JP6474420B2 - 組換えタンパク質のグリコシル化を調節するためのn−グリコシル化経路制御因子の過剰発現 - Google Patents
組換えタンパク質のグリコシル化を調節するためのn−グリコシル化経路制御因子の過剰発現 Download PDFInfo
- Publication number
- JP6474420B2 JP6474420B2 JP2016549077A JP2016549077A JP6474420B2 JP 6474420 B2 JP6474420 B2 JP 6474420B2 JP 2016549077 A JP2016549077 A JP 2016549077A JP 2016549077 A JP2016549077 A JP 2016549077A JP 6474420 B2 JP6474420 B2 JP 6474420B2
- Authority
- JP
- Japan
- Prior art keywords
- cell
- culture
- protein
- cells
- recombinant protein
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 title claims description 56
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 title claims description 56
- 230000004988 N-glycosylation Effects 0.000 title claims description 13
- 230000037361 pathway Effects 0.000 title claims description 10
- 230000002018 overexpression Effects 0.000 title description 6
- 230000013595 glycosylation Effects 0.000 title description 5
- 238000006206 glycosylation reaction Methods 0.000 title description 5
- 210000004027 cell Anatomy 0.000 claims description 270
- 108090000623 proteins and genes Proteins 0.000 claims description 116
- 102000004169 proteins and genes Human genes 0.000 claims description 97
- 238000004113 cell culture Methods 0.000 claims description 85
- 238000000034 method Methods 0.000 claims description 85
- 238000004519 manufacturing process Methods 0.000 claims description 70
- 230000010412 perfusion Effects 0.000 claims description 63
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 claims description 38
- 101150093077 Mgat2 gene Proteins 0.000 claims description 33
- 101150030635 Mgat1 gene Proteins 0.000 claims description 30
- 210000004962 mammalian cell Anatomy 0.000 claims description 27
- -1 mannose glycan Chemical class 0.000 claims description 15
- 239000000203 mixture Substances 0.000 claims description 15
- 108060003951 Immunoglobulin Proteins 0.000 claims description 14
- 102000018358 immunoglobulin Human genes 0.000 claims description 14
- 238000009472 formulation Methods 0.000 claims description 13
- 230000002829 reductive effect Effects 0.000 claims description 11
- 241000894007 species Species 0.000 claims description 8
- 241000699802 Cricetulus griseus Species 0.000 claims description 7
- 238000010899 nucleation Methods 0.000 claims description 7
- 210000001672 ovary Anatomy 0.000 claims description 6
- 239000004017 serum-free culture medium Substances 0.000 claims description 6
- 108091006020 Fc-tagged proteins Proteins 0.000 claims description 5
- 101100491335 Caenorhabditis elegans mat-2 gene Proteins 0.000 claims description 4
- 102100040428 Chitobiosyldiphosphodolichol beta-mannosyltransferase Human genes 0.000 claims description 4
- 101000891557 Homo sapiens Chitobiosyldiphosphodolichol beta-mannosyltransferase Proteins 0.000 claims description 4
- 238000012258 culturing Methods 0.000 claims description 4
- 239000012528 membrane Substances 0.000 claims description 4
- DINOPBPYOCMGGD-VEDJBHDQSA-N Man(a1-2)Man(a1-2)Man(a1-3)[Man(a1-2)Man(a1-3)[Man(a1-2)Man(a1-6)]Man(a1-6)]Man(b1-4)GlcNAc(b1-4)GlcNAc Chemical compound O[C@@H]1[C@@H](NC(=O)C)C(O)O[C@H](CO)[C@H]1O[C@H]1[C@H](NC(C)=O)[C@@H](O)[C@H](O[C@H]2[C@H]([C@@H](O[C@@H]3[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O[C@@H]3[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O[C@@H]3[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)[C@H](O)[C@@H](CO[C@@H]3[C@H]([C@@H](O[C@@H]4[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O4)O[C@@H]4[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O4)O)[C@H](O)[C@@H](CO[C@@H]4[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O4)O[C@@H]4[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O4)O)O3)O)O2)O)[C@@H](CO)O1 DINOPBPYOCMGGD-VEDJBHDQSA-N 0.000 claims description 3
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 claims description 3
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical group CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 claims description 3
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 claims description 3
- 102100033782 UDP-galactose translocator Human genes 0.000 claims description 3
- 108010075920 UDP-galactose translocator Proteins 0.000 claims description 3
- 229950006780 n-acetylglucosamine Drugs 0.000 claims description 3
- 230000003796 beauty Effects 0.000 claims description 2
- 238000001471 micro-filtration Methods 0.000 claims description 2
- 238000000108 ultra-filtration Methods 0.000 claims description 2
- 101710186714 2-acylglycerol O-acyltransferase 1 Proteins 0.000 claims 1
- 102100022622 Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase Human genes 0.000 claims 1
- 125000003047 N-acetyl group Chemical group 0.000 claims 1
- 108090000992 Transferases Proteins 0.000 claims 1
- 102000004357 Transferases Human genes 0.000 claims 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 claims 1
- 229960002442 glucosamine Drugs 0.000 claims 1
- 235000018102 proteins Nutrition 0.000 description 83
- 239000006143 cell culture medium Substances 0.000 description 31
- 108090000765 processed proteins & peptides Proteins 0.000 description 31
- 239000000427 antigen Substances 0.000 description 28
- 108091007433 antigens Proteins 0.000 description 28
- 102000036639 antigens Human genes 0.000 description 28
- 150000001413 amino acids Chemical class 0.000 description 26
- 229920001184 polypeptide Polymers 0.000 description 26
- 102000004196 processed proteins & peptides Human genes 0.000 description 26
- 239000002609 medium Substances 0.000 description 25
- 210000000601 blood cell Anatomy 0.000 description 24
- 235000001014 amino acid Nutrition 0.000 description 23
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 22
- 229940024606 amino acid Drugs 0.000 description 22
- 230000014509 gene expression Effects 0.000 description 22
- 230000012010 growth Effects 0.000 description 22
- 230000003698 anagen phase Effects 0.000 description 21
- 230000027455 binding Effects 0.000 description 17
- 102000025171 antigen binding proteins Human genes 0.000 description 16
- 108091000831 antigen binding proteins Proteins 0.000 description 16
- 230000008859 change Effects 0.000 description 16
- 230000001965 increasing effect Effects 0.000 description 15
- 108020004999 messenger RNA Proteins 0.000 description 13
- 230000010261 cell growth Effects 0.000 description 12
- 239000000306 component Substances 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 229960001230 asparagine Drugs 0.000 description 11
- 239000012634 fragment Substances 0.000 description 11
- 235000015097 nutrients Nutrition 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 102000005962 receptors Human genes 0.000 description 11
- 108020003175 receptors Proteins 0.000 description 11
- 238000001890 transfection Methods 0.000 description 11
- 238000010923 batch production Methods 0.000 description 10
- 238000002474 experimental method Methods 0.000 description 10
- 239000003446 ligand Substances 0.000 description 10
- 239000013598 vector Substances 0.000 description 10
- 108020004459 Small interfering RNA Proteins 0.000 description 9
- 239000012526 feed medium Substances 0.000 description 9
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 8
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 8
- 239000001963 growth medium Substances 0.000 description 8
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 7
- 230000007704 transition Effects 0.000 description 7
- 102000004127 Cytokines Human genes 0.000 description 6
- 108090000695 Cytokines Proteins 0.000 description 6
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 6
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 229940072221 immunoglobulins Drugs 0.000 description 6
- 150000007523 nucleic acids Chemical class 0.000 description 6
- 238000011084 recovery Methods 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 6
- 230000035899 viability Effects 0.000 description 6
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 5
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 108010022394 Threonine synthase Proteins 0.000 description 5
- 239000005557 antagonist Substances 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 102000004419 dihydrofolate reductase Human genes 0.000 description 5
- 239000013604 expression vector Substances 0.000 description 5
- 150000004676 glycans Chemical class 0.000 description 5
- 229960000485 methotrexate Drugs 0.000 description 5
- 108020004707 nucleic acids Proteins 0.000 description 5
- 102000039446 nucleic acids Human genes 0.000 description 5
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 5
- UWYZHKAOTLEWKK-UHFFFAOYSA-N 1,2,3,4-tetrahydroisoquinoline Chemical compound C1=CC=C2CNCCC2=C1 UWYZHKAOTLEWKK-UHFFFAOYSA-N 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 210000004102 animal cell Anatomy 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical compound NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 4
- 238000012136 culture method Methods 0.000 description 4
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 4
- 238000010195 expression analysis Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 230000004927 fusion Effects 0.000 description 4
- 239000003102 growth factor Substances 0.000 description 4
- 229960002591 hydroxyproline Drugs 0.000 description 4
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 4
- 230000001939 inductive effect Effects 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 150000002632 lipids Chemical class 0.000 description 4
- 230000002503 metabolic effect Effects 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 4
- 239000006152 selective media Substances 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 230000014616 translation Effects 0.000 description 4
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 description 3
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 3
- 239000002028 Biomass Substances 0.000 description 3
- 229940123587 Cell cycle inhibitor Drugs 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 3
- 102000003886 Glycoproteins Human genes 0.000 description 3
- 108090000288 Glycoproteins Proteins 0.000 description 3
- 101001031613 Homo sapiens Fibroleukin Proteins 0.000 description 3
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 3
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 3
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 description 3
- 102000014128 RANK Ligand Human genes 0.000 description 3
- 108010025832 RANK Ligand Proteins 0.000 description 3
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 238000007792 addition Methods 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 230000022131 cell cycle Effects 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 238000009295 crossflow filtration Methods 0.000 description 3
- 230000001186 cumulative effect Effects 0.000 description 3
- 235000018417 cysteine Nutrition 0.000 description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 3
- 229960003067 cystine Drugs 0.000 description 3
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- UHBYWPGGCSDKFX-VKHMYHEASA-N gamma-carboxy-L-glutamic acid Chemical compound OC(=O)[C@@H](N)CC(C(O)=O)C(O)=O UHBYWPGGCSDKFX-VKHMYHEASA-N 0.000 description 3
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 3
- 239000012510 hollow fiber Substances 0.000 description 3
- 229940088597 hormone Drugs 0.000 description 3
- 235000003642 hunger Nutrition 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 230000000977 initiatory effect Effects 0.000 description 3
- 239000012139 lysis buffer Substances 0.000 description 3
- 239000003550 marker Substances 0.000 description 3
- 201000000050 myeloid neoplasm Diseases 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 230000037351 starvation Effects 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000004114 suspension culture Methods 0.000 description 3
- 229940104230 thymidine Drugs 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 3
- DQLHSFUMICQIMB-VIFPVBQESA-N (2s)-2-amino-3-(4-methylphenyl)propanoic acid Chemical compound CC1=CC=C(C[C@H](N)C(O)=O)C=C1 DQLHSFUMICQIMB-VIFPVBQESA-N 0.000 description 2
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- OGNSCSPNOLGXSM-UHFFFAOYSA-N 2,4-diaminobutyric acid Chemical compound NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 2
- OYIFNHCXNCRBQI-UHFFFAOYSA-N 2-aminoadipic acid Chemical compound OC(=O)C(N)CCCC(O)=O OYIFNHCXNCRBQI-UHFFFAOYSA-N 0.000 description 2
- BRMWTNUJHUMWMS-UHFFFAOYSA-N 3-Methylhistidine Natural products CN1C=NC(CC(N)C(O)=O)=C1 BRMWTNUJHUMWMS-UHFFFAOYSA-N 0.000 description 2
- PECYZEOJVXMISF-UHFFFAOYSA-N 3-aminoalanine Chemical compound [NH3+]CC(N)C([O-])=O PECYZEOJVXMISF-UHFFFAOYSA-N 0.000 description 2
- CMUHFUGDYMFHEI-QMMMGPOBSA-N 4-amino-L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N)C=C1 CMUHFUGDYMFHEI-QMMMGPOBSA-N 0.000 description 2
- PZNQZSRPDOEBMS-QMMMGPOBSA-N 4-iodo-L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(I)C=C1 PZNQZSRPDOEBMS-QMMMGPOBSA-N 0.000 description 2
- 229940117976 5-hydroxylysine Drugs 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 2
- 101150013553 CD40 gene Proteins 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 2
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 101800003838 Epidermal growth factor Proteins 0.000 description 2
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 2
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 2
- 108700039887 Essential Genes Proteins 0.000 description 2
- 102100038647 Fibroleukin Human genes 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 2
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 2
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 2
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 2
- 108090000144 Human Proteins Proteins 0.000 description 2
- 102000003839 Human Proteins Human genes 0.000 description 2
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 108010064600 Intercellular Adhesion Molecule-3 Proteins 0.000 description 2
- 102100037871 Intercellular adhesion molecule 3 Human genes 0.000 description 2
- 102000008070 Interferon-gamma Human genes 0.000 description 2
- 108010074328 Interferon-gamma Proteins 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 2
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 2
- 102000004557 Interleukin-18 Receptors Human genes 0.000 description 2
- 108010017537 Interleukin-18 Receptors Proteins 0.000 description 2
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 2
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 2
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 description 2
- 108010002386 Interleukin-3 Proteins 0.000 description 2
- 102100039064 Interleukin-3 Human genes 0.000 description 2
- 102000010787 Interleukin-4 Receptors Human genes 0.000 description 2
- 108010038486 Interleukin-4 Receptors Proteins 0.000 description 2
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 description 2
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 2
- 102000004058 Leukemia inhibitory factor Human genes 0.000 description 2
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 2
- 239000012098 Lipofectamine RNAiMAX Substances 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 102100034256 Mucin-1 Human genes 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- DTERQYGMUDWYAZ-ZETCQYMHSA-N N(6)-acetyl-L-lysine Chemical compound CC(=O)NCCCC[C@H]([NH3+])C([O-])=O DTERQYGMUDWYAZ-ZETCQYMHSA-N 0.000 description 2
- JDHILDINMRGULE-LURJTMIESA-N N(pros)-methyl-L-histidine Chemical compound CN1C=NC=C1C[C@H](N)C(O)=O JDHILDINMRGULE-LURJTMIESA-N 0.000 description 2
- BNQSTAOJRULKNX-UHFFFAOYSA-N N-(6-acetamidohexyl)acetamide Chemical compound CC(=O)NCCCCCCNC(C)=O BNQSTAOJRULKNX-UHFFFAOYSA-N 0.000 description 2
- JJIHLJJYMXLCOY-BYPYZUCNSA-N N-acetyl-L-serine Chemical compound CC(=O)N[C@@H](CO)C(O)=O JJIHLJJYMXLCOY-BYPYZUCNSA-N 0.000 description 2
- PYUSHNKNPOHWEZ-YFKPBYRVSA-N N-formyl-L-methionine Chemical compound CSCC[C@@H](C(O)=O)NC=O PYUSHNKNPOHWEZ-YFKPBYRVSA-N 0.000 description 2
- KSPIYJQBLVDRRI-UHFFFAOYSA-N N-methylisoleucine Chemical compound CCC(C)C(NC)C(O)=O KSPIYJQBLVDRRI-UHFFFAOYSA-N 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 102000003982 Parathyroid hormone Human genes 0.000 description 2
- 108090000445 Parathyroid hormone Proteins 0.000 description 2
- 239000001888 Peptone Substances 0.000 description 2
- 108010080698 Peptones Proteins 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 108010077895 Sarcosine Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 2
- 108010000449 TNF-Related Apoptosis-Inducing Ligand Receptors Proteins 0.000 description 2
- 102000002259 TNF-Related Apoptosis-Inducing Ligand Receptors Human genes 0.000 description 2
- 102100036922 Tumor necrosis factor ligand superfamily member 13B Human genes 0.000 description 2
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 description 2
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 description 2
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 2
- LFTYTUAZOPRMMI-CFRASDGPSA-N UDP-N-acetyl-alpha-D-glucosamine Chemical compound O1[C@H](CO)[C@@H](O)[C@H](O)[C@@H](NC(=O)C)[C@H]1OP(O)(=O)OP(O)(=O)OC[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(NC(=O)C=C2)=O)O1 LFTYTUAZOPRMMI-CFRASDGPSA-N 0.000 description 2
- LFTYTUAZOPRMMI-UHFFFAOYSA-N UNPD164450 Natural products O1C(CO)C(O)C(O)C(NC(=O)C)C1OP(O)(=O)OP(O)(=O)OCC1C(O)C(O)C(N2C(NC(=O)C=C2)=O)O1 LFTYTUAZOPRMMI-UHFFFAOYSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 238000004115 adherent culture Methods 0.000 description 2
- 230000001464 adherent effect Effects 0.000 description 2
- 102000013529 alpha-Fetoproteins Human genes 0.000 description 2
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- JCZLABDVDPYLRZ-AWEZNQCLSA-N biphenylalanine Chemical compound C1=CC(C[C@H](N)C(O)=O)=CC=C1C1=CC=CC=C1 JCZLABDVDPYLRZ-AWEZNQCLSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 150000001720 carbohydrates Chemical group 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000010960 commercial process Methods 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 238000011143 downstream manufacturing Methods 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 229940116977 epidermal growth factor Drugs 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000005090 green fluorescent protein Substances 0.000 description 2
- 239000000122 growth hormone Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 238000002013 hydrophilic interaction chromatography Methods 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 230000016784 immunoglobulin production Effects 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000000411 inducer Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 239000000199 parathyroid hormone Substances 0.000 description 2
- 229960001319 parathyroid hormone Drugs 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 235000019319 peptone Nutrition 0.000 description 2
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical compound OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 2
- 229920001451 polypropylene glycol Polymers 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 210000001236 prokaryotic cell Anatomy 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000004062 sedimentation Methods 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 150000008163 sugars Chemical group 0.000 description 2
- 230000000153 supplemental effect Effects 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 description 2
- 239000003104 tissue culture media Substances 0.000 description 2
- 239000011573 trace mineral Substances 0.000 description 2
- 235000013619 trace mineral Nutrition 0.000 description 2
- 238000003151 transfection method Methods 0.000 description 2
- 230000001131 transforming effect Effects 0.000 description 2
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- GMKMEZVLHJARHF-UHFFFAOYSA-N (2R,6R)-form-2.6-Diaminoheptanedioic acid Natural products OC(=O)C(N)CCCC(N)C(O)=O GMKMEZVLHJARHF-UHFFFAOYSA-N 0.000 description 1
- YOWXRBVUSFLFJO-ZETCQYMHSA-N (2S)-7-amino-2-(methylamino)heptanoic acid Chemical compound CN[C@@H](CCCCCN)C(=O)O YOWXRBVUSFLFJO-ZETCQYMHSA-N 0.000 description 1
- NMDDZEVVQDPECF-LURJTMIESA-N (2s)-2,7-diaminoheptanoic acid Chemical compound NCCCCC[C@H](N)C(O)=O NMDDZEVVQDPECF-LURJTMIESA-N 0.000 description 1
- PIVJVCRQCUYKNZ-HNNXBMFYSA-N (2s)-2-(benzylazaniumyl)-3-phenylpropanoate Chemical compound C([C@@H](C(=O)O)NCC=1C=CC=CC=1)C1=CC=CC=C1 PIVJVCRQCUYKNZ-HNNXBMFYSA-N 0.000 description 1
- BVAUMRCGVHUWOZ-ZETCQYMHSA-N (2s)-2-(cyclohexylazaniumyl)propanoate Chemical compound OC(=O)[C@H](C)NC1CCCCC1 BVAUMRCGVHUWOZ-ZETCQYMHSA-N 0.000 description 1
- FYMNTAQFDTZISY-QMMMGPOBSA-N (2s)-2-amino-3-[4-(diaminomethylideneamino)phenyl]propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N=C(N)N)C=C1 FYMNTAQFDTZISY-QMMMGPOBSA-N 0.000 description 1
- VEVRNHHLCPGNDU-MUGJNUQGSA-N (2s)-2-amino-5-[1-[(5s)-5-amino-5-carboxypentyl]-3,5-bis[(3s)-3-amino-3-carboxypropyl]pyridin-1-ium-4-yl]pentanoate Chemical compound OC(=O)[C@@H](N)CCCC[N+]1=CC(CC[C@H](N)C(O)=O)=C(CCC[C@H](N)C([O-])=O)C(CC[C@H](N)C(O)=O)=C1 VEVRNHHLCPGNDU-MUGJNUQGSA-N 0.000 description 1
- WAMWSIDTKSNDCU-ZETCQYMHSA-N (2s)-2-azaniumyl-2-cyclohexylacetate Chemical compound OC(=O)[C@@H](N)C1CCCCC1 WAMWSIDTKSNDCU-ZETCQYMHSA-N 0.000 description 1
- OPOTYPXOKUZEKY-QMMMGPOBSA-N (2s)-2-nitramido-3-phenylpropanoic acid Chemical compound [O-][N+](=O)N[C@H](C(=O)O)CC1=CC=CC=C1 OPOTYPXOKUZEKY-QMMMGPOBSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- RIJNIVWHYSNSLQ-UHFFFAOYSA-N 2-(2,3-dihydro-1h-inden-2-ylazaniumyl)acetate Chemical compound C1=CC=C2CC(NCC(=O)O)CC2=C1 RIJNIVWHYSNSLQ-UHFFFAOYSA-N 0.000 description 1
- LRHRHAWNXCGABU-UHFFFAOYSA-N 2-(cyclopentylazaniumyl)acetate Chemical compound OC(=O)CNC1CCCC1 LRHRHAWNXCGABU-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- RDFMDVXONNIGBC-UHFFFAOYSA-N 2-aminoheptanoic acid Chemical compound CCCCCC(N)C(O)=O RDFMDVXONNIGBC-UHFFFAOYSA-N 0.000 description 1
- JUQLUIFNNFIIKC-UHFFFAOYSA-N 2-aminopimelic acid Chemical compound OC(=O)C(N)CCCCC(O)=O JUQLUIFNNFIIKC-UHFFFAOYSA-N 0.000 description 1
- OFYAYGJCPXRNBL-UHFFFAOYSA-N 2-azaniumyl-3-naphthalen-1-ylpropanoate Chemical compound C1=CC=C2C(CC(N)C(O)=O)=CC=CC2=C1 OFYAYGJCPXRNBL-UHFFFAOYSA-N 0.000 description 1
- YXDGRBPZVQPESQ-QMMMGPOBSA-N 4-[(2s)-2-amino-2-carboxyethyl]benzoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(C(O)=O)C=C1 YXDGRBPZVQPESQ-QMMMGPOBSA-N 0.000 description 1
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 description 1
- 108010054404 Adenylyl-sulfate kinase Proteins 0.000 description 1
- 102100021266 Alpha-(1,6)-fucosyltransferase Human genes 0.000 description 1
- 101710146120 Alpha-(1,6)-fucosyltransferase Proteins 0.000 description 1
- 102100034608 Angiopoietin-2 Human genes 0.000 description 1
- 108010048036 Angiopoietin-2 Proteins 0.000 description 1
- 102000005666 Apolipoprotein A-I Human genes 0.000 description 1
- 108010059886 Apolipoprotein A-I Proteins 0.000 description 1
- 102100029470 Apolipoprotein E Human genes 0.000 description 1
- 101710095339 Apolipoprotein E Proteins 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N Arginine Chemical compound OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 108010028006 B-Cell Activating Factor Proteins 0.000 description 1
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 1
- 102100023995 Beta-nerve growth factor Human genes 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 1
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 101710167766 C-type lectin domain family 11 member A Proteins 0.000 description 1
- 102100032528 C-type lectin domain family 11 member A Human genes 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 108010008629 CA-125 Antigen Proteins 0.000 description 1
- 102000007269 CA-125 Antigen Human genes 0.000 description 1
- 102100024217 CAMPATH-1 antigen Human genes 0.000 description 1
- 108010046080 CD27 Ligand Proteins 0.000 description 1
- 102100027207 CD27 antigen Human genes 0.000 description 1
- 108010017987 CD30 Ligand Proteins 0.000 description 1
- 102100032912 CD44 antigen Human genes 0.000 description 1
- 108010065524 CD52 Antigen Proteins 0.000 description 1
- 102100025221 CD70 antigen Human genes 0.000 description 1
- 102000055006 Calcitonin Human genes 0.000 description 1
- 108060001064 Calcitonin Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 102000013602 Cardiac Myosins Human genes 0.000 description 1
- 108010051609 Cardiac Myosins Proteins 0.000 description 1
- 102000001327 Chemokine CCL5 Human genes 0.000 description 1
- 108010055166 Chemokine CCL5 Proteins 0.000 description 1
- 102100022641 Coagulation factor IX Human genes 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 108010028773 Complement C5 Proteins 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 1
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 1
- 102000010170 Death domains Human genes 0.000 description 1
- 108050001718 Death domains Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 108010082495 Dietary Plant Proteins Proteins 0.000 description 1
- 108700021041 Disintegrin Proteins 0.000 description 1
- 102000001301 EGF receptor Human genes 0.000 description 1
- 108060006698 EGF receptor Proteins 0.000 description 1
- 102100029722 Ectonucleoside triphosphate diphosphohydrolase 1 Human genes 0.000 description 1
- 108010067770 Endopeptidase K Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 102000003951 Erythropoietin Human genes 0.000 description 1
- 108090000394 Erythropoietin Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- 108010076282 Factor IX Proteins 0.000 description 1
- 108010054218 Factor VIII Proteins 0.000 description 1
- 102000001690 Factor VIII Human genes 0.000 description 1
- 108010054265 Factor VIIa Proteins 0.000 description 1
- 102000003972 Fibroblast growth factor 7 Human genes 0.000 description 1
- 108090000385 Fibroblast growth factor 7 Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 1
- 102100035233 Furin Human genes 0.000 description 1
- 108090001126 Furin Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 102400000321 Glucagon Human genes 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- 108010017544 Glucosylceramidase Proteins 0.000 description 1
- 102000004547 Glucosylceramidase Human genes 0.000 description 1
- IKAIKUBBJHFNBZ-LURJTMIESA-N Gly-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)CN IKAIKUBBJHFNBZ-LURJTMIESA-N 0.000 description 1
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 108010054017 Granulocyte Colony-Stimulating Factor Receptors Proteins 0.000 description 1
- 102100039622 Granulocyte colony-stimulating factor receptor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 108010092372 Granulocyte-Macrophage Colony-Stimulating Factor Receptors Proteins 0.000 description 1
- 102000016355 Granulocyte-Macrophage Colony-Stimulating Factor Receptors Human genes 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 101150001754 Gusb gene Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 108010058597 HLA-DR Antigens Proteins 0.000 description 1
- 102000006354 HLA-DR Antigens Human genes 0.000 description 1
- 108010036449 HLA-DR10 antigen Proteins 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 1
- 102100021866 Hepatocyte growth factor Human genes 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 1
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 1
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 1
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 1
- 101001012447 Homo sapiens Ectonucleoside triphosphate diphosphohydrolase 1 Proteins 0.000 description 1
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 description 1
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 1
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 1
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 1
- YZJSUQQZGCHHNQ-UHFFFAOYSA-N Homoglutamine Chemical compound OC(=O)C(N)CCCC(N)=O YZJSUQQZGCHHNQ-UHFFFAOYSA-N 0.000 description 1
- LCWXJXMHJVIJFK-UHFFFAOYSA-N Hydroxylysine Natural products NCC(O)CC(N)CC(O)=O LCWXJXMHJVIJFK-UHFFFAOYSA-N 0.000 description 1
- GRRNUXAQVGOGFE-UHFFFAOYSA-N Hygromycin-B Natural products OC1C(NC)CC(N)C(O)C1OC1C2OC3(C(C(O)C(O)C(C(N)CO)O3)O)OC2C(O)C(CO)O1 GRRNUXAQVGOGFE-UHFFFAOYSA-N 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 1
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 1
- 102000012745 Immunoglobulin Subunits Human genes 0.000 description 1
- 108010079585 Immunoglobulin Subunits Proteins 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 1
- 102000014429 Insulin-like growth factor Human genes 0.000 description 1
- 108010008212 Integrin alpha4beta1 Proteins 0.000 description 1
- 102100025390 Integrin beta-2 Human genes 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000003814 Interleukin-10 Human genes 0.000 description 1
- 108090000174 Interleukin-10 Proteins 0.000 description 1
- 102000004559 Interleukin-13 Receptors Human genes 0.000 description 1
- 108010017511 Interleukin-13 Receptors Proteins 0.000 description 1
- 102000004556 Interleukin-15 Receptors Human genes 0.000 description 1
- 108010017535 Interleukin-15 Receptors Proteins 0.000 description 1
- 102000004554 Interleukin-17 Receptors Human genes 0.000 description 1
- 108010017525 Interleukin-17 Receptors Proteins 0.000 description 1
- 102000004388 Interleukin-4 Human genes 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 108010002616 Interleukin-5 Proteins 0.000 description 1
- 102000000743 Interleukin-5 Human genes 0.000 description 1
- 102000010781 Interleukin-6 Receptors Human genes 0.000 description 1
- 108010038501 Interleukin-6 Receptors Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 102000000704 Interleukin-7 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 102000004890 Interleukin-8 Human genes 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 description 1
- QUOGESRFPZDMMT-UHFFFAOYSA-N L-Homoarginine Natural products OC(=O)C(N)CCCCNC(N)=N QUOGESRFPZDMMT-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- 108010092694 L-Selectin Proteins 0.000 description 1
- AGPKZVBTJJNPAG-UHNVWZDZSA-N L-allo-Isoleucine Chemical compound CC[C@@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-UHNVWZDZSA-N 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 1
- QUOGESRFPZDMMT-YFKPBYRVSA-N L-homoarginine Chemical compound OC(=O)[C@@H](N)CCCCNC(N)=N QUOGESRFPZDMMT-YFKPBYRVSA-N 0.000 description 1
- XIGSAGMEBXLVJJ-YFKPBYRVSA-N L-homocitrulline Chemical compound NC(=O)NCCCC[C@H]([NH3+])C([O-])=O XIGSAGMEBXLVJJ-YFKPBYRVSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- 102100033467 L-selectin Human genes 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 description 1
- 102000003959 Lymphotoxin-beta Human genes 0.000 description 1
- 108090000362 Lymphotoxin-beta Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 101710132836 Membrane primary amine oxidase Proteins 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 108090000143 Mouse Proteins Proteins 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 101000845218 Mus musculus Thymic stromal lymphopoietin Proteins 0.000 description 1
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 1
- NTNWOCRCBQPEKQ-YFKPBYRVSA-N N(omega)-methyl-L-arginine Chemical compound CN=C(N)NCCC[C@H](N)C(O)=O NTNWOCRCBQPEKQ-YFKPBYRVSA-N 0.000 description 1
- OLNLSTNFRUFTLM-UHFFFAOYSA-N N-ethylasparagine Chemical compound CCNC(C(O)=O)CC(N)=O OLNLSTNFRUFTLM-UHFFFAOYSA-N 0.000 description 1
- YPIGGYHFMKJNKV-UHFFFAOYSA-N N-ethylglycine Chemical compound CC[NH2+]CC([O-])=O YPIGGYHFMKJNKV-UHFFFAOYSA-N 0.000 description 1
- 108010065338 N-ethylglycine Proteins 0.000 description 1
- AKCRVYNORCOYQT-YFKPBYRVSA-N N-methyl-L-valine Chemical compound CN[C@@H](C(C)C)C(O)=O AKCRVYNORCOYQT-YFKPBYRVSA-N 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 108090000742 Neurotrophin 3 Proteins 0.000 description 1
- 102100029268 Neurotrophin-3 Human genes 0.000 description 1
- 102100023050 Nuclear factor NF-kappa-B p105 subunit Human genes 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 102000004140 Oncostatin M Human genes 0.000 description 1
- 108090000630 Oncostatin M Proteins 0.000 description 1
- 239000012570 Opti-MEM I medium Substances 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 108010035042 Osteoprotegerin Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000015795 Platelet Membrane Glycoproteins Human genes 0.000 description 1
- 108010010336 Platelet Membrane Glycoproteins Proteins 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 239000005700 Putrescine Substances 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 108090000244 Rat Proteins Proteins 0.000 description 1
- 108010038036 Receptor Activator of Nuclear Factor-kappa B Proteins 0.000 description 1
- 102000010498 Receptor Activator of Nuclear Factor-kappa B Human genes 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 102100034201 Sclerostin Human genes 0.000 description 1
- 108050006698 Sclerostin Proteins 0.000 description 1
- 101150083863 Slc35a3 gene Proteins 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 102100039024 Sphingosine kinase 1 Human genes 0.000 description 1
- 108010039445 Stem Cell Factor Proteins 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 102000019197 Superoxide Dismutase Human genes 0.000 description 1
- 108010012715 Superoxide dismutase Proteins 0.000 description 1
- 108010000499 Thromboplastin Proteins 0.000 description 1
- 102000002262 Thromboplastin Human genes 0.000 description 1
- 102000036693 Thrombopoietin Human genes 0.000 description 1
- 108010041111 Thrombopoietin Proteins 0.000 description 1
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 1
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 102000046299 Transforming Growth Factor beta1 Human genes 0.000 description 1
- 102000011117 Transforming Growth Factor beta2 Human genes 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 101800002279 Transforming growth factor beta-1 Proteins 0.000 description 1
- 101800000304 Transforming growth factor beta-2 Proteins 0.000 description 1
- 102100032100 Tumor necrosis factor ligand superfamily member 8 Human genes 0.000 description 1
- 102100032236 Tumor necrosis factor receptor superfamily member 11B Human genes 0.000 description 1
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 1
- 206010054094 Tumour necrosis Diseases 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 238000012452 Xenomouse strains Methods 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- 229960003697 abatacept Drugs 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- SNEIUMQYRCDYCH-UHFFFAOYSA-N acetylarginine Chemical compound CC(=O)NC(C(O)=O)CCCN=C(N)N SNEIUMQYRCDYCH-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 description 1
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 1
- 229940024142 alpha 1-antitrypsin Drugs 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- QWCKQJZIFLGMSD-UHFFFAOYSA-N alpha-aminobutyric acid Chemical compound CCC(N)C(O)=O QWCKQJZIFLGMSD-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229940124277 aminobutyric acid Drugs 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 235000021120 animal protein Nutrition 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 229950001863 bapineuzumab Drugs 0.000 description 1
- 210000000270 basal cell Anatomy 0.000 description 1
- 229960004669 basiliximab Drugs 0.000 description 1
- 229960003270 belimumab Drugs 0.000 description 1
- 229940000635 beta-alanine Drugs 0.000 description 1
- 150000001576 beta-amino acids Chemical class 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 239000012496 blank sample Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 229960002874 briakinumab Drugs 0.000 description 1
- 229960003735 brodalumab Drugs 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 1
- 229960004015 calcitonin Drugs 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229960001838 canakinumab Drugs 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 229920006317 cationic polymer Polymers 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 238000012777 commercial manufacturing Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 1
- 229950007276 conatumumab Drugs 0.000 description 1
- 239000003636 conditioned culture medium Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 230000001120 cytoprotective effect Effects 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- AIUDWMLXCFRVDR-UHFFFAOYSA-N dimethyl 2-(3-ethyl-3-methylpentyl)propanedioate Chemical class CCC(C)(CC)CCC(C(=O)OC)C(=O)OC AIUDWMLXCFRVDR-UHFFFAOYSA-N 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 229960002224 eculizumab Drugs 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940105423 erythropoietin Drugs 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- 229950004912 etrolizumab Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 229960004222 factor ix Drugs 0.000 description 1
- 229940012414 factor viia Drugs 0.000 description 1
- 229960000301 factor viii Drugs 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 108700014844 flt3 ligand Proteins 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 235000021588 free fatty acids Nutrition 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 101150023212 fut8 gene Proteins 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- 229940044627 gamma-interferon Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 108060003196 globin Proteins 0.000 description 1
- 102000018146 globin Human genes 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- 108010015792 glycyllysine Proteins 0.000 description 1
- 229960001743 golimumab Drugs 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 102000056133 human AOC3 Human genes 0.000 description 1
- 102000051284 human FGL2 Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 210000004754 hybrid cell Anatomy 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- QJHBJHUKURJDLG-UHFFFAOYSA-N hydroxy-L-lysine Natural products NCCCCC(NO)C(O)=O QJHBJHUKURJDLG-UHFFFAOYSA-N 0.000 description 1
- GRRNUXAQVGOGFE-NZSRVPFOSA-N hygromycin B Chemical compound O[C@@H]1[C@@H](NC)C[C@@H](N)[C@H](O)[C@H]1O[C@H]1[C@H]2O[C@@]3([C@@H]([C@@H](O)[C@@H](O)[C@@H](C(N)CO)O3)O)O[C@H]2[C@@H](O)[C@@H](CO)O1 GRRNUXAQVGOGFE-NZSRVPFOSA-N 0.000 description 1
- 229940097277 hygromycin b Drugs 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 230000014726 immortalization of host cell Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 150000002484 inorganic compounds Chemical class 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 108010043603 integrin alpha4beta7 Proteins 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- RGXCTRIQQODGIZ-UHFFFAOYSA-O isodesmosine Chemical compound OC(=O)C(N)CCCC[N+]1=CC(CCC(N)C(O)=O)=CC(CCC(N)C(O)=O)=C1CCCC(N)C(O)=O RGXCTRIQQODGIZ-UHFFFAOYSA-O 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 150000002605 large molecules Chemical class 0.000 description 1
- 238000012007 large scale cell culture Methods 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229950008001 matuzumab Drugs 0.000 description 1
- 239000012092 media component Substances 0.000 description 1
- 238000011177 media preparation Methods 0.000 description 1
- 239000012533 medium component Substances 0.000 description 1
- 239000013028 medium composition Substances 0.000 description 1
- 210000003593 megakaryocyte Anatomy 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- GMKMEZVLHJARHF-SYDPRGILSA-N meso-2,6-diaminopimelic acid Chemical compound [O-]C(=O)[C@@H]([NH3+])CCC[C@@H]([NH3+])C([O-])=O GMKMEZVLHJARHF-SYDPRGILSA-N 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- SXTAYKAGBXMACB-UHFFFAOYSA-N methionine S-imide-S-oxide Natural products CS(=N)(=O)CCC(N)C(O)=O SXTAYKAGBXMACB-UHFFFAOYSA-N 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000007837 multiplex assay Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 229940053128 nerve growth factor Drugs 0.000 description 1
- 229940032018 neurotrophin 3 Drugs 0.000 description 1
- 229940043504 normal human immunoglobulins Drugs 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 235000021232 nutrient availability Nutrition 0.000 description 1
- XXUPLYBCNPLTIW-UHFFFAOYSA-N octadec-7-ynoic acid Chemical compound CCCCCCCCCCC#CCCCCCC(O)=O XXUPLYBCNPLTIW-UHFFFAOYSA-N 0.000 description 1
- 230000009437 off-target effect Effects 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 229960000402 palivizumab Drugs 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 229940066779 peptones Drugs 0.000 description 1
- 229960002087 pertuzumab Drugs 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920001993 poloxamer 188 Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 1
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000013587 production medium Substances 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 238000001498 protein fragment complementation assay Methods 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 238000005086 pumping Methods 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 108091006084 receptor activators Proteins 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000011218 seed culture Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000002639 sodium chloride Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000010473 stable expression Effects 0.000 description 1
- 238000003153 stable transfection Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229940126622 therapeutic monoclonal antibody Drugs 0.000 description 1
- YSMODUONRAFBET-WHFBIAKZSA-N threo-5-hydroxy-L-lysine Chemical compound NC[C@@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-WHFBIAKZSA-N 0.000 description 1
- 229960000187 tissue plasminogen activator Drugs 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- 230000002463 transducing effect Effects 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229920000428 triblock copolymer Polymers 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 238000003211 trypan blue cell staining Methods 0.000 description 1
- 229960004914 vedolizumab Drugs 0.000 description 1
- 231100000747 viability assay Toxicity 0.000 description 1
- 238000003026 viability measurement method Methods 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 229950008250 zalutumumab Drugs 0.000 description 1
- 229950009002 zanolimumab Drugs 0.000 description 1
- JPZXHKDZASGCLU-LBPRGKRZSA-N β-(2-naphthyl)-alanine Chemical compound C1=CC=CC2=CC(C[C@H](N)C(O)=O)=CC=C21 JPZXHKDZASGCLU-LBPRGKRZSA-N 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/005—Glycopeptides, glycoproteins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1048—Glycosyltransferases (2.4)
- C12N9/1051—Hexosyltransferases (2.4.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y204/00—Glycosyltransferases (2.4)
- C12Y204/01—Hexosyltransferases (2.4.1)
- C12Y204/01101—Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase (2.4.1.101)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y204/00—Glycosyltransferases (2.4)
- C12Y204/01—Hexosyltransferases (2.4.1)
- C12Y204/01143—Alpha-1,6-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase (2.4.1.143)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
- C07K2317/41—Glycosylation, sialylation, or fucosylation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y204/00—Glycosyltransferases (2.4)
- C12Y204/01—Hexosyltransferases (2.4.1)
- C12Y204/01094—Protein N-acetylglucosaminyltransferase (2.4.1.94)
Description
本出願は、参照により本明細書に組み込まれる2014年1月29日に出願された米国仮特許出願第61/933,192号の利益を主張する。
本明細書で用いるとき、「a」及び「an」という用語は、特に別段の指示がない限り、1つまたは複数を意味する。さらに、文脈により必要とされない限り、単数形の用語は複数形を含み、複数形の用語は単数形を含むものとする。一般に、本明細書に記載の、細胞及び組織培養、分子生物学、免疫学、微生物学、遺伝学及びタンパク質及び核酸化学及びハイブリダイゼーションに関連して使用される命名法、及びこれらの技法は、当技術分野において周知のものであり、一般に使用される。
組換えタンパク質産生中、所望の密度まで細胞が増殖し、次いで細胞の生理学的状態が、増殖停止した、より多くの細胞を作成する代わりに目的の組み換えタンパク質を産生するために細胞がエネルギーと基質を使用する高生産性状態に切り替えられる、制御システムを有することが望ましい。この目的を達成するための様々な方法が存在し、それには温度変化及びアミノ酸飢餓、ならびに細胞周期阻害剤または細胞死を引き起こさずに細胞増殖を停止することができる他の分子の使用が含まれる。
本発明の方法は、目的の組換えタンパク質を発現する細胞を培養するために使用することができる。発現された組換えタンパク質は、回復及び/または採取されることができる培養培地に分泌され得る。加えて、タンパク質は、そのような培養物または成分から(例えば、培養培地から)、既知のプロセス及び商業的供給者から入手可能な製品を使用して精製または部分的に精製されることができる。次いで、精製されたタンパク質は「製剤化」されることができ、これは、緩衝液が交換され、滅菌され、バルク包装され、及び/または最終使用者用に包装されることを意味する。医薬組成物に好適な製剤には、Remington’s Pharmaceutical Sciences,第18版.1995,Mack Publishing Company,Easton,PAに記載の製剤が含まれる。
低レベルの高マンノース型(HM)発現を歴史的に呈した組換えヒト抗体(MAb A)を発現する、モノクローナル抗体産生チャイニーズハムスター卵巣(CHO)細胞株をsiRNA実験に使用した。細胞株は、DHFRベースの選択を使用してクローン的に誘導された;通例の培養については、細胞は、懸濁液内、メトトレキサート(MTX)を含有する選択培地内で培養された。培養物は、通気性125mLまたは250mLエルレンマイヤー振とうフラスコ(Corning Life Sciences、マサチューセッツ州ローウェル)または50mL通気性スピンチューブ(TPP、スイス、トラザーディンゲン)内で36℃、5%CO2及び85%相対湿度で維持された。エルレンマイヤーフラスコを、120rpm、25mm軌道直径で大容量自動式CO2インキュベーター(Thermo Fisher Scientific、マサチューセッツ州ウォルサム)内で振とうし、スピンチューブを225rpm、50mm軌道直径で大容量ISF4−Xインキュベーター(Kuhner AG、スイス、バーゼル)内で振とうした。
正規化率=(目的の遺伝子のMFI)/(ハウスキーピング遺伝子のMFI)
各標的RNAについてのアッセイの感受性は、シグナルがバックグラウンドを3標準偏差超えた標的濃度として定義される、検出限界(LOD)を決定することにより評価された。変動係数(CV)は、アッセイの精度を測定し、標準偏差と平均値の比率である。
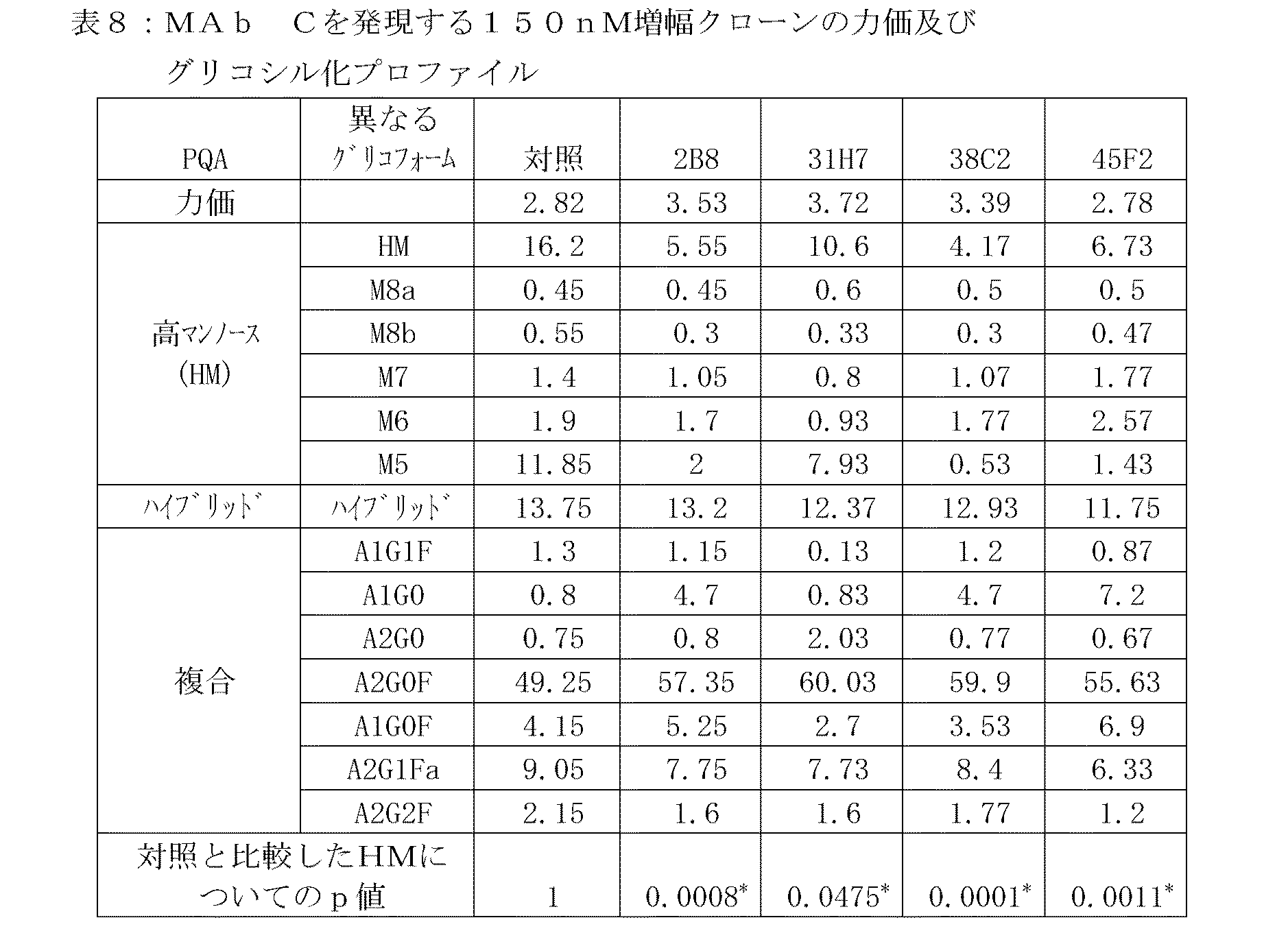
高レベル(すなわち、10%超)の高マンノース型グリカンを歴史的に呈した組換えヒト抗体(MAb B)を発現する、モノクローナル抗体産生チャイニーズハムスター卵巣(CHO)細胞株をトランスフェクション実験に使用した。細胞株は、DHFRベースの選択を使用してクローン的に誘導された;通例の培養については、細胞は懸濁液内、MTXを含有する選択培地内で培養された。培養物は、通気性125mLまたは250mLエルレンマイヤー振とうフラスコ(Corning Life Sciences、マサチューセッツ州ローウェル)または50mL通気性スピンチューブ(TPP、スイス、トラザーディンゲン)内で、実質的に前述の通りに維持された。
CHO宿主細胞を、Mgat1、またはMgat2のいずれかで個別にトランスフェクトした、またはMgat1及びMgat2発現ベクターの両方で同時トランスフェクトした。選択培地において80%を超える生存能までこれらの細胞を回復させた後、これらを、フローサイトメトリーを使用して単一細胞クローニングした。合計で291個のクローンを、Mgat1及びMgat2の遺伝子の発現について解析した。これらの中で、良好な増殖及び生存能に基づいて、歴史的に低レベル(すなわち5%未満)の高マンノース型グリカンを伴うヒトモノクローナル抗体(MAb A)を発現する組換えCHO細胞株において検出されるレベルを超えるMgat1及びMgat2レベルを発現する48個のクローンを選択した。
Claims (25)
- 哺乳類細胞培養により産生される組換えタンパク質の高マンノース型グリコフォーム含量を低減させる方法であって、
N−グリコシル化経路に関与するタンパク質を過剰発現するように哺乳類宿主細胞をトランスフェクトすることと、ここで、前記N−グリコシル化経路に関与する前記タンパク質は、N−アセチル−グルコサミン転移酵素−1(Mgat1によりコードされる);N−アセチル−グルコサミン転移酵素−2(Mgat2によりコードされる);及びそれらの組み合わせからなる群より選択され、
前記哺乳類宿主細胞を培養して、哺乳類細胞培養を確立することと
を含み、
前記哺乳類宿主細胞は、少なくとも1つの組換えタンパク質を発現し、
前記組換えタンパク質の前記高マンノース型グリコフォーム含量は、N−グリコシル化経路に関与するタンパク質を過剰発現するようにトランスフェクトされる前の前記哺乳類宿主細胞により産生される前記組換えタンパク質と比較して、低減されており、
前記組換えタンパク質の前記高マンノース型グリコフォーム含量は、10%以下である、方法。 - 前記N−グリコシル化経路に関与する2つ以上のタンパク質を発現するように前記哺乳類宿主細胞をトランスフェクトし、前記タンパク質が、Mgat1及びMgat2;Mgat1及びSlc35a2によりコードされるUDP−ガラクトース輸送体;Mgat2及びSlc35a2;並びにMgat1、Mgat2及びSlc35a2からなる群から選択される、請求項1に記載の方法。
- 前記哺乳類宿主細胞が、Mgat1及びMgat2を発現するようトランスフェクトされている、請求項1又は2に記載の方法。
- 前記哺乳類宿主細胞が組換えタンパク質を発現するように予めトランスフェクトされており、N−グリコシル化経路に関与するタンパク質を発現するようにその後トランスフェクトされる、請求項1〜3のいずれか1項に記載の方法。
- 前記哺乳類宿主細胞が最初にN−グリコシル化経路に関与するタンパク質でトランスフェクトされ、その後組換えタンパク質を発現するようにトランスフェクトされる、請求項1〜3のいずれか1項に記載の方法。
- 前記組換えタンパク質が、抗体Fc領域を含むタンパク質である、請求項1〜5のいずれか1項に記載の方法。
- 前記組換えタンパク質が、Fc融合タンパク質、抗体、免疫グロブリン、及びペプチボディー(peptibody)からなる群より選択される、請求項6に記載の方法。
- N結合型グリコシル化に関与するタンパク質を過剰発現するようにトランスフェクトされている前記細胞内で発現される前記組換えタンパク質の高マンノース型グリコフォーム含量が、N結合型グリコシル化に関与するタンパク質を過剰発現するようにトランスフェクトされる前の前記哺乳類宿主細胞により産生される組換えタンパク質の高マンノース型グリコフォーム含量と比較して低減されている、請求項1〜7のいずれか1項に記載の方法。
- 前記高マンノース型グリカン種が、マンノース5(Man5);マンノース6(Man6);マンノース7(Man7);マンノース8(マンノース8a及び8b(Man8a及び8b)を含む);Man8a及び8b、及びマンノース9(Man9);又はそれらの組み合わせからなる群より選択される、請求項8に記載の方法。
- 前記組換えタンパク質の前記高マンノース型グリコフォーム含量が、5%以下である、請求項8又は9に記載の方法。
- 前記哺乳類細胞培養が、流加培養、灌流培養、又はそれらの組み合わせである、請求項1〜10のいずれか1項に記載の方法。
- 前記培養が、交互接線流(ATF)を用いて灌流される、請求項11に記載の方法。
- 灌流が前記細胞培養の1日目又はその前後から9日目又はその前後に開始される、請求項12に記載の方法。
- 灌流が前記細胞培養の3日目又はその前後から7日目又はその前後に開始される、請求項12に記載の方法。
- 前記細胞が産生期に達したときに灌流が開始される、請求項12〜14のいずれか1項に記載の方法。
- 灌流が限外濾過膜又は精密濾過膜を用いる交互接線流により達成される、請求項15に記載の方法。
- 前記細胞培養が流加により維持される、請求項11に記載の方法。
- 前記培養が産生中に3回供給を受ける、請求項17に記載の方法。
- 前記培養が、2日目と4日目の間の日、5日目と7日目の間の日、及び8日目と10日目の間の日に供給を受ける、請求項18に記載の方法。
- 前記培養が産生中に4回供給を受ける、請求項17に記載の方法。
- 前記培養が、2日目と4日目の間の日、5日目と6日目の間の日、7日目と8日目の間の日、及び8日目と10日目の間又はそれ以降に供給を受ける、請求項20に記載の方法。
- 前記哺乳類細胞培養が、無血清培養培地中、少なくとも0.5×106から3.0×106細胞/mLで前記バイオリアクターに播種することにより確立される、請求項12〜21のいずれか1項に記載の方法。
- 前記哺乳類細胞がチャイニーズハムスター卵巣(CHO)細胞である、請求項1〜22のいずれか1項に記載の方法。
- 前記方法が、前記組換えタンパク質を回収することをさらに含む、請求項12〜23のいずれか1項に記載の方法。
- 前記回収された組換えタンパク質が精製され、医薬的に許容され得る製剤に製剤化される、請求項24に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461933137P | 2014-01-29 | 2014-01-29 | |
| US61/933,137 | 2014-01-29 | ||
| PCT/US2014/069744 WO2015116315A1 (en) | 2014-01-29 | 2014-12-11 | Overexpression of n-glycosylation pathway regulators to modulate glycosylation of recombinant proteins |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019011871A Division JP6797950B2 (ja) | 2014-01-29 | 2019-01-28 | 組換えタンパク質のグリコシル化を調節するためのn−グリコシル化経路制御因子の過剰発現 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2017505620A JP2017505620A (ja) | 2017-02-23 |
| JP2017505620A5 JP2017505620A5 (ja) | 2017-11-30 |
| JP6474420B2 true JP6474420B2 (ja) | 2019-02-27 |
Family
ID=53757626
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016549077A Active JP6474420B2 (ja) | 2014-01-29 | 2014-12-11 | 組換えタンパク質のグリコシル化を調節するためのn−グリコシル化経路制御因子の過剰発現 |
| JP2019011871A Active JP6797950B2 (ja) | 2014-01-29 | 2019-01-28 | 組換えタンパク質のグリコシル化を調節するためのn−グリコシル化経路制御因子の過剰発現 |
| JP2020190707A Active JP7034236B2 (ja) | 2014-01-29 | 2020-11-17 | 組換えタンパク質のグリコシル化を調節するためのn-グリコシル化経路制御因子の過剰発現 |
| JP2022029077A Pending JP2022068365A (ja) | 2014-01-29 | 2022-02-28 | 組換えタンパク質のグリコシル化を調節するためのn-グリコシル化経路制御因子の過剰発現 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019011871A Active JP6797950B2 (ja) | 2014-01-29 | 2019-01-28 | 組換えタンパク質のグリコシル化を調節するためのn−グリコシル化経路制御因子の過剰発現 |
| JP2020190707A Active JP7034236B2 (ja) | 2014-01-29 | 2020-11-17 | 組換えタンパク質のグリコシル化を調節するためのn-グリコシル化経路制御因子の過剰発現 |
| JP2022029077A Pending JP2022068365A (ja) | 2014-01-29 | 2022-02-28 | 組換えタンパク質のグリコシル化を調節するためのn-グリコシル化経路制御因子の過剰発現 |
Country Status (14)
| Country | Link |
|---|---|
| US (3) | US10227627B2 (ja) |
| EP (1) | EP3099806A1 (ja) |
| JP (4) | JP6474420B2 (ja) |
| KR (3) | KR102519540B1 (ja) |
| CN (2) | CN106170554B (ja) |
| AU (3) | AU2014380174B2 (ja) |
| BR (1) | BR112016017606A2 (ja) |
| CA (1) | CA2938079C (ja) |
| CL (1) | CL2016001900A1 (ja) |
| EA (2) | EA202190244A3 (ja) |
| IL (3) | IL282517B (ja) |
| MX (2) | MX2016009911A (ja) |
| SG (1) | SG10201912081RA (ja) |
| WO (1) | WO2015116315A1 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101936049B1 (ko) | 2015-10-15 | 2019-01-08 | (주)알테오젠 | IgG Fc 도메인을 가지는 융합 단백질의 생산방법 |
| US20200131518A1 (en) * | 2017-03-14 | 2020-04-30 | Amgen Inc. | Control of total afucosylated glycoforms of antibodies produced in cell culture |
| CN109576212B (zh) * | 2018-12-14 | 2021-06-22 | 杭州奕安济世生物药业有限公司 | 高密度接种培养中种子细胞的培养方法及其应用 |
| CA3216655A1 (en) | 2021-04-23 | 2022-10-27 | Amgen Inc. | Anti-tslp antibody compositions and uses thereof |
| CN113684322B (zh) * | 2021-09-09 | 2024-01-23 | 上海药明生物技术有限公司 | 一种降低抗体类蛋白高聚甘露糖型水平的方法 |
| CN114525252B (zh) * | 2022-03-10 | 2023-12-01 | 广州源井生物科技有限公司 | 一种提高mda-mb-231单细胞克隆形成率的单克隆增强培养基与培养方法及其应用 |
| WO2024092064A1 (en) | 2022-10-26 | 2024-05-02 | Amgen Inc. | Anti-tslp antibody compositions and uses thereof |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6936694B1 (en) | 1982-05-06 | 2005-08-30 | Intermune, Inc. | Manufacture and expression of large structural genes |
| AU588819B2 (en) | 1984-10-29 | 1989-09-28 | Immunex Corporation | Cloning of human granulocyte-macrophage colony stimulating factor gene |
| US5672502A (en) | 1985-06-28 | 1997-09-30 | Celltech Therapeutics Limited | Animal cell culture |
| US4968607A (en) | 1987-11-25 | 1990-11-06 | Immunex Corporation | Interleukin-1 receptors |
| US5075222A (en) | 1988-05-27 | 1991-12-24 | Synergen, Inc. | Interleukin-1 inhibitors |
| AU643427B2 (en) | 1988-10-31 | 1993-11-18 | Immunex Corporation | Interleukin-4 receptors |
| US5395760A (en) | 1989-09-05 | 1995-03-07 | Immunex Corporation | DNA encoding tumor necrosis factor-α and -β receptors |
| EP1132471A3 (de) | 1989-09-12 | 2001-11-28 | F. Hoffmann-La Roche Ag | TNF-bindende Proteine |
| US6204363B1 (en) | 1989-10-16 | 2001-03-20 | Amgen Inc. | Stem cell factor |
| US5149792A (en) | 1989-12-19 | 1992-09-22 | Amgen Inc. | Platelet-derived growth factor B chain analogs |
| US5272064A (en) | 1989-12-19 | 1993-12-21 | Amgen Inc. | DNA molecules encoding platelet-derived growth factor B chain analogs and method for expression thereof |
| US5350683A (en) | 1990-06-05 | 1994-09-27 | Immunex Corporation | DNA encoding type II interleukin-1 receptors |
| AU651596B2 (en) | 1990-06-05 | 1994-07-28 | Immunex Corporation | Type II interleukin-1 receptors |
| GB9014932D0 (en) | 1990-07-05 | 1990-08-22 | Celltech Ltd | Recombinant dna product and method |
| WO1994004679A1 (en) | 1991-06-14 | 1994-03-03 | Genentech, Inc. | Method for making humanized antibodies |
| JPH05244982A (ja) | 1991-12-06 | 1993-09-24 | Sumitomo Chem Co Ltd | 擬人化b−b10 |
| WO1994010308A1 (en) | 1992-10-23 | 1994-05-11 | Immunex Corporation | Methods of preparing soluble, oligomeric proteins |
| US5981713A (en) | 1994-10-13 | 1999-11-09 | Applied Research Systems Ars Holding N.V. | Antibodies to intereleukin-1 antagonists |
| NZ311982A (en) | 1995-06-29 | 1999-08-30 | Immunex Corp | Tnf related apoptosis inducing ligand (trail), a method for producing them and associated antibodies |
| US6613544B1 (en) | 1995-12-22 | 2003-09-02 | Amgen Inc. | Osteoprotegerin |
| US6096728A (en) | 1996-02-09 | 2000-08-01 | Amgen Inc. | Composition and method for treating inflammatory diseases |
| US6271349B1 (en) | 1996-12-23 | 2001-08-07 | Immunex Corporation | Receptor activator of NF-κB |
| US6235883B1 (en) | 1997-05-05 | 2001-05-22 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| GB9722131D0 (en) | 1997-10-20 | 1997-12-17 | Medical Res Council | Method |
| US6337072B1 (en) | 1998-04-03 | 2002-01-08 | Hyseq, Inc. | Interleukin-1 receptor antagonist and recombinant production thereof |
| WO2001036637A1 (en) | 1999-11-17 | 2001-05-25 | Immunex Corporation | Receptor activator of nf-kappa b |
| US6544424B1 (en) | 1999-12-03 | 2003-04-08 | Refined Technology Company | Fluid filtration system |
| GB9928787D0 (en) | 1999-12-03 | 2000-02-02 | Medical Res Council | Direct screening method |
| GB0025144D0 (en) | 2000-10-13 | 2000-11-29 | Medical Res Council | Concatenated nucleic acid sequences |
| US20040202995A1 (en) | 2003-04-09 | 2004-10-14 | Domantis | Nucleic acids, proteins, and screening methods |
| US20050186577A1 (en) * | 2004-02-20 | 2005-08-25 | Yixin Wang | Breast cancer prognostics |
| PL1720972T3 (pl) * | 2004-03-05 | 2014-06-30 | Dpx Holdings Bv | Proces hodowli komórek poprzez ciągłą perfuzję i zmienny przepływ styczny |
| BRPI0516011A (pt) | 2004-09-24 | 2008-08-19 | Amgen Inc | moléculas fc modificadas |
| US8053238B2 (en) | 2005-10-31 | 2011-11-08 | Unhwa Corporation | Isolated population of plant single cells and method of preparing the same |
| US20070190057A1 (en) * | 2006-01-23 | 2007-08-16 | Jian Wu | Methods for modulating mannose content of recombinant proteins |
| US20100145032A1 (en) * | 2007-01-18 | 2010-06-10 | Suomen Punainen Risti, Veripalelu | Novel carbohydrate profile compositions from human cells and methods for analysis and modification thereof |
| US20100221823A1 (en) * | 2007-06-11 | 2010-09-02 | Amgen Inc. | Method for culturing mammalian cells to improve recombinant protein production |
| TW201028431A (en) * | 2008-10-31 | 2010-08-01 | Lonza Ag | Novel tools for the production of glycosylated proteins in host cells |
| US9034341B2 (en) * | 2009-04-20 | 2015-05-19 | Transtech Pharma, Llc | Control of RAGE fusion protein glycosylation and RAGE fusion protein compositions |
| US20130196410A1 (en) * | 2010-03-05 | 2013-08-01 | Alnylam Pharmaceuticals, Inc | Compositions and methods for modifying the glycosylation pattern of a polypeptide |
| MX2012011648A (es) * | 2010-04-07 | 2012-11-29 | Momenta Pharmaceuticals Inc | Glicanos de alta manosa. |
| TW201209160A (en) * | 2010-04-27 | 2012-03-01 | Lonza Ag | Improved glycosylation of proteins in host cells |
| US9133493B2 (en) | 2011-04-21 | 2015-09-15 | Amgen Inc. | Method for culturing mammalian cells to improve recombinant protein production |
-
2014
- 2014-12-11 JP JP2016549077A patent/JP6474420B2/ja active Active
- 2014-12-11 IL IL282517A patent/IL282517B/en unknown
- 2014-12-11 SG SG10201912081RA patent/SG10201912081RA/en unknown
- 2014-12-11 KR KR1020227019897A patent/KR102519540B1/ko active IP Right Grant
- 2014-12-11 CN CN201480074585.XA patent/CN106170554B/zh active Active
- 2014-12-11 CA CA2938079A patent/CA2938079C/en active Active
- 2014-12-11 AU AU2014380174A patent/AU2014380174B2/en active Active
- 2014-12-11 MX MX2016009911A patent/MX2016009911A/es active IP Right Grant
- 2014-12-11 EP EP14821424.0A patent/EP3099806A1/en active Pending
- 2014-12-11 EA EA202190244A patent/EA202190244A3/ru unknown
- 2014-12-11 BR BR112016017606A patent/BR112016017606A2/pt not_active Application Discontinuation
- 2014-12-11 KR KR1020217028516A patent/KR102410393B1/ko active IP Right Grant
- 2014-12-11 WO PCT/US2014/069744 patent/WO2015116315A1/en active Application Filing
- 2014-12-11 EA EA201691532A patent/EA037565B1/ru unknown
- 2014-12-11 KR KR1020167021676A patent/KR102301034B1/ko active IP Right Grant
- 2014-12-11 CN CN202110902965.XA patent/CN113774084B/zh active Active
-
2016
- 2016-07-24 IL IL246914A patent/IL246914B/en active IP Right Grant
- 2016-07-26 CL CL2016001900A patent/CL2016001900A1/es unknown
- 2016-07-29 MX MX2020012356A patent/MX2020012356A/es unknown
-
2018
- 2018-09-13 US US16/130,879 patent/US10227627B2/en active Active
- 2018-11-15 AU AU2018264100A patent/AU2018264100B2/en active Active
-
2019
- 2019-01-28 JP JP2019011871A patent/JP6797950B2/ja active Active
- 2019-01-29 US US16/261,311 patent/US10655156B2/en active Active
- 2019-11-24 IL IL270861A patent/IL270861B/en active IP Right Grant
-
2020
- 2020-05-15 US US16/875,832 patent/US10907186B2/en active Active
- 2020-09-23 AU AU2020239681A patent/AU2020239681B2/en active Active
- 2020-11-17 JP JP2020190707A patent/JP7034236B2/ja active Active
-
2022
- 2022-02-28 JP JP2022029077A patent/JP2022068365A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7034236B2 (ja) | 組換えタンパク質のグリコシル化を調節するためのn-グリコシル化経路制御因子の過剰発現 | |
| US11299760B2 (en) | Use of monensin to regulate glycosylation of recombinant proteins | |
| US10106829B2 (en) | Overexpression of N-glycosylation pathway regulators to modulate glycosylation of recombinant proteins |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171019 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20171019 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180724 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20180720 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20181019 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20181101 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20181204 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20181213 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190129 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6474420 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |