JP6154397B2 - ピペリジニルナフチル酢酸 - Google Patents
ピペリジニルナフチル酢酸 Download PDFInfo
- Publication number
- JP6154397B2 JP6154397B2 JP2014546433A JP2014546433A JP6154397B2 JP 6154397 B2 JP6154397 B2 JP 6154397B2 JP 2014546433 A JP2014546433 A JP 2014546433A JP 2014546433 A JP2014546433 A JP 2014546433A JP 6154397 B2 JP6154397 B2 JP 6154397B2
- Authority
- JP
- Japan
- Prior art keywords
- fluoro
- naphthalen
- acetic acid
- piperidin
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- DOKPLNBYACXXGU-UHFFFAOYSA-N 2-naphthalen-1-yl-2-piperidin-1-ium-1-ylacetate Chemical compound C=1C=CC2=CC=CC=C2C=1C(C(=O)O)N1CCCCC1 DOKPLNBYACXXGU-UHFFFAOYSA-N 0.000 title 1
- -1 methanesulfonyl phenyl Chemical group 0.000 claims description 142
- 150000001875 compounds Chemical class 0.000 claims description 122
- 125000000217 alkyl group Chemical group 0.000 claims description 48
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 39
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 33
- 229910052736 halogen Inorganic materials 0.000 claims description 20
- 150000002367 halogens Chemical class 0.000 claims description 16
- 150000003839 salts Chemical class 0.000 claims description 15
- 125000003545 alkoxy group Chemical group 0.000 claims description 12
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 11
- 239000001257 hydrogen Substances 0.000 claims description 11
- 229910052739 hydrogen Inorganic materials 0.000 claims description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 10
- 125000001188 haloalkyl group Chemical group 0.000 claims description 10
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 10
- 125000001072 heteroaryl group Chemical group 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 8
- 208000023504 respiratory system disease Diseases 0.000 claims description 7
- ZURONWOPCWYMDD-UHFFFAOYSA-N 2-[6-fluoro-4-[1-(3-methylsulfonylphenyl)sulfonylpiperidin-4-yl]naphthalen-2-yl]acetic acid Chemical compound CS(=O)(=O)C1=CC=CC(S(=O)(=O)N2CCC(CC2)C=2C3=CC(F)=CC=C3C=C(CC(O)=O)C=2)=C1 ZURONWOPCWYMDD-UHFFFAOYSA-N 0.000 claims description 6
- 208000006673 asthma Diseases 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 230000002265 prevention Effects 0.000 claims description 6
- NVDVYLYYBDSMAG-UHFFFAOYSA-N 2-[4-[1-(2-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1Cl NVDVYLYYBDSMAG-UHFFFAOYSA-N 0.000 claims description 5
- LHVHVRSLNGBUDP-UHFFFAOYSA-N 2-[4-[1-[(2,6-difluorophenyl)carbamoyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1C(=O)NC1=C(F)C=CC=C1F LHVHVRSLNGBUDP-UHFFFAOYSA-N 0.000 claims description 5
- CWKBNTTUEYAUQT-UHFFFAOYSA-N 2-[4-[1-[(2-aminophenyl)methylsulfonyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)CC1=CC=CC=C1N CWKBNTTUEYAUQT-UHFFFAOYSA-N 0.000 claims description 5
- UXOOPOKKWZBMNJ-UHFFFAOYSA-N 2-[4-[1-[(3-chlorophenyl)methylsulfonyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)CC1=CC=CC(Cl)=C1 UXOOPOKKWZBMNJ-UHFFFAOYSA-N 0.000 claims description 5
- AFJLRFUSZSJLRO-UHFFFAOYSA-N 2-[4-[1-[(4-chlorophenyl)methylsulfonyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)CC1=CC=C(Cl)C=C1 AFJLRFUSZSJLRO-UHFFFAOYSA-N 0.000 claims description 5
- FQVXIERYNXXING-UHFFFAOYSA-N 2-[4-[1-[3,5-bis(trifluoromethyl)phenyl]sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 FQVXIERYNXXING-UHFFFAOYSA-N 0.000 claims description 5
- KNSXVORBWILQAR-UHFFFAOYSA-N 2-[6-fluoro-3-methyl-4-(1-pyrrolidin-1-ylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)N1CCCC1 KNSXVORBWILQAR-UHFFFAOYSA-N 0.000 claims description 5
- XRZQBKFLJZTYSN-UHFFFAOYSA-N 2-[6-fluoro-3-methyl-4-[1-[(2-nitrophenyl)methylsulfonyl]piperidin-4-yl]naphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)CC1=CC=CC=C1[N+]([O-])=O XRZQBKFLJZTYSN-UHFFFAOYSA-N 0.000 claims description 5
- FFWFICYQAYWEOC-UHFFFAOYSA-N 2-[6-fluoro-4-[1-(2-methoxyphenyl)sulfonylpiperidin-4-yl]-3-methylnaphthalen-2-yl]acetic acid Chemical compound COC1=CC=CC=C1S(=O)(=O)N1CCC(C=2C3=CC(F)=CC=C3C=C(CC(O)=O)C=2C)CC1 FFWFICYQAYWEOC-UHFFFAOYSA-N 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- GOEDZNUWHOXYOF-UHFFFAOYSA-N 2-[4-[1-(2,4-dichlorobenzoyl)piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1C(=O)C1=CC=C(Cl)C=C1Cl GOEDZNUWHOXYOF-UHFFFAOYSA-N 0.000 claims description 4
- ANQZCLZRCHDYIZ-UHFFFAOYSA-N 2-[4-[1-(2,4-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)C1=CC=C(Cl)C=C1Cl ANQZCLZRCHDYIZ-UHFFFAOYSA-N 0.000 claims description 4
- GFORKNAMGSZGRL-UHFFFAOYSA-N 2-[4-[1-(2,5-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)C1=CC(Cl)=CC=C1Cl GFORKNAMGSZGRL-UHFFFAOYSA-N 0.000 claims description 4
- OXXQWDYOVAZRMJ-UHFFFAOYSA-N 2-[4-[1-(2,6-dichlorobenzoyl)piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1C(=O)C1=C(Cl)C=CC=C1Cl OXXQWDYOVAZRMJ-UHFFFAOYSA-N 0.000 claims description 4
- VESZEWODJZQNMH-UHFFFAOYSA-N 2-[4-[1-(2,6-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)C1=C(Cl)C=CC=C1Cl VESZEWODJZQNMH-UHFFFAOYSA-N 0.000 claims description 4
- IQGANGUTSFQBQE-UHFFFAOYSA-N 2-[4-[1-(2-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1Cl IQGANGUTSFQBQE-UHFFFAOYSA-N 0.000 claims description 4
- UVLLQCHNYUAHEP-UHFFFAOYSA-N 2-[4-[1-(benzenesulfonyl)piperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1 UVLLQCHNYUAHEP-UHFFFAOYSA-N 0.000 claims description 4
- GWJVJXNVOLNBND-UHFFFAOYSA-N 2-[4-[1-[(2,4-dichlorophenyl)carbamoyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1C(=O)NC1=CC=C(Cl)C=C1Cl GWJVJXNVOLNBND-UHFFFAOYSA-N 0.000 claims description 4
- BUZXFELDPJZHCX-UHFFFAOYSA-N 2-[4-[1-[(2-chlorophenyl)carbamoyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1C(=O)NC1=CC=CC=C1Cl BUZXFELDPJZHCX-UHFFFAOYSA-N 0.000 claims description 4
- BAWKSUNQUIPSMX-UHFFFAOYSA-N 2-[6-fluoro-3-methyl-4-(1-methylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C1CCN(S(C)(=O)=O)CC1 BAWKSUNQUIPSMX-UHFFFAOYSA-N 0.000 claims description 4
- NCVIYDFWBYBHKO-UHFFFAOYSA-N 2-[6-fluoro-4-(1-pyridin-2-ylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=N1 NCVIYDFWBYBHKO-UHFFFAOYSA-N 0.000 claims description 4
- FSOAWLRBHNVEMA-UHFFFAOYSA-N 2-[6-fluoro-4-(1-pyridin-3-ylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CN=C1 FSOAWLRBHNVEMA-UHFFFAOYSA-N 0.000 claims description 4
- QPFMSGNYQMFRRC-UHFFFAOYSA-N 2-[6-fluoro-4-(1-pyridin-4-ylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=NC=C1 QPFMSGNYQMFRRC-UHFFFAOYSA-N 0.000 claims description 4
- 125000000068 chlorophenyl group Chemical group 0.000 claims description 4
- 125000006501 nitrophenyl group Chemical group 0.000 claims description 4
- JIWCZCFFAKTYIX-UHFFFAOYSA-N 2-[4-(1-benzylsulfonylpiperidin-4-yl)-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)CC1=CC=CC=C1 JIWCZCFFAKTYIX-UHFFFAOYSA-N 0.000 claims description 3
- KRCYNLFPONXZCX-UHFFFAOYSA-N 2-[4-(1-cyclopentylsulfonylpiperidin-4-yl)-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)C1CCCC1 KRCYNLFPONXZCX-UHFFFAOYSA-N 0.000 claims description 3
- OFCRRHOGCRBAKT-UHFFFAOYSA-N 2-[4-[1-(2,5-difluorobenzoyl)piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1C(=O)C1=CC(F)=CC=C1F OFCRRHOGCRBAKT-UHFFFAOYSA-N 0.000 claims description 3
- USLDTFLWLWTNIJ-UHFFFAOYSA-N 2-[4-[1-(3-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC(Cl)=C1 USLDTFLWLWTNIJ-UHFFFAOYSA-N 0.000 claims description 3
- KTCVCRWYGGSUDM-UHFFFAOYSA-N 2-[4-[1-(4-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=C(Cl)C=C1 KTCVCRWYGGSUDM-UHFFFAOYSA-N 0.000 claims description 3
- BMFNCADIGSOVGK-UHFFFAOYSA-N 2-[6-fluoro-4-[1-(4-methylphenyl)sulfonylpiperidin-4-yl]naphthalen-2-yl]acetic acid Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1CCC(C=2C3=CC(F)=CC=C3C=C(CC(O)=O)C=2)CC1 BMFNCADIGSOVGK-UHFFFAOYSA-N 0.000 claims description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 3
- BHDIOFCMVILJRW-UHFFFAOYSA-N 2-[4-[1-(2,4-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=C(Cl)C=C1Cl BHDIOFCMVILJRW-UHFFFAOYSA-N 0.000 claims description 2
- SWFZXUBGXJGAKB-UHFFFAOYSA-N 2-[4-[1-(2,5-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC(Cl)=CC=C1Cl SWFZXUBGXJGAKB-UHFFFAOYSA-N 0.000 claims description 2
- HBRZEDBLVAFGLF-UHFFFAOYSA-N 2-[4-[1-(3,5-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetic acid Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)O)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC(Cl)=CC(Cl)=C1 HBRZEDBLVAFGLF-UHFFFAOYSA-N 0.000 claims description 2
- CYKXTPZURWDJQW-UHFFFAOYSA-N 2-[6-fluoro-3-methyl-4-[1-[3-(trifluoromethyl)phenyl]sulfonylpiperidin-4-yl]naphthalen-2-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C=C2C=CC(F)=CC2=C1C(CC1)CCN1S(=O)(=O)C1=CC=CC(C(F)(F)F)=C1 CYKXTPZURWDJQW-UHFFFAOYSA-N 0.000 claims description 2
- 208000009079 Bronchial Spasm Diseases 0.000 claims description 2
- 208000014181 Bronchial disease Diseases 0.000 claims description 2
- 206010006482 Bronchospasm Diseases 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 2
- 208000019693 Lung disease Diseases 0.000 claims description 2
- 239000013543 active substance Substances 0.000 claims description 2
- 230000001684 chronic effect Effects 0.000 claims description 2
- 125000004188 dichlorophenyl group Chemical group 0.000 claims description 2
- 125000004212 difluorophenyl group Chemical group 0.000 claims description 2
- 239000003814 drug Substances 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 239000011737 fluorine Substances 0.000 claims description 2
- 230000000414 obstructive effect Effects 0.000 claims description 2
- 125000003944 tolyl group Chemical group 0.000 claims description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 324
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 201
- 239000000243 solution Substances 0.000 description 178
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 159
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 156
- 239000000203 mixture Substances 0.000 description 147
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 137
- 239000011541 reaction mixture Substances 0.000 description 100
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 96
- 238000004949 mass spectrometry Methods 0.000 description 91
- 239000007787 solid Substances 0.000 description 87
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 64
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 64
- 230000002829 reductive effect Effects 0.000 description 64
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 59
- 238000006243 chemical reaction Methods 0.000 description 51
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 50
- 239000002904 solvent Substances 0.000 description 48
- 238000005481 NMR spectroscopy Methods 0.000 description 46
- 239000000284 extract Substances 0.000 description 40
- 238000010898 silica gel chromatography Methods 0.000 description 39
- 239000012267 brine Substances 0.000 description 37
- 229910052757 nitrogen Inorganic materials 0.000 description 37
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 37
- 239000011734 sodium Substances 0.000 description 34
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 32
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 32
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 31
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 27
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 27
- 239000012043 crude product Substances 0.000 description 27
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 27
- 239000012044 organic layer Substances 0.000 description 26
- 239000000460 chlorine Substances 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 239000000543 intermediate Substances 0.000 description 22
- 238000000034 method Methods 0.000 description 21
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 20
- 239000007864 aqueous solution Substances 0.000 description 20
- 239000002585 base Substances 0.000 description 19
- 238000001914 filtration Methods 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 18
- 239000002244 precipitate Substances 0.000 description 18
- 238000000746 purification Methods 0.000 description 18
- 238000010992 reflux Methods 0.000 description 18
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 17
- QGKDWJMOEPSJMD-UHFFFAOYSA-N methyl 2-(6-fluoro-3-methyl-4-piperidin-4-ylnaphthalen-2-yl)acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C1CCNCC1 QGKDWJMOEPSJMD-UHFFFAOYSA-N 0.000 description 16
- KXKVLQRXCPHEJC-UHFFFAOYSA-N methyl acetate Chemical compound COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 150000004702 methyl esters Chemical class 0.000 description 15
- 239000000725 suspension Substances 0.000 description 15
- QONDGSHRRNTIRE-UHFFFAOYSA-N methyl 2-(6-fluoro-4-piperidin-4-ylnaphthalen-2-yl)acetate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C1CCNCC1 QONDGSHRRNTIRE-UHFFFAOYSA-N 0.000 description 14
- 239000012299 nitrogen atmosphere Substances 0.000 description 14
- 102000009389 Prostaglandin D receptors Human genes 0.000 description 13
- 108050000258 Prostaglandin D receptors Proteins 0.000 description 13
- 230000007062 hydrolysis Effects 0.000 description 13
- 238000006460 hydrolysis reaction Methods 0.000 description 13
- 238000005406 washing Methods 0.000 description 13
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- 238000003556 assay Methods 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 239000003643 water by type Substances 0.000 description 11
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 10
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 10
- 210000004241 Th2 cell Anatomy 0.000 description 9
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 9
- 239000003153 chemical reaction reagent Substances 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 238000003818 flash chromatography Methods 0.000 description 9
- 239000003960 organic solvent Substances 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 9
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- CUYKAIDJBBVRKC-UHFFFAOYSA-N methyl 2-(6-fluoro-3-methyl-4-piperidin-4-ylnaphthalen-2-yl)acetate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C1CCNCC1 CUYKAIDJBBVRKC-UHFFFAOYSA-N 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 125000005843 halogen group Chemical group 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 6
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Natural products O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- 239000012442 inert solvent Substances 0.000 description 6
- 230000018711 interleukin-13 production Effects 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 125000006413 ring segment Chemical group 0.000 description 6
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 6
- 238000003828 vacuum filtration Methods 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- PSAGPCOTGOTBQB-UHFFFAOYSA-N 4-hydroxynaphthalene-1-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=C(O)C2=C1 PSAGPCOTGOTBQB-UHFFFAOYSA-N 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 125000003118 aryl group Chemical group 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 5
- 150000002894 organic compounds Chemical class 0.000 description 5
- 229910052763 palladium Inorganic materials 0.000 description 5
- 150000003335 secondary amines Chemical class 0.000 description 5
- 229940124530 sulfonamide Drugs 0.000 description 5
- 150000003456 sulfonamides Chemical class 0.000 description 5
- 0 **N(CC1)CCC1c1c(cc(*)cc2)c2cc(CC(O)=O)c1* Chemical compound **N(CC1)CCC1c1c(cc(*)cc2)c2cc(CC(O)=O)c1* 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- 239000012981 Hank's balanced salt solution Substances 0.000 description 4
- 102000003816 Interleukin-13 Human genes 0.000 description 4
- 108090000176 Interleukin-13 Proteins 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 4
- XSUXDJHVNPNNFJ-UHFFFAOYSA-N OBOC=C Chemical compound OBOC=C XSUXDJHVNPNNFJ-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 238000006600 Stobbe condensation reaction Methods 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 208000026935 allergic disease Diseases 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzenecarboxaldehyde Natural products O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 229910002091 carbon monoxide Inorganic materials 0.000 description 4
- 238000005810 carbonylation reaction Methods 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 239000000975 dye Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- SXWRTZOXMUOJER-UHFFFAOYSA-N hydron;piperidin-4-one;chloride;hydrate Chemical compound O.Cl.O=C1CCNCC1 SXWRTZOXMUOJER-UHFFFAOYSA-N 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- UFLUXAPXBPAUOH-UHFFFAOYSA-N methyl 2-[6-fluoro-3-methyl-4-(trifluoromethylsulfonyloxy)naphthalen-2-yl]acetate Chemical compound C1=C(F)C=C2C(OS(=O)(=O)C(F)(F)F)=C(C)C(CC(=O)OC)=CC2=C1 UFLUXAPXBPAUOH-UHFFFAOYSA-N 0.000 description 4
- XNZPLUGIVNLLOF-UHFFFAOYSA-N methyl 2-[6-fluoro-4-(trifluoromethylsulfonyloxy)naphthalen-2-yl]acetate Chemical compound C1=C(F)C=CC2=CC(CC(=O)OC)=CC(OS(=O)(=O)C(F)(F)F)=C21 XNZPLUGIVNLLOF-UHFFFAOYSA-N 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 238000007363 ring formation reaction Methods 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical group COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 4
- 239000001384 succinic acid Substances 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 4
- GFUUVLQCTGDFOU-UHFFFAOYSA-N (6-fluoro-3-methyl-4-phenylmethoxynaphthalen-2-yl)methanol Chemical compound CC1=C(CO)C=C2C=CC(F)=CC2=C1OCC1=CC=CC=C1 GFUUVLQCTGDFOU-UHFFFAOYSA-N 0.000 description 3
- CAIPGSXZOYHXGC-UHFFFAOYSA-N (6-fluoro-4-phenylmethoxynaphthalen-2-yl)methanol Chemical compound C=12C=C(F)C=CC2=CC(CO)=CC=1OCC1=CC=CC=C1 CAIPGSXZOYHXGC-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- LJXDPWOOCPRIHJ-UHFFFAOYSA-N 3-(chloromethyl)-7-fluoro-1-phenylmethoxynaphthalene Chemical compound C12=CC(F)=CC=C2C=C(CCl)C=C1OCC1=CC=CC=C1 LJXDPWOOCPRIHJ-UHFFFAOYSA-N 0.000 description 3
- UOQXIWFBQSVDPP-UHFFFAOYSA-N 4-fluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C=C1 UOQXIWFBQSVDPP-UHFFFAOYSA-N 0.000 description 3
- IIKCSJAYQCTXKN-UHFFFAOYSA-N 6-fluoro-4-hydroxy-3-methylnaphthalene-2-carboxylic acid Chemical compound FC1=CC=C2C=C(C(O)=O)C(C)=C(O)C2=C1 IIKCSJAYQCTXKN-UHFFFAOYSA-N 0.000 description 3
- GJOYQDWKFISCDW-UHFFFAOYSA-N 6-fluoro-4-hydroxynaphthalene-2-carboxylic acid Chemical compound C1=C(F)C=CC2=CC(C(=O)O)=CC(O)=C21 GJOYQDWKFISCDW-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 206010012438 Dermatitis atopic Diseases 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 210000001744 T-lymphocyte Anatomy 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 238000007605 air drying Methods 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 201000008937 atopic dermatitis Diseases 0.000 description 3
- 210000003651 basophil Anatomy 0.000 description 3
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 210000003979 eosinophil Anatomy 0.000 description 3
- 238000005886 esterification reaction Methods 0.000 description 3
- VDDXQSUSMHZCLS-UHFFFAOYSA-N ethenyl trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC=C VDDXQSUSMHZCLS-UHFFFAOYSA-N 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 238000004896 high resolution mass spectrometry Methods 0.000 description 3
- 238000007327 hydrogenolysis reaction Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 239000012280 lithium aluminium hydride Substances 0.000 description 3
- JTRJTQLGBOZEAJ-UHFFFAOYSA-N methyl 2-[6-fluoro-4-[1-[(2-methoxyphenyl)carbamoyl]piperidin-4-yl]-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1C(=O)NC1=CC=CC=C1OC JTRJTQLGBOZEAJ-UHFFFAOYSA-N 0.000 description 3
- GKFXRSUWILKQJF-UHFFFAOYSA-N methyl 6-fluoro-4-hydroxynaphthalene-2-carboxylate Chemical compound C1=C(F)C=CC2=CC(C(=O)OC)=CC(O)=C21 GKFXRSUWILKQJF-UHFFFAOYSA-N 0.000 description 3
- KIFQAWRSZHYZOJ-UHFFFAOYSA-N methyl 6-fluoro-4-phenylmethoxynaphthalene-2-carboxylate Chemical compound C=12C=C(F)C=CC2=CC(C(=O)OC)=CC=1OCC1=CC=CC=C1 KIFQAWRSZHYZOJ-UHFFFAOYSA-N 0.000 description 3
- 150000002790 naphthalenes Chemical class 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 3
- 235000011056 potassium acetate Nutrition 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- BHMBVRSPMRCCGG-OUTUXVNYSA-N prostaglandin D2 Chemical compound CCCCC[C@H](O)\C=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O BHMBVRSPMRCCGG-OUTUXVNYSA-N 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- WUBVEMGCQRSBBT-UHFFFAOYSA-N tert-butyl 4-(trifluoromethylsulfonyloxy)-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(OS(=O)(=O)C(F)(F)F)=CC1 WUBVEMGCQRSBBT-UHFFFAOYSA-N 0.000 description 3
- VNMMPOSHGPZEHJ-UHFFFAOYSA-N tert-butyl 4-[7-fluoro-3-(2-methoxy-2-oxoethyl)naphthalen-1-yl]piperidine-1-carboxylate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C1CCN(C(=O)OC(C)(C)C)CC1 VNMMPOSHGPZEHJ-UHFFFAOYSA-N 0.000 description 3
- YFWQFKUQVJNPKP-UHFFFAOYSA-N tert-butyl 4-iodopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(I)CC1 YFWQFKUQVJNPKP-UHFFFAOYSA-N 0.000 description 3
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- YNLSJGPULHOKPM-UHFFFAOYSA-N (4-hydroxynaphthalen-1-yl) acetate Chemical compound C1=CC=C2C(OC(=O)C)=CC=C(O)C2=C1 YNLSJGPULHOKPM-UHFFFAOYSA-N 0.000 description 2
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- DIOHEXPTUTVCNX-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenyl-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=CC=C1 DIOHEXPTUTVCNX-UHFFFAOYSA-N 0.000 description 2
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- VSRXYLYXIXYEST-KZTWKYQFSA-N 13,14-dihydro-15-ketoprostaglandin D2 Chemical compound CCCCCC(=O)CC[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O VSRXYLYXIXYEST-KZTWKYQFSA-N 0.000 description 2
- HFFXLYHRNRKAPM-UHFFFAOYSA-N 2,4,5-trichloro-n-(5-methyl-1,2-oxazol-3-yl)benzenesulfonamide Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C(=CC(Cl)=C(Cl)C=2)Cl)=N1 HFFXLYHRNRKAPM-UHFFFAOYSA-N 0.000 description 2
- BWZVCCNYKMEVEX-UHFFFAOYSA-N 2,4,6-Trimethylpyridine Chemical compound CC1=CC(C)=NC(C)=C1 BWZVCCNYKMEVEX-UHFFFAOYSA-N 0.000 description 2
- BXCOSWRSIISQSL-UHFFFAOYSA-N 2,5-dichlorobenzenesulfonyl chloride Chemical compound ClC1=CC=C(Cl)C(S(Cl)(=O)=O)=C1 BXCOSWRSIISQSL-UHFFFAOYSA-N 0.000 description 2
- 125000004098 2,6-dichlorobenzoyl group Chemical group O=C([*])C1=C(Cl)C([H])=C([H])C([H])=C1Cl 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- IOMMJBPMFXYWOT-UHFFFAOYSA-N 2-[6-fluoro-4-[1-[(2-methoxyphenyl)carbamoyl]piperidin-4-yl]-3-methylnaphthalen-2-yl]acetic acid Chemical compound COC1=CC=CC=C1NC(=O)N1CCC(C=2C3=CC(F)=CC=C3C=C(CC(O)=O)C=2C)CC1 IOMMJBPMFXYWOT-UHFFFAOYSA-N 0.000 description 2
- KMVZDSQHLDGKGV-UHFFFAOYSA-N 2-chlorobenzenesulfonyl chloride Chemical compound ClC1=CC=CC=C1S(Cl)(=O)=O KMVZDSQHLDGKGV-UHFFFAOYSA-N 0.000 description 2
- WXUAQHNMJWJLTG-UHFFFAOYSA-N 2-methylbutanedioic acid Chemical compound OC(=O)C(C)CC(O)=O WXUAQHNMJWJLTG-UHFFFAOYSA-N 0.000 description 2
- QIMMUPPBPVKWKM-UHFFFAOYSA-N 2-methylnaphthalene Chemical compound C1=CC=CC2=CC(C)=CC=C21 QIMMUPPBPVKWKM-UHFFFAOYSA-N 0.000 description 2
- VSWICNJIUPRZIK-UHFFFAOYSA-N 2-piperideine Chemical class C1CNC=CC1 VSWICNJIUPRZIK-UHFFFAOYSA-N 0.000 description 2
- MTJGVAJYTOXFJH-UHFFFAOYSA-N 3-aminonaphthalene-1,5-disulfonic acid Chemical compound C1=CC=C(S(O)(=O)=O)C2=CC(N)=CC(S(O)(=O)=O)=C21 MTJGVAJYTOXFJH-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- LLTHKSPQEZBOKI-UHFFFAOYSA-N C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=NC=CC=C1)F)=O Chemical compound C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=NC=CC=C1)F)=O LLTHKSPQEZBOKI-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 229910010082 LiAlH Inorganic materials 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 101150003085 Pdcl gene Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- XYBSFZVFZVMUPZ-UHFFFAOYSA-N [Zn]C1CCNCC1 Chemical compound [Zn]C1CCNCC1 XYBSFZVFZVMUPZ-UHFFFAOYSA-N 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 230000009285 allergic inflammation Effects 0.000 description 2
- 230000007815 allergy Effects 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000003935 benzaldehydes Chemical class 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000006184 cosolvent Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- NFOQJNGQQXICBY-UHFFFAOYSA-N dimethyl 2-methylbutanedioate Chemical compound COC(=O)CC(C)C(=O)OC NFOQJNGQQXICBY-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 2
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- GRWIABMEEKERFV-UHFFFAOYSA-N methanol;oxolane Chemical compound OC.C1CCOC1 GRWIABMEEKERFV-UHFFFAOYSA-N 0.000 description 2
- YWGIIMRGSVMHLE-UHFFFAOYSA-N methyl 2-(6-fluoro-3-methyl-4-phenylmethoxynaphthalen-2-yl)acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1OCC1=CC=CC=C1 YWGIIMRGSVMHLE-UHFFFAOYSA-N 0.000 description 2
- PBZPAABJBJOXQD-UHFFFAOYSA-N methyl 2-(6-fluoro-4-phenylmethoxynaphthalen-2-yl)acetate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1OCC1=CC=CC=C1 PBZPAABJBJOXQD-UHFFFAOYSA-N 0.000 description 2
- YIROATHMTKWGAM-UHFFFAOYSA-N methyl 2-[4-[1-(2,4-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)C1=CC=C(Cl)C=C1Cl YIROATHMTKWGAM-UHFFFAOYSA-N 0.000 description 2
- BFYCBCZYGQHZBD-UHFFFAOYSA-N methyl 2-[4-[1-(2,5-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)C1=CC(Cl)=CC=C1Cl BFYCBCZYGQHZBD-UHFFFAOYSA-N 0.000 description 2
- CKIOHOLYJQNVQB-UHFFFAOYSA-N methyl 2-[4-[1-(2,6-dichlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)C1=C(Cl)C=CC=C1Cl CKIOHOLYJQNVQB-UHFFFAOYSA-N 0.000 description 2
- UOXIAVAPOBHWEV-UHFFFAOYSA-N methyl 2-[4-[1-(2-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1Cl UOXIAVAPOBHWEV-UHFFFAOYSA-N 0.000 description 2
- UXHXWVJBTGKTIF-UHFFFAOYSA-N methyl 2-[4-[1-[(2-chlorophenyl)carbamoyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1C(=O)NC1=CC=CC=C1Cl UXHXWVJBTGKTIF-UHFFFAOYSA-N 0.000 description 2
- UJZWAGYCGHPRTD-UHFFFAOYSA-N methyl 2-[6-fluoro-3-methyl-4-[1-[(2-nitrophenyl)methylsulfonyl]piperidin-4-yl]naphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)CC1=CC=CC=C1[N+]([O-])=O UJZWAGYCGHPRTD-UHFFFAOYSA-N 0.000 description 2
- CJEHYEBMBJKYMZ-UHFFFAOYSA-N methyl 6-fluoro-3-methyl-4-phenylmethoxynaphthalene-2-carboxylate Chemical compound CC=1C(C(=O)OC)=CC2=CC=C(F)C=C2C=1OCC1=CC=CC=C1 CJEHYEBMBJKYMZ-UHFFFAOYSA-N 0.000 description 2
- ZNBMIDXYOLMKBB-UHFFFAOYSA-N methyl 6-fluoro-4-hydroxy-3-methylnaphthalene-2-carboxylate Chemical compound C1=C(F)C=C2C(O)=C(C)C(C(=O)OC)=CC2=C1 ZNBMIDXYOLMKBB-UHFFFAOYSA-N 0.000 description 2
- 125000004492 methyl ester group Chemical group 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 2
- 229960003081 probenecid Drugs 0.000 description 2
- BHMBVRSPMRCCGG-UHFFFAOYSA-N prostaglandine D2 Natural products CCCCCC(O)C=CC1C(CC=CCCCC(O)=O)C(O)CC1=O BHMBVRSPMRCCGG-UHFFFAOYSA-N 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- JQJOGAGLBDBMLU-UHFFFAOYSA-N pyridine-2-sulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=N1 JQJOGAGLBDBMLU-UHFFFAOYSA-N 0.000 description 2
- CDRNYKLYADJTMN-UHFFFAOYSA-N pyridine-3-sulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CN=C1 CDRNYKLYADJTMN-UHFFFAOYSA-N 0.000 description 2
- NHMOJCOXIZRTRR-UHFFFAOYSA-N pyridine-4-sulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=NC=C1 NHMOJCOXIZRTRR-UHFFFAOYSA-N 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 230000006103 sulfonylation Effects 0.000 description 2
- 238000005694 sulfonylation reaction Methods 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- VVDCRJGWILREQH-UHFFFAOYSA-N tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC(B2OC(C)(C)C(C)(C)O2)=C1 VVDCRJGWILREQH-UHFFFAOYSA-N 0.000 description 2
- BXZYLVKQZSMICF-UHFFFAOYSA-N tert-butyl 4-[7-fluoro-3-(2-methoxy-2-oxoethyl)-2-methylnaphthalen-1-yl]piperidine-1-carboxylate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C1CCN(C(=O)OC(C)(C)C)CC1 BXZYLVKQZSMICF-UHFFFAOYSA-N 0.000 description 2
- JMDFDKCEZCMHPS-UHFFFAOYSA-N tert-butyl 4-[7-fluoro-3-(2-methoxy-2-oxoethyl)naphthalen-1-yl]-3,6-dihydro-2h-pyridine-1-carboxylate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C1=CCN(C(=O)OC(C)(C)C)CC1 JMDFDKCEZCMHPS-UHFFFAOYSA-N 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 150000003672 ureas Chemical class 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- LXTGNVLBPVVMSL-UHFFFAOYSA-N (3-chlorophenyl)methanesulfonyl chloride Chemical compound ClC1=CC=CC(CS(Cl)(=O)=O)=C1 LXTGNVLBPVVMSL-UHFFFAOYSA-N 0.000 description 1
- DBJRPJSDYFDWPV-UHFFFAOYSA-N (4-chlorophenyl)methanesulfonyl chloride Chemical compound ClC1=CC=C(CS(Cl)(=O)=O)C=C1 DBJRPJSDYFDWPV-UHFFFAOYSA-N 0.000 description 1
- HWSQVCFFAPYCCM-UHFFFAOYSA-N (4-phenylmethoxynaphthalen-1-yl) acetate Chemical compound C12=CC=CC=C2C(OC(=O)C)=CC=C1OCC1=CC=CC=C1 HWSQVCFFAPYCCM-UHFFFAOYSA-N 0.000 description 1
- WXUAQHNMJWJLTG-VKHMYHEASA-N (S)-methylsuccinic acid Chemical compound OC(=O)[C@@H](C)CC(O)=O WXUAQHNMJWJLTG-VKHMYHEASA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- YXHDLKWTPVMIOH-UHFFFAOYSA-N 1,3-difluoro-2-isocyanatobenzene Chemical compound FC1=CC=CC(F)=C1N=C=O YXHDLKWTPVMIOH-UHFFFAOYSA-N 0.000 description 1
- XMWGTKZEDLCVIG-UHFFFAOYSA-N 1-(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1 XMWGTKZEDLCVIG-UHFFFAOYSA-N 0.000 description 1
- NOHQUGRVHSJYMR-UHFFFAOYSA-N 1-chloro-2-isocyanatobenzene Chemical compound ClC1=CC=CC=C1N=C=O NOHQUGRVHSJYMR-UHFFFAOYSA-N 0.000 description 1
- SUVCZZADQDCIEQ-UHFFFAOYSA-N 1-isocyanato-2-methoxybenzene Chemical compound COC1=CC=CC=C1N=C=O SUVCZZADQDCIEQ-UHFFFAOYSA-N 0.000 description 1
- SXJYSIBLFGQAND-UHFFFAOYSA-N 1-isocyanato-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC(N=C=O)=C1 SXJYSIBLFGQAND-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- WUEPMEXUMQVEGN-UHFFFAOYSA-N 2,2,2-trifluoroacetic acid;2,2,2-trifluoro-n-[2-[2-[(2,2,2-trifluoroacetyl)amino]ethylamino]ethyl]acetamide Chemical compound OC(=O)C(F)(F)F.FC(F)(F)C(=O)NCCNCCNC(=O)C(F)(F)F WUEPMEXUMQVEGN-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- OLBJNSPBWLCTOT-UHFFFAOYSA-N 2,4-dichloro-1-isocyanatobenzene Chemical compound ClC1=CC=C(N=C=O)C(Cl)=C1 OLBJNSPBWLCTOT-UHFFFAOYSA-N 0.000 description 1
- FDTPBIKNYWQLAE-UHFFFAOYSA-N 2,4-dichlorobenzenesulfonyl chloride Chemical compound ClC1=CC=C(S(Cl)(=O)=O)C(Cl)=C1 FDTPBIKNYWQLAE-UHFFFAOYSA-N 0.000 description 1
- RLRUKKDFNWXXRT-UHFFFAOYSA-N 2,5-difluorobenzoyl chloride Chemical compound FC1=CC=C(F)C(C(Cl)=O)=C1 RLRUKKDFNWXXRT-UHFFFAOYSA-N 0.000 description 1
- WGGKQIKICKLWGN-UHFFFAOYSA-N 2,6-dichlorobenzenesulfonyl chloride Chemical compound ClC1=CC=CC(Cl)=C1S(Cl)(=O)=O WGGKQIKICKLWGN-UHFFFAOYSA-N 0.000 description 1
- JBLIDPPHFGWTKU-UHFFFAOYSA-N 2,6-dichlorobenzoyl chloride Chemical compound ClC(=O)C1=C(Cl)C=CC=C1Cl JBLIDPPHFGWTKU-UHFFFAOYSA-N 0.000 description 1
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- GYOBZOBUOMDRRN-UHFFFAOYSA-N 2-methoxybenzenesulfonyl chloride Chemical compound COC1=CC=CC=C1S(Cl)(=O)=O GYOBZOBUOMDRRN-UHFFFAOYSA-N 0.000 description 1
- VIBOGIYPPWLDTI-UHFFFAOYSA-N 2-naphthylacetic acid Chemical class C1=CC=CC2=CC(CC(=O)O)=CC=C21 VIBOGIYPPWLDTI-UHFFFAOYSA-N 0.000 description 1
- VMZCDNSFRSVYKQ-UHFFFAOYSA-N 2-phenylacetyl chloride Chemical compound ClC(=O)CC1=CC=CC=C1 VMZCDNSFRSVYKQ-UHFFFAOYSA-N 0.000 description 1
- BTRCVKADYDVSLI-UHFFFAOYSA-N 3,5-bis(trifluoromethyl)benzenesulfonyl chloride Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(S(Cl)(=O)=O)=C1 BTRCVKADYDVSLI-UHFFFAOYSA-N 0.000 description 1
- RJSQINMKOSOUGT-UHFFFAOYSA-N 3,5-dichlorobenzenesulfonyl chloride Chemical compound ClC1=CC(Cl)=CC(S(Cl)(=O)=O)=C1 RJSQINMKOSOUGT-UHFFFAOYSA-N 0.000 description 1
- SLWMKNVXZCAXDA-UHFFFAOYSA-N 3-(chloromethyl)-7-fluoro-2-methyl-1-phenylmethoxynaphthalene Chemical compound CC1=C(CCl)C=C2C=CC(F)=CC2=C1OCC1=CC=CC=C1 SLWMKNVXZCAXDA-UHFFFAOYSA-N 0.000 description 1
- ONCAZCNPWWQQMW-UHFFFAOYSA-N 3-(trifluoromethyl)benzenesulfonyl chloride Chemical compound FC(F)(F)C1=CC=CC(S(Cl)(=O)=O)=C1 ONCAZCNPWWQQMW-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OINWZUJVEXUHCC-UHFFFAOYSA-N 3-chlorobenzenesulfonyl chloride Chemical compound ClC1=CC=CC(S(Cl)(=O)=O)=C1 OINWZUJVEXUHCC-UHFFFAOYSA-N 0.000 description 1
- CQEFJGDLGWJIRV-UHFFFAOYSA-N 3-methylsulfonylbenzenesulfonyl chloride Chemical compound CS(=O)(=O)C1=CC=CC(S(Cl)(=O)=O)=C1 CQEFJGDLGWJIRV-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- ZLYBFBAHAQEEQQ-UHFFFAOYSA-N 4-chlorobenzenesulfonyl chloride Chemical compound ClC1=CC=C(S(Cl)(=O)=O)C=C1 ZLYBFBAHAQEEQQ-UHFFFAOYSA-N 0.000 description 1
- 239000012114 Alexa Fluor 647 Substances 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- CHWWNXZHXXMKNV-UHFFFAOYSA-N C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=CC(=CC(=C1)C(F)(F)F)C(F)(F)F)F)=O Chemical compound C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=CC(=CC(=C1)C(F)(F)F)C(F)(F)F)F)=O CHWWNXZHXXMKNV-UHFFFAOYSA-N 0.000 description 1
- FKQPKQWSFUBRSL-UHFFFAOYSA-N C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=CC(=CC=C1)S(=O)(=O)C)F)=O Chemical compound C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=CC(=CC=C1)S(=O)(=O)C)F)=O FKQPKQWSFUBRSL-UHFFFAOYSA-N 0.000 description 1
- PZUJQPFWIDNMQT-UHFFFAOYSA-N C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=CC=NC=C1)F)=O Chemical compound C(C)(C)N(CC)C(C)C.COC(CC1=CC2=CC=C(C=C2C(=C1)C1CCN(CC1)S(=O)(=O)C1=CC=NC=C1)F)=O PZUJQPFWIDNMQT-UHFFFAOYSA-N 0.000 description 1
- MPWUJGAPYDCLDW-UHFFFAOYSA-N COC(=O)C1=CC2=CC=C(C=C2C(=C1C)O)F.COC(=O)C1=CC2=CC=C(C=C2C(=C1C)OCC1=CC=CC=C1)F Chemical compound COC(=O)C1=CC2=CC=C(C=C2C(=C1C)O)F.COC(=O)C1=CC2=CC=C(C=C2C(=C1C)OCC1=CC=CC=C1)F MPWUJGAPYDCLDW-UHFFFAOYSA-N 0.000 description 1
- JMTYIPBKOAOFNY-UHFFFAOYSA-N COC(CC1=CC2=CC=C(C=C2C(=C1)OCC1=CC=CC=C1)F)=O.COC(CC1=CC2=CC=C(C=C2C(=C1)O)F)=O Chemical compound COC(CC1=CC2=CC=C(C=C2C(=C1)OCC1=CC=CC=C1)F)=O.COC(CC1=CC2=CC=C(C=C2C(=C1)O)F)=O JMTYIPBKOAOFNY-UHFFFAOYSA-N 0.000 description 1
- KBNZIHNAARKBOC-UHFFFAOYSA-N COC(CC1=CC2=CC=C(C=C2C(=C1C)C1CCNCC1)F)=O.COC(CC1=CC2=CC=C(C=C2C(=C1C)C1CCN(CC1)S(=O)(=O)CC1=C(C=CC=C1)[N+](=O)[O-])F)=O Chemical compound COC(CC1=CC2=CC=C(C=C2C(=C1C)C1CCNCC1)F)=O.COC(CC1=CC2=CC=C(C=C2C(=C1C)C1CCN(CC1)S(=O)(=O)CC1=C(C=CC=C1)[N+](=O)[O-])F)=O KBNZIHNAARKBOC-UHFFFAOYSA-N 0.000 description 1
- KKAOLJKHDMXWRX-UHFFFAOYSA-N Cc1c(CC(O)=O)cc(ccc(F)c2)c2c1C(CC1)CCN1C(Nc1cccc(C(F)(F)F)c1)=O Chemical compound Cc1c(CC(O)=O)cc(ccc(F)c2)c2c1C(CC1)CCN1C(Nc1cccc(C(F)(F)F)c1)=O KKAOLJKHDMXWRX-UHFFFAOYSA-N 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- MUXOBHXGJLMRAB-UHFFFAOYSA-N Dimethyl succinate Chemical compound COC(=O)CCC(=O)OC MUXOBHXGJLMRAB-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 101001002657 Homo sapiens Interleukin-2 Proteins 0.000 description 1
- 101001002709 Homo sapiens Interleukin-4 Proteins 0.000 description 1
- GRRNUXAQVGOGFE-UHFFFAOYSA-N Hygromycin-B Natural products OC1C(NC)CC(N)C(O)C1OC1C2OC3(C(C(O)C(O)C(C(N)CO)O3)O)OC2C(O)C(CO)O1 GRRNUXAQVGOGFE-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- XBTXHHLXXIDYHS-UHFFFAOYSA-N OC(=O)C(F)(F)F.OC(=O)C(F)(F)F.COC(=O)Cc1cc(C2CCNCC2)c2cc(F)ccc2c1 Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F.COC(=O)Cc1cc(C2CCNCC2)c2cc(F)ccc2c1 XBTXHHLXXIDYHS-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 239000005662 Paraffin oil Substances 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102100024218 Prostaglandin D2 receptor 2 Human genes 0.000 description 1
- 241000508269 Psidium Species 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- 239000005708 Sodium hypochlorite Substances 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 201000009961 allergic asthma Diseases 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000003185 calcium uptake Effects 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 150000001722 carbon compounds Chemical class 0.000 description 1
- 230000006315 carbonylation Effects 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002975 chemoattractant Substances 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000011340 continuous therapy Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HZVKYZHPDGEECE-UHFFFAOYSA-N cyclopentanesulfonyl chloride Chemical compound ClS(=O)(=O)C1CCCC1 HZVKYZHPDGEECE-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000005959 diazepanyl group Chemical group 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000005050 dihydrooxazolyl group Chemical group O1C(NC=C1)* 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- QGFRSPMTVBPWNK-UHFFFAOYSA-N dilithium;oxygen(2-);hydrate Chemical compound [Li+].[Li+].O.[O-2] QGFRSPMTVBPWNK-UHFFFAOYSA-N 0.000 description 1
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical group C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 1
- KZTYYGOKRVBIMI-UHFFFAOYSA-N diphenyl sulfone Chemical group C=1C=CC=CC=1S(=O)(=O)C1=CC=CC=C1 KZTYYGOKRVBIMI-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 235000013312 flour Nutrition 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 102000055277 human IL2 Human genes 0.000 description 1
- 102000055229 human IL4 Human genes 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 235000011167 hydrochloric acid Nutrition 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- GRRNUXAQVGOGFE-NZSRVPFOSA-N hygromycin B Chemical compound O[C@@H]1[C@@H](NC)C[C@@H](N)[C@H](O)[C@H]1O[C@H]1[C@H]2O[C@@]3([C@@H]([C@@H](O)[C@@H](O)[C@@H](C(N)CO)O3)O)O[C@H]2[C@@H](O)[C@@H](CO)O1 GRRNUXAQVGOGFE-NZSRVPFOSA-N 0.000 description 1
- 229940097277 hygromycin b Drugs 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- JILPJDVXYVTZDQ-UHFFFAOYSA-N lithium methoxide Chemical compound [Li+].[O-]C JILPJDVXYVTZDQ-UHFFFAOYSA-N 0.000 description 1
- FZRNJOXQNWVMIH-UHFFFAOYSA-N lithium;hydrate Chemical compound [Li].O FZRNJOXQNWVMIH-UHFFFAOYSA-N 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- AGJSNMGHAVDLRQ-IWFBPKFRSA-N methyl (2s)-2-[[(2s)-2-[[(2s)-2-[[(2r)-2-amino-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,3-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoate Chemical compound SC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(=O)OC)CC1=CC=C(O)C(C)=C1C AGJSNMGHAVDLRQ-IWFBPKFRSA-N 0.000 description 1
- YJCMJJGSUMEXIK-UHFFFAOYSA-N methyl 2-(6-fluoro-4-hydroxy-3-methylnaphthalen-2-yl)acetate Chemical compound C1=C(F)C=C2C(O)=C(C)C(CC(=O)OC)=CC2=C1 YJCMJJGSUMEXIK-UHFFFAOYSA-N 0.000 description 1
- QOMVYQPPZSXVGL-UHFFFAOYSA-N methyl 2-(6-fluoro-4-hydroxynaphthalen-2-yl)acetate Chemical compound C1=C(F)C=CC2=CC(CC(=O)OC)=CC(O)=C21 QOMVYQPPZSXVGL-UHFFFAOYSA-N 0.000 description 1
- WEVSIJNEAVZQOX-UHFFFAOYSA-N methyl 2-[4-(1-cyclopentylsulfonylpiperidin-4-yl)-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)C1CCCC1 WEVSIJNEAVZQOX-UHFFFAOYSA-N 0.000 description 1
- OYTYIAFWTGUTFY-UHFFFAOYSA-N methyl 2-[4-[1-(2-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1Cl OYTYIAFWTGUTFY-UHFFFAOYSA-N 0.000 description 1
- LQPYGFFKSBNCEP-UHFFFAOYSA-N methyl 2-[4-[1-(3-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC(Cl)=C1 LQPYGFFKSBNCEP-UHFFFAOYSA-N 0.000 description 1
- XJSGHGYTXHBNKW-UHFFFAOYSA-N methyl 2-[4-[1-(4-chlorophenyl)sulfonylpiperidin-4-yl]-6-fluoronaphthalen-2-yl]acetate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=C(Cl)C=C1 XJSGHGYTXHBNKW-UHFFFAOYSA-N 0.000 description 1
- JSVMURMKEMJUGA-UHFFFAOYSA-N methyl 2-[4-[1-(benzenesulfonyl)piperidin-4-yl]-6-fluoronaphthalen-2-yl]acetate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1 JSVMURMKEMJUGA-UHFFFAOYSA-N 0.000 description 1
- UWOXYAXAWYFVKK-UHFFFAOYSA-N methyl 2-[4-[1-[(2,4-dichlorophenyl)carbamoyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1C(=O)NC1=CC=C(Cl)C=C1Cl UWOXYAXAWYFVKK-UHFFFAOYSA-N 0.000 description 1
- NNCZNMDTWBFAAZ-UHFFFAOYSA-N methyl 2-[4-[1-[(2,6-difluorophenyl)carbamoyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1C(=O)NC1=C(F)C=CC=C1F NNCZNMDTWBFAAZ-UHFFFAOYSA-N 0.000 description 1
- HLHJLKKHYVLLRW-UHFFFAOYSA-N methyl 2-[4-[1-[(2-aminophenyl)methylsulfonyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)CC1=CC=CC=C1N HLHJLKKHYVLLRW-UHFFFAOYSA-N 0.000 description 1
- KZHHVENGXBUOPT-UHFFFAOYSA-N methyl 2-[4-[1-[(3-chlorophenyl)methylsulfonyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)CC1=CC=CC(Cl)=C1 KZHHVENGXBUOPT-UHFFFAOYSA-N 0.000 description 1
- OQCRPNQZPZLQCS-UHFFFAOYSA-N methyl 2-[4-[1-[(4-chlorophenyl)methylsulfonyl]piperidin-4-yl]-6-fluoro-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)CC1=CC=C(Cl)C=C1 OQCRPNQZPZLQCS-UHFFFAOYSA-N 0.000 description 1
- PPWFQFFQUALTCR-UHFFFAOYSA-N methyl 2-[6-fluoro-3-methyl-4-(1-methylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C1CCN(S(C)(=O)=O)CC1 PPWFQFFQUALTCR-UHFFFAOYSA-N 0.000 description 1
- ZIMOYXJPGRVNAS-UHFFFAOYSA-N methyl 2-[6-fluoro-3-methyl-4-(1-pyrrolidin-1-ylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)N1CCCC1 ZIMOYXJPGRVNAS-UHFFFAOYSA-N 0.000 description 1
- QKIONPVFQNBWLU-UHFFFAOYSA-N methyl 2-[6-fluoro-4-(1-pyridin-4-ylsulfonylpiperidin-4-yl)naphthalen-2-yl]acetate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=NC=C1 QKIONPVFQNBWLU-UHFFFAOYSA-N 0.000 description 1
- SNSBXLNERSEQMP-UHFFFAOYSA-N methyl 2-[6-fluoro-4-[1-(2-methoxyphenyl)sulfonylpiperidin-4-yl]-3-methylnaphthalen-2-yl]acetate Chemical compound CC=1C(CC(=O)OC)=CC2=CC=C(F)C=C2C=1C(CC1)CCN1S(=O)(=O)C1=CC=CC=C1OC SNSBXLNERSEQMP-UHFFFAOYSA-N 0.000 description 1
- CPCPEHFPOZBFAC-UHFFFAOYSA-N methyl 2-[6-fluoro-4-[1-(3-methylsulfonylphenyl)sulfonylpiperidin-4-yl]naphthalen-2-yl]acetate Chemical compound C=12C=C(F)C=CC2=CC(CC(=O)OC)=CC=1C(CC1)CCN1S(=O)(=O)C1=CC=CC(S(C)(=O)=O)=C1 CPCPEHFPOZBFAC-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 238000001682 microtransfer moulding Methods 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- WWECJGLXBSQKRF-UHFFFAOYSA-N n,n-dimethylformamide;methanol Chemical class OC.CN(C)C=O WWECJGLXBSQKRF-UHFFFAOYSA-N 0.000 description 1
- VGKONPUVOVVNSU-UHFFFAOYSA-N naphthalen-1-yl acetate Chemical compound C1=CC=C2C(OC(=O)C)=CC=CC2=C1 VGKONPUVOVVNSU-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000005961 oxazepanyl group Chemical group 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- HBEQXAKJSGXAIQ-UHFFFAOYSA-N oxopalladium Chemical compound [Pd]=O HBEQXAKJSGXAIQ-UHFFFAOYSA-N 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- OAHKWDDSKCRNFE-UHFFFAOYSA-N phenylmethanesulfonyl chloride Chemical compound ClS(=O)(=O)CC1=CC=CC=C1 OAHKWDDSKCRNFE-UHFFFAOYSA-N 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- XUWHAWMETYGRKB-UHFFFAOYSA-N piperidin-2-one Chemical compound O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940127293 prostanoid Drugs 0.000 description 1
- 150000003814 prostanoids Chemical class 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- DVECLMOWYVDJRM-UHFFFAOYSA-N pyridine-3-sulfonic acid Chemical compound OS(=O)(=O)C1=CC=CN=C1 DVECLMOWYVDJRM-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- JRIQCVWTPGVBBH-UHFFFAOYSA-N pyrrolidine-1-sulfonyl chloride Chemical compound ClS(=O)(=O)N1CCCC1 JRIQCVWTPGVBBH-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000019254 respiratory burst Effects 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LFKDJXLFVYVEFG-UHFFFAOYSA-N tert-butyl carbamate Chemical group CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/20—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by singly bound oxygen or sulphur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/92—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with a hetero atom directly attached to the ring nitrogen atom
- C07D211/96—Sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pulmonology (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
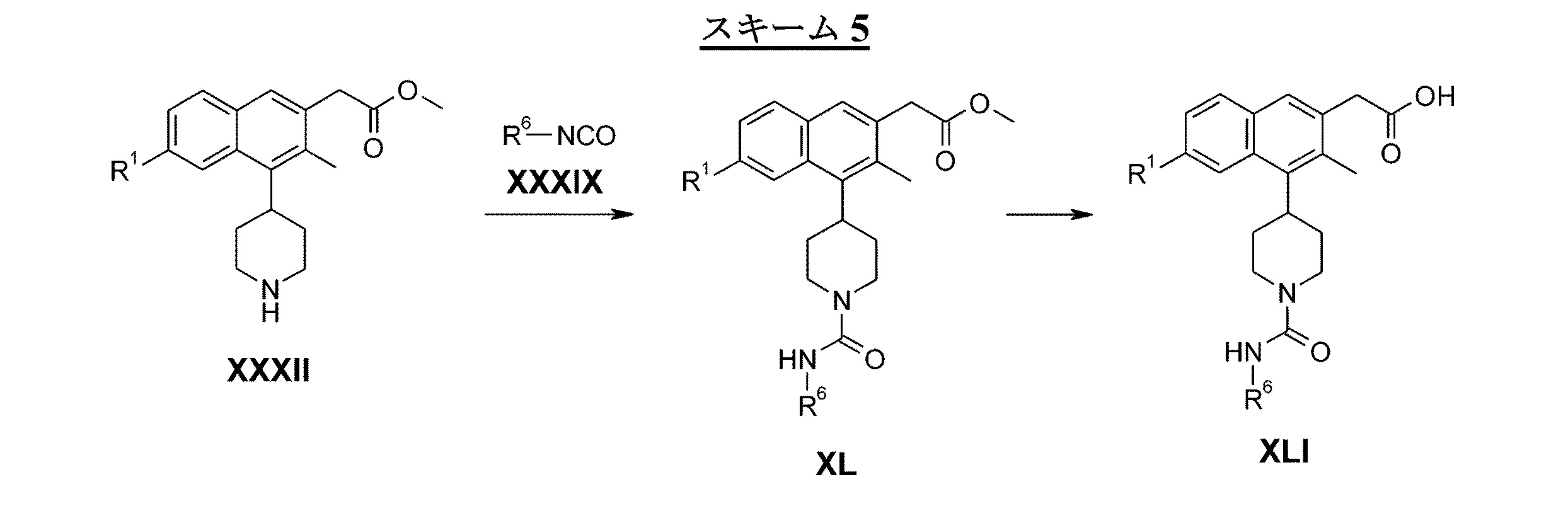
[式中、
Xは、−SO2−又は−C(O)−であり;
R1は、ハロゲンであり;
R2は、− 非置換であるか、又は低級アルキル、ハロゲン、アルコキシ、−SO2−低級アルキルもしくはハロアルキルで独立に一もしくは二置換されている、フェニル、
− 非置換ヘテロアリール、
− 非置換であるか、又は非置換フェニル又はハロゲン、NO2もしくはNH2で置換されているフェニルで置換されている、低級アルキル、
− 非置換シクロアルキル、
− 非置換ヘテロシクロアルキル、あるいは
− NH−フェニル(該フェニルは、非置換であるか、又はハロゲン、低級アルキル、ハロアルキル、−SO2−低級アルキルもしくはアルコキシで置換されている)であり;そして
R3は、低級アルキル又は水素である]で示される化合物又はその薬学的に許容しうる塩を提供する。
[式中、
Xは、−SO2−又は−C(O)−であり;
R1は、ハロゲンであり;
R2は、− 非置換であるか、又は低級アルキル、ハロゲン、アルコキシ、−SO2−低級アルキルもしくはハロアルキルで独立に一もしくは二置換されている、フェニル、
− 非置換ヘテロアリール、
− 非置換であるか、又は非置換フェニル又はハロゲン、NO2もしくはNH2で置換されているフェニルで置換されている、低級アルキル、
− 非置換シクロアルキル、
− 非置換ヘテロシクロアルキル、あるいは
− NH−フェニル(該フェニルは、非置換であるか、又はハロゲン、低級アルキル、ハロアルキル、−SO2−低級アルキルもしくはアルコキシで置換されている)であり;そして
R3は、低級アルキル又は水素である]
で示される化合物又はその薬学的に許容しうる塩を提供する。
[4−(1−ベンゼンスルホニル−ピペリジン−4−イル)−6−フルオロ−ナフタレン−2−イル]−酢酸;
{6−フルオロ−4−[1−(トルエン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(ピリジン−2−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(ピリジン−3−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(ピリジン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(3−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(4−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(3−メタンスルホニル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(3,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(3,5−ビス−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
[6−フルオロ−4−(1−メタンスルホニル−ピペリジン−4−イル)−3−メチル−ナフタレン−2−イル]−酢酸;
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,6−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
[6−フルオロ−3−メチル−4−(1−フェニルメタンスルホニル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸;
{6−フルオロ−4−[1−(2−メトキシ−ベンゼンスルホニル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(3−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(4−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル−ナフタレン−2−イル}−酢酸;
[4−(1−シクロペンタンスルホニル−ピペリジン−4−イル)−6−フルオロ−3−メチル−ナフタレン−2−イル]−酢酸;
{6−フルオロ−3−メチル−4−[1−(ピロリジン−1−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{4−[1−(2−アミノ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,4−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,6−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,5−ジフルオロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
[6−フルオロ−3−メチル−4−(1−フェニルアセチル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸;
{4−[1−(2,6−ジフルオロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,4−ジクロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2−クロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(2−メトキシ−フェニルカルバモイル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸;又は
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−フェニルカルバモイル)−ピペリジン−4−イル−ナフタレン−2−イル}−酢酸
である。
ある特定の例示的実施態様が、本明細書において示され、そして説明されるが、それらは、本発明の範囲を限定するようにみなされるべきではなく、単にその例示及び代表としてみなされるべきである。本発明の化合物を、本明細書において概して記載される方法に従い、及び/又は当業者にとって利用可能な方法によって、適切な出発材料を使用して調製することができる。
(6−フルオロ−4−トリフルオロメタンスルホニルオキシ−ナフタレン−2−イル)−酢酸メチルエステルの調製法
2−[1−(4−フルオロ−フェニル)−メタ−(E)−イリデン]−コハク酸
tert−ブタノール(2mL)中の4−フルオロベンズアルデヒド(1.0g、8.05mmol)及びコハク酸ジメチル(1.41g、9.66mmol)の溶液を、30分以内に、tert−ブタノール(8mL)中のカリウムtert−ブトキシド(0.994g、8.86mmol)の還流混合物に滴下した。還流を、全ての出発材料が消費されたとTLC分析によって実証されるまで続けた。反応温度を、室温まで温め、そしてtert−ブタノールを、真空下で除去した。残留物を、1.0N HCl水溶液(10mL)に溶解し、そして酢酸エチルで抽出した(2×5mL)。合わせた有機抽出物を、無水硫酸ナトリウムで乾燥させ、そして真空下で濃縮して、油状物を得た。この粗生成物を、メタノール(10mL)に溶解した。2.0NのNaOH水溶液(10mL)を、溶液に室温で加え、そして混合物を、一晩還流した。得られた懸濁液を濃縮し、そして得られた残留物を、水(10mL)に溶解した。得られた溶液を、酢酸エチルで洗浄した(2×10mL)。水層を、濃HClの滴下で酸性化(pH約2)した。得られた混合物を、酢酸エチルで抽出した(3×10mL)。合わせた有機抽出物を、無水硫酸ナトリウムで乾燥させ、そして真空下で濃縮して、粗残留物を得た。エーテル/ヘキサンからの結晶化により、2−[1−(4−フルオロ−フェニル)−メタ−(E)−イリデン]−コハク酸(0.74g、41%)を白色の固体として得た。MS C11H8FO4[(M-H)-]についての計算値:223, 実測値 223.1.
トリフルオロメタンスルホン酸(0.5mL)中の2−[1−(4−フルオロ−フェニル)−メタ−(E)−イリデン]−コハク酸(0.100g、0.44mmol)の溶液を、室温で16時間攪拌した。得られた混合物を、連続的に撹拌しながら注意深く氷冷水に注いで、固体沈殿物を得て、これを濾過し、水で洗浄し、そして真空下で乾燥させて、6−フルオロ−4−ヒドロキシ−ナフタレン−2−カルボン酸(0.065g、71%)を黄色の固体として得た。この粗生成物は、さらに精製することなく次の工程に進むのに十分に純粋であった。
MeOH(10mL)中の6−フルオロ−4−ヒドロキシ−ナフタレン−2−カルボン酸(0.500g、2.42mmol)の溶液に、0℃で塩化チオニル(0.265mL、3.63mmol)を滴下し、そして得られた混合物を、5時間還流した。メタノールを、減圧下で留去し、そして残留物を、水(5mL)で希釈し、そして酢酸エチルで抽出した(2×10mL)。合わせた有機抽出物を、水(5mL)、続いてブライン(5mL)で洗浄し、乾燥させ、そして減圧下で濃縮して、粗生成物を得た。シリカゲルカラムクロマトグラフィー(ヘキサン中5%酢酸エチル)により、6−フルオロ−4−ヒドロキシ−ナフタレン−2−カルボン酸メチルエステル(0.150g、28%)を明黄色の固体として得た。MS C12H10FO3[(M+H)+]についての計算値:221, 実測値221.2.
脱水DMF(40mL)中の6−フルオロ−4−ヒドロキシ−ナフタレン−2−カルボン酸メチルエステル(3.7g、16.8mmol)の溶液に、窒素下、室温でK2CO3(3.25g、23.5mmol)、臭化ベンジル(3.44g、20.2mmol)及びBu4NI(0.02g)を同時に加えた。反応混合物を、室温で3時間攪拌した。混合物を、水(60mL)で希釈し、そして酢酸エチルで抽出した(2×100mL)。合わせた有機抽出物を、水(30mL)、ブライン(30mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、そして真空下で濃縮して、粗生成物を得た。シリカゲルカラムクロマトグラフィー(ヘキサン中2%〜5%酢酸エチル)により、4−ベンジルオキシ−6−フルオロ−ナフタレン−2−カルボン酸メチルエステル(4.6g、88%)をオフホワイトの固体として得た。MS C19H16FO3[(M+H)+]についての計算値:311, 実測値311.0.
脱水THF(20mL)中のLiAlH4(1.76g、46.4mmol)の懸濁液に、窒素下、0℃でTHF(30mL)中の4−ベンジルオキシ−6−フルオロ−ナフタレン−2−カルボン酸メチルエステル(4.80g、15.5mmol)の溶液を滴下した。反応混合物を、室温で3時間攪拌した。反応混合物を、HClの水溶液(50%;50mL)の添加を通してクエンチし、そして得られた混合物を、celiteベッドを通して濾過した。濾液を、酢酸エチルで抽出した(2×100mL)。合わせた有機抽出物を、水(30mL)、続いてブライン(30mL)で洗浄し、乾燥させ、そして真空下で濃縮して、(4−ベンジルオキシ−6−フルオロ−ナフタレン−2−イル)−メタノール(3.6g、82.2%)を得た。粗生成物は、さらに精製することなく次の工程に進むのに十分に純粋であった。
脱水THF(160mL)中のトリフェニルホスフィン(37.2g、142mmol)の溶液に、CCl4(50mL)を加えた。反応混合物を、10分間攪拌し、そして(4−ベンジルオキシ−6−フルオロ−ナフタレン−2−イル)−メタノール(20.0g、70.8mmol)を、固体として窒素下、室温で導入した。得られた溶液を、2時間還流した。THFを、減圧下で留去し、そして残留した物質を、水(100mL)で希釈し、そして酢酸エチルで抽出した(2×150mL)。合わせた有機抽出物を、水(50mL)、ブライン(50mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、そして真空下で濃縮した。得られた粗生成物を、シリカゲルカラムクロマトグラフィー(100〜200メッシュ、ヘキサン中5%酢酸エチル)で精製して、化合物1−ベンジルオキシ−3−クロロメチル−7−フルオロ−ナフタレン(19.3g、90.6%)を白色の固体として得た。
2:1 THF−MeOH(300mL)中の1−ベンジルオキシ−3−クロロメチル−7−フルオロ−ナフタレン(19.3g、64.2mmol)の攪拌溶液に、室温でK2CO3(9.75g、70.6mmol)及びPdCl2(PPh3)2(2.25g、3.2mmol)を加えた。溶液を脱気し(アルゴンで5分間パージすることによって)、そして一酸化炭素下(バルーン圧)、室温で一晩攪拌した。溶媒を、減圧下で留去し、そして得られた粗残留物を、水(150mL)で希釈し、そして酢酸エチルで抽出した(2×200mL)。合わせた有機抽出物を、水(50mL)、ブライン(50mL)で洗浄し、無水硫酸ナトリウムで乾燥させ、そして真空下で濃縮して、粗生成物を得た。シリカゲルクロマトグラフィー(100〜200メッシュ、ヘキサン中5%酢酸エチル)により、(4−ベンジルオキシ−6−フルオロ−ナフタレン−2−イル)−酢酸メチルエステル(18.5g、89%)を白色の固体として得た。MS C20H17FO3[(M+H)+]についての計算値:325, 実測値325.2.
メタノール(200mL)中の(4−ベンジルオキシ−6−フルオロ−ナフタレン−2−イル)−酢酸メチルエステル(18.5g、57.0mmol)の攪拌溶液に、10%パラジウム担持炭(2.78g)を加えた。得られた混合物を、水素のバルーン下で一晩激しく攪拌した。反応混合物を、celiteを通して濾過した。濾液を、真空下で濃縮して、粗生成物を得た。シリカゲルクロマトグラフィー(10%酢酸エチル−ヘキサン)により、(6−フルオロ−4−ヒドロキシ−ナフタレン−2−イル)−酢酸メチルエステル(10.24g、76%)を白色の固体として得た。MS C13H12FO3[(M+H)+]についての計算値:235, 実測値235.2.
氷浴中で0℃に冷却した、ジクロロメタン(20mL)中の(6−フルオロ−4−ヒドロキシ−ナフタレン−2−イル)−酢酸メチルエステル(350mg、1.5mmol)及びトリフルオロメタンスルホン酸無水物(506mg、1.8mmol)の混合物に、ピリジン(0.6mL)を滴下した。反応混合物を、0℃で2時間攪拌し、そして次に真空下で濃縮した。残留物を、酢酸エチル(50mL)に溶解した。得られた溶液を、水、1N 塩酸水溶液、水及びブラインで洗浄し、次に硫酸ナトリウムで乾燥させ、濾過し、そして真空下で濃縮した。残留物を、カラムクロマトグラフィー(ヘキサン中0〜30%酢酸エチル)により精製して、(6−フルオロ−4−トリフルオロメタンスルホニルオキシ−ナフタレン−2−イル)−酢酸メチルエステル(450mg、82%)を明黄色の固体として得た。
4−オキソ−ピペリジン−1−カルボン酸tert−ブチルエステル
1,4−ジオキサン(500mL)中の4−ピペリドン一水和物塩酸塩(Sigma-Aldrichから入手可能;50g、326mmol)の攪拌溶液に、水(170mL)中の重炭酸ナトリウム(60g、714mmol)の溶液を加え、続いて室温で二炭酸ジ−tert−ブチル(97mL、422mmol)を滴下した。反応混合物を、室温で30分間、そして次に70℃で一晩攪拌した。反応混合物を濃縮して、ジオキサンを除去し、そして残留混合物を、酢酸エチルで抽出した(3×500mL)。合わせた有機抽出物を、ブラインで洗浄し、そして次に減圧下で蒸発させて、4−オキソ−ピペリジン−1−カルボン酸tert−ブチルエステル(60.21g、93%)を白色の固体として得て、これを、さらに精製することなく次の工程で直接使用した。
n−ブチルリチウム(1.9M;30.6mL、58mmol)を、窒素下、約−75℃でTHF(60mL)中のジイソプロピルアミン(4.6mL、32.6mmol)の攪拌溶液に滴下した。反応温度を、約0℃にし、そして混合物を、この温度で1時間攪拌した。反応混合物を、−75℃に冷却し、そしてTHF中の4−オキソ−ピペリジン−1−カルボン酸tert−ブチルエステル(10.0g、50.2mmol)の溶液を加えた。混合物を、−75℃で1時間攪拌し、そして次にN−フェニルビス(トリフルオロメタンスルホンイミド)(19.7g、55.1mmol)を加えた。混合物を、室温まで温まるにまかせ、そして一晩攪拌した。反応混合物を、酢酸エチルで抽出し、そして抽出物を、水で洗浄した。有機抽出物を、Na2SO4で乾燥させ、濾過し、蒸発させて、そしてアルミナカラムのクロマトグラフィーにより精製して、4−トリフルオロメタンスルホニルオキシ−3,6−ジヒドロ−2H−ピリジン−1−カルボン酸tert−ブチルエステル(8g、48%)を得た。1H NMR (300 MHz, CDCl3) δ ppm: 5.75 (s, 1 H), 4.03 (s, 2 H), 3.60 - 3.63 (m, 2 H), 1.46 (s, 9 H); MS C11H17F3NO5S[(M+H)+]の計算値332, 実測値332.5.
4−トリフルオロメタンスルホニルオキシ−3,6−ジヒドロ−2H−ピリジン−1−カルボン酸tert−ブチルエステル(先に記載したようにして調製することもできる;900mg、2.7mmol)、ビス(ピナコラト)ジボロン(Sigma-Aldrichから入手可能;760mg、3mmol)及び酢酸カリウム(799mg、8.1mmol)及びジオキサン(15mL)の混合物を、アルゴンで30分間脱気した。ジクロロメタン(Sigma-Aldrichから入手可能;66.5mg、0.08mmol)との複合体、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロ−パラジウム(II)及び1,1’−ビス(ジフェニルホスフィノ)フェロセン(45mg、0.08mmol)を加え、そして混合物を、アルゴンで10分間脱気した。混合物を、80〜90℃で一晩加熱し、室温まで冷えるにまかせ、そして次にceliteを通して濾過した。濾液を、減圧下で蒸発させて、そして残留物を、カラムクロマトグラフィーにより精製して、4−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−3,6−ジヒドロ−2H−ピリジン−1−カルボン酸tert−ブチルエステル(500mg、60%)を得た。1H NMR (300 MHz, DMSO-d6) δ ppm: 6.38 (br s, 1 H), 3.82 - 3.90 (m, 2 H), 2.05 - 2.10 (m, 2 H), 1.40 (s, 9 H), 1.20 (s, 12 H); MS C16H29BNO4 [(M+H)+]の計算値310, 実測値310.2.
4−(7−フルオロ−3−メトキシカルボニルメチル−ナフタレン−1−イル)−3,6−ジヒドロ−2H−ピリジン−1−カルボン酸tert−ブチルエステル
無水炭酸カリウム(0.40g、2.9mmol)を、圧力管内の、脱水DMF−MeOH(4:1、5mL)中の化合物(6−フルオロ−4−トリフルオロメタンスルホニルオキシ−ナフタレン−2−イル)−酢酸メチルエステル(先に記載したようにして調製することができる;0.37g、1mmol)及び4−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−3,6−ジヒドロ−2H−ピリジン−1−カルボン酸tert−ブチルエステル(0.30g、0.97mmol)の攪拌溶液に加えた。混合物を、アルゴンで15分間バブリングすることによって脱酸素した。ジクロロメタン(Sigma-Aldrichから入手可能;80mg、0.1mmol)との複合体、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)を、アルゴン下で加えた。混合物を、12時間加熱還流し、そして次に冷却した。酢酸エチル(20mL)を加え、そして混合物を、celiteパッドを通して濾過した。Celiteを、酢酸エチルで洗浄し、そして合わせた濾液を、減圧下で濃縮した。残留物を、溶離剤として5%酢酸エチル/ヘキサンを使用したシリカゲルカラムクロマトグラフィー(100〜200メッシュ)により精製して、4−(7−フルオロ−3−メトキシカルボニルメチル−ナフタレン−1−イル)−3,6−ジヒドロ−2H−ピリジン−1−カルボン酸tert−ブチルエステル(0.21g、54%)を得た。1H NMR (300 MHz, DMSO-d6) δ ppm: 7.78 (dd, J = 6.6, 4.4 Hz, 1 H), 7.64 (s, 1 H), 7.51 (dd, J = 7.1, 1.1 Hz, 1 H), 7.21 - 7.26 (m, 2 H), 5.77 (s, 1 H), 4.12 (s, 2 H), 3.70 - 3.76 (m, 7 H), 2.45 - 2.50 (m, 2 H), 1.52 (m, 9 H); MS C23H27FNO4[(M+H)+]の計算値 400, 実測値400.2.
10%パラジウム担持炭(0.09g)を、Parr振盪容器内で、エタノール(30mL)中の4−(7−フルオロ−3−メトキシカルボニルメチル−ナフタレン−1−イル)−3,6−ジヒドロ−2H−ピリジン−1−カルボン酸tert−ブチルエステル(これを先に記載したように調製することもできる;0.64g、1.6mmol)の脱酸素溶液に加えた。混合物を、60psiで16時間水素化し、そして次にceliteのパッドを通して濾過した。Celiteを、エタノール(2×5mL)で洗浄し、そして合わせた濾液を、減圧下で濃縮した。残留物を、溶離剤として20%酢酸エチル/ヘキサンを使用したシリカゲルカラムクロマトグラフィー(100〜200メッシュ)により精製して、4−(7−フルオロ−3−メトキシカルボニルメチル−ナフタレン−1−イル)−ピペリジン−1−カルボン酸tert−ブチルエステル(0.40g、62%)を粘着性の無色の固体として得た。MS C23H29FNO4[(M+H)+]の計算値402, 実測値402.4.
トリフルオロ酢酸(2.9mL、38mmol)を、窒素下、脱水CH2Cl2(25mL)中の4−(7−フルオロ−3−メトキシカルボニルメチル−ナフタレン−1−イル)−ピペリジン−1−カルボン酸tert−ブチルエステル(これを先に記載したように調製することもできる;1.05g、2.6mmol)の攪拌溶液に0℃で加えた。溶液を、室温で16時間攪拌し、そして次に溶媒を、減圧下で蒸発させた。残留物を、ジエチルエーテルでトリチュレートし(3×5mL)、そして真空下で乾燥させて、(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(0.80g、74%)を白色の固体として得た。MS C18H21FNO2[(M+H)+]の計算値302, 実測値302.4.
2−[1−(4−フルオロ−フェニル)−メタ−(E)−イリデン]−3−メチル−コハク酸
トルエン(150mL)中の水素化ナトリウム(パラフィン油中60%、31g、775mmol)の懸濁液に、トルエン(150mL)中の4−フルオロベンズアルデヒド(30g、242mmol)及びメチルコハク酸ジメチル(58g、362mmol)の溶液を窒素下、0℃で1時間かけて加えた。反応を、室温でのメタノールの滴下によって開始し、そして室温で2時間攪拌した。反応を、0℃での2.0N NaOH水溶液(300mL)のゆっくりとした添加によってクエンチした。得られた混合物を、110℃で4時間攪拌した。次に混合物を、室温に冷却し、そして水層を、水(300mL)で希釈し、そしてEt2Oで洗浄した(2×300mL)。水相を、氷水浴内で冷却した。濃HClの添加に続いて、酢酸エチルで抽出した(2×100mL)。合わせた有機抽出物を、水(50mL)、続いてブライン(50mL)で洗浄した。有機相を、無水硫酸ナトリウムで乾燥させ、そして濃縮した。残留物を、酢酸エチル−ヘキサンから結晶化して、2−[1−(4−フルオロ−フェニル)−メタ−(E)−イリデン]−3−メチルコハク酸を得た。先の手順を、別量の4−フルオロベンズアルデヒド(30g、242mmol)を使用して繰返した。2つの反応の生成物を合わせて、2−[1−(4−フルオロ−フェニル)−メタ−(E)−イリデン]−3−メチルコハク酸を淡黄色の固体(28g、全体で24%)として得た。MS C12H12FO4[(M+H)+]の計算値239, 実測値239.2.
トリフルオロメタンスルホン酸(140mL)中の2−[1−(4−フルオロ−フェニル)−メタ−(E)−イリデン]−3−メチルコハク酸(28g、118mmol)の溶液を、室温で16時間攪拌した。得られた混合物を、連続撹拌しながら氷冷水に注意深く注いで、固体沈殿物を得て、これを濾過し、水で洗浄し、そして真空下で乾燥させて、6−フルオロ−4−ヒドロキシ−3−メチル−ナフタレン−2−カルボン酸(28g、>100%粗)を黄色の固体として得た。この粗生成物を、さらに精製することなく次の工程で使用した。MS C12H8FO3[(M-H)-]の計算値219, 実測値218.9.
MeOH(240mL)中の粗6−フルオロ−4−ヒドロキシ−3−メチル−ナフタレン−2−カルボン酸(28g、約118mmol)の0℃溶液に、濃硫酸(18.9mL、382mmol)を滴下した。次に反応混合物を、室温に温め、そして一晩還流した。この後、メタノールを、減圧下で留去し、そして粗混合物を、酢酸エチルで希釈した。この溶液を、飽和NaHCO3水溶液で洗浄した。有機相を、Na2SO4で乾燥させ、濾過し、そして減圧下で濃縮して、粗生成物を得た。シリカゲルカラムクロマトグラフィー(6%酢酸エチル−ヘキサン)により、6−フルオロ−4−ヒドロキシ−3−メチル−ナフタレン−2−カルボン酸メチルエステル(14.8g、54%)を明黄色の固体として得た。MS C13H12FO3[(M+H)+]の計算値235, 実測値235.2.
脱水DMF(250mL)中の6−フルオロ−4−ヒドロキシ−3−メチル−ナフタレン−2−カルボン酸メチルエステル(21.7g、92.7mmol)の溶液に、窒素下、室温でK2CO3(17.9g、130mmol)、臭化ベンジル(13mL、111mmol)及びBu4NI(0.250g)を加えた。反応混合物を、室温で3時間攪拌した。反応混合物を、水で希釈し、そして酢酸エチルで抽出した。合わせた有機抽出物を、水、ブラインで洗浄し、無水硫酸ナトリウムで乾燥させ、そして濃縮して、粗生成物を得て、これを、シリカゲルカラムクロマトグラフィー(2〜5%酢酸エチル−ヘキサン)を使用して精製して、4−ベンジルオキシ−6−フルオロ−3−メチル−ナフタレン−2−カルボン酸メチルエステル(25.4g、84%)をオフホワイトの固体として得た。MS C20H18FO3[(M+H)+] の計算値325, 実測値 325.1.
脱水THF(120mL)中のLiAlH4(8.8g、235mmol)の懸濁液に、窒素下、0℃でTHF(180mL)中の4−ベンジルオキシ−6−フルオロ−3−メチル−ナフタレン−2−カルボン酸メチルエステル(25.4g、78.4mmol)の溶液を滴下した。反応混合物を、室温で3時間攪拌した。この後、反応混合物を、0℃に冷却し、そして冷水(10mL)、続いて15% NaOH溶液(10mL)そして追加の水の添加によって、注意深くクエンチした。得られた溶液を、1時間攪拌し、次に、焼結ガラス漏斗を通して濾過した。フィルターパッドを、THF(50mL)で洗浄した。合わせた濾液を、Na2SO4で乾燥させ、濾過し、そして濃縮して、(4−ベンジルオキシ−6−フルオロ−3−メチル−ナフタレン−2−イル)−メタノール(21.5g、92%、粗)を白色の固体として得た。粗生成物を、さらに精製することなく次の工程で使用した。MS C19H16FO2[(M-H)-]の計算値295, 実測値294.9.
脱水THF(190mL)中のトリフェニルホスフィン(41.6g、159mmol)の溶液に、CCl4(59mL)を加えた。反応混合物を、10分間攪拌し、そして(4−ベンジルオキシ−6−フルオロ−3−メチル−ナフタレン−2−イル)−メタノール(21.5g、72.6mmol)を、窒素下、室温で固体として導入した。得られた溶液を、2時間還流した。溶媒を、減圧下で留去し、そして残留物を、水で希釈した。得られた混合物を、酢酸エチルで2回抽出した。合わせた有機抽出物を、水及びブラインで洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、そして濃縮した。シリカゲルカラムクロマトグラフィー(100〜200メッシュ、ヘキサン中5%酢酸エチル)により、1−ベンジルオキシ−3−クロロメチル−7−フルオロ−2−メチル−ナフタレン(18.5g、81%)をオフホワイトの固体として得た。
THF−メタノール混合物(2:3;500mL)中の1−ベンジルオキシ−3−クロロメチル−7−フルオロ−2−メチル−ナフタレン(18.5g、58.9mmol)の攪拌溶液に、K2CO3(8.94g、64.7mmol)及びPdCl2(PPh3)2(2.06g、2.96mmol)を室温で加えた。溶液を、アルゴンで5分間パージすることによって脱気した。反応混合物を、一酸化炭素のバルーン下、室温で一晩攪拌した。この後、反応の進行を、TLCによりモニタリングした(ヘキサン中5%酢酸エチル)。反応混合物を濃縮し、そして得られた粗残留物を、水で希釈し、そして酢酸エチルで抽出した。合わせた有機抽出物を、水及びブラインで洗浄し、無水硫酸ナトリウムで乾燥させ、濾過し、そして濃縮して、粗生成物を得た。シリカゲルクロマトグラフィー(100〜200メッシュ、5%酢酸エチル−ヘキサン)により、(4−ベンジルオキシ−6−フルオロ−3−メチル−ナフタレン−2−イル)−酢酸メチルエステル(15.8g、80%)を淡黄色の固体として得た。MS C21H20FO3[(M+H)+]の計算値339, 実測値339.0.
MeOH(150mL)中の(4−ベンジルオキシ−6−フルオロ−3−メチル−ナフタレン−2−イル)−酢酸メチルエステル(15.8g、46.7mmol)の攪拌溶液に、10%パラジウム担持炭(2.4g)を加えた。得られた混合物を、水素のバルーン下、一晩激しく攪拌した。反応混合物を、celiteを通して濾過した。濾液を濃縮して、粗生成物を得て、これを、シリカゲルクロマトグラフィー(ヘキサン中10%酢酸エチル)により精製して、(6−フルオロ−4−ヒドロキシ−3−メチル−ナフタレン−2−イル)−酢酸メチルエステル(9.5g、82%)を白色の固体として得た。MS C14H13FO3[(M+H)+]の計算値249, 実測値249.1.
塩化メチレン(500mL)中の(6−フルオロ−4−ヒドロキシ−3−メチル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;12.2g、49.1mmol)の明黄色の溶液を、氷−アセトン浴を使用して0℃に冷却した。ピリジン(5.17mL、63.9mmol)を加え、そして次にトリフルオロメタンスルホン酸無水物(20.8g、73.7mmol)を、40分間かけて冷溶液に滴下した。得られた明黄色の溶液を、室温に温める前に、0℃で2時間攪拌した。反応混合物を、室温でさらに30分間攪拌した。混合物を、水(300mL)でクエンチし、そして2つの層を分離した。水層を、ジクロロメタン(200mL)で抽出した。合わせた有機層を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、そして濃縮して、粗生成物を明黄色の固体として得た。粗生成物を、加熱しながらジクロロメタン(約50mL)に溶解し、そして次に混合物を、ヘキサン(約100mL)で希釈した。一部の溶媒を、ヒートガンで加熱することによって除去した。得られた明褐色の溶液を、冷凍庫内に15時間保管した。白色の固体が沈殿し、これを濾過によって回収し、そしてヘキサンで洗浄した。風乾後、(6−フルオロ−3−メチル−4−トリフルオロメタンスルホニルオキシ−ナフタレン−2−イル)−酢酸メチルエステル(14.32g、77%)を単離した。1H NMR (400 MHz, DMSO-d6) δ ppm 7.83 (dd, J=9.03, 5.52 Hz, 1 H), 7.75 (s, 1 H), 7.65 (dd, J=10.29, 2.51 Hz, 1 H), 7.31 (td, J=8.60, 2.38 Hz, 1 H), 3.85 (s, 2 H), 3.74 (s, 3 H).
(4−(7−フルオロ−3−メトキシカルボニルメチル−2−メチル−ナフタレン−1−イル)−ピペリジン−1−カルボン酸tert−ブチルエステル
100mL容量の追加の漏斗、ゴム製セプタム及びL字型アダプターを備えた、オーブン乾燥させた三口の50mL容量丸底フラスコに、窒素下で、亜鉛末(1.31g、20.0mmol)及び塩化リチウム(848mg、20.0mmol)を入れた。固体を攪拌し、そして混合物を、高真空下、171℃で1.5時間加熱した。フラスコを、室温に冷却し、窒素を充填し戻し、そして脱水テトラヒドロフラン(2.0mL)を、灰色の混合物に加えた。反応フラスコに温度計を装備した。反応混合物に、1,2−ジブロモエタン(376mg、2.00mmol)を加え、そして懸濁液を、ガス発生及び起泡に至るまで、ヒートガンで穏やかに加熱した。ガス発生が完了すると、混合物を、50℃まで冷えるにまかせ、そして再度加熱した。この方法を3回繰り返して、確実に亜鉛末を完全に活性化させた。活性亜鉛末を、クロロトリメチルシラン(217mg、2.00mmol)で処理し、そして懸濁液を、室温で15分間攪拌した。得られた混合物を、室温でテトラヒドロフラン(5.0mL)中の4−ヨードピペリジン−1−カルボン酸tert−ブチルエステル(3.112g、10.0mmol)の溶液で処理した。添加後、反応混合物を、ヒートガンで加熱して、反応を開始し、そして温度が68℃に上昇した。50℃に冷却した後、それを60℃(油浴温度)に再度加熱し、そして1.5時間攪拌した。反応混合物を、室温に冷却し、そしてテトラヒドロフラン(5mL)で希釈し、そして撹拌を停止して、過剰な亜鉛末を3時間かけて沈降させた。
ジクロロメタン(10mL)中の(4−(7−フルオロ−3−メトキシカルボニルメチル−2−メチル−ナフタレン−1−イル)−ピペリジン−1−カルボン酸tert−ブチルエステル(200mg、0.48mmol)の黄色の溶液に、窒素下、室温で過剰量のトリフルオロ酢酸(1.1g、742μL、9.6mmol)を加えた。得られた暗褐色の溶液を、窒素下、室温で2時間攪拌した。溶媒を、真空下で除去し、そして残留物(約270mg)を、フラッシュクロマトグラフィー(40g ISCOカラム、0〜100%酢酸エチル/ヘキサン、次に20% MeOH/ジクロロメタン)により精製して、(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩を明黄色の固体(110mg、53%)として得た。MS C19H23FNO2[(M+H)+]の計算値316, 実測値316.1.
飽和重炭酸ナトリウム水溶液(10mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(50mg、0.116mmol)の混合物を、室温で数分間攪拌した。得られた溶液を、酢酸エチルで抽出した(2×15mL)。合わせた有機層を、無水Na2SO4で乾燥させ、濾過し、そして濃縮して、(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(36mg、97%)を得た。
実施例1
[4−(1−ベンゼンスルホニル−ピペリジン−4−イル)−6−フルオロ−ナフタレン−2−イル]−酢酸
[4−(1−ベンゼンスルホニル−ピペリジン−4−イル)−6−フルオロ−ナフタレン−2−イル]−酢酸メチルエステル
ジイソプロピルエチルアミン(90μL、0.52mmol)を、テトラヒドロフラン(6mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;100mg、0.24mmol)の溶液に加えた。塩化フェニルスルホニル(Sigma-Aldrichから入手可能;43μL、0.34mmol)を加え、そして混合物を、室温で一晩攪拌した。溶媒を、減圧下で蒸発させた。水及び酢酸エチルを加え、そして有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、シリカゲルカラムクロマトグラフィー(100〜200メッシュ)により精製して、[4−(1−ベンゼンスルホニル−ピペリジン−4−イル)−6−フルオロ−ナフタレン−2−イル]−酢酸メチルエステル(64mg、60%)を得た。MS C24H25FNO4S [(M+H)+]の計算値:442, 実測値 442.2.
THF(8mL)中の[4−(1−ベンゼンスルホニル−ピペリジン−4−イル)−6−フルオロ−ナフタレン−2−イル]−酢酸メチルエステル(90mg、0.2mmol)の攪拌溶液に、水(2mL)中の水酸化リチウム一水和物(26mg、0.6mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、[4−(1−ベンゼンスルホニル−ピペリジン−4−イル)−6−フルオロ−ナフタレン−2−イル]−酢酸(35mg、40%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.36 (br s, 1 H), 7.92 - 7.98 (m, 1 H), 7.88 (d, J=11.2 Hz, 1 H), 7.81 (d, J=7.8 Hz, 2 H), 7.76 (d, J=6.8 Hz, 1 H), 7.65 - 7.73 (m, 3 H), 7.33 - 7.42 (m, 2 H), 3.82 (d, J=11.7 Hz, 2 H), 3.71 (s, 2 H), 1.90 (d, J=11.2 Hz, 2 H), 1.69 - 1.82 (m, 2 H); MS C23H23FNO4S [(M-H)-]の計算値:426, 実測値426.2.
{6−フルオロ−4−[1−(トルエン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
{6−フルオロ−4−[1−(トルエン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化p−トルエンスルホニル(Sigma-Aldrichから入手可能;103mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システム(Biotage purification system)を使用し、12%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{6−フルオロ−4−[1−(トルエン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(76mg、46%)を白色の固体として得た。MS C24H25FNO4S [(M+H)+]の計算値:456, 実測値456.4.
THF(4mL)中の{6−フルオロ−4−[1−(トルエン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(70mg、0.2mmol)の攪拌溶液に、水(1mL)中の水酸化リチウム一水和物(32mg、0.76mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、[{6−フルオロ−4−[1−(トルエン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(52mg、77%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.37 (br s, 1 H), 7.94 (dd, J=8.8, 6.4 Hz, 1 H), 7.88 (d, J=10.3 Hz, 1 H), 7.66 - 7.73 (m, 3 H), 7.50 (d, J=8.3 Hz, 2 H), 7.33 - 7.42 (m, 2 H), 3.79 (d, J=12.2 Hz, 2 H), 3.71 (s, 2 H), 1.90 (d, J=11.7 Hz, 2 H), 1.69 - 1.82 (m, 1 H); MS C24H25FNO4S [(M-H)-]の計算値:440, 実測値440.4.
{6−フルオロ−4−[1−(ピリジン−2−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
塩化ピリジン−2−スルホニル
2−メルカプトピリジン(Sigma-Aldrichから入手可能;0.25g、2.25mmol)を、H2SO4(8mL)に加え、そして混合物を、約−5℃に冷却した。次亜塩素酸ナトリウム(10%溶液;17mL)を加え、そして反応混合物を、約0℃で30分間攪拌した。水(10mL)を加え、そして混合物を、CH2Cl2で抽出した(3×20mL)。合わせた有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させて、粗塩化ピリジン−2−スルホニル(0.23g、58%)を得て、これを、さらに精製することなく次の工程で直接使用した。
ジイソプロピルエチルアミン(0.4mL、2.3mmol)を、0℃でCH2Cl2(6mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;100mg、0.24mmol)の溶液に加えた。溶液を、室温で30分間攪拌し、そして次に、CH2Cl2(1mL)中の塩化ピリジン−2−スルホニル(これを先に記載したように調製することもできる;213mg、1.2mmol)の溶液を加えた。混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、25%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{6−フルオロ−4−[1−(ピリジン−2−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(32mg、30%)を白色の固体として得た。MS C23H24FN2O4S [(M+H)+]の計算値:443, 実測値443.3.
THF(4mL)中の{6−フルオロ−4−[1−(ピリジン−2−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(30mg、0.07mmol)の攪拌溶液に、水(1mL)中の水酸化リチウム一水和物(14mg、0.33mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{6−フルオロ−4−[1−(ピリジン−2−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(20mg、69%)を褐色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.38 (br s, 1 H), 8.84 (d, J=3.9 Hz, 1 H) 8.12 - 8.20 (m, 1 H) 7.85 - 8.02 (m, 3 H) 7.76 (dd, J=7.6, 4.6 Hz, 1 H) 7.69 (s, 1 H) 7.39 (t, J=8.6 Hz, 1 H) 7.33 (s, 1 H) 3.91 (d, J=11.7 Hz, 2 H) 3.72 (s, 2 H) 2.99 (t, J=11.7 Hz, 3 H) 1.89 (d, J=12.7 Hz, 2 H) 1.63 - 1.78 (m, 2 H); MS C22H22FN2O4S [(M+H)+]の計算値:429, 実測値429.2.
{6−フルオロ−4−[1−(ピリジン−3−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
塩化ピリジン−3−スルホニル
五塩化リン(1.57g、7.5mmol)を、トルエン(5mL)中のピリジン−3−スルホン酸(1.00g、6.3mmol)の攪拌溶液に加えた。混合物を、16時間加熱還流し、そして次に室温に冷却した。混合物を、celiteを通して濾過し、そしてceliteを、ベンゼンで洗浄した(2×10mL)。合わせた濾液を、減圧下で濃縮して、粗塩化ピリジン−3−スルホニル(0.70g、100%)を得て、これを、さらに精製することなく次の工程で直接使用した。
ジイソプロピルエチルアミン(0.3mL、1.7mmol)を、0℃でCH2Cl2(6mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加えた。溶液を、室温で30分間攪拌し、そして次に、CH2Cl2(1mL)中の塩化ピリジン−3−スルホニル(これを先に記載したように調製することもできる;320mg、1.8mmol)の溶液を加えた。混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、30%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{6−フルオロ−4−[1−(ピリジン−3−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(85mg、53%)を粘着性の白色の固体として得た。MS C23H24FN2O4S [(M+H)+]の計算値:443, 実測値443.2.
THF(6mL)中の{6−フルオロ−4−[1−(ピリジン−3−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(85mg、0.19mmol)の攪拌溶液に、水(1.5mL)中の水酸化リチウム一水和物(40mg、0.95mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{6−フルオロ−4−[1−(ピリジン−3−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(60mg、73%)を白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.38 (br s, 1 H), 8.87 - 9.04 (m, 1 H), 8.21 (d, J=7.1 Hz, 1 H), 7.77 - 7.99 (m, 2 H), 7.68 (br. s., 2 H), 7.35 (br. s., 2 H), 3.91 (d, J=11.6 Hz, 2 H), 3.71 (s, 2 H), 1.96 (d, J=12.1 Hz, 3 H), 1.78 (d, J=11.6 Hz, 3 H); MS C22H22FN2O4S [(M+H)+]の計算値:429, 実測値429.2.
{6−フルオロ−4−[1−(ピリジン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
塩化ピリジン−4−スルホニル
4−メルカプトピリジン(Sigma-Aldrichから入手可能;1.10g、1mmol)を、H2SO4(7.5mL)に加え、そして混合物を、約−10℃に冷却した。塩素ガスを、溶液に通して45分間バブリングし、そして次に炭酸カルシウム(1.0g)をゆっくりと加えた。反応混合物を、CHCl3(20mL)に加え、そして混合物を、−10℃に冷却した。追加の炭酸カルシウム(7.0g)を少量ずつ加えた。有機層をデカントし、そして残留物を、CHCl3で洗浄した(2×20mL)。合わせた有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させて、粗塩化ピリジン−4−スルホニルを得て、これを、さらに精製することなく次の工程で直接使用した。
ジイソプロピルエチルアミン(0.3mL、1.7mmol)を、0℃でCH2Cl2(6mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;100mg、0.24mmol)の溶液に加えた。溶液を、室温で30分間攪拌し、そして次にCH2Cl2(1mL)中の塩化ピリジン−4−スルホニル(これを先に記載したように調製することもできる;213mg、1.2mmol)の溶液を加えた。混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。残留物を、シリカゲルクロマトグラフィーにより精製して、{6−フルオロ−4−[1−(ピリジン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(45mg、42%)を橙色の液体として得た。MS C23H24FN2O4S [(M+H)+]の計算値:443, 実測値443.4.
THF(4mL)中の{6−フルオロ−4−[1−(ピリジン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(これを先に記載したように調製することもできる;44.5mg、0.1mmol)の攪拌溶液に、水(1mL)中の水酸化リチウム一水和物(20mg、0.48mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{6−フルオロ−4−[1−(ピリジン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(12mg)を黄色の固体として得た。LCMS分析が、この物質の純度が42%であることを示した。MS C22H22FN2O4S [(M+H)+]の計算値:429, 実測値429.2.
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化2−クロロベンゼンスルホニル(Sigma-Aldrichから入手可能;114mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、10%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(94mg、55%)を白色の固体として得た。MS C24H24ClFNO4S [(M+H)+]の計算値:476, 実測値476.2.
THF(4mL)中の{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(90mg、0.19mmol)の攪拌溶液に、水(1mL)中の水酸化リチウム一水和物(40mg、0.95mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸(40mg、46%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.36 (br. s., 1 H), 8.03 (d, J=7.8 Hz, 1 H), 7.88 - 7.99 (m, 2 H), 7.67 - 7.79 (m, 3 H), 7.61 (t, J=8.8 Hz, 1 H), 7.40 (dt, J=8.7, 1.7 Hz, 1 H), 7.35 (s, 1 H), 3.88 (d, J=12.2 Hz, 2 H), 3.72 (s, 2 H), 3.46 (t, J=11.7 Hz, 1 H), 3.04 (t, J=11.7 Hz, 2 H), 1.88 - 1.96 (m, 2 H), 1.64 - 1.79 (m, 2 H); MS C23H22ClFNO4S [(M-H)-]の計算値:462, 実測値462.2.
{4−[1−(3−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸
{4−[1−(3−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.3mL、1.7mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化3−クロロベンゼンスルホニル(Sigma-Aldrichから入手可能;110mg、0.52mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、10%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{4−[1−(3−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(97mg、56%)を白色の固体として得た。MS C24H24ClFNO4S [(M+H)+] の計算値: 476, 実測値 476.4.
THF(4mL)中の{4−[1−(3−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(90mg、0.19mmol)の攪拌溶液に、水(1mL)中の水酸化リチウム一水和物(40mg、0.95mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{4−[1−(3−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸(57mg、65%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.34 (br. s., 1 H), 7.95 (dd, J=9.0, 6.1 Hz, 1 H), 7.84 - 7.92 (m, 2 H), 7.71 - 7.83 (m, 3 H), 7.69 (s, 1 H), 7.34 - 7.42 (m, 2 H), 3.83 (d, J=11.2 Hz, 2 H), 3.27 - 3.34 (m, 7 H), 2.63 (t, J=11.2 Hz, 2 H), 1.88 - 1.96 (m, 2 H), 1.69 - 1.83 (m, 2 H); MS C23H22ClFNO4S [(M-H)+]の計算値:462, 実測値462.2.
{4−[1−(4−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸
{4−[1−(4−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化4−クロロベンゼンスルホニル(Sigma-Aldrichから入手可能;114mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、10%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{4−[1−(4−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(86mg、50%)を白色の固体として得た。MS C24H24ClFNO4S [(M+H)+]の計算値:476, 実測値476.4.
THF(4mL)中の{4−[1−(4−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(80mg、0.17mmol)の攪拌溶液に、水(1mL)中の水酸化リチウム一水和物(35mg、0.83mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{4−[1−(4−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸(57mg、65%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1 H), 7.95 (dd, J=9.0, 6.1 Hz, 1 H), 7.88 (d, J=10.3 Hz, 1 H), 7.76 - 7.85 (m, 4 H), 7.68 (s, 1 H), 7.34 - 7.41 (m, 2 H), 3.80 (d, J=11.2 Hz, 2 H), 3.71 (s, 2 H), 2.59 (t, J=11.2 Hz, 2 H), 1.87 - 1.94 (m, 3 H), 1.73 - 1.82 (m, 2 H); MS C23H20ClFNO4S [(M-H)-]の計算値:460, 実測値460.3.
{6−フルオロ−4−[1−(3−メタンスルホニル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
{6−フルオロ−4−[1−(3−メタンスルホニル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化3−メチルスルホニルベンゼンスルホニル(Matrix Scientificから入手可能;138mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、20%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{6−フルオロ−4−[1−(3−メタンスルホニル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(120mg、64%)をオフホワイトの固体として得た。MS C25H27FNO6S2 [(M+H)+]の計算値:520, 実測値520.2.
THF(6mL)中の{6−フルオロ−4−[1−(3−メタンスルホニル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(115mg、0.22mmol)の攪拌溶液に、水(1.5mL)中の水酸化リチウム一水和物(46mg、1.1mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{6−フルオロ−4−[1−(3−メタンスルホニル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(90mg、80%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.35 (br s, 1 H), 8.34 (d, J=7.8 Hz, 1 H), 8.23 (s, 1 H), 8.18 (d, J=7.8 Hz, 1 H), 7.98 - 8.04 (m, 1 H), 7.95 (dd, J=9.3, 6.4 Hz, 1 H), 7.86 (d, J=12.7 Hz, 1 H), 7.69 (s, 1 H), 7.35 - 7.41 (m, 2 H), 3.87 (d, J=10.3 Hz, 2 H), 3.71 (s, 2 H), 3.39 (s, 3 H), 2.66 (d, J=10.3 Hz, 2 H), 1.89 - 1.96 (m, 2 H), 1.73 - 1.83 (m, 1 H); MS C24H25FNO6S2[(M-H)-]の計算値:506, 実測値506.2.
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化2,5−ジクロロベンゼンスルホニル(Sigma-Aldrichから入手可能;133mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、10%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(126mg、68%)を粘着性のオフホワイトの固体として得た。MS C24H23Cl2FNO4S [(M+H)+]の計算値:510, 実測値510.2.
THF(6mL)中の{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(120mg、0.24mmol)の攪拌溶液に、水(1.5mL)中の水酸化リチウム一水和物(50mg、1.2mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸(105mg、90%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.38 (br s, 1 H), 7.96 - 8.01 (m, 2 H), 7.94 (d, J=4.4 Hz, 1 H), 7.80 - 7.83 (m, 2 H), 7.69 (s, 1 H), 7.41 (t, J=8.8 Hz, 1 H), 7.36 (s, 1 H), 3.91 (d, J=12.2 Hz, 2 H), 3.72 (s, 2 H), 3.08 (t, J=11.5 Hz, 2 H), 1.90-1.96 (m, 2 H), 1.67 - 1.77 (m, 2 H); MS C23H19Cl2FNO4S [(M-H)-]の計算値:494, 実測値494.3.
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化2,4−ジクロロベンゼンスルホニル(Sigma-Aldrichから入手可能;133mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、10%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(120mg、65%)を粘着性のオフホワイトの固体として得た。MS C24H23Cl2FNO4S [(M+H)+]の計算値:510, 実測値510.2.
THF(6mL)中の{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(115mg、0.23mmol)の攪拌溶液に、水(1.5mL)中の水酸化リチウム一水和物(50mg、1.2mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸(100mg、89%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.35 (br s, 1 H), 8.02 (d, J=8.3 Hz, 1 H), 7.96 - 8.00 (m, 2 H), 7.87 - 7.95 (m, 1 H), 7.68 - 7.73 (m, 2 H), 7.34 - 7.44 (m, 2 H), 3.87 (d, J=12.2 Hz, 2 H), 3.72 (s, 2 H), 3.04 (t, J=11.5 Hz, 2 H), 1.89 - 1.96 (m, 2 H), 1.67 - 1.78 (m, 2 H); MS C23H19Cl2FNO4S [(M-H)-]の計算値:494, 実測値494.3.
{4−[1−(3,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸
{4−[1−(3,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化3,5−ジクロロベンゼンスルホニル(Sigma-Aldrichから入手可能;133mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、10%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{4−[1−(3,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(78mg、42%)を粘着性のオフホワイトの固体として得た。MS C24H23Cl2FNO4S [(M+H)+]の計算値:510, 実測値510.2.
THF(6mL)中の{4−[1−(3,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(75mg、0.15mmol)の攪拌溶液に、水(1.5mL)中の水酸化リチウム一水和物(30mg、0.7mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{4−[1−(3,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸(60mg、82%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.35 (br s, 1 H), 8.10 (s, 1 H), 7.86 - 8.01 (m, 2 H), 7.82 (s, 2 H), 7.69 (s, 1 H), 7.31 - 7.44 (m, 2 H), 3.85 (d, J=12.2 Hz, 2 H), 3.72 (s, 2 H), 2.62 - 2.73 (m, 3 H), 1.93 (d, J=12.2 Hz, 2 H), 1.69 - 1.83 (m, 2 H); MS C23H19Cl2FNO4S [(M-H)-]の計算値:494, 実測値494.2.
{4−[1−(3,5−ビス−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸
{4−[1−(3,5−ビス−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル
ジイソプロピルエチルアミン(0.6mL、3.4mmol)を、0℃でテトラヒドロフラン(5mL)中の(6−フルオロ−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;150mg、0.36mmol)の溶液に加え、そして混合物を、室温で30分間攪拌した。塩化3,5−ビス(トリフルオロメチル)ベンゼンスルホニル(Sigma-Aldrichから入手可能;169mg、0.54mmol)を加え、そして混合物を、室温で16時間攪拌した。溶媒を、減圧下で蒸発させた。酢酸エチルを加え、そして混合物を、NaHCO3水溶液(5mL)及び水(5mL)で洗浄した。有機層を、Na2SO4で乾燥させ、濾過し、そして減圧下で蒸発させた。残留物を、Biotage精製システムを使用し、10%酢酸エチル/ヘキサンで溶離するシリカゲルクロマトグラフィーにより精製して、{4−[1−(3,5−ビス−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(87mg、42%)をオフホワイトの固体として得た。MS C26H23F7NO4S [(M+H)+]の計算値:578, 実測値578.4.
THF(4mL)中の{4−[1−(3,5−ビス−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸メチルエステル(80mg、0.14mmol)の攪拌溶液に、水(1mL)中の水酸化リチウム一水和物(30mg、0.7mmol)の溶液を加え、そして反応混合物を、室温で24時間攪拌した。反応混合物を、減圧下で濃縮し、そしてジエチルエーテルで洗浄した(3×5mL)。洗液を廃棄した。水(5mL)を、残留物に加え、そして混合物を、塩酸の水溶液(2N)で酸性化し(pH約2〜3)、そして酢酸エチルで抽出した(3×5mL)。合わせた有機抽出物を、Na2SO4で乾燥させ、そして減圧下で蒸発させて、{4−[1−(3,5−ビス−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸(65mg、83%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.33 (br s, 1 H), 8.64 (s, 1 H), 8.38 (s, 2 H), 7.91 - 8.01 (m, 1 H), 7.84 (d, J=12.7 Hz, 1 H), 7.69 (s, 1 H), 7.31 - 7.44 (m, 2 H), 3.94 (d, J=11.7 Hz, 2 H), 3.71 (s, 2 H), 2.68 (d, J=11.2 Hz, 2 H), 1.88 - 1.98 (m, 2 H), 1.77 (d, J=8.3 Hz, 2 H); MS C25H21F7NO4S [(M+H)+]の計算値:564, 実測値564.0.
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
THF(6mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;108mg、0.34mmol)及び塩化2−クロロベンゼンスルホニル(145mg、0.69mmol)の溶液に、窒素雰囲気下、0℃で過剰量のN,N−ジイソプロピルエチルアミン(179μL、1.0mmol)を加えた。得られた明褐色の溶液を、2時間かけてゆっくりと室温まで温まるにまかせ、そして次に窒素雰囲気下、室温で15時間攪拌した。反応混合物を、水及びブライン溶液で希釈し、そして有機化合物を、酢酸エチルで抽出した(2×50mL)。合わせた抽出物を、ブライン溶液で洗浄し、そして無水硫酸マグネシウムで乾燥させた。濾過及び濃縮により、褐色の粗油状物を得て、これを、フラッシュクロマトグラフィー(40g ISCOカラム、0〜60%酢酸エチル/ヘキサンで溶離する)により精製して、{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(110mg、65%)をオフホワイトの固体として得た。MS C25H26ClFNO4S [(M+H)+]の計算値490, 実測値490.0.
THF(8.0mL)中の{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(108mg、0.23mmol)の透明で、ほぼ無色の溶液に、水(2.0mL)中の水酸化リチウム一水和物(106mg、4.4mmol)の溶液を加えた。混合物を、ヒートガンで加熱して、透明な溶液を得た。得られた溶液を、室温で15時間攪拌した。溶媒を、真空下で除去した。残留物を、水(約20mL)で希釈し、そして混合物を、1.0N HClで中和した。沈殿した白色の固体を、濾過によって回収し、そして水及びヘキサンで洗浄した。風乾後、{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(93mg、87%)を、白色の固体として単離した。1H NMR (400 MHz, DMSO-d6) δ ppm 12.42 (br. s, 1 H), 8.07 (d, J=7.53 Hz, 2 H), 783 - 7.96 (m, 1 H), 7.65 - 7.82 (m, 3 H), 7.56 - 7.64 (m, 1 H), 7.28 - 7.40 (m, 1 H), 3.91 (d, J=11.29 Hz, 4 H), 3.76 ( br. s, 3 H), 3.05 (br. s, 2 H), 2.36 (s, 3 H), 1.68 (d, J=12.3 Hz, 2 H). MS C24H24ClFNO4S [(M+H)+]の計算値476, 実測値476.1.
[6−フルオロ−4−(1−メタンスルホニル−ピペリジン−4−イル)−3−メチル−ナフタレン−2−イル]−酢酸
[6−フルオロ−4−(1−メタンスルホニル−ピペリジン−4−イル)−3−メチル−ナフタレン−2−イル]−酢酸メチルエステル
THF(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;82mg、0.26mmol)及び塩化メタンスルホニル(59.6mg、0.52mmol)の溶液に、窒素雰囲気下、0℃でN,N−ジイソプロピルエチルアミン(227μL、1.3mmol)を加えた。得られた懸濁液を、2時間の間、室温まで温まるにまかせ、そして次に窒素雰囲気下、15時間攪拌した。反応混合物を、水及びブライン溶液で希釈し、そして有機化合物を、酢酸エチルで抽出した(2×75mL)。合わせた有機抽出物を、ブライン溶液で洗浄し、そして無水MgSO4で乾燥させ、濾過し、そして濃縮して、褐色の粗油状物を得て、これを、フラッシュクロマトグラフィー(40g ISCOカラム、0〜100%酢酸エチル/ヘキサンで溶離する)により精製して、[6−フルオロ−4−(1−メタンスルホニル−ピペリジン−4−イル)−3−メチル−ナフタレン−2−イル]−酢酸メチルエステル(56mg、55%)をオフホワイトの固体として得た。MS C20H25FNO4S [(M+H)+]の計算値394, 実測値393.9.
THF(6.0mL)中の[6−フルオロ−4−(1−メタンスルホニル−ピペリジン−4−イル)−3−メチル−ナフタレン−2−イル]−酢酸メチルエステル(51mg、0.13mmol)の溶液に、水(1.5mL)中の水酸化リチウム一水和物(62.1mg、2.6mmol)の溶液を加えた。得られた溶液を、室温で15時間攪拌した。溶媒を、真空下で除去した。残留物を、水(約20mL)で希釈し、そして1.0N HCl水溶液で中和して、懸濁液を形成した。生成物を、酢酸エチルに抽出した(2×50mL)。合わせた抽出物を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、そして濃縮して、粗固体を得て、これを、ジクロロメタン及びヘキサンでトリチュレートした。固体を、濾過によって回収し、そしてヘキサンで洗浄した。風乾後、[6−フルオロ−4−(1−メタンスルホニル−ピペリジン−4−イル)−3−メチル−ナフタレン−2−イル]−酢酸(48mg、94%)を、白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.4 (br. s, 1 H), 7.91 (dd, J=8.91, 6.65 Hz, 2 H), 7.70 (s, 1 H), 7.36 (t, J=8.41 Hz, 1 H), 3.78 (br. s, 6 H), 2.95 (s, 3 H), 2.26 - 2.47 (m, 6 H), 1.75 (d, J=12.05 Hz, 2 H). MS C19H23FNO4S [(M+H)+]の計算値380, 実測値380.0.
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
THF(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;110mg、0.35mmol)及び塩化2,4−ジクロロベンゼンスルホニル(171mg、0.7mmol)の0℃溶液に、N,N−ジイソプロピルエチルアミン(183μL、1.05mmol)を加えた。得られた明黄色の溶液を、2時間の間、室温まで温まるにまかせ、そして次にそれを、窒素下、室温で15時間攪拌した。反応混合物を、水及びブライン溶液で希釈し、そして有機化合物を、酢酸エチルに抽出した(2×50mL)。合わせた抽出物を、ブライン溶液で洗浄し、そしてMgSO4で乾燥させた。濾過及び濃縮により粗生成物を得て、これを、フラッシュクロマトグラフィー(40g ISCOカラム、ヘキサン中0〜60%酢酸エチルで溶離する)により精製して、{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(119mg、65%)をオフホワイトの固体として得た。MS C20H25FNO4S [(M+H)+]の計算値524, 実測値523.8.
THF(8.0mL)中の{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(109mg、0.208mmol)の無色の溶液に、室温で、水(2.0mL)中の過剰量の水酸化リチウム(100mg、4.18mmol)の溶液を加えた。得られた明黄色の溶液を、15時間攪拌した。反応混合物を濃縮し、そしてその水溶液を、水で希釈し、次に1.0N HCl水溶液で中和した。得られた懸濁液を、酢酸エチルに抽出した(2×40mL)。合わせた抽出物を、ブラインで洗浄し、そして無水MgSO4で乾燥させ、濾過し、そして濃縮して、{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(105mg、99%)を明黄色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.32 (br. s, 1 H), 8.06 (d, J=8.5 Hz, 1 H), 7.98 (d, J=2.0 Hz, 1 H), 7.88 (dd, J=8.5, 6.8 Hz, 1 H), 7.64 - 7.82 (m, 3 H), 7.25 - 7.39 (m, 1 H), 3.82- 3.94 (m, 2 H), 3.76 (br. s, 2 H), 3.47 - 3.72 (m, 1 H), 3.07 (br. s, 2 H), 2.36 (s, 3 H), 2.09 - 2.46 (m, 2 H), 1.68 (d, J=12.0 Hz, 2 H). MS C24H23Cl2FNO4S [(M+H)+]の計算値510, 実測値510.1.
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化2,5−ジクロロベンゼンスルホニル(116mg、0.474mmol)を加えた。0℃で約15分間攪拌した後、明黄色の反応混合物を、室温まで温まるにまかせ、そして次に窒素下、室温で一晩攪拌した。この時間の間に、いくらかの溶媒が蒸発した。塩化メチレン(約10mL)及び水(約25mL)を加えた。得られた混合物を、塩化メチレンで抽出した(2×25mL)。合わせた抽出物を、ブライン溶液で洗浄し、次に濃縮して、粗生成物を得て、これを、シリカゲルプラグからの溶離(ヘキサン中20%酢酸エチル)により精製して、{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(52mg、42%)をオフホワイトの固体として得た。MS C25H25Cl2FNO4S [(M+H)+]の計算値:524, 実測値524.0.
THF(8.0mL)中の{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(52mg、0.099mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(42.4mg、1.0mmol)の溶液を加えた。反応混合物を、窒素下、室温で一晩攪拌した。この後、反応混合物を、水(約20mL)で希釈し、そして次に1.0N HCl水溶液(2mL)で酸性化した。得られた混合物を、酢酸エチルで抽出し(2×25mL)、次に合わせた抽出物を、ブライン(30mL)で洗浄した。有機相を、無水MgSO4で乾燥させ、濾過し、そして濃縮して、{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(47mg、93%)を黄色の膜状物として得た。MS C24H23Cl2FNO4S [(M+H)+]の計算値:510, 実測値510.
{4−[1−(2,6−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,6−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化2,6−ジクロロベンゼンスルホニル(116mg、0.47mmol)を加えた。0℃で約25分間攪拌した後、明黄色の反応混合物を、室温まで温まるにまかせ、そして次に窒素下、室温で6.5時間攪拌した。この時間の間に、いくらかの溶媒が蒸発していた。塩化メチレン(約10mL)及び水(約25mL)を加え、そして混合物を、塩化メチレンで抽出した(2×25mL)。合わせた抽出物を、ブライン溶液で洗浄し、無水MgSO4で乾燥させ、濾過し、濃縮し、そしてシリカゲルクロマトグラフィー(25%酢酸エチル/ヘキサンで溶離する)により精製して、{4−[1−(2,6−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(96mg、77%)を明黄色の生成物として得た。MS C25H25Cl2FNO4S [(M+H)+]の計算値:524, 実測値524.1.
THF(8.0mL)中の{4−[1−(2,6−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(96mg、0.183mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(87.6mg、2.1mmol)の溶液を加えた。反応混合物を、窒素下、室温で一晩攪拌した。この後、反応混合物を、水(約20mL)で希釈し、そして次に1.0N HCl水溶液(3.66mL)で酸性化した。得られた混合物を、酢酸エチルで抽出し(2×25mL)、次に合わせた抽出物を、ブライン(30mL)で洗浄した。有機相を、無水MgSO4で乾燥させ、濾過し、そして濃縮して、{4−[1−(2,6−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(63mg、67%)を黄色の固体として得た。MS C24H23Cl2FNO4S [(M+H)+]の計算値:510, 実測値510.1.
[6−フルオロ−3−メチル−4−(1−フェニルメタンスルホニル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸
[6−フルオロ−3−メチル−4−(1−フェニルメタンスルホニル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化α−トルエンスルホニル(90.4mg、0.474mmol)を加えた。0℃で約10分間撹拌した後、明黄色の反応混合物を、室温まで温まるにまかせ、そして次に窒素下、室温で4時間攪拌した。この時間の間に、いくらかの溶媒が蒸発していた。反応混合物を、水(約25mL)で希釈し、そして塩化メチレンで抽出した(3×25mL)。合わせた有機層を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、濃縮し、そしてシリカゲルクロマトグラフィー(33%酢酸エチル/ヘキサンで溶離する)により精製して、[6−フルオロ−3−メチル−4−(1−フェニルメタンスルホニル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸メチルエステル(88mg、79%)を得た。MS C26H29FNO4S [(M+H)+]の計算値:470, 実測値470.2.
THF(8.0mL)中の[6−フルオロ−3−メチル−4−(1−フェニルメタンスルホニル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸メチルエステル(82mg、0.175mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(83.8mg、2mmol)の溶液を加えた。反応混合物を、窒素下、室温で一晩攪拌した。この後、反応混合物を、水(約20mL)で希釈し、そして次に1.0N HCl水溶液(3.5mL)で酸性化した。得られた白色の沈殿物を、真空濾過によって回収し、ヘキサンで洗浄し、そして次に、さらに真空下で乾燥させて、[6−フルオロ−3−メチル−4−(1−フェニルメタン−スルホニル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸(50mg、63%)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.43 (br. s., 1 H), 7.90 (dd, J=9.03, 6.53 Hz, 1 H), 7.80 - 8.13 (m, 1 H), 7.69 (s, 1 H), 7.31 - 7.53 (m, 6 H), 4.49 (br. s., 2 H), 3.66 - 3.85 (m, 4 H), 3.46 - 3.57 (m, 1 H), 2.87 - 3.20 (m, 2 H), 2.41 (br. s., 3 H), 2.13 - 2.37 (m, 2 H), 1.61 - 1.71 (m, 2 H). MS C25H27FNO4S [(M+H)+]の計算値:456, 実測値456.2.
{6−フルオロ−4−[1−(2−メトキシ−ベンゼンスルホニル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸
{6−フルオロ−4−[1−(2−メトキシ−ベンゼンスルホニル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化2−メトキシベンゼンスルホニル(98mg、0.474mmol)を加えた。0℃で約10分間攪拌した後、明黄色の反応混合物を、室温まで温まるにまかせ、そして次に窒素下、室温で4時間攪拌した。この時間の間に、いくらかの溶媒が蒸発していた。さらなる塩化メチレンを加えた。反応混合物を、水(約25mL)で希釈し、そして塩化メチレンで抽出した(3×25mL)。合わせた有機層を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(20〜33%酢酸エチル/ヘキサン)により、{6−フルオロ−4−[1−(2−メトキシ−ベンゼンスルホニル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(64mg、55%)を得た。MS C26H29FNO5S [(M+H)+] の計算値:486, 実測値486.2.
THF(8.0mL)中の{6−フルオロ−4−[1−(2−メトキシ−ベンゼンスルホニル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(64mg、0.132mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(63.3mg、1.5mmol)の溶液を加えた。反応混合物を、窒素下、室温で一晩攪拌した。この後、反応混合物を濃縮した。水(約20mL)を加え、そして混合物を、1.0N HCl水溶液(2.7mL)で酸性化した。得られた白色の沈殿物を、真空濾過によって回収し、ヘキサンで洗浄し、そして次にさらに真空下で乾燥させて、{6−フルオロ−4−[1−(2−メトキシ−ベンゼンスルホニル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸(51.5mg、83%)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.45 (br. s, 1 H), 8.04 (br. s, 1 H), 7.85 - 7.92 (m, 1 H), 7.78 - 7.84 (m, 1 H), 7.67 (s, 2 H), 7.29 - 7.37 (m, 2 H), 7.13 (t, J=8.00 Hz, 1 H), 3.94 (s, 3 H), 3.82 - 3.92 (m, 2 H), 3.70 - 3.80 (m, 2 H), 2.82 - 3.02 (m, 2 H), 2.35 (s, 3 H), 2.13 - 2.32 (m, 1 H), 1.60 - 1.71 (m, 2 H). MS C25H27FNO5S [(M+H)+]の計算値:472, 実測値472.2.
{4−[1−(3−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(3−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化(3−クロロ−フェニル)−メタンスルホニル(106.7mg、0.474mmol)を加えた。反応混合物を、0℃で10分間攪拌し、次にそれを、室温に温め、そして室温で一晩攪拌した。反応混合物を、水で希釈した。得られた混合物を、酢酸エチルで抽出した。有機相を、MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中20〜25%酢酸エチル)により、{4−[1−(3−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(52.4mg、44%)を無色の膜状物として得た。MS C26H28ClFNO4S [(M+H)+]の計算値:504, 実測値504.1.
THF(8.0mL)中の{4−[1−(3−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(52.4mg、0.104mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(49.8mg、2.08mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下、一晩攪拌した。溶媒を蒸発させた。水(約25mL)を加え、そして混合物を、1.0N HCl水溶液(2.1mL)で酸性化した。得られた白色の沈殿物を、濾過によって回収し、水及びヘキサンで洗浄し、そして高真空下で乾燥させた。この粗生成物を、分取逆相HPLCを使用してさらに精製し、{4−[1−(3−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(9mg、18%)を白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.44 (br. s, 1 H), 7.95 - 8.12 (m, 1 H), 7.90 (dd, J=9.03, 6.53 Hz, 1 H), 7.65 - 7.74 (m, 2 H), 7.55 (s, 1 H), 7.46 (d, J=1.00 Hz, 2 H), 7.36 (t, J=9.00 Hz, 1 H), 4.54 (br. s., 2 H), 4.13 (dd, J=5.52, 3.51 Hz, 1 H), 3.78 (br. s., 4 H), 3.48 - 3.65 (m, 1 H), 2.91 - 3.22 (m, 3 H), 2.42 (br. s., 5 H), 1.69 (m, 2 H). MS C25H26ClFNO4S [(M+H)+]の計算値:490, 実測値490.2.
{4−[1−(4−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(4−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化(4−クロロ−フェニル)−メタンスルホニル(106.7mg、0.474mmol)を加えた。反応混合物を、0℃で10分間攪拌し、次にそれを、室温に温め、そして室温で4時間攪拌した。反応混合物を、水(25mL)で希釈した。得られた混合物を、酢酸エチルで抽出した(2×25mL)。有機相を、MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中20〜25%酢酸エチル)により、{4−[1−(4−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(28.5mg、24%)を無色の膜状物として得た。MS C26H28ClFNO4S [(M+H)+]の計算値:504, 実測値504.2.
THF(8.0mL)中の{4−[1−(4−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(28.5mg、0.057mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(27.3mg、1.14mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下、一晩攪拌した。溶媒を蒸発させた。水(約25mL)を加え、そして混合物を、1.0N HCl水溶液(1.1mL)で酸性化した。得られた白色の沈殿物を、濾過によって回収し、水及びヘキサンで洗浄し、そして高真空下で乾燥させた。この粗生成物を、分取逆相HPLCを使用してさらに精製し、{4−[1−(4−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(11.2mg、40%)を白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.42 (br. s, 1 H), 7.90 (dd, J=9.00, 6.50 Hz, 1 H), 7.78 - 8.11 (m, 1 H), 7.69 (s, 1 H), 7.50 (s, 4 H), 7.36 (td, J=9.00, 2.50 Hz, 1 H), 4.53 (br. s, 2 H), 3.64 - 3.86 (m, 5 H), 2.91 - 3.19 (m, 3 H), 2.41 (br. s., 3 H), 2.15 - 2.35 (br. s, 1 H), 1.61 - 1.73 (m, 2 H). MS C25H25ClFNO4S [(M+H)+]の計算値:490, 実測値490.1.
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル−ナフタレン−2−イル}−酢酸
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化3−(トリフルオロメチル)ベンゼンスルホニル(76μL、0.474mmol)を加えた。0℃で約10分間撹拌した後、明黄色の反応混合物を、室温まで温まるにまかせ、そして次に室温で一晩攪拌した。水(約25mL)を加え、そして混合物を、塩化メチレンで抽出した(2×30mL)。合わせた有機層を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中25〜33%酢酸エチル)により、{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(104mg、84%)を得た。MS C26H26F4NO4S [(M+H)+]の計算値:524, 実測値524.1.
THF(8.0mL)中の{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(104.2mg、0.199mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(95.3mg、2.3mmol)の溶液を加えた。反応混合物を、窒素下、室温で一晩攪拌した。この後、反応混合物を濃縮した。水(約25mL)を加え、そして混合物を、1.0N HCl水溶液(4mL)で酸性化した。得られた白色の沈殿物を、真空濾過によって回収し、水及びヘキサンで洗浄し、そして次に真空下でさらに乾燥させて、{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(92.2mg、91%)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.43 (br. s, 1 H), 8.18 - 8.20 (m, 1 H), 8.16 - 8.18 (m, 1 H), 8.07 (s, 1 H), 7.97 (t, J=7.80 Hz, 1 H), 7.84 - 7.91 (m, 1 H), 7.67 (s, 1 H), 7.33 (t, J=9.00 Hz, 1 H), 3.92 (br. s., 2 H), 3.74 (s, 2 H), 3.41 - 3.70 (m, 2 H), 2.57 - 2.84 (m, 2 H), 2.31 (s, 3 H), 2.40 - 2.28 (m, 1 H), 1.69 (m, 2 H). MS C25H22F4NO4S [(M-H)-]の計算値:508, 実測値508.1.
[4−(1−シクロペンタンスルホニル−ピペリジン−4−イル)−6−フルオロ−3−メチル−ナフタレン−2−イル]−酢酸
[4−(1−シクロペンタンスルホニル−ピペリジン−4−イル)−6−フルオロ−3−メチル−ナフタレン−2−イル]−酢酸メチルエステル
塩化メチレン(10.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化シクロペンタンスルホニル(62.5μL、0.474mmol)を加えた。反応混合物を、0℃で20分間攪拌し、次にそれを、室温に温め、そして室温で一晩攪拌した。反応混合物を、水(25mL)で希釈した。得られた混合物を、塩化メチレンで抽出した(2×30mL)。有機相を、MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中33%酢酸エチル)により、[4−(1−シクロペンタンスルホニル−ピペリジン−4−イル)−6−フルオロ−3−メチル−ナフタレン−2−イル]−酢酸メチルエステル(64.8mg、61%)を得た。MS C24H31FNO4S [(M+H)+]の計算値:448, 実測値448.2.
THF(8.0mL)中の[4−(1−シクロペンタンスルホニル−ピペリジン−4−イル)−6−フルオロ−3−メチル−ナフタレン−2−イル]−酢酸メチルエステル(64.8mg、0.145mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(69.5mg、1.7mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下で3時間攪拌した。溶媒を蒸発させた。水(20mL)を加え、そして混合物を、1.0N HCl水溶液(3mL)で酸性化した。得られた白色の沈殿物を、濾過によって回収し、水及びヘキサンで洗浄し、そして高真空下で乾燥させた。この粗生成物を、分取逆相HPLCを使用してさらに精製し、[4−(1−シクロペンタン−スルホニル−ピペリジン−4−イル)−6−フルオロ−3−メチル−ナフタレン−2−イル]−酢酸(19mg、30%)を白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.43 (br. s., 1 H), 7.97 - 8.17 (m, 1 H), 7.90 (dd, J=9.03, 6.53 Hz, 1 H), 7.69 (s, 1 H), 7.35 (t, J=7.53 Hz, 1 H), 3.78 (br. s., 6 H), 3.11 (br. s., 2 H), 2.42 (br. s., 4 H), 1.96 - 2.08 (m, 2 H), 1.80 - 1.93 (m, 2 H), 1.53 - 1.78 (m, 6 H). MS C23H29FNO4S [(M+H)+]の計算値:434, 実測値434.2.
{6−フルオロ−3−メチル−4−[1−(ピロリジン−1−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
{6−フルオロ−3−メチル−4−[1−(ピロリジン−1−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(10.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の0℃溶液に、塩化ピロリジン−1−スルホニル(80.4mg、0.474mmol)を加えた。反応混合物を、0℃で25分間攪拌し、そして次に室温に温め、そして一晩攪拌した。この期間の間に、溶媒は蒸発乾固した。追加の塩化メチレン(10mL)を、朝に加えた。反応混合物を、水(25mL)で希釈した。得られた混合物を、酢酸エチルで抽出した(2×30mL)。有機相を、MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中33%酢酸エチル)により、{6−フルオロ−3−メチル−4−[1−(ピロリジン−1−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(86.5mg、81%)を得た。MS C23H30FN2O4S [(M+H)+]の計算値:449, 実測値449.2.
THF(8.0mL)中の{6−フルオロ−3−メチル−4−[1−(ピロリジン−1−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(86.5mg、0.193mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(92.4mg、2.2mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下で一晩攪拌した。溶媒を蒸発させた。水(20mL)を加え、そして混合物を、1.0N HCl水溶液(4mL)で酸性化した。得られた白色の沈殿物を、濾過によって回収し、水及びヘキサンで洗浄し、そして高真空下で乾燥させて、{6−フルオロ−3−メチル−4−[1−(ピロリジン−1−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(55.7mg、66%)をオフホワイトの固体として得た。1H NMR (400 MHz, DMSO-d6) δ 12.44 (br. s., 1 H), 8.07 (br. s, 1 H), 7.90 (dd, J= 9.0, 6.5 Hz, 1 H), 7.69 (s, 1 H), 7.35 (td, J=8.5, 2.0 Hz, 1 H), 3.78 (br. s., 4 H), 3.22 - 3.43 (m, 5 H), 3.09 (br. s., 2 H), 2.42 (br. s., 3 H), 1.81 - 1.97 (m, 4 H), 1.67 (bd, 2 H). MS C22H28FN2O4S [(M+H)+]の計算値:434, 実測値434.2.
{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル
THF(30mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;560mg、1.78mmol)及び塩化(2−ニトロフェニル)メタンスルホニル(837mg、3.55mmol)の明褐色の溶液を、窒素下、0℃に冷却し、そして次にN,N−ジイソプロピルエチルアミン(930μL、5.34mmol)を加えた。得られた明褐色の溶液を、温度0℃で2時間攪拌し、そして次に室温まで温まるにまかせ、そして36時間攪拌した。反応混合物を、水及びブラインで希釈し、そして有機化合物を、酢酸エチルに抽出した(2×100mL)。合わせた有機層を、ブラインで洗浄し、そしてMgSO4で乾燥させ、濾過し、そして濃縮した。粗生成物を、フラッシュクロマトグラフィー(120g ISCOカラム 0〜60%酢酸エチル/ヘキサンで溶離する)を使用して精製し、{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(645mg、71%)を明黄色の固体として得た。1H NMR (400 MHz, クロロホルム-d) δ 8.07 - 8.1 (m, 1 H), 7.85 (br. s, 1 H), 7.55 (dd, J= 8.8, 6.3 Hz, 1 H), 7.67 - 7.72 (m, 2 H), 7.56 - 7.64 (m, 2 H), 7.16 - 7.24 (m, 1 H), 4.83 (s, 2 H), 3.89 (d, J= 10.0 Hz, 2 H), 3.82 (br. s, 2 H), 3.72 (s, 3 H), 3.43 - 3.63 (m, 1 H), 2.90 (t, , J= 11.5 Hz, 2 H), 2.32 - 2.71 (m, 5 H), 1.77 (d, , J=13.8 Hz, 2 H). MS C26H27FN2O6S [(M+H)+]の計算値514, 実測値515.2.
THF(4mL)中の{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(100mg、0.194mmol)の明黄色の溶液に、室温で水(1mL)中の水酸化リチウム(93.1mg、3.89mmol)の溶液を加えた。得られた明褐色の溶液を、室温で15時間攪拌した。THFを、真空下で蒸発させて、そして塩基性水層を、1.0N HClで中和した。得られた固体を、濾紙を使用して濾過し、そして水及びヘキサンで洗浄した。白色の固体(82mg)を、酢酸エチル(約5mL)に加熱しながら溶解し、そして混合物を、ヘキサンで希釈した。希釈の間に、いくらかの沈殿物が形成された。冷蔵庫内での冷却により、さらなる固体を沈殿させた。{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(88mg、90%)を、白色の固体として単離した。1H NMR (400 MHz, DMSO-d6) δ 12.44 (br. s, 1 H), 8.08 (d, J=8.03 Hz, 1 H), 7.90 (dd, J=8.91, 6.65 Hz, 1 H), 7.74 - 7.86 (m, 3 H), 7.64 - 7.73 (m, 2 H), 7.36 (t, J=8.03 Hz, 1 H), 4.97 (s, 2 H), 3.52 - 3.90 (m, 7 H), 2.42 (br. s, 3 H), 1.60-184 (m, 4 H). MS C25H25FN2O6S [(M+H)+]の計算値500, 実測値501.1.
{4−[1−(2−アミノ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2−アミノ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(505mg、0.981mmol)、亜鉛末(325メッシュ)(642mg、9.82mmol)及び塩化アンモニウム(787mg、14.7mmol)の混合物を、窒素下、メタノール(10mL)及び水(5mL)と合わせて、懸濁液を作り出した。ヒートガンを使用した適度な加熱を使用して、より多くの出発材料を溶液にし、そしてさらなるメタノール(5mL)を加えた。懸濁液を、2時間攪拌し、その時点で、LCMS及びTLC分析(1:1 ヘキサン−酢酸エチル)分析は、未反応の出発材料及びより極性の大きい生成物の存在を示した。別の20当量の亜鉛末(1.02g)を加え、そして反応混合物を、2時間攪拌し、この期間の間、ヒートガンでいくらか加熱した。TLC分析は、反応が完了しなかったことを示した。別の30当量の塩化アンモニウム(1.4g)を加え、そして得られた懸濁液を、加熱マントルで40〜50℃に加熱した。反応混合物を、室温に冷却し、そして亜鉛を濾別し、そしてメタノールで洗浄した。濾液を濃縮し、そして残留物を、水で希釈した。懸濁液を、酢酸エチルで抽出し(2×75mL)、そして合わせた抽出物を、ブラインで洗浄し、MgSO4で乾燥させ、濾過し、そして濃縮した。フラッシュクロマトグラフィー(80g ISCOカラム、0〜70%酢酸エチル/ヘキサンで溶離する)により、{4−[1−(2−アミノ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(385mg、81%)を白色の固体として得た。MS C26H30FN2O4S [(M+H)+]の計算値484, 実測値485.2.
THF(8.0mL)中の{4−[1−(2−アミノ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(178mg、0.367mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(176mg、7.35mmol)の溶液を加えた。得られた溶液を、窒素下、15時間攪拌した。溶媒を、真空下で除去し、そして水溶液を、水で希釈し、そしてpHが5〜6の間になるまで1.0N HClでゆっくり中和した。得られた沈殿した固体を、濾過によって回収し、そして水及びヘキサンで洗浄した。空気中で乾燥させた後、{4−[1−(2−アミノ−フェニルメタン−スルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(159mg、92%)を、オフホワイトの固体として単離した。1H NMR (400 MHz, DMSO-d6) δ 12.44 (br. s, 1 H), 7.86 (dd, J=8.85, 6.59 Hz, 1 H), 7.61 (s, 1 H), 7.31 (t, J=7.91 Hz, 1 H), 7.18 (d, J=7.35 Hz, 1 H), 6.99 - 7.12 (m, 2 H), 6.68 - 6.78 (m, 1 H), 6.53 - 6.67 (m, 1 H), 5.14 (br. s, 2 H), 4.28 - 4.51 (m, 4 H), 3.76 (d, J=10.93 Hz, 2 H), 3.61 (br. s, 5 H), 2.41 (br. s, 3 H), 1.66 (d, J=11.49 Hz, 2 H). MS C25H26FN2O4S [(M-H)-]の計算値469, 実測値469.1.
{4−[1−(2,4−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,4−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
ジクロロメタン(10mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;110mg、0.35mmol)及び塩化2,4−ジクロロベンゾイル(110mg、0.53mmol)の明褐色の溶液に、窒素雰囲気下、室温でN,N−ジイソプロピルエチルアミン(183μL、1.05mmol)を加えた。得られた明褐色の溶液を、15時間攪拌した。反応混合物を、水及びブラインで希釈し、そして得られた混合物を、酢酸エチルで抽出した(2×50mL)。合わせた有機抽出物を、ブライン溶液で洗浄し、MgSO4で乾燥させ、濾過し、そして濃縮して、粗生成物を得た。フラッシュクロマトグラフィー(40g ISCOカラム、0〜60%酢酸エチル/ヘキサンで溶離する)により、{4−[1−(2,4−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(111mg、65%)を白色の固体として得た。MS C26H25Cl2FNO3[(M+H)+]の計算値488, 実測値488.0.
THF(8.0mL)中の{4−[1−(2,4−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(102mg、0.21mmol)の無色の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(100mg、4.2mmol)の溶液を加えた。得られた無色の溶液を、15時間攪拌した。溶媒を除去し、そして水溶液を、水での希釈後、1.0N HCl水溶液で中和した。得られた白色の沈殿物を、濾過によって回収し、そしてヘキサンで洗浄した。空気中で乾燥させた後、粗生成物 62mgを、白色の固体として単離した。粗生成物を、加熱しながら酢酸エチル(約3mL)に溶解し、そして得られた溶液を、ヘキサン(約20mL)で希釈した。溶液を、冷凍庫内に3日間保管し、そして沈殿した固体を、濾過によって回収し、そしてヘキサンで洗浄した。空気中で乾燥させた後、{4−[1−(2,4−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(48mg、48%)を、オフホワイトの固体として単離した。1H NMR (400 MHz, DMSO-d6) δ ppm 12.42 (br. s, 1 H), 7.85 - 7.97 (m, 1 H), 7.81 (br. s, 1 H), 7.75 (s, 1 H), 7.69 (s, 1 H), 7.51 - 7.63 (m, 1 H), 7.41 - 7.50 (m, 1 H), 7.35 (t, J=8.03 Hz, 1 H), 4.71 (br. s, 1 H), 3.78 (br. s, 4 H), 3.40 (br. s, 2 H), 2.42 (br. s, 3 H), 1.77 (br. s, 2 H), 1.60 (d, J=9.79 Hz, 2 H). MS C25H23Cl2FNO3[(M+H)+]の計算値474, 実測値474.1.
{4−[1−(2,6−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,6−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(10.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の溶液に、塩化2,6−ジクロロベンゾイル(74.4mg、0.355mmol)を加えた。反応混合物を、室温で3時間攪拌した。水(約25mL)を加え、そして混合物を、塩化メチレンで抽出した(2×25mL)。合わせた有機層を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過しそして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中25%酢酸エチル)により、{4−[1−(2,6−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(100mg、86%)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 7.85 - 8.20 (m, 1 H), 7.87 - 7.96 (m, 1 H), 7.71 (s, 1 H), 7.55 - 7.66 (m, 2 H), 7.48 (s, 1 H), 7.31 - 7.40 (m, 1 H), 4.67 - 4.81 (m, 1 H), 3.74 - 3.97 (m, 3 H), 3.63 (s, 3 H), 3.35 - 3.44 (m, 2 H), 3.02 - 3.21 (m, 1 H), 2.40 (s, 2 H), 2.17 - 2.31 (m, 1 H), 1.58 - 1.84 (m, 2 H).
THF(8.0mL)中の{4−[1−(2,6−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(96mg、0.197mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(94.2mg、2.2mmol)の溶液を加えた。反応混合物を、室温で一晩攪拌した。この後、反応混合物を濃縮した。水(約20mL)を加え、そして混合物を、1.0N HCl水溶液(3.9mL)で酸性化した。得られた白色の沈殿物を、真空濾過によって回収して、ヘキサンで洗浄し、そして次に真空下でさらに乾燥させて、{4−[1−(2,6−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(66mg、71%)を得た。1H NMR (300 MHz, DMSO-d6) δ ppm 7.84 - 7.97 (m, 2 H), 7.68 (s, 1 H), 7.55 - 7.66 (m, 2 H), 7.44 - 7.53 (m, 1 H), 7.34 (s, 1 H), 4.69 - 4.80 (m, 1 H), 3.76 (br. s., 2 H), 3.32 (br. s., 6 H), 2.43 (s, 2 H), 1.57 - 1.85 (m, 2 H). MS C25H23Cl2FNO3[(M+H)+]の計算値:474, 実測値474.1.
{4−[1−(2,5−ジフルオロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,5−ジフルオロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(10.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の溶液に、塩化2,5−ジフルオロベンゾイル(62.7mg、0.355mmol)を加えた。反応混合物を、室温で3時間攪拌した。水(約25mL)を加え、そして混合物を、塩化メチレンで抽出した(2×25mL)。合わせた有機層を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中25%酢酸エチル)により、{4−[1−(2,5−ジフルオロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(73mg、68%)を得た。
THF(8.0mL)中の{4−[1−(2,5−ジフルオロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(73mg、0.160mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(76.6mg、1.8mmol)の溶液を加えた。反応混合物を、室温で一晩攪拌した。この後、反応混合物を濃縮した。水(約20mL)を加え、そして混合物を、1.0N HCl水溶液(3.2mL)で酸性化した。得られた白色の沈殿物を、真空濾過によって回収し、ヘキサンで洗浄し、そして次に真空下でさらに乾燥させて、{4−[1−(2,5−ジフルオロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(57.5mg、81%)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.46 (br. s, 1 H), 7.80 - 8.20 (m, 1 H), 7.90 (dd, J=9.00, 6.50 Hz, 1 H), 7.69 (s, 1 H), 7.31 - 7.52 (m, 4 H), 4.62 - 4.77 (m, 1 H), 3.68 - 4.08 (m, 4 H), 3.48 - 3.60 (m, 1 H), 2.95 - 3.21 (m, 1 H), 2.43 (br. s., 3 H), 2.11 - 2.34 (m, 2 H), 1.57 - 1.82 (m, 2 H). MS C25H23F3NO3[(M+H)+]の計算値:442, 実測値442.2.
[6−フルオロ−3−メチル−4−(1−フェニルアセチル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸
[6−フルオロ−3−メチル−4−(1−フェニルアセチル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸メチルエステル
塩化メチレン(10.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステルトリフルオロ酢酸塩(これを先に記載したように調製することもできる;102mg、0.237mmol)及びN,N−ジイソプロピルエチルアミン(124μL、0.711mmol)の溶液に、塩化フェニルアセチル(47μL、0.355mmol)を加えた。反応混合物を、室温で4時間攪拌した。水(約25mL)を加え、そして混合物を、塩化メチレンで抽出した(2×25mL)。合わせた有機層を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、そして濃縮した。シリカゲルクロマトグラフィー(ヘキサン中25〜33%酢酸エチル)により、[6−フルオロ−3−メチル−4−(1−フェニルアセチル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸メチルエステル(77mg、75%)を得た。MS C27H29FNO3[(M+H)+]の計算値:434, 実測値434.2.
THF(8.0mL)中の[6−フルオロ−3−メチル−4−(1−フェニルアセチル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸メチルエステル(77mg、0.178mmol)の溶液に、水(2mL)中の水酸化リチウム一水和物(74.7mg、1.8mmol)の溶液を加えた。反応混合物を、室温で一晩攪拌した。この後、反応混合物を濃縮した。水(約20mL)を加え、そして混合物を、1.0N HCl水溶液(3.1mL)で酸性化した。得られた白色の沈殿物を、真空濾過によって回収し、水及びヘキサンで洗浄し、そして次に真空下でさらに乾燥させて、[6−フルオロ−3−メチル−4−(1−フェニルアセチル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸(52.5mg、70%)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.5 (br. s, 1 H), 7.88 (dd, J=9.00, 6.50 Hz, 1 H), 7.66 (s, 1 H), 7.46 - 7.61 (m, 1 H), 7.27 - 7.43 (m, 5 H), 7.18 - 7.26 (m, 1 H), 4.48 - 4.74 (m, 1 H), 4.00 - 4.20 (m, 1 H), 3.57 - 3.99 (m, 6 H), 2.65 - 2.97 (m, 1 H), 2.11 - 2.46 (m, 3 H), 1.70 - 2.10 (m, 1 H), 1.42 - 1.69 (m, 2 H). MS C26H27FNO3 [(M+H)+]の計算値:420, 実測値420.2.
{4−[1−(2,6−ジフルオロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,6−ジフルオロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(8.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;75.5mg、0.239mmol)の溶液に、窒素下、室温で無希釈イソシアン酸2,6−ジフルオロフェニル(74.1mg、0.478mmol)を加えた。得られた明褐色の溶液を、一晩攪拌した。反応混合物を、水(約20mL)で希釈し、そして得られた混合物を、塩化メチレンで抽出した(2×30mL)。合わせた有機層を、飽和NaHCO3水溶液で洗浄し、無水Na2SO4で乾燥させ、濾過し、次に濃縮して、黄色を帯びた膜状物にした。シリカゲルクロマトグラフィー(ヘキサン中33%酢酸エチル)により、{4−[1−(2,6−ジフルオロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(95.4mg、85%)を無色の膜状物として得た。MS C26H26F3N2O3[(M+H)+]の計算値:471, 実測値471.2.
THF(8.0mL)中の{4−[1−(2,6−ジフルオロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(95.4mg、0.203mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(97.2mg、2.3mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下、室温で一晩攪拌した。次に、溶媒を除去し、そして残留物を、水(25mL)と合わせた。混合物を、1.0N HClで中和した。得られた白色の沈殿物を、濾過によって回収し、そして高真空下、乾燥させて、{4−[1−(2,6−ジフルオロ−フェニル−カルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(59.2mg、64%)を白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.46 (br. s, 1 H), 8.38 (br. s, 1 H), 7.90 (dd, J=9.00, 6.40 Hz, 1 H), 7.75 - 7.85 (m, 1 H), 7.65 - 7.74 (m, 1 H), 7.24 - 7.42 (m, 2 H), 7.14 (m, 2 H), 4.17 - 4.38 (m, 2 H), 3.79 (m, 3 H), 2.99 - 3.24 (m, 2 H), 2.44 (s, 3 H), 2.13 - 2.36 (m, 1 H), 1.57 - 1.73 (m, 2 H). MS C25H24F3N2O3[(M+H)+]の計算値:457, 実測値457.2.
{4−[1−(2,4−ジクロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2,4−ジクロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
メタノール(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;47.2mg、0.150mmol)の溶液に、窒素下、室温で無希釈イソシアン酸2,4−ジクロロフェニル(56.4mg、0.300mmol)を加えた。得られた明褐色の溶液を、室温で15分間攪拌した。反応混合物を、水(約25mL)で希釈し、そして得られた混合物を、酢酸エチルで抽出した(2×30mL)。合わせた有機層を、飽和NaHCO3水溶液で洗浄し、無水Na2SO4で乾燥させ、濾過し、そして濃縮して、無色の膜状物にした。シリカゲルクロマトグラフィー(ヘキサン中20〜25%酢酸エチル)により、{4−[1−(2,4−ジクロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(74.8mg、99%)を無色の膜状物として得た。MS C26H26Cl2FN2O3[(M+H)+]の計算値:503, 実測値503.1.
THF(8.0mL)中の{4−[1−(2,4−ジクロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(74.8mg、0.149mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(71.4mg、1.7mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下、室温で一晩攪拌した。溶媒を蒸発させた。水(約20mL)を加え、そして混合物を、1.0N HCl(30mL)で中和した。得られた白色の沈殿物を、濾過によって回収し、そして高真空下で一晩乾燥させて、{4−[1−(2,4−ジクロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(57.8mg、80%)を白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.5 (br. s, 1 H), 8.46 (br. s, 1 H), 7.90 (dd, J=8.80, 6.50 Hz, 1 H), 7.76 - 7.86 (m, 1 H), 7.68 (m, 2 H), 7.50 (d, J=8.78 Hz, 1 H), 7.40 (dd, J=8.70, 2.30 Hz, 1 H), 7.30 - 7.37 (m, 1 H), 4.20 - 4.32 (m, 2 H), 3.68 - 3.97 (m, 4 H), 3.02 - 3.24 (m, 2 H), 2.43 (s, 3 H), 2.16 - 2.40 (m, 2 H), 1.61 - 1.70 (m, 2 H). MS C25H24Cl2FN2O3[(M+H)+]の計算値:489, 実測値489.1.
{4−[1−(2−クロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸
{4−[1−(2−クロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
メタノール(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;153mg、0.485mmol)の溶液を、窒素下、室温で無希釈1−クロロ−2−イソシアナトベンゼン(81.9mg、0.534mmol)に加えた。得られた明褐色の溶液を、15時間加熱還流し、この時点で、LCMS分析は、かなりの出発材料の存在を示した。還流を週末にかけて続けた。LCMS分析は、反応進行においてほとんど変化を示さなかった。反応混合物に、室温でN,N−ジイソプロピルエチルアミン(93.2μL、0.534mmol)を加え、そして次に反応混合物を、24時間再度灌流した。かなりの出発材料が依然として残った。反応混合物を、水(25mL)で希釈し、そして得られた混合物を、酢酸エチルで抽出した(2×50mL)。合わせた抽出物を、ブラインで洗浄し、無水MgSO4で乾燥させ、濾過し、そして濃縮して、粗生成物を半固体(230mg)として得た。フラッシュクロマトグラフィー(40g ISCOカラム、0〜100%酢酸エチル/ヘキサン、そして最終的には25% MeOH/ジクロロメタンで溶離する)により、{4−[1−(2−クロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(10.5mg、4.6%)を粘性油状物として得た。MS C26H27ClFN2O3[(M+H)+]の計算値469, 実測値469.0.
THF(2.0mL)中の{4−[1−(2−クロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(10mg、0.021mmol)の溶液に、室温で水(0.5mL)中の水酸化リチウム一水和物(10.2mg、0.426mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下、室温で15時間攪拌した。溶媒を除去し、そして水層を、1.0N HClで中和した。得られた白色の固体を、濾過によって回収し、水及びヘキサンで洗浄し、そして空気中で乾燥させて、{4−[1−(2−クロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸(4.5mg、46%)を白色の固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.42 (br. s, 1 H), 7.9 (dd, J=8.91, 6.65 Hz, 1 H), 7.69 (s, 1 H), 7.48 (d, J=8.03 Hz, 2 H), 7.25 - 7.42 (m, 3 H), 7.09 - 7.23 (m, 1 H), 4.27 (d, J=10.79 Hz, 2 H), 3.79 (br. s, 4 H), 2.43 (s, 4 H), 1.65 (d, J=12.05 Hz, 3 H), 1.23 (s, 2 H). MS C25H25ClFN2O3[(M+H)+]の計算値455, 実測値455.0.
{6−フルオロ−4−[1−(2−メトキシ−フェニルカルバモイル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸
{6−フルオロ−4−[1−(2−メトキシ−フェニルカルバモイル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(8.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;83.6mg、0.265mmol)の溶液に、窒素下、室温でイソシアン酸2−メトキシフェニル(71μL、0.53mmol)を加えた。得られた明褐色の溶液を、室温で10分間攪拌した。反応混合物を、水(約25mL)で希釈し、そして得られた混合物を、塩化メチレンで抽出した(2×30mL)。合わせた有機層を、飽和NaHCO3水溶液で洗浄し、無水Na2SO4で乾燥させ、濾過し、そして濃縮して、明黄色の膜状物にした。フラッシュクロマトグラフィー(ヘキサン中0〜60%酢酸エチル)により、{6−フルオロ−4−[1−(2−メトキシ−フェニルカルバモイル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(107.6mg、87%)を無色の膜状物として得た。MS C27H30FN2O4 [(M+H)+]の計算値:465, 実測値465.3.
THF(8.0mL)中の{6−フルオロ−4−[1−(2−メトキシ−フェニルカルバモイル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸メチルエステル(107.6mg、0.232mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(111.1mg、2.6mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下、室温で一晩攪拌した。溶媒を蒸発させた。水(約25mL)を加え、そして混合物を、1.0N HCl水溶液(4.6mL)で酸性化した。得られた白色の沈殿物を、濾過によって回収し、そして高真空下で乾燥させて、{6−フルオロ−4−[1−(2−メトキシ−フェニルカルバモイル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸(86.4mg、83%)を白色の粉末として得た。1H NMR (300 MHz, DMSO-d6) δ ppm 12.45 (b. s, 1 H), 7.90 (dd, J=9.23, 6.59 Hz, 1 H), 7.78 (br. s., 1 H), 7.65 - 7.72 (m, 2 H), 7.35 (t, J=8.01 Hz, 1 H), 6.99 - 7.06 (m, 2 H), 6.86 - 6.95 (m, 1 H), 4.23 (m, 2 H), 3.82 (s, 3 H), 3.78 (br. s., 2 H), 3.32 (br. s., 3 H), 3.09 (br. s., 2 H), 2.43 (s, 3 H), 1.66 (m, 2 H). MS C26H28FN2O4[(M+H)+]の計算値:451, 実測値451.2.
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−フェニルカルバモイル)−ピペリジン−4−イル−ナフタレン−2−イル}−酢酸
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−フェニルカルバモイル)−ピペリジン−4−イル−ナフタレン−2−イル}−酢酸メチルエステル
塩化メチレン(6.0mL)中の(6−フルオロ−3−メチル−4−ピペリジン−4−イル−ナフタレン−2−イル)−酢酸メチルエステル(これを先に記載したように調製することもできる;94mg、0.298mmol)の溶液に、窒素下、室温でイソシアン酸トリフルオロ−m−トリル(82μL、0.596mmol)を加えた。得られた明褐色の溶液を、室温で40分間攪拌した。反応混合物を、水(約25mL)で希釈し、そして得られた混合物を、塩化メチレンで抽出した(3×25mL)。合わせた有機層を、飽和NaHCO3水溶液で洗浄し、無水Na2SO4で乾燥させ、濾過し、そして濃縮して、橙黄色の膜状物にした。シリカゲルクロマトグラフィー(ヘキサン中20〜25%酢酸エチル)により、{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−フェニルカルバモイル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(97mg、65%)を無色の膜状物として得た。MS C27H27F4N2O3[(M+H)+]の計算値:503, 実測値503.2.
THF(8.0mL)中の{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−フェニルカルバモイル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸メチルエステル(97mg、0.193mmol)の溶液に、室温で水(2.0mL)中の水酸化リチウム一水和物(92.4mg、2.2mmol)の溶液を加えた。得られた溶液を、窒素雰囲気下、4時間攪拌した。溶媒を蒸発させた。水(約25mL)を加え、そして混合物を、1.0N HCl水溶液(3.9mL)で酸性化した。得られた白色の沈殿物を、濾過によって回収し、水及びヘキサンで洗浄し、そして高真空下で乾燥させて、{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−フェニルカルバモイル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸(65.8mg、70%)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 12.50 (br. s, 1 H), 8.97 (br. s, 1 H), 8.00 - 8.22 (m, 1 H), 7.97 (s, 1 H), 7.90 (dd, J=9.10, 6.50 Hz, 1 H), 7.78 (d, J=8.50 Hz, 1 H), 7.66 - 7.73 (m, 1 H), 7.49 (t, J=8.30 Hz, 1 H), 7.30 - 7.38 (m, 1 H), 7.28 (d, J=8.30 Hz, 1 H), 4.28 - 4.36 (m, 2 H), 3.71 - 3.85 (m, 3 H), 3.01 - 3.18 (m, 2 H), 2.43 (s, 3 H), 2.17 - 2.34 (m, 1 H), 1.64 - 1.73 (m, 2 H). MS C26H25F4N2O3[(M+H)+]の計算値:489, 実測値489.2.
化合物の活性及び使用
式Iの化合物は、有益な薬理学的特性を持つ。該化合物は、CRTH2受容体においてアンタゴニストであり、その受容体に関連する疾患及び障害、例えば喘息を処置するのに有用でありうるということが見出された。CRTH2受容体アンタゴニストとしての本化合物の活性は、以下の生物学的アッセイによって証明される。
細胞培養条件:Gα16で予めトランスフェクトしておいたCHO−K1細胞を、その後、ヒトCRTH2受容体及びネオマイシン耐性遺伝子でトランスフェクトした。800μg/mL G418(ジェネティシン)での選択に続いて、個々のクローンを、抗ヒトCRTH2 IgGでの染色に基づいて、それらの受容体発現についてアッセイし、続いてCa2+フラックスアッセイにおいて13,14−ジヒドロ−15−ケトプロスタグランジンD2(DK−PDG2)(リガンド)に対するそれらの応答についてアッセイした。次に陽性クローンを、限界希釈クローニングによってクローン化した。トランスフェクトした細胞を、10%ウシ胎仔血清、2mM グルタミン、100U/mL ペニシリン/100μg/mL ストレプトマイシン、200μg/mL ヒグロマイシンB、及び800μg/mL G418(ジェネティシン)を補充したHam’s F−12培地中で培養した。細胞を、トリプシン−EDTA(トリプシン−エチレンジアミンテトラ酢酸)で収集し、そしてViaCount(登録商標)試薬(Guava Technologies, Inc.から。これは、試薬使用者が生存細胞と非生存細胞とを区別できるようにする2種類のDNA結合色素を含有する)を使用してカウントした。細胞懸濁液容量を、完全増殖培地で2.5×105細胞/mLに調整した。50μLのアリコートを、BD Falcon(商標)384ウェル黒色/透明マイクロプレート(Becton, Dickinson and Companyの一部門である、BD Biosciencesより)に分配し、そしてマイクロプレートを、37℃のCO2インキュベーター内に一晩置いた。次の日、マイクロプレートを、アッセイに使用した。
色素(MDS Analytical Technologies and MDS Inc.の一部門であるMolecular Devicesからの、FLIPR(登録商標)カルシウム3アッセイキットから)を含有する添加緩衝液を、1ボトルの内容物を20mM HEPES(4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸)及び2.5mM プロベネシドを含有するハンクス平衡塩類溶液 200mLに溶解することによって調製した。増殖培地を、細胞プレートから取り除き、そして20mM HEPES、0.05% BSA及び2.5mM プロベネシドを含有するハンクス平衡塩類溶液(HBSS) 25μLを各ウェルに加え、続いてマルチドロップディスペンサー(Multidrop dispenser)を使用して、希釈された色素 25μLを加えた。次に、プレートを37℃で1時間インキュベートした。
2型ヘルパーT(Th2)細胞における13,14−ジヒドロ−15−ケトプロスタグランジンD2(DK−PGD2)−誘導IL−13生成の阻害を適用して、化合物細胞効力を評価した。
Claims (21)
- 式(I):
[式中、
Xは、−SO2−又は−C(O)−であり;
R1は、ハロゲンであり;
R2は、− 非置換であるか、又はC 1 −C 7 アルキル、ハロゲン、アルコキシ、−SO2−C 1 −C 7 アルキルもしくはハロアルキルで独立に一もしくは二置換されている、フェニル、
− 非置換ヘテロアリール、
− 非置換であるか、又は非置換フェニル又はハロゲン、NO2もしくはNH2で置換されているフェニルで置換されている、C 1 −C 7 アルキル、
− 非置換シクロアルキル、
− 非置換ヘテロシクロアルキル、あるいは
− NH−フェニル(該フェニルは、非置換であるか、又はハロゲン、C 1 −C 7 アルキル、ハロアルキル、−SO2−C 1 −C 7 アルキルもしくはアルコキシで置換されている)であり;そして
R3は、C 1 −C 7 アルキル又は水素である]で示される化合物又はその薬学的に許容しうる塩。 - Xが−SO2−である、請求項1記載の化合物。
- Xが−C(O)−である、請求項1記載の化合物。
- R1がフッ素である、請求項1〜3のいずれか一項記載の化合物。
- Xが−SO2−であり、そしてR2が、非置換であるか、又はC 1 −C 7 アルキル、ハロゲン、アルコキシ、−SO2−C 1 −C 7 アルキルもしくは−CF3で独立に一もしくは二置換されている、フェニル;非置換ヘテロアリール;あるいは、非置換であるか、又は非置換フェニル又はハロゲン、NO2もしくはNH2で置換されているフェニルで置換されている、C 1 −C 7 アルキルである、請求項1又は2のいずれか一項記載の化合物。
- Xが−SO2−であり、そしてR2が、非置換シクロアルキル;非置換ヘテロシクロアルキル;又は、NH−フェニル(該フェニルは、非置換であるか、又はハロゲン、C 1 −C 7 アルキル、ハロアルキル、−SO2−C 1 −C 7 アルキルもしくはアルコキシで置換されている)である、請求項1又は2のいずれか一項記載の化合物。
- R2が、フェニル、メチルフェニル、クロロフェニル、メタンスルホニルフェニル、ジクロロフェニル、ジフルオロフェニル、トリフルオロメチルフェニル、ビス−トリフルオロメチルフェニル又はメトキシフェニルである、請求項1〜5のいずれか一項記載の化合物。
- R2がピリジニルである、請求項1〜4のいずれか一項記載の化合物。
- R2が、メチル、又はフェニル、クロロフェニル、ニトロフェニルもしくはアミノフェニルで置換されているメチルである、請求項1〜4のいずれか一項記載の化合物。
- R2がシクロペンチルである、請求項1〜4のいずれか一項記載の化合物。
- R2がピロリジニルである、請求項1〜4のいずれか一項記載の化合物。
- R2が、−NH−ジフルオロフェニル、−NH−ジクロロフェニル、−NH−クロロフェニル、−NH−メトキシフェニル又は−NH−トリフルオロメチルフェニルである、請求項1〜4のいずれか一項記載の化合物。
- R3がメチルである、請求項1〜12のいずれか一項記載の化合物。
- R3が水素である、請求項1〜12のいずれか一項記載の化合物。
- 化合物が以下:
[4−(1−ベンゼンスルホニル−ピペリジン−4−イル)−6−フルオロ−ナフタレン−2−イル]−酢酸;
{6−フルオロ−4−[1−(トルエン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(ピリジン−2−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(ピリジン−3−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(ピリジン−4−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(3−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(4−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(3−メタンスルホニル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(3,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(3,5−ビス−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−ナフタレン−2−イル}−酢酸;
{4−[1−(2−クロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
[6−フルオロ−4−(1−メタンスルホニル−ピペリジン−4−イル)−3−メチル−ナフタレン−2−イル]−酢酸;
{4−[1−(2,4−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,5−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,6−ジクロロ−ベンゼンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
[6−フルオロ−3−メチル−4−(1−フェニルメタンスルホニル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸;
{6−フルオロ−4−[1−(2−メトキシ−ベンゼンスルホニル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(3−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(4−クロロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−ベンゼンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
[4−(1−シクロペンタンスルホニル−ピペリジン−4−イル)−6−フルオロ−3−メチル−ナフタレン−2−イル]−酢酸;
{6−フルオロ−3−メチル−4−[1−(ピロリジン−1−スルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{6−フルオロ−3−メチル−4−[1−(2−ニトロ−フェニルメタンスルホニル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸;
{4−[1−(2−アミノ−フェニルメタンスルホニル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,4−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,6−ジクロロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,5−ジフルオロ−ベンゾイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
[6−フルオロ−3−メチル−4−(1−フェニルアセチル−ピペリジン−4−イル)−ナフタレン−2−イル]−酢酸;
{4−[1−(2,6−ジフルオロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2,4−ジクロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{4−[1−(2−クロロ−フェニルカルバモイル)−ピペリジン−4−イル]−6−フルオロ−3−メチル−ナフタレン−2−イル}−酢酸;
{6−フルオロ−4−[1−(2−メトキシ−フェニルカルバモイル)−ピペリジン−4−イル]−3−メチル−ナフタレン−2−イル}−酢酸;又は
{6−フルオロ−3−メチル−4−[1−(3−トリフルオロメチル−フェニルカルバモイル)−ピペリジン−4−イル]−ナフタレン−2−イル}−酢酸
である、請求項1記載の化合物。 - 治療有効量の請求項1〜15のいずれか一項記載の化合物及び薬学的に許容しうる担体を含む、医薬組成物。
- 治療活性物質としての使用のための、請求項1〜15のいずれか一項記載の化合物。
- 呼吸障害の治療又は予防のための請求項16記載の医薬組成物。
- 呼吸障害の治療又は予防のための医薬の製造のための、請求項1〜15のいずれか一項記載の化合物の使用。
- 呼吸障害の治療又は予防のための、請求項1〜15のいずれか一項記載の化合物。
- 慢性閉塞性肺障害(COPD)、喘息及び気管支痙攣より選択される呼吸障害を処置するための、請求項16記載の医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161569313P | 2011-12-12 | 2011-12-12 | |
| US61/569,313 | 2011-12-12 | ||
| PCT/EP2012/074882 WO2013087544A1 (en) | 2011-12-12 | 2012-12-10 | Piperidinyl naphthylacetic acids |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2015504863A JP2015504863A (ja) | 2015-02-16 |
| JP6154397B2 true JP6154397B2 (ja) | 2017-06-28 |
Family
ID=47427287
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014546433A Expired - Fee Related JP6154397B2 (ja) | 2011-12-12 | 2012-12-10 | ピペリジニルナフチル酢酸 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US8691993B2 (ja) |
| EP (1) | EP2791111B1 (ja) |
| JP (1) | JP6154397B2 (ja) |
| KR (1) | KR20140103319A (ja) |
| CN (1) | CN103958468B (ja) |
| BR (1) | BR112014013958A8 (ja) |
| CA (1) | CA2855940A1 (ja) |
| HK (1) | HK1200453A1 (ja) |
| MX (1) | MX350862B (ja) |
| RU (1) | RU2628083C2 (ja) |
| WO (1) | WO2013087544A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2624379T3 (es) | 2011-12-21 | 2017-07-14 | Idorsia Pharmaceuticals Ltd | Derivados de heterociclilo y su uso como moduladores del receptor de prostaglandina D2 |
| WO2014006585A1 (en) | 2012-07-05 | 2014-01-09 | Actelion Pharmaceuticals Ltd | 1-phenyl-substituted heterocyclyl derivatives and their use as prostaglandin d2 receptor modulators |
| CA2889922C (en) | 2012-11-06 | 2016-01-19 | Evolution Engineering Inc. | Fluid pressure pulse generator and method of using same |
| CA2895346C (en) | 2012-12-17 | 2018-10-23 | Evolution Engineering Inc. | Downhole telemetry signal modulation using pressure pulses of multiple pulse heights |
| US10753201B2 (en) | 2012-12-17 | 2020-08-25 | Evolution Engineering Inc. | Mud pulse telemetry apparatus with a pressure transducer and method of operating same |
| EP3000961A1 (en) | 2012-12-17 | 2016-03-30 | Evolution Engineering Inc. | Method of operating a mud pulse telemetry apparatus with a pressure transducer |
| US9670774B2 (en) | 2014-06-27 | 2017-06-06 | Evolution Engineering Inc. | Fluid pressure pulse generator for a downhole telemetry tool |
| CA2895681A1 (en) | 2014-06-27 | 2015-12-27 | Evolution Engineering Inc. | Fluid pressure pulse generator for a downhole telemetry tool |
| CA2895680A1 (en) | 2014-06-27 | 2015-12-27 | Evolution Engineering Inc. | Fluid pressure pulse generator for a downhole telemetry tool |
| WO2020006557A1 (en) * | 2018-06-29 | 2020-01-02 | Adonia Papathanassiu | Compositions and methods of using itaconic acid derivatives |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4443462A (en) * | 1979-08-06 | 1984-04-17 | Merrell Dow Pharmaceuticals Inc. | Antipsychotic 4-(naphthalenyloxy)piperidine derivatives |
| MX2007010215A (es) * | 2005-02-24 | 2007-11-07 | Millennium Pharm Inc | Antagonistas del receptor pgd2 para el tratamiento de enfermedades inflamatorias. |
| EP2407453A1 (en) * | 2005-09-27 | 2012-01-18 | Shionogi & Co., Ltd. | PGD2 receptor antagonist |
| JP2009539881A (ja) * | 2006-06-09 | 2009-11-19 | アイコス コーポレイション | Dp−2アンタゴニストとしての置換フェニル酢酸 |
| WO2010045276A2 (en) * | 2008-10-16 | 2010-04-22 | Cara Therapeutics, Inc. | Azabenzimidazolones |
| US8621184B1 (en) | 2008-10-31 | 2013-12-31 | Netapp, Inc. | Effective scheduling of producer-consumer processes in a multi-processor system |
| US8188090B2 (en) | 2008-11-17 | 2012-05-29 | Hoffman-La Roche Inc. | Naphthylacetic acids |
| DK2346819T3 (da) * | 2008-11-17 | 2013-05-13 | Hoffmann La Roche | Naphthyleddikesyre |
-
2012
- 2012-12-06 US US13/706,497 patent/US8691993B2/en not_active Expired - Fee Related
- 2012-12-10 CN CN201280057114.9A patent/CN103958468B/zh not_active Expired - Fee Related
- 2012-12-10 RU RU2014127143A patent/RU2628083C2/ru not_active IP Right Cessation
- 2012-12-10 KR KR1020147019166A patent/KR20140103319A/ko not_active Application Discontinuation
- 2012-12-10 EP EP12805511.8A patent/EP2791111B1/en not_active Not-in-force
- 2012-12-10 JP JP2014546433A patent/JP6154397B2/ja not_active Expired - Fee Related
- 2012-12-10 MX MX2014006468A patent/MX350862B/es active IP Right Grant
- 2012-12-10 CA CA2855940A patent/CA2855940A1/en not_active Abandoned
- 2012-12-10 BR BR112014013958A patent/BR112014013958A8/pt not_active Application Discontinuation
- 2012-12-10 WO PCT/EP2012/074882 patent/WO2013087544A1/en active Application Filing
-
2015
- 2015-01-28 HK HK15100955.6A patent/HK1200453A1/xx not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| US8691993B2 (en) | 2014-04-08 |
| BR112014013958A2 (pt) | 2017-06-13 |
| MX350862B (es) | 2017-09-19 |
| MX2014006468A (es) | 2014-09-01 |
| CA2855940A1 (en) | 2013-06-20 |
| WO2013087544A1 (en) | 2013-06-20 |
| BR112014013958A8 (pt) | 2017-06-13 |
| JP2015504863A (ja) | 2015-02-16 |
| CN103958468B (zh) | 2017-03-01 |
| EP2791111A1 (en) | 2014-10-22 |
| RU2628083C2 (ru) | 2017-08-14 |
| CN103958468A (zh) | 2014-07-30 |
| EP2791111B1 (en) | 2017-01-18 |
| RU2014127143A (ru) | 2016-02-10 |
| KR20140103319A (ko) | 2014-08-26 |
| US20130150407A1 (en) | 2013-06-13 |
| HK1200453A1 (en) | 2015-08-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6154397B2 (ja) | ピペリジニルナフチル酢酸 | |
| EP2358677B1 (en) | Naphthylacetic acids used as crth2 antagonists or partial agonists | |
| ES2378755T3 (es) | �?cidos aminotetrahidroindazoloacéticos | |
| JP5302398B2 (ja) | 置換アミノテトラリン | |
| AU2013254657B2 (en) | Novel compounds | |
| JP5394488B2 (ja) | アミノテトラヒドロインダゾロ酢酸 | |
| JP6654574B2 (ja) | Cb2アゴニストとして有用なピリジン−2−アミド | |
| CA2654927A1 (en) | Substituted phenyl acetic acids as dp-2 antagonists | |
| CA2732210A1 (en) | Monoaryl aminotetralines | |
| MX2011005200A (es) | Acidos naftilaceticos. | |
| CN110753690A (zh) | 吡啶衍生物 | |
| US9000044B2 (en) | Substituted naphthylacetic acids | |
| US8470884B2 (en) | Alkenyl naphthylacetic acids | |
| ES2273278T3 (es) | Sulfonamidas de hidroxamato como inhibidores de la escision de cd23. | |
| KR20050018938A (ko) | Ccr-3 수용체 길항제 ix로서 2,5-치환된 피리미딘유도체 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20150312 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20151209 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20160630 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160830 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20161111 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170118 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170224 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20170509 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20170601 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6154397 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |