JP4865129B2 - タンパク質キナーゼ阻害剤を設計するための新規の方法 - Google Patents
タンパク質キナーゼ阻害剤を設計するための新規の方法 Download PDFInfo
- Publication number
- JP4865129B2 JP4865129B2 JP2000593770A JP2000593770A JP4865129B2 JP 4865129 B2 JP4865129 B2 JP 4865129B2 JP 2000593770 A JP2000593770 A JP 2000593770A JP 2000593770 A JP2000593770 A JP 2000593770A JP 4865129 B2 JP4865129 B2 JP 4865129B2
- Authority
- JP
- Japan
- Prior art keywords
- src
- inhibitors
- inhibitor
- peptide
- protein kinase
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000000034 method Methods 0.000 title abstract description 42
- 239000003909 protein kinase inhibitor Substances 0.000 title description 27
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 title description 17
- 230000000694 effects Effects 0.000 claims abstract description 57
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 19
- 230000002401 inhibitory effect Effects 0.000 claims abstract description 15
- 239000005483 tyrosine kinase inhibitor Substances 0.000 claims abstract description 10
- 201000011510 cancer Diseases 0.000 claims abstract description 8
- 201000004681 Psoriasis Diseases 0.000 claims abstract description 3
- 230000005965 immune activity Effects 0.000 claims abstract 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 40
- 150000001875 compounds Chemical class 0.000 claims description 40
- 229910052739 hydrogen Inorganic materials 0.000 claims description 26
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 22
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims description 22
- 239000001257 hydrogen Substances 0.000 claims description 20
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 claims description 16
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 claims description 15
- 102000001332 SRC Human genes 0.000 claims description 4
- 108060006706 SRC Proteins 0.000 claims description 4
- 229910052731 fluorine Inorganic materials 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 206010023230 Joint stiffness Diseases 0.000 claims description 2
- 239000003814 drug Substances 0.000 claims description 2
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 claims 1
- 239000003937 drug carrier Substances 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- 239000008194 pharmaceutical composition Substances 0.000 claims 1
- 208000024891 symptom Diseases 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 230
- 108090000765 processed proteins & peptides Proteins 0.000 abstract description 90
- 102000001253 Protein Kinase Human genes 0.000 abstract description 68
- 108060006633 protein kinase Proteins 0.000 abstract description 68
- 201000001320 Atherosclerosis Diseases 0.000 abstract 1
- 101150001535 SRC gene Proteins 0.000 description 128
- 238000003556 assay Methods 0.000 description 69
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 63
- 230000027455 binding Effects 0.000 description 59
- 239000000758 substrate Substances 0.000 description 53
- 102000008130 Cyclic AMP-Dependent Protein Kinases Human genes 0.000 description 52
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 48
- 230000005764 inhibitory process Effects 0.000 description 46
- 125000000524 functional group Chemical group 0.000 description 45
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 44
- 102100037787 Protein-tyrosine kinase 2-beta Human genes 0.000 description 36
- 230000036515 potency Effects 0.000 description 36
- 150000001408 amides Chemical class 0.000 description 35
- 210000004027 cell Anatomy 0.000 description 35
- 230000003197 catalytic effect Effects 0.000 description 34
- 125000001624 naphthyl group Chemical group 0.000 description 30
- 108010001127 Insulin Receptor Proteins 0.000 description 27
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 26
- 102000003746 Insulin Receptor Human genes 0.000 description 26
- 238000006243 chemical reaction Methods 0.000 description 26
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 24
- 125000001041 indolyl group Chemical group 0.000 description 23
- 230000003993 interaction Effects 0.000 description 23
- 238000012360 testing method Methods 0.000 description 23
- 238000013461 design Methods 0.000 description 22
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 20
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 20
- 230000002860 competitive effect Effects 0.000 description 18
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 18
- JOGDFBLWRNQZRO-UHFFFAOYSA-N methyl 5-hydroxy-1h-indole-2-carboxylate Chemical compound OC1=CC=C2NC(C(=O)OC)=CC2=C1 JOGDFBLWRNQZRO-UHFFFAOYSA-N 0.000 description 18
- 229910052799 carbon Inorganic materials 0.000 description 16
- 230000003389 potentiating effect Effects 0.000 description 16
- 238000003786 synthesis reaction Methods 0.000 description 16
- 230000015572 biosynthetic process Effects 0.000 description 14
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 13
- 125000005620 boronic acid group Chemical group 0.000 description 13
- 235000019439 ethyl acetate Nutrition 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- 229940122924 Src inhibitor Drugs 0.000 description 12
- -1 hydroxide ions Chemical class 0.000 description 12
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 11
- 239000013078 crystal Substances 0.000 description 11
- 230000002209 hydrophobic effect Effects 0.000 description 11
- 230000001965 increasing effect Effects 0.000 description 11
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 11
- 238000012216 screening Methods 0.000 description 11
- 239000000243 solution Substances 0.000 description 11
- 102000004190 Enzymes Human genes 0.000 description 10
- 108090000790 Enzymes Proteins 0.000 description 10
- 150000001412 amines Chemical class 0.000 description 10
- 125000004429 atom Chemical group 0.000 description 10
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 10
- 230000007423 decrease Effects 0.000 description 10
- 238000011161 development Methods 0.000 description 10
- 229940043355 kinase inhibitor Drugs 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- SVSKFMJQWMZCRD-MCDZGGTQSA-L MgADP Chemical compound [Mg+2].C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP(O)([O-])=O)[C@@H](O)[C@H]1O SVSKFMJQWMZCRD-MCDZGGTQSA-L 0.000 description 9
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 9
- 102100023038 WD and tetratricopeptide repeats protein 1 Human genes 0.000 description 9
- WUVFAMRFBRHSQW-UHFFFAOYSA-N chromen-2-imine Chemical group C1=CC=C2OC(=N)C=CC2=C1 WUVFAMRFBRHSQW-UHFFFAOYSA-N 0.000 description 9
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 230000012010 growth Effects 0.000 description 9
- 238000000021 kinase assay Methods 0.000 description 9
- 239000000370 acceptor Substances 0.000 description 8
- 230000003834 intracellular effect Effects 0.000 description 8
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 8
- 238000005556 structure-activity relationship Methods 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- 210000004881 tumor cell Anatomy 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 7
- 238000005859 coupling reaction Methods 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 229940043437 protein kinase A inhibitor Drugs 0.000 description 7
- 239000011541 reaction mixture Substances 0.000 description 7
- 229960001153 serine Drugs 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 238000007792 addition Methods 0.000 description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- 239000007795 chemical reaction product Substances 0.000 description 6
- 230000008878 coupling Effects 0.000 description 6
- 238000010168 coupling process Methods 0.000 description 6
- 150000004702 methyl esters Chemical class 0.000 description 6
- 230000003278 mimic effect Effects 0.000 description 6
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 6
- 230000026731 phosphorylation Effects 0.000 description 6
- 238000006366 phosphorylation reaction Methods 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 108010087686 src-Family Kinases Proteins 0.000 description 6
- 102000009076 src-Family Kinases Human genes 0.000 description 6
- XXCSQHMDTJAWNX-PVKGJVGDSA-N (2s)-n-[(2s)-1-[[(2s)-1-[[(2s,3r)-1-[[(2s)-1-[[(2s)-4-amino-1-[[(2s)-1-[[(2s)-1-[[(2s)-1-amino-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]- Chemical compound N([C@@H](CCC(N)=O)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CO)C(N)=O)[C@@H](C)O)C(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CCCCN XXCSQHMDTJAWNX-PVKGJVGDSA-N 0.000 description 5
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 5
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 5
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 5
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 5
- 150000008064 anhydrides Chemical class 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 150000002475 indoles Chemical class 0.000 description 5
- 238000000302 molecular modelling Methods 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- BIMHWDJKNOMNLD-UHFFFAOYSA-N 5-Hydroxyindole-2-carboxylic acid Chemical compound OC1=CC=C2NC(C(=O)O)=CC2=C1 BIMHWDJKNOMNLD-UHFFFAOYSA-N 0.000 description 4
- CHCDEEJBYZUZEY-UHFFFAOYSA-N 5-hydroxy-n-[(3-methoxyphenyl)methyl]-1h-indole-2-carboxamide Chemical compound COC1=CC=CC(CNC(=O)C=2NC3=CC=C(O)C=C3C=2)=C1 CHCDEEJBYZUZEY-UHFFFAOYSA-N 0.000 description 4
- LMIQERWZRIFWNZ-UHFFFAOYSA-N 5-hydroxyindole Chemical compound OC1=CC=C2NC=CC2=C1 LMIQERWZRIFWNZ-UHFFFAOYSA-N 0.000 description 4
- PVKSNHVPLWYQGJ-KQYNXXCUSA-N AMP-PNP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)NP(O)(O)=O)[C@@H](O)[C@H]1O PVKSNHVPLWYQGJ-KQYNXXCUSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 229910015845 BBr3 Inorganic materials 0.000 description 4
- 102000001301 EGF receptor Human genes 0.000 description 4
- 108060006698 EGF receptor Proteins 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- CKRZKMFTZCFYGB-UHFFFAOYSA-N N-phenylhydroxylamine Chemical compound ONC1=CC=CC=C1 CKRZKMFTZCFYGB-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 4
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 4
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 239000012300 argon atmosphere Substances 0.000 description 4
- 239000004305 biphenyl Substances 0.000 description 4
- 235000010290 biphenyl Nutrition 0.000 description 4
- 238000004113 cell culture Methods 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- LVCDXCQFSONNDO-UHFFFAOYSA-N n-benzylhydroxylamine Chemical compound ONCC1=CC=CC=C1 LVCDXCQFSONNDO-UHFFFAOYSA-N 0.000 description 4
- 210000002997 osteoclast Anatomy 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 230000011664 signaling Effects 0.000 description 4
- 238000010898 silica gel chromatography Methods 0.000 description 4
- 239000007790 solid phase Substances 0.000 description 4
- 238000010189 synthetic method Methods 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical class C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 3
- YELLAPKUWRTITI-UHFFFAOYSA-N 3,5-dihydroxynaphthalene-2-carboxylic acid Chemical compound C1=CC(O)=C2C=C(O)C(C(=O)O)=CC2=C1 YELLAPKUWRTITI-UHFFFAOYSA-N 0.000 description 3
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 3
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 3
- HCUARRIEZVDMPT-UHFFFAOYSA-N Indole-2-carboxylic acid Chemical class C1=CC=C2NC(C(=O)O)=CC2=C1 HCUARRIEZVDMPT-UHFFFAOYSA-N 0.000 description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 description 3
- 229930012538 Paclitaxel Natural products 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 108010060350 arginyl-arginyl-leucyl-isoleucyl-glutamyl-aspartyl-alanyl-glutamyl-tyrosyl-alanyl-alanyl-arginyl-glycine Proteins 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 3
- 229960004316 cisplatin Drugs 0.000 description 3
- 238000006880 cross-coupling reaction Methods 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 229960004679 doxorubicin Drugs 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 229960005420 etoposide Drugs 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 230000009036 growth inhibition Effects 0.000 description 3
- 108010046629 kemptamide Proteins 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- QOMUVNDHVNXTLJ-UHFFFAOYSA-N methyl 5-dibenzylphosphoryl-1h-indole-2-carboxylate Chemical compound C=1C=C2NC(C(=O)OC)=CC2=CC=1P(=O)(CC=1C=CC=CC=1)CC1=CC=CC=C1 QOMUVNDHVNXTLJ-UHFFFAOYSA-N 0.000 description 3
- DFQICHCWIIJABH-UHFFFAOYSA-N naphthalene-2,7-diol Chemical compound C1=CC(O)=CC2=CC(O)=CC=C21 DFQICHCWIIJABH-UHFFFAOYSA-N 0.000 description 3
- 239000006225 natural substrate Substances 0.000 description 3
- 230000036963 noncompetitive effect Effects 0.000 description 3
- 239000012038 nucleophile Substances 0.000 description 3
- 229960001592 paclitaxel Drugs 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 229940080469 phosphocellulose Drugs 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 239000012656 protein kinase A inhibitor Substances 0.000 description 3
- 108010060709 protein kinase inhibitor peptide (5-24) Proteins 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- JHFAEUICJHBVHB-UHFFFAOYSA-N 1h-indol-2-ol Chemical class C1=CC=C2NC(O)=CC2=C1 JHFAEUICJHBVHB-UHFFFAOYSA-N 0.000 description 2
- VFHUJFBEFDVZPJ-UHFFFAOYSA-N 1h-indole-2-carboxamide Chemical class C1=CC=C2NC(C(=O)N)=CC2=C1 VFHUJFBEFDVZPJ-UHFFFAOYSA-N 0.000 description 2
- HCUARRIEZVDMPT-UHFFFAOYSA-M 1h-indole-2-carboxylate Chemical compound C1=CC=C2NC(C(=O)[O-])=CC2=C1 HCUARRIEZVDMPT-UHFFFAOYSA-M 0.000 description 2
- IXWOUPGDGMCKGT-UHFFFAOYSA-N 2,3-dihydroxybenzaldehyde Chemical compound OC1=CC=CC(C=O)=C1O IXWOUPGDGMCKGT-UHFFFAOYSA-N 0.000 description 2
- CWLKGDAVCFYWJK-UHFFFAOYSA-N 3-aminophenol Chemical compound NC1=CC=CC(O)=C1 CWLKGDAVCFYWJK-UHFFFAOYSA-N 0.000 description 2
- 238000004679 31P NMR spectroscopy Methods 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- DMLARLNVEPLLMB-UHFFFAOYSA-N 5-hydroxy-n-[(3-hydroxyphenyl)methyl]-1h-indole-2-carboxamide Chemical compound OC1=CC=CC(CNC(=O)C=2NC3=CC=C(O)C=C3C=2)=C1 DMLARLNVEPLLMB-UHFFFAOYSA-N 0.000 description 2
- HOSGXJWQVBHGLT-UHFFFAOYSA-N 6-hydroxy-3,4-dihydro-1h-quinolin-2-one Chemical group N1C(=O)CCC2=CC(O)=CC=C21 HOSGXJWQVBHGLT-UHFFFAOYSA-N 0.000 description 2
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 2
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 2
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 2
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 2
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 2
- 125000002842 L-seryl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])O[H] 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 2
- BZQFBWGGLXLEPQ-UHFFFAOYSA-N O-phosphoryl-L-serine Natural products OC(=O)C(N)COP(O)(O)=O BZQFBWGGLXLEPQ-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 108010026479 Src peptide Proteins 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 238000000423 cell based assay Methods 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 230000010307 cell transformation Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000001311 chemical methods and process Methods 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 239000000356 contaminant Substances 0.000 description 2
- MLIREBYILWEBDM-UHFFFAOYSA-N cyanoacetic acid Chemical compound OC(=O)CC#N MLIREBYILWEBDM-UHFFFAOYSA-N 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000004807 desolvation Methods 0.000 description 2
- 229950006137 dexfosfoserine Drugs 0.000 description 2
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 2
- 229940000406 drug candidate Drugs 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- RBUSDPZPBBBAKR-UHFFFAOYSA-N ethyl 5,6-dimethoxy-1h-indole-2-carboxylate Chemical compound COC1=C(OC)C=C2NC(C(=O)OCC)=CC2=C1 RBUSDPZPBBBAKR-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 210000002744 extracellular matrix Anatomy 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000011905 homologation Methods 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- WMYBSXNKEVGXGX-UHFFFAOYSA-N methyl 5-methyl-1h-indole-2-carboxylate Chemical compound CC1=CC=C2NC(C(=O)OC)=CC2=C1 WMYBSXNKEVGXGX-UHFFFAOYSA-N 0.000 description 2
- 238000000324 molecular mechanic Methods 0.000 description 2
- LTMYAMBXTBIZMH-UHFFFAOYSA-N n-(1-amino-3-methyl-1-oxopentan-2-yl)-5-hydroxy-1h-indole-2-carboxamide Chemical compound OC1=CC=C2NC(C(=O)NC(C(C)CC)C(N)=O)=CC2=C1 LTMYAMBXTBIZMH-UHFFFAOYSA-N 0.000 description 2
- 150000002790 naphthalenes Chemical class 0.000 description 2
- 229930014626 natural product Natural products 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 2
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical group OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 2
- CDRPUGZCRXZLFL-OWOJBTEDSA-N piceatannol Chemical compound OC1=CC(O)=CC(\C=C\C=2C=C(O)C(O)=CC=2)=C1 CDRPUGZCRXZLFL-OWOJBTEDSA-N 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 238000011533 pre-incubation Methods 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical group C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 108700026239 src Genes Proteins 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 150000003460 sulfonic acids Chemical class 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 125000002827 triflate group Chemical group FC(S(=O)(=O)O*)(F)F 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- IYKLZBIWFXPUCS-VIFPVBQESA-N (2s)-2-(naphthalen-1-ylamino)propanoic acid Chemical group C1=CC=C2C(N[C@@H](C)C(O)=O)=CC=CC2=C1 IYKLZBIWFXPUCS-VIFPVBQESA-N 0.000 description 1
- GRRIMVWABNHKBX-UHFFFAOYSA-N (3-methoxyphenyl)methanamine Chemical compound COC1=CC=CC(CN)=C1 GRRIMVWABNHKBX-UHFFFAOYSA-N 0.000 description 1
- OZPPBQMEFADOEU-QAXPSLGGSA-N (4s)-4-[[2-[[(2s)-2-[[(2s,3s)-2-acetamido-3-methylpentanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]acetyl]amino]-5-[[(2s)-1-amino-1-oxo-3-phenylpropan-2-yl]amino]-5-oxopentanoic acid Chemical compound C([C@H](NC(=O)[C@@H](NC(C)=O)[C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)C1=CC=C(O)C=C1 OZPPBQMEFADOEU-QAXPSLGGSA-N 0.000 description 1
- QRDAPCMJAOQZSU-KQQUZDAGSA-N (e)-3-[4-[(e)-3-(3-fluorophenyl)-3-oxoprop-1-enyl]-1-methylpyrrol-2-yl]-n-hydroxyprop-2-enamide Chemical compound C1=C(\C=C\C(=O)NO)N(C)C=C1\C=C\C(=O)C1=CC=CC(F)=C1 QRDAPCMJAOQZSU-KQQUZDAGSA-N 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- OXDSKEQSEGDAFN-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenylmethanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)NC1=CC=CC=C1 OXDSKEQSEGDAFN-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- USASUEYARBQNBM-UHFFFAOYSA-N 1h-indol-2-ylboronic acid Chemical compound C1=CC=C2NC(B(O)O)=CC2=C1 USASUEYARBQNBM-UHFFFAOYSA-N 0.000 description 1
- XAWPKHNOFIWWNZ-UHFFFAOYSA-N 1h-indol-6-ol Chemical compound OC1=CC=C2C=CNC2=C1 XAWPKHNOFIWWNZ-UHFFFAOYSA-N 0.000 description 1
- BMIBJCFFZPYJHF-UHFFFAOYSA-N 2-methoxy-5-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound COC1=NC=C(C)C=C1B1OC(C)(C)C(C)(C)O1 BMIBJCFFZPYJHF-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- IXPJMUMWKOSEMB-UHFFFAOYSA-N 3,6-dihydroxynaphthalene-2-carboxylic acid Chemical compound C1=C(O)C=C2C=C(O)C(C(=O)O)=CC2=C1 IXPJMUMWKOSEMB-UHFFFAOYSA-N 0.000 description 1
- QMWOUSYSNFCKAZ-UHFFFAOYSA-N 3,7-dihydroxynaphthalene-2-carboxylic acid Chemical compound OC1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 QMWOUSYSNFCKAZ-UHFFFAOYSA-N 0.000 description 1
- JNZYADHPGVZMQK-UHFFFAOYSA-N 3-(aminomethyl)phenol Chemical group NCC1=CC=CC(O)=C1 JNZYADHPGVZMQK-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- MENAYYMPBRSAAE-AWEZNQCLSA-N 3-[[5-[[(2s)-1-carboxy-3-oxopropan-2-yl]carbamoyl]pyridin-2-yl]methylsulfamoyl]benzoic acid Chemical compound N1=CC(C(=O)N[C@@H](CC(=O)O)C=O)=CC=C1CNS(=O)(=O)C1=CC=CC(C(O)=O)=C1 MENAYYMPBRSAAE-AWEZNQCLSA-N 0.000 description 1
- XFXOLBNQYFRSLQ-UHFFFAOYSA-N 3-amino-2-naphthoic acid Chemical compound C1=CC=C2C=C(C(O)=O)C(N)=CC2=C1 XFXOLBNQYFRSLQ-UHFFFAOYSA-N 0.000 description 1
- 229940018563 3-aminophenol Drugs 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- NLMQHXUGJIAKTH-UHFFFAOYSA-N 4-hydroxyindole Chemical compound OC1=CC=CC2=C1C=CN2 NLMQHXUGJIAKTH-UHFFFAOYSA-N 0.000 description 1
- NAEYBZOUAJOJON-UHFFFAOYSA-N 5,6-dihydroxy-1h-indole-2-carboxamide Chemical compound OC1=C(O)C=C2NC(C(=O)N)=CC2=C1 NAEYBZOUAJOJON-UHFFFAOYSA-N 0.000 description 1
- HIHOEGPXVVKJPP-JTQLQIEISA-N 5-fluoro-2-[[(1s)-1-(5-fluoropyridin-2-yl)ethyl]amino]-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyridine-3-carbonitrile Chemical compound N([C@@H](C)C=1N=CC(F)=CC=1)C(C(=CC=1F)C#N)=NC=1NC=1C=C(C)NN=1 HIHOEGPXVVKJPP-JTQLQIEISA-N 0.000 description 1
- VLDZAMRHGMFFFB-UHFFFAOYSA-N 5-hydroxy-1h-indole-2-carboxamide Chemical class OC1=CC=C2NC(C(=O)N)=CC2=C1 VLDZAMRHGMFFFB-UHFFFAOYSA-N 0.000 description 1
- KAUQJMHLAFIZDU-UHFFFAOYSA-N 6-Hydroxy-2-naphthoic acid Chemical compound C1=C(O)C=CC2=CC(C(=O)O)=CC=C21 KAUQJMHLAFIZDU-UHFFFAOYSA-N 0.000 description 1
- SZTDBOWPIYUEOJ-UHFFFAOYSA-N 6-hydroxy-1h-indole-2-carboxamide Chemical compound C1=C(O)C=C2NC(C(=O)N)=CC2=C1 SZTDBOWPIYUEOJ-UHFFFAOYSA-N 0.000 description 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 1
- 102000004452 Arginase Human genes 0.000 description 1
- 108700024123 Arginases Proteins 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 208000006386 Bone Resorption Diseases 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical group [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- XHZFGHRGDFVDLU-UHFFFAOYSA-N COC(Cc(cc1)ccc1NC(c1cc(cc(cc2)O)c2[nH]1)=O)=O Chemical compound COC(Cc(cc1)ccc1NC(c1cc(cc(cc2)O)c2[nH]1)=O)=O XHZFGHRGDFVDLU-UHFFFAOYSA-N 0.000 description 1
- 101100314454 Caenorhabditis elegans tra-1 gene Proteins 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical class NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- VWDXGKUTGQJJHJ-UHFFFAOYSA-N Catenarin Natural products C1=C(O)C=C2C(=O)C3=C(O)C(C)=CC(O)=C3C(=O)C2=C1O VWDXGKUTGQJJHJ-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 108010049894 Cyclic AMP-Dependent Protein Kinases Proteins 0.000 description 1
- 108091000126 Dihydroorotase Proteins 0.000 description 1
- 102100034581 Dihydroorotase Human genes 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 239000010282 Emodin Substances 0.000 description 1
- RBLJKYCRSCQLRP-UHFFFAOYSA-N Emodin-dianthron Natural products O=C1C2=CC(C)=CC(O)=C2C(=O)C2=C1CC(=O)C=C2O RBLJKYCRSCQLRP-UHFFFAOYSA-N 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- YOOXNSPYGCZLAX-UHFFFAOYSA-N Helminthosporin Natural products C1=CC(O)=C2C(=O)C3=CC(C)=CC(O)=C3C(=O)C2=C1O YOOXNSPYGCZLAX-UHFFFAOYSA-N 0.000 description 1
- 101000727826 Homo sapiens Tyrosine-protein kinase RYK Proteins 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- JDAMFKGXSUOWBV-WHFBIAKZSA-N L-isoleucinamide Chemical compound CC[C@H](C)[C@H](N)C(N)=O JDAMFKGXSUOWBV-WHFBIAKZSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 239000007993 MOPS buffer Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 238000006219 Matteson homologation reaction Methods 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- LQQYARMJQBHKQQ-UHFFFAOYSA-N N-[3-chloro-4-[(6-methoxypyrazolo[1,5-a]pyridine-3-carbonyl)amino]phenyl]-1-[1-(3-cyano-3-methylbutanoyl)piperidin-4-yl]-5-fluoroindazole-3-carboxamide Chemical compound COc1ccc2c(cnn2c1)C(=O)Nc1ccc(NC(=O)c2nn(C3CCN(CC3)C(=O)CC(C)(C)C#N)c3ccc(F)cc23)cc1Cl LQQYARMJQBHKQQ-UHFFFAOYSA-N 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- HDQHFRDOKRVFDW-UHFFFAOYSA-N O.OBO Chemical class O.OBO HDQHFRDOKRVFDW-UHFFFAOYSA-N 0.000 description 1
- WSDRAZIPGVLSNP-UHFFFAOYSA-N O.P(=O)(O)(O)O.O.O.P(=O)(O)(O)O Chemical group O.P(=O)(O)(O)O.O.O.P(=O)(O)(O)O WSDRAZIPGVLSNP-UHFFFAOYSA-N 0.000 description 1
- NNOZTDIONUPHDY-UHFFFAOYSA-N OBO.OP(O)=O Chemical compound OBO.OP(O)=O NNOZTDIONUPHDY-UHFFFAOYSA-N 0.000 description 1
- AQSIOUCMQSBQEM-UHFFFAOYSA-N OC1=Cc2c(O)cccc2CC1=C=O Chemical class OC1=Cc2c(O)cccc2CC1=C=O AQSIOUCMQSBQEM-UHFFFAOYSA-N 0.000 description 1
- ALVZWOMDFUWPNZ-UHFFFAOYSA-N OC1=Cc2ccc(O)cc2CC1=C=O Chemical compound OC1=Cc2ccc(O)cc2CC1=C=O ALVZWOMDFUWPNZ-UHFFFAOYSA-N 0.000 description 1
- CMWQAVAGNOFRMR-UHFFFAOYSA-N Oc1ccc(CCNC(c2cc(cc(cc3)O)c3[nH]2)=O)cc1 Chemical compound Oc1ccc(CCNC(c2cc(cc(cc3)O)c3[nH]2)=O)cc1 CMWQAVAGNOFRMR-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108010058765 Oncogene Protein pp60(v-src) Proteins 0.000 description 1
- 108010055723 PDGF receptor tyrosine kinase Proteins 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 229910002666 PdCl2 Inorganic materials 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 108700020978 Proto-Oncogene Proteins 0.000 description 1
- 102000052575 Proto-Oncogene Human genes 0.000 description 1
- 102100027384 Proto-oncogene tyrosine-protein kinase Src Human genes 0.000 description 1
- 101710122944 Proto-oncogene tyrosine-protein kinase Src Proteins 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 101710100968 Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- NTGIIKCGBNGQAR-UHFFFAOYSA-N Rheoemodin Natural products C1=C(O)C=C2C(=O)C3=CC(O)=CC(O)=C3C(=O)C2=C1O NTGIIKCGBNGQAR-UHFFFAOYSA-N 0.000 description 1
- 102000014400 SH2 domains Human genes 0.000 description 1
- 108050003452 SH2 domains Proteins 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 206010043458 Thirst Diseases 0.000 description 1
- 102100029759 Tyrosine-protein kinase RYK Human genes 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 239000003875 Wang resin Substances 0.000 description 1
- 102100023870 YLP motif-containing protein 1 Human genes 0.000 description 1
- NERFNHBZJXXFGY-UHFFFAOYSA-N [4-[(4-methylphenyl)methoxy]phenyl]methanol Chemical compound C1=CC(C)=CC=C1COC1=CC=C(CO)C=C1 NERFNHBZJXXFGY-UHFFFAOYSA-N 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 108010004174 acetyl-isoleucyl-tyrosyl-glycyl-glutamyl-phenylalaninamide Proteins 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 150000001299 aldehydes Chemical group 0.000 description 1
- 150000008431 aliphatic amides Chemical class 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000000746 allylic group Chemical group 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- ZXKINMCYCKHYFR-UHFFFAOYSA-N aminooxidanide Chemical compound [O-]N ZXKINMCYCKHYFR-UHFFFAOYSA-N 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 230000009925 apoptotic mechanism Effects 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000003305 autocrine Effects 0.000 description 1
- 102000004441 bcr-abl Fusion Proteins Human genes 0.000 description 1
- JPYQFYIEOUVJDU-UHFFFAOYSA-N beclamide Chemical compound ClCCC(=O)NCC1=CC=CC=C1 JPYQFYIEOUVJDU-UHFFFAOYSA-N 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- 125000001743 benzylic group Chemical group 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 230000024279 bone resorption Effects 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 238000000205 computational method Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000039 congener Substances 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- VASFLQKDXBAWEL-UHFFFAOYSA-N emodin Natural products OC1=C(OC2=C(C=CC(=C2C1=O)O)O)C1=CC=C(C=C1)O VASFLQKDXBAWEL-UHFFFAOYSA-N 0.000 description 1
- RHMXXJGYXNZAPX-UHFFFAOYSA-N emodin Chemical compound C1=C(O)C=C2C(=O)C3=CC(C)=CC(O)=C3C(=O)C2=C1O RHMXXJGYXNZAPX-UHFFFAOYSA-N 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 150000003948 formamides Chemical class 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000000799 fusogenic effect Effects 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 229930182480 glucuronide Natural products 0.000 description 1
- 150000008134 glucuronides Chemical class 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 210000002768 hair cell Anatomy 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000012203 high throughput assay Methods 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- GTTBQSNGUYHPNK-UHFFFAOYSA-N hydroxymethylphosphonic acid Chemical group OCP(O)(O)=O GTTBQSNGUYHPNK-UHFFFAOYSA-N 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 108091006086 inhibitor proteins Proteins 0.000 description 1
- 238000013101 initial test Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000009878 intermolecular interaction Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 238000005567 liquid scintillation counting Methods 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- GLCZQTLCVLVFGV-UHFFFAOYSA-N methyl 4-methoxy-1h-indole-2-carboxylate Chemical compound C1=CC=C2NC(C(=O)OC)=CC2=C1OC GLCZQTLCVLVFGV-UHFFFAOYSA-N 0.000 description 1
- OPUUCOLVBDQWEY-UHFFFAOYSA-N methyl 6-methoxy-1h-indole-2-carboxylate Chemical compound C1=C(OC)C=C2NC(C(=O)OC)=CC2=C1 OPUUCOLVBDQWEY-UHFFFAOYSA-N 0.000 description 1
- 125000004492 methyl ester group Chemical group 0.000 description 1
- 150000005217 methyl ethers Chemical class 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 238000000329 molecular dynamics simulation Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- NSBIQPJIWUJBBX-UHFFFAOYSA-N n-methoxyaniline Chemical compound CONC1=CC=CC=C1 NSBIQPJIWUJBBX-UHFFFAOYSA-N 0.000 description 1
- UTWYCVYPCQKRDR-UHFFFAOYSA-N n-methyl-n-phenylhydroxylamine Chemical compound CN(O)C1=CC=CC=C1 UTWYCVYPCQKRDR-UHFFFAOYSA-N 0.000 description 1
- MTSNDBYBIZSILH-UHFFFAOYSA-N n-phenylquinazolin-4-amine Chemical group N=1C=NC2=CC=CC=C2C=1NC1=CC=CC=C1 MTSNDBYBIZSILH-UHFFFAOYSA-N 0.000 description 1
- HUMMCEUVDBVXTQ-UHFFFAOYSA-N naphthalen-1-ylboronic acid Chemical compound C1=CC=C2C(B(O)O)=CC=CC2=C1 HUMMCEUVDBVXTQ-UHFFFAOYSA-N 0.000 description 1
- KPTRDYONBVUWPD-UHFFFAOYSA-N naphthalen-2-ylboronic acid Chemical compound C1=CC=CC2=CC(B(O)O)=CC=C21 KPTRDYONBVUWPD-UHFFFAOYSA-N 0.000 description 1
- NXPPAOGUKPJVDI-UHFFFAOYSA-N naphthalene-1,2-diol Chemical group C1=CC=CC2=C(O)C(O)=CC=C21 NXPPAOGUKPJVDI-UHFFFAOYSA-N 0.000 description 1
- 230000017066 negative regulation of growth Effects 0.000 description 1
- 108010087904 neutravidin Proteins 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- RQKYHDHLEMEVDR-UHFFFAOYSA-N oxo-bis(phenylmethoxy)phosphanium Chemical compound C=1C=CC=CC=1CO[P+](=O)OCC1=CC=CC=C1 RQKYHDHLEMEVDR-UHFFFAOYSA-N 0.000 description 1
- 150000002926 oxygen Chemical class 0.000 description 1
- 235000019629 palatability Nutrition 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- PKUBGLYEOAJPEG-UHFFFAOYSA-N physcion Natural products C1=C(C)C=C2C(=O)C3=CC(C)=CC(O)=C3C(=O)C2=C1O PKUBGLYEOAJPEG-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000005588 protonation Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 238000012289 standard assay Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid Chemical compound NS(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- FGTJJHCZWOVVNH-UHFFFAOYSA-N tert-butyl-[tert-butyl(dimethyl)silyl]oxy-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)O[Si](C)(C)C(C)(C)C FGTJJHCZWOVVNH-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000036962 time dependent Effects 0.000 description 1
- 231100000041 toxicology testing Toxicity 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 230000031143 xenobiotic glucuronidation Effects 0.000 description 1
Images













Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/48—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving transferase
- C12Q1/485—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving transferase involving kinase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2500/00—Screening for compounds of potential therapeutic value
- G01N2500/04—Screening involving studying the effect of compounds C directly on molecule A (e.g. C are potential ligands for a receptor A, or potential substrates for an enzyme A)
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- General Engineering & Computer Science (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Peptides Or Proteins (AREA)
- Indole Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
Description
本出願は、1999年1月13日に出願され
た米国仮特許出願第60/115643号の恩典を主張するものである。
【0002】
発明の背景
タンパク質キナーゼは、ATPからタンパク質およびペプチドにおけるSer/ThrまたはTyrの側鎖水酸基へのγリン酸基転移を触媒し、様々な重要な細胞機能、おそらく最も顕著にはシグナルトランスダクション、分化および増殖の制御に密接に関与している大きな酵素群である。ヒトの体には約2000の異なるタンパク質キナーゼがあると推定されており(Hunter、1987、1994、Hanks & Hunter、1995)、これらはそれぞれ特定のタンパク質/ペプチド基質をリン酸化するが、いずれも高度に保存されたポケット内の同じ第2の基質ATPに結合する。
【0003】
様々な公知のタンパク質キナーゼの阻害剤は、十分な選択性と許容できるインビボでの薬理学的性質がこのような阻害剤に組み込まれうるとするならば、様々な治療への適用例を有すると考えられる(Levitzki、1996a)。おそらく、タンパク質キナーゼ阻害剤の最も有望な治療的使用の可能性は、抗癌剤としての使用である。タンパク質チロシンキナーゼ(「PTK」)阻害剤のこのような適用の可能性が、最近多くの総説で強調されている(例えば、Lawrence & Hiu、1998、Kolibaba & Druker、1997、Showalter & Kraker、1997、Patrick & Heimbrook、1996、Groundwaterら、1996、Levitzki、1995)。この適用の基礎は部分的には、公知の癌遺伝子産物の約50%はPTKであり、そのキナーゼ活性は細胞形質転換を引き起こすことが判明している(Yamamoto、1993)という事実に基づいている。
【0004】
PTKは、膜受容体PTK(例えば、成長因子受容体PTK)および非受容体PTK(例えば、プロト癌遺伝子産物のsrcファミリー)の2つの範疇に分類することができる。非受容体PTKのsrcファミリーには少なくとも9つのメンバーがあり、pp60c-src(以下、単に「src」と呼ぶ)が約300アミノ酸の触媒ドメインが高度に保存されているファミリーの原型PTKである(Ruddら、1993、Courtneidge、1994)。srcの過剰活性化が、大腸癌(Maoら、1997、Talamontiら、1993)、乳癌(Luttrellら、1994)、肺癌(Mazurenkoら、1992)、膀胱癌(Fanningら、1992)および皮膚癌(Barnekovら、1987)ならびに胃癌(Takeshimaら、1991)、有毛細胞白血病(Lynchら、1993)および神経芽細胞腫(Bjelfmanら、1990)を含む、いくつかのヒト癌で報告されている。膜貫通型受容体(例えばEGFRおよびp185HER2/Neu)から細胞内部への過刺激細胞増殖シグナルも、srcを通過するようである(Maoら、1997、Parsons & Parsons、1997、Bjorgeら、1996)、Taylor & Shalloway、1996)。したがって、srcの過剰活性化(突然変異なし)がヒト腫瘍の多くの重要なタイプの腫瘍イニシエーション、進行、および転移に関与しているため、最近、srcは癌治療の普遍的な標的であると提唱されている(Levitzki、1996)。
【0005】
様々なタンパク質キナーゼの阻害剤に対する大きな、また成長しつつある可能性を考慮して、有用な阻害剤を得るための様々なアプローチが必要とされている。PTK阻害剤の発見の状態(Lawrence & Niu、1988、Showalter & Kraker、1997、Patrick & Heimbrook、1996、Groundwaterら、1996、Buddeら、1995、Levitzki & Gazit、1995)が広く再調査されている。非ペプチドタンパク質キナーゼ阻害剤の特定においては無作為スクリーニングによる努力が功を奏したが、これらの大半は高度に保存されたATP結合部位に結合する。そのような非ペプチドATP競合的阻害剤のうち、注目に値する最近の一例は4-アニリノキナゾリンおよびその類縁体で、これらは上皮成長因子受容体PTK(EGFRTK)に対して有効であることが明らかにされている(例えば、Rewcastleら、1996)。このクラスの阻害剤は他の6つのPTK(srcを含む)と比べてEGFRPTKに選択的であると報告された(Fryら、1994)が、すべてATPに結合する残りの2000のタンパク質キナーゼのほとんど、ならびにATP、ADP、GTP、GDP等を利用する体内の多くの他のタンパク質に対する影響がどのようなものであるかは不明である。したがって、費用のかかる動物毒性試験やヒト臨床試験後にのみ見いだされる、ATPを模したPTK阻害剤からの副作用の可能性は、いまだに重大な問題である。また、このクラスの化合物は素晴らしい発見であり、さらに探究が行われているが、これらはいかなる所望のPTK、例えば、この場合にはsrcの非ペプチド阻害剤を得るための合理的かつ一般的解決にはならない。ATP競合的PTK阻害剤が、単離酵素アッセイ法で用いられるμMレベルではなく、細胞内ATPのmMレベルと競合する場合に示す、固有の3桁の効力低下に加え、これらの阻害剤に伴うインビボでの不十分な特異性のリスクが、他の研究者らによって認められている(例えば、Lawrence & Niu、1998、Hankeら、1996、Kelloffら、1996参照)。
【0006】
より古く、より広範に研究された非ペプチドPTK阻害剤の1つのクラスはエルブスタチンおよび関連するチロホスチンである(総説参照)。このクラスの阻害剤は受容体PTKに対して活性で、その阻害の様式は複雑であるが、活性部位のペプチド基質特異部位領域における結合には関与しないようである(Hsuら、1992、Posnerら、1994)。さらに、これらは非天然アッセイ金属Mn2+を天然Mg2+に置き換えると、単離PTKに対して不活性で(Hsuら、1992)、化学的に不安定であり(Buddeら、1995、Ramdasら、1995および1994)、またタンパク質を架橋することにより正常および腫瘍細胞に対して細胞毒性を示すること(Stanwellら、1995および1996)、ならびにPTKの阻害ではなく、ミトコンドリアを破壊することにより細胞増殖を阻害すること(Burgerら、1995)が知られている。
【0007】
タンパク質キナーゼ分野への重要な貢献は、熱安定阻害剤タンパク質由来の20残基ペプチドであるPKI(5〜24)に結合したセリンキナーゼcAMP依存性タンパク質キナーゼ(「PKA」)、およびMg2ATPによるX線構造研究である(Taylorら、1993)。この構造研究は、タンパク質キナーゼは単一の祖先タンパク質キナーゼから進化したため、PKAがタンパク質キナーゼの全ファミリーの原型であると考えられることから、特に価値がある。PKAと他のセリンおよびチロシンキナーゼの配列アライメントにより、保存された約260残基の触媒コアと、このコア内の高度に保存された11残基が特定された(Taylorら、1993)。本明細書において提唱される研究のために特に重要な2つの高度に保存された残基は、基質のOHと相互作用するとされている一般塩基Asp-166と、セリンキナーゼではLys-168、チロシンキナーゼではArg(Knightonら、1993)で、これはATPのγリン酸基と相互作用してこのリン酸基転移の触媒作用を助けるとされている。さらに別の2つの重要なPKA結晶構造が報告されており(Madhusudanら、1994)、1つはPKI Ala 21がSerで置換されている(それにより基質となっている)三元のPKA:ADP:PKI(5〜24)複合体、もう1つはPKI Ala 21がホスホセリン(最終産物阻害剤)で置換されている二元のPKA:PKI(5〜24)複合体についてである。三元複合体では、Asp-166にH-結合を供与し、Lys168の側鎖からH-結合を受容するセリンのOHが認められる。二元複合体では、Lys-168の側鎖との塩橋であって、Asp-166のカルボキシル基のH-結合距離内の塩橋を形成するホスホセリンのリン酸基が認められる。これらの構造は、先に提唱されたAsp-166およびLys-168の触媒メカニズムにおける役割を支持している。
【0008】
PKAのX線構造は、酵素が2つのローブからなり、小さいローブはATPに結合し、大きいローブはペプチド基質に結合することを示している。触媒作用はローブの間の裂け目で起きる。PKAを用いた結晶学および溶液構造試験により、酵素が基質と結合すると、「開いた」形状から「閉じた」触媒として活性な形状への大きなコンフォメーションの変化を受けることが示された(Coxら、1994)。これらコンフォメーションの変化は、基質の結合に伴い2つのローブ間の裂け目を閉じ、リン酸基を直接転移させるためにATPのγリン酸基およびSerのOHをより接近させると推定される。
【0009】
しかし、タンパク質キナーゼ阻害剤はなお、治療に用いる際に望まれる特異性および効力が不足している。癌、乾癬、関節硬化症を含むいくつかの異なる疾患においてタンパク質キナーゼが重要な役割を果たし、免疫系の活性調節に役割を果たすことから、特定のタンパク質キナーゼ阻害剤が必要である。本発明は、より強力で、標的とする経路により特異的な、タンパク質キナーゼ阻害剤を設計するための新規なアプローチを提供する。
【0010】
発明の概要
本発明はタンパク質キナーゼ阻害剤を同定するための方法を提供する。タンパク質キナーゼの触媒残基に結合するための1つまたは複数の官能基を有する第一のモジュールを非ペプチド骨格を提供する第二のモジュールに組み合わせる。タンパク質キナーゼ活性を阻害する第一および第二のモジュールの組み合わせを選択する。
【0011】
本発明はまた、タンパク質キナーゼを阻害する方法も提供する。タンパク質キナーゼを、タンパク質キナーゼの触媒残基に結合するための官能基を有する第一のモジュールと非ペプチド骨格を提供する第二のモジュールとを含む化合物に接触させる。第一と第二のモジュールの組み合わせがタンパク質キナーゼ活性を阻害する。
【0012】
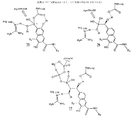
さらなる態様において、本発明は下記の式を有する非ペプチドタンパク質キナーゼ阻害剤を提供する:
【化10】

【0013】
本発明はまた、下記の式を有する非ペプチドタンパク質チロシンキナーゼ阻害剤も提供する:
【化11】
【0014】
さらにもう一つの態様において、本発明は患者のタンパク質キナーゼ阻害剤に反応性の状態を治療する方法を提供する。タンパク質キナーゼ阻害剤を患者に投与する。タンパク質キナーゼ阻害剤は、タンパク質キナーゼの触媒残基に結合するための官能基を有する第一のモジュールと、非ペプチド骨格を提供する第二のモジュールとを有する。第一と第二のモジュールの組み合わせが患者のタンパク質キナーゼ活性を阻害する。
【0015】
発明の詳細な説明
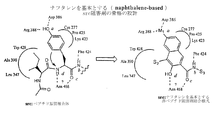
本発明はタンパク質キナーゼ阻害剤を同定するための方法を提供する。非ペプチドPTK阻害剤開発のための一般的モジュール方法を図1に示す。基本的に、タンパク質キナーゼの触媒残基に結合するための1つまたは複数の官能基を有する第一のモジュールを非ペプチド骨格を提供する第二のモジュールに組み合わせる。次いで、タンパク質キナーゼ活性を阻害する第一と第二のモジュールの組み合わせを選択する。段階1は、すでに得られていたタンパク質キナーゼ阻害剤の情報から始める、すなわち、PKAまたはsrcの基質特異性部位に結合するペンタペプチド骨格は、保存された触媒残基MgATPまたはMgADPと相互作用するための様々な合理的に設計された官能基(すなわち、モジュール「M1」または「第一のモジュール」)を配置するためにすでに用いられていた。このようにして好ましい官能基の選択が行われ、段階1の最初のM1モジュールとして用いられた。これらのM1官能基は段階1においてsrc阻害のための有望な非ペプチド骨格を同定するために用いられている。M1付属物のみを有するこれら裸の非ペプチド骨格は、結合親和性が低く、PTKの間で相対的に非選択的であると予想された。段階1のレベルで選択性に欠けることは、別のPTKに適用することができる一般的方法の開発のためには有益と見なされる。したがって、段階1で特定される一連の非ペプチド骨格は、これらをすべて新しいPTK標的を用いて再度スクリーニングし、よりよいものを段階2および3に進めることによって、別のPTKに用いるために再循環させることができる。段階1からのこれら裸の骨格の効力は、1つまたは2つの最初の特異的因子(Sn)を連結させることによって十分高められ、骨格の非ATP競合性の妥当性確認ができ、合理的に導かれるコンビナトリアルケミストリーを用いたさらなる効力増強に適したものとなる。段階1で特定された有望なsrc非ペプチドM2(第二のモジュール)骨格は段階2に進み、srcに対する効力ならびにATPと比較しての非競合的結合において1桁から2桁の上昇を示した。
【0016】
資源集中的コンビナトリアルライブラリー合成に着手し、段階3の試験を行う前に、段階2のレベルで骨格の妥当性確認を行うことが、次の3つの理由から重要である:1)特異的因子(Sn)側鎖を付加するための化学反応を開発するため。2)これらの阻害剤はATP競合的ではないことを調べるため。3)src:阻害剤複合体の研究モデル(これにより、一連の個々の選択的因子Snを段階3の集中ライブラリーに含めるために、合理的に導かれた選択ができるとの確信が得られる)に基づいて予期されるとおり、効力は側鎖Snの性質および連結点に反応することを調べるため。
【0017】
選択的因子(Sn)を有する多くのM1官能基(および非常に近い類縁体M1')をコンビナトリアルケミストリーおよびハイスループットスクリーニングを通じて実験的に評価するため、段階3では特定のPTKに対する高い効力と特異性が予期される。効力および選択性は、別の特異的因子(図1の任意のSn参照)を付加することにより、必要があればさらに高めることができる。段階2から選択されたsrc阻害剤の1つは、すでに固相樹脂に結合されており、現在段階3に続くコンビナトリアルライブラリーへと開発中である。
【0018】
段階1〜3のそれぞれで、IRTK:ペプチド:AMP-PNP結晶構造、src:ペプチド複合体のモデル、および特定の骨格を基本とする個々の阻害剤ファミリーとのsrc複合体のモデルを用いた分子モデリング試験を定性的指標として用いる。これらのモデリング試験は、以下に詳述するとおり、阻害剤を設計する指標として、ここまでは非常に有用であった。このようにして構造に基づく設計とコンビナトリアルケミストリー法とを組み合わせることによって協力効果が得られ、分離して用いられる場合のこれらの方法の主な個々の欠点はもう一方の長所によって対処される。構造に基づく設計の主な欠点は、リガンド結合親和性を定量的に予測するのが困難な点であり、これは溶媒和とエントロピーとの複雑な影響によって特にむずかしいものとなっている(Ajay & Murcko、1995)。構造に基づく設計の主な長所は、どのようなタイプの分子が良いリガンドとなる可能性を有しているかを予測できることである。構造に基づく設計は分子のサイズや形のおおまかな境界(タンパク質は柔軟性を有することがあり、このことを考慮に入れる必要がある)を決定することができ、ならびに疎水性のH-結合およびイオン性相互作用が起こりやすい位置を示すことができる。他方、コンビナトリアルケミストリーの主な欠点は、薬物サイズの分子(すなわち、分子量が約500以下)のための「分子空間」が大きすぎて、この分子空間すべてを妥当なサイズのコンビナトリアルライブラリーにおいて高密度でカバーして調べられるとは考えられない。最近推定された、炭素、窒素、酸素、およびイオウ(Hの他に)だけから選ばれた30個までの原子を含む、可能な化合物の数は1060個である(Bohacekら、1996)。この数は典型的な薬物分子の分子量の範囲で、これでも他の原子、例えばハロゲンにより提供される分散性は含まれていない。したがって、特定の薬物候補が位置する可能性のある分子空間の領域を特定するためには、さらに制約を用いる必要がある。構造に基づく設計は、良いリガンドである確率の高い分子のタイプを特定することにより、調べるべき分子空間の大きさを劇的に縮小することができる。これらの「集中的」コンビナトリアルライブラリーのどのメンバーが実際に最も密接に結合するリガンドであるかを定量的に予測できない問題(すなわち、定量の問題)は、効率の良いコンビナトリアル合成とライブラリーのハイスループット試験を用いることによって解決される。
【0019】
これまでペプチドを基本とするセリンおよびチロシンキナーゼ阻害剤を設計しようとする際に、保存された触媒残基との相互作用のためのタンパク質キナーゼ阻害剤モジュールM1を設計するための簡便な定性的モデルとしてPKAが用いられた。PKAについては、いかなる他のタンパク質キナーゼよりも多くの構造および動態に関する情報が得られている。
【0020】
Mg2ATPおよび偽基質(すなわち、OHがHに置換されている)ペプチド阻害剤(PKI 5〜24アミド)と複合したPKAの結晶構造が解析されており(Zhengら、1993)、この阻害剤のP 0 Alaに近接する活性部位相互作用を図2に示している。
【0021】
この結晶構造から、Mg2ATPはPKAの小さい方のローブに結合し、20残基の偽基質ペプチド阻害剤は大きい方のローブに結合し、酵素の全体のコンフォメーションは閉じて(すなわち、2つのローブが接触している)活性化された状態であることが明らかである。複合体におけるP 0 Ala側鎖の炭素と近くの重原子との距離は図1にAoで示している。これらの距離は、Ala側鎖は周囲の原子とファンデルワールス接触距離内であることを示し、また、付加されているバルキーなM1官能基にはAla側鎖までほとんどスペースがないことを示している。しかし、PKAは開いたり、閉じたり、および中間のコンフォメーションを取れる柔軟性の酵素であり(Coxら、1994)、これらのより開いたコンフォメーションによってATPのγリン酸基が阻害剤のAlaから後退し、それにより付加されたM1官能基のための結合空隙を作ることになると考えられる。さらに、PKAはMgATPと同等の親和性でMgADPと結合し(Whitehouseら、1983)、細胞内のATP/ADP比は通常10/1である(Albertsら、1994)。したがって、平衡状態では、細胞タンパク質キナーゼは約10%がMgADPと結合した状態であり、酵素のこの形状も阻害剤の標的となって、すべての酵素が触媒サイクルからPKA:MgADP:阻害剤の不活性複合体へと排出されうる。
【0022】
PKA触媒残基Asp-166およびLys-168はすべてのセリンキナーゼで完全に保存されており、チロシンキナーゼはLys-168がArgで置換されている点だけが異なっている(Taylorら、1993)ため、M1として役立ち、全タンパク質キナーゼファミリーに対する阻害剤を開発するために広く有用となる阻害剤官能基の選択範囲を標的とするために、この活性部位の領域を近接するMgATPまたはMgADPと共に選択した。ヌクレオチドに隣接する活性部位の領域にM1を向けることにより、非ペプチド阻害剤の配向点が提供されて、阻害剤はATP/ADP結合と必ずしも競合せずにペプチド結合特異性部位へと伸びることができる。
【0023】
M1として利用しうる官能基の選択は、この活性部位領域は非常に高度に保存されているが、それぞれの特定のタンパク質キナーゼは活性部位のコンフォメーションや近接する残基にわずかな差があるため、この選択範囲の中で幾分異なる嗜好性を示すと考えられる。さらに、M1のこの選択範囲の中の優先順位は、M1モジュールが異なる非ペプチド骨格に付加しているため、幾分変動することもある。この予測は、各非ペプチド骨格が各個々のタンパク質キナーゼおよび各特定の選択的因子(Sn)側鎖群と幾分異なる配向で結合する可能性に基づいている。ペンタペプチド骨格は、このような大きいペプチド骨格の結合配向が系列を通じて非常に一貫性があって予測しやすく(すなわち、X線で観察したものと非常に類似している)、且つ試験される各M1官能基を意図されたとおり保存触媒残基に隣接して置くと確実に予測できるため、M1官能基の最初のスクリーニングのために選ばれた。したがって、この以前のペプチドを基本とする研究の目標は、非ペプチド骨格の最初のスクリーニングのため(段階1)だけでなく、最終の非ペプチドコンビナトリアルライブラリーにおいて非常に近い類縁体を通してさらに拡大し、それによって他の側鎖と同時に最適化することができる最初のM1側鎖群として(段階3)も用いることができるM1官能基の集合を特定することであった。
【0024】
PKA活性部位のこの保存触媒領域における候補M1官能基のモデリングを行うため、図3に示すとおり、シリコングラフィクス(Silicone Graphics)ワークステーションでSYBYL分子モデリングパッケージ(Tripos)を用いてPKA三元構造におけるP 0 Ala位上にこれらの基を構築した。
【0025】
MgATPおよび阻害剤が結合したPKAのより「開いた」コンフォメーションの結晶構造は得られなかったため、最初のモデリング試験は単にATPのγリン酸基を除去することにより、図2に示した三元複合体由来のPKAのMgADP結合型に対して実施した。最初のモデリング試験は、合成および試験前にタンパク質キナーゼファミリーのために興味深いと思われるM1官能基を同定するための定性的指標を提供するために用いた。自由エネルギー摂動法(Free Energy Perturbation)法などの、結合自由エネルギーを定量的に予測するための最も進んだコンピューターアルゴリズムは、コンピューターによる集中的方法で、この時点では専門家以外の者がルーチンに用いるには実用的でない。最先端の方法であっても、サンプリングが難しく、分子力場/パラメーターが不十分で、水中の静電学の理解が不完全であるために、不正確になることもある(Ajay & Mureko、1995)。厳密性が低い(より使いやすい)コンピューター法は、結合親和性の定量的予測を行う際に、特にM1結合に関与しているような複数の極性でイオン性の相互作用を扱う場合には、信頼性が低い傾向にある。
【0026】
シリコングラフィクスワークステーションを用い、妥当な時間で分子力学の計算を行うために、ペプチド阻害剤とMg2ADPと共にPKAの活性部位を取り囲んでいる2層の残基をPKA三元構造から切り取った。次いで、M1官能基をP 0 Ala側鎖に付加し、PKA活性部位:Mg2ADP:改変ペプチド阻害剤複合体全体を、SYBYLを用いて適当な形式電荷を割当て、ガスティガーマーシリー(Gasteiger Marsili)点電荷を計算した後、距離に依存する誘電率でTripos力場を用いて300回の分子力学最小化にかけた。最大反復回数300の設定は複合体にいかなる重大なひずみがあってもこれを除去するのに十分で、収束に達しない場合にも全体の構造を開始X線構造から著しく「ずれ」させることはなかった。次いで、これらの最小化した複合体を画像で評価し、付加した個々のM1官能基が保存触媒残基および/またはMg2ADPと好ましい相互作用をするかどうかを調べた。この画像評価では、他の標準的相互作用評価の中でも特に、原子−原子の距離を測定して、水素結合およびイオン性相互作用が好ましく形成されているかどうかを調べた。
【0027】
個々のM1官能基と保存触媒残基またはMg2ADPとの間の好ましい分子間相互作用は、必ずしも新規の阻害剤について増強された結合親和性が認められるということを意味しているのではない。極性M1官能基と極性PKA活性部位残基の両方の好ましくない脱溶媒作用(ならびに複合体エントロピー作用)はこの分析には含まれず、付加されたM1官能基が意図されたとおり保存触媒残基および/またはMg2ADP(またはMgATP)と相互作用するとしても、改変された阻害剤は対応するP 0 Ala阻害剤よりも効力が低い可能性があると言ってもよい点まで正味の結合親和性を低下させる可能性がある。この脱溶媒作用に伴う不都合によって結合親和性に正味の増大が見られない場合でも、これらのM1官能基は非ペプチド阻害剤類縁体をタンパク質キナーゼ活性部位における正しい位置に置くための配向基として有用である。これらの極性官能基は保存触媒残基およびMgADP/MgATPに隣接して受容されることが示された能力(ペンタペプチド骨格に適切につながれて)に基づいて特別に設計され、選択されたため、これらを活性部位内の別の位置に置くと(骨格が活性部位に有利に結合している一方で、これらの官能基が大量の溶媒へと伸びることができないようにつながれていると仮定して)、結合親和性が低下する結果となりやすい。段階1で特定のM1官能基が非ペプチド骨格を正しく配置しないと、段階2における合理的に連結している最初の特異的因子によって効力を改善しようとする試みは失敗に終わりやすいと考えられる。
【0028】
文献中にはタンパク質キナーゼアッセイ法でADPを追加しているものは存在しない。文献における典型的なPKAアッセイ法(Glassら、1989)を、細胞における自然な1/10の比を反映させるため、ATP濃度の10%のADPを加えることによって改変した。このタンパク質キナーゼアッセイ法を以下「文献模倣」アッセイ法と呼ぶ。これはPKAならびにsrcに対して用いられてきた。文献および市販のタンパク質キナーゼアッセイ法の試験から、研究所ごと、会社ごとに一貫性に乏しく、これらのアッセイ法はすべて公知の細胞内の条件とはかなり異なる物理化学的条件を用いていることが明らかとなった。細胞内タンパク質キナーゼの阻害が薬物発見の最終目標であるため、細胞内で知られている全体の細胞質ゾルの物理化学的条件をより厳密に模擬する新しいタンパク質キナーゼアッセイ法を開発した。これらの「細胞模倣」タンパク質キナーゼアッセイ法の開発を、どの形のタンパク質キナーゼが所与の阻害剤に最も良く結合するかを調べるための新規な方法(STAIRe法)と共に、本明細書に記載する。データは、細胞模倣アッセイ法における新規の非ペプチドsrc阻害剤の活性を、LA25 src形質転換細胞株で得られたものと相関させて収集した(下記参照)。
【0029】
これら2つのアッセイ条件を、図3に示すとおりに設計したペンタペプチドを基本とするPKA阻害剤のいくつかに適用し、表1に示す結果を得た。同じアッセイ条件を同様に設計したペンタペプチドを基本とするsrc阻害剤にも適用し、表2に示す結果を得た。
【0030】
表1のPKA阻害剤の大多数に対して選択した標準的ペンタペプチド配列は、図1に示す結晶構造が解明された時の、PKAに結合していたペプチド阻害剤の偽基質配列からのものであった。表2のsrcに用いた標準的ペンタペプチド配列、Ac-Ile-AA-Gly-Glu-Phe-NH2は、Nair、Kimら、1995に記載されていた。PKA阻害剤を調製するために用いた化学反応のいくつかは、Nair、LeeおよびHangauer、1995に記載されている。いくつかのsrc阻害剤を開発するために用いた合成法はLaiら、1998に記載されている。
【0031】
表1および表2の一連の結果から、セリンキナーゼPKAおよびPTK srcはいずれも、様々な大きい極性M1官能基をP 0リン酸化部位に適応させうることが明らかである。さらに、STAIRe法(Choiら、1996参照)を用いて、スルファミン酸阻害剤8、および関連する阻害剤は、MgATP(MgADPやヌクレオチドではなく)も結合している場合に実際に最も良く結合することが示された。このことは、タンパク質キナーゼについて一般的に認められている反応機構において、これらの阻害剤はリン酸基転移後、MgADPと同時に結合しなければならない「最終産物阻害剤」1および12の類縁体であるため、幾分驚く結果であった。
【0032】
これらの結果は、PKAとsrcはいずれも構造的に非常に類似の阻害剤に対し、大きく異なる結合親和性を示しうることも明らかにしている。例えば、スルファミン酸PKA阻害剤8(表1)のKiは、文献模倣アッセイ条件(L)下で0.16μMであるのに対し、等配電子のスルホンアミド7では1,875分の1の効力(Ki=300μM)である。スルファミン酸阻害剤8は最終産物リン酸基阻害剤1とも等配電子であるが、文献模倣アッセイ条件(31倍)および細胞模倣(C)アッセイ条件(108倍)のいずれでもはるかに密接に結合する。基質Serと同様の位置にある酸素原子の有益な効果を、ホスホン酸塩2とリン酸塩1との比較、およびエーテル6とリン酸塩1との比較により示している。この酸素原子はセリン様OH側鎖としても配置して、結合を増強する(2を3Aおよび4Aと比較されたい)ことができ、よりセリンに類似の4Aの方が活性が高い。ジアステレオマー阻害剤3AまたはBおよび4AまたはBの活性の差から、M1設計において企図されたとおり活性部位である触媒残基Asp-166との特異的相互作用が実際に起こっていることが示唆される(図3)。
【0033】
【表1】
PKAペンタペプチド骨格に付加している初期M1スクリーニング結果
src阻害の結果(表2)より、最終産物阻害剤12は、PKA最終産物阻害剤1と同様、文献模倣アッセイ条件からイオン強度がより高い細胞模倣アッセイ条件へと変えることで、活性が低下することが明らかである。しかし、極性M1官能基を有するPKA阻害剤はすべて、細胞模倣アッセイ条件下では活性が低かったが、3つのsrc阻害剤14、15および17は、これらのイオン強度がより高いアッセイ条件下でもその活性を維持していた。また、ヒドロキシホスホン酸塩src阻害剤13(RおよびSジアステレオマーの混合物)はPKA阻害剤3Aの類縁体で、いずれもおおざっぱに言えば、文献模倣アッセイ条件下ではそれぞれ対応する最終産物阻害剤12および1と同じ活性範囲である。ホスホン酸塩src阻害剤13の側鎖長を炭素原子1個分短縮する(同時に連結されたOHを必ず除去する)ことにより、活性が改善された14(PKA阻害剤3と4の比較と同様)となり、より重要なことに、細胞模倣アッセイ条件下で活性が同等となる。16〜19のsrc結果(特に17、下記の非ペプチドsrc阻害剤に付加した類似のα-トリカルボン酸M1類縁体参照)からも、類似のアミドが非ペプチドsrc阻害剤と共に調査するための有用なM1官能基であることが示唆される。
【0035】
非ペプチドsrc阻害剤の中にはsrcに対して二重の効果を持つものもあることが部分的原因となって、ペプチド骨格を基本とする化合物よりも非ペプチド阻害剤の方が好まれる。例えば、ホスホン酸塩阻害剤14は活性部位に競合的に結合してsrcを阻害するだけでなく、SH2に結合し、それにより分子内自己阻害メカニズムを開放することによって、srcを活性化する(Xuら、1997)。この相反する効果により14のIC50曲線は普通とは異なり、低阻害剤濃度ではsrcは滑らかな用量−反応様式で(最初のより強いSH2結合により)刺激され(最大70%まで)、続いてより高濃度では典型的なIC50阻害曲線を示す(活性部位の低い親和性遮断による)。このペンタペプチド阻害剤の相反する活性化作用により、実際よりも活性部位阻害剤として効力が低いように見え、このペンタペプチド骨格に付加しているM1基を正確に評価するのが困難になる。しかし、srcペンタペプチド骨格によって特定されたより良いM1基でも、活性部位の触媒領域に適応するはずで、したがって意図されたとおり、現在行っている非ペプチドsrc阻害剤試験のための有用な配向基である。PKAはSH2ドメインを有していないため、この複雑な問題はPKAペンタペプチド阻害剤のM1試験データを解釈する際の因子にはならない。
【0036】
【表2】
SRCペンタペプチド骨格に付加している初期M1スクリーニング結果
【0037】
調査した次のクラスのM1官能基はボロン酸基であった。この官能基は次のいくつかの理由によりM1として興味をそそられる候補である:1)非イオン状態で存在しうるため、細胞膜を通過しての非ペプチド阻害剤の受動的吸収を阻止しない。2)平面で三角形のボロン酸は、空の2p軌道を通じ、触媒Aspカルボキシル基またはATP/ADP末端リン酸基酸素などのタンパク質キナーゼ活性部位にアニオンが存在する、可逆的な四面体の共有結合ボロン酸塩複合体を形成すると考えられる(ボロン酸のよく知られた性質、LoomisおよびDurst、1992参照)。この活性部位求核体とボロン酸塩複合体を形成する能力は、セリンプロテアーゼの遅結合阻害剤を開発するために広く利用されており(例えば、KettnerおよびShenvi、1984参照)、求核性のセリンOHはボロン酸の空の2p軌道と共有結合を形成して四面体のボロン酸塩複合体となる(例えば、Skordalakesら、1997参照)。また、ボロン酸と尿素NH2との分子内複合体が、ジヒドロオロターゼの遷移状態類縁阻害剤を調製するために用いられた(Kinderら、1990)。3)ボロン酸はルイス酸としてはたらき、水中で水または水酸化物イオンとボロン酸塩複合体を形成することにより四面体水和物に変換される。したがって、これらのボロン酸水和物が図2で提唱された模擬リン酸基およびM1モジュールとして機能する可能性もある。この水和性はBaggioら(1997)が利用し、水和ボロン酸はアルギナーゼに対する遷移状態類縁阻害剤の官能基として機能した。これらの研究者は、阻害された複合体をX線により評価し、水和ボロン酸官能基が活性部位の触媒Glu-277カルボキシル側鎖と2つの水素結合を形成し、他の水和ボロン酸のOHの1つが活性部位の2つの触媒Mn2+と相互作用することを明らかにした。これらの結合相互作用は、タンパク質キナーゼ活性部位で提唱されているもの、すなわち、触媒Asp側鎖カルボキシル基とのH-結合および活性部位Mg2+との相互作用(図2参照)と非常に類似である。4)タンパク質キナーゼ阻害剤のためのボロン酸の使用は過去に調査されたことはない。
【0038】
ペンタペプチドを基本としたPKA阻害剤の分野において、ボロン酸官能基がM1モジュールの候補として表3に示す4つの阻害剤21〜24を用いて調製され、試験されている(これらの化合物を調製するために用いたいくつかの化学反応については、HsiaoおよびHangauer、1998参照)。
【0039】
【表3】
ボロン酸含有ペプチド阻害剤によるPKA阻害結果
L=文献模倣アッセイ条件
C=細胞模倣アッセイ条件
Inh=阻害
Sti=刺激
【0040】
これらのボロン酸含有PKA阻害剤を試験する際に、表3に示す時間依存性阻害の調査中に、対応するペンタペプチド偽基質阻害剤20を内部対照として含めた。文献模倣アッセイ条件下、予備インキュベーションせずに得られた最初の結果より、鎖長が最短のL-アミノ酸21は偽基質阻害剤20と同じ親和性(すなわち、Ki約9μM)で結合していることが示唆された。この側鎖の鎖長が伸びるにしたがい(23、次いで24)、結合親和性は低下するようであった。非天然アミノ酸の立体化学を21のLから22のDに反転させると、結合親和性は3倍に上がるようであった。この結合における改善は、21のボロン酸のOHはL-ホモセリンと同じ鎖長であるが、天然の基質L-セリンは鎖長が炭素1個分短いことから起こると考えられる。PKA三元構造によるモデリングの結果より、α-炭素の立体化学を21のLから22のDに反転させ、次いで触媒残基(Asp-166およびArg-168)に隣接する天然基質L-セリンのOHの位置をより厳密に模して側鎖を配置することにより、ボロン酸のOHが幾分後退させられることが示された。モデリングの結果は、続いて、PKAをこれらの阻害剤と共に競合するペプチド基質(ケンプタミド:LRRASLG-NH2)を加えずに4時間までインキュベートすることにより、21と22の両方が基質として機能し、D-ジアステレオマーである22がより速やかにリン酸化されるという知見によって裏付けられた。
【0041】
これらのボロン酸阻害剤も基質になるという事実は、細胞模倣条件下、予備インキュベーションを行った場合と行わない場合の両方で得られた、大きく変形したIC50曲線によってより明らかになった(PKAとsrcはいずれも、文献模倣条件下よりも細胞模倣条件下でより活性が高い酵素である)。これらの結果を得るために用いたアッセイ法において、P32リン酸化ケンプタミド生成物(γ-P32 ATPから生成した25)を基質インキュベーション期間の終わりに3つのカチオン基(2つのArgとN末端)を介してホスホセルロースフィルターペーパーに結合させることによって単離し、次いでペーパー上に単離されたリン酸化生成物のレベルを液体シンチレーション計数によって測定した(cpm)。ボロン酸阻害剤21〜24もその配列中に2つのArgを有するため、ケンプタミド以外にこれらもホスホセルロースペーパーに結合することになる(正電荷が1つ少ないため、一貫して、または完全に結合はしないが)。したがって、阻害剤として分析した場合、生成したリン酸化ケンプタミドの量だけではなく、同時に生成したリン酸化阻害剤(例えば、下記の26参照)の量も計数された。正味の結果は、場合によってはより高い阻害剤濃度では正味の「刺激」を示す変形したIC50曲線が得られるということである。Dジアステレオマー22が細胞模倣条件下、PKAと共に4時間予備インキュベートすると、最も大きい見かけの「刺激」を示し(71%)、次いでLジアステレオマー21(19%)、その次が1炭素同族体の23(5%)で、これら3つはすべてPKAの基質であることを示している(表3)。これら「阻害剤」の潜在的な基質としての挙動によって、現在のアッセイ法ではその阻害抗力の正確な測定は不可能となる。しかし、データから、ボロン酸官能基をCH2基だけで同族体化することにより(ボロン酸非ペプチドsrc阻害剤による同族体化も実施してもよい)、結合親和性および基質として機能する能力が低下するようである。
【化12】
ボロン酸阻害剤21および22は図4に示すとおり、同じであるがケンプタミドを加えないアッセイ法を行い、反応を様々な時点で停止させることによってPKAの基質であることが明らかにされた。グラフはそれぞれのリン酸化の速度およびレベルを示しており、ケンプタミドなどの標準的L-Ser基質と同様に、時間の経過に伴い最初の速度動態の典型的消失が認められる(基質の枯渇と最終産物の阻害による)。図示した21と22の比較を、cpmを直接比較できるよう、等しいボロン酸基質濃度で、等しい細胞模倣アッセイ溶液を用いて、同じアッセイで実施した。グラフより、D異性体22では1時間以内に最初の速度動態が消失したが、L異性体21では直線性が失われるのが幾分遅いようで、出発物質の消費が遅いことが示唆された。ボロン酸部分がPKAによってリン酸化されるのは驚くべき知見であったが、生成したホスホン酸−ボロン酸混合無水物(例えば26)が非常に安定で、pH7.2/37℃のアッセイインキュベーションに耐え、10%TCAを用いた酸による反応停止後、ホスホセルロースペーパーに結合させ、このホスホセルロースペーパーを25mMリン酸で洗浄(X3)することにより単離されたことは、さらに驚くべきことである。ホスホン酸およびボロン酸の混合無水物に対してSTN基礎構造検索を行い、823°Kでエタンからアセトアルデヒドへ部分酸化するための固体表面含浸触媒としてのホウ酸およびリン酸から生成された類似の推定(証明されてはいない)無水物に関する実験および理論的計算についての文献を3つだけ見いだした(ZhanpeisovおよびOtsuka、1992、Otsukaら、1992、Murakamiら、1990)。しかし、この非常に変わった無水物について、固体表面から遊離して合成、単離、または特徴付けがなされたことはない。したがって、これはタンパク質キナーゼ阻害剤設計のための新規な酵素反応および化学的実体である。
【0043】
前述のM1をつないだsrcおよびPKAペンタペプチド骨格の評価より、段階1(図1)に示した可能な非ペプチド骨格のスクリーニングに用いることができる様々な配向M1基が特定された。src非ペプチド骨格スクリーニングのために利用できる可能性があるM1のメニューから、最初にボロン酸(22から)、ホスホン酸塩(14から)、およびスルファミン酸(8から)が選択された。これらの中で、ボロン酸M1基が段階1の非ペプチド骨格スクリーニングに有効であることが証明された。
【0044】
ATPと競合しない非ペプチドsrc阻害剤の設計に利用できる、最も有用な結晶構造は、天然のsrc構造およびIRTK:ペプチド:AMP-PNP三元構造である。以下で論じるすべてのモデリング研究では、SYBYL分子モデリングソフトウェアパッケージをシリコングラフィクスワークステーション上で用いる。
【0045】
srcおよびIRTK構造は非ペプチド骨格の設計およびコンビナトリアルライブラリーにおける定性的指標としてのみ用いられるため、過去のPKAモデリング研究と同様に天然のsrcおよびIRTK三元構造から周囲の残基の2層に加えて活性部位を切り取った。比較相同モデリング法を用いてsrc構造上にsrc残基配列424〜418を構築する指標とするために、IRTK:ペプチド:AMP-PNP三元構造活性部位領域を鋳型構造として用いた(HutchinsおよびGreer、1991参照)。これらの残基は天然src結晶構造では乱雑で、そのためX線では見ることができなかった。これらは、いくつかのモデリング研究で重要なペプチド基質のためのP+1からP+3結合部位の形成を助けるため、再導入した。IRTK三元構造における類似の残基はX線で見られ、結合したペプチド基質と直接相互作用する。事実、この配列の位置に秩序を与え、X線で見られるようにするのは、おそらく結合したペプチド基質と考えられる。次いで、IRTK三元構造を鋳型に用いて、srcペンタペプチド基質Ac-Ile-Tyr-Gly-Glu-Phe-NH2(Nairら、1995)をsrc活性部位に再度結合させた。次いで、この複合体を部分的にきれいにするため、手動でわずかな調整を行い、すべての水素原子を加え、適当な形式および部分電荷(ガスティガーマーシリー(Gasteiger Marsili)法により計算)を加え、次いで過去のPKAモデリング法と同様、複合体全体をTripos力場を用いて300回の分子力学最小化にかけた。このモデリングした複合体を図5に図示している。このsrc:ペプチドおよびsrc:阻害剤モデルにおけるいかなる不正確性も、数と多様性をコンビナトリアルケミストリーの様式でsrcモデル活性部位における特定の結合領域の構造に対する不確定性のレベルに合わせておおよそ調整している、一連の側鎖を実験的に評価することによって適合させる(下記参照)。
【0046】
図5に示すとおり、srcに組み込まれた残基424〜418は、基質主鎖(IRTKペプチド基質と同様)とのベータシート型水素結合相互作用を通じて、P+1からP+3基質残基Gly-Glu-Phe-NH2とそれぞれ相互作用する。Lys 423は2つの重要な相互作用に関わっている:1)βおよびγCH2は疎水性結合相互作用に関わっているTyrフェニル環のP 0の上に折り重なっている、2)この側鎖の残りのCH2-CH2-NH3+は示されたとおりP+2 Glu側鎖と塩橋を形成するよう伸びている。P 0 Tyr疎水性結合ポケットの残りはフェニル環の下のPro 425およびフェニル環の上のCys 277側鎖の一部によって形成される。ペプチドsrc基質の大きなコンビナトリアルライブラリーを用いて、Songyangら(1995)は、P+1位に対して最も一般的に選ばれる側鎖はGlyと、続いてGluであることを見いだした。本発明のモデルにより、P+1 Glu側鎖は図5に示すとおり、近接するArg 469と塩橋を形成する可能性がある。過去に、研究者らはP+2位にGluだけが選ばれることを見いだし、本発明のモデルはこの側鎖がLys 423側鎖と塩橋を形成することを示している。P+3位において、Pheが非常に強く好まれ、モデルはこの側鎖がPhe 424側鎖とスタッキング相互作用を形成することを示している。P-1位において、SongyangらはIleが最も好ましい残基であり、続いてValおよびLeuの順であることを見いだした。モデルはP-1側鎖を結合するための疎水性ポケットが主にTrp 428、Ala 390およびLeu 347によって形成されることを示している。酵素は反応が起こる部位に近いこの領域でより重大な相互作用を形成することが多いため、P 0 Tyr主側鎖は触媒として能力のある複合体の活性部位と強く相互作用(水素結合であるが)すると予想される。IRTK三元複合体は、P 0 Tyr NHまたはカルボニルのいずれとも良好な水素結合を示さない。IRTK構造におけるこの相互作用に最も近い候補残基は、側鎖のNH2がTyrのカルボニル酸素から3.71Åの距離にあるAsn 1215である。背景および重要性の項で述べた4つの残基を用いてIRTK三元構造をsrcの天然構造に重ねると、src構造のAsn 468は類似のIRTKのAsn 1215と非常に近い位置にあることが判明した。このことは、この保存された残基が重要な役割を果たしており、触媒として活性な複合体においては基質のP 0 NHおよびカルボニルにやや近く(すなわち、約1Å)移動して図5に示す水素結合相互作用を形成する可能性があることを示唆している。最後に、触媒性のArg 388およびAsp 386はsrcモデルにおいて、γリン酸基のATPからTyr OHへの転移を触媒するために正しく配置されている。
【0047】
このように、src:ペプチド基質複合体は、実験的に評価する新しい骨格を選ぶ前に、すべて適当に連結されたM1官能基を有する、可能な非ペプチド骨格のモデルとして、また特異的因子の好ましい置換位置を決めるために用いることができる。IRTK:ペプチド:AMP-PNP三元構造も、これら可能な骨格および好ましい置換位置のモデルとして用いることができる。これらの骨格は、図1に示す方法に従い、適当な特異的因子を用いてさらに開発することにより、選択的PTK阻害剤の開発のための広範な有用性を有する。
【0048】
このsrc:ペプチド基質モデルで評価した第一の非ペプチド骨格はナフタレン骨格であった。これは、ATPと競合しない非ペプチドPTK阻害剤のために二環式芳香族骨格を初めて用いた例である。この目的のためのナフタレン骨格の有用性は、IRTKおよびEGF受容体PTKの非ペプチド阻害剤を開発することによって示された(Sapersteinら、1989)。IRTK三元複合体は続いて、この骨格をsrc阻害に適合させるために用いられた(Marsiljeら、2000参照)。まず、図6に示すとおり、原子a〜dのペプチド基質への最小自乗フィッティングを行うことによって、ナフタレン骨格をsrc活性部位に合体させた。このようにして、ナフタレン骨格は図6の矢印によって示される環化および基質Tyr NHの置換基として付加されたOHによって、ペプチド基質に関連づけられる。これは、マーシルジェ(Marsilje)2000に記載のこの骨格をIRTK構造に合体させるために用いられた工程と、本質的に同じ工程である。次いで、ペプチド基質を活性部位から除去し、様々なM1官能基や特異的因子S2およびS3を示されたとおりに骨格に付加し、次いで複合体を個々に300回の最小化にかけた。結合様式を図7に示すイソキノリンおよびインドール骨格を設計するためにも、この同じ工程を用いた。
【0049】
これらのモデリングした複合体すべてにおいて、選択的因子S2は基質P-1 Ile側鎖と同じポケット内で結合しうる様々な疎水性側鎖からなり、選択的因子S3はペプチド基質結合部位のP+1からP+3領域で結合しうる様々な分子断片からなる(図5)。M1が結合する活性部位領域はすべてのタンパク質キナーゼで高度に保存されているため、過去にペプチド骨格を用いて特定されたM1官能基の小さいメニューを骨格の示された位置に連結するための最初のM1基とした。2つの選択的因子結合部位のうち、S2のための疎水性結合腔の構造は、S3のためのP+1からP+3結合領域よりも、高い信頼性をもって知られている。これは、S3結合部位は部分的には比較相同モデリングによって構築されたのに対し、S2部位は天然srcについてX線で求められた構造からほとんど変わっていないからである。これらのM1、S2およびS3についてモデリングされた結合部位における信頼性のレベルにばらつきがあることを考慮して、コンビナトリアルライブラリーにおける側鎖の多様性と数が最も多いのはS3部位で、続いてS2部位およびM1の順になるようにコンビナトリアルライブラリーの多様性を調整する。
【0050】
有望な非ペプチド骨格を実験的に同定するためのM1官能基を用いたsrc結果を表4に示している。表4のデータから、いくつかの結論が導き出せる:1)骨格に連結された適当なM1官能基によって、低いが測定可能な阻害効力を得ることができる(例えば27および38)。2)このタイプのスクリーニングにとって1mMの阻害剤濃度は望まれるよりも高いが、100μMは低すぎる。M1基を有する骨格のスクリーニングは500μMで最適に実施される。3)PKAペンタペプチド骨格(22、表3および8、表1)またはsrcペンタペプチド骨格(14、表2)を用いてそれぞれ特定されたボロン酸、スルファミン酸、およびホスホン酸M1官能基は、モデルとなるナフタレン阻害剤:src複合体で特定されたM1の好ましい位置である、ナフタレン環の2位に置いた場合(それぞれ27、28および30)に測定可能な活性を示す(図6)。ボロン酸またはホスホン酸M1基を1位に動かす(32または33)と、活性が低下した。4)PKAペンタペプチド骨格上で活性が低かった、関連するM1スルホンアミド官能基(7および9、表1)は、ナフタレン骨格の2位(31)または1位(34)に付加した場合にも活性が低い。ナフタレン2位のスルホン酸類縁体(29)は、たとえ1mMでも完全に不活性である。5)ボロン酸M1基をナフタレンの2位と類似の位置に配置した場合は、ナフタレン骨格は著しい活性低下なしにベンゾフラン(35)またはベンゾチオフェン(36)骨格に置き換えることができる。6)ボロン酸M1基がイソキノリン(37)またはインドール(38)骨格のモデリング結果によって示された位置に付加した場合も、活性化合物を提供する(図7)。しかし、インドール骨格はイソキノリン骨格よりも明らかに好ましく、Asn 468の水素結合を与える能力(図7参照)がより高い活性のために重要であることが示唆される(これはイソキノリンのプロトン化を必要とするが、隣接する電子吸引性のエステル基にとっては好ましくない)。この結論は、ペプチド基質が水素結合供与ペプチド結合NHを類似の位置に配置しうる(図6)と考えることによって、また同等に配置されたフェノールのOH(図6)が効力を改善する(フェノールのOHは受容体よりもはるかに良好なH結合供与体である)という知見によっても支持される。8)他のM1基と直接比較すると、ボロン酸基は優れている(例えば、27対28〜31、38対39)。9)ビフェニル骨格をsrcおよびIRTK活性部位内にモデリングし、この骨格の有望な結合様式が認められた。コンビナトリアルライブラリーがビフェニル骨格で作製され(Paviaら、1996参照)、モデリング結果は有望なものであった。したがって、ボロン酸M1基を有するパラ(40)およびメタ(41)異性体を評価した。両ビフェニル化合物は最良のナフタレンボロン酸(27)と同等の効力を示し、したがって、さらに評価し、開発するためのもう一つの骨格構造(2つのフェニル環は平面ではない)を提供する。
【0051】
M1基だけが付加している裸の骨格は結合親和性が低いことが多いため、2-ナフタレンボロン酸およびスルファミン酸阻害剤のIC50およびKiを求めて、典型的な用量/反応IC50曲線を確実に得られるようにした。この分析から、より強力な阻害剤で見られる典型的な形状の用量/反応曲線が得られた。これらの単純な阻害剤のIC50およびKiから、ボロン酸阻害剤27がスルファミン酸類縁体28よりも効力が高く、Kiは約554μMであることも確認された。
【0052】
これらの単純な阻害剤についてさらに開発を進める前に、これらに向けられた次の問題はその阻害の様式、特にATP競合的阻害剤であるかどうかということであった。ナフタレン阻害剤27および28の場合、ATP濃度が3段階で1mMまで上昇する間のIC50を測定した。比較として、ペンタペプチドホスホン酸src阻害剤14(表2)のIC50も測定した。これらの阻害剤のいずれかがATPと競合していたならば、そのIC50はATP濃度に比例して上昇するはずである(すなわち、破線)。示されるように、3つの阻害剤のIC50はすべて、ATP濃度が上昇しても本質的に一定値を保ち、これらがATP競合的阻害剤ではないことが証明された。非常に類似しているが、費用がはるかに低く抑えられる(市販のsrcは高価である)分析を、インドールボロン酸阻害剤38で実施した。この場合、38の阻害%を、阻害剤濃度は一定の500μMであるが、ATP濃度は200、500および1,000μMに漸増させて測定した。ここでも、阻害剤の効力はATP濃度が上昇しても低下することはなく、38も非ATP競合的であることが示された。
【0053】
段階1で得られた最初の結果から、この方法でさらに開発を進めるための有望な骨格を特定できることが示唆される。段階1の最大の不確定性は、このようにして特定された骨格の中に、先のモデリング評価によって示唆された様式で結合していないものがあることである。これは本質的には「偽陽性」の問題である。これら「偽陽性」は段階2で、モデリングされた複合体を指標に用いて結合改善について評価されれば、落とされると考えられる。偽陽性結果の中には、M1基だけが付加した基本骨格が容易に得られるため、段階1で許容されるものもある。さらなる阻害剤開発のために、新しい骨格を段階2および段階3に進める必要があるたびに、段階1に戻ることもできる。段階1を繰り返すたびに、得られた最良のM1を用いることができる。現在のところ、ボロン酸M1基は裸の骨格に付加して測定可能な活性を示す能力が証明されているため、この基が好ましい。ボロン酸M1基はまた、次の理由により保存触媒残基との共有および非共有結合のための複数の興味深い可能性を提供する:ボロン酸M1基は、1)水和できる、2)電子に富む活性部位原子とホウ酸塩複合体を形成しうる、および/または3)リン酸化され、次いで活性部位の求核物質と反応するか、もしくは別の非共有相互作用に関与することができる。表4のデータから、段階2の最初の試みとして、ナフタレンおよびインドール骨格をM2として選択した(ビフェニル骨格も好ましい骨格である)。また、srcペプチド基質においてP 0 チロシンをナフチルアラニンおよび類縁体で都合よく置換することができ(例えば、Alfaro-Lopezら、1998)、ナフタレンおよび関連する骨格がP 0部位に結合しうるという知見をさらに支持していることは言及に値する。
【0054】
【表4】
初期の段階1の結果:細胞模倣アッセイ法におけるSRC阻害%
ボロン酸M1基を有するナフタレンおよびインドール骨格(すなわち、表4の27と38)の結果を比較して、インドールの水素結合供与NHおよび/または隣接するエステル基が抗力増強の原因と考えられる。したがって、段階2における最初の試みの1つは、モデリング結果(図6)により示唆されたナフタレン骨格の隣接する位置に水酸基およびアミド(S2も共に)を付加することであった。インドール骨格については、1つの優先事項は、S2特異的因子によって効力が増大しうるかどうかを見るために、いくつかのアミド類縁体を調製することであった(図7)。これら最初の類縁体の合成を容易にするために、ボロン酸M1基をOHで一時的に置換した。OH基は触媒残基との相互作用によってリン酸化速度が加速される天然の基質M1であるため、OH基もM1基に要求される触媒残基との相互作用をすることが知られている。いくつかの最初の類縁体で得られた結果を表5に示しており、非ATP競合的であると報告されている文献に記載の2つのsrc阻害剤50および51と、細胞模倣srcアッセイ法で突き合わせて比較した結果も併せて示している。これらの結果のいくつかと、別の類縁体が、マーシルジェ(Marsilje)2000に記載されている。
【0056】
イミノクロメン骨格はナフタレン骨格と密接に関連しており、その結合様式はモデル(図6)に基づき非常に類似していると予想されるため、阻害剤50および類縁体(Huangら、1995)は特に興味が持たれる。一部にはこの密接な類似性により、表5に示すとおりヒドロキシアニリンのナフタレンおよびインドール骨格とのアミドを試験した。また、これらのヒドロキシアニリンアミド類縁体によるsrc活性部位でのモデリング試験から、この水酸基はペプチド基質と同様にsrcのPhe 424〜Ala 422骨格のペプチド結合との水素結合相互作用に関与しうることが示された(図5参照)。これらのモデリング試験から、同族のヒドロキシベンジルアミドも活性で、より重要なことに、付加している側鎖がP-1側鎖ポケットで結合する(例えば、図5のArg 469に)ための置換位置(すなわち、ベンジル炭素)を提供することも示された。
【0057】
表5のデータより、次の結論を導き出すことができる:1)ナフタレンおよびインドール骨格の両方でアミドによる伸長部分を付け加えると、これらの骨格がsrc活性部位で結合したモデルによって予想されるとおり、効力を高めることができる(42対43-メタおよび47対48の場合に約5倍)。2)ナフタレン骨格のアミドの隣に水酸基を付加すると、srcモデルによって予想されるとおり効力を高めることができ(43-メタ対44の場合に約5倍)、また、Asn 468がこのOHと水素結合することも示唆される。3)M1のOH基をsrcモデルにおいて最良と予想される位置から隣の位置に移動させると、効力が1桁低下する(43-メタから45)。4)ヒドロキシアニリン伸長部分のレジオ化学に対し、インドール骨格はナフタレン骨格よりも反応性が低い(48対43)。5)ナフタレンおよびインドール骨格では、ヒドロキシベンジルアミドを用いることによって生じる1炭素同族体化は許容される(46対43および49対48)。6)ナフタレン骨格の2つのM1ヒドロキシレジオ異性体はいずれも非ATP競合的である(Marsilje 2000参照)。7)メチルヒドロキシアニリンおよびヒドロキシベンジルアミド阻害剤はすべて活性が低いことが判明し、アミド伸長部分の水酸基は水素結合供与体として機能していることが示唆された。この点に関しては、もう一つのsrcペプチド基質コンビナトリアルライブラリー試験において、SerおよびThrがP+2位の最も好ましい残基の2つであると特定され(Alfaro-Lopezら、1998)、アミド伸長部分のOHにとってモデリング試験で示唆されたPhe424〜Ala 422ペプチド結合以外に他の結合の可能性があることが示唆されることは言及に値する。8)過去に文献に開示された最も強力な非ATP競合的非ペプチドsrc阻害剤(50)は、細胞模倣アッセイ条件下で試験した場合、報告されたほどの効力を示さず(Huangら、1995によって報告されたIC50=118nMに対し、100μMでわずか30%の阻害)、最も類似の阻害剤(50対45)を含むいくつかの今回の阻害剤(特に43-メタ)よりも効力が低い。彼らのアッセイ(Huangら、1995)で50のヒドロキシレジオ異性体について報告された構造活性相関(SAR)も、関連するナフタレン阻害剤で得られたSARに対応していない。例えば、彼らのアッセイ法では、最も強力なナフタレン阻害剤43-メタのイミノクロメン類縁体は50の効力の230分の1である。イミノクロメン骨格に比べてナフタレン骨格の重要な利点は、ナフタレン骨格ではP-1疎水性部位に接近するための非常に望ましいS2特異的因子を付加することができる(図6参照)が、イミノクロメン骨格では同様の位置は環の酸素原子によって占有されているため、置換することができない点である。
【0058】
【表5】
初期の段階2の結果:細胞模倣アッセイ法におけるSRC阻害%
src細胞模倣アッセイ法における阻害剤の効力を、他の文献上の非ATP非ペプチドsrc阻害剤に対してさらに較正することができる。別の2つの例は、エルブスタチン類縁体の「チロホスチン」ファミリー(LawrenceおよびNiu、1998参照)の一員である51(ST 638、Shiraishiら、1989)および天然生成物のPTK阻害剤ピセアタノール52(Thakkarら、1993)である。細胞模倣アッセイ法では、これら公知の阻害剤はすべて報告されているよりも効力が低く、このアッセイ法は高い効力を得ることに関して特に要求がきびしいことが示唆される。すべての阻害剤について完全なIC50曲線を作るには市販のsrcは高価すぎるため、src阻害剤の最初の試験は単一の濃度を用いて(三回繰り返して)実施する。しかし、IC50用量反応曲線は直線ではなく、100μMでの約50%の阻害と100μMでの約90%の阻害との間の差は実際には2倍ではなく10倍である(例えば、45対43-メタ)ことは述べておくべきであろう。したがって、文献のsrc阻害剤50〜52は、現在の最も強力な阻害剤43-メタに比べて1桁以上活性が低いのである。
【0060】
これらの阻害剤の効力を報告している文献で見られる矛盾、PKA阻害剤を用いて以前に述べられたアッセイ条件の感受性、および多くの研究所および市販のタンパク質キナーゼアッセイキット間に一貫性がないことが、この分野で見逃されていたが、非常に重大な問題である。本発明によって製造される阻害剤は他のアッセイ条件下でも効力が高いかもしれないが、全細胞または組織アッセイ法に進むための化合物を選ぶ前に阻害剤を評価するための最初の効力および順位指標として、細胞内の物理化学的条件をできる限り模した細胞模倣アッセイ法を用いるべきである。以下にさらに詳細に述べるとおり、ここまでに細胞模倣アッセイ法から得られた最も強力なナフタレンを基本とする阻害剤(すなわち、43-メタ、IC50=18μMおよびKi=10μM)は、pp60v-src刺激細胞増殖の特異的阻止においても有効で、IC50は同程度の約25μMである。これは、細胞模倣srcアッセイ法が予測的であるというだけでなく、このクラスのナフタレンを基本とする阻害剤は容易に細胞膜を通過して細胞内srcを阻害することも示唆している。
【0061】
前述のいくつかのナフタレンおよびインドール阻害剤の類縁体は、図6および図7に示すとおり、M1 OHの代わりのボロン酸M1基および/またはsrc P-1部位で結合するために連結されたS2疎水性特異的因子を用いて調製することができる。次いで、ナフタレンおよびインドール骨格を、以下に述べるとおりに段階3に進めることができる。段階1からの新しい骨格で段階2を繰り返すごとに、以前の骨格で見いだされた最良の選択的因子S2および/またはS3を段階3のコンビナトリアルライブラリーで用いることになる。M1、S2およびS3の最適な組み合わせは各骨格で異なると考えられるとはいえ、先の関連した骨格(例えばナフタレンからインドール骨格に移る場合)で最適であると判明したものは、新しい骨格による段階2のよりよい初期特異的因子として用いるのに適していると考えられる。src阻害剤のため、または後の別の治療的に重要なPTK阻害剤のために、十分な効力、選択性および適当な薬学的性質が得られるまで他の骨格を試す必要があるごとに、同じ工程を繰り返すことになる。
【0062】
ナフタレン阻害剤を調製するために用いるいくつかの化学反応がマーシルジェ(Marsilje)2000に記載されている。表5のsrc阻害剤において、M1水酸基の代わりにボロン酸官能基を連結するために、アリールトリフラート(Ishiyamaら、1997)またはハロゲン化アリール(Ishiyama、1995)のいずれかを市販のジボロンのピナコールエステルと結合させることができる、Pd(0)触媒によるクロスカップリング法を用いた。最近完了した実例を図8に示す。
【0063】
図8に示す例は、M1位の立体障害が小さい方のOHを選択的にトリフラート化することができ、これはトリフラートを除去して56とし、続いて置換パターンを1H NMRで検証することにより証明されたことを示している。次いで、モノトリフラート53を示されたとおり所望のボロン酸55に変換した。同じ反応順序が、表5の阻害剤45に対応する42のレジオ異性体にも都合よく作用する。図9に示す合成模式図に従って、疎水性S2選択的因子をナフタレン骨格に連結することができる。
【0064】
96穴プレート様式でナフタレン骨格のコンビナトリアルライブラリーを合成するために、ナフタレン化学反応を固相に変換することができる。44で表されるナフタレンレジオ異性体は、マーシルジェ(Marsilje)2000に記載のとおり、市販の3,5-ジヒドロキシ-2-ナフトイン酸から容易に得られるため、ここまではこの活性が低い化合物でモデル反応を行ってきた。これまでに成功したモデル反応を図10に示している。
【0065】
これらのモデル反応は、ナフタレン骨格をワン(Wang)樹脂に結合し(63)、次いでトリフラートへの化学反応(この場合、ボロン酸エステル64へのPd(0)触媒によるクロスカップリング)後、温和な条件下での切断(65)を実施することが可能であることを示している。63のエステルは、鹸化した後カップリング反応を行い、S3選択的因子を含むアミドを形成することもできる。
【0066】
ナフタレン骨格は現在のところ、コンビナトリアルライブラリーで調査すべき3つの多様性を示す部位、M1、S2およびS3を提供する。以前の研究(Paviaら、1996)で用いられたものと類似の96穴プレート反応器による固相コンビナトリアルケミストリー。前述した理由により、S3について最も数が多く、多様な側鎖が用いられ、次にS2、そしてM1の順となる。前述の合成モデルに基づき、これらのライブラリーを調製するための1つの可能な全合成方法を図11に示している。
【0067】
もちろん、このルートで問題が生じた場合は、多くの他の可能性がある。例えば、67を得るための光延反応の収率が低すぎる(付加された隣接アリル基の立体的過密状態の増大によるが、おそらく図10で得られた92%の充填を考慮すれば問題ではない)場合、アシルスルホンアミド「セーフティキャッチ」リンカー(Backesら、1996)を用いて、骨格をOHではなくカルボキシル基によって樹脂につなぎ、最後にアミドを形成する(過剰のアミンは切断後に酸性樹脂を通して濾過することにより除去することができる)こともできる。同様に、ベンジルエーテル存在下でアルケンの還元(67から68)が望まれるが、問題がある場合には、他のリンカーおよび/または樹脂を用いることもできる。図11で提唱されている化学反応の第一の用途は、ボロン酸M1基を含み、適所にアリル側鎖を持たない96個のアミドのライブラリーを単純に調製することで、これら2つの複雑化要因は最初は問題とならず、最も有望なS3因子を速やかに同定することができる。
【0068】
srcモデルのP-1部位への候補側鎖のモデリング(図6)に基づき、また対応するウィッティヒ試薬を調製するために必要なハロゲン化物が市販されているかどうかに基づき、少なくとも14のS2疎水性側鎖(直鎖、分枝、および環状を含む)がさらなる試験のために特定される(対応するアルケンも調査する場合には28)。下記のS3選択的因子として可能性がある基を提供する、少なくとも96の市販のアミンが利用できる:1)炭化水素(4)、2)水素結合受容体を含むアルキル基(4)、3)水素結合受容体および供与体の両方を含むアルキル基(19)、4)水素結合受容体および供与体を含むアルキル/アリール基(25)、5)アリール水素結合受容体および供与体(10)、6)複素環水素結合受容体および供与体(20)、7)カチオン基を含む側鎖(4)、8)アニオン基を含む側鎖(9)、およびsrc活性の内部対照としての阻害剤43-メタからの3-アミノフェノール側鎖。srcモデルにおけるこの結合部位の不確定性レベルが高いことにより、ここではライブラリーの過剰な偏りが生じないよう、S3のために広範囲のアミンが含まれた。
【0069】
ほとんど同じ様式で、インドール骨格をコンビナトリアルライブラリーに作製することができる。この場合、類似のMinsunobu反応が知られている(BhagwatおよびGude、1994)ため、ワン(Wang)(または他の)樹脂につなぐための連結点としてインドールのNHを用いる。置換インドールの合成のために多くの合成法が開発され、S2疎水性側鎖を含めるためのルートが設計されている(図7)(Ezquerraら、1996)。
【0070】
中間体69からの反応2で形成されるトリフラート官能基(図11)は、アミンに変換し(Wolfeら、1997)、次いで図12に示す反応順序に従って一連のアミドまたは他のアミン誘導体に変換することができる。事実、トリフラートは用途の広い合成ハンドルで、他の官能基に変換することもできる。
【0071】
アミン72が得られれば、既知のM1(例えば、表5のsrc阻害剤28からのスルファミン酸および表3の17のアミド−酸)を、このより発展した骨格と共に評価することができ、いくつかの新しいアミン誘導体可能なM1として評価する。例えば、構造73に示す水和トリカルボニルアミドM1基(およびその非水和前駆体)を、この合成法を通じて得ることができ(Laiら、1996参照)、保存触媒残基との様々な興味深い相互作用を形成することができる。
【0072】
前述のモデリング法に従い、図13に示すボロン酸M1基の一連の水酸基含有類縁体を、srcおよびIRTK活性部位でモデリングし、興味深い可能性の一部として図示した相互作用/結合様式を見いだした。ボロン酸をリン酸化することにより、別の興味深い可能性が得られる(例えば、得られた混合無水物と活性部位求核体との反応を通じての自殺型阻害)。Tyr様フェニル環上に別の水酸基があることが必要不可欠で、多くのPTK阻害剤(例えば、表5のピセアタノール52)では一般的で、PKAホスホン酸阻害剤では側鎖上で有益であることが示された(例えば、表1の2対3および4)。したがって、図13に示すとおり、ボロン酸阻害剤のM1の設計に1つまたは複数のOHを付加することは、効力を著しく増強することがある。これらのOH基はまた、触媒AspおよびArgを超えてボロン酸側鎖を伸長し、PKAボロン酸同族体の場合におそらく生じている、炭化水素でそれらを覆うことに伴う不都合はない(表3の23および24)。ヒドロキシボロン酸76および77への可能な1つのルートは、Mattesonのキラルなボロン酸エステルによる同族体化法を用いることである(Mattesonら、1987、1988および1990参照)。
【0073】
本発明の好ましい態様において、第一のモジュールは第一のモジュールをペプチド骨格に連結することによって製造される。タンパク質キナーゼの触媒残基に優先的に結合する1つまたは複数の官能基を同定する。さらに、第一のモジュールを第二のモジュールに組み合わせて、ペプチド骨格を第二のモジュールで置換する。
【0074】
好ましい第一のモジュールは、ボロン酸、水酸基、ホスホン酸、スルファミン酸、グアニジノ基、カルボン酸、アルデヒド、アミド、およびヒドロキシメチルホスホン酸などの官能基を有する。本発明の化合物は、第一のモジュール内に複数の官能基を有していてもよい。より好ましいモジュールはボロン酸基、水酸基、またはアミド基である。さらにより好ましいアミド基は隣接するトリカルボニルアミドである。
【0075】
好ましい第二のモジュールは、インドール、ナフタレン、ビフェニル、イソキノリン、ベンゾフラン、およびベンゾチオフェンを含む。より好ましい第二のモジュールは、インドールまたはナフタレンである。本発明のいくつかの態様において、複数の第一のモジュールが第二のモジュールに結合していてもよい。加えて、第一のモジュールは、第一のモジュールを第二のモジュールに連結する1個から3個の炭素原子を含む直鎖を有していてもよい。もう一つの態様において、直鎖中の炭素原子の1個が窒素、酸素、または硫黄原子で置換されていてもよい。
【0076】
本発明の方法および化合物は、いかなるタンパク質キナーゼにも広く適用可能である。好ましいタンパク質キナーゼはタンパク質チロシンキナーゼおよびタンパク質セリンキナーゼである。好ましいタンパク質チロシンキナーゼは、pp60c-src、p56lck、ZAPキナーゼ、血小板由来成長因子受容体チロシンキナーゼ、Bcr-Abl、VEGF(血管内皮成長因子)受容体チロシンキナーゼ、ならびに表皮成長因子受容体チロシンキナーゼ、および表皮成長因子受容体様チロシンキナーゼである。より好ましいタンパク質チロシンキナーゼはpp60c-srcである。好ましいセリンタンパク質キナーゼには、MAP(マイトジェン活性化タンパク質)キナーゼ、タンパク質キナーゼC、およびCDK(サイクリン依存性タンパク質キナーゼ)が含まれる。
【0077】
本発明の方法は、第一および第二のモジュールの組み合わせに1つまたは複数の特異性側鎖因子を付加することをさらに含みうる。特異性側鎖は阻害剤の効力および特異性を高めることができる。
【0078】
有望な第二のモジュールが特定されれば、方法のすべての段階を繰り返す必要はない。むしろ、第一のモジュール、特異性側鎖、または2つの組み合わせを改変し、元の阻害剤を改善しうる、すなわち、改変されていない第一の阻害剤と比べたときに、タンパク質キナーゼ活性を阻害する能力が増大された阻害剤しうる。
【0079】
本発明の方法は、タンパク質キナーゼへのATPの結合の阻害によって作用しないタンパク質キナーゼ阻害剤を優先的に提供するよう設計されている。タンパク質キナーゼの阻害剤は強力であってよいが、特異性に欠けることが多く、したがって、良い薬剤候補物とならないことが多い。したがって、タンパク質キナーゼ活性を阻害するが、タンパク質キナーゼへのATP結合を阻害しないか、または弱く阻害するだけのタンパク質キナーゼ阻害剤が好ましい。
【0080】
本発明はまた、タンパク質キナーゼ活性を阻害する能力について化合物を試験する方法も提供する。化合物は請求項1のように製造される。タンパク質キナーゼの活性は、阻害剤の存在下、タンパク質キナーゼを発現する細胞で見られるのと同じ温度、pH、イオン強度、モル浸透圧濃度、および遊離マグネシウム濃度で測定される。タンパク質キナーゼ活性のレベルを阻害剤不在下でのタンパク質キナーゼからの活性レベルと比較する。生理学的条件を模倣するこのようなアッセイシステムは、最も適切な阻害データを提供する。アッセイ法は自動アッセイシステムで実施されうる。さらに、多くの候補を速やかにスクリーニングするために、アッセイ法をコンビナトリアルケミストリー法と組み合わせてもよい。
【0081】
96穴プレートコンビナトリアルライブラリーの初期srcスクリーニングのための細胞模倣アッセイ条件に、Pierce 96穴プレート非放射性ELISA PTKアッセイ法が適用されうる。このハイスループットアッセイ法には、市販の96穴プレート中でNeutrAvidinコーティングされたウェルに連結できるようにビオチン化されていること以外は同様のRR-SRCペプチド基質が用いられる。このハイスループット阻害アッセイ法は、ウェルにあらかじめ結合したRR-SRC基質と共にsrcをインキュベートした後、その抗ホスホチロシン抗体(PY20)-セイヨウワサビペルオキシダーゼ(HRP)複合体およびそのHRP基質を加えることによって、生成されたホスホ-RR-SRCのレベルを、96穴プレートUV測定器によるHRP生成物のレベル測定を通じて定量することができる。標準的ロースループットP32-ATP放射能アッセイ法が用いられてきたが、96穴プレート様式が好ましく、可能ならば特に非放射能アッセイ法が好ましい。非常に強力なsrc阻害剤が開発されるに伴い、タンパク質キナーゼアッセイ法のパネルを約6つまでの市販のタンパク質キナーゼ(ほとんどがPTK)で細胞模倣タンパク質キナーゼアッセイ条件を用いて設定し、特異性の初期評価のためにパネル全体でこれらの阻害剤を試験することができる。適当な時期に、さらなる共同研究を通じて、細胞培養物およびインビボにおいて全部の約2000のタンパク質キナーゼに関するより完全な特異性評価を実施する必要があると考えられる。
【0082】
正常ラット腎(NRK)細胞およびこの細胞系統の温度感受性pp60v-src形質転換体(LA25)を用いた1組の並列細胞ベースアッセイ法において、活性src阻害剤を試験することができる。LA25形質転換体は、pp60v-srcの活性化により「許容」温度33℃で足場非依存的成長および血清非依存的成長をするが、pp60v-srcが活性化されない「非許容」温度40℃ではしない(Liら、1996)。許容温度と非許容温度の両方においてsrc阻害剤を試験するためにこの一対の密接に関連した細胞系統を用いることにより、異なる制御機構または一般的毒性作用によって、所与のsrc阻害剤がsrcシグナル伝達経路の特異的遮断に起因する細胞の成長を阻止しているかどうかを決定することができる。この一対の細胞系統における非ペプチドsrc阻害剤43-メタ(表5)の初期試験の結果を図14に示す。
【0083】
このグラフに示されるとおり、許容温度33℃におけるLA25細胞の成長は、対照としての非許容温度40℃でのLA25細胞の成長に比べて、43-メタの濃度25μMで約50%阻害される。濃度が400μMまで上昇するに従い、NRK非形質転換細胞、非許容温度40℃でのLA25細胞および許容温度33℃(しかし、pp60v-srcは43-メタによって完全に阻害された)でのLA20細胞の基本的成長が低下しないだけでなく、実際には幾分上昇する(多分、この化合物のsrc関連でない活性による)ことから、43-メタに細胞毒性がないことは明らかである。可溶化のために低濃度のDMSOで43-メタの溶液を調製したため、DMSO対照もまた同じ濃度で試験した。25μM周辺に中心がある、より完全な用量/反応曲線が作製されると考えられる。
【0084】
もう一つの態様において、本発明はタンパク質キナーゼを阻害する方法を提供する。タンパク質キナーゼを、タンパク質キナーゼの触媒残基に結合するための官能基を有する第一のモジュールと非ペプチド骨格を提供する第二のモジュールとを有する化合物に接触させる。第一および第二のモジュールの組み合わせがタンパク質キナーゼの活性を阻害する。
【0085】
本発明の好ましい非ペプチドタンパク質チロシンキナーゼ阻害剤は以下の式を有する:
【化13】
【0086】
もう一つの好ましい非ペプチドタンパク質チロシンキナーゼ阻害剤は以下の式を有する:
【化14】
【0087】
さらにもう一つの好ましい非ペプチドタンパク質チロシンキナーゼは以下の式を有する:
【化15】
本発明は、患者におけるタンパク質キナーゼ阻害剤反応性の症状を治療する方法をさらに提供する。タンパク質キナーゼ阻害剤の有効量を患者に投与する。タンパク質キナーゼ阻害剤は、タンパク質キナーゼの触媒残基に結合するための官能基を有する第一のモジュールと、非ペプチド骨格を提供する第二のモジュールとを有し、第一および第二のモジュールの組み合わせがタンパク質キナーゼ活性を阻害する。
【0089】
最後に、有望なsrc阻害剤は初期のヒト腫瘍組織アッセイ法において、特に他の公知の抗癌剤との相乗作用をさがすためにスクリーニングされうる。
【0090】
実施例
実施例1−pp60c-srcチロシンキナーゼの非Atp競合的ヒドロキシナフタレン誘導体阻害剤の設計、合成および活性
自己阻害ヒトIRTK触媒ドメインの結晶構造(Hubbardら、1994)を用いて、定性的分子モデリング試験を実施し(SYBYL(商標)、6.4、Tripos Inc.、セントルイス)、ナフタレン環をIRTK Tyr 1,162に重ね合わせた。Tyr 1,162を含むIRTK領域は活性部位に折り返され、Tyr 1,162はペプチド基質のリン酸化可能なTyrと同様に配置されて、それによりチロシンキナーゼを自己阻害していた。この重ね合わせにより、アミドのカルボニルはTyr 1,162のカルボニルを模してナフタレン環の2位に配置されるべきであり、水酸基はTyr 1,162の水酸基を模して6位に配置されるべきである(模式図1)ことが示された。これらのモデリング試験から、3位の水酸基がTyr 1,162のNHを模倣できることもまた示された。
【化16】
これらの設計概念を実験的に試験するために、2位のカルボニル基をメチルエステルまたは一連のアミドのいずれかとして付加した(表6)。付加されたヒドロキシN-フェニルアミド側鎖を含むイミノクロメン類縁体で観察されたpp60c-src阻害剤の効力の増大(Huangら、1995)に基づき、ヒドロキシN-フェニル(X=0)およびベンジル(X=1)アミドを選択した。6-ヒドロキシ基が除去されたか、または移動した類縁体も調製し、モデリング試験から予想されたような効力の低下が起こるかどうかを調べた。
【0092】
一連の2-カルボニル-3,5-ジヒドロキシナフタレン阻害剤(1a、2a〜2d、2i〜2l、2o〜2p)および2-カルボニル-3,7-ジヒドロキシナフタレン阻害剤(1c、2t〜2u)を、それぞれ市販(Aldrich)の3,5-ジヒドロキシ-2-ナフトイン酸および3,7-ジヒドロキシ-2-ナフトイン酸から合成した。それぞれの酸出発物質を、HClガスであらかじめ飽和させたメタノール中で、48時間還流させることにより、メチルエステル1aおよび1cを得た。それぞれのカルボン酸を市販(AldrichまたはLancaster)のアミンと、2つのうちの一方の方法を用いてカップリングさせることにより、アミド(2a〜2d、2i〜2l、2o〜2p、2t〜2u)を合成した。第一の方法には、フローエンによって記載された(Froyen、1997)NBS/PPh3法を用いた。第二の方法は、カップリング試薬としてIIDQ(Aldrich)を用いた。カルボン酸をまず、1.0当量のIIDQと無水DMF中、室温で24時間反応させた。次いで、それぞれのアミン(2.0当量)をニートに(neat)加え、反応混合物を80℃で2〜6時間加熱した。水処理後、シリカゲルクロマトグラフィおよびCH2Cl2/ヘキサンからの沈殿に続き、必要があればプレパラティブC-18 RP-HPLC(CH3CN/H2O)により精製を行った。ベンジルアミンは、その対応する水酸基保護メチルエーテルとしてのみ市販されていた。したがって、アミド生成後、DCM中、6当量のBBr3で、-78℃で1分間の後、室温で1時間処理することにより脱保護した。
【0093】
【表6】
ヒドロキシナフタレン誘導体および選択された公知の阻害剤のpp60c-src阻害活性a,b,c
b新しい化合物はすべて、プロトンNMR、EIまたはFAB(+)MSで特徴付け、TLCで純粋であった。
cN/A=適用されない。n.t.=試験していない。
dATP-競合的
【0094】
一連の2-カルボニル,3,6-ジヒドロキシナフタレン阻害剤(1b、2e〜2h、2m〜2n、2q〜2s)を3,6-ジヒドロキシ-2-ナフトイン酸6から前述の方法を用いて合成した。開発された中間体6の合成は市販の2,7-ジヒドロキシナフタレン3(Aldrich)を出発物質として模式図2に示す。
【化17】
化合物1dを3-アミノ-2-ナフトイン酸(Aldrich)からDCM中、TMS-ジアゾメタンとの室温での反応により合成した。化合物2vは6-ヒドロキシ-2-ナフトイン酸(Aldrich)からフローエン記載(Froyen、1997)のアミド化法を用いて合成した。
【0096】
キナーゼアッセイ条件が、測定した阻害活性に影響することが明らかにされている(Lawrenceら、1998)。したがって、新しく設計されたpp60c-src阻害剤類の相対的効力を正確に求めるために、以前に報告された4つの非ATP競合的PTK阻害剤の阻害活性も試験した。ピセアタノール、ST-638、およびチロホスチンA47は市販されており(SigmaまたはCalbiochem)、一連の公知の非ATP競合的PTK阻害剤の代表であるため、これらを選んだ。チロシンキナーゼp56lckで分析したところ、エモジン(Calbiochem)はATP競合的である。以前は、報告されている中で最も強力な非ATP競合的pp60c-src阻害剤はイミノクロメン9TAであった(Huangら、1995)。イミノクロメン9TAは市販されていなかったため、3-アミノフェノールを対応するTBDMSエーテルに変換することによる新規なルートを用いて合成した(1.1当量TBDMS-Cl、1.1当量DIEA、5mol%DMAP、DMF、室温24時間、71%)。得られたアニリンを2.0当量のシアノ酢酸を用いてカップリングさせた(1.1当量EDCl、1.1当量TEA、DMF、75℃18時間、70%)。得られたアミドと1.2当量の2,3-ジヒドロキシベンズアルデヒドとの縮合(触媒量のピペリジン、無水EtOH、60℃2時間)の後、脱保護(1.1当量TBAF、THF、15分、全収率43%)して、フラッシュクロマトグラフィ(DCM:MeOH=10:1)による精製後の、基本的なFAB(+)MSおよび1H NMRが良好であるようなイミノクロメン9TAを得た。
【0097】
化合物1a〜dおよび2a〜2vについて表6に示した阻害活性を、精製した全長ヒト組換えpp60c-srcを用いて測定した。試験する化合物の数が多く、それに伴う費用がかかるため、それらの順位効力をまず一定の阻害剤濃度(100μM)で測定した。モデリング試験によって予想されたとおり、IRTK Tyr 1,162の水酸基との類似性に基づき、エステル(1b、47%対1a、5%および1c、19%)およびアミド(例えば、2f、89%対2b、51%および2t、68%)の両方でナフタレンの水酸基を5または7位の炭素よりも6位の炭素に配置する方が好ましい結果が見られた。水酸基をナフタレンの3位の炭素に連結すると(Tyr 1,162 NHの模倣)効力が改善されるとの予測も確認された(2f、89%対2v、45%)。最後に、阻害剤を2位でアミドとして伸長する(ペプチド結合の模倣)ことにより、効力をさらに改善することができるとの予測も同じく確認された(例えば、2f、89%対1b、47%)。
【0098】
表6のデータは、ナフタレンの水酸基を最適な6位から隣の5位に移動させると、アミド側鎖の1つまたは複数の水酸基の最適な並行配置に関して、構造活性特性が異なってくることを示している(例えば、2f/2g対2b/2c)。同じく注目すべきは、化合物2i〜2nにおけるアミド側鎖水酸基の対応するメトキシ基への置換である。N-フェニルアミド(2i〜2j)の場合、対応するヒドロキシアミド(2b〜2c)と比較した活性は、N-ベンジルアミド(2k〜2n対2o〜2q、2s)の場合ほど低下しなかった。このことから、ベンジル誘導体では、アミド側鎖水酸基が水素結合供与体として酵素と相互作用するか、またはメトキシ基が大きすぎて結合部位に合わないかのいずれかであることが示唆される。
【0099】
5位の炭素と6位の炭素への水酸基配置の選択性に関するより定量的な分析を、それぞれ2f(16μM)と2b(150μM)のIC50を比較することによって行う。これらの結果からも、予期されたとおり、約90%から約50%への阻害%低下は効力の桁違いの差を表していることが確認された。同様に、約50%から約10%への阻害%低下は、別の、効力に桁違いの差があることを示していると考えられる。
【0100】
この一連の阻害剤群由来の最も強力な阻害剤である化合物2fを、表6の、過去に報告された5つのPTK阻害剤と直接比較することにより、これらのアッセイ条件下では2fの効力が1〜2桁高いことが示される。興味深いことに、イミノクロメン9TAは過去にpp60c-srcに対して118nMのIC50を有すると報告され(Huangら、1995)、最も強力な公知の非ATP競合的pp60c-src阻害剤であったが、今回のアッセイ条件では100μMで30%の阻害しか認められなかった。これらの結果は、タンパク質キナーゼ阻害剤を等しいアッセイ条件下で比較することの重要性を再度強調するものである(Lawrenceら、1998)。
【0101】
これらの試験の目標は、ATPと競合しない非ペプチドpp60c-src阻害剤を得ることであった。したがって、2fおよび2bによる一定の阻害剤濃度でのpp60c-srcの阻害%を、細胞を模した1mMレベルまでの[ATP]の上昇の関数として測定した。[ATP]は阻害%にほとんど影響をおよぼさなかったため、2fおよび2bはいずれもATPに関して非競合的阻害剤である。阻害剤濃度を一定に保ちながら、200μM、500μMおよび1,000μMのATP濃度を用いて阻害%を測定した。阻害剤がATPと直接競合する場合、阻害%への影響に関して、全体で5倍というこの[ATP]上昇は、阻害剤濃度の5分の1への低下に等しい。したがって、ATPとの直接の競合が起こっているならば、この実験で用いた固定阻害剤濃度の1/5の濃度では、阻害%はIC50用量反応曲線(200μM ATPで得た)で観察された値まで低下するはずである。阻害剤2f(25μMで固定)については、200μM、500μMおよび1,000μM ATPでそれぞれ62%(±5)、54%(±3)および50%(±1)の阻害が観察されたが、ATPとの直接の競合が起こっていたならば、1,000μM ATPでの阻害のレベルは約20%まで低下するはずであった。キナーゼの多くが高価であるために、他の研究者も同じ目的のために[ATP]上昇の関数として阻害効力を測定している(Sapersteinら、1989;Burkeら、1993;Davisら、1992;Faltynekら、1995;およびSawutzら、1996)。
【0102】
要約すると、構造に基づく設計により、ATPと競合しない一連のヒドロキシナフタレンpp60c-src非ペプチド阻害剤が製造された。細胞を基準とするアッセイ法、ならびに様々なアッセイ条件下での詳細な動態試験におけるこの一連の化合物の結果はやがて報告されると考えられる。ナフタレン骨格からインドール骨格へのこれらの設計概念の拡張を、以下に報告する。
【0103】
実施例2−pp60c-srcチロシンキナーゼの非ATP競合的ヒドロキシインドール誘導体阻害剤の設計、合成および活性
前述の実施例において、ナフタレン骨格を用いた一連のpp60c-src阻害剤の構造に基づく設計が記載された。これらの化合物は細胞環境において、ATP基質部位に比べて、ペプチド基質部位の方により高い選択性および有効性をもって結合するよう設計された。この実施例は、インドール骨格を基本とする一連のpp60c- src阻害剤へのこれらの設計概念の拡張を示す。ここでも、自己阻害インスリン受容体PTK(IRTK)の結晶構造を用いて、定性的分子モデリング試験を実施したが、この場合はインドール環をIRTK Tyr 1,162に重ね合わせた。この重ね合わせにより、インドールのNHはTyr 1,162のNHを模倣することができ、カルボニルと水酸基はそれぞれTyr 1,162のカルボニルとOHを模して2位と5位に配置されるべきであることが示された(模式図1)。ナフタレン骨格の場合の6員環に対し、模式図1に示すように、より小さいインドールの5員環へとTyr 1,162を環化させたと考えると(Karniら、1999)、OHの最適な位置はナフタレン骨格お6位の炭素からインドール骨格の5位の炭素に移動する。
【化18】
前の実施例で報告した類似のナフタレンを基本とするヒドロキシフェニルアミドで観察されたpp60c-src阻害剤効力の上昇に基づいて、ヒドロキシフェニル/ベンジル側鎖を含むそれぞれのインドールアミド誘導体2d〜f、2j〜l(表7)を選択した。対応するメチルエーテル2a〜c、g〜i、vが合成前駆体である。表7に示す他の類縁体は、側鎖の範囲を水酸基/メトキシ基以外に拡張するために調製され、インドールおよびナフタレン骨格の両方で広く調査された。
【0105】
水酸基またはメトキシ基側鎖のみを含むインドールアミドを図示したとおりに合成した。
【化19】
2-インドールカルボン酸誘導体、メトキシフェニルアミン(1.1当量、Aldrich、LancasterまたはFluka)、およびカップリング試薬PyBOP(ベンゾトリアゾール-1-イロキシ)トリピロリジノ-ホスホニウム-ヘキサフルオロホスフェート)(1当量、Fluka)を無水DMFに溶解した。溶液をアルゴン雰囲気下0℃に冷却し、次いでジイソプロピルエチルアミン(DIEA、3当量)を加えた。反応混合物を0℃で1分間、続いて室温で1時間撹拌した。後処理後、残渣をシリカゲルクロマトグラフィで精製した。
【0107】
望ましい場合には、メチルエーテルを三臭化ホウ素(1M DCM溶液、Aldrich)で切断した。インドールアミドメチルエーテルを無水DCMに懸濁し、アルゴン雰囲気下-78℃に冷却した。出発物質の各ヘテロ原子1個につき1当量と過剰に1当量のBBr3を加えた。得られた暗赤色の溶液を-78℃で30分、続いて室温で1時間〜2時間撹拌した。反応を水で停止(10分)させた後、後処理を行った。
【表7】
ヒドロキシインドール誘導体のpp60c-src阻害活性a,b,c
b化合物はすべて、プロトンNMR、EIまたはFAB(+)MSで特徴付け、TLCで純粋であった。
cN/A=適用されない。
【0108】
この合成経路を用い、一連の5-ヒドロキシインドールアミド阻害剤2a〜m、y、zを5-ヒドロキシ-2-インドールカルボン酸から調製した。4-および6-ヒドロキシインドールアミド(それぞれ2x、u)を、それぞれ4-メトキシ-2-インドールカルボン酸メチルおよび6-メトキシ-2-インドールカルボン酸メチルから合成した。5,6-ジヒドロキシインドールアミド2tを5,6-ジメトキシインドール-2-カルボン酸エチルから調製した。エステルを1N NaOH中で1時間超音波処理することにより、カップリングのための対応するカルボン酸を得た。デス-ヒドロキシインドールアミド2v、wをインドール-2-カルボン酸から合成した。インドール出発物質はすべて、市販のものであった(AldrichまたはLancaster)。
【0109】
フルオロ阻害剤2r、sは対応するフルオロフェニルアミン(Aldrich)から同様に調製した。対応するアミノカルボン酸(Aldrich)からアミド側鎖にエステルまたはカルボン酸を含む阻害剤2n〜qを調製した。側鎖カルボン酸をまずメチルエステルとして保護し(HClであらかじめ飽和させておいた無水MeOH、還流、1日)、その後PyBOPカップリング(前述)を行い、次いで望ましい場合には鹸化してカルボン酸に戻した。
【0110】
メチルエステル1aは、HClガスであらかじめ飽和させておいた無水メタノール中のカルボン酸溶液を一晩還流させることによって調製した。エチルエステル1bは、前述のとおり、5,6-ジメトキシインドール-2-カルボン酸エチルのBBr3脱保護により調製した。表7に記載の阻害剤はすべて、シリカゲルクロマトグラフィにより精製した。
【0111】
マーシルジェ(Marsilje)2000と同様、この一連のpp60c-src阻害剤の順位活性を、まず一定の阻害剤濃度で測定した(表7)。今回の一連のインドール阻害剤、前の一連のナフタレン阻害剤、および文献記載の5つの非ATP競合的PTK阻害剤(引用論文を参照のこと)で同じ阻害剤濃度(100μM)を用いた。これにより、全体に等しいアッセイ条件下で59個の化合物の有効な順位比較を行うことができた。
【0112】
モデリング試験により、インドール骨格の5位の炭素の水酸基が最適であると予想された。5-ヒドロキシインドール阻害剤2k(74%)と類似の6-ヒドロキシインドール阻害剤2u(56%)および4-ヒドロキシインドール阻害剤2x(60%)の比較によりこの予想が確認されたが、選択性は強くない。5位の炭素の水酸基が活性を改善する(水酸基がないものに比べて)との予想は、5-ヒドロキシインドール阻害剤2l(54%)と対応するデス-ヒドロキシ阻害剤2w(36%)との比較によって確認される。
【0113】
前のナフタレン阻害剤に基づいて予想されたとおり、インドール阻害剤を2位の炭素でのアリールアミドとして伸長することにより、効力が改善された。例えば、メタ-ヒドロキシベンジルアミドインドール2kは100μMで74%の阻害を示したが、類似のメチルエステル1aは500μMで40%の阻害を示すにすぎなかった。興味深いことに、5,6-ジヒドロキシエチルエステル1b(28%)と対応するアリールアミド2t(26%)の比較により、6位の炭素に2つ目の水酸基が同時に存在すると、そうでなければ好ましいメタ-ヒドロキシベンジルアミド側鎖によって通常提供される効力増強が妨げられることが示された。このアミド側鎖は、5-水酸基が単独で存在する場合には今回の一連の阻害剤中最も良く(2k、74%)、6-水酸基が単独で存在する場合にもまだ良好な阻害を示す(2u、56%)。また、2個の水酸基が5位と6位に同時に存在すると、アミド側鎖がない場合に耐容性が高いと考えられる(1b対1aおよび2e)。このデータより、2つ目の水酸基の存在によってインドール骨格の結合配向性に変化が起こり、異なるアミド側鎖が好まれることが示唆される。炭素4〜7位の側鎖(水酸基の官能基置換を含む(Laiら、1999))とアミド側鎖との最適な組み合わせを、現在調査中である。
【0114】
一般に、表7のデータによって判明したインドール骨格構造活性相関(「SAR」)は、ナフタレン骨格について先に報告したものと類似している。いずれの場合にも、モデリング試験で特定されたとおり、骨格上の水酸基をTyr 1,162のOHと同様に配置すると、最も高い効力が得られた。いずれの場合にも、この水酸基を隣接する炭素の1つに移動させると効力が低下したが、劇的な低下ではなかった。Tyr 1,162のペプチド結合を模倣するために、モデリング試験で特定された位置で両骨格をアリールアミドにょり伸長すると、効力が改善された。いずれの骨格においても、アミド側鎖の水酸基をメトキシ基で置換すると、通常は効力が低下し、側鎖がより長いベンジルアミドの場合には低下の程度が大きかった(例えば、2e、30%対2b、21%に比べて2k、74%対2h、7%)。これら2つの骨格のSARにおける主な相違は、5-ヒドロキシインドール骨格ではより長いm-ヒドロキシベンジルアミド側鎖が好ましい(2k、74%対2e、30%)のに対し、類似の3,6-ジヒドロキシナフタレン骨格ではより短いm-ヒドロキシアニリン由来のアミド側鎖が好ましいことである。5-ヒドロキシインドール骨格は本質的には、より短いアミド側鎖上の水酸基の位置について偏向性を示すことはなかった(2d〜f)が、より長いヒドロキシベンジルアミド側鎖では、メタ位に有意に偏向した(2〜l)。3,6-ジヒドロキシナフタレン骨格の場合、反対の現象が認められた。
【0115】
インドール骨格でより長いアミド側鎖についての好ましい構造をさらに調査するため、追加の分子モデリング試験を実施した。以前の報告から最も活性の高いナフタレン阻害剤3を鋳型として用い、その上に類似のインドール阻害剤2eと、同族体化したインドール阻害剤2kを重ね合わせた。ナフタレン阻害剤3の3つの最も重要な側鎖官能基は、両方の阻害剤群の合理的設計およびSARに基づき、6-水酸基(H-結合供与体および受容体)、3-水酸基からの水素(H-結合供与体)、および側鎖水酸基(H-結合受容体)であると考えられる。模式図3において、両方の阻害剤群のこの3点ファルマコフォアモデルをアステリスクにより同定する。
【化20】
2eおよび2kを3に重ね合わせるために、SYBYL(商標)(6.4、Tripos、セントルイス)内の「マルチフィット」、エネルギー最小化および「フィット原子」機能を順に用いた。この全フィッティング段階を、骨格ファルマコフォアOおよびH(100)を最適に重ね合わせることを最も強調し、続いて側鎖のO(10)、その次に介在するアミド結合(1)を適当に重ね合わせるよう選んだバネ定数(マルチフィット)と重量(フィット原子)で実施した。「マルチフィット」段階が最大のファルマコフォアフィットのためのコンフォメーションを調整し、続く最小化によって最も近い局部最少エネルギーのコンフォメーションが得られ、最後に「フィット原子」段階により、これら最小化されたコンフォメーションの最良のファルマコフォアの重ね合わせが得られた。予想されたとおり、2eと2k両方の骨格ファルマコフォアOおよびHは、対応する3の原子上に近く、類似の状態で重ね合わされた(すべて約0.50Å以内)。しかし、2eおよび2kの側鎖ファルマコフォアOは、対応する3のOへの重ね合わせにおいて著しく異なり、ずれはそれぞれ1.8Åとわずか0.08Åであった。この2kと3との間の3つの鍵となるファルマコフォア部位が密接に一致していることが、効力の差がわずか2.4倍しかない(I50はそれぞれ38μMと16μM)ことの説明となる。
【0117】
もう一つの炭素原子によりアミド側鎖を伸長すると活性が低下した(2m、21%対2l、54%)。メチル基を2kのベンジル炭素に、いずれの立体化学でも付加すると、活性が大きく低下した(2y、15%および2z、13%対2k、74%)。側鎖水酸基(パラ位)をカルボン酸アニオンで置換する(2n、0%対2l、54%および2p、7%対2f45%)と活性が低下したが、対応するメチルエステル(それぞれ2o、11%および2q、32%)の効力低下はそれよりも小さいものであった。重要なことに、側鎖水酸基をフッ素で置換しても、効力はほぼ維持された(2s、57%対2k、74%および2r、21%対2e、30%)。したがって、フルオロ類縁体2sでは、可能性のある第II相代謝(例えば、グルクロニド生成)のために水酸基は1つしか残っておらず、この残っている水酸基が今回の置換のための標的となる(Laiら、1998)。
【0118】
前述の例(Marsilje2000)と同じ方法を用いて、今回の一連のインドール阻害剤の中で最も強力な阻害剤(2k)のATP競合について、阻害剤濃度を一定に保ったまま[ATP]を増大させて阻害%を測定することにより分析した。[ATP]は阻害%にあまり影響をおよぼさなかった(2k:45μMでの[ATP]:200μMまたは1,000μMの阻害%はそれぞれ46%および41%)ため、2kはこれらのアッセイ条件下ではATPに関して非競合的である。
【0119】
要約すると、非ATP競合的pp60c-src阻害剤の開発のために、インドール骨格が設計され、初期SARが実施された。今回の一連の阻害剤の中で最良のインドール骨格阻害剤の効力は、最良のナフタレン骨格阻害剤の効力とほぼ同等であることが判明した。2kが45μMで、[ATP]が200μMまたは1,000μMの場合の阻害%はそれぞれ46%および41%であった。
【0120】
実施例3−インドール誘導体タンパク質キナーゼ阻害剤の合成
下記にインドール由来のタンパク質キナーゼ阻害剤の合成および試験の結果を示す。4つの反応模式図を示し、続いてこれらそれぞれの反応模式図の最終生成物を調製するための詳細な実験方法を別々に示す。これらの最終生成物は、インドールを基本骨格とするチロシンキナーゼ阻害剤の例で、好ましいR基による合成を示している(ボロン酸、模式図1;OH、模式図2;脂肪族アミド伸長部分、模式図3;およびホスホン酸、模式図4)。
【化21】
5-ヒドロキシ-2-インドールカルボン酸メチル(1)
3.50gの5-ヒドロキシ-2-インドールカルボン酸をHClガスであらかじめ飽和させた無水MeOHに溶解し、48時間還流させた。減圧濃縮し、AcCNで3回粉砕して、残存する酸を除去した。EtOAcを用い、シリカプラグを通して濾過し、ベースライン混入物を除去した。4.32gを回収した(定量的収率)。TLC Rf=0.78(EtOAc) 1H NMR(DMSO-d6):3.82(s、3H)、6.78(d、J=8.8Hz、1H)、6.88(s、1H)、6.93(s、1H)、7.23(d、J=8.8Hz、1H)、8.90(s、1H)、11.62(s、1H)FAB(+)MS m/e 191.9(M+1)。
【0122】
5-[(トリフルオロメチル)スルホニルオキシ]インドール-2-カルボン酸メチル(2)
3.24g(17mmol)の5-ヒドロキシ-2-インドールカルボン酸メチル(1)および6.67g(18.7mmol)のn-フェニルトリフルオロメタンスルホンアミドに150mlの無水DCMを0℃で加えた。2.6mlのトリエチルアミンを滴下し、この時点で澄明な黄色溶液が生成した。0℃で1時間撹拌後、室温まで戻し、2時間撹拌した。減圧濃縮し、シリカゲルカラム(EtOAc/ヘキサン=1/1)を通して精製した。4.69g(86%)を回収した。TLC Rf=0.63(EtOAc/ヘキサン=1/1)。HPLC Rf=20.879 1H NMR(DMSO-d6):3.87(s、3H)、7.25(s、1H)、7.31(d、J=9.2Hz、1H)、7.55(d、J=9.2Hz、1H)、7.80(s、1H)、12.34(s、1H)FAB(+)MS m/e 323.1(M+1)
【0123】
5-メチルインドール-2-カルボン酸メチル,4,4,5,5-テトラメチル-1,3,2-ジオキサボロランメチル(3)
500mg 1.55mmolの5-[(トリフルオロメチル)スルホニルオキシ]インドール-2-カルボン酸メチル(2)、37.9mg(0.05mmol)のPdCl2(dppf)、432mg(1.7mmol)のビスピナコラートジボロン、454.8mg(4.65mmol)の酢酸カリウム、および25.7mg(0.05mmol)dppfをフラスコに加え、40℃で2時間減圧乾燥した。20mlの無水ジオキサンを加え、80℃で終夜加熱した。Pd黒が沈殿し、反応混合物は黒色に変わった。触媒をろ去し、シリカプラグを通過させて、ベースライン不純物を除去した。TLC Rf=0.51(EtOAc/ヘキサン=1/4) 粗生成物を次の反応に用いた。
【0124】
5-ボロニルインドール-2-カルボン酸メチル(4)
391.2mg(1.3mmol)の5-メチルインドール-2-カルボン酸メチル,4,4,5,5-テトラメチル-1,3,2-ジオキサボロランメチル(3)をEtOAcに溶解した。0.25ml(2.6mmol)のジエタノールアミンを加え、反応混合物を室温で終夜撹拌した。生成した白色沈殿物を濾過し、1N HCl中で超音波処理した。得られた白色沈殿物を濾過し、MeOHに溶解し、減圧濃縮した。36.6mg(13%)を回収した。HPLC Rf=13.912 1H NMR(DMSO-d6):3.85(s、3H)、7.15(s、1H)、7.36(d、J=8.4Hz、1H)、7.67(d、J=8.4Hz、1H)、7.87(s、1H)、8.14(s、1H)、11.91(s、1H)
【化22】
(5-ヒドロキシインドール-2-イル)-N-[(3-メトキシフェニル)メチル]カルボキシアミド(5)
2.00g(11.3mmol)の5-ヒドロキシ-2-インドールカルボン酸、1.6ml(12.4mmol)の3-メトキシベンジルアミン、および5.87g(11.3mmol)のPyBOPを10mlの無水DMFに溶解した。0℃に冷却し、5.9ml(33.9mmol)のDIEAを加えた。0℃で5分間撹拌し、室温に戻して1時間放置した。2.83g(収率85%)を回収した。TLC Rf=0.34(EtOAc/ヘキサン=1/1) 1H NMR(DMSO-d6):3.70(s、3H)、4.43(d、J=4.4Hz、2H)、6.69(d、J=8.8Hz、1H)、6.78(d、J=7.7Hz、1H)、6.83(s、1H)、6.86(s、1H)、6.94(s、1H)、7.20(m、3H)、8.92(t、J=4.4Hz、1H)、11.36(s、1H) FAB(+)MS m/e 297.3(M+1)
【0126】
(5-ヒドロキシインドール-2-イル)-N-[(3-ヒドロキシフェニル)メチル]カルボキシアミド(6)
200mg(0.67mmol)の(5-ヒドロキシインドール-2-イル)-N-[(3-メトキシフェニル)メチル]カルボキシアミド(5)に20mlの無水DCMを加え、アルゴン雰囲気下-78℃に冷却した。4.0ml(4.0mmol、6当量)のBBr3を加えた。5分間-78℃に維持した後、室温まで戻した。室温に90分間置いた後、H2Oで反応停止し、10分間撹拌した。EtOAcと混合して希釈し、NaHCO3および食塩水で洗浄した。有機層をMgSO4で乾燥し、減圧濃縮した。シリカプラグを通過させ、ベースライン混入物を除去した。Xmg(収率80%)を回収した。TLC Rf=0.21(EtOAc/ヘキサン=1/1) 1H NMR(DMSO-d6):4.38(d、J=4.8Hz、2H)、6.59(d、J=8.8Hz、1H)、6.71(m、3H)、6.83(d、J=1.8Hz、1H)、6.94(s、1H)、7.08(dd、J=7.7Hz、1H)、7.19(d、J=8.8Hz、1H)、8.84(t、J=5.9Hz)、11.28(s、1H)。FAB(+)MS m/e 283.2(M+1)。
【化23】
N-(1-カルバモイル-2-メチルブチル)(5-ヒドロキシインドール-2-イル)カルボキシアミド(7)
100mg(0.56mmol)の5-ヒドロキシ-2-インドールカルボン酸、103.4mg(0.62mmol、1.1当量)のL-イソロイシンアミド、および291mg(0.56mmol、1当量)のPyBOPをすべて1mlの無水DMFに溶解した。溶液を0℃に冷却し、0.3ml(1.68mmol、3当量)のDIEAを加えた。反応混合物を0℃で1分間、次いで室温で1時間撹拌した。反応混合物を次いでEtOAcで希釈し、1N HClで3回と飽和NaHCO3で3回洗浄した。有機層をMgSO4で乾燥し、減圧濃縮して166.7mg(収率91%)を得た。TLC Rf=0.08(EtOAc/ヘキサン=1/1) 1H NMR(DMSO-d6):0.83(m、6H)、1.15(m、2H)、1.68(m、1H)、1.83(m、1H)、4.29(t、J=8.8Hz、1H)、6.69(d、J=8.5Hz、1H)、6.83(s、1H)、7.01(s、1H)、7.06(s、1H)、7.19(d、J=8.4Hz、1H)、7.48(s、1H)、8.00(d、9.2Hz、1H)、8.76(s、1H)、11.3(s、1H)。FAB(+)MS m/e 290.1(M+1)
【化24】
5-ジベンジルホスホリルインドール-2-カルボン酸メチル(8)
200mg(0.62mmol)の5-[(トリフルオロメチル)スルホニルオキシ]インドール-2-カルボン酸メチル(2)、195.8mg(0.74mmol、1.2当量)のジベンジルホスファイト、0.14ml(0.81mmol、1.3当量)のDIEA、および35.7mg(0.03mmol、5mol%)のPd(PPh3)4をすべてアルゴン雰囲気下で無水AcCNに溶解した。反応混合物を80℃で終夜加熱した。溶媒を減圧下で除去し、標題化合物をシリカゲルクロマトグラフィにより単離した。130mg(収率50%)。TLC Rf=0.28(EtOAc/ヘキサン=1/1)。1H NMR(DMSO-d6):3.85(s、3H)、4.98〜5.01(m、4H)、7.28〜7.32(m、11H)、7.53〜7.55(m、2H)、8.17(d、J=14.6Hz、1H) 31P NMR(DMSO-d6):23.89
【0129】
5-ホスホノールインドール-2-カルボン酸メチル
5-ジベンジルホスホリルインドール-2-カルボン酸メチル(8)(125mg)を10mlのMeOHに溶解した。20mgのPd-Cを加え、混合物をParr装置内で終夜水素添加した。触媒をろ去し、溶媒を減圧下で除去した。72.5mg(収率73%)を得た。TLC Rf=EtOAc中でベースライン。1H NMR(DMSO-d6):3.84(s、3H)、7.24(s、1H)、7.44〜7.49(m、2H)、8.01(d、J=14.3Hz、1H)、12.11(s、1H) 31P NMR(DMSO-d6):17.22
【0130】
この実施例におけるエステル化合物は、エステルをアミドに変換すること、および/または追加の特異的因子を加えることによって、効力を増強することができた。
【0131】
実施例4−さらなるインドール誘導体タンパク質キナーゼ阻害剤の合成
いくつかのさらなるインドール阻害剤の合成を以下に示す。これらの合成により、pp60c-srcおよび他のチロシンキナーゼに対する効力がより強い化合物が得られる。続いて、効力を高めるために、メチルエステル基を様々なアミド誘導体に変換することができる。
【化25】
実施例5−Src阻害剤の毒性
多くの最近の文献が、重大な毒性をもたらさない、広く有用な癌治療へのアプローチとして、pp60c-src(Src)を標的化を支持している。例えば、EGF受容体PTKシグナル伝達の増大を示すか、または関連のHer-2/neu受容体を過剰発現する腫瘍は、構成的にSrcを活性化し、腫瘍の侵襲性を促進する。これらの細胞においてSrcを阻害すると、増殖の停止が誘導され、アポトーシスが引き起こされ、形質転換された表現型が戻る(Karniら、1999)。異常に上昇したSrcの活性は、形質転換細胞の足場非依存的な増殖を可能にすることが知られている。これは明らかに、細胞外マトリックスシグナル伝達が分裂促進的なシグナル伝達と協調する様式でFAK/Src経路におけるSrc活性を上昇させ、それにより通常は活性化されているアポトーシスメカニズムを阻止するという事実によって引き起こされる。したがって、細胞外マトリックスから解放されることにより通常は活性化されているアポトーシスメカニズムが誘導されると考えられるため、腫瘍細胞におけるFAK/Src阻害はアポトーシスを誘導すると考えられる(Hisanoら、1997)。加えて、Src阻害によりVEGF mRNA発現の低下が認められ、これらのSrc阻害された細胞系統由来の腫瘍は、脈管形成性の発生の低下を示した(Ellisら、1998)。
【0133】
Src阻害の潜在的毒性の問題は、非常に有望な結果によって対処された。例えば、マウスにおけるSrc遺伝子のノックアウトは、たった1つの異常、すなわち破骨細胞が波状縁を形成できず、したがって骨再吸収をしない破骨細胞をもたらした。しかし、これらのマウスにおける破骨細胞の骨再吸収機能は、キナーゼ欠損Src遺伝子を挿入することによって救済された(Schwartzbergら、1997)。Srcタンパク質の存在は破骨細胞必須シグナル伝達複合体における他のPTK(破骨細胞の機能を維持するのに必須である)を動員し、活性化するのに十分であることが明らかな為、これにより、既知の毒性のみを誘発することなく、Srcキナーゼ活性がインビボで阻害されうることが示唆された。
【0134】
前述された腫瘍(Levitzki、1996)に加えて、Srcが過剰に活性化されることが判明したヒト腫瘍の数が増加しているため、Srcは癌治療のための「万能」標的であると提唱されている。引用された文献やその他の文献に基づき、癌治療のためのSrc阻害の潜在的恩典は、4倍になったように思われる。すなわち、1)オートクライン成長因子ループ効果などにより引き起こされる無制御の細胞増殖の阻害、2)細胞マトリックスからの解放によるアポトーシス誘発が原因の転移の阻害、3)VEGFレベル低下を通じての腫瘍脈管形成の阻害、4)低毒性。
【0135】
初期非ペプチドSrc阻害剤は、4つの異なる一連の細胞培養アッセイ法においても非常に有望な結果を示している。1)NIHの60腫瘍細胞パネルアッセイ法では、前立腺の系統を含む腫瘍細胞株に対し、広い活性(Src阻害剤に対して期待されるとおり)が示された。例えば、阻害剤の3つはNIH前立腺癌細胞株に対して下記の増殖阻害IC50を示した:TOM 2-32(PC-3、15μM;DU-145、38μM)、TOM 2-47(PC-3、19μM)、KLM 2-31(PC-3、39μM;DU-145、>100μM)。2)v-Src形質転換正常ラット腎細胞株(LA25)において、TOM 2-47およびTOM 2-32は非形質転換親細胞の正常な増殖は阻害せずに、v-Src誘導細胞増殖を特異的に阻止した。この結果より、阻害剤が正常な細胞には影響をおよぼさないが、Src誘導細胞形質転換の阻害において有効であることが示された。3)3名の異なる患者由来の卵巣腫瘍およびもう1名の患者由来の腹部癌における抗癌剤エトポシド、タキソール、ドキソルビシンおよびシスプラチンに対するSrc阻害剤。すべての例で、Src阻害剤は公知の抗癌剤と少なくとも同程度に有効、典型的にはより有効であり、試験した最低用量(3μM)で完全な有効性が認められた。代表的な例として、タキソールおよびドキソルビシン(この特定の腫瘍細胞培養物においては、これらはエトポシドおよびシスプラチンよりも有効であった)と前述の3つのSrc阻害剤との、腫瘍N015からの卵巣腫瘍細胞を用いた比較を図15Aに示す。4)正常なヒト線維芽細胞の増殖阻害についてもSrc阻害剤を試験し、正常な細胞増殖の阻害が全く見られなかった(サブコンフルエントとコンフルエント両方;代わりに若干の増殖促進が観察された)が、このことはこれらの阻害剤が10倍高い濃度であっても正常細胞には毒性でないことを示している。データの例を図15Bに示す。
【0136】
全体として、ここまでに得られた細胞データは、Srcに期待すること、すなわち、通常の細胞毒性がほとんどまたは全くない多くの癌細胞株に対する広い活性を示している。
【0137】
予備的Src阻害剤は、そこからより強力で選択性が高い阻害剤を設計することができる先導構造である。天然産物チロシンキナーゼ阻害剤ダムナカンサル(Faltynekら、1995)のペプチド競合的結合部位を調べるために、チロシンキナーゼ結晶構造の利用に加えて、この阻害剤を用いた分子モデリング試験を実施することができる。これらの追加のモデリング試験により、ダムナカンサルの鍵となるファルマコフォア成分が新しい阻害剤に組み込まれている、Src阻害剤の」さらなる類縁体を設計することが可能となる。報告通りにこれらの合成が行われ、単離Srcの試験が行われる(Marsilje 2000)。
【0138】
本明細書において好ましい態様を示し、詳細に述べてきたが、本発明の精神から逸脱することなく様々な修正、追加、置換などが加えられることが当業者には明らかであり、これらはしたがって、特許請求の範囲に定義されるとおり、本発明の範囲内であると考えられる。
【0139】
文献の引用
本明細書に引用されている以下の参考文献は、これによって参照として本明細書に組み入れられている。
【図面の簡単な説明】
【図1】 非ペプチドタンパク質キナーゼ阻害剤を開発するためのモジュール方法を示す図である。段階1は有望な非ペプチド骨格を同定するための1つまたは複数の第一のモジュール(「M1」)を利用する。段階2は特異的因子を加えることによって効力を増強する。この段階において骨格の妥当性を確認する。阻害剤が非ATP競合的であるかどうかも調べることができる。段階3では、コンビナトリアルライブラリーを用いて効力および選択性をさらに増強し、M1および特異的因子を最適化する。
【図2】 (PKA):Mg2ATP:偽基質阻害剤のX線構造を示す図を提供する。
【図3】 保存されたタンパク質キナーゼ触媒領域に結合するための一般的モジュールM1の設計を提供する。
【図4】 ボロン酸「阻害剤」21および22がPKAの基質となることを示す。
【図5】 モデルsrc活性部位におけるsrc基質Ac-Ile-Tyr-Glu-Phe-NH2の結合相互作用を示す。
【図6】 ナフタレンを基本とするsrc阻害剤の骨格の設計を示す図である。
【図7】 イソキノリンおよびインドールを基本とするsrc阻害剤の骨格の設計を示す。
【図8】 マーシルジェ(Marsilje)2000に記載のナフタレン阻害剤を調製するために用いる化学反応の一例を提供する。表5からのsrc阻害剤において、アリールトリフラート(Ishiyamaら、1997)またはハロゲン化アリール(Ishiyama、1995)のいずれかを市販のジボロンピナコールエステルと結合させることができるPd(0)触媒クロスカップリング法を用いて、M1水酸基の代わりにボロン酸官能基を導入することができる。
【図9】 ナフタレン骨格に疎水性のS2選択的因子を連結させるために従うことができる合成スキームを示す。
【図10】 ナフタレン化学による良好なモデル反応を示す図であり、96穴プレート様式でこの骨格のコンビナトリアルライブラリーを合成するための調製において固相に変換することができる。この化学反応は、44で表される活性が低いナフタレンレジオ異性体がマーシルジェ(Marsilje)2000に記載のとおり市販の3,5-ジヒドロキシ-2-ナフトエ酸から容易に得られるため、この化合物で実施している。
【図11】 コンビナトリアルライブラリーにおけるナフタレン骨格を改変するための可能な方法を提供する。
【図12】 中間体69からの反応2で生成したトリフラート官能基(図11)の、アミン(Wolfeら、1997)、次いで一連のアミドまたは他のアミン誘導体への変換を示す。
【図13】 前述のモデリング法に従い、図13に示すボロン酸M1基の一連のヒドロキシ含有類縁体がsrcおよびIRTK(インスリン受容体タンパク質チロシンキナーゼ)活性部位においてモデリングされ、いくつかの興味深い可能性として図示された相互作用/結合様式が見いだされたことを示す。
【図14】 LA25およびNRK細胞株における非ペプチドsrc阻害剤43-メタ(表5)の試験からの結果を示す。
【図15】 図15Aは、腫瘍N015由来の卵巣腫瘍細胞を用いた、タキソールおよびドキソルビシン(これらは、この腫瘍培養細胞においてはエトポシドおよびシスプラチンよりも有効である)と前述の3つのSrc阻害剤との比較を示す。図15Bは、正常ヒト線維芽細胞の増殖阻害に対するsrc阻害剤の試験からの結果を示す。正常細胞では増殖阻害は認められず(準融合性と融合性の両方;代わりにいくらかの増殖促進が見られた)、これらの阻害剤は10倍の濃度でさえ正常な細胞には非毒性であることが示される。図15Cは、src阻害剤TOM 2-32、TOM 2-47、およびKLM 2-31の構造を提供する。
Claims (12)
- 下記の式を有する化合物:
- 化合物が、インドール化合物
より選択される、請求項1記載の化合物。 - R1がHである、請求項1記載の化合物。
- R3がHである、請求項1記載の化合物。
- R4がCH2である、請求項1記載の化合物。
- R5がHである、請求項1記載の化合物。
- R6がOHである、請求項1記載の化合物。
- R7がHである、請求項1記載の化合物。
- 下記の構造を有する化合物:
- 請求項1または3〜9のいずれか一項記載の化合物および少なくとも1つの薬学的に許容される担体を含む、薬学的組成物。
- タンパク質チロシンキナーゼ阻害剤に反応性の症状の治療のための薬物の製造における、請求項1または3〜9のいずれか一項記載の化合物の使用であって、ここで化合物はチロシンキナーゼの活性を阻害し、症状が癌、乾癬、関節硬化症、および免疫系の活性より選択される、使用。
- タンパク質チロシンキナーゼがpp60c-srcである、請求項11記載の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11564399P | 1999-01-13 | 1999-01-13 | |
| US60/115,643 | 1999-01-13 | ||
| PCT/US2000/000803 WO2000042213A1 (en) | 1999-01-13 | 2000-01-13 | A novel method for designing protein kinase inhibitors |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010151851A Division JP2010279363A (ja) | 1999-01-13 | 2010-07-02 | タンパク質キナーゼ阻害剤を設計するための新規の方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2002534132A JP2002534132A (ja) | 2002-10-15 |
| JP2002534132A5 JP2002534132A5 (ja) | 2007-04-12 |
| JP4865129B2 true JP4865129B2 (ja) | 2012-02-01 |
Family
ID=22362611
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000593770A Expired - Lifetime JP4865129B2 (ja) | 1999-01-13 | 2000-01-13 | タンパク質キナーゼ阻害剤を設計するための新規の方法 |
| JP2010151851A Pending JP2010279363A (ja) | 1999-01-13 | 2010-07-02 | タンパク質キナーゼ阻害剤を設計するための新規の方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010151851A Pending JP2010279363A (ja) | 1999-01-13 | 2010-07-02 | タンパク質キナーゼ阻害剤を設計するための新規の方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US7901894B2 (ja) |
| EP (1) | EP1147214B1 (ja) |
| JP (2) | JP4865129B2 (ja) |
| AT (1) | ATE494388T1 (ja) |
| AU (1) | AU3208200A (ja) |
| BR (1) | BR0007532A (ja) |
| CA (1) | CA2360581A1 (ja) |
| DE (1) | DE60045474D1 (ja) |
| MX (1) | MXPA01007099A (ja) |
| WO (1) | WO2000042213A1 (ja) |
Families Citing this family (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1147214B1 (en) | 1999-01-13 | 2011-01-05 | The Research Foundation Of State University Of New York | A novel method for designing protein kinase inhibitors |
| WO2002012189A1 (fr) | 2000-08-09 | 2002-02-14 | Mitsubishi Pharma Corporation | Composes amide bicycliques condenses et utilisations medicales associees |
| HU227197B1 (en) | 2000-10-24 | 2010-10-28 | Richter Gedeon Nyrt | Nmda receptor antagonist carboxylic acid amide derivatives and pharmaceutical compositions containing them |
| CA2450807A1 (en) * | 2001-06-14 | 2003-08-14 | Yissum Research Development Company Of The Hebrew University Of Jerusale M | Non-myeloablative tolerogenic treatment with tyrphostins |
| US7041280B2 (en) | 2001-06-29 | 2006-05-09 | Genzyme Corporation | Aryl boronate functionalized polymers for treating obesity |
| US6858592B2 (en) | 2001-06-29 | 2005-02-22 | Genzyme Corporation | Aryl boronic acids for treating obesity |
| EP1444204A4 (en) * | 2001-10-22 | 2009-11-04 | Univ New York State Res Found | INHIBITORS OF PROTEIN KINASES AND PROTEINS PHOSPHATASES, METHODS OF IDENTIFICATION AND METHODS OF USE THEREOF |
| US7005445B2 (en) | 2001-10-22 | 2006-02-28 | The Research Foundation Of State University Of New York | Protein kinase and phosphatase inhibitors and methods for designing them |
| US8143210B2 (en) | 2002-02-14 | 2012-03-27 | The Board Of Trustees Of The Leland Stanford Junior University | Enzyme treatment of foodstuffs for celiac sprue |
| DK1572127T4 (da) | 2002-02-14 | 2014-11-24 | Univ Leland Stanford Junior | Enzymbehandling af fødevarer til cøliaki |
| GB0205175D0 (en) | 2002-03-06 | 2002-04-17 | Astrazeneca Ab | Chemical compounds |
| GB0205165D0 (en) | 2002-03-06 | 2002-04-17 | Astrazeneca Ab | Chemical compounds |
| GB0205176D0 (en) | 2002-03-06 | 2002-04-17 | Astrazeneca Ab | Chemical compounds |
| GB0205170D0 (en) | 2002-03-06 | 2002-04-17 | Astrazeneca Ab | Chemical compounds |
| GB0205162D0 (en) | 2002-03-06 | 2002-04-17 | Astrazeneca Ab | Chemical compounds |
| GB0205166D0 (en) | 2002-03-06 | 2002-04-17 | Astrazeneca Ab | Chemical compounds |
| GB2403474B (en) * | 2002-04-18 | 2006-10-11 | Inst Med Molecular Design Inc | Amide derivatives |
| WO2004045392A2 (en) | 2002-11-20 | 2004-06-03 | The Board Of Trustees Of The Leland Stanford Junior University | Diagnostic method for celiac sprue |
| CA2487247A1 (en) * | 2002-05-14 | 2003-11-27 | The Board Of Trustees Of The Leland Stanford Junior University | Drug therapy for celiac sprue |
| US7579313B2 (en) | 2003-11-18 | 2009-08-25 | The Board Of Trustees Of The Leland Stanford Junior University | Transglutaminase inhibitors and methods of use thereof |
| WO2006108270A1 (en) | 2005-04-11 | 2006-10-19 | Pharmagap Inc. | Inhibitors of protein kinases and uses thereof |
| US7838542B2 (en) * | 2006-06-29 | 2010-11-23 | Kinex Pharmaceuticals, Llc | Bicyclic compositions and methods for modulating a kinase cascade |
| JO3598B1 (ar) * | 2006-10-10 | 2020-07-05 | Infinity Discovery Inc | الاحماض والاسترات البورونية كمثبطات اميد هيدروليز الحامض الدهني |
| WO2008059370A2 (en) | 2006-11-17 | 2008-05-22 | Pfizer Japan Inc. | Substituted bicyclocarboxyamide compounds |
| NZ580425A (en) | 2007-03-16 | 2011-12-22 | Univ Leland Stanford Junior | Combination enzyme therapy for digestion of dietary gluten |
| AU2009233711B2 (en) | 2008-04-09 | 2015-02-12 | Infinity Pharmaceuticals, Inc | Inhibitors of fatty acid amide hydrolase |
| KR100963102B1 (ko) * | 2009-03-16 | 2010-06-14 | 호서대학교 산학협력단 | 표피성장인자 수용체 타이로신 키나제 저해제의 탐색방법, 및 그에 의해서 탐색된 저해제 |
| US11673914B2 (en) | 2009-11-10 | 2023-06-13 | Allegro Pharmaceuticals, LLC | Peptide therapies for reduction of macular thickening |
| US9896480B2 (en) | 2009-11-10 | 2018-02-20 | Allegro Pharmaceuticals, Inc. | Integrin receptor antagonists and their methods of use |
| CN102791296B (zh) | 2009-11-10 | 2019-08-16 | 急速制药公司 | 用于抑制细胞粘附至rgd结合位点或引导诊断剂或治疗剂至rgd结合位点的组合物和方法 |
| WO2011097233A1 (en) | 2010-02-03 | 2011-08-11 | Infinity Pharmaceuticals, Inc. | Fatty acid amide hydrolase inhibitors |
| ES2949357T3 (es) | 2016-03-09 | 2023-09-28 | Raze Therapeutics Inc | Inhibidores de 3-fosfoglicerato deshidrogenasa y usos de los mismos |
| DK3426243T3 (da) | 2016-03-09 | 2021-07-19 | Raze Therapeutics Inc | 3-phosphoglyceratdehydrogenase-inhibitorer og anvendelser deraf |
| JP6865296B2 (ja) * | 2017-03-14 | 2021-04-28 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Phgdh阻害薬としてのトシルアセタートベース化合物及びその誘導体 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997029091A1 (en) * | 1996-02-09 | 1997-08-14 | Phytera Symbion Aps | Balanol analogues |
| WO1998007695A1 (en) * | 1996-08-23 | 1998-02-26 | Sugen, Inc. | Indolinone combinatorial libraries and related products and methods for the treatment of disease |
Family Cites Families (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1261835A (en) | 1984-08-20 | 1989-09-26 | Masaaki Toda | (fused) benz(thio)amides |
| JPH01132579A (ja) | 1987-11-19 | 1989-05-25 | Ss Pharmaceut Co Ltd | インドール誘導体 |
| GB8827189D0 (en) | 1988-11-21 | 1988-12-29 | Fujisawa Pharmaceutical Co | 2(1h)-quinolinone compounds processes for preparation thereof & pharmaceutical composition comprising same |
| DK0507861T3 (ja) | 1989-12-28 | 1997-03-03 | Upjohn Co | |
| US5552534A (en) | 1991-08-22 | 1996-09-03 | The Trustees Of The University Of Pennsylvania | Non-Peptide peptidomimetics |
| DE4307883A1 (en) | 1992-03-12 | 1993-09-23 | Westarp Martin Egon Dr Med | Use of anti-retroviral substances - to treat motor-neuronal diseases |
| US5705585A (en) | 1993-06-30 | 1998-01-06 | Arqule, Inc. | Aminimide-containing molecules and materials as molecular recognition agents |
| CZ283965B6 (cs) | 1992-08-06 | 1998-07-15 | Warner-Lambert Company | 2-thioindolové, 2-indolinthionové a polysulfidové sloučeniny, 2-selenoindolové, 2-indolinselenonové a selenidové sloučeniny a farmaceutické prostředky na jejich bázi |
| US6011175A (en) | 1993-05-18 | 2000-01-04 | University Of Pittsburgh | Inhibition of farnesyltransferase |
| US5532167A (en) | 1994-01-07 | 1996-07-02 | Beth Israel Hospital | Substrate specificity of protein kinases |
| US5712171A (en) * | 1995-01-20 | 1998-01-27 | Arqule, Inc. | Method of generating a plurality of chemical compounds in a spatially arranged array |
| FR2733995B1 (fr) | 1995-05-09 | 1997-07-25 | Inst Nat Sante Rech Med | Inhibiteurs de l'inactivation de neuropeptides endogenes notamment la cholecystokinine, leurs procedes de preparation leur utilisation comme medicaments et procede de criblage de medicaments |
| CA2342471C (en) | 1995-06-06 | 2002-10-29 | Judith L. Treadway | Heterocyclecarbonylmethyl amine intermediates |
| US6297269B1 (en) | 1995-06-06 | 2001-10-02 | Pfizer Inc. | Substituted n-(indole-2-carbonyl-) amides and derivatives as glycogen phosphorylase inhibitors |
| US5648378A (en) | 1995-06-07 | 1997-07-15 | Research Corporation Technologies, Inc. | 2-iminochromene derivatives as inhibitors of protein tyrosine kinase |
| CA2231509C (en) | 1995-09-11 | 2008-07-08 | Osteoarthritis Sciences, Inc. | Protein tyrosine kinase inhibitors for treating osteoarthritis |
| US5952322A (en) | 1996-12-05 | 1999-09-14 | Pfizer Inc. | Method of reducing tissue damage associated with non-cardiac ischemia using glycogen phosphorylase inhibitors |
| EP0946587A2 (en) | 1996-12-16 | 1999-10-06 | Fujisawa Pharmaceutical Co., Ltd. | New amide compounds |
| CN1251577A (zh) | 1997-02-21 | 2000-04-26 | 武田药品工业株式会社 | 稠环化合物及其生产和应用 |
| US6420338B1 (en) | 1997-06-13 | 2002-07-16 | New York University Medical Center | Inhibition of the Src kinase family pathway as a method of treating HBV infection and hepatocellular carcinoma |
| GB9718913D0 (en) | 1997-09-05 | 1997-11-12 | Glaxo Group Ltd | Substituted oxindole derivatives |
| WO1999034018A1 (en) * | 1997-12-24 | 1999-07-08 | The Regents Of The University Of California | Methods of using chemical libraries to search for new kinase inhibitors |
| AU3363599A (en) * | 1998-03-26 | 1999-10-18 | Max-Planck Institut Fur Biochemie | Heterocyclic families of compounds for the modulation of tyrosine protein kinase |
| WO1999054309A1 (en) | 1998-04-23 | 1999-10-28 | Takeda Chemical Industries, Ltd. | Naphthalene derivatives, their production and use |
| WO1999059973A1 (en) | 1998-05-15 | 1999-11-25 | Guilford Pharmaceuticals Inc. | Carboxamide compounds, compositions, and methods for inhibiting parp activity |
| US7070936B1 (en) | 1999-01-13 | 2006-07-04 | The Research Foundation Of State University Of New York | Method for designing protein kinase inhibitors |
| EP1147214B1 (en) | 1999-01-13 | 2011-01-05 | The Research Foundation Of State University Of New York | A novel method for designing protein kinase inhibitors |
| JP2003508382A (ja) | 1999-08-27 | 2003-03-04 | スージェン・インコーポレーテッド | リン酸模倣体およびホスファターゼ阻害剤を用いる治療方法 |
| US6921763B2 (en) | 1999-09-17 | 2005-07-26 | Abbott Laboratories | Pyrazolopyrimidines as therapeutic agents |
| HN2001000008A (es) | 2000-01-21 | 2003-12-11 | Inc Agouron Pharmaceuticals | Compuesto de amida y composiciones farmaceuticas para inhibir proteinquinasas, y su modo de empleo |
| AU2868601A (en) | 2000-01-27 | 2001-08-07 | Ribotargets Ltd | Biaryl compounds, their preparation and their use in therapy |
| UA73976C2 (uk) | 2000-02-15 | 2005-10-17 | Суджен, Інк. | Заміщені піролом 2-індолінони, фармацевтична композиція, спосіб модуляції каталітичної активності протеїнкінази та спосіб лікування або профілактики захворювання, пов'язаного з протеїнкіназою |
| KR100423899B1 (ko) | 2000-05-10 | 2004-03-24 | 주식회사 엘지생명과학 | 세포 증식 억제제로 유용한 1,1-디옥소이소티아졸리딘을갖는 인다졸 |
| US6414013B1 (en) | 2000-06-19 | 2002-07-02 | Pharmacia & Upjohn S.P.A. | Thiophene compounds, process for preparing the same, and pharmaceutical compositions containing the same background of the invention |
| SI1294724T1 (sl) | 2000-06-26 | 2006-08-31 | Pfizer Prod Inc | Pirolo(2,3-d)primidinske spojine kot imunosupresivna sredstva |
| JP2004502775A (ja) | 2000-07-07 | 2004-01-29 | ノボ ノルディスク アクティーゼルスカブ | 非ヌクレオシド逆転写阻害剤抗レトロウイルス療法をモニターするための手段および方法 |
| US6803362B2 (en) | 2001-03-09 | 2004-10-12 | Ortho-Mcneil Pharmaceutical Inc. | Heterocyclic compounds |
| KR100429841B1 (ko) | 2001-07-19 | 2004-05-04 | 삼성전자주식회사 | Grin 렌즈를 구비하는 광 기록헤드 |
| EP1444204A4 (en) | 2001-10-22 | 2009-11-04 | Univ New York State Res Found | INHIBITORS OF PROTEIN KINASES AND PROTEINS PHOSPHATASES, METHODS OF IDENTIFICATION AND METHODS OF USE THEREOF |
| US7005445B2 (en) | 2001-10-22 | 2006-02-28 | The Research Foundation Of State University Of New York | Protein kinase and phosphatase inhibitors and methods for designing them |
| PL370766A1 (en) | 2001-12-04 | 2005-05-30 | F.Hoffmann-La Roche Ag | Substituted 2-amino-cycloalkanecarboxamides and their use as cysteine protease inhibitors |
| EP1494663A1 (en) | 2002-04-10 | 2005-01-12 | SmithKline Beecham Corporation | Derivatives of 1-(oxoaminoacetyl) pentylcarbamate as cathepsin k inhibitors for the treatment of bone loss |
| EP1501514B1 (en) | 2002-05-03 | 2012-12-19 | Exelixis, Inc. | Protein kinase modulators and methods of use |
| AU2003258491A1 (en) | 2002-09-05 | 2004-03-29 | Neurosearch A/S | Amide derivatives and their use as chloride channel blockers |
| CA2501611A1 (en) | 2002-10-11 | 2004-04-22 | Astrazeneca Ab | 1,4-disubstituted piperidine derivatives and their use as 11-betahsd1 inhibitors |
| CA2510471A1 (en) | 2002-12-19 | 2004-07-08 | Neurogen Corporation | Substituted biphenyl-4-carboxylic acid arylamide analogues as capsaicin receptor modulators |
| CA2541299A1 (en) | 2003-10-07 | 2005-04-14 | Renovis, Inc. | Amide compounds as ion channel ligands and uses thereof |
| WO2005035551A2 (en) | 2003-10-08 | 2005-04-21 | Incyte Corporation | Inhibitors of proteins that bind phosphorylated molecules |
| MX2007001540A (es) | 2004-08-10 | 2007-04-23 | Incyte Corp | Compuestos amido y sus usos como farmaceuticos. |
| US8211919B2 (en) | 2005-09-02 | 2012-07-03 | Astellas Pharma Inc. | Amide derivatives as rock inhibitors |
| US7838542B2 (en) * | 2006-06-29 | 2010-11-23 | Kinex Pharmaceuticals, Llc | Bicyclic compositions and methods for modulating a kinase cascade |
-
2000
- 2000-01-13 EP EP00909899A patent/EP1147214B1/en not_active Expired - Lifetime
- 2000-01-13 MX MXPA01007099A patent/MXPA01007099A/es active IP Right Grant
- 2000-01-13 DE DE60045474T patent/DE60045474D1/de not_active Expired - Lifetime
- 2000-01-13 CA CA002360581A patent/CA2360581A1/en not_active Abandoned
- 2000-01-13 AT AT00909899T patent/ATE494388T1/de not_active IP Right Cessation
- 2000-01-13 BR BR0007532-9A patent/BR0007532A/pt active Search and Examination
- 2000-01-13 AU AU32082/00A patent/AU3208200A/en not_active Abandoned
- 2000-01-13 WO PCT/US2000/000803 patent/WO2000042213A1/en active Search and Examination
- 2000-01-13 JP JP2000593770A patent/JP4865129B2/ja not_active Expired - Lifetime
-
2005
- 2005-10-27 US US11/261,858 patent/US7901894B2/en not_active Expired - Lifetime
-
2010
- 2010-07-02 JP JP2010151851A patent/JP2010279363A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997029091A1 (en) * | 1996-02-09 | 1997-08-14 | Phytera Symbion Aps | Balanol analogues |
| WO1998007695A1 (en) * | 1996-08-23 | 1998-02-26 | Sugen, Inc. | Indolinone combinatorial libraries and related products and methods for the treatment of disease |
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA01007099A (es) | 2002-03-27 |
| US7901894B2 (en) | 2011-03-08 |
| DE60045474D1 (de) | 2011-02-17 |
| US20060089401A1 (en) | 2006-04-27 |
| JP2002534132A (ja) | 2002-10-15 |
| EP1147214A1 (en) | 2001-10-24 |
| EP1147214A4 (en) | 2008-08-27 |
| ATE494388T1 (de) | 2011-01-15 |
| AU3208200A (en) | 2000-08-01 |
| EP1147214B1 (en) | 2011-01-05 |
| CA2360581A1 (en) | 2000-07-20 |
| BR0007532A (pt) | 2001-11-20 |
| WO2000042213A1 (en) | 2000-07-20 |
| JP2010279363A (ja) | 2010-12-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4865129B2 (ja) | タンパク質キナーゼ阻害剤を設計するための新規の方法 | |
| JP4623962B2 (ja) | タンパク質キナーゼおよびホスファターゼ阻害剤、それらを設計する方法、ならびにそれらを使用する方法 | |
| Zhou et al. | Development of selective mono or dual PROTAC degrader probe of CDK isoforms | |
| US8088768B2 (en) | Protein kinase and phosphatase inhibitors | |
| US11370770B2 (en) | 3-arylindazoles as selective MEK4 inhibitors | |
| WO2020234103A1 (en) | Identification and use of kras inhibitors | |
| Wang et al. | Design, synthesis and biological evaluation of tetrahydroquinoline-based reversible LSD1 inhibitors | |
| Zhao et al. | Discovery of novel BTK PROTACs for B-Cell lymphomas | |
| Liu et al. | Synthesis and SAR of derivatives based on 2-biarylethylimidazole as bombesin receptor subtype-3 (BRS-3) agonists for the treatment of obesity | |
| Zhan et al. | Design, synthesis, and biological evaluation of novel highly selective polo-like kinase 2 inhibitors based on the tetrahydropteridin chemical scaffold | |
| Lesuisse et al. | Discovery of the first non-ATP competitive IGF-1R kinase inhibitors: Advantages in comparison with competitive inhibitors | |
| CA3172987A1 (en) | Small molecule inhibitors of oncogenic chd1l with preclinical activity against colorectal cancer | |
| Liu et al. | Design, synthesis and biological evaluation of pyridine derivatives as selective SHP2 inhibitors | |
| Li et al. | Novel pyrrolo [2, 1-c][1, 4] benzodiazepine-3, 11-dione (PBD) derivatives as selective HDAC6 inhibitors to suppress tumor metastasis and invasion in vitro and in vivo | |
| AboulMagd et al. | Saccharomonosporine A inspiration; synthesis of potent analogues as potential PIM kinase inhibitors | |
| Wang et al. | Discovery of a new class of valosine containing protein (VCP/P97) inhibitors for the treatment of non-small cell lung cancer | |
| US7070936B1 (en) | Method for designing protein kinase inhibitors | |
| Xu et al. | Design and synthesis of Grp94 selective inhibitors based on Phe199 induced fit mechanism and their anti-inflammatory effects | |
| KR101789934B1 (ko) | 상피세포성장인자수용체 변이체를 가진 종양 진단 및 종양 성장 억제 활성을 갖는 신규 화합물 및 이를 포함하는 의학적 용도 | |
| Liu et al. | Design, synthesis, and bioactivity study on Lissodendrins B derivatives as PARP1 inhibitor | |
| US20200405707A1 (en) | Diazinyl amino acridines and medical uses thereof | |
| Fang et al. | Novel 7-formyl-naphthyridyl-ureas derivatives as potential selective FGFR4 inhibitors: Design, synthesis, and biological activity studies | |
| KR100874209B1 (ko) | 3,10-다이하이드로-1H-[1,4]다이아제피노[5,6-b]인돌-2-온 유도체 및 약학적으로 허용 가능한 염, 그의 제조방법및 그의 용도 | |
| WO2022272313A1 (en) | Modulators of histone acetyltransferase 1 and methods of treatment thereof | |
| Jones | The design and synthesis of novel epigenetic modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070112 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070112 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20090305 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100106 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100401 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100408 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100702 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110525 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110812 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110908 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110909 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20111020 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20111110 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20141118 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4865129 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |