JP6505705B2 - 経口投与用製剤の製造方法 - Google Patents
経口投与用製剤の製造方法 Download PDFInfo
- Publication number
- JP6505705B2 JP6505705B2 JP2016536250A JP2016536250A JP6505705B2 JP 6505705 B2 JP6505705 B2 JP 6505705B2 JP 2016536250 A JP2016536250 A JP 2016536250A JP 2016536250 A JP2016536250 A JP 2016536250A JP 6505705 B2 JP6505705 B2 JP 6505705B2
- Authority
- JP
- Japan
- Prior art keywords
- microparticles
- nanoparticles
- solution
- solvent
- cellulose
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images
























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4535—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom, e.g. pizotifen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4168—1,3-Diazoles having a nitrogen attached in position 2, e.g. clonidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/428—Thiazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/554—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one sulfur as ring hetero atoms, e.g. clothiapine, diltiazem
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
- A61K9/1694—Processes resulting in granules or microspheres of the matrix type containing more than 5% of excipient
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5036—Polysaccharides, e.g. gums, alginate; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5138—Organic macromolecular compounds; Dendrimers obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5192—Processes
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Optics & Photonics (AREA)
- Nanotechnology (AREA)
- Biomedical Technology (AREA)
- Physics & Mathematics (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
a)a1)溶解形態の上記1又は複数の治療剤と、
a2)溶解形態の上記1又は複数のポリマーと、
a3)(i)水と完全に混和可能であり、上記1又は複数の治療剤に対する溶媒であるとともに、上記1又は複数のポリマーに対する溶媒である少なくとも1つの溶媒S1と、(ii)溶媒S1と完全に混和可能であり、水と部分的に混和可能である少なくとも1つの溶媒S2とを含有する溶媒混合物と、
を含有する溶液を提供する工程と、
b)工程a)で提供された上記溶液を撹拌しながら、工程a)で提供された上記溶液の体積の少なくとも2倍の体積の水性界面活性剤溶液を工程a)で提供された上記溶液に添加する工程と、
c)溶媒S1及び溶媒S2を上記水性界面活性剤溶液へと抽出することにより、上記ナノ粒子及び/又はミクロ粒子を形成させる工程と、
を含む、方法に関する。
(i)セルロースエーテル及びセルロースエステル、
(ii)メタクリル酸と1又は複数の(メタ)アクリル酸エステルとのコポリマー、
(iii)2以上の(メタ)アクリル酸エステルのコポリマー、
及びそれらの混合物から選択される1又は複数のポリマーを含有する。上記マトリクスは、更なるマトリクス成分として、他のポリマー及び/又は非ポリマーの賦形剤を含有してもよい。典型的には、マトリクス中の上に定義される1又は複数のポリマー(i)〜(iii)の含量は、マトリクスの総重量ベースで(すなわち、治療剤及び任意のコーティング剤の重量を除いて)50重量%以上、好ましくは70重量%以上、より好ましくは80重量%以上、特に90重量%以上である。さらに、上に定義される1又は複数のポリマー(i)〜(iii)は上記マトリクス中の唯一のポリマーであることが好ましい。上記マトリクスは、ポリマー(i)〜(iii)から選択される1又は複数のポリマーからなることがより好ましく、上記ナノ粒子及び/又はミクロ粒子は、ポリマー(i)〜(iii)から選択される1若しくは複数のポリマー及び治療剤からなること、又はポリマー(i)〜(iii)から選択される1若しくは複数のポリマー、治療剤、及びポリマー(i)〜(iii)から選択されるポリマーの表面に塗布されるコーティング剤からなることが特に好ましい。

(i)ナノ粒子及び/又はミクロ粒子と1又は複数の薬学的に許容可能な賦形剤とを合わせる工程、
(ii)ヒト又は動物の患者による飲み込みが可能な固体形態、特に錠剤又はカプセル剤の医薬製剤又は医薬品を提供する工程、
を含んでもよい。
a)a1)溶解形態の上記1又は複数の治療剤と、
a2)溶解形態の上記1又は複数のポリマーと、
a3)(i)水と完全に混和可能であり、上記1又は複数の治療剤に対する溶媒であるとともに、上記1又は複数のポリマーに対する溶媒である少なくとも1つの溶媒S1と、(ii)溶媒S1と完全に混和可能であり、水と部分的に混和可能である少なくとも1つの溶媒S2とを含有する溶媒混合物と、
を含有する溶液を提供する工程と、
b)工程a)で提供された上記溶液を撹拌しながら、工程a)で提供された上記溶液の体積の少なくとも2倍の体積の水性界面活性剤溶液を工程a)で提供された上記溶液に添加する工程と、
c)溶媒S1及び溶媒S2を上記水性界面活性剤溶液へと抽出することにより、上記ナノ粒子及び/又はミクロ粒子を形成させる工程と、
を含む、方法。
(i)ナノ粒子及び/又はミクロ粒子と1又は複数の薬学的に許容可能な賦形剤とを合わせる工程、
(ii)ヒト又は動物の患者による飲み込みが可能な固体形態の医薬製剤又は医薬品を提供する工程、
を含む、項29に記載の方法。
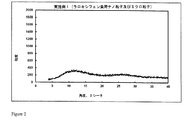
2.0gのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。500mgのラロキシフェン塩酸塩を含有する1.5mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2500rpmで10分間、及び3000rpmで6分間に亘りポリマー溶液中に分散させる。
1.0gのポリマーEudragit S100及び1.0gのポリマーEudragit FS 30Dを酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのラロキシフェン塩酸塩を含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。
750mg gのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))10mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのラロキシフェン塩酸塩を含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40(体積/体積))10ml中のポリマーエチルセルロース250mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用いて3000rpmで更に10分間ポリマー溶液中に分散させる。
1500mg gのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)(60/40%(体積/体積))の混合物10mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのラロキシフェン塩酸塩を含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40%(体積/体積))10ml中のポリマーエチルセルロース500mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用いて3000rpmで更に10分間ポリマー溶液中に分散させる。
3.0gのポリマーEudragit RS100を酢酸エチルとジメチルスルホキシド(DMSO)(60/40%(体積/体積))の混合物15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。500mgのラロキシフェン塩酸塩を含有する1.5mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit RS100を10mlの酢酸エチルに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit RS100を10mlの酢酸エチルに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit RS100を10mlの酢酸エチルに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで30分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit RS100を10mlの酢酸エチルに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのTHF溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit RS100を10mlの酢酸エチルに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのアセトン溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit L100−55を酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで5分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit L100−55を酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで5分間に亘り上記ポリマー溶液中に分散させる。
2.0gのポリマーEudragit L100を酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで7分間及び3000rpmで11分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで7分間及び3000rpmで11分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーエチルセルロースを酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーHPMC AS_HGのポリマーを酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーCAPを酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアトルバスタチンCa塩三水和物を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100をギ酸イソプロピルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのロスバスタチンCa塩を含有する1.0mLのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーエチルセルロースを酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのロスバスタチンCa塩を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に上記ポリマー溶液中に分散させる。
1.5gのポリマーEudragit S100を酢酸エチルとDMSOとの混合物(60/40(体積/体積))10mlに溶解し、ポリマー溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのロスバスタチンCa塩を含有する1.5mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで7分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40(体積/体積))10ml中のポリマーエチルセルロース500mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、3000rpmで更に8分間に亘りポリマー溶液中に分散させる。
1.5gのポリマーEudragit S100及び500mgのHPMC AS−HGを酢酸エチルとDMSOとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのロスバスタチンCa塩を含有する1.5mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。
1.5gのポリマーEudragit S100を酢酸エチルとDMSOとの混合物(60/40(体積/体積))10mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのロスバスタチンCa塩を含有する1.5mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40(体積/体積))10ml中のポリマーエチルセルロース500mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、3000rpmで更に10分間に亘りポリマー溶液中に分散させる。
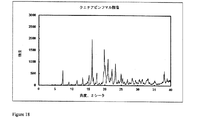
2.5gのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(70/30(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。500mgのクエチアピンフマル酸塩を含有する2.0mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで13分間、及び3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーHPMCP HP 55Sを酢酸エチルとDMSOとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクエチアピンフマル酸塩を含有するDMSO溶液2.0mlを機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
0.5gのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))5mlに溶解し、1.5gのHPMC HP 55Sを酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))10mlに溶解する。両方のポリマー溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクエチアピンフマル酸塩を含有するDMSO溶液2.0mlを機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。
0.5gのEudragitポリマーFS30Dを酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))5mlに溶解する。1.5gのポリマーHPMCP HP55Sを酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))10mlに溶解し、両方の溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクエチアピンフマル酸塩を含有する2.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーCAPを酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクエチアピンフマル酸塩を含有する2.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
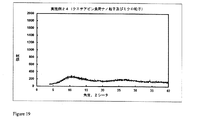
2.0gのポリマーHPMCP HP 55Sを酢酸エチルとジメチルスルホキシド(DMSO)との混合物(70/30(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのデュロキセチン塩酸塩を含有する3.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで22分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit L100−55を酢酸エチルとDMSOとの混合物(70/30(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。700mgのデュロキセチン塩酸塩を含有する1.2mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで25分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとDMSOとの混合物(70/30(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのデュロキセチン塩酸塩を含有する1.0mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとグリコフロールとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのデュロキセチン塩酸塩を含有する1.0mlのグリコフロール溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。
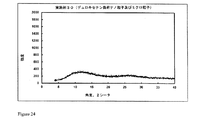
2.0gのポリマーEudragit S100を酢酸エチルとDMSOとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアセナピンマイレン塩酸塩を含有するDMSO溶液1.0mlを機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとDMSOとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアセナピンマイレン塩を含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとNMPとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアリピプラゾールを含有する1.0mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit L100−55を酢酸エチルとNMPとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのアリピプラゾールを含有する1.0mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとDMSOとの混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。500mgのラロキシフェン塩酸塩を含有する1.5mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで20分間に亘りポリマー溶液中に分散させる。
1.0gのポリマーエチルセルロースS100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのラロキシフェン塩酸塩及び100mgのポリマーHPMC AS−HGを含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで15分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(70/30(体積/体積))15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクエチアピンフマル酸塩及び100mgのポリマーHPMC AS−HGを含有する1.0mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで13分間、及び3000rpmで15分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーエチルセルロースを酢酸エチルとジメチルスルホキシド(DMSO)との混合物15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクエチアピンフマル酸塩及び100mgのポリマーHPMC AS−HGを含有する1.0mlのNMP溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで13分間、及び3000rpmで15分間に亘りポリマー溶液中に分散させる。
2.0gのポリマーエチルセルロースを酢酸エチルとジメチルスルホキシド(DMSO)との混合物15mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクエチアピンフマル酸塩及び100mgのポリマーHPMC AS−HGを含有する2mlのジメチルスルホキシド(DMSO)1.0mlを機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて2000rpmで13分間、及び3000rpmで15分間に亘りポリマー溶液中に分散させる。
1500mgのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))10mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのクロニジン塩酸塩を含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40(体積/体積))10ml中のポリマーエチルセルロース500mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用いて3000rpmで更に10分間ポリマー溶液中に分散させる。
1500mgのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))10mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのカルベジロールを含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40(体積/体積))10ml中のポリマーエチルセルロース500mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用いて3000rpmで更に10分間ポリマー溶液中に分散させる。
1500mgのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))10mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのカンデサルタンシレテキシルを含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40(体積/体積))10ml中のポリマーエチルセルロース500mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用いて3000rpmで更に10分間ポリマー溶液中に分散させる。
1500mgのポリマーEudragit S100を酢酸エチルとジメチルスルホキシド(DMSO)との混合物(60/40(体積/体積))10mlに溶解し、溶液を二重壁のガラス容器(内側の高さ16.0cm、内径3.4cm)に移す。350mgのプラミペキソールを含有する1.0mlのDMSO溶液を機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用い、室温にて3000rpmで10分間に亘りポリマー溶液中に分散させる。さらに、酢酸エチルとDMSOとの混合物(60/40(体積/体積))10ml中のポリマーエチルセルロース500mgを添加した後、機械撹拌装置(2.5cmの溶解機ディスクを備えた、ドイツのVMA-Getzmann GmbH製Dispermat FT)を用いて3000rpmで更に10分間ポリマー溶液中に分散させる。
Claims (11)
- ナノ粒子及び/又はミクロ粒子を製造する方法であって、該粒子が、セルロースエーテル、セルロースエステル、メタクリル酸と1又は複数の(メタ)アクリル酸エステルとのコポリマー、2以上の(メタ)アクリル酸エステルのコポリマー、及びそれらの混合物から選択される1又は複数のポリマーを含有するマトリクス中に非結晶形態で分散された1又は複数の治療剤を含有し、前記方法が、
a)a1)溶解形態の前記1又は複数の治療剤と、
a2)溶解形態の前記1又は複数のポリマーと、
a3)(i)水と完全に混和可能であり、前記1又は複数の治療剤に対する溶媒であるとともに、前記1又は複数のポリマーに対する溶媒である少なくとも1つの溶媒S1と、(ii)溶媒S1と完全に混和可能であり、水と部分的に混和可能である少なくとも1つの溶媒S2とを含有する溶媒混合物と、
を含有する溶液を提供する工程と、
b)工程a)で提供された前記溶液を撹拌しながら、工程a)で提供された前記溶液の体積の少なくとも2倍の体積の水性界面活性剤溶液を工程a)で提供された前記溶液に添加する工程と、
c)溶媒S1及び溶媒S2を前記水性界面活性剤溶液へと抽出することにより、前記ナノ粒子及び/又はミクロ粒子を形成させる工程と、
を含み、
前記溶媒S1は、アセトン、ジメチルスルホキシド(DMSO)、N−メチルピロリドン(NMP)、グリコフロール、テトラヒドロフラン、エタノール、及び2以上のそれらの混合物から選択され、
前記溶媒S2は、C1〜C3酢酸アルキル、C1〜C3ギ酸アルキル、炭酸ジメチル、エチルメチルケトン、及び2以上のそれらの混合物から選択される、方法。 - 前記ナノ粒子及び/又はミクロ粒子の前記マトリクスが、メチルセルロース、エチルセルロース、プロピルセルロース、エチルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシエチルエチルセルロース、セルロースアセテートフタレート(CAP)、セルロースアセテート、ヒドロキシプロピルメチルセルロースフタレート(HPMCP)、ヒドロキシプロピルメチルセルロースアセテートスクシネート(HPMC AS)、及びそれらの混合物から選択されるセルロースエーテル又はセルロースエステルを含有する、請求項1に記載の方法。
- 前記ナノ粒子及び/又はミクロ粒子の前記マトリクスが、メタクリル酸と、ポリ(メタクリル酸−コ−メタクリル酸メチル)、ポリ(メタクリル酸−コ−アクリル酸メチル)、ポリ(メタクリル酸−コ−アクリル酸エチル)、ポリ(メタクリル酸−コ−メタクリル酸エチル)、ポリ(アクリル酸メチル−コ−メタクリル酸メチル−コ−メタクリル酸)、及びそれらの混合物から選択される1又は複数の(メタ)アクリル酸エステルとのコポリマーを含有する、請求項1又は2に記載の方法。
- 前記ナノ粒子及び/又はミクロ粒子の前記マトリクスが、ポリ(アクリル酸エチル−コ−メタクリル酸メチル)、ポリ(アクリル酸エチル−コ−メタクリル酸メチル−コ−トリメチルアンモニオエチルメタクリレートクロリド)、又はそれらの混合物から選択される2以上の(メタ)アクリル酸エステルのコポリマーを含有する、請求項1〜3のいずれか1項に記載の方法。
- 前記治療剤がラロキシフェン、アトルバスタチン、ロスバスタチン、クエチアピン、デュロキセチン、アセナピン、アリピプラゾール、メトトレキサート、グルカゴン、バソプレッシン、アレンドロン酸、クロドロン酸、エゼチミブ、バルサルタン、オルメサルタン、テルミサルタン、イルベサルタン、カンデサルタン、ボセンタン、シタラビン、ドキソルビシン、イリノテカン、ジルチアゼム、ベラパミル、コルチゾール、エストラジオール、プロゲステロン、タモキシフェン、モルヒネ、ブプレノルフィン、セレギリン、リセドロン酸、テルブタリン、チルドロン酸、ゾレドロン酸、ジプラシドン、ナロキソン、パミドロン酸、エチドロン酸、アルベンダゾール、アミドリン、カルベジロール、パクリタキセル、ドセタキセル、メサラジン、ブデソニド、プレドニソン、パラセタモール、デキサメタゾン、オメプラゾール、リスペリドン、L−ドーパ、ジクロフェナク、メトプロロール、ブレオマイシン、ペリンドプリル、トランドラプリル、ラミプリル、シラザプリル、モエキシプリル、スピラプリル、フルオロウラシル、イブプロフェン、ニフェジピン、オンダンセトロン、リバスチグミン、シンバスタチン、ロサルタン、エプロサルタン、リシノプリル、及びカプトプリルから、又は適宜、それらの薬学的に許容可能な塩形態から選択される、請求項1〜4のいずれか1項に記載の方法。
- 前記治療剤が、ラロキシフェン、アトルバスタチン、ロスバスタチン、クエチアピン、デュロキセチン、アリピプラゾール、及びアセナピン、又はそれらの薬学的に許容可能な塩形態から選択される、請求項5に記載の方法。
- 工程a)で提供された前記溶液中の前記1又は複数のポリマーの濃度が、工程a)で提供された前記溶液の総重量ベースで1重量%〜40重量%である、請求項1〜6のいずれか1項に記載の方法。
- 前記水性界面活性剤溶液中の前記界面活性剤の濃度が、前記界面活性剤の総体積ベースで0.1%(重量/体積)〜30%(重量/体積)の範囲であり、前記界面活性剤溶液が6以下のpH値を有する、請求項1〜7のいずれか1項に記載の方法。
- 前記水性界面活性剤溶液が溶解した塩又は溶解した糖を含有する、請求項1〜8のいずれか1項に記載の方法。
- 前記ナノ粒子及び/又はミクロ粒子が多糖により形成されたコーティングを持ち、前記方法が工程c)で形成されるナノ粒子及び/又はミクロ粒子と多糖の溶液とを接触させ、前記溶媒を除去して前記コーティングを形成する工程を更に含む、請求項1〜9のいずれか1項に記載の方法。
- 前記多糖が、キトサン、アルギン酸塩、ペクチン、イヌリン、グアーガム、デキストラン、コンドロイチン硫酸塩、ヒアルロン酸、及び2以上のそれらの組合せから選択される、請求項10に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13195835 | 2013-12-05 | ||
| EP13195835.7 | 2013-12-05 | ||
| PCT/EP2014/076455 WO2015082562A1 (en) | 2013-12-05 | 2014-12-03 | Process for the production of drug formulations for oral administration |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2016539953A JP2016539953A (ja) | 2016-12-22 |
| JP2016539953A5 JP2016539953A5 (ja) | 2017-12-28 |
| JP6505705B2 true JP6505705B2 (ja) | 2019-04-24 |
Family
ID=49759055
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016536250A Expired - Fee Related JP6505705B2 (ja) | 2013-12-05 | 2014-12-03 | 経口投与用製剤の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20160303102A1 (ja) |
| EP (1) | EP3076951B1 (ja) |
| JP (1) | JP6505705B2 (ja) |
| KR (1) | KR20160093611A (ja) |
| CA (1) | CA2928772A1 (ja) |
| WO (1) | WO2015082562A1 (ja) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011006012A1 (en) | 2009-07-08 | 2011-01-13 | Charleston Laboratories Inc. | Pharmaceutical compositions |
| EP3307245A1 (en) | 2015-06-11 | 2018-04-18 | Alrise Biosystems GmbH | Process for the preparation of drug loaded microparticles |
| US10736855B2 (en) | 2016-02-25 | 2020-08-11 | Dexcel Pharma Technologies Ltd. | Compositions comprising proton pump inhibitors |
| CA3055170A1 (en) | 2016-03-04 | 2017-09-08 | Charleston Laboratories, Inc. | Pharmaceutical compositions |
| CA3019384A1 (en) * | 2016-03-31 | 2017-10-05 | Tuttifoodi B.V. | Granules |
| GB201609222D0 (en) | 2016-05-25 | 2016-07-06 | F2G Ltd | Pharmaceutical formulation |
| WO2018115001A1 (en) | 2016-12-20 | 2018-06-28 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system containing asenapine |
| WO2018115010A1 (en) | 2016-12-20 | 2018-06-28 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system containing asenapine and polysiloxane or polyisobutylene |
| ES2881783T3 (es) | 2017-06-26 | 2021-11-30 | Lts Lohmann Therapie Systeme Ag | Sistema terapéutico transdérmico que contiene asenapina y polímero de acrílico y silicona |
| TR201719919A2 (tr) * | 2017-12-08 | 2019-06-21 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | The pharmaceutical combination comprising raloxifene and aripiprazole |
| KR102066402B1 (ko) * | 2017-12-22 | 2020-01-15 | 대화제약 주식회사 | 이리노테칸 또는 그의 약제학적으로 허용가능한 염을 포함하는 경구투여용 약제학적 조성물 |
| MX2020014286A (es) | 2018-06-20 | 2021-03-25 | Lts Lohmann Therapie Systeme Ag | Sistema terapeutico transdermico que contiene asenapina. |
| CN108938588B (zh) * | 2018-08-16 | 2021-06-22 | 广州维奥康药业科技有限公司 | 一种依折麦布片 |
| US11819503B2 (en) | 2019-04-23 | 2023-11-21 | F2G Ltd | Method of treating coccidioides infection |
| KR102185475B1 (ko) * | 2019-06-20 | 2020-12-02 | 대화제약 주식회사 | 이리노테칸 자유 염기를 포함하는 경구투여용 약학 조성물 |
| BR112022013996A2 (pt) | 2019-10-31 | 2022-09-06 | Evonik Operations Gmbh | Forma de dosagem farmacêutica ou nutracêutica, nano ou micropartículas e processo para preparar nano ou micropartículas |
| WO2022033743A1 (en) | 2020-08-14 | 2022-02-17 | Re-Organic As | Method to disperse nano-cellulose in polymers, and derived products |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5102668A (en) * | 1990-10-05 | 1992-04-07 | Kingaform Technology, Inc. | Sustained release pharmaceutical preparation using diffusion barriers whose permeabilities change in response to changing pH |
| US5650173A (en) * | 1993-11-19 | 1997-07-22 | Alkermes Controlled Therapeutics Inc. Ii | Preparation of biodegradable microparticles containing a biologically active agent |
| US5792477A (en) * | 1996-05-07 | 1998-08-11 | Alkermes Controlled Therapeutics, Inc. Ii | Preparation of extended shelf-life biodegradable, biocompatible microparticles containing a biologically active agent |
| US6350786B1 (en) * | 1998-09-22 | 2002-02-26 | Hoffmann-La Roche Inc. | Stable complexes of poorly soluble compounds in ionic polymers |
| DE60039802D1 (de) * | 1999-02-10 | 2008-09-25 | Pfizer Prod Inc | Vorrichtung mit matrixgesteuerter Wirkstofffreisetzung |
| US7838037B2 (en) * | 1999-11-17 | 2010-11-23 | Tagra Biotechnologies Ltd. | Method of microencapsulation |
| US6824822B2 (en) * | 2001-08-31 | 2004-11-30 | Alkermes Controlled Therapeutics Inc. Ii | Residual solvent extraction method and microparticles produced thereby |
| ATE361057T1 (de) * | 2000-12-21 | 2007-05-15 | Alrise Biosystems Gmbh | Verfahren umfassend einen induzierten phasenübergang zur herstellung von hydrophobe wirkstoffe enthaltenden mikropartikeln |
| JP5123452B2 (ja) * | 2001-09-19 | 2013-01-23 | 帝三製薬株式会社 | 徐放性外用剤 |
| EP1469830A2 (en) * | 2002-02-01 | 2004-10-27 | Pfizer Products Inc. | Method for making homogeneous spray-dried solid amorphous drug dispersions using pressure nozzles |
| DE60222734T2 (de) * | 2002-03-15 | 2008-07-17 | Alrise Biosystems Gmbh | Mikropartikel und Verfahren zur deren Herstellung |
| KR20060033859A (ko) * | 2003-04-30 | 2006-04-20 | 데비오 팜 소시에떼 아노님 | 생식선 자극 호르몬 방출 호르몬을 이용한 방법 및 조성물 |
| WO2008075320A2 (en) * | 2006-12-21 | 2008-06-26 | Ranbaxy Laboratories Limited | Antilipidemic pharmaceutical compositions and process for preparation thereof |
| CN102014910A (zh) * | 2008-03-07 | 2011-04-13 | 美国辉瑞有限公司 | 在无食物条件下施用齐拉西酮的方法、剂型和试剂盒 |
| JP4856752B2 (ja) * | 2009-11-30 | 2012-01-18 | ホソカワミクロン株式会社 | 薬物含有ナノ粒子の製造方法 |
| US8927619B2 (en) * | 2011-12-21 | 2015-01-06 | Jorg Thomas Wilken | Color-stabilized iodopropynyl butylcarbamate |
| CN102579362B (zh) * | 2012-02-23 | 2014-09-10 | 浙江工业大学 | 一种非洛地平缓释微球及其制备方法 |
| CN102579368B (zh) * | 2012-03-28 | 2013-07-10 | 广州万泽医药科技有限公司 | 美托洛尔缓释微球、缓释药用组合物及其制备方法 |
-
2014
- 2014-12-03 US US15/101,873 patent/US20160303102A1/en not_active Abandoned
- 2014-12-03 KR KR1020167013757A patent/KR20160093611A/ko not_active Application Discontinuation
- 2014-12-03 JP JP2016536250A patent/JP6505705B2/ja not_active Expired - Fee Related
- 2014-12-03 CA CA2928772A patent/CA2928772A1/en not_active Abandoned
- 2014-12-03 EP EP14808594.7A patent/EP3076951B1/en active Active
- 2014-12-03 WO PCT/EP2014/076455 patent/WO2015082562A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| EP3076951A1 (en) | 2016-10-12 |
| US20160303102A1 (en) | 2016-10-20 |
| KR20160093611A (ko) | 2016-08-08 |
| WO2015082562A1 (en) | 2015-06-11 |
| JP2016539953A (ja) | 2016-12-22 |
| EP3076951B1 (en) | 2020-09-30 |
| CA2928772A1 (en) | 2015-06-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6505705B2 (ja) | 経口投与用製剤の製造方法 | |
| US10034854B2 (en) | Pharmaceutical composition with improved bioavailability | |
| TWI706793B (zh) | 包含紫杉烷之非晶形固體分散體、包含其之錠劑及其等之製備方法 | |
| AU2014221630B2 (en) | Suspension for oral administration comprising amorphous tolvaptan | |
| CN1244796A (zh) | 具有外涂一种杀真菌剂和一种聚合物的核心的药丸 | |
| JP2007504266A (ja) | ジプラシドンの持続放出剤形 | |
| WO2012013088A1 (zh) | 决奈达隆固体分散体及其制备方法 | |
| CN102655856A (zh) | 纳米粒状奥美沙坦酯组合物、其制备方法及含有它们的药物组合物 | |
| ES2929730T3 (es) | Dispersión sólida | |
| JP6666352B2 (ja) | デュタステリド含有固体分散体及びこれを含む組成物 | |
| EP2701689B1 (en) | Pharmaceutical compositions of raltegravir, methods of preparation and use thereof | |
| CA2854612C (en) | Delivery systems for improving oral bioavailability of fenobam, its hydrates, and salts | |
| WO2020246526A1 (ja) | キサンチンオキシダーゼ阻害剤含有腸溶性製剤 | |
| WO2007064084A1 (en) | Granules containing pranlukast and processes for the preparation thereof | |
| TWI520752B (zh) | 決奈達隆固體分散體及其製備方法 | |
| WO2007064083A1 (en) | Spray-dried granules and processes for the preparation thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20160805 |
|
| RD01 | Notification of change of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7426 Effective date: 20160805 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171115 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20171115 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20180718 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180814 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20181113 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190226 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190327 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6505705 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |