JP4299517B2 - 糖脂質代謝異常症治療剤 - Google Patents
糖脂質代謝異常症治療剤 Download PDFInfo
- Publication number
- JP4299517B2 JP4299517B2 JP2002260534A JP2002260534A JP4299517B2 JP 4299517 B2 JP4299517 B2 JP 4299517B2 JP 2002260534 A JP2002260534 A JP 2002260534A JP 2002260534 A JP2002260534 A JP 2002260534A JP 4299517 B2 JP4299517 B2 JP 4299517B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- galactosidase
- compound
- active ingredient
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images

Landscapes
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Description
【発明の属する技術分野】
本発明は糖脂質代謝異常症処置剤、特にβ-ガラクトシダーゼ遺伝子の変異に起因する糖脂質代謝異常症の処置剤に関する。
【0002】
【従来の技術】
【非特許文献1】
Nature Medicine, 5(1), 112-115(1999)
【非特許文献2】
Bioorganic and Medical Chemistry, 10, 1967-1972(2002)
糖脂質代謝異常症としては、従来GM1ガングリオシドーシス、モルキオB病、クラッベ病、ファブリー病、ゴーシェ病、テイサックス病、サンドホフ病などが知られている。これらの疾病は各種の糖分解酵素が変異を起こして酵素活性を失った結果生じる疾病である。その中でもGM1ガングリオシドーシス、モルキオB病、クラッベ病はβ-ガラクトシダーゼが変異して生ずる疾病であるが、未だ有効な医薬は開発されていない。
【0003】
ところで、ファブリー病はα-ガラクトシダーゼの変異によって生ずる疾病であるが、この疾病はα-ガラクトシダーゼ阻害剤が有効な治療薬となる可能性のあることが【非特許文献1】には記載されている。上記酵素阻害剤が酵素の活性を取り戻すのは、細胞内で発現した変異酵素タンパク質を強力な酵素阻害剤が低濃度で安定化させるというメカニズムによると考えられる。
【0004】
【発明が解決しようとする課題】
酵素阻害剤が変異酵素タンパク質を安定化させ得るのであれば、β-ガラクトシダーゼ遺伝子が変異して惹起される疾病の治療薬又は予防薬として、β-ガラクトシダーゼ阻害剤が有効である可能性が高い。
しかし、ヒトのβ-ガラクトシダーゼを特異的に且つ強力に阻害するβ-ガラクトシダーゼ阻害剤を利用した有用な糖脂質代謝異常症処置剤は未だ得られていない。
【0005】
【課題を解決するための手段】
本発明者らは上記課題に鑑み、β-ガラクトシダーゼ活性を特異的に阻害する特定の糖アナログが、遺伝子変異により酵素活性を減退乃至失ったヒト由来の変異β-ガラクトシダーゼの酵素活性を再生することを見いだし、本発明を完成した。
すなわち、本発明の要旨は下記一般式(1)で表されるカルバ糖アミン誘導体を有効成分として含有する糖脂質代謝異常症処置剤である。
【0006】
【化3】

但し、R1はアルキル基を示し、R2、R3、R4及びR5はそれぞれ独立にヒドロキシル基又は置換基を有するヒドロキシル基である。
【0008】
【発明の実施の形態】
以下、発明の実施の形態により、本発明をより詳細に説明する。
本発明は、上記一般式(1)により表されるカルバ糖アミン誘導体を有効成分として含有する糖脂質代謝異常症処置剤(以下「本発明薬剤」とも記載する)である。
【0009】
ここで「糖脂質代謝異常症処置剤」とは、β-ガラクトシダーゼが変異して生ずる糖脂質代謝異常症(例えばGM1ガングリオシドーシス、モルキオB病、クラッベ病等)が発症した後にその症状を治癒又は緩和するための「治療剤」、及びその発症を予防するための「予防剤」を含む概念である。
【0010】
上記一般式(1)においてR1はアルキル基を示し、該アルキル基としては例えば炭素数1〜23の直鎖又は分枝を有するアルキル基が挙げられ、特に炭素数2〜20の直鎖のアルキル基が好ましい。特に本発明薬剤はスフィンゴ糖脂質の代謝系の異常によって生じる疾病へ適用されるため、その有効成分はスフィンゴ糖脂質のアナログとなることが好ましい。すなわち、一般式(1)中、R1のアルキル基は、生体内に主に存在する炭素数2〜18のアルキル基を有するスフィンゴ糖脂質のアナログとなる、炭素数2〜18のアルキル基であることがもっとも好ましい。
【0011】
R2、R3、R4、及びR5はヒドロキシル基又は置換基を有するヒドロキシル基を示すが、特にヒドロキシル基が好ましい。ここで、R3、R4、R5、及びR6のヒドロキシル基の置換基としては、アラルキル基(ベンジル基、フェネチル基、α-メチルベンジル基等)、シリル基(トリメチルシリル基、トリエチルシリル基、トリイソプロピルシリル(TIPS)基、t-ブチルジフェニルシリル(TBDPS)基、t-ブチルジメチルシリル(TBDMS)基等)、アルカノイル基(アセチル基、ブチリル基等)、アロイル基(ベンゾイル基、トルオイル基、ナフトイル基等)、アルコキシアルキル基(メトキシメチル(MOM)基等)、アラルキルオキシアルキル基(ベンジルオキシメチル(BOM)基等)等が例示される。
【0012】
尚、本発明薬剤の有効成分であるカルバ糖アミン誘導体は擬似糖の1種であるため、本明細書中における炭素番号の付与は、ヘキソースの例に従って下記構造式(2)に示す方法によって記載する。
【0013】
【化4】
また、一般式(1)は5a-カルバ-D-ヘキソピラノースの(5-5a)不飽和誘導体と見なせるため、1位に結合した置換アミノ基(NHR1)が六員環に対して上側にあるものをβ型、反対側にあるものをα型とすると、本発明薬剤の有効成分であるカルバ糖アミン誘導体はβ型である。本発明薬剤の有効成分であるカルバ糖アミン誘導体は、下記一般式(3)に示す物質が本発明薬剤の最も好ましい有効成分である。
【0015】
【化5】
ただし、n=0〜22である。
【0017】
この物質の調製は後述の実施例に記載の方法によって行うことができる。
式(1)で示されるカルバ糖アミン誘導体は、ヒト由来の正常なβ−ガラクトシダーゼに対して特異的且つ強い阻害活性を有すると同時に、変異して酵素活性を失ったヒト由来β−ガラクトシダーゼの活性を生体内において再生する作用を有するため、β−ガラクトシダーゼ遺伝子変異に基づく糖脂質代謝異常症の優れた処置剤となりうる。
【0018】
尚、本発明薬剤の有効成分であるカルバ糖アミン誘導体が有するβ-ガラクトシダーゼ阻害活性は、β-ガラクトシダーゼと基質が存在する溶液中に、被検物質を添加し、酵素活性を本発明物質無添加の場合と比較することで、算出することが可能である。
【0019】
上記本発明薬剤は、錠剤、カプセル剤、液剤、注射剤、顆粒剤、散剤、リポ化剤、吸入散剤等、経口投与、注射等の投与ルート、目的、対象等に応じて製剤化することができる。本発明薬剤中における有効成分の濃度は特に限定はされないが、0.001〜5%とするのが好ましい。例えば本発明薬剤を経口投与用の液剤とする場合には、0.01%(W/V)以上とするのが好ましく、0.03%〜0.2%(W/V)とするのが最も好ましい。また、筋注又は静注用の注射剤とする場合には0.01%(W/V)以上とするのが好ましく、0.03%(W/V)以上とするのが最も好ましい。
【0020】
【実施例】
以下、本発明を実施例により具体的に説明する。カルバ糖アミン誘導体(R1=(CH2)7CH3:N-オクチル-5a-カルバ-α-L-arabino-ヘキソ-5(5a)-エノピラノシルアミン)の合成
【0021】
(1)N-オクチル-N-tert-ブトキシカルボニル-1-デオキシ-2,3;4,6-di-O-イソプロピリデン- 5a-カルバ-β-D-xylo-ヘキソ-5(5a)-エノピラノシルアミン(化合物1)の合成
【0022】
【化6】
式中「Boc」はtert-ブトキシカルボニル基を示す。以下同様である。
【0024】
Bioorganic & Medicinal Chemistry Letters, 6 (1996) 929-932に記載された方法で調製したN-オクチル-2,3:4,6-Di-O-イソプロピリジン-5a-カルバ-β-D-xylo-ヘキソ-5(5a)-エノピラノシルアミン(644mg、1.75mmol)のジクロロメタン溶液(13ml)にトリエチルアミン(0.97mL,7.01mmol)とdi-tart-ブチルジカルボネート(0.81mL、3.50mmol)を添加し、1.5時間室温で攪拌した。トリエチルアミン(0.97mL、7.01mmol)tert-ブチルジカルボネート(0.81mL、3.50mmol)を加えた後、更に1時間攪拌を継続した。反応混液を180mLの酢酸エチルで希釈した後、溶液を60mLの炭酸水素ナトリウム溶液、その後60mLの食塩水で2回洗浄し、その後乾燥して濃縮した。
【0025】
粗精製物はシリカゲルクロマトグラフィー(50g、濃度勾配溶出、1:11→1:8 酢酸エチル/ヘキサン)で精製し、無色の油状物質として化合物1を得た(807mg、収率99%)。
【0026】
TLC:Rf=0.49(1:5 酢酸エチル/トルエン)
[α]22 D:−61°(c=0.90 クロロホルム)
IR(Neat):υ(cm-1)=2960(CH3)、2930及び2855(CH2)、1695(アミド)
1H NMR(300MHz、(CD3)2SO, 110℃)
δ=5.22(br s, 1H, H-5a), 4.59 (br d, 1H, J4,3=9.3Hz, 4-H), 4.57(br d, 1H, J1,2=9.3Hz, 1-H), 4.40 and 4.13 (2d, each 1H, Jgem=13.7Hz, 6,6-H), 3.74 (dd, 1H, J2,3=9.3Hz, 2-H), 3.59 (dd, 1H, 3-H), 3.11 and 2.93 (2m, each 1H, NCH2), 1.54-1.18 (m, 12H, 6×CH2), 1.47, 1.37, 1.36 and 1.29 (4s, each 3H, 2×CMe2), 1.40 (s, 9H, t-Bu), 0.86 (t, 3H, J=6.6Hz, CH2CH3)
Anal. Calcd for C26H45NO6: C,66.78; H,9.70; N, 3.00.
Found: C66.57; H, 10.10; N, 3.11.
【0027】
(2)N-オクチル-N-tert-ブトキシカルボニル-1-デオキシ-5a-カルバ-β-D-xylo-ヘキソ-5(5a)-エノピラノシルアミン(化合物2)の合成
【0028】
【化7】
化合物1(768mg、1.64mmol)と60%の酢酸溶液(16mL)を60℃で30分間攪拌し、濃縮した。残渣をエタノールで3回エバポレートした。粗精製物はシリカゲル(47g)によりカラムクロマトグラフィー(濃度勾配溶出法、1:10→1:4 エタノール/トルエン)により精製し(539mg、収率85%)、無色の油状物質として得た。
【0030】
TLC:Rf=0.54(1:5 メタノール/クロロホルム)
[α]20 D:−87°(c=1.10 メタノール)
IR(KBr-Disk):υ(cm-1)=3445(OH)、2960(CH3)、2930及び2855(CH2)
1H NMR(270MHz、1:2 CD3OD/CDCl3)
δ=5.42(br s, 1H, H-5a), 4.38-4.05 (m, 2H, H-1, H-4), 4.20 and 4.10 (2d, each 1H, Jgem=13.4Hz, 6,6-H), 3.72 (dd, 1H, J1,2=9.2Hz, J2,3=9.5Hz, 2-H), 3.31 (dd, 1H, J2,3=9.5Hz, J3,4=8.0Hz, 3-H), 3.26 (m, 1H, 1'a-H), 3.01 (m, 1H, 1'b-H), 1.73-1.18 (m, 12H, CH2×6: H-2'-H-7'), 1.47 (s, 9H, t-Bu: Boc), 0.89 (t, 3H, J=6.4Hz, CH2CH3)
【0031】
(3)N-オクチル- N-tert-ブトキシカルボニル-4,6-O-ベンジリデン-5a-カルバ-β-D-xylo-ヘキソ-5(5a)-エノピラノシルアミン(化合物3)の合成
【0032】
【化8】
化合物2(242mg, 0.624mmol)のDMF溶液(6mL)にα,α-ジメトキシトルエン(112μL, 0.746mmol)、p-トルエンスルホン酸・H2O(12mg, 0.063mmol)を添加し、減圧条件下45℃で3.5時間攪拌した。更にα,α-ジメトキシトルエン(50μL, 0.333mmol)を添加し、更に2時間攪拌した。反応混液を60mLの酢酸エチルで希釈し、この溶液を20mLの水、次に20mLの飽和炭酸水素ナトリウム水溶液、更に20mLの水で洗浄し、硫酸ナトリウムで乾燥し、ろ過後濃縮した。この濃縮物を薄層クロマトグラフィーで分析した結果、2種類の成分が検出された(Rf 0.33及び0.74:アセトン/トルエン、1:5; Rf 0.05及び0.70:酢酸エチル/トルエン、1:5)。
【0034】
粗精製物をシリカゲルクロマトグラフィー(16g、濃度勾配溶出法、1:24→1:3酢酸エチル/トルエン)を行い、無色の油状物質である2,3:4,6-di-O-ベンジリデン化化合物(化合物4:最初の溶出画分:159mg、収率45%)及び無色の油状物質である化合物3を得た(化合物3:164mg、収率55%)。
【0035】
化合物4を5mLのメタノールに溶解した後、0℃でp-トルエンスルホン酸一水和物(3mg、0.016mmol)を添加し、10分間攪拌した後、トリエチルアミンを滴下して中和し、濃縮した。粗精製物をシリカゲルクロマトグラフィー(7g、1:7アセトン/トルエン)で精製し、化合物3を得た(110mg、化合物2からの直接の反応生成物と合わせた収率82%)。
【0036】
TLC:Rf=0.33(1:5 アセトン/トルエン)
[α]22 D:−57°(c=0.97 クロロホルム)
IR(neat):υ(cm-1)=3420(OH)、2960(CH3)、2925及び2855(CH2)、1695(アミド)
1H NMR(300MHz、(CD3)2SO、110℃)
δ=7.46-7.27(m, 5H, Ph), 5.65 (s, 1H, CHPh), 5.31 (br s, 1H, 5a-H)、4.39(br s、2H、6,6-H), 4.36 (br d, 1H, J3,4=7.6Hz, 4-H), 4.16 (br s, 1H, 1-H), 3.64 (dd, 1H, J1,2= J2,3=9.3Hz, 2-H), 3.52 (dd, 1H, 3-H), 3.25-2.80 (m, 2H, NCH2), 1.57-1.17 (m, 12H, 6×CH2), 1.39 (s, 9H, CMe3), 0.87 (t, 3H, J=6.2Hz, CH2CH3).
元素分析
計算値 C27H41NO6・0.5H2O: C,66.92; H,8.73; N, 2.89.
実測値: C, 66.94; H, 8.85; N, 2.91.
【0037】
(4)N-オクチル-N-tert-ブトキシカルボニル-4,6-O-ベンジリデン-2,3-di-O-メトキシメチル-5a-カルバ-β-D-xylo-ヘキソ-5(5a)-エノピラノシルアミン(化合物5)の合成
【0038】
【化9】
化合物3(269mg、0.566mmol)の1,2-ジクロロエタン溶液(7mL)にクロロメチルエーテル(0.43mL、5.66mmol)及びN,N-ジイソプロピルエチルアミン(1.97mL、11.31mmol)を添加し、60℃で3時間攪拌した。反応混液を60mLのクロロホルムで希釈し、30mLの1mol/L塩酸、30mLの飽和炭酸水素ナトリウム水溶液、30mLの水で順に洗浄し、濃縮した。粗精製物をシリカゲルクロマトグラフィー(20g、濃度勾配溶出、1:14→1:9 酢酸エチル/トルエン)を行い、化合物5を無色油状物質として得た(315mg、収率99%)。
【0040】
TLC:Rf=0.44(1:5 酢酸エチル/トルエン)
[α]21 D:−126°(c=1.10 クロロホルム)
IR(neat):υ(cm-1)=2960(CH3)、2925及び2855(CH2)、1695(アミド)
1H NMR(300MHz、(CD3)2SO、110℃)
δ=7.45-7.28(m, 5H, Ph), 5.70 (s, 1H, CHPh), 5.38 (br s, 1H, 5a-H)、4.79 and 4.77(2d, each 1H, Jgem=6.1Hz)and 4.75 and 4.63 (2d, each 1H, Jgem=6.3Hz)(2×OCH2), 4.57 (br d, 1H, J3,4=7.7Hz, 4-H), 4.43 (br s, 2H, 6,6-H), 4.27 (br s, 1H, 1-H), 3.99 (dd, 1H, J1,2=9.3Hz, J2,3=10.0Hz, 2-H), 3.78 (dd, 1H, 3-H), 3.28-3.27 (2s, each 3H, 2×OMe), 3.07-2.87 (m, 2H, NCH2), 1.62-1.20 (m, 12H, 6×CH2), 1.39 (s, 9H, t-Bu), 0.87 (t, 3H, J=6.5Hz, CH2CH3)
元素分析
計算値 C31H49NO8: C,66.05; H,8.76; N, 2.48.
実測値: C, 65.80; H, 8.99; N, 2.42.
【0041】
(5)N-オクチル-N-tert-ブトキシカルボニル-2,3-di-O-メトキシメチル-5a-カルバ-β-D-xylo-ヘキソ-5(5a)-エノピラノシルアミン(化合物6)の合成
【0042】
【化10】
化合物5(315mg、0.559mmol)の60%酢酸水溶液(8mL)を60℃で2時間攪拌して濃縮した。残渣をエタノールを用いて3回エバポレートした後、トルエンを用いて3回エバポレートした。化合物6の粗精製物をシリカゲルクロマトグラフィー(20g、濃度勾配溶出、1:6→1:5 アセトン/トルエン)で無色の油状物質として得た(216mg、収率83%)。
【0044】
TLC:Rf=0.13(1:5 アセトン/トルエン)
[α]20 D:−141°(c=1.11 クロロホルム)
IR(neat):υ(cm-1)=3440(OH)、2955(CH3)、2925及び2855(CH2)、1695(アミド)
1H NMR(300MHz、(CD3)2SO、110℃)
δ=5.32 (br s, 1H, H-5a)、4.83 and 4.76(2d, each 1H, Jgem=5.6Hz, CH2:MOM), 4.73 and 4.61 (2d, each 1H, Jgem=5.9Hz, CH2:MOM), 4.23 (br s, 1H, 1-H), 4.10 (br s, 1H, 4-H), 3.99 (br s, 2H, 6,6-H), 3.84 (dd, 1H, J1,2=8.8Hz, J2,3=9.5Hz, 2-H), 3.52 (dd, 1H, J2,3=9.5Hz, J3,4=7.8Hz, 3-H), 3.36 and 3.26 (2s, each 3H, 2×CH3: MOM), 3.26-2.83 (m, 2H, H-1'a, 1'b-H), 1.63-1.17 (m, 12H, CH2×6: H-2'-H-7'), 1.38 (s, 9H, t-Bu: BOC), 0.86 (t, 3H, J=6.1Hz, CH2CH3)
【0045】
(6)N-オクチル- N-tert-ブトキシカルボニル-2,3-di-O-メトキシメチル-6-O-tert-ブチルジメチルシリル-5a-カルバ-β-D-xylo-ヘキソ-5(5a)-エノピラノシルアミン(化合物7)の合成
【0046】
【化11】
化合物6(159mg、0.334mmol)のN,N'-ジメチルホルムアミド(以下「DMF」と記載する)溶液(5mL)にイミダゾール(91mg、1.34mmol)及びtert-ブチルクロロジメチルシラン(100mg、0.670mmol)を添加し、室温で1時間攪拌した。反応混液を60mLの酢酸エチルで希釈し、20mLの水で3回洗浄し、硫酸ナトリウムで乾燥、ろ過後、濃縮した。化合物7の粗精製物は、シリカゲルクロマトグラフィーグラフィー(20g、1:6 酢酸エチル/トルエン)により無色の油状物質として得られた(194mg、収率98%)。
【0048】
TLC:Rf=0.24(1:5 酢酸エチル/トルエン)
[α]23 D:−101°(c=0.95 クロロホルム)
IR(neat):υ(cm-1)=3445(OH)、2955(CH3)、2925及び2855(CH2)、1695(アミド)
1H NMR(300MHz、(CD3)2SO、110℃)
δ=5.36 (br s, 1H, 5a-H)、4.83 and 4.76(2d, each 1H, Jgem=6.1Hz)and 4.73 and 4.60 (2d, each 1H, Jgem=6.3Hz)(2×OCH2), 4.21 and 4.11(ABq, Jgem=13.9Hz, 6,6-H), 4.12-4.04 (m, 2H, 1-H, 4-H), 3.87 (dd, 1H, J1,2=9.3Hz, J2,3=9.9Hz, 2-H), 3.53 (dd, 1H, J3,4=7.9Hz, 3-H), 3.35 and 3.26 (2s, each 3H, 2×OCH3), 3.05-2.88 (m, 2H, NCH2), 1.60-1.20 (m, 12H, 6×CH2), 1.38 (s, 9H, OCMe3), 0.89 (s, 9H, CCMe3), 0.86 (t, 3H, J=6.8Hz, CH2CH3), 0.05 (s, 6H, SiMe2).
元素分析
計算値 C30H59NO8Si: C,61.08; H,10.08; N, 2.37.
実測値: C, 60.82; H, 10.38; N, 2.45.
【0049】
(7)N-オクチル- N-tert-ブトキシカルボニル-2,3-di-O-メトキシメチル-6-O-tert-ブチルジメチルシリル-5a-カルバ-α-L-arabino-ヘキソ-5(5a)-エノピラノシルアミン(化合物8)の合成
【0050】
【化12】
化合物7(75mg、0.127mmol)のジクロロメタン溶液(2mL)に粉末状の4Åモレキュラーシーブ(75mg)とピリジニウムクロロクロメート(41mg、0.190mmol)を加え、室温で1時間攪拌した。ピリジニウムクロロクロメート(41mg、0.190mmol)を更に加え、1時間更に攪拌した。セライトフィルターを通した後、短いシリカゲルカラムに通した(溶出液:ジエチルエーテル)。溶出液をエバポレートし、粗精製のケトンを得た後、ケトン体をテトラヒドロフラン(以下「THF」と記載する)(0.7mL)に溶解し、1mol/L(以下「mol/L」を「M」とも記載する)リチウム-tri-sec-ブチル水素化ホウ素/THF溶液(0.51mL、0.51mmol)で、アルゴン雰囲気下-78℃で30分間処理した。反応混液を0℃まで暖め、飽和塩化アンモニウムにより反応を停止した。この反応混液に硫酸マグネシウムを添加した後、セライトろ過した。溶出液をエバポレートし、シリカゲルクロマトグラフィー(7g、濃度勾配溶出、1:9→1:6 酢酸エチル/トルエン)により無色の油状物質として化合物8を得た(49.1mg、収率66%)。
【0052】
TLC:Rf=0.16(1:5 酢酸エチル/トルエン)
[α]23 D:−68°(c=1.125 クロロホルム)
IR(neat):υ(cm-1)=3460(OH)、2955(CH3)、2930及び2855(CH2)、1695(アミド)
1H NMR(300MHz、(CD3)2SO、110℃)
δ=5.36 (br s, 1H, 5a-H)、4.71 and 4.59(2d, each 1H, Jgem=5.6Hz,)and 4.75-4.66 (m, 2H)(2×OCH2), 4.38 (br s, 1H, 1-H), 4.19 and 4.12 (2d, each 1H, Jgem=13.2Hz, 6,6-H), 4.13-4.02 (m, 2H, 2-H, 4-H), 3.46 (dd, 1H, J2,3=10.3Hz, J3,4=2.0Hz, 3-H), 3.33 and 3.25 (2s, each 3H, 2×OM2), 3.10-2.86 (m, 2H, 1',1'-H), 1.62-1.16 (m, 12H, 6×CH2), 1.39 (s, 9H, CCMe3), 0.89 (s, 9H, SiCMe3), 0.86 (t, 3H, J=7.3Hz, CH2CH3), 0.05 (s, 6H, SiMe2).
元素分析
計算値 C30H59NO8Si: C,61.08; H,10.08; N, 2.37.
実測値: C, 60.80; H, 10.37; N, 2.53.
【0053】
(8)N-オクチル-5a-カルバ-α-L-arabino-ヘキソ-5(5a)-エノピラノシルアミン(一般式(1)(R1=オクチル基、R2、R3、R4、R5=ヒドロキシル基)のカルバ糖アミン誘導体)の合成
【0054】
【化13】
THF(0.5mL)に溶解した化合物8(29mg、0.049mmol)に4mol/L塩酸(1.5mL)を添加し、65℃で1.5時間攪拌し、濃縮した。残渣をエタノールで3回エバポレートした。生成物をイオン交換カラム(Dowex 50-X2(H+))により精製した。溶出には1%メタノール性アンモニウムを用い、式(1)のカルバ糖アミン誘導体(以下「有効成分1」とも記載する)を白色固体として得た(12.8mg、収率91%)。
【0056】
融点:126〜128℃
TLC:Rf=0.76(35:60:5メタノール/クロロホルム/水)
[α]23 D:+16°(c=0.64 メタノール)
IR(KBr-Disk):υ(cm-1)=3485(OH)、3250(アミン)、2960(CH3)、2925及び2855(CH2)
1H NMR(300MHz、1:2 CD3OD/CHCl3)
δ=5.73 (d, 1H, J1,5a =1.8Hz、5a-H)、4.16 (d, 1H, J3,4=4.2Hz, 4-H), 4.16 (br s, 2H, 6,6-H), 3.64 (dd, 1H, J1,2=8.1Hz, J2,3=10.0Hz, 2-H), 3.48 (dd, 1H, 3-H), 3.12 (dd, 1H, 1-H), 2.78(ddd, 1H, J1'a,2'=7.4Hz, Jgem=11.2Hz)and 2.57 (ddd, 1H)(NCH2), 1.64-1.21(m, 12H, 6×CH2), 0.89 (t, 3H, J=6.7Hz, CH2CH3).
元素分析
計算値 C15H29NO4: C,62.69; H,10.17; N, 4.87.
実測値: C, 62.67; H, 10.47; N, 5.01.
【0057】
<2>ガラクトシダーゼ阻害活性の測定
ヒト正常β-ガラクトシダーゼ(GP8(Brain Dev., 23(5), 284-287, 2001))cDNAを導入しSV40ウイルス遺伝子で不死化したノックアウトマウス皮膚線維芽細胞を37℃、5%CO2、10%牛胎児血清(FCS)を含むダルベッコの改変イーグル培地(DMEM:ギブコ社製)で培養した。細胞が8割コンフルエントになった状態で10mM NH4Clを含む FCS非添加DMEMに培地交換し24時間培養した後、培養上清を回収し、10mM リン酸ナトリウム緩衝液(pH=6.5)で4時間透析した。その後、VIVAPORE-20(Vivapore社製)で濃縮し酵素源として使用した。蛍光基質として4-メチルウンベリフェリル-β-D-ガラクトピラノシドを使用し、被検物質の存在下又は非存在下で、37℃、30分間の酵素活性を測定した(表1)。被検物質の最終濃度は、0.05、0.125、0.25、0.5、1、2.5、5μMとした。
【0058】
【表1】
<3>変異β-ガラクトシダーゼ活性の測定
β-ガラクトシダーゼ欠損ノックアウトマウスの培養皮膚繊維芽細胞にSV40ウイルス遺伝子を導入して不死化し、pSV2neoと発現ベクターに組み込まれた酵素遺伝子を同時に導入し、モデル細胞を樹立した(Brain Dev., 23(5), 284-287, 2001)。用いた遺伝子は、ヒト正常(GP8)および変異β-ガラクトシダーゼ遺伝子Y316C、G123R(乳児型GM1-ガングリオシドーシス)、R201C(若年型GM1-ガングリオシドーシス)、I51T、T82M、R201H、P263S、R457Q(成人型GM1-ガングリオシドーシス)、W273L、Y83H、R482H、R482C(モルキオB病)である(Hum. Genet., 93(2), 109-114, 1994)。
【0060】
対照物質として、0.5mM N-(n-ブチル)-デオキシガラクトノジリマイシン(NB-DGJ:The Journal of Biological Chemistry 269, 27108-27114 (1994))を使用し、有効成分1は0.2μmol/lで使用した。
【0061】
具体的には、有効成分1を含む細胞培養液(10%FCS DMEM)で、モデル細胞を4日間培養した後、細胞を回収して細胞浮遊液のタンパク量が一定になるように希釈した後、細胞希釈液の酵素活性を測定した。蛍光基質として4-メチルウンベリフェリル-β-D-ガラクトピラノシドを使用し、37℃、30分間の酵素活性を測定した(表2)。その結果、T82M以外で酵素活性の増大が観察され、R201C、R201H、R457Q、W273L、Y83Hなどの遺伝子の変異による酵素活性の喪失に対しては、対照と比して150%以上に酵素活性を増強し、かつ何れも有効成分1で高い活性を示していることから特に本発明薬剤としての有用性を示唆する結果が得られた。
【0062】
【表2】
<4>有効成分1の組織移行性及び活性回復への効果
本発明薬剤を経口投与した際に、有効成分が組織に移行してβ-ガラクトシダーゼ活性の回復に効果を示すことを確認するために1週間の投与実験をマウスを用いて行なった。
【0064】
すなわちβ-ガラクトシダーゼ欠損ノックアウトマウスに、ヒト変異β-ガラクトシダーゼ遺伝子R201Cを導入して作り出した組換えマウス(3ヶ月令)に対して、有効成分1を1mM含有する水道水を飲料水として1週間、継続して投与した。1週間経過した後、マウスを安楽死させ、大脳、小脳、心臓、肺、肝臓、脾臓、腎臓、筋肉(大腿筋)、及び血漿を採取した。また同一個体に於ける投与前後のβ-ガラクトシダーゼ活性の変化を確認するために尾の組織を使用した。各臓器をホモジナイザーを用いて氷冷下で破砕した後、細胞破砕液を8,000×gで30分間遠心分離処理を行なった。その後、上清画分を回収し、酵素溶液とした。この酵素溶液のタンパク質量をブラッドフォード法により測定し、β-ガラクトシダーゼ活性の測定を行なった。β-ガラクトシダーゼ活性の測定は、蛍光基質として4-メチルウンベリフェリル-β-D-ガラクトピラノシドを使用し、37℃、30分間の酵素活性を測定した。各々対照として有効成分1を含まない水道水を用いて1週間飼育した上記組換えマウスを用い、さらに同一個体の飼育前後に於ける酵素活性の変化を調べるために、実験群と同様に尾を用いた(陰性対照)。各群とも3匹の個体を用いてその平均値を求めた。なお、いずれのマウスは1日に約5mlの飲料水を飲んだため、1日あたり約1.4mg、一週間で約10.2mgの有効成分1を経口で摂取したことになる。
【0065】
その結果、陰性対照においては同一個体の飼育前後で全くβ-ガラクトシダーゼ活性に変化が見られなかったのに対し(表3:尾(非投与群))、実験群においては同一個体の飼育前後で8倍近くまでβ-ガラクトシダーゼ活性が回復していることが判明した(表3:尾(投与群))。
【0066】
また、各臓器とも、実験群においてβ-ガラクトシダーゼ活性が大幅に増強されていることが明かとなり、最もβ-ガラクトシダーゼ活性の増強が低かった肝臓に於いて4.7倍、最も高い増強を示した脾臓に於いては17.1倍を示した。実験群においては平均9.1倍にβ-ガラクトシダーゼ活性が増強されていることが明かとなった。特に、大脳、小脳に於いても高いβ-ガラクトシダーゼ活性増強効果が得られていることから、本発明薬剤が中枢神経系の神経疾患治療剤として極めて有用であることが示された。β-ガラクトシダーゼの変異によって生ずる疾病は、本来β-ガラクトシダーゼによって分解されるべき物質の蓄積によって生ずるが、大脳や小脳で得られた活性(50nmol/mgタンパク質/30分)程度でも十分に疾病の症状を緩和し、症状の発現を防ぐことができる。なお、何れの実験群に於いてもマウスの死亡例は見られず、実験期間を通じて極めて健康状態が良好であったことから、有効成分1は生体に対して安全性が高いことが示唆された。
【0067】
【表3】
【発明の効果】
本発明により新規な糖脂質代謝異常症治療剤が提供される。
【図面の簡単な説明】
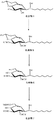
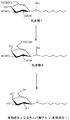
【図1】 本発明薬剤の好ましい有効成分である化合物の合成過程を示すスキーム中、出発物質から化合物3の合成ルートを示す図である。
【図2】 本発明薬剤の好ましい有効成分である化合物の合成過程を示すスキーム中、化合物3から化合物7の合成ルートを示す図である。
【図3】 本発明薬剤の好ましい有効成分である化合物の合成過程を示すスキーム中、化合物7から目的物質の合成ルートを示す図である。
Claims (3)
- 下記一般式(1)で表されるカルバ糖アミン誘導体を有効成分とするβ−ガラクトシダーゼ遺伝子の変異に起因する糖脂質代謝異常症の処置剤。
但し、R1はアルキル基を示し、R2、R3、R4及びR5はそれぞれ独立にヒドロキシル基又はアラルキル基、シリル基、アルカノイル基、アロイル基、アルコキシアルキル基若しくはアラルキルオキシアルキル基から選択される置換基を有するヒドロキシル基である。 - R1は炭素数1〜23の直鎖又は分枝を有するアルキル基であり、R2、R3、R4及びR5がそれぞれヒドロキシル基である請求項1記載の処置剤。
- 経口投与用製剤であることを特徴とする請求項1又は2記載の処置剤。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002260534A JP4299517B2 (ja) | 2001-09-07 | 2002-09-05 | 糖脂質代謝異常症治療剤 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001-272776 | 2001-09-07 | ||
| JP2001272776 | 2001-09-07 | ||
| JP2002260534A JP4299517B2 (ja) | 2001-09-07 | 2002-09-05 | 糖脂質代謝異常症治療剤 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2003155229A JP2003155229A (ja) | 2003-05-27 |
| JP2003155229A5 JP2003155229A5 (ja) | 2005-10-27 |
| JP4299517B2 true JP4299517B2 (ja) | 2009-07-22 |
Family
ID=26621877
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002260534A Expired - Fee Related JP4299517B2 (ja) | 2001-09-07 | 2002-09-05 | 糖脂質代謝異常症治療剤 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4299517B2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018200618A1 (en) * | 2017-04-25 | 2018-11-01 | Amicus Therapeutics, Inc. | Novel compositions for preventing and/or treating degenerative disorders of the central nervous system and/or lysosomal storage disorders |
-
2002
- 2002-09-05 JP JP2002260534A patent/JP4299517B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2003155229A (ja) | 2003-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8211945B2 (en) | Carba-sugar amine derivative and glycolipid metabolic disorder treating agent containing the same as active ingredient | |
| TWI584808B (zh) | 抗氧化發炎調節劑:c-17同系齊墩果酸衍生物 | |
| CA2998681C (en) | C4-modified oleanolic acid derivatives for inhibition of il-17 and other uses | |
| JP7036792B2 (ja) | スピロ-ラクタムnmda受容体修飾因子およびその使用 | |
| AU2018217236A1 (en) | Synthesis of polycyclic-carbamoylpyridone compounds | |
| EP2039361A2 (en) | Use of immunomodulatory compounds | |
| EA019263B1 (ru) | Антиоксидативные модуляторы воспаления: производные олеанолевой кислоты с аминовыми и другими модификациями на с-17 | |
| JP3947229B2 (ja) | デプシペプチドおよびこれを有効成分とする医薬 | |
| JP2003520783A (ja) | ウイルス感染を二重ターゲティングおよびガン細胞をターゲティングするための組成物および方法 | |
| JPH05178820A (ja) | 含フッ素ビタミンd3類縁体およびこれらを有効成分とする医薬組成物 | |
| JPH05509327A (ja) | 抗炎症および抗ウイルス活性を有する蛋白質キナーゼcモジュレーター | |
| WO2005063788A1 (ja) | ベンズイミダゾール誘導体及びその医薬用途 | |
| JP4299517B2 (ja) | 糖脂質代謝異常症治療剤 | |
| LU86491A1 (fr) | Nouveaux disaccharides,leur preparatione et leur utilisation comme medicaments | |
| JP2001213858A (ja) | スフィンゴシン誘導体 | |
| JPH0925235A (ja) | 1−o−アシルグリセロール−2,3−ホスフェート誘導体を有効成分とするがん転移抑制剤 | |
| JP7082977B2 (ja) | レスベラトロールのシリル化誘導体および神経変性疾患、神経疾患、または炎症性疾患におけるその使用 | |
| JP3688337B2 (ja) | ピリピロペン誘導体 | |
| JP4057264B2 (ja) | カルバ糖アミン誘導体 | |
| JP5788653B2 (ja) | 糖鎖修飾ヌクレオチド及びその使用 | |
| JP2023518399A (ja) | ファルネソイドx受容体アゴニストの結晶質形態 | |
| JPH0692986A (ja) | 置換ヌクレオシド誘導体、その製造法およびそれを含む医薬組成物 | |
| JPH07188205A (ja) | α−グルコシダーゼ阻害剤 | |
| Kobayashi et al. | Syntheses of 2, 6-dideoxy-6-fluoro-2-[(3R and 3S)-3-hydroxytetradecanamido]-3-O-[(3R)-3-(tetradecanoyloxy)-tetradecanoyl]-d-glucopyranose 4-(dihydrogen phosphate) and 2-deoxy-2-[(3R and 3S)-3-hydroxytetradecanamido]-3-O-[(3R)-3-(tetradecanoyloxy) tetradecanoyl]-α-d-glucopyranosyl fluoride 4-(dihydrogen phosphate): fluorosugar analogues of GLA-60 | |
| CN114805454A (zh) | α-半乳糖神经酰胺类化合物及其制备方法和用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050825 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050825 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090220 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090319 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090409 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090417 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120424 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120424 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130424 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130424 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130424 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140424 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |