好ましい実施の形態では、全てのX1、X2、及びX3基が酸素であり、従って化合物Iは、式P(OR1)(OR2)(OR3)(但し、R1R2及びR3の定義は、以下に記載されたものである。)のホスファイトである。
本発明に従えば、R1、R2、R3は、それぞれ独立して、同一又は異なる有機基(organic radical)である。R1、R2、R3は、それぞれ独立して、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、i−ブチル、s−ブチル、t−ブチル等の好ましくは1〜10個の炭素原子を有するアルカリ基、フェニル、o−トリル、m−トリル、p−トリル、1−ナフチル、2−ナフチル等のアリール基、又は1,1’−ビフェノール、1,1’−ビナフトール等の、好ましくは1〜20個の炭素原子を有するヒドロカルビルである。R1、R2及びR3基は互いに直接的に結合していても良く、すなわち、中心の燐原子を介して結合している場合に限られない。R1、R2及びR3基が互いに直接的に結合していないことが好ましい。
好ましい実施の形態では、R1、R2及びR3基は、フェニル、o−トリル、m−トリル、及びp−トリルから成る群から選ばれる基である。特に好ましい実施の形態では、R1、R2及びR3基の最大2個がフェニル基であるべきである。
他の好ましい実施の形態では、R1、R2及びR3基の最大2個がo−トリル基であるべきである。
使用して良い特に好ましい化合物は、式Ia、
(o−トリル−O−)w(m−トリル−O−)x(p−トリル−O−)y(フェニル−O−)zP (Ia)
(但し、w、x、y、zがそれぞれ自然数であり、及び次の定義が適用される:w+x+y+z=3 and w、z≦2。)の化合物である。
このような化合物Iaは、例えば、(p−トリル−O−)(フェニル−O−)2P、(m−トリル−O−)(フェニル−O−)2P、(o−トリル−O−)(フェニル−O−)2P、(p−トリル−O−)2(フェニル−O−)P、(m−トリル−O−)2(フェニル−O−)P、(o−トリル−O−)2(フェニル−O−)P、(m−トリル−O−)(p−トリル−O−)(フェニル−O−)P、(o−トリル−O−)(p−トリル−O−)(フェニル−O−)P、(o−トリル−O−)(m−トリル−O−)(フェニル−O−)P、(p−トリル−O−)3P、(m−トリル−O−)(p−トリル−O−)2P、(o−トリル−O−)(p−トリル−O−)2P、(m−トリル−O−)2(p−トリル−O−)P、(o−トリル−O−)2(p−トリル−O−)P、(o−トリル−O−)(m−トリル−O−)(p−トリル−O−)P、(m−トリル−O−)3P、(o−トリル−O−)(m−トリル−O−)2P、(o−トリル−O−)2(m−トリル−O−)P、又はこのような化合物の混合物である。
例えば、原油(crude oil)の蒸留加工物中に得られるm−クレゾール及びp−クレゾールを、特にモル比が2:1の割合で含む混合物を、燐トリクロリド等の燐トリハロゲン化物と反応させることによって、(m−トリル−O−)3P、(m−トリル−O−)2(p−トリル−O−)P、(m−トリル−O−)(p−トリル−O−)2P、及び(p−トリル−O−)3Pの混合物が得られる。
他の同様な好ましい実施の形態では、燐配位子は、DE−A19953058に詳細に記載されたホスファイトで、このホスファイトは式Ib
P(O−R1)x(O−R2)y(O−R3)z(O−R4)p (Ib)
(但し、
R1:燐原子を芳香族系に結合させる酸素原子に対してo−位に、C1−C18−アルキル置換基を有する芳香族基、又は燐原子を芳香族系に結合させる酸素原子に対してo−位に、芳香族置換基を有する芳香族基、又は燐原子を芳香族系に結合させる酸素原子に対してo−位に、融解芳香族系(fused aromatic system)を有する芳香族基、
R2:燐原子を芳香族系に結合させる酸素原子に対してm−位に、C1−C18−アルキル置換基を有する芳香族基、又は燐原子を芳香族系に結合させる酸素原子に対してm−位に、芳香族置換基を有する芳香族基、又は燐原子を芳香族系に結合させる酸素原子に対してm−位に、融解芳香族系(fused aromatic system)を有する芳香族基で、芳香族基は、燐原子を芳香族系に結合させる酸素原子に対してo−位に水素原子を帯びている、
R3:燐原子を芳香族系に結合させる酸素原子に対してp−位に、C1−C18−アルキル置換基を有する芳香族基、又は燐原子を芳香族系に結合させる酸素原子に対してp−位に、芳香族置換基を有する芳香族基で、芳香族基は、燐原子を芳香族系に結合させる酸素原子に対してo−位に水素原子を帯びている、
R4:R1、R2及びR3以外の置換基を、燐原子を芳香族系に結合させる酸素原子に対してo−、m−及びp−位に帯びている芳香族基で、芳香族基は、燐原子を芳香族系に結合させる酸素原子に対してo−位に水素原子を帯びている、
x:1又は2
y、z、p:それぞれが独立して0、1又は2(但し、x+y+z=3である。)
である。)のホスファイトである。
式Ibの好ましいホスファイトは、DE−A19953058から得ることができる。R1基は、o−トリル、o−エチルフェニル、o−n−プロピルフェニル、o−イソプロピルフェニル、o−n−ブチルフェニル、o−sec−ブチルルフェニル、o−tert−ブチルルフェニル、(o−フェニル)フェニル又は1−ナフチル基が有利である。
好ましいR2基は、m−トリル、m−エチルフェニル、m−n−プロピルフェニル、m−イソプロピルフェニル、m−n−ブチルフェニル、m−sec−ブチルフェニル、m−tert−ブチルフェニル、(m−フェニル)フェニル又は2−ナフチル基である。
有利なR3基は、p−トリル、p−エチルフェニル、p−n−プロピルフェニル、p−イソプロピルフェニル、p−n−ブチルフェニル、p−sec−ブチルフェニル、p−tert−ブチルフェニル又は(p−フェニル)フェニル基である。
R4基は、フェニルが好ましい。pはゼロが好ましい。化合物Ib中の指数x、y、z及びpに関し、以下のものが可能である。
式Ibの好ましいホスファイトは、pがゼロであり、及びR1、R2及びR3がそれぞれ独立して、o−イソプロピルフェニル、m−トリル及びp−トリルであり、及びR4がフェニルであるものである。
式Ibの好ましいホスファイトは、R1がo−イソプロピルフェニル基、R2がm−トリル基、及びR3がp−トリル基で、上述した表に記載した指数を有するものであり、また、R1がo−トリル基、R2がm−トリル基、及びR3がp−トリル基で、上述した表に記載した指数を有するものであり、追加的に、R1が1−ナフチル基、R2がm−トリル基、及びR3がp−トリル基で、上述した表に記載した指数を有するものであり、及び、R1がo−トリル基、R2が2−ナフチル基、及びR3がp−トリル基で、上述した表に記載した指数を有するものでもあり、そして、最後に、R1がo−イソプロピルフェニル基、R2が2−ナフチル基、及びR3がp−トリル基で、上述した表に記載した指数を有するものであり、及びこれらホスファイトの混合物でもある。
式Ibのホスファイトは、以下の工程
a)燐トリハロゲン化物とR1OH、R2OH、R3OH及びR4OH又はこれらの混合物からなる群から選ばれるアルコールとを反応させ、ジハロホスホラスモノエステルを得る工程、
b)上記ジハロホスホラスモノエステルとR1OH、R2OH、R3OH及びR4OH又はこれらの混合物からなる群から選ばれるアルコールとを反応させ、モノハロホスホラスジエステルを得る工程、及び
c)上記モノハロホスホラスジエステルとR1OH、R2OH、R3OH及びR4OH又はこれらの混合物からなる群から選ばれるアルコールとを反応させ、式Ibのホスファイトを得る工程、
によって得ても良い。
反応は、別々になった3工程で行なってよい。同様に、3工程の内の2工程を結合しても良い、すなわち、a)とb)、又はb)とc)を結合しても良い。この代わりに、全工程a)、b)及びc)を一緒に結合しても良い。
R1OH、R2OH、R3OH及びR4OH又はこれらの混合物からなる群から選ばれるアルコールの適当なパラメーターと量は、いくつかの簡単な予備実験によって容易に決定され得る。
有用な燐トリハロゲン化物は、原則として全ての燐トリハロゲン化物であり、好ましくは、使用されるハロゲン化物がCl、Br、I、特にCl及びこれらの混合物のものである。種々の同一又は異なる(燐トリハロゲン化物としての)ハロゲン−置換したホスフィンの混合物を使用することも可能である。PCl3が特に好ましい。ホスファイトIbの製造における反応条件と後処理(workup)の更なる詳細は、DE−A19953058から得ることができる。
ホスファイトIbは、配位子として、異なるホスファイトIbの混合物の状態で使用されても良い。このような混合物は、例えば、ホスファイトIbの製造において得ても良い。
しかしながら、多座、特に二座の燐配位子が好ましい。従って、使用される配位子は、好ましくは、式II、
(但し、
X11、X12、X13、X21、X22、X23が、それぞれ独立して、酸素又は単結合であり、
R11、R12が、それぞれ独立して同一又は異なる、分離した又は橋状結合した有機基であり、
R21、R22が、それぞれ独立して同一又は異なる、分離した又は橋状結合した有機基であり、
Yが、橋状結合基である)を有している。
本発明において、化合物IIは、単一化合物又は上述した式の異なる化合物の混合物である。
好ましい実施の形態では、X11、X12、X13、X21、X22、X23は、それぞれ酸素であって良い。このような場合、橋状結合した基Yは、ホスファイト基に結合される。
他の好ましい実施の形態では、X11及びX12は、それぞれ酸素であり、及びX13が単結合であり、又はX11及びX13がそれぞれ酸素であり、及びX12が単結合であり、従って、X11、X12及びX13に囲まれた燐原子が、ホスファイトの中心原子である。このような場合、X21、X22及びX23が、それぞれ酸素、又はX21とX22がそれぞれ酸素でX23が単結合で良く、又はX21とX23がそれぞれ酸素でX22が単結合で良く、又はX23が酸素でX21とX22がそれぞれ単結合で良く、又はX21が酸素でX22とX23がそれぞれ単結合で良く、又はX21、X22及びX23が、それぞれ単結合で良く、従って、X21、X22及びX23に囲まれた燐原子が、ホスファイト、ホスホナイト、ホスフィナイト、又はホスフィン、好ましくはホスホナイトの中心原子である。
他の好ましい実施の形態では、X13が酸素であり、及びX11とX12がそれぞれ単結合で良く、又は、X11が酸素であり、及びX12とX13がそれぞれ単結合で良く、従って、X11、X12及びX13に囲まれた燐原子が、ホスホナイトの中心原子である。このような場合、X21、X22及びX23がそれぞれ酸素で良く、又はX23が酸素であり、及びX21とX22がそれぞれ単結合で良く、又は、X21が酸素であり、及びX22とX23がそれぞれ単結合で良く、又は、X21、X22及びX23がそれぞれ単結合で良く、従って、X21、X22及びX23に囲まれた燐原子が、ホスファイト、ホスフィナイト、又はホスフィン、好ましくはホスフィナイトの中心原子である。
他の好ましい実施の形態では、X11、X12及びX13がそれぞれ単結合で良く、従って、X11、X12及びX13囲まれた燐原子がホスフィンの中心原子である。このような場合、X21、X22及びX23がそれぞれ酸素、又はX21、X22及びX23がそれぞれ単結合で良く、従って、X21、X22及びX23に囲まれた燐原子がホスファイト、又はホスフィン、好ましくはホスフィンの中心原子である。
橋状結合した基Yは、例えば、C1−C4−アリール、フッ素、塩素、臭素等のハロゲン、トリフルオロメチル等のハロゲン化アルキル、フェニル等のアリールによって置換されたアリール基であることが好ましく、又は好ましくは6個〜20個の炭素原子を芳香族系に有する無置換のアリール基、特にピロカテコール、ビス(フェノール)又はビス(ナフトール)が好ましい。
R11及びR12基は、それぞれ独立して、同一又は異なる有機基であって良い。有利なR11及びR12基はアリール基であり、このアリール基は6個〜10個の炭素原子を有していることが好ましく、このアリール基は、無置換又は、特にC1−C4−アリキル、フッ素、塩素、臭素等のハロゲン、トリフルオロメチル等のハロゲン化アルキル、フェニル等のアリール、又は無置換のアリール基によって単一置換、又は多置換されたアリール基であって良い。
R21及びR22基は、それぞれ独立して、同一又は異なる有機基であって良い。有利なR21及びR22基は、好ましくは6個〜10個の炭素原子を有するアリール基であり、アリール基は、無置換又は、特にC1−C4−アリール、フッ素、塩素、臭素等のハロゲン、トリフルオロメチル等のハロゲン化アルキル、フェニル等のアリール、又は無置換のアリール基によって置換されたアリール基である。
R11及びR12基は、それぞれが分離されて良く、又橋状結合して良い。R21及びR22基も、それぞれが分離されて良く、又橋状結合して良い。R11、R12、R21及びR22基は、それぞれが分離されて良く、2種が橋状結合し、そして2種が分離されて良く、又は4種全てが橋状結合して良く、その方法は上述のものである。
特に好ましい実施の形態では、有用な化合物は、US5723641に記載された式I、II、III、IV、及びVのものである。特に好ましい実施の形態では、有用な化合物は、US5512696に記載された式I、II、III、IV、V、VI、及びVIIのものであり、特に、実施例1〜31に使用されている化合物である。特に好ましい実施の形態では、有用な化合物は、US5821378に記載された式I、II、III、IV、V、VI、VII、VIII、IX、X、XI、XII、XIII、XIV及びXVのものであり、特に実施例1〜73に使用されている化合物である。
特に好ましい実施の形態では、有用な化合物は、US5512695に記載された式I、II、III、IV、V及びVIのものであり、特に、実施例1〜6に使用された化合物である。特に好ましい実施の形態では、有用な化合物は、US5981772に記載された式I、II、III、IV、V、VI、VII、VIII、IX、X、XI、XII、XIII及びXIVのものであり、特に実施例1〜66に使用された化合物である。
特に好ましい実施の形態では、有用な化合物は、US6127567に記載された化合物であり、及び実施例1〜29に使用された化合物である。特に好ましい実施の形態では、有用な化合物は、US6020516に記載された式I、II、III、IV、V、VI、VII、VIII、IX及びXのものであり、特に実施例1〜33に記載された化合物である。特に好ましい実施の形態では、有用な化合物は、US5959135に記載された化合物であり、及び実施例1〜13に使用された化合物である。
特に好ましい実施の形態では、有用な化合物は、US5847191に記載された式I、II及びIIIのものである。特に好ましい実施の形態では、有用な化合物は、US5523453に記載されたものであり、特に同文献に式1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20及び21で説明された化合物である。特に好ましい実施の形態では、有用な化合物は、WO01/14392に記載されたものであり、好ましくは、同文献に式V、VI、VII、VIII、IX、X、XI、XII、XIII、XIV、XV、XVI、XVII、XXI、XXII、XXIIIで説明された化合物である。
特に好ましい実施の形態では、有用な化合物は、WO98/27054に記載された化合物である。特に好ましい実施の形態では、有用な化合物は、WO99/13983に記載された化合物である。特に好ましい実施の形態では、有用な化合物は、WO99/64155に記載された化合物である。
特に好ましい実施の形態では、有用な化合物は、ドイツ特許出願DE10038037に記載されたものである。特に好ましい実施の形態では、有用な化合物は、ドイツ特許出願DE10046025に記載されたものである。特に好ましい実施の形態では、有用な化合物は、ドイツ特許出願DE10150285に記載されたものである。
特に好ましい実施の形態では、有用な化合物は、ドイツ特許出願DE10150286に記載されたものである。特に好ましい実施の形態では、有用な化合物は、ドイツ特許出願DE10207165に記載されたものである。本発明の更に特に好ましい実施の形態では、有用な燐キレート配位子は、US2003/0100442A1に記載されたものである。
本発明の更に特に好ましい実施の形態では、有用な燐キレート配位子は、ドイツ特許出願の参照番号DE10350999.2(10.30.2003)に記載されたものであるが、同文献は、先の優先日を有しており、しかし本出願の優先日の時点では発行されていない。
上記化合物I、Ia、Ib及びII及びその製造は本質的に公知である。使用する燐配位子は、化合物I、Ia、Ib及びIIの少なくとも2種を含む混合物でも良い。
本発明に従う方法の特に好ましい実施の形態では、ニッケル(0)錯体の燐配位子及び/又は遊離(free) 燐配位子は、トリトリルホスファイト、二座の燐キレート配位子及び式Ib、
P(O−R1)x(O−R2)y(O−R3)z(O−R4)p (Ib)
(但し、R1、R2、及びR3が、それぞれ独立してo−イソプロピルフェニル、m−トリル及びp−トリルから選ばれ、R4が、フェニルであり;xが1又は2であり、及びy、z、pが、x+y+z+p=3を条件として、それぞれ独立して0、1又は2である)で表されるホスファイト、及びこれらの混合物から選ばれるものである。
本発明に従う方法の工程(a)は、当業者に公知である適切な如何なる装置ででも行なうことができる。反応のための有用な装置は、例えば、Kirk−Othmer,Encyclopedia of Chemical Technology,4th Ed.Vol.20,John Wiley&Sons,New York 1996, 1040〜1055頁に記載されているような通常の装置であり、この装置は、例えば、攪拌タンク反応器、ループ反応器、ガス循環反応器、バブルカラム反応器、又はチューブ反応器等のものであり、各場合において、所望により反応熱を除去する装置を有していても良い。反応は、2台(基)又は3台等の複数の装置で行なっても良い。
本発明に従う方法の好ましい実施の形態では、有利な反応器は、逆混合(backmixing)特性を有するもの、又は逆混合特性を有する反応器群に見出される。逆混合特性を有する有利な反応器群は、シアン化水素の計量に関し、クロス流モード(crossflow mode)で運転されるものであることがわかった。
シアン化水素は、溶媒の存在下又は不存在下に行なわれて良い。溶媒が使用される場合、溶媒は液体であり、そして与えられた反応温度及び反応圧力において、不飽和化合物及び少なくとも1種の触媒に対して不活性であるべきである。通常、使用する溶媒は、炭化水素、例えばベンゼン、又はキシレン、又はニトリル、例えば、アセトニトリル、又はベンゾニトリルである。しかしながら、溶媒として配位子を使用することが好ましい。
反応は、非連続方式(バッチ方式)、連続方式、又は準非連続方式で操作して良い。
シアン化水素処理反応は、装置に全ての反応物を充填して行なって良い。しかしながら、装置に触媒、不飽和有機化合物、及び所望により溶媒が充填されることが好ましい。気体状シアン化水素は、反応混合物の表面に浮くか、又は反応混合物に通されることが好ましい。装置に充填する別の手順は、装置に触媒、シアン化水素、及び所望により、溶媒を充填し、そして反応混合物に徐々に不飽和化合物を計量導入することである。この代わりに、反応物を反応器に導入し、そして反応混合物が反応温度とされ、この温度で、シアン化水素が液体状態で混合物に加えられることも可能である。更に、反応温度に加熱する前にシアン化水素を加えても良い。反応は、温度、雰囲気、反応温度等に関して通常のシアン化水素処理の条件で行なわれる。
シアン化水素処理を、1段階以上の工程で連続して行なうことが好ましい。複数の工程が使用される場合、直列状に連結された工程が好ましい。この場合、製造物は、ある工程から次の工程へと直接的に移される。シアン化水素は、最初の工程に直接的に、又は個々の工程間に供給されて良い。
本発明に従う工程が準非連続操作方式(準バッチ方式)で行なわれる場合、最初に触媒化合物を充填し、そして1,3−ブタジエンを反応器に充填し、この間にシアン化水素が反応時間にわたって、反応混合物に計量導入されることが好ましい。
反応は、好ましくは、0.1〜500MPaの絶対圧、より好ましくは、0.5〜50MPaの絶対圧、特に1〜5MPaの絶対圧で行なわれる。反応は、好ましくは、273K〜473Kの温度、より好ましくは、313K〜423Kの温度、特に333K〜393Kの温度で行なわれる。液体反応相の有利な平均滞留時間は、0.001〜100時間の範囲、好ましくは0.05〜20時間の範囲、より好ましくは0.1〜5時間の範囲に見出され、この時間は、各場合において反応器ごとものものである。
ある実施の形態において、反応は、気体相の存在下に、及び所望により固体懸濁相の存在下に、液体相中で行なわれて良い。出発材料、シアン化水素、及び1,3−ブタジエンは、それぞれ、液体又は気体状態で計量導入されて良い。
更なる実施の形態では、反応は、液体相中で行なわれ、この場合、反応器内の圧力は、1,3−ブタジエン、シアン化水素、及び少なくとも1種の触媒等の全供給原料が、液体状態で計量導入されるものであり、そして、反応混合物内の液相中に存在するものである。固体の懸濁相は、反応混合物中に存在して良く、及び少なくとも1種の触媒と共に計量導入しても良いが、触媒は、例えば特にニッケル(II)を含む触媒組成物の分解生成物から成るものである。
工程(a)において、流れ1が得られるが、流れ1は、3−ペンテンニトリル、2−メチル−3−ブテンニトリル、少なくとも1種の触媒、及び未転化の1,3−ブタジエン、及び未転化のシアン化水素の残留物をも含むものである。この流れ1は、好ましくは、次の組成を有する、すなわち、1〜80質量%、より好ましくは5〜50質量%の、少なくとも1種の触媒、0.1〜50質量%、より好ましくは1〜25質量%の1,3−ブタジエン、1〜80質量%、より好ましくは10〜50質量%のペンテンニトリル(ペンテンニトリルは、トランス−3−ペンテンニトリル、2−メチル−3−ブテンニトリル及び別のペンテンニトリル異性体をも含む)、及び0.1質量ppm〜10質量%、より好ましくは10質量ppm〜1質量%のシアン化水素を有するが、これら濃度は各場合において流れ1の全組成に対してのものである。
3−ペンテンニトリル、2−メチル−3−ブテンニトリル、少なくとも1種の触媒、及び未転化の1,3−ブタジエンを含む流れ1は、次に、工程(b)において蒸留装置K1に移される。この蒸留装置内で、流れ1が蒸留され、塔頂生成物として高濃度1,3−ブタジエン流2が得られ、そして3−ペンテンニトリル、少なくとも1種の触媒及び2−メチル−3−ブテンニトリルを含む塔底生成物として、低濃度1,3−ブタジエン流3が得られる。
本発明に従う方法の工程(b)は、当業者にとって公知である如何なる適切な装置でも行なうことができる。蒸留のために適切な装置は、例えば、Kirk−Othmer,Encyclopetia of Chemical Technology,4th Ed.,Vol.8,John Wiley&Sons,New York,1996,334−348頁に記載されているように、篩トレイカラム、バブルキャップトレイカラム、構造化パッキング又はランダムパッキングを有するカラム等のもの、又は落下フィルム(falling-film)蒸発器、薄厚フィルム蒸発器、フラッシュ蒸発器、多相らせんチューブ蒸発器、自然循環蒸発器、又は強制循環フラッシュ蒸発器等の、単段階の蒸発器である。蒸留は、2台又は3台等の複数の装置で行なって良く、好ましくは単一の装置で行なって良い。
本発明に従う好ましい実施の形態では、蒸留装置内に、構造化パッキング(structured packing)を有するカラムが存在し、そして、カラムは2〜60段の間、より好ましくは3〜40段の間、特に4〜20段の間に分離段(separation stage)を形成するものである。
本発明に従う特に好ましい実施の形態では、工程(b)の蒸留装置と連結した少なくとも1段階の蒸発器段階が、次のように形成される、すなわち、蒸発させるべき材料が、例えば、落下フィルム蒸発器、多相らせんチューブ蒸発器、薄厚フィルム蒸発器又はショートパス蒸発器による、蒸発器表面における材料の短い接触時間、及び蒸発器表面の非常に低い温度によって達成されるように、非常に小さな熱的損失しか受けないように形成される。
本発明に従う方法の好ましい実施の形態では、工程(b)で操作される蒸留装置は、分割されたカラム底部を有し、この蒸発装置の操作において、流れ3より通常数倍大きい循環流が、蒸発装置の蒸留カラムの第1のカラム底部から導かれ、しかし、蒸発器からの液体排出流が第1のカラム底部に直接的に戻されず、この代わりに第2のカラム底部に集められ(ここで、第2のカラム底部は、第1のカラム底部から分離されている)、第2のカラム底部から流れ3が得られ、そして、残っている過剰の蒸発装置循環流が、第1のカラム底部にオーバーフローされ、第2のカラム底部からの流れ3として、混合物(この混合物は、第1のカラム底部から取り出された蒸発器循環流と比較して低沸点物が消耗している)を得る。使用した蒸発器は、落下フィルム(falling-film)蒸発器が好ましい。
本発明に従う、更に好ましい実施の形態では、工程(b)における1台(基)以上の蒸留装置の底部領域内の液相の平均滞留時間が、共に10時間未満、より好ましくは5時間未満、特に1時間未満で、蒸留が行なわれる。
本発明に従う方法の更なる好ましい実施の形態では、蒸留装置の塔頂(頂部)における濃縮が、塔頂流出物の側流(substream)を濃縮器に戻す(flush back)ようにして行なわれる。
本発明に従う方法の更なる好ましい実施の形態では、蒸留が直接濃縮器で行なわれて良く、これにより濃縮がカラム区分で行なわれ、そしてカラム区分には、好ましくは、構造化カラムパッキング、このパッキングの下部に位置する収集カップ(collecting cup)、収集カップからの液体排出部、(液体の取出に付随して、ポンプと熱交換器を有する)ポンプ循環装置、及び循環においてポンプ汲出しされた液体を、収集カップ上のパッキングに供給するための少なくとも1台の装置が設けられている。
工程(b)で使用された蒸留装置K1は、ストリッピング区分を有する蒸留カラムを含み、そして蒸留カラムは、好ましくは2〜60段、好ましくは3〜40段、特に4〜20段の理論段(theoretical plate)を有している。
工程(a)における部分的な転化であるにもかかわらず、1,3−ブタジエンに関して非常に高い工程収率を達成するために、高濃度1,3−ブタジエン流2を工程(a)に再循環させることが好ましい。流れ2の工程(a)への再循環は、所望により部分的なもののみであっても良い。
更なる実施の形態では、工程(b)の蒸留において、工程(a)での反応に更に必要とされる1,3−ブタジエンを、カラムの塔頂領域(頂部領域)又は流れ2に加えて良い。
更なる実施の形態では、加えられた1,3−ブタジエンは、tert−ブチルピロカテコール又は2,6−ジ−tert−ブチル−パラ−クレゾール等の安定剤を含み、これらは「Ullmann’s Encyclopedia Of Lndustrial Chemistry, 6th Edition, 2000 Electronic Release, chapter Butadiene−6. Stabilization,Storage and Transportation」の記載に従うものである。
本発明に従う好ましい実施の形態では、工程(a)で直接使用された1,3−ブタジエン、又は工程(b)で加えられた1,3−ブタジエンの何れか、及び流れ2を介して工程(a)に運ばれた1,3−ブタジエンは、水分が除去されたものであり、及び存在する場合には、安定剤が除去されたものであり、これら除去は、孔径が10オングストローム未満の分子ふるいに接触させることにより行なわれ、又はアルミナに接触させることによって行なわれる。
更なる好ましい実施の形態では、この方法で使用された1,3−ブタジエン、すなわち、工程(a)で直接的に使用された1,3−ブタジエン又は流れ2に供給された1,3−ブタジエンは、安定剤を有していない。そしてこの場合、1,3−ブタジエンの重合を防止するため、特にポップコーンポリマー核(nuclei)の成長を制限するために、圧力条件を適切に選択して、工程(b)の蒸留装置の塔頂領域における濃縮温度が293K未満に維持される。
市販の1,3−ブタジエンは、シス−2−ブテンを相当量で含む。
1−ブテンは、ニッケル(0)触媒を使用した1,3−ブタジエンのシアン化水素処理の副生成物として形成される。
シス−2−ブテン及び1−ブテンは両方とも、本発明に従う方法の1,3−ブタジエンの循環内に、再循環効率の良好性に依存して蓄積される。1,3−ブタジエンが完全に再循環される程、この蓄積は早期に顕著になる。
従って流れ2は、トランス−2−ブテン、シス−2−ブテン及び1−ブテンの合計を、好ましくは50質量%未満、より好ましくは25質量%未満、特に15質量%未満及び好ましくは1質量%以上、より好ましくは2.5質量%以上、特に5質量%以上含むように製造されることが好ましい。
ブテン異性体の蓄積を所望の値に制限する1方法として、工程(a)に再循環される流れ(2)からの側流を排出することが挙げられる。これは、場合によっては1,3−ブタジエンを損失することになる。この損失の理由は、一方では、循環流2内のシス−2−ブテン含有量が非常に高くなるということがなく、しかし他方において、この排出において1,3−ブタジエンが常時排出されることが不可避であるからである。流れ2は、好ましくは、気体状態で取り出される。
ブタジエン循環(サイクル)からシス−2−ブテンを除去する別の方法として、本発明に従い、蒸留装置K1を次のように操作することが例示される。すなわち、流れ1の供給の下方で分離段を作動させ、及び流れ3内で1,3−ブタジエンに対してシス−2−ブテンの蓄積を許容するように蒸留装置K1を操作することである。好ましい実施の形態として以下に説明するように、流れ2から排出する代わりに、工程(c)において、流れ3から生成された流れ4bの状態で排出する方法がある。
排出は気体状態が好ましい。
工程(b)における絶対圧は、好ましくは、0.001〜100バール、より好ましくは0.01〜10バール、特に0.5〜5バールである。蒸留は、蒸留装置の塔底内の温度が、好ましくは30〜140℃、より好ましくは50〜130℃、特に60〜120℃の範囲で行なわれる。蒸留は、蒸留装置の塔頂における濃縮温度が、好ましくは−50〜140℃、より好ましくは−15〜60℃、特に5〜45℃の範囲で行なわれる。本発明に従う方法の特に好ましい実施の形態では、上述した温度範囲が、蒸留装置の塔頂及び塔底の両方で維持される。
蒸留装置の塔頂における還流比は、流れ2が1〜1000ppm、より好ましくは5〜500ppm、特に10〜200ppmの2メチル−3−ブテンニトリルを含むように調整されることが好ましい。
工程(b)において、塔頂生成物として高濃度−1,3−ブタジエン流2が得られ、そして塔底生成物として低濃度−1,3−ブタジエン流3が得られる。高濃度−1,3−ブタジエン又は低濃度−1,3−ブタジエンという流れの呼称は、工程(b)で使用される流れ1の1,3−ブタジエン含有量に基づいている。
本発明に従う好ましい実施の形態では、高濃度−1,3−ブタジエン流2は、合計で、50〜100質量%、より好ましくは80〜100質量%、特に85〜99質量%の1,3−ブタジエンと、ブタジエン異性体とを含み、及び合計で0〜50質量%、より好ましくは0〜20質量%、特に10質量ppm〜1質量%のペンテンニトリル異性体を含み、ここで、実質的には2−メチル−3−ブテンニトリル及びトランス−3−ペンテンニトリルが流れ2内に存在する。
本発明に従う好ましい実施の形態では、低濃度−1,3−ブタジエン流3は、それぞれが流れ3の全組成に対して、合計で、0〜50質量%、より好ましくは1〜30質量%、特に2〜20質量%の1,3−ブタジエンとブタジエン異性体を含む。本発明に従う特に好ましい実施の形態では、上述した1,3−ブタジエンの濃度内容は、流れ2と流れ3の両方で達成される。
工程(b)で得られ、及び1,3−ブタジエンを含む流れ2は、流れ2が工程(a)に再循環される前に、所望により濃縮されることが好ましい。濃縮は、例えば、濃縮器を使用した間接的な熱除去によって行われる。
この代わりに、工程(b)において、蒸留カラムの精留区分で、流れが蒸留装置K1の側部排出部(側部取出部)において沸騰状態で得られ、濃縮器上で間接的な熱除去によって濃縮され、冷却された流れを得、そして蒸留装置K1の塔頂に再循環され、そして流れ2’が濃縮の前又は後に取り出され、そして、流れ2の代わりに流れ2’が工程(a)に再循環される。
安定剤が流れ2’に加えられないことが好ましい。経済的な使用を目的として、結果として得られた流れ2’を工程(a)に再循環して良い。
流れ2’は、その使用の前には、流れ2と同等であるとみなされる。従って、流れ2の説明は、流れ2’にも該当し、その逆も同様である。
工程(b)から生じ、及び3−ペンテンニトリル、少なくとも1種の触媒、及び2−メチル−3−ブテンニトリルを含む低濃度−1,3−ブタジエン流3は、次に工程(c)で蒸留装置に運ばれる。この蒸留装置内で、流れ3が蒸留されて、1,3−ブタジエンを含む流れ4を塔頂生成物として得、3−ペンテンニトリル及び2−メチル−3−ブテンニトリルを含む流れ5をカラムの側部排出部にて得、少なくとも1種の触媒を含む流れ6を、塔底生成物として得る。
本発明に従う工程(c)は、当業者にとって公知である適切な何れの装置を使用しても良い。この蒸留のために適切な装置は、例えば、Kirk−Othmer,Encyclopedia of Chemical Technology, 4th Ed.,Vol.8,John Wiley&Sons,New York,1996,334−348頁に記載されているように、篩トレーカラム、バブルキャップトレーカラム、構造化パッキング又はランダムパッキングを有するカラム、又は落下フィルム蒸発器、薄厚フィルム蒸発器、フラッシュ蒸発器、多相らせん状チューブ蒸発器、自然循環蒸発器、又は強制循環フラッシュ蒸発器等の単段階蒸発器等のものである。蒸留は、2台(基)又は3台等の複数台の装置で行なって良いが、1台の装置で行なうことが好ましい。
特に好ましい実施の形態では、工程(c)で選択された蒸留装置は、ストリッピング区分を有する少なくとも1台の蒸留装置であり、より好ましくは1個のみのストリッピング区分を有する1台のみの蒸留カラムである。
蒸留装置は、構造化パッキングを備えていることが好ましく、構造化パッキングは、2〜50段、より好ましくは3〜40段、特に4〜30段の理論段(理論分離段:theoretical plate)を発生させるものである。
本発明に従う方法の特に好ましい実施の形態では、工程(c)における蒸留装置に結合する、少なくとも一の蒸発器工程が導入され、この導入は、蒸発するべき材料が非常に小さな熱的損失しか受けないように行なわれ、熱的損傷を非常に小さくすることは、例えば落下フィルム蒸発器、多相らせん状チューブ蒸発器、薄厚蒸発器又はショートパス蒸発器による、蒸発器表面における材料の短い接触時間、及び蒸発器表面の非常に低い温度によって達成されるものである。
本発明に従う方法の更なる好ましい実施の形態では、工程(c)における蒸留装置の塔底領域の液相の平均滞留時間が、合わせて10時間未満、より好ましくは5時間未満、特に1時間未満で蒸留が行なわれる。
本発明に従う方法の特に好ましい実施の形態では、工程(b)と(c)における蒸留装置の塔底領域の液相の平均残留時間が、共に10時間未満、より好ましくは5時間未満、特に1時間未満で、蒸留が行なわれる。
工程(c)における絶対圧は、好ましくは0.001〜10バール、より好ましくは0.010〜1バール、特に0.020〜0.5バールである。蒸留は、蒸留装置の塔底の温度が、好ましくは30〜140℃、より好ましくは40〜130℃、特に50〜120℃で行なわれる。蒸留は、蒸留装置の塔頂での濃縮温度が、好ましくは−20〜140℃、より好ましくは−10〜80℃、特に−5〜60℃の範囲で行なわれる。本発明に従う方法の特に好ましい実施の形態では、上述した温度範囲は、蒸留装置の塔頂と塔底で維持される。
工程(c)の蒸留において、塔頂生成物として流れ4が得られる。この流れ4は、好ましくは合計で、50〜100質量%、より好ましくは80〜100質量%、特に90〜99.9質量%の1,3−ブタジエンとブテン異性体を含み、及びまた、合計で0〜50質量%、より好ましくは0〜20質量%、特に10質量ppm〜10質量%のペンテンニトリル異性体を含み、ここで、2−メチル−3−ブテンニトリル、及びトランス−3−ペンテンニトリルが流れ4内に実質的に存在する。
本発明に従う方法の好ましい実施の形態では、流れ4は、蒸留装置の塔頂の少なくとも1台の濃縮器内で気体状態で得られ、そして工程(c)の蒸留装置の蒸気流からのペンテンニトリル成分が、圧力及び温度等が上述した範囲の濃縮条件で、少なくとも1台の濃縮器内で、少なくとも部分的に濃縮され、そしてペンテンニトリル及び1,3−ブタジエン 及びブテン異性体をも含む流れとして、少なくとも部分的に液体の状態でカラム内に再循環される。
本発明に従う方法に使用される1,3−ブタジエンの工程収率を増加させるために、流れ4を、直接的又は間接的に工程(a)に再循環させることが好ましい。流れ4を工程(a)に間接的に再循環させるということは、流れ4が、最初に工程(b)の蒸留装置K1に再循環され、そして次に流れ2を介して工程(a)再循環されることを意味する。
特に好ましい流れ4の間接的な再循環について、蒸留条件に依存して流れ4に存在して良いペンテンニトリル成分が、好ましくは流れ4から除去されるが、この除去は、流れ4を工程(b)の蒸留装置に再循環させることによって行なわれ、そして最終的に流れ4の1,3−ブタジエン、及びブテン異性体の含有分だけが、流れ2を介して工程(a)に再循環される。流れ4の再循環は、条件によっては部分的のみのものであって良い。流れ4が再循環される前に、流れ4がこの方法の目的のために1以上の操作、例えば高圧への加圧に付されても良い。
本発明に従う一実施形態では、流れ4が、工程(b)の蒸留装置K1内で遅延することなく、又は遅延して部分的に再循環され(流れ4a)、そして側流4bが、排出のために、流れ4から液体又は気体状態で取り出される。流れ4は、ブテン異性体を高い割合で含み、そして従って、流れ2よりもブタジエン含有量が少なく、従って、ブタジエンの強制排出量は少なくなり、そして工程収率が高くなり、及びブテン異性体の含有量が上記に有利なものであるとして記載したレベルに維持され得るので、この実施の形態は特に有利である。
再循環された流れ4又は4a中のトランス−2−ブテン、シス−2−ブテン及び1−ブテンの合計での含有量は、好ましくは2質量%以上、より好ましくは10質量%以上、特に15質量%以上、及び好ましくは80質量%未満、より好ましくは70質量%未満、特に50質量%未満である。
流れ4が得られる前に、二トリル含有化合物は、蒸留装置K2の蒸気流の複数段階での濃縮によって量が低減されることが好ましい。
工程(c)における蒸留装置K2で得られた流れ4又は4aは、好ましくは、蒸気の状態で取り出され、そして加圧器V1で加圧され、そして圧力が上昇する。これにより、加圧流4又は4aが得られる。
この加圧流4又は4aは、濃縮によって液体化されることが好ましい。これにより液化流4又は4aが形成される。
このような圧縮流4及び/又は液化流4は、後に工程(b)の蒸留装置K1に再循環される。
特に好ましい実施の形態では、流れ4又は流れ4aは、工程(b)の蒸留装置の分割カラム塔底の還流区分に導入される。
その使用の前には、流れ4aは流れ4と等しいと考えられる。従って、流れ4の記載は、同様に流れ4aにも該当し、その逆もあてはまる。工程(c)では、流れ4に加え、流れ5を含み、流れ5はカラムの側部排出部で得られる。この流れ5は、3−ペンテンニトリル及び2−メチル−3−ブテンニトリルを、他のペンテンニトリル異性体及び1,3−ブタジエン及びブテン異性体の残留成分に加えて含んでいる。流れ5内の3−ペンテンニトリル及び2−メチル−3−ブテンニトリルの割合は、合計で、好ましくは80〜100質量%、より好ましくは85〜99.998質量%、特に90〜99.9質量%(それぞれ、流れ5に対してのものである)である。流れ5内の1,3−ブタジエン及びブテン異性体の割合は、好ましくは、0〜20質量%、より好ましくは10質量ppm〜5質量%、特に50質量ppm〜2質量%(それぞれ、流れ5に対してのものである)である。流れ5は蒸気の状態で取り出されることが好ましい。
蒸留装置の側部排出部は、好ましくは流れ3の供給点の下部に配置され、より好ましくは流れ3の供給点の下部の、1〜20、特に2〜10の蒸留分離段に配置される。
工程(c)で得られた塔底生成物は流れ6であり、流れ6は、少なくとも1種の触媒を含み、及びトランス−3−ペンテンニトリル及び2−メチル−3−ブテンニトリルも含む。流れ6内のペンテンニトリル異性体の割合は、それぞれが流れ6に対して、合計で、好ましくは0.1〜80質量%、より好ましくは5〜50質量%、特に10〜40質量%である。
これに加え、流れ6が部分的にシアン化水素処理の工程(a)に再循環することが特に好ましい。再循環された触媒は、部分的に再生処理することが可能であるが、再生処理は、例えばドイツ特許出願DE…表題:「共沸混合物を乾燥させたニッケル(II)ハロゲン化物の使用方法」(BASF AG(B03/0484))に記載されたものである。
本発明に従う方法の好ましい実施の形態では、この再循環された流れ6内の2−メチル−3−ブテンニトリルの含有量は、10質量%未満、より好ましくは5質量%未満、特に1質量%未満である。これは、流れ5の取出箇所と流れ6の取出箇所との間に十分な蒸留分離段を設けることによって達成される。
好ましい実施の形態では触媒に作用する熱負荷(thermal stress)は、塔底温度を140℃を超えないようにすることにより低く維持することができ、このことは適切な圧力条件によって確保される。
これに加え、工程(c)からの流れ6を、他のシアン化水素処理、例えば3−ペンテンニトリルのシアン化水素処理のための触媒流として、全部又は部分的に使用することができる。触媒流6が3−ペンテンニトリルのシアン化水素処理に使用された場合、この触媒流6内の2−メチル−3−ブテンニトリルの含有量が非常に低く、そして上述した値を超えないことも好ましい。
更なる好ましい実施の形態では、工程(a)への触媒流全体のペンテンニトリル含有量を、上述した範囲内に制御するために、新しい(fresh)触媒流が工程(c)の蒸留装置内に導入される。
本発明に従う方法の更なる好ましい実施の形態では、触媒の排出量と、従って補充するために必要とされる新しい触媒の量が調整されるが、この量は、それぞれ触媒循環に対して、触媒循環中のメチルグルタロニトリルの含有量が、50質量%を超えないように、より好ましくは20質量%を超えないように、特に10質量%を超えないように行なわれ、そしてこの調整は、再生処理に排出される特定の触媒流を、メチルグルタロニトリルの抑制効果を最小限にした状態で、(存在する)ニッケル(0)の排出部に得るためのものである。
本発明に従う方法の更なる好ましい実施の形態では、触媒排出の量と、従って補充するために必要とされる新しい触媒の量が調整されるが、この調整は触媒循環中のニッケル(0)錯体の含有量が、それぞれ触媒循環に対して、及びそれぞれ金属ニッケル(0)として計算して、0.05質量%未満にならないように、より好ましくは0.1質量%未満にならないように、特に0.2質量%未満にならないように行なわれ、そして、この調整は、ニッケル(0)が損失されるにもかかわらず、工程(a)での反応の間、又は工程(b)と(c)での蒸留工程での間、特に工程(a)での反応の間、シアン化水素処理の触媒活性を確実なものにするためである。
本発明に従う方法の更なる好ましい実施の形態では、工程(a)で得られた流れ1を、工程(b)を除外した状態で工程(c)に直接的に移行させることができる。
この後、流れ5は工程(d)で別の蒸留装置に移される。この蒸留装置において、流れ5が蒸留され、2−メチル−3−ブテンニトリルを含む流れ7及び3−ペンテンニトリルを含む流れ8を得る。流れ7は、蒸留装置の塔頂で得られ、流れ8は蒸留装置の塔底(底部)で得られる。
本発明に従う方法の特に好ましい実施の形態では、場合により気体状の側部取出しとして得られる流れ5は、気体状態で工程(d)の蒸留装置に移行され、そして工程(d)の蒸留装置内の流れ5の供給箇所での圧力が、工程(c)の蒸留装置内の流れ5の側部排出部の箇所での圧力よりも小さいか又は等しいものとなる。
工程(d)の圧力が任意に選択され、そして気体流5が、所望により(c)の取出箇所よりも高い圧力に加圧されるか、又は濃縮により液化されるか、及び所望により工程(d)に供給するためにポンプで輸送されるという方法の変形例は本発明の範囲から除外されるものではない。
本発明に従う方法の工程(d)は、当業者に公知である適切な如何なる装置で行って良い。この蒸留に適切な装置は、例えば、Kirk−Othmer,Encyclopedia of Chemical Technology,4th Ed.,Vol.8,John Wiley&Sons,New York,1996,334−348頁に記載されている篩トレイカラム、バブルキャップトレイカラム、構造化パッキング又はランダムパッキングを有するカラム等のもの、又は落下フィルム蒸発器、薄厚フィルム蒸発器、フラッシュ蒸発器、多相らせんチューブ蒸発器、自然循環蒸発器、又は強制循環フラッシュ蒸発器等の単段階の蒸発器である。蒸留は、2台又は3台等の複数の装置で行なって良いが、一台の装置で行うことが好ましい。
カラムは構造化パッキングを含むことが好ましい。構造化パッキングは、5〜100段、より好ましくは10〜80段、特に15〜50段の理論段を発生させることが好ましい。
工程(d)の圧力は、好ましくは0.001〜100バール、より好ましくは0.01〜20バール、特に0.05〜2バールである。蒸留は、蒸留装置の塔底の温度が、好ましくは30〜250℃、より好ましくは50〜200℃、特に60〜180℃で行なわれる。
蒸留は、蒸留装置の塔頂における濃縮温度が、好ましくは−50〜250℃、より好ましくは0〜180℃、特に15〜160℃の範囲で行なわれる。本発明に従う方法の特に好ましい実施の形態では、上述した温度範囲が、蒸留装置の塔頂と塔底の両方で維持される。本発明に従う方法の一実施形態では、工程(d)で得られた流れ7は、DE−A−102004671に従う異性体化に供給される。
本発明に従う方法の一実施形態では、工程(d)で得られた流れ7が工程(a)及び/又は工程(b)に再循環され、そして工程(a)の反応条件又は工程(b)の塔底における液相の滞留時間が、2−メチル−3−ブテンニトリルがトランス−3−ペンテンニトリルに、少なくとも部分的に転化されるように選択される。
本発明に従う、更なる好ましい実施の形態では、流れ7が、工程(d)の蒸留装置内の側部取出流として得られ、そして、この蒸留カラムの得られた塔頂生成物は、2−メチル−3−ブテンニトリルに加え、実質的に(Z)−2−メチル−2−ブテンニトリル及びある場合では、1,3−ブタジエン及びブテン異性体、及びまたビニルシクロヘキセン及びエチリデンシクロヘキセンをも含む流れである。従って流れ7が、塔頂流よりも2−メチル−3−ブテンニトリルを多く含むので、この実施の形態は有利である。
流れ7内のトランス−3−ペンテンニトリルの含有量は、好ましくは0〜50質量%であり、より好ましくは100質量ppm〜20質量%、特に1〜15質量%である。流れ8内の2−メチル−3−ブテンニトリルは、好ましくは0〜10質量%、より好ましくは5ppm〜5質量%、特に50ppm〜1質量%である。
本発明に従う方法は、総合工程(integrated process)において3−ペンテンニトリル及び2−メチル−3−ブテンニトリルの製造を可能とし、この総合工程は、1,3−ブタジエン流及び触媒流の実質的に全範囲に亘って循環が可能であるために、供給原料に対する高い工程収率を有するものである。触媒流からの1,3−ブタジエン及びペンテンニトリル異性体の蒸留による除去に必要な温度と圧力条件は、次のように選択される。すなわち、第1に、本方法が、工業的に実施可能な滞留時間で、工業的規模で行なわれる場合に、塔底蒸発器の温度が低いことであり、そして触媒の損傷を起こさない程に低いことが優先され、そして、第2に、特定の蒸留工程の塔頂生成物の濃縮が、製造規模での熱除去が許容可能な経済的コストで行ない得る温度で優先的に行なわれるということである。
本発明を以下に示す実施例を用いて詳細に説明する。
実施例において、以下の略号が使用される。
BD:1,3−ブタジエン
TBP:tert−ブチルピロカテコール
C2BU:シス−2−ブテン
T3PN:トランス−3−ペンテンニトリル
2M3BN:2−メチル−3−ブテンニトリル
Z2M2BN:(Z)−2−メチル−2−ブテンニトリル
E2M2BN:(E)−2−メチル−2−ブテンニトリル
MGN:メチルグルタロニトリル、及び
ADN:アジポニトリル
HCN:シアン化水素
CAT:触媒
REG:再生
実施例1:
図1を用いて実施例1を説明する。
実施例1では、ニッケル(0)をベースにした、配位子の混合物との触媒組成物がBDのシアン化水素処理(ヒドロシアノ化:hydrocyanation)に使用されている。シアン化水素処理のための配位子混合物は、約60モル%のトリ(m/p−トリル)ホスファイト、及び40モル%のキレートホスホナイト1を含む。
工程(a)では、以下の流れが25lの容量を有するループ反応器R1内に導入され、そしてループ反応器R1は、ノズル、衝動変換チューブ、外部ポンプ作動の循環及び反応熱のエネルギーを除去するためにポンプ作動の循環装置に配置された熱交換器が設けられており、そして流れは357Kに加熱される。
(1)蒸留によって水分が除去された、10kg/hの液体の、安定化されていないシアン化水素、
(2)0.25質量%のC2BUを含む22kg/hの市販のBD(BDは、水とTBP安定化剤を除去するために、アルミナと接触させることにより処理されている)、
(3)工程(b)のカラムK1からの再循環された8kg/hのBD(流れ2)で、従って、反応器R1に供給される得られたBDの全量は、90質量%のBD、5質量%のC2BU、及び5質量%の1−ブテンを含む、30kg/hの流れである。
(4)この実施例において以下に記載したように、カラムK2からの流れ6aとして得られた21kg/hのニッケル(0)触媒溶液。
反応器R1(63kg/h)から取り出された流れ1は、合計で11質量%のBD及びC2BUを含み(79%のBDを転化したことになる)、そしてまた、合計で63質量%のペンテンニトリル、31質量%のT3PN、29質量%の2M3BN、微量のシス−3−ペンテンニトリル、トランス−2−ペンテンニトリル、シス−2−ペンテンニトリル、4−ペンテンニトリル、及び少量のZ2M2BN及びE2M2BN、及びまた触媒成分、及び触媒分解成分及びメチルグルタロニトリルを含む。
工程(b)において、流れ1が蒸留カラムK1に供給されるが、蒸留カラムK1は、精留区分とストリッピング区分とで操作されるもので、そして落下フィルム蒸発器と分割されたカラム塔底が設けられており、そしてカラム内部は10段の理論段(theoretical plate)を発生させる構造化パッキングを有している。カラムK1は、塔頂にて直接濃縮器を使用して操作され、直接濃縮器は、以下の構成のカラム区分からなるものであり、すなわちカラム区分は、構造化パッキングが設けられ、そして総合収集カップ(total collecting cup)、ポンプ作動の循環、及び外部熱交換器を有している。カラムK1は、塔頂圧力が、絶対圧力で2.0バール、塔頂温度が288K、及び塔底取出し温度が363Kで操作される。
初めに述べたように反応器R1への再循環流として計量されたれ2が、カラムK1の塔頂を介して得られる。カラムK1の塔頂での還流割合は、流れ2が約100ppmの2M3BNを含むように調整される。
2.9質量%のBD、4.6質量%のC2BU、67質量%のペンテンニトリル、及び追加的に触媒成分を含む、59kg/hの流れ3がカラムK1の塔底を介して得られる。供給物と比較して、BDに対するC2BUの濃縮が明確である。
工程(c)において、流れ3が蒸留カラムK2に導入されるが、蒸留カラムK2は、ストリッピングモードで操作され、そして落下フィルム蒸発器、ポスト濃縮器を有する塔頂濃縮器を備え、及びカラム内部には、10段の理論段を発生させる構造化パッキングを有している。カラムは、絶対圧で150mバールの塔頂圧力、塔頂温度329K、及び塔底取出し温度373Kで操作される。カラムの蒸気流れは、308Kで部分的に濃縮され、そしてポスト濃縮器を使用して263Kで処理される。従って、2M3BNの量と他のペンテンニトリルの量とが低減された流れ4が、コンプレッサーV1で絶対圧1.2バールで圧縮(加圧)される。圧縮された気体流は、279Kで大部分が濃縮され、流れ4a(5kg/h)が得られ、そして側流4b(約50l(STP)/h、44質量%のC2BUを含む)が気体状態で処理される。流れ4aは、カラムK1の分割カラム塔底部の還流区分に、液体状態で再循環される。
カラムK2の気体状態での側部取出しにおいて、約50ppmのBD、46質量%の2M3BN、及び48質量%のT3PN、及びまた、より少い範囲でのE2M2BN及びZ2M2BNを、他のペンテンニトリル異性体に加えて含む流れ5(40kg/h)が得られる。側部排出部の位置は、次のように選択される。すなわち、蒸留区分の側部排出部の下部において、塔底を介して得られた流れ6内の2M3BN成分が、T3PNと比較して低減されるように選択される。
カラムK2内には、13kg/hの触媒流10が導入され、そして、流れ10は、合計で73質量%のペンテンニトリル、0.5質量%のNi(0)、18質量%の配位子混合物、及び約5質量%のADNを含んでいる。
カラムK2の塔底を介して触媒流6が得られるが、触媒流6は、0.5質量%のNi(0)、約100ppmの2M3BN、及び35質量%の残留ペンテンニトリルを含んでいる。流れ6は部分的(流れ6a)に反応器R1(21kg/h)に再循環される。他の部分(流れ6b:5.4kg/h)は、例えば、DE−A−10351002に記載されている再生処理(REG)に供給されるが、これは、再生処理の後、例えば、DE−A−102004683に従う3−ペンテンニトリルのシアン化水素処理の実施例1に使用するためのものである。
工程(d)において、流れ5が蒸留カラムK3に導入されるが、蒸留カラムK3には、循環蒸発器及び塔頂濃縮器が設けられ、及び30段の理論段を発生させる構造化パッキングも設けられている。カラムK3は、絶対圧で180ミリバールの塔頂圧力、塔頂温度345K、及び塔底取出し温度363Kで操作される。
39kg/hの流れ9がカラムK3に導入されるが、流れ9は、54質量%のT3PN、23質量%の2M3BN及び16質量%のZ2M2BN、及びまた、少量の他のペンテンニトリル異性体を含んでいる。流れ9は、例えばDE−A−102004004671の実施例1に記載されているように、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化のための工程からの再循環ペンテンニトリル流として得て良い。
カラムK3の塔頂を介して、40kg/hの流れ7が得られるが、流れ7は、10質量%のT3PN、68質量%の2M3BN、16質量%のZ2M2BN、及びまた合計で0.1質量%のBDとC2BUを含んでいる。ドイツ特許出願DE…(表題:線状ペンテンニトリルの製造方法、BASF AG(B03/0436)の実施例1に記載されているように、この流れを、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化のための工程に供給して良い。
カラムK3の塔底を介して、39kg/hの流れ8が得られるが、流れ8は合計で、97質量%のT3PN、C3PN、及び4PN、及びまた、約100ppmの2M3BN、及び約1質量%のE2M2BNを含んでいる。
実施例1は、シアン化水素処理工程で、1,3−ブタジエンを実質的に全部回収することが可能な方法を示している。実施例1において、第1にストリッピング区分を有するカラムK1の操作により、第2に蒸発器V1での除去流(purge stream)4bの排出により、ブタジエンサイクル中のシス−2−ブテンの蓄積が達成され、そして、流れ4b(約50l(STP)/h)は、シス−2−ブテンを約40体積%含んでいる。
実施例1に見られる1,3−ブタジエンの損失は、実施例2と比較して少なく、ここで、実施例2は、カラムK1がストリッピング区分なしで操作され、そして蓄積を制限するのに必要な除去流がカラムK1の塔部で流れ2bとして取り出されるものである(330l(STP)/h)(シス−2−ブテンは7質量%だけであり、そして1,3−ブタジエンは92質量%であり、経済的に相当な損失になる)。
実施例2
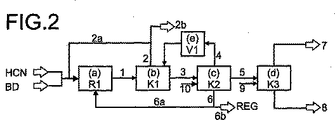
図2を用いて実施例2を説明する。
実施例2において、配位子としてキレートホスホナイト1とのニッケル(0)錯体をベースにした触媒組成物をシアン化水素処理に使用した。
工程(a)において、以下に示す流れがループ反応器R1(容量25l)に導入された(ここで、反応器R1は、ノズル、衝動変換チューブ、外部ポンプ作動の循環、及び反応熱を除去するためにポンプ作動循環装置に配置された熱交換器を備えており、そして357Kに加熱される。)。
(1)蒸留により水を除去した、10kg/hの液体の、安定化していない(unstabilized)シアン化水素、
(2)水分を10ppm未満の濃度にまで除去するために、分子篩に接触させて処理した、0.25質量%のC2BUを含む22kg/hの市販のBD、
(3)工程(b)のカラムK1からの8kg/hの再循環BD(流れ2a)で、従って、反応器R1に供給される、得られたBDの全量は、90質量%のBD、8質量%のC2BU、及び2質量%の1−ブテンを含む、30kg/hの流れである。
(4)この実施例において以下に記載したように、カラムK2からの流れ6aとして得られた21kg/hのニッケル(0)触媒溶液。
反応器R1(63kg/h)から取り出された流れ1は、合計で13質量%のBD及びC2BUを含み(79%のBDを転化したことになる)、そしてまた、合計で63質量%のペンテンニトリル、31質量%のT3PN、29質量%の2M3BN、微量のシス−3−ペンテンニトリル、トランス−2−ペンテンニトリル、シス−2−ペンテンニトリル、4−ペンテンニトリル、及び少量のZ2M2BN及びE2M2BN、及びまた触媒成分、及び触媒分解生成物及びMGNを含む。
工程(b)において、流れ1が蒸留カラムK1に供給されるが、蒸留カラムK1は、精留区分で操作されるもので、そして落下フィルム蒸発器と分割されたカラム塔底部が設けられており、そしてカラム内部は2段の理論段(theoretical plate)を有している。カラムK1は、塔頂にてカラム区分から成る直接濃縮器を使用して操作され、カラム区分には、ランダムパッキングが設けられ、そして総合収集カップ、ポンプ作動の循環、及び外部熱交換器を有している。カラムK1は、塔頂圧力が、絶対圧力で2.0バール、頂部温度が290K、及び塔底取出し温度が363Kで操作される。
カラムK1の塔頂において、濃縮器を用いた循環流から流れ2が得られ、流れ2は初めに述べたように、反応器R1への再循環流2aとして部分的に計量される。カラムK1の塔頂における還流割合は、流れ2が約100ppmの2M3BNを含むように調整される。
カラムK1の塔頂濃縮器から取出された気体状の流れは、排出流2b(約330l(STP)/h)であり、排出流2bは、92質量%のブタジエン、及び7質量%のシス−2−ブテン、及び少量の1−ブテンをも含んでいる。排出流の量は、ブタジエン再循環流2aが、合計で約10質量%の2−ブテン異性体と1−ブテンを含むものである。
カラムK1の塔底を介して、59kg/hの流れ3が得られ、流れ3は4.1質量%のBD、3.9質量%のC2BU、67質量%のペンテンニトリル、及び追加的に触媒成分をも含む。
工程(c)において、流れ3が蒸留カラムK2に導入され、そして蒸留カラムK2は、ストリッピングモードで操作され、及び落下フィルム蒸発器、ポスト濃縮器を有する塔頂濃縮器を備え、そしてカラム内部は、10段の理論段を発生させる構造化パッキングを有するものである。カラムは、絶対圧で150ミリバールの塔頂圧力、塔頂温度354K、及び塔底取出し温度371Kで操作される。
カラムの蒸気流は288Kで部分的に濃縮され、そしてポスト濃縮器を使用して263Kで処理される。従って、気体流4(5kg/h)は2M3BNと他のペンテンニトリルの量が低下し、46質量%のブタジエン、45質量%のシス−2−ブテン、及び約5質量%のペンテンニトリル異性体が、加圧器V1内で2.0バール以上の絶対圧に加圧され、この加圧は、蒸発器の加圧側とカラムK1との圧力差が、加圧気体流を気体状態でカラムK1に戻すのに十分であるように行なわれる。
カラムK2の側部気体排出部において、流れ5(40kg/h)が得られ、流れ5は、約50ppmのBD、46質量%の2M3BN、及び48質量%のT3PN、及びまた少量のE2M2BM及びZ2M2BNを他のペンテンニトリル異性体に加えて含んでいる。側部排出部の位置は、塔底を介して得られた流れ6内の2M3BN成分が、側部排出部の下部のストリッピング区分内で、T3PNと比較して、量が低下(消費:deplete)するように選択される。
カラムK2内には、13kg/hの触媒流10が導入され、触媒流10は、合計で73質量%のペンテンニトリル、0.5質量%のNi(0)、18質量%の配位子混合物、及び約5質量%のADNを含んでいる。
カラムK2の塔底を介して触媒流6(27kg/h)が得られ、触媒流6は1.0質量%のNi(0)、約2000ppmの2M3BN、及び合計で35質量%の残留ペンテンニトリルを含んでいる。流れ6は部分的(流れ6a)に反応器R1に再循環(21kg/h)される。他の部分(流れ6b:5.4kg/h)は、例えばDE−A−10351002に記載された再生処理(REG)に供給されて良い。
工程(d)において、流れ5が蒸留カラムK3に導入されるが、蒸留カラムK3は、循環蒸発器、及び塔頂濃縮器、及び30段の理論段を発生させる構造化パッキングをも備えている。カラムK3は、絶対圧で180ミリバールの塔頂圧力、塔頂温度345K、及び塔底取出し温度363Kで操作される。
39kg/hの流れ9がカラムK3に導入されるが、流れ9は、54質量%のT3PN、23質量%の2M3BN、及び16質量%のZ2M2BN、及び少量の別のペンテンニトリル異性体をも含む。流れ9は、例えば、DE−A−102004004671の実施例1に記載されているように、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化の工程からの再循環ペンテンニトリル流として得て良い。
カラムK3の塔頂を介して40kg/hの流れ7が得られるが、流れ7は10質量%のT3PN、68質量%の2M3BN、16質量%のZ2M2BN、及び合計で0.1質量%のBD及びC2BUをも含む。この流れは、DE−A−102004004671の実施例1に記載されているように、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化に供給されて良い。
カラムK3の底部を介して、39kg/hの流れ8が得られ、ここで流れ8は、合計で97質量%のT3PN、C3PNと4PN、及びまた約100ppmの2M3BN及び約1質量%のE2M2BNを含んでいる。
実施例3において、カラムK1がストリッピング区分を備えた場合には、実施例2に類似した方法で、明らかにREGの損失が少ない流れ2bが許容されなければならい。これは、1,3−ブタジエンの代わりに、実質的にシス−2−ブテンがカラムK2を介してカラムK3に排出されるからである。
実施例3
図2を用いて、実施例3を同様に説明する。
実施例3では、配位子としてキレートホスホナイト1とのニッケル(0)錯体をベースにした触媒組成物がシアン化水素処理に使用されている。
工程(a)において、以下に示す流れがループ反応器R1(容量25l)に導入され、そしてループ反応器はノズル、衝動変換チューブ(impulse exchange tube)、外部ポンプ作動の循環、及び反応熱を除去するためにポンプ作動の循環装置に配置された熱交換器を備えており、そして357Kに加熱される。
(5)蒸留により水を除去した、10kg/hの液体の、安定化していないシアン化水素、
(6)水分を10ppm未満の濃度にまで除去するために、分子篩に接触させて処理した、0.25質量%のC2BUを含む22kg/h市販のBD、
(7)工程(b)のカラムK1からの8kg/hの再循環BD(流れ2a)で、従って、反応器R1に供給される、得られたBDの全量は、90質量%のBD、4質量%のC2BU、及び6質量%の1−ブテンを含む、30kg/hの流れである。
(8)この実施例において以下に記載したように、カラムK2からの流れ6aとして得られた21kg/hのニッケル(0)触媒溶液。
反応器R1(63kg/h)から取り出された流れ1は、合計で13質量%のBD及びC2BUを含み(79%のBDを転化したことになる)、そしてまた、合計で63質量%のペンテンニトリル、31質量%のT3PN、29質量%の2M3BN、微量のシス−3−ペンテンニトリル、トランス−2−ペンテンニトリル、シス−2−ペンテンニトリル、4−ペンテンニトリル、及び少量のZ2M2BN及びE2M2BN、及びまた触媒成分、及び触媒分解生成物及びMGNを含む。
工程(b)において、流れ1が蒸留カラムK1に供給されるが、蒸留カラムK1は、精留及びストリッピング区分で操作されるもので、そして落下フィルム蒸発器と分割されたカラム塔底が設けられており、そしてカラム内部は10段の理論段を発生させる構造化パッキングを有している。カラムK1は、塔頂にてカラム区分から成る直接濃縮器を使用して操作され、カラム区分には、構造化パッキングが設けられ、そして総合収集カップ(total collecting cup)、ポンプ作動の循環、及び外部熱交換器を有している。カラムK1は、塔頂圧力が、絶対圧力で2.0バール、塔頂温度が288K、及び塔底取出し温度が363Kで操作される。
カラムK1の塔頂での濃縮器を用いた循環流から流れ2が得られ、流れ2は初めに述べたように、反応器R1への再循環流2aとして部分的に計量される。カラムK1の塔頂における還流割合は、流れ2が約100ppmの2M3BNを含むように調整される。
カラムK1の塔頂濃縮器から取出された気体状の流れは、排出流2b(約55l(STP)/h)で、排出流2bは、93質量%のブタジエン、及び3質量%のシス−2−ブテン、及び少量の1−ブテンをも含んでいる。排出流の量は、ブタジエン再循環流2aが、合計で約10質量%の2−ブテン異性体及び1−ブテンを含むものである。
カラムK1の底部を介して、59kg/hの流れ3が得られ、流れ3は2.2質量%のBD、6.3質量%のC2BU、67質量%のペンテンニトリル、及び追加的に触媒成分をも含む。
工程(c)において、流れ3が蒸留カラムK2に導入され、そして蒸留カラムK2は、ストリッピングモードで操作され、及び落下フィルム蒸発器、ポスト濃縮器を有する塔頂濃縮器を備え、そしてカラム内部は、10段の理論段を発生させる構造化パッキングを有するものである。カラムは、絶対圧で150ミリバールの塔頂圧力、塔頂温度354K、及び塔底取出し温度371Kで操作される。
カラムの蒸気流は313Kで部分的に濃縮され、そしてポスト濃縮器を使用して263Kで処理される。気体流れ4(5kg/h)は2M3BNと他のペンテンニトリルの量が低下し(消耗され)、23質量%のブタジエン、66質量%のシス−2−ブテン、及び約5質量%のペンテンニトリル異性体が、加圧器V1内で2.0バール以上の絶対圧に加圧され、この加圧は、蒸発器の加圧側とカラムK1との圧力差が、加圧気体流を気体状態でカラムK1に導入して戻すのに十分であるように行なわれる。
カラムK2の側部気体排出部において、流れ5(40kg/h)が得られ、流れ5は、約200ppmのBD、46質量%の2M3BN、及び48質量%のT3PN、及びまた少量のE2M2BM及びZ2M2BNを他のペンテンニトリル異性体に加えて含んでいる。側部排出部の位置は、塔底を介して得られた流れ6内の2M3BN成分が、側部排出部の下方のストリッピング区分内で、T3PNと比較して、量が低下(消耗)するように選択される。
カラムK2内には、13kg/hの触媒流10が導入され、触媒流10は、合計で73質量%のペンテンニトリル、0.5質量%のNi(0)、18質量%の配位子混合物、及び約5質量%のADNを含んでいる。
カラムK2の底部を介して触媒流6が得られ、触媒流6は1.0質量%のNi(0)、約2000ppmの2M3BN、及び合計で35質量%の残留ペンテンニトリルを含んでいる。流れ6は部分的(流れ6a)に反応器R1に再循環(21kg/h)される。他の部分(流れ6b:5.4kg/h)は、例えばDE−A−10351002に記載された再生処理(REG)に供給されて良い。
工程(d)において、流れ5が蒸留カラムK3に導入されるが、蒸留カラムK3は、循環蒸発器、及び塔頂濃縮器、及び30段の理論段を発生させる構造化パッキングをも備えている。カラムK3は、絶対圧で180ミリバールの塔頂圧力、塔頂温度345K、及び塔底取出し温度363Kで操作される。
39kg/hの流れ9がカラムK3に導入されるが、流れ9は、54質量%のT3PN、23質量%の2M3BN、及び16質量%のZ2M2BN、及び少量の別のペンテンニトリル異性体をも含む。流れ9は、例えば、DE−A−102004004671の実施例1に記載されているように、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化の工程からの再循環ペンテンニトリル流として得て良い。
カラムK3の塔頂を介して40kg/hの流れ7が得られるが、流れ7は10質量%のT3PN、68質量%の2M3BN、16質量%のZ2M2BN、及び約0.1質量%のBD及び約1.5質量%のC2BUをも含む。この流れは、DE−A−102004004の実施例1に記載されているように、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化に供給されて良い。
カラムK3の底部を介して、39kg/hの流れ8が得られ、流れ8は、合計で97質量%のT3PN、C3PNと4PN、及びまた約100ppmの2M3BN及び約1質量%のE2M2BNを含んでいる。DE−A−102004004683に従う実施例1の3−ペンテンニトリルのシアン化水素処理に記載されているように、流れ8は、3−ペンテンニトリルのアジポニトリルへのシアン化水素処理のための工程に使用可能である。
比較例
図3を使用して比較例を説明する。
比較例においては、配位子としてホスファイトを有するニッケル(0)錯体をベースにした触媒組成物がシアン化水素処理に使用される。
工程(a)において、以下に示す流れが、それぞれ容量が12lの2台(基)の反応器R1a及びR1bから構成される装置に導入された(ここで、反応器R1a及びR1bは、それぞれノズル、衝動変換チューブ、外部ポンプ作動の循環、及び反応熱を除去するためにポンプ作動循環装置に配置された熱交換器を備えており、そして363Kに加熱される。)。
(1)蒸留により水を除去した、安定化していない6kg/hの液体のシアン化水素(R1aへのもの)、
(2)蒸留により水を除去した、安定化していない6kg/hの液体のシアン化水素(R1bへのもの、
(3)R1aへの25kg/hの市販のBD(0.25質量%のC2BUを含んでいる)で、BDは、水分とTBP安定剤を除去するために、アルミナに接触させて処理されている、
(4)工程(b)のカラムK1からのR1aへの2kg/hの再循環BD(流れ2)で、従って、反応器R1に供給される、得られたBDの全量は、98質量%のBD、及び合計で2質量%のC2BUと1−ブテンを含む、27kg/hの流れである。
(5)この実施例において以下に記載したように、カラムK2からの流れ6aとして得られた14kg/hのニッケル(0)触媒溶液(R1aへのもの)。
反応器R1bから取り出された流れ1(54kg/h)は、合計で4質量%のBD及びC2BUを含み(94%のBDを転化したことになる)、そしてまた、合計で74質量%のペンテンニトリル、33質量%のT3PN、37質量%の2M3BN、微量のシス−3−ペンテンニトリル、トランス−2−ペンテンニトリル、シス−2−ペンテンニトリル、4−ペンテンニトリル、及び少量のZ2M2BN及びE2M2BN、及びまた触媒成分、及び触媒分解生成物及びメチルグルタロニリルを含む。
工程(b)において、流れ1が蒸留カラムK1に供給されるが、蒸留カラムK1は、精留カラムとして操作されるもので、そして落下フィルム蒸発器が設けられており、そしてカラム内部は4段の理論段を発生させる構造化パッキングを有している。カラムK1は、塔頂にてカラム区分から成る直接濃縮器を使用して操作され、カラム区分には、ランダムパッキングが設けられ、そして総合収集カップ、ポンプ作動の循環、及び外部熱交換器を有している。カラムK1は、塔頂圧力が、絶対圧力で0.8バール、塔頂温度が263K、及び塔底取出し温度が393Kで操作される。
カラムK1の塔頂を介して流れ2が得られ、流れ2は初めに述べたように、再循環流として反応器R1aへと計量される。カラムK1の塔頂における還流割合は、流れ2が0.1質量%の2M3BNを含むように調整される。
カラムK1の底部を介して、52kg/hの流れ3が得られ、流れ3は0.3質量%のBD、0.1質量%のC2BU、76質量%のペンテンニトリル、及び追加的に触媒成分をも含む。
工程(c)において、流れ3が蒸留カラムK2に導入され、そして蒸留カラムK2は、ストリッピングモードで操作され、及び落下フィルム蒸発器、ポスト濃縮器を有する塔頂濃縮器を備え、そしてカラム内部は、4段の理論段を発生させる構造化パッキングを有するものである。カラムは、絶対圧で70ミリバールの塔頂圧力、塔頂温度333K、及び塔底取出し温度373Kで操作される。
カラムK2の気体状態での塔頂取出しにおいて、0.4質量%のBD、54質量%の2M3BN、及び42質量%のT3PN、及びまた、より少い範囲でのE2M2BN及びZ2M2BNを、他のペンテンニトリル異性体に加えて含む流れ5(40kg/h)が得られる。カラムK2内には、3kg/hの触媒流4が導入され、触媒流4は、合計で45質量%のペンテンニトリル、1.5質量%のNi(0)及びキレート配位子を含んでおり、Ni(0)及びキレート配位子混合物は、例えば、ニッケル(0)(シクロオクタジエニル)2錯体をキレートホスファイト2と反応させて得られるものである。
カラムK2の塔底を介して触媒流6が得られるが、触媒流6は、1.2質量%のNi(0)、0.3質量%の2M3BN、及び17質量%の残留ペンテンニトリルを含んでいる。流れ6は部分的(流れ6a)に反応器R1(14kg/h)に再循環される。他の部分(流れ6b:3.8kg/h)は、例えば、DE−A−10351002に記載されている再生処理(REG)に供給されるが、これは、再生処理の後、例えば3−ペンテンニトリルのシアン化水素処理に使用して良く、又は所望により本発明に従う方法のブタジエンのシアン化水素処理に再循環されて良い。
工程(d)において、流れ5が蒸留カラムK3に導入されるが、蒸留カラムK3には、循環蒸発器及び塔頂濃縮器が設けられ、及び45段の理論段を発生させる構造化パッキングも設けられている。カラムK3は、絶対圧で1.0バールの塔頂圧力、塔頂温度395K、及び塔底取出し温度416Kで操作される。
24kg/hの流れ9がカラムK3に導入されるが、流れ9は、70質量%のT3PN、14質量%の2M3BN及び7質量%のZ2M2BN、及びまた、少量の他のペンテンニトリル異性体を含んでいる。流れ9は、例えばDE−A−102004004671の実施例2に記載されているように、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化のための工程からの再循環ペンテンニトリル流として得て良い。
カラムK3の頂部を介して、30kg/hの流れ7が得られるが、流れ7は、1質量%のT3PN、85質量%の2M3BN、8質量%のZ2M2BN、及びまた合計で3質量%のBDとC2BUを含んでいる。カラムK3の還流割合は、オーバーヘッドにて1質量%の3−ペンテンニトリルが得られるように調整される。この流れは、例えばDE−A−102004004671の実施例2に記載されているように、2−メチル−3−ブテンニトリルの3−ペンテンニトリルへの異性体化のための工程に供給して良い。
カラムK3を介して、38kg/hの流れ8が得られるが、流れ8は合計で、97質量%のT3PN、C3PN、及び4PN、及びまた、約10ppmの2M3BN、及び約2質量%のE2M2BN、及び少量のメチルグルタロニトリルを含んでいる。流れ8は、DE−A−102004004683の実施例2に記載されているように、3−ペンテンニトリルのアジポニトリルへのシアン化水素処理の工程に供給して良い。
比較例では、1,3−ブタジエンが再循環する蒸留工程K1及びK2における2工程でのブタジエン除去がなく、1,3−ブタジエンが再加圧されることがなく、又はストリッピングカラムとして蒸留工程K1での操作をすることなく、そして、実施例1〜3に示された値に近似した1,3−ブタジエンの損失割合を達成するために、好ましい温度と圧力を使用することが困難になることが明確になっている。従って、カラムK1における1,3−ブタジエン再循環を全面的に行なうために必要な温度(実施例1〜3で90℃であったものが、この代わりに120℃)が、熱的に敏感なキレート配位子及びニッケル錯体(触媒損失に)に導入されることになり、そしてこの導入は、ホスファイト、又はホスホナイトが触媒損失に使用されるか否かとは無関係に行なわれる。1,3−ブタジエンの量を約0.5質量%にまで低下させるために、塔底温度が120℃にて必要な約0.8バールの圧力が、(1,3−ブタジエンを濃縮し、そして1,3−ブタジエンを液体状態で反応器に再循環させるために)塔頂濃縮器において−10℃という非常に低い温度に導入される。比較例での、この温度レベルでの濃縮の熱除去は、例えば、実施例1では熱の除去が冷却水を使用して可能であることと比較して、非常に複雑なものとなる。