JP4391237B2 - リガンド発見のための方法 - Google Patents
リガンド発見のための方法 Download PDFInfo
- Publication number
- JP4391237B2 JP4391237B2 JP2003547631A JP2003547631A JP4391237B2 JP 4391237 B2 JP4391237 B2 JP 4391237B2 JP 2003547631 A JP2003547631 A JP 2003547631A JP 2003547631 A JP2003547631 A JP 2003547631A JP 4391237 B2 JP4391237 B2 JP 4391237B2
- Authority
- JP
- Japan
- Prior art keywords
- target
- ligand
- compound
- elongation factor
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *CCNC(*)=O Chemical compound *CCNC(*)=O 0.000 description 4
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/62—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
- C07D333/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D333/70—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B20/00—Methods specially adapted for identifying library members
- C40B20/08—Direct analysis of the library members per se by physical methods, e.g. spectroscopy
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B30/00—Methods of screening libraries
- C40B30/04—Methods of screening libraries by measuring the ability to specifically bind a target molecule, e.g. antibody-antigen binding, receptor-ligand binding
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6845—Methods of identifying protein-protein interactions in protein mixtures
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Analytical Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Microbiology (AREA)
- General Physics & Mathematics (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Food Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pathology (AREA)
- Peptides Or Proteins (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Other Investigation Or Analysis Of Materials By Electrical Means (AREA)
Description
薬物発見プロセスは、通常、引き続く医化学最適化のための適度な親和性リード(lead)を同定するための、化合物ライブラリーの大規模な機能的スクリーニングで開始する。しかし、目的の標的の全てがこのようなスクリーニングに耐えられるわけではない。いくつかの場合において、ハイスループットスクリーニングに耐えられるアッセイが、利用可能ではない。他の場合において、標的は、複数の結合様式を有し得、その結果、このようなスクリーニングの結果が曖昧であり、そして解釈が困難である。なお他の場合において、ハイスループットアッセイのためのアッセイ条件は、人為結果を受けやすいようなものである。その結果、必ずしも機能的スクリーニングに依存しない、リガンドの発見のための代替の方法が必要とされる。
本発明は、結合技術を使用する、リガンド発見のための方法に関する。
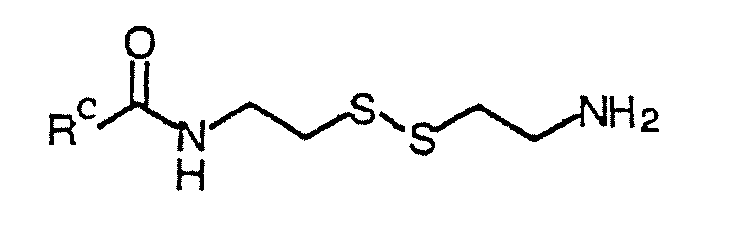
mは、0、1、または2であり;そして
nは、1または2である。
a)標的タンパク質に結合する、式RDSSR1の第一の化合物を同定する工程;
b)標的タンパク質に結合する、式RESSR1の第二の化合物を同定する工程;および
c)RDおよびREを含む結合体化合物を形成する工程であって、ここで、RDおよびR1は、各々独立して、C1〜C20の非置換脂肪族、C1〜C20の置換脂肪族、非置換アリール、および置換アリールであり;そしてR1は、非置換C1〜C10脂肪族、置換C1〜C10脂肪族、非置換アリールである。この方法の特定の実施形態において、標的に結合する第二の化合物の同定は、第一の化合物の存在下で行われる。
mは、0、1、または2であり;そして
nは、1または2である。
a)目的の部位またはその近くで共有結合を形成し得るか、または金属に配位し得る、アンカー基を有する標的を提供する工程;
b)この標的を伸長因子と接触させて、これによって、標的−伸長因子複合体を形成する工程であって、ここで、この伸長因子は、共有結合を形成するかまたは金属に配位するかのいずれかである、第一の官能基、および共有結合を形成し得る第二の官能基を含む、工程;
c)この標的−伸長因子複合体を、第二の官能基と共有結合を形成し得る基を含む候補リガンドと接触させる工程;
d)標的伸長因子複合体と候補リガンドとの間で、共有結合を形成させる工程;ならびに
e)標的−伸長因子−リガンド結合体中に存在する、候補リガンドを同定する工程。
a)反応性の求核基を、目的の部位またはその近くに有する標的を提供する工程;
b)この標的を伸長因子と接触させて、これによって、標的−伸長因子複合体を形成する工程であって、ここで、この伸長因子は、標的における求核基と反応して共有結合を形成する、第一の官能基、およびジスルフィド結合を形成し得る、第二の官能基を含む、工程;
c)この標的−伸長因子複合体を、ジスルフィド結合を形成し得るリガンド候補物と接触させる工程;
d)標的−伸長因子複合体とリガンド候補物との間でジスルフィド結合を形成する工程であって、これによって、標的−伸長因子−リガンド結合体を形成する、工程;ならびに
e)この標的−伸長因子−リガンド結合体中に存在する、リガンド候補物を同定する工程。
本発明は、目的の標的における選択された部位に結合し得るリガンドを同定するための、迅速かつ効率的な方法を提供する。本明細書中の方法によって同定されるリガンド自体が、例えば、新規治療薬剤、酵素インヒビター、標識化合物、診断剤、タンパク質精製のための親和性薬剤などの開発のための、リード化合物としての用途を見出す。
本明細書中で使用される用語の定義としては、以下が挙げられる:
用語「脂肪族」または「非置換脂肪族」とは、直鎖、分枝鎖、環状、または多環式の炭化水素をいい、そしてアルキル部分、アルケニル部分、アルキニル部分、シクロアルキル部分、シクロアルケニル部分、およびシクロアルキニル部分が挙げられる。
本発明は、リガンド発見のための新規の方法を提供し、この方法は、「結合」と呼ばれるプロセスによる。潜在的なリガンドは、標的に共有結合されるかまたは「結合」され、続いて同定される。前に示したように、本発明の1つの局面において、本方法は、以下の工程を包含する:
a)目的の部位またはその近くに化学的反応性基を含む標的を、化学的反応性基と共有結合を形成し得る化合物と接触させる工程;
b)標的と化合物との間に共有結合を形成し、それによって標的化合物結合体を形成する工程;および
c)標的化合物結合体を同定する工程。
a)目的の部位またはその近くに化学的反応性基を含む標的を得る工程;
b)化学的反応性基に共有結合し得る複数の化合物と標的を合わせ、ここで少なくとも1つの化合物が、標的と共有結合を形成する工程;および
c)標的−化合物結合体において共有結合を形成した化合物を同定する工程。
a)ジスルフィド結合を形成し得る標的タンパク質とリガンド候補(これもまたジスルフィド結合を形成し得る)とを接触させる工程;
b)標的タンパク質とリガンド候補との間にジスルフィド結合を形成し、それによって標的タンパク質−リガンド結合体を形成する工程;および
c)標的タンパク質−リガンド結合体中に存在するリガンドを同定する工程、
を包含する。
a)ジスルフィド結合を形成し得る標的タンパク質と、やはりジスルフィド結合を形成し得るリガンド候補物とを接触させる工程;
b)この標的タンパク質とリガンド候補物との間でジスルフィド結合を形成して、それにより、標的タンパク質−リガンド結合体を形成する工程;
c)この標的タンパク質−リガンド結合体と、還元剤とを接触させる工程;および
d)この標的タンパク質−リガンド結合体の量を所望の量に減少させるために還元剤の濃度を決定する工程。
a)ジスルフィド結合を形成し得る標的タンパク質と、やはりジスルフィド結合を形成し得る較正化合物とを接触させる工程;
b)この標的タンパク質と較正化合物との間でジスルフィド結合を形成して、それにより、標的タンパク質−較正化合物結合体を形成する工程;
c)この標的タンパク質−較正化合物結合体と、還元剤とを接触させる工程;および
d)この標的タンパク質−リガンド結合体の量を所望の量に減少させるために必要な還元剤の濃度を決定する工程。
*1213結晶は、1非対称単位につき1つのモノマーを含む。P63形態は、生物学的に関連したホモダイマーを含む。
†括弧中の値は、最高の分解能ビン(resolution bin)である。
$Rsym(l)=Σhki|lhki(lhki)|/Σhkilhki(ここでlhkiは、反射hkiの強度である)
§Rcryst=Σhki||Fobs|−|Fcalc||/|Fobs|(ここでFobsおよびFcalcは、それぞれ、細分に使用されるデータについての観察および計算された構造因子である)
¶Rtree=は、Σhki||Fobs|−|Fcalc||/|Fobs|(ここでFobsおよびFcalcは、それぞれ、細分から省略されたデータの10%についての観察および計算された構造因子である)
重大なことには、N−トシル−D−プロリン部分の位置は、3つの場合全てにおいて非常に類似している(タンパク質中の全てのCα炭素について0.11〜0.56Åと比較して、0.55〜1.88ÅのRMSD)。N−トシル−D−プロリン置換基が、きっちりと重複している一方で、アルキル−ジスルフィド結合が異なるシステイン残基に由来するこの部分に集中するという事実は、N−トシル−D−プロリン部分(結合ではなく)が、結合決定因子であるという概念を支持する。
a)標的タンパク質に結合する第1の化合物を同定する工程;
b)この標的タンパク質に結合する第2の化合物を同定する工程;ならびに
c)第1の化合物および第2の化合物を、リンカーエレメントを介して結合して、標的タンパク質に結合する結合体分子を形成する工程。好ましい実施形態において、この結合体分子は、第1の化合物または第2の化合物いずれか単独よりも高い結合親和性で標的タンパク質に結合する。
a)標的タンパク質に結合する第1の化合物を同定する工程;
b)この標的タンパク質に結合した第1の化合物の存在下で、この標的タンパク質に結合する第2の化合物を同定する工程;ならびに
c)この第1の化合物および第2の化合物を、リンカーエレメントを介して結合して、この標的タンパク質に結合する結合体分子を形成する工程。図7は、この方法の模式的例示である。第1の結合実験において、その結合決定因子RDが同定される。一旦RDが同定されると、第2の反応性システインが導入されるかまたはマスクが外されるかのいずれかであり、結合決定因子REを同定するための結合実験は、結合決定因子RDの存在下で起こる。これら2つの結合決定因子RDおよびREは、その後結合されて、その標的タンパク質に結合する結合体分子を形成する。
a)共有結合を形成し得る固定基(anchoring group)を有する標的を提供するか、または目的の部位もしくはその部位の近くにある金属を配位結合する工程;
b)この標的と、エクステンダーとを接触させ、それにより標的−エクステンダー錯体を形成する工程であって、ここでこのエクステンダーは、共有結合を形成するかまたは金属を配位結合させるかのいずれかである第1の官能基、および共有結合を形成し得る第2の官能基を含む、工程;
c)この標的−エクステンダー錯体と、第2の官能基と共有結合を形成し得る基を含む候補配位子とを接触させる工程;
d)この標的−エクステンダー錯体とこの候補配位子との間で共有結合を形成しする工程;ならびに
e)この標的−エクステンダー−配位子結合体中に存在する候補配位子を同定する工程。
a)目的の部位にまたはその近くに反応性求核基を有する標的を提供する工程;および
b)この標的と、エクステンダーとを接触させる工程であって、それによって、標的−エクステンダー錯体を形成する工程であって、ここでこのエクステンダーは、標的中の求核基と反応して共有結合を形成する第1の官能基およびジスルフィド結合を形成し得る第2の官能基を含む、工程;
c)この標的−エクステンダー錯体を、ジスルフィド結合を形成し得る配位子候補物と接触させる工程;
d)この標的−エクステンダー錯体とこの配位子候補物との間でジスルフィド結合を形成し、それによって、標的−エクステンダー−配位子結合体を形成する工程;ならびに
e)この標的−エクステンダー−配位子結合体中に存在する配位子候補物を同定する工程。必要に応じて、この標的を、還元剤の存在下で配位子候補物と接触させる。
未改変すなわち「野生型」のE.coli TS酵素のいくつかの変異体を作製し、(TS遺伝子が排除された)E.coli株χ2913において過剰発現させ、そして精製した。このχ2913株は、チミジン補充を必要とする。なぜなら、(欠失した)TS遺伝子は、生存のために必須であるからである。第一の変異体は、活性部位のシステインがセリンで置き換えられたものである(C146Sと略記される)。第二の変異体および第三の変異体は、C146S変異に加えて、活性部位に導入された非ネイティブなシステインを含む。第二の変異体は、残基143において、ロイシンの代わりにシステインを含み、そしてC146S/L143Cと示される。第三の変異体は、残基147において、ヒスチジンの代わりにシステインを含み、そしてC146S/H147Cと示される。他の変異体としては、D169C、W83C、およびI79Cが挙げられ、これらでは、活性部位のシステイン(C146)を維持した。
ジスルフィド含有ライブラリーメンバーを、市販のカルボン酸およびモノ−N−(tert−ブトキシカルボニル)−保護シスタミン(モノ−BOC−シスタミン)から、Parlowおよび共同研究者(Mol.Diversity 1:266−269(1995))の方法を適用することによって作製した。簡単には、260μmolの各カルボン酸を、ポリスチレン樹脂上の130μmo1当量の4−ヒドロキシ−3−ニトロベンゾフェノン上に、N,N−ジメチルホルムアミド(「DMF」)中の1,3−ジイソプロピルカルボジイミド(「DIC」)を使用して固定した。室温で4時間後、この樹脂をDMF(2×)、ジクロロメタン(DCM、3×)、およびテトラヒドロフラン(「THF」、1×)でリンスして、未結合の酸およびDICを除去した。THF中66μmolのモノ−BOC保護シスタミンでのアミド形成を介して、この酸を樹脂から切断した。周囲温度で12時間の反応後、溶媒をエバポレートし、そしてBOC基を、DCM中80%のトリフルオロ酢酸(「TFA」)を使用して、各ジスルフィドの結合していない半分から除去した。この生成物を、HPLC−MSによって特徴付け、そして実質的に純粋な生成物を、さらなる精製なしで使用した。合計530の化合物を、この方法論を使用して作製した。
N−トシル−プロリン誘導体を、以下のように合成した。プロリンメチルエステル塩酸塩を、4−(クロロスルホニル)安息香酸および炭酸ナトリウムと、水中で反応させた。この生成物を、トリフルオロ酢酸ペンタフルオロフェニルおよびピリジンと、N,N−ジメチルホルムアミド中で反応させることによって、ペンタフルオロフェニルエステルに転換し、そしてフラッシュクロマトグラフィーによって精製した。次いで、この活性化エステルを、グルタメート(または試験される任意の他のアミノ酸)のメチルエステルで、トリエチルアミンおよびジクロロメタンの存在下で反応させ、この生成物を、フラッシュクロマトグラフィーによって精製し、そしてこのメチルエステルを水中で水酸化リチウムで加水分解した。その最終生成物を逆相HPLCで精製し、そして凍結乾燥した。
ジスルフィドライブラリースクリーニングを、以下のように行った。代表的な実験において、8〜15のジスルフィド含有化合物のライブラリーを含む1μlのDMSO溶液を、49μlのタンパク質含有緩衝液に添加した。これらの化合物を、各々が独特の分子量を有するように選択した。理想的には、これらの分子量は、解析が明瞭であるように、少なくとも10原子量単位(amu)異なる。8〜15のジスルフィド含有化合物のプールを、代表的に、解析の容易さのために使用したが、より大きいプールを使用し得る。タンパク質は、約15μMの濃度で存在し、ジスルフィドライブラリーメンバーの各々は、約0.2mMで存在し、従って、全てのジスルフィドライブラリーメンバーの合計濃度は、約2mMである。スクリーニングを、25mMのリン酸カリウム(pH7.5)および1mMの2−メルカプトエタノールを含有する緩衝液中で行なったが、他の緩衝液および還元剤を使用し得る。これらの反応を、周囲温度で少なくとも30分間平衡させた。これらの条件は、タンパク質が質量分析系中でイオン化されることの容易さ(以下を参照のこと)、特定のシステインの反応性などに依存して、かなり変動し得る。TSの場合、上記条件で満足であることが見出された。酸素または外来の金属イオンを排除するために、特別の努力を払わなかった;これらの反応の時間スケールにおいて、ジスルフィド交換を容易にするために十分な遊離チオールが存在する。
結晶を、非共有結合複合体については1mMの化合物を結晶化緩衝液に含めたことを例外として、Perryら,Proteins 8:315−333(1990)に以前に記載されたようにして、成長させた。データ収集の前に、結晶を、70%の飽和(NH4)2SO4、20%のグリセロール、50mMのK2HPO4(pH7.0)を含む溶液に移した。非共有結合N−トシル−D−プロリン複合体については、10mMの化合物を、浸漬溶液に添加した;他の複合体については、1mMの化合物を含めた。回折データを、Rigaku RU−3R発生器およびR−axis−IV検出器を使用して−170℃で収集し、そしてd*TREKを使用して処理した。これらの結晶は、以前に記載された構造(I2 13形態についてPDBコード1TJS、およびP6 3形態について2TSC)と同形であったので、精製を、REFMAC(CCP4)を使用する剛体精製によって開始した。このタンパク質モデルを、INSIGHT−II(MSI,San Diego)において構築した化合物モデルを使用して調整し、そしてPROTIN(CCP4)辞書を、MAKEDIC(CCP4)を使用して作製した。位置因子および個々の等方性温度因子での精製を、示される解像範囲における全ての反射を使用して、REFMAC(CCP4)を用いて実施した。溶媒分子を、ARPP(CCP4)を使用して自動的に配置し、そして介在する特徴がFo−Fc差マップに残らなくなるまで、精製を続けた。PDB登録番号は、ネイティブのC146結合N−トシル−D−プロリン、L143C結合N−トシル−D−プロリン、N−トシル−D−プロリン遊離酸浸漬、グルタメート−N−トシル−D−プロリン浸漬、およびグルタメート−N−トシル−D−プロリンβ−アラニン結晶についてそれぞれ、1F4B、1F4C、1F4D、1F4E、1F4F、である。
選択されたN−トシル−D−プロリン化合物を、結合を使用して、一連のリガンド候補物として最適化および試験した。TSに結合したN−トシル−D−プロリンの結晶構造に基づいて、フェニル環から離れたメチル基は、誘導体化点として使用するための有望な位置にあった。スキーム1は、88の異なるアルデヒド(ここで、R5は、非置換アリールまたは置換アリールから選択される)および6つの異なるリンカーを使用して、誘導体を合成するために使用された一般的方法を説明する。
この実施例は、化合物13の合成のための1つの実施形態を記載する。一般的な反応スキームは、スキーム2に概説される。
アセチルスルファニル−酢酸ペンタフルオロフェニルエステル(1.6g、5.3mmol)およびH−Asp(OtBu)−OH(1g、5.3mmol)を、20mlの乾燥ジクロロメタン(DCM)中で混合した。次いで、1.6mlのトリエチルアミン(11.5mmol)を添加し、そしてこの反応を、周囲温度で3.5時間進行させた。次いで、有機層を、3×15mlの1M炭酸ナトリウムで抽出し、合わせた水性画分を、100mlの1M硫酸水素ナトリウムで酸性化し、そして3×30mlの酢酸エチルで抽出した。次いで、合わせた有機画分を、30mlの1M硫酸水素ナトリウム、30mlの5M NaClでリンスし、硫酸ナトリウムで乾燥し、濾過し、そして減圧下でエバポレートして、1.97gの24をほぼ無色のシロップとして得、これを、さらに精製せずに使用した。MW=305(実測値306、M+l)。
遊離酸24を、10mlの乾燥テトラヒドロフラン(THF)に溶解し、0℃に冷却し、そして0.58mlのN−メチル−モルホリン(5.3mmol)および0.69mlのイソブチルクロロホルメートで処理した。濃厚な白色沈澱が即座に形成し、そして30分後、この反応物を、ガラスフリットに通して濾過し、そしてさらに10mlのTHFを含む新たなフラスコに移した。その一方で、1−メチル−3−ニトロ−1−ニトロソグアニジン(2.3g、15.6mmol)を、7.4mlの40%水性KOHおよび25mlのジエチルエーテルと45分間0℃で反応させることによって、ジアゾメタンを調製した。次いで、黄色のエーテル層を、混合無水物を含有する反応物中にデカンテーションし、そして165分間かけて周囲温度にゆっくりと温めながら、この反応を進行させた。この反応物を、8℃に冷却し、そして1.5mlのジオキサン中4N HCl(合計6mmol)を滴下した。これにより、かなりの気泡が生じ、そして黄色の溶液は無色になった。周囲温度に次第に温めながら、この反応を2時間進行させ、次いで、1mlの氷酢酸でクエンチした。その溶媒を減圧下で除去し、そしてその残渣を、75mlの酢酸エチルに再溶解し、2×50mlの飽和重炭酸ナトリウム、50mlの5M NaClでリンスし、硫酸ナトリウムで乾燥し、濾過し、そして乾燥するまでエバポレートし、その後、90:10のクロロホルム:酢酸エチルを使用するフラッシュクロマトグラフィーによって精製して、0.747gの25を淡黄色油状物(2.2mmol、23から42%)として得た。計算値MW=337.7、実測値338(M+1)。
クロロメチルケトン25(0.25g、0.74mmol)を、5mlの乾燥N,N−ジメチルホルムアミド(DMF)に溶解し、これに、0.17gの2,6−ジクロロ安息香酸(0.89mmol)および0.107gのKF(1.84mmol)を添加した。この反応を、周囲温度で19時間進行させ、この時点で、これを75mlの酢酸エチルで希釈し、2×50mlの飽和重炭酸ナトリウム、50mlの1M硫酸水素ナトリウム、50mlの5M NaClでリンスし、硫酸ナトリウムで乾燥し、濾過し、そして減圧下で乾燥して、黄色シロップを得、これは、HPLC−MSによって、約75%の生成物26および25%の未反応25であることがわかった。これを、さらに精製せずに使用した。計算値MW=492.37、実測値493(M+l)。
生成物26を、10mlの乾燥DCMに溶解し、0℃に冷却し、そして9mlのトリフルオロ酢酸(TFA)で処理した。次いで、この反応物を氷浴から取り除き、そして1時間かけて、周囲温度に温めた。溶媒を減圧下で除去し、そしてその残渣を、DCMへの再溶解およびエバポレーションを2回行って、残留TFAを除去した。粗製生成物13を、逆相高圧液体クロマトグラフィーによって精製して、101.9mg(0.234mmol、25から32%)の白色吸湿性粉末を得た。計算値MW=436.37、実測値437(M+1)。これを、ジメチルスルホキシド(DMSO)に溶解して、50mMのストック溶液を得た。
a.本実施例は、特定の伸長因子(化合物32(これをカスパーゼ3についての結合実験において用いた))についての1つの実施形態を記載する。一般的なスキームをスキーム3に記載する。
本実施例は、化合物14の合成についての1つの実施形態を記載する。一般的な反応スキームを、スキーム4に概説する。
本実施例は、カスパーゼ3との結合において使用される伸長因子40の合成についての1つの実施形態を記載し、ここで、チオールが、この酵素のプライム部位に指向される。一般的な反応スキームを、スキーム5に概説する。
本実施例は、伸長因子13を用いるカスパーゼ3の改変を説明する。カスパーゼ3を、クローン化し、過剰発現し、そして標準技術を用いて精製する。2mlの0.2mg/ml溶液に10μlの50mM化合物13を添加し、そして反応を周囲温度で3.5時間続けた。3.5時間の時点での質量分析は、カスパーゼ3大サブユニットの完全な改変を示した(MW 16861、計算値16860)。チオエステルを、PBS緩衝液中で緩衝化された0.2mlの0.5Mヒドロキシルアミンを添加することにより脱保護し、そして反応を18時間続けた。18時間の時点で、大サブユニットは、質量16819(計算値16818)を有した。タンパク質を、Ultrafree 5 MWCOユニット中で濃縮し、そして緩衝液をNap−5カラムを用いて0.1MTES pH7.5に交換した。
カスパーゼ3の結晶を、20℃で懸滴拡散蒸着法(hanging drop vapor diffusion method)を用いて成長させた。等量のタンパク質溶液(10mM Tris pH8.5中の5〜10mg/mlの以前に改変したタンパク質)を100mMクエン酸ナトリウム、pH5.9、4%グリセロール、10〜20%PEG6000および10mMDTTを含む貯蔵溶液と混合する。小さな斜方形板は、通常1〜2週間後に現れる。斜方形板は、2か月後、約200×200×20μmの最大サイズに達する。データを集める前に、結晶を25%グリセロールを含む貯蔵溶液に短時間浸し、次いで液体窒素中で瞬間凍結する。
本実施例は、化合物50の合成についての一つの実施形態を説明する。一般的な反応スキームは、スキーム6に概説される。
i)化合物47を、化合物27(97%)の代わりに5−(メタンスルホニル)チオフェン−2−カルボン酸を用いて出発することを除いて、実施例8bの手順により調製した。ES(+)MS m/e=372(M+H)。
本実施例は、以下に示される化合物51の合成のための一実施形態を示す。
本実施例は、化合物54の合成のための一実施形態を示す。一般的な反応スキームを、図7中で概説する。
本実施例は、化合物56の合成のための一実施形態を示す。一般的な反応スキームを、図8中で概説する。
本実施例は、以下に示される化合物57の合成のための一実施形態を記載する。
本実施例は、以下に示される化合物58の合成のための一実施形態を記載する。
本実施例は、以下に示される化合物59の合成のための一実施形態を記載する。
本実施例は、以下に示される化合物60の合成のための一実施形態を記載する。
本実施例は、以下に示される化合物61の合成のための一実施形態を記載する。
Claims (8)
- 以下:
a)共有結合を形成し得るアンカー基を有するか、または目的の部位にて、もしくは目的の部位の近傍にて金属を配位結合する能力を有するアンカー基を有する、標的を提供する工程;
b)該標的を、伸長因子と接触させ、それにより、標的−伸長因子複合体を形成させる工程であって、ここで、該伸長因子が、第一の官能基および第二の官能基を含み、該第一の官能基は、共有結合を形成するかまたは金属を配位結合するかのいずれかであり、そして該第二の官能基が共有結合を形成する能力を有する、工程;
c)該標的−伸長因子複合体を候補リガンドと接触させる工程であって、該候補リガンドが、該第二の官能基と共有結合を形成する能力を有する基を含む、工程;
d)該標的−伸長因子複合体と該候補リガンドとの間に共有結合を形成させる工程;および
e)該標的−伸長因子−リガンド結合体中に存在する該候補リガンドを同定する工程、
を包含し、
該伸長因子は、以下の式:
該第一の官能基は、
該第二の官能基は、−SR’である、
方法。 - 前記アンカー基が、反応性求電子基、反応性求核基、および金属配位部位からなる群より選択される、請求項1に記載の方法。
- 以下:
a)目的の部位にて、もしくは目的の部位の近傍にて反応性求核基を有する標的を提供する工程;
b)該標的を、伸長因子と接触させ、それにより、標的−伸長因子複合体を形成させる工程であって、ここで、該伸長因子が、第一の官能基および第二の官能基を含み、該第一の官能基は、該標的における該求核基と反応して共有結合を形成し、そして該第二の官能基がジスルフィド結合を形成する能力を有する、工程;
c)該標的−伸長因子複合体を候補リガンドと接触させる工程であって、該候補リガンドが、ジスルフィド結合を形成する能力を有する、工程;
d)該標的−伸長因子複合体と該候補リガンドとの間にジスルフィド結合を形成し、それにより、標的−伸長因子−リガンド結合体を形成させる工程;ならびに
e)該標的−伸長因子−リガンド結合体中に存在する該候補リガンドを同定する工程、
を包含し、
該伸長因子が、以下:
該第一の官能基は、
該第二の官能基は、−SR’である、
方法。 - 前記標的上の前記反応性求核基がチオールまたはマスクされたチオールである、請求項3に記載の方法。
- 請求項1または3に記載の方法であって、前記標的−伸長因子−リガンド結合体中に存在する候補リガンドが、質量分析法により同定される、方法。
- 請求項1または3に記載の方法であって、前記標的−伸長因子−リガンド結合体中に存在する候補リガンドが、標識プローブを使用することにより同定される、方法。
- 請求項1または3に記載の方法であって、前記標的−伸長因子−リガンド結合体中に存在する候補リガンドが、クロマトグラフィーにより同定される、方法。
- 請求項1または3に記載の方法であって、前記標的−伸長因子−リガンド結合体中に存在する候補リガンドが、表面プラズモン共鳴を使用することにより同定される、方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/990,421 US6919178B2 (en) | 2000-11-21 | 2001-11-21 | Extended tethering approach for rapid identification of ligands |
| US10/121,216 US6998233B2 (en) | 1998-06-26 | 2002-04-10 | Methods for ligand discovery |
| PCT/US2002/013061 WO2003046200A2 (en) | 2001-11-21 | 2002-04-24 | Methods for ligand discovery |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009020721A Division JP2009109515A (ja) | 2001-11-21 | 2009-01-30 | リガンド発見のための方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005510574A JP2005510574A (ja) | 2005-04-21 |
| JP2005510574A5 JP2005510574A5 (ja) | 2005-12-22 |
| JP4391237B2 true JP4391237B2 (ja) | 2009-12-24 |
Family
ID=26819229
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003547631A Expired - Fee Related JP4391237B2 (ja) | 2001-11-21 | 2002-04-24 | リガンド発見のための方法 |
| JP2009020721A Withdrawn JP2009109515A (ja) | 2001-11-21 | 2009-01-30 | リガンド発見のための方法 |
| JP2011040912A Pending JP2011140497A (ja) | 2001-11-21 | 2011-02-25 | リガンド発見のための方法 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009020721A Withdrawn JP2009109515A (ja) | 2001-11-21 | 2009-01-30 | リガンド発見のための方法 |
| JP2011040912A Pending JP2011140497A (ja) | 2001-11-21 | 2011-02-25 | リガンド発見のための方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20050142614A1 (ja) |
| EP (2) | EP1939625A3 (ja) |
| JP (3) | JP4391237B2 (ja) |
| AU (2) | AU2002254725A1 (ja) |
| CA (1) | CA2464094C (ja) |
| IL (1) | IL161409A0 (ja) |
| MX (1) | MXPA04004814A (ja) |
| NZ (1) | NZ532579A (ja) |
| RU (1) | RU2295518C2 (ja) |
| WO (1) | WO2003046200A2 (ja) |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7214487B2 (en) | 1998-06-26 | 2007-05-08 | Sunesis Pharmaceuticals, Inc. | Methods for identifying compounds that modulate enzymatic activities by employing covalently bonded target-extender complexes with ligand candidates |
| US6919178B2 (en) * | 2000-11-21 | 2005-07-19 | Sunesis Pharmaceuticals, Inc. | Extended tethering approach for rapid identification of ligands |
| US6335155B1 (en) * | 1998-06-26 | 2002-01-01 | Sunesis Pharmaceuticals, Inc. | Methods for rapidly identifying small organic molecule ligands for binding to biological target molecules |
| WO2003081210A2 (en) | 2002-03-21 | 2003-10-02 | Sunesis Pharmaceuticals, Inc. | Identification of kinase inhibitors |
| KR20070053214A (ko) * | 2004-08-26 | 2007-05-23 | 니콜라스 피라말 인디아 리미티드 | 신규 생분해성 링커를 함유하는 프로드럭 |
| DK2526933T3 (en) | 2006-09-22 | 2015-05-18 | Pharmacyclics Inc | Inhibitors of Bruton's tyrosine kinase |
| SG10202107066WA (en) * | 2007-03-28 | 2021-07-29 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US20120101114A1 (en) | 2007-03-28 | 2012-04-26 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| CA2730930C (en) | 2008-07-16 | 2015-01-13 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase for the treatment of solid tumors |
| JP2012501654A (ja) * | 2008-09-05 | 2012-01-26 | アビラ セラピューティクス, インコーポレイテッド | 不可逆的インヒビターの設計のためのアルゴリズム |
| US9556426B2 (en) | 2009-09-16 | 2017-01-31 | Celgene Avilomics Research, Inc. | Protein kinase conjugates and inhibitors |
| JP2013516422A (ja) | 2009-12-30 | 2013-05-13 | アビラ セラピューティクス, インコーポレイテッド | タンパク質のリガンド−指向性共有的修飾 |
| CN102939110A (zh) | 2010-04-21 | 2013-02-20 | 诺沃—诺迪斯克有限公司 | 蛋白质的选择性修饰 |
| CA3007787C (en) | 2010-06-03 | 2020-03-10 | Pharmacyclics Llc | The use of inhibitors of bruton's tyrosine kinase (btk) |
| JP2014520863A (ja) | 2011-07-13 | 2014-08-25 | ファーマサイクリックス,インク. | Bruton型チロシンキナーゼの阻害剤 |
| US8377946B1 (en) | 2011-12-30 | 2013-02-19 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| KR20210033067A (ko) | 2012-06-04 | 2021-03-25 | 파마싸이클릭스 엘엘씨 | 브루톤 타이로신 키나아제 저해제의 결정 형태 |
| CN104704129A (zh) | 2012-07-24 | 2015-06-10 | 药品循环公司 | 与对布鲁顿酪氨酸激酶(btk)抑制剂的抗性相关的突变 |
| EA201590855A1 (ru) | 2012-11-15 | 2015-11-30 | Фармасайкликс, Инк. | Соединения пирролопиримидина как ингибиторы киназ |
| JP6800750B2 (ja) | 2013-08-02 | 2020-12-16 | ファーマサイクリックス エルエルシー | 固形腫瘍の処置方法 |
| US9415050B2 (en) | 2013-08-12 | 2016-08-16 | Pharmacyclics Llc | Methods for the treatment of HER2 amplified cancer |
| BR112016006978A2 (pt) | 2013-09-30 | 2017-08-01 | Pharmacyclics Llc | inibidores de tirosina quinase de bruton |
| US9885086B2 (en) | 2014-03-20 | 2018-02-06 | Pharmacyclics Llc | Phospholipase C gamma 2 and resistance associated mutations |
| AU2015296215A1 (en) | 2014-08-01 | 2017-03-23 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| KR20170033358A (ko) | 2014-08-07 | 2017-03-24 | 파마싸이클릭스 엘엘씨 | 브루톤 티로신 키나아제 저해제의 신규한 제제 |
| IL315294A (en) | 2015-03-03 | 2024-10-01 | Pharmacyclics Llc | Pharmaceutical formulations of bruton's tyrosine kinase inhibitor |
| JP7697280B2 (ja) * | 2021-06-15 | 2025-06-24 | 株式会社オートネットワーク技術研究所 | 車載制御装置、イーサネットスイッチおよび機器設定方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US5367058A (en) * | 1993-08-25 | 1994-11-22 | Becton, Dickinson And Company | Modified antibodies with increased affinity |
| US5361058A (en) * | 1993-11-02 | 1994-11-01 | Gould Electronics Inc. | Time delay fuse |
| DE19610103A1 (de) * | 1996-03-15 | 1997-09-18 | Basf Ag | Cycloalkylderivate und ihre Synthese an fester Phase |
| WO1999041216A1 (en) * | 1998-02-17 | 1999-08-19 | Chembridge Corporation | Supports for solid state chemical reactions and method of use thereof |
| US6344330B1 (en) * | 1998-03-27 | 2002-02-05 | The Regents Of The University Of California | Pharmacophore recombination for the identification of small molecule drug lead compounds |
| CA2430234C (en) * | 2000-11-21 | 2008-02-12 | Sunesis Pharmaceuticals, Inc. | An extended tethering approach for rapid identification of ligands |
-
2002
- 2002-04-24 MX MXPA04004814A patent/MXPA04004814A/es active IP Right Grant
- 2002-04-24 IL IL16140902A patent/IL161409A0/xx unknown
- 2002-04-24 AU AU2002254725A patent/AU2002254725A1/en not_active Abandoned
- 2002-04-24 EP EP08006555A patent/EP1939625A3/en not_active Withdrawn
- 2002-04-24 JP JP2003547631A patent/JP4391237B2/ja not_active Expired - Fee Related
- 2002-04-24 RU RU2004119036/04A patent/RU2295518C2/ru not_active IP Right Cessation
- 2002-04-24 EP EP02723967A patent/EP1446496A4/en not_active Withdrawn
- 2002-04-24 CA CA2464094A patent/CA2464094C/en not_active Expired - Fee Related
- 2002-04-24 WO PCT/US2002/013061 patent/WO2003046200A2/en not_active Ceased
- 2002-04-24 NZ NZ532579A patent/NZ532579A/en not_active IP Right Cessation
-
2005
- 2005-02-09 US US11/054,754 patent/US20050142614A1/en not_active Abandoned
-
2009
- 2009-01-30 JP JP2009020721A patent/JP2009109515A/ja not_active Withdrawn
- 2009-07-22 AU AU2009202965A patent/AU2009202965A1/en not_active Abandoned
-
2011
- 2011-02-25 JP JP2011040912A patent/JP2011140497A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| IL161409A0 (en) | 2004-09-27 |
| US20050142614A1 (en) | 2005-06-30 |
| MXPA04004814A (es) | 2004-08-11 |
| WO2003046200A3 (en) | 2003-07-24 |
| JP2005510574A (ja) | 2005-04-21 |
| EP1446496A4 (en) | 2005-03-30 |
| EP1939625A3 (en) | 2008-09-03 |
| CA2464094A1 (en) | 2003-06-05 |
| NZ532579A (en) | 2006-11-30 |
| JP2011140497A (ja) | 2011-07-21 |
| CA2464094C (en) | 2013-07-02 |
| JP2009109515A (ja) | 2009-05-21 |
| AU2009202965A1 (en) | 2009-08-13 |
| RU2004119036A (ru) | 2005-05-27 |
| RU2295518C2 (ru) | 2007-03-20 |
| WO2003046200A2 (en) | 2003-06-05 |
| EP1446496A2 (en) | 2004-08-18 |
| EP1939625A2 (en) | 2008-07-02 |
| AU2002254725A1 (en) | 2003-06-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4391237B2 (ja) | リガンド発見のための方法 | |
| JP2013116121A (ja) | リガンド発見のための方法 | |
| JP3836791B2 (ja) | リガンドの迅速な同定のための拡張されたテザー化アプローチ | |
| AU2002225731A1 (en) | An extended tethering approach for rapid identification of ligands | |
| US20120077711A1 (en) | Novel Ligands and Libraries of Ligands | |
| US6919178B2 (en) | Extended tethering approach for rapid identification of ligands | |
| CA2719579C (en) | Methods of chemotype evolution | |
| JP2004537594A (ja) | ジスルフィドリガンドおよびチオスルホネートリガンド、ならびにこれらのリガンドを含むライブラリー | |
| ZA200403668B (en) | Methods for ligand discovery | |
| AU2005203048B2 (en) | An extended tethering approach for rapid identification of ligands | |
| HK1063844B (en) | An extended tethering approach for rapid identification of ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050413 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050413 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080730 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20081029 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20081106 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20081126 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20081203 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20081222 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090106 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090130 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090317 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090715 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20090724 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090827 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090901 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090928 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20091007 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121016 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131016 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |