JP6953494B2 - ヘプタデカトリエン側鎖のヒドロキノンの合成方法 - Google Patents
ヘプタデカトリエン側鎖のヒドロキノンの合成方法 Download PDFInfo
- Publication number
- JP6953494B2 JP6953494B2 JP2019193894A JP2019193894A JP6953494B2 JP 6953494 B2 JP6953494 B2 JP 6953494B2 JP 2019193894 A JP2019193894 A JP 2019193894A JP 2019193894 A JP2019193894 A JP 2019193894A JP 6953494 B2 JP6953494 B2 JP 6953494B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound represented
- reaction
- compound
- preparing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title description 18
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 title description 16
- DQKNHNPHGVWPJF-UHFFFAOYSA-N heptadeca-1,3,5-triene Chemical group CCCCCCCCCCCC=CC=CC=C DQKNHNPHGVWPJF-UHFFFAOYSA-N 0.000 title description 3
- 230000002194 synthesizing effect Effects 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims description 96
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 19
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 18
- 238000002360 preparation method Methods 0.000 claims description 18
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 14
- 238000006243 chemical reaction Methods 0.000 claims description 14
- 238000003381 deacetylation reaction Methods 0.000 claims description 13
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 12
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 claims description 12
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 claims description 10
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 10
- 230000006196 deacetylation Effects 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 10
- 150000005671 trienes Chemical group 0.000 claims description 10
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 claims description 8
- 238000007239 Wittig reaction Methods 0.000 claims description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 6
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 6
- 238000006192 iodination reaction Methods 0.000 claims description 6
- ABDKAPXRBAPSQN-UHFFFAOYSA-N veratrole Chemical compound COC1=CC=CC=C1OC ABDKAPXRBAPSQN-UHFFFAOYSA-N 0.000 claims description 6
- GTQHJCOHNAFHRE-UHFFFAOYSA-N 1,10-dibromodecane Chemical compound BrCCCCCCCCCCBr GTQHJCOHNAFHRE-UHFFFAOYSA-N 0.000 claims description 5
- 238000007254 oxidation reaction Methods 0.000 claims description 4
- 235000009518 sodium iodide Nutrition 0.000 claims description 4
- 230000029936 alkylation Effects 0.000 claims description 3
- 238000005804 alkylation reaction Methods 0.000 claims description 3
- 230000017858 demethylation Effects 0.000 claims description 3
- 238000010520 demethylation reaction Methods 0.000 claims description 3
- 230000001590 oxidative effect Effects 0.000 claims description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 3
- -1 sodium hydrocarbon Chemical class 0.000 claims description 3
- 239000004215 Carbon black (E152) Substances 0.000 claims description 2
- 230000001335 demethylating effect Effects 0.000 claims description 2
- 229930195733 hydrocarbon Natural products 0.000 claims description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N o-dimethylbenzene Natural products CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 2
- 239000011734 sodium Substances 0.000 claims description 2
- 229910052708 sodium Inorganic materials 0.000 claims description 2
- 239000000047 product Substances 0.000 description 14
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- 238000005481 NMR spectroscopy Methods 0.000 description 12
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 11
- 239000000203 mixture Substances 0.000 description 11
- 239000000243 solution Substances 0.000 description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 9
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Substances C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 8
- 239000000741 silica gel Substances 0.000 description 8
- 229910002027 silica gel Inorganic materials 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- 150000005206 1,2-dihydroxybenzenes Chemical class 0.000 description 4
- OHBQPCCCRFSCAX-UHFFFAOYSA-N 1,4-Dimethoxybenzene Chemical compound COC1=CC=C(OC)C=C1 OHBQPCCCRFSCAX-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N Butyraldehyde Chemical compound CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 4
- 102000003964 Histone deacetylase Human genes 0.000 description 4
- 108090000353 Histone deacetylase Proteins 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 229910010082 LiAlH Inorganic materials 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000012467 final product Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 239000012265 solid product Substances 0.000 description 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000010189 synthetic method Methods 0.000 description 3
- 150000005208 1,4-dihydroxybenzenes Chemical class 0.000 description 2
- RQFUZUMFPRMVDX-UHFFFAOYSA-N 3-Bromo-1-propanol Chemical compound OCCCBr RQFUZUMFPRMVDX-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical group CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 101710183280 Topoisomerase Proteins 0.000 description 2
- 244000044283 Toxicodendron succedaneum Species 0.000 description 2
- 229940041181 antineoplastic drug Drugs 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- ZAXKASRBECJCPQ-YTXTXJHMSA-N (1e,3e)-hepta-1,3-dien-1-ol Chemical compound CCC\C=C\C=C\O ZAXKASRBECJCPQ-YTXTXJHMSA-N 0.000 description 1
- CJZFOZYXWDHMQD-MQQKCMAXSA-N (2E,4E)-7-iodohepta-2,4-diene Chemical compound C\C=C\C=C\CCI CJZFOZYXWDHMQD-MQQKCMAXSA-N 0.000 description 1
- OGQVROWWFUXRST-FNORWQNLSA-N (3e)-hepta-1,3-diene Chemical compound CCC\C=C\C=C OGQVROWWFUXRST-FNORWQNLSA-N 0.000 description 1
- AHYDCPUKEAXCKZ-UHFFFAOYSA-M 3-hydroxypropyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCCO)C1=CC=CC=C1 AHYDCPUKEAXCKZ-UHFFFAOYSA-M 0.000 description 1
- QDNKORPVEZWCAT-UHFFFAOYSA-N CC(Oc(cc1)cc(CCCCCCCCCCBr)c1OC(C)=O)=O Chemical compound CC(Oc(cc1)cc(CCCCCCCCCCBr)c1OC(C)=O)=O QDNKORPVEZWCAT-UHFFFAOYSA-N 0.000 description 1
- UEZRYNCKFMOHTF-UHFFFAOYSA-N CC(Oc(cc1CCCCCCCCCC=O)ccc1OC(C)=O)=O Chemical compound CC(Oc(cc1CCCCCCCCCC=O)ccc1OC(C)=O)=O UEZRYNCKFMOHTF-UHFFFAOYSA-N 0.000 description 1
- PNCOGQJUSXDRGF-UHFFFAOYSA-N COC(=O)C1=CC=CC=C1.C1=CC=C(C=C1)P(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound COC(=O)C1=CC=CC=C1.C1=CC=C(C=C1)P(C1=CC=CC=C1)C1=CC=CC=C1 PNCOGQJUSXDRGF-UHFFFAOYSA-N 0.000 description 1
- 101100537937 Caenorhabditis elegans arc-1 gene Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- PCBOWMZAEDDKNH-HOTGVXAUSA-N [4-(trifluoromethoxy)phenyl]methyl (3as,6as)-2-(3-fluoro-4-sulfamoylbenzoyl)-1,3,3a,4,6,6a-hexahydropyrrolo[3,4-c]pyrrole-5-carboxylate Chemical compound C1=C(F)C(S(=O)(=O)N)=CC=C1C(=O)N1C[C@H]2CN(C(=O)OCC=3C=CC(OC(F)(F)F)=CC=3)C[C@@H]2C1 PCBOWMZAEDDKNH-HOTGVXAUSA-N 0.000 description 1
- 230000000397 acetylating effect Effects 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000001336 alkenes Chemical group 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 1
- TUZARBGEVAGZNZ-UHFFFAOYSA-N benzene-1,4-diol;1,4-dimethoxybenzene Chemical compound OC1=CC=C(O)C=C1.COC1=CC=C(OC)C=C1 TUZARBGEVAGZNZ-UHFFFAOYSA-N 0.000 description 1
- 230000000850 deacetylating effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 230000026045 iodination Effects 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- CMSYDJVRTHCWFP-UHFFFAOYSA-N triphenylphosphane;hydrobromide Chemical compound Br.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 CMSYDJVRTHCWFP-UHFFFAOYSA-N 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
Images


Landscapes
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
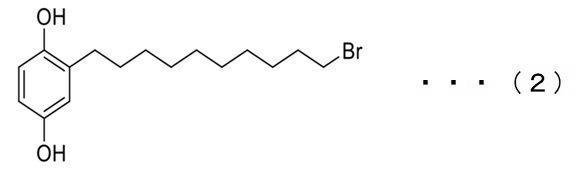
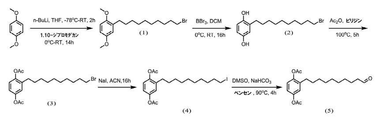
1,4−ジメトキシベンゼン(10g、72.4mmol)およびn−ブチルリチウム(5.8g、90.5mmol)を丸底フラスコに投入した。次に、溶媒のテトラヒドロフラン(200mL)を入れた後、丸底フラスコを低温反応器に入れ、−78℃で1時間攪拌した。その後、室温に移して1時間攪拌した。これとは別に追加で一口フラスコを準備し、1,10−ジブロモデカンを秤量した(65.17g、217.2mmol)。次に、この1,10−ジブロモデカンをテトラヒドロフラン(100mL)に溶解させて、この溶液を元の溶液にゆっくりと滴下し、室温で14時間攪拌した。その後、シリカゲルカラムで精製することにより、17gの2−(10'−ブロモインドリル)−1,4−ジメトキシベンゼン(式(1)に示す化合物)を生成物として回収した。収率は65%であった。
Claims (7)
- ジメトキシベンゼンのアルキル化および脱メチル化により式(3)に示す化合物を調製するステップ(a)と、
- 前記ヘプタジエン−1−トリフェニルホスホニウムヨージドは、式(10)に示す化合物である、請求項1に記載の製造方法。
- 前記ヘプタデカトリエン側鎖のヒドロキノンを調製するステップ(c)は、前記式(5)に示す化合物と、前記式(10)に示す化合物とのウィッティヒ反応によって式(11)に示す化合物を調製する請求項2記載の製造方法。
- 前記ステップ(c)の脱アセチル化反応は、前記式(11)に示す化合物を、室温でメタノール中のナトリウムメトキシドと反応させることによって式(12)に示す化合物を調製することにより行われる請求項3に記載の製造方法。
- 前記ステップ(a)は、n−ブチルリチウムにより活性化されたジメトキシベンゼンが1,10−ジブロモデカンと反応することによって式(1)に示す化合物を調製するステップと、
- 前記ステップ(b)のヨウ素化反応は、式(3)に示す化合物に対してヨウ化ナトリウムを加えて式(4)に示す化合物を調製することにより行われる請求項5に記載の製造方法。
- 前記ステップ(b)の酸化反応は、前記式(4)に示す化合物をジメチルスルホキシドおよびベンゼンと混合し、炭化水素ナトリウムを添加することによって式(5)に示す化合物を調製することにより行われる請求項6に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019193894A JP6953494B2 (ja) | 2019-10-25 | 2019-10-25 | ヘプタデカトリエン側鎖のヒドロキノンの合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019193894A JP6953494B2 (ja) | 2019-10-25 | 2019-10-25 | ヘプタデカトリエン側鎖のヒドロキノンの合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021066695A JP2021066695A (ja) | 2021-04-30 |
| JP6953494B2 true JP6953494B2 (ja) | 2021-10-27 |
Family
ID=75636809
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019193894A Active JP6953494B2 (ja) | 2019-10-25 | 2019-10-25 | ヘプタデカトリエン側鎖のヒドロキノンの合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6953494B2 (ja) |
-
2019
- 2019-10-25 JP JP2019193894A patent/JP6953494B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2021066695A (ja) | 2021-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2015531404A (ja) | ビタミンk2のmk−7型の調整方法 | |
| CN114920769A (zh) | 一种硅甲基菲衍生物及其制备方法 | |
| CN108821975A (zh) | 一种含环外双键的氢化菲类衍生物及其制备方法 | |
| TW200821281A (en) | Process for production of benzaldehyde compound | |
| JP6953494B2 (ja) | ヘプタデカトリエン側鎖のヒドロキノンの合成方法 | |
| CN101323567B (zh) | 制备肉桂酸酯及其衍生物的方法 | |
| US11078144B2 (en) | Process for synthesizing of hydroquinone derivatives with heptadecatrienyl side chain | |
| CN102180773B (zh) | 一种白藜芦醇的制备方法 | |
| EP3805190B1 (en) | Process for synthesizing of hydroquinone derivatives with heptadecatrienyl side chain | |
| WO2018205299A1 (zh) | 4,5-二取代-1-氢-吡咯(2,3-f)喹啉-2,7,9-三羧酸酯化合物及应用 | |
| CN109651437B (zh) | 一种手性氮磷配体及其制备方法,及一种拆分消旋薄荷醇的方法 | |
| CN100509729C (zh) | 一种合成白藜芦醇的方法 | |
| CN103288605B (zh) | 考布他丁的合成方法 | |
| JPS60185750A (ja) | ベンジルアルコ−ル誘導体の新規製法 | |
| KR101477058B1 (ko) | 피리딘 유도체의 제조방법 | |
| KR100669167B1 (ko) | 코엔자임 Qn의 제조방법 및 그의 중간체 | |
| CN100588643C (zh) | 一种含有二取代金刚烷基的维甲酸类化合物制备方法 | |
| CN116813525B (zh) | 一种多乙酰基取代的氧化吲哚类化合物的合成方法 | |
| JP7553927B2 (ja) | ビリベルジン又はその誘導体の製造方法 | |
| CN109336748B (zh) | 抗nash药物中间体双醛基厚朴酚的制备方法 | |
| KR20250158663A (ko) | 시코닌 및 그 유도체의 합성방법 | |
| KR102285494B1 (ko) | 할로겐이 치환된 다이메틸칼콘 유도체 및 이의 제조방법 | |
| CN118420440A (zh) | 一种反式白藜芦醇的制备方法 | |
| CN109232249B (zh) | 一种多取代苯甲酸酯的制备方法 | |
| CN117362160A (zh) | 一种二苯乙烯衍生物z构型异构体的合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20191025 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20201201 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210219 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210317 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20210914 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20210929 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6953494 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |