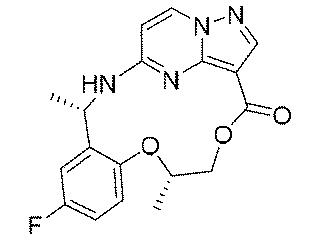
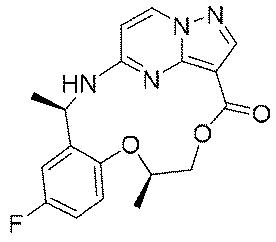
本発明の1つの目的は、優れたチロシンキナーゼ阻害活性を有する新規化合物又はその薬学的に許容される塩、溶媒和物、活性代謝物、多形体、同位体標識物、異性体又はプロドラッグを提供することである。
本発明の他の目的は、医薬組成物を提供することである。
本発明の別の目的は、新規化合物又はその薬学的に許容される塩、溶媒和物、活性代謝物、多形体、同位体標識物、異性体又はプロドラッグの用途を提供することである。
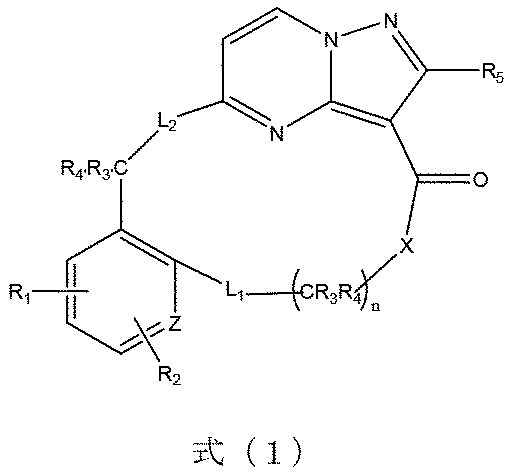
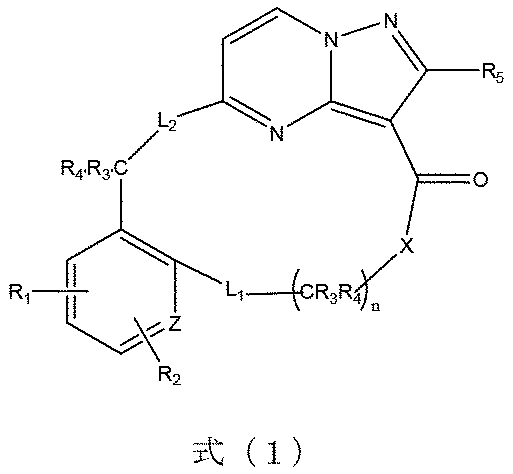
本発明は、式(1)で示される化合物又はその薬学的に許容される塩、溶媒和物、活性代謝物、多形体、同位体標識物、異性体又はプロドラッグを提供し、
式(1)において、Xは、-O-、-S-又は-CR
aR
b-から選ばれ、R
a、R
bは、それぞれ独立して置換若しくは無置換の水素、ハロゲン、C
1~8アルキル基、C
1~8アルコキシ基、C
1~8ハロゲン化アルキル基、C
3~8シクロアルキル基、C
3~8複素環基、C
6~20アリール基、C
5~20ヘテロアリール基、ヒドロキシ基、メルカプト基、カルボキシル基、エステル基、アシル基、アミノ基、アミド基、スルホニル基、シアノ基から選ばれるか、又は、CR
aR
bは共同して3~10員のシクロアルキル基又は少なくとも1つのヘテロ原子を含有する3~10員の複素環基を形成し、L
1は、-O-、-S-、-S(=O)-、-S(=O)
2-、-NR
6-から選ばれるか、又は、単結合であり、L
2は、-O-、-S-、-S(=O)-、-S(=O)
2-又は-NR
6-から選ばれ、R
1、R
2、R
5は、それぞれ独立して置換若しくは無置換の水素、ハロゲン、C
1~8アルキル基、C
1~8アルコキシ基、C
1~8ハロゲン化アルキル基、C
3~8シクロアルキル基、C
3~8複素環基、C
6~20アリール基、C
5~20ヘテロアリール基、ヒドロキシ基、メルカプト基、カルボキシル基、エステル基、アシル基、アミノ基、アミド基、スルホニル基又はシアノ基から選ばれ、各C原子上の置換基R
3、R
4は、それぞれ独立して置換若しくは無置換の水素、ハロゲン、C
1~8アルキル基、C
1~8アルコキシ基、C
1~8ハロゲン化アルキル基、C
3~8シクロアルキル基、C
3~8複素環基、C
6~20アリール基、C
5~20ヘテロアリール基、ヒドロキシ基、メルカプト基、カルボキシル基、エステル基、アシル基、アミノ基、アミド基、スルホニル基、シアノ基から選ばれるか、又は、X基と共に3~10員のシクロアルキル基、少なくとも1つのヘテロ原子を含有する3~10員の複素環基又は少なくとも1つのヘテロ原子を含有する5~10員のヘテロアリール基を形成するか、又は、R
3、R
4は、それぞれ独立して前記C原子と隣接する大員環の環原子とを結合する単結合である。
R3’、R4’は、それぞれ独立して置換若しくは無置換の水素、ハロゲン、C1~8アルキル基、C1~8アルコキシ基、C1~8ハロゲン化アルキル基、C3~8シクロアルキル基、C3~8複素環基、C6~20アリール基、C5~20ヘテロアリール基、ヒドロキシ基、メルカプト基、カルボキシル基、エステル基、アシル基、アミノ基、アミド基、スルホニル基、シアノ基から選ばれるか、又は、結合しているC及L2と共に少なくとも1つのヘテロ原子を含有する3~10員の複素環基又は少なくとも1つのヘテロ原子を含有する5~10員のヘテロアリール基を形成し、R6は、置換若しくは無置換の水素、ハロゲン、C1~8アルキル基、C1~8アルコキシ基、C1~8ハロゲン化アルキル基、C3~8シクロアルキル基、C3~8複素環基、C6~20アリール基、C5~20ヘテロアリール基、ヒドロキシ基、メルカプト基、カルボキシル基、エステル基、アシル基、アミノ基、アミド基、スルホニル基又はシアノ基から選ばれ、Zは、環原子としてC又はヘテロ原子を表し、nは、1~10の整数を表し、前記基の置換基は、ハロゲン、C1~8アルキル基、C1~8ハロゲン化アルキル基、C1~8アルコキシ基、C3~8シクロアルキル基、C3~8複素環基、C6~20アリール基、C5~20ヘテロアリール基、ヒドロキシ基、メルカプト基、カルボキシル基、エステル基、アシル基、アミノ基、アミド基、スルホニル基又はシアノ基から選ばれ、前記ヘテロ原子は、N、O又はSから選ばれる。
選択的に、式(1)における不飽和単環は、他の類似した構造で置換されていてもよく、例えば、環において1つ又は複数のO、S、Nなどのヘテロ原子を追加してもよい。例えば、式(1)に示される環原子「Z」は、Nを表すことができる。
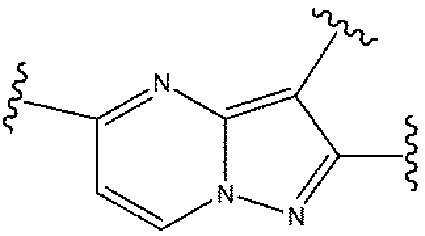
選択的に、式(1)における2員のヘテロアリール基
は、他の類似した構造で置換されていてもよく、例えば、N原子の置換位置を変更したり、環において1つ又は複数のN原子を追加又は減少したりしてもよい。好ましくは、
又は
で置換されている。
上記の式(1)において、Ra、Rb、R1、R2、R3、R4、R3’、R4’、R5、R6及びその選択可能な置換基で表される基は、以下の基を含むが、これらに限定されない。
水素は、-Hで表され、二重水素、三重水素などの同位体で置換されていてもよい。
ハロゲンは、フッ素、塩素、臭素、ヨウ素を含んでもよい。
C1~8アルキル基は、メチル基、エチル基、n-プロピル基、イソプロピル基、2-メチル-l-プロピル基、2-メチル-2-プロピル基、2-メチル-1-ブチル基、3-メチル-l-ブチル基、2-メチル-3-ブチル基、2,2-ジメチル-1-プロピル基、2-メチル-1-ペンチル基、3-メチル-1-ペンチル基、4-メチル-l-ペンチル基、2-メチル-2-ペンチル基、3-メチル-2-ペンチル基、4-メチル-2-ペンチル基、2,2-ジメチル-l-ブチル基、3,3-ジメチル-1-ブチル基、2-エチル-1-ブチル基、n-ブチル基、イソブチル基、s-ブチル基、t-ブチル基、n-ペンチル基、イソペンチル基、ネオペンチル基、t-ペンチル基、ヘキシル基、ヘプチル基、オクチル基などを含んでもよい。
C1~8アルコキシ基は、-OC1~8アルキル基で表され、その中のC1~8アルキル基に含まれる基は、上記と同義である。例えば、C1~8アルコキシ基は、メトキシ基、エトキシ基、n-プロポキシ基、イソプロポキシ基、n-ブトキシ、イソブトキシ基、s-ブトキシ基、t-ブトキシ基などを含んでもよい。
C1~8ハロゲン化アルキル基は、C1~8アルキル基における任意の数の水素原子がハロゲンで置換された基で表され、その中のC1~8アルキル基、ハロゲン基に含まれる基は、上記と同義である。例えば、C1~8ハロゲン化アルキル基は、-CF3などを含んでもよい。
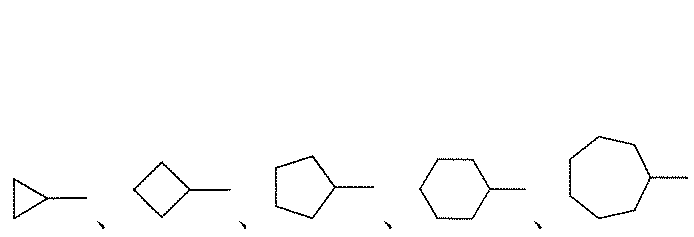
C
3~8シクロアルキル基は、非芳香族の飽和炭素環で表され、単環炭素環(1つの環を有する)及二環炭素環(2つの環を有する)を含み、例えば、C
3~8シクロアルキル基は、
などを含んでもよい。
C3~8複素環基は、C3~8シクロアルキル基における任意の数の環原子がO、S、N、P、Siなどのヘテロ原子で置換された基で表され、その中のC3~8シクロアルキル基に含まれる基は、上記と同義である。例えば、C3~8複素環基は、オキシラニル(oxiranyl)、チイラン基(Ethylene sulfide)、アジリジニル基(aziridinyl)、アゼチジニル基、オキセタニル基(oxetanyl)、チエタニル基(thietanyl)、テトラヒドロフラニル基、ピロリジニル基、オキサゾリジニル基、テトラヒドロピラゾリル基、ピロリニル基、ジヒドロフラニル基、ジヒドロチエニル基、ピペリジニル基、テトラヒドロピラニル基、テトラヒドロチオピラニル基、モルホリニル基、ピペラジニル基、ジヒドロピリジル基、テトラヒドロピリジル基、ジヒドロピラニル基、テトラヒドロピラニル基、ジヒドロチオピラニル基、アゼパニル基、オキセパニル基、チエパニル基、オキサ-アザビシクロ[2.2.1]ヘプチル基、アザスピロ[3.3]ヘプチル基などを含んでもよい。
C6~20アリール基は、単環式アリール基、二環式アリール基又はそれ以上の環のアリール基を含んでもよく、例えば、フェニル基、ビフェニル基、ナフチル基、フェナントリル基、アントリル基、アズレニル基などを含んでもよい。
C5~20ヘテロアリール基は、環原子として任意の数のO、S、N、P、Siなどのヘテロ原子を含有する不飽和基を表すことができる。例えば、C5~20ヘテロアリール基は、ピロリル基、フラニル基、チエニル基、イミダゾリル基、オキサゾリル基、ピラゾリル基、ピリジル基、ピリミジニル基、ピラジニル基、キノリジニル基、イソキノリジニル基、テトラゾリル基、トリアゾイル基、トリアジニル基、ベンゾフラニル基、ベンゾチオフェニル基、インドリル基、イソインドリル基などを含んでもよい。
ヒドロキシ基は、-OHで表される。メルカプト基は、-SHで表される。カルボキシル基は、-COOHで表される。エステル基は、-COOR’で表され、R’は、式(1)に記載された置換基と同義であってもよく、例えばC1~8アルキル基で置換されているエステル基は、-COOC1~8アルキル基で表され、その中のC1~8アルキル基に含まれる基は、上記と同義である。
アシル基は、-COR’で表され、R’は、式(1)に記載された置換基と同義であってもよく、例えばC1~8アルキル基で置換されているアシル基は、-COC1~8アルキル基で表され、その中のC1~8アルキル基に含まれる基は、上記と同義である。
アミノ基は、-NH2、-NHR’又は-N(R’)2で表され、R’は、式(1)に記載された置換基と同義であってもよく、例えばC1~8アルキル基で置換されているアミノ基は、-NHC1~8アルキル基又は-N(C1~8アルキル基)2で表され、その中のC1~8アルキル基に含まれる基は、上記と同義である。
アミド基は、-COアミノ基で表され、その中のアミノ基は、上記と同義である。
スルホニル基は、-S(O)2R’で表され、R’は、式(1)に記載された置換基と同義であってもよく、例えばC1~8アルキル基で置換されているスルホニル基は、-S(O)2C1~8アルキル基で表され、その中のC1~8アルキル基に含まれる基は、上記と同義である。
シアノ基は、-CNで表される。
上記の定義において、炭素原子の数が変化する場合、上記の定義は、炭素原子の数のみの変化に応じて変化し、その基の種類の定義に影響を与えない。例えば、「C1~5アルキル基」は、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、s-ブチル基、t-ブチル基、n-ペンチル基、イソペンチル基、ネオペンチル基などの前記「C1~8アルキル基」の定義における炭素原子の数が1~5のすべての基を含んでもよい。
さらに、上記の式(1)において、Ra、Rb、R1、R2、R3、R4、R3’、R4’、R5、R6及びその選択可能な置換基で表される基は、水素、二重水素、フッ素、塩素、臭素、メチル基、エチル基、プロピル基、イソプロピル基、n-ブチル基、s-ブチル基、t-ブチル基、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、-CN、-CF3、-NH2、-NH(C1~4アルキル基)、-N(C1~4アルキル基)2、-CO2C1~4アルキル基、-CO2H、-NHC(O)C1~4アルキル基、-SO2C1~4アルキル基、-C(O)NH2、-C(O)NH(C1~4アルキル基)、-C(O)N(C1~4アルキル基)2、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、ピロリジニル基、ピラゾリル基、ピペリジニル基、ピリジル基、ピペラジニル基、トリアジニル基、フラニル基、チオフラニル基、モルホリニル基、チオモルホリニル基、フェニル基、ナフチル基、ビフェニル基、ターフェニル基などを含むが、これらに限定されない。
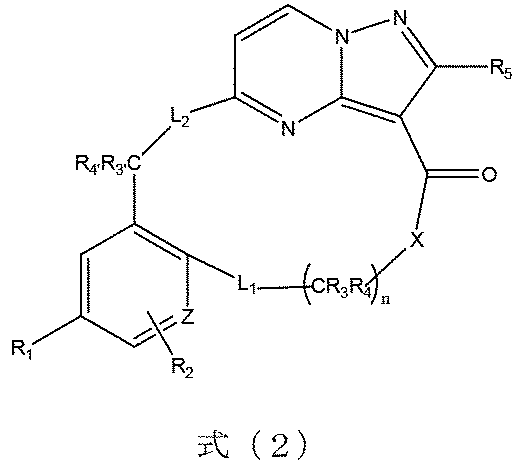
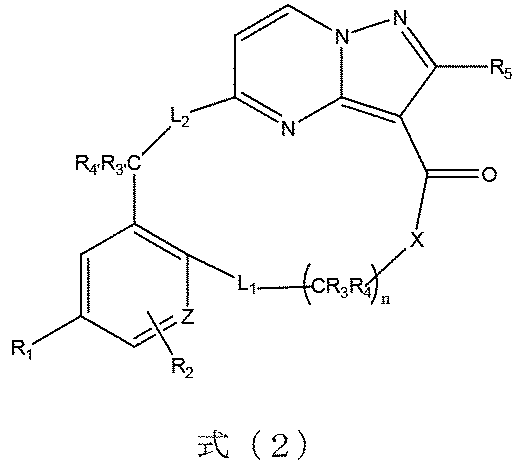
本発明の一実施形態によれば、前記化合物は、式(2)に示すようなものであり、
式(2)において、R
1は、フッ素又は臭素から選ばれる。
本発明の一実施形態によれば、式(1)又は式(2)におけるR2は、水素、フッ素又は臭素から選ばれる。
本発明の一実施形態によれば、式(1)又は式(2)におけるL1は、-O-、-S-から選ばれるか、又は、単結合である。本明細書における単結合とは、L1基がないことを意味し、即ち、X基から最も遠い-(CR3R4)-におけるCは、芳香族環単位に直接に結合されている。
本発明の一実施形態によれば、式(1)又は式(2)におけるL2は、-NR6-から選ばれる。
本発明の一実施形態によれば、式(1)又は式(2)におけるnは、2、3、4、5又は6の整数を表す。
本発明の一実施形態によれば、式(1)又は式(2)において、C原子上の置換基R3、R4がそれぞれ独立してX基と共にシクロアルキル基、複素環基又はヘテロアリール基を形成することは、X基と隣接する-(CR3R4)-基においてR3又はR4が結合するC及びX基と共に形成することである。
本発明の一実施形態によれば、式(1)又は式(2)において、R3、R4がそれぞれ独立して前記C原子と隣接する大員環の環原子とを結合する単結合である場合、隣接する大員環の環原子は、それに隣接する他の-(CR3R4)-基における環原子Cであってもよく、それに隣接するX基における環原子であってもよい。好ましい実施形態において、X基に近接する-(CR3R4)-基におけるR3、R4は、それぞれ独立して前記C原子と前記X基とを結合する単結合であってもよく、即ち、R3及び/又はR4は、C原子とX中心原子(即ち-CRaRb-におけるC)とを結合する単結合である。例えば、R3、R4のうちの1つが単結合である場合、X基は、隣接する-(CR3R4)-基と二重結合を形成し、R3、R4のうちの2つが単結合である場合、X基は、隣接する-(CR3R4)-基と三重結合を形成する。
同様に、大員環における隣接する2つの-(CR3R4)-基の間、C、C原子の間には、置換若しくは無置換の二重結合又は三重結合が形成されてもよい。
本発明の一実施形態によれば、式(1)又は式(2)において、各C原子上の置換基R3、R4は、それぞれ独立して置換若しくは無置換の水素、ハロゲン、C1~5アルキル基、C1~5アルコキシ基、C1~5ハロゲン化アルキル基、C3~6シクロアルキル基から選ばれるか、又は、R3、R4は、それぞれ独立して前記C原子と隣接する大員環の環原子とを結合する単結合である。
本発明の一実施形態によれば、式(1)又は式(2)において、R3’、R4’は、それぞれ独立して置換若しくは無置換の水素、ハロゲン、C1~5アルキル基、C1~5アルコキシ基、C1~5ハロゲン化アルキル基、C3~6シクロアルキル基から選ばれか、又は、結合されるC及L2と共に少なくとも1つのヘテロ原子を含有する4~8員の複素環基を形成する。好ましくは、ここで複素環基は、ピロリジニル基又はピペリジニル基であってもよく、その中のNがL2に由来する。
本発明の一実施形態によれば、式(1)又は式(2)において、-CR3R4-基又は-CR3’R4’-基における大員環原子C又は置換基におけるCは、異なる基に応じて1つ又は複数のキラル中心を生成し、本発明は、すべての光学異性体及ラセミ体を含む。
本発明の一実施形態によれば、式(1)又は式(2)において、R5は、置換若しくは無置換の水素、ハロゲン、C1~5アルキル基、C1~5アルコキシ基、C1~5ハロゲン化アルキル基、C3~6シクロアルキル基、ヒドロキシ基、メルカプト基、カルボキシル基、アミノ基又はシアノ基から選ばれる。
本発明の一実施形態によれば、式(1)又は式(2)において、R6は、置換若しくは無置換の水素、ハロゲン、C1~5アルキル基、C1~5アルコキシ基、C1~5ハロゲン化アルキル基又はC3~6シクロアルキル基から選ばれる。
本発明の一実施形態によれば、上記の実施形態における選択可能な置換基は、フッ素、臭素、-CN、-OH、-CF3、-NH2、-NH(C1~4アルキル基)、-N(C1~4アルキル基)2、-CO2C1~4アルキル基、-CO2H、-NHC(O)C1~4アルキル基、-SO2C1~4アルキル基、-C(O)NH2、-C(O)NH(C1~4アルキル基)、-C(O)N(C1~4アルキル基)2、C1~5アルキル基、C3~6シクロアルキル基、C3~6複素環基、C6~10アリール基又はC5~10ヘテロアリール基から選ばれる。
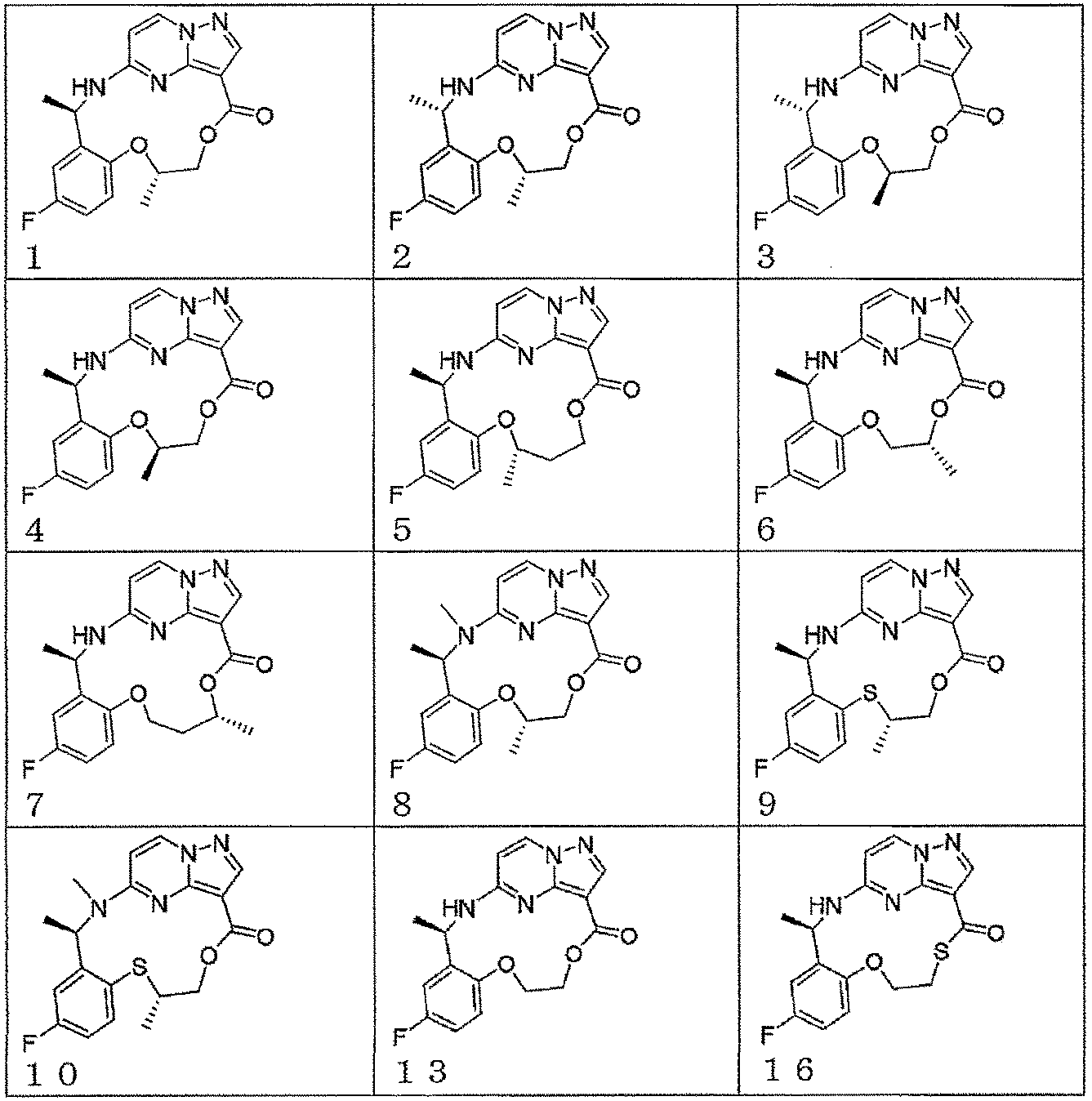
本発明の一実施形態によれば、前記化合物は、以下の構造から選ばれる。
本発明は、以上の技術案のいずれかに記載の化合物又はその薬学的に許容される塩、溶媒和物、活性代謝物、多形体、同位体標識物、異性体又はプロドラッグ、及び薬学的に許容される担体を含む医薬組成物をさらに提供する。
前記医薬組成物は、経口剤形、非経口投与剤形、外用剤形及び直腸投与剤形を含むが、これらに限定されない。例えば、前記医薬組成物は、経口錠剤、カプセル、丸剤、粉剤、徐放性製剤、溶液及び懸濁液、非経口注射用の滅菌溶液、懸濁液若しくは乳液、外用の軟膏剤、クリーム、ゲル剤など、又は直腸投与に用いられる坐剤であってもよい。
前記医薬組成物は、前記化合物又はその薬学的に許容される塩、溶媒和物、活性代謝物、多形体、同位体標識物、異性体又はプロドラッグと併用する他の活性成分又は薬物をさらに含んでもよい。
本発明は、チロシンキナーゼが介在する疾患を治療するための薬物の調製における、上記の化合物又はその薬学的に許容される塩、溶媒和物、活性代謝物、多形体、同位体標識物、異性体又はプロドラッグ、及び上記の医薬組成物の使用をさらに提供する。
さらに、前記チロシンキナーゼは、ALK、ROS1、TRKA、TRKB、TRKC、JAK2、SRC、FYN、LYN、YES、FGR、FAK、AXL、ARK5から選ばれる1種又は複数種である。
さらに、前記チロシンキナーゼが介在する疾患は、癌、疼痛、神経疾患、自己免疫疾患及び炎症を含む。
さらに、前記チロシンキナーゼが介在する癌は、肺癌、結腸直腸癌、乳癌、卵巣癌、甲状腺癌、前立腺癌、肝細胞癌、腎細胞癌、胃と食道癌、胆管癌、神経膠腫、膠芽腫、頭頸部癌、炎症性筋線維芽細胞腫、血管肉腫、類上皮血管内皮腫、未分化大細胞型リンパ腫などを含んでもよい。
さらに、前記チロシンキナーゼが介在する疼痛は、癌性疼痛、化学療法による疼痛、神経疼痛、損傷による疼痛又は他の由来の疼痛を含む、いずれかの由来又は病因による疼痛であってもよい。
さらに、前記チロシンキナーゼが介在する自己免疫疾患は、関節リウマチ、シェーグレン症候群(Sjogren syndrome)、I型糖尿病、全身性エリテマトーデスなどを含む。
さらに、前記チロシンキナーゼが介在する神経疾患は、アルツハイマー症(Alzheimer’s Disease)、パーキンソン病(Parkinson’s Disease)、筋萎縮性側索硬化症、ハンチントン病(Huntington’s disease)などを含む。
さらに、前記チロシンキナーゼが介在する炎症疾患は、動脈硬化症、アレルギー、感染又は損傷による炎症などを含む。
本発明によって提供されるジアリール大員環化合物及びその医薬組成物は、顕著なチロシンキナーゼ阻害活性を有し、腫瘍の薬剤耐性を克服でき、血液脳関門を突破でき、優れた薬物動態学的性質及び極めてよい経口バイオアベイラビリティをさらに有し、比較的少ない投与量で施用でき、これにより、患者の治療コスト及び可能な毒性副作用を低減でき、このため、大きな適用の潜在力を有する。
詳細
別途定義されない限り、本発明の全ての技術及び科学用語は、請求項の主題が属する分野の当業者に一般的に理解されるものと同じ意味を有する。特別な説明がない限り、本明細書に引用されるすべての特許、特許出願、公開資料は、何れもその全体を参照によって本明細書に組み込まれている。本明細書において商品名が現れる場合、対応する商品又はその活性成分を意味する。
なお、上記の簡単な説明及び以下の詳細な説明は例示的なものであり、解釈に用いられるものに過ぎず、本発明の主題を限定するものではない。本願では、特に明記されていない限り、本明細書及び添付する特許請求の範囲に記載の単数形式は、言及されたものの複数の形式も含む。なお、特別な説明がない限り、使用される「又は」、「或いは」は、「及び/又は」を意味する。また、使用される用語「含む」及びその他の形態、例えば「含む」、「有する」及び「含有する」は、いずれも非限定的な説明である。
標準化学用語の定義は参考文献(Carey and Sundberg、Advanced Organic Chemistry 4th Ed,Vol A(2000)and B(2001)、Plenum Press、New Yorkを含む)に記載されている。特別な説明がない限り、質量分析法、NMR、HPLC、蛋白質化学、生化学、組換えDNA技術、薬理学的方法といったこの分野の技術範囲内にある常法が使用される。別途定義されない限り、本明細書において分析化学、有機合成化学、医薬及び医薬化学などの化学に関連する命名及び実験室の操作と技術は当業者に知られている。標準的な技術は、化学合成、化学分析、医薬品の調製、製剤、送達、及び患者に対する治療において使用されることができる。標準的な技術は、組換えDNA、オリゴヌクレオチドの合成、組織培養及び形質変換(例えば電気穿孔法(エレクトロポレーション)、リポソームトランスフェクション)において使用されることができる。例えば、メーカより提供された説明書付きのキットを使用するか、又は当分野における公知の方法、又は本発明に記載の方法により反応及び精製技術を実施することができる。一般的に、前記技術及びステップは、当該技術分野においてよく知られている従来の方法、及び種々の一般的な文献又はより具体的な文献に記載された従来の方法により実施されることができ、これらの文献は、本発明において引用及び議論されている。
置換基が左から右に書かれた通常の化学式で明記される場合、構造式を右から左に書く場合に得られる化学的に等価な置換基もそれらの置換基に含まれ、例えば-CH2O-は-OCH2-に相当する。
用語「置換」は、特定の原子の原子価状態が正常で置換後の化合物が安定である場合、特定の原子上の任意の1つ又は複数の水素原子が置換基で置換されていることを意味する。置換基がオキソ(即ち=O)である場合、2つの水素原子が置換されていることを意味し、芳香族基ではオキソ基で置換されることはない。
化合物の組成又は構造において任意の変数(例えばR)は1回以上現れる場合、いずれにおいてその変数の定義は独立している。このため、例えば、基が0~2個のRで置換されている場合、前記基は、多くとも2個のRで置換されていてもよく、且つ、いずれの場合においても、Rは独立した選択肢がある。なお、置換基及び/又はその変形体の組み合わせは、このような組み合わせが安定な化合物を形成する場合にのみ許容される。
本明細書に使用されるCm~nは、この部分においてm~n個の炭素原子を有することを意味する。例えば、前記「C1~8」基は、この部分において1~8個の炭素原子を有し、即ち、基が1つの炭素原子、2つの炭素原子、3つの炭素原子…8つの炭素原子を含むことを意味する。このため、例えば、「C1~8アルキル基」は、1~8個の炭素原子を有するアルキル基を意味し、即ち、前記アルキル基は、メチル基、エチル基、プロピル基、イソプロピル基、n-ブチル基、イソブチル基、s-ブチル基、t-ブチル基、オクチル基などから選ばれる。本明細書における数字の範囲は、例えば「1~8」は、所定の範囲内の各整数を意味し、例えば「1~8個の炭素原子」は、前記基が1つの炭素原子、2つの炭素原子、3つの炭素原子、4つの炭素原子、5つの炭素原子、6つの炭素原子、7つの炭素原子又は8つの炭素原子を有してもよいことを意味する。
用語「員」は、環を構成する骨格原子の数を意味する。例えば、ピリジンが六員環であり、ピロールが五員環である。
用語「薬学的に許容される」は、それらの化合物、材料、組成物及び/又は剤形について、信頼できる医学的判断の範囲内において、人や動物の組織と接触して使用するのに適しているが、過度の毒性、刺激性、過敏性反応又はその他の問題又は合併症がなく、合理的な利益/リスク比に見合ったものである。
用語「医薬組成物」は、薬学的に許容される化学成分又は試薬を少なくとも1つ混合した、任意の生物活性化合物を意味し、前記薬学的に許容される化学成分又は試薬は「担体」であり、化合物を細胞又は組織に導入することに寄与し、安定化剤、希釈剤、懸濁剤、増粘剤及び/又は賦形剤を含むが、これらに限定されない。
用語「薬学的に許容される塩」は、所定の化合物の遊離酸及び遊離塩基の生物学的効果を保持し、且つ生物学又は他の点で悪影響がない塩を意味する。特別な説明がない限り、本発明における塩として、金属塩、アンモニウム塩、有機塩基と形成した塩、無機酸と形成した塩、有機酸と形成した塩、塩基性又は酸性アミノ酸と形成した塩などが挙げられる。金属塩の非限定的な例は、例えばナトリウム塩、カリウム塩などのアルカリ金属の塩、例えばカルシウム塩、マグネシウム塩、バリウム塩などのアルカリ土類金属の塩、アルミニウム塩などを含むが、これらに限定されない。有機塩基と形成した塩の非限定的な例は、トリメチルアミン、トリエチルアミン、ピリジン、メチルピリジン、2,6-ジメチルピリジン、エタノールアミノ、ジエタノールアミン、トリエタノールアミン、シクロヘキシルアミン、ジシクロヘキシルアミンなどと形成した塩を含むが、これらに限定されない。無機酸と形成した塩の非限定的な例は、塩酸、臭化水素酸、硝酸、硫酸、リン酸などと形成した塩を含むが、これらに限定されない。有機酸と形成した塩の非限定的な例は、蟻酸、酢酸、トリフルオロ酢酸、フマル酸、シュウ酸、リンゴ酸、マレイン酸、酒石酸、クエン酸、コハク酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸などと形成した塩を含むが、これらに限定されない。塩基性アミノ酸と形成した塩の非限定的な例は、アルギニン、リジン、オルニチンなどと形成した塩を含むが、これらに限定されない。酸性アミノ酸と形成した塩の非限定的な例は、アスパラギン酸、グルタミン酸などと形成した塩を含むが、これらに限定されない。
薬学的に許容される塩は、酸根又は塩基性基を含む親化合物から通常の化学的方法によって合成されることができる。通常の場合、このような塩の調製方法は、水又は有機溶媒又は両者の混合物において、遊離酸又はアルカリの形態のこれらの化合物と化学量論的に適切なアルカリ又は酸とを反応させて調製する。一般的に、エーテル、酢酸エチル、エタノール、イソプロパノール又はアセトニトリルなどの非水媒体が好ましい。
用語「溶媒和物」は、本発明における1つの化合物と1つ又は複数の溶媒分子とから形成される物理的な集合体を意味し、この物理的な集合体は、様々な程度のイオン及び例えば水素結合などの共有結合を含む。この溶媒和物が分離可能であることが確認されており、例えば、結晶の格子に1つ又は複数の溶媒分子が混在している場合、「溶媒和物」は溶媒相と分離可能な溶媒和物の2つの部分を含む。対応する溶媒和物には、エタノール溶媒和物、メタノール溶媒和物などを含む多くの例がある。「水和物」は、水(H2O)分子を溶媒とする溶媒和物を意味する。本発明における1つ又は複数の化合物は、いずれも任意に溶媒和物として調製することができる。溶媒和物の調製はよく知られており、例えば、M.Cairaなど,J.PharmaceuticalSci.,93(3),601-611(2004)に抗真菌薬であるフルコナゾールの溶媒和物の調製、即ち酢酸エチルと水を用いた調製が記載されている。E.C.vanTonderなど,AAPS PharmSciTech.,5(1),article 12(2004)及びA.L.Binghamなど,Chem.Commun.,603-604(2001)にも、溶媒和物や水和物の類似した調製方法が記載されている。典型的且つ非限定的な調製過程は、常温より高い温度で本発明の化合物を必要量の理想溶媒(有機溶媒、水又はそれらの混合溶媒)に溶解し、降温し、放置し晶析させ、その後、標準的な方法で結晶を分離及び抽出する。I.R.分光分析技術によって、結晶において溶媒和物(水和物)を形成する溶媒(水)の存在を確認することができる。
用語「活性代謝物」は、化合物の代謝の時に形成される当該化合物の活性誘導体を意味する。
用語「多形体」は、異なる結晶格子の形態で存在する本発明の化合物を意味する。
用語「同位体標識物」は、同位体標識を有する本発明の化合物を意味する。例えば本発明の化合物における同位体は、H、C、N、O、P、F、Sなどの元素の様々な同位体、例えば2H、3H、13C、14C、15N、18O、17O、31P、32P、35S、18F及び36Sを含んでもよい。
用語「薬学的に許容されるプロドラッグ」又は「プロドラッグ」は、本発明の化合物の任意の薬学的に許容される塩、エステル、エステルの塩又は他の誘導体を意味し、受容体に投与された後に本発明の化合物又は薬学的活性を有するその代謝物又は残基を直接又は間接的に提供することができる。特に好ましい誘導体又はプロドラッグは、患者に投与される場合に本願の化合物のバイオアベイラビリティを向上させるか(例えば、経口化合物をより容易に血液中に吸収させることができる)、又は親化合物の生物学的器官や作用部位(例えば脳やリンパ系など)への送達を促進することができる化合物である。通常の操作又は体内で親化合物に分解できるような修飾方式により、化合物に存在する官能基を修飾し、プロドラッグを調製することができる。様々なプロドラッグの形態は当分野でよく知られているものである。T.HiguchiとV.StellaのPro-drugs as Novel Delivery Systems(1987)Vol.14 of the A.C.S. Symposium Series,Bioreversible Carriers in Drug Design,(1987)Edward B.Roche,ed.,American Pharmaceutical Association及びPergamon Pressに提示されたプロドラッグに関する議論を参照する。Design of Prodrugs,Bundgaard,A.Ed.,Elseview,1985 and Method in Enzymology,Widder,K.et al.,Ed.;Academic,1985,vol.42,p.309-396;Bundgaard,H.“Design and Application of Prodrugs”in A Textbook of Drug Design and Development,Krosgaard-Larsen and H.Bundgaard,Ed.,1991,第5章、113-191頁;及びBundgaard,H.,Advanced Drug Delivery Review,1992,8,1-38という文献は、引用により本明細書に組み込まれている。
用語「立体異性体」は、分子内の原子の異なる空間的な配列方式によって生成される異性体を意味する。本発明の化合物は、不斉又はキラル中心及び二重結合などの構造を含むので、光学異性体、幾何異性体、互変異性体、アトロプ異性体などの様々な異性体の形態を含み得る。これらの異性体及びそれらの単一異性体、ラセミ体などは、いずれも本発明の範囲に含まれる。例えば、光学異性体については、キラル分離、キラル合成、キラル試薬又はその他の通常の技術により光学活性な(R)-及び(S)-異性体及びDとL異性体を調製することができる。例えば、適当な光学活性物質(例えばキラルアルコールやモッシャー(Mosher`s)の酸塩化物)と反応してジアステレオマーに変換し、これを分離して対応する単一異性体に変換する(例えば加水分解)ことができる。また、例えば、カラムにより分離することもできる。
本明細書における「医薬組成物」は、薬剤分野でよく知られている方法で調製することができ、局所的又は全身的な治療が必要か否か及び治療される領域に応じて、様々な経路により投与又は施用することができる。局所(例えば、鼻、膣、直腸投与を含む経皮、皮膚、眼、粘膜)、肺(例えば、噴霧器により粉末又はエアロゾル剤を気管内や鼻内に吸入又は吹入させる)、経口又は非経口で投与する。非経口投与には、静脈内、動脈内、皮下、腹腔内又は筋肉内の注射又は注入、又は鞘内や脳室内などの頭蓋内の投与が含まれる。1回高用量で非経口投与するか、又は例えば連続注入ポンプで投与してもよい。本明細書の医薬組成物は、錠剤、丸剤、散剤、トローチ剤、サシェ(sachets)、カシェ(cachets)剤、エリキシル剤、懸濁剤、乳剤、液剤、シロップ、エアロゾル剤(固体又は液体溶媒に溶解);活性化合物を例えば10重量%と高い割合で含有する軟膏剤、軟と硬ゼラチンカプセル、坐剤、滅菌注射溶液及び無菌包装粉末などの形を含むが、これらに限定されない。
本明細書の医薬組成物は、単位剤形で調製することができ、各投与量は、約0.1~1000mg、通常約5~1000mg、さらに約100~500mgの活性成分を含んでもよい。用語「単位剤形」は、ヒト患者と他の哺乳動物に適している、物理的に離散した単一投与量単位であり、各単位は、適切な薬物担体と混合された、計算により所望の治療効果を産生できる所定量の活物質を含有する。
用語「個体」は、疾患、病症又は病状などに罹患している個体を意味し、哺乳動物と非哺乳動物とを含む。哺乳動物の実施例として、哺乳綱のあらゆるメンバーを含むが、これらに限定されなく、ヒト、ヒト以外の霊長類動物(例えばチンパンジー、その他の猿類及びサル)、例えばウシ、ウマ、ヒツジ、ヤギ、ブタなどの家畜、例えばウサギ、イヌ、ネコなどの家庭動物、げっ歯類の動物、例えばラット、マウス、モルモットなどを含む実験動物が挙げられる。
用語「治療」とその他の類似の同義語は、疾患又は病症の症状を寛解、軽減又は改善すること、及び、その他の症状を予防すること、症状に繋がる潜在的な代謝要因を改善又は予防すること、疾患又は病症を抑制すること、例えば、疾患又は病症の発展を阻止すること、疾患又は病症を寛解すること、疾患又は病症を好転させること、疾患又は病症による症状を寛解すること、或いは疾患又は病症の症状を中止することを含み、また、当該用語は予防の目的を含むこともできる。当該用語は治療効果及び/又は予防効果を得ることをさらに含む。前記治療効果は、治療される潜在的な疾患を治癒又は改善することを意味する。なお、潜在的な疾患に関連する1種又は複数種の生理症状に対する治癒又は改善も治療効果であり、例えば、患者は依然として潜在的な疾患の影響を受ける可能性があるものの、患者の状況の改善が見られる。予防効果について、特定の疾患に罹患するリスクがある患者に前記組成物又は化合物を投与し、又は、疾患が未だ診断されていないものの、当該疾患の1つ又は複数の生理症状が現れた患者に前記組成物又は化合物を投与することができる。
用語「必要な治療効果を得る投与量」又は「治療有効量」は、施用後に治療される疾患又は病症の1つ又は複数の症状をある程度寛解するのに十分な少なくとも1種の薬剤又は化合物の量を意味する。その結果は、兆候、症状又は病因の低減及び/又は寛解であってもよいし、又は生物系の任意のその他の所望変化であってもよい。例えば用量漸増試験の技術を使用して任意の個体の症例に最適な有効量を測定することができる。実際的に投与される化合物、医薬組成物又は薬剤の量は、通常、治療される病症、選択される投与経路、投与される実際の化合物、個々の患者の年齢、体重及び反応、ならびに患者の症状の重症度を含む関連する状況に応じて医師によって決定される。
薬用組成物における本発明の化合物の割合又は濃度は、一定でなくてもよく、投与量、化学特性(例えば疎水性)、投与経路などを含む様々な要因に依存する。本発明の化合物は、非経口投与のために、例えば約0.1~10%w/vの当該化合物を含む緩衝生理水溶液で提供することができる。典型的な投与量の範囲は、約1μg/kg~約1g/kg体重/日である。いくつかの実施形態において、投与量の範囲は、約0.01mg/kg~約100mg/kg体重/日である。投与量は、疾患又は病症の種類及び進行度、具体的な患者の一般的健康状態、選択される化合物の相対的な生物学的効果、賦形剤及びその投与経路などのような変数に依存する可能性がある。
用語「施用」は、化合物又は組成物を生物学的作用が行われる所望サイトまで送達できる方法を意味する。これらの方法は、経口投与、経十二指腸投与、非経口注射(静脈内、皮下、腹腔内、筋肉内、動脈内に注射若しくは注入することを含む)、外用、及び経直腸投与を含むが、これらに限定されない。本明細書に記載の化合物及び方法に使用される施用技術、例えばGoodman及びGilman,The Pharmacological Basis of Therapeutics,currented.;Pergamon;and Remington’s,Pharmaceutical Sciences(current edition),Mack Publishing Co.,Easton,Paに検討されたものは、当業者によく知られている。
用語「IC50」は、このような効果を測定する分析において最大効果を50%阻害する効果が得られることを意味する。
以下、本発明の目的、技術案と利点をさらに明確にするために、本発明の例示的な実施例の技術案についてさらに説明する。
本発明は、下記の方法により本発明に記載の化合物を調製することができる。以下の方法及び実施例は、これらの方法を説明するためのものである。これらの流れ及び実施例は、任意の方式により本発明を限定するものとして解釈されるべきではない。当業者に知られている標準的な合成技術を使用して本明細書に記載されるような化合物を合成し、又は当該分野で知られている方法と本明細書に記載される方法とを組み合わせて使用することもできる。
本発明の実施例の化学反応は、本発明の化学変化及びそれに必要な試薬及び材料に適した適切な溶媒中で行われる。本発明の化合物を得るために、当業者が既存の実施形態に基づいて合成ステップ又は反応の流れを修正又は選択する必要がある場合がある。
本分野の任意の合成経路計画における重要な考慮事項の1つは、反応性官能基(例えば、本発明におけるアミノ基)のために適切な保護基を選択することである。訓練を受けた当業者にとって、Greene及びWutsの(Protective Groups In Organic Synthesis,Wiley and Sons,1991)はこの方面において権威的である。本発明に引用されている参考文献のすべては、その全体を本発明に組み込まれている。
本分野で知られている任意の適切な方法に従って本明細書に記載された反応をモニタリングすることができる。例えば、核磁気共鳴スペクトル(例えば1H又は13C)、赤外スペクトル、分光光度測定(例えばUV-可視光)、質量分析法などの広いスペクトル法、又は、例えば高速液体クロマトグラフィー(HPLC)又は薄層クロマトグラフィーなどのクロマトグラフィーにより生成物の形成をモニタリングすることができる。
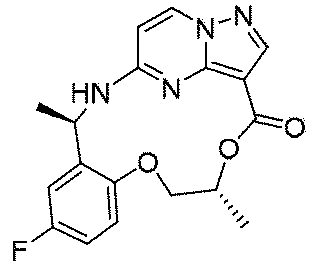
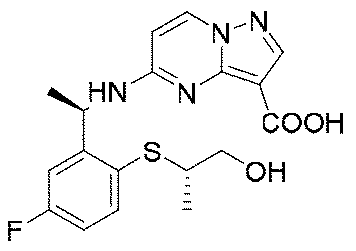
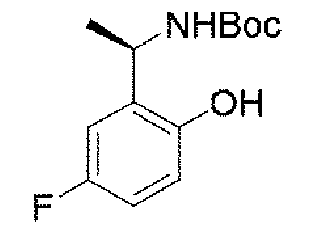
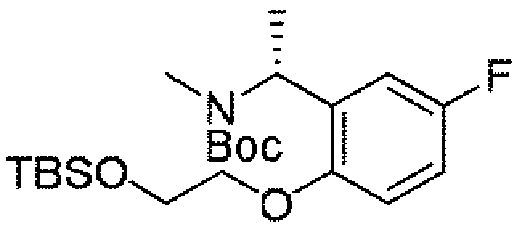
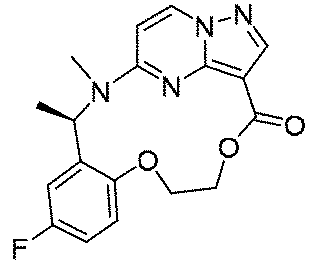
実施例1(1 3 E,1 4 E,3R,6S)-4 5 -フルオロ-3,6-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン((1 3 E,1 4 E,3R,6S)-4 5 -fluoro-3,6-dimethyl-5,8-dioxa-2-aza-1(5,3)-pyrazolo[1,5-a]pyrimidina-4(1,2)-benzenacyclononaphan-9-one)
合成経路は、図1に示すようである。
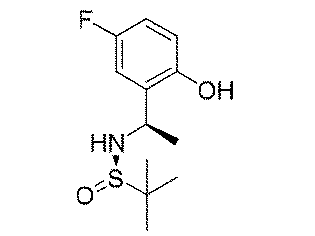
ステップA:(E)-N-(5-フルオロ-2-ヒドロキシベンジリデン)t-ブチルスルフィンアミド
5.8g(41.4mmol、1.0eq)の5-フルオロ-2-ヒドロキシベンズアルデヒド及び5.0g(41.4mmol、1eq)の(R)-t-ブチルスルフィンアミドを100mLのジクロロメタンに加え、磁気攪拌下で21.5g(66.3mmol、1.6eq)の炭酸セシウムを加えて終夜反応させた。反応終了後、吸引濾過し、ジクロロメタンで濾過ケーキを洗い流した。濾液を濃縮して得られた生成物をそのまま次のステップに用いた(8.5g、収率=85%)。
ステップB:N-((R)-1-(5-フルオロ-2-ヒドロキシフェニル)エチル)t-ブチルスルフィンアミド
24g(100mmol、1.0eq)の(E)-N-(5-フルオロ-2-ヒドロキシベンジリデン)t-ブチルスルフィンアミドを300mLのテトラヒドロフランに溶解して-65℃に降温し、N2雰囲気下で臭化メチルマグネシウム100mL(3N、3.0eq)をゆっくりと加え、反応温度を-50℃以下に保った。添加終了後、ゆっくりと室温まで上昇し、終夜反応させた。TLCで反応終了をモニタリングし、氷浴で500mLの水を加えて反応を徐々にクエンチした。500mLの酢酸エチルで抽出し、有機相を水及び飽和食塩水で2回順次洗浄し、有機相を無水硫酸ナトリウムにより乾燥し、蒸発乾固し、残留物をシリカゲルカラムクロマトグラフィーにより精製して生成物を得た(8.5g、収率=33%)。
1HNMR(400MHz、CDCl3)δ9.02(s,1H),6.79(d,J=4.0Hz,1H),6.65-6.51(m,1H),6.50-6.41(m,1H),5.04(d,J=8.0Hz,1H),4.45-4.30(m,1H),1.53(d,J=8.0Hz,3H),1.29(s,9H).
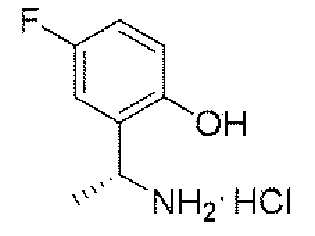
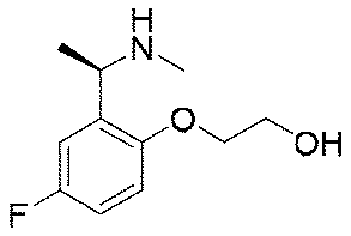
ステップC:(R)-2-(1-アミノエチル)-4-フルオロフェノール塩酸塩
8.5g(32.8mmol、1.0eq)のN-((R)-1-(5-フルオロ-2-ヒドロキシフェニル)エチル)t-ブチルスルフィンアミドを100mLのジオキサン(4N)溶液に溶解し、室温で3時間反応させた。TLCで反応終了をモニタリングし、溶媒を蒸発乾固し、残留物に100mLの酢酸エチルを加えて叩解し、吸引濾過し、酢酸エチルで洗い流し、固体を収集し、乾燥して目標生成物を得た(5.4g、収率=87%)。
ステップD:(R)-5-((1-(5-フルオロ-2-ヒドロキシフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-蟻酸エチル
5.3g(27.9mmol、1.0eq)の(R)-2-(1-アミノエチル)-4-フルオロフェノール塩酸塩、6.29g(27.9mmol、1.0eq)の5-クロロピラゾロ[1,5-a]ピリミジン-3-蟻酸エチル、12mL(167mmol、6.0eq)のジイソプロピルエチルアミンを60mLのN,N-ジメチルホルムアミドに溶解し、体系を120℃まで昇温して5時間反応させた。TLCで反応終了をモニタリングし、反応液を濃縮して蒸発乾固し、100mLの水、200mLの酢酸エチルを加えて抽出し、有機相を水及び飽和食塩水で2回順次洗浄し、有機相を無水硫酸ナトリウムにより乾燥し、蒸発乾固し、残留物をシリカゲルカラムクロマトグラフィーにより精製して生成物を得た(2.9g、収率=31%)。
1HNMR(400MHz,CDCl3)δ9.17(brs,1H),8.24(s,1H),8.14(d,J=8.0Hz,1H),6.93-6.90(m,2H),6.83(d,J=8.0Hz,1H),6.13(d,J=8.0Hz,1H),5.81(d,J=8.0Hz,1H),5.64(t,J=8.0Hz,1H),4.42(q,J=8.0Hz,2H),1.61(d,J=8.0Hz,3H),1.41(t,3H).
ステップE:(R)-2-ヒドロキシプロピルピバレート((R)-2-hydroxypropyl pivalate)
7.6g(100mmol、1.0eq)の(R)-プロパン-1,2-ジオール及び16mL(200mmol、2.0eq)のピリジンを80mLのジクロロメタンに溶解し、氷浴で反応系に12.6g(100mmol、1.0eq)のピバロイルクロリドを徐々に滴下した。滴下終了後、室温まで昇温して終夜反応させた。TLCで反応終了をモニタリングし、反応液を濃縮して蒸発乾固し、100mLの水及び250mLの酢酸エチルを加えて抽出し、有機相を水及び飽和食塩水で2回順次洗浄し、有機相を無水硫酸ナトリウムにより乾燥し、蒸発乾固し、残留物をシリカゲルカラムクロマトグラフィーにより精製して生成物を得た(10.5g、収率=66%)。
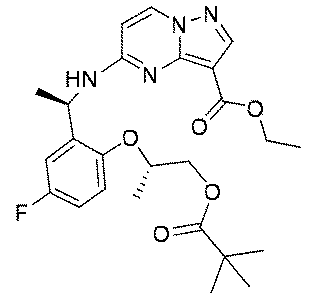
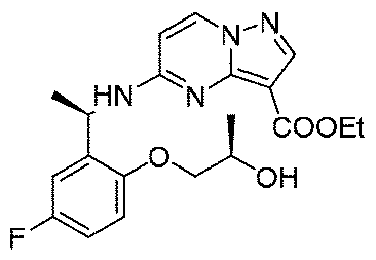
2.0g(5.8mmol、1.0eq)の(R)-5-((1-(5-フルオロ-2-ヒドロキシフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-蟻酸エチル、1.39g(8.7mmol、1.5eq)の(R)-2-ヒドロキシプロピルピバレート、3.04g(11.6mmol、2.0eq)のトリフェニルホスフィンを35mLのジクロロメタンに溶解し、氷浴で反応系に2.02g(11.6mmol、2.0eq)のアゾジカルボン酸ジイソプロピルを徐々に滴下した。添加終了後、室温まで昇温して終夜反応させた。TLCで反応終了をモニタリングし、100mLの水及び250mLの酢酸エチルを加えて抽出し、有機相を水及び飽和食塩水で2回順次洗浄し、有機相を無水硫酸ナトリウムにより乾燥し、蒸発乾固し、残留物をシリカゲルカラムクロマトグラフィーにより精製して生成物を得た(2.1g、収率=75%)。
1HNMR(400MHz,CDCl3)δ8.25(s,1H),8.16(d,J=8.0Hz,1H),7.10(d,J=8.0Hz,1H),6.90(d,J=8.0Hz,2H),6.08(d,J=8.0Hz,1H),5.24(s,1H),4.74-4.65(m,1H),4.38(q,J=8.0Hz,2H),4.28-4.13(m,3H),1.56(d,J=8.0Hz,3H),1.40(t,J=8.0Hz,3H),1.25(d,J=8.0Hz,3H),1.20(t,9H).
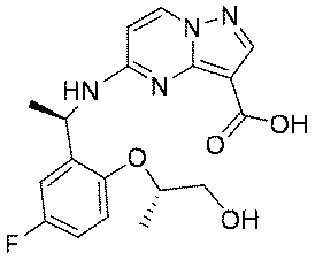
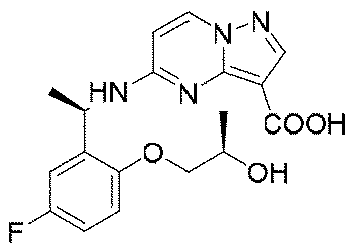
1.5g(3.1mmol、1.0eq)の化合物11を10mLのメタノールに溶解し、氷浴で3.1mL(10N、10.0eq)の水酸化ナトリウム水溶液を加えた。添加終了後、室温まで昇温して2時間反応させた。TLCで反応終了をモニタリングし、濃縮して溶媒を除去し、1Nの塩酸水溶液を加えてpHを中性に調整し、吸引濾過して固体を収集し、200mLの水で濾過ケーキを洗い流し、乾燥して目標生成物12を得た(0.72g、収率=63%)。
1HNMR(400MHz,DMSO)δ11.50(brs,1H),8.55(d,J=9.2Hz,1H),8.28(d,J=9.2Hz,1H),8.10(s,1H),7.15-6.94(m,3H),6.45(d,J=9.2Hz,1H),5.61(d,J=8.0Hz,1H),4.52-4.35(m,1H),3.68-3.42(m,2H),1.43(d,J=8.0Hz,3H),1.24(d,J=8.0Hz,3H).
ステップH:(1
3E,1
4E,3R,6S)-4
5-フルオロ-3,6-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン((1
3E,1
4E,3R,6S)-4
5-fluoro-3,6-dimethyl-5,8-dioxa-2-aza-1(5,3)-pyrazolo[1, 5 -a]pyrimidina-4(1,2)-benzenacyclononaphan-9-one)
150mg(0.40mmol、1.0eq)の化合物12、489mg(2.0mmol、5.0eq)の2,4,6-トリクロロベンゾイルクロリド、280mg(2.4mmol、6.0eq)のトリエチルアミンを10mLのテトラヒドロフランに溶解し、室温で30min攪拌反応させた。その後、上記の反応液を488mg(4.0mmol、10.0eq)の4-ジメチルアミノピリジンのトルエン溶液500mLに徐々に滴下し、この反応液を100℃まで昇温して2時間撹拌し、その後、室温まで降温して終夜反応させた。TLCで反応終了をモニタリングし、濃縮して溶媒を除去し、残留物に30mLの水、50mLの酢酸エチルを加えて抽出し、有機相を水及び飽和食塩水で2回順次洗浄し、有機相を無水硫酸ナトリウムにより乾燥し、蒸発乾固し、残留物をシリカゲルカラムクロマトグラフィーにより精製して生成物を得た(23mg、収率=16%)。
LC-MS:m/z=357[M+H]+.
1HNMR(400MHz,CDCl3)δ8.26-8.20(m,2H),7.01(d,J=8.0Hz,1H),6.87-6.69(m,2H),6.19(d,J=4.0Hz,1H),6.01-5.85(m,1H),5.55(s,1H),4.89(dd,J=8.0,4.0Hz,1H),4.62(s,1H),4.13(t,J=8.0Hz,1H),1.63-1.52(m,6H).
実施例2(1
3
E,1
4
E,3S,6S)-4
5
-フルオロ-3,6-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン((1
3
E,1
4
E,3S,6S)-4
5
-fluoro-3,6-dimethyl-5,8-dioxa-2-aza-1(5,3)-pyrazolo[1,5-a]pyrimidina-4(1,2)-benzenacyclononaphan-9-one)
実施例1に記載されたステップを参照して目標化合物(20mg、15%)を調製した。LC-MS:m/z=357[M+H]+.
実施例3(1
3
E,1
4
E,3S,6R)-4
5
-フルオロ-3,6-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン((1
3
E,1
4
E,3S,6R)-4
5
-fluoro-3,6-dimethyl-5,8-dioxa-2-aza-1(5,3)-pyrazolo [1,5-a]pyrimidina-4(1,2)-benzenacyclononaphan-9-one)
実施例1に記載されたステップを参照して目標化合物を調製した(31mg、22%)。LC-MS:m/z=357[M+H]+.
実施例4(1
3
E,1
4
E,3R,6R)-4
5
-フルオロ-3,6-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン((1
3
E,1
4
E,3R,6R)-4
5
-fluoro-3,6-dimethyl-5,8-dioxa-2-aza-1(5,3)-pyrazolo[1,5-a]pyrimidina-4(1,2)-benzenacyclononaphan-9-one)
実施例1に記載されたステップを参照して目標化合物を調製した(35mg、24%)。LC-MS:m/z=357[M+H]+.
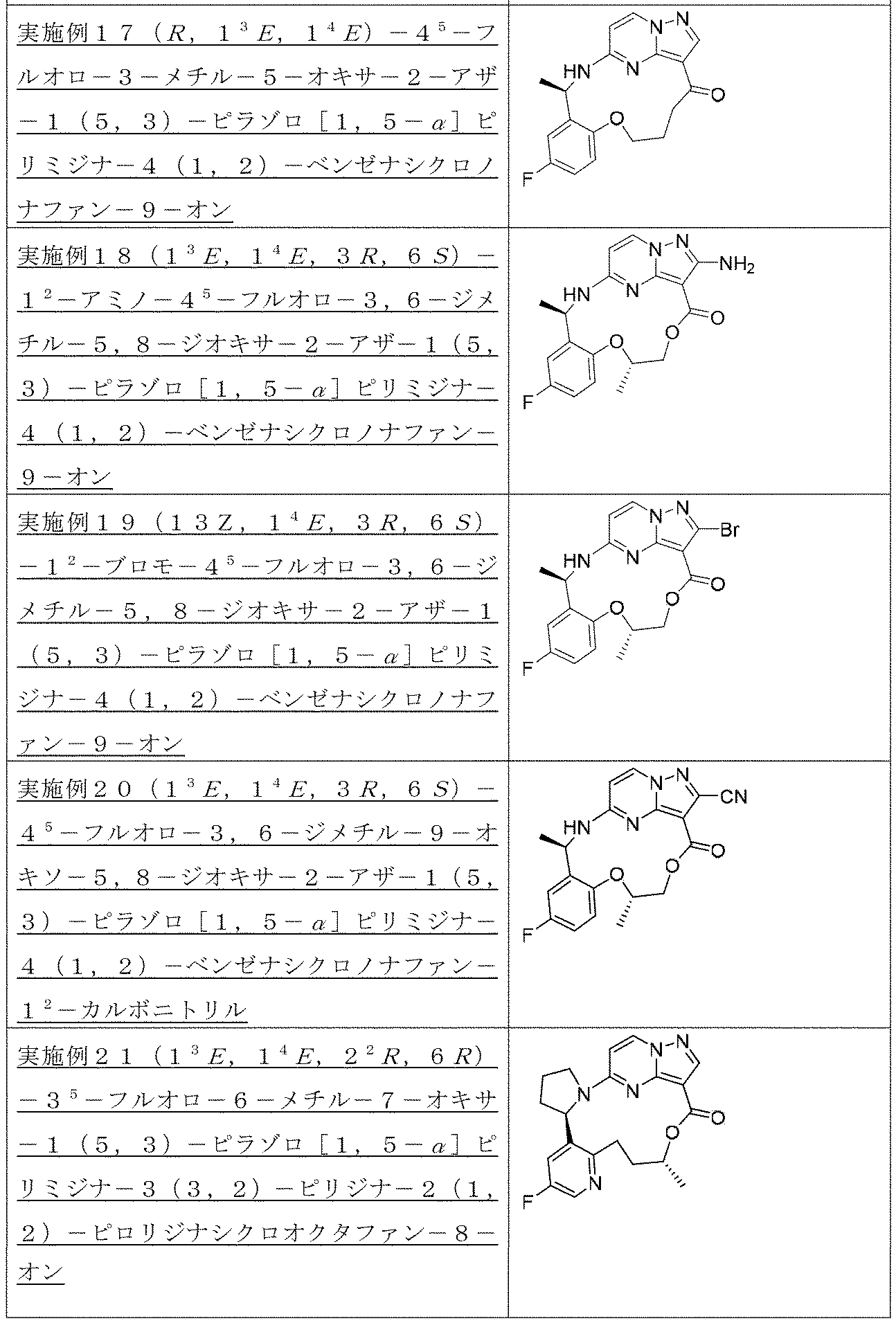
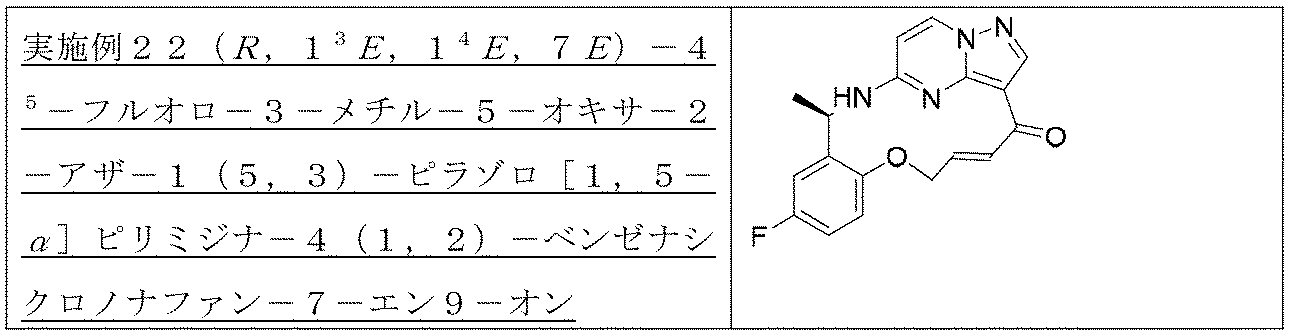
以下の化合物は、実施例1を参照して類似した方法により合成される。
実施例6(1 3 E,1 4 E,3R,7R)-4 5 -フルオロ-3,7-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
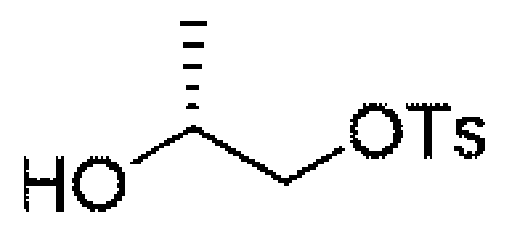
ステップA:(R)-2-ヒドロキシプロピル-4-メチルベンゼンスルホン酸
2.08g(27.3mmol、1.0eq)の(R)-1,2-プロピレングリコールのジクロロメタン溶液にトリエチルアミン8.3g(81.9mmol、3.0eq)を加え、その後、4-メチルベンゼンスルホニルクロリド5.2g(27.34mmol、1.0eq)を加え、さらに触媒量のDMAPを加え、室温で終夜反応させ、水を加え、ジクロロメタンで3回抽出し、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(2.1g、収率=33%)。
1HNMR(400MHz,CDCl3)δ7.85(d,J=8.2Hz,2H),7.40(d,J=8.0Hz,2H),4.16-3.98(m,2H),3.92-3.87(m,1H),2.50(s,3H),1.20(d,J=6.4Hz,3H).
ステップB:5-(((R)-1-(5-フルオロ-2-((R)-2-ヒドロキシプロポキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチル
680mg(1.97mmol、1.0eq)の(R)-5-((1-(5-フルオロ-2-ヒドロキシフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-蟻酸エチルのDMF溶液に炭酸カリウム1.36g(9.85mmol、5.0eq)を加え、その後、500mg(2.17mmol、1.1eq)の(R)-2-ヒドロキシプロピル-4-メチルベンゼンスルホン酸を加え、添加終了後、オイルバスで80℃に加熱して4時間反応させ、室温まで冷却し、水を加え、酢酸エチルで3回抽出し、有機相を合わせ、水で3回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(278mg、収率=35%)。
LC-MS:m/z=403[M+H]+.
ステップC:5-(((R)-1-(5-フルオロ-2-((R)-2-ヒドロキシプロポキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸
実験操作について、実施例13におけるステップCを参照する。
LC-MS:m/z=375[M+H]+.
ステップD:(1
3E,1
4E,3R,7R)-4
5-フルオロ-3,7-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
実験操作について、実施例13におけるステップDを参照する。
1HNMR(400MHz,CDCl3)δ8.24(s,1H),8.18(d,J=7.2Hz,1H),7.01(d,J=9.0Hz,1H),6.88-6.86(m,1H),6.79(s,1H),6.17-6.15(m,2H),5.62(s,1H),5.52(s,1H),4.37(d,J=10.4Hz,1H),4.08(d,J=10.0Hz,1H),1.71(d,J=6.4Hz,3H),1.57(d,J=7.0Hz,3H).LC-MS:m/z=357[M+H]+.
実施例9(1
3
E,1
4
E,3R,6S)-4
5
-フルオロ-3,6-ジメチル-8-オキサ-5-チア-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
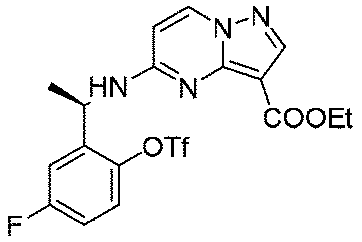
ステップA:(R)-5-((1-(5-フルオロ-2-(((トリフルオロメチル)スルホニル)オキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチル
氷浴で0.71g(2.06mmol、1.0eq)の(R)-5-((1-(5-フルオロ-2-ヒドロキシフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-蟻酸エチルのジクロロメタン溶液にピリジン489mg(3.18mmol、3.0eq)を加え、その後、トリフルオロメタンスルホン酸無水物872mg(3.09mmol、1.5eq)を加え、室温で攪拌し続けて2時間反応させ、水を加え、ジクロロメタンで3回抽出し、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(785mg、収率=80%)。
LC-MS:m/z=477[M+H]+.
ステップB:(R)-1-((t-ブチルジメチルシリル)オキシ)プロパン-2-イル4-メチルベンゼンスルホン酸
実験操作は、実施例6における的ステップAを参照する。
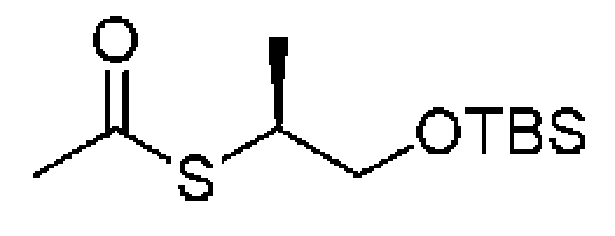
ステップC:(S)-S-(1-((t-ブチルジメチルシリル)オキシ)プロパン-2-イル)エタンチオール
2.0g(5.8mmol、1.0eq)の(S)-S-(1-((t-ブチルジメチルシリル)オキシ)プロパン-2-イル)エタンチオールのDMF溶液にチオ酢酸カリウム796mg(6.96mmol、1.2eq)を加え、添加終了後、オイルバスで60℃に加熱して攪拌し続けて4時間反応させ、室温まで冷却し、水を加え、酢酸エチルで3回抽出し、さらに水で3回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(1.0g、収率=69%)。
ステップD:(S)-1-((t-ブチルジメチルシリル)オキシ)プロパン-2-チオール
氷浴で1.0g(4.02mmol、1.0eq)の(S)-S-(1-((t-ブチルジメチルシリル)オキシ)プロパン-2-イル)エタンチオールのメタノール/水(5:1)溶液に水酸化ナトリウム241mg(6.03mmol、1.5eq)を加え、攪拌し続けて0.5時間反応させ、減圧蒸留してメタノールを除去し、ジクロロメタンで3回抽出し、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(700mg、収率=84%)。
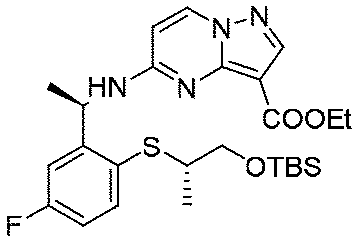
ステップE:5-(((R)-1-(2-(((S)-1-((t-ブチルジメチルシリル)オキシ)プロパン-2-イル)チオ)-5-フルオロフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-蟻酸エチル
646mg(1.35mmol、1.0eq)の(R)-5-((1-(5-フルオロ-2-(((トリフルオロメチル)スルホニル)オキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチルのジオキサン溶液に700mg(3.39mmol、2.5eq)の(S)-1-((t-ブチルジメチルシリル)オキシ)プロパン-2-チオール、156mg(0.27mmol、0.2eq)のキサントホス(Xantphos)、1.3mL(8.1mmol、6.0eq)のDIEA及び124mg(0.135mmol、0.1eq)のPd2(dba)3を加え、N2で3回置換し、オイルバスで120℃に加熱して終夜反応させ、室温まで冷却し、水を加え、酢酸エチルで3回抽出し、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(390mg、収率=54%)。
LC-MS:m/z=533[M+H]+.
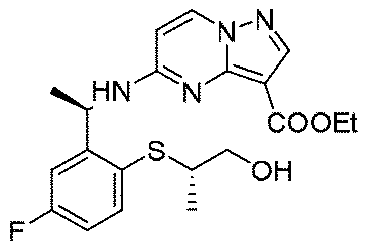
ステップF:5-(((R)-1-(5-フルオロ-2-(((S)-1-ヒドロキシプロパン-2-イル)チオ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチル
実験操作について、実施例13におけるステップBを参照する。
LC-MS:m/z=419[M+H]+.
ステップG:5-(((R)-1-(5-フルオロ-2-(((S)-1-ヒドロキシプロパン-2-イル)チオ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸
実験操作について、実施例13におけるステップCを参照する。
LC-MS:m/z=391[M+H]+.
ステップH:(1
3E,1
4E,3R,6S)-4
5-フルオロ-3,6-ジメチル-8-オキサ-5-チア-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
実験操作について、実施例13におけるステップDを参照する。
1HNMR(400MHz,CDCl3)δ8.36-8.24(m,2H),7.49-7.47(m,1H),7.13-6.94(m,2H),6.28(d,J=7.6Hz,1H),6.12-6.01(m,1H),5.66(s,1H),5.03(dd,J=11.6,3.8Hz,1H),3.87(t,J=11.2Hz,1H),3.50-3.49(m,1H),1.74(d,J=7.2Hz,3H),1.50(d,J=7.0Hz,3H).
LC-MS:m/z=373[M+H]+.
実施例13(R,1
3
E,1
4
E)-4
5
-フルオロ-3-メチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
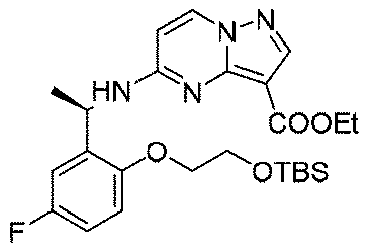
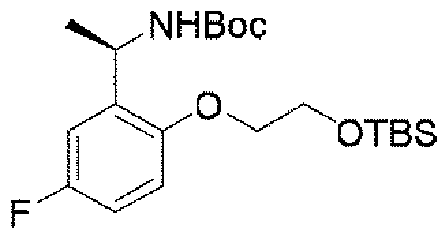
ステップA:(R)-5-((1-(2-(2-((t-ブチルジメチルシリル)オキシ)エトキシ)-5-フルオロフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチル
600mg(1.74mmol、1.0eq)の(R)-5-((1-(5-フルオロ-2-ヒドロキシフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-蟻酸エチル、614mg(2.48mmol、2.0eq)の(R)-2-ヒドロキシプロピルピバレート及び911mg(3.48mmol、2.0eq)のトリフェニルホスフィンを35mLのジクロロメタンに溶解し、氷浴で反応系に703mg(3.48mmol、2.0eq)のアゾジカルボン酸ジイソプロピルを徐々に滴下した。添加終了後、室温まで昇温して終夜反応させた。TLCで反応終了をモニタリングし、減圧蒸留して濃縮した残留物をシリカゲルカラムクロマトグラフィーにより精製して生成物を得た(719mg、収率=82%)。
LC-MS:m/z=503[M+H]+
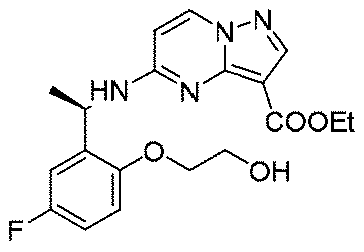
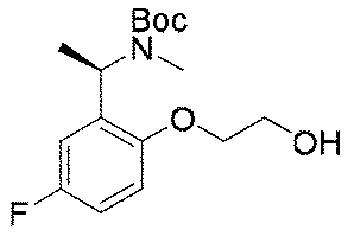
ステップB:(R)-5-((1-(5-フルオロ-2-(2-ヒドロキシエトキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチル
氷浴で719mg(1.43mmol、1.0eq)の(R)-5-((1-(2-(2-((t-ブチルジメチルシリル)オキシ)エトキシ)-5-フルオロフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチルのテトラヒドロフラン溶液にフッ化テトラブチルアンモニウム・三水和物900mg(2.86mmol、2.0eq)を加えた。添加終了後、室温まで昇温して1.5時間反応を続け、水を加え、酢酸エチルで3回抽出し、有機相を合わせ、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(519mg、収率=93%)。
LC-MS:m/z=389[M+H]+.
ステップC:(R)-5-((1-(5-フルオロ-2-(2-ヒドロキシエトキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸
519mg(1.34mmol、1.0eq)の(R)-5-((1-(5-フルオロ-2-(2-ヒドロキシエトキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチルのメタノール/水(5:1)溶液に水酸化ナトリウム1.6g(40.2mmol、40.0eq)を加えた。添加終了後、オイルバスで80℃に加熱して5時間反応を続けた。室温まで冷却し、減圧蒸留して溶媒を除去して残渣を得て、1N塩酸溶液でpHを5に調整し、酢酸エチルで2回抽出し、有機相を合わせ、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮して生成物を得た(482mg、収率=100%)。
LC-MS:m/z=361[M+H]+.
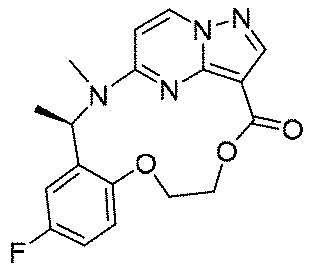
ステップD:(R,1
3E,1
4E)-4
5-フルオロ-3-メチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
150mg(0.42mmol、1.0eq)の(R)-5-((1-(5-フルオロ-2-(2-ヒドロキシエトキシ)フェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸のジクロロメタン溶液に1-エチル-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(0.84mmol、2.0eq)を加え、その後、4-ジメチルアミノピリジン(5mg、0.1eq)を加え、オイルバスで加熱しながら、5時間還流反応させ、室温まで冷却し、水を加え、ジクロロメタンで3回抽出し、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(60mg、収率=42%)。
1HNMR(400MHz,CDCl3)δ8.26(s,1H),8.20(d,J=7.4Hz,1H),7.04(dd,J=9.0,2.6Hz,1H),6.95-6.78(m,2H),6.21(d,J=7.4Hz,1H),6.14-6.01(m,1H),5.85(d,J=5.9Hz,1H),5.06(d,J=11.8Hz,1H),4.62-4.59(m,1H),4.49-4.45(m,1H),4.39-4.19(m,2H),1.56(d,J=7.0Hz,3H).LC-MS:m/z=343[M+H]+.
実施例23(R,1
3
E,1
4
E)-4
5
-フルオロ-2,3-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
ステップA:(R)-1-(5-フルオロ-2-ヒドロキシフェニル)エチル)カルバミン酸t-ブチル
3.7g(19.3mmol、1.0eq)の(R)-2-(1-アミノエチル)-4-フルオロフェノール塩酸塩のジクロロメタン溶液にトリエチルアミン5.4mL(38.6mmol、2.0eq)を加え、その後、ジ-t-ブチルジカーボネート4.6g(21.2mmol、1.1eq)を滴下し,滴下終了後、室温で2時間反応を続け、減圧蒸留して濃縮し、残渣を得て、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(4.9g、収率=100%)。
LC-MS:m/z=256[M+H]+.
ステップB:(R)-1-(2-(2-((t-ブチルジメチルシリル)オキシ)エトキシ)-5-フルオロフェニル)エチル)カルバミン酸t-ブチル
600mg(2.35mmol、1.0eq)のDMF溶液に炭酸カリウム1.3g(9.4mmol、4.0eq)を加え、その後、1.1g(4.7mmol、2.0eq)の(2-ブロモエトキシ)-t-ブチルジメチルシランを加え、オイルバスで80℃に加熱して4時間反応を続け、室温まで冷却し、水を加え、酢酸エチルで3回抽出し、有機相を合わせ、水で3回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(670mg、収率=69%)。
LC-MS:m/z=414[M+H]+.
ステップC:(R)-(1-(2-(2-((t-ブチルジメチルシリル)オキシ)エトキシ)-5-フルオロフェニル)エチル)(メチル)カルバミン酸t-ブチル
氷浴で670mg(1.62mmol、1.0eq)の(R)-1-(2-(2-((t-ブチルジメチルシリル)オキシ)エトキシ)-5-フルオロフェニル)エチル)カルバミン酸t-ブチルのDMF溶液に水素化ナトリウム194mg(4.86mmol、3.0eq)を加え、添加終了後、当該温度で攪拌し続けて15分間反応させ、その後、CH3I460mg(3.24mmol、2.0eq)を滴下し、滴下終了後、2時間反応を続け、水を加え、酢酸エチルで3回抽出し、有機相を合わせ、水で3回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(692mg、収率=100%)。
LC-MS:m/z=428[M+H]+.
ステップD:(R)-1-(5-フルオロ-2-(2-ヒドロキシエトキシ)フェニル)エチル)(メチル)カルバミン酸t-ブチル
実験操作について、実施例13におけるステップBを参照する。
1HNMR(400MHz,CDCl3)δ7.12-6.94(m,2H),6.84-6.80(mHz,1H),5.92(bRs,1H),4.63(bRs,1H),4.15-4.13(m,1H),3.92-3.88(m,3H),2.55(s,3H),1.53(s,9H),1.46(d,J=7.0Hz,3H).LC-MS:m/z=314[M+H]+.
ステップE:(R)-2-(4-フルオロ-2-(1-(メチルアミノ)エチル)フェノキシ)エタン-1-オール
実験操作について、実施例24におけるステップCを参照する。
LC-MS:m/z=214[M+H]+.
ステップF:(R)-5-((1-(5-フルオロ-2-(2-ヒドロキシエトキシ)フェニル)エチル)(メチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチル
実験操作について、実施例1におけるステップDを参照する。
LC-MS:m/z=403[M+H]+.
ステップG:(R)-5-((1-(5-フルオロ-2-(2-ヒドロキシエトキシ)フェニル)エチル)(メチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸
実験操作について、実施例13におけるステップCを参照する。
LC-MS:m/z=375[M+H]+.
ステップH:(R,1
3E,1
4E)-4
5-フルオロ-2,3-ジメチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
実験操作について、実施例13におけるステップDを参照する。
1HNMR(400MHz,CDCl3)δ8.39-8.25(m,2H),7.01-6.69(m,4H),6.42(d,J=7.8Hz,1H),5.0-5.07(m,1H),4.58-5.56(m,1H),4.44-4.42(m,1H),4.17-4.15(m,1H),3.43(s,3H),1.62(d,J=7.4Hz,3H).LC-MS:m/z=357[M+H]+.
実施例24(R,1
3
E,1
4
E)-4
5
,6,6-トリフルオロ-3-メチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
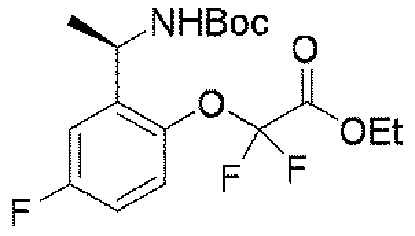
ステップA:(R)-2-(2-(1-((t-ブトキシカルボニル)アミノ)エチル)-4-フルオロフェノキシ)-2,2-ジフルオロ酢酸エチル
700mg(2.75mmol、1.0eq)のN,N-ジメチルホルムアミド溶液にブロモジフルオロ酢酸エチル1.4g(6.86mmol、2.5eq)を加え、その後、DBU1.06g(6.86mmol、2.5eq)を加え、オイルバスで70℃に加熱して4時間反応させ、室温まで冷却し、水を加え、酢酸エチルで3回抽出し、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(720mg、収率=69%)。
1HNMR(400MHz,CDCl3)δ7.33-7.23(m,1H),7.10-7.07(m,1H),6.97-5.93(m,1H),5.19(s,1H),4.98(s,1H),4.45(q,J=7.0Hz,2H),1.49-1.39(m,15H).
ステップB:(R)-1-(2-(1,1-ジフルオロ-2-ヒドロキシエトキシ)-5-フルオロフェニル)エチル)カルバミン酸t-ブチル
氷浴で720mg(1.9mmol、1.0eq)の(R)-2-(2-(1-((t-ブトキシカルボニル)アミノ)エチル)-4-フルオロフェノキシ)-2,2-ジフルオロ酢酸エチルのテトラヒドロフラン溶液に水素化アルミニウムリチウム160mg(4.2mmol、2.2eq)を加え、添加終了後、当該温度で2時間反応を続け、水を加えて反応をクエンチし、酢酸エチルで3回抽出し、飽和食塩水で1回洗浄し、無水硫酸ナトリウムで乾燥し、減圧蒸留して濃縮し、シリカゲルカラムクロマトグラフィーにより精製して生成物を得た(450mg、収率=71%)。
1HNMR(400MHz,CDCl3)δ7.36-7.33(m,1H),7.14-6.94(m,2H),5.27(s,1H),4.87(bRs,2H),4.05-3.97(m,2H),1.51-1.41(m,12H).
ステップC:(R)-2-(2-(1-アミノエチル)-4-フルオロフェノキシ)-2,2-ジフルオロメチル-1-アルコール
450mg(1.34mmol、1eq)のジクロロメタン溶液にトリフルオロ酢酸4mLを加え、添加終了後、室温で攪拌し続けて3時間反応させ、減圧蒸留して濃縮し、残渣を得て、飽和炭酸水素ナトリウム溶液を加えて塩基性に調整し、酢酸エチルで3回抽出し、無水硫酸ナトリウムで乾燥し、減圧濃縮して生成物を得た(315mg、収率=100%)。
LC-MS:m/z=236[M+H]+.
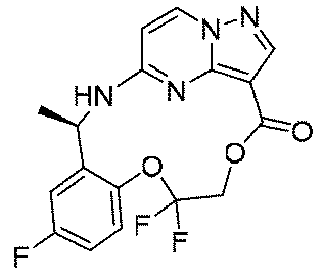
ステップD:(R)-5-((1-(2-(1,1-ジフルオロ-2-ヒドロキシエトキシ)-5-フルオロフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸エチル
実験操作について、実施例1におけるステップDを参照する。
LC-MS:m/z=425[M+H]+.
ステップE:(R)-5-((1-(2-(1,1-ジフルオロ-2-ヒドロキシエトキシ)-5-フルオロフェニル)エチル)アミノ)ピラゾロ[1,5-a]ピリミジン-3-カルボン酸
実験操作について、実施例13におけるステップCを参照する。
LC-MS:m/z=397[M+H]+.
ステップF:(R,1
3E,1
4E)-4
5,6,6-トリフルオロ-3-メチル-5,8-ジオキサ-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
実験操作について、実施例13におけるステップDを参照する。
1HNMR(400MHz,CDCl3)δ8.27-8.25(m,2H),7.23(s,1H),7.08(dd,J=8.8,3.0Hz,1H),6.94-6.90(m,1H),6.26(d,J=7.5Hz,1H),5.85(bRs,1H),5.57(bRs,1H),5.01-4.93(m,1H),4.82-4.77(m,1H),1.58(d,J=7.0Hz,3H).LC-MS:m/z=379[M+H]+.
実施例25(R,1
3
E,1
4
E)-4
5
-フルオロ-3-メチル-8-オキサ-5-チア-2-アザ-1(5,3)-ピラゾロ[1,5-α]ピリミジナ-4(1,2)-ベンゼナシクロノナファン-9-オン
実施例9の合成を参照する。
1HNMR(400MHz,CDCl3)δ8.33-8.20(m,2H),7.44-7.42(m,1H),7.06-7.04(m,1H),6.96-6.94(m,1H),6.25(d,J=7.4Hz,1H),6.17-5.98(m,1H),5.64(s,1H),5.02-5.00(m,1H),4.21-4.06(m,1H),3.78-3.76(m,1H),3.33-3.18(m,1H),1.49(d,J=7.0Hz,3H).LC-MS:m/z=359[M+H]+.
効果の評価
1.化合物のTRKA、TRKB、TRKC及びROS1に対する酵素阻害活性(IC50)の検出
移動度シフト検出技術(Mobility shift assay)によりTRKA、TRKB、TRKC及びROS1キナーゼに対して化合物の阻害活性の選別を行う。この選別プラットフォームは、マイクロ流体チップ技術に基づくMSAを中核とし、キャピラリー電気泳動の基本理念をマイクロ流体環境に適用する。実験用基質は、蛍光標識付きのポリペプチドであり、反応系における酵素の触媒作用によって生成物に変換し、それに応じてその電荷も変化した。MSAは、基質と生成物の電荷の違いによって両者を分離して、それぞれ検出を行う。
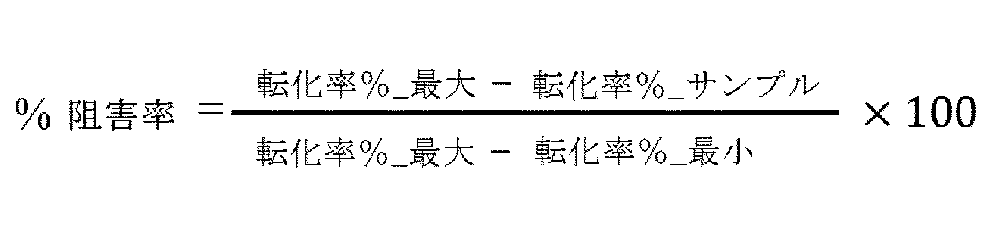
操作方法は、以下のように簡単に説明する。化合物粉末を100%DMSO(Sigma、Cat.D8418-1L)に十分に溶解し、10mMのストック溶液を調製した。化合物の初期実験濃度を100nMとし、3倍段階希釈して、10つの濃度を設定し、複数のウェルにて検出した。TRKA、TRKB及びTRKCキナーゼターゲットの場合、LOXO-101(Selleckchem、Cat.S7960)を陽性対照化合物とし、ROS1ターゲットの場合、スタウロスポリン(Selleckchem、Cat.S1421)を陽性対照化合物とした。段階希釈された化合物及び終濃度が2.5nM/2.55nM/2.5nM/0.3nMのTRKA/TRKB/TRKC/ROS1キナーゼ(Carna、Cat.08-186/08-187/08-197/08-163)をOptiplate-384Fウェルプレート(PerkinElmer、Cat.6007270)に均一に混合した後、室温で10分間インキュベートした。その後、終濃度が47.8μM/71.2μM/44.4μM/26.7μMのATP及び3μMのキナーゼ基質22(GLBiochem、Cat.112393)を加え、均一に混合した後、それぞれ室温で30分間/40分間/20分間/20分間反応させた。停止液を加えて酵素反応を停止させた後、Caliper EZ ReaderIIで転化率を読み取った。
転化率%_サンプル:サンプルの転化率;転化率%_最小:陰性対照ウェルの平均値、酵素活性なしのウェルの転化率の示度を表す。転化率%_最大:陽性対照ウェルの平均比、化合物による阻害なしのウェルの転化率の示度を表す。
濃度のlog値をX軸とし、阻害百分率をY軸とし、解析ソフトGraphPad Prism5で量・反応曲線(dose response curve)をフィッティングすることにより、各化合物の酵素活性阻害の強さを示すIC50値を得た。
2.化合物のROS1-G2032Rに対する酵素阻害活性(IC50)の検出
Lance Ultra(Perkin Elmer、CR97-100)の原理を利用してROS1-G2032Rキナーゼ活性検出プラットフォームを構築し、化合物活性の測定を行った。
化合物粉末を100%DMSO(Sigma、Cat.D8418)に溶解し、10mMのストック溶液を調製した。化合物の初期実験濃度を1000nMとし、3倍段階希釈して、11つの濃度を設定し、複数のウェルにて検出した。TPX-0005(WuXi AppTec.)を陽性対照化合物とした。段階希釈された化合物と終濃度が0.016nMのROS1-G2032Rキナーゼ(Abcam、Cat.ab206012)、50nMのLANCE Ultra ULight-polyGT peptide(PerkinElmer、Cat.TRF0100-M)及び2.6μMのATP(Sigma、Cat.A7699)をOptiplate-384Fウェルプレート(PerkinElmer、Cat.6007299)に均一に混合した後、室温で60分間インキュベートした。40mMのEDTAを5μl加えて5分間反応を停止させ、その後、終濃度が2nMのユーロピウム-抗ホスホチロシン(Europium-anti-phosphotyrosine)(PT66)(PerkinElmer、Cat.AD0069)を加え、十分に混合して室温で60分間反応させた。EnVisionTM(PerkinElmer、#2014)によりLANCEシグナルを取得した(励起光320nm、発射光665nm)。化合物のIC50値は、XLFIT5(IDBS公司)のソフトによって計算された。
3.化合物のTRKA及びALK-L1196Mに対する酵素阻害活性(IC50)の検出
CisbioのHTRF(Cisbio、Cat.08-529)の原理に基づくTRKA及びALK-L1196Mキナーゼ活性検出プラットフォームにより化合物活性の測定を行った。化合物粉末を100%DMSO(Sigma、Cat.D8418)に溶解し、10mMのストック溶液を調製した。それぞれ化合物の初期実験濃度を100nM及び10000nMとし、3倍段階希釈して、11つの濃度を設定し、複数のウェルにて検出した。RXDX-101(WuXi AppTec.)及びCrizotinib(WuXi AppTec.)を陽性対照化合物とした。
段階希釈された化合物、終濃度が0.5nMのTRKA(Carna、Cat.08-186)/ALK-L1196Mキナーゼ(Carna、Cat.08-529)、0.3μM/1μMのTK基質-ビオチン及び90μM/30μMのATP(Sigma、Cat.A7699)をOptiplate-384Fウェルプレート(PerkinElmer、Cat.6007299)に均一に混合した後、室温で90分間/120分間インキュベートした。終濃度が0.67nMのEu標識TK-抗体及び50nMのストレプトアビジン標識XL-665を加え、十分に混合して室温で60分間反応させた。Envision(PerkinElmer、#2014)において蛍光値(励起光320nm、発射光665nm)を読み取った。化合物のIC50値は、XLFIT5(IDBS公司)ソフトによって計算された。
4.化合物のTRK融合及びその変異安定細胞株の増殖に対する阻害作用(IC50)の検出
6つの細胞株の成長に対する化合物の阻害作用を検出するために、それぞれLOXO-101(Selleckchem、Cat.S7960)及びTPX-0005(Selleckchem、Cat.S8583)を対照化合物として使用した。この実験に使用された細胞株は、Ba/F3 LMNA-NTRK1-WT、Ba/F3 LMNA-NTRK1-G595R、Ba/F3 ETV6-NTRK2-WT、Ba/F3 ETV6-NTRK2-G639R、Ba/F3ETV6-NTRK2-G639R及びBa/F3 ETV6-NTRK3-G623Rであった。6つの細胞株における最高試験薬物濃度は、それぞれ1μM、1μM、10μM、100μM、1μM及び10μMの9つの濃度であり、3.16倍希釈した。
実験方法は、以下のように簡単に説明した。対数増殖期の細胞を収穫し、細胞生存率が90%以上であることを確保した。3000細胞/ウェルで96-ウェル細胞培養プレート(Corning(登録商標),Cat#3603)に敷き、37℃、5%CO2、湿度95%で細胞を終夜培養した。細胞が接種された96ウェルプレートの各ウェルに薬物溶液を加え、薬物濃度につき3ウェルずつ設定し、引き続き72時間培養した後、CellTiter-Glo(登録商標)キット(Promega、Cat#G7572)で検出を行った。先ず、CTG試薬を解凍して細胞プレートを室温に平衡化した。各ウェルに同体積のCTG溶液を加えて、シェーカーで5分間振盪して細胞を溶解させた。ルミネセンスシグナルを安定させるように、室温で細胞プレートを20分間放置した。SpectraMaxマルチラベルマイクロプレート検出器(MD、2104-0010A)によりルミネセンス値を読み取った。その後、GraphPad Prism5.0ソフトを使用してデータを分析し、非線形S曲線回帰によりデータをフィッティングして投与量-効果曲線を得ることにより、IC50値を計算した。
細胞の生存率(%)=(Lum被験薬物-Lum培養液対照)/(Lum細胞対照-Lum培養液対照)×100%
*マークは、同じバッチの実験結果を表す;#マークは、同じバッチの実験結果を表す。
5.細胞レベルでの化合物の相乗効果
インキュベーター(37℃、5%CO2)においてH1975細胞(L858R及びT790M二重変異)を1640培地+10%FBS(ウシ胎児血清)及び1%P/S(ペニシリン/ストレプトマイシン)で培養した。化合物の検出において、H1975細胞を各ウェルあたり3000個/195μLの濃度で96ウェルプレート(Corning)に敷き、化合物を10mMから、3倍段階希釈して11つの濃度を設定し、各濃度につき4μLを96μLの1640培地に加えて25×化合物に希釈し、その後、5μLを195μLの細胞培養液(DMSO終濃度は0.1%、v/v)に加え、72時間処理後、35μLのCellTiter-Blue(登録商標)(Promegaから購入)を加え、説明書の操作フローに従ってFlex Station3(Molecular Device)で蛍光シグナルを測定し、GraphPad Prism5.0により化合物の細胞増殖に対する阻害を示すIC50値を計算した。ChouとTalalayによる組み合わせ指数により薬物併用効果を計算し、組み合わせ指数(CoI)値として、0.9≦CI≦1.1が相加作用を示し、0.8≦CI<0.9が低度の相乗作用を示し、0.6≦CI<0.8が中等度の相乗作用を示し、0.4≦CI<0.6が高度の相乗作用を示し、0.2≦CI<0.4が強い相乗作用を示す。
実験結果から、実施例の化合物をAZD9291と併用する場合、EGFR二重変異細胞H1975(L858RとT790M二重変異)に対して中等度から高度の相乗作用(実施例1、CI=0.53-0.67)を奏し、実施例の化合物とEGFR阻害剤との併用によりEGFR耐性を克服できることが示された。
6.薬物動態学実験
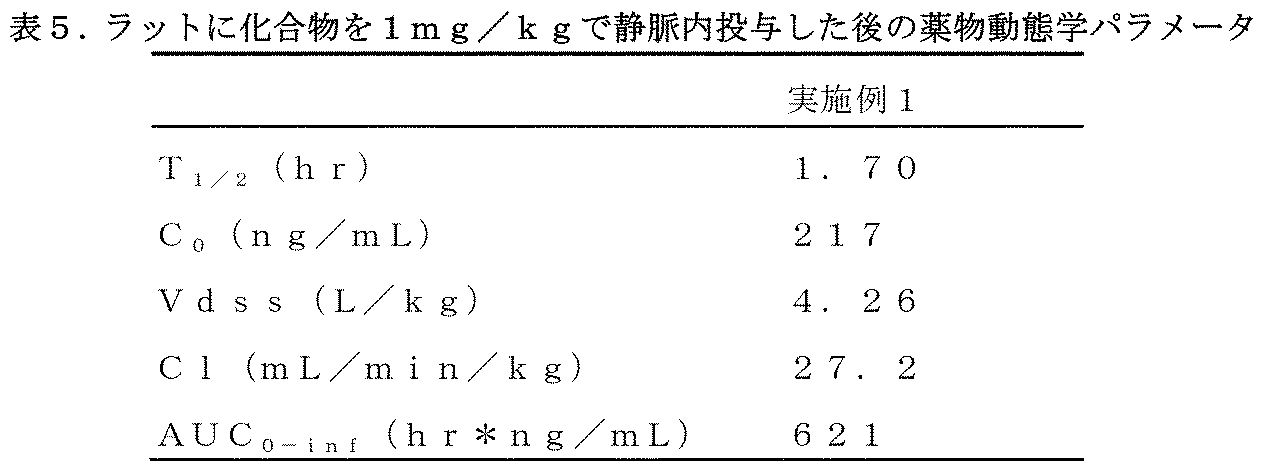
雄SDラットを3匹ずつ群分けてそれぞれ実施例1の化合物(5mg/kg)及びTPX-0005(5mg/kg)を胃内に単回経口投与し、実施例1の化合物(1mg/kg)を静脈内注射した。実験前に動物を終夜絶食させ、投与前10時間から投与後4時間までの間は絶食とした。経口投与群は、投与から0.25、0.5、1、2、4、8及び24時間後に採血し、静脈内注射群は、注射から0.083、0.25、0.5、1、2、4、8及び24時間後に採血した。小動物用麻酔器によりイソフルランで麻酔した後、眼底静脈叢から全血0.3mLを採取し、ヘパリン抗凝固管に入れ、サンプルを4℃、4000rpmで5min遠心し、血漿を遠心チューブに移して、分析まで-80℃で保存した。血漿サンプルをタンパク質沈殿法で抽出し、LC/MS/MSで抽出液を分析した。薬物動態学の実験結果を表2、表3に示す。
以上の結果から、本発明の実施例1の化合物は、従来の化合物TPX-0005よりも経口薬物動態学的性能が優れており、且つ、実施例1の経口バイオアベイラビリティが100%に達したことが分かる。同様の方法で本発明の他の実施例の化合物を実験したところ、得られた薬物動態学的性能もTPX-0005より優れていた。
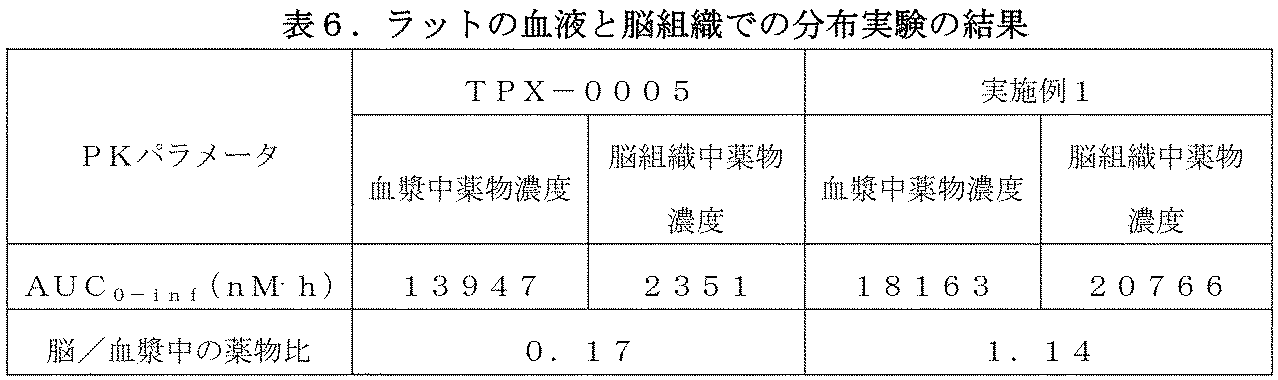
7.血液脳分布実験
雄SDラットを12匹ずつ群分けてそれぞれ実施例の化合物(10mg/kg)を胃内に単回経口投与した。実験前に動物を終夜絶食させ、投与前10時間から投与後4時間までの間は絶食とした。各ラット群は、投与から0.5、1、4及び12時間後に致死させて血と脳組織を採取し、サンプルを処理した後、4℃、4000rpmで5min遠心し、血漿を遠心チューブに移して、分析まで-80℃で保存した。血漿サンプルをタンパク質沈殿法で抽出し、LC/MS/MSで抽出液を分析した。
本発明を説明するために、本発明の好ましい実施形態を開示したが、当業者は、特許請求の範囲により限定される本発明の構想及び範囲から逸脱することなく、本発明に対して様々な修正、追加及び置換を行うことができることを理解すべきである。