JP4444690B2 - 水素化処理触媒前駆体およびその製造方法並びに精製炭化水素油の製造方法 - Google Patents
水素化処理触媒前駆体およびその製造方法並びに精製炭化水素油の製造方法 Download PDFInfo
- Publication number
- JP4444690B2 JP4444690B2 JP2004052227A JP2004052227A JP4444690B2 JP 4444690 B2 JP4444690 B2 JP 4444690B2 JP 2004052227 A JP2004052227 A JP 2004052227A JP 2004052227 A JP2004052227 A JP 2004052227A JP 4444690 B2 JP4444690 B2 JP 4444690B2
- Authority
- JP
- Japan
- Prior art keywords
- hydrotreating catalyst
- catalyst precursor
- oil
- mass
- producing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images



















Landscapes
- Catalysts (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
Description
60質量%以上の複合酸化物成分を含む担体にEDTAを含む含浸液を含浸する工程と;
80〜220℃で乾燥処理する工程と;を含み、
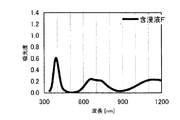
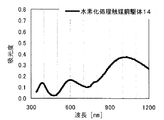
得られた水素化処理触媒前駆体の電子スペクトルが980〜1030nmおよび580〜620nmに吸収極大ピークを有することを特徴とする水素化処理触媒前駆体の製造方法が提供される。本発明において、好ましくは、EDTAを含む含浸液の電子スペクトルが980〜1030nmおよび580〜620nmに吸収極大ピークを有する。この電子スペクトルはNi―EDTA錯体によるものと同定される。本発明では、このNi―EDTA錯体を前駆体に残留させるために80〜220℃で乾燥処理する工程を含むが、高温焼成工程は行わない。また、好ましくは、水素化処理触媒前駆体の細孔直径50〜1000nmの範囲の細孔容積が0.01〜0.5mL/gである。
前記担体が複合酸化物成分を60質量%以上含み、
タングステンの含有量が5〜50質量%であり、ニッケルの含有量が0.1〜10質量%であり、
さらに、水素化処理触媒前駆体がEDTAを含むことにより、980〜1030nmおよび580〜620nmに電子スペクトルの吸収極大ピークを呈することを特徴とする水素化処理触媒前駆体が提供される。
本発明の水素化処理触媒前駆体に用いられる担体は、複合酸化物の粒子およびその粒子間に存在するバインダー部分からなり、複合酸化物を60質量%以上含む。好ましくは、複合酸化物は、担体中に65〜95質量%、さらには70〜90質量%含まれる。複合酸化物が60質量%未満では、複合酸化物がもつ特性が触媒反応に十分反映されない。また、好ましくは、担体の細孔直径50〜1000nmの範囲の細孔容積は、0.01〜0.5mL/g、より好ましくは、0.02〜0.3mL/gである。細孔直径50nm以上の細孔は、マクロポアと呼ばれ、反応分子の拡散を促進し、炭化水素油の反応の促進に好ましいが、担体の細孔直径50〜1000nmの範囲の細孔容積が0.5mL/gより大きいと、水素化処理触媒前駆体の機械的強度が低下したり、水素化処理触媒のかさ密度が低下して単位体積当たりの触媒性能が低下するので好ましくない。マクロポア特性は、水銀圧入法を用いて測定でき、水銀の接触角を140゜、表面張力を480dynes/cmとし、全ての細孔は円筒形であると仮定して算出できる。また、担体は、好ましくは、いわゆるメソポアの細孔特性について、中央細孔直径が4〜20nmであり、さらに好ましくは4〜15nmである。メソポア細孔特性は、窒素ガス吸着法によって測定され、BJH法などによって細孔容積と細孔直径の関係を算出することができる。また、中央細孔直径は、窒素ガス吸着法において相対圧0.9667の条件で得られる細孔容積をVとするとき、細孔直径の大きい側からの累積細孔容積がV/2となる細孔直径をいう。また、担体の比表面積は、好ましくは100〜1000m2/gであり、さらに好ましくは150〜800m2/gである。
本発明でいう複合酸化物とは、固体酸性を有する複合酸化物である。例えば、二元複合酸化物では、K. Shibata, T. Kiyoura, J. Kitagawa, K. Tanabe, Bull. Chem. Soc. Jpn.,
46, 2985 (1973)にて酸性発現が確認されているものをはじめ数多くのものが知られているが、シリカ−アルミナ、シリカ−チタニア、シリカ−ジルコニア、シリカ−マグネシアが好ましく用いられる。三元複合酸化物としては、シリカ−アルミナ−チタニア、シリカ−アルミナ−ジルコニアが好ましく用いられる。また、本発明でいう複合酸化物には、固体酸性を有するゼオライトを含む。ゼオライトとしては、フォージャサイトX型ゼオライト、フォージャサイトY型ゼオライト、βゼオライト、モルデナイト型ゼオライト、ZSM系ゼオライト(ZSM−4、5、8、11、12、20、21、23、34、35、38、46等がある)、MCM−41,MCM−22、MCM−48、SSZ−33、UTD−1、CIT−5、VPI−5、TS−1、TS−2等が本発明における複合酸化物として使用でき、特に、Y型ゼオライト、安定化Y型ゼオライト、βゼオライト、モルデナイト型ゼオライトまたはMCM−22が好ましい。また、ゼオライトは、プロトン型またはアンモニウムイオン型が好ましい。本発明の水素化処理触媒前駆体の担体は、これら複合酸化物を合計量で60質量%以上含む。また、複合酸化物の凝集粒子の平均直径が、好ましくは15μm以下、特には1〜10μmであることが好ましい。
本発明の水素化処理触媒前駆体に用いられる担体に使用されるバインダーは、アルミナ、シリカおよびチタニアから選ばれる1種または2種以上から構成され、特にアルミナが好ましい。アルミナとしては、α−アルミナ、β−アルミナ、γ−アルミナ、δ−アルミナ等の種々のアルミナを使用することができるが、多孔質で高比表面積であるアルミナが好ましく、中でもγ−アルミナが適している。アルミナの純度は、約98質量%以上、好ましくは約99質量%以上のものが適している。アルミナ中の不純物としては、SO4 2−、Cl−、Fe2O3、Na2O等が挙げられるが、これらの不純物はできるだけ少ないことが望ましく、不純物全量で2質量%以下、より好ましくは1質量%以下である。アルミナをバインダーとする場合の原料は、アルミニウム水酸化物および/または水和酸化物からなる粉体(以下、単にアルミナ粉体ともいう)、特には、擬ベーマイトなどのベーマイト構造を有するアルミニウム水和酸化物を用いることが好ましい。
担体の製造方法は特に限定されないが、複合酸化物粉体とバインダー成分を混練し、成形した後、乾燥、焼成して担体とすることが好ましい。混練には、一般に触媒調製に用いられている混練機を用いることができる。通常は原料を投入し、水を加えて攪拌羽根で混合するような方法が好適に用いられるが、原料および添加物の投入順序など特に限定はない。混練の際には通常水を加えるが、原料がスラリー状の場合などには特に水を加える必要はない。混練時の温度や混練時間は、原料となる複合酸化物、バインダー成分により異なるが、好ましい細孔構造が得られる条件であれば、特に制限はない。また、上述の好ましい担体特性を得たり、生産性を改善するために、硝酸などの酸やアンモニアなどの塩基、セルロースエーテル類やポリビニルアルコールのような水溶性高分子化合物、セラミックス繊維などを加えて混練しても構わない。
本発明の水素化処理触媒前駆体は、5〜50質量%のタングステンおよび0.1〜10質量%のニッケルを含む。好ましくは、タングステンを8〜40質量%、さらには10〜30質量%、特には12〜25質量%、ニッケルを0.5〜8質量%、さらには1〜6質量%、特には1.5〜5質量%含む。また、タングステン含有量に対するニッケル含有量は、モル比で、好ましくは0.1〜1、さらには0.2〜0.8、特には0.25〜0.75であることが好ましい。好ましくは、本発明の水素化処理触媒前駆体は、リンまたはホウ素を0.1〜10質量%含む。リンを含ませる場合、より好ましくは、0.2〜5質量%である。また、タングステン含有量に対するリン含有量は、モル比で、好ましくは0.02〜0.5、さらには0.02〜0.3、一層好ましくは0.08〜0.2、特には0.1〜0.18であることが好ましい。
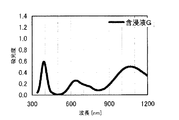
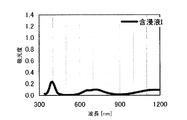
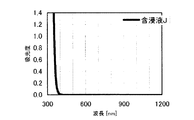
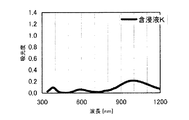
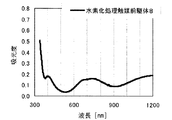
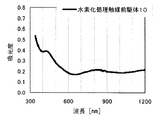
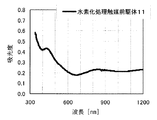
本発明の水素化処理触媒前駆体は、その電子スペクトルが980〜1030nmおよび580〜620nmに吸収極大ピークを有する。この吸収極大ピークは、Ni2+イオンのd電子の遷移に起因する配位子場吸収帯によるもので、含浸液に添加したEDTAがニッケルと錯体を形成した形態で水素化処理触媒前駆体中に存在していることを示す特徴であり、本発明の水素化処理触媒前駆体が水素化処理触媒として高い性能を示すために不可欠である。可視・近赤外領域には、水素化処理触媒前駆体に含まれるニッケルおよびEDTA以外の他の成分に起因する吸収や他の成分とニッケルイオンとの相互作用に起因する吸収等のピークやショルダーが上記の吸収極大ピーク以外に観測されうるが、980〜1030nmおよび580〜620nmに吸収極大ピークがあれば構わない。また、340〜1200nmの領域の電子スペクトルにおいて、本発明の水素化処理触媒前駆体は、好ましくは、おおよそ450〜520nmでの吸収が最も小さくなり、980〜1030nmおよび580〜620nmに吸収極大波長での吸光度は、500nmでの吸光度の1.5倍以上、より好ましくは2倍以上である。さらに、好ましくは、980〜1030nmの吸収極大波長の吸光度は、580〜620nmの吸収極大波長の吸光度よりも大きい。
本発明の水素化処理触媒前駆体は、好ましくは、細孔直径50〜1000nmの範囲の細孔容積が0.01〜0.5mL/gであり、さらに好ましくは、0.02〜0.3mL/gである。また、好ましくは、メソポアの中央細孔直径が、4〜20nmであり、さらに好ましくは4〜15nmである。さらに、好ましくは、比表面積が、30〜800m2/gであり、一層好ましくは50〜600m2/gである。
水素化処理触媒前駆体には、タングステン、ニッケルおよびEDTAを含ませることが不可欠であり、水素化処理触媒前駆体が上述の電子スペクトルを示すような製造方法であれば特に制限はないが、タングステン、ニッケルまたはEDTA、所望であればリンやホウ素を含む1種または2種以上の含浸液を調製し、その含浸液を担体に含浸させ、乾燥処理することによって製造することが好ましい。含浸液を担体に含浸させる工程においては、同一の含浸液を複数回に分けて含浸させてよい。また、含浸液を担体に含浸させた後、乾燥処理を施して、その後に同じ含浸液または別の含浸液をさらに含浸させてよく、含浸と乾燥処理を繰り返すことにより、より多くの金属成分等を水素化処理触媒前駆体に含ませることができる。乾燥処理は、所望の成分をすべて含ませた後においても、逐次含浸液を含浸させる途中の段階においても、220℃を越えない温度で行うことが好ましい。220℃を超える温度で処理すると、EDTAが形成している錯体あるいはEDTAが分解して、水素化処理触媒としたときの性能が低下してしまうので好ましくない。したがって、より好ましくは、200℃以下の温度で乾燥処理を行うとよい。乾燥処理は、風乾、熱風乾燥、加熱乾燥、減圧乾燥、凍結乾燥等の種々の乾燥方法により行うことができ、空気中で行っても不活性ガス雰囲気下で行ってもよい。乾燥処理する時間は、処理温度が上述の範囲内であれば、任意に設定してよいが、0.1〜100時間の範囲であることが好ましい。含浸液を担体に含浸させる方法に特に制限はないが、スプレー、浸漬などによる含浸法やイオン交換法等が好適に用いられる。担体や水素化処理触媒前駆体製造途上の乾燥物(水素化処理触媒前駆体中間品)の吸水量に相当する液量の含浸液を含浸させるポアフィリング法が特に好ましい。
本発明の水素化処理触媒は、水素化処理触媒前駆体を硫化処理することによって製造される。通常、硫化処理は、水素化処理触媒前駆体を、本発明の精製炭化水素油の製造方法に用いる反応装置中に充填した後に行われる。この硫化処理は、約75〜400℃、好ましくは約100〜350℃で、常圧あるいはそれ以上の水素分圧の水素雰囲気下、硫黄化合物を含む石油蒸留物、それに硫黄含有化合物を添加したもの、あるいは硫化水素を用いて行う。石油蒸留物に硫黄含有化合物を添加して用いる場合の硫黄含有化合物は、硫化処理条件下で分解して硫化水素に転化し得るものであれば特に限定はないが、好ましくは、チオール類、二硫化炭素、チオフェン類、ジメチルスルフィド、ジメチルジスルフィドおよび種々のポリスルフィド類である。水素化処理触媒前駆体を反応装置に充填した後、硫化処理を開始する前に、水素化処理触媒前駆体に付着した水分を除去するための乾燥処理を行ってもよい。この乾燥処理は、水素または不活性ガスの雰囲気下で、常圧あるいはそれ以上の圧力でガスを流通させ、常温〜220℃、好ましくは150℃以下で行う。220℃を超える高温で乾燥処理を行うと、水素化処理触媒前駆体に含まれるEDTAが形成している錯体あるいはEDTAが分解して、水素化処理触媒としての性能が低下してしまうので好ましくない。
本発明の水素化処理触媒は、水素化精製(水素化脱硫、水素化脱窒素、芳香族水素化、オレフィン水素化)触媒機能、水素化分解触媒機能、水素化異性化触媒機能を有するので、原料炭化水素油と反応条件を適切に選択することにより、水素化処理触媒を水素の存在下で原料炭化水素油と接触させることで、様々な精製炭化水素油を製造することができる。得られる精製炭化水素油は、軽油、灯油、ジェット燃料、ガソリン、LPG、重油等の燃料、燃料電池システム用燃料、潤滑油、溶剤等やそれらの基材として、好ましく用いることができる。また、接触改質原料油や接触分解原料油等、石油精製プロセス原料油として好ましく用いることができる。
本発明の精製炭化水素油の製造方法における好ましい実施形態の1つとして、250℃以上の沸点を有する留分を主成分とする留分の水素化分解処理による精製軽質留分の製造方法が挙げられる。本発明の精製炭化水素油の製造方法によれば、減圧軽油留分を原料炭化水素油とし、これを本発明の水素化処理触媒で水素化処理することによって、精製炭化水素油として硫黄分が10質量ppm以下、好ましくは5質量ppm以下、さらに好ましくは1質量ppm以下の精製軽質留分を製造することができる。ここでいう精製軽質留分とは、精製軽油留分、精製灯油留分、精製ナフサ留分を指す。
本発明の精製炭化水素油の製造方法における好ましい実施形態の1つとして、ノルマルパラフィンを主成分とする原料炭化水素油を本発明の水素化処理触媒で水素化分解処理および水素化異性化処理することによる精製軽油留分の製造方法が挙げられる。
本発明の精製炭化水素油の製造方法における好ましい実施形態の1つとして、軽油留分の水素化精製処理による低硫黄軽油の製造方法が挙げられる。この実施形態の精製炭化水素油の製造方法によれば、軽油留分を原料炭化水素油とし、これを水素化精製処理することによって、精製炭化水素油として硫黄分が50質量ppm以下、好ましくは10質量ppm以下、さらに好ましくは5質量ppm以下の低硫黄軽油を製造することができる。
本発明の精製炭化水素油の製造方法における好ましい実施形態の1つとして、粗精製軽油留分の水素化精製処理による低硫黄軽油の製造方法が挙げられる。この実施形態の低硫黄軽油の製造方法は、原料炭化水素油が、モリブデンを含む水素化精製触媒を水素の存在下で軽油留分と接触させる粗精製工程で得られる粗精製軽油留分であり、精製炭化水素油が硫黄分10質量ppm以下の低硫黄軽油である低硫黄軽油の製造方法である。この実施形態によれば、硫黄分が10質量ppm以下、好ましくは5質量ppm以下、さらに好ましくは1質量ppm以下の低硫黄軽油を製造することができる
乾燥担体基準でシリカアルミナ80質量%およびアルミナ20質量%からなるような配合比で、シリカアルミナ粉体および擬ベーマイト粉体を混合し、硝酸水溶液を添加して混練し、呼び寸法1/20インチ三つ葉型のダイスを通して押出成形した後、乾燥し、空気気流下、600℃で1時間焼成することで担体Aを調製した。シリカアルミナ粉体としては、シリカ/アルミナモル比4.4、平均粒経8.8μm、強熱減量15.3質量%の粉体を用いた。担体Aのメソポア構造を窒素吸着法で分析したところ、細孔容積は0.626mL/g、比表面積は469m2/g、中央細孔径は47Åであった。担体Aのマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.166mL/g、細孔直径500〜5000Åのマクロポア容積は0.089mL/gであった。
関東化学株式会社製アンモニア水(28%)9.0gを約20mLのイオン交換水に加えて攪拌した。このとき、溶液のpHは11.9であった。ここに、EDTA(関東化学株式会社製鹿特級)14.0g(0.0479mol)を加えて攪拌し、EDTAを溶解させた。このとき、溶液のpHは5.48であった。この溶液に硝酸ニッケル六水和物(関東化学株式会社製鹿特級)29.51g(0.1014mol)を加えて攪拌し、青緑色の均一な溶液を得た。このとき、溶液のpHは0.8であった。この溶液にAMT水溶液(日本無機化学工業株式会社製MW−2、W濃度693g/L)47.26mLを添加して、均一な含浸液A約130mLを得た。含浸液の電子スペクトルは、他の例で得られた含浸液との関係で後述する。この溶液のpHは0.93であった。この含浸液Aを担体Aの吸水率に合わせてイオン交換水で希釈し、全量の60%を担体A100gに対してスプレー含浸して得られたペレットを130℃で24時間乾燥した。残りの含浸液をペレットの吸水率に合わせてイオン交換水で希釈して乾燥させたペレットにさらに含浸して得られたペレットを130℃で24時間乾燥し、水素化処理触媒前駆体1を得た。水素化処理触媒前駆体1のマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.085mL/g、細孔直径500〜5000Åのマクロポア容積は0.042mL/gであった。水素化処理触媒前駆体1の電子スペクトルは、他の例で得られた水素化処理触媒前駆体との関係で後述する。
実施例1で得た含浸液Aにリン酸(関東化学株式会社製特級、リン酸含有率85質量%)3.11gを加えて攪拌し、含浸液Bを得た。この溶液のpHは、1.10であった。この含浸液Bを、実施例1と同様に希釈し、2回に分けて担体Aに含浸させ、乾燥して水素化処理触媒前駆体2を得た。
関東化学株式会社製リン酸(85%)3.11gを約60mLのイオン交換水に加えて加熱攪拌した。10分後、ここにEDTA(関東化学株式会社製鹿特級)14.0g(0.0479mol)を加えて60〜70℃で1時間加熱攪拌した。このとき、EDTAはスラリー状であった。この溶液に炭酸ニッケル(日本化学産業株式会社製)13.23g(0.101mol)を加えて攪拌し、青色の均一な溶液を得た。この溶液にAMT水溶液(日本無機化学工業株式会社製MW−2、W濃度693g/L)47.26mLを添加して、含浸液Cを得た。この溶液のpHは、3.51であった。この含浸液Cを、実施例1と同様に、希釈し、2回に分けて担体Aに含浸させ、乾燥して水素化処理触媒前駆体3を得た。水素化処理触媒前駆体3のマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.091mL/g、細孔直径500〜5000Åのマクロポア容積は0.050mL/gであった。
担体Aの吸水率に合わせてAMT水溶液(日本無機化学工業株式会社製MW−2、W濃度693g/L)47.26mLをイオン交換水で希釈し、全量を担体A100gに対してスプレー含浸して得られたペレットを130℃で24時間乾燥した。これを水素化処理触媒前駆体中間品Aとした。関東化学株式会社製特級硝酸(硝酸含有率60〜61質量%)2.0gを約60mLのイオン交換水に加えて加熱攪拌した。10分後、ここにEDTA(関東化学株式会社製 鹿特級 製品名)14.0g(0.0479mol)を加えて60〜70℃で1時間加熱攪拌した。このとき、EDTAはスラリー状であった。この溶液に炭酸ニッケル(日本化学産業株式会社製)13.85g(0.105mol)を加えて攪拌し、青色の均一な含浸液Dを得た。この溶液のpHは、5.90であった。この含浸液Dを、水素化処理触媒前駆体中間品Aの吸水率に合わせてイオン交換水で希釈して水素化処理触媒前駆体中間品Aのペレットに含浸して得られたペレットを130℃で24時間乾燥し、水素化処理触媒前駆体4を得た。
〔水素化処理触媒前駆体5の製造〕
実施例1で得た水素化処理触媒前駆体1を、ロータリーキルンで、500℃で30分焼成して水素化処理触媒前駆体5を得た。水素化処理触媒前駆体5のマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.113mL/g、細孔直径500〜5000Åのマクロポア容積は0.058mL/gであった。
〔水素化処理触媒前駆体6の製造〕
複合酸化物成分を添加することなく、擬ベーマイト粉に硝酸水溶液を添加して混練し、呼び寸法1/20インチ三つ葉型のダイスを通して押出成形した後、乾燥し、600℃で1時間焼成することで、複合酸化物成分を添加していないアルミナ担体である担体Bを得た。担体Bのメソポア構造を窒素吸着法で分析したところ、細孔容積は0.651mL/g、比表面積は256m2/g、中央細孔径は81Åであった。担体Bのマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.003mL/g、細孔直径500〜5000Åのマクロポア容積は0.003mL/gであった。実施例1で用いた担体Aの代わりに、担体Bを用いること以外は、実施例1と同様にして、含浸液Aを用いて、水素化処理触媒前駆体6を得た。水素化処理触媒前駆体6のマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.003mL/g、細孔直径500〜5000Åのマクロポア容積は0.003mL/gであった。
〔水素化処理触媒前駆体7の製造〕
実施例1の含浸液Aの調製にEDTAを用いる代わりに、CyDTA一水和物16.6g(関東化学株式会社製鹿特級、0.0479mol)を用いて含浸液Eを調製した。含浸液EのpHは、1.26であった。実施例1で含浸液Aを用いる代わりに含浸液Eを用いること以外は実施例1と同様にして、担体Aを用いて水素化処理触媒前駆体7を得た。水素化処理触媒前駆体7のマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.102mL/g、細孔直径500〜5000Åのマクロポア容積は0.041mL/gであった。
〔水素化処理触媒前駆体8の製造〕
関東化学株式会社製特級硝酸(硝酸含有率60〜61質量%)2.0gを約60mLのイオン交換水に加えて加熱攪拌した。10分後、ここに炭酸ニッケル(日本化学産業株式会社製)13.23g(0.111mol)を加えて60〜70℃で1時間加熱攪拌した。このとき、混合物はスラリー状であった。ここにクエン酸一水和物(関東化学株式会社製特級)16.17g(0.077mol)を加えて60〜70℃で10分間加熱攪拌し、青色の均一な含浸液Fを得た。この溶液のpHは、2.74であった。この含浸液Fを、実施例4記載の水素化処理触媒前駆体中間品Aの吸水率に合わせてイオン交換水で希釈して、水素化処理触媒前駆体中間品Aのペレットに含浸して得られたペレットを130℃で24時間乾燥し、水素化処理触媒前駆体8を得た。
〔水素化処理触媒前駆体9の製造〕
関東化学株式会社製特級硝酸(硝酸含有率60〜61質量%)2.0gを約60mLのイオン交換水に加えて加熱攪拌した。10分後、ここにNTA(関東化学株式会社製 鹿特級)9.81g(0.0513mol)を加えて60〜70℃で1時間加熱攪拌した。このとき、混合物はスラリー状であった。ここに炭酸ニッケル(日本化学産業株式会社製)13.23g(0.101mol)を加えて、さらにクエン酸一水和物(関東化学製特級)3.00g(0.0143mol)を加えて60〜70℃で10分間加熱攪拌し、青色の均一な含浸液Gを得た。この溶液のpHは、2.78であった。この含浸液Gを、実施例4記載の水素化処理触媒前駆体中間品Aの吸水率に合わせてイオン交換水で希釈して、水素化処理触媒前駆体中間品Aのペレットに含浸して得られたペレットを130℃で24時間乾燥し、水素化処理触媒前駆体9を得た。水素化処理触媒前駆体9のマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.094mL/g、細孔直径500〜5000Åのマクロポア容積は0.045mL/gであった。
〔水素化処理触媒前駆体10の製造〕
実施例3で得た水素化処理触媒前駆体3を、ロータリーキルンで、500℃で30分焼成して水素化処理触媒前駆体10を得た。水素化処理触媒前駆体9のマクロポア構造を水銀圧入法で分析したところ、細孔直径500〜10000Åのマクロポア容積は0.107mL/g、細孔直径500〜5000Åのマクロポア容積は0.059mL/gであった。
〔水素化処理触媒前駆体11の製造〕
AMT水溶液(日本無機化学工業株式会社製MW−2、W濃度693g/L)47.26mLを担体Aの吸水率に合わせてイオン交換水で希釈した含浸液を、担体A100gに対してスプレー含浸して得られたペレットを130℃で24時間乾燥した。硝酸ニッケル六水和物(関東化学株式会社製鹿特級)29.51g(0.1014mol)イオン交換水に溶解してペレットの吸水率に合わせて希釈した含浸液Hをペレットに含浸して得られたペレットを130℃で24時間乾燥し、ロータリーキルンで500℃で30分焼成して、水素化処理触媒前駆体11を得た。
〔水素化処理触媒前駆体12の製造〕
この参考例では、タングステンおよびEDTAを使用せずに含浸液を調製し、水素化処理触媒前駆体を製造した。硝酸ニッケル六水和物(関東化学株式会社製特級)6.26g(0.0215mol)をイオン交換水に溶解してペレットの担体Aの吸水率に合わせて希釈した含浸液Iを、担体A30gに対してスプレー含浸して得られたペレットを130℃で24時間乾燥して、水素化処理触媒前駆体12を得た。
〔水素化処理触媒前駆体13の製造〕
この参考例では、ニッケルおよびEDTAを使用せずに含浸液を調製し、水素化処理触媒前駆体を製造した。AMT水溶液(日本無機化学工業株式会社製MW−2、W濃度693g/L)13.2mLをイオン交換水に溶解して担体Aの吸水率に合わせて希釈した含浸液Jを、担体A30gに対してスプレー含浸して得られたペレットを130℃で24時間乾燥して、水素化処理触媒前駆体13を得た。
〔水素化処理触媒前駆体14の製造〕
この参考例では、タングステンを使用せずに含浸液を調製し、水素化処理触媒前駆体を製造した。エチレンジアミン四酢酸二ナトリウムニッケル(東京化成工業株式会社製)9.88g(0.0215mol)をイオン交換水に溶解して担体Aの吸水率に合わせて希釈した含浸液Kを、担体A30gに対してスプレー含浸して得られたペレットを130℃で24時間乾燥して、水素化処理触媒前駆体14を得た。
〔水素化処理触媒前駆体15の製造〕
この参考例では、タングステンを使用せずに含浸液を調製し、水素化処理触媒前駆体を製造した。関東化学株式会社製アンモニア水(28%)2.7gを約10mLのイオン交換水に加えて攪拌した。このとき、溶液のpHは11.9であった。この溶液に、エチレンジアミン四酢酸二ナトリウムニッケル(東京化成工業株式会社製)9.88g(0.0215mol)を加えて攪拌して溶解させ、イオン交換水を加えて担体Aの吸水率に合わせて希釈した含浸液Lを、担体A30gに対してスプレー含浸して得られたペレットを130℃で24時間乾燥して、水素化処理触媒前駆体15を得た。

実施例1で得られた水素化処理触媒前駆体1を10mL、固定床流通式反応装置に充填し、水素圧力5MPa、10L/hで水素を流通させながら室温から150℃まで昇温し、150℃で2時間、水素を流通させた。その後、以下の手順で水素化処理触媒前駆体1を硫化処理して、硫化された水素化処理触媒とした。硫化剤(市販軽油に1質量%の二硫化炭素を混合したもの)を水素圧力5MPa、水素/オイル比500NL/L、LHSV
2.0h−1、150℃の条件下で2時間通油した。その後、温度以外の条件を一定として硫化剤と水素の供給を継続し、20℃/hで230℃まで昇温して、4時間、230℃で一定とした。その後さらに、17.5℃/hで300℃まで昇温して、11時間、300℃で一定とした。この後、この硫化処理された水素化処理触媒を用いて軽油留分の水素化精製反応を行った。
実施例1で得られた水素化処理触媒前駆体1の代わりに比較例1〜7で得られた水素化処理触媒前駆体5〜11を用いること以外は、実施例5と同様の方法で硫化処理して、硫化された水素化処理触媒とし、実施例5と同様の条件で水素化精製反応を行った。得られた生成油の硫黄分を表3及び4に示す。
1塔目の反応器での反応で得られる中間生成油をサンプリングできる二塔式固定床流通式反応装置の1塔目の反応器に水素化処理触媒前駆体16を50mL、2塔目の反応器に水素化処理触媒前駆体1を50mL充填し、水素圧力5.0MPa、40L/hで水素を流通させながら2時間で室温から120℃まで昇温した。その後、以下の手順で水素化処理触媒前駆体1を硫化処理して、硫化された水素化処理触媒とした。硫化剤(市販軽油に1質量%の二硫化炭素を混合したもの)を水素圧力5.0MPa、水素/オイル比200NL/L、LHSV2.0h−1、120℃の条件下で2時間通油した。その後、温度以外の条件を一定として硫化剤と水素の供給を継続し、27.5℃/hで230℃まで昇温して、4時間、230℃で一定とした。その後さらに、42.5℃/hで300℃まで昇温して、7時間、300℃で一定とした。この後、この硫化処理された水素化処理触媒を用いて軽油留分Aの水素化精製反応を行った。水素圧力5.0MPa、水素/原料油供給比200NL/L、反応器1と2触媒充填量合計に対するLHSV1.5h−1および反応器1および2の温度をいずれも350℃として反応を行った。得られた生成油の硫黄分は7質量ppmであった。中間生成油の硫黄分は614質量ppmであった。
水素化処理触媒前駆体1の代わりに、水素化処理触媒前駆体5を用いること以外は実施例8と同様にして、反応器1および反応器2の温度をいずれも350℃として軽油留分Aの水素化精製反応を行ったところ、得られた生成油の硫黄分は12質量ppmであった。
用いた反応装置の概略フローを図3に示す。本反応装置は、反応器1および反応器2の2つの反応器を備え、その間に高圧分離槽3とストリッパー4を備え、反応器2は高圧分離槽5、ミスト分離槽6及びストリッパー7に連結されており、それらは配管16〜42で連結されている。反応器1および2に対する水素供給は、各々、配管14および配管29、30から行われる。原料油は、配管13,15を通じて反応器1に送られる。ストリッパー4には配管23から水素ガスを供給して、ストリッパー4内に滞留する液体と気液接触させることができる。高圧分離槽3およびストリッパー4からは、各々、配管21および配管24を通して水素化精製反応で生成した硫化水素やアンモニアを含むガスを反応系外に(オフガス)除去することができる。ストリッパー4から取り出された液体は、配管26〜28,31を通じて反応器2に供給される。反応器2で水素化処理された反応混合物は、高圧分離槽5およびミスト分離槽6で気液分離され、液体成分がストリッパー7に送られてストリッピングされた後に、生成油として取り出される。
水素化処理触媒前駆体1の代わりに、水素化処理触媒前駆体5を用いること以外は実施例9と同様にして、反応器1、反応器2、高圧分離槽3およびストリッパー4の温度をいずれも330℃として軽油留分Aの水素化精製反応を行ったところ、配管38から得られた生成油の硫黄分は24質量ppmであった。
水素化処理触媒前駆体を粉砕した後、直径30mmの円板上に加圧成形したものを測定試料とし、大型試料室積分球付属装置を備えた株式会社日立製作所製U−3410型自記分光光度計を用いて、340〜1200nmの波長範囲を白色板をレファレンスとして、反射法で電子スペクトルを測定した。なお、スキャンスピードは、340〜800nm範囲を120nm/分、800〜1200nm範囲を240nm/分とした。
含浸液を内厚0.5mmの石英セルに入れ、株式会社日立製作所製U−3410型自記分光光度計を用いて、340〜1200nmの波長範囲を水をレファレンスとして電子スペクトルを測定した。なお、スキャンスピードは、340〜800nm範囲を120nm/分、800〜1200nm範囲を240nm/分とした。
日機装株式会社製MICROTRAC粒度分析計を用い、湿式測定法で測定した。これは、粉体を水中に分散させ、流れる凝集粒子群にレーザー光を照射し、その前方散乱光により粒度分析を行うものである。
水銀圧入法による細孔特性の測定には、Micromeritics社製AutoPore9200型測定器を用いた。窒素ガス吸着法による細孔特性の測定には、Micromeritics社製ASAP2400型測定器を用いた。
軽油留分の硫黄分の測定は、理学電機工業株式会社製ZSX101e型蛍光X線分析装置を用いて行った。
ミスト分離槽、7: ストリッパー、8〜12: 開閉バルブ、 13〜42: 配管
Claims (8)
- 5〜50質量%のタングステンおよび0.1〜10質量%のニッケルを含む水素化処理触媒前駆体の製造方法であって、
60質量%以上の複合酸化物成分を含む担体にEDTAを含む含浸液を含浸する工程と;
80〜220℃で乾燥処理する工程と;を含み、
得られた水素化処理触媒前駆体の電子スペクトルが980〜1030nmおよび580〜620nmに吸収極大ピークを有することを特徴とする水素化処理触媒前駆体の製造方法。 - 前記EDTAを含む含浸液の電子スペクトルが980〜1030nmおよび580〜620nmに吸収極大ピークを有することを特徴とする請求項1に記載の水素化処理触媒前駆体の製造方法。
- 水素化処理触媒前駆体の細孔直径50〜1000nmの範囲の細孔容積が0.01〜0.5mL/gであることを特徴とする請求項1または2に記載の水素化処理触媒前駆体の製造方法。
- 請求項1〜3のいずれか一項に記載の製造方法で得られた水素化処理触媒前駆体を硫化処理することを特徴とする水素化処理触媒の製造方法。
- 担体にタングステンおよびニッケルを含浸して含む水素化処理触媒前駆体であって、
前記担体が複合酸化物成分を60質量%以上含み、
タングステンの含有量が5〜50質量%であり、ニッケルの含有量が0.1〜10質量%であり、
さらに、水素化処理触媒前駆体がEDTAを含むことにより、980〜1030nmおよび580〜620nmに電子スペクトルの吸収極大ピークを呈することを特徴とする水素化処理触媒前駆体。 - 請求項5に記載の水素化処理触媒前駆体を、硫化処理した後、水素の存在下で原料炭化水素油と接触させることを特徴とする精製炭化水素油の製造方法。
- 前記原料炭化水素油が、軽油留分を水素の存在下でモリブデンを含む水素化精製触媒と接触させる粗精製工程で得られる粗精製軽油留分であり、前記精製炭化水素油が硫黄分10質量ppm以下の低硫黄軽油である請求項6に記載の精製炭化水素油の製造方法。
- 前記原料炭化水素油が、前記粗精製工程の反応混合物を気液分離して得られる粗精製軽油留分であり、前記精製炭化水素油が硫黄分10質量ppm以下の低硫黄軽油である請求項7に記載の精製炭化水素油の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004052227A JP4444690B2 (ja) | 2004-02-26 | 2004-02-26 | 水素化処理触媒前駆体およびその製造方法並びに精製炭化水素油の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004052227A JP4444690B2 (ja) | 2004-02-26 | 2004-02-26 | 水素化処理触媒前駆体およびその製造方法並びに精製炭化水素油の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005238128A JP2005238128A (ja) | 2005-09-08 |
| JP4444690B2 true JP4444690B2 (ja) | 2010-03-31 |
Family
ID=35020426
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004052227A Expired - Lifetime JP4444690B2 (ja) | 2004-02-26 | 2004-02-26 | 水素化処理触媒前駆体およびその製造方法並びに精製炭化水素油の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4444690B2 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007070394A2 (en) | 2005-12-14 | 2007-06-21 | Advanced Refining Technologies Llc | Method of making hydroprocessing catalyst |
| US9187702B2 (en) * | 2009-07-01 | 2015-11-17 | Chevron U.S.A. Inc. | Hydroprocessing catalyst and method of making the same |
| CN102166520B (zh) * | 2010-02-25 | 2013-03-27 | 中国石油天然气股份有限公司 | 加氢精制催化剂 |
| CN102166521B (zh) * | 2010-02-25 | 2013-03-27 | 中国石油天然气股份有限公司 | 一种加氢精制催化剂制备方法 |
| KR101854737B1 (ko) * | 2017-04-07 | 2018-05-04 | 서울대학교 산학협력단 | 촉매 및 촉매 제조 방법 |
| US11332834B2 (en) | 2014-10-21 | 2022-05-17 | Seoul National University R&Db Foundation | Catalyst and manufacturing method thereof |
| CN117753445A (zh) * | 2022-09-19 | 2024-03-26 | 中国石油化工股份有限公司 | Ag改性加氢催化剂及其制法和应用 |
| CN116371451B (zh) * | 2023-04-14 | 2024-05-17 | 西安交通大学 | 一种适用于甲烷干重整的铈掺杂镍基催化剂及其制备方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1043069B1 (en) * | 1999-04-08 | 2005-05-25 | Albemarle Netherlands B.V. | Process for sulphiding a hydrotreating catalyst comprising an organic compound comprising N and carbonyl |
| JP2001198471A (ja) * | 2000-01-18 | 2001-07-24 | Univ Tohoku | 水素化処理触媒製造用含浸液および水素化処理触媒の製造方法 |
| WO2002004117A1 (en) * | 2000-07-12 | 2002-01-17 | Akzo Nobel N.V. | Process for preparing an additive-based mixed metal catalyst |
| JP4643805B2 (ja) * | 2000-07-28 | 2011-03-02 | 日本ケッチェン株式会社 | 重質炭化水素油の水素化処理触媒および水素化処理方法 |
| WO2003002253A1 (fr) * | 2001-06-27 | 2003-01-09 | Japan Energy Corporation | Procede servant a preparer un catalyseur d'hydroraffinage |
| JP4202064B2 (ja) * | 2002-07-24 | 2008-12-24 | 篤 石原 | 水素化脱硫触媒の製造方法 |
| JP4680520B2 (ja) * | 2004-02-26 | 2011-05-11 | Jx日鉱日石エネルギー株式会社 | 低硫黄軽油の製造方法および環境対応軽油 |
-
2004
- 2004-02-26 JP JP2004052227A patent/JP4444690B2/ja not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| JP2005238128A (ja) | 2005-09-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2022202785B2 (en) | Middle distillate hydrocracking catalyst with a base extrudate having a high nanopore volume | |
| CN105247016B (zh) | 润滑油基础油的制造方法 | |
| WO2003000410A1 (en) | Catalyst for hydrogenation treatment of gas oil and method for preparation thereof, and process for hydrogenation treatment of gas oil | |
| CN109196077A (zh) | 升级重油的系统和方法 | |
| KR20120040150A (ko) | 윤활유 기유의 제조 방법 및 윤활유 기유 | |
| JP2023093493A (ja) | 高い酸点分布を有する安定化yゼオライトを高含有する中間留分水素化分解触媒 | |
| JP2025000619A (ja) | 高いナノ細孔の安定化yゼオライトを含有する中間留分水素化分解触媒 | |
| JP3662495B2 (ja) | 水素化分解触媒及びその製造方法並びに水素化分解方法 | |
| JP2011068728A (ja) | 炭化水素油及び潤滑油基油の製造方法 | |
| RU2609834C1 (ru) | Катализатор, способ его приготовления и способ гидрооблагораживания дизельных дистиллятов | |
| JP4444690B2 (ja) | 水素化処理触媒前駆体およびその製造方法並びに精製炭化水素油の製造方法 | |
| JP2002239385A (ja) | 炭化水素油用水素化処理触媒の製造方法及び炭化水素油の水素化処理方法 | |
| JP4658491B2 (ja) | 環境対応軽油の製造方法 | |
| RU2468864C1 (ru) | Катализатор, способ его приготовления и способ гидрооблагораживания дизельных дистиллятов | |
| RU2607925C1 (ru) | Катализатор и способ гидрооблагораживания дизельных дистиллятов | |
| JP7659552B2 (ja) | 脱油アスファルトを水素化処理するための方法およびシステム | |
| CN103059976A (zh) | 一种生产优质低凝柴油的方法 | |
| JP2015168688A (ja) | 軽油基材の製造方法 | |
| KR102327495B1 (ko) | 고활성 수소화처리 촉매 | |
| TW201435076A (zh) | 烴油的氫化脫硫催化劑 | |
| JP2014111233A (ja) | 炭化水素油の水素化脱硫触媒 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20061116 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20090615 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100112 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100114 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4444690 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130122 Year of fee payment: 3 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130122 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130122 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140122 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |