JP4749334B2 - 組換えアデノウイルス用パッケージング細胞 - Google Patents
組換えアデノウイルス用パッケージング細胞 Download PDFInfo
- Publication number
- JP4749334B2 JP4749334B2 JP2006530273A JP2006530273A JP4749334B2 JP 4749334 B2 JP4749334 B2 JP 4749334B2 JP 2006530273 A JP2006530273 A JP 2006530273A JP 2006530273 A JP2006530273 A JP 2006530273A JP 4749334 B2 JP4749334 B2 JP 4749334B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- sequence
- sequences
- genome
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images





















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
- C12N15/861—Adenoviral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10322—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10341—Use of virus, viral particle or viral elements as a vector
- C12N2710/10343—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10351—Methods of production or purification of viral material
- C12N2710/10352—Methods of production or purification of viral material relating to complementing cells and packaging systems for producing virus or viral particles
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/38—Vector systems having a special element relevant for transcription being a stuffer
Description
本発明は、分子及び細胞生物学の分野、更に詳しくは、パッケージング細胞、及び組換えアデノウイルスのバッチの生成のためのその使用に関する。
複製欠損組換えアデノウイルスは、例えば、遺伝子治療において、及びワクチン接種目的のために有用である。それらは、通常、アデノウイルスのE1-領域を欠き、及び従ってE1配列を提供する補完細胞(complementing cells)において増殖する。パッケージング細胞は、細胞において包まれ、組換えウイルス粒子を形成するベクターの複製のために必要なすべての情報を提供する。パッケージング細胞の例は、293細胞であり、それは、アデノウイルス5のゲノムのヌクレオチド1-4344を含み(Louis(ルイス)等、1997年)、それは、5'逆方向末端反復(ITR、inverted terminal repeat)、パッケージング信号、E1A及びE1Bのコード配列及びpIXコード配列を含む。パッケージング細胞におけるアデノウイルス配列及びベクター間の重複(overlap)は、これらの配列間の相同組換えを導くことがあり、結果として、複製適格性アデノウイルス(RCA、replication competent adenovirus)の生成をもたらす[Lochmuller(ロッホミュラー)等、1994年]。この問題は株細胞(cell line)及びベクターからなるパッケージングシステムを用いることによって解決されており、それらは、かかる重複配列(overlapping sequences)を欠くことによって互いに適合される[Fallaux(ファラオー)等、1998年; US patent(米国特許)第5,994,128号明細書]。かかる適用において特に有用な株細胞の1種の例は、PER.C6(R)(商標)株細胞である[米国特許第5,994,128号明細書; Nichols(ニカルズ)等、2002年]。
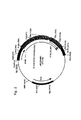
図1はpIG.E1Aのマップである。図2はpIG.ElAB21のマップである。図3はpElBのマップである。図4はpCC.ElAのマップである。図5はpCC.ElAB21のマップである。図6はpCC101のマップである。図7はpCC105のマップである。図8はpCC.55Kcolのマップである。図9はpIG.ElBのマップである。図10はpCC.ElBcolのマップである。図11はpEC.ElBのマップである。図12はpSC.55Kのマップである。
図13は一次細胞(primary cells、初代培養細胞)の形質転換に適切な大分子の設計の例である。
I: スペーサーDNAによって隣接される(flanked)E1A及びE1B蛋白質(A及びB、それぞれの)のためのコード領域を有する単一のコスミドベクターの図式的な提示であり、RE=スペーサーDNA又はE1配列において存在しない制限酵素認識部位であり、
II: スペーサーDNAによって隣接されるいずれかのE1A(A)又はE1B(B)のコード領域を持つ2種の別々の核酸(例えば、プラスミド又はコスミドベクター)の図式的な表示であり、
III: 一次細胞の宿主細胞ゲノムへの、Iからの分子でのか、又はIIの2種の分子での形質転換、及び統合(integration、組込み)後に起こるかもしれない状態である。宿主ゲノムにおいて、挿入物(insert)はスペーサーDNA、及び逆方向にされる配向(inverted orientation)におけるその断片/それらの断片群の第2のコピーによって分けられるE1A領域及びE1B領域で構成される。J: 逆方向反復の間の接合部(junction)。これらの配列の1部分(矢によって示される)が組換えアデノウイルスにおいて組み換えられる場合、ゲノムは包まれるには余りにも大きくなる。ITR: 逆方向末端反復、Φ: パッケージング信号、FI: 組換えウイルス中に挿入される断片で、ここには逆方向反復配列を含む。逆方向反復の存在は、その後に必要なウイルスゲノムからの他の配列を欠失させる工程に役立つかもしれないが、逆方向反復配列の存在は、組み換えられるウイルスの左端の鏡像重複(mirror-imaged duplication)をウイルスゲノムの複製の間にもたらし、及び再びスペーサーDNAのために包まれるには余りにも大きなゲノム(重複される断片、DF)を生じさせる。
図14はpIG.ElA.ElBのマップである。図15はpCC200のマップである。図16はpCC205のマップである。図17はpdys44のマップである。図18はp44-l.ccElAのマップである。
図19は生じる株細胞からの蛋白質溶解物(lysates)のウエスタンブロットである。レーンは、1: HER01-B-71、2: HER01-H-86、3: HER01-H-87、4: HER01-H-88、5: HER01-H-89、6: HER01-B-90、7: HER01 pn06細胞、8: PER.C6細胞である。
図20は形質転換細胞クローン、-: 陰性対照(control); MOF: 蛍光の平均; B-71、B-90、H-86、H-87、H-88、H-89: 生じる形質転換細胞クローンを用いるE1-欠失アデノウイルスベクターの相補性(complementation)の例示である。
図21は、形質移入(トランスフェクション)のために用いるAd5.E1構築物の例3における図式的概観である。ゲノムDNAを消化するため、及び探査子(プローブ)断片を生じさせるために用いる制限部位を示す。E1A及びE1BのDNA断片をElA-含有断片の5'端で番号付けが開始されるヌクレオチドと仮想的に連結(linked)する。
図22は本発明の若干の具体例の図式的な提示である(寸法通りではない)。調節配列(Regulating sequences)(プロモータ及びポリA信号のようなもの)は描いていない。
Aは、アデノウイルスゲノムのE1-領域の自然な配置であり: E1Aに次いでE1Bがあり、それはE1B-19K及びE1B-55Kからなる。スペーサー配列はE1A及びE1Bの間に存在しない。従来技術のパッケージング細胞は、DNAをこの配置において導入することによって構成され、及び従ってまた、E1領域をそれらのゲノムにおけるこの配置において持つ。
Bは、本発明に従う2種の最も単純な可能な配置であり、そこでは、スペーサー[又はスタッファ(詰めもの、stuffer)]がE1領域において存在する。1は、E1A及びElB-19Kの間に導入されるスタッファ、2は、E1B-19K及びE1B-55Kの間に導入されるスタッファである。理論的根拠は、E1A、E1B-19K及びE1B-55Kの間の距離を、自然な状況に比べて増加させ、これらの3種の読み枠の単一アデノウイルス粒子への同時取込みの可能性を低下させるか、又は全体を妨げる(RCA及びHDEPを妨げる)。スタッファは、本発明に従う株細胞を得るには、少なくとも4kbであるべきであり、前記細胞はアデノウイルスのE1A及びE1B-55K及びE1B-19Kのコード配列をそのゲノムにおいて備えており、前記細胞は核酸配列のストレッチ(伸張)を欠き、そこでは、前記E1A及び双方のE1Bのコード配列が4kb未満によって前記ゲノムにおいて分けられることを特徴とする。読み枠の次数(程度、order of)は、それらが、ゲノムにおいて、E1A、E1B-19K及びE1B-55Kの発現があるような方法ですべて存在する限り、問題にならない。したがって、次数は、ElA-ElB/19k-ElB/55k、ElB/19k-ElB/55k-ElA、ElB/55k-ElB/19k-ElA、ElB/55k-ElA-ElB/19k又はElB19k-ElA-ElB55kでよい。ElB/19k及びElB/55kが隣接し、それらが単一の転写単位としてか、又は別々の転写単位として存在してよいとき、これは、本発明にとって重大ではない。双方の配置(1及び2)は本発明にかかるこの間隔基準(spacing criterion)を満たす株細胞を生じさせることができる。少なくとも6、8、10、15又は34.5kbのスタッファ長は、すべての3種の読み枠を持つが、核酸配列のストレッチを欠く本発明に従う細胞を生じさせることができ、そこでは、前記E1A及び双方のE1Bのコード配列が6、8、10、15又は34.5kb未満によってそれらのゲノムにおいて分けられる。このことは、E1A+E1B-19K+E1B-55Kを含有する単一の断片が、互いに、生じる細胞から由来すべきとき、この断片が、少なくとも7kb(スタッファ4kb)、9kb(スタッファ6kb)、等、少なくとも37.5kb(34.5kbのスタッファ)までで、Aにおける状況とは対照的であるべきで、そこでは、これらの配列は、かかる構成物(E1A+E1B-19K+E1B-55Kの読み枠が互いに約3kbを備える)を用いて生じるパッケージング細胞のゲノムの単に3kbに過ぎない断片において見出すことができる。
Cは、Bの原理の拡張であり: Bにおいて示すような断片が互いに隣接して統合し得るので、スタッファがまた、本発明の好適例においてE1Aの前及び/又はE1Bの後に位置される(E1Bの第1の反復によるE1Aの第2の反復への近接近を妨げる)。得られるパッケージング細胞のゲノムを示すが、ここでは、スタッファDNAを備えるE1領域の2種のコピーが、互いに隣接してタンデム(直列、tandem)反復において統合されるときに示される。本発明にかかるこれらのパッケージング細胞は、それらのゲノムE1A及びE1B-19K及びE1B-55Kにおいて統合するが、核酸のストレッチをそれらのゲノムにおいて含んでおらず、そこでは、前記E1A及び双方のE1Bのコード配列は4kb未満、等によって、スタッファ長に依存して分けられる[この例においては、E1A及びE1B-19Kの間(第1番(no. 1))のか、又はE1B-19K及びE1B-55Kの間(第2番(no. 2))の少なくとも4kb、及び少なくともE1Aの上流の2kb及びE1B-55Kの下流の2kb(直接に隣接して統合されるとき互いに4 kb)であるべきである]。
本発明は、アデノウイルスのE1A及びE1Bの配列を備えるパッケージング細胞を生じさせる方法を記載し、そこでは、E1A及び少なくとも1種のE1Bのコード領域が互いから細胞のゲノムにおいて分けられる。この目的のために、少なくとも3種の具体例を提供する。第1の具体例において、E1A及びE1Bを前駆細胞において異なる時点で導入することができ、E1A及びE1Bが同じ染色体位置上に共-統合(co-integrate)される機会を減らす。第2の具体例では、同じ遺伝子座への共-統合の機会は、E1A及びE1Bのコード配列を前駆細胞中に、2種の異なる分子上で、さもなければ、これらの配列間の相同組換えを導くことがある重複配列を欠くものにおいて導入することによって減らす。第3の具体例では、E1A及び少なくとも1種のE1Bのコード領域を1種の核酸分子に基づいて導入し、そこでは、これらの配列がある所定の距離によって分けられる。第3の具体例において、また、単一の核酸分子の代わりに、2種又はそれよりも多い種類の核酸分子を導入することができ、それらは、これらの2種又はそれよりも多い種類の分子が、1種の分子、例えば、連結、組換え、及び同様のものによって形成される場合に、E1A及び少なくとも1種のE1Bのコード領域が所定の距離によって分けられる規準を満たすことは明らかである。得られるパケージング細胞は、E1A及び双方のE1Bのコード領域が互いから距離を隔てて存在し、及び従って、そのようなパッケージング細胞において増殖する組換えアデノウイルスのゲノム中に組み込まれるE1A及び双方のE1Bのコード配列の機会を減少させることで特徴付けられる。
複製適格性アデノウイルス(RCA)は、ヘルパーアデノウイルスについての必要性を伴わない細胞において複製が可能なアデノウイルス粒子である。‘HDEP’(ヘルパー依存性E1-含有粒子)は、本明細書において用いるように、アデノウイルスからの少なくともE1A及びE1B-55Kのコード配列を備えるウイルス粒子として規定され、前記粒子は、機能的なE1A及び/又はE1B-55Kを欠く細胞において‘ヘルパーウイルス’の不存在下に複製されることがない。ヘルパーウイルスは、野生型又は突然変異体のアデノウイルスでよく、しかしまたHDEPにおいて欠ける機能を提供する任意のアデノウイルスベクターであってよく、及び従ってパッケージング細胞上で増殖する(E1欠損の)望ましい生成物の組換えアデノウイルスによって提供することができる。E1A及びE1B 55Kの配列はアデノウイルスの増殖のために要求される。HDEPは通常、更にElB-19Kのコード配列を含む。HDEPにおいて存在するE1配列は、組換えE1-欠損アデノウイルスを補うことができるべきで、それは、HDEPのためのヘルパーウイルスとして働くことができる。HDEPは、パッケージング細胞におけるE1配列及びベクターの間の重複における相同組換えによって生じることがあるが(Murakami等、2002年)、今回、いくつかの調査から予想に反して分かるように、それは、パッケージング細胞における配列及びベクターの間の実質的な相同/重複の不存在下に、即ち、本明細書に言及する非-相同的組換えのようなプロセスによっても生じさせることができる(例えば、Murakami等、HDEPの可能な構造についての2004年を参照)。したがって、本明細書で用いるように、‘実質的な重複’は、相同組換えについて十分な重複として規定される。本発明で‘実質的な重複の不存在’として言及する具体例における任意の場合に、この条件は、重複が、50種のヌクレオチド(nt)以下、好ましくは40nt未満、より一層好ましくは30nt未満、更により一層好ましくは20nt未満、更により一層好ましくは10nt未満で存在し、最も好ましくはまったく重複がない場合に、満たされると考えられる。HDEPの形成の機構は現在、十分には理解されないが、その中で、E1配列がパッケージング細胞のゲノムから由来することは明らかである。E1配列の組換えアデノウイルスバッチにおける存在は望まれておれず、それはRCA又はHDEPの形態である。本発明はHDEPを生じさせる機会を最小にする方法及び手段を提供する。加えて、細胞を調製するための方法及び得られる細胞を提供し、そこでは、細胞をE1-欠損組換えアデノウイルスベクターの相補性のために用いるとき、ベクター及び本発明にかかる補完細胞の染色体性に位置付けられるE1配列の間の実質的な配列重複の不存在下に、組換えアデノウイルス中へのElA及びE1Bのコード配列の一緒の導入、及び従ってHDEPの形成を妨げる染色体配置において、前記細胞はE1A及びE1Bを持つ。この目的のため、本発明の1具体例に従う細胞の染色体配置はE1A及び少なくとも1種のE1Bのコード領域が新しい細胞のゲノムにおいて分かれるようなものである。逆方向反復がアデノウイルス配列の欠失を刺激することによってHDEPの形成に寄与すると思われるので、別の具体例において、本発明は、E1A及びE1Bのコード配列を備える細胞を提供し、そこでは、E1配列が前記パッケージング細胞のゲノムにおいて逆方向反復配置で存在しない。
組換えアデノウイルスは、株細胞から>20kbのDNAを取り込むことができ、その中でそれは増殖し、及び効率的なパッケージングを可能にするアデノウイルスの最大ゲノム長はおよそ38kbである(アデノウイルスのタイプ5系ベクターのものについて;この形態(figure)は血清型に若干依存することがある)。この特性を、本発明にかかる新しい補完細胞を調製するのに用い、前記細胞はE1A及びE1Bのコード配列を備え、及び前記細胞は、前記E1A及び少なくとも1種のE1Bの配列が、少なくとも4kb、好ましくは少なくとも10kb、より一層好ましくは少なくとも34.5kbによって前記ゲノムにおいて分けられることを特徴とする。このことは、かかる細胞のゲノムが、核酸配列のストレッチを欠き、そこでは、前記E1A及び双方のE1Bのコード配列が、4、10又は34.5kbのそれぞれ未満によって分けられることを意味する(例えば、図式的表示としての図22参照)。本発明で述べるように、2種の配列が少なくとも所定の距離によって細胞のゲノムにおいて分けられるのは明らかであり、この要件は、2種の配列が別々の染色体上に存在するときに、同様に満たされるとみなされる。したがって、2種の配列が異なる染色体にある具体例には、本明細書において用いるように、その意味の範囲内での‘少なくともxのkb離れて(apart)’又は‘少なくともxのkbによって分けられる’が明白に包含される。かかる具体例は、この技術の熟練者(当業者)に既知の方法、例えば、インシトゥハイブリダイゼーションにおける蛍光(FISH)によって簡易に調べることができ、それを用いて所定の配列が存在する染色体及び/又は染色体位置を位置付けることができる。
本発明に従う細胞は、アデノウイルスElA及びE1B-55K及びE1B-19Kのコード配列を前駆体のか又は祖先の細胞に導入することによって生じさせることができる(従って細胞はその中へのE1配列の導入によって前駆体か又は祖先の細胞から導かれる)。本発明によれば、前記E1A及び少なくとも1種のE1Bのコード配列は、前記前躯体細胞中に非連鎖の(unlinked)様式において導入されるべきものである。この意味合いで用いるような‘非連鎖’は、アデノウイルスにおいて見出されるように、E1A及びE1Bの自然な配置、即ち互いに直接隣接する配置を称するように意図されるものとは異なる(例えば、図14を参照し、そこでは、自然の-即ち、ElA及びE1Bの‘連結した’-配置がプラスミドのpIG.E1A.E1Bにおいて存在するとして示される)。本発明に先立っては、そのゲノムの中にアデノウイルスE1A及び双方のE1Bのコード配列を備えるパッケージング細胞を得るのを望む当業者が、ElA及び少なくとも1種のE1Bの配列を非連鎖の様式において導入することを熟慮しないことに注目すべきであり、その理由は:a)それがより一層多くの作業、及びそれ故により一層不便であり、及びb)双方のElA及びE1Bの配列が不死化のために求められるからである。以前の調査では、E1A及びElBの蛋白質の形質転換の能力に洞察力を注ぐものは、E1A及び少なくとも1種のE1Bのコード配列の導入によって確立されたクローンが導かれることを例証している(van den Elsen(ファン・デン・エルセン)等、1982年; Gallimore等、1986年; Jochemsen(ヨッヒェンセン)等、1987年; Rao(ラオ)等、1992年; Nevels(ニーベルス)等、2001年)。しかし、これらのプラスミドの同時導入、及び/又は導入配列、例えば、プラスミド配列、プロモータ及び/又はポリアデニル化信号のような調節配列の間の重複、及びE1B-55K及びE1B-19Kの重複配列は、同じ遺伝子座でのゲノムにおけるE1A及び双方のE1Bのコード領域の共-統合をもたらし得る。実際、かかる共-統合はvan den Elsen等によって観察された(1982年)。ある場合に(Jochemsen等、1987年)、仔ラット腎臓(BRK)細胞は、まずE1Aで形質移入され、及び次いで採集したクローンをE1Bプラスミドで形質移入し、E1A発現におけるE1B発現に及ぼす影響を調査した。明らかに、この調査は細胞の特定の種類において、及び本出願に記載の発明に無関係な理由のために行われた。本発明に先立っては、パッケージング細胞を生成する目的のために、E1A及び少なくとも1種のE1Bのコード配列を別々に形質移入する理由は存在しなかった。組換えアデノウイルスのゲノム及びパッケージング細胞の間の実質的な重複の不存在下でのHDEPの形成は、今までには記述されていない。さらに、当業者は、実際、例えば、ヒト細胞のようなBRK-誘導細胞以外を得るためにこの方法を用いることを断念させられ、その理由は、Gallomore等(1986年)により、ヒト細胞が単にE1Aだけの発現によってしか形態学的に形質転換されず、及びほとんどのクローンがインシトゥで、又は分離後直ちに死滅することを記述したからである。唯一の場合、クローンをもたらすAdl2(亜群(subgroup)A)のE1A発現プラスミドが形質移入後114日に確認され、及びクローンが皿(dish)で大いに増殖した(overgrown)後に採集できたに過ぎない。明らかに、当業者はE1A及び少なくとも1種のE1Bの遺伝子を前躯体細胞中に導入することに対して、従来技術に基づく別の時点では動機付けられない。細胞のゲノムにおいて分けられるElA及び少なくとも1種のE1Bのコード配列を持つ細胞を得るためにかかる方法を提供することは、本発明の長所である。一定の局面では、本発明にかかる細胞は、好ましくは、仔ラット腎臓(BRK)細胞でない。1種の好適な局面では、本発明にかかる細胞は、好ましくはヒト細胞である。好適な局面では、細胞はHER細胞から導かれる。細胞は、ここではE1配列を本発明に従って導入するが、前記導入の前には、好ましくは、アデノウイルスのE1A及びE1Bのコード配列をそれらのゲノムにおいて含まず、及び配列の導入によってのみ、それらのゲノムにおけるE1配列が得られ、それによって、不死化してきて、及び欠失をE1領域において有する組換えアデノウイルスをパッケージングできることは明らかである。例えば、国際公開第WO03/031633号パンフレットは、Adl1 E1B-55kの293細胞への導入を記述し:これは明らかに本発明の利点をもたらさず、その理由は、293において既に存在するようなE1配列が元の配置において残り、及びE1AのORFから4kb未満離れてE1BのORFを持つからである。対照的に、本発明は、アデノウイルスのE1A及びE1B-19K及びE1B-55Kのコード配列を細胞のゲノムにおいて持つ細胞を生じさせる方法、並びにこれらの方法から得られる細胞を提供し、それによって、E1A及び少なくとも1種のE1Bのコード配列を備える核酸配列が、互いに十分に分けられ、細胞のゲノムからのすべてのE1A及びE1Bの配列が単一のアデノウイルス粒子のゲノム中に同時に導入されるのを妨げる。
1種の局面において、本発明はアデノウイルスのE1A蛋白質及び少なくとも1種のアデノウイルスのE1B蛋白質を暗号化する核酸配列を備える組換え分子を提供するが、E1A蛋白質を暗号化する前記核酸配列及び少なくとも1種のE1B蛋白質を暗号化する前記核酸配列が、少なくとも4kb、好ましくは少なくとも10kb、より一層好ましくは少なくとも34.5kbによって分けられることで特徴付けられる。かかる分子は、本発明に従う方法において用い、本発明に従う細胞を生じさせることができる。かかる分子は、プラスミド、コスミド、細菌性人工染色体、YAC、及び同様のもののような、当業者に既知の種々の形態を採ることができる。
HDEPを、パッケージング細胞と組換えアデノウイルスとの間の実質的な重複の不存在下に生じさせる機構が何であろうと、第1の工程は、この処理において、パッケージング細胞のゲノムからのE1配列のウイルスゲノムへの統合の事象であり得る。これらの余分な配列について適応させるためには、ウイルスはその後にアデノウイルス配列を削除しなければならない(Murakami等、2002年)。この工程は、逆方向反復が存在するときにより一層効率的となり得る(Steinwaerder(シュタインブエルデ)等、1999年)。PER.C6(R)株細胞は、そのゲノム中にpIG.E1A.E1Bプラスミドのいくらかの反復を含み、それが株細胞の生成のために用いられ、そのいくつかで、反復が互いに関して逆方向配向にある。したがって、E1領域の逆方向反復のPER.C6(R)株細胞のゲノムにおける存在は、HDEPの発生の頻度に影響を及ぼすことがある。HDEP粒子の形成がまた、他のアデノウイルスのパッケージング株細胞において起こるが、かかる株細胞において、古典的なRCAの出現のために検出されなくなることは注目すべきである。E1配列のパッケージング株細胞での逆方向反復配向における不存在は、組換えアデノウイルスがかかるパッケージング細胞上で増殖するとき、より一層低い頻度でのか、又は更に完全に不存在なHDEPの生成をもたらし得る。
別の局面において、本発明は、本発明に従う細胞において組換えアデノウイルスを生成するための方法を提供する。これは、この技術において既知のシステム上で生産されるバッチと比べて、著しく減少させた頻度のHDEPを持つ、好ましくはまったくHDEPのない、組換えアデノウイルスのバッチを発生させる。特に好適な具体例では、前記組換えアデノウイルスの生成のために用いるベクター及びパッケージング細胞は、実質的な配列重複を欠き(即ち、好ましくは、10nt未満、又はより一層好ましくは、まったく重複がない)、及び好ましくは重複を持たず、それによって、パッケージング細胞におけるベクター及び配列の間の相同組換えの機会を最小にし、E1-含有粒子(HDEP、RCA)を持たないウイルスバッチをもたらす。
本発明の実行では、他に示す場合を除き、通常の技術の分子生物学、細胞生物学、及び組換えDNAを採用するが、それらはこの技術の熟練の範囲内である。例えば、Sambrook(サムブルック)、Fritsch(フリッチェ)及びManiatis(マニアティス)の、Molecular Cloning(分子クローニング):A Laboratory Manual(実験室マニュアル)、第2版(2nd edition)、1989年; Current Protocols in Molecular Biology(分子生物学における最新プロトコル)、Ausubel(オースベル) FM等、eds(編)、1987年;the series Methods in Enzymology(酵素学における方法論のシリーズ)(Academic Press, Inc.(アカデミック・プレス社); PCR2:A Practical Approach(実用的な手法)、MacPherson(マクファーソン) MJ、Hams(ハムス)BD、Taylor(ティラー)GR、編、1995年参照。
アデノウイルスのE1遺伝子による一次細胞の完全な形態学的な形質転換は、ElA及びE1Bの領域によって暗号化される蛋白質の組み合わされた活性の成果である。溶菌感染において、及び形質転換において、異なるE1蛋白質の役割が広く調査されている(Zantema(ザンテマ)及びvan der Eb(ファン・デル・エバ)、1995年;White(ホワイト)、1995年、1996年において評論されている)。アデノウイルスE1A蛋白質は一次細胞の形質転換のために必須である。アポトーシスの随伴性誘導(concomitant induction)は、双方のE1B-19K及びE1B-55Kによってであるが、異なる機構によって妨げられる。ElA領域はいくつかの蛋白質を暗号化するが、全体の領域は通常E1Aのコード領域と称され、及びすべての具体例において、本明細書で用いるように、‘E1Aのコード配列’は、すべてのE1A蛋白質を暗号化する配列に言及する。
構築物pIG.E1Aを、pIG.E1A.E1BのHincIIを用いる消化によって作製し、次いで、得られる5kbの断片を、ゲルからQIAEX(キアエックス) IIのゲル抽出キット(Qiagen(キアゲン社))を製造者の指示に従って用いて精製した。単離した断片の再連結及びSTBL2適格性細胞への形質転換(Invitrogen(インビトロゲン))は、pIG.E1Aを与えた(図1)。この構築物はAd5ゲノムからのnt. 459から1578までを含む(Genbank Acc. No. M73260)。
構築物pIG.ElAをXbaI及びHpaIを用いて消化し、及び得られる4.8kbの断片を、ゲルから上述のように単離した。構築物のpIG.ElA.ElBを、BsrGIを用いて消化し、Klenow(クレノウ)酵素(New England Biolabs(ニューイングランド・バイオラブズ社))を用いて処置し、5'の突出末端を平滑端に(鈍く、blunt)した。次いで、DNAを、QIAquick(キアクイック) PCRの精製キット(Qiagen)を製造者の指示に従って用いて精製し、及びその後XbaIを用いて消化した。得られる913bpの断片は、E1A及びE1B19Kのコード配列の3'部分を含み、ゲルから、記載したように単離した。2種の単離断片の連結及びDH5α-Tlr細胞(Invitrogen)への形質転換は、構築物pIG.E1AB21を与えた(図2)。この構築物は、このようにして、Ad5ゲノムからのnt. 459から2253までを含み(Genbank Acc. No. M73260)、それによって、E1A配列はPGKプロモータによって導かれ、及びE1BプロモータはElB-19K遺伝子を駆動する。
Ad5-ElB領域を含む構築物を、次いで、次のようにして生じさせた。最初に、PCR断片を、プライマの5ElBfor-1: 5'-CGG AAT TCG GCG TGT TAA ATG GGG CG-3' (SEQ.ID.NO. 2)、及び5ElB-rev: 5'-TAG CAG GCG ATT CTT GTG TC-3' (SEQ.ID.NO. 3)を用い、pIG.E1A.E1BのDNAを鋳型として、及びPwo DNAポリメラーゼ(Roche(ロシュ社))を製造者の指示に従ってDMSOの3%の終濃度と共に用いて生じさせた。増幅プログラムは、94℃で2分間であり、30回のサイクルが続き(94℃で30秒間、50℃で30秒間及び72℃で1分間)及び72℃で10分間によって終わった。得られる481bpの増幅された断片を、QIAquick PCR精製キット(Qiagen)を用いて精製し、及びPCRのクローニングキット(Stratagene(ストラタジーン社))からのpCR-ScriptAmpのベクターにSrfI酵素の存在下、製造者の指示に従って連結させた。(平滑端)連結は、挿入物の2種の配向をベクターにおいて与え、その1種は、挿入物の(5')端におけるEcoRI部位及びベクターにおけるEcoRI部位の間の最も大きな断片を有し、任意に選ばれた。このことで、構成物pCR5Bをもたらした。標的配列の正しい増幅が(KpnI及びEcoRIの部位間で)、配列決定によって確認された。
pCR5BをKpnI及びEcoRIで消化し、及び415bpの断片を、QIAquickゲル抽出キット(Qiagen)を用いて、ゲルから単離した。構築物pIG.E1A.E1Bを次いで、EcoRI及びKpnIで消化し、及び得られる5.2kbのベクター断片をゲルから上述のように単離した。単離した断片の連結及びDH5α-Tlr細胞(Invitrogen)への形質転換は、構築物pElBをもたらした(図3)。この構築物におけるE1B配列はAd5ゲノムからのnt.1642から3510までを構成する。
ElA配列を、次のプライマを用いて増幅した: 5ElA-For: 5'-CCG AAT TCG ATC GTG TAG TG-3'(SEQ.ID.NO. 4)及び5ElA-rev: 5'-CGG GAT CCA TTT AAC ACG CCA TGC AAG-3'(SEQ.ID.NO. 5)。反応を、pIG.ElA.E1Bの鋳型DNA上で、Pwo DNAポリメラーゼ(Roche)を製造者の指示に従うが、3%DMSOの終濃度と共に用いて行った。PCRプログラムは、94℃で2分間、次いで30回のサイクルのもの(94℃で30秒間、58℃で30秒間及び72℃で120秒間)及び72℃で8分間によって終わらせる設定にした。得られる1.2kbの断片は、EcoRI(5')及びBamHI(3')の部位によって隣接される、Ad5からのE1A配列(Genbank Acc. No. M73260におけるようなヌクレオチド459から1655まで)を含む。
pCC100(以下参照)をXbaIで消化し、及び得られる線状断片を、上述のようにQIAEX IIゲル抽出キットを用いてゲルから精製した。リンカは、オリゴヌクレオチドX-SM-1: 5'-CTAGGTCGACCAATTG-3'(SEQ.ID.NO. 7)と共にX-SM-2: 5'-CTAGCAATTGGTCGAC-3'(SEQ.ID.NO. 8)をアニーリングすることによって調製した。これに、1μgの各オリゴヌクレオチドを、2μLの10×NEB2の緩衝液(NEB)及び20μLの終容量までのmilliQ(ミリキュウ)H2Oと混合した。混合物を98℃で位置させ、及びPCR機械において4℃にまで緩徐に冷却した(2℃/分の冷却速度)。次いで、アニールしたリンカを、単離したXbaI消化断片に、4×モル過剰のリンカを断片に対して使用して連結した。連結DNAを、QIAquick PCR精製キット(Qiagen)を製造者の指示に従って用いて精製し、及びXbaIで消化して自己-連結(self-ligated)ベクターDNAを除去した。XbaI酵素の熱-不活化の後、次いで、混合物を、pCC101(図6)をもたらすDH5α-Tlrの適格性細胞を形質転換するのに用いた。
構築物pCC271(WO02/40665号に記述される)を、EcoRI及びPstIで消化し、及び3kbのベクター断片を上述のようにゲルから単離した。リンカを、オリゴヌクレオチドEcoPst-3: 5'-AAT TGA TAT CGA ATT CGC CGA GCT CGT AAG CTT GGA TCC CTG CA-3'(SEQ.ID. 9)と共にオリゴヌクレオチドEcoPst-4: 5'-GGG ATC CAA GCT TAC GAG CTC GGC GAA TTC GAT ATC-3'(SEQ.ID.NO. 10)のアニーリングによって調製した。ここでは、オリゴヌクレオチドを、上述のように混合し、及び98℃で2分間、65℃で30分間及び室温で2時間のインキュベーションによってアニールした。次いで、単離したベクター断片を、過剰にアニールしたオリゴに連結し、及び構築物pCC100をもたらすDH5α-Tlrの適格性細胞中に形質転換した。
プラスミドpBR322(GenBank J01749.1)をEcoRIで消化し、及び次いで、Klenow酵素で平滑端化し、次に、QIAquick PCR精製キット(Qiagen)で精製した。NheIでの第2の消化の後、4150bpのベクター断片を、アガロースゲルからQIAEX IIゲル抽出キット(Qiagen)を用いて単離した。並行して、構築物pCC100を、BsaAIで消化し、Klenow酵素で平滑端化し、及びQIAquick PCR精製キット(Qiagen)で精製し、次に、XbaIでの第2の消化及び350bp断片のアガロースゲルからの単離を行った。次に、断片を、化学的に適格性のSTBL-2細胞(Invitrogen)中に連結し、及び形質転換し、pCC200をもたらした(図15)。
構築物pCC105は、ヒトPGKのプロモータ、及びヒトCOL1A2遺伝子から導かれるポリ-アデニル化の配列を含む。最初に、COL1A2ポリ-アデニル化配列(Natalizio(ナタリジオ)等、2002年)を、組換えTaqポリメラーゼ(Invitrogen)及びプライマCOL1A2F: 5'-CAG CTA GCC TGC AGG AAG TAT GCA GAT TAT TTG-3'(SEQ.ID.NO.11)及びCOLlA2R-sal: 5'-ACA CGT CGA CGG CTG GTAGAG ATG C-3'(SEQ.ID.NO. 12)を用い、著者等によって記載するようにヒトゲノムDNAからPCRによって増幅した。
構築物pCC205を、SalI及びEcoRIでのpCC200の消化によって、次いで、4225ntのベクター断片のアガロースゲルからの、QIAEX IIゲル抽出キット(Qiagen)を用いる精製によって作製した。並行して、310ntのCOL1A2のpolyA(ポリA)を、構築物pCC105から、EcoRI及びSalIでの消化によって、次いで、ゲル電気泳動、及びQIAEX IIゲル抽出キットを用いる断片の精製によって単離した。次いで、2種の断片を、等モル量において連結し、及びDH5α-Tlrに対して形質転換し、pCC205(図16)をもたらした。
ベクターpCC205を、Ad5 ElB-55k蛋白質を発現するプラスミドの構築物のために用いる。これには、PCR生成物を、次のプライマを用いて生じさせる: 55KforE: 5'-GGA ATT CGC CAC CAT GGA GCG AAG AAA CCC ATC TGA-3'(SEQ.ID.NO. 13)及び55KrevB: 5'-gga tcc TCA ATC TGT ATC TTC ATC GCT AGA GCC-3'(SEQ.ID.NO. 14)。PCRを、Pwo DNAポリメラーゼを、製造者のプロトコルに従って3%DMSOの存在下に実行する。増幅を、pIG.ElA.ElB上で行い、及びプログラムを、94℃で2分間、次に30回のサイクルのもの(94℃で30秒間、60℃で30秒間、及び72℃で90秒間)、及び72℃で8分間によって終わるように設定する。得られる1510bpの増幅した断片は、Ad5配列からのE1B-55K配列のnt.の2019から3510までを含む。断片を、QIAquick PCR精製キット(Qiagen)を用いて精製し、EcoRI及びBamHIで消化し、次に、切断端(cleaved end)を除去するためにゲル電気泳動する。次いで、この断片を、アガロースゲルから、GeneCleanIIキット(Bio101)を用いて精製し、及びpCC205に連結し、これをまた、EcoRI及びBamHIで消化し、及びアガロースゲルから、上述のように単離する。連結混合物を、化学的に適格性のSTBL2細胞(Invitrogen)に対して形質転換し、構築物pCC.55Kcol(図8)をもたらす。
PCR断片を、Pwo DNAポリメラーゼ(Roche)を製造者の指示に従って3%DMSOの存在下に用いて生じさせた。次のプライマを増幅反応において用いた: 5ElBstart: 5'-GGA ATT CCT CAT GGA GGC TTG GG-3'(SEQ.ID.NO. 15)及び5ElBrev2: 5'-GTG TCT CAC AAC CGC TCT C-3'(SEQ.ID.NO. 16)。増幅をpIG.E1A.E1B上で行い、及びプログラムを94℃で2分間、次に5回のサイクルのもの(94℃で30秒間、56℃で30秒間及び72℃で60秒間)に設定し、これらのサイクルに次いで別の35回のサイクルを行い(94℃で30秒間、60℃で30秒間及び72℃で60秒間)、及び68℃で8分間によって終わらせた。次いで、390ntのPCRの断片をKpnI及びEcoRIを用いて消化し、347ntの断片をもたらし、それをアガロースゲルからQIAEX IIゲル抽出キット(Qiagen)を用いて単離した。
双方の19K及び55KのAd5 E1Bコード配列を運ぶプラスミドの構築のために、プラスミドpCC.55KcolをEcoRI及びKpnIで消化する。5970ntのベクター断片をゲル電気泳動によって単離し、及びアガロースゲルから上述のようにGeneClean kitII(Bio101)を用いて精製する。次いで、構築物pIG.E1Bをまた、KpnI及びEcoRIで消化し、及び347ntの断片を、ゲルから単離し、及びQIAquickゲル抽出キット(Qiagen)を用いて精製する。この挿入物及び単離されたpCC.55Kcolのベクター断片の連結及びSTBL2細胞への形質転換は、構築物pCC.ElBcolを与える(図10)。この構築物はAd5ゲノム配列からのnt.の1711から3510までを含む。
ヒト伸長因子(Human Elongation Factor)1-αプロモータ(EF1-α)を、プラスミドpEF/myc/nuc(Invitrogen)から、EcoRI及びPmlIでの消化によって単離する。消化後、Klenow酵素で平滑端化し、及び次いで、1183ntのEF1-αプロモータ断片をゲル電気泳動によって単離し、及びGeneCleanIIキット(Bio101)で精製する。並行して、ベクターpCC.ElBcolをBstXIで消化し、次いで、平滑端(the ends blunt)を作製するためにT4 DNAポリメラーゼ処置を行い、及びPCR精製カラム(Qiagen)上で精製する。次いで、第2の消化をEcoRVで実行し、次いで、ゲル電気泳動を行う。次に、5827ntのベクター断片を、GeneCleanIIキット(Biol0l)を用いて精製する。双方のEF1-α断片及びベクター断片を、等モル量において一緒に連結し、及び化学的に適格性のSTBL-2細胞(invitrogen)に対して形質転換し、これは、プラスミドpCC.ElBcolにおけるような同じAd5-ElB配列を含むプラスミドpEC.ElB(図11)をもたらす。
SV40のプロモータを、pEF/myc/nucプラスミドDNA(Invitrogen)から、組換えTaq DNAポリメラーゼ(Invitrogen)及び次のプライマを用いることによって増幅する: SV40.forS: 5'-CAA CTA GTA CAT GTG GAA TGT GTG TGT TCA GTT AGG-3'(SEQ.ID.NO. 17)及びSV40.RevERI: 5'-GGA ATT CAG CTT TTT GCA AAA GCC TAG G-3'(SEQ.ID.NO. 18)。増幅プログラムを、94℃で2分間、次に、5回のサイクルのもの(94℃で30秒間、48℃で30秒間、及び72℃で45秒間)、次いで25回の追加のサイクルのもの(94℃で30秒間、58℃で30秒間、及び72℃で45秒間)に設定し、及び68℃での8分間によって終わらせた。得られる357bpの増幅された断片(SV40配列GenBank Acc. No. J02400からのヌクレオチドの266から-71まで)を、アガロースゲルから、QIAEX IIゲル抽出キット(Qiagen)を用いて単離した。次いで、この断片を、PCR TOPO4bluntのクローニングキット(Invitrogen)を用いて、pCR-TOPO中にクローン化した。配列確認の後、357bpの挿入物を、TOPOベクターから、EcoRI及びSpeIでの消化によって単離し、及びアガロースゲルから、QIAEX IIゲル抽出キット(Qiagen)を用いて単離した。並行して、プラスミドpCC.55Kcol(pBR322-バックボーン(幹)を持つもの)を、AvrII及びEcoRIで消化し、次いでゲル電気泳動する。5508ntのベクター断片をゲルから上述のように単離し、及び等モル量において、消化されたPCR断片に連結する。連結混合物を、化学的に適格性のSTBL-2細胞中に形質転換し、pCC.55Kcolでのような同じAd5 E1B-55K配列を含むプラスミドpSC.55K(図12)をもたらす。
先の例で記載するように、安定して形質転換された細胞クローンを、一次細胞から、E1A及びE1Bについての別々のプラスミドを用いて生じさせることが可能である。しかし、DNA断片を一緒に形質移入するとき、まだ、形質移入された断片が互いに近接近する機会があるが、この機会は著しい配列重複の不存在によって減少する。30kbよりも少ないnon(非)-E1配列を有する、E1A及びElBの発現カセットを用いる、染色体におけるそれらの間への組込みの可能性は、大きな一片(large pieces)の非-(El-)コードDNA配列(所謂、‘スペーサー’断片)を用いる異なるE1発現カセットを隣接させることによって避けることができる。ElA-搬送断片が、発生したパッケージング細胞のゲノムにおいてElB-含有断片に対して隣に統合される場合、大きな側方配列が十分な距離を創成すべきであり、E1A及びE1Bの暗号化配列が、前記パッケージング細胞における組換えアデノウイルスベクターの増殖に際し、同じ組換えベクター分子中に再び組み入れられる機会を排除する。用いることができる大きなスペーサーDNA断片の非制限的な例は、例えば、ヒトのジストロフィン遺伝子又はヒトのApo-E1遺伝子から導かれる大きな配列である。他のスペーシング(間隔空け)分子をまた、非-ヒト供給源からさえも、用いることができる。好ましくは、側方配列は、機能的蛋白質のためのコード領域を含ず、及び従ってイントロン配列から導くことができる。また、E1A及びE1Bの配列は、異なる分子上にある必要はないが、コスミドのような同じ大きな分子上に置かれてもよい。これらのE1A及びE1Bの搬送分子の例を図13に与える。好適例では、E1A及びE1Bのコード領域間の距離は>34.5kbであり、その理由は、そうするとウイルスへの2種の領域への共-挿入(co-insertion)が包まれるには余りにも大きなゲノムをもたらすからである。この状態は、例えば、発現カセットを概略中間部分において持つ、およそ40kbの2種の別々のコスミド断片が用いられる場合に達する。しかし、逆方向反復構造の形態におけるE1がHDEPの生成の頻度に貢献すると仮定すると、そのとき、E1A及びE1Bのカセットがおよそ22 kb(図13Iにおけるように)の全長の単一のコスミド断片上に位置付けられる構築物は、第2のコピーが第1のものに対して隣の逆方向配向における細胞のゲノムにおいて位置付けられることになるときでさえ十分である。次いで、鏡像構造の完全な統合はまた、>38kbの断片を包含する。また、逆方向反復の存在がHDEP形成の頻度を増加させる場合、E1A及びE1Bの2種の完全なコピーを含むDNA断片が≦20kbの長さを持つときでさえ、多数コピー(multiple copies)がダイレクト(直列)反復(配列)として統合される状態は、より一層問題が少ないと考えられる。逆方向反復の不存在下では、組換えウイルスにおいて残るスペース(空間)よりも大きな任意のE1-含有断片(即ち、38kbから組換えウイルスの実際のゲノム長を減じるもの)の組換えウイルスへの統合は、それに、逐次工程として本質的なウイルス配列の部分を削除するように強いて、パッケージング及び従って増殖を確かにする。相同組換えを介して起こる最初のHDEPゲノムの分析(Murakami等、2002年)は、ウイルス配列の欠失が可能であることを示した。
この例は、本発明に従う新しい株細胞を、大きなスタッファDNAによって隣接されるElA発現カセットを持つ第1のDNA断片、及びプラスミドDNAによって隣接されるE1B発現カセットを持つ第2のDNA断片の共-形質移入によって生じさせることを記載する。

Byrd(バード) P, Brown(ブラウン) KW, Gallimore(ガルリモア) PH. 1982年. Malignant transformation of human embryo retinoblasts by cloned adenovirus 12 DNA(クローン化アデノウイルス12 DNAによるヒト胚網膜芽細胞の悪性形質転換). Nature(ネイチァー) 298: 69-71.
Byrd PJ, Grand(グランド) RJA, Gallimore PH. 1988年. Differential transformation of primary human embryo retinal cells by adenovirus E1 regions and combinations of ElA + ras(アデノウイルスE1領域及びE1A+rasの組合せによる一次ヒト胚網膜の示差形質転換). Oncogene(オンコジーン) 2: 477-484.
Fallaux(ファラオー) FJ, Bout(ブート) A, van der Velde(ファン・デル・フェルデ) I, van den Wollenberg(ファン・デル・ウォーレンバーグ) DJM, Hehir(ヘイール) KM, Keegan(キーガン) J, Auger(オーガー) C, Cramer(クラマー) SJ, van Ormondt(ファン・オーモンド) H, van der Eb(ファン・デル・エバ) A, Valerio(ヴァレリオ) D, Hoeben(ホーベン) RC. 1998年. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses(新ヘルパー細胞及び対応初期領域1-欠失アデノウイルスベクターが複製-適格性アデノウイルスの生成を妨げる). Hum Gene Ther(ヒューマン・ジーン・セラピー) 9: 1909-1917.
Farkas(ファーカス) G, Udvardy(オドバルディ) A. 1992年. Sequence of scs and scs' Drosophila DNA fragments with boundary function in the control of gene expression(遺伝子発現の調節における境界機能を有するscs及びscs'のショウジョウバエDNA断片の配列). Nucleic Acids Res(ヌクレイック・アシッド・リサーチ). 20: 2604.
Gallimore, P. H., Byrd, P. J., Whittaker(ホイッタカー), J. L.及びGrand, R. J. A. (1985年). Properties of rat cells transformed by DNA plasmids containing adenovirus type 12 E1 DNA or specific fragments of the E1 region: comparison of transforming frequencies(アデノウイルスタイプ12のE1 DNA又はE1領域の特定断片を含有するDNAプラスミドによって形質転換されるラット細胞の特性: 形質転換頻度の比較). Cancer Res(キャンサー・リサーチ)., 45, p2670-2680.
Gallimore, P. H., Grand, R. J. A.及びByrd, P. J. (1986年). Transformation of human embryo retinoblasts with simian virus 40, adenovirus and ras oncogenes(サルウイルス40、アデノウイルス及びras発癌遺伝子を用いるヒト胚網膜芽細胞の形質転換). AntiCancer Res(アンチキャンサー・リサーチ). 6, p499-508.
Graham(グラハム) FL, Smiley(スマイリー) J, Russell(ラッセル) WC, Nairn(ネアン) R. 1977年. Characteristics of a human cell line transformed by DNA from human adenovirus type 5(ヒトアデノウイルスタイプ5からのDNAによって形質転換されたヒト株細胞の特徴付け). J Gen Virol(ジャーナル・オブ・ジェネラル・バイロロジー) 36: 59-72.
Jochemsen(ヨッヒェンセン) AG, Peltenburg(ペルテンブルク) LTC, te Pas(テ・パス) MFW, de Wit(デ・ウィット) CM, Bos(ボス) JL, van der Eb AJ. 1997年. Activation of adenovirus 5 E1A transcription by region E1B in transformed primary rat cells(形質転換一次ラット細胞における領域E1Bによるアデノウイルス5のE1A転写の活性化). EMBO J(ジャーナル). 6: 3399-3405.
Kwaks(クワクス) TH, Barnett(バーネット) P, Hemrika(ヘムリカ) W, Siersma(シーアスマ) T, Sewalt(シューワルト) RG, Satijn(サティジン) DP, Brons(ブロンス) JF, Van Blokland(ファン・ブロックランド) R, Kwakman(クバクマン) P, Kruckeberg(クルックベルク) AL, Kelder(ケルダー) A, Otte(オテ) AP. 2003年. Identification of anti-repressor elements that confer high and stable protein production in mammalian cells(哺乳類細胞における高くて安定な蛋白質を与える抗-阻止要素の確認). Nature Biotech(ネイチャー・バイオテクノロジー) 21: 553-558 (+誤植(corrigendum) volume 21 number 7(第21巻、第7号), july(7月) 2003年, p. 822).
Kellum(ケラム) R, Schedl(シェーデル) P. 1991年. A position-effect assay for boundaries of higher order chromosomal domains(より一層高い程度の染色体ドメインの境界についての位置-効果アッセイ). Cell(セル) 64: 941-950.
Lochmuller(ロッホミュラー), H., Jani(ジャニ), A., Huard(ユアール), J., Prescott(プレスコット), M., Simoneau(シモノー), M., Massie(マッシー), B., Karpath(カルパト), G.及びAcsadi(アクサディ), G.(1994年) Emergence of early region 1-containing replication-competent adenovirus in stocks of replication-defective adenovirus recombinants (dEl+dE3) during multiple passages in 293 cells(複製-欠損アデノウイルス組換え体(dE1+dE3)の293細胞における多数継代の間の貯蔵物での初期領域1-含有複製-適格性アデノウイルスの出現). Human Gene Therapy, 5, 1485-1491.
Louis(ルイス) N, Evelegh(イブレグ) C, Graham(グラハム) FL. 1997年. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line(ヒトアデノウイルスタイプ5形質転換293株細胞からの細胞性-ウイルス接合部のクローン化及び配列決定). Virol(バイロロジー). 233: 423-429.
Murakami(ムラカミ) P, Pungor(プンゴア) E, Files(ファイルズ) J, Do L(ドエル), van Rijnsoever(ラインソエバー) R, Vogels(フォーゲルス) R, Bout(ブート) A, McCaman(マクケイマン) M. 2002年. A single short stretch of homology between adenoviral vector and packaging cell line can give rise to cytopathic effect-inducing, helper-dependent E1-positive particles(アデノウイルスベクター及びパッケージング株細胞の間の相同性の単一短ストレッチが細胞変性効果-誘導、ヘルパー-依存性E1-陽性粒子を生じさせ得る). Hum Gene Ther 13: 909-920.
Murakami P, Havenga(ハベンガ) M, Fawaz(ファバツ) F, Vogels R, Marzio(マルチオ) G, Pungor E, Files J, Do L, Goudsmit(ハウトスミット) J, McCaman M. 2004年. Common structure of rare replication-deficient E1-positive particles in adenoviral vector batches(アデノウイルスベクターのバッチにおける稀な複製-欠損E1-陽性粒子の共通構造). J Virol(ジャーナル・オブ・バイロロジー) 78: 6200-6208.
Nevels(ニーベルス) M, Tauber(タウバー) B, Spruss(シュプラス) T, Wolf(ウォルフ) H, Dobner(ドプナー) T. 2001年. "Hit-and-Run" transformation by adenovirus oncogenes(アデノウイルスオンコジーンによる“ヒット-アンド-ラン(奇襲攻撃)”形質転換). J Virol. 75: 3089-3094.
Nichols(ニカルズ) WW, Lardenoije(ラルデノイジェ) R, Ledwith(レッドウィズ) BJ, Brouwer(ブラウエル) K, Manam(マナーム) S, Vogels R, Kaslow(カスロウ) D, Zuidgeest(スッドゲスト) D, Bett(ベト) AJ, Chen(チェン) L, van der Kaaden(カーデン) M, Galloway(ギャロウェー) SM, Hill(ヒル) RB, Machotka(マホトカ) SV, Anderson(アンダーソン) CA, Lewis(ルイス) J, Martinez(マルチネス) D, Lebron(ルブロン) J, Russo(ラッソ) C, Valerio D, Bout A. Propagation of adenoviral vectors: use of PER.C6 cells(アデノウイルスベクターの増殖: PER.C6細胞の使用). In Adenoviral vectors for gene therapy(遺伝子治療のためのアデノウイルスベクターにおいて), (Ed.(編集) Curiel(クリエル) D. T.及びDouglas(ダグラス), J. T.). Pub(出版). Academic Press(アカデミック・プレス社), 2002年.
Rao(ラオ), L., Debbas(デバス), M., Sabbatini(サッバティーニ), P., Hockenbery(ホッケンベリー), D., Korsmeyer(コルスマイヤー), S.及びWhite(ホワイト), E. (1992年). The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins(アデノウイルスE1A蛋白質がアポトーシスを誘導し、それがE1B 19-kDa及びBcl-2の蛋白質によって抑制される). Proc. Natl. Acad. Sci. USA(プロシーディングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシーズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ) 89, p7742-7746.
Steinwaerder(シュタインブエルデ) DS, Carlson(カールスン) CA, Lieber(リーバー) A. 1999年. Generation of adenovirus vectors devoid of all viral genes by recombination between inverted repeats(逆方向反復の間の組換えによるすべてのウイルス遺伝子を欠くアデノウイルスベクターの生成). J. Virol. 73: 9303-9313.
Van den Elsen(ファン・デン・エルセン) P, de Pater(デ・パテール) S, Houweling(ハウエリング) A, van der Veer(ファン・デル・ビア) J, van der Eb A. 1982年. The relationship between region Ela and Elb of human adenoviruses in cell transformation(細胞形質転換におけるヒトアデノウイルスの領域E1a及びE1bの間の関係). Gene(ジーン) 18: 175-185.
White, E. (1995年). Regulation of p53-dependent apoptosis by E1A and E1B(E1A及びE1Bによるp53-依存アポトーシスの調節). In: The molecular repertoire of adenoviruses III(アデノウイルスIIIの分子レパートリーにおける). Eds(編集). Doerfler(ドルフラー), W.及びBohm(ボーム), P.. Springer-Verlag(シュプリンガー出版) Berlin(ベルリン) Heidelberg(ハイデルベルク) 1995年, p33-58.
White, E. (1996年). Life, death, and the pursuit of apoptosis(生命、死、及びアポトーシスの探求). Genes Dev(ジーンズ・アンド・デベロップメント). 10(1), p1-15.
Zantema(ザンテマ), A.及びvan der Eb, A. J. (1995年). Modulation of gene expression by adenovirus transformation(アデノウイルス形質転換による遺伝子発現の調節). In: The molecular repertoire of adenoviruses III(アデノウイルスIIIの分子レパートリーにおける). Eds(編集). Doerfler, W. and Bohm, P. Springer-Verlag Berlin Heidelberg 1995年, p1-23.
Claims (5)
- 細胞のゲノム中に統合されるアデノウイルスのE1AおよびE1B−19KおよびE1B−55Kのコード配列をもち、前記アデノウイルス配列が前記細胞において発現されるようにそれらの配列が調節配列の制御下にある細胞を調製する方法であって、前駆体細胞中に、次の
E1AおよびE1B−19KおよびE1B−55Kのコード配列を備え、前記E1Aおよび少なくとも1種のE1Bコード配列が少なくとも8kbによって分けられた単一の核酸分子であるもの、またはE1AおよびE1B−19KおよびE1B−55Kのコード配列を備える2つまたはそれよりも多くの核酸分子で、前記2つまたはそれよりも多くの核酸分子は末端間結合によって単一分子を形成するとき、前記E1Aおよび少なくとも1種のE1Bコード配列が少なくとも8kbによって分けられたもの
を導入するステップを含む、細胞の調製方法。 - 前記E1Aおよび少なくとも1種のE1Bコード配列は少なくとも10kbによって分けられた、請求項1記載の方法。
- 前記E1Aおよび少なくとも1種のE1Bコード配列は少なくとも34.5kbによって分けられた、請求項2記載の方法。
- E1欠損アデノウイルスベクターを、ヘルパー依存性E1含有粒子を生じさせずに補うことが可能な、請求項1−3のいずれか一項記載の方法によって得られた、細胞。
- E1領域において欠失をもつ組換えアデノウイルスのバッチを生成するための方法であって、次のステップ
a)前記組換えアデノウイルスまたはそのゲノムを、前記組換えアデノウイルスの欠失E1配列を補うことが可能なアデノウイルスのE1配列を備えた細胞中に導入すること、
b)前記細胞を培養すること、および前記組換えアデノウイルスを収集すること
を含み、
前記細胞は請求項4記載の細胞であることを特徴とする、方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EPPCT/EP03/50679 | 2003-10-02 | ||
| EP0350679 | 2003-10-02 | ||
| PCT/EP2004/052428 WO2005033320A1 (en) | 2003-10-02 | 2004-10-04 | Packaging cells for recombinant adenovirus |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2007507216A JP2007507216A (ja) | 2007-03-29 |
| JP2007507216A5 JP2007507216A5 (ja) | 2007-11-22 |
| JP4749334B2 true JP4749334B2 (ja) | 2011-08-17 |
Family
ID=34400432
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006530273A Expired - Fee Related JP4749334B2 (ja) | 2003-10-02 | 2004-10-04 | 組換えアデノウイルス用パッケージング細胞 |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US7816104B2 (ja) |
| EP (1) | EP1670925B1 (ja) |
| JP (1) | JP4749334B2 (ja) |
| KR (1) | KR101203817B1 (ja) |
| CN (2) | CN102174536B (ja) |
| AU (1) | AU2004278517B2 (ja) |
| BR (1) | BRPI0414670A (ja) |
| CA (1) | CA2538025C (ja) |
| DK (1) | DK1670925T3 (ja) |
| EA (1) | EA010924B1 (ja) |
| HK (1) | HK1084974A1 (ja) |
| HR (1) | HRP20130616T1 (ja) |
| IL (2) | IL174489A (ja) |
| NZ (1) | NZ545501A (ja) |
| SI (1) | SI1670925T1 (ja) |
| WO (1) | WO2005033320A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6492169B1 (en) | 1999-05-18 | 2002-12-10 | Crucell Holland, B.V. | Complementing cell lines |
| NZ545501A (en) | 2003-10-02 | 2010-12-24 | Crucell Holland Bv | Packaging cells for recombinant adenovirus |
| US20090110695A1 (en) * | 2006-03-27 | 2009-04-30 | Menzo Jans Emko Havenga | Compositions Comprising a Recombinant Adenovirus and an Adjuvant |
| US20100263066A1 (en) * | 2007-04-30 | 2010-10-14 | Medtronic, Inc | Inert dna sequences for efficient viral packaging and methods of use |
| MX2011004292A (es) * | 2008-11-03 | 2011-05-31 | Crucell Holland Bv | Metodo para la produccion de vectores adenovirales. |
| KR101427200B1 (ko) * | 2011-11-24 | 2014-08-07 | 주식회사 바이로메드 | 아데노바이러스 생산 신규 세포주 및 그의 용도 |
| AU2012343981B2 (en) | 2011-11-28 | 2017-09-07 | Janssen Vaccines & Prevention B.V. | Influenza virus vaccines and uses thereof |
| CA2867950C (en) * | 2012-03-22 | 2023-02-21 | Crucell Holland B.V. | Vaccine against rsv |
| WO2018194438A1 (ko) | 2017-04-21 | 2018-10-25 | (주)지뉴인텍 | 비복제 아데노 바이러스 생산 세포주 및 이의 제조방법 |
| CN108342362A (zh) * | 2018-01-31 | 2018-07-31 | 武汉枢密脑科学技术有限公司 | 一种用于扩增重组犬腺病毒cav2的稳定细胞系mdck及其构建方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002040665A2 (en) * | 2000-11-15 | 2002-05-23 | Crucell Holland B.V. | Complementing cell lines |
| WO2003031633A2 (de) * | 2001-10-04 | 2003-04-17 | Probiogen Ag | Adenovirales vektorsystem |
| WO2005010146A2 (en) * | 2003-07-03 | 2005-02-03 | Cell Genesys, Inc. | Adenoviral e1a/e1b complementing cell line |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4889806A (en) * | 1987-04-15 | 1989-12-26 | Washington University | Large DNA cloning system based on yeast artificial chromosomes |
| US5610053A (en) | 1993-04-07 | 1997-03-11 | The United States Of America As Represented By The Department Of Health And Human Services | DNA sequence which acts as a chromatin insulator element to protect expressed genes from cis-acting regulatory sequences in mammalian cells |
| US5834256A (en) * | 1993-06-11 | 1998-11-10 | Cell Genesys, Inc. | Method for production of high titer virus and high efficiency retroviral mediated transduction of mammalian cells |
| US5820868A (en) | 1993-12-09 | 1998-10-13 | Veterinary Infectious Disease Organization | Recombinant protein production in bovine adenovirus expression vector system |
| EP1548118A2 (en) | 1994-06-10 | 2005-06-29 | Genvec, Inc. | Complementary adenoviral vector systems and cell lines |
| JPH10509880A (ja) | 1994-11-28 | 1998-09-29 | ジェネティック セラピー, インコーポレイテッド | 組織特異的複製のためのベクター |
| CA2206683A1 (en) | 1994-12-12 | 1996-06-20 | Genetic Therapy, Inc. | Improved adenoviral vectors and producer cells |
| DE69638058D1 (de) * | 1995-06-15 | 2009-11-26 | Crucell Holland Bv | Verpackungssysteme für humane rekombinante Adenoviren zur Gentherapie |
| SI1533380T1 (sl) | 1999-04-15 | 2010-03-31 | Crucell Holland Bv | Proizvajanje rekombinantnega proteina v človeški celici ki obsega vsaj en protein E adenovirusa |
| US6558948B1 (en) | 1999-11-23 | 2003-05-06 | Stefan Kochanek | Permanent amniocytic cell line, its production and use for the production of gene transfer vectors |
| EP1103610A1 (en) | 1999-11-26 | 2001-05-30 | Introgene B.V. | Production of vaccines from immortalised mammalian cell lines |
| IL159674A0 (en) | 2001-07-04 | 2004-06-20 | Chromagenics Bv | Dna sequences comprising gene transcription regulatory qualities and methods for detecting and using such dna sequences |
| NZ545501A (en) | 2003-10-02 | 2010-12-24 | Crucell Holland Bv | Packaging cells for recombinant adenovirus |
-
2004
- 2004-10-04 NZ NZ545501A patent/NZ545501A/en not_active IP Right Cessation
- 2004-10-04 CN CN2011100542020A patent/CN102174536B/zh not_active Expired - Fee Related
- 2004-10-04 BR BRPI0414670-0A patent/BRPI0414670A/pt not_active Application Discontinuation
- 2004-10-04 SI SI200432052T patent/SI1670925T1/sl unknown
- 2004-10-04 WO PCT/EP2004/052428 patent/WO2005033320A1/en active Application Filing
- 2004-10-04 DK DK04791140.9T patent/DK1670925T3/da active
- 2004-10-04 AU AU2004278517A patent/AU2004278517B2/en not_active Ceased
- 2004-10-04 KR KR1020067006263A patent/KR101203817B1/ko active IP Right Grant
- 2004-10-04 CN CN2004800286959A patent/CN1863918B/zh not_active Expired - Fee Related
- 2004-10-04 EA EA200600693A patent/EA010924B1/ru not_active IP Right Cessation
- 2004-10-04 JP JP2006530273A patent/JP4749334B2/ja not_active Expired - Fee Related
- 2004-10-04 EP EP04791140.9A patent/EP1670925B1/en active Active
- 2004-10-04 CA CA2538025A patent/CA2538025C/en not_active Expired - Fee Related
-
2006
- 2006-03-20 US US11/384,850 patent/US7816104B2/en not_active Expired - Fee Related
- 2006-03-22 IL IL174489A patent/IL174489A/en active IP Right Grant
- 2006-06-22 HK HK06107148.0A patent/HK1084974A1/xx not_active IP Right Cessation
-
2010
- 2010-07-14 US US12/804,179 patent/US8114637B2/en not_active Expired - Fee Related
-
2013
- 2013-07-02 HR HRP20130616TT patent/HRP20130616T1/hr unknown
- 2013-07-18 IL IL227543A patent/IL227543A/en active IP Right Grant
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002040665A2 (en) * | 2000-11-15 | 2002-05-23 | Crucell Holland B.V. | Complementing cell lines |
| WO2003031633A2 (de) * | 2001-10-04 | 2003-04-17 | Probiogen Ag | Adenovirales vektorsystem |
| WO2005010146A2 (en) * | 2003-07-03 | 2005-02-03 | Cell Genesys, Inc. | Adenoviral e1a/e1b complementing cell line |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2004278517A1 (en) | 2005-04-14 |
| US20100311172A1 (en) | 2010-12-09 |
| US7816104B2 (en) | 2010-10-19 |
| JP2007507216A (ja) | 2007-03-29 |
| IL174489A0 (en) | 2006-08-01 |
| EP1670925B1 (en) | 2013-05-01 |
| US20060183232A1 (en) | 2006-08-17 |
| BRPI0414670A (pt) | 2006-11-21 |
| IL174489A (en) | 2013-10-31 |
| DK1670925T3 (da) | 2013-07-08 |
| IL227543A (en) | 2015-08-31 |
| US8114637B2 (en) | 2012-02-14 |
| SI1670925T1 (sl) | 2013-10-30 |
| CN102174536A (zh) | 2011-09-07 |
| KR20060095557A (ko) | 2006-08-31 |
| CN1863918B (zh) | 2011-03-30 |
| WO2005033320A1 (en) | 2005-04-14 |
| AU2004278517B2 (en) | 2010-04-01 |
| CA2538025C (en) | 2012-07-17 |
| NZ545501A (en) | 2010-12-24 |
| CN1863918A (zh) | 2006-11-15 |
| HRP20130616T1 (en) | 2013-08-31 |
| KR101203817B1 (ko) | 2012-11-23 |
| EA200600693A1 (ru) | 2006-10-27 |
| CA2538025A1 (en) | 2005-04-14 |
| EP1670925A1 (en) | 2006-06-21 |
| HK1084974A1 (en) | 2006-08-11 |
| CN102174536B (zh) | 2013-02-13 |
| EA010924B1 (ru) | 2008-12-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8114637B2 (en) | Packaging cells for recombinant adenovirus | |
| US9228205B2 (en) | Complementing cell lines | |
| Hardy et al. | Construction of adenovirus vectors through Cre-lox recombination | |
| US5922576A (en) | Simplified system for generating recombinant adenoviruses | |
| JP4495588B2 (ja) | 安定なアデノウイルスベクターおよびその増殖方法 | |
| AU2003271737B2 (en) | Means and methods for the production of adenovirus vectors | |
| EP1226264B9 (en) | Cell lines and constructs useful in production of e1-deleted adenoviruses in absence of replication competent adenovirus | |
| US20020119942A1 (en) | Packaging systems for human recombinant adenovirus to be used in gene therapy | |
| AU2002218569A1 (en) | Complementing cell lines | |
| Ruzsics et al. | Transposon-Assisted Cloning and Traceless Mutagenesis of Adenoviruses: Development of a Novel Vector Based on SpeciesD | |
| Youil et al. | Comparative analysis of the effects of packaging signal, transgene orientation, promoters, polyadenylation signals, and E3 region on growth properties of first-generation adenoviruses | |
| US8709778B2 (en) | Method of adenoviral vector synthesis | |
| Ng et al. | Adenoviral vector construction I: mammalian systems | |
| Class et al. | Patent application title: COMPLEMENTING CELL LINES Inventors: Ronald Vogels (Linschoten, NL) Menzo Jans Emco Havenga (Alphen A/d Rijn, NL) Majid Mehtall (Plobsheim, FR) | |
| Cultured | Epstein-Barr Virus Hybrid− An Adenovirus |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071001 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20071001 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20071001 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100817 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20101115 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20101122 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20101216 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20101224 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110214 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110510 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110517 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4749334 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140527 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |