JP6404215B2 - Syk阻害剤としてのピロロ[2,3−b]ピラジン - Google Patents
Syk阻害剤としてのピロロ[2,3−b]ピラジン Download PDFInfo
- Publication number
- JP6404215B2 JP6404215B2 JP2015527883A JP2015527883A JP6404215B2 JP 6404215 B2 JP6404215 B2 JP 6404215B2 JP 2015527883 A JP2015527883 A JP 2015527883A JP 2015527883 A JP2015527883 A JP 2015527883A JP 6404215 B2 JP6404215 B2 JP 6404215B2
- Authority
- JP
- Japan
- Prior art keywords
- pyrrolo
- pyrazine
- tert
- butyl
- carboxamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000003112 inhibitor Substances 0.000 title description 8
- WMHRXFNTQPIYDT-UHFFFAOYSA-N pyrrolo[2,3-b]pyrazine Chemical compound C1=C[N]C2=NC=CC2=N1 WMHRXFNTQPIYDT-UHFFFAOYSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 123
- 125000000217 alkyl group Chemical group 0.000 claims description 71
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 17
- 239000003814 drug Substances 0.000 claims description 16
- 125000003545 alkoxy group Chemical group 0.000 claims description 15
- 201000010099 disease Diseases 0.000 claims description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 14
- 150000003839 salts Chemical class 0.000 claims description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims description 12
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 12
- 238000011282 treatment Methods 0.000 claims description 12
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 10
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 8
- 208000006673 asthma Diseases 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 8
- 229910052805 deuterium Inorganic materials 0.000 claims description 8
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 206010012601 diabetes mellitus Diseases 0.000 claims description 7
- 150000001408 amides Chemical class 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 6
- 206010028980 Neoplasm Diseases 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 208000026278 immune system disease Diseases 0.000 claims description 5
- GMDSTVTWCURGLX-UHFFFAOYSA-N 2-[3-(1-aminoethyl)anilino]-n-tert-butyl-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC(N)C1=CC=CC(NC=2N=C3C(C(=O)NC(C)(C)C)=CNC3=NC=2)=C1 GMDSTVTWCURGLX-UHFFFAOYSA-N 0.000 claims description 4
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 4
- 208000029462 Immunodeficiency disease Diseases 0.000 claims description 4
- 108010025020 Nerve Growth Factor Proteins 0.000 claims description 4
- 102000007072 Nerve Growth Factors Human genes 0.000 claims description 4
- 239000002260 anti-inflammatory agent Substances 0.000 claims description 4
- 229940121363 anti-inflammatory agent Drugs 0.000 claims description 4
- 230000001028 anti-proliverative effect Effects 0.000 claims description 4
- 201000011510 cancer Diseases 0.000 claims description 4
- 238000002512 chemotherapy Methods 0.000 claims description 4
- 239000003085 diluting agent Substances 0.000 claims description 4
- 239000002955 immunomodulating agent Substances 0.000 claims description 4
- 239000003018 immunosuppressive agent Substances 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- NABZXOUEDLQIKE-UHFFFAOYSA-N n-tert-butyl-2-(2,4-dimethylanilino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=CC(C)=CC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 NABZXOUEDLQIKE-UHFFFAOYSA-N 0.000 claims description 4
- BMODYWDWHTWCPV-UHFFFAOYSA-N n-tert-butyl-2-(2-fluoro-4-methylanilino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound FC1=CC(C)=CC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 BMODYWDWHTWCPV-UHFFFAOYSA-N 0.000 claims description 4
- OPDIWJLWMXZILR-UHFFFAOYSA-N n-tert-butyl-2-(3-methylsulfonylanilino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CNC2=NC=C1NC1=CC=CC(S(C)(=O)=O)=C1 OPDIWJLWMXZILR-UHFFFAOYSA-N 0.000 claims description 4
- MXWBPVUOAUXKGW-UHFFFAOYSA-N n-tert-butyl-2-(4-methylanilino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=CC(C)=CC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 MXWBPVUOAUXKGW-UHFFFAOYSA-N 0.000 claims description 4
- OZGPACKEDNQGSG-UHFFFAOYSA-N n-tert-butyl-2-(4-methylsulfonylanilino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CNC2=NC=C1NC1=CC=C(S(C)(=O)=O)C=C1 OZGPACKEDNQGSG-UHFFFAOYSA-N 0.000 claims description 4
- DKKNFTQTTHRINS-UHFFFAOYSA-N n-tert-butyl-2-[2-(dimethylcarbamoyl)anilino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CN(C)C(=O)C1=CC=CC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 DKKNFTQTTHRINS-UHFFFAOYSA-N 0.000 claims description 4
- WDFIRAASCCQJKG-UHFFFAOYSA-N n-tert-butyl-2-[3-(1-hydroxyethyl)anilino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC(O)C1=CC=CC(NC=2N=C3C(C(=O)NC(C)(C)C)=CNC3=NC=2)=C1 WDFIRAASCCQJKG-UHFFFAOYSA-N 0.000 claims description 4
- ZUNPGFQPXCBQLG-UHFFFAOYSA-N n-tert-butyl-3-methyl-2-(3-methylsulfonylanilino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=NC=2NC=C(C(=O)NC(C)(C)C)C=2N=C1NC1=CC=CC(S(C)(=O)=O)=C1 ZUNPGFQPXCBQLG-UHFFFAOYSA-N 0.000 claims description 4
- 239000003900 neurotrophic factor Substances 0.000 claims description 4
- 210000000056 organ Anatomy 0.000 claims description 4
- 229940124597 therapeutic agent Drugs 0.000 claims description 4
- 208000024827 Alzheimer disease Diseases 0.000 claims description 3
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 3
- 208000011231 Crohn disease Diseases 0.000 claims description 3
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 3
- 201000004681 Psoriasis Diseases 0.000 claims description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 3
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 3
- 201000008937 atopic dermatitis Diseases 0.000 claims description 3
- 208000010928 autoimmune thyroid disease Diseases 0.000 claims description 3
- 208000032839 leukemia Diseases 0.000 claims description 3
- 206010025135 lupus erythematosus Diseases 0.000 claims description 3
- 201000006417 multiple sclerosis Diseases 0.000 claims description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- 238000002689 xenotransplantation Methods 0.000 claims description 3
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims 4
- 125000006620 amino-(C1-C6) alkyl group Chemical group 0.000 claims 2
- 125000001475 halogen functional group Chemical group 0.000 claims 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims 1
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 claims 1
- 229940121354 immunomodulator Drugs 0.000 claims 1
- 230000002584 immunomodulator Effects 0.000 claims 1
- 229960003444 immunosuppressant agent Drugs 0.000 claims 1
- 230000001861 immunosuppressant effect Effects 0.000 claims 1
- 238000002054 transplantation Methods 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 290
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 279
- 239000000203 mixture Substances 0.000 description 211
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 181
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 155
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 144
- 239000007787 solid Substances 0.000 description 142
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 98
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 86
- 238000000034 method Methods 0.000 description 85
- 239000011541 reaction mixture Substances 0.000 description 73
- -1 aliphatic aldehydes Chemical class 0.000 description 72
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 69
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 68
- 239000000243 solution Substances 0.000 description 65
- 238000004587 chromatography analysis Methods 0.000 description 62
- 239000000377 silicon dioxide Substances 0.000 description 59
- 230000008569 process Effects 0.000 description 56
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 50
- 239000000908 ammonium hydroxide Substances 0.000 description 50
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 45
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 43
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 37
- 239000012267 brine Substances 0.000 description 36
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 36
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 34
- 102000000551 Syk Kinase Human genes 0.000 description 30
- 108010016672 Syk Kinase Proteins 0.000 description 30
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 30
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical group C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 28
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 21
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 21
- 239000000706 filtrate Substances 0.000 description 20
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 20
- 235000019341 magnesium sulphate Nutrition 0.000 description 20
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 20
- 230000002829 reductive effect Effects 0.000 description 20
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 19
- LFWWXJQDBSQIPS-UHFFFAOYSA-N 2-bromo-n-tert-butyl-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=C(Br)N=C2C(C(=O)NC(C)(C)C)=CN(COCC[Si](C)(C)C)C2=N1 LFWWXJQDBSQIPS-UHFFFAOYSA-N 0.000 description 19
- 239000000284 extract Substances 0.000 description 18
- 150000003857 carboxamides Chemical class 0.000 description 17
- 239000004480 active ingredient Substances 0.000 description 16
- 238000001914 filtration Methods 0.000 description 16
- 238000000746 purification Methods 0.000 description 16
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 15
- 238000009472 formulation Methods 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- 239000000047 product Substances 0.000 description 15
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 14
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 14
- 229910000024 caesium carbonate Inorganic materials 0.000 description 14
- 239000000725 suspension Substances 0.000 description 14
- 125000003118 aryl group Chemical group 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- 125000005843 halogen group Chemical group 0.000 description 12
- 239000012044 organic layer Substances 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 108091000080 Phosphotransferase Proteins 0.000 description 11
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 102000020233 phosphotransferase Human genes 0.000 description 11
- 239000000843 powder Substances 0.000 description 11
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 125000001072 heteroaryl group Chemical group 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 9
- 0 CC(c1cc(Nc2nc(C(C(NC(C)(C)C)=O)=C[*+]3)c3nc2)ccc1)N Chemical compound CC(c1cc(Nc2nc(C(C(NC(C)(C)C)=O)=C[*+]3)c3nc2)ccc1)N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 239000004615 ingredient Substances 0.000 description 8
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 125000001424 substituent group Chemical group 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 7
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 7
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000007864 aqueous solution Substances 0.000 description 7
- 239000012300 argon atmosphere Substances 0.000 description 7
- 125000003710 aryl alkyl group Chemical group 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 230000004968 inflammatory condition Effects 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 7
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- RQFGYCXSMQFACA-UHFFFAOYSA-N 2-bromo-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carbaldehyde Chemical compound BrC1=CN=C2N(COCC[Si](C)(C)C)C=C(C=O)C2=N1 RQFGYCXSMQFACA-UHFFFAOYSA-N 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 229910004298 SiO 2 Inorganic materials 0.000 description 5
- 125000002947 alkylene group Chemical group 0.000 description 5
- 229910052786 argon Inorganic materials 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 235000015165 citric acid Nutrition 0.000 description 5
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 125000003884 phenylalkyl group Chemical group 0.000 description 5
- 239000002953 phosphate buffered saline Substances 0.000 description 5
- 125000003373 pyrazinyl group Chemical group 0.000 description 5
- 125000003226 pyrazolyl group Chemical group 0.000 description 5
- 125000004076 pyridyl group Chemical group 0.000 description 5
- 125000000714 pyrimidinyl group Chemical group 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 239000007921 spray Substances 0.000 description 5
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- WLAMTCFJXMIERW-UHFFFAOYSA-N 2-[(2-bromo-3-methylpyrrolo[2,3-b]pyrazin-5-yl)methoxy]ethyl-trimethylsilane Chemical compound N1=C(Br)C(C)=NC2=C1C=CN2COCC[Si](C)(C)C WLAMTCFJXMIERW-UHFFFAOYSA-N 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 4
- 241000400611 Eucalyptus deanei Species 0.000 description 4
- 239000007821 HATU Substances 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 108010090804 Streptavidin Proteins 0.000 description 4
- 230000001363 autoimmune Effects 0.000 description 4
- CELPHAGZKFMOMR-UHFFFAOYSA-N azanium;dichloromethane;methanol;hydroxide Chemical compound [NH4+].[OH-].OC.ClCCl CELPHAGZKFMOMR-UHFFFAOYSA-N 0.000 description 4
- 210000003719 b-lymphocyte Anatomy 0.000 description 4
- 239000011324 bead Substances 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 230000002950 deficient Effects 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 125000004438 haloalkoxy group Chemical group 0.000 description 4
- 125000001188 haloalkyl group Chemical group 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 125000000842 isoxazolyl group Chemical group 0.000 description 4
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- QRIASSRYSIATDH-UHFFFAOYSA-N n-tert-butyl-2-(1-methyl-6,7-dihydro-5h-pyrazolo[4,3-b]pyridin-4-yl)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2NC=C(C(=O)NC(C)(C)C)C2=NC(N2C=3C=NN(C=3CCC2)C)=C1 QRIASSRYSIATDH-UHFFFAOYSA-N 0.000 description 4
- HIVDHCDMABRACE-UHFFFAOYSA-N n-tert-butyl-2-(2-methyl-6,7-dihydro-5h-pyrazolo[4,3-b]pyridin-4-yl)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2NC=C(C(=O)NC(C)(C)C)C2=NC(N2C3=CN(N=C3CCC2)C)=C1 HIVDHCDMABRACE-UHFFFAOYSA-N 0.000 description 4
- SNEAMYQJSXHMMW-UHFFFAOYSA-N n-tert-butyl-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound S1N=C(C)C=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 SNEAMYQJSXHMMW-UHFFFAOYSA-N 0.000 description 4
- 239000007922 nasal spray Substances 0.000 description 4
- 229940097496 nasal spray Drugs 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 239000011550 stock solution Substances 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 239000000829 suppository Substances 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 238000013268 sustained release Methods 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- 125000001113 thiadiazolyl group Chemical group 0.000 description 4
- 125000000335 thiazolyl group Chemical group 0.000 description 4
- 239000002562 thickening agent Substances 0.000 description 4
- SSMZHWLBKAFZMR-UHFFFAOYSA-N (2-bromo-5h-pyrrolo[2,3-b]pyrazin-7-yl)methanol Chemical compound C1=C(Br)N=C2C(CO)=CNC2=N1 SSMZHWLBKAFZMR-UHFFFAOYSA-N 0.000 description 3
- ZRTGHKVPFXNDHE-UHFFFAOYSA-N (3-methyl-1,2-thiazol-5-yl)azanium;chloride Chemical compound [Cl-].CC=1C=C([NH3+])SN=1 ZRTGHKVPFXNDHE-UHFFFAOYSA-N 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 3
- XGCAODDQVDBQFA-UHFFFAOYSA-N 1-methyl-4,5,6,7-tetrahydropyrazolo[4,3-b]pyridine Chemical compound N1CCCC2=C1C=NN2C XGCAODDQVDBQFA-UHFFFAOYSA-N 0.000 description 3
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 3
- ROLYQILMGFXZHD-UHFFFAOYSA-N 2-[(5-methylpyridin-2-yl)amino]-n-propan-2-yl-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)C)=CNC2=NC=C1NC1=CC=C(C)C=N1 ROLYQILMGFXZHD-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- GBTGEWOBXMUQJH-UHFFFAOYSA-N 2-bromo-3-chloro-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carbaldehyde Chemical compound BrC1=C(Cl)N=C2N(COCC[Si](C)(C)C)C=C(C=O)C2=N1 GBTGEWOBXMUQJH-UHFFFAOYSA-N 0.000 description 3
- HSMNZAAANMEUSY-UHFFFAOYSA-N 2-bromo-3-chloro-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1=C(Br)C(Cl)=NC2=C1C=CN2 HSMNZAAANMEUSY-UHFFFAOYSA-N 0.000 description 3
- AVKOBWDVWAMCBD-UHFFFAOYSA-N 2-bromo-3-chloro-5h-pyrrolo[2,3-b]pyrazine-7-carbaldehyde Chemical compound N1=C(Br)C(Cl)=NC2=C1C(C=O)=CN2 AVKOBWDVWAMCBD-UHFFFAOYSA-N 0.000 description 3
- BVECEHJYNSLRKL-UHFFFAOYSA-N 2-bromo-3-methyl-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carbaldehyde Chemical compound N1=C(Br)C(C)=NC2=C1C(C=O)=CN2COCC[Si](C)(C)C BVECEHJYNSLRKL-UHFFFAOYSA-N 0.000 description 3
- AIVGPLDAXQMUBJ-UHFFFAOYSA-N 2-bromo-3-methyl-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound N1=C(Br)C(C)=NC2=C1C(C(O)=O)=CN2COCC[Si](C)(C)C AIVGPLDAXQMUBJ-UHFFFAOYSA-N 0.000 description 3
- DKJPISUROVINRP-UHFFFAOYSA-N 2-bromo-3-methyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1=C(Br)C(C)=NC2=C1C=CN2 DKJPISUROVINRP-UHFFFAOYSA-N 0.000 description 3
- TZANAKVXISCMLH-UHFFFAOYSA-N 2-bromo-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound BrC1=CN=C2N(COCC[Si](C)(C)C)C=C(C(O)=O)C2=N1 TZANAKVXISCMLH-UHFFFAOYSA-N 0.000 description 3
- HCYBNYLZWLXOHD-UHFFFAOYSA-N 2-bromo-n-(1-hydroxy-2-methylpropan-2-yl)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=C(Br)N=C2C(C(=O)NC(C)(CO)C)=CN(COCC[Si](C)(C)C)C2=N1 HCYBNYLZWLXOHD-UHFFFAOYSA-N 0.000 description 3
- IREVUSISKVVJOR-UHFFFAOYSA-N 2-bromo-n-tert-butyl-3-chloro-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound ClC1=C(Br)N=C2C(C(=O)NC(C)(C)C)=CN(COCC[Si](C)(C)C)C2=N1 IREVUSISKVVJOR-UHFFFAOYSA-N 0.000 description 3
- LMKWUPAUGHIUOF-UHFFFAOYSA-N 2-methyl-4,5,6,7-tetrahydropyrazolo[4,3-b]pyridine Chemical compound N1CCCC2=NN(C)C=C21 LMKWUPAUGHIUOF-UHFFFAOYSA-N 0.000 description 3
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 3
- KMOICDJWYSOXRT-UHFFFAOYSA-N 3,5-dibromo-6-chloropyrazin-2-amine Chemical compound NC1=NC(Cl)=C(Br)N=C1Br KMOICDJWYSOXRT-UHFFFAOYSA-N 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- RTTBBADOMOOPTQ-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1h-pyrazolo[4,3-b]pyridine Chemical compound C1CCNC2=C1NN=C2 RTTBBADOMOOPTQ-UHFFFAOYSA-N 0.000 description 3
- FWKJVZYRJQREKF-UHFFFAOYSA-N 5-bromo-6-chloro-3-(2-trimethylsilylethynyl)pyrazin-2-amine Chemical compound C[Si](C)(C)C#CC1=NC(Br)=C(Cl)N=C1N FWKJVZYRJQREKF-UHFFFAOYSA-N 0.000 description 3
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo(3.3.1)nonane Chemical compound C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 3
- ZKHQWZAMYRWXGA-KQYNXXCUSA-N Adenosine triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-N 0.000 description 3
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 3
- 208000023275 Autoimmune disease Diseases 0.000 description 3
- GWRSZJVZKMVGRW-UHFFFAOYSA-O CC(C)(C)NC(c1c[n](COCC[SH+](C)(C)C)c(nc2)c1nc2Br)=O Chemical compound CC(C)(C)NC(c1c[n](COCC[SH+](C)(C)C)c(nc2)c1nc2Br)=O GWRSZJVZKMVGRW-UHFFFAOYSA-O 0.000 description 3
- 239000005973 Carvone Substances 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 239000003810 Jones reagent Substances 0.000 description 3
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 3
- 240000007472 Leucaena leucocephala Species 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 3
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 3
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 3
- JWPNRJMHUPVNIZ-UHFFFAOYSA-N [2-bromo-3-chloro-5-(hydroxymethyl)pyrrolo[2,3-b]pyrazin-7-yl]methanol Chemical compound ClC1=C(Br)N=C2C(CO)=CN(CO)C2=N1 JWPNRJMHUPVNIZ-UHFFFAOYSA-N 0.000 description 3
- REUFRRWFHBFBDR-UHFFFAOYSA-N [2-bromo-5-(hydroxymethyl)pyrrolo[2,3-b]pyrazin-7-yl]methanol Chemical compound C1=C(Br)N=C2C(CO)=CN(CO)C2=N1 REUFRRWFHBFBDR-UHFFFAOYSA-N 0.000 description 3
- QQIRAVWVGBTHMJ-UHFFFAOYSA-N [dimethyl-(trimethylsilylamino)silyl]methane;lithium Chemical compound [Li].C[Si](C)(C)N[Si](C)(C)C QQIRAVWVGBTHMJ-UHFFFAOYSA-N 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- AUALQMFGWLZREY-UHFFFAOYSA-N acetonitrile;methanol Chemical compound OC.CC#N AUALQMFGWLZREY-UHFFFAOYSA-N 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 125000003282 alkyl amino group Chemical group 0.000 description 3
- 239000007900 aqueous suspension Substances 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 230000005754 cellular signaling Effects 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 210000003979 eosinophil Anatomy 0.000 description 3
- 235000019634 flavors Nutrition 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 125000004404 heteroalkyl group Chemical group 0.000 description 3
- 210000003630 histaminocyte Anatomy 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 230000002519 immonomodulatory effect Effects 0.000 description 3
- 229940125721 immunosuppressive agent Drugs 0.000 description 3
- 208000027866 inflammatory disease Diseases 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000012669 liquid formulation Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 3
- 235000019796 monopotassium phosphate Nutrition 0.000 description 3
- 239000012452 mother liquor Substances 0.000 description 3
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 3
- LYDBEEVIHIWCIK-UHFFFAOYSA-N n-(1-hydroxy-2-methylpropan-2-yl)-2-[(5-methylpyridin-2-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=CC(C)=CC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)CO)C2=N1 LYDBEEVIHIWCIK-UHFFFAOYSA-N 0.000 description 3
- IPKHDQZNIUVOMW-UHFFFAOYSA-N n-tert-butyl-2-(pyridin-3-ylamino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CNC2=NC=C1NC1=CC=CN=C1 IPKHDQZNIUVOMW-UHFFFAOYSA-N 0.000 description 3
- BTMFTOKULDCEBW-UHFFFAOYSA-N n-tert-butyl-2-[(1,3-dimethylpyrazol-4-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=NN(C)C=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 BTMFTOKULDCEBW-UHFFFAOYSA-N 0.000 description 3
- KFCSHAIHPNJEHP-UHFFFAOYSA-N n-tert-butyl-2-[(1-methylpyrazol-3-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CN1C=CC(NC=2N=C3C(C(=O)NC(C)(C)C)=CNC3=NC=2)=N1 KFCSHAIHPNJEHP-UHFFFAOYSA-N 0.000 description 3
- WQVAPHCMGOOMKB-UHFFFAOYSA-N n-tert-butyl-2-[(1-methylpyrazol-4-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NN(C)C=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 WQVAPHCMGOOMKB-UHFFFAOYSA-N 0.000 description 3
- ANMGGNMIYKBQII-UHFFFAOYSA-N n-tert-butyl-2-[(2-methoxypyrimidin-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NC(OC)=NC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 ANMGGNMIYKBQII-UHFFFAOYSA-N 0.000 description 3
- CZEMSIGWEBSIMZ-UHFFFAOYSA-N n-tert-butyl-2-[(2-methylpyrimidin-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NC(C)=NC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 CZEMSIGWEBSIMZ-UHFFFAOYSA-N 0.000 description 3
- YUUBIYZWTLTWIP-UHFFFAOYSA-N n-tert-butyl-2-[(2-methylsulfonylpyridin-4-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CNC2=NC=C1NC1=CC=NC(S(C)(=O)=O)=C1 YUUBIYZWTLTWIP-UHFFFAOYSA-N 0.000 description 3
- YQMYGDZDKVKCMB-UHFFFAOYSA-N n-tert-butyl-2-[(3-methyl-1,2,4-thiadiazol-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=NSC(NC=2N=C3C(C(=O)NC(C)(C)C)=CNC3=NC=2)=N1 YQMYGDZDKVKCMB-UHFFFAOYSA-N 0.000 description 3
- UFPBWPYHBZNKQD-UHFFFAOYSA-N n-tert-butyl-2-[(3-methyl-1,2-oxazol-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound O1N=C(C)C=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 UFPBWPYHBZNKQD-UHFFFAOYSA-N 0.000 description 3
- RESGPJGRBGGBAJ-UHFFFAOYSA-N n-tert-butyl-2-[(5-methylpyrazin-2-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NC(C)=CN=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 RESGPJGRBGGBAJ-UHFFFAOYSA-N 0.000 description 3
- FLNUTOVGNFXLNP-UHFFFAOYSA-N n-tert-butyl-2-[(5-methylsulfonylpyridin-3-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CNC2=NC=C1NC1=CN=CC(S(C)(=O)=O)=C1 FLNUTOVGNFXLNP-UHFFFAOYSA-N 0.000 description 3
- VXKVTHRQKWDSKU-UHFFFAOYSA-N n-tert-butyl-2-[(6-methylpyridin-3-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NC(C)=CC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 VXKVTHRQKWDSKU-UHFFFAOYSA-N 0.000 description 3
- YVQCRCRRJDSFBX-UHFFFAOYSA-N n-tert-butyl-2-[(6-methylsulfonylpyridin-3-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CNC2=NC=C1NC1=CC=C(S(C)(=O)=O)N=C1 YVQCRCRRJDSFBX-UHFFFAOYSA-N 0.000 description 3
- MAFLUZXIYFUXOK-UHFFFAOYSA-N n-tert-butyl-2-[methyl(pyridin-3-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C=1N=C2NC=C(C(=O)NC(C)(C)C)C2=NC=1N(C)C1=CC=CN=C1 MAFLUZXIYFUXOK-UHFFFAOYSA-N 0.000 description 3
- ACHNXKUPTZZINC-UHFFFAOYSA-N n-tert-butyl-3-chloro-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound S1N=C(C)C=C1NC(C(=N1)Cl)=NC2=C1NC=C2C(=O)NC(C)(C)C ACHNXKUPTZZINC-UHFFFAOYSA-N 0.000 description 3
- VSGAZGIZKBAKIE-UHFFFAOYSA-N n-tert-butyl-3-methyl-2-(3-methylsulfonylanilino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=NC=2N(COCC[Si](C)(C)C)C=C(C(=O)NC(C)(C)C)C=2N=C1NC1=CC=CC(S(C)(=O)=O)=C1 VSGAZGIZKBAKIE-UHFFFAOYSA-N 0.000 description 3
- SBNJPXLAAWDUFI-UHFFFAOYSA-N n-tert-butyl-3-methyl-2-[(1-methylpyrazol-4-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=NC=2NC=C(C(=O)NC(C)(C)C)C=2N=C1NC=1C=NN(C)C=1 SBNJPXLAAWDUFI-UHFFFAOYSA-N 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 3
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 3
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 3
- 229960002218 sodium chlorite Drugs 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 239000012730 sustained-release form Substances 0.000 description 3
- VUEOIIWQDOZFFL-UHFFFAOYSA-N tert-butyl 1,5,6,7-tetrahydropyrazolo[4,3-b]pyridine-4-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCCC2=C1C=NN2 VUEOIIWQDOZFFL-UHFFFAOYSA-N 0.000 description 3
- IUIBGUOQHXLBMT-UHFFFAOYSA-N tert-butyl 1-methyl-6,7-dihydro-5h-pyrazolo[4,3-b]pyridine-4-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCCC2=C1C=NN2C IUIBGUOQHXLBMT-UHFFFAOYSA-N 0.000 description 3
- MUBUESHCCFFCBN-UHFFFAOYSA-N tert-butyl 2-bromo-3-chloropyrrolo[2,3-b]pyrazine-5-carboxylate Chemical compound BrC1=C(Cl)N=C2N(C(=O)OC(C)(C)C)C=CC2=N1 MUBUESHCCFFCBN-UHFFFAOYSA-N 0.000 description 3
- UVRXHABLVFPICV-UHFFFAOYSA-N tert-butyl 2-methyl-6,7-dihydro-5h-pyrazolo[4,3-b]pyridine-4-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCCC2=NN(C)C=C21 UVRXHABLVFPICV-UHFFFAOYSA-N 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- AXTGDCSMTYGJND-UHFFFAOYSA-N 1-dodecylazepan-2-one Chemical compound CCCCCCCCCCCCN1CCCCCC1=O AXTGDCSMTYGJND-UHFFFAOYSA-N 0.000 description 2
- OKPCAONVUKBJJQ-UHFFFAOYSA-N 1-methylpyrazol-4-amine;hydrochloride Chemical compound Cl.CN1C=C(N)C=N1 OKPCAONVUKBJJQ-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 2
- CZZZABOKJQXEBO-UHFFFAOYSA-N 2,4-dimethylaniline Chemical compound CC1=CC=C(N)C(C)=C1 CZZZABOKJQXEBO-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- ROHOBSZPCYKALU-UHFFFAOYSA-N 2-[(2-bromopyrrolo[2,3-b]pyrazin-5-yl)methoxy]ethyl-trimethylsilane Chemical compound BrC1=CN=C2N(COCC[Si](C)(C)C)C=CC2=N1 ROHOBSZPCYKALU-UHFFFAOYSA-N 0.000 description 2
- UGXIDQBJMLDFQA-UHFFFAOYSA-N 2-[(5-methylpyridin-2-yl)amino]-n-propan-2-yl-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)C)=CN(COCC[Si](C)(C)C)C2=NC=C1NC1=CC=C(C)C=N1 UGXIDQBJMLDFQA-UHFFFAOYSA-N 0.000 description 2
- MXNVONAUOWULLW-UHFFFAOYSA-N 2-bromo-3-chloro-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound BrC1=C(Cl)N=C2N(COCC[Si](C)(C)C)C=C(C(O)=O)C2=N1 MXNVONAUOWULLW-UHFFFAOYSA-N 0.000 description 2
- KUSRFAMMTFGMEO-UHFFFAOYSA-N 2-bromo-n-tert-butyl-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=C(Br)N=C2C(C(=O)NC(C)(C)C)=CNC2=N1 KUSRFAMMTFGMEO-UHFFFAOYSA-N 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- AWBLTDFTDYGFGJ-UHFFFAOYSA-N 3,5-dibromo-6-methylpyrazin-2-amine Chemical compound CC1=NC(N)=C(Br)N=C1Br AWBLTDFTDYGFGJ-UHFFFAOYSA-N 0.000 description 2
- CUYKNJBYIJFRCU-UHFFFAOYSA-N 3-aminopyridine Chemical compound NC1=CC=CN=C1 CUYKNJBYIJFRCU-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- IEBONVRIICHJFI-UHFFFAOYSA-N 3-methyl-2-[(1-methylpyrazol-4-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound CC1=C(N=C2C(=N1)N(C=C2C(=O)O)COCC[Si](C)(C)C)NC=2C=NN(C2)C IEBONVRIICHJFI-UHFFFAOYSA-N 0.000 description 2
- XWEKPEOAJIRVLG-UHFFFAOYSA-N 3-methyl-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound S1N=C(C)C=C1NC(C(=N1)C)=NC2=C1NC=C2C(O)=O XWEKPEOAJIRVLG-UHFFFAOYSA-N 0.000 description 2
- GZIVCOBWIGEBFW-UHFFFAOYSA-N 5-bromo-6-methyl-3-(2-trimethylsilylethynyl)pyrazin-2-amine Chemical compound CC1=NC(N)=C(C#C[Si](C)(C)C)N=C1Br GZIVCOBWIGEBFW-UHFFFAOYSA-N 0.000 description 2
- CMBSSVKZOPZBKW-UHFFFAOYSA-N 5-methylpyridin-2-amine Chemical compound CC1=CC=C(N)N=C1 CMBSSVKZOPZBKW-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 230000003844 B-cell-activation Effects 0.000 description 2
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 2
- FIPWRIJSWJWJAI-UHFFFAOYSA-N Butyl carbitol 6-propylpiperonyl ether Chemical compound C1=C(CCC)C(COCCOCCOCCCC)=CC2=C1OCO2 FIPWRIJSWJWJAI-UHFFFAOYSA-N 0.000 description 2
- PRGJEHOJEWOIRC-UHFFFAOYSA-N CC(C)(C)NC(c(c1n2)c[n](COCC[Si+](C)(C)C)c1ncc2Br)=O Chemical compound CC(C)(C)NC(c(c1n2)c[n](COCC[Si+](C)(C)C)c1ncc2Br)=O PRGJEHOJEWOIRC-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical group OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- DHCLVCXQIBBOPH-UHFFFAOYSA-N Glycerol 2-phosphate Chemical compound OCC(CO)OP(O)(O)=O DHCLVCXQIBBOPH-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 2
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- YIVDQOPEMLYPQR-UHFFFAOYSA-N N-tert-butyl-2-(2,4-dimethylanilino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound Cc1ccc(Nc2cnc3n(COCC[Si](C)(C)C)cc(C(=O)NC(C)(C)C)c3n2)c(C)c1 YIVDQOPEMLYPQR-UHFFFAOYSA-N 0.000 description 2
- UMUAOXWYXNZIAE-UHFFFAOYSA-N N-tert-butyl-2-(2-fluoro-4-methylanilino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC2=C(C=C(C=C2)C)F)COCC[Si](C)(C)C UMUAOXWYXNZIAE-UHFFFAOYSA-N 0.000 description 2
- VTNQVXDYKWGDMS-UHFFFAOYSA-N N-tert-butyl-2-(3-methylsulfonylanilino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC2=CC(=CC=C2)S(=O)(=O)C)COCC[Si](C)(C)C VTNQVXDYKWGDMS-UHFFFAOYSA-N 0.000 description 2
- RKMPSEDNHMWOAE-UHFFFAOYSA-N N-tert-butyl-2-(4-methylanilino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound Cc1ccc(Nc2cnc3n(COCC[Si](C)(C)C)cc(C(=O)NC(C)(C)C)c3n2)cc1 RKMPSEDNHMWOAE-UHFFFAOYSA-N 0.000 description 2
- DHRSPEVTVNNWFL-UHFFFAOYSA-N N-tert-butyl-2-(pyridin-3-ylamino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC(C)(C)NC(=O)c1cn(COCC[Si](C)(C)C)c2ncc(Nc3cccnc3)nc12 DHRSPEVTVNNWFL-UHFFFAOYSA-N 0.000 description 2
- ZPVDHOMAYKATBV-UHFFFAOYSA-N N-tert-butyl-2-[(2-methylpyrimidin-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC=2C=NC(=NC2)C)COCC[Si](C)(C)C ZPVDHOMAYKATBV-UHFFFAOYSA-N 0.000 description 2
- GOVJCGOLHJQLOV-UHFFFAOYSA-N N-tert-butyl-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC2=CC(=NS2)C)COCC[Si](C)(C)C GOVJCGOLHJQLOV-UHFFFAOYSA-N 0.000 description 2
- VCHQEXLJWLEWNT-UHFFFAOYSA-N N-tert-butyl-2-[(5-methylpyrazin-2-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC2=NC=C(N=C2)C)COCC[Si](C)(C)C VCHQEXLJWLEWNT-UHFFFAOYSA-N 0.000 description 2
- PJQRAMJKBGFJHT-UHFFFAOYSA-N N-tert-butyl-2-[(5-methylpyridin-2-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC2=NC=C(C=C2)C)COCC[Si](C)(C)C PJQRAMJKBGFJHT-UHFFFAOYSA-N 0.000 description 2
- BZFOKJXFFBGREQ-UHFFFAOYSA-N N-tert-butyl-2-[(6-methylpyridin-3-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC=2C=NC(=CC2)C)COCC[Si](C)(C)C BZFOKJXFFBGREQ-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 2
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 2
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 125000004658 aryl carbonyl amino group Chemical group 0.000 description 2
- 125000005129 aryl carbonyl group Chemical group 0.000 description 2
- 125000004657 aryl sulfonyl amino group Chemical group 0.000 description 2
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000012259 ether extract Substances 0.000 description 2
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 2
- RCBVKBFIWMOMHF-UHFFFAOYSA-L hydroxy-(hydroxy(dioxo)chromio)oxy-dioxochromium;pyridine Chemical compound C1=CC=NC=C1.C1=CC=NC=C1.O[Cr](=O)(=O)O[Cr](O)(=O)=O RCBVKBFIWMOMHF-UHFFFAOYSA-L 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 238000000021 kinase assay Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 239000002324 mouth wash Substances 0.000 description 2
- 229940051866 mouthwash Drugs 0.000 description 2
- TXXHDPDFNKHHGW-UHFFFAOYSA-N muconic acid Chemical group OC(=O)C=CC=CC(O)=O TXXHDPDFNKHHGW-UHFFFAOYSA-N 0.000 description 2
- OKDQKPLMQBXTNH-UHFFFAOYSA-N n,n-dimethyl-2h-pyridin-1-amine Chemical compound CN(C)N1CC=CC=C1 OKDQKPLMQBXTNH-UHFFFAOYSA-N 0.000 description 2
- STUYEBWPURSWQG-UHFFFAOYSA-N n-(1-hydroxy-2-methylpropan-2-yl)-2-(pyridin-3-ylamino)-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(CO)C)=CNC2=NC=C1NC1=CC=CN=C1 STUYEBWPURSWQG-UHFFFAOYSA-N 0.000 description 2
- QSFJISQOQIHBTF-UHFFFAOYSA-N n-(1-hydroxy-2-methylpropan-2-yl)-2-[(1-methylpyrazol-4-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NN(C)C=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)CO)C2=N1 QSFJISQOQIHBTF-UHFFFAOYSA-N 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- HAXWCVYMCBYQNX-UHFFFAOYSA-N n-tert-butyl-2-(1-methyl-6,7-dihydro-5h-pyrazolo[4,3-b]pyridin-4-yl)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2N(COCC[Si](C)(C)C)C=C(C(=O)NC(C)(C)C)C2=NC(N2C=3C=NN(C=3CCC2)C)=C1 HAXWCVYMCBYQNX-UHFFFAOYSA-N 0.000 description 2
- ZPTZQPGGHINOFK-UHFFFAOYSA-N n-tert-butyl-2-(2-methyl-6,7-dihydro-5h-pyrazolo[4,3-b]pyridin-4-yl)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2N(COCC[Si](C)(C)C)C=C(C(=O)NC(C)(C)C)C2=NC(N2C3=CN(N=C3CCC2)C)=C1 ZPTZQPGGHINOFK-UHFFFAOYSA-N 0.000 description 2
- IGTYWDPITPIOQY-UHFFFAOYSA-N n-tert-butyl-2-(4-methylsulfonylanilino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CN(COCC[Si](C)(C)C)C2=NC=C1NC1=CC=C(S(C)(=O)=O)C=C1 IGTYWDPITPIOQY-UHFFFAOYSA-N 0.000 description 2
- YWEWXZOIGPIVLC-UHFFFAOYSA-N n-tert-butyl-2-[(1,5-dimethylpyrazol-4-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NN(C)C(C)=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 YWEWXZOIGPIVLC-UHFFFAOYSA-N 0.000 description 2
- OBKUVCDXQVBADF-UHFFFAOYSA-N n-tert-butyl-2-[(1-ethylpyrazol-4-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NN(CC)C=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 OBKUVCDXQVBADF-UHFFFAOYSA-N 0.000 description 2
- ODBLCERRTRAZGE-UHFFFAOYSA-N n-tert-butyl-2-[(2-methoxypyrimidin-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NC(OC)=NC=C1NC1=CN=C(N(COCC[Si](C)(C)C)C=C2C(=O)NC(C)(C)C)C2=N1 ODBLCERRTRAZGE-UHFFFAOYSA-N 0.000 description 2
- IXKCQPSBOIMIIK-UHFFFAOYSA-N n-tert-butyl-2-[(3-methyl-1,2-oxazol-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound O1N=C(C)C=C1NC1=CN=C(N(COCC[Si](C)(C)C)C=C2C(=O)NC(C)(C)C)C2=N1 IXKCQPSBOIMIIK-UHFFFAOYSA-N 0.000 description 2
- ADGYBXSZVFJGDQ-UHFFFAOYSA-N n-tert-butyl-2-[(5-methyl-1h-pyrazol-3-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1C(C)=CC(NC=2N=C3C(C(=O)NC(C)(C)C)=CNC3=NC=2)=N1 ADGYBXSZVFJGDQ-UHFFFAOYSA-N 0.000 description 2
- ULTSNMDITVYLTG-UHFFFAOYSA-N n-tert-butyl-2-[(5-methylpyridin-2-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=CC(C)=CC=C1NC1=CN=C(NC=C2C(=O)NC(C)(C)C)C2=N1 ULTSNMDITVYLTG-UHFFFAOYSA-N 0.000 description 2
- VZNNSEKPIYTPRD-UHFFFAOYSA-N n-tert-butyl-2-[3-(1-hydroxyethyl)anilino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC(O)C1=CC=CC(NC=2N=C3C(C(=O)NC(C)(C)C)=CN(COCC[Si](C)(C)C)C3=NC=2)=C1 VZNNSEKPIYTPRD-UHFFFAOYSA-N 0.000 description 2
- RCTNOSBSMYKCOH-UHFFFAOYSA-N n-tert-butyl-2-[methyl(pyridin-3-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C=1N=C2N(COCC[Si](C)(C)C)C=C(C(=O)NC(C)(C)C)C2=NC=1N(C)C1=CC=CN=C1 RCTNOSBSMYKCOH-UHFFFAOYSA-N 0.000 description 2
- AWZSAYAHOBQALB-UHFFFAOYSA-N n-tert-butyl-3-chloro-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound S1N=C(C)C=C1NC(C(=N1)Cl)=NC2=C1N(COCC[Si](C)(C)C)C=C2C(=O)NC(C)(C)C AWZSAYAHOBQALB-UHFFFAOYSA-N 0.000 description 2
- QKDCLDFGNSMNAO-UHFFFAOYSA-N n-tert-butyl-3-methyl-2-[(1-methylpyrazol-4-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=NC=2N(COCC[Si](C)(C)C)C=C(C(=O)NC(C)(C)C)C=2N=C1NC=1C=NN(C)C=1 QKDCLDFGNSMNAO-UHFFFAOYSA-N 0.000 description 2
- UEKCRFGIAVALQB-UHFFFAOYSA-N n-tert-butyl-3-methyl-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound S1N=C(C)C=C1NC(C(=N1)C)=NC2=C1N(COCC[Si](C)(C)C)C=C2C(=O)NC(C)(C)C UEKCRFGIAVALQB-UHFFFAOYSA-N 0.000 description 2
- 210000003928 nasal cavity Anatomy 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- RZXMPPFPUUCRFN-UHFFFAOYSA-N p-toluidine Chemical compound CC1=CC=C(N)C=C1 RZXMPPFPUUCRFN-UHFFFAOYSA-N 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000008024 pharmaceutical diluent Substances 0.000 description 2
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 2
- 229960005235 piperonyl butoxide Drugs 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920006316 polyvinylpyrrolidine Polymers 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical group OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- KWMLJOLKUYYJFJ-GASJEMHNSA-N (2xi)-D-gluco-heptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C(O)=O KWMLJOLKUYYJFJ-GASJEMHNSA-N 0.000 description 1
- JSBRHLNQBYRNGE-UHFFFAOYSA-N (7,7-dimethyl-2-oxo-1-bicyclo[2.2.1]heptanyl)methanesulfonic acid 4-methylbicyclo[2.2.2]oct-2-ene-1-carboxylic acid Chemical compound CC12CCC(CC1)(C=C2)C(O)=O.CC1(C)C2CCC1(CS(O)(=O)=O)C(=O)C2 JSBRHLNQBYRNGE-UHFFFAOYSA-N 0.000 description 1
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- CSNIZNHTOVFARY-UHFFFAOYSA-N 1,2-benzothiazole Chemical compound C1=CC=C2C=NSC2=C1 CSNIZNHTOVFARY-UHFFFAOYSA-N 0.000 description 1
- KTZQTRPPVKQPFO-UHFFFAOYSA-N 1,2-benzoxazole Chemical compound C1=CC=C2C=NOC2=C1 KTZQTRPPVKQPFO-UHFFFAOYSA-N 0.000 description 1
- QFMZQPDHXULLKC-UHFFFAOYSA-N 1,2-bis(diphenylphosphino)ethane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCP(C=1C=CC=CC=1)C1=CC=CC=C1 QFMZQPDHXULLKC-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- AXLKRXWNAFFPDB-UHFFFAOYSA-N 1,3-dimethylpyrazol-4-amine Chemical compound CC1=NN(C)C=C1N AXLKRXWNAFFPDB-UHFFFAOYSA-N 0.000 description 1
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 description 1
- MOUVQIJWMDHVDL-UHFFFAOYSA-N 1,5-dimethylpyrazol-4-amine;dihydrochloride Chemical compound Cl.Cl.CC1=C(N)C=NN1C MOUVQIJWMDHVDL-UHFFFAOYSA-N 0.000 description 1
- QPKNDHZQPGMLCJ-UHFFFAOYSA-N 1-(3-aminophenyl)ethanol Chemical compound CC(O)C1=CC=CC(N)=C1 QPKNDHZQPGMLCJ-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- QOVXVNPYUBGBDP-UHFFFAOYSA-N 1-ethylpyrazol-4-amine;hydrochloride Chemical compound Cl.CCN1C=C(N)C=N1 QOVXVNPYUBGBDP-UHFFFAOYSA-N 0.000 description 1
- LBGSWBJURUFGLR-UHFFFAOYSA-N 1-methylpyrazol-4-amine Chemical compound CN1C=C(N)C=N1 LBGSWBJURUFGLR-UHFFFAOYSA-N 0.000 description 1
- JVVRJMXHNUAPHW-UHFFFAOYSA-N 1h-pyrazol-5-amine Chemical compound NC=1C=CNN=1 JVVRJMXHNUAPHW-UHFFFAOYSA-N 0.000 description 1
- AMFYRKOUWBAGHV-UHFFFAOYSA-N 1h-pyrazolo[4,3-b]pyridine Chemical compound C1=CN=C2C=NNC2=C1 AMFYRKOUWBAGHV-UHFFFAOYSA-N 0.000 description 1
- YRAJNWYBUCUFBD-UHFFFAOYSA-N 2,2,6,6-tetramethylheptane-3,5-dione Chemical compound CC(C)(C)C(=O)CC(=O)C(C)(C)C YRAJNWYBUCUFBD-UHFFFAOYSA-N 0.000 description 1
- XXKHDSGLCLCFSC-UHFFFAOYSA-N 2,3-diphenylphenol Chemical compound C=1C=CC=CC=1C=1C(O)=CC=CC=1C1=CC=CC=C1 XXKHDSGLCLCFSC-UHFFFAOYSA-N 0.000 description 1
- DJWDAKFSDBOQJK-UHFFFAOYSA-N 2,5-diazabicyclo[2.2.2]octane Chemical compound C1NC2CCC1NC2 DJWDAKFSDBOQJK-UHFFFAOYSA-N 0.000 description 1
- UDSAJFSYJMHNFI-UHFFFAOYSA-N 2,6-diazaspiro[3.3]heptane Chemical compound C1NCC11CNC1 UDSAJFSYJMHNFI-UHFFFAOYSA-N 0.000 description 1
- XPCYNFMKSKIPKD-UHFFFAOYSA-N 2-(2-trimethylsilylethoxymethyl)-5H-pyrrolo[2,3-b]pyrazine-7-carbaldehyde Chemical compound C[Si](CCOCC=1N=C2C(=NC=1)NC=C2C=O)(C)C XPCYNFMKSKIPKD-UHFFFAOYSA-N 0.000 description 1
- KDFCFWVYIOLVTH-UHFFFAOYSA-N 2-[(2-bromo-1-tert-butyl-2H-pyrrolo[2,3-b]pyrazin-5-yl)methoxy]ethyl-trimethylsilane Chemical compound BrC1N(C2=C(N=C1)N(C=C2)COCC[Si](C)(C)C)C(C)(C)C KDFCFWVYIOLVTH-UHFFFAOYSA-N 0.000 description 1
- AUNIQBYZVOKWJK-UHFFFAOYSA-N 2-[1-(2-hydroxyethyl)piperazin-2-yl]ethanesulfonic acid Chemical compound OCCN1CCNCC1CCS(O)(=O)=O AUNIQBYZVOKWJK-UHFFFAOYSA-N 0.000 description 1
- JTPXVCKCLBROOJ-UHFFFAOYSA-N 2-amino-6-chloropyrazine Chemical compound NC1=CN=CC(Cl)=N1 JTPXVCKCLBROOJ-UHFFFAOYSA-N 0.000 description 1
- JQPPBPUSGNPFIS-UHFFFAOYSA-N 2-amino-n-tert-butyl-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=C(N)N=C2C(C(=O)NC(C)(C)C)=CN(COCC[Si](C)(C)C)C2=N1 JQPPBPUSGNPFIS-UHFFFAOYSA-N 0.000 description 1
- KTKMLXBEBKGQGL-UHFFFAOYSA-N 2-bromo-5h-pyrrolo[2,3-b]pyrazine Chemical compound BrC1=CN=C2NC=CC2=N1 KTKMLXBEBKGQGL-UHFFFAOYSA-N 0.000 description 1
- KTQCGYNMGWTOHT-UHFFFAOYSA-N 2-bromo-5h-pyrrolo[2,3-b]pyrazine-7-carbaldehyde Chemical compound BrC1=CN=C2NC=C(C=O)C2=N1 KTQCGYNMGWTOHT-UHFFFAOYSA-N 0.000 description 1
- ZYEDXUGCYDTQLE-UHFFFAOYSA-N 2-bromo-n-propan-2-yl-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=C(Br)N=C2C(C(=O)NC(C)C)=CN(COCC[Si](C)(C)C)C2=N1 ZYEDXUGCYDTQLE-UHFFFAOYSA-N 0.000 description 1
- YTQPYLUAJXFOHP-UHFFFAOYSA-N 2-bromo-n-tert-butyl-3-methyl-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C(Br)C(C)=NC2=C1C(C(=O)NC(C)(C)C)=CN2COCC[Si](C)(C)C YTQPYLUAJXFOHP-UHFFFAOYSA-N 0.000 description 1
- ZQEXBVHABAJPHJ-UHFFFAOYSA-N 2-fluoro-4-methylaniline Chemical compound CC1=CC=C(N)C(F)=C1 ZQEXBVHABAJPHJ-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical group C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- HKHRENFWPKWVML-UHFFFAOYSA-N 2-methoxypyrimidin-5-amine Chemical compound COC1=NC=C(N)C=N1 HKHRENFWPKWVML-UHFFFAOYSA-N 0.000 description 1
- VGPRNGGSKJHOFE-UHFFFAOYSA-N 2-methylpyrimidin-5-amine Chemical compound CC1=NC=C(N)C=N1 VGPRNGGSKJHOFE-UHFFFAOYSA-N 0.000 description 1
- AVGNTKARJQHJRN-UHFFFAOYSA-N 2-methylsulfonylpyridin-4-amine Chemical compound CS(=O)(=O)C1=CC(N)=CC=N1 AVGNTKARJQHJRN-UHFFFAOYSA-N 0.000 description 1
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- LKDJYZBKCVSODK-UHFFFAOYSA-N 3,8-diazabicyclo[3.2.1]octane Chemical compound C1NCC2CCC1N2 LKDJYZBKCVSODK-UHFFFAOYSA-N 0.000 description 1
- XLZYKTYMLBOINK-UHFFFAOYSA-N 3-(4-hydroxybenzoyl)benzoic acid Chemical compound OC(=O)C1=CC=CC(C(=O)C=2C=CC(O)=CC=2)=C1 XLZYKTYMLBOINK-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- CNAIMMQZMLBREW-UHFFFAOYSA-N 3-bromo-5-methylsulfonylpyridine Chemical compound CS(=O)(=O)C1=CN=CC(Br)=C1 CNAIMMQZMLBREW-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- DJKUIGPCSNRFRK-UHFFFAOYSA-N 3-methyl-1,2,4-thiadiazol-5-amine Chemical compound CC1=NSC(N)=N1 DJKUIGPCSNRFRK-UHFFFAOYSA-N 0.000 description 1
- FNXYWHTZDAVRTB-UHFFFAOYSA-N 3-methyl-1,2-oxazol-5-amine Chemical compound CC=1C=C(N)ON=1 FNXYWHTZDAVRTB-UHFFFAOYSA-N 0.000 description 1
- ALGTUXWSZCDSKH-UHFFFAOYSA-N 3-methyl-2-(3-methylsulfonylanilino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound CS(=O)(=O)C=1C=C(C=CC1)NC=1N=C2C(=NC1C)N(C=C2C(=O)O)COCC[Si](C)(C)C ALGTUXWSZCDSKH-UHFFFAOYSA-N 0.000 description 1
- SWERHPHLAMCUJG-UHFFFAOYSA-N 3-methyl-2-(3-methylsulfonylanilino)-5H-pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound CS(=O)(=O)C=1C=C(C=CC1)NC=1N=C2C(=NC1C)NC=C2C(=O)O SWERHPHLAMCUJG-UHFFFAOYSA-N 0.000 description 1
- DOEKKXDEPSNZKV-UHFFFAOYSA-N 3-methyl-2-[(1-methylpyrazol-4-yl)amino]-5H-pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound CC1=C(N=C2C(=N1)NC=C2C(=O)O)NC=2C=NN(C2)C DOEKKXDEPSNZKV-UHFFFAOYSA-N 0.000 description 1
- LGSCIMCJGYVHPY-UHFFFAOYSA-N 3-methyl-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxylic acid Chemical compound CC1=C(N=C2C(=N1)N(C=C2C(=O)O)COCC[Si](C)(C)C)NC2=CC(=NS2)C LGSCIMCJGYVHPY-UHFFFAOYSA-N 0.000 description 1
- MBNPJRQKQLLRIS-UHFFFAOYSA-N 3-methylsulfonylaniline Chemical compound CS(=O)(=O)C1=CC=CC(N)=C1 MBNPJRQKQLLRIS-UHFFFAOYSA-N 0.000 description 1
- 125000006201 3-phenylpropyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- RJWBTWIBUIGANW-UHFFFAOYSA-N 4-chlorobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Cl)C=C1 RJWBTWIBUIGANW-UHFFFAOYSA-N 0.000 description 1
- XJEVFFNOMKXBLU-UHFFFAOYSA-N 4-methylsulfonylaniline Chemical compound CS(=O)(=O)C1=CC=C(N)C=C1 XJEVFFNOMKXBLU-UHFFFAOYSA-N 0.000 description 1
- QJMYXHKGEGNLED-UHFFFAOYSA-N 5-(2-hydroxyethylamino)-1h-pyrimidine-2,4-dione Chemical compound OCCNC1=CNC(=O)NC1=O QJMYXHKGEGNLED-UHFFFAOYSA-N 0.000 description 1
- FYTLHYRDGXRYEY-UHFFFAOYSA-N 5-Methyl-3-pyrazolamine Chemical compound CC=1C=C(N)NN=1 FYTLHYRDGXRYEY-UHFFFAOYSA-N 0.000 description 1
- ZNQOALAKPLGUPH-UHFFFAOYSA-N 5-methylpyrazin-2-amine Chemical compound CC1=CN=C(N)C=N1 ZNQOALAKPLGUPH-UHFFFAOYSA-N 0.000 description 1
- UAOOJJPSCLNTOP-UHFFFAOYSA-N 6-methylpyrazin-2-amine Chemical compound CC1=CN=CC(N)=N1 UAOOJJPSCLNTOP-UHFFFAOYSA-N 0.000 description 1
- UENBBJXGCWILBM-UHFFFAOYSA-N 6-methylpyridin-3-amine Chemical compound CC1=CC=C(N)C=N1 UENBBJXGCWILBM-UHFFFAOYSA-N 0.000 description 1
- IAQVUJPCFHWOGZ-UHFFFAOYSA-N 6-methylsulfonylpyridin-3-amine Chemical compound CS(=O)(=O)C1=CC=C(N)C=N1 IAQVUJPCFHWOGZ-UHFFFAOYSA-N 0.000 description 1
- AMKGKYQBASDDJB-UHFFFAOYSA-N 9$l^{2}-borabicyclo[3.3.1]nonane Chemical compound C1CCC2CCCC1[B]2 AMKGKYQBASDDJB-UHFFFAOYSA-N 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- OBMZMSLWNNWEJA-XNCRXQDQSA-N C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 Chemical compound C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 OBMZMSLWNNWEJA-XNCRXQDQSA-N 0.000 description 1
- RMPLNQWYNMRKDF-UHFFFAOYSA-N C=[n-](ccc1n2)c1nc(Cl)c2Br Chemical compound C=[n-](ccc1n2)c1nc(Cl)c2Br RMPLNQWYNMRKDF-UHFFFAOYSA-N 0.000 description 1
- DTIUMVGSYUDDID-UHFFFAOYSA-N CC(C)(C)N(C)C(c1c[nH]c(nc2)c1nc2Nc1nc(C)n[s]1)=O Chemical compound CC(C)(C)N(C)C(c1c[nH]c(nc2)c1nc2Nc1nc(C)n[s]1)=O DTIUMVGSYUDDID-UHFFFAOYSA-N 0.000 description 1
- RSMNRVRWXWNXDT-UHFFFAOYSA-N CC(C)(C)NC(c1c[nH]c(nc2)c1nc2N(C)c1n[n](C)cc1)=O Chemical compound CC(C)(C)NC(c1c[nH]c(nc2)c1nc2N(C)c1n[n](C)cc1)=O RSMNRVRWXWNXDT-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Chemical group OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Chemical group OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 102000009438 IgE Receptors Human genes 0.000 description 1
- 108010073816 IgE Receptors Proteins 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical group OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- TXXHDPDFNKHHGW-CCAGOZQPSA-N Muconic acid Chemical group OC(=O)\C=C/C=C\C(O)=O TXXHDPDFNKHHGW-CCAGOZQPSA-N 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- AFBPFSWMIHJQDM-UHFFFAOYSA-N N-methyl-N-phenylamine Natural products CNC1=CC=CC=C1 AFBPFSWMIHJQDM-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- XBPRIWSOQQALDQ-UHFFFAOYSA-N N-tert-butyl-2-[(1-methylpyrazol-4-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC=2C=NN(C2)C)COCC[Si](C)(C)C XBPRIWSOQQALDQ-UHFFFAOYSA-N 0.000 description 1
- QIIAJYMXWOFKLM-UHFFFAOYSA-N N-tert-butyl-2-[(3-methyl-1,2,4-thiadiazol-5-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC2=NC(=NS2)C)COCC[Si](C)(C)C QIIAJYMXWOFKLM-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 101710176384 Peptide 1 Proteins 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 102000014400 SH2 domains Human genes 0.000 description 1
- 108050003452 SH2 domains Proteins 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 239000012317 TBTU Substances 0.000 description 1
- QYTDEUPAUMOIOP-UHFFFAOYSA-N TEMPO Chemical group CC1(C)CCCC(C)(C)N1[O] QYTDEUPAUMOIOP-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 1
- OCAFBWCIHWDTKE-UHFFFAOYSA-N [dimethylamino-(3-oxidobenzotriazol-3-ium-1-yl)methylidene]-dimethylazanium Chemical compound C1=CC=C2[N+](=C(N(C)C)N(C)C)N=[N+]([O-])C2=C1 OCAFBWCIHWDTKE-UHFFFAOYSA-N 0.000 description 1
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000010398 acute inflammatory response Effects 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 229960001456 adenosine triphosphate Drugs 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000004471 alkyl aminosulfonyl group Chemical group 0.000 description 1
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000005466 alkylenyl group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 125000005100 aryl amino carbonyl group Chemical group 0.000 description 1
- 125000005141 aryl amino sulfonyl group Chemical group 0.000 description 1
- 125000005116 aryl carbamoyl group Chemical group 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 208000037979 autoimmune inflammatory disease Diseases 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- AZWXAPCAJCYGIA-UHFFFAOYSA-N bis(2-methylpropyl)alumane Chemical compound CC(C)C[AlH]CC(C)C AZWXAPCAJCYGIA-UHFFFAOYSA-N 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 230000009460 calcium influx Effects 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- KYKAJFCTULSVSH-UHFFFAOYSA-N chloro(fluoro)methane Chemical compound F[C]Cl KYKAJFCTULSVSH-UHFFFAOYSA-N 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229940061631 citric acid acetate Drugs 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000000306 component Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- 125000005117 dialkylcarbamoyl group Chemical group 0.000 description 1
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 239000013024 dilution buffer Substances 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- BFMYDTVEBKDAKJ-UHFFFAOYSA-L disodium;(2',7'-dibromo-3',6'-dioxido-3-oxospiro[2-benzofuran-1,9'-xanthene]-4'-yl)mercury;hydrate Chemical compound O.[Na+].[Na+].O1C(=O)C2=CC=CC=C2C21C1=CC(Br)=C([O-])C([Hg])=C1OC1=C2C=C(Br)C([O-])=C1 BFMYDTVEBKDAKJ-UHFFFAOYSA-L 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical group CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008393 encapsulating agent Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 125000002587 enol group Chemical group 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- FDLDWKIEWAWOSL-UHFFFAOYSA-N ethyl acetate;2-methylpentane Chemical compound CCCC(C)C.CCOC(C)=O FDLDWKIEWAWOSL-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000000174 gluconic acid Chemical group 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Chemical group 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- ZFGMDIBRIDKWMY-PASTXAENSA-N heparin Chemical compound CC(O)=N[C@@H]1[C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O[C@@H]1O[C@@H]1[C@@H](C(O)=O)O[C@@H](O[C@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@@H](O[C@@H]3[C@@H](OC(O)[C@H](OS(O)(=O)=O)[C@H]3O)C(O)=O)O[C@@H]2O)CS(O)(=O)=O)[C@H](O)[C@H]1O ZFGMDIBRIDKWMY-PASTXAENSA-N 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004634 hexahydroazepinyl group Chemical group N1(CCCCCC1)* 0.000 description 1
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- ZMPXGSYLKFAGOV-UHFFFAOYSA-N hydron;3-methylsulfonylaniline;chloride Chemical compound Cl.CS(=O)(=O)C1=CC=CC(N)=C1 ZMPXGSYLKFAGOV-UHFFFAOYSA-N 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 239000005414 inactive ingredient Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000011812 mixed powder Substances 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 235000019426 modified starch Nutrition 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- XXZNFWHGOMHWCO-UHFFFAOYSA-N n,n-diethylthiohydroxylamine Chemical compound CCN(S)CC XXZNFWHGOMHWCO-UHFFFAOYSA-N 0.000 description 1
- YAFSQBDMABSKEB-UHFFFAOYSA-N n-(1-hydroxy-2-methylpropan-2-yl)-2-(pyridin-3-ylamino)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(CO)C)=CN(COCC[Si](C)(C)C)C2=NC=C1NC1=CC=CN=C1 YAFSQBDMABSKEB-UHFFFAOYSA-N 0.000 description 1
- KJRGMCQCTQQPGI-UHFFFAOYSA-N n-(1-hydroxy-2-methylpropan-2-yl)-2-[(1-methylpyrazol-4-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound C1=NN(C)C=C1NC1=CN=C(N(COCC[Si](C)(C)C)C=C2C(=O)NC(C)(C)CO)C2=N1 KJRGMCQCTQQPGI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- DBGFNLVRAFYZBI-UHFFFAOYSA-N n-methylpyridin-3-amine Chemical compound CNC1=CC=CN=C1 DBGFNLVRAFYZBI-UHFFFAOYSA-N 0.000 description 1
- XNSWEUWIESIGDY-UHFFFAOYSA-N n-tert-butyl-2-[(1,3-dimethylpyrazol-4-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound CC1=NN(C)C=C1NC1=CN=C(N(COCC[Si](C)(C)C)C=C2C(=O)NC(C)(C)C)C2=N1 XNSWEUWIESIGDY-UHFFFAOYSA-N 0.000 description 1
- DOECTFNXFJFXGU-UHFFFAOYSA-N n-tert-butyl-2-[(6-methylsulfonylpyridin-3-yl)amino]-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound N1=C2C(C(=O)NC(C)(C)C)=CN(COCC[Si](C)(C)C)C2=NC=C1NC1=CC=C(S(C)(=O)=O)N=C1 DOECTFNXFJFXGU-UHFFFAOYSA-N 0.000 description 1
- PAENRTZWYSNKHE-UHFFFAOYSA-N n-tert-butyl-3-methyl-2-[(3-methyl-1,2-thiazol-5-yl)amino]-5h-pyrrolo[2,3-b]pyrazine-7-carboxamide Chemical compound S1N=C(C)C=C1NC(C(=N1)C)=NC2=C1NC=C2C(=O)NC(C)(C)C PAENRTZWYSNKHE-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000021766 negative regulation of B cell proliferation Effects 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 102000037979 non-receptor tyrosine kinases Human genes 0.000 description 1
- 108091008046 non-receptor tyrosine kinases Proteins 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical group CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Chemical group CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000004533 oil dispersion Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Chemical group OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- YWAKXRMUMFPDSH-UHFFFAOYSA-N pentene Chemical compound CCCC=C YWAKXRMUMFPDSH-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- LBJQKYPPYSCCBH-UHFFFAOYSA-N spiro[3.3]heptane Chemical compound C1CCC21CCC2 LBJQKYPPYSCCBH-UHFFFAOYSA-N 0.000 description 1
- 239000008117 stearic acid Chemical group 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- 239000005061 synthetic rubber Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- FVPBCJLOGUNOKW-UHFFFAOYSA-N tert-butyl N-[1-[3-[[7-(tert-butylcarbamoyl)-5-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyrazin-2-yl]amino]phenyl]ethyl]carbamate Chemical compound C(C)(C)(C)NC(=O)C1=CN(C2=NC=C(N=C21)NC=2C=C(C=CC2)C(C)NC(OC(C)(C)C)=O)COCC[Si](C)(C)C FVPBCJLOGUNOKW-UHFFFAOYSA-N 0.000 description 1
- QZEKBDHFLADZKW-UHFFFAOYSA-N tert-butyl n-[1-(3-aminophenyl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)NC(C)C1=CC=CC(N)=C1 QZEKBDHFLADZKW-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Diabetes (AREA)
- Neurosurgery (AREA)
- Pulmonology (AREA)
- Psychiatry (AREA)
- Dermatology (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Hospice & Palliative Care (AREA)
- Rheumatology (AREA)
- Transplantation (AREA)
- Oncology (AREA)
- Obesity (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Enzymes And Modification Thereof (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
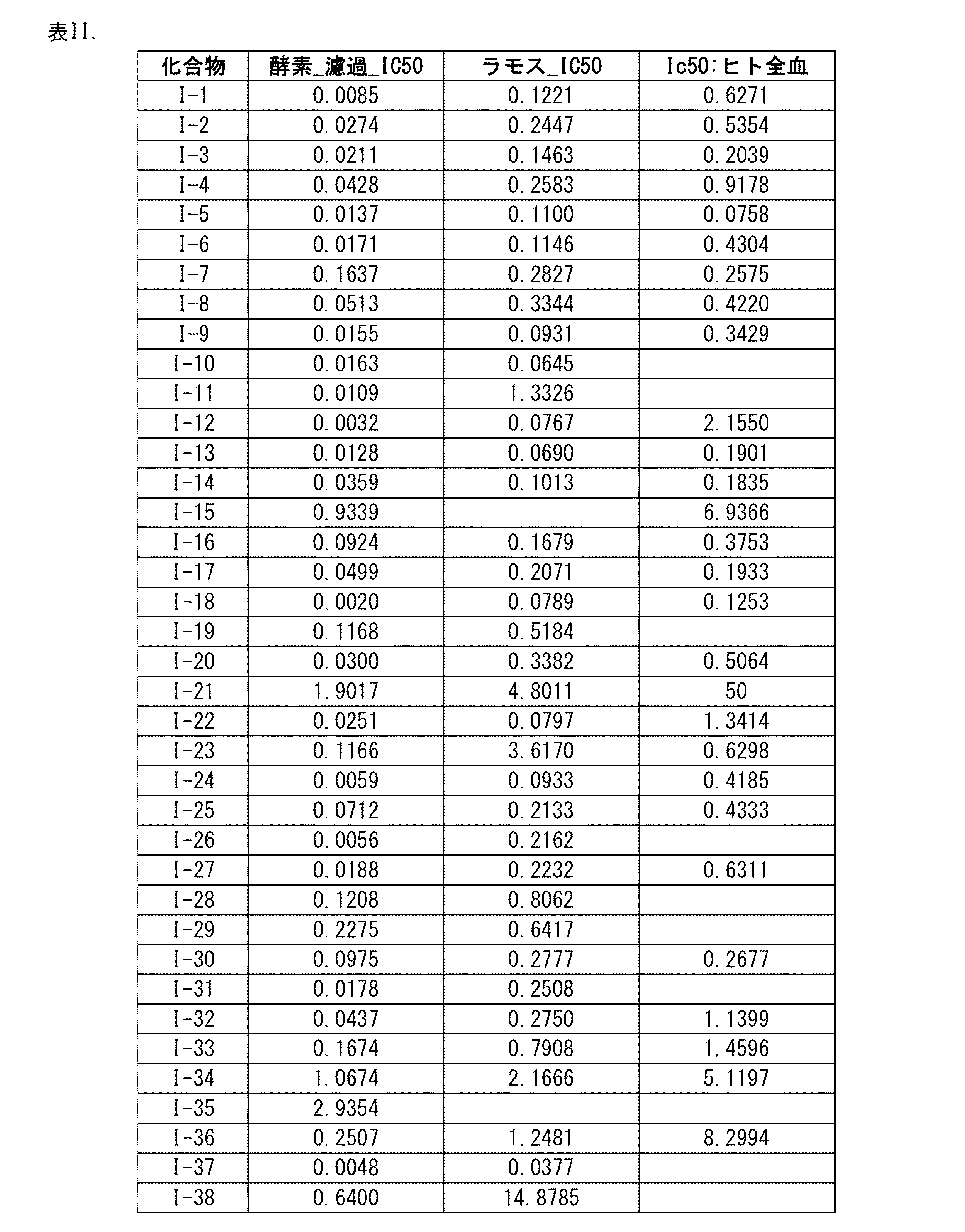
R1は、H、ハロ、又は低級アルキルであり;
R2は、低級アルキル又は低級ヒドロキシアルキルであり;
R3は、H又は低級アルキルであり;
Aは、一又は複数のA’で置換されていてもよい、単環式又は二環式ヘテロアリール(好ましくは、ピリジニル、ピリミジル、ピラゾリル、イソオキサゾリル、チアジアゾリル、チアゾリル若しくはピラジニル)又はフェニルであり;且つ各A’は、独立して、低級アルキル、ハロ、低級アルキルスルホニル、低級アルキルアミド、アミド、低級ヒドロキシアルキル、低級アルコキシ、アミノ低級アルキル、又は重水素である]の化合物;又はその薬学的に許容される塩を提供する。
ここで使用される「a」又は「an」実体なる語句は、その実体の一又は複数を指す;例えば、化合物(a compound)は、一若しくは複数の化合物又は少なくとも一の化合物を指す。同様に、「a」(又は「an」)、「一又は複数」、及び「少なくとも一」なる用語は、本明細書では互換的に使用され得る。
例はまた、例えば、3,8−ジアザ−ビシクロ[3.2.1]オクタン、2,5−ジアザ−ビシクロ[2.2.2]オクタン又はオクタヒドロ−ピラジノ[2,1−c][1,4]オキサジンなどの二環式であってもよい。
本願は、式I、II又はIII
R1は、H、ハロ、又は低級アルキルであり;
R2は、低級アルキル又は低級ヒドロキシアルキルであり;
R3は、H又は低級アルキルであり;
Aは、一又は複数のA’で置換されていてもよい、単環式又は二環式ヘテロアリール(好ましくは、ピリジニル、ピリミジル、ピラゾリル、イソオキサゾリル、チアジアゾリル、チアゾリル若しくはピラジニル)又はフェニルであり;且つ各A’は、独立して、低級アルキル、ハロ、低級アルキルスルホニル、低級アルキルアミド、アミド、低級ヒドロキシアルキル、低級アルコキシ、アミノ低級アルキル、又は重水素である]の化合物;又はその薬学的に許容される塩を提供する。
R1は、H、ハロ、又は低級アルキルであり;
R2は、低級アルキル又は低級ヒドロキシアルキルであり;
R3は、H又は低級アルキルであり;
Aは、一又は複数のA’で置換されていてもよい、単環式又は二環式ヘテロアリール(好ましくは、ピリジニル、ピリミジル、ピラゾリル、イソオキサゾリル、チアジアゾリル、チアゾリル若しくはピラジニル)又はフェニルであり;且つ各A’は、独立して、低級アルキル、ハロ、低級アルキルスルホニル、アミド、低級ヒドロキシアルキル、低級アルコキシ、アミノ低級アルキル、又は重水素である]の化合物;又はその薬学的に許容される塩を提供する。
N-tert-ブチル-2-(5-メチルピリジン-2-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-(1-ヒドロキシ-2-メチルプロパン-2-イル)-2-(5-メチルピリジン-2-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(1-メチル-1H-ピラゾール-3-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-イソプロピル-2-(5-メチルピリジン-2-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(1-メチル-1H-ピラゾール-4-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(6-メチルピリジン-3-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2-メチルピリミジン-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(5-メチルピラジン-2-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-メチル-1H-ピラゾール-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(ピリジン-3-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2-フルオロ-4-メチルフェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(p-トリルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-(メチルスルホニル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(1-エチル-1H-ピラゾール-4-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2-(ジメチルカルバモイル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(1,5-ジメチル-1H-ピラゾール-4-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-(1-ヒドロキシ-2-メチルプロパン-2-イル)-2-(1-メチル-1H-ピラゾール-4-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-メチルイソチアゾール-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2,4-ジメチルフェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(4-(メチルスルホニル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(メチル(ピリジン-3-イル)アミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-メチルイソオキサゾール-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(6-(メチルスルホニル)ピリジン-3-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-(1-ヒドロキシエチル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2-メトキシピリミジン-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-(1-ヒドロキシ-2-メチルプロパン-2-イル)-2-(ピリジン-3-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
2-(3-(1-アミノエチル)フェニルアミノ)-N-tert-ブチル-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2-(メチルスルホニル)ピリジン-4-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(5-(メチルスルホニル)ピリジン-3-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(1,3-ジメチル-1H-ピラゾール-4-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-メチル-1,2,4-チアジアゾール-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2-メチル-6,7-ジヒドロ-2H-ピラゾロ[4,3-b]ピリジン-4(5H)-イル)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(1-メチル-6,7-ジヒドロ-1H-ピラゾロ[4, 3-b]ピリジン-4(5H)-イル)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
3-メチル-2-(1-メチル-1H-ピラゾール-4-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボン酸 tert-ブチルアミド;
2-(3-メタンスルホニル-フェニルアミノ)-3-メチル-5H-ピロロ[2,3-b]ピラジン-7-カルボン酸 tert-ブチルアミド;
3-メチル-2-(3-メチル-イソチアゾール-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボン酸 tert-ブチルアミド;
2-フェニルアミノ(D5)-5H-ピロロ[2,3-b]ピラジン-7-カルボン酸 tert-ブチルアミド;及び
N-tert-ブチル-3-クロロ-2-(3-メチルイソチアゾール-5-イルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミドからなる群より選択される化合物を提供する。
一般スキーム
以下の一般スキームにおいて、R2は、低級アルキル又は低級ヒドロキシアルキルであり得;Aは、一又は複数のA’で置換されていてもよい、単環式又は二環式ヘテロアリール又はフェニルであり得;各A’は、独立して、低級アルキル、ハロ、低級アルキルスルホニル、アミド、低級ヒドロキシアルキル、低級アルコキシ、アミノ低級アルキル、又は重水素であり得る。
本発明の化合物は、多種多様な経口投与投薬形態及び担体で製剤化され得る。経口投与は、錠剤、コーティング錠剤、糖衣錠剤、硬質及び軟質ゼラチンカプセル、溶液、乳液、シロップ又は懸濁液の形態であり得る。本発明の化合物は、投与経路のうちでも、連続(点滴)局所非経口、筋肉内、静脈内、皮下、経皮(これは浸透増強剤を含み得る)、頬側、鼻腔、吸入及び坐剤投与を含む、他の投与経路によって投与される場合、効果的である。好ましい投与方法は、一般には、苦痛の程度及び活性成分に対する患者の反応に応じて調整され得る簡便な1日投与レジメンを使用する経口投与である。
様々な経路による送達のための薬学的調合剤は、以下の表に示すとおりに製剤化される。表中に使用する場合の「活性成分」又は「活性化合物」とは、一又は複数の式Iの化合物を意味する。
約0.025〜0.5パーセントの活性化合物を含む数種の水性懸濁液を、鼻腔スプレー製剤として調製する。この製剤は、例えば、微結晶セルロース、カルボキシメチルセルロースナトリウム、デキストロースなどの不活性成分を含んでもよい。pHを調整するために塩酸を添加してもよい。鼻腔スプレー製剤は、典型的には1回の作動当たり約50〜100マイクロリットルの製剤を送達する、鼻腔スプレー計量ポンプによって送達してもよい。典型的な投与スケジュールは、4〜12時間ごとに2〜4回のスプレーである。
本明細書で記載される化合物は、キナーゼ阻害剤、特にSYK阻害剤である。これらの阻害剤は、哺乳動物における、キナーゼ阻害に応答性の一又は複数の疾病(SYK阻害及び/又はB細胞増殖の阻害に応答性の疾病を含む)の治療に有用であり得る。いかなる特定の理論にも拘束されることを望むことなく、本発明の化合物とSYKとの相互作用は、SYK活性の阻害、したがって、これらの化合物の薬学的有用性をもたらすと考えられている。したがって、本発明は、SYK活性の阻害に応答性の疾病を有し、及び/又はB細胞増殖を阻害する、哺乳動物、例えば、ヒトを治療するための方法であって、このような疾病を有する哺乳動物に、本明細書で提供される少なくとも一種の化学的実体の有効量を投与することを含む方法を含む。有効濃度は、例えば、化合物の血中濃度をアッセイすることによって実験的に、又は生物学的利用能を計算することによって、理論的に確定されてもよい。SYKに加えて影響を受け得る他のキナーゼには、他のチロシンキナーゼ及びセリン/スレオニンキナーゼが含まれるが、これらに限定されない。
一般に使用される略語は:アセチル(Ac)、アゾ−ビス−イソブチリルニトリル(AIBN)、大気(Atm)、9−ボラビシクロ[3.3.1]ノナン(9−BBN又はBBN)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(BINAP)、tert−ブトキシカルボニル(Boc)、ジ−tert−ブチルピロカーボネート又はboc無水物(BOC2O)、ベンジル(Bn)、ブチル(Bu)、ケミカルアブストラクト登録番号(CASRN)、ベンジルオキシカルボニル(CBZ又はZ)、カルボニル ジイミダゾール(CDI)、1,4−ジアザビシクロ[2.2.2]オクタン(DABCO)、ジエチルアミノ硫黄トリフルオリド(DAST)、ジベンジリデンアセトン(dba)、1,5−ジアザビシクロ[4.3.0]ノナ−5−エン(DBN)、1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン(DBU)、N,N’−ジシクロヘキシルカルボジイミド(DCC)、1,2−ジクロロエタン(DCE)、ジクロロメタン(DCM)、2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(DDQ)、ジエチル アゾジカルボキシレート(DEAD)、ジ−イソ−プロピルアゾジカルボキシレート(DIAD)、ジ−イソ−ブチルアルミナムヒドリド(DIBAL又はDIBAL−H)、ジ−イソ−プロピルエチルアミン(DIPEA)、N,N−ジメチル アセトアミド(DMA)、4−N,N−ジメチルアミノピリジン(DMAP)、N,N−ジメチルホルムアミド(DMF)、ジメチル スルホキシド(DMSO)、1,1’−ビス−(ジフェニルホスフィノ)エタン(dppe)、1,1’−ビス−(ジフェニルホスフィノ)フェロセン(dppf)、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩(EDCI)、2−エトキシ−1−エトキシカルボニル−1,2−ジヒドロキノリン(EEDQ)、エチル(Et)、酢酸エチル(EtOAc)、エタノール(EtOH)、2−エトキシ−2H−キノリン−1−カルボン酸 エチル エステル(EEDQ)、ジエチル エーテル(Et2O)、エチル イソプロピル エーテル(EtOiPr)、O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’N’−テトラメチルウロニウム ヘキサフルオロホスファート 酢酸(HATU)、酢酸(HOAc)、1−N−ヒドロキシベンゾトリアゾール(HOBt)、高圧液体クロマトグラフィー(HPLC)、イソ−プロパノール(IPA)、イソプロピルマグネシウムクロリド(iPrMgCl)、ヘキサメチル ジシラザン(HMDS)、液体クロマトグラフィー質量分光法(LCMS)、リチウム ヘキサメチル ジシラザン(LiHMDS)、メタ−クロロペロキシ安息香酸(m−CPBA)、メタノール(MeOH)、融点(mp)、MeSO2−(メシル又はMs)、メチル(Me)、アセトニトリル(MeCN)、m−クロロ過安息香酸(MCPBA)、質量スペクトル(ms)、メチル t−ブチル エーテル(MTBE)、メチル テトラヒドロフラン(MeTHF)、N−ブロモスクシンイミド(NBS)、n−ブチルリチウム(nBuLi)、N−カルボキシ無水物(NCA)、N−クロロスクシンイミド(NCS)、N−メチルモルホリン(NMM)、N−メチルピロリドン(NMP)、ピリジニウム クロロクロマート(PCC)、ジクロロ−((ビス−ジフェニルホスフィノ)フェロセニル)パラジウム(II)(Pd(dppf)Cl2)、酢酸パラジウム(II)(Pd(OAc)2)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(Pd2(dba)3)、ピリジニウム ジクロメート(PDC)、フェニル(Ph)、プロピル(Pr)、イソ−プロピル(i−Pr)、1平方インチ当たりポンド(psi)、ピリジン(pyr)、1,2,3,4,5−ペンタフェニル−1’−(ジ−tert−ブチルホスフィノ)フェロセン(Q−Phos)、室温(周囲温度、rt又はRT)、sec−ブチルリチウム(sBuLi)、tert−ブチルジメチルシリル又はt−BuMe2Si(TBDMS)、テトラ−n−ブチルアンモニウム フルオリド(TBAF)、トリエチルアミン(TEA又はEt3N)、2,2,6,6−テトラメチルピペリジン 1−オキシル(TEMPO)、トリフラート又はCF3SO2−(Tf)、トリフルオロ酢酸(TFA)、1,1’−ビス−2,2,6,6−テトラメチルヘプタン−2,6−ジオン(TMHD)、O−ベンゾトリアゾル−1−イル−N,N,N’,N’−テトラメチルウロニウム テトラフルオロボラート(TBTU)、薄層クロマトグラフィー(TLC)、テトラヒドロフラン(THF)、トリメチルシリル又はMe3Si(TMS)、p−トルエンスルホン酸一水和物(TsOH又はpTsOH)、4−Me−C6H4SO2−又はトシル(Ts)、及びN−ウレタン−N−カルボキシ無水物(UNCA)を含む。ノルマル(n)、イソ(i−)、第二級(sec−)、第三級(tert−)及びネオを含む慣用の命名法による名称は、アルキル部分とともに使用される場合、それらの通常の意味を有する。(J. Rigaudy and D. P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford)
別段の記載がない限り、融点(すなわち、MP)を含む全ての温度は、摂氏(℃)である。示された及び/又は所望された生成物を生成する反応が、初期に添加された二種の反応試薬の組合せから必ずしも直接生じるとは限らないことがある、すなわち、示された及び/又は所望された生成物の形成を最終的にもたらす混合物中で生成される一又は複数の中間体が存在してもよいことを理解するべきである。先の略語は、調製及び実施例で使用してもよい。名称は全て、Autonom又はChemDrawを使用して作成した。
実施例1
手順1
2−ブロモ−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルバルデヒド
工程1
(2−ブロモ−7−ヒドロキシメチル−ピロロ[2,3−b]ピラジン−5−イル)−メタノール
(2−ブロモ−5H−ピロロ[2,3−b]ピラジン−7−イル)−メタノール
2−ブロモ−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルバルデヒド
工程1
2−ブロモ−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン
2−ブロモ−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルバルデヒド
2−ブロモ−5−(2−トリメチルシラニルエトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸
2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
2−ブロモ−N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
N−tert−ブチル−2−(5−メチルピリジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
5−メチルピリジン−2−アミン(38.0mg、351μmol)、2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(100mg、234μmol)(R)−(+)−2,2’−ビス(ジフェニルホスフィノ)−1,1’ビナフチル(7.28mg、11.7μmol)、酢酸パラジウム(II)(13.1mg、58.5μmol)及びナトリウムtert−ブトキシド(56.2mg、585μmol)の混合物に、DMF(1mL)及びトルエン(500μL)を添加し、混合物をマイクロ波で140℃で20分間加熱した。生成された濃色の混合物を水に注ぎ、酢酸エチルで抽出した。有機層を混ぜ合わせ、ブラインで洗浄し、乾燥させ(硫酸マグネシウム)、濾過して、クロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(5−メチルピリジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(62mg、136μmol、58%)をオフホワイト色の固体として得た。MS (EI/CI) m/z: 455.3 [M+H]。
N−tert−ブチル−2−(5−メチルピリジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(162mg、356μmol)が入ったジクロロメタン(3mL)の溶液に、トリフルオロ酢酸(813mg、549μL、7.13mmol)を添加し、混合物を室温で16時間撹拌した。次いで、混合物を真空で濃縮し、得られた残留物をジクロロメタン(3mL)、メタノール(1mL)及び水酸化アンモニウム(0.6mL)に溶解し、混合物を室温で30分間撹拌した。形成された黄色の沈殿物を濾過により収集し、乾燥させた。母液をクロマトグラフィー(シリカ、5−25%、ジクロロメタン中メタノール:ジクロロメタンが1:4の溶液)で精製し、固体と混ぜ合わせてN−tert−ブチル−2−(5−メチルピリジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(77mg、237μmol、67%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.37 (s, 1 H), 9.88 (s, 1 H), 8.38 (s, 1 H), 8.13 (d, J=2.5 Hz, 1 H), 8.04 (d, J=3.2 Hz, 1 H), 7.88 (d, J=8.5 Hz, 1 H), 7.74 (s, 1 H), 7.51 (dd, J=8.2, 2.3 Hz, 1 H), 2.25 (s, 3 H), 1.47 (s, 9 H); MS (EI/CI) m/z: 325.1 [M+H]。
N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(5−メチルピリジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
5−メチルピリジン−2−アミン(72.1mg、666μmol)、2−ブロモ−N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(197mg、444μmol)、(R)−(+)−2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(13.8mg、22.2μmol)、酢酸パラジウム(II)(24.9mg、111μmol)とナトリウムtert−ブトキシド(107mg、1.11mmol)の混合物に、DMF(2mL)及びトルエン(1mL)を添加し、次いでマイクロ波で140℃で20分間加熱した。生成された濃色の混合物を水に注ぎ、酢酸エチル(3x)で抽出した。有機層を混ぜ合わせ、ブライン(2x)で洗浄し、硫酸マグネシウム上で乾燥させ、クロマトグラフィー(シリカ、ヘキサン中65−100%の酢酸エチル)で精製し、N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(5−メチルピリジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(66mg、140μmol、32%)を淡黄色の固体として得た。MS (EI/CI) m/z: 471.4。
N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(5−メチルピリジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(66mg、140μmol)が入ったジクロロメタン(1.5mL)の溶液に、トリフルオロ酢酸(320mg、216μL、2.8mmol)を添加し、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮し、次いで残留物をジクロロメタン(2mL)、メタノール(0.5mL)及び水酸化アンモニウム(0.3mL)に溶解した。室温で30分間撹拌した後、混合物を真空で濃縮し、エーテル及び水で粉砕した。黄色の沈殿物を濾過により収集し、水、次いでエーテルで洗浄し、N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(5−メチルピリジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(31mg、91.1μmol、65%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δ ppm: 12.38 (s, 1 H), 9.86 (s, 1 H), 8.39 (s, 1 H), 8.12 (d, J=2.5 Hz, 1 H), 8.04 (d, J=3.3 Hz, 1 H), 7.95 (d, J=8.4 Hz, 1 H), 7.74 (s, 1 H), 7.54 (dd, J=8.6, 2.3 Hz, 1 H), 5.06 (s, 1 H), 3.58 (s, 2 H), 2.25 (s, 3 H), 1.39 (s, 6 H); MS (EI/CI) m/z: 341.1 [M+H]。
N−tert−ブチル−2−(1−メチル−1H−ピラゾール−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(200mg、468μmol)、1−メチル−1H−ピラゾール−3−アミン(68.2mg、702μmol)、BINAP(14.6mg、23.4μmol)、酢酸パラジウム(II)(26.3mg、117μmol)及びナトリウムtert−ブトキシド(112mg、1.17mmol)の混合物に、DMF(1.01mL)及びトルエン(503μL)を添加し、混合物をマイクロ波で140℃で20分間加熱した。反応混合物を冷却し、次いで水で希釈し、酢酸エチル中に抽出した。有機層を混ぜ合わせ、ブラインで洗浄し、硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中20−100%の酢酸エチル)で精製し、N−tert−ブチル−2−(1−メチル−1H−ピラゾール−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(140mg、316μmol、67%)を黄色の固体として得た。MS (EI/CI) m/z: 444.3。
N−tert−ブチル−2−(1−メチル−1H−ピラゾール−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(140mg、316μmol)が入ったジクロロメタン(3.5mL)の溶液に、トリフルオロ酢酸(720mg、486μL、6.31mmol)を添加し、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮し、得られた残留物をジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.65mL)に溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いでエーテル及び水で粉砕した。固体を濾過により収集し、真空で乾燥させ、N−tert−ブチル−2−(1−メチル−1H−ピラゾール−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(44mg、140μmol、45%)を褐色の固体として得た。1H NMR (400 MHz, メタノール-d)δppm: 8.07 (s, 1 H), 7.93 (s, 1 H), 7.81 (s, 1 H), 7.35 (d, J=2.2 Hz, 1 H), 6.32 (d, J=2.2 Hz, 1 H), 3.67 (s, 3 H), 1.35 (s, 9 H); MS (EI/CI) m/z: 314.1 [M+H]。
N−イソプロピル−2−(5−メチルピリジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
2−ブロモ−N−イソプロピル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、363μmol)、5−メチルピリジン−2−アミン(58.9mg、544μmol)、BINAP(11.3mg、18.1μmol)、酢酸パラジウム(II)(20.4mg、90.7μmol)及びナトリウムtert−ブトキシド(87.2mg、907μmol)の混合物に、DMF(780μL)及びトルエン(390μL)を添加し、混合物をマイクロ波で140℃で20分間加熱した。混合物を冷却し、次いで水及び酢酸エチルで希釈した。混合物を酢酸エチル(3x)で抽出し、次いで有機抽出物を混ぜ合わせ、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、クロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−イソプロピル−2−(5−メチルピリジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(90mg、204μmol、56%)をオフホワイトの固体として得た。MS (EI/CI) m/z: 441.3 [M+H]。
N−イソプロピル−2−(5−メチルピリジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(90mg、204μmol)が入ったジクロロメタン(2.5mL)の溶液に、トリフルオロ酢酸(466mg、315μL、4.09mmol)を添加し、室温で16時間撹拌した。反応混合物を真空で濃縮し、得られた残留物をジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.45mL)に溶解した。室温に置いて3時間後、混合物を真空で濃縮し、次いで水及びエーテルで粉砕した。固体を濾過により収集し、N−イソプロピル−2−(5−メチルピリジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(23mg、74.1μmol、36%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.40 (s, 1 H), 9.92 (s, 1 H), 8.43 (s, 1 H), 8.14 (s, 1 H), 8.09 (d, J=3.2, 1 H), 7.86 (s, 1 H), 7.85 (d, J=15.1 Hz, 1 H), 7.55 (dd, J=8.5, 2.3 Hz, 1 H), 4.16 (m, 1 H), 2.26 (s, 3 H), 1.25 (d, J=6.5, 6 H); MS (EI/CI) m/z: 311.1 [M+H]。
N−tert−ブチル−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、1−メチル−1H−ピラゾール−4−アミン塩酸塩(70.3mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)の混合物に、DMF(1mL)及びトルエン(500μL)を添加した。マイクロ波で140℃で20分間加熱した後、反応混合物を冷却し、水で希釈し、酢酸エチル(3x)で抽出した。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させ、クロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(89mg、201μmol、57%)を黄色の固体として得た。MS (EI/CI) m/z: 314.1 [M+H]。
N−tert−ブチル−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(89mg、201μmol)が入ったジクロロメタン(3mL)の溶液に、トリフルオロ酢酸(458mg、309μL、4.01mmol)を添加し、反応混合物を室温で16時間撹拌した。混合物を真空で濃縮し、次いでジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.45mL)に溶解し、混合物を室温で1時間撹拌した。真空で濃縮後、混合物を水で粉砕し、濾過して固体を収集した。固体をエーテルで洗浄して乾燥させ、N−tert−ブチル−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(58mg、185μmol、92%)を褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.18 (s, 1 H), 9.11 (s, 1 H), 7.90 (d, J=3.2, 1 H), 7.86 (s, 1 H), 7.85 (s, 1 H), 7.72 (s, 1 H), 7.55 (s, 1 H), 3.80 (s, 3 H), 1.43 (s, 9 H); MS (EI/CI) m/z: 314.1 [M+H]。
N−tert−ブチル−2−(6−メチルピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、6−メチルピリジン−3−アミン(56.9mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を冷却し、次いで水で希釈し、酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(6−メチルピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(57mg、125μmol、36%)を褐色の固体として得た。MS (EI/CI) m/z: 455.2 [M+H]。
N−tert−ブチル−2−(6−メチルピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(55mg、121μmol)が入ったジクロロメタン(1.9mL)の溶液に、トリフルオロ酢酸(276mg、186μL、2.42mmol)を添加し、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮し、次いで得られた残留物をジクロロメタン(2mL)、メタノール(1mL)及び水酸化アンモニウム(0.25mL)に溶解し、混合物を室温で1時間撹拌した。混合物を濃縮して水で粉砕し、次いで濾過し、固体を得、これをエーテルで洗浄して乾燥させ、N−tert−ブチル−2−(6−メチルピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(33mg、102μmol、84%)を褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.23 (s, 1 H), 9.41 (s, 1 H), 8.60 (d, J=2.3, 1 H), 7.91 (m, 3 H), 7.59 (s, 1 H), 7.06 (d, J=8.5, 1 H), 2.31 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z: 325.1 [M+H]。
N−tert−ブチル−2−(2−メチルピリミジン−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、2−メチルピリミジン−5−アミン(57.4mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中40−80%の酢酸エチル)で精製し、N−tert−ブチル−2−(2−メチルピリミジン−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(78mg、171μmol、49%)を褐色の固体として得た。MS (EI/CI) m/z: 456.2 [M+H]。
N−tert−ブチル−2−(2−メチルピリミジン−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(78mg、171μmol)が入ったジクロロメタン(2.5mL)の溶液に、トリフルオロ酢酸(390mg、264μL、3.42mmol)を添加し、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮し、得られた残留物をジクロロメタン(2.5mL)、メタノール(1.2mL)及び水酸化アンモニウム(0.35mL)で希釈し、混合物を室温で1時間撹拌した。反応混合物を真空で濃縮し、次いで水で粉砕した。固体を濾過により収集して水で洗浄し、N−tert−ブチル−2−(2−メチルピリミジン−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(43mg、132μmol、77%)を褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.31 (s, 1 H), 9.57 (s, 1 H), 8.90 (s, 2 H), 7.96 (d, J=3.3, 1 H), 7.92 (s, 1 H), 7.53 (s, 1 H), 2.46 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z: 326.1 [M+H]。
N−tert−ブチル−2−(5−メチルピラジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、5−メチルピラジン−2−アミン(57.4mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)を添加した混合物をマイクロ波で140℃で20分間加熱した。反応混合物を、水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(5−メチルピラジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(72mg、158μmol、45%)をオフホワイトの固体として得た。MS (EI/CI) m/z: 456.2 [M+H]。
N−tert−ブチル−2−(5−メチルピラジン−2−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(72mg、158μmol)が入ったジクロロメタン(2.5mL)の溶液に、トリフルオロ酢酸(360mg、243μL、3.16mmol)を添加して、室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(2.5mL)、メタノール(1.2mL)及び水酸化アンモニウム(0.35mL)で希釈し、混合物を室温で1時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集し、水及びエーテルで洗浄し、N−tert−ブチル−2−(5−メチルピラジン−2−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(26mg、79.9μmol、51%)を淡緑色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.45 (s, 1 H), 10.12 (s, 1 H), 9.17 (d, J=1.5, 1 H), 8.40 (s, 1 H), 8.21 (s, 1 H), 8.10 (d, J=3.2, 1 H), 7.73 (s, 1 H), 2.42 (s, 3 H), 1.46 (s, 9 H); MS (EI/CI) m/z: 326.1 [M+H]。
N−tert−ブチル−2−(3−メチル−1H−ピラゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、3−メチル−1H−ピラゾール−5−アミン(51.1mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中50−100%の酢酸エチル)で精製し、N−tert−ブチル−2−(3−メチル−1H−ピラゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(27mg、60.9μmol、17.3%)を褐色の固体として得た。MS (EI/CI) m/z: 444.3 [M+H]。
N−tert−ブチル−2−(3−メチル−1H−ピラゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(25mg、56.4μmol)が入ったジクロロメタン(1mL)の溶液に、トリフルオロ酢酸(129mg、86.8μL、1.13mmol)を添加し、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(1mL)、メタノール(0.5mL)及び水酸化アンモニウム(0.15mL)で希釈して、室温で1時間撹拌した。クロマトグラフィー(シリカ、20−50%[10%メタノール/ジクロロメタン/0.5%水酸化アンモニウム]/ジクロロメタン)で精製し、N−tert−ブチル−2−(3−メチル−1H−ピラゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(10mg、31.9μmol、57%)を淡褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.19 (s, 1 H), 10.12 (s, 1 H), 9.53 (s, 1 H), 8.07 (s, 1 H), 7.93 (d, J=3.0, 1 H), 7.81 (s, 1 H), 6.33 (s, 1 H), 2.19 (s, 3 H), 1.45 (s, 9 H); MS (EI/CI) m/z: 314.1 [M+H]。
N−tert−ブチル−2−(ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、ピリジン−3−アミン(49.5mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(44mg、99.9μmol、28.5%)を黄色の固体として得た。MS (EI/CI) m/z: 311.1 [M+H]。
N−tert−ブチル−2−(ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(44mg、99.9μmol)が入ったジクロロメタン(1.6mL)の溶液に、トリフルオロ酢酸(228mg、154μL、2.00mmol)を添加して、反応混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(1.6mL)、メタノール(0.8mL)及び水酸化アンモニウム(0.2mL)で希釈して、室温で1時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集して水及びエーテルで洗浄し、N−tert−ブチル−2−(ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(24mg、77.3μmol、77%)を褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.26 (s, 1 H), 9.54 (s, 1 H), 8.77 (d, J=2.5, 1 H), 8.04 (d, J=4.6, 1 H), 7.98 (d, J=8.5, 1 H), 7.92 (d, J=3.3, 1 H), 7.91 (s, 1 H), 7.57 (s, 1 H), 7.19 (m, 1 H), 1.31 (s, 9 H); MS (EI/CI) m/z: 311.1 [M+H]。
N−tert−ブチル−2−(2−フルオロ−4−メチルフェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、2−フルオロ−4−メチルアニリン(65.9mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、20−50%の酢酸エチル/ヘキサン)で精製し、N−tert−ブチル−2−(2−フルオロ−4−メチルフェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(87mg、184μmol、53%)をオフホワイトの固体として得た。MS (EI/CI) m/z: 470.1 [M-H]。
N−tert−ブチル−2−(2−フルオロ−4−メチルフェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(87mg、184μmol)が入ったジクロロメタン(3mL)の溶液に、トリフルオロ酢酸(421mg、284μL、3.69mmol)を添加して、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.4mL)で希釈して、室温で1時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集して、水及びエーテルで洗浄し、N−tert−ブチル−2−(2−フルオロ−4−メチルフェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(58mg、170μmol、92%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.27 (s, 1 H), 8.97 (s, 1 H), 8.10 (s, 1 H), 7.96 (d, J=2.3, 1 H), 7.81 (t, J=8.6, 1 H), 7.70 (s, 1 H), 7.10 (d, J=12.4, 1 H), 6.95 (d, J=8.0, 1 H), 2.29 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z: 342.1 [M+H]。
N−tert−ブチル−2−(p−トリルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、p−トルイジン(56.4mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(84.3mg、877μmol)の混合物を、マイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中20−45%の酢酸エチル)で精製し、N−tert−ブチル−2−(p−トリルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(80mg、116μmol、33%)を褐色のゴムとして得た。MS (EI/CI) m/z: 454.4 [M+H]。
N−tert−ブチル−2−(p−トリルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(80mg、176μmol)が入ったジクロロメタン(3mL)の溶液に、トリフルオロ酢酸(402mg、272μL、3.53mmol)を添加して、混合物を室温で16時間撹拌した。反応混合物を真空で濾過して、得られた残留物をジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.4mL)で希釈して、室温で1時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集し、水及びエーテルで洗浄した。クロマトグラフィー(シリカ、ジクロロメタン中20−40%の、メタノール:ジクロロメタン:水酸化アンモニウムが10:89.5:0.5の溶液)で精製し、N−tert−ブチル−2−(p−トリルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(17mg、52.6μmol、30%)を淡黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.26 (s, 1 H), 9.34 (s, 1 H), 7.97 (s, 1 H), 7.96 (d, J=3.3, 1 H), 7.76 (s, 1H), 7.57 (t, J=8.5, 2 H), 7.09 (d, J=8.1, 2 H), 2.26 (s, 3 H), 1.44 (s, 9 H); MS (EI/CI) m/z: 324.1 [M+H]。
N−tert−ブチル−2−(3−(メチルスルホニル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、3−(メチルスルホニル)アニリン塩酸塩(109mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(101mg、1.05mmol、当量:3)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中20−50%の酢酸エチル)で精製し、N−tert−ブチル−2−(3−(メチルスルホニル)フェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(61mg、118μmol、34%)を褐色のゴムとして得た。(EI/CI) m/z: 518.3 [M+H]。
N−tert−ブチル−2−(3−(メチルスルホニル)フェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(61mg、118μmol)が入ったジクロロメタン(1.8mL)の溶液に、トリフルオロ酢酸(269mg、182μL、2.36mmol)を添加して、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(1.8mL)、メタノール(0.9mL)及び水酸化アンモニウム(0.25mL)で希釈して、室温で1時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集し、水及びエーテルで洗浄した。クロマトグラフィー(シリカ、勾配未確認 ジクロロメタン/メタノール/0.5%水酸化アンモニウム)で精製し、N−tert−ブチル−2−(3−(メチルスルホニル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(26mg、67.1μmol、57%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.42 (s, 1 H), 9.87 (s, 1 H), 8.33 (d, J=8.2, 1 H), 8.05 (d, J=3.4, 1 H), 8.03 (s, 1H), 7.79 (t, J=2.0, 1 H), 7.68 (s, 1 H), 7.54 (t, J=8.0, 1 H), 7.47 (d, J=7.7, 1 H), 3.21 (s, 3 H), 1.42 (s, 9 H); MS (EI/CI) m/z: 388.2 [M+H]。
N−tert−ブチル−2−(1−エチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、1−エチル−1H−ピラゾール−4−アミン塩酸塩(51.8mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(101mg、1.05mmol、当量:3)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中40−80%の酢酸エチル)で精製し、N−tert−ブチル−2−(1−エチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(82mg、179μmol、51%)を黄色のゴムとして得た。(EI/CI) m/z: 458.4 [M+H]。
N−tert−ブチル−2−(1−エチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(82mg、179μmol)が入ったジクロロメタン(2.7mL)の溶液に、トリフルオロ酢酸(409mg、276μL、3.58mmol)を添加し、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.4mL)で希釈して、室温で1時間撹拌した。反応混合物を濃縮し、次いでクロマトグラフィー(シリカ、15−65%ジクロロメタン/[10%ジクロロメタン/メタノール/0.5%水酸化アンモニウム])で精製し、N−tert−ブチル−2−(1−エチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(33mg、101μmol、56%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.16 (s, 1 H), 9.06 (s, 1 H), 7.90 (d, J=3.3, 1 H), 7.85 (s, 1 H), 7.84 (s, 1H), 7.73 (s, 1 H), 7.59 (s, 1 H), 4.08 (q, J=7.3, 2 H), 1.42 (s, 9 H), 1.36 (t, J=7.3, 3 H); MS (EI/CI) m/z: 328.1 [M+H]。
N−tert−ブチル−2−(2−(ジメチルカルバモイル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)が入ったジクロロメタン(5.5mL)の溶液に、トリフルオロ酢酸(800mg、541μL、7.02mmol)を添加して、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(5mL)、メタノール(2.5mL)及び水酸化アンモニウム(0.7mL)で希釈して、室温で1時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集して、水及びエーテルで洗浄し、2−ブロモ−N−tert−ブチル−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(80mg、269μmol、77%)をオフホワイトの固体として得た。
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(80mg、269μmol)、2−アミノ−N,N−ジメチルベンズアミド塩酸塩(81.0mg、404μmol)、BINAP(8.38mg、13.5μmol)、酢酸パラジウム(II)(15.1mg、67.3μmol)及びナトリウムtert−ブトキシド(77.6mg、808μmol、当量:3)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(15−45%ジクロロメタン/[10%ジクロロメタン/メタノール/0.5%水酸化アンモニウム])で精製し、N−tert−ブチル−2(2(ジメチルカルバモイル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(16mg、42.1μmol、16%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.25 (s, 1 H), 8.77 (s, 1 H), 8.06 (s, 1 H), 7.96 (d, J=3.0, 1 H), 7.91 (d, J=7.9, 1 H), 7.78 (s, 1H), 7.38 (t, J=7.7, 1 H), 7.30 (d, J=7.5, 1 H), 7.11 (t, J=7.5, 1 H), 2.88 (s, 3 H), 2.80 (s, 3 H), 1.27 (s, 9 H); MS (EI/CI) m/z: 381.2 [M+H]。
N−tert−ブチル−2−(1,5−ジメチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、1,5−ジメチル−1H−ピラゾール−4−アミン二塩酸塩(96.9mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(118mg、1.23mmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(ヘキサン中50−100%の酢酸エチル)で精製し、N−tert−ブチル−2−(1,5−ジメチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(45mg、98.3μmol、28%)を黄色のゴムとして得た。(EI/CI) m/z: 458.3 [M+H]。
N−tert−ブチル−2−(1,5−ジメチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(45mg、98.3μmol)が入ったジクロロメタン(1.5mL)の溶液に、トリフルオロ酢酸(224mg、152μL、1.97mmol)を添加し、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(1.5mL)、メタノール(0.75mL)及び水酸化アンモニウム(0.2mL)で希釈して、室温で2時間撹拌した。反応混合物を濃縮し、次いでクロマトグラフィー(シリカ、ジクロロメタン中10−45%の、メタノール:ジクロロメタンが1:4の溶液)で精製し、N−tert−ブチル−2−(1,5−ジメチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(18mg、55.0μmol、56%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.10 (s, 1 H), 8.39 (s, 1 H), 7.87 (s, 1 H), 7.86 (s, 1 H), 7.85 (d, J=3.3, 1 H), 3.71 (s, 3 H), 2.16 (s, 3 H), 1.30 (s, 9 H); MS (EI/CI) m/z: 328.2 [M+H]。
N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、338μmol)、1−メチル−1H−ピラゾール−4−アミン塩酸塩(67.8mg、526μmol)、BINAP(10.5mg、16.9μmol)、酢酸パラジウム(II)(19.0mg、84.6μmol)及びナトリウムtert−ブトキシド(81.3mg、846μmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ジクロロメタン中20−65%の、メタノール:ジクロロメタン:水酸化アンモニウムが10:89.5:0.5の溶液)で精製し、N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(28mg、60.9μmol、18%)淡黄色の固体として得た。(EI/CI) m/z: 460.3 [M+H]。
N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(26mg、56.6μmol)が入ったジクロロメタン(1mL)に、トリフルオロ酢酸(129mg、87.2μL、1.13mmol)を添加して、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(1mL)、メタノール(0.5mL)及び水酸化アンモニウム(0.15mL)で希釈して、室温で2時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集して水及びエーテルで洗浄し、N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(12mg、36.4μmol、64%)を褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.19 (s, 1 H), 9.16 (s, 1 H), 8.01 (s, 1 H), 7.91 (d, J=3.0, 1 H), 7.86 (s, 1H), 7.70 (s, 1 H), 7.49 (s, 1 H), 5.15 (t, J=5.7, 1 H), 3.82 (s, 3 H), 3.57 (d, J=5.9, 2 H), 1.37 (s, 9 H); MS (EI/CI) m/z: 330.2 [M+H]。
N−tert−ブチル−2−(3−メチルイソチアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1.2mL)及びトルエン(600μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、3−メチルイソチアゾール−5−アミン塩酸塩(79.3mg、526μmol)、BINAP(10.9mg、17.5μmol)、酢酸パラジウム(II)(19.7mg、87.7μmol)及びナトリウムtert−ブトキシド(101mg、1.05mmol、当量:3)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中50−100%の酢酸エチル)で精製し、N−tert−ブチル−2−(3−メチルイソチアゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(32mg、69.5μmol、20%)を黄色の固体として得た。(EI/CI)m/z: 461.3 [M+H]。
N−tert−ブチル−2−(3−メチルイソチアゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(32mg、69.5μmol)が入ったジクロロメタン(1.2mL)の溶液に、トリフルオロ酢酸(158mg、107μL、1.39mmol)を添加して、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(1.2mL)、メタノール(0.6mL)及び水酸化アンモニウム(0.15mL)で希釈して、室温で2時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集して水及びエーテルで洗浄し、N−tert−ブチル−2−(3−メチルイソチアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(18mg、54.5μmol、78.4%)を褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.55 (s, 1 H), 11.39 (s, 1 H), 8.15 (d, J=3.2, 1 H), 8.11 (s, 1 H), 7.37 (s, 1H), 6.70 (s, 1 H), 2.33 (s, 3 H), 1.55 (s, 9 H); MS (EI/CI) m/z: 331.1 [M+H]。
N−tert−ブチル−2−(2,4−ジメチルフェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
DMF(1mL)及びトルエン(500μL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(200mg、468μmol)、2,4−ジメチルアニリン(85.1mg、526μmol)、BINAP(14.6mg、23.4μmol)、酢酸パラジウム(II)(26.3mg、117μmol)及びナトリウムtert−ブトキシド(112mg、1.17mmol)の混合物をマイクロ波で140℃で20分間加熱した。反応混合物を水で希釈し、次いで酢酸エチル中に抽出した(3x)。混合有機抽出物をブラインで洗浄し、次いで硫酸マグネシウム上で乾燥させた。クロマトグラフィー(シリカ、ヘキサン中20−60%の酢酸エチル)で精製し、N−tert−ブチル−2−(2,4−ジメチルフェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(82mg、175μmol、37.5%)を褐色のゴムとして得た。(EI/CI) m/z: 468.4 [M+H]。
N−tert−ブチル−2−(2,4−ジメチルフェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(82mg、175μmol)が入ったジクロロメタン(2.7mL)の溶液に、トリフルオロ酢酸(400mg、270μL、3.51mmol)を添加して、混合物を室温で16時間撹拌した。反応混合物を真空で濃縮して、得られた残留物をジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.4mL)で希釈して、室温で1時間撹拌した。混合物を濃縮して、水で粉砕した。固体を濾過により収集して水及びエーテルで洗浄し、N−tert−ブチル−2−(2,4−ジメチルフェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(7mg、20.7μmol、12%)を黄色の固体として得た。1H NMR (400 MHz, CDCl-3-d)δppm: 9.19 (s, 1 H), 8.12 (d, J=3.0, 1 H), 7.95 (s, 1 H), 7.86 (s, 1H), 7.50 (d, J=7.8, 1 H), 7.15 (s, 1 H), 7.08 (d, J=8.2, 1 H), 6.33 (s, 1 H), 2.38 (s, 3 H), 2.34 (s, 1 H), 1.49 (s, 9 H); MS (EI/CI) m/z: 338.2 [M+H]。
N−tert−ブチル−2−(4−(メチルスルホニル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(125mg、292μmol)、4−(メチルスルホニル)アニリン(75.1mg、439μmol)、キサントホス(50.8mg、87.7μmol)、Pd2(dba)3(26.8mg、29.2μmol)及び炭酸セシウム(191mg、585μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、セライトを通して濾過し、濾液を真空で濃縮した。クロマトグラフィー(シリカ、ヘキサン中20−75%の酢酸エチル)で精製し、N−tert−ブチル−2−(4−(メチルスルホニル)フェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(90mg、174μmol、59%)を淡黄色の固体として得た。(EI/CI) m/z: 518.3 [M+H]。
N−tert−ブチル−2−(4−(メチルスルホニル)フェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(90mg、174μmol)をジクロロメタン(3mL)に溶解し、トリフルオロ酢酸(396mg、268μL、3.48mmol)で処理した。室温で16時間撹拌した後、混合物を真空で濃縮して、次いでジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.35mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で希釈し、粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、N−tert−ブチル−2−(4−(メチルスルホニル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(65mg、168μmol、97%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.55 (s, 1 H), 10.12 (s, 1 H), 8.19 (d, J=2.8, 1 H), 8.16 (s, 1H), 7.95 (d, J=8.8, 2 H), 7.88 (d, J=8.8, 2 H), 7.75 (s, 1 H), 3.22 (s, 3 H), 1.54 (s, 9 H); MS (EI/CI) m/z: 388.2 [M+H]。
N−tert−ブチル−2−(メチル(ピリジン−3−イル)アミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(125mg、292μmol)、N−メチルピリジン−3−アミン(47.4mg、439μmol)、キサントホス(50.8mg、87.7μmol)、Pd2(dba)3(26.8mg、29.2μmol)及び炭酸セシウム(191mg、585μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却して、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、酢酸エチルが入ったヘキサン)で精製し、N−tert−ブチル−2−(メチル(ピリジン−3−イル)アミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(38mg、83.6μmol、29%)を黄色のゴムとして得た。(EI/CI) m/z: 455.4 [M+H]。
N−tert−ブチル−2−(メチル(ピリジン−3−イル)アミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(38mg、83.6μmol)をジクロロメタン(1.5mL)に溶解し、トリフルオロ酢酸(191mg、129μL、1.67mmol)で処理した。16時間後、混合物を濃縮して、残留物をジクロロメタン(1.5mL)、メタノール(0.75mL)及び水酸化アンモニウム(0.2mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を濃縮し、次いで水で粉砕して濾過し、固体を得た。これをエーテルで洗浄し、次いで乾燥させ、N−tert−ブチル−2−(メチル(ピリジン−3−イル)アミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(16mg、49.3μmol、59%)を淡黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.33 (s, 1 H), 8.64 (d, J=2.5, 1 H), 8.40 (dd, J=4.7, 1.2, 1 H), 8.05 (d, J=3.1, 1 H), 7.96 (s, 1H), 7.92 (s, 1 H), 7.82 (m, 1 H), 7.46 (m, 1 H), 3.50 (s, 3 H), 1.32 (s, 9 H); MS (EI/CI) m/z: 325.1 [M+H]。
N−tert−ブチル−2−(3−メチルイソオキサゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(125mg、292μmol)、3−メチルイソオキサゾール−5−アミン(43.0mg、439μmol)、キサントホス(50.8mg、87.7μmol)、Pd2(dba)3(26.8mg、29.2μmol)及び炭酸セシウム(191mg、585μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中20−75%の酢酸エチル)で精製し、N−tert−ブチル−2−(3−メチルイソオキサゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(33mg、74.2μmol、25.4%)を黄色の固体として得た。(EI/CI) m/z: 455.3 [M+H]。
N−tert−ブチル−2−(3−メチルイソオキサゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(33mg、74.2μmol)をジクロロメタン(1.3mL)に溶解し、トリフルオロ酢酸(169mg、114μL、1.48mmol)で処理した。混合物を室温で16時間撹拌し、次いで濃縮して、ジクロロメタン(1.3mL)、メタノール(0.65mL)及び水酸化アンモニウム(0.2mL)に再溶解し、室温で1時間撹拌した。混合物を濃縮して、水で希釈し、粉砕して濾過した。得られた固体をエーテルで洗浄して乾燥させ、N−tert−ブチル−2−(3−メチルイソオキサゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(16mg、45.8μmol、62%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm:12.53 (s, 1 H), 11.06 (s, 1 H), 8.14 (d, J=3.0, 1 H), 8.09 (s, 1 H), 7.57 (s, 1H), 6.18 (s, 1 H), 2.17 (s, 3 H), 1.48 (s, 9 H); MS (EI/CI) m/z: 315.1 [M+H]。
N−tert−ブチル−2−(6−(メチルスルホニル)ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2.4mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、6−(メチルスルホニル)ピリジン−3−アミン(90.7mg、526μmol)、キサントホス(60.9mg、105μmol)、Pd2(dba)3(32.1mg、35.1μmol)及び炭酸セシウム(229mg、702μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中20−95%の酢酸エチル)で精製し、N−tert−ブチル−2−(6−(メチルスルホニル)ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(97mg、187μmol、53%)を黄色の固体として得た。(EI/CI) m/z: 458.3 [M+H]。
N−tert−ブチル−2−(6−(メチルスルホニル)ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(97mg、187μmol)をジクロロメタン(2.88mL)に溶解し、トリフルオロ酢酸(426mg、288μL、3.74mmol)で処理した。混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.5mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、N−tert−ブチル−2−(6−(メチルスルホニル)ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(71mg、183μmol、98%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.51 (s, 1 H), 10.26 (s, 1 H), 8.99 (d, J=2.5, 1 H), 8.37 (dd, J=8.4, 2.7, 1 H), 8.14 (s, 1H), 8.11 (s, 1 H), 7.92 (d, J=8.7, 1 H), 7.62 (s, 1 H), 3.21 (s, 3 H), 1.45 (s, 9 H); MS (EI/CI) m/z: 389.2 [M+H]。
N−tert−ブチル−2−(3−(1−ヒドロキシエチル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、1−(3−アミノフェニル)エタノール(72.2mg、526μmol)、キサントホス(60.9mg、105μmol)、Pd2(dba)3(32.1mg、35.1μmol)及び炭酸セシウム(229mg、702μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中40−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(3−(1−ヒドロキシエチル)フェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(143mg、148μmol、42%)を黄色の固体として得た。(EI/CI) m/z: 519.3 [M+H]。
N−tert−ブチル−2−(3−(1−ヒドロキシエチル)フェニルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(143mg、296μmol)をジクロロメタン(4.5mL)に溶解し、トリフルオロ酢酸(674mg、456μL、5.91mmol)で処理した。混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(4mL)、メタノール(3mL)及び水酸化アンモニウム(0.7mL)に再溶解し、混合物を室温で2時間撹拌した。反応混合物を濃縮し、次いでクロマトグラフィー(シリカ、メタノールが入ったジクロロメタン)で精製し、N−tert−ブチル−2−(3−(1−ヒドロキシエチル)フェニルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(55mg、156μmol、53%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.29 (s, 1 H), 9.44 (s, 1 H), 8.00 (s, 1 H), 7.99 (s, 1 H), 7.98 (d, J=2.8, 1 H), 7.78 (s, 1 H), 7.19 (m, 2 H), 6.91 (d, J=7.5, 1 H), 5.17 (d, J=4.0, 1 H), 4.70 (m, 1 H), 3.38 (q, J=7.2, 1 H), 1.43 (s, 9 H), 1.34 (d. J=6.3, 3 H), 1.09 (t, J=7.2, 1 H); MS (EI/CI) m/z: 354.2 [M+H]。
N−tert−ブチル−2−(2−メトキシピリミジン−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、2−メトキシピリミジン−5−アミン(65.9mg、526μmol)、キサントホス(60.9mg、105μmol)、Pd2(dba)3(32.1mg、35.1μmol)及び炭酸セシウム(229mg、702μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中40−80%の酢酸エチル)で精製し、N−tert−ブチル−2−(2−メトキシピリミジン−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(99mg、210μmol、60%)を黄色の固体として得た。(EI/CI) m/z: 484.3 [M+H]。
N−tert−ブチル−2−(2−メトキシピリミジン−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(100mg、212μmol)をジクロロメタンに溶解し、トリフルオロ酢酸(484mg、327μL、4.24mmol)で処理した。混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.45mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、N−tert−ブチル−2−(2−メトキシピリミジン−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(64mg、187μmol、88%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.35 (s, 1 H), 9.48 (s, 1 H), 8.86 (s, 2 H), 8.01 (s, 1 H), 7.97 (s, 1H), 7.63 (s, 1 H), 3.88 (s, 3 H), 1.38 (s, 9 H); MS (EI/CI) m/z: 342.1 [M+H]。
N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、338μmol)、ピリジン−3−アミン(47.8mg、507μmol)、キサントホス(58.7mg、101μmol)、Pd2(dba)3(31.0mg、33.8μmol)及び炭酸セシウム(220mg、677μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中勾配40−100%の酢酸エチル、次いでジクロロメタン/メタノール)で精製し、N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(32mg、59.6μmol、18%)を淡黄色の固体として得た。(EI/CI) m/z: 472.3 [M+H]。
N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(32mg、70.1μmol)をジクロロメタン(1.2mL)に溶解し、トリフルオロ酢酸(160mg、108μL、1.4mmol)で処理した。混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(1mL)、メタノール(0.5mL)及び水酸化アンモニウム(0.2mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、N−(1−ヒドロキシ−2−メチルプロパン−2−イル)−2−(ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(21mg、64.3μmol、92%)を淡黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.39 (s, 1 H), 9.66 (s, 1 H), 8.85 (d, J=2.5, 1 H), 8.21 (m, 1 H), 8.16 (dd, J=4.7, 1.6, 1 H), 8.04 (s, 1 H), 8.03 (s, 1 H), 7.67 (s, 1 H), 7.33 (m, 1 H), 5.05 (t, J=5.6, 1 H), 3.58 (d, J=5.9, 2 H), 1.36 (s, 6 H); MS (EI/CI) m/z: 327.1 [M+H]。
2−(3−(1−アミノエチル)フェニルアミノ)−N−tert−ブチル−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、tert−ブチル 1−(3−アミノフェニル)エチルカルバメート(124mg、526μmol)、キサントホス(60.9mg、105μmol)、Pd2(dba)3(32.1mg、35.1μmol)及び炭酸セシウム(229mg、702μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィーで精製し(シリカ、ヘキサン中20−40%の酢酸エチル)で精製し、tert−ブチル1−(3−(7−(tert−ブチルカルバモイル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−2−イルアミノ)フェニル)エチルカルバメート(150mg、257μmol、73%)を黄色のゴムとして得た。(EI/CI) m/z: 583.5 [M+H]。
tert−ブチル 1−(3−(7−(tert−ブチルカルバモイル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−2−イルアミノ)フェニル)エチルカルバメート(150mg、257μmol)が入ったジクロロメタン(4mL)の溶液に、トリフルオロ酢酸(587mg、397μL、5.15mmol)を添加し、混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(4mL)、メタノール(2mL)及び水酸化アンモニウム(0.7mL)に再溶解し、混合物を室温で1時間撹拌した。反応混合物を濃縮し、次いでクロマトグラフィー(シリカ、6−10%メタノール/ジクロロメタン)で精製し、2−(3−(1−アミノエチル)フェニルアミノ)−N−tert−ブチル−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(64mg、182μmol、71%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 9.44 (s, 1 H), 8.01 (s, 1 H), 7.99 (d, J=7.9, 1 H), 7.98 (s, 1 H), 7.77 (s, 1H), 7.22 (t, J=7.8, 1 H), 7.20 (s, 1 H), 6.98 (d, J=7.8, 1 H), 4.05 (q, J=6.1, 1 H), 1.43 (s, 9 H), 1.32 (d, J=6.5, 3 H); MS (EI/CI) m/z: 353.2 [M+H]。
N−tert−ブチル−2−(2−(メチルスルホニル)ピリジン−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(120mg、281μmol)、2−(メチルスルホニル)ピリジン−4−アミン(55mg、319μmol)、キサントホス(48.7mg、84.2μmol)、Pd2(dba)3(25.7mg、28.1μmol)及び炭酸セシウム(183mg、562μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中35−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(2−(メチルスルホニル)ピリジン−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(127mg、245μmol、87%)を淡黄色のゴムとして得た。(EI/CI) m/z: 519.1 [M+H]。
N−tert−ブチル−2−(2−(メチルスルホニル)ピリジン−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(127mg、245μmol)が入ったジクロロメタンの溶液に、トリフルオロ酢酸(558mg、377μL、4.9mmol)を添加し、混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(4mL)、メタノール(2mL)及び水酸化アンモニウム(0.7mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、N−tert−ブチル−2−(2−(メチルスルホニル)ピリジン−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(94mg、242μmol、99%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.51 (s, 1 H), 10.36 (s, 1 H), 8.39 (d, J=5.9, 1 H), 8.10 (s, 1 H), 8.01 (s, 1H), 7.99 (dd, J=5.3, 2.3, 1 H), 7.83 (d, J=2.1, 1 H), 7.52 (s, 1 H), 3.15 (s, 3 H), 1.35 (s, 9 H); MS (EI/CI) m/z: 389.1 [M+H]。
N−tert−ブチル−2−(5−(メチルスルホニル)ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−アミノ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(92mg、253μmol)、3−ブロモ−5−(メチルスルホニル)ピリジン(77.7mg、329μmol)、キサントホス(43.9mg、75.9μmol)、Pd2(dba)3(23.2mg、25.3μmol)及び炭酸セシウム(165mg、506μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(5−(メチルスルホニル)ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(125mg、241μmol、95%)を黄色のゴムとして得た。(EI/CI) m/z: 519.3 [M+H]。
N−tert−ブチル−2−(5−(メチルスルホニル)ピリジン−3−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(125mg、241μmol)が入ったジクロロメタンの溶液に、トリフルオロ酢酸(550mg、371μL、4.82mmol)を添加し、混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(3.7mL)、メタノール(1.6mL)及び水酸化アンモニウム(0.5mL)に再溶解し混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、N−tert−ブチル−2−(5−(メチルスルホニル)ピリジン−3−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(92mg、237μmol、98%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.38 (s, 1 H), 9.90 (s, 1 H), 9.40 (d, J=2.5, 1 H), 8.55 (d, J=1.9, 1 H), 8.06 (t, J=2.2, 1H), 8.01 (s, 1 H), 7.96 (s, 1 H), 7.54 (s, 1 H), 3.22 (s, 3 H), 1.31 (s, 9 H); MS (EI/CI) m/z: 389.1 [M+H]。
N−tert−ブチル−2−(1,3−ジメチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、1,3−ジメチル−1H−ピラゾール−4−アミン(58.5mg、526μmol)、キサントホス(60.9mg、105μmol)、Pd2(dba)3(32.1mg、35.1μmol)及び炭酸セシウム(229mg、702μmol)の混合物をマイクロ波で150℃で20分間加熱した。混合物を冷却し、次いでセライトのパッドを通して濾過した。濾液を真空で濃縮し、次いでクロマトグラフィー(シリカ、ヘキサン中25−100%の酢酸エチル)で精製し、N−tert−ブチル−2−(1,3−ジメチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(89mg、194μmol、55%)を黄色のゴムとして得た。(EI/CI) m/z: 458.3 [M+H]。
N−tert−ブチル−2−(1,3−ジメチル−1H−ピラゾール−4−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(89mg、194μmol)が入ったジクロロメタン(3mL)の溶液に、トリフルオロ酢酸(443mg、300μL、3.89mmol)を添加し、混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(3mL)、メタノール(1.5mL)及び水酸化アンモニウム(0.45mL)に再溶解し、混合物を室温で2時間撹拌した。反応混合物を濃縮し、次いでクロマトグラフィー(シリカ、ジクロロメタン中5−8%のメタノール)で精製し、N−tert−ブチル−2−(1,3−ジメチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(40mg、122μmol、63%)を淡褐色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.13 (s, 1 H), 8.44 (s, 1 H), 7.91 (s, 1 H), 7.87 (d, J=3.2, 1 H), 7.82 (s, 1 H), 7.81 (s, 1 H), 3.71 (s, 3 H), 2.09 (s, 3 H), 1.35 (s, 9 H); MS (EI/CI) m/z: 328.1 [M+H]。
N−tert−ブチル−2−(3−メチル−1,2,4−チアジアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(150mg、351μmol)、3−メチル−1,2,4−チアジアゾール−5−アミン(60.6mg、526μmol)、キサントホス(60.9mg、105μmol)、Pd2(dba)3(32.1mg、35.1μmol)及び炭酸セシウム(229mg、702μmol)の混合物を、マイクロ波で150℃で20分間加熱した。クロマトグラフィー(シリカ、ヘキサン中30−70%の酢酸エチル)で精製し、N−tert−ブチル−2−(3−メチル−1,2,4−チアジアゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(128mg、277μmol、79%)を橙色の固体として得た。(EI/CI) m/z: 462.3 [M+H]。
N−tert−ブチル−2−(3−メチル−1,2,4−チアジアゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(128mg、277μmol)が入ったジクロロメタン(4.3mL)の溶液に、トリフルオロ酢酸(632mg、427μL、5.55mmol)を添加し、混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(4mL)、メタノール(2mL)及び水酸化アンモニウム(0.6mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、N−tert−ブチル−2−(3−メチル−1,2,4−チアジアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(90mg、272μmol、98%)を淡黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.80 (s, 1 H), 12.57 (s, 1 H), 8.35 (s, 1 H), 8.34 (s, 1 H), 7.34 (s, 1 H), 2.53 (s, 3 H), 1.62 (s, 9 H); MS (EI/CI) m/z: 332.1 [M+H]。
N−tert−ブチル−2−(2−メチル−6,7−ジヒドロ−2H−ピラゾロ[4,3−b]ピリジン−4(5H)−イル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
工程1
4,5,6,7−テトラヒドロ−1H−ピラゾロ[4,3−b]ピリジン
tert−ブチル 6,7−ジヒドロ−1H−ピラゾロ[4,3−b]ピリジン−4(5H)−カルボキシレート
tert−ブチル 2−メチル−6,7−ジヒドロ−2H−ピラゾロ[4,3−b]ピリジン−4(5H)−カルボキシレート及びtert−ブチル 1−メチル−6,7−ジヒドロ−1H−ピラゾロ[4,3−b]ピリジン−4(5H)−カルボキシレート
2−メチル−4,5,6,7−テトラヒドロ−2H−ピラゾロ[4,3−b]ピリジン
N−tert−ブチル−2−(2−メチル−6,7−ジヒドロ−2H−ピラゾロ[4,3−b]ピリジン−4(5H)−イル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
N−tert−ブチル−2−(2−メチル−6,7−ジヒドロ−2H−ピラゾロ[4,3−b]ピリジン−4(5H)−イル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
N−tert−ブチル−2−(1−メチル−6,7−ジヒドロ−1H−ピラゾロ[4,3−b]ピリジン−4(5H)−イル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
工程1
1−メチル−4,5,6,7−テトラヒドロ−1H−ピラゾロ[4,3−b]ピリジン
N−tert−ブチル−2−(1−メチル−6,7−ジヒドロ−1H−ピラゾロ[4,3−b]ピリジン−4(5H)−イル)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
N−tert−ブチル−2−(1−メチル−6,7−ジヒドロ−1H−ピラゾロ[4,3−b]ピリジン−4(5H)−イル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
3−メチル−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸 tert−ブチルアミド
工程1
3,5−ジブロモ−6−メチル−ピラジン−2−イルアミン
5−ブロモ−6−メチル−3−トリメチルシラニルエチニル−ピラジン−2−イルアミン
2−ブロモ−3−メチル−5H−ピロロ[2,3−b]ピラジン
MS (EI/CI) m/z: 212 [M+H]。
2−ブロモ−3−メチル−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b] ピラジン
1H NMR (400 MHz, DMSO-d6)δ ppm -0.11 (s, 9 H), 0.82 (m, 2 H), 2.67 (s, 3 H), 3.51 (m, 2 H), 5.59 (s, 2 H), 6.67 (d, J=3.5 Hz, 1 H), 8.01 (d, J=3.5 Hz, 1 H)。
2−ブロモ−3−メチル−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b] ピラジン−7−カルバルデヒド
2−ブロモ−3−メチル−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b] ピラジン−7−カルボン酸
2−ブロモ−3−メチル−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b] ピラジン−7−カルボン酸 tert−ブチルアミド
MS (EI/CI) m/z: 441 [M+H]
3−メチル−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸 tert−ブチルアミド
3−メチル−2−(1−メチル−1H−ピラゾール−4−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド
2−(3−メタンスルホニル−フェニルアミノ)−3−メチル−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド
工程1
2−(3−メタンスルホニル−フェニルアミノ)−3−メチル−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸 tert−ブチルアミド
2−(3−メタンスルホニル−フェニルアミノ)−3−メチル−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸 tert−ブチルアミド
3−メチル−2−(3−メチル−イソチアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド
工程13−メチル−2−(3−メチル−イソチアゾール−5−イルアミノ)−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド
3−メチル−2−(3−メチル−イソチアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド
2−フェニルアミノ(D5)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド
ジオキサン(2mL)に入った2−ブロモ−N−tert−ブチル−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド(125mg、292μmol)、アニリン−D5(43.1mg、439μmol)、キサントホス(50.8mg、87.7μmol)、Pd2(dba)3(26.8mg、29.2μmol)及び炭酸セシウム(191mg、585μmol)の混合物を、マイクロ波で150℃で20分間加熱した。反応混合物をセライトのパッドを通して濾過し、次いで濾液を真空で濃縮して、クロマトグラフィー(シリカ、ヘキサン中20−60%の酢酸エチル)で精製し、2−フェニルアミノ(D5)−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド(40mg、90.0μmol、31%)を淡黄色の固体として得た。1H NMR (400 MHz, CDCl3-d)δppm: 8.15 (s, 1 H), 7.99 (s, 1 H), 7.93 (s, 1 H), 6.70 (s, 1 H), 5.63 (s, 2 H), 3.57 (t, J= 8.2, 2 H), 1.57 (s, 9 H), 0.96 (t, J=8.1, 2 H), 0.01 (s, 9 H)。
2−フェニルアミノ(D5)−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド(40mg、90.0μmol)が入ったジクロロメタン(1.5mL)の溶液に、トリフルオロ酢酸(205mg、139μL、1.8mmol)を添加し、混合物を室温で16時間撹拌した。混合物を濃縮し、次いでジクロロメタン(1.5mL)、メタノール(0.75mL)及び水酸化アンモニウム(0.2mL)に再溶解し、混合物を室温で1時間撹拌した。混合物を真空で濃縮し、次いで水で粉砕して濾過した。得られた固体をエーテルで洗浄し、次いで乾燥させ、2−フェニルアミノ(D5)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸tert−ブチルアミド(24mg、76.3μmol、85%)を黄色の固体として得た。1H NMR (400 MHz, DMSO-d)δppm: 12.31 (s, 1 H), 9.47 (s, 1 H), 8.00 (s, 1 H), 7.99 (s, 1 H), 7.76 (s, 1 H), 1.44 (s, 9 H); MS (EI/CI) m/z: 315.1 [M+H]。
N−tert−ブチル−3−クロロ−2−(3−メチルイソチアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
2−ブロモ−N−tert−ブチル−3−クロロ−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
3,5−ジブロモ−6−クロロ−ピラジン−2−イルアミン
5−ブロモ−6−クロロ−3−トリメチルシラニルエチニル−ピラジン−2−イルアミン
2−ブロモ−3−クロロ−5H−ピロロ[2,3−b]ピラジン
2−ブロモ−3−クロロ−ピロロ[2,3−b]ピラジン−5−カルボン酸tert−ブチル エステル
(2−ブロモ−3−クロロ−7−ヒドロキシメチル−ピロロ[2,3−b]ピラジン−5−イル)−メタノール
2−ブロモ−3−クロロ−5H−ピロロ[2,3−b]ピラジン−7−カルバルデヒド
注:Jone’s試薬の調製:CrO3(13.66g)が入った水溶液(20mL)を0℃に冷却し、濃H2SO4(11.5mL)をゆっくりと添加した。得られる溶液の体積が50mLとなるように水を添加して、Jone’s試薬を得た。
2−ブロモ−3−クロロ−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルバルデヒド
2−ブロモ−3−クロロ−5−(2−トリメチルシラニル−エトキシメチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボン酸
2−ブロモ−N−tert−ブチル−3−クロロ−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
N−tert−ブチル−3−クロロ−2−(3−メチルイソチアゾール−5−イルアミノ)−5−((2−(トリメチルシリル)エトキシ)メチル)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
N−tert−ブチル−3−クロロ−2−(3−メチルイソチアゾール−5−イルアミノ)−5H−ピロロ[2,3−b]ピラジン−7−カルボキサミド
SYKアッセイ情報
脾臓チロシンキナーゼ(SYK)阻害のIC50の決定:
SYKキナーゼアッセイは、96ウェルプレートフォーマットに適合した標準的なキナーゼアッセイである。このアッセイは、96−ウェルフォーマット中、10の半対数希釈及び40μLの反応容量を示した8試料によるIC50決定のために実施する。このアッセイは、天然に存在するホスホアクセプターコンセンサス配列に由来する、N−末端ビオチン化ペプチド基質(ビオチン−11aaDY*E)への放射標識化33PγATPの取込みを測定する。リン酸化生成物をEDTAとの反応の停止及びストレプトアビジンコートビーズの添加時に検出した。代表的な結果は、上の表IIにある。
ストレプトアビジンコートビーズ:ストレプトアビジンSepharose TM、50mM EDTA/PBS希釈(1:100)中懸濁液5.0mL(Amersham、カタログ番号17−5113−01)
化合物:100%ジメチルスルホキシド(DMSO)中10mM、最終濃度:10%DMSO中0.003〜100μMの化合物。
酵素:SYK RPA精製-、脾臓チロシンキナーゼの不完全コンストラクトaa360−635、ストック溶液1mg/mL、MW:31.2KDa、最終濃度:0.0005μM
ペプチド1:ビオチン化ペプチドは、天然に存在するホスホ−アクセプター-コンセンサス配列に由来する(ビオチン−EPEGDYEEVLE)、QCBからの特注品、ストック溶液20mM、最終濃度:5.0μM
ATP:アデノシン−5’−三リン酸20mM(ROCHEカタログ番号:93202720)、最終濃度:20μM
バッファー:HEPES:2−ヒドロキシエチル ピペラジン−2−エタンスルホン酸(Sigma、カタログ番号:H−3375)最終濃度:50mM HEPES pH7.5
BSA:ウシ血清アルブミン第V画分、脂肪酸を含まない(Roche Diagnostics GmbH、カタログ番号:9100221)、0.1%の最終濃度に希釈。
EDTA:EDTAストック溶液500mM、(GIBCO、カタログ番号:15575−038)最終濃度:0.1mM
DTT:1,4−ジチオスレイトール(Roche Diagnostics GmbH、カタログ番号:197777)、最終濃度:1mM
MgCl2 x 6H2O:MERCK、カタログ番号:105833.1000、最終濃度:10mM
アッセイ希釈バッファー(ADB):50mM HEPES、0.1mM EGTA、0.1mM バナジン酸Na、0.1mM β−グリセロリン酸、10mM MgCl2、1mM DTT、0.1% BSA、pH7.5
ビーズ洗浄バッファー:2M NaCl+1%リン酸を含む、10g/L PBS(リン酸緩衝生理食塩水)。
40μL容量において、26μLのADBで希釈した精製済組換えヒトSYK360−635[0.5nM]を、4μLの10X濃度の試験化合物([10%]DMSO中[通常100μM−0.003μM])と混合し、この混合物を室温で10分間インキュベートした。
阻害%=100/(1+(IC50/阻害剤濃度)n)
IC50は、XLfitソフトウェア(ID Business Solu-tion Ltd., Guilford, Surrey, UK)で非線形曲線フィットを用いて計算した。
RPMI−1640中で、ヒトB細胞リンパ腫細胞株ラモスを10%ウシ胎児血清(Invitrogen)と培養した。メーカーの指示に従い(Beckton Dickinson Calcium Kit、カタログ番号80500)、細胞をアッセイプレートへ播種し、30μLの染料ロードバッファーを添加することにより、カルシウム染料でロードした。次いで、刺激前に、細胞を連続希釈化合物で30分間処理した。基線蛍光は、約20秒間で記録され、その後ラモス細胞に対しマウス抗ヒトIgM(10μg/mL、クローンM2E6、Antibody Solutions Inc.)で刺激し、各ウェルの最大蛍光計数を記録した。各ウェルに対する基線の倍率変化として定義される、基線計数により分けられた最大計数をIC50計算に使用した。
ヒト血液を健康なボランティアからヘパリンナトリウムを含むバキュテナー(BD Biosciences,San Joe, CA)中へ収集した。試験化合物をDMSO中に懸濁させ、9の半対数連続希釈液を作製した。アッセイ中化合物の濃度は、0.5%であった。100μLの全血を化合物と一緒に30分間予備インキュベートし、次いでヤギF(ab’)2抗ヒトIgM(50μg/mL、Southern Biotech)で20時間刺激した。20時間のインキュベーションの最後で、試料を蛍光色素コンジュゲート抗体、PEマウス抗ヒトCD20及びAPCマウス抗ヒトCD69(BD Biosciences)と一緒に30分間インキュベートした。次いで、試料をLyse溶液(BD)で溶解し、2%ウシ胎児血清(FBS)を含むPBSで洗浄した。蛍光シグナルをフローサイトメーターLSRII(BD)で取得し、データをFlow Joで解析した。活性化(CD69hi)B細胞リンパ球(CD20+)のパーセントは、基準ガイドラインとして非刺激(負対照)及び刺激(正対照)ウェルを使用して決定した。阻害パーセントを計算し、IC50曲線を、シグモイド曲線フィッティングを用いたグラフパッドプリズム(GraphPad Prism)ソフトウェアを使用して構築した。
Claims (13)
- 式I
[上式中:
R1は、Hであり;
R2は、t−ブチルであり;
R3は、H又はC1−6アルキルであり;
Aは、一又は複数のA’で置換されていてもよい、フェニルであり;且つ各A’は、独立して、C1−6アルキル、ハロ、C1−6アルキルスルホニル、アミド、C1−6ヒドロキシアルキル、C1−6アルコキシ、アミノC1−6アルキル、又は重水素である]の化合物;
又はその薬学的に許容される塩。 - R3がC1−6アルキルである、請求項1に記載の化合物。
- R3がHである、請求項1に記載の化合物。
- 各A’が、独立して、C1−6アルキル、ハロ、C1−6アルキルスルホニル、C1−6ヒドロキシアルキル、C1−6アルコキシ、アミノC1−6アルキル又は重水素である、請求項1から3の何れか一項に記載の化合物。
- A’がC1−6アルキルである、請求項1から4の何れか一項に記載の化合物。
- A’がC1−6アルキルスルホニルである、請求項1から5の何れか一項に記載の化合物。
- A’がC1−6アルキル又はC1−6アルキルスルホニルである、請求項1から6の何れか一項に記載の化合物。
- N-tert-ブチル-2-(2-フルオロ-4-メチルフェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(p-トリルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-(メチルスルホニル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2-(ジメチルアミノカルボニル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(2,4-ジメチルフェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(4-(メチルスルホニル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
N-tert-ブチル-2-(3-(1-ヒドロキシエチル)フェニルアミノ)-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
2-(3-(1-アミノエチル)フェニルアミノ)-N-tert-ブチル-5H-ピロロ[2,3-b]ピラジン-7-カルボキサミド;
2-(3-メタンスルホニル-フェニルアミノ)-3-メチル-5H-ピロロ[2,3-b]ピラジン-7-カルボン酸 tert-ブチルアミド;及び
2-フェニルアミノ(D5)-5H-ピロロ[2,3-b]ピラジン-7-カルボン酸 tert-ブチルアミド
からなる群より選択される、請求項1に記載の化合物。 - 少なくとも一種の薬学的に許容される担体、賦形剤又は希釈剤と混合された、請求項1から8の何れか一項に記載の化合物を含む薬学的組成物。
- 化学療法若しくは抗増殖剤、抗炎症剤、免疫調節剤若しくは免疫抑制剤、神経栄養因子、心臓血管病を治療するための薬剤、糖尿病を治療するための薬剤及び免疫不全疾患を治療するための薬剤から選択される追加の治療剤を更に含む、請求項9に記載の薬学的組成物。
- Sykに関連する疾患の治療に有用な医薬の製造のための、請求項1から8の何れか一項に記載の化合物の使用。
- Sykに関連する疾患の治療において使用するための、請求項1から8の何れか一項に記載の化合物。
- 狼瘡、多発性硬化症、関節リウマチ、乾癬、I型糖尿病、臓器移植による合併症、異種移植、糖尿病、がん、喘息、アトピー性皮膚炎、自己免疫甲状腺疾患、潰瘍性大腸炎、クローン病、アルツハイマー病及び白血病を含む免疫疾患の治療において使用するための、請求項1から8の何れか一項に記載の化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261691384P | 2012-08-21 | 2012-08-21 | |
| US61/691,384 | 2012-08-21 | ||
| PCT/EP2013/067233 WO2014029732A1 (en) | 2012-08-21 | 2013-08-19 | Pyrrolo[2,3-b]pyrazines as syk inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2015531766A JP2015531766A (ja) | 2015-11-05 |
| JP6404215B2 true JP6404215B2 (ja) | 2018-10-10 |
Family
ID=49000939
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015527883A Expired - Fee Related JP6404215B2 (ja) | 2012-08-21 | 2013-08-19 | Syk阻害剤としてのピロロ[2,3−b]ピラジン |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US9334278B2 (ja) |
| EP (1) | EP2888262B1 (ja) |
| JP (1) | JP6404215B2 (ja) |
| KR (1) | KR20150043323A (ja) |
| CN (1) | CN104507945B (ja) |
| AR (1) | AR092173A1 (ja) |
| BR (1) | BR112015002262A2 (ja) |
| CA (1) | CA2876543A1 (ja) |
| HK (1) | HK1206354A1 (ja) |
| MX (1) | MX2015002181A (ja) |
| RU (1) | RU2656853C2 (ja) |
| TW (1) | TWI639601B (ja) |
| WO (1) | WO2014029732A1 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10111882B2 (en) | 2016-09-14 | 2018-10-30 | Gilead Sciences, Inc. | SYK inhibitors |
| TW201822764A (zh) | 2016-09-14 | 2018-07-01 | 美商基利科學股份有限公司 | Syk抑制劑 |
| WO2018195471A1 (en) | 2017-04-21 | 2018-10-25 | Gilead Sciences, Inc. | Syk inhibitors in combination with hypomethylating agents |
| CN114364798A (zh) | 2019-03-21 | 2022-04-15 | 欧恩科斯欧公司 | 用于治疗癌症的Dbait分子与激酶抑制剂的组合 |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2024156938A1 (en) | 2023-01-28 | 2024-08-02 | Orion Corporation | Cbl-b inhibitors |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CL2007002617A1 (es) * | 2006-09-11 | 2008-05-16 | Sanofi Aventis | Compuestos derivados de pirrolo[2,3-b]pirazin-6-ilo; composicion farmaceutica que comprende a dichos compuestos; y su uso para tratar inflamacion de las articulaciones, artritis reumatoide, tumores, linfoma de las celulas del manto. |
| WO2009106444A1 (en) * | 2008-02-25 | 2009-09-03 | F. Hoffmann-La Roche Ag | Pyrrolopyrazine kinase inhibitors |
| EP2250172B1 (en) * | 2008-02-25 | 2011-08-31 | F. Hoffmann-La Roche AG | Pyrrolopyrazine kinase inhibitors |
| MX2010008198A (es) * | 2008-02-25 | 2010-08-23 | Hoffmann La Roche | Inhibidores de cinasa de pirrolopirazina. |
| RU2012152352A (ru) * | 2010-05-20 | 2014-06-27 | Ф. Хоффманн-Ля Рош Аг | ПРОИЗВОДНЫЕ ПИРРОЛО[2,3-b]ПИРАЗИН-7-КАРБОКСАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK И SYK |
-
2013
- 2013-08-19 BR BR112015002262A patent/BR112015002262A2/pt not_active Application Discontinuation
- 2013-08-19 CN CN201380035821.2A patent/CN104507945B/zh not_active Expired - Fee Related
- 2013-08-19 US US14/421,084 patent/US9334278B2/en not_active Expired - Fee Related
- 2013-08-19 WO PCT/EP2013/067233 patent/WO2014029732A1/en active Application Filing
- 2013-08-19 KR KR20157003641A patent/KR20150043323A/ko not_active Application Discontinuation
- 2013-08-19 CA CA2876543A patent/CA2876543A1/en not_active Abandoned
- 2013-08-19 MX MX2015002181A patent/MX2015002181A/es unknown
- 2013-08-19 RU RU2015107426A patent/RU2656853C2/ru not_active IP Right Cessation
- 2013-08-19 EP EP13750700.0A patent/EP2888262B1/en not_active Not-in-force
- 2013-08-19 JP JP2015527883A patent/JP6404215B2/ja not_active Expired - Fee Related
- 2013-08-20 TW TW102129887A patent/TWI639601B/zh not_active IP Right Cessation
- 2013-08-20 AR ARP130102940A patent/AR092173A1/es unknown
-
2015
- 2015-07-23 HK HK15107036.4A patent/HK1206354A1/xx not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| CN104507945B (zh) | 2018-03-23 |
| MX2015002181A (es) | 2015-05-07 |
| KR20150043323A (ko) | 2015-04-22 |
| AR092173A1 (es) | 2015-03-25 |
| US20150218170A1 (en) | 2015-08-06 |
| WO2014029732A1 (en) | 2014-02-27 |
| RU2656853C2 (ru) | 2018-06-07 |
| CN104507945A (zh) | 2015-04-08 |
| HK1206354A1 (en) | 2016-01-08 |
| EP2888262A1 (en) | 2015-07-01 |
| US9334278B2 (en) | 2016-05-10 |
| BR112015002262A2 (pt) | 2019-12-10 |
| TW201412743A (zh) | 2014-04-01 |
| EP2888262B1 (en) | 2018-12-19 |
| TWI639601B (zh) | 2018-11-01 |
| CA2876543A1 (en) | 2014-02-27 |
| RU2015107426A (ru) | 2016-10-10 |
| JP2015531766A (ja) | 2015-11-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2627661C2 (ru) | Соединения пиридазинамида и их применение в качестве ингибиторов тирозинкиназы селезенки (syk) | |
| JP6404215B2 (ja) | Syk阻害剤としてのピロロ[2,3−b]ピラジン | |
| JP5823616B2 (ja) | ブルトンチロシンキナーゼインヒビター | |
| JP5859640B2 (ja) | ブルトンチロシンキナーゼ阻害剤 | |
| EP2229390A1 (en) | Novel imidazoý1,2-a¨pyridine and imidazoý1,2-b¨pyridazine derivatives | |
| US8742098B2 (en) | Inhibitors of Bruton's tyrosine kinase | |
| JP6109193B2 (ja) | チエノピリミジン化合物 | |
| US9469644B2 (en) | Inhibitors of Bruton's tyrosine kinase | |
| CA2881070A1 (en) | Inhibitors of bruton's tyrosine kinase | |
| JP2015531781A (ja) | ブルトン型チロシンキナーゼの阻害剤 | |
| JP6089124B2 (ja) | ブルトン型チロシンキナーゼの阻害薬 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20160816 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170608 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170620 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170919 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171220 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180123 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180420 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180720 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180814 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20180912 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6404215 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |