JP6777542B2 - 置換尿素化合物の合成のためのプロセス - Google Patents
置換尿素化合物の合成のためのプロセス Download PDFInfo
- Publication number
- JP6777542B2 JP6777542B2 JP2016548135A JP2016548135A JP6777542B2 JP 6777542 B2 JP6777542 B2 JP 6777542B2 JP 2016548135 A JP2016548135 A JP 2016548135A JP 2016548135 A JP2016548135 A JP 2016548135A JP 6777542 B2 JP6777542 B2 JP 6777542B2
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- heteroaryl
- heterocyclyl
- aryl
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000000034 method Methods 0.000 title claims description 49
- 230000008569 process Effects 0.000 title claims description 35
- 230000015572 biosynthetic process Effects 0.000 title claims description 26
- 238000003786 synthesis reaction Methods 0.000 title description 11
- 150000003672 ureas Chemical class 0.000 title description 4
- -1 carbamoyl halide Chemical class 0.000 claims description 85
- 150000001875 compounds Chemical class 0.000 claims description 44
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 42
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 36
- 239000002904 solvent Substances 0.000 claims description 33
- 239000004202 carbamide Substances 0.000 claims description 22
- 150000003839 salts Chemical class 0.000 claims description 17
- LEQAOMBKQFMDFZ-UHFFFAOYSA-N glyoxal Chemical compound O=CC=O LEQAOMBKQFMDFZ-UHFFFAOYSA-N 0.000 claims description 10
- 238000004519 manufacturing process Methods 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 7
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 6
- 239000002243 precursor Substances 0.000 claims description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 5
- 229940015043 glyoxal Drugs 0.000 claims description 5
- 125000002883 imidazolyl group Chemical group 0.000 claims description 5
- 150000007522 mineralic acids Chemical class 0.000 claims description 5
- 150000001299 aldehydes Chemical class 0.000 claims description 4
- 239000000908 ammonium hydroxide Substances 0.000 claims description 4
- 229910052801 chlorine Inorganic materials 0.000 claims description 4
- 229910052731 fluorine Inorganic materials 0.000 claims description 4
- 229910052740 iodine Inorganic materials 0.000 claims description 4
- 125000006501 nitrophenyl group Chemical group 0.000 claims description 4
- 230000003647 oxidation Effects 0.000 claims description 4
- 238000007254 oxidation reaction Methods 0.000 claims description 4
- 125000005843 halogen group Chemical group 0.000 claims description 2
- 239000007800 oxidant agent Substances 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 description 136
- 125000000623 heterocyclic group Chemical group 0.000 description 102
- 125000003118 aryl group Chemical group 0.000 description 83
- 125000001072 heteroaryl group Chemical group 0.000 description 82
- 125000003545 alkoxy group Chemical group 0.000 description 66
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 47
- 239000000203 mixture Substances 0.000 description 41
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 40
- 239000007787 solid Substances 0.000 description 36
- 229910052736 halogen Inorganic materials 0.000 description 35
- 150000002367 halogens Chemical class 0.000 description 35
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 33
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 31
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 30
- 239000000243 solution Substances 0.000 description 28
- 238000006243 chemical reaction Methods 0.000 description 26
- 239000000543 intermediate Chemical class 0.000 description 26
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical class C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 25
- 239000000725 suspension Substances 0.000 description 24
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 23
- 239000000047 product Substances 0.000 description 23
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 235000013877 carbamide Nutrition 0.000 description 20
- 125000005842 heteroatom Chemical group 0.000 description 20
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 20
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 18
- 125000005553 heteroaryloxy group Chemical group 0.000 description 17
- 125000005844 heterocyclyloxy group Chemical group 0.000 description 17
- 125000004430 oxygen atom Chemical group O* 0.000 description 17
- 125000004432 carbon atom Chemical group C* 0.000 description 16
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 16
- 125000004104 aryloxy group Chemical group 0.000 description 15
- 238000005755 formation reaction Methods 0.000 description 15
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 15
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 14
- 239000002002 slurry Substances 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- 125000000753 cycloalkyl group Chemical group 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- 238000013459 approach Methods 0.000 description 12
- 150000002430 hydrocarbons Chemical group 0.000 description 12
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 12
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 12
- 239000011541 reaction mixture Substances 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 11
- 125000002619 bicyclic group Chemical group 0.000 description 11
- 229910052739 hydrogen Inorganic materials 0.000 description 11
- 125000001424 substituent group Chemical group 0.000 description 11
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 10
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 9
- 125000002252 acyl group Chemical group 0.000 description 9
- 125000002950 monocyclic group Chemical group 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 8
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 8
- 238000004128 high performance liquid chromatography Methods 0.000 description 8
- 239000001257 hydrogen Substances 0.000 description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 229930195734 saturated hydrocarbon Natural products 0.000 description 8
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 description 7
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 7
- 125000004663 dialkyl amino group Chemical group 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- DTMTYKUCZFYAEU-UHFFFAOYSA-N n-methylcyclopentanamine;hydrochloride Chemical compound Cl.CNC1CCCC1 DTMTYKUCZFYAEU-UHFFFAOYSA-N 0.000 description 7
- 239000007858 starting material Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- 238000002844 melting Methods 0.000 description 6
- 230000008018 melting Effects 0.000 description 6
- SUXDLDZKUQQCNZ-UHFFFAOYSA-N n-cyclopentyl-n-methylcarbamoyl chloride Chemical compound ClC(=O)N(C)C1CCCC1 SUXDLDZKUQQCNZ-UHFFFAOYSA-N 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 5
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 150000001335 aliphatic alkanes Chemical class 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- JMFVWNKPLURQMI-UHFFFAOYSA-N cyclopentyl carbamate Chemical compound NC(=O)OC1CCCC1 JMFVWNKPLURQMI-UHFFFAOYSA-N 0.000 description 5
- 238000004821 distillation Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000012280 lithium aluminium hydride Substances 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 description 4
- AMSITKGZVUXDQM-UHFFFAOYSA-N 5-(3-nitrophenyl)-1h-imidazole Chemical compound [O-][N+](=O)C1=CC=CC(C=2NC=NC=2)=C1 AMSITKGZVUXDQM-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 4
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 4
- 125000000304 alkynyl group Chemical group 0.000 description 4
- 235000011114 ammonium hydroxide Nutrition 0.000 description 4
- 230000021235 carbamoylation Effects 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 235000019439 ethyl acetate Nutrition 0.000 description 4
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 4
- 239000012948 isocyanate Substances 0.000 description 4
- 150000002513 isocyanates Chemical class 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 4
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 4
- 239000012452 mother liquor Substances 0.000 description 4
- GKKCIDNWFBPDBW-UHFFFAOYSA-M potassium cyanate Chemical compound [K]OC#N GKKCIDNWFBPDBW-UHFFFAOYSA-M 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 238000006722 reduction reaction Methods 0.000 description 4
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 3
- 125000004916 (C1-C6) alkylcarbonyl group Chemical group 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- 102100029111 Fatty-acid amide hydrolase 1 Human genes 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 125000003282 alkyl amino group Chemical group 0.000 description 3
- 125000005129 aryl carbonyl group Chemical group 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 239000003610 charcoal Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 108010046094 fatty-acid amide hydrolase Proteins 0.000 description 3
- 150000002460 imidazoles Chemical class 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 150000003335 secondary amines Chemical class 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 239000011550 stock solution Substances 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- 125000006625 (C3-C8) cycloalkyloxy group Chemical group 0.000 description 2
- ARKIFHPFTHVKDT-UHFFFAOYSA-N 1-(3-nitrophenyl)ethanone Chemical compound CC(=O)C1=CC=CC([N+]([O-])=O)=C1 ARKIFHPFTHVKDT-UHFFFAOYSA-N 0.000 description 2
- CXOWYJMDMMMMJO-UHFFFAOYSA-N 2,2-dimethylpentane Chemical compound CCCC(C)(C)C CXOWYJMDMMMMJO-UHFFFAOYSA-N 0.000 description 2
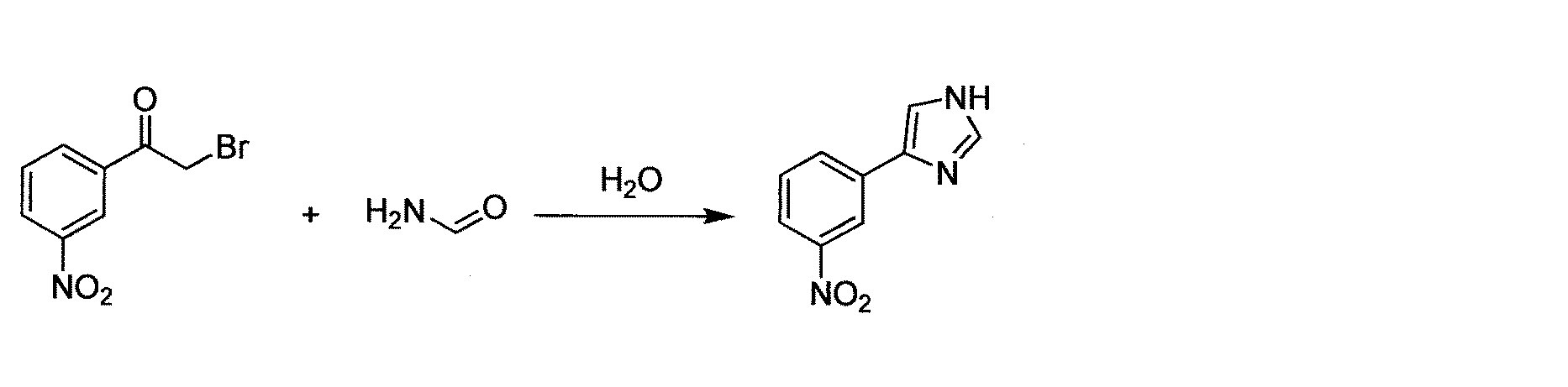
- GZHPNIQBPGUSSX-UHFFFAOYSA-N 2-bromo-1-(3-nitrophenyl)ethanone Chemical compound [O-][N+](=O)C1=CC=CC(C(=O)CBr)=C1 GZHPNIQBPGUSSX-UHFFFAOYSA-N 0.000 description 2
- MEAVLRNBGURMFI-UHFFFAOYSA-N 2-imidazol-1-ylacetamide Chemical group NC(=O)CN1C=CN=C1 MEAVLRNBGURMFI-UHFFFAOYSA-N 0.000 description 2
- WEGYGNROSJDEIW-UHFFFAOYSA-N 3-Acetylpyridine Chemical compound CC(=O)C1=CC=CN=C1 WEGYGNROSJDEIW-UHFFFAOYSA-N 0.000 description 2
- AORMDLNPRGXHHL-UHFFFAOYSA-N 3-ethylpentane Chemical compound CCC(CC)CC AORMDLNPRGXHHL-UHFFFAOYSA-N 0.000 description 2
- VLJXXKKOSFGPHI-UHFFFAOYSA-N 3-methylhexane Chemical compound CCCC(C)CC VLJXXKKOSFGPHI-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 101100203596 Caenorhabditis elegans sol-1 gene Proteins 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 230000021523 carboxylation Effects 0.000 description 2
- 238000006473 carboxylation reaction Methods 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical compound NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 2
- 239000008367 deionised water Substances 0.000 description 2
- 229910021641 deionized water Inorganic materials 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- CBIKNPNOQWAFBJ-UHFFFAOYSA-N ethyl n-cyclopentylcarbamate Chemical compound CCOC(=O)NC1CCCC1 CBIKNPNOQWAFBJ-UHFFFAOYSA-N 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 229940011051 isopropyl acetate Drugs 0.000 description 2
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 2
- KKTBUCVHSCATGB-UHFFFAOYSA-N n-methylcyclopentanamine Chemical compound CNC1CCCC1 KKTBUCVHSCATGB-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 125000003107 substituted aryl group Chemical group 0.000 description 2
- ZISSAWUMDACLOM-UHFFFAOYSA-N triptane Chemical compound CC(C)C(C)(C)C ZISSAWUMDACLOM-UHFFFAOYSA-N 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 description 1
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 description 1
- 125000006728 (C1-C6) alkynyl group Chemical group 0.000 description 1
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 1
- KPKNTUUIEVXMOH-UHFFFAOYSA-N 1,4-dioxa-8-azaspiro[4.5]decane Chemical compound O1CCOC11CCNCC1 KPKNTUUIEVXMOH-UHFFFAOYSA-N 0.000 description 1
- 125000001088 1-naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- MWVMYAWMFTVYED-UHFFFAOYSA-N 2,3,4,5-tetrahydro-1h-3-benzazepine Chemical compound C1CNCCC2=CC=CC=C21 MWVMYAWMFTVYED-UHFFFAOYSA-N 0.000 description 1
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 1
- 125000001216 2-naphthoyl group Chemical group C1=C(C=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- YFOKBFRTGLSZLU-UHFFFAOYSA-N 3-(1h-imidazol-5-yl)pyridine Chemical compound N1C=NC=C1C1=CC=CN=C1 YFOKBFRTGLSZLU-UHFFFAOYSA-N 0.000 description 1
- MCSXGCZMEPXKIW-UHFFFAOYSA-N 3-hydroxy-4-[(4-methyl-2-nitrophenyl)diazenyl]-N-(3-nitrophenyl)naphthalene-2-carboxamide Chemical compound Cc1ccc(N=Nc2c(O)c(cc3ccccc23)C(=O)Nc2cccc(c2)[N+]([O-])=O)c(c1)[N+]([O-])=O MCSXGCZMEPXKIW-UHFFFAOYSA-N 0.000 description 1
- GAMYYCRTACQSBR-UHFFFAOYSA-N 4-azabenzimidazole Chemical compound C1=CC=C2NC=NC2=N1 GAMYYCRTACQSBR-UHFFFAOYSA-N 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical class OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- 125000005915 C6-C14 aryl group Chemical group 0.000 description 1
- GNEQAVHVVYOSGJ-UHFFFAOYSA-N CCCCC(C)C.CCCCC(C)C Chemical compound CCCCC(C)C.CCCCC(C)C GNEQAVHVVYOSGJ-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical group NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 0 Cc1c(*)[n]c(*)n1 Chemical compound Cc1c(*)[n]c(*)n1 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 240000001812 Hyssopus officinalis Species 0.000 description 1
- 235000010650 Hyssopus officinalis Nutrition 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 150000001200 N-acyl ethanolamides Chemical class 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241000907661 Pieris rapae Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- PNNCWTXUWKENPE-UHFFFAOYSA-N [N].NC(N)=O Chemical compound [N].NC(N)=O PNNCWTXUWKENPE-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 150000008062 acetophenones Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 239000012296 anti-solvent Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000005959 diazepanyl group Chemical group 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 239000002621 endocannabinoid Substances 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000003784 fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000020169 heat generation Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004871 hexylcarbonyl group Chemical group C(CCCCC)C(=O)* 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000005935 hexyloxycarbonyl group Chemical group 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 125000006328 iso-butylcarbonyl group Chemical group [H]C([H])([H])C([H])(C(*)=O)C([H])([H])[H] 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 125000004674 methylcarbonyl group Chemical group CC(=O)* 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- BZWYKPFEWYJQJZ-UHFFFAOYSA-N n-cyclohexyl-n-methylcarbamoyl chloride Chemical compound ClC(=O)N(C)C1CCCCC1 BZWYKPFEWYJQJZ-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 125000001148 pentyloxycarbonyl group Chemical group 0.000 description 1
- LIGACIXOYTUXAW-UHFFFAOYSA-N phenacyl bromide Chemical compound BrCC(=O)C1=CC=CC=C1 LIGACIXOYTUXAW-UHFFFAOYSA-N 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- BSCCSDNZEIHXOK-UHFFFAOYSA-N phenyl carbamate Chemical class NC(=O)OC1=CC=CC=C1 BSCCSDNZEIHXOK-UHFFFAOYSA-N 0.000 description 1
- PWXJULSLLONQHY-UHFFFAOYSA-N phenylcarbamic acid Chemical class OC(=O)NC1=CC=CC=C1 PWXJULSLLONQHY-UHFFFAOYSA-N 0.000 description 1
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004346 phenylpentyl group Chemical group C1(=CC=CC=C1)CCCCC* 0.000 description 1
- 125000004344 phenylpropyl group Chemical group 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 125000006308 propyl amino group Chemical group 0.000 description 1
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- ILVXOBCQQYKLDS-UHFFFAOYSA-N pyridine N-oxide Chemical compound [O-][N+]1=CC=CC=C1 ILVXOBCQQYKLDS-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical class OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Chemical group 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 238000009331 sowing Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 150000003413 spiro compounds Chemical class 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical group 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000005942 tetrahydropyridyl group Chemical group 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 125000005247 tetrazinyl group Chemical group N1=NN=NC(=C1)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical class CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- PRXNKYBFWAWBNZ-UHFFFAOYSA-N trimethylphenylammonium tribromide Chemical compound Br[Br-]Br.C[N+](C)(C)C1=CC=CC=C1 PRXNKYBFWAWBNZ-UHFFFAOYSA-N 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/44—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of carboxylic acids or esters thereof in presence of ammonia or amines, or by reduction of nitriles, carboxylic acid amides, imines or imino-ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/68—Preparation of compounds containing amino groups bound to a carbon skeleton from amines, by reactions not involving amino groups, e.g. reduction of unsaturated amines, aromatisation, or substitution of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/04—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups from amines with formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
- C07D233/61—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms not forming part of a nitro radical, attached to ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
ここで、式IIa'の中間体は、R5およびR6の誘導体R6-C(=O)CH2R5を酸化してグリオキサール中間体R6-C(=O)(C=O)R5を形成し、これを水酸化アンモニウムおよびアルデヒドR8CHOで処理し、式IIa'の中間体を設けることによって調製される、
式中、
式Aの化合物の調製の場合、R6は、NH2CONH-フェニル、またはニトロフェニル、アミノフェニル、もしくは尿素形成後にNH2CONH-フェニル基に変換可能なこの部分のアミノ保護アミノフェニル前駆体であり、R5はHであり、R1はメチルであり、かつR2はシクロペンチルである;そして、
式IIaの化合物の調製の場合、R1およびR2は、それぞれ独立して、H、C1-20アルキル、C1-6アルコキシ、アリール、ヘテロアリール、部分的または完全飽和のヘテロシクリル、C3-10シクロアルキル、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、C3-10シクロアルキルC1-6アルキル、R1a、ハロゲン、OH、OR1a、OCOR1a、SH、SR1a、SCOR1a、NH2、NHR1a、NHSO2NH2、NHSO2R1a、NR1aCOR1b、NHCOR1a、NR1aR1b、COR1a、CSR1a、CN、COOH、COOR1a、CONH2、CONHOH、CONHR1a、CONHOR1a、SO2R1a、SO3H、SO2NH2、CONR1aR1b、SO2NR1aR1bから選択され得る(式中、R1aおよびR1bは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、または、R1aおよびR1bは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)、
式中、R1またはR2が、C1-20アルキル、アルコキシ、アリール、ヘテロアリール、ヘテロシクリル、C3-10シクロアルキル、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、C3-10シクロアルキルC1-6アルキル、または1つもしくは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合により、R1c、ハロゲン、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、アリールC1-6アルコキシ、ヘテロアリールC1-6アルコキシ、ヘテロシクリルC1-6アルコキシ、C1-6アルキルアミノ、C1-6ジアルキルアミノ、C1-10アルキル、OH、OR1c、OCOR1c、SH、SR1c、SCOR1c、NH2、NO2、NHR1c、NHSO2NH2、NHSO2R1c、NR1cCOR1d、NHC(NH)NH2、NHCOR1c、NR1cR1d、COR1c、CSR1c、CN、COOH、COOR1c、CONH2、CONHOH、CONHR1c、CONHOR1c、C(NOH)NH2、CONR1cR1d、SO2R1c、SO3H、SO2NH2、SO2NR1cR1dから選択される1つまたは複数の基で置換されてもよい(式中、R1cおよびR1dは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR1cおよびR1dは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)、
式中、R1またはR2の置換基が、C1-10アルキル、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、アリールC1-6アルコキシ、ヘテロアリールC1-6アルコキシ、ヘテロシクリルC1-6アルコキシ、C1-6アルキルアミノ、C1-6ジアルキルアミノ、C1-6アルキル、C3-8シクロアルキル、または1つもしくは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合によりR1e、ハロゲン、C1-10アルキル、OH、OR1e、OCOR1e、SH、SR1e、SCOR1e、NH2、NO2、NHR1e、NHSO2NH2、NHSO2R1e、NR1eCOR1f、NHC(NH)NH2、NHCOR1e、NR1eR1f、COR1e、CSR1e、CN、COOH、COOR1e、CONH2、CONHOH、CONHR1e、CONHOR1e、C(NOH)NH2、CONR1eR1f、SO2R1e、SO3H、SO2NH2、SO2NR1eR1fから選択される1つまたは複数の基で置換されてもよい(式中、R1eおよびR1fは。独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR1eおよびR1fは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)、
但し、R1およびR2の両方ともHである場合は除く;
あるいは、
R1およびR2は、それらが結合されるNと一緒になってヘテロアリールまたはヘテロシクリル基を形成し得て、これらはそれぞれ、場合により、1つまたは複数の酸素原子またはアリール、ヘテロアリール、部分的または完全飽和のヘテロシクリル、C3-8シクロアルキル、C1-6アルキル、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、C3-8シクロアルキルC1-6アルキル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、R2a、ハロゲン、OH、OR2a、OCOR2a、SH、SR2a、SCOR2a、NH2、NO2、NHR2a、NHSO2NH2、NHSO2R2a、NR2aCOR2b、NHC(NH)NH2、NHCOR2a、NR2aR2b、COR2a、CSR2a、CN、COOH、COOR2a、CONH2、CONHOH、CONHR2a、CONHOR2a、C(NOH)NH2、CONR2aR2b、SO2R2a、SO3H、SO2NH2、SO2NR2aR2bから選択される1つまたは複数の基で置換されてもよい(式中、R2aおよびR2bは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR2aおよびR2bは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)、
ここで、R1およびR2が一緒になって形成されるヘテロアリールまたはヘテロシクリルの置換基が、アリール、ヘテロアリール、ヘテロシクリル、C3-8シクロアルキル、C1-6アルキル、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、C3-8シクロアルキルC1-6アルキル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、または1つまたは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合により、ハロゲン、ヒドロキシル、C1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、C3-8シクロアルキル、C1-4アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、C3-8シクロアルキルオキシ、アリールC1-4アルコキシ、ヘテロアリールC1-6アルコキシ、ヘテロシクリルC1-4アルコキシ、C3-8シクロアルキルC1-4アルコキシ、R2c、OR2c、OCOR2c、SH、SR2c、SCOR2c、NH2、NO2、NHR2c、NHSO2NH2、NHSO2R2c、NR2cCOR2d、NHC(NH)NH2、NHCOR2c、NR2cR2d、COR2c、CSR2c、CN、COOH、COOR2c、CONH2、CONHOH、CONHR2c、CONHOR2c、C(NOH)NH2、CONR2cR2d、SO2R2c、SO3H、SO2NH2、SO2NR2cR2dから選択される1つまたは複数の基で置換されてもよい(式中、R2cおよびR2dは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR2cおよびR2dは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)、
ここで、R1およびR2が一緒になって形成されるヘテロアリールまたはヘテロシクリルの置換基が、C1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、C3-8シクロアルキル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、C3-8シクロアルキルオキシ、アリールC1-4アルコキシ、ヘテロアリールC1-4アルコキシ、ヘテロシクリルC1-4アルコキシ、C3-8シクロアルキルC1-4アルコキシ、または1つもしくは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合により、C1-4アルコキシ、R2e、ハロゲン、OH、OR2e、OCOR2e、SH、SR2e、SCOR2e、NH2、NO2、NHR2e、NHSO2NH2、NHSO2R2e、NR2eCOR2f、NHC(NH)NH2、NR2eR2f、NHCOR2e、COR2e、CSR2e、CN、COOH、COOR2e、CONH2、CONHOH、CONHR2e、CONHOR2e、C(NOH)NH2、CONR2eR2f、SO2R2e、SO3H、SO2NH2、SO2NR2eR2fから選択される1つまたは複数の基で置換されてもよい(式中、R2eおよびR2fは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR2eおよびR2fは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る);
R5は、それが結合するCと一緒になって、これにより再構成された式IIaにおいて二重結合を有するカルボニル基を形成するか、または、R5は、H、C1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、C3-8シクロアルキル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、R5a、ハロゲン、OH、OR5a、SH、SR5a、OCOR5a、SCOR5a、NH2、NO2、NHR5a、NHSO2NH2、NHSO2R5a、NR5aCOR5b、NHCOR5a、NHC(NH)NH2、NR5aR5b、COR5a、CSR5a、CN、COOH、COOR5a、CONH2、CONHOH、CONHR5a、CONHOR5a、C(NOH)NH2、CONR5aR5b、SO2R5a、SO3H、SO2NH2、SO2NR5aR5bから選択される(式中、R5aおよびR5bは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR5aおよびR5bは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)
ここで、R5が、C1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、C1-6アルキル、C3-8シクロアルキル、または1つもしくは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合により、ハロゲン、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、R5c、C1-6アルキル、OH、OR5c、OCOR5c、SH、SR5c、SCOR5c、NH2、NO2、NHR5c、NHSO2NH2、NHSO2R5c、NR5cCOR5d、NHCOR5c、NHC(NH)NH2、NR5cR5d、COR5c、CSR5c、CN、COOH、COOR5c、CONH2、CONHOH、CONHR5c、CONHOR5c、C(NOH)NH2、CONR5cR5d、SO2R5c、SO3H、SO2NH2、SO2NR5cR5dから選択される1つまたは複数の基で置換されてもよい(式中、R5cおよびR5dは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR5cおよびR5dは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)
ここで、R5の置換基が、C1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、C3-8シクロアルキル、または1つもしくは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合により、ハロゲン、R5e、C1-6アルキル、OH、OR5e、OCOR5e、SH、SR5e、SCOR5e、NH2、NO2、NHR5e、NHSO2NH2、NHSO2R5e、NR5eCOR5f、NHCOR5e、NHC(NH)NH2、NR5eR5f、COR5e、CSR5e、CN、COOH、COOR5e、CONH2、CONHOH、CONHR5e、CONHOR5e、C(NOH)NH2、CONR5eR5f、SO2R5e、SO3H、SO2NH2、SO2NR5eR5fから選択される1つまたは複数の基で置換されてもよい(式中、R5eおよびR5fは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR5eおよびR5fは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る);
R6は、C1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、R6a、ハロゲン、OH、OR6a、SH、SR6a、OCOR6a、SCOR6a、NH2、NO2、NHR6a、NHSO2NH2、NHSO2R6a、NR6aCOR6b、NHCOR6a、NHC(NH)NH2、NR6aR6b、COR6a、CSR6a、CN、COOH、COOR6a、CONH2、CONHOH、CONHR6a、CONHOR6a、C(NOH)NH2、CONR6aR6b、SO2R6a、SO3H、SO2NH2、SO2NR6aR6bから選択される(式中、R6aおよびR6bは、独立してC1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR6aおよびR6bは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)
ここで、R6がヘテロアリールまたはヘテロシクリルである場合、これらの部分はそれぞれ、場合により1つまたは複数の酸素原子で置換されてもよく、そしてR6がC1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、C3-8シクロアルキル、または1つもしくは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合により、ハロゲン、R6c、C1-6アルキル、C1-6アルキニル、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、アリールC1-6アルコキシ、ヘテロアリールC1-6アルコキシ、ヘテロシクリルC1-6アルコキシ、OH、OR6c、OCOR6c、SH、SR6c、SCOR6c、NH2、NO2、NHR6c、NHSO2NH2、NHC(NH)NH2、NHSO2R6c、NR6cCOR6d、NHCOR6c、NR6cR6d、COR6c、CSR6c、CN、COOH、COOR6c、CONH2、CONHR6c、CONHOR6c、CONHOH、C(NOH)NH2、CONR6cR6d、SO2R6c、SO3H、SO2NH2、SO2NR6cR6dから選択される1つまたは複数の基で置換されてもよい(式中、R6cおよびR6dは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR6cおよびR6dは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る)
ここで、R6の置換基がヘテロアリールまたはヘテロシクリルである場合、これらの部分はそれぞれ、場合により1つまたは複数の酸素原子で置換されてもよく、または、R6の置換基がC1-6アルキル、C1-6アルキニル、アリール、ヘテロアリール、ヘテロシクリル、C1-6アルコキシ、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、アリールC1-6アルキル、ヘテロアリールC1-6アルキル、ヘテロシクリルC1-6アルキル、アリールC1-6アルコキシ、ヘテロアリールC1-6アルコキシ、ヘテロシクリルC1-6アルコキシ、C3-8シクロアルキル、または1つもしくは複数のこれらの部分を含有する基である場合、これらの部分はそれぞれ、場合により、ハロゲン、R6e、C1-6アルキル、C1-4アルコキシ、OH、OR6e、OCOR6e、SH、SR6e、SCOR6e、NH2、NO2、NHR6e、NHSO2NH2、NHC(NH)NH2、NHSO2R6e、NR6eCOR6f、NHCOR6e、NR6eR6f、COR6e、CSR6e、CN、COOH、COOR6e、CONH2、CONHOH、CONHR6e、CONHOR6e、C(NOH)NH2、CONR6eR6f、SO2R6e、SO3H、SO2NH2、SO2NR6eR6fから選択される1つまたは複数の基で置換されてもよい(式中、R6eおよびR6fは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択されるか、またはR6eおよびR6fは、それらが結合するヘテロ原子と一緒になってヘテロシクリルを形成し得る);かつ、
R8は、水素またはC1-6アルキル、アリール、ヘテロアリール、ヘテロシクリル、およびC3-8シクロアルキルから選択される基である(式中、これらの部分はそれぞれ、場合により、ハロゲン、ヒドロキシ、C1-6アルキル、C1-6ハロアルキル、C1-6アルコキシ、アリール、ヘテロアリール、ヘテロシクリル、アリールオキシ、ヘテロアリールオキシ、ヘテロシクリルオキシ、およびアシルから選択される1つまたは複数の基で置換されてもよい)、
ここで、R8の置換基が、C1-6アルキル、C1-6アルコキシ、またはアシルである場合、これらの部分はそれぞれ、場合により、ハロゲン、ヒドロキシ、C1-6アルコキシ、ヘテロシクリル(場合により、ハロゲン、C1-6アルキル、C1-6ハロアルキル、およびヘテロアリール、C1-6アルキルアミノ、およびC1-6ジアルキルアミノから選択される1〜3個の基で置換される)から選択される1つまたは複数の基で置換されてもよい、
ここで、R8の置換基が、アリール、ヘテロアリール、ヘテロシクリル、アリールオキシ、ヘテロアリールオキシ、またはヘテロシクリルオキシである場合、これらの部分はそれぞれ、場合により、ハロゲン、ヒドロキシ、C1-6アルコキシ、C1-6アルキルアミノ、C1-6ジアルキルアミノ、アリール(場合により、ハロゲンおよびC1-6ハロアルキル、およびアシルから選択される1〜3個の基で置換される)から選択される1つまたは複数の基で置換されてもよい)、
前記プロセス。
(1)ホルミル(すなわち-CHO);
(2)C1-6アルキルカルボニロキシ;
(3)C1-6アルキルカルボニル;
(4)C6-10アリールカルボニル;
(5)カルボキシル(すなわち-CO2H);
(6)C1-6アルキルカルバモイル;
(7)カルバモイル(すなわち-CONH2);および、
(8)C1-6アルコキシカルボニル。
(1)ホルミル(すなわち-CHO);
(2)場合により、ハロゲン、ヒドロキシ、およびアリールから選択される1〜3個の基で置換されたC1-6アルキルカルボニロキシ;
(3)場合により、ハロゲン、ヒドロキシ、およびアリールから選択される1〜3個の基で置換されたC1-6アルキルカルボニル;
(4)場合により、ハロゲン、ヒドロキシ、C1-6アルキル、およびC1-6アルコキシから選択される1〜3個の基で置換されたC6-10アリールカルボニル;
(5)カルボキシル(すなわちCO2H);
(6)場合により、ハロゲン、ヒドロキシ、およびアリールから選択される1〜3個の基で置換されたC1-6アルキルカルバモイル;
(7)カルバモイル;並びに、
(8)場合により、ハロゲン、ヒドロキシ、およびアリールから選択される1〜3個の基で置換されたC1-6アルコキシカルボニル。
ここで、R6の置換基が、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、ヘテロシクリル、または1つまたは複数のこれらの部分を含有する基である場合、これらの部分のぞれぞれは、場合により、OR6c、OH、およびCONH2(式中、R6cおよびR6dは、独立して、C1-6アルキル、置換C1-6アルキル、アリール、ヘテロアリール、C3-8シクロアルキル、およびヘテロシクリルから選択される)から選択される1つまたは複数の基で置換されてもよく、そして、R6の置換基が、ヘテロアリールまたはヘテロシクリルである場合、これらの部分のぞれぞれは、場合により1つまたは複数の酸素原子で置換されてもよい。
場合により、ハロゲン、ヒドロキシ、およびC1-6アルコキシ(場合により、ヒドロキシ、C1-6アルキル、C1-6アルキルアミノ、およびC1-6ジアルキルアミノから選択される1〜3個の基で置換される)から選択される基で置換されたC1-6アルキル;
場合により、ハロゲン、ヒドロキシ、C1-6アルキル、C1-6アルコキシ、C1-6ハロアルキル、アシル、およびアリール(場合によりハロゲン、C1-6ハロアルキル、およびアシルから選択される1〜3個の基で置換される)で置換されたアリール;
場合により、ハロゲン、ヒドロキシ、C1-6アルコキシ、アシル、およびC1-6アルキル(場合により、ヘテロシクリルおよびヘテロアリール(それぞれ場合によりC1-6アルキル、およびヘテロアリールC1-6ハロアルキルから選択される1〜3個の基で更に置換される)から選択される1つまたは複数の基で置換される)で置換されたヘテロアリール;
場合により、ハロゲン、ヒドロキシ、C1-6アルコキシ、およびアシルから選択される1つまたは複数の基で置換されたヘテロシクリル;並びに
場合により、ハロゲン、ヒドロキシ、C1-6アルコキシ、およびアシルから選択される1つまたは複数の基で置換されたC3-8シクロアルキル。
該プロセスは、R5およびR6の誘導体R6-C(=O)CH2R5を酸化してグリオキサール中間体R6-C(=O)(C=O)R5を形成し、これを水酸化アンモニウムおよびアルデヒドR8CHOで処理し、式IIa'の中間体を設けることを含み、
(式中、R5、R6、およびR8は第1の態様に関し規定した通りである)
ここで、式IIa'の中間体が4-(3-ピリジル)イミダゾールである場合、式IIa'の中間体は、ジクロロメタンを用いる反応混合物から抽出されない、
前記プロセスを提供する。
1.合成法
記載した化合物の合成に用いる方法を、下記のスキームによって説明する。全ての化合物および中間体は、核磁気共鳴(NMR)によって特徴づけた。これらの化合物を調製する際に用いる出発物質および試薬は、市場から入手可能であるか、または当業者にとって明らかな方法で調製できる。
NMT=以下(not more than)
NLT=以上(not less than)
LOD=乾燥減量(loss on drying)
SM=出発物質(starting materisl)
以下のスキームにおける室温とは、20℃〜25℃の範囲の温度を意味する。
(1H, 600 MHz, 20℃, CDCl3) δH: 8.83 (1H, t, J = 2Hz), 8.49 (1H, ddd, J = 1.0, 2.3, 8.2 Hz), 8.34 (1H, ddd, J = 1.0, 1.7, 7.8 Hz), 7.75 (1H, t, J = 8.1 Hz), 4.49 (2H, s).
(13C, 150 MHz, 20℃, CDCl3) δC: 189.3, 148.5, 135.1, 134.4, 130.2, 128.1, 123.8, 29.9.
融点(mp): 90-91℃
(1H, 600 MHz, 20℃, DMSO) δH:12.37 (1H, s, br), 8.58 (1H, mt, J = 2.0 Hz), 8.21 (1H, ddd, J = 1.0, 1.6, 7.8 Hz), 8.02 (1H, ddd, J = 1.0, 2.5, 8.2 Hz), 7.88 (1H, dd, J = 1.2 Hz), 7.79 (1H, dd, J = 1.1 Hz), 7.64 (1H, t, J = 8.1 Hz).
(13C, 150 MHz, 20℃, DMSO) δC:148.4, 137.9, 136.8, 136.6, 130.5, 130.0, 120.5, 118.3, 114.6.
融点:221℃(度)
(1H, 600 MHz, 20℃, CDCl3) δH:4.65 (1H, m), 3.0, 2.93 (3H, 2 singlets), 1.92 (2H, m), 1.73 (2H, m), 1.59 (4H, m).
(13C, 150 MHz, 20℃, CDCl3) δC:149.7, 149.3, 61.1, 59.5, 33.1, 31.1, 28.8, 28.5, 24.0.
(1H, 600 MHz, 20℃, CDCl3) δH:8.63 (1H, mt, J = 2.0 Hz), 8.16 (1H, ddd, J = 1.0, 1.6, 7.8 Hz), 8.14 (1H, ddd, J = 1.0, 2.3, 8.2 Hz), 7.96 (1H, d, J = 1.3 Hz), 7.65 (1H, dd, J = 1.3 Hz), 7.58 (1H, t, J = 8.1 Hz), 4.45 (1H, m), 3.03 (3H, s), 1.98 (2H, m), 1.80 (2H, m), 1.73 (2H, m), 1.66 (2H, m).
(13C, 150 MHz, 20℃, CDCl3) δC:151.3, 148.7, 140.1, 137.3, 134.9, 130.9, 129.7, 122.1, 119.9, 114.6, 59.4, 31.3, 28.9, 24.4.
融点:121-122℃.
(1H, 600 MHz, 20℃, DMSO) δH:8.06 (1H, d, J = 1.3 Hz), 7.77 (1H, d, J = 1.1 Hz), 7.08 (1H, t, J = 1.9 Hz), 7.0 (1H, t, J = 7.8 Hz), 6.98 (1H, md, J = 7.7 Hz), 6.45 (1H, ddd, J = 1.2, 2.3, 7.7 Hz), 5.07 (2H, s), 4.37 (1H, m), 2.92 (3H, s), 1.87 (2H, m), 1.68 (4H, m), 1.53 (2H, m).
(13C, 150 MHz, 20℃, DMSO) δC:151.2, 148.8, 141.4, 137.3, 133.8, 129.0, 113.7, 112.9, 112.8, 110.4, 58.4, 31.2, 28.2, 24.0.
融点:108-109℃.
(13C, 150 MHz, 20℃, DMSO) δC:156, 151.1, 140.9, 140.8, 137.5, 133.7, 128.9, 117.9, 116.6, 114.2, 114.2, 58.4, 31.2, 28.2, 24.
(1H, 600 MHz, 20℃, DMSO) δH:8.55 (1H, s), 8.09 (1H, d, J = 1.2 Hz), 7.86 (1H, d, J = 1.2 Hz), 7.85 (1H, t, J = 1.8 Hz), 7.35 (1H, md), 7.34 (1H, md), 7.22 (1H, t, J = 7.8 Hz), 5.84 (2H, s), 4.36 (1H, m), 2.93 (3H, s), 1.87 (2H, m), 1.69 (4H, m), 1.54 (2H, m).
以下、式Aの化合物を調製するのに有用な式IIa'の中間体の製造を示す。
1-(3-ニトロフェニル)エタノン(50 g, 303 mmol)をDMSO(150ml)中に含む溶液に、臭化水素(48%, 86ml, 757 mmol)を、内部温度を30℃未満に保ちつつ、30分かけて滴下により20℃で添加した。注:この添加は発熱性である。反応混合物を、DMSの蒸留が終わるまで(約1時間)、70℃(内部温度)に加熱した。
上記の実施例2では、THFを、イミダゾールとシクロペンチル(メチル)カルバミン酸クロリド間の尿素形成反応の溶媒として使用する。本質的にピリジンからなる溶媒を使用することによって収率が改善され得ることが発見された。
式Aの化合物の合成の改良例を以下に示す。
モル収率: 61.1%
品質範囲: HPLCにより95.2%の面積
モル収率: 74%
品質範囲: HPLCにより99.6%の面積
モル収率: 100%
品質範囲: NMRにより>95%
モル収率: 92%
品質範囲: HPLCにより>99%
モル収率: 96%
品質範囲: HPLCにより>99%
モル収率: 定量的と仮定
品質範囲: HPLCにより94%
モル収率: 2ステージにより推定60%のモル収率
品質範囲: HPLCにより99.2%
シクロペンチルアミン(58.1ml, 587 mmol)を2-MeTHF(200mL)中に含む0℃の溶液に、3M水酸化ナトリウム(300ml, 900 mmol)、そして滴下によりエチルクロロギ酸エステル(67.3ml, 705 mmol)を30分かけてそれぞれ添加した。得られた二相混合物を室温にて完了するまで撹拌した。反応混合物を、2-MeTHF(150mL)で希釈した。得られた混合物を室温にて10分撹拌し、その後分離させた。有機層を2-MeTHF(100mL)で抽出した。合わせた有機層を水(5vol)、0.5M HCl(3vol)、その後水で洗浄し、その後減圧下で濃縮した。エチルシクロペンチルカルバメート(100g)が無色の油状物として得られた(DMSOにおけるNMR 1Hにより、2-MeTHFが混入した純粋な化合物であることが示された。純度は90.1%w/w)。更なる精製をせずに次のステップに使用した。
LAH(31,0 g, 816 mmol, 2.5eq)を乾燥THF(350ml)中に含む懸濁液に、エチルシクロペンチルカルバメート(57 g, 326 mmol, 1eq)を乾燥THF(100ml)中に含む溶液を、内部温度を20℃未満に保ちつつ、15℃(ジャケット温度)で1時間かけて添加した(注:ガスの発生が見られ、レートの添加によって制御した)。得られたスラリーをゆっくりと45℃(ジャケット温度)まで加熱した(注:内部反応温度が急上昇し始め、溶媒還流するまで(内部温度69℃)行い、その後発泡した)。強い発熱を制御するために、ジャケット温度を速やかに0℃に冷却した。30分以内に、温度を冷却し、ジャケット温度を55℃に設定した。その後、灰色のスラリーを3時間55℃(ジャケット温度)で撹拌した。LC/MSでは、出発物質の残存は示されなかった。
Claims (5)
- 式A:
のプロセスであって、該プロセスは、式IIa':
ここで、式IIa'の中間体は、R5およびR6の誘導体R6-C(=O)CH2R5を酸化してグリオキサール中間体R6-C(=O)(C=O)R5を形成し、これを水酸化アンモニウムおよびアルデヒドR8CHOで処理し、式IIa'の中間体を設けることによって調製される、
式中、
R6は、NH2CONH-フェニル、またはニトロフェニル、アミノフェニル、もしくは尿素形成後にNH2CONH-フェニル基に変換可能なこの部分のアミノ保護アミノフェニル前駆体であり、R5はHであり、R8はHであり、R1はメチルであり、かつR2はシクロペンチルである、
前記プロセス。 - 前記R5およびR6の誘導体の酸化は無機酸を使用する、請求項1に記載のプロセス。
- 前記無機酸はHX(式中、Xはハロゲン原子である)である、請求項2に記載のプロセス。
- 前記無機酸はHClまたはHBrである、請求項3に記載のプロセス。
- 前記R5およびR6の誘導体の酸化における溶媒および酸化剤としてDMSOを使用する、請求項1〜4のいずれか1項に記載のプロセス。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1401198.5 | 2014-01-24 | ||
| GBGB1401198.5A GB201401198D0 (en) | 2014-01-24 | 2014-01-24 | Process for the syntheis of substituted urea compounds |
| PCT/PT2015/000009 WO2015112036A2 (en) | 2014-01-24 | 2015-01-23 | Processes for the synthesis of substituted urea compounds |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2017505309A JP2017505309A (ja) | 2017-02-16 |
| JP2017505309A5 JP2017505309A5 (ja) | 2018-03-01 |
| JP6777542B2 true JP6777542B2 (ja) | 2020-10-28 |
Family
ID=50287496
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016548135A Expired - Fee Related JP6777542B2 (ja) | 2014-01-24 | 2015-01-23 | 置換尿素化合物の合成のためのプロセス |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US20170001962A1 (ja) |
| EP (1) | EP3097082B1 (ja) |
| JP (1) | JP6777542B2 (ja) |
| CN (1) | CN105934431B (ja) |
| CA (1) | CA2936192A1 (ja) |
| ES (1) | ES2763312T3 (ja) |
| GB (1) | GB201401198D0 (ja) |
| PT (1) | PT3097082T (ja) |
| RU (1) | RU2760719C2 (ja) |
| WO (1) | WO2015112036A2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4118081A1 (en) | 2020-03-27 | 2023-01-18 | Landos Biopharma, Inc. | Plxdc2 ligands |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1316280A (en) | 1919-09-16 | Glenn h | ||
| DE3330192A1 (de) * | 1983-08-20 | 1985-03-07 | Basf Ag, 6700 Ludwigshafen | 4-alkylimidazol-derivate, ihre herstellung und verwendung |
| GB8334210D0 (en) * | 1983-12-22 | 1984-02-01 | Roussel Lab Ltd | Imidazo(1 2-c)pyrimidines |
| US4859691A (en) * | 1987-07-08 | 1989-08-22 | Ciba-Geigy Corporation | Certain 1,2-benzisoxazole derivatives |
| US5852192A (en) * | 1992-03-11 | 1998-12-22 | Dr. Karl Thomae Gmbh | Cyclic urea derivatives, pharmaceutical compositions containing these compounds and processes for preparing them |
| DK1474425T3 (da) * | 2002-01-07 | 2006-09-25 | Eisai Co Ltd | Deazapuriner og anvendelser deraf |
| WO2006047167A2 (en) | 2004-10-21 | 2006-05-04 | Janssen Pharmaceutica, N.V. | 9 alkyl and 9 alkylidenyl 6-0 alkyl-11, 12 carbamate ketolide antimicrobials |
| EP2151442A3 (en) * | 2008-08-07 | 2011-04-06 | Chemi SPA | Process for preparing temozolomide |
| MX342128B (es) | 2008-12-24 | 2016-09-14 | Bial - Portela & C A S A | Compuestos farmaceuticos. |
| EA020151B1 (ru) * | 2009-10-23 | 2014-09-30 | Эли Лилли Энд Компани | Ингибиторы akt и фармацевтические составы, их содержащие |
| BR112013001721A2 (pt) | 2010-07-29 | 2016-05-31 | Bial Portela & Ca Sa | processo de síntese de compostos de ureia substituída |
| WO2014017936A2 (en) | 2012-07-24 | 2014-01-30 | Bial- Portela & Ca, S.A. | Urea compounds and their use as enzyme inhibitors |
| JP2015528013A (ja) | 2012-07-27 | 2015-09-24 | ビアル−ポルテラ エ コンパニア,ソシエダッド アノニマ | 置換ウレア化合物の合成方法 |
-
2014
- 2014-01-24 GB GBGB1401198.5A patent/GB201401198D0/en not_active Ceased
-
2015
- 2015-01-23 WO PCT/PT2015/000009 patent/WO2015112036A2/en not_active Ceased
- 2015-01-23 EP EP15706552.5A patent/EP3097082B1/en active Active
- 2015-01-23 PT PT157065525T patent/PT3097082T/pt unknown
- 2015-01-23 CN CN201580005554.3A patent/CN105934431B/zh not_active Expired - Fee Related
- 2015-01-23 JP JP2016548135A patent/JP6777542B2/ja not_active Expired - Fee Related
- 2015-01-23 RU RU2016133457A patent/RU2760719C2/ru active
- 2015-01-23 CA CA2936192A patent/CA2936192A1/en not_active Abandoned
- 2015-01-23 US US15/113,619 patent/US20170001962A1/en not_active Abandoned
- 2015-01-23 ES ES15706552T patent/ES2763312T3/es active Active
-
2019
- 2019-07-16 US US16/512,872 patent/US11078163B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| WO2015112036A2 (en) | 2015-07-30 |
| EP3097082A2 (en) | 2016-11-30 |
| PT3097082T (pt) | 2020-01-10 |
| WO2015112036A3 (en) | 2015-09-17 |
| RU2016133457A (ru) | 2018-03-01 |
| US20200140391A1 (en) | 2020-05-07 |
| JP2017505309A (ja) | 2017-02-16 |
| RU2760719C2 (ru) | 2021-11-29 |
| ES2763312T3 (es) | 2020-05-28 |
| CN105934431A (zh) | 2016-09-07 |
| CN105934431B (zh) | 2020-11-17 |
| US11078163B2 (en) | 2021-08-03 |
| RU2016133457A3 (ja) | 2018-09-28 |
| GB201401198D0 (en) | 2014-03-12 |
| EP3097082B1 (en) | 2019-10-02 |
| CA2936192A1 (en) | 2015-07-30 |
| US20170001962A1 (en) | 2017-01-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5543555B2 (ja) | 4−アミノ−2−(2,6−ジオキソピペリジン−3−イル)イソインドリン−1,3−ジオン化合物を調製するための方法 | |
| JP2017522306A (ja) | 3−(3−クロロ−1h−ピラゾール−1−イル)ピリジンの製造方法 | |
| EP2346850A1 (en) | A method for the preparation of dabigatran and its intermediates | |
| EP2707367B1 (en) | Methods for preparing naphthyridines | |
| TW201609694A (zh) | 用於製備3-(3-氯-1h-吡唑-1-基)吡啶的方法(一) | |
| CN104662002A (zh) | 取代脲类化合物的合成方法 | |
| JP6777542B2 (ja) | 置換尿素化合物の合成のためのプロセス | |
| JP2025026621A (ja) | シス-(-)-フロシノピペリドールの製造方法 | |
| JP5017101B2 (ja) | 不斉四置換炭素原子含有化合物の製法 | |
| KR101686087B1 (ko) | 광학 활성을 갖는 인돌린 유도체 또는 이의 염의 신규 제조 방법 | |
| KR100850558B1 (ko) | 아토르바스타틴의 효율적인 제조방법 | |
| JP6169721B2 (ja) | パピローマウイルスの治療で用いることができるヒドラジンの合成方法 | |
| AU2011221383B2 (en) | Processes for the preparation of 4-amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione compounds | |
| JPS59139383A (ja) | (±)−4−オキソ−1,2,3,6,7,11b−ヘキサヒドロ−4H−ピラジノ〔2,1−a〕イソキノリン誘導体の製造方法 | |
| ITMI20111672A1 (it) | Metodo efficiente per la preparazione di dronedarone cloridrato |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180122 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180122 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20180920 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20181002 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20181227 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190402 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190618 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20190917 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20200317 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200715 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20200717 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20200813 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200908 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20201008 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6777542 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |