JP6190875B2 - 脱ハロゲン化水素によるc3−c7(ヒドロ)フルオロアルケンを製造する方法 - Google Patents
脱ハロゲン化水素によるc3−c7(ヒドロ)フルオロアルケンを製造する方法 Download PDFInfo
- Publication number
- JP6190875B2 JP6190875B2 JP2015509495A JP2015509495A JP6190875B2 JP 6190875 B2 JP6190875 B2 JP 6190875B2 JP 2015509495 A JP2015509495 A JP 2015509495A JP 2015509495 A JP2015509495 A JP 2015509495A JP 6190875 B2 JP6190875 B2 JP 6190875B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- hydro
- alumina
- metal
- nitrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/25—Preparation of halogenated hydrocarbons by splitting-off hydrogen halides from halogenated hydrocarbons
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/02—Boron or aluminium; Oxides or hydroxides thereof
- B01J21/04—Alumina
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- B01J21/066—Zirconium or hafnium; Oxides or hydroxides thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the alkali- or alkaline earth metals or beryllium
- B01J23/04—Alkali metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/26—Chromium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/615—100-500 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0201—Impregnation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
この例は、高ナトリウムアルミナ担持ジルコニウム触媒を用いたHFO−1234zeへのHFC−245faの脱フッ化水素を記載する。
オキシ塩化ジルコニウム(IV)八水和物(10.4467g)の無水エタノール(463g)溶液で50ml(39.2925g)のアルミナ球体(直径が5mm及び表面積が310m2/gでナトリウム含有量が2340ppmのものをAldrichから入手)を一晩含浸させた。次いでエタノールを真空下で留去し、最後に触媒を再び真空下150℃で乾燥させた。製造された状態では、触媒のZr充填量は名目上7.5%wt/wt(触媒の実際のZr充填量を測定すると1.81%wt/wtであることが分かった)であり、含浸した触媒の表面積は251.33m2/gであった。
Zr/アルミナ触媒を、未希釈の245fa供給物を用いて大気圧で試験した。大気試験装置は反応管を2本備えており、それぞれ独立したHF、有機物、及び窒素供給物を有していた。この研究のため、各反応管に上に記載の触媒を充填した(反応器A5ml及び反応器B2ml)。両方の反応器の触媒をまず窒素下(60ml/分)200℃で72時間乾燥させた。次いで、これらを、窒素中で希釈したHF(30ml/分のHF及び20ml/分のN2)により200℃で1時間、前フッ素化した。この時点で窒素流を停止し、触媒を未希釈のHFで処理し、温度を200℃から450℃に40℃/時で上昇させた。これらの条件をさらに1時間維持した後、触媒を200℃に冷却した。次いでHF流を停止し、窒素流(60ml/分)と置き換え、反応器のオフガスにさらなるHFが検出されなくなった時点で触媒は使用できる状態となった。
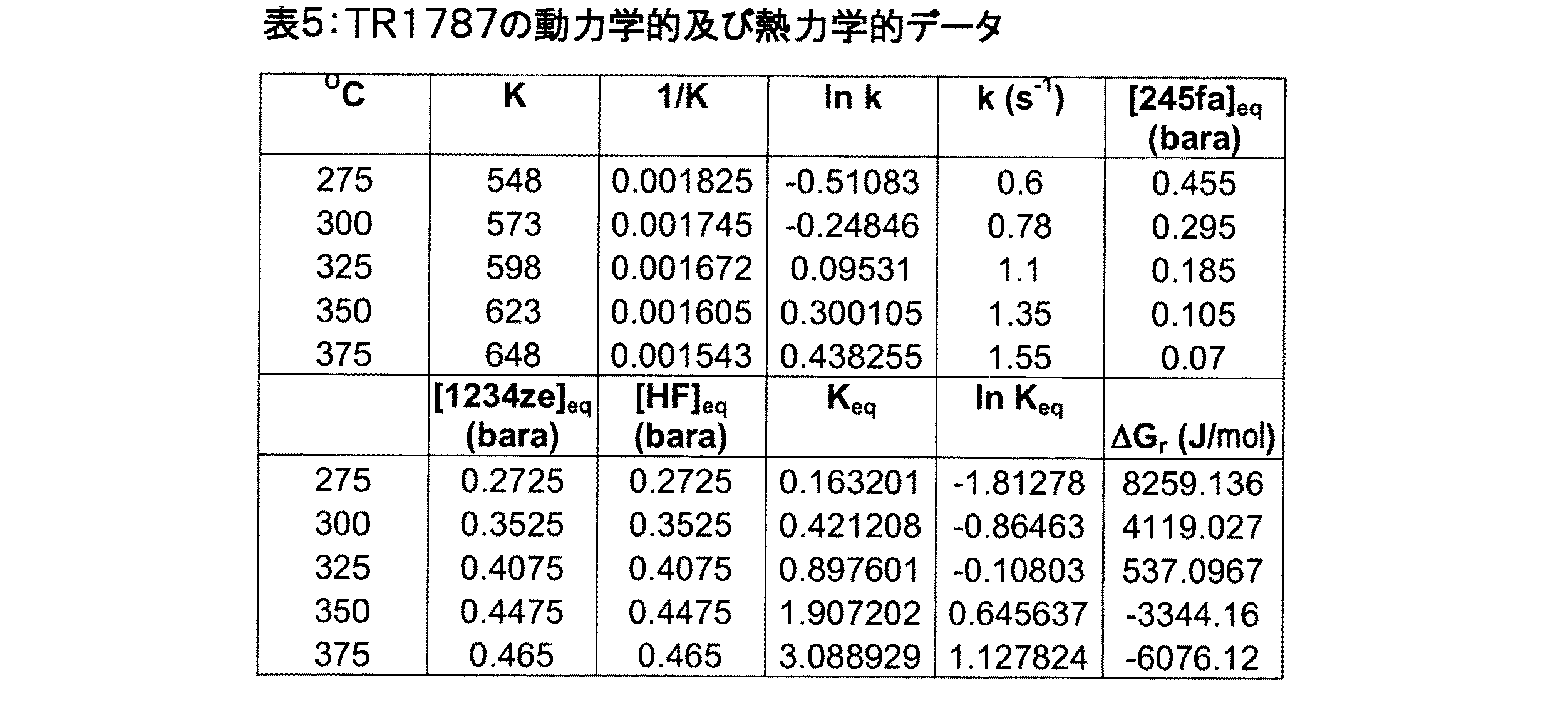
この実験の目的は、温度及び接触時間の変化に対する反応の応答を測定し、根底にある反応の動力学及び熱力学を調査すべくデータを使用することであった。反応器を分化条件に近似させるため、比較的少量の触媒を投入し、接触時間を短くした。表2にデータをまとめる。
・活性化エネルギー(Eact)=55.7kJ/mol
・Ln頻度因子(Ln A)=10.756
・反応熱(dHr)=+74.76kJ/mol
・反応のエントロピー(dSr)=+123.17J/mol.K
この例に用いた触媒は、245faの脱フッ化水素用の、ナトリウムを111ppm含有し表面積が140.6m2/gであるアルミナ担持クロミア触媒(TR1787と表示する)であった。
ナトリウム含有量が2170〜2410ppmであるアルミナ(ASM Catalysts LLC)にCr(H2O)6Cl3を含浸させてアルミナ担持クロミア触媒を製造した。よって、塩化クロム(III)6水和物24gにDI水を17ml加えた。この混合物を水浴中で80℃まで加熱し、次いで冷却した後にASMアルミナを50.2g加えた。アルミナを一晩含浸させたが、含浸は均一ではなく、水がすべて吸収されたので、DI水をさらに13ml加え、触媒を再び一晩放置した。触媒を真空下90℃で乾燥させた。製造された状態では触媒のクロムIII充填量は名目上9.36%wt/wtであった。TR1816と表示される得られた触媒を空気中でか焼するか又は窒素中でか焼した。
名目上の触媒のZr充填量が7.5%wt/wtであるさらなるアルミナ担持ジルコニア触媒(TR1817と表示する)を製造した(触媒の実際のZr充填量を測定し、1.68%wt/wtであることが分かった)。使用したアルミナは、ASM Catalysts LLCから入手し、ナトリウム含有量が2170〜2410ppmであった。例2及び3に関して上に記載したのと同じ触媒試験条件を用いて、この触媒は、1234zeへの245faの脱フッ化水素に関する活性化エネルギーが51.1kJ/molであることが測定された。これは、例1に記載の異なる供給源から得られたアルミナ担持ジルコニア触媒とよく一致している。
1)30℃で試料を導入し、200ml/分の窒素で炉を5分間パージ
2)パージ流を100ml/分まで1分間減少
3)100ml/分の窒素下20℃/分で550℃まで試料を加熱
4)パージ流を50ml/分まで減少させ、試料を550℃で15分間保持
5)25ml/分の窒素下550から200℃まで30℃/分で試料を冷却
6)200℃及び25ml/分の窒素で5分間試料を安定化
7)気体流を10ml/分の空気に変更し、系を5分間安定化
8)試料温度を10ml/分の空気下550℃まで15℃/分で上昇
9)10ml/分の空気下550℃で18分間滞留
本発明による低ナトリウム含有量のアルミナ担持ジルコニア触媒を2種類製造し、1234zeへの245faの脱フッ化水素を触媒するこれらの活性について、比較例1、例4、及びさらなる比較例7の高ナトリウム含有量のアルミナ担持ジルコニア触媒と比較した。
例2及び比較例3を参照して、アルミナ担持クロミア触媒をさらに2種類製造し、1234zeへの245faの脱フッ化水素を触媒するこれらの活性について比較した。
例8及び9で使用したのと同じアルミナ担持クロミア触媒を、1234yfへの245cbの脱フッ化水素を触媒するこれらの活性について比較した。
例8〜11で使用したのと同じアルミナ担持クロミア触媒を、1234yfへの245ebの脱フッ化水素を触媒するこれらの活性について比較した。触媒はいずれも60ml/分の窒素と共に200℃で1時間、続いて60ml/分の窒素と共に360℃で1時間乾燥させて、触媒をか焼した。60ml/分の窒素で希釈した30ml/分のHFを300℃で1時間触媒上に流した。窒素のスイッチをオフにし、30ml/分のHFを360℃で1時間流した。
例8〜13で使用したのと同じアルミナ担持クロミア触媒を、1243zfへの254fbの脱フッ化水素を触媒するこれらの活性について比較した。
Claims (18)
- アルミナに担持された金属酸化物を含んでなる触媒の存在下でC3−7ヒドロ(ハロ)フルオロアルカンを脱ハロゲン化水素することを含んでなる、C3−7(ヒドロ)フルオロアルケンを製造する方法であって、前記触媒のナトリウム含有量が800ppm未満であり、前記金属酸化物の金属がCr、Zr、Nb、Ta、V、Mo又はCoから選択される遷移金属である、方法。
- 前記触媒のナトリウム含有量が500ppm未満である、請求項1に記載の方法。
- 前記触媒のナトリウム含有量が150ppm未満である、請求項2に記載の方法。
- 前記触媒が、前記触媒の全重量に対して前記アルミナ担体を少なくとも60重量%含んでなる、請求項1〜3のいずれか一項に記載の方法。
- 前記触媒が、前記触媒の全重量に対して前記アルミナ担体を少なくとも70重量%含んでなる、請求項4に記載の方法。
- 前記金属がZr又はCrである、請求項1〜5のいずれか一項に記載の方法。
- 前記触媒が、前記触媒の全重量に対して前記金属酸化物を40重量%以下含んでなる、請求項1〜6のいずれか一項に記載の方法。
- 前記触媒が、前記触媒の全重量に対して前記金属酸化物を30重量%以下含んでなる、請求項7に記載の方法。
- 前記触媒が、少なくとも1種の追加金属又は前記追加金属の化合物をさらに含んでなり、前記追加金属はZn、Zr、Cr、In、Co及びその混合物から選択される、請求項1〜8のいずれか一項に記載の方法。
- 前記触媒が、前記触媒の全重量に対して前記少なくとも1種の追加金属又は前記追加金属の化合物を20重量%以下含んでなる、請求項9に記載の方法。
- 前記触媒が、前記触媒の全重量に対して前記少なくとも1種の追加金属又は前記追加金属の化合物を10重量%以下含んでなる、請求項10に記載の方法。
- 添加したフッ化水素(HF)の非存在下で前記C3−7ヒドロ(ハロ)フルオロアルカンを脱ハロゲン化水素することを含んでなる、請求項1〜11のいずれか一項に記載の方法。
- 50〜400℃の温度及び30bara以下の圧力で実施される、請求項1〜12のいずれか一項に記載の方法。
- ヒドロ(ハロ)フルオロプロパンを脱ハロゲン化水素することを含んでなる、(ヒドロ)フルオロプロペンを製造するための請求項1〜13のいずれか一項に記載の方法。
- 製造された(ヒドロ)フルオロプロペンが、トリフルオロプロペン、テトラフルオロプロペン、及びペンタフルオロプロペンから選択される、請求項14に記載の方法。
- 1,1,1,3,3−ペンタフルオロプロパン(HFC−245fa)を脱フッ化水素することを含んでなる、1,3,3,3−テトラフルオロプロペン(HFO−1234ze)を製造するための請求項14又は15に記載の方法。
- 1,1,1,2,2−ペンタフルオロプロパン(HFC−245cb)及び/又は1,1,1,2,3−ペンタフルオロプロパン(HFC−245eb)を脱フッ化水素することを含んでなる、2,3,3,3−テトラフルオロプロペン(HFO−1234yf)を製造するための請求項14又は15に記載の方法。
- 1,1,1,2,3,3−ヘキサフルオロプロパン(HFC−236ea)を脱フッ化水素することを含んでなる、1,2,3,3,3−ペンタフルオロプロペン(HFO−1225ye)を製造するための請求項14又は15に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1207666.7 | 2012-05-02 | ||
| GB201207666A GB201207666D0 (en) | 2012-05-02 | 2012-05-02 | Process |
| PCT/GB2013/051129 WO2013164618A1 (en) | 2012-05-02 | 2013-05-01 | Process for preparing a c3-c7 (hydro) fluoroalkene by dehydrohalogenation |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2015515973A JP2015515973A (ja) | 2015-06-04 |
| JP6190875B2 true JP6190875B2 (ja) | 2017-08-30 |
Family
ID=46330672
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015509495A Active JP6190875B2 (ja) | 2012-05-02 | 2013-05-01 | 脱ハロゲン化水素によるc3−c7(ヒドロ)フルオロアルケンを製造する方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10167242B2 (ja) |
| EP (1) | EP2846907B1 (ja) |
| JP (1) | JP6190875B2 (ja) |
| CN (2) | CN104271235A (ja) |
| ES (1) | ES2725824T3 (ja) |
| GB (1) | GB201207666D0 (ja) |
| HK (1) | HK1204303A1 (ja) |
| IN (1) | IN2014MN02182A (ja) |
| WO (1) | WO2013164618A1 (ja) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110240535B (zh) * | 2014-08-14 | 2022-09-13 | 科慕埃弗西有限公司 | 通过脱氟化氢来制备E-1,3,3,3-四氟丙烯(HFC-1234ze)的方法 |
| JP6328589B2 (ja) | 2015-05-29 | 2018-05-23 | ダイキン工業株式会社 | 含フッ素オレフィンの製造方法 |
| CN104945221B (zh) * | 2015-06-11 | 2017-07-18 | 浙江衢州巨新氟化工有限公司 | 一种联产2,3,3,3‑四氟丙烯和1,3,3,3‑四氟丙烯的方法 |
| GB2540427B (en) | 2015-07-17 | 2017-07-19 | Mexichem Fluor Sa De Cv | Process for the preparation of 2,3,3,3-tetrafluoropropene (1234yf) |
| CN106316777B (zh) * | 2016-08-17 | 2019-01-08 | 巨化集团技术中心 | 一种2,3,3,3-四氟丙烯的制备方法 |
| CN110013865A (zh) * | 2019-05-23 | 2019-07-16 | 浙江师范大学 | 一种用于HFC-245fa裂解制备HFO-1234ze的催化剂及其制备方法 |
| CN111484391B (zh) * | 2020-03-19 | 2023-04-21 | 山东东岳化工有限公司 | 由六氟丙烯制备1,2,3,3,3-五氟丙烯的方法 |
| US11274069B2 (en) | 2020-08-13 | 2022-03-15 | L'Air Liquide, Société Anonyme pour l'Etude et l'Exploitation des Procédés Georges Claude | Mono-substituted cyclopentadienes and metal cyclopentadienyl complexes and synthesis methods thereof |
| US12083493B2 (en) | 2021-03-04 | 2024-09-10 | American Air Liquide, Inc. | Selective adsorption of halocarbon impurities containing cl, br and i in fluorocarbons or hydrofluorocarbons using adsorbent supported metal oxide |
| CN114634395B (zh) * | 2022-03-25 | 2024-05-28 | 浙江工业大学 | 一种由四氟乙烯制备2,3,3,3-四氟丙烯的方法 |
| US20240376032A1 (en) * | 2023-05-12 | 2024-11-14 | Honeywell International Inc. | Method for reducing 1,1,1,2,2-pentafluoropropane (hfc-245cb) in a process for producing trans-1,3,3,3-tetrafluoropropene (hfo-1234ze(e)) |
Family Cites Families (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1868869A (en) | 1928-11-28 | 1932-07-26 | Aluminum Co Of America | Adsorbent material and method of producing the same |
| US2015593A (en) | 1932-02-24 | 1935-09-24 | Aluminum Co Of America | Manufacture of adsorbents |
| US2499675A (en) | 1945-12-13 | 1950-03-07 | Phillips Petroleum Co | Preparation of pelleted alumina catalyst |
| US3201199A (en) | 1962-03-06 | 1965-08-17 | Aluminum Lab Ltd | Method of producing low soda content alumina |
| NL293754A (ja) * | 1962-06-14 | |||
| SU507551A1 (ru) | 1974-10-01 | 1976-03-25 | Ташкентский Государственный Университет Им.В.И.Ленина | Способ получени винилфторида |
| JPS6140229A (ja) | 1984-07-19 | 1986-02-26 | イー・アイ・デユポン・デ・ニモアス・アンド・カンパニー | 2,3‐ジクロロブタンから2‐クロロブテンを製造する方法 |
| WO1989010341A1 (fr) | 1988-04-28 | 1989-11-02 | Showa Denko Kabushiki Kaisha | Procede de production d'un compose d'organofluor |
| US5036036A (en) * | 1989-06-13 | 1991-07-30 | E. I. Du Pont De Nemours And Company | Chromium oxide catalyst composition |
| CA2019913A1 (en) | 1990-06-08 | 1991-12-08 | Michael S. Bolmer | Production of vinylidene fluoride from 1,1-difluoroethane |
| FR2669022B1 (fr) | 1990-11-13 | 1992-12-31 | Atochem | Procede de fabrication du tetrafluoro-1,1,1,2-ethane. |
| EP0726243A1 (en) | 1992-06-05 | 1996-08-14 | Daikin Industries, Limited | Method for manufacturing 1,1,1,2,3-pentafluoropropene and method for manufacturing 1,1,1,2,3-pentafluoropropane |
| US5986151A (en) | 1997-02-05 | 1999-11-16 | Alliedsignal Inc. | Fluorinated propenes from pentafluoropropane |
| JP3886229B2 (ja) | 1997-11-11 | 2007-02-28 | セントラル硝子株式会社 | 1,3,3,3−テトラフルオロプロペンの製造法 |
| US6124510A (en) | 1998-07-21 | 2000-09-26 | Elf Atochem North America, Inc. | 1234ze preparation |
| US7230146B2 (en) | 2003-10-27 | 2007-06-12 | Honeywell International Inc. | Process for producing fluoropropenes |
| US7563936B2 (en) | 2006-10-27 | 2009-07-21 | Honeywell International Inc | Processes for geometric isomerization of halogenated olefins |
| US7592494B2 (en) * | 2003-07-25 | 2009-09-22 | Honeywell International Inc. | Process for the manufacture of 1,3,3,3-tetrafluoropropene |
| US8530708B2 (en) | 2003-07-25 | 2013-09-10 | Honeywell International Inc. | Processes for selective dehydrohalogenation of halogenated alkanes |
| US9255046B2 (en) | 2003-07-25 | 2016-02-09 | Honeywell International Inc. | Manufacturing process for HFO-1234ze |
| WO2005037431A1 (en) | 2003-10-14 | 2005-04-28 | E.I. Dupont De Nemours And Company | Chromium oxide compositions containing zinc, their preparation, and their use as catalysts and catalyst precursors |
| US8318627B2 (en) | 2005-08-10 | 2012-11-27 | Sd Lizenzverwertungsgesellschaft Mbh & Co. Kg | Process for preparation of a catalyst carrier |
| US8664455B2 (en) | 2008-08-08 | 2014-03-04 | Honeywell International Inc. | Process to manufacture 2-chloro-1,1,1,2-tetrafluoropropane (HCFC-244bb) |
| SI3336074T1 (sl) | 2006-01-03 | 2025-09-30 | Honeywell International Inc. | Postopek za proizvodnjo fluoriranih organskih spojin |
| JP4693811B2 (ja) * | 2006-06-13 | 2011-06-01 | セントラル硝子株式会社 | 1,3,3,3−テトラフルオロプロペンの製造方法 |
| CN101466656B (zh) * | 2006-06-13 | 2013-07-03 | 中央硝子株式会社 | 用于生产1,3,3,3-四氟丙烯的方法 |
| US7982073B2 (en) * | 2006-07-13 | 2011-07-19 | E. I. Du Pont De Nemours And Company | Catalytic production processes for making tetrafluoropropenes and pentafluoropropenes |
| US8067650B2 (en) | 2006-08-24 | 2011-11-29 | Honeywell International Inc. | Process for the production of HFO trans-1234ze from HFC-245fa |
| US7485760B2 (en) | 2006-08-24 | 2009-02-03 | Honeywell International Inc. | Integrated HFC trans-1234ze manufacture process |
| WO2008030440A2 (en) | 2006-09-05 | 2008-03-13 | E. I. Du Pont De Nemours And Company | Process to manufacture 2,3,3,3-tetrafluoropropene |
| US7420094B2 (en) | 2006-09-05 | 2008-09-02 | E.I. Du Pont De Nemours And Company | Catalytic isomerization processes of 1,3,3,3-tetrafluoropropene for making 2,3,3,3-tetrafluoropropene |
| CN101522597B (zh) | 2006-10-03 | 2013-05-29 | 墨西哥化学阿玛科股份有限公司 | 通过脱卤化氢制备c3-c6(氢)氟烯烃的方法 |
| GB0706978D0 (en) | 2007-04-11 | 2007-05-16 | Ineos Fluor Holdings Ltd | Process |
| US8258355B2 (en) | 2007-07-25 | 2012-09-04 | Honeywell International Inc. | Processes for preparing 1,1,2,3-tetrachloropropene |
| US20100197980A1 (en) | 2007-08-16 | 2010-08-05 | E.I. Du Pont De Nemours And Company | Catalytic Isomerization Between E and Z Isomers of 1,2,3,3,3 Pentafluoropropene Using Aluminum Catalyst |
| US8513473B2 (en) | 2007-10-10 | 2013-08-20 | Central Glass Company, Limited | Method for producing trans-1,3,3,3-tetrafluoropropene |
| JP2009091301A (ja) | 2007-10-10 | 2009-04-30 | Central Glass Co Ltd | シス−1,2,3,3,3−ペンタフルオロプロペンの製造方法 |
| JP2009097301A (ja) | 2007-10-19 | 2009-05-07 | Daiwa House Ind Co Ltd | 減衰機能を備えた転がり免震支承装置 |
| US8070975B2 (en) | 2008-02-26 | 2011-12-06 | Honeywell International Inc. | Azeotrope-like composition of 2-chloro-1,1,1,2-tetrafluoropropane (HCFC-244bb) and hydrogen fluoride (HF) |
| FR2929273B1 (fr) * | 2008-03-28 | 2017-05-26 | Arkema France | Procede de preparation de composes fluores. |
| FR2929272B1 (fr) * | 2008-03-28 | 2010-04-09 | Arkema France | Procede pour la preparation du 2,3,3,3-tetrafluoro-1-propene |
| GB0808836D0 (en) * | 2008-05-15 | 2008-06-18 | Ineos Fluor Ltd | Process |
| US8053612B2 (en) | 2008-05-30 | 2011-11-08 | Honeywell International Inc. | Process for dehydrochlorinating 1,1,1,2-tetrafluoro-2-chloropropane to 2,3,3,3-tetrafluoropropene in the presence of an alkali metal-doped magnesium oxyfluoride catalyst and methods for making the catalyst |
| EP2318339B1 (en) * | 2008-07-30 | 2012-09-05 | Daikin Industries, Ltd. | Process for preparing 2,3,3,3-tetrafluoropropene |
| ES2507575T3 (es) | 2008-08-22 | 2014-10-15 | Daikin Industries, Ltd. | Procedimiento para preparar 2,3,3,3-tetrafluoropropeno |
| FR2935700B1 (fr) | 2008-09-11 | 2013-05-10 | Arkema France | Procede de preparation de composes trifluores et tetrafluores |
| US8487145B2 (en) | 2009-06-03 | 2013-07-16 | E I De Pont De Nemours And Company | Catalysts and process to manufacture 2,3,3,3-tetrafluoropropene |
| ES2643322T3 (es) | 2009-06-03 | 2017-11-22 | The Chemours Company Fc, Llc | Proceso de fabricación del 2,3,3,3-tetrafluoropropeno |
| CN106316776A (zh) * | 2010-02-12 | 2017-01-11 | 大金工业株式会社 | 用于制备含氟烯烃的方法 |
| US8263817B2 (en) | 2010-07-06 | 2012-09-11 | E I Du Pont De Nemours And Company | Synthesis of 1234YF by selective dehydrochlorination of 244BB |
| US9334207B2 (en) | 2010-09-03 | 2016-05-10 | Honeywell International Inc. | Integrated process to coproduce trans-1-chloro-3,3,3-trifluoropropene, trans-1,3,3,3-tetrafluoropropene, and 1,1,1,3,3-pentafluoropropane |
| JP5790438B2 (ja) | 2011-11-21 | 2015-10-07 | セントラル硝子株式会社 | トランス−1−クロロ−3,3,3−トリフルオロプロペンの製造方法 |
| FR2989374A1 (fr) | 2012-04-11 | 2013-10-18 | Arkema France | Procede de preparation d'un compose olefinique sous forme d'un isomere specifique. |
| JP6251992B2 (ja) | 2012-06-29 | 2017-12-27 | セントラル硝子株式会社 | シス−1,3,3,3−テトラフルオロプロペンの製造方法 |
| WO2014134821A1 (en) | 2013-03-08 | 2014-09-12 | Honeywell International Inc. | Low gwp heat transfer compositions including co2 |
-
2012
- 2012-05-02 GB GB201207666A patent/GB201207666D0/en not_active Ceased
-
2013
- 2013-05-01 HK HK15104714.0A patent/HK1204303A1/xx unknown
- 2013-05-01 CN CN201380023119.4A patent/CN104271235A/zh active Pending
- 2013-05-01 CN CN201910085349.2A patent/CN109942367A/zh active Pending
- 2013-05-01 US US14/398,252 patent/US10167242B2/en active Active
- 2013-05-01 EP EP13727313.2A patent/EP2846907B1/en active Active
- 2013-05-01 IN IN2182MUN2014 patent/IN2014MN02182A/en unknown
- 2013-05-01 ES ES13727313T patent/ES2725824T3/es active Active
- 2013-05-01 JP JP2015509495A patent/JP6190875B2/ja active Active
- 2013-05-01 WO PCT/GB2013/051129 patent/WO2013164618A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| EP2846907A1 (en) | 2015-03-18 |
| HK1204303A1 (en) | 2015-11-13 |
| EP2846907B1 (en) | 2019-03-27 |
| WO2013164618A1 (en) | 2013-11-07 |
| US10167242B2 (en) | 2019-01-01 |
| IN2014MN02182A (ja) | 2015-09-04 |
| ES2725824T3 (es) | 2019-09-27 |
| JP2015515973A (ja) | 2015-06-04 |
| CN104271235A (zh) | 2015-01-07 |
| US20150126786A1 (en) | 2015-05-07 |
| GB201207666D0 (en) | 2012-06-13 |
| CN109942367A (zh) | 2019-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6190875B2 (ja) | 脱ハロゲン化水素によるc3−c7(ヒドロ)フルオロアルケンを製造する方法 | |
| JP5722623B2 (ja) | 金属フッ化物触媒上でのハロゲンおよび水素を有するアルケンの製造 | |
| CN101687737B (zh) | 通过卤代烃的催化脱卤化氢制备氟化烯烃 | |
| US9340473B2 (en) | Catalytic gas phase fluorination | |
| US10988423B2 (en) | Gas-phase catalytic fluorination with chromium catalysts | |
| JP5905967B2 (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| MX2013008409A (es) | Fluoracion catalitica en fase gas. | |
| JP6334741B2 (ja) | ハロゲン化アルカンの脱ハロゲン化水素化によるハロゲン化アルケンの調製のための方法 | |
| JP2009108049A (ja) | トランス−1,3,3,3−テトラフルオロプロペンの製造方法 | |
| EP3060537B1 (en) | Process for the isomerisation of c3-7 (hydro)(halo)fluoroalkenes | |
| CN101980993B (zh) | 制备氟化合物的方法 | |
| CN107922295A (zh) | 由1,2‑二氯‑3,3,3‑三氟丙烯制备2‑氯‑3,3,3‑三氟丙烯的新方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20150910 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20160225 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160303 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160603 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20161124 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170223 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170227 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20170710 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20170807 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6190875 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |