JP 2020529034A JP 7288904 B2 JP7288904 B2 JP 7288904B2
- Authority
- JP
- Japan
- Prior art keywords
- oxy
- fluoro
- mmol
- group
- piperidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images

























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/94—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
一方、EGFR遺伝子は、肺がん等でエクソン20挿入変異が起こることが知られておりその結果、EGFR蛋白質に1アミノ酸残基から4アミノ酸残基の挿入変異が見られるが、頻度が多いのは3アミノ酸残基の挿入変異(V769_D770ins.ASV、D770_N771ins.SVD、H773_V774ins.NPH等)である(非特許文献6-8)。
HER2(human epidermal growth factor receptor 2)は、ヒト上皮細胞増殖因子受容体2型関連がん遺伝子として同定された代表的な増殖因子受容体型のがん遺伝子産物のひとつであり、分子量185kDaのチロシンキナーゼドメインを持つ膜貫通型受容体蛋白である(非特許文献9)。
HER2(neu,ErbB-2)はEGFRファミリーのひとつであり、ホモダイマー或は他のEGFR受容体であるHER1(EGFR,ErbB-1)、HER3(ErbB-3)、HER4(ErbB-4)とのヘテロダイマー形成によって細胞内チロシン残基が自己リン酸化されて活性化することにより、正常細胞及びがん細胞において細胞の増殖・分化・生存に重要な役割を果たしていることが知られている。HER2は乳がん、胃がん、卵巣がん等様々ながん種において過剰発現している(非特許文献10-15)。HER2遺伝子は、エクソン20挿入変異が肺がん、乳がん、膀胱がん、卵巣がん等で起こることが知られており、1アミノ酸残基から4アミノ酸残基の挿入による変異であり、頻度が多いのは4アミノ酸残基の挿入変異(A775_G776 ins. YVMA, E770_A771 ins.AYVM, A771_Y772 ins.YVMA, Y772_A775dup)である(非特許文献6、7、16)。
(1)一般式(I)

R1は、1乃至3個のハロゲン原子にて置換されていてもよいC1-C3アルキル基を示し、
R2は、下記式(II)

Xは、下記A群より独立に選ばれる1または2個の置換基を有していてもよいアミノ基、下記A群より独立に選ばれる1または2個の置換基を有していてもよいピロリジニル基、下記A群より独立に選ばれる1または2個の置換基を有していてもよいアゼチジニル基、またはモルホリル基を示し、
R3は、ハロゲン原子を示し、
R4は、水素原子またはハロゲン原子を示し、
R5は、ベンゼン環、チアゾール環、またはハロゲン原子及びC1-C3アルキル基からなる群より独立に選ばれる1または2個の置換基を有していてもよいピラゾール環を示し、
R6は、下記B群から独立に選ばれる1または2個の置換基を有していてもよいオキサジアゾリル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいトリアゾリル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいピリジル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいピリミジル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいチアジアゾリル基、-CO-N(Y)(Z)、または-CH2-CO-N(Y)(Z)を示し、
YおよびZは、それぞれ独立に、水素原子、1乃至3個のハロゲン原子で置換されていてもよいC1-C6アルキル基、または1乃至3個のハロゲン原子で置換されていてもよいC1-C3アルキル基で置換されたC3-C6シクロアルキル基を示すか、または
Y、Zおよびそれらが結合する窒素原子が一緒になって4乃至6員の含窒素飽和複素環を形成してもよい]
で表される化合物またはその製薬上許容される塩。
A群:ハロゲン原子、C1-C3アルキル基、C1-C3アルコキシ基、テトラヒドロフリル基
B群:ハロゲン原子、C1-C3アルキル基、C3-C6シクロアルキル基、C1-C3アルコキシ基
(3)R2が、上記式(II)で示される基である、(1)または(2)に記載の化合物またはその製薬上許容される塩。
(4)R3が、塩素原子またはフッ素原子であり、R4が、水素原子である、(1)乃至(3)のいずれか1に記載の化合物またはその製薬上許容される塩。
(5)R5が、ベンゼン環、またはピラゾール環である、(1)乃至(4)のいずれか1に記載の化合物またはその製薬上許容される塩。
(6)R6が、メチル基で置換されていてもよいオキサジアゾリル基、フッ素原子及びメチル基からなる群から独立に選ばれる1または2個の置換基で置換されていてもよいピリジル基、または-CO-NH-Yであり、
Yが、1乃至3個のフッ素原子で置換されていてもよいtert-ブチル基である、(1)乃至(5)のいずれか1に記載の化合物またはその製薬上許容される塩。
(7)下記群から選ばれるいずれか1つの化合物またはその製薬上許容される塩。
N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド、
3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド、
1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、
1-{4-[(4-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、
1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン、および
1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン。
(9)N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド メタンスルホン酸塩。
(10)3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド、またはその製薬上許容される塩。
(11)3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド 1,5-ナフタレンジスルホン酸塩。
(12)1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、またはその製薬上許容される塩。
(13)1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン メタンスルホン酸塩。
(14)1-{4-[(4-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、またはその製薬上許容される塩。
(15)1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン、またはその製薬上許容される塩。
(16)1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン ベンゼンスルホン酸塩。
(17)1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 酒石酸塩。
(18)1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩。
(19)1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン、またはその製薬上許容される塩。
(21)1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 1,5-ナフタレンジスルホン酸塩。
(22)1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩。
(23)3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミドの結晶であって、CuKα放射線を用いた粉末X線回折において、5.78±0.2、15.48±0.2、16.38±0.2、17.24±0.2、19.28±0.2、19.90±0.2、20.42±0.2、20.82±0.2、22.04±0.2および24.50±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(24)3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド 1,5-ナフタレンジスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、5.74±0.2、10.32±0.2、11.58±0.2、14.62±0.2、14.94±0.2、18.72±0.2、19.60±0.2、20.84±0.2、22.96±0.2および26.42±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(25)1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オンの結晶であって、CuKα放射線を用いた粉末X線回折において、4.26±0.2、8.66±0.2、13.64±0.2、14.34±0.2、14.98±0.2、17.60±0.2、19.08±0.2、22.10±0.2、23.02±0.2および25.88±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(26)1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン メタンスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、3.56±0.2、7.24±0.2、15.02±0.2、16.84±0.2、17.68±0.2、20.26±0.2、21.88±0.2、22.92±0.2、25.76±0.2および27.08±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(27)1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オンの結晶であって、CuKα放射線を用いた粉末X線回折において、8.08±0.2、10.32±0.2、12.90±0.2、13.48±0.2、13.82±0.2、15.44±0.2、19.76±0.2、23.60±0.2、24.24±0.2および25.90±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(29)1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 酒石酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、3.58±0.2、14.50±0.2、16.50±0.2、24.28±0.2、24.70±0.2、24.98±0.2、25.76±0.2、26.12±0.2、26.60±0.2および27.40±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(30)1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、7.36±0.2、8.74±0.2、13.62±0.2、15.32±0.2、16.32±0.2、17.56±0.2、19.02±0.2、19.44±0.2、21.28±0.2および25.02±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(31)1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オンの結晶であって、CuKα放射線を用いた粉末X線回折において、8.14±0.2、10.56±0.2、13.10±0.2、15.16±0.2、15.50±0.2、15.92±0.2、19.30±0.2、20.18±0.2、23.92±0.2および25.54±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(33)1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 1,5-ナフタレンジスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、8.50±0.2、13.98±0.2、15.56±0.2、16.94±0.2、18.28±0.2、19.52±0.2、20.04±0.2、25.16±0.2、25.44±0.2および26.10±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(34)1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、5.34±0.2、7.32±0.2、7.86±0.2、8.68±0.2、13.56±0.2、16.26±0.2、17.50±0.2、19.36±0.2、21.22±0.2および24.90±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(35)N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド メタンスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、6.72±0.2、8.38±0.2、11.10±0.2、13.62±0.2、16.28±0.2、17.92±0.2、19.02±0.2、21.66±0.2、22.40±0.2および25.64±0.2から選択される回折角度(2θ)に、少なくとも5つのピークを有する結晶。
(36)(1)乃至(22)のいずれか1つに記載の化合物またはその製薬上許容される塩、または(23)乃至(35)のいずれか1つに記載の結晶を有効成分として含有する医薬組成物。
(37)(1)乃至(22)のいずれか1つに記載の化合物またはその製薬上許容される塩、または(23)乃至(35)のいずれか1つに記載の結晶を有効成分として含有する、エクソン20挿入変異を有するEGFRチロシンキナーゼおよび/またはエクソン20挿入変異を有するHER2チロシンキナーゼ阻害剤。
(39)腫瘍が肺がん、乳がん、膀胱がん、または卵巣がんである、(38)に記載の抗腫瘍剤。
(40)腫瘍が、EGFRチロシンキナーゼのエクソン20挿入変異および/またはHER2チロシンキナーゼのエクソン20挿入変異を有する腫瘍である、(38)または(39)に記載の抗腫瘍剤。
(41)(1)乃至(22)のいずれか1つに記載の化合物またはその製薬上許容される塩、または(23)乃至(35)のいずれか1つに記載の結晶を投与することを特徴とする、腫瘍の治療方法。
(42)腫瘍が肺がん、乳がん、膀胱がん、または卵巣がんである、(41)に記載の治療方法。
(43)腫瘍が、EGFRチロシンキナーゼのエクソン20挿入変異および/またはHER2チロシンキナーゼのエクソン20挿入変異を有する腫瘍である、(41)または(42)に記載の治療方法。
(44)腫瘍の治療のための(1)乃至(22)のいずれか1つに記載の化合物またはその製薬上許容される塩、または(23)乃至(35)のいずれか1つに記載の結晶。
(45)腫瘍が肺がん、乳がん、膀胱がん、または卵巣がんである、(44)に記載の化合物またはその製薬上許容される塩、または結晶。
(46)腫瘍が、EGFRチロシンキナーゼのエクソン20挿入変異および/またはHER2チロシンキナーゼのエクソン20挿入変異を有する腫瘍である、(44)または(45)に記載の化合物またはその製薬上許容される塩、または結晶。
(47)腫瘍を治療することに用いるための医薬組成物の製造における、(1)乃至(22)のいずれか1つに記載の化合物またはその製薬上許容される塩、または(23)乃至(35)のいずれか1つに記載の結晶の使用。
(48)腫瘍が肺がん、乳がん、膀胱がん、または卵巣がんである、(47)に記載の使用。
(49)腫瘍が、EGFRチロシンキナーゼのエクソン20挿入変異および/またはHER2チロシンキナーゼのエクソン20挿入変異を有する腫瘍である、(47)または(48)に記載の使用。
Yが、1個のフッ素原子で置換されていてもよいtert-ブチル基である。





別の態様におけるR1、R2、R3、R4、R5およびR6の好適な組み合わせは、R1が、メチル基であり、R2が、上記式(II)で示される基であり、R3が、フッ素原子であり、R4が、水素原子であり、R5が、ベンゼン環であり、R6が、メチル基で置換されていてもよいオキサジアゾリル基、または-CO-NH-C(CH3)3である。
別の態様におけるR1、R2、R3、R4、R5およびR6の好適な組み合わせは、R1が、メチル基であり、R2が、上記式(II)で示される基であり、R3が、フッ素原子であり、R4が、水素原子であり、R5が、ピラゾール環であり、R6が、フッ素原子及びメチル基からなる群から独立に選ばれる1または2個の置換基で置換されていてもよいピリジル基、または-CO-NH-Yであり、
Yが、1個のフッ素原子で置換されていてもよいtert-ブチル基である。
本発明において、結晶とは、その内部構造が三次元的に構成原子や分子の規則正しい繰り返しで出来てなる固体をいい、そのような規則正しい内部構造を持たない無定形の固体、または非晶質体とは区別される。
本発明において、結晶には、一般式(I)で表される化合物の結晶、一般式(I)で表される化合物の水和物結晶、一般式(I)で表される化合物の溶媒和物結晶、一般式(I)で表される化合物の製薬上許容される塩の結晶、一般式(I)で表される化合物の製薬上許容される塩の水和物結晶、及び一般式(I)で表される化合物の製薬上許容される塩の溶媒和物結晶が包含される。
本発明の水和物結晶は、例えば、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、3.0、3.1、3.2、3.3、3.4、3.5、3.6、3.7、3.8、3.9、4.0、4.1、4.2、4.3、4.4、4.5、4.6、4.7、4.8、4.9または5.0水和物の形をとることができ、湿度により水和水の増減が生じることがある。
本発明においては、一般式(I)で表される化合物及びその製薬上許容される塩が結晶形態であることは、偏光顕微鏡による観察、粉末X線結晶分析、または、単結晶X線回析測定を利用することによって確認することができる。さらに、結晶の特徴を予め測定しておいた各指標に基づくデータと比較することにより、その結晶のタイプを特定することもできる。よって、本発明の好ましい態様によれば、本発明による結晶は、このような測定手段を利用して結晶であることが確認できるものである。
本発明の結晶(以下、それぞれ「本発明実施例64の結晶」、「本発明実施例65の結晶」、「本発明実施例66の結晶」、「本発明実施例67の結晶」、「本発明実施例68の結晶」、「本発明実施例69の結晶」、「本発明実施例70の結晶」、「本発明実施例71の結晶」、「本発明実施例72の結晶」、「本発明実施例73の結晶」、「本発明実施例74の結晶」、「本発明実施例75の結晶」、「本発明実施例76の結晶」ということがある)は、医薬の製造に用いられる原薬の結晶として安定的に供給することが可能で、吸湿性または安定性に優れるものである。これらの結晶形の相違は、特に、粉末X線回折によって区別される。
本発明実施例64の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.78±0.2、15.48±0.2、16.38±0.2、17.24±0.2、19.28±0.2、19.90±0.2、20.42±0.2、20.82±0.2、22.04±0.2および24.50±0.2にピークを有するものである。
本発明実施例64の結晶は、例えば、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9または1.0水和物の形をとることができる。好ましくは0.2水和物である。
本発明実施例65の結晶は、例えば、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4または1.5水和物の形をとることができる。好ましくは1.0水和物である。
本発明実施例66の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)8.08±0.2、10.32±0.2、12.90±0.2、13.48±0.2、13.82±0.2、15.44±0.2、19.76±0.2、23.60±0.2、24.24±0.2および25.90±0.2にピークを有するものである。
本発明実施例66の結晶は、例えば、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、または2.0水和物の形をとることができる。好ましくは1.5水和物である。
本発明実施例67の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)8.14±0.2、10.56±0.2、13.10±0.2、15.16±0.2、15.50±0.2、15.92±0.2、19.30±0.2、20.18±0.2、23.92±0.2および25.54±0.2にピークを有するものである。
本発明実施例67の結晶は、例えば、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4または1.5水和物の形をとることができる。好ましくは1.0水和物である。
本発明実施例68の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)6.72±0.2、8.38±0.2、11.10±0.2、13.62±0.2、16.28±0.2、17.92±0.2、19.02±0.2、21.66±0.2、22.40±0.2および25.64±0.2にピークを有するものである。
本発明実施例68の結晶は、例えば、2.5、2.6、2.7、2.8、2.9、3.0、3.1、3.2、3.3、3.4または3.5水和物の形をとることができる。好ましくは3.0水和物である。
本発明実施例69の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.74±0.2、10.32±0.2、11.58±0.2、14.62±0.2、14.94±0.2、18.72±0.2、19.60±0.2、20.84±0.2、22.96±0.2および26.42±0.2にピークを有するものである。
本発明実施例69の結晶は、例えば、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9または1.0水和物の形をとることができる。好ましくは0.2水和物である。
本発明実施例70の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)3.56±0.2、7.24±0.2、15.02±0.2、16.84±0.2、17.68±0.2、20.26±0.2、21.88±0.2、22.92±0.2、25.76±0.2および27.08±0.2にピークを有するものである。
本発明実施例70の結晶は、例えば、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4または1.5水和物の形をとることができる。好ましくは1.0水和物である。
本発明実施例71の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)6.22±0.2、12.16±0.2、13.60±0.2、16.26±0.2、18.50±0.2、19.58±0.2、20.58±0.2、21.06±0.2、23.30±0.2および25.76±0.2にピークを有するものである。
本発明実施例71の結晶は、例えば、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9または2.0水和物の形をとることができる。好ましくは1.4水和物である。
本発明実施例72の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)3.58±0.2、14.50±0.2、16.50±0.2、24.28±0.2、24.70±0.2、24.98±0.2、25.76±0.2、26.12±0.2、26.60±0.2および27.40±0.2にピークを有するものである。
本発明実施例72の結晶は、例えば、3.5、3.6、3.7、3.8、3.9、4.0、4.1、4.2、4.3、4.4または4.5水和物の形をとることができる。好ましくは3.8水和物である。
本発明実施例73の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)7.36±0.2、8.74±0.2、13.62±0.2、15.32±0.2、16.32±0.2、17.56±0.2、19.02±0.2、19.44±0.2、21.28±0.2および25.02±0.2にピークを有するものである。
本発明実施例73の結晶は、例えば、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9または2.0水和物の形をとることができる。好ましくは1.2水和物である。
本発明実施例74の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.28±0.2、5.98±0.2、7.70±0.2、8.28±0.2、10.64±0.2、12.60±0.2、13.48±0.2、16.68±0.2、17.66±0.2および20.80±0.2にピークを有するものである。
本発明実施例74の結晶は、例えば、3.5、3.6、3.7、3.8、3.9、4.0、4.1、4.2、4.3、4.4または4.5水和物の形をとることができる。好ましくは4.0水和物である。
本発明実施例75の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)8.50±0.2、13.98±0.2、15.56±0.2、16.94±0.2、18.28±0.2、19.52±0.2、20.04±0.2、25.16±0.2、25.44±0.2および26.10±0.2にピークを有するものである。
本発明実施例75の結晶は、例えば、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4または2.5水和物の形をとることができる。好ましくは2.0水和物である。
本発明実施例75の結晶は、例えば、1/2、一、3/2または二 1,5-ナフタレンジスルホン酸塩の形をとることができる。好ましくは1/2 1,5-ナフタレンジスルホン酸塩である。
本発明実施例76の結晶は、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.34±0.2、7.32±0.2、7.86±0.2、8.68±0.2、13.56±0.2、16.26±0.2、17.50±0.2、19.36±0.2、21.22±0.2および24.90±0.2にピークを有するものである。
本発明実施例76の結晶は、例えば、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4または2.5水和物の形をとることができる。好ましくは2.0水和物である。
本明細書では、「治療する」及びその派生語は、がんを発症した患者において、がんの臨床症状の寛解、緩和及び/又は悪化の遅延を意味する。
HER2はヒト上皮細胞増殖因子受容体2型関連がん遺伝子として同定された代表的な増殖因子受容体型のがん遺伝子産物のひとつであり、分子量185kDaのチロシンキナーゼドメインを持つ膜貫通型受容体蛋白である。HER1(EGFR,ErbB-1)、HER2(neu,ErbB-2)、HER3(ErbB-3)、HER4(ErbB-4)からなるEGFRファミリーのひとつであり、ホモ或は他のEGFRであるHER1、HER3、又はHER4とのヘテロダイマー形成により細胞内チロシン残基が自己リン酸化されて活性化することにより、正常細胞及び腫瘍細胞において細胞の増殖・分化・生存に重要な役割を果たすことが知られている。
本発明の、EGFRチロシンキナーゼおよび/またはHER2チロシンキナーゼの阻害効果は、当業者に通常用いられるキナーゼ阻害活性を測定する方法により測定可能である。
[A法]
一般式(I)で表される化合物において、R2が、式(II)である化合物(a15)は、以下に示される方法によって製造することができる。それぞれの工程は、反応基質ならびに反応生成物に影響を与えない限りにおいて、必ずしも以下に示す順序に従って実施する必要は無い。

A-2工程は、化合物a3のカルボキシ基をエステルにて保護し化合物a4を得る工程である。本工程におけるエステル化は、例えば、N,N-ジメチルジメチルホルムアミド等の溶媒中、炭酸カリウム等の塩基存在下、ヨウ化メチル等のアルキル化剤を作用させることにより実施することができる。
A-3工程は、化合物a4のフッ素原子をヒドロキシ基へと変換し化合物a5を得る工程である。本工程は、例えば、ジメチルスルホキシド等の溶媒中、炭酸カリウム等の塩基存在下、N-ヒドロキシアセトアミドを作用させ、加熱することで実施することができる。
A-4工程は、化合物a5から、ヒドロキシ基のアルキル化により、化合物a7を得る工程である。本工程は、例えば、N,N-ジメチルジメチルホルムアミド等の溶媒中、炭酸カリウムまたは水素化ナトリウム等の塩基存在下、ヨウ化メチル等のアルキル化剤a6を作用させることで実施することができる。また本工程は、テトラヒドロフラン等の溶媒中、トリフェニルホスフィン等のホスフィンおよびアゾジカルボン酸ビス(2-メトキシエチル)等のアゾジカルボン酸エステル共存下、アルコールa6を作用させる、光延反応によっても実施することができる。また、一旦ヒドロキシ基に脱離基を有する置換基を導入した後に、再度置換反応を行うことで化合物a7を得ることもできる。
A-5工程は、化合物a7を化合物a8へと変換する工程である。本工程における還元反応は、例えば、エタノール等の溶媒中、パラジウム炭素等の貴金属触媒を用いた接触水素還元、またはメタノール等の溶媒中、酢酸等の酸存在下、亜鉛等の金属を用いたベシャンプ(Bechamp)還元により実施することができる。
A-7工程は、化合物a9に脱離基を導入し化合物a10へと変換する工程である。本工程における脱離基の導入は、例えば、N,N-ジメチルホルムアミド等の溶媒中、塩化チオニル等のハロゲン化剤を作用させ加熱することにより実施することができる。
A-8工程は、化合物a10のアミノ基を脱保護して化合物a11を得る工程である。本工程における脱保護反応は、アミノ基の脱保護に通常使用される方法を用いることができる。この除去方法(試薬、溶媒、反応条件)は、使用した保護基に応じた脱保護方法であって、さらに、反応基質および反応生成物に影響を与えない方法であれば、いかなる方法も使用することができる。
A-9工程は、化合物a11のアミノ基をアシル化することにより、化合物a13を得る工程である。本工程におけるアシル化反応は、例えば、酸塩化物、酸無水物等のアシル化剤の使用や、縮合剤を用いたカルボン酸成分との縮合により、反応条件に応じた溶媒、反応条件であれば、いかなる方法も使用することができる。
A-10工程は、化合物a13に対して化合物a14を導入し、化合物a15を得る工程である。本工程は、反応基質に応じた溶媒、反応条件であれば、いかなる方法も使用できる。例えば、化合物a14または化合物a14へと変換可能な合成中間体を基質として、塩酸やトリフルオロ酢酸等の酸共存下で実施することができ、炭酸カリウムや炭酸セシウム等の塩基存在下においても実施することができる。または、それらの添加をしない場合においても、例えば、2-プロパノールやジメチルスルホキシド等の溶媒中にて実施することができる。
A-11工程は、化合物a9に対して化合物a14を導入し、化合物a16を得る工程である。本工程における求核置換反応は、化合物a14または化合物a14へと変換可能な合成中間体を基質として、例えば、アセトニトリル等の溶媒中、1,8-ジアザビシクロ[5,4,0]-7-ウンデセン等の塩基共存下、1H-ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスファート等のホスホニウム系縮合剤を作用させることにより実施することができる。
A-10またはA-11工程において、化合物a14へと変換可能な合成中間体を基質として用いた際は、反応基質ならびに反応生成物に影響を与えない任意の合成段階で、後述する一般式(III)で示す化合物の製造方法を用いることにより、化合物a15へと変換することができる。
一般式(I)で表される化合物においてR2が-NH-CO-CH=CH-CH2-Xである化合物(b11)は下記の方法に従って製造することができる。それぞれの工程は、反応基質ならびに反応生成物に影響を与えない限りにおいて、必ずしも以下に示す順序に従って実施する必要は無い。

B-1工程は、化合物b1から、求核置換反応により、化合物b3を得る工程である。本工程における求核置換反応は、例えば、N,N-ジメチルホルムアミド、テトラヒドロフラン等の溶媒中、水素化ナトリウム等の塩基存在下、アルコールb2を作用させ加熱することにより実施することができる。
B-2工程は、化合物b3に脱離基を導入し化合物b4へと変換する工程である。本工程はA-7工程と同様の条件で実施することができる。
B-3工程は、化合物b4に対する求核置換反応により、化合物b5を導入し、化合物b6を得る工程である。本工程は、化合物b5または化合物b5へと変換可能な合成中間体を基質として、A-10工程と同様の条件で実施することができる。
B-3工程において化合物b5へと変換可能な合成中間体を基質として用いた際は、反応基質ならびに反応生成物に影響を与えない任意の合成段階で、後述する一般式(III)で示す化合物の製造方法により、化合物b11へと変換することができる。
B-4工程は、化合物b6を化合物b7へと変換する工程である。本工程は、A-5工程と同様の条件で実施することができる。
B-5工程は、化合物b7と化合物b8を縮合し、直接化合物b11、または化合物b11の合成前駆体b9を得る工程である。本工程におけるアシル化反応は、反応基質に応じた溶媒、反応条件であれば、いかなる方法も使用できる。例えば、酸塩化物、酸無水物等のアシル化剤によるアシル化、または、縮合剤を用いたカルボン酸との縮合反応を用いることができる。
B-6工程は、化合物b9に置換反応を行い、化合物b11を得る工程である。本工程における置換反応は、例えば、N,N-ジメチルホルムアミド等の溶媒中、N,N-ジイソプロプルエチルアミン等の塩基存在下、所望のアミンb10を作用させることにより実施することができる。
次に、一般式(III)で示される化合物の製造方法を示す。

反応部位ごとの一般的製造方法を順に示すが、それぞれの工程は、反応基質ならびに反応生成物に影響を与えない限りにおいて、必ずしも以下に示す順序に従って実施する必要は無い。また、A法およびB法における任意の工程で、一般式(III)で示される化合物へ変換可能な合成中間体を化合物a9、a10、a11、a13、またはb4等に導入した化合物においても、反応基質ならびに反応生成物に影響を与えない限りにおいて、以下の工程を任意の順序で実施することができる。
一般式(III)で示される化合物において、ビアリールエーテル部位は、C法にて製造することができる。

C-1工程は、化合物c1と化合物c2から化合物c3を得る工程、または化合物c4と化合物c5から化合物c3を得る工程である。本工程における求核置換反応は、例えば、ジメチルスルホキシド、N,N-ジメチルホルムアミド、またはテトラヒドロフラン等の溶媒中、炭酸セシウム、炭酸カリウム、またはN,N-ジイソプロピルエチルアミン等の塩基を、基質に応じた反応温度で作用させることにより実施することができる。
化合物c3は、C-2工程によっても得ることができる。C-2工程は化合物c4と化合物c5とのウルマン(Ullmann)型カップリングにより、化合物c3を得る工程である。本工程におけるカップリング反応は、例えば、ブチロニトリル、1,4-ジオキサン等の溶媒中、炭酸セシウム、または炭酸カリウム等の塩基を用い、trans-N,N'-ジメチルシクロヘキサン-1,2-ジアミン、またはN,N-ジメチルグリシン等の配位子共存下、ヨウ化銅(I)等の触媒を作用させ加熱することにより実施することができる。
一般式(I)で示される化合物においてR6が-CO-N(Y)(Z)または-CH2-CO-N(Y)(Z)である場合、R6部位はD法にて製造することができる。


または一般式(IV)で示される基へ変換可能な置換基を示す。Rd2は、例えば、エチル基、tert-ブチル基等のカルボキシ基の保護基を示す。Rd3、およびRd4は、Y、Z、Yへ変換可能な置換基、またはZへ変換可能な置換基を示す。Y、およびZは、前記(1)に記載の定義と同義である。]
D-1工程は、化合物d1を化合物d2へと変換する工程である。本工程は、例えば、Rd2がエチル基等の場合、テトラヒドロフラン等の溶媒中、水酸化ナトリウム水溶液等の塩基を作用させることにより実施することができる。Rd2がtert-ブチル基の場合には、ジクロロメタン等の溶媒中、トリフルオロ酢酸等の酸を作用させることにより実施することができる。
D-2工程は、化合物d2と化合物d3を縮合させ、化合物d4を得る工程である。本工程におけるアミド化は、例えば、N,N-ジメチルホルムアミド等の溶媒中、N,N-ジイソプロピルエチルアミン等の塩基共存下、1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ[4,5-b]ピリジニウム3-オキシドヘキサフルオロホスファート等の縮合剤を作用させることにより実施することができる。
D-3工程は、化合物d1を還元して化合物d5を得る工程である。本工程における還元反応は、例えば、ジクロロメタン等の溶媒中、水素化ジイソブチルアルミニウム等の還元剤を作用させることにより実施することができる。
D-5工程は、化合物d6に対し、置換反応によりシアノ基を導入し、化合物d7を得る工程である。本工程は、例えば、ジメチルスルホキシド等の溶媒中、シアン化ナトリウム等を作用させ加熱することにより実施することができる。
D-6工程は、化合物d7を化合物d8へと変換する工程である。本工程における加水分解は、例えば、エタノール等の溶媒中、水酸化ナトリウム水溶液等の塩基を作用させ加熱することにより実施することができる。
D-7工程は、化合物d8と化合物d3を縮合させ、化合物d9を得る工程である。本工程は、D-2工程と同様の条件で実施することができる。
D-8工程は、化合物d10を化合物d11を用いてアルキル化し、化合物d12を得る工程である。本工程におけるアルキル化は、例えば、N,N-ジメチルホルムアミド等の溶媒中、炭酸カリウム等の塩基存在下、クロロ酢酸 tert-ブチル等のアルキル化剤を作用させることにより実施することができる。
D-9工程は、化合物d12から化合物d8を得る工程である。本工程は、D-1工程と同様の条件で実施することができる。
D-10工程は、化合物d10を化合物d13を用いてアルキル化し、化合物d9を得る工程である。本工程におけるアルキル化は、例えば、N,N-ジメチルホルムアミド等の溶媒中、炭酸カリウム等の塩基存在下、N-tert-ブチル-2-クロロアセトアミド等のアルキル化剤を作用させることにより実施することができる。
一般式(I)で示される化合物において、R5がトリアゾール環である場合、R5-R6部位はE法にて製造することができる。

E-1工程は、薗頭反応により化合物e1にアルキニル基を導入する工程である。本工程における薗頭反応は、例えば、トリエチルアミン等の溶媒中、ビス(トリフェニルホスフィン)パラジウム(II)等のパラジウム触媒およびヨウ化銅(I)等の銅触媒共存下、化合物e2を添加して加熱することにより実施することができる。
E-2工程は、末端アセチレンを脱保護し化合物e4を得る工程である。本工程における脱保護反応は、末端アセチレンの脱保護に通常用いられる方法により実施することができる。例えば、Pgがトリメチルシリル基の場合、メタノール等の溶媒中、炭酸カリウム等の塩基を作用させることにより実施することができる。
E-3工程は、化合物e4とアジド化合物との1,3-双極子付加環化反応により、化合物e6を得る工程である。本工程における1,3-双極子付加環化反応は、例えば、アセトニトリル等の溶媒中、アジ化ナトリウム等のアジド源、化合物e5(例えば、ヨードメタン等のアルキル化剤)、ヨウ化銅(I)等の触媒を加熱下作用させることにより実施することができる。
一般式(I)で示される化合物において、R5が、1,3,4-オキサジアゾール環または1,3,4-チアジアゾール環である場合、R5-R6部位はF法にて製造することができる。

F-1工程は、化合物f1にヒドラジンを作用させて化合物f2を得る工程である。本工程は、例えば、エタノール等の溶媒中、ヒドラジン一水和物を加熱下作用させることで実施することができる。
F-2工程は、化合物f2をアシル化し、化合物f5を得る工程である。本工程は、例えば、ジクロロメタン等の溶媒中、化合物f3、または化合物f4を作用させることで実施することができる。
F-3工程は、化合物f1から化合物f6を得る工程である。本工程は、D-1工程と同様の条件で実施することができる。
F-4工程は、化合物f6と化合物f7を作用させ、化合物f5を得る工程である。本工程は、D-2工程と同様の条件で実施することができる。
F-5工程は、化合物f5からパール-クノール(Paal-Knorr)型の環化反応により化合物f8を得る工程である。本工程において、1,3,4-オキサジアゾール環は、例えば、ジクロロメタン等の溶媒中、トリエチルアミンの共存下、p-トルエンスルホニルクロリドを作用させ加熱することにより得ることができる。1,3,4-チアジアゾール環については、例えば、トルエン等の溶媒中、ローソン試薬を作用させ加熱することにより得ることができる。
一般式(I)で示される化合物において、R5がピラゾール環であり、R6が-CO-N(Y)(Z)である場合、R6部位はG法にて製造することができる。


または、一般式(V)で示される基へ変換可能な置換基を示す。Rg2、Rg3はY、Z、Yへ変換可能な置換基、またはZへ変換可能な置換基を示す。Y、Zは前記(1)に記載の定義と同義である。]
G-1工程は、化合物g1を化合物g2へと変換する工程である。本工程における窒素原子の保護は、通常使用される方法を用いることができる。
G-2工程は、化合物g2のヒドロキシ基を修飾し、化合物g3を得る工程である。本工程は、C-1工程と同様の条件で実施することができる。
G-3工程は、化合物g3の窒素原子を脱保護し、化合物g4へと変換する工程である。本工程における窒素原子の脱保護は、通常使用される方法を用いることができる。
G-4工程は、化合物g4と化合物g5を反応させ、化合物g6へと変換する工程である。本工程におけるカルバモイル化は、例えば、1,2-ジクロロエタン等の溶媒中、トリエチルアミン等の塩基共存下、加熱することで実施することができる。
G-5工程は、化合物g7を化合物g5へと変換する工程である。本工程における酸アジド化、続くクルチウス(Curtius)転位反応は、例えば、トルエン等の溶媒中、トリエチルアミン等の塩基存在下、ジフェニルリン酸アジドを作用させ加熱することにより実施することができる。
G-6工程は、化合物g4とクロロぎ酸4-ニトロフェニルを反応させ、化合物g8を得る工程である。本工程におけるウレタン化は、例えば、ジクロロメタン等の溶媒中、トリエチルアミン等の塩基を共存させることにより実施することができる。
G-7工程は、化合物g8に化合物g9を反応させ、化合物g6へと変換する工程である。本工程におけるカルバモイル化は、例えば、ジクロロメタン等の溶媒中、トリエチルアミン等の塩基を共存させることにより実施することができる。
一般式(I)で示される化合物において、R5が下記の化合物h2である場合、R5部位はH法にて製造することができる。

H-1工程は、化合物h1の4位をフッ素化し、化合物h2を得る工程である。本工程におけるフッ素化は、例えば、アセトニトリル等の溶媒中、N-フルオロ-N'-(クロロメチル)トリエチレンジアミン ビス(テトラフルオロボラート)を作用させ、加熱することにより実施することができる。
[I法]
一般式(I)で表される化合物において、R5が下記の化合物i3である場合、R5-R6部位はI法にて製造することができる。

I-1工程は、化合物i1と化合物i2の求核置換反応により、化合物i3を得る工程である。本工程における求核置換反応は、例えば、ジメチルスルホキシド等の溶媒中、炭酸セシウム等の塩基存在下で、実施することができる。
I-2工程は、化合物i1と化合物i4とのウルマン(Ullmann)型カップリングにより、化合物i3を得る工程である。本工程におけるカップリング反応は、例えば、Lgがブロモ基の場合、ジメチルスルホキシド、トルエン等の溶媒中、炭酸カリウム等の塩基、trans-N,N'-ジメチルシクロヘキサン-1,2-ジアミン、N,N-ジメチルグリシン等の配位子共存下、ヨウ化銅(I)等の金属触媒を作用させ、加熱することにより実施することができる。
[J法]
一般式(I)で示される化合物において、R5が、下記の化合物j3である場合、R5-R6部位は、J法にて製造することができる。

J-1工程は、化合物j1と化合物j2から化合物j3を得る工程である。本工程におけるカップリング反応は、化合物j1と化合物j2を、例えば、含水1,2-ジメトキシエタン等の溶媒中、炭酸セシウム等の塩基、テトラキス(トリフェニルホスフィン)パラジウム(0)、[1,1'-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物等の金属触媒存在下、加熱することにより実施することができる。
J-2工程は、化合物j1から化合物j4を得る工程である。本工程におけるカップリング反応は、化合物j1を、例えば、1,4-ジオキサン等の溶媒中、酢酸カリウム等の塩基、[1,1'-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物等の金属触媒存在下、ビス(ピナコラト)ジボロンを加熱下作用させることにより実施することができる。
J-3工程は、化合物j4と化合物j5から化合物j3を得る工程である。本工程におけるカップリング反応は、化合物j4と化合物j5を、例えば、含水1,2-ジメトキシエタン等の溶媒中、炭酸セシウム等の塩基、テトラキス(トリフェニルホスフィン)パラジウム(0)、[1,1'-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物等の金属触媒存在下、加熱することにより実施することができる。
[K法]
一般式(III)で示される化合物における芳香族アミン部位は、K法にて製造することができる。


または一般式(VI)で示される基へ変換可能な置換基を示す。]
K-1工程は、化合物k1のアミノ基を脱保護して化合物k2を得る工程である。本工程における脱保護反応は、アミノ基の脱保護に通常使用される方法により実施することができる。例えば、Pg1がtert-ブトキシカルボニル基の場合、ジクロロメタン等の溶媒中、トリフルオロ酢酸等の酸を作用させることにより実施することができる。
K-2工程は、化合物k2のアミノ基を保護して化合物k1を得る工程である。本工程におけるアミノ基の保護は、アミノ基の保護に通常使用される方法により実施することができる。例えば、Pg1がtert-ブトキシカルボニル基の場合、テトラヒドロフラン等の溶媒中、二炭酸ジ-tert-ブチル等を作用させることにより実施することができる。
K-3工程は、化合物k3から化合物k2を得る工程である。本工程は、A-5工程と同様の条件で実施することができる。
K-4工程は、化合物k4から化合物k5を得る工程である。本工程における脱保護反応は、カルボキシ基の脱保護に通常使用される方法により実施することができる。例えば、Pg2がtert-ブチル基の場合、ジクロロメタン等の溶媒中、トリフルオロ酢酸等の酸を作用させることにより実施することができる。
K-5工程は、化合物k5から化合物k1を得る工程である。本工程における酸アジド化、続くクルチウス(Curtius)転位反応を経るカルバメート化は、例えば、Pg1がtert-ブトキシカルボニル基の場合、トルエン等の溶媒中、トリエチルアミン等の塩基存在下、化合物k5にジフェニルリン酸アジドを作用させ加熱してイソシアン酸エステルを得た後に、tert-ブタノールを加熱下作用させることにより実施することができる。
上記の方法で製造された化合物は、公知の方法、例えば、抽出、沈殿、蒸留、クロマトグラフィー、分別再結晶、再結晶等により単離、精製することができる。
プロトン核磁気共鳴スペクトル (1H-NMR) は、日本電子社製400 MHz、あるいは、バリアン社製400 MHz 核磁気共鳴装置を用いて測定した。スペクトルデータの表記は、意義のあるピークについて示しており、化学シフト (テトラメチルシランを標準物質とした相対ppm (δ) として示した)、プロトン数、ピーク分裂の多重度 (s: 一重線; d: 二重線; t: 三重線; q: 四重線; m: 多重線; br: ブロードなどと示した)、ならびに、明示できる場合はスピン結合定数をJ値 (単位はHz) として示した。
質量スペクトル (MS m/z) は、電子スプレーイオン化法 (ESI) あるいは、大気圧化学イオン化法 (APCI) を用いて測定した。質量スペクトルのデータは、逆相高速液体クロマトグラフィーカラム (Agilent システム; カラム: Develosil Combi-RP-5, 2.0×50 mm、Cadenza CD-C18, 3.0×75 mm、あるいはZORBAX SB-C18, 1.8 μm, 2.1×50 mm; 溶媒: 0.1%ギ酸含有アセトニトリル/水系、あるいは0.01%トリフルオロ酢酸含有アセトニトリル/水系) を通過後の最大イオン化ピーク(ほとんどの場合に最大UV吸収ピークと一致) について示した。
シリカゲルカラムクロマトグラフィーは、市販のパック済みカラムと自動分取精製装置 (バイオタージ社製 SP1、山善社製 EPCLC-W-Prep2XY、昭光サイエンス社製 Purif-α2等) を用いて行い、移動相に用いた複数の溶媒種のみを記述した。溶出は薄層クロマトグラフィー (TLC) による観察下に行い、TLCプレートとしてメルク社製のシリカゲル60 F254または60 NH2 F254s、和光純薬工業社製のNH2シリカゲル60 F254プレートもしくは富士シリシア化学社製 CHROMATOREX NH TLCを、展開溶媒としてはカラムクロマトグラフィーに用いた移動相を、検出方法としてはUV検出器もしくは呈色試薬を、それぞれ採用した。
分取薄層クロマトグラフィー (PTLC) は、メルク社製のシリカゲル60 F254プレート、和光純薬工業社製のシリカゲル70 PF254プレート、NH2シリカゲル60 F254プレートを用いて行ない、移動相に用いた複数の溶媒種のみを記述した。
分取高速液体クロマトグラフィー (分取HPLC) は、野村化学社製逆相用カラム (Develosil Combi-RP-5) を用いて行い、移動相に0.1%ギ酸含有アセトニトリル/水系を用いた。
これらクロマトグラフィーに用いた溶媒量や溶媒比率、その変換タイミング、及びグラジエント方法は示さないが、ここで用いた精製・分離方法は通常の化学合成の知識・技術をもってすれば容易に再現できると考えられる。
機種: Rigaku Rint TTR-III
サンプルホルダー: 無反射試料ホルダー
試料: 適量
X線発生条件: 50 kV, 300 mA
波長: 1.54 Å (銅のKα線)
測定温度:室温
走査速度: 20°/min
走査範囲: 2~40°
サンプリング幅: 0.02°
サンプル調製: 数mgの結晶をスパーテルで採取し、無反射試料ホルダーにのせ、薬包紙で平たくした。その後、上述の条件にてピークパターンを解析した。
実施例で用いる略号は、次のような意義を有する。
Ac: アセチル基、Bn: ベンジル基、Boc: tert-ブトキシカルボニル基、tBu: tert-ブチル基、Cbz: ベンジルオキシカルボニル基、CDCl3: 重クロロホルム、CD3OD: 重メタノール、DMSO: ジメチルスルホキシド、Et: エチル基、Me: メチル基、Ms: メタンスルホニル基、TMS: トリメチルシリル基。

1H-NMR (DMSO-D6) δ: 1.82-2.01 (2H, m), 2.08-2.26 (2H, m), 3.02-3.34 (4H, m), 4.03 (3H, s), 4.95-5.08 (1H, m), 7.52 (1H, s), 7.57 (1H, s), 8.71-8.96 (1H, m), 8.91 (1H, s). MS m/z: 294 [M+H]+.

1H-NMR (CDCl3) δ: 1.93-2.15 (4H, m), 3.55-3.67 (1H, m), 3.76-3.95 (3H, m), 4.05 (3H, s), 4.80-4.89 (1H, m), 5.73 (1H, dd, J = 10.9, 1.8 Hz), 6.32 (1H, dd, J = 17.0, 1.8 Hz), 6.63 (1H, dd, J = 17.0, 10.9 Hz), 7.36 (1H, s), 7.46 (1H, s), 8.88 (1H, s). MS m/z: 348 [M+H]+.

1H-NMR (CDCl3) δ: 1.48 (9H, s), 1.76-1.79 (2H, m), 2.04-2.06 (2H, m), 3.22-3.28 (2H, m), 3.84-3.88 (5H, m), 4.61-4.67 (1H, m), 6.09 (1H, br s), 7.14 (1H, s), 7.49 (1H, s). MS m/z: 297 [M-Boc+H]+.

1H-NMR (CDCl3) δ: 1.47 (12H, br s), 1.83 (2H, br s), 1.93 (2H, br s), 3.39 (2H, br s), 3.69 (2H, br s), 3.90 (3H, br s), 4.15 (2H, br s), 4.60 (1H, br s), 7.11 (1H, br s), 7.43 (1H, br s). MS m/z: 425 [M+H]+.
同様の方法により対応するハロゲン化アルキルを用い以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.47 (9H, s), 1.79-1.85 (2H, m), 1.95-1.96 (2H, m), 2.26 (3H, s), 3.34-3.40 (2H, m), 3.70-3.72 (2H, m), 3.91 (3H, s), 4.60-4.64 (1H, m), 5.28 (2H, s), 7.11 (1H, s), 7.57 (1H, s). MS m/z: 357 [M+H]+.

1H-NMR (CDCl3) δ: 1.47 (9H, s), 1.83-1.84 (2H, m), 1.95-1.97 (2H, m), 3.34-3.40 (2H, m), 3.70-3.72 (2H, m), 3.92 (3H, s), 4.64-4.65 (1H, m), 5.77 (2H, d, J = 53.5 Hz), 7.13 (1H, s), 7.79 (1H, s).

1H-NMR (CDCl3) δ: 1.49-1.51 (12H, br m), 1.87 (2H, br s), 1.96 (2H, br s), 3.39 (2H, br s), 3.74 (2H, br s), 4.20 (2H, br s), 4.63 (1H, br s), 7.15 (1H, br s), 7.67 (1H, br s), 7.99 (1H, br s), 10.69 (1H, br s). MS m/z: 390 [M+H]+.

1H-NMR (CDCl3) δ: 1.48 (9H, br s), 1.85 (2H, br s), 2.00 (2H, br s), 3.37 (2H, br s), 3.76 (2H, br s), 4.68 (1H, br s), 5.86 (2H, d, J = 52.7 Hz), 7.48 (1H, br s), 7.71 (1H, br s), 8.02 (1H, br s), 10.94 (1H, br s). MS m/z: 394 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 4.05 (3H, s), 7.45 (1H, s), 8.31 (1H, s), 8.54 (1H, s). MS m/z: 240 [M+H]+.
[参考例B1] 3-(ベンジルオキシ)-N'-プロパノイルベンゾヒドラジド

1H-NMR (DMSO-D6) δ: 1.06 (3H, t, J = 7.6 Hz), 2.19 (2H, q, J = 7.6 Hz), 5.17 (2H, s), 7.21 (1H, dd, J = 7.9, 1.8 Hz), 7.32-7.36 (1H, m), 7.38-7.43 (3H, m), 7.51-7.44 (4H, m), 9.84 (1H, s), 10.27 (1H, s). MS m/z: 299 [M+H]+.
[参考例B2] 2-[3-(ベンジルオキシ)フェニル]-5-エチル-1,3,4-オキサジアゾール

1H-NMR (CDCl3) δ: 1.44 (3H, t, J = 7.6 Hz), 2.96 (2H, q, J = 7.6 Hz), 5.14 (2H, s), 7.13 (1H, dd, J = 8.5, 2.4 Hz), 7.32-7.37 (1H, m), 7.39-7.47 (5H, m), 7.61-7.64 (1H, m), 7.68-7.67 (1H, m). MS m/z: 281 [M+H]+.
[参考例B3] 3-(5-エチル-1,3,4-オキサジアゾール-2-イル)フェノール

1H-NMR (CDCl3) δ: 1.45 (3H, t, J = 7.7 Hz), 2.97 (2H, q, J = 7.7 Hz), 6.48 (1H, s), 7.05 (1H, dd, J = 8.2, 2.7 Hz), 7.38 (1H, t, J = 7.9 Hz), 7.53 (1H, d, J = 7.3 Hz), 7.95 (1H, s). MS m/z: 191 [M+H]+.
[参考例B4] 3-ヒドロキシ-1H-ピラゾール-1-カルボン酸tert-ブチル

1H-NMR (CDCl3) δ: 1.63 (9H, s), 5.90 (1H, d, J = 3.1 Hz), 7.82 (1H, br s). MS m/z: 129 [M-tBu+H]+
[参考例C1] 3-(3-クロロ-4-ニトロフェノキシ)安息香酸メチル

1H-NMR (CDCl3) δ: 3.94 (3H, s), 6.91-6.96 (1H, m), 7.07 (1H, d, J = 2.4 Hz), 7.28-7.32 (1H, m), 7.54 (1H, t, J = 8.2 Hz), 7.73-7.76 (1H, m), 7.94-8.01 (2H, m).
[参考例C2] 2-フルオロ-4-[3-(メトキシカルボニル)フェノキシ]安息香酸

1H-NMR (CDCl3) δ: 3.93 (3H, s), 6.71 (1H, dd, J = 11.6, 2.4 Hz), 6.78-6.84 (1H, m), 7.29-7.33 (1H, m), 7.52 (1H, t, J = 7.9 Hz), 7.75-7.77 (1H, m), 7.92-7.95 (1H, m), 8.01 (1H, t, J = 8.5 Hz).
[参考例C3] 2-クロロ-4-(3-エチニルフェノキシ)-1-ニトロベンゼン

1H-NMR (CDCl3) δ: 3.15 (1H, s), 6.93 (1H, dd, J = 9.2, 2.4 Hz), 7.05-7.11 (2H, m), 7.20 (1H, s), 7.38-7.42 (2H, m), 7.98 (1H, d, J = 9.2 Hz).
[参考例C4] 3-(4-アミノ-3-フルオロフェノキシ)安息香酸エチル

1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.3 Hz), 3.66 (2H, br s), 4.38 (2H, q, J = 7.3 Hz), 6.68-6.84 (3H, m), 7.16 (1H, dt, J = 8.9, 1.8 Hz), 7.39 (1H, t, J = 8.2 Hz), 7.61-7.63 (1H, m), 7.74-7.78 (1H, m).
[参考例C5] (3-{4-[(tert-ブトキシカルボニル)アミノ]-3-フルオロフェノキシ}フェニル)酢酸メチル

1H-NMR (CDCl3) δ: 1.53 (9H, s), 3.60 (2H, s), 3.70 (3H, s), 6.58 (1H, br s), 6.73-7.06 (5H, m), 7.25-7.32 (1H, m), 7.99 (1H, br s). MS m/z: 276 [M-Boc+H]+.
同様の方法により対応するハロベンゼンから以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.45 (3H, t, J = 7.5 Hz), 2.97 (2H, q, J = 7.5 Hz), 6.70 (1H, d, J = 11.6 Hz), 7.27-7.30 (1H, m), 7.63 (1H, t, J = 7.9 Hz), 7.79-7.78 (1H, m), 8.00 (1H, d, J= 7.9 Hz), 8.31 (1H, d, J = 7.9 Hz). MS m/z: 364 [M+H]+.
[参考例 C8] 3-(2-クロロ-5-フルオロ-4-ニトロフェノキシ)-1H-ピラゾール-1-カルボン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.65 (9H, s), 6.19 (1H, d, J = 3.0 Hz), 7.39 (1H, d, J = 11.5 Hz), 8.09 (1H, d, J = 3.0 Hz), 8.25 (1H, d, J = 7.9 Hz). MS m/z: 258 [M-Boc+H]+
同様の方法により対応するハロベンゼンおよび中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.39 (3H, t, J = 7.0 Hz), 1.53 (9H, s), 4.38 (2H, q, J = 7.0 Hz), 6.69 (1H, br s), 7.04-7.16 (2H, m), 7.72 (1H, s), 8.06-8.23 (1H, m). MS m/z: 383 [M+H]+.
同様の方法により対応するブロモチアゾールから以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.63 (9H, s), 3.63 (2H, br s), 5.89 (1H, d, J = 3.0 Hz), 6.74 (1H, t, J = 9.1 Hz), 6.80-6.85 (1H, m), 6.91 (1H, dd, J = 11.5, 2.4 Hz), 7.94 (1H, d, J = 3.0 Hz). MS m/z: 238 [M-tBu+H]+.
[参考例D2] 3-(4-アミノ-2-クロロ-5-フルオロフェノキシ)-1H-ピラゾール-1-カルボン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.62 (9H, s), 5.89 (1H, d, J = 3.0 Hz), 6.84 (1H, d, J = 9.1 Hz), 7.01 (1H, d, J = 10.9 Hz), 7.93 (1H, d, J = 3.0 Hz). MS m/z: 272 [M-tBu+H]+.
[参考例D3] 3-(4-{[(ベンジルオキシ)カルボニル]アミノ}-3-フルオロフェノキシ)-1H-ピラゾール-1-カルボン酸tert-ブチル

1H-NMR (CDCl3) δ: 1.63 (9H, s), 5.22 (2H, s), 5.92-5.99 (1H, m), 6.80 (1H, s), 6.94-7.04 (2H, m), 7.31-7.48 (5H, m), 7.91-8.17 (2H, m). MS m/z: 450 [M+Na]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 6.04 (1H, d, J = 2.4 Hz), 7.13 (1H, dd, J = 9.1, 3.0 Hz), 7.29 (1H, d, J = 3.0 Hz), 7.57 (1H, d, J = 2.4 Hz), 7.99 (1H, d, J = 9.1 Hz), 9.64 (1H, br s). MS m/z: 240 [M+H]+.
[参考例E2] 3-(2-クロロ-5-フルオロ-4-ニトロフェノキシ)-1H-ピラゾール

1H-NMR (CDCl3) δ: 6.11 (1H, d, J = 2.4 Hz), 7.13 (1H, d, J = 12.1 Hz), 7.60 (1H, d, J = 2.4 Hz), 8.26 (1H, d, J = 7.9 Hz), 9.67 (1H, br s). MS m/z: 258 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.52 (9H, s), 5.87 (1H, d, J = 2.4 Hz), 6.59 (1H, br s), 6.88-6.96 (2H, m), 7.43 (1H, d, J = 2.4 Hz), 7.98 (1H, br s), 9.79 (1H, br s). MS m/z: 294 [M+H]+.
[参考例F1] N-tert-ブチル-3-(2-クロロ-5-フルオロ-4-ニトロフェノキシ)-1H-ピラゾール-1-カルボキサミド

1H-NMR (CDCl3) δ: 1.46 (9H, s), 6.15 (1H, d, J = 3.0 Hz), 6.70 (1H, s), 7.10 (1H, d, J = 11.5 Hz), 8.23 (1H, d, J = 3.0 Hz), 8.28 (1H, d, J = 7.3 Hz). MS m/z: 258 [M-CONHtBu+H]+.
同様の方法により対応する中間体およびイソシアン酸エステルから以下の化合物を合成した。


1H-NMR (CDCl3) δ: 6.39 (1H, d, J = 3.0 Hz), 7.46-7.53 (3H, m), 8.26-8.30 (2H, m), 8.34-8.39 (2H, m).
[参考例F5] 3-(2-クロロ-5-フルオロ-4-ニトロフェノキシ)-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド

1H-NMR (CDCl3) δ: 1.47 (6H, d, J = 1.8 Hz), 4.51 (2H, d, J = 47.4 Hz), 6.17 (1H, d, J = 3.0 Hz), 6.78 (1H, br s), 7.13 (1H, d, J = 11.5 Hz), 8.22 (1H, d, J = 3.0 Hz), 8.28 (1H, d, J = 7.9 Hz).
同様の方法により対応するアミンから以下の化合物を合成した。


1H-NMR (CDCl3) δ: 6.15 (1H, d, J = 3.0 Hz), 6.69 (1H, br s), 7.10 (1H, d, J = 11.5 Hz), 8.23 (1H, d, J = 3.0 Hz), 8.28 (1H, d, J = 7.3 Hz).
[参考例G1] 3-{4-[(tert-ブトキシカルボニル)アミノ]-3-フルオロフェノキシ}安息香酸メチル

1H-NMR (CDCl3) δ: 1.53 (9H, s), 3.90 (3H, s), 6.59 (1H, s), 6.73-6.82 (2H, m), 7.16-7.22 (1H, m), 7.36-7.43 (1H, m), 7.62 (1H, s), 7.78 (1H, d, J = 7.9 Hz), 8.02 (1H, s).
[参考例G2] 3-{4-[(tert-ブトキシカルボニル)アミノ]-3-フルオロフェノキシ}安息香酸

1H-NMR (DMSO-D6) δ: 1.46 (9H, s), 6.83-6.89 (1H, m), 7.05 (1H, dd, J = 11.6, 2.4 Hz), 7.31 (1H, dd, J = 7.9, 2.4 Hz), 7.42-7.44 (1H, m), 7.49-7.60 (2H, m), 7.71 (1H, d, J = 7.3 Hz), 8.96 (1H, s), 13.18 (1H, s).
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.49 (9H, s), 4.72 (2H, s), 5.96-6.03 (1H, m), 7.09-7.16 (1H, m), 7.27-7.30 (1H, m), 7.42-7.47 (1H, m), 7.95-8.00 (1H, m). MS m/z: 298 [M-tBu+H]+.
[参考例G5] [3-(3-クロロ-4-ニトロフェノキシ)-1H-ピラゾール-1-イル]酢酸

1H-NMR (DMSO-D6) δ: 4.92 (2H, s), 6.12 (1H, d, J = 2.4 Hz), 7.23 (1H, dd, J = 9.1, 2.4 Hz), 7.42 (1H, t, J = 2.4 Hz), 7.81 (1H, d, J = 2.4 Hz), 8.17 (1H, d, J = 9.1 Hz), 13.15 (1H, br s). MS m/z: 298 [M+H]+.
[参考例G6] (2-フルオロ-4-{[4-(ヒドロキシメチル)-1,3-チアゾール-2-イル]オキシ}フェニル)カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.53 (9H, s), 2.20 (1H, t, J = 5.5 Hz), 4.58 (2H, d, J = 5.5 Hz), 6.65-6.79 (2H, m), 7.01-7.14 (2H, m), 8.13 (1H, s). MS m/z: 341 [M+H]+.
[参考例G7] (4-{[4-(シアノメチル)-1,3-チアゾール-2-イル]オキシ}-2-フルオロフェニル)カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.53 (9H, s), 3.73 (2H, s), 6.55-6.76 (1H, m), 6.83 (1H, s), 6.98-7.14 (2H, m), 8.07-8.23 (1H, m). MS m/z: 294 [M-tBu+H]+.
[参考例G8] [2-(4-アミノ-3-フルオロフェノキシ)-1,3-チアゾール-4-イル]酢酸塩酸塩

MS m/z: 269 [M+H]+.
[参考例H1] {4-[3-(tert-ブチルカルバモイル)フェノキシ]-2-フルオロフェニル}カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.46 (9H, s), 1.53 (9H, s), 5.89 (1H, s), 6.59 (1H, s), 6.74-6.81 (2H, m), 7.06-7.11 (1H, m), 7.33-7.44 (3H, m), 8.01 (1H, s).
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.28 (9H, s), 4.53 (2H, s), 5.23 (2H, s), 5.88 (1H, d, J = 2.4 Hz), 6.07 (1H, s), 6.84 (1H, s), 6.99 (1H, d, J = 10.9 Hz), 7.33 (1H, d, J = 2.4 Hz), 7.34-7.45 (5H, m), 8.28 (1H, br s). MS m/z: 475 [M+H]+.
[参考例I1] N-tert-ブチル-2-[3-(3-クロロ-4-ニトロフェノキシ)-4-フルオロ-1H-ピラゾール-1-イル]アセトアミド

1H-NMR (CDCl3) δ: 1.34 (9H, s), 4.53 (2H, s), 5.74 (1H, br s), 7.13 (1H, dd, J = 9.1, 2.4 Hz), 7.28 (1H, d, J = 2.4 Hz), 7.48 (1H, d, J = 4.3 Hz), 8.00 (1H, d, J = 9.1 Hz). MS m/z: 371 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 4.17 (3H, s), 6.97 (1H, dd, J = 8.9, 2.7 Hz), 7.06 (1H, dd, J = 7.9, 2.4 Hz), 7.10 (1H, d, J = 2.4 Hz), 7.47-7.53 (1H, m), 7.59-7.61 (1H, m), 7.70 (1H, d, J= 7.9 Hz), 7.78 (1H, s), 7.99 (1H, d, J= 9.2 Hz). MS m/z: 331 [M+H]+.
[参考例J2] (2-フルオロ-4-{3-[(トリメチルシリル)エチニル]フェノキシ}フェニル)カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 0.24 (9H, s), 1.53 (9H, s), 6.58 (1H, br s), 6.74-6.81 (2H, m), 6.94-6.98 (1H, m), 7.03-7.05 (1H, m), 7.18-7.27 (2H, m), 8.00 (1H, br s).
[参考例J3] [4-(3-エチニルフェノキシ)-2-フルオロフェニル]カルバミン酸tert-ブチル

1H-NMR (CDCl3) δ: 1.53 (9H, d, J = 1.2 Hz), 6.59 (1H, br s), 6.74-6.83 (2H, m), 6.96-7.01 (1H, m), 7.06-7.09 (1H, m), 7.20-7.32 (3H, m), 8.01 (1H, br s).
[参考例J4] {2-フルオロ-4-[3-(1-メチル-1H-1,2,3-トリアゾール-4-イル)フェノキシ]フェニル}カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.53 (9H, s), 4.14 (3H, s), 6.59 (1H, br s), 6.78-6.86 (2H, m), 6.94-6.98 (1H, m), 7.36-7.44 (2H, m), 7.56-7.60 (1H, m), 7.72 (1H, s), 8.00 (1H, br s).
[参考例K1] (2-フルオロ-4-{[1-(2-フルオロピリジン-4-イル)-1H-ピラゾール-3-イル]オキシ}フェニル)カルバミン酸ベンジル
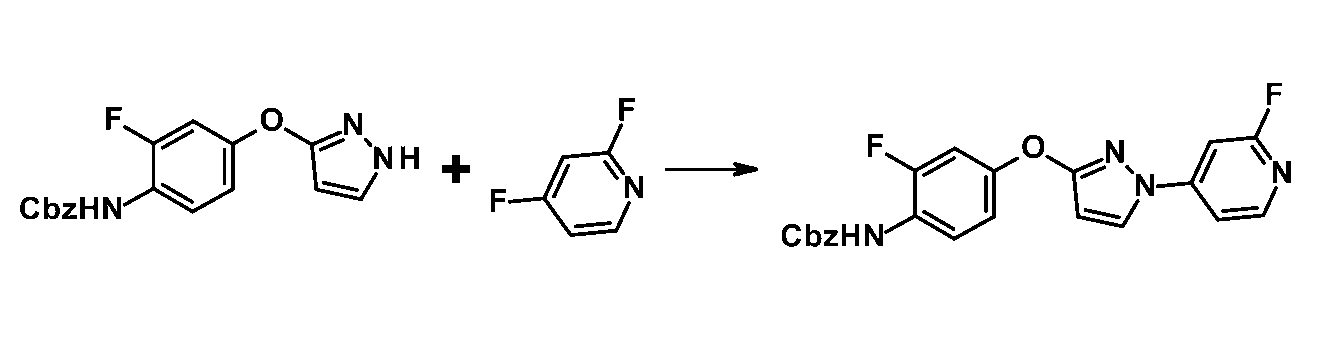
1H-NMR (CDCl3) δ: 5.23 (2H, s), 6.12 (1H, d, J = 3.1 Hz), 6.85 (1H, br s), 7.00-7.07 (2H, m), 7.13-7.18 (1H, m), 7.34-7.44 (6H, m), 7.92 (1H, d, J = 3.1 Hz), 8.10 (1H, br s), 8.22 (1H, d, J = 6.1 Hz). MS m/z: 423 [M+H]+.
[参考例K2] (2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}フェニル)カルバミン酸 ベンジル

1H-NMR (CDCl3) δ: 2.55 (3H, d, J = 2.5 Hz), 5.23 (2H, s), 6.06 (1H, d, J = 3.1 Hz), 6.82 (1H, br s), 6.96-7.06 (2H, m), 7.32-7.46 (5H, m), 7.69 (1H, dd, J = 10.1, 2.1 Hz), 7.84 (1H, d, J = 3.1 Hz), 8.07 (1H, br s), 8.59 (1H, d, J = 1.8 Hz).
同様の方法により対応するブロモ体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.54 (9H, s), 4.07 (3H, s), 6.02-6.07 (1H, m), 6.57-6.66 (1H, m), 6.94-7.06 (2H, m), 7.71-7.77 (1H, m), 8.04 (1H, br s), 8.80 (2H, s). MS m/z: 402 [M+H]+.
同様の方法により対応するブロモ体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 3.65 (2H, s), 3.97 (3H, s), 5.99 (1H, d, J = 2.5 Hz), 6.74-6.80 (1H, m), 6.84-6.89 (1H, m), 6.93-6.99 (2H, m), 7.16 (1H, dd, J = 5.8, 2.1 Hz), 7.85 (1H, d, J = 2.5 Hz), 8.16 (1H, d, J = 5.5 Hz). MS m/z: 301 [M+H]+.
[参考例L1] (2-フルオロ-4-{[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1,3-チアゾール-2-イル]オキシ}フェニル)カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.34 (12H, s), 1.53 (9H, s), 6.65 (1H, s), 6.99-7.10 (2H, m), 7.50 (1H, s), 8.09 (1H, s).
[参考例L2] (2-フルオロ-4-{[4-(2-メトキシピリミジン-5-イル)-1,3-チアゾール-2-イル]オキシ}フェニル)カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.54 (9H, s), 4.05 (3H, s), 6.70 (1H, br s), 7.00 (1H, s), 7.09-7.20 (2H, m), 8.09-8.23 (1H, m), 8.90 (2H, s). MS m/z: 419 [M+H]+.
同様の方法により対応するブロモ体から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 1.35 (9H, s), 6.74 (1H, dd, J = 8.5, 1.8 Hz), 6.90-7.29 (3H, m), 7.34-7.44 (2H, m), 7.55 (1H, d, J = 7.9 Hz), 7.79 (1H, s).
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 4.17 (3H, s), 6.69-6.86 (3H, m), 6.94-6.99 (1H, m), 7.31-7.34 (1H, m), 7.36-7.41 (1H, m), 7.49 (1H, d, J = 7.9 Hz), 7.74 (1H, s).
類似した方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 2.54 (3H, d, J = 2.5 Hz), 3.78 (2H, br s), 6.81 (1H, t, J = 9.2 Hz), 6.95-6.99 (1H, m), 7.03-7.09 (2H, m), 7.71-7.77 (1H, m), 8.72 (1H, t, J = 1.5 Hz). MS m/z: 320 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 3.89 (3H, s), 3.97 (2H, br s), 6.74-6.85 (2H, m), 7.00 (1H, d, J = 2.4 Hz), 7.13-7.16 (1H, m), 7.37 (1H, t, J = 7.9 Hz), 7.54-7.58 (1H, m), 7.71-7.74 (1H, m). MS m/z: 278 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


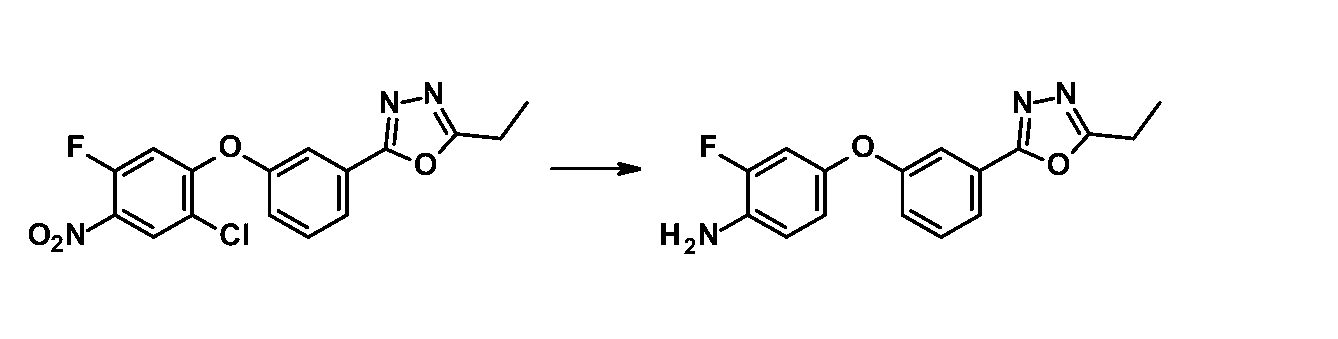
1H-NMR (CDCl3) δ: 1.43 (3H, t, J = 7.7 Hz), 2.95 (2H, q, J = 7.7 Hz), 3.66 (2H, br s), 6.71 (1H, dd, J = 8.2, 2.1 Hz), 6.76-6.82 (2H, m), 7.09-7.12 (1H, m), 7.43 (1H, t, J = 8.2 Hz), 7.56-7.59 (1H, m), 7.72 (1H, d, J = 8.2 Hz). MS m/z: 300 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (CDCl3) δ: 1.27 (6H, d, J = 6.7 Hz), 3.66 (2H, br s), 4.04-4.14 (1H, m), 5.86 (1H, d, J= 3.0 Hz), 6.64-6.71 (1H, m), 6.73-6.82 (2H, m), 6.89 (1H, dd, J = 11.5, 2.4 Hz), 8.08 (1H, d, J = 3.0 Hz). MS m/z: 279 [M+H]+.
[参考例O1] 3-{4-[(tert-ブトキシカルボニル)アミノ]-3-クロロフェノキシ}安息香酸メチル

1H-NMR (CDCl3) δ: 1.51 (9H, s), 3.88 (3H, s), 6.88-6.93 (2H, m), 7.02 (1H, d, J = 3.1 Hz), 7.14-7.17 (1H, m), 7.38 (1H, t, J = 7.9 Hz), 7.56-7.60 (1H, m), 7.73-7.79 (1H, m), 8.10 (1H, d, J = 9.2 Hz).
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.54 (9H, s), 4.08 (2H, s), 6.85-7.16 (4H, m), 7.30-7.49 (4H, m), 8.13 (1H, d, J = 9.1 Hz). MS m/z: 378 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.54 (9H, s), 2.61 (3H, s), 6.87-7.01 (2H, m), 7.05-7.17 (2H, m), 7.46 (1H, t, J = 8.2 Hz), 7.56-7.60 (1H, m), 7.75-7.81 (1H, m), 8.15 (1H, d, J = 9.1 Hz). MS m/z: 402 [M+H]+.
[参考例O6] {2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]フェニル}カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.54 (9H, s), 2.61 (3H, s), 6.62 (1H, br s), 6.78-6.87 (2H, m), 7.15 (1H, dd, J = 8.2, 2.7 Hz), 7.46 (1H, t, J = 7.9 Hz), 7.57-7.61 (1H, m), 7.76-7.80 (1H, m), 7.96-8.13 (1H, m). MS m/s 386 [M+H]+.
[参考例O7] (2-フルオロ-4-{3-[5-(プロパン-2-イル)-1,3,4-オキサジアゾール-2-イル]フェノキシ}フェニル)カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.45 (6H, d, J = 7.3 Hz), 1.53 (9H, s), 3.21-3.33 (1H, m), 6.61 (1H, br s), 6.77-6.87 (2H, m), 7.10-7.16 (1H, m), 7.42-7.49 (1H, m), 7.62-7.66 (1H, m), 7.76-7.82 (1H, m), 7.97-8.11 (1H, m). MS m/z: 414 [M+H]+.
[参考例O8] [4-(3-{[2-(シクロプロピルカルボニル)ヒドラジニル]カルボニル}フェノキシ)-2-フルオロフェニル]カルバミン酸tert-ブチル

1H-NMR (CDCl3) δ: 0.86-0.91 (2H, m), 1.05-1.09 (2H, m), 1.50-1.55 (1H, m), 1.53 (9H, s), 6.60 (1H, br s), 6.76-6.81 (2H, m), 7.15 (1H, dd, J = 7.3, 2.4 Hz), 7.38-7.43 (2H, m), 7.51 (1H, d, J = 9.2 Hz), 8.02 (1H, br s), 8.57-8.61 (1H, m), 8.85-8.81 (1H, m). MS m/z: 374 [M-tBu+H]+.
[参考例O9] {4-[3-(5-シクロプロピル-1,3,4-オキサジアゾール-2-イル)フェノキシ]-2-フルオロフェニル}カルバミン酸tert-ブチル

MS m/z: 412 [M+H]+.
[参考例O10] {2-フルオロ-4-[3-(5-メチル-1,3,4-チアジアゾール-2-イル)フェノキシ]フェニル}カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.53 (9H, s), 2.82 (3H, s), 6.61 (1H, br s), 6.76-6.89 (2H, m), 7.04-7.12 (1H, m), 7.42 (1H, t, J = 7.9 Hz), 7.56-7.58 (1H, m), 7.62-7.66 (1H, m), 8.04 (1H, br s). MS m/z: 402 [M+H]+.
[参考例O11] (4-{[4-(5-シクロプロピル-1,3,4-オキサジアゾール-2-イル)-1,3-チアゾール-2-イル]オキシ}-2-フルオロフェニル)カルバミン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.07-1.31 (4H, m), 1.54 (9H, s), 2.13-2.28 (1H, m), 6.65-6.78 (1H, m), 7.03-7.19 (2H, m), 7.55-7.69 (1H, m), 8.09-8.22 (1H, m).
[参考例P1] 2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリン

1H-NMR (CDCl3) δ: 2.61 (3H, s), 3.66 (2H, br s), 6.68-6.84 (3H, m), 7.10-7.13 (1H, m), 7.43 (1H, t, J = 7.9 Hz), 7.53-7.57 (1H, m), 7.69-7.74 (1H, m). MS m/s 286 [M+H]+.
参考例P1の前半工程と同様の方法により対応する中間体から以下の化合物を合成した。

3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}安息香酸メチル

1H-NMR (CDCl3) δ: 1.88-2.11 (4H, m), 3.49-3.62 (1H, m), 3.67-3.77 (1H, m), 3.82-4.04 (2H, m), 3.91 (3H, s), 4.02 (3H,s), 4.67-4.76 (1H, m), 5.72 (1H, dd, J = 10.7, 1.8 Hz), 6.31 (1H, dd, J = 17.1, 1.8 Hz), 6.63 (1H, dd, J = 17.1, 10.7 Hz), 6.84-6.94 (2H, m), 7.15-7.19 (1H, m), 7.21-7.33 (3H, m), 7.45 (1H, t, J = 8.2 Hz), 7.68-7.71 (1H, m), 7.83 (1H, d, J = 7.3 Hz), 8.33-8.41 (1H, m), 8.68 (1H, s). MS m/z: 573 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (CDCl3) δ: 1.48 (9H, s), 1.54 (3H, t, J = 7.0 Hz), 1.85-1.88 (2H, m), 1.95-1.98 (2H, m), 2.62 (3H, s), 3.34-3.40 (2H, m), 3.74-3.78 (2H, m), 4.23 (2H, q, J= 7.0 Hz), 4.62-4.63 (1H, m), 6.89-6.94 (2H, m), 7.19-7.95 (5H, m), 7.51 (1H, J = 7.9 Hz), 7.81 (1H, d, J = 7.9 Hz), 8.28 (1H, t, J = 8.8 Hz), 8.63 (1H, s). MS m/z: 657 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (DMSO-D6) δ: 1.59-1.78 (2H, m), 1.98-2.11 (2H, m), 3.24-3.54 (2H, m), 3.84-3.99 (2H, m), 3.94 (3H, s), 4.72-4.82 (1H, m), 5.69 (1H, dd, J = 10.3, 2.4 Hz), 6.12 (1H, dd, J = 17.0, 2.4 Hz), 6.86 (1H, dd, J = 17.0, 10.3 Hz), 6.97 (1H, dd, J = 8.8, 2.1 Hz), 7.16 (1H, dd, J = 11.2, 2.7 Hz), 7.22 (1H, s), 7.41 (1H, dd, J = 8.8, 2.1 Hz), 7.48-7.64 (3H, m), 7.76 (1H, d, J = 7.9 Hz), 7.89 (1H, s), 8.36 (1H, s), 9.45 (1H, br, s), 13.15 (1H, br, s). MS m/z: 559 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 1.60-1.78 (2H, m), 1.91 (3H, s), 1.99-2.11 (2H, m), 3.19-3.56 (2H, m), 3.84-3.99 (2H, m), 3.94 (3H, s), 4.72-4.81 (1H, m), 5.69 (1H, dd, J= 10.6, 2.4 Hz), 6.12 (1H, dd, J= 16.7, 2.4 Hz), 6.86 (1H, dd, J= 16.7, 10.6 Hz), 6.91-6.97 (1H, m), 7.13 (1H, dd, J = 11.2, 2.7 Hz), 7.21 (1H, s), 7.35 (1H, dd, J = 7.6, 2.1 Hz), 7.49-7.62 (3H, m), 7.70 (1H, d, J = 7.9 Hz), 7.89 (1H, s), 8.36 (1H, s), 9.44 (1H, s), 9.91 (1H, s), 10.38 (1H, s).
[参考例S1] 4-{[4-(4-{[1-(tert-ブチルカルバモイル)-1H-ピラゾール-3-イル]オキシ}-2-フルオロアニリノ)-7-ヒドロキシキナゾリン-6-イル]オキシ}ピペリジン-1-カルボン酸 tert-ブチル

1H-NMR (CD3OD) δ: 1.45 (9H, s), 1.48 (9H, s), 1.72-1.90 (2H, m), 2.03-2.17 (2H, m), 3.21-3.40 (2H, m), 3.85-3.97 (2H, m), 4.73-4.86 (1H, m), 6.17 (1H, d, J = 3.1 Hz), 7.06-7.24 (2H, m), 7.13 (1H, s), 7.19 (1H, br s), 7.56 (1H, t, J = 8.6 Hz), 7.85 (1H, s), 7.90 (1H, s), 8.19 (1H, d, J = 3.1 Hz), 8.36 (1H, s). MSm/z: 636 [M+H]+.
[参考例S2] 4-({4-(4-{[1-(tert-ブチルカルバモイル)-1H-ピラゾール-3-イル]オキシ}-2-フルオロアニリノ)-7-[(2H3)メチルオキシ]キナゾリン-6-イル}オキシ)ピペリジン-1-カルボン酸 tert-ブチル

を粗生成物として得た。
[参考例T1] 7-エトキシ-N-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]フェニル}-6-[(ピペリジン-4-イル)オキシ]キナゾリン-4-アミン

同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (DMSO-D6) δ: 3.63 (3H, s), 3.75 (2H, s), 4.10 (3H, s), 6.85-7.20 (5H, m), 7.32-7.59 (3H, m), 8.79 (1H, s), 9.37 (1H, s), 11.17 (1H, br s). MS m/z: 479 [M+H]+.
[参考例U2] N-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]フェニル}-7-メトキシ-6-ニトロキナゾリン-4-アミン

1H-NMR (DMSO-D6) δ: 2.58 (3H, s), 4.07 (3H, s), 7.01 (1H, d, J= 9.1 Hz), 7.20 (1H, d, J = 10.9 Hz), 7.39 (1H, d, J = 7.3 Hz), 7.49 (1H, s), 7.60-7.54 (2H, m), 7.67 (1H, t, J = 7.6 Hz), 7.80 (1H, d, J= 7.9 Hz), 8.56 (1H, s), 9.19 (1H, s), 10.18 (1H, s). MS m/z: 489 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。





1H-NMR (DMSO-D6) δ: 1.00-1.23 (4H, m), 2.23-2.36 (1H, m), 4.11 (3H, s), 7.38-7.46 (1H, m), 7.54 (1H, s), 7.63-7.74 (2H, m), 8.06-8.10 (1H, m), 8.79 (1H, s), 9.35 (1H, s). MS m/z: 522 [M+H]+.
[参考例U11] 3-{3-フルオロ-4-[(7-メトキシ-6-ニトロキナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボン酸tert-ブチル

1H-NMR (CDCl3) δ: 1.63 (9H, s), 4.09 (3H, s), 6.07 (1H, d, J = 3.0 Hz), 7.01-7.11 (2H, m), 7.41 (1H, s), 7.68-7.75 (1H, m), 8.03 (1H, d, J= 3.0 Hz), 8.20 (1H, t, J = 8.8 Hz), 8.56 (1H, s), 8.74 (1H, s). MS m/z: 497 [M+H]+.
[参考例U12]
7-フルオロ-N-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]フェニル}-6-ニトロキナゾリン-4-アミン

1H-NMR (DMSO-D6) δ: 2.58 (3H, s), 7.01 (1H, dd, J= 9.2, 2.4 Hz), 7.21 (1H, dd, J= 11.6, 2.4 Hz), 7.38-7.41 (1H, m), 7.54-7.59 (2H, m), 7.67 (1H, t, J = 7.9 Hz), 7.79-7.85 (2H, m), 8.60 (1H, br s), 9.50-9.54 (1H, m), 10.58 (1H, br s). MS m/z: 477 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.30 (9H, s), 3.44 (2H, s), 4.04 (3H, s), 4.33 (2H, br s), 6.43 (1H, br s), 6.61 (1H, s), 6.95 (1H, s), 7.11-7.36 (4H, m), 8.62 (1H, s), 8.75 (1H, t, J = 8.9 Hz). MS m/z: 497 [M+H]+.
[参考例V1] (3-{3-フルオロ-4-[(7-メトキシ-6-ニトロキナゾリン-4-イル)アミノ]フェノキシ}フェニル)酢酸

1H-NMR (DMSO-D6) δ: 3.63 (2H, s), 4.07 (3H, s), 6.81-7.18 (5H, m), 7.31-7.60 (3H, m), 8.55 (1H, s), 9.19 (1H, s), 10.15 (1H, s), 12.38 (1H, br s). MS m/z: 465 [M+H]+.
[参考例V2] N-tert-ブチル-2-(3-{3-フルオロ-4-[(7-メトキシ-6-ニトロキナゾリン-4-イル)アミノ]フェノキシ}フェニル)アセトアミド

1H-NMR (CDCl3) δ: 1.30 (9H, s), 3.46 (2H, s), 4.09 (3H, s), 5.29 (1H, s), 6.76-7.09 (5H, m), 7.28-7.36 (1H, m), 7.41 (1H, s), 7.98-8.13 (1H, m), 8.58 (1H, s), 8.71 (1H, s). MS m/z: 520 [M+H]+.
[参考例V3] N-tert-ブチル-2-{3-フルオロ-4-[(7-メトキシ-6-ニトロキナゾリン-4-イル)アミノ]フェノキシ}-1,3-チアゾール-4-カルボキサミド

1H-NMR (CDCl3) δ: 1.45 (9H, s), 4.11 (3H, s), 6.91 (1H, br s), 7.21-7.23 (2H, m), 7.46 (1H, s), 7.57 (1H, br s), 7.64 (1H, s), 8.50 (1H, s), 8.55 (1H, t, J = 8.9 Hz), 8.81 (1H, s). MS m/z: 513 [M+H]+.
[参考例W1] 7-エトキシ-N-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]フェニル}-6-ニトロキナゾリン-4-アミン

1H-NMR (CDCl3) δ: 1.56 (3H, t, J = 6.9 Hz), 2.62 (3H, s), 4.33 (2H, q, J= 6.9 Hz), 6.91-6.95 (2H, m), 7.24 (1H, dd, J = 8.5, 2.4 Hz), 7.40 (1H, s), 7.52 (1H, t, J = 7.9 Hz), 7.64 (1H, br s), 7.71 (1H, s), 7.83 (1H, d, J = 7.9 Hz), 8.28 (1H, q, J = 9.8 Hz), 8.51 (1H, d, J = 2.4 Hz), 8.76 (1H, s). MS m/z: 503 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.31 (9H, s), 3.46 (2H, s), 4.03 (3H, s), 4.31 (2H, br s), 5.22 (1H, br s), 6.82-7.05 (6H, m), 7.13 (1H, br s), 7.20 (1H, s), 7.33 (1H, t, J = 8.0 Hz), 8.50 (1H, t, J = 8.9 Hz), 8.59 (1H, s). MS m/z: 490 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。




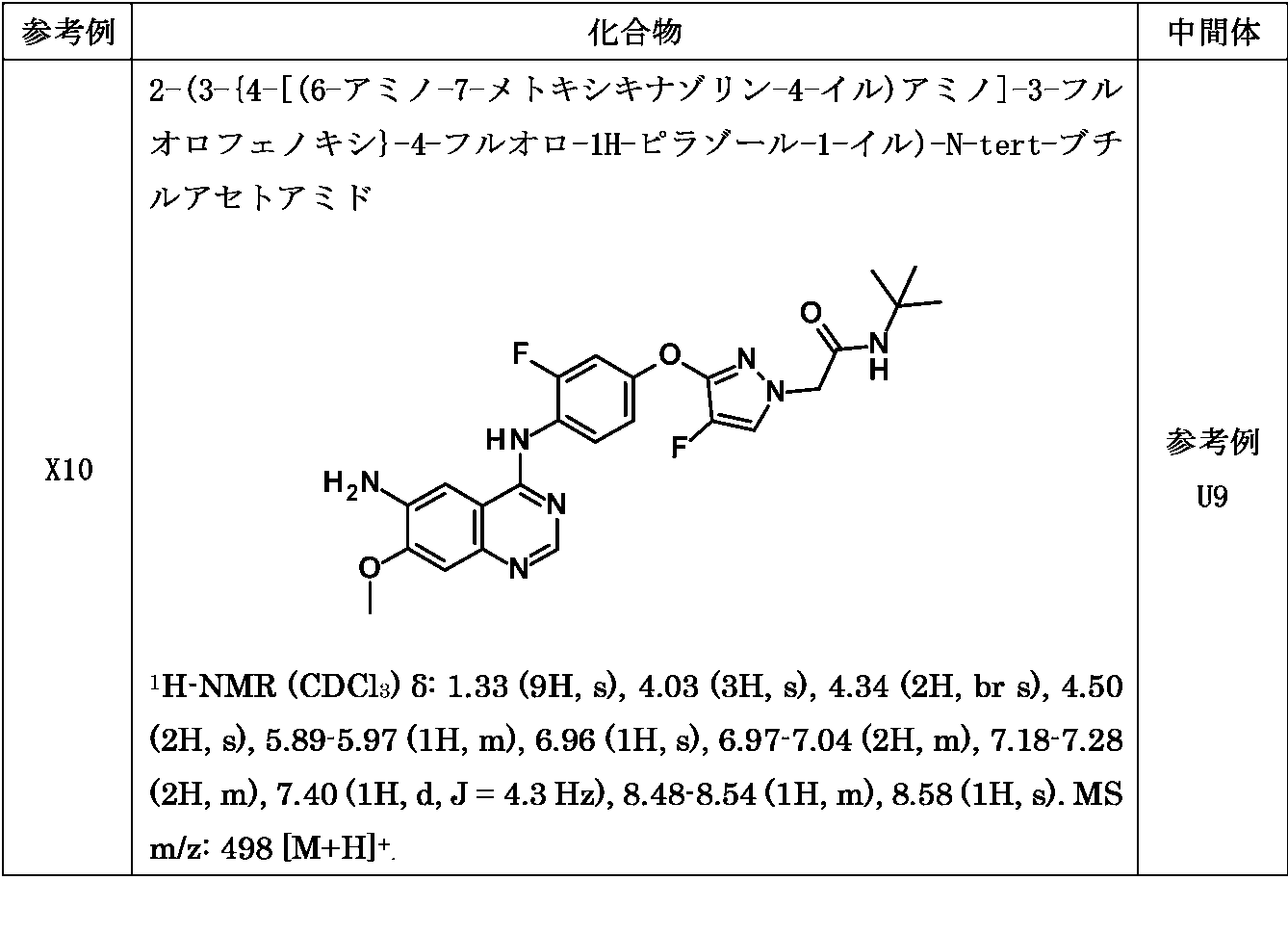
2-{4-[(6-アミノ-7-メトキシキナゾリン-4-イル)アミノ]-3-フルオロフェノキシ}-N-tert-ブチル-1,3-チアゾール-4-カルボキサミド

1H-NMR (CDCl3) δ: 1.45 (9H, s), 4.04 (3H, s), 4.34 (2H, br s), 6.95 (2H, s), 7.16-7.18 (2H, m), 7.22 (1H, s), 7.25 (1H, d, J = 3.7 Hz), 7.61 (1H, s), 8.63 (1H, s), 8.80 (1H, t, J= 9.2 Hz). MS m/z: 483 [M+H]+.
類似した方法により対応する中間体から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 1.03-1.22 (4H, m), 2.22-2.37 (1H, m), 3.97 (3H, s), 5.41 (2H, s), 7.09 (1H, s), 7.27-7.35 (2H, m), 7.51-7.59 (1H, m), 7.65-7.74 (1H, m), 8.04-8.07 (1H, m), 8.24-8.28 (1H, m), 9.24 (1H, s). MS m/z: 492 [M+H]+.
[参考例X14] 3-{4-[(6-アミノ-7-メトキシキナゾリン-4-イル)アミノ]-3-フルオロフェノキシ}-1H-ピラゾール-1-カルボン酸 tert-ブチル

1H-NMR (CDCl3) δ: 1.65 (9H, s), 4.03 (3H, s), 4.31 (2H, br s), 5.98-6.04 (1H, m), 6.92-7.23 (5H, m), 7.99-8.03 (1H, m), 8.53-8.64 (2H, m). MS m/z: 467 [M+H]+.
[参考例Y1] (2E)-N-[4-(4-{3-[2-(tert-ブチルアミノ)-2-オキソエチル]フェノキシ}-2-フルオロアニリノ)-7-メトキシキナゾリン-6-イル]-4-クロロブタ-2-エンアミド

1H-NMR (CDCl3) δ: 1.31 (9H, s), 3.47 (2H, s), 4.09 (3H, s), 4.24-4.34 (2H, m), 5.23 (1H, br s), 6.30-6.45 (1H, m), 6.75-7.20 (6H, m), 7.30-7.40 (2H, m), 7.51 (1H, br s), 8.13-8.26 (2H, m), 8.66 (1H, s), 9.15 (1H, s). MS m/z: 592 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。





1H-NMR (CDCl3) δ: 1.65 (9H, s), 2.32 (6H, s), 3.14-3.20 (2H, m), 4.06 (3H, s), 6.03 (1H, m), 6.18-6.29 (1H, m), 6.97-7.12 (3H, m), 7.23-7.29 (1H, m), 7.53 (1H, s), 7.99-8.03 (1H, m), 8.12-8.31 (2H, m), 8.65 (1H, s), 9.16 (1H, s). MS m/z: 578 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 2.43 (6H, s), 3.36-3.47 (2H, m), 4.01 (3H, s), 5.97-6.02 (1H, m), 6.56-7.11 (4H, m), 7.22-7.50 (2H, m), 7.71-7.78 (1H, m), 8.37 (1H, s), 8.90 (1H, s), 9.57-9.89 (2H, m), 12.48 (1H, s). MS m/z: 478 [M+H]+.
[参考例Z4] (3-{3-クロロ-4-[(6-{[(2E)-4-(ジメチルアミノ)ブタ-2-エノイル]アミノ}-7-メトキシキナゾリン-4-イル)アミノ]フェノキシ}-4-フルオロ-1H-ピラゾール-1-イル)酢酸

1H-NMR (CD3OD) δ: 2.93 (6H, s), 3.95-4.03 (2H, m), 4.10 (3H, s), 4.76 (2H, s), 6.73-6.85 (1H, m), 6.88-7.01 (1H, m), 7.11-7.19 (1H, m), 7.24 (1H, s), 7.32 (1H, s), 7.58-7.64 (1H, m), 7.71-7.77 (1H, m), 8.41 (1H, s), 9.07 (1H, s). MS m/z: 570 [M+H]+.
[実施例1] N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}ベンズアミド

1H-NMR (CDCl3) δ: 1.47 (9H, s), 1.89-2.09 (4H, m), 3.50-3.79 (2H, m), 3.82-4.00 (2H, m), 4.01 (3H, s), 4.68-4.77 (1H, m), 5.72 (1H, dd, J = 10.4, 1.8 Hz), 5.93 (1H, s), 6.31 (1H, dd, J = 17.1, 1.8 Hz), 6.63 (1H, dd, J = 17.1, 10.4 Hz), 6.84-6.92 (2H, m), 7.11-7.20 (2H, m), 7.22-7.31 (2H, m), 7.38-7.48 (3H, m), 8.30-8.37 (1H, m), 8.67 (1H, s). MS m/z: 614 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (CDCl3) δ: 1.92-2.10 (4H, m), 3.50-3.61 (1H, m), 3.66-3.77 (1H, m), 3.84-3.99 (2H, m), 4.02 (3H, s), 4.15 (3H, s), 4.67-4.75 (1H, m), 5.72 (1H, dd, J= 10.4, 1.8 Hz), 6.31 (1H, dd, J= 17.1, 1.8 Hz), 6.63 (1H, dd, J= 17.1, 10.4 Hz), 7.02 (1H, dd, J= 7.9, 2.4 Hz), 7.09 (1H, dd, J= 8.9, 2.7 Hz), 7.18 (1H, d, J= 2.4 Hz), 7.24 (1H, s), 7.31 (1H, s), 7.40-7.45 (1H, m), 7.51-7.53 (1H, m), 7.55 (1H, s), 7.60 (1H, d, J = 7.9 Hz), 7.76 (1H, s), 8.55 (1H, d, J= 9.2 Hz), 8.69 (1H, s). MS m/z: 612 [M+H]+.
[実施例4] N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド

1H-NMR (CDCl3) δ: 1.46 (9H, s), 1.90-2.07 (4H, m), 3.49-3.60 (1H, m), 3.68-3.79 (1H, m), 3.84-3.97 (2H, m), 4.02 (3H, s), 4.67-4.75 (1H, m), 5.72 (1H, dd, J = 10.4, 1.8 Hz), 5.99 (1H, d, J = 3.1 Hz), 6.31 (1H, dd, J = 17.1, 1.8 Hz), 6.63 (1H, dd, J = 17.1, 10.4 Hz), 6.81 (1H, s), 7.01-7.08 (2H, m), 7.17 (1H, s), 7.22 (1H, s), 7.31 (1H, s), 8.13 (1H, d, J = 2.4 Hz), 8.39-8.46 (1H, m), 8.69 (1H, s). MS m/z: 604 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (DMSO-D6) δ: 1.66-1.78 (2H, m), 1.98-2.09 (2H, m), 3.44-3.56 (2H, m), 3.83-3.90 (2H, m), 3.90 (3H, s), 3.94 (3H, s), 4.71-4.81 (1H, m), 5.66 (1H, dd, J = 10.4, 2.1 Hz), 6.09 (1H, dd, J = 16.6, 2.1 Hz), 6.36 (1H, d, J = 2.8 Hz), 6.80 (1H, dd, J = 16.6, 10.4 Hz), 7.10-7.17 (2H, m), 7.22 (1H, s), 7.26 (1H, dd, J = 11.3, 2.8 Hz), 7.37-7.45 (1H, m), 7.56 (1H, t, J = 8.9 Hz), 7.90 (1H, s), 8.21 (1H, d, J = 6.1 Hz), 8.35 (1H, s), 8.65 (1H, d, J = 2.8 Hz), 9.31 (1H, s). MS m/z: 612 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (CDCl3) δ: 1.92-2.11 (4H, m), 2.62 (3H, s), 3.52-3.61 (1H, m), 3.67-3.78 (1H, m), 3.85-4.00 (2H, m), 4.03 (3H, s), 4.67-4.75 (1H, m), 5.72 (1H, dd, J = 10.9, 1.8 Hz), 6.31 (1H, dd, J = 17.0, 1.8 Hz), 6.63 (1H, dd, J = 17.0, 10.9 Hz), 7.10 (1H, dd, J = 9.2, 3.1 Hz), 7.17-7.25 (3H, m), 7.32 (1H, s), 7.50 (1H, t, J = 7.9 Hz), 7.58 (1H, s), 7.64-7.69 (1H, m), 7.81 (1H, d, J = 7.9 Hz), 8.62-8.73 (2H, m). MS m/z: 613 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (CDCl3) δ: 1.89-2.09 (4H, m), 2.62 (3H, s), 3.51-3.61 (1H, m), 3.68-3.79 (1H, m), 3.82-3.99 (2H, m), 4.02 (3H, s), 4.68-4.76 (1H, m), 5.68-5.75 (1H, m), 6.26-6.35 (1H, m), 6.63 (1H, dd, J = 16.8, 10.7 Hz), 6.89-6.97 (2H, m), 7.17-7.33 (4H, m), 7.51 (1H, t, J = 7.9 Hz), 7.67-7.70 (1H, m), 7.81 (1H, d, J = 7.9 Hz), 8.40 (1H, t, J = 9.5 Hz), 8.69 (1H, s). MS m/z: 597 [M+H]+.
[実施例18] 1-{4-[(7-エトキシ-4-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}キナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン

1H-NMR (CDCl3) δ: 1.54 (3H, t, J = 7.0 Hz), 1.98-2.01 (4H, m), 2.62 (3H, s), 3.59-3.61 (1H, m), 3.86-3.88 (3H, m), 4.24 (2H, q, J = 7.0 Hz), 4.68-4.71 (1H, m), 5.72 (1H, dd, J = 10.3, 1.8 Hz), 6.31 (1H, dd, J = 16.7, 1.8 Hz), 6.63 (1H, dd, J = 16.7, 10.3 Hz), 6.91-6.94 (2H, m), 7.21-7.29 (4H, m), 7.50 (1H, t, J = 8.2 Hz), 7.68 (1H, t, J = 2.1 Hz), 7.81 (1H, d, J = 7.9 Hz), 8.40-8.47 (1H, m), 8.68 (1H, s). MS m/z: 611 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。



1H-NMR (CDCl3) δ: 1.24-1.30 (2H, m), 1.40-1.45 (2H, m), 1.91-2.08 (4H, m), 3.52-3.62 (1H, m), 3.69-3.80 (1H, m), 3.84-3.98 (2H, m), 4.02 (3H, s), 4.66-4.75 (1H, m), 5.72 (1H, dd, J = 10.9, 1.8 Hz), 6.06 (1H, d, J = 3.0 Hz), 6.31 (1H, dd, J = 16.4, 1.8 Hz), 6.62 (1H, dd, J = 16.4, 10.9 Hz), 7.04-7.10 (2H, m), 7.18-7.23 (2H, m), 7.32 (1H, s), 7.40 (1H, br s), 8.14 (1H, d, J = 3.0 Hz), 8.42-8.49 (1H, m), 8.70 (1H, s). MS m/z: 656 [M+H]+.
同様の方法により対応するカルボン酸から以下の化合物を合成した。

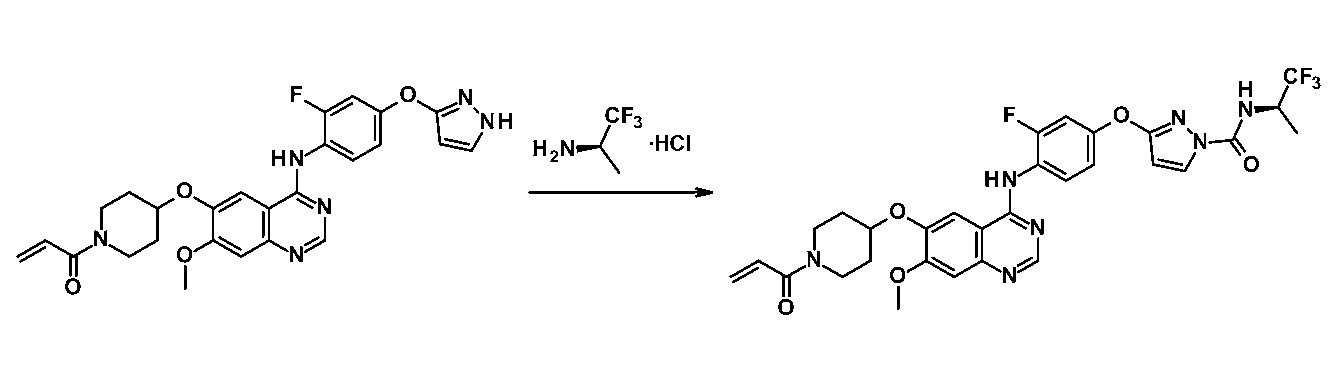
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 7.3 Hz), 1.91-2.08 (4H, m), 3.52-3.61 (1H, m), 3.69-3.80 (1H, m), 3.84-3.98 (2H, m), 4.02 (3H, s), 4.63-4.75 (2H, m), 5.72 (1H, dd, J= 10.5, 1.8 Hz), 6.07 (1H, d, J= 3.0 Hz), 6.31 (1H, dd, J = 16.7, 1.8 Hz), 6.63 (1H, dd, J= 16.7, 10.5 Hz), 6.94-7.01 (1H, m), 7.05-7.10 (2H, m), 7.17-7.23 (2H, m), 7.32 (1H, s), 8.15 (1H, d, J = 3.0 Hz), 8.43-8.51 (1H, m), 8.69 (1H, s). MS m/z: 644 [M+H]+.
同様の方法により対応する中間体およびアミンから以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 1.66-1.79 (2H, m), 1.96-2.08 (2H, m), 2.46-2.48 (3H, m), 3.43-3.54 (2H, m), 3.82-3.92 (2H, m), 3.94 (3H, s), 4.71-4.80 (1H, m), 5.66 (1H, dd, J = 10.4, 2.4 Hz), 6.09 (1H, dd, J = 17.1, 2.4 Hz), 6.34 (1H, d, J = 2.4 Hz), 6.80 (1H, dd, J = 17.1, 10.4 Hz), 7.09-7.15 (1H, m), 7.19-7.27 (2H, m), 7.55 (1H, t, J= 8.9 Hz), 7.90 (1H, s), 8.00-8.08 (1H, m), 8.34 (1H, s), 8.56 (1H, d, J = 2.4 Hz), 8.79 (1H, d, J = 1.8 Hz), 9.29 (1H, s). MS m/z: 614 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 1.66-1.79 (2H, m), 1.96-2.08 (2H, m), 3.43-3.55 (2H, m), 3.82-3.92 (2H, m), 3.95 (3H, s), 4.71-4.80 (1H, m), 5.61-5.71 (1H, m), 6.03-6.13 (1H, m), 6.41-6.44 (1H, m), 6.80 (1H, dd, J = 16.6, 10.4 Hz), 7.11-7.32 (3H, m), 7.57 (1H, t, J = 8.6 Hz), 7.76-7.81 (1H, m), 7.86-7.93 (2H, m), 8.35 (1H, s), 8.44 (1H, d, J = 5.5 Hz), 8.70-8.75 (1H, m), 9.31 (1H, s). MS m/z: 616 [M+H]+.
[実施例30] 1-(4-{[4-(2-フルオロ-4-{[1-(2-フルオロピリジン-4-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン

1H-NMR (DMSO-D6) δ: 1.64-1.77 (2H, m), 1.98-2.08 (2H, m), 3.42-3.55 (2H, m), 3.82-3.92 (2H, m), 3.94 (3H, s), 4.72-4.81 (1H, m), 5.67 (1H, dd, J = 10.4, 2.5 Hz), 6.10 (1H, dd, J = 16.6, 2.5 Hz), 6.45 (1H, d, J = 2.5 Hz), 6.82 (1H, dd, J = 16.6, 10.4 Hz), 7.16 (1H, dd, J = 8.6, 2.5 Hz), 7.22 (1H, s), 7.29 (1H, dd, J = 11.0, 2.5 Hz), 7.49-7.60 (2H, m), 7.74 (1H, d, J = 5.5 Hz), 7.90 (1H, s), 8.29 (1H, d, J = 6.1 Hz), 8.35 (1H, s), 8.74 (1H, d, J = 2.5 Hz), 9.35 (1H, br s). MS m/z: 600 [M+H]+.
[実施例31] 1-(4-{[4-(4-{[1-(6-シクロプロピルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}-2-フルオロアニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン

1H-NMR (DMSO-D6) δ: 0.92-1.00 (4H, m), 1.64-1.79 (2H, m), 1.97-2.08 (2H, m), 2.11-2.20 (1H, m), 3.43-3.54 (2H, m), 3.81-3.91 (2H, m), 3.94 (3H, s), 4.71-4.79 (1H, m), 5.66 (1H, dd, J = 10.4, 2.5 Hz), 6.09 (1H, dd, J = 16.6, 2.5 Hz), 6.29 (1H, d, J = 2.5 Hz), 6.80 (1H, dd, J = 16.6, 10.4 Hz), 7.10 (1H, dd, J = 9.2, 2.5 Hz), 7.18-7.24 (2H, m), 7.39 (1H, d, J = 8.6 Hz), 7.53 (1H, t, J = 8.6 Hz), 7.90 (1H, s), 7.99 (1H, dd, J = 8.3, 2.8 Hz), 8.34 (1H, s), 8.44-8.48 (1H, m), 8.82 (1H, d, J = 2.5 Hz), 9.29 (1H, s). MS m/z: 622 [M+H]+.
類似した方法により対応するブロモ体から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 1.35 (6H, d, J = 7.4 Hz), 1.65-1.79 (2H, m), 1.95-2.09 (2H, m), 3.22-3.33 (1H, m), 3.44-3.55 (2H, m), 3.82-3.92 (2H, m), 3.95 (3H, s), 4.72-4.80 (1H, m), 5.66 (1H, dd, J = 10.4, 2.5 Hz), 6.09 (1H, dd, J = 16.9, 2.5 Hz), 6.80 (1H, dd, J = 16.9, 10.4 Hz), 7.23 (1H, s), 7.31-7.37 (1H, m), 7.50-7.58 (1H, m), 7.69 (1H, t, J = 8.6 Hz), 7.91 (1H, s), 8.06 (1H, s), 8.38 (1H, s), 9.38 (1H, s). MS m/z: 632 [M+H]+.
[実施例34] 1-(4-{[4-(4-{[4-(6-エチルピリジン-3-イル)-1,3-チアゾール-2-イル]オキシ}-2-フルオロアニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン

1H-NMR (DMSO-D6) δ: 1.25 (3H, t, J = 7.4 Hz), 1.67-1.79 (2H, m), 1.97-2.08 (2H, m), 2.78 (2H, q, J = 7.4 Hz), 3.42-3.55 (2H, m), 3.82-3.92 (2H, m), 3.95 (3H, s), 4.72-4.81 (1H, m), 5.66 (1H, dd, J = 10.7, 2.5 Hz), 6.09 (1H, dd, J = 16.6, 2.5 Hz), 6.80 (1H, dd, J = 16.6, 10.7 Hz), 7.23 (1H, s), 7.28-7.40 (2H, m), 7.50-7.57 (1H, m), 7.63-7.75 (2H, m), 7.91 (1H, s), 8.08 (1H, dd, J = 8.0, 2.5 Hz), 8.38 (1H, s), 8.94 (1H, d, J = 2.5 Hz), 9.37 (1H, s). MS m/z: 627 [M+H]+.
同様の方法により対応するボロン酸から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 1.67-1.79 (2H, m), 1.97-2.08 (2H, m), 2.44-2.56 (3H, m), 3.44-3.55 (2H, m), 3.82-3.92 (2H, m), 3.95 (3H, s), 4.72-4.81 (1H, m), 5.66 (1H, dd, J = 10.4, 2.1 Hz), 6.09 (1H, dd, J = 16.6, 2.1 Hz), 6.80 (1H, dd, J = 16.6, 10.4 Hz), 7.23 (1H, s), 7.27-7.39 (2H, m), 7.50-7.57 (1H, m), 7.68 (1H, t, J = 8.6 Hz), 7.73 (1H, s), 7.91 (1H, s), 8.06 (1H, dd, J = 8.0, 2.5 Hz), 8.38 (1H, s), 8.91 (1H, d, J = 1.8 Hz), 9.37 (1H, s). MS m/z: 613 [M+H]+.
同様の方法により対応するボロン酸から以下の化合物を合成した。


1H-NMR (DMSO-D6) δ: 2.19 (6H, s), 2.58 (3H, s), 3.08 (2H, d, J= 5.5 Hz), 4.02 (3H, s), 6.59 (1H, d, J= 15.3 Hz), 6.79 (1H, dt, J = 15.3, 5.5 Hz), 6.97-7.00 (1H, m), 7.15-7.18 (1H, m), 7.27 (1H, s), 7.37-7.40 (1H, m), 7.53 (1H, t, J = 8.9 Hz), 7.58 (1H, s), 7.67 (1H, t, J= 7.9 Hz), 7.79 (1H, d, J = 7.9 Hz), 8.40 (1H, s), 8.93 (1H, s), 9.68 (1H, s), 9.71 (1H, s). MS m/z: 570 [M+H]+.
同様の方法により対応する中間体から以下の化合物を合成した。





1H-NMR (DMSO-D6) δ: 1.21-1.28 (6H, m), 2.20 (6H, s), 3.05-3.15 (2H, m), 4.01 (3H, s), 4.44 (2H, d, J= 47.8 Hz), 4.65 (2H, s), 6.52-6.64 (1H, m), 6.71-6.86 (1), 7.06-7.16 (1H, m), 7.23-7.32 (2H, m), 7.44-7.52 (1H, m), 7.93-8.07 (2H, m), 8.33 (1H, s), 8.91 (1H, s), 9.62-9.76 (2H, m). MS m/z: 643 [M+H]+.
[実施例49] (2E)-N-[4-(4-{3-[2-(tert-ブチルアミノ)-2-オキソエチル]フェノキシ}-2-フルオロアニリノ)-7-メトキシキナゾリン-6-イル]-4-(モルホリン-4-イル)ブタ-2-エンアミド

1H-NMR (DMSO-D6) δ: 1.23 (9H, s), 2.36-2.44 (4H, m), 3.09-3.20 (2H, m), 3.34-3.44 (2H, m), 3.55-3.68 (4H, m), 4.01 (3H, s), 6.53-6.65 (1H, m), 6.72-6.89 (2H, m), 6.93-7.11 (4H, m), 7.26 (1H, s), 7.31-7.53 (2H, m), 7.71 (1H, s), 8.37 (1H, s), 8.90 (1H, s), 9.60-9.74 (2H, m). MS m/z: 643 [M+H]+.
同様の方法により対応する中間体およびアミンから以下の化合物を合成した。




1H-NMR (CD3OD) δ: 1.35 (9H, s), 1.95-2.35 (2H, m), 2.50-2.62 (1H, m), 2.68-2.87 (1H, m), 2.95-3.16 (2H, m), 3.39-3.48 (2H, m), 4.09 (3H, s), 4.62 (2H, s), 5.10-5.36 (1H, m), 6.50-6.65 (1H, m), 6.97-7.07 (1H, m), 7.12-7.20 (1H, m), 7.24 (1H, s), 7.31-7.36 (1H, m), 7.57-7.89 (3H, m), 8.33 (1H, s), 9.01 (1H, s). MS m/z: 669 [M+H]+.
同様の方法により対応する中間体およびアミンから以下の化合物を合成した。




実施例5に記載の化合物 (7.95 g, 12.8 mmol) に酢酸エチル (64 mL)、ヘキサン (32 mL) を室温で加えた。析出した結晶をろ取し、ヘキサン/酢酸エチル=2:1の混合溶媒にて洗浄後、60℃で5時間減圧乾燥して表記結晶を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 59.61%、H: 5.42%、N: 15.60%、F: 5.93%
表49に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例65] 1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オンの結晶
実施例13に記載の化合物 (18.0 g, 29.4 mmol) に2-プロパノール (120 mL) を加え、50℃で30分間攪拌した後、水 (120 mL) を加え、50℃で15分間、室温にて1時間攪拌した。析出した結晶をろ取し、60℃で減圧乾燥して表記結晶を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 61.01%、H: 5.12%、N: 13.21%、Cl: 5.76%
表50に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例66] 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オンの結晶
実施例27に記載の化合物 (143 g, 233 mmol) に2-プロパノール (1 L) を加え攪拌後、減圧下2-プロパノールを留去した。さらにこの操作を3回繰り返した後、残留物に2-プロパノール (1 L) を加え攪拌した。析出した結晶をろ取、2-プロパノールにて洗浄し、表記結晶を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 59.88%、H: 5.06%、N: 15.32%、F: 5.99%
表51に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例67] 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オンの結晶
実施例28に記載の化合物を (24.77 g, 41.59 mmol) にエタノール (100 mL) を加え、得られた懸濁液を超音波照射下30分間攪拌した後、析出した結晶をろ取、エタノール (50 mL) で洗浄し、40℃にて終夜乾燥して表記結晶を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 62.45%、H: 5.71%、N: 15.21%、F: 3.01%
表52に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例68] N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド メタンスルホン酸塩
実施例4に記載の化合物 (20.19 mg, 32.34 μmol) に1.0 mol/Lのメタンスルホン酸水溶液 (34.0 μL, 1.05eq.)、水 (168μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定を行った。
元素分析実測値; C: 50.87%、H: 5.25%、N: 12.78%、F: 4.14%、S: 4.06%
表53に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例69] 3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド 1,5-ナフタレンジスルホン酸塩
実施例5に記載の化合物 (301.21 mg, 484.55 μmol) にエタノール (753 μL)、0.802 mol/Lの1,5-ナフタレンジスルホン酸水溶液 (317μL, 0.525eq.)、水 (436 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 56.14%、H: 5.02%、N: 13.00%、F: 5.31%、S: 3.62%
表54に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例70] 1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン メタンスルホン酸塩
実施例13に記載の化合物 (20.13 mg, 31.90 μmol) に1.000 mol/Lのメタンスルホン酸水溶液 (33.5μL, 1.05eq.)、水 (168 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 55.71%、H: 4.79%、N: 11.62%、Cl: 4.97%、S: 3.36%
表55に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例71] 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン ベンゼンスルホン酸塩
実施例27に記載の化合物 (20.72 mg, 33.77 μmol) にエタノール (41μL)、0.998 mol/Lのベンゼンスルホン酸水溶液 (35.5 μL, 1.05eq.)、水 (130 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 57.22%、H: 4.79%、N: 12.30%、F: 4.84%、S: 4.01%
表56に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例72] 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 酒石酸塩
実施例27に記載の化合物 (20.21 mg, 32.94 μmol) にエタノール (40 μL)、1.000 mol/Lの酒石酸水溶液 (40 μL, 1.05eq.)、水 (127 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 51.96%、H: 5.01%、N: 11.64%、F: 4.58%
表57に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例73] 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩
実施例27に記載の化合物 (20.57 mg, 33.52 μmol) にエタノール (41 μL)、1.000 mol/Lのクエン酸水溶液 (35.2 μL, 1.05eq.)、水 (129 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 55.11%、H: 4.79%、N: 11.92%、F: 4.55%
表58に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例74] 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 塩酸塩
実施例28に記載の化合物 (20.24 mg, 31.56 μmol) にエタノール (40 μL)、1.004 mol/Lの塩酸 (33.0 μL, 1.05eq.)、水 (129 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 54.64%、H: 5.59%、N: 13.91%、Cl: 4.43%、F: 2.72%
表59に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例75] 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 1,5-ナフタレンジスルホン酸塩
実施例28に記載の化合物 (20.08 mg, 31.31 μmol) にエタノール (40 μL)、1.000 mol/Lの1,5-ナフタレンジスルホン酸水溶液 (32.9 μL, 1.05eq.)、水 (128 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 57.41%、H: 4.82%、N: 12.57%、F: 2.64%、S: 4.35%
表60に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

[実施例76] 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩
実施例28に記載の化合物 (20.23 mg, 31.55 μmol) にエタノール (40 μL)、1.000 mol/Lのクエン酸水溶液 (33.1 μL, 1.05eq.)、水 (129 μL) を室温で加えた。40℃で終夜攪拌した後、室温で約30分間攪拌し、析出した結晶をろ取し、室温で終夜乾燥して表記化合物を得た。本結晶について、粉末X線回折測定および元素分析を行った。
元素分析実測値; C: 55.53%、H: 5.20%、N: 12.22%、F: 2.97%
表61に粉末X線回折スペクトルにおける回折角 (2θ)、格子面間隔 (d値)、及び相対強度を記載する。

細胞増殖抑制試験
Ba/F3-Mock、Ba/F3-EGFR WT、Ba/F3-EGFR ins. ASV、及びBa/F3-EGFR ins. SVD細胞株は、10% FBS含有RPMI 1640に1.5 ug/mL puromycin 添加の培養液をベースとして培養し、これにBa/F3-Mockは5 ng/mL IL-3を、Ba/F3-EGFR WTはEGF 100 ng/mLをそれぞれ添加して、37℃、5% CO2に設定したCO2インキュベータにて培養した。希釈調製後の検体を、Echo555(Labcyte Inc.)を用いて384穴組織培養用プレートに播種し、Ba/F3-Mock、Ba/F3-EGFR WT、及びBa/F3-EGFR ins. SVDは 400 cells/well、Ba/F3-EGFR ins. ASVは200 cells/wellとなるように細胞を播種し(day 0)、さらに3日間培養した。化合物添加当日(day 0)ならびに化合物添加3日後(day 3)にCellTiter-Glo(登録商標) 2.0(Promega Corporation.)を用いてATP量を測定し、細胞量の指標とした(N=4)。試験時はpuromycin 無添加の培養液を用いた。EXCELを用いて、day 0からday 3までの細胞増殖を50%抑制する濃度(GI50)を算出した。結果を表62に示す。



抗腫瘍試験
実施例4、5、13、27、28、33、40、49、及び55に記載の化合物について、マウスproB細胞由来Ba/F3細胞に遺伝子導入して作製したBa/F3-EGFR ins. ASVあるいはBa/F3-HER2 ins. YVMAを皮下移植して抗腫瘍試験を実施した。
各細胞はリン酸緩衝生理食塩水を用いて1×108cells/mLになるよう懸濁し、調製した細胞懸濁液をヌードマウス(雌性、6週齢)の皮下に0.1 mL移植した。使用するマウスの平均推定腫瘍体積が100-300 mm3に達した時点で、推定腫瘍体積値による群分けを行い、同日から強制経口投与を開始した。投与は1日1回あるいは2回、10 mL/kgの薬液で行った。
各化合物は0.5%メチルセルロース液(0.5% MC) または2当量メタンスルホン酸添加0.5% MCで懸濁、あるいは20% PEG400/3% Tween80/DWにて溶解した。経時的に腫瘍の長径(mm) および短径 (mm) を電子デジタルノギスで計測し、以下に示す計算式(1)により推定腫瘍体積を算出して図を作成した。また経時的に小動物用自動天秤を用いて体重(g) を測定し、以下に示す計算式(2) により体重変化率 (Body weight change %) を算出して化合物投与の体重への影響を検討すると共に、直近の体重測定結果を投与量算出に用いた。
各個体の推定腫瘍体積 = An×Bn2/2
An:n日目の腫瘍の長径
Bn:n日目の腫瘍の短径
Body Weight Change(%) = 各個体の体重変化率の平均値・・・(2)
各個体の体重変化率 = (1-BWn/BWs)×100
BWn:n日目の体重
BWs:投与開始日の体重
結果を図1~12に示す。
Claims (46)
- 一般式(I)

[式(I)中、
R1は、1乃至3個のハロゲン原子にて置換されていてもよいC1-C3アルキル基を示し、
R2は、下記式(II)

または、-NH-CO-CH=CH-CH2-Xを示し、
Xは、下記A群より独立に選ばれる1または2個の置換基を有していてもよいアミノ基、下記A群より独立に選ばれる1または2個の置換基を有していてもよいピロリジニル基、下記A群より独立に選ばれる1または2個の置換基を有していてもよいアゼチジニル基、またはモルホリル基を示し、
R3は、ハロゲン原子を示し、
R4は、水素原子またはハロゲン原子を示し、
R5は、ベンゼン環、チアゾール環、またはハロゲン原子及びC1-C3アルキル基からなる群より独立に選ばれる1または2個の置換基を有していてもよいピラゾール環を示し、
R6は、下記B群から独立に選ばれる1または2個の置換基を有していてもよいオキサジアゾリル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいトリアゾリル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいピリジル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいピリミジル基、下記B群から独立に選ばれる1または2個の置換基を有していてもよいチアジアゾリル基、-CO-N(Y)(Z)、または-CH2-CO-N(Y)(Z)を示し、
YおよびZは、それぞれ独立に、水素原子、1乃至3個のハロゲン原子で置換されていてもよいC1-C6アルキル基、または1乃至3個のハロゲン原子で置換されていてもよいC1-C3アルキル基で置換されたC3-C6シクロアルキル基を示すか、または
Y、Zおよびそれらが結合する窒素原子が一緒になって4乃至6員の含窒素飽和複素環を形成してもよい]
で表される化合物またはその製薬上許容される塩。
A群:ハロゲン原子、C1-C3アルキル基、C1-C3アルコキシ基、テトラヒドロフリル基
B群:ハロゲン原子、C1-C3アルキル基、C3-C6シクロアルキル基、C1-C3アルコキシ基 - R1が、メチル基である、請求項1に記載の化合物またはその製薬上許容される塩。
- R2が、上記式(II)で示される基である、請求項1または2に記載の化合物またはその製薬上許容される塩。
- R3が、塩素原子またはフッ素原子であり、R4が、水素原子である、請求項1乃至3のいずれか1項に記載の化合物またはその製薬上許容される塩。
- R5が、ベンゼン環、またはピラゾール環である、請求項1乃至4のいずれか1項に記載の化合物またはその製薬上許容される塩。
- R6が、メチル基で置換されていてもよいオキサジアゾリル基、フッ素原子及びメチル基からなる群から独立に選ばれる1または2個の置換基で置換されていてもよいピリジル基、または-CO-NH-Yであり、
Yが、1乃至3個のフッ素原子で置換されていてもよいtert-ブチル基である、請求項1乃至5のいずれか1項に記載の化合物またはその製薬上許容される塩。 - 下記群から選ばれるいずれか1つの化合物またはその製薬上許容される塩。
N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド、
3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド
、
1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、
1-{4-[(4-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、
1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン、および
1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン。 - N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド、またはその製薬上許容される塩。
- N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド メタンスルホン酸塩。
- 3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド、またはその製薬上許容される塩。
- 3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド 1,5-ナフタレンジスルホン酸塩。
- 1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、またはその製薬上許容される塩。
- 1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン メタンスルホン酸塩。
- 1-{4-[(4-{2-フルオロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン、またはその製薬上許容される塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン、またはその製薬上許容される塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン ベンゼンスルホン酸塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 酒石酸塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン、またはその製薬上許容される塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 塩酸塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 1,5-ナフタレンジスルホン酸塩。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩。
- 3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミドの結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.78±0.2、15.48±0.2、16.38±0.2、17.24±0.2、19.28±0.2、19.90±0.2、20.42±0.2、20.82±0.2、22.04±0.2および24.50±0.2にピークを有する結晶。
- 3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-N-(1-フルオロ-2-メチルプロパン-2-イル)-1H-ピラゾール-1-カルボキサミド 1,5-ナフタレンジスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.74±0.2、10.32±0.2、11.58±0.2、14.62±0.2、14.94±0.2、18.72±0.2、19.60±0.2、20.84±0.2、22.96±0.2および26.42±0.2にピークを有する結晶。
- 1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オンの結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)4.26±0.2、8.66±0.2、13.64±0.2、14.34±0.2、14.98±0.2、17.60±0.2、19.08±0.2、22.10±0.2、23.02±0.2および25.88±0.2にピークを有する結晶。
- 1-{4-[(4-{2-クロロ-4-[3-(5-メチル-1,3,4-オキサジアゾール-2-イル)フェノキシ]アニリノ}-7-メトキシキナゾリン-6-イル)オキシ]ピペリジン-1-イル}プロパ-2-エン-1-オン メタンスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)3.56±0.2、7.24±0.2、15.02±0.2、16.84±0.2、17.68±0.2、20.26±0.2、21.88±0.2、22.92±0.2、25.76±0.2および27.08±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オンの結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)8.08±0.2、10.32±0.2、12.90±0.2、13.48±0.2、13.82±0.2、15.44±0.2、19.76±0.2、23.60±0.2、24.24±0.2および25.90±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン ベンゼンスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)6.22±0.2、12.16±0.2、13.60±0.2、16.26±0.2、18.50±0.2、19.58±0.2、20.58±0.2、21.06±0.2、23.30±0.2および25.76±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 酒石酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)3.58±0.2、14.50±0.2、16.50±0.2、24.28±0.2、24.70±0.2、24.98±0.2、25.76±0.2、26.12±0.2、26.60±0.2および27.40±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(5-フルオロ-6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)7.36±0.2、8.74±0.2、13.62±0.2、15.32±0.2、16.32±0.2、17.56±0.2、19.02±0.2、19.44±0.2、21.28±0.2および25.02±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オンの結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)8.14±0.2、10.56±0.2、13.10±0.2、15.16±0.2、15.50±0.2、15.92±0.2、19.30±0.2、20.18±0.2、23.92±0.2および25.54±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 塩酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.28±0.2、5.98±0.2、7.70±0.2、8.28±0.2、10.64±0.2、12.60±0.2、13.48±0.2、16.68±0.2、17.66±0.2および20.80±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン 1,5-ナフタレンジスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)8.50±0.2、13.98±0.2、15.56±0.2、16.94±0.2、18.28±0.2、19.52±0.2、20.04±0.2、25.16±0.2、25.44±0.2および26.10±0.2にピークを有する結晶。
- 1-(4-{[4-(2-フルオロ-4-{[1-(6-メチルピリジン-3-イル)-1H-ピラゾール-3-イル]オキシ}アニリノ)-7-メトキシキナゾリン-6-イル]オキシ}ピペリジン-1-イル)プロパ-2-エン-1-オン クエン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)5.34±0.2、7.32±0.2、7.86±0.2、8.68±0.2、13.56±0.2、16.26±0.2、17.50±0.2、19.36±0.2、21.22±0.2および24.90±0.2にピークを有する結晶。
- N-tert-ブチル-3-{3-フルオロ-4-[(7-メトキシ-6-{[1-(プロパ-2-エノイル)ピペリジン-4-イル]オキシ}キナゾリン-4-イル)アミノ]フェノキシ}-1H-ピラゾール-1-カルボキサミド メタンスルホン酸塩の結晶であって、CuKα放射線を用いた粉末X線回折において、回折角度(2θ)6.72±0.2、8.38±0.2、11.10±0.2、13.62±0.2、16.28±0.2、17.92±0.2、19.02±0.2、21.66±0.2、22.40±0.2および25.64±0.2にピークを有する結晶。
- 請求項1乃至22のいずれか1項に記載の化合物またはその製薬上許容される塩、または請求項23乃至35のいずれか1項に記載の結晶を有効成分として含有する医薬組成物。
- 請求項1乃至22のいずれか1項に記載の化合物またはその製薬上許容される塩、または請求項23乃至35のいずれか1項に記載の結晶を有効成分として含有する、エクソン20挿入変異を有するEGFRチロシンキナーゼおよび/またはエクソン20挿入変異を有するHER2チロシンキナーゼ阻害剤。
- 請求項1乃至22のいずれか1項に記載の化合物またはその製薬上許容される塩、または請求項23乃至35のいずれか1項に記載の結晶を有効成分として含有する抗腫瘍剤。
- 腫瘍が肺がん、乳がん、膀胱がん、または卵巣がんである、請求項38に記載の抗腫瘍剤。
- 腫瘍が、EGFRチロシンキナーゼのエクソン20挿入変異および/またはHER2チロシンキナーゼのエクソン20挿入変異を有する腫瘍である、請求項38または39に記載の抗腫瘍剤。
- 腫瘍の治療のための請求項1乃至22のいずれか1項に記載の化合物またはその製薬上許容される塩、または請求項23乃至35のいずれか1項に記載の結晶。
- 腫瘍が肺がん、乳がん、膀胱がん、または卵巣がんである、請求項41に記載の化合物またはその製薬上許容される塩、または結晶。
- 腫瘍が、EGFRチロシンキナーゼのエクソン20挿入変異および/またはHER2チロシンキナーゼのエクソン20挿入変異を有する腫瘍である、請求項41または42に記載の化合物またはその製薬上許容される塩、または結晶。
- 腫瘍を治療することに用いるための医薬組成物の製造における、請求項1乃至22のいずれか1項に記載の化合物またはその製薬上許容される塩、または請求項23乃至35のいずれか1項に記載の結晶の使用。
- 腫瘍が肺がん、乳がん、膀胱がん、または卵巣がんである、請求項44に記載の使用。
- 腫瘍が、EGFRチロシンキナーゼのエクソン20挿入変異および/またはHER2チロシンキナーゼのエクソン20挿入変異を有する腫瘍である、請求項44または45に記載の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018127829 | 2018-07-04 | ||
| JP2018127829 | 2018-07-04 | ||
| PCT/JP2019/026483 WO2020009156A1 (ja) | 2018-07-04 | 2019-07-03 | ビアリールエーテル型キナゾリン誘導体 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JPWO2020009156A1 JPWO2020009156A1 (ja) | 2021-07-15 |
| JPWO2020009156A5 JPWO2020009156A5 (ja) | 2022-05-26 |
| JP7288904B2 true JP7288904B2 (ja) | 2023-06-08 |
Family
ID=69059535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020529034A Active JP7288904B2 (ja) | 2018-07-04 | 2019-07-03 | ビアリールエーテル型キナゾリン誘導体 |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US20230025510A1 (ja) |
| EP (1) | EP3822263A4 (ja) |
| JP (1) | JP7288904B2 (ja) |
| KR (1) | KR20210029165A (ja) |
| CN (1) | CN112334460A (ja) |
| AU (1) | AU2019297889A1 (ja) |
| BR (1) | BR112020027064A2 (ja) |
| CA (1) | CA3105602A1 (ja) |
| CO (1) | CO2021000800A2 (ja) |
| IL (1) | IL279858A (ja) |
| MX (1) | MX2021000014A (ja) |
| PH (1) | PH12021550003A1 (ja) |
| SG (1) | SG11202013218QA (ja) |
| TW (1) | TW202012391A (ja) |
| WO (1) | WO2020009156A1 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020262998A1 (ko) * | 2019-06-26 | 2020-12-30 | 한미약품 주식회사 | 항 종양 활성을 갖는 신규 퀴나졸린 유도체 및 이를 포함하는 약학 조성물 |
| WO2022033410A1 (zh) * | 2020-08-10 | 2022-02-17 | 上海和誉生物医药科技有限公司 | 一种egfr抑制剂及其制备方法和应用 |
| CN115803326B (zh) * | 2020-11-23 | 2024-03-26 | 上海和誉生物医药科技有限公司 | Egfr抑制剂及其制备方法与在药学上的应用 |
| CN113264888A (zh) * | 2021-03-04 | 2021-08-17 | 江苏康可得生物技术股份有限公司 | 酪氨酸激酶抑制剂及其药物应用 |
| WO2022194265A1 (zh) * | 2021-03-19 | 2022-09-22 | 北京赛特明强医药科技有限公司 | 一种喹唑啉类化合物、组合物及其应用 |
| PE20240327A1 (es) | 2021-04-13 | 2024-02-22 | Nuvalent Inc | Heterociclos con sustitucion amino para tratar canceres con mutaciones de egfr |
| TW202404590A (zh) * | 2022-04-05 | 2024-02-01 | 美商纜圖藥品公司 | Egfr抑制劑 |
| CN117384162A (zh) * | 2022-05-17 | 2024-01-12 | 浙江文达医药科技有限公司 | 选择性her2抑制剂 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000508657A (ja) | 1996-04-12 | 2000-07-11 | ワーナー―ランバート・コンパニー | チロシンキナーゼの不可逆的阻害剤 |
| JP2007501854A (ja) | 2003-05-27 | 2007-02-01 | ファイザー・プロダクツ・インク | 受容体型チロシンキナーゼ阻害薬としてのキナゾリン類およびピリド[3,4−d]ピリミジン類 |
| JP2010529115A (ja) | 2007-06-05 | 2010-08-26 | ハンミ ファーム. シーオー., エルティーディー. | 癌細胞成長抑制用新規アミド誘導体 |
| WO2018094225A1 (en) | 2016-11-17 | 2018-05-24 | Board Of Regents, The University Of Texas System | Compounds with anti-tumor activity against cancer cells bearing egfr or her2 exon 20 mutations |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013173254A1 (en) * | 2012-05-14 | 2013-11-21 | Dawei Zhang | Bicyclic compounds as kinases inhibitors |
| CN103965120B (zh) * | 2013-01-25 | 2016-08-17 | 上海医药集团股份有限公司 | 喹啉及喹唑啉衍生物、制备方法、中间体、组合物及应用 |
| WO2020262998A1 (ko) * | 2019-06-26 | 2020-12-30 | 한미약품 주식회사 | 항 종양 활성을 갖는 신규 퀴나졸린 유도체 및 이를 포함하는 약학 조성물 |
-
2019
- 2019-07-02 TW TW108123293A patent/TW202012391A/zh unknown
- 2019-07-03 WO PCT/JP2019/026483 patent/WO2020009156A1/ja active Application Filing
- 2019-07-03 CA CA3105602A patent/CA3105602A1/en active Pending
- 2019-07-03 US US17/257,238 patent/US20230025510A1/en not_active Abandoned
- 2019-07-03 KR KR1020207038066A patent/KR20210029165A/ko active Search and Examination
- 2019-07-03 MX MX2021000014A patent/MX2021000014A/es unknown
- 2019-07-03 JP JP2020529034A patent/JP7288904B2/ja active Active
- 2019-07-03 EP EP19830650.8A patent/EP3822263A4/en not_active Withdrawn
- 2019-07-03 SG SG11202013218QA patent/SG11202013218QA/en unknown
- 2019-07-03 AU AU2019297889A patent/AU2019297889A1/en not_active Abandoned
- 2019-07-03 BR BR112020027064-4A patent/BR112020027064A2/pt not_active Application Discontinuation
- 2019-07-03 CN CN201980044514.8A patent/CN112334460A/zh active Pending
-
2020
- 2020-12-30 IL IL279858A patent/IL279858A/en unknown
-
2021
- 2021-01-03 PH PH12021550003A patent/PH12021550003A1/en unknown
- 2021-01-25 CO CONC2021/0000800A patent/CO2021000800A2/es unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000508657A (ja) | 1996-04-12 | 2000-07-11 | ワーナー―ランバート・コンパニー | チロシンキナーゼの不可逆的阻害剤 |
| JP2007501854A (ja) | 2003-05-27 | 2007-02-01 | ファイザー・プロダクツ・インク | 受容体型チロシンキナーゼ阻害薬としてのキナゾリン類およびピリド[3,4−d]ピリミジン類 |
| JP2010529115A (ja) | 2007-06-05 | 2010-08-26 | ハンミ ファーム. シーオー., エルティーディー. | 癌細胞成長抑制用新規アミド誘導体 |
| WO2018094225A1 (en) | 2016-11-17 | 2018-05-24 | Board Of Regents, The University Of Texas System | Compounds with anti-tumor activity against cancer cells bearing egfr or her2 exon 20 mutations |
Non-Patent Citations (1)
| Title |
|---|
| Proceedings of the National Academy of Sciences of the United States of America,2006年,Vol. 103, No. 20,pp. 7817-7822,ISSN 0027-8424 |
Also Published As
| Publication number | Publication date |
|---|---|
| SG11202013218QA (en) | 2021-02-25 |
| BR112020027064A2 (pt) | 2021-05-25 |
| WO2020009156A1 (ja) | 2020-01-09 |
| EP3822263A4 (en) | 2022-03-16 |
| CA3105602A1 (en) | 2020-01-09 |
| AU2019297889A1 (en) | 2021-02-18 |
| TW202012391A (zh) | 2020-04-01 |
| PH12021550003A1 (en) | 2021-09-20 |
| US20230025510A1 (en) | 2023-01-26 |
| MX2021000014A (es) | 2021-03-09 |
| CO2021000800A2 (es) | 2021-02-08 |
| JPWO2020009156A1 (ja) | 2021-07-15 |
| IL279858A (en) | 2021-03-01 |
| EP3822263A1 (en) | 2021-05-19 |
| CN112334460A (zh) | 2021-02-05 |
| KR20210029165A (ko) | 2021-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7288904B2 (ja) | ビアリールエーテル型キナゾリン誘導体 | |
| CA2924705C (en) | Quinazoline derivative and preparation method therefor | |
| KR102438302B1 (ko) | 리신 특이적 데메틸라제-1의 억제제 | |
| EP2857404B1 (en) | IMIDAZO[1,2-b]PYRIDAZINE DERIVATIVES AS KINASE INHIBITORS | |
| RU2664055C2 (ru) | Дигидронафтиридины и родственные соединения, подходящие в качестве ингибиторов киназ для лечения пролиферативных заболеваний | |
| JP7050093B2 (ja) | 置換5員および6員複素環式化合物、その調製方法、薬剤の組み合わせおよびその使用 | |
| EA027563B1 (ru) | Пиридопиразины, обладающие противораковой активностью через ингибирование fgfr киназ | |
| EP3590941B1 (en) | Urea-substituted aromatic ring-linked dioxane-quinazoline and -linked dioxane-quinoline compounds, preparation method therefor and use thereof | |
| CA2954060A1 (en) | Inhibitors of lysine specific demethylase-1 | |
| EP1559715B1 (en) | N-{2-chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl}-n'-(5-methyl-3-isoxazolyl)urea salt in crystalline form | |
| WO2014100533A1 (en) | NOVEL SUBSTITUTED IMIDAZOLES AS CASEIN KINASE 1 δ/ε INHIBITORS | |
| CA3059283A1 (en) | C5-anilinoquinazoline compounds and their use in treating cancer | |
| EP2662367B1 (en) | Novel indole or indazole derivative or salt thereof | |
| EA031671B1 (ru) | Твердые формы 2-(трет-бутиламино)-4-((1r,3r,4r)-3-гидрокси-4-метилциклогексиламино)пиримидин-5-карбоксамида, его композиции и способы его применения | |
| KR20230043887A (ko) | 피리다진 유도체 유리 염기의 결정 형태, 및 이의 제조 방법 및 이의 용도 | |
| JP7168245B2 (ja) | 三環式化合物 | |
| EP3760633B1 (en) | Oxazino-quinazoline and oxazino-quinazoline type compound, preparation method therefor, and uses thereof | |
| CN111303128B (zh) | 一种多环类化合物、其制备方法及应用 | |
| WO2022067063A1 (en) | Mutant selective egfr inhibitors and methods of use thereof | |
| WO2022197577A1 (en) | Heteroaroyl amides as lrrk2 inhibitors, pharmaceutical compositions, and uses thereof | |
| CN113811300A (zh) | Tead转录因子的新型小分子抑制剂 | |
| TW202115023A (zh) | 新型細胞凋亡訊號調節激酶1抑制劑 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220518 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220518 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20230330 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230510 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230522 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230529 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7288904 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |