JP5009621B2 - チオール開裂し得るマイトマイシンコンジュゲート - Google Patents
チオール開裂し得るマイトマイシンコンジュゲート Download PDFInfo
- Publication number
- JP5009621B2 JP5009621B2 JP2006532557A JP2006532557A JP5009621B2 JP 5009621 B2 JP5009621 B2 JP 5009621B2 JP 2006532557 A JP2006532557 A JP 2006532557A JP 2006532557 A JP2006532557 A JP 2006532557A JP 5009621 B2 JP5009621 B2 JP 5009621B2
- Authority
- JP
- Japan
- Prior art keywords
- mitomycin
- lipid
- liposomes
- drug
- conjugate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 title claims abstract description 320
- 229960004857 mitomycin Drugs 0.000 title claims abstract description 222
- 238000003776 cleavage reaction Methods 0.000 title abstract description 10
- 230000007017 scission Effects 0.000 title abstract description 10
- 229930192392 Mitomycin Natural products 0.000 title description 4
- 125000003396 thiol group Chemical class [H]S* 0.000 title description 2
- 239000002502 liposome Substances 0.000 claims abstract description 161
- 150000002632 lipids Chemical class 0.000 claims abstract description 66
- 125000004396 dithiobenzyl group Chemical group 0.000 claims abstract description 23
- 230000002209 hydrophobic effect Effects 0.000 claims abstract description 21
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 claims description 73
- 239000000203 mixture Substances 0.000 claims description 61
- 235000012000 cholesterol Nutrition 0.000 claims description 36
- 238000011282 treatment Methods 0.000 claims description 15
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 claims description 12
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 claims description 12
- 239000000232 Lipid Bilayer Substances 0.000 claims description 11
- 150000001982 diacylglycerols Chemical class 0.000 claims description 8
- 150000003904 phospholipids Chemical class 0.000 claims description 8
- 238000010348 incorporation Methods 0.000 claims description 5
- 150000003335 secondary amines Chemical group 0.000 claims description 3
- 239000003814 drug Substances 0.000 abstract description 105
- 229940079593 drug Drugs 0.000 abstract description 95
- 150000001875 compounds Chemical class 0.000 abstract description 24
- 238000000034 method Methods 0.000 abstract description 24
- 229940002612 prodrug Drugs 0.000 abstract description 21
- 239000000651 prodrug Substances 0.000 abstract description 21
- 238000001727 in vivo Methods 0.000 abstract description 14
- 239000003638 chemical reducing agent Substances 0.000 abstract description 9
- 230000001988 toxicity Effects 0.000 abstract description 5
- 231100000419 toxicity Toxicity 0.000 abstract description 5
- 230000004044 response Effects 0.000 abstract description 4
- 238000007079 thiolysis reaction Methods 0.000 abstract 1
- 210000004027 cell Anatomy 0.000 description 72
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 67
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 67
- 235000018417 cysteine Nutrition 0.000 description 67
- 241000699670 Mus sp. Species 0.000 description 60
- 206010028980 Neoplasm Diseases 0.000 description 53
- 238000009472 formulation Methods 0.000 description 41
- 230000006870 function Effects 0.000 description 31
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 30
- 238000012360 testing method Methods 0.000 description 29
- 238000002360 preparation method Methods 0.000 description 26
- 239000000243 solution Substances 0.000 description 26
- 238000006243 chemical reaction Methods 0.000 description 24
- 230000012010 growth Effects 0.000 description 22
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 21
- 241001465754 Metazoa Species 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical group CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- 231100000135 cytotoxicity Toxicity 0.000 description 17
- 230000003013 cytotoxicity Effects 0.000 description 17
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 16
- 239000010432 diamond Substances 0.000 description 16
- 238000011081 inoculation Methods 0.000 description 16
- 229920001223 polyethylene glycol Polymers 0.000 description 16
- 210000004881 tumor cell Anatomy 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- 241000699666 Mus <mouse, genus> Species 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 238000004128 high performance liquid chromatography Methods 0.000 description 12
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- 238000000338 in vitro Methods 0.000 description 11
- 201000011510 cancer Diseases 0.000 description 10
- 229940124597 therapeutic agent Drugs 0.000 description 10
- 150000003573 thiols Chemical class 0.000 description 10
- 239000002202 Polyethylene glycol Substances 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- 229960004679 doxorubicin Drugs 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 239000000523 sample Substances 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 230000015556 catabolic process Effects 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 238000001802 infusion Methods 0.000 description 8
- 229920000642 polymer Polymers 0.000 description 8
- 241000700159 Rattus Species 0.000 description 7
- 239000011248 coating agent Substances 0.000 description 7
- 238000000576 coating method Methods 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 7
- 238000011534 incubation Methods 0.000 description 7
- 238000001990 intravenous administration Methods 0.000 description 7
- 108090000765 processed proteins & peptides Proteins 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- -1 thiocarbonate Chemical compound 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000002246 antineoplastic agent Substances 0.000 description 6
- 230000017531 blood circulation Effects 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 229920001477 hydrophilic polymer Polymers 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 230000036457 multidrug resistance Effects 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 241001460678 Napo <wasp> Species 0.000 description 5
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 230000010261 cell growth Effects 0.000 description 5
- 210000002683 foot Anatomy 0.000 description 5
- 125000003827 glycol group Chemical group 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 229920001184 polypeptide Polymers 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 238000003908 quality control method Methods 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 210000003371 toe Anatomy 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- DRSHXJFUUPIBHX-UHFFFAOYSA-N COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 Chemical compound COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 DRSHXJFUUPIBHX-UHFFFAOYSA-N 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 4
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 229930182558 Sterol Natural products 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 238000002512 chemotherapy Methods 0.000 description 4
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 4
- 229960004630 chlorambucil Drugs 0.000 description 4
- 238000011026 diafiltration Methods 0.000 description 4
- 239000003640 drug residue Substances 0.000 description 4
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 4
- 238000001125 extrusion Methods 0.000 description 4
- 229960002949 fluorouracil Drugs 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 4
- 150000003432 sterols Chemical class 0.000 description 4
- 235000003702 sterols Nutrition 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- 231100000331 toxic Toxicity 0.000 description 4
- 230000002588 toxic effect Effects 0.000 description 4
- PORPENFLTBBHSG-MGBGTMOVSA-N 1,2-dihexadecanoyl-sn-glycerol-3-phosphate Chemical group CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCC PORPENFLTBBHSG-MGBGTMOVSA-N 0.000 description 3
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 3
- RBTBFTRPCNLSDE-UHFFFAOYSA-N 3,7-bis(dimethylamino)phenothiazin-5-ium Chemical compound C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 RBTBFTRPCNLSDE-UHFFFAOYSA-N 0.000 description 3
- 239000004254 Ammonium phosphate Substances 0.000 description 3
- 101150065749 Churc1 gene Proteins 0.000 description 3
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 3
- 102100038239 Protein Churchill Human genes 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 235000021355 Stearic acid Nutrition 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 229910000148 ammonium phosphate Inorganic materials 0.000 description 3
- 235000019289 ammonium phosphates Nutrition 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 230000004087 circulation Effects 0.000 description 3
- 208000029742 colonic neoplasm Diseases 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 239000008367 deionised water Substances 0.000 description 3
- 229910021641 deionized water Inorganic materials 0.000 description 3
- MNNHAPBLZZVQHP-UHFFFAOYSA-N diammonium hydrogen phosphate Chemical compound [NH4+].[NH4+].OP([O-])([O-])=O MNNHAPBLZZVQHP-UHFFFAOYSA-N 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 229960005420 etoposide Drugs 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229960000907 methylthioninium chloride Drugs 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical group CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000012265 solid product Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000008117 stearic acid Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000004580 weight loss Effects 0.000 description 3
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 2
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 2
- 0 *CC1CCCC1 Chemical compound *CC1CCCC1 0.000 description 2
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- LVNGJLRDBYCPGB-UHFFFAOYSA-N 1,2-distearoylphosphatidylethanolamine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(COP([O-])(=O)OCC[NH3+])OC(=O)CCCCCCCCCCCCCCCCC LVNGJLRDBYCPGB-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- KEPSVHAYVNZDDW-UHFFFAOYSA-N 3-(2,3-dihydroxypropyldisulfanyl)propane-1,2-diol Chemical compound OCC(O)CSSCC(O)CO KEPSVHAYVNZDDW-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- 229930003347 Atropine Natural products 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- 108010017384 Blood Proteins Proteins 0.000 description 2
- 102000004506 Blood Proteins Human genes 0.000 description 2
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 206010059866 Drug resistance Diseases 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 2
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 101150066553 MDR1 gene Proteins 0.000 description 2
- HYFMSAFINFJTFH-UHFFFAOYSA-N Mitomycin-A Natural products O=C1C(OC)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)N2CC2NC21 HYFMSAFINFJTFH-UHFFFAOYSA-N 0.000 description 2
- FIWILGQIZHDAQG-UHFFFAOYSA-N NC1=C(C(=O)NCC2=CC=C(C=C2)OCC(F)(F)F)C=C(C(=N1)N)N1N=C(N=C1)C1(CC1)C(F)(F)F Chemical compound NC1=C(C(=O)NCC2=CC=C(C=C2)OCC(F)(F)F)C=C(C(=N1)N)N1N=C(N=C1)C1(CC1)C(F)(F)F FIWILGQIZHDAQG-UHFFFAOYSA-N 0.000 description 2
- 102000002933 Thioredoxin Human genes 0.000 description 2
- OIRDTQYFTABQOQ-UHTZMRCNSA-N Vidarabine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O OIRDTQYFTABQOQ-UHTZMRCNSA-N 0.000 description 2
- JLPULHDHAOZNQI-JLOPVYAASA-N [(2r)-3-hexadecanoyloxy-2-[(9e,12e)-octadeca-9,12-dienoyl]oxypropyl] 2-(trimethylazaniumyl)ethyl phosphate Chemical class CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCC\C=C\C\C=C\CCCCC JLPULHDHAOZNQI-JLOPVYAASA-N 0.000 description 2
- 229960004150 aciclovir Drugs 0.000 description 2
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 150000001263 acyl chlorides Chemical class 0.000 description 2
- 229930013930 alkaloid Natural products 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 2
- 229960000396 atropine Drugs 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 229920001429 chelating resin Polymers 0.000 description 2
- 229940044683 chemotherapy drug Drugs 0.000 description 2
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 230000000887 hydrating effect Effects 0.000 description 2
- 229960004716 idoxuridine Drugs 0.000 description 2
- 239000002198 insoluble material Substances 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 231100001231 less toxic Toxicity 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229920001427 mPEG Polymers 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- HYFMSAFINFJTFH-NGSRAFSJSA-N mitomycin A Chemical compound O=C1C(OC)=C(C)C(=O)C2=C1[C@@H](COC(N)=O)[C@]1(OC)N2C[C@@H]2N[C@@H]21 HYFMSAFINFJTFH-NGSRAFSJSA-N 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 2
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 2
- 229960002340 pentostatin Drugs 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 2
- 150000003905 phosphatidylinositols Chemical class 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 235000013772 propylene glycol Nutrition 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 239000013062 quality control Sample Substances 0.000 description 2
- 229960001404 quinidine Drugs 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 208000016691 refractory malignant neoplasm Diseases 0.000 description 2
- 230000003307 reticuloendothelial effect Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- YNQYZBDRJZVSJE-UHFFFAOYSA-M sodium;2,3-dihydroxypropyl 2,3-di(octadecanoyloxy)propyl phosphate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC(=O)OCC(COP([O-])(=O)OCC(O)CO)OC(=O)CCCCCCCCCCCCCCCCC YNQYZBDRJZVSJE-UHFFFAOYSA-M 0.000 description 2
- 239000012086 standard solution Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 108060008226 thioredoxin Proteins 0.000 description 2
- 229940094937 thioredoxin Drugs 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- 229960003636 vidarabine Drugs 0.000 description 2
- 229960002555 zidovudine Drugs 0.000 description 2
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical group C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 206010065553 Bone marrow failure Diseases 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 206010012335 Dependence Diseases 0.000 description 1
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 238000000729 Fisher's exact test Methods 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 108010090054 Membrane Glycoproteins Proteins 0.000 description 1
- 102000012750 Membrane Glycoproteins Human genes 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 206010034133 Pathogen resistance Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- 239000004695 Polyether sulfone Substances 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 101710167005 Thiol:disulfide interchange protein DsbD Proteins 0.000 description 1
- 206010070863 Toxicity to various agents Diseases 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- QNEPTKZEXBPDLF-JDTILAPWSA-N [(3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-yl] carbonochloridate Chemical compound C1C=C2C[C@@H](OC(Cl)=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 QNEPTKZEXBPDLF-JDTILAPWSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 201000005188 adrenal gland cancer Diseases 0.000 description 1
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 229920006187 aquazol Polymers 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000003012 bilayer membrane Substances 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 229920000547 conjugated polymer Polymers 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 150000002009 diols Chemical group 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 238000002296 dynamic light scattering Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 125000005313 fatty acid group Chemical group 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 239000012510 hollow fiber Substances 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 238000011221 initial treatment Methods 0.000 description 1
- 229910052816 inorganic phosphate Inorganic materials 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 238000001906 matrix-assisted laser desorption--ionisation mass spectrometry Methods 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- PJYWOULTKKMXAA-UHFFFAOYSA-N methyl (2-aminoethyldisulfanyl)formate Chemical compound COC(=O)SSCCN PJYWOULTKKMXAA-UHFFFAOYSA-N 0.000 description 1
- AGJSNMGHAVDLRQ-IWFBPKFRSA-N methyl (2s)-2-[[(2s)-2-[[(2s)-2-[[(2r)-2-amino-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,3-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoate Chemical compound SC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(=O)OC)CC1=CC=C(O)C(C)=C1C AGJSNMGHAVDLRQ-IWFBPKFRSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- OAOSXODRWGDDCV-UHFFFAOYSA-N n,n-dimethylpyridin-4-amine;4-methylbenzenesulfonic acid Chemical compound CN(C)C1=CC=NC=C1.CC1=CC=C(S(O)(=O)=O)C=C1 OAOSXODRWGDDCV-UHFFFAOYSA-N 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- UQDHUTAFUQLDFX-UHFFFAOYSA-N phenyl(sulfanyl)methanol Chemical compound OC(S)C1=CC=CC=C1 UQDHUTAFUQLDFX-UHFFFAOYSA-N 0.000 description 1
- 125000001095 phosphatidyl group Chemical group 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 229920002432 poly(vinyl methyl ether) polymer Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 229920005604 random copolymer Polymers 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000006748 scratching Methods 0.000 description 1
- 230000002393 scratching effect Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000007447 staining method Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 238000003239 susceptibility assay Methods 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 239000002676 xenobiotic agent Substances 0.000 description 1
- 230000002034 xenobiotic effect Effects 0.000 description 1
Images






























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/095—Sulfur, selenium, or tellurium compounds, e.g. thiols
- A61K31/105—Persulfides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/543—Lipids, e.g. triglycerides; Polyamines, e.g. spermine or spermidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6905—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion
- A61K47/6911—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion the form being a liposome
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Dispersion Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
HARRISON’S PRINCIPLES OF INTERNAL MEDICINE、Wilson他編集、第12版、パート11、1592頁、1991 Endicott,J.およびLing,V.、Annu.Rev.Biochem.、58:137−171、(1989) Gottesman,M.M.、Cancer Research、53:747−754(1993) Gottesman,M.他、Current Opinion in Genetics and Development、6:610−617(1996) Endicott,J.およびLing,V.、Annu.Rev.Biochem.、58:137−171、(1989) Maeda H.他、J.Controlled Release、65(1−2):271(2000)
従って、本発明の目的は、遊離形態の薬剤に比べて毒性が低くかつ多剤耐性細胞が取り込み得るマイトマイシンCリポソーム製剤を提供することにある。即ち、マイトマイシンCはこれを遊離形態で投与すると多剤耐性細胞の中に蓄積されないが、それを本明細書に記述するリポソーム製剤の中に取り込ませたプロドラッグコンジュゲートの形態で投与すると、それは前記細胞の中に蓄積し得る。

で表されるコンジュゲートで構成させたリポソーム組成物の形態で供給することを含んで成る。
で表されるコンジュゲートを生じさせるが、このコンジュゲートでは、マイトマイシンCのアジリジン部分の中の第二級アミンが前記ジチオベンジルとマイトマイシンCの間のウレタン結合を形成している。
で表されるコンジュゲートで構成させたリポソーム組成物の形態で投与することを含んで成る。
(発明の詳細な説明)
I. 定義
語句「リポソーム脂質二重層の中に取り込ませるに適した疎水性部分」は、リポソーム脂質二重層の疎水性二重層領域と一体化し得る疎水性部分を含んで成る材料のいずれかを意味する。そのような疎水性部分は典型的に脂質であり、それには、疎水性脂質尾部と親水性極性頭部を有する両親媒性脂質、例えば燐脂質およびジアシルグリセロールなどが含まれる。また、トリグリセリド、ステロール、燐脂質、ジアシルグリセロール、ステロールおよびトリグリセリドの誘導体、そして天然源に由来するか或は合成的に作られた他の脂質も考えられる。
II.コンジュゲート組成物および製造方法
本発明は、1つの面において、形態:
で表されるコンジュゲートを包含する。前記疎水性部分Lは典型的に脂質、例えばジアシルグリセロール、ステロール、燐脂質、このような脂質の誘導体、天然に存在する他の脂質およびそれらの合成類似物などである。
で表されるコンジュゲートを生じさせることができる。
III. コンジュゲート含有リポソームの調製
本発明の方法では、小胞形成性脂質とマイトマイシンCプロドラッグコンジュゲートで構成させたリポソーム組成物の形態でマイトマイシンCプロドラッグコンジュゲートを提供する。リポソームは多様な治療目的で用いられる密封型脂質小胞であり、特に、リポソームを全身投与することによって治療薬を標的領域または細胞に運ばせる。特に、親水性重合体鎖、例えばポリエチレングリコール(PEG)などの表面被膜を有するリポソームが薬剤担体として望ましい、と言うのは、そのようなリポソームが示す血液循環寿命の方が重合体被膜を持たないリポソームが示すそれよりも長いからである。そのような重合体は血中蛋白質に対するバリヤーとして働くことで前記蛋白質と当該リポソームの結合を防止しかつ当該リポソームがマクロファージおよび他の細網内皮系細胞によって認識されて吸収および除去されるのを妨害するからである。
IV. コンジュゲート含有リポソームのインビトロ特徴付け
A. インビトロ薬剤放出
実施例4A−4Bに記述するようにしてリポソームを調製して、それらを還元剤と接触させた後にマイトマイシンCが放出される速度をインビトロで測定することで、それらの特徴付けを行った。インビトロ研究では、試験媒体にシステインを典型的には約150μMの濃度で添加することで還元条件を誘発した。インビボの内因性還元条件は脂質−DTB−薬剤コンジュゲートがチオール開裂分解を起こして当該薬剤が放出されるに充分であり得ることは理解されるであろう。更に、適切な還元剤、例えばシステインまたはグルタチオンなどを投与することでインビボの還元条件を人工的に誘発することも可能であると考えている。
B. インビトロ細胞毒性
脂質−DTB−マイトマイシンCコンジュゲート(化合物XVIII)を含有させたリポソームがインビトロで示す細胞毒性の評価をM−109細胞、即ちマウス肺癌株を用いて実施した。実施例6に記述するように、M109細胞を遊離マイトマイシンCの存在下またはジステアロイル−DTB−マイトマイシンCコンジュゲート含有リポソームの存在下でインキュベートした。実施例6Aに示すモル比を用いて実施例4A−4Bに記述するようにして調製したリポソームに試験を受けさせた。そのようなコンジュゲートのチオール開裂分解およびマイトマイシンCの放出を起こさせる目的で試験細胞のいくつかにシステインを150μM、500μMおよび1000μMの濃度で添加した。
C. インビボ薬物速度
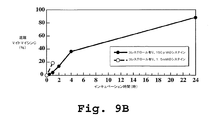
ラットを用いてコレステロール含有リポソームおよびコレステロール無しリポソーム製剤がインビボで示す薬物速度を測定した。実施例7に記述するようにして、動物にマイトマイシンCを遊離形態でか或は本発明に従う脂質−DTB−マイトマイシンCコンジュゲート形態でリポソームの中に取り込ませた形態で約0.1mg/mLの量で1回ボーラス静脈注入することで前記動物を処置した。注入後に血液サンプルを採取してマイトマイシンCの量を分析した。その結果を図13A−13Bに示す。
材料
あらゆる材料を商業的に適切な製造供給元、例えばAldrich Corporationなどから入手した。
[実施例1]
A. パラ−ジアシルジグリセロールジチオベンズアルコール
この反応を図1に示す。化合物IIおよびIIIの調製ではSnyder,W.R.、Journal of Lipid Research、28:949(1987)の手順に従った。
収量:630mg、63%。1HNMR(CD3OD、360MHz)δ 2.77、2.95(2xd、CH2OH、2H)、3.59(M、SCH2、2H)、3.87(m、CH、1H)ppm。
分析 理論値 測定値
炭素 70.93% 70.67%
水素 10.50% 10.41%
硫黄 8.25% 8.31%
B. オルソジグリセロールジチオベンズアルコール
rac−3,3’−ジチオビス(1,2−プロパンジステアロイル)(化合物III)(200mg、0.156ミリモル)の溶体をトルエン(30mL)に溶解させた後、氷浴の中に置いた。塩化スルフリル(39μl、0.47ミリモル)をピペットでフラスコの中に入れた後、その混合物を冷氷浴温度で30分間撹拌した。次に、前記フラスコを室温に置いて更に30分間撹拌した。ロータリーエバポレーターを用いて余分な塩化スルフリルを除去した。この反応フラスコに新しい分量(20mL)のトルエンを加えた後、氷浴の上に置いた。これに、2−メルカプトベンズアルコール(48mg、35ミリモル)をトルエンに入れることで生じさせた溶液をゆっくりした速度で加えた。5時間の反応時間後、ロータリーエバポレーターを用いて溶媒全部を蒸発させて乾固させた。前記反応フラスコに温かい酢酸エチル(10mL)を加えることで固体を溶解させた後、不溶物を濾過で除去した。その酢酸エチル溶液にエーテルを50mL加えることで沈澱を起こさせた後、固体状生成物(オルソ−ジアシル−ジグリセロール−ジチオベンザルアルコール)を濾過で集めた。この過程を2回繰り返した。その固体をP2O5の上に置いて真空下で乾燥させた。収率:75%、190mg。1HNMR:(CDCl3、360MHz)δ 0.86(t、CH3、6H)、1.25(s、脂質、56H)、1.58(m、CH2CH2(CO)O、4H)、2.28(2xt、CH2(CO)O、4H)、2.91(d、CH2S、2H)、4.14および4.35(2xd、脂質のCH2CH、2H)、4.86(s、CH2、bz、2H)、5.26(m、脂質のCHCH2)、7.31(m、芳香族、2H)、7.48および7.75(d、芳香族、2H)ppm。
[実施例2]
この反応を図3Aに示す。
[実施例4]
A. コレステロール含有リポソーム
1. リポソーム調製
1mLの乾燥エタノールに60−65℃でHSPCを59mg、コレステロールを14.4mg、mPEG−DSPEを17.4mgおよびパラ−ジステアロイル−DTB−マイトマイシンCを7.4mg(60/30/5/5のモル比)加えた後、溶解するまで、ほぼ10分間混合した。
2. 押出し
前記リポソームをテフロンが内張りされているステンレス鋼製容器の中に入っているポリカーボネート製フィルターカートリッジに通して制御した様式で押出すことで大きさを所望の平均粒径に合わせた。この押出し工程全体、即ち6−8時間に渡って、そのリポソーム懸濁液を63−65℃に保持した。
3. 透析濾過(Diafiltration)
前記リポソーム懸濁液からエタノールを透析濾過で除去した。ヒスチジン(10mM)と塩化ナトリウム(150mM)を無菌水に溶解させることでヒスチジン/塩化ナトリウム溶液を調製した。この溶液のpHを約7に調整した。この溶液を0.22μmのDuraporeフィルターに通して濾過した。このリポソーム懸濁液を前記ヒスチジン/塩化ナトリウム溶液で約1:1(体積/体積)の比率に希釈した後、ポリスルホン製中空繊維限外濾過装置に通す透析濾過を行った。前記ヒスチジン/塩化ナトリウム溶液に対する体積交換を8回実施することでエタノールを除去した。この過程の流体温度を約20−30℃に保持した。全透析濾過時間は約4.5時間であった。
4. 無菌濾過
前記リポソーム懸濁液を33−38℃に加熱した後、0.2μmのGelman Suporポリエーテルスルホン製フィルターに通して濾過した。全濾過時間は約10分間であった。
HSPCとmPEG−DSPEとパラ−ジステアロイル−DTB−マイトマイシンCが90/5/5のモル比の脂質組成を用いてリポソームをこの上に記述したようにして調製した。具体的には、1mLのエタノールにHSPCを88.5mg、mPEG−DSPE(PEGのMWが2000ダルトン)を17.9mgおよび前記コンジュゲートを7.3mg溶解させた。各処理段階後にリポソームの大きさ、脂質および薬剤の濃度、そして外部の懸濁用媒体に入っている遊離マイトマイシンCの濃度を測定した。
実施例4A−4Bに記述したようにして調製したリポソームを0.6Mのオクタイルグルコピラノシドで希釈した。前記リポソームを150mMのシステインの存在下37℃でインキュベートした。サンプルを時間ゼロ、30分、1時間、2時間、4時間および24時間の時に取り出した。3.5x5cmのWater Symmetry C8カラムを用いたHPLCで20μLの体積を分析した。流量を1mL/分にしそして可動相の勾配を下記の如くにした:
出発 10%MEOH 90% 10mM NaPO4、pH=7
5分 25%MEOH 75% 10mM NaPO4、pH=7
10分 25%MEOH 75% 10mM NaPO4、pH=7
15分 100%MEOH −
25分 100%MEOH −
30分 10% 90% 10mM NaPO4、pH=7
35分 10%MEOH 90% 10mM NaPO4、pH=7
[実施例6]
A. リポソーム調製
実施例4A−4Bに記述した如く調製したリポソームはHSPC/mPEG−DSPE/ジステアロイル−DTB−マイトマイシンC(90/5/5)またはHSPC/コレステロール/mPEG−DSPE/ジステアロイル−DTB−マイトマイシンC(90/45/5/5)で構成されていた。これらのリポソーム製剤を0.45μmのセルロース膜に通して無菌濾過したが、押出しで大きさを小さくすることは行わなかった。リポソーム形成後、リポソームをイソプロパノールに10−20倍の希釈度で溶解させて360nmの所の吸光度を用いてマイトマイシンCの濃度を測定しかつ無機燐酸塩検定を用いて燐脂質濃度を測定した。
B. 化学療法剤感受性検定および増殖速度測定
遊離マイトマイシンCまたはリポソームの中に取り込ませたジステアロイル−DTB−マイトマイシンCコンジュゲートの形態のマイトマイシンCが示す細胞毒性効果を以前に記述されたメチレンブルー染色方法[Horowitz,A.T.他、Biochim.Biophys.Acta、1109:203−209(1992)]に若干の修飾を受けさせた方法を用いて比色分析で検定した。この検定が完了した時点で細胞を固定しそしてメチレンブルー染色検定を用いて評価した。
[実施例7]
A. リポソーム製剤
コレステロール含有リポソームおよびコレステロール無しリポソームを実施例5Aおよび5Bに記述した如く調製した。
B. 動物
8匹のラットを無作為に下記の如き処置群に分けた:
C. 血漿中のマイトマイシンCを測定するHPLC方法
1. 溶液調製
1Lの容量フラスコに脱イオン水を充填してこれに燐酸アンモニウムを1.321g入れることを通して、燐酸アンモニウム含有量が10mMの緩衝剤水溶液(pH=7)を調製した。前記混合物を撹拌しながらo−燐酸を用いてpHを7.0に調整した。この緩衝液を使用前に0.45μmのナイロン製フィルターに通して濾過した。
2. 標準溶液および品質制御サンプルの調製
2種類の個々別々の重量のマイトマイシンCおよびマイトマイシンCコンジュゲートを標準サンプルおよび品質制御サンプルとして調製した。マイトマイシンCおよびマイトマイシンCコンジュゲートを1mgづつ計り取って個別に1mLの希釈剤(クロロホルムが20%でメタノールが80%の混合物)に溶解させた。両方の化合物の原液の濃度を1mg/mLにした。標準サンプルおよび品質制御サンプルに関して5μg/mLから100μg/mLの濃度を得る目的で数種の希釈液を希釈状態で作成した。
3. サンプル調製
900μLのメタノールを用いて100μLの血漿サンプルを変性させた後、3,000rpmの遠心分離に10分間かけた。300μL分量の上澄み液を注入用インサートが300μL入っているHPLC用瓶に移した。
4. クロマトグラフィー条件
4.6mmx5cmのSupelco(商標)C−8(5μ)カラムを用いた。可動相Aを10mMの燐酸アンモニウム(pH7)にした。可動相Bをメタノールにした。流量を1mM/分にしそして360nmの紫外線で検出した。注入体積を40μLにしそして典型的な実行時間は15分間であった。勾配プログラムは下記の通りであった:
[実施例8]
スパソゲン(Spathogen)の無い特殊な施設の中に10週令のメスBALB/cマウスを維持した。M109細胞またはM109R細胞をインビトロ懸濁液の状態で増殖させた。50μL(106個の細胞)をマウスの右後足蹠に注入した。試験終了時まで足蹠の厚みをカリパスで測定し、試験が終了した時点でマウスを屠殺し、最終的な腫瘍数を記録し、そして対照および腫瘍を接種した足蹠を足関節の高さの所で切断して重量を測定した。正常な足蹠および腫瘍を有する足蹠の間の重量の差として腫瘍重量を推定した。1グループ当たりの最終的な腫瘍罹病率の差が統計学的に有意であるか否かを分割表およびフィッシャーの直接確率検定で分析した。その結果を図15A−15Bおよび図16A−16B、図18、図19A−19Cに示す。
Claims (5)
- P−糖蛋白質を発現する多剤耐性細胞の治療のためのリポソーム組成物であって、小胞形成性脂質と1から30モルパーセントの範囲の一般形態:
で表されるコンジュゲートで形成されているリポソームを含んで成る組成物。 - マイトマイシンCがウレタン結合で共有結合している請求項1記載の組成物。
- Lがコレステロール、ジアシルグリセロールおよび燐脂質から成る群から選択される請求項1記載の組成物。
- 前記コンジュゲートが構造:
で表されるコンジュゲートを形成するように前記ジチオベンジル部分と共有結合しているマイトマイシンCを含んで成る請求項1記載の組成物。 - R4の第二級アミン部分が前記ジチオベンジルとマイトマイシンCの間のウレタン結合を形成している請求項4記載の組成物。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US46707003P | 2003-04-30 | 2003-04-30 | |
| US60/467,070 | 2003-04-30 | ||
| US10/714,085 US7303760B2 (en) | 1999-04-23 | 2003-11-14 | Method for treating multi-drug resistant tumors |
| US10/714,085 | 2003-11-14 | ||
| PCT/US2004/013820 WO2004110497A2 (en) | 2003-04-30 | 2004-04-29 | Mitomycin conjugates cleavable by thiols |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007502323A JP2007502323A (ja) | 2007-02-08 |
| JP5009621B2 true JP5009621B2 (ja) | 2012-08-22 |
Family
ID=33555221
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006532557A Expired - Fee Related JP5009621B2 (ja) | 2003-04-30 | 2004-04-29 | チオール開裂し得るマイトマイシンコンジュゲート |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7303760B2 (ja) |
| EP (1) | EP1617873B1 (ja) |
| JP (1) | JP5009621B2 (ja) |
| KR (1) | KR20060033711A (ja) |
| AT (1) | ATE541589T1 (ja) |
| AU (1) | AU2004247004B2 (ja) |
| CA (1) | CA2524179C (ja) |
| ES (1) | ES2380620T3 (ja) |
| WO (1) | WO2004110497A2 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7303760B2 (en) * | 1999-04-23 | 2007-12-04 | Alza Corporation | Method for treating multi-drug resistant tumors |
| AU769425B2 (en) * | 1999-04-23 | 2004-01-29 | Alza Corporation | Conjugate having a cleavable linkage for use in a liposome |
| US7238368B2 (en) * | 1999-04-23 | 2007-07-03 | Alza Corporation | Releasable linkage and compositions containing same |
| FR2906533B1 (fr) | 2006-09-28 | 2013-02-22 | Pf Medicament | Procede de generation d'anticorps actifs contre un antigene de resistance,anticorps obtenus par ledit procede et leurs utilisations |
| US9937261B2 (en) | 2014-06-09 | 2018-04-10 | Lipomedix Pharmaceuticals Ltd. | Combination therapy comprising a liposomal prodrug of mitomycin C and radiotherapy |
| US20180071253A1 (en) * | 2015-03-17 | 2018-03-15 | Lipomedix Pharmaceuticals Ltd. | Methods for the treatment of bladder cancer |
| WO2018089481A1 (en) * | 2016-11-08 | 2018-05-17 | Mallinckrodt Llc | Mitomycin c prodrug liposome formulations and uses thereof |
| EP3908261A1 (en) | 2019-01-11 | 2021-11-17 | Lipomedix Pharmaceuticals Ltd. | Liposome composition comprising liposomal prodrug of mitomycin c and method of manufacture |
| JPWO2022050369A1 (ja) * | 2020-09-04 | 2022-03-10 |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| JPS5240B2 (ja) | 1973-12-17 | 1977-01-05 | ||
| GB8430252D0 (en) | 1984-11-30 | 1985-01-09 | Beecham Group Plc | Compounds |
| US4766106A (en) | 1985-06-26 | 1988-08-23 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| US4917888A (en) | 1985-06-26 | 1990-04-17 | Cetus Corporation | Solubilization of immunotoxins for pharmaceutical compositions using polymer conjugation |
| JPH01113391A (ja) | 1987-10-24 | 1989-05-02 | Kyowa Hakko Kogyo Co Ltd | マイトマイシン誘導体 |
| ZA886812B (en) | 1987-11-23 | 1989-07-26 | Bristol Myers Co | Anti-tumor prodrugs |
| US4952394A (en) | 1987-11-23 | 1990-08-28 | Bristol-Myers Company | Drug-monoclonal antibody conjugates |
| US5103556A (en) | 1988-05-05 | 1992-04-14 | Circon Corporation | Method of manufacturing an electrohydraulic probe |
| US4902502A (en) | 1989-01-23 | 1990-02-20 | Cetus Corporation | Preparation of a polymer/interleukin-2 conjugate |
| TW211015B (ja) | 1990-01-11 | 1993-08-11 | Nippon Shinyaku Co Ltd | |
| PT706373E (pt) * | 1992-03-23 | 2000-11-30 | Univ Georgetown | Taxol encapsulado num liposoma e um metodo |
| WO1994005259A1 (en) * | 1992-09-02 | 1994-03-17 | Georgetown University | Method of encapsulating anthracycline glycosides in liposomes |
| US5395619A (en) | 1993-03-03 | 1995-03-07 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| JPH09268190A (ja) | 1996-04-02 | 1997-10-14 | Sagami Chem Res Center | マイトマイシンc誘導体及び非受容体型チロシンキナーゼ阻害剤 |
| TW520297B (en) | 1996-10-11 | 2003-02-11 | Sequus Pharm Inc | Fusogenic liposome composition and method |
| JP2001503396A (ja) | 1996-10-11 | 2001-03-13 | アルザ コーポレイション | 治療用リポソーム組成物および方法 |
| JPH1160499A (ja) | 1997-08-22 | 1999-03-02 | Hiroshi Maeda | 抗腫瘍剤 |
| SE513149C2 (sv) | 1997-12-05 | 2000-07-17 | Katarina Edwards | Läkemedelsdistributionssystem med tvåstegsmålsökning, till specifika celler eller vävnad och till dess cellkärna |
| US6180095B1 (en) | 1997-12-17 | 2001-01-30 | Enzon, Inc. | Polymeric prodrugs of amino- and hydroxyl-containing bioactive agents |
| EE05214B1 (et) | 1998-04-28 | 2009-10-15 | Applied Research Systems Ars Holding N.V. | Polool-IFN- β konjugaat, selle valmistamismeetod ja farmatseutiline kompositsioon |
| JP2002542386A (ja) | 1999-04-23 | 2002-12-10 | アルザ・コーポレーション | 放出可能な結合及びこれを含む組成物 |
| AU769425B2 (en) | 1999-04-23 | 2004-01-29 | Alza Corporation | Conjugate having a cleavable linkage for use in a liposome |
| US7112337B2 (en) | 1999-04-23 | 2006-09-26 | Alza Corporation | Liposome composition for delivery of nucleic acid |
| US7303760B2 (en) | 1999-04-23 | 2007-12-04 | Alza Corporation | Method for treating multi-drug resistant tumors |
| AU7868400A (en) | 1999-10-08 | 2001-04-23 | Alza Corporation | Neutral-cationic lipid for nucleic acid and drug delivery |
| WO2002026265A2 (en) | 2000-09-29 | 2002-04-04 | Schering Corporation | Pegylated interleukin-10 |
| US7260330B2 (en) | 2002-11-04 | 2007-08-21 | The Boeing Company | Optical communication system using correlation receiver |
-
2003
- 2003-11-14 US US10/714,085 patent/US7303760B2/en not_active Expired - Lifetime
-
2004
- 2004-04-29 JP JP2006532557A patent/JP5009621B2/ja not_active Expired - Fee Related
- 2004-04-29 WO PCT/US2004/013820 patent/WO2004110497A2/en active Application Filing
- 2004-04-29 AT AT04751275T patent/ATE541589T1/de active
- 2004-04-29 EP EP04751275A patent/EP1617873B1/en not_active Expired - Lifetime
- 2004-04-29 KR KR1020057020690A patent/KR20060033711A/ko not_active Application Discontinuation
- 2004-04-29 ES ES04751275T patent/ES2380620T3/es not_active Expired - Lifetime
- 2004-04-29 CA CA2524179A patent/CA2524179C/en not_active Expired - Lifetime
- 2004-04-29 AU AU2004247004A patent/AU2004247004B2/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| US7303760B2 (en) | 2007-12-04 |
| ES2380620T3 (es) | 2012-05-16 |
| WO2004110497A3 (en) | 2005-03-24 |
| JP2007502323A (ja) | 2007-02-08 |
| AU2004247004A1 (en) | 2004-12-23 |
| CA2524179A1 (en) | 2004-12-23 |
| WO2004110497A2 (en) | 2004-12-23 |
| EP1617873A2 (en) | 2006-01-25 |
| CA2524179C (en) | 2012-06-19 |
| EP1617873B1 (en) | 2012-01-18 |
| AU2004247004B2 (en) | 2010-10-14 |
| ATE541589T1 (de) | 2012-02-15 |
| KR20060033711A (ko) | 2006-04-19 |
| US20040161455A1 (en) | 2004-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4558952B2 (ja) | リポソームにおいての使用のための開裂可能な結合を有する複合体 | |
| EP2558072B1 (en) | Prodrug compositions, prodrug nanoparticles, and methods of use thereof | |
| JP3074733B2 (ja) | 脂肪乳剤 | |
| Nguyen et al. | Recent trends in bioresponsive linker technologies of prodrug-based self-assembling nanomaterials | |
| EP0482185A1 (en) | Cell internalizable conjugates and complexes including intracellularly cleavable moieties | |
| KR20240141854A (ko) | 알파 및 감마-d 폴리글루타메이트화 항엽산 및 이의 용도 | |
| US20080057026A1 (en) | Amphiphilic Star-Like Or Scorpion-Like Macromolecules, Various Compositions And Uses Threof | |
| JP5009621B2 (ja) | チオール開裂し得るマイトマイシンコンジュゲート | |
| CA2491661A1 (en) | The use of isocyanate linkers to make hydrolyzable active agent biopolymer conjugates | |
| ES2329374T3 (es) | Formulaciones farmaceuticas que utilizan esfingolipidos de cadena corta y usos de las mismas. | |
| US8304565B2 (en) | PEG-lipid conjugates for liposomes and drug delivery | |
| JP2002542386A (ja) | 放出可能な結合及びこれを含む組成物 | |
| KR20050071641A (ko) | 리포솜 | |
| WO2024108173A1 (en) | Prodrug strategy that enhances efficacy and lowers systemic toxicity of mertansine | |
| OA19187A (en) | Polyglutamated antifolates and uses thereof. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20070309 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070326 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20070309 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20081028 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100914 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20101116 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20101124 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20111101 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120301 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20120302 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20120405 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120501 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120531 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5009621 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150608 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |