JP6419498B2 - 多価ヒドロキシ樹脂及び当該樹脂の製造方法。 - Google Patents
多価ヒドロキシ樹脂及び当該樹脂の製造方法。 Download PDFInfo
- Publication number
- JP6419498B2 JP6419498B2 JP2014186890A JP2014186890A JP6419498B2 JP 6419498 B2 JP6419498 B2 JP 6419498B2 JP 2014186890 A JP2014186890 A JP 2014186890A JP 2014186890 A JP2014186890 A JP 2014186890A JP 6419498 B2 JP6419498 B2 JP 6419498B2
- Authority
- JP
- Japan
- Prior art keywords
- resin
- aromatic monomer
- formula
- crosslinking agent
- epoxy resin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images






Landscapes
- Laminated Bodies (AREA)
- Phenolic Resins Or Amino Resins (AREA)
- Epoxy Resins (AREA)
- Structures Or Materials For Encapsulating Or Coating Semiconductor Devices Or Solid State Devices (AREA)
Description
[1]複数のフェノール性水酸基を有する芳香族モノマーと、以下の式(I)

で表される架橋剤とを反応させて得られる多価ヒドロキシ樹脂であって、
前記多価ヒドロキシ樹脂が、以下の式(II)
で表される構造を有する成分を樹脂全体に対して70%以上含有することを特徴する、該多価ヒドロキシ樹脂;
[2]前記芳香族モノマーが、ジヒドロキシベンゼンである、[1]に記載の多価ヒドロキシ樹脂;
[3]前記芳香族モノマーが、レゾルシンである、[1]に記載の多価ヒドロキシ樹脂;
[4]前記式(I)で表される架橋剤が、オルソヒドロキシベンズアルデヒド、ベンズアルデヒド、又はトルアルデヒドである、[1]〜[3]のいずれか1に記載の多価ヒドロキシ樹脂
を提供するものである。
[5][1]〜[4]のいずれか1に記載の多価ヒドロキシ樹脂をエポキシ化してなる、エポキシ樹脂;
[6][5]に記載のエポキシ樹脂と硬化剤とを含む、熱硬化性成形材料;
[7]前記硬化剤がフェノール樹脂系硬化剤である、[6]に記載の熱硬化性成形材料;
[8][1]〜[4]のいずれか1に記載の多価ヒドロキシ樹脂、及び[5]に記載のエポキシ樹脂よりなる群から選択される1種以上を含有する、熱硬化性成形材料;
[9][6]〜[8]のいずれか1に記載の熱硬化性成形材料よりなる、半導体封止材;
[10][6]〜[8]のいずれか1に記載の熱硬化性成形材料を用いて半導体素子を封止してなる、半導体装置;
[11][6]〜[8]のいずれか1に記載の熱硬化性成形材料よりなる、積層板
を提供するものである。
[12]複数のフェノール性水酸基を有する芳香族モノマーと、以下の式(I)
で表される架橋剤とを溶媒中で反応させる工程、及び、
反応溶液からの析出生成物を分離して以下の式(II)
で表される構造を有する成分を樹脂全体に対して70%以上含有する多価ヒドロキシ樹脂を得る工程を含む、多価ヒドロキシ樹脂の製造方法;
[13]前記芳香族モノマーが、ジヒドロキシベンゼンである、[12]に記載の製造方法;
[14]前記芳香族モノマーが、レゾルシンである、[12]に記載の製造方法;
[15]前記式(I)で表される架橋剤が、オルソヒドロキシベンズアルデヒド、ベンズアルデヒド、又はトルアルデヒドである、[12]〜[14]のいずれか1に記載の製造方法;
[16]前記芳香族モノマーと前記架橋剤を、芳香族モノマー/架橋剤=0.50〜1.20のモル比で反応させる、[12]〜[15]のいずれか1に記載の製造方法;
[17]前記芳香族モノマーと前記架橋剤との反応が、25〜150℃の温度で行われる、[12]〜[16]のいずれか1に記載の製造方法;
[18]前記溶媒が、水、メタノール、又は水−メタノール混合溶媒である、[12]〜[17]のいずれか1に記載の製造方法;
[19]前記芳香族モノマーと前記架橋剤との反応が、臭化水素酸、塩酸、硫酸、リン酸、シュウ酸、トリフルオロ酢酸、p−トルエンスルホン酸、メタンスルホン酸、三フッ化ホウ素、塩化アルミニウム、塩化鉄、及び塩化亜鉛よりなる群から選択される触媒の存在下で行われる、[12]〜[18]のいずれか1に記載の製造方法
を提供するものである。
本発明の1つの実施態様は、複数のフェノール性水酸基を有する芳香族モノマーと、以下の式(I)
前記多価ヒドロキシ樹脂が、以下の式(II)
本発明の多価ヒドロキシ樹脂は、以下の製造方法によって得ることができ、当該方法も本発明に含まれる。すなわち、本発明の製造方法は、複数のフェノール性水酸基を有する芳香族モノマーと、上記式(I)で表される架橋剤とを溶媒中で反応させる工程、及び、反応溶液からの析出生成物を分離して上記式(II)で表される構造を有する成分を樹脂全体に対して70%以上含有する多価ヒドロキシ樹脂を得る工程を含む、多価ヒドロキシ樹脂の製造方法である。
本発明の多価ヒドロキシ樹脂の水酸基をエポキシ化剤でエポキシ化することによって、熱硬化性成形材料に用いられるエポキシ樹脂を得ることができる。
本発明のさらなる態様は、上記多価ヒドロキシ樹脂又は当該樹脂をエポキシ化したエポキシ樹脂組成物を含む熱硬化性成形材料である。当該熱硬化性成形材料は、a)本発明の多価ヒドロキシ樹脂をエポキシ化して得たエポキシ樹脂と任意の硬化剤を配合し、又はb)硬化剤として本発明の多価ヒドロキシ樹脂を用い、任意のエポキシ樹脂と配合し、加熱混合等をすることによって調製することができる。
後述する合成例で得られた樹脂について、分子量Mw、分散度Mw/Mn、軟化点、溶融粘度を以下の方法で測定した。
(1)分子量Mw、分散度Mw/Mn
ゲル浸透クロマトグラフィー(GPC)測定装置:東ソー社製HLC8120GPC
カラム:TSKgel G3000H+G2000H+G2000H
(2)軟化点
JIS K 6910に従って軟化点を測定した。
(3)溶融粘度
150℃に設定した粘度計(ブルックフィールド社製CAP2000 VISCOMETER)により150℃における溶融粘度を測定した。
(4)ガラス転移温度
樹脂成形物を幅10.0mm×長さ55.0mm×厚さ2.0mmに加工し、粘弾性スペクトロメーター(セイコーインスツルメンツ社製DMS 110)を用いて2℃/分の昇温速度で30℃〜350℃の範囲で測定した。
(5)熱分解温度
樹脂成形物を示差熱熱重量同時測定装置(セイコーインスツルメンツ社製TG/DTA6300)により、エアー雰囲気下で熱重量減量を測定し、熱分解開始温度を求めた。
(6)熱膨張率
樹脂成形物を幅2.0mm×長さ2.0mm×厚さ2.0mmに加工し、熱機械的分析装置(日立ハイテク社製TMA7100)を用いて10°/分の昇温速度で30〜350℃の範囲で測定した。
[合成例1]
樹脂A(レゾルシンとオルソヒドロキシベンズアルデヒドとの反応)
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に水440.0g、レゾルシン220.0g(2.0mol)、48%臭化水素酸40.0gを仕込み65℃まで昇温した。そこにオルソヒドロキシベンズアルデヒド183.0g(1.5mol)を2時間かけて滴下し、その後2時間反応を行った。次いで、50%NaOHで中和し、析出した樹脂をろ過後、乾燥させ多価ヒドロキシ樹脂Aを得た。得られた樹脂におけるベンゼン環5核体以上の構造はGPCで解析を行い98.6%であり(図1)、分子量Mw870、分散度Mw/Mnは1.033であった。また、水酸基当量は68g/eqであった。
樹脂B(レゾルシンとパラヒドロキシベンズアルデヒドとの反応)
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に水440.0g、レゾルシン220.0g(2.0mol)、48%臭化水素酸40.0gを仕込み65℃まで昇温した。そこにパラヒドロキシベンズアルデヒド183.0g(1.5mol)を2時間かけて滴下し、その後2時間反応を行った。次いで、50%NaOHで中和し、析出した樹脂をろ過後、乾燥させ多価ヒドロキシ樹脂Bを得た。得られた樹脂におけるベンゼン環5核体以上の構造はGPCで解析を行い97.9%であり(図2)、分子量Mw951、分散度Mw/Mnは1.055であった。また、水酸基当量は69g/eqであった。
樹脂C(レゾルシンとベンズアルデヒドとの反応)
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に水220.0g、レゾルシン220.0g(2.0mol)、48%臭化水素酸40.0gを仕込み80℃まで昇温した。そこにベンズアルデヒド159.0g(1.5mol)を2時間かけて滴下し、その後2時間反応を行った。次いで、水220.0g添加後、50%NaOHで中和し、析出した樹脂をろ過後、乾燥させ多価ヒドロキシ樹脂Cを得た。得られた樹脂におけるベンゼン環5核体以上の構造はGPCで解析を行い98.4%であり(図3)、分子量Mw821、分散度Mw/Mnは1.039であった。また、水酸基当量は88g/eqであった。
樹脂D(レゾルシンとトルアルデヒドとの反応)
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に水110.0g、メタノール110.0g、レゾルシン220.0g(2.0mol)、48%臭化水素酸40.0gを仕込み80℃まで昇温した。そこにパラトルアルデヒド180.0g(1.5mol)を2時間かけて滴下し、その後2時間反応を行った。次いで、水220.0g添加後、50%NaOHで中和し、析出した樹脂をろ過後、乾燥させ多価ヒドロキシ樹脂Dを得た。得られた樹脂におけるベンゼン環5核体以上の構造はGPCで解析を行い93.8%であり(図4)、分子量Mw897、分散度Mw/Mnは1.084であった。また、水酸基当量は93g/eqであった。
樹脂E(レゾルシンとアニスアルデヒドとの反応)
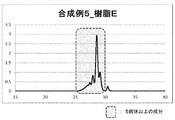
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に水110.0g、メタノール110.0g、レゾルシン220.0g(2.0mol)、48%臭化水素酸40.0gを仕込み80℃まで昇温した。そこにパラアニスアルデヒド204.0g(1.5mol)を2時間かけて滴下し、その後2時間反応を行った。次いで、水220.0g添加後、50%NaOHで中和し、析出した樹脂をろ過後、乾燥させ多価ヒドロキシ樹脂Eを得た。得られた樹脂におけるベンゼン環5核体以上の構造はGPCで解析を行い96.1%であり(図5)、分子量Mw984、分散度Mw/Mnは1.110であった。また、水酸基当量は99g/eqであった。
[合成例6]
樹脂Aのエポキシ化
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に多価ヒドロキシ樹脂Aを68.0g、DMSO136.0g、エピクロロヒドリン462.5g(5.0mol)を仕込み65℃まで昇温した。50%NaOHを発熱に注意しながら1時間かけて滴下した。その後65℃で1時間、80℃に昇温し2時間反応をおこなった。次いで、80℃で減圧下、過剰のエピクロルヒドリン等の溶剤を留去した。残留物にメチルイソブチルケトン204.0g加え溶解し、70℃にまで昇温した。そこに50%の水酸化ナトリウム水溶液2.0gを加え、1時間反応を行なった後、洗浄水が中性になるまで水洗を行ない、得られた溶液を150℃減圧下でメチルイソブチルケトン等を留去することにより多価エポキシ樹脂Aを得た。GPC測定における分子量はMw1609、分散度はMw/Mn1.191であった。また、エポキシ当量は162g/eq、軟化点は94.4℃、180℃における溶融粘度は3.5Pであった。
樹脂Bのエポキシ化
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に多価ヒドロキシ樹脂Bを69.0g、DMSO138.0g、エピクロロヒドリン462.5g(5.0mol)を仕込み65℃まで昇温した。50%NaOHを発熱に注意しながら1時間かけて滴下した。その後65℃で1時間、80℃に昇温し2時間反応をおこなった。次いで、80℃で減圧下、過剰のエピクロルヒドリン等の溶剤を留去した。残留物にメチルイソブチルケトン207.0g加え溶解し、70℃にまで昇温した。そこに50%の水酸化ナトリウム水溶液2.0gを加え、1時間反応を行なった後、洗浄水が中性になるまで水洗を行ない、得られた溶液を150℃減圧下でメチルイソブチルケトン等を留去することにより多価エポキシ樹脂Bを得た。GPC測定における分子量はMw1781、分散度はMw/Mn1.189であった。また、エポキシ当量は168g/eq、軟化点は95.3℃、180℃における溶融粘度は4.3Pであった。
樹脂Cのエポキシ化
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に多価ヒドロキシ樹脂Cを88.0g、DMSO176.0g、エピクロロヒドリン462.5g(5.0mol)を仕込み65℃まで昇温した。50%NaOHを発熱に注意しながら1時間かけて滴下した。その後65℃で1時間、80℃に昇温し2時間反応をおこなった。次いで、80℃で減圧下、過剰のエピクロルヒドリン等の溶剤を留去した。残留物にメチルイソブチルケトン264.0g加え溶解し、70℃にまで昇温した。そこに50%の水酸化ナトリウム水溶液2.0gを加え、1時間反応を行なった後、洗浄水が中性になるまで水洗を行ない、得られた溶液を150℃減圧下でメチルイソブチルケトン等を留去することにより多価エポキシ樹脂Cを得た。GPC測定における分子量はMw1299、分散度はMw/Mn1.176であった。また、エポキシ当量は200g/eq、軟化点は117.9℃、180℃における溶融粘度は12.4Pであった。
樹脂Dのエポキシ化
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に多価ヒドロキシ樹脂Dを93.0g、DMSO186.0g、エピクロロヒドリン462.5g(5.0mol)を仕込み65℃まで昇温した。50%NaOHを発熱に注意しながら1時間かけて滴下した。その後65℃で1時間、80℃に昇温し2時間反応をおこなった。次いで、80℃で減圧下、過剰のエピクロルヒドリン等の溶剤を留去した。残留物にメチルイソブチルケトン279.0g加え溶解し、70℃にまで昇温した。そこに50%の水酸化ナトリウム水溶液2.0gを加え、1時間反応を行なった後、洗浄水が中性になるまで水洗を行ない、得られた溶液を150℃減圧下でメチルイソブチルケトン等を留去することにより多価エポキシ樹脂Dを得た。GPC測定における分子量はMw1226、分散度はMw/Mn1.157であった。また、エポキシ当量は205g/eq、軟化点は99.0℃、180℃における溶融粘度は4.5Pであった。
樹脂Eのエポキシ化
温度計、攪拌機、冷却管を備えた内容量1Lのガラス製反応容器に多価ヒドロキシ樹脂Eを99.0g、DMSO198.0g、エピクロロヒドリン462.5g(5.0mol)を仕込み65℃まで昇温した。50%NaOHを発熱に注意しながら1時間かけて滴下した。その後65℃で1時間、80℃に昇温し2時間反応をおこなった。次いで、80℃で減圧下、過剰のエピクロルヒドリン等の溶剤を留去した。残留物にメチルイソブチルケトン297.0g加え溶解し、70℃にまで昇温した。そこに50%の水酸化ナトリウム水溶液2.0gを加え、1時間反応を行なった後、洗浄水が中性になるまで水洗を行ない、得られた溶液を150℃減圧下でメチルイソブチルケトン等を留去することにより多価エポキシ樹脂Eを得た。GPC測定における分子量はMw1475、分散度はMw/Mn1.154であった。また、エポキシ当量は216g/eq、軟化点は85.9℃、180℃における溶融粘度は3.1Pであった。
各実施例において、エポキシ樹脂及び硬化剤は、エポキシ樹脂中のエポキシ基当量と、硬化剤中の水酸基当量との当量比が1となるように配合量を設定した。使用した他の原料の詳細を以下に示す。
硬化剤:フェノールノボラック樹脂(群栄化学工業社製、製品名「PSM−4261」)
充填剤:球状シリカ(龍森社製、製品名「MSR−2212」)
硬化促進剤:トリフェニルホスフィン
離型剤:カルナバワックス(日本ワックス社製)
Claims (17)
- 複数のフェノール性水酸基を有する芳香族モノマーと、以下の式(I)
で表される架橋剤とを反応させて得られる多価ヒドロキシ樹脂をエポキシ化してなるエポキシ樹脂であって、
前記多価ヒドロキシ樹脂が、以下の式(II)
で表される構造を有する成分を樹脂全体に対して70%以上含有することを特徴する、該エポキシ樹脂。 - 前記芳香族モノマーが、ジヒドロキシベンゼンである、請求項1に記載のエポキシ樹脂。
- 前記芳香族モノマーが、レゾルシンである、請求項1に記載のエポキシ樹脂。
- 前記式(I)で表される架橋剤が、オルソヒドロキシベンズアルデヒド、ベンズアルデヒド、又はトルアルデヒドである、請求項1〜3のいずれか1に記載のエポキシ樹脂。
- 請求項1〜4のいずれか1に記載のエポキシ樹脂と硬化剤とを含む、熱硬化性成形材料。
- 前記硬化剤がフェノール樹脂系硬化剤である、請求項5に記載の熱硬化性成形材料。
- 請求項5又は6に記載の熱硬化性成形材料よりなる、半導体封止材。
- 請求項5又は6に記載の熱硬化性成形材料を用いて半導体素子を封止してなる、半導体装置。
- 請求項5又は6に記載の熱硬化性成形材料よりなる、積層板。
- 複数のフェノール性水酸基を有する芳香族モノマーと、以下の式(I)
で表される架橋剤とを溶媒中で反応させる工程、
反応溶液からの析出生成物を分離して以下の式(II)
で表される構造を有する成分を樹脂全体に対して70%以上含有する多価ヒドロキシ樹脂を得る工程、及び
前記多価ヒドロキシ樹脂の水酸基をエポキシ化剤でエポキシ化する工程
を含む、エポキシ樹脂の製造方法。 - 前記芳香族モノマーが、ジヒドロキシベンゼンである、請求項10に記載の製造方法。
- 前記芳香族モノマーが、レゾルシンである、請求項10に記載の製造方法。
- 前記式(I)で表される架橋剤が、オルソヒドロキシベンズアルデヒド、ベンズアルデヒド、又はトルアルデヒドである、請求項10〜12のいずれか1に記載の製造方法。
- 前記芳香族モノマーと前記架橋剤を、芳香族モノマー/架橋剤=0.50〜1.20のモル比で反応させる、請求項10〜13のいずれか1に記載の製造方法。
- 前記芳香族モノマーと前記架橋剤との反応が、25〜150℃の温度で行われる、請求項10〜14のいずれか1に記載の製造方法。
- 前記溶媒が、水、メタノール、又は水−メタノール混合溶媒である、請求項10〜15のいずれか1に記載の製造方法。
- 前記芳香族モノマーと前記架橋剤との反応が、臭化水素酸、塩酸、硫酸、リン酸、シュウ酸、トリフルオロ酢酸、p−トルエンスルホン酸、メタンスルホン酸、三フッ化ホウ素、塩化アルミニウム、塩化鉄、及び塩化亜鉛よりなる群から選択される触媒の存在下で行われる、請求項10〜16のいずれか1に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014186890A JP6419498B2 (ja) | 2014-09-12 | 2014-09-12 | 多価ヒドロキシ樹脂及び当該樹脂の製造方法。 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014186890A JP6419498B2 (ja) | 2014-09-12 | 2014-09-12 | 多価ヒドロキシ樹脂及び当該樹脂の製造方法。 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016060751A JP2016060751A (ja) | 2016-04-25 |
| JP6419498B2 true JP6419498B2 (ja) | 2018-11-07 |
Family
ID=55795712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014186890A Active JP6419498B2 (ja) | 2014-09-12 | 2014-09-12 | 多価ヒドロキシ樹脂及び当該樹脂の製造方法。 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6419498B2 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201806995A (zh) * | 2016-04-06 | 2018-03-01 | 迪愛生股份有限公司 | 酚醛清漆型樹脂之製造方法 |
| KR102260811B1 (ko) | 2018-12-26 | 2021-06-03 | 삼성에스디아이 주식회사 | 하드마스크 조성물, 하드마스크 층 및 패턴 형성 방법 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62201832A (ja) * | 1978-10-17 | 1987-09-05 | Kanegafuchi Chem Ind Co Ltd | フエノ−ル性化合物と芳香族アルデヒドとの反応生成物及びその製造方法 |
| JP2014157169A (ja) * | 2011-06-24 | 2014-08-28 | Nissan Chem Ind Ltd | ポリヒドロキシベンゼンノボラック樹脂を含むレジスト下層膜形成組成物 |
-
2014
- 2014-09-12 JP JP2014186890A patent/JP6419498B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016060751A (ja) | 2016-04-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6406847B2 (ja) | 変性多価ヒドロキシ樹脂、エポキシ樹脂、エポキシ樹脂組成物及びその硬化物 | |
| JP6091295B2 (ja) | エポキシ樹脂組成物及びその硬化物 | |
| JP5682928B2 (ja) | フェノール樹脂、エポキシ樹脂及びその硬化物 | |
| JP5931234B2 (ja) | エポキシ樹脂組成物の製造方法 | |
| JPWO2014065152A1 (ja) | エポキシ樹脂組成物、エポキシ樹脂硬化物の製造方法、及び半導体装置 | |
| JP2017095524A (ja) | エポキシ樹脂、エポキシ樹脂組成物、及びその硬化物 | |
| JP6605828B2 (ja) | 多価ヒドロキシ樹脂、エポキシ樹脂、それらの製造方法、エポキシ樹脂組成物及びその硬化物 | |
| JP6462295B2 (ja) | 変性多価ヒドロキシ樹脂、エポキシ樹脂、エポキシ樹脂組成物及びその硬化物 | |
| JP5000191B2 (ja) | カルバゾール骨格含有樹脂、カルバゾール骨格含有エポキシ樹脂、エポキシ樹脂組成物及びその硬化物 | |
| JP6799370B2 (ja) | 多価ヒドロキシ樹脂、エポキシ樹脂、それらの製造方法、エポキシ樹脂組成物及びその硬化物 | |
| TWI648317B (zh) | Phenolic resin, phenol resin mixture, epoxy resin, epoxy resin composition and hardened materials thereof | |
| JP6139997B2 (ja) | エポキシ樹脂、エポキシ樹脂組成物、及びその硬化物 | |
| JP6124865B2 (ja) | 多価ヒドロキシ樹脂、エポキシ樹脂、それらの製造方法、エポキシ樹脂組成物及びその硬化物 | |
| JP6419498B2 (ja) | 多価ヒドロキシ樹脂及び当該樹脂の製造方法。 | |
| JPWO2003068837A1 (ja) | インドール樹脂、エポキシ樹脂及びこれらの樹脂を含む樹脂組成物 | |
| JP2010229304A (ja) | フェノール樹脂、該樹脂の製造方法及び該樹脂を含むエポキシ樹脂組成物、ならびにその硬化物 | |
| JP2014210860A (ja) | エポキシ樹脂、エポキシ樹脂組成物およびその硬化物 | |
| JP5841947B2 (ja) | エポキシ樹脂組成物及び硬化物 | |
| JP6292925B2 (ja) | エポキシ樹脂組成物及びその硬化物 | |
| JP2017061665A (ja) | カリックスアレーン系化合物、組成物、その製造方法、エポキシ樹脂用硬化剤、エポキシ樹脂組成物、積層板、半導体封止材及び半導体装置 | |
| JP4667753B2 (ja) | エポキシ樹脂の製造方法、エポキシ樹脂組成物及び硬化物 | |
| JP5579300B2 (ja) | エポキシ樹脂、エポキシ樹脂組成物及びその硬化物 | |
| WO2023162693A1 (ja) | エポキシ樹脂、多価ヒドロキシ樹脂、エポキシ樹脂組成物、及びエポキシ樹脂硬化物並びに多価ヒドロキシ樹脂の製造方法 | |
| WO2015060307A1 (ja) | フェノール樹脂、エポキシ樹脂、エポキシ樹脂組成物、プリプレグ、およびその硬化物 | |
| JP2022118926A (ja) | ジヒドロキシ化合物、エポキシ樹脂、その製造方法、それを用いたエポキシ樹脂組成物及び硬化物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20170407 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20180202 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180313 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180424 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180925 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20181010 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6419498 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |