JP4230228B2 - Dye-sensitized photoelectric conversion element - Google Patents
Dye-sensitized photoelectric conversion element Download PDFInfo
- Publication number
- JP4230228B2 JP4230228B2 JP2003007360A JP2003007360A JP4230228B2 JP 4230228 B2 JP4230228 B2 JP 4230228B2 JP 2003007360 A JP2003007360 A JP 2003007360A JP 2003007360 A JP2003007360 A JP 2003007360A JP 4230228 B2 JP4230228 B2 JP 4230228B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- photoelectric conversion
- dye
- conversion element
- ring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000006243 chemical reaction Methods 0.000 title claims description 64
- 125000001424 substituent group Chemical group 0.000 claims description 73
- 150000001875 compounds Chemical class 0.000 claims description 68
- 239000000975 dye Substances 0.000 claims description 67
- 239000004065 semiconductor Substances 0.000 claims description 66
- 239000010419 fine particle Substances 0.000 claims description 35
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 25
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 21
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 19
- 125000000217 alkyl group Chemical group 0.000 claims description 16
- 125000004432 carbon atom Chemical group C* 0.000 claims description 12
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 7
- 125000003545 alkoxy group Chemical group 0.000 claims description 6
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- 125000001624 naphthyl group Chemical group 0.000 claims description 5
- 239000000434 metal complex dye Substances 0.000 claims description 4
- 239000004408 titanium dioxide Substances 0.000 claims description 2
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 claims 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims 2
- 125000004663 dialkyl amino group Chemical group 0.000 claims 1
- 239000012860 organic pigment Substances 0.000 claims 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 123
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 35
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 31
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 28
- 239000010409 thin film Substances 0.000 description 28
- 125000000623 heterocyclic group Chemical group 0.000 description 27
- -1 benzoindolenine Chemical compound 0.000 description 25
- 150000004945 aromatic hydrocarbons Chemical group 0.000 description 24
- 239000000243 solution Substances 0.000 description 23
- 230000015572 biosynthetic process Effects 0.000 description 22
- 238000003786 synthesis reaction Methods 0.000 description 22
- 239000003792 electrolyte Substances 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 15
- 238000010521 absorption reaction Methods 0.000 description 14
- 238000000034 method Methods 0.000 description 14
- 239000000758 substrate Substances 0.000 description 14
- 125000002252 acyl group Chemical group 0.000 description 13
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 13
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical group C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 12
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 12
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 12
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 11
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- 125000003118 aryl group Chemical group 0.000 description 10
- 229910052736 halogen Inorganic materials 0.000 description 10
- 238000002844 melting Methods 0.000 description 10
- 230000008018 melting Effects 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 9
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical group C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 8
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 8
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 8
- 150000002367 halogens Chemical class 0.000 description 8
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 239000000203 mixture Substances 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 7
- 125000003277 amino group Chemical group 0.000 description 7
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 7
- 150000002500 ions Chemical class 0.000 description 7
- 239000002244 precipitate Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 6
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 6
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 6
- 125000005843 halogen group Chemical group 0.000 description 6
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 6
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 6
- 239000002002 slurry Substances 0.000 description 6
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 230000005525 hole transport Effects 0.000 description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Inorganic materials [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- ODIRBFFBCSTPTO-UHFFFAOYSA-N 1,3-selenazole Chemical compound C1=C[se]C=N1 ODIRBFFBCSTPTO-UHFFFAOYSA-N 0.000 description 4
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical compound C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 4
- JGRMXPSUZIYDRR-UHFFFAOYSA-N 2-(4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl)acetic acid Chemical compound OC(=O)CN1C(=O)CSC1=S JGRMXPSUZIYDRR-UHFFFAOYSA-N 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 4
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 4
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 4
- CUFNKYGDVFVPHO-UHFFFAOYSA-N azulene Chemical compound C1=CC=CC2=CC=CC2=C1 CUFNKYGDVFVPHO-UHFFFAOYSA-N 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 235000019416 cholic acid Nutrition 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- 239000008151 electrolyte solution Substances 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 4
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 4
- 239000011630 iodine Substances 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 description 4
- 239000011259 mixed solution Substances 0.000 description 4
- 239000012046 mixed solvent Substances 0.000 description 4
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 4
- 239000000049 pigment Substances 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 239000002798 polar solvent Substances 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 229930192474 thiophene Natural products 0.000 description 4
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 4
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 3
- AIGNCQCMONAWOL-UHFFFAOYSA-N 1,3-benzoselenazole Chemical compound C1=CC=C2[se]C=NC2=C1 AIGNCQCMONAWOL-UHFFFAOYSA-N 0.000 description 3
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 3
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 3
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 3
- RVBUGGBMJDPOST-UHFFFAOYSA-N 2-thiobarbituric acid Chemical compound O=C1CC(=O)NC(=S)N1 RVBUGGBMJDPOST-UHFFFAOYSA-N 0.000 description 3
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 3
- OOWFYDWAMOKVSF-UHFFFAOYSA-N 3-methoxypropanenitrile Chemical compound COCCC#N OOWFYDWAMOKVSF-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 239000004380 Cholic acid Substances 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 150000004703 alkoxides Chemical class 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- HNYOPLTXPVRDBG-UHFFFAOYSA-N barbituric acid Chemical compound O=C1CC(=O)NC(=O)N1 HNYOPLTXPVRDBG-UHFFFAOYSA-N 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 3
- 229960002471 cholic acid Drugs 0.000 description 3
- QZHPTGXQGDFGEN-UHFFFAOYSA-N chromene Chemical compound C1=CC=C2C=C[CH]OC2=C1 QZHPTGXQGDFGEN-UHFFFAOYSA-N 0.000 description 3
- 238000009833 condensation Methods 0.000 description 3
- 230000005494 condensation Effects 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 229910052738 indium Inorganic materials 0.000 description 3
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- YLGYACDQVQQZSW-UHFFFAOYSA-N n,n-dimethylprop-2-enamide Chemical compound CN(C)C(=O)C=C YLGYACDQVQQZSW-UHFFFAOYSA-N 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 125000003396 thiol group Chemical group [H]S* 0.000 description 3
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 3
- QJGKCQWQNOPAMG-UHFFFAOYSA-N 1,4-dithiophen-2-ylbutane-1,4-dione Chemical compound C=1C=CSC=1C(=O)CCC(=O)C1=CC=CS1 QJGKCQWQNOPAMG-UHFFFAOYSA-N 0.000 description 2
- JAPREJVTTCXZBR-UHFFFAOYSA-N 1-(dimethylamino)penta-1,4-dien-3-one Chemical compound CN(C)C=CC(=O)C=C JAPREJVTTCXZBR-UHFFFAOYSA-N 0.000 description 2
- QKPVEISEHYYHRH-UHFFFAOYSA-N 2-methoxyacetonitrile Chemical compound COCC#N QKPVEISEHYYHRH-UHFFFAOYSA-N 0.000 description 2
- UGWULZWUXSCWPX-UHFFFAOYSA-N 2-sulfanylideneimidazolidin-4-one Chemical compound O=C1CNC(=S)N1 UGWULZWUXSCWPX-UHFFFAOYSA-N 0.000 description 2
- MCXOBQMSPQWOMF-UHFFFAOYSA-N 3-sulfanyl-1,3-oxazolidin-2-one Chemical compound SN1CCOC1=O MCXOBQMSPQWOMF-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 2
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical group C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 2
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- KXNQKOAQSGJCQU-UHFFFAOYSA-N benzo[e][1,3]benzothiazole Chemical compound C1=CC=C2C(N=CS3)=C3C=CC2=C1 KXNQKOAQSGJCQU-UHFFFAOYSA-N 0.000 description 2
- WMUIZUWOEIQJEH-UHFFFAOYSA-N benzo[e][1,3]benzoxazole Chemical compound C1=CC=C2C(N=CO3)=C3C=CC2=C1 WMUIZUWOEIQJEH-UHFFFAOYSA-N 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- MLIREBYILWEBDM-UHFFFAOYSA-N cyanoacetic acid Chemical compound OC(=O)CC#N MLIREBYILWEBDM-UHFFFAOYSA-N 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 2
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical compound C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 description 2
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000002612 dispersion medium Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000010304 firing Methods 0.000 description 2
- RMBPEFMHABBEKP-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2C3=C[CH]C=CC3=CC2=C1 RMBPEFMHABBEKP-UHFFFAOYSA-N 0.000 description 2
- 229910052733 gallium Inorganic materials 0.000 description 2
- 239000003349 gelling agent Substances 0.000 description 2
- 150000002366 halogen compounds Chemical class 0.000 description 2
- 150000002391 heterocyclic compounds Chemical class 0.000 description 2
- WJRBRSLFGCUECM-UHFFFAOYSA-N hydantoin Chemical compound O=C1CNC(=O)N1 WJRBRSLFGCUECM-UHFFFAOYSA-N 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 238000007654 immersion Methods 0.000 description 2
- 125000002462 isocyano group Chemical group *[N+]#[C-] 0.000 description 2
- 125000001810 isothiocyanato group Chemical group *N=C=S 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- DZVCFNFOPIZQKX-LTHRDKTGSA-M merocyanine Chemical compound [Na+].O=C1N(CCCC)C(=O)N(CCCC)C(=O)C1=C\C=C\C=C/1N(CCCS([O-])(=O)=O)C2=CC=CC=C2O\1 DZVCFNFOPIZQKX-LTHRDKTGSA-M 0.000 description 2
- 229910044991 metal oxide Inorganic materials 0.000 description 2
- 150000004706 metal oxides Chemical class 0.000 description 2
- ANGDWNBGPBMQHW-UHFFFAOYSA-N methyl cyanoacetate Chemical compound COC(=O)CC#N ANGDWNBGPBMQHW-UHFFFAOYSA-N 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 229910052758 niobium Inorganic materials 0.000 description 2
- 239000010955 niobium Substances 0.000 description 2
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 2
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N o-biphenylenemethane Natural products C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 description 2
- PPXGQLMPUIVFRE-UHFFFAOYSA-N penta-2,4-dienal Chemical group C=CC=CC=O PPXGQLMPUIVFRE-UHFFFAOYSA-N 0.000 description 2
- 125000002080 perylenyl group Chemical group C1(=CC=C2C=CC=C3C4=CC=CC5=CC=CC(C1=C23)=C45)* 0.000 description 2
- CSHWQDPOILHKBI-UHFFFAOYSA-N peryrene Natural products C1=CC(C2=CC=CC=3C2=C2C=CC=3)=C3C2=CC=CC3=C1 CSHWQDPOILHKBI-UHFFFAOYSA-N 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 150000004032 porphyrins Chemical class 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 2
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 2
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical compound O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- KIWUVOGUEXMXSV-UHFFFAOYSA-N rhodanine Chemical compound O=C1CSC(=S)N1 KIWUVOGUEXMXSV-UHFFFAOYSA-N 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 230000001235 sensitizing effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 235000009518 sodium iodide Nutrition 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 2
- 125000000858 thiocyanato group Chemical group *SC#N 0.000 description 2
- 229910052718 tin Inorganic materials 0.000 description 2
- 239000011135 tin Substances 0.000 description 2
- 229910052719 titanium Inorganic materials 0.000 description 2
- 239000010936 titanium Substances 0.000 description 2
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 2
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- RUDATBOHQWOJDD-UHFFFAOYSA-N (3beta,5beta,7alpha)-3,7-Dihydroxycholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)CC2 RUDATBOHQWOJDD-UHFFFAOYSA-N 0.000 description 1
- QGKMIGUHVLGJBR-UHFFFAOYSA-M (4z)-1-(3-methylbutyl)-4-[[1-(3-methylbutyl)quinolin-1-ium-4-yl]methylidene]quinoline;iodide Chemical compound [I-].C12=CC=CC=C2N(CCC(C)C)C=CC1=CC1=CC=[N+](CCC(C)C)C2=CC=CC=C12 QGKMIGUHVLGJBR-UHFFFAOYSA-M 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- ISHFYECQSXFODS-UHFFFAOYSA-M 1,2-dimethyl-3-propylimidazol-1-ium;iodide Chemical compound [I-].CCCN1C=C[N+](C)=C1C ISHFYECQSXFODS-UHFFFAOYSA-M 0.000 description 1
- 125000000355 1,3-benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- JBOIAZWJIACNJF-UHFFFAOYSA-N 1h-imidazole;hydroiodide Chemical compound [I-].[NH2+]1C=CN=C1 JBOIAZWJIACNJF-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 1
- ADSOSINJPNKUJK-UHFFFAOYSA-N 2-butylpyridine Chemical compound CCCCC1=CC=CC=N1 ADSOSINJPNKUJK-UHFFFAOYSA-N 0.000 description 1
- GRWPYGBKJYICOO-UHFFFAOYSA-N 2-methylpropan-2-olate;titanium(4+) Chemical compound [Ti+4].CC(C)(C)[O-].CC(C)(C)[O-].CC(C)(C)[O-].CC(C)(C)[O-] GRWPYGBKJYICOO-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- WDGCBNTXZHJTHJ-UHFFFAOYSA-N 2h-1,3-oxazol-2-id-4-one Chemical group O=C1CO[C-]=N1 WDGCBNTXZHJTHJ-UHFFFAOYSA-N 0.000 description 1
- AGIJRRREJXSQJR-UHFFFAOYSA-N 2h-thiazine Chemical compound N1SC=CC=C1 AGIJRRREJXSQJR-UHFFFAOYSA-N 0.000 description 1
- XNTYYYINMGRBQW-ZEZONBOOSA-N 3,12-Diketocholanic acid Chemical compound C([C@H]1CC2)C(=O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)C(=O)C1 XNTYYYINMGRBQW-ZEZONBOOSA-N 0.000 description 1
- WFFZGYRTVIPBFN-UHFFFAOYSA-N 3h-indene-1,2-dione Chemical group C1=CC=C2C(=O)C(=O)CC2=C1 WFFZGYRTVIPBFN-UHFFFAOYSA-N 0.000 description 1
- QNGVNLMMEQUVQK-UHFFFAOYSA-N 4-n,4-n-diethylbenzene-1,4-diamine Chemical compound CCN(CC)C1=CC=C(N)C=C1 QNGVNLMMEQUVQK-UHFFFAOYSA-N 0.000 description 1
- YSHMQTRICHYLGF-UHFFFAOYSA-N 4-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=NC=C1 YSHMQTRICHYLGF-UHFFFAOYSA-N 0.000 description 1
- IMZSHPUSPMOODC-UHFFFAOYSA-N 5-oxo-1-phenyl-4h-pyrazole-3-carboxylic acid Chemical compound O=C1CC(C(=O)O)=NN1C1=CC=CC=C1 IMZSHPUSPMOODC-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- KYNSBQPICQTCGU-UHFFFAOYSA-N Benzopyrane Chemical group C1=CC=C2C=CCOC2=C1 KYNSBQPICQTCGU-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- DLYVTEULDNMQAR-SRNOMOOLSA-N Cholic Acid Methyl Ester Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@H](C)CCC(=O)OC)[C@@]2(C)[C@@H](O)C1 DLYVTEULDNMQAR-SRNOMOOLSA-N 0.000 description 1
- 238000003512 Claisen condensation reaction Methods 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 239000004985 Discotic Liquid Crystal Substance Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-O Imidazolium Chemical compound C1=C[NH+]=CN1 RAXXELZNTBOGNW-UHFFFAOYSA-O 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- PZXRCGGBZOBKKH-UHFFFAOYSA-N OC1=CC=C(O)C(=O)C1=O Chemical class OC1=CC=C(O)C(=O)C1=O PZXRCGGBZOBKKH-UHFFFAOYSA-N 0.000 description 1
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical compound C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 239000004695 Polyether sulfone Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920000265 Polyparaphenylene Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 239000012327 Ruthenium complex Substances 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000000980 acid dye Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000000274 adsorptive effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 229910021417 amorphous silicon Inorganic materials 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 150000008430 aromatic amides Chemical group 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- 125000005129 aryl carbonyl group Chemical group 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- 125000000440 benzylamino group Chemical group [H]N(*)C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- VTJUKNSKBAOEHE-UHFFFAOYSA-N calixarene Chemical compound COC(=O)COC1=C(CC=2C(=C(CC=3C(=C(C4)C=C(C=3)C(C)(C)C)OCC(=O)OC)C=C(C=2)C(C)(C)C)OCC(=O)OC)C=C(C(C)(C)C)C=C1CC1=C(OCC(=O)OC)C4=CC(C(C)(C)C)=C1 VTJUKNSKBAOEHE-UHFFFAOYSA-N 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000005521 carbonamide group Chemical group 0.000 description 1
- SKOLWUPSYHWYAM-UHFFFAOYSA-N carbonodithioic O,S-acid Chemical compound SC(S)=O SKOLWUPSYHWYAM-UHFFFAOYSA-N 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- RUDATBOHQWOJDD-BSWAIDMHSA-N chenodeoxycholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)CC1 RUDATBOHQWOJDD-BSWAIDMHSA-N 0.000 description 1
- 229960001091 chenodeoxycholic acid Drugs 0.000 description 1
- QQVDYSUDFZZPSU-UHFFFAOYSA-M chloromethylidene(dimethyl)azanium;chloride Chemical compound [Cl-].C[N+](C)=CCl QQVDYSUDFZZPSU-UHFFFAOYSA-M 0.000 description 1
- 239000002812 cholic acid derivative Substances 0.000 description 1
- 150000001842 cholic acids Chemical class 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 150000004700 cobalt complex Chemical class 0.000 description 1
- 229920001940 conductive polymer Polymers 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- PDZKZMQQDCHTNF-UHFFFAOYSA-M copper(1+);thiocyanate Chemical compound [Cu+].[S-]C#N PDZKZMQQDCHTNF-UHFFFAOYSA-M 0.000 description 1
- 150000003983 crown ethers Chemical class 0.000 description 1
- 229910021419 crystalline silicon Inorganic materials 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 150000001923 cyclic compounds Chemical class 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 150000003997 cyclic ketones Chemical class 0.000 description 1
- CFBGXYDUODCMNS-UHFFFAOYSA-N cyclobutene Chemical compound C1CC=C1 CFBGXYDUODCMNS-UHFFFAOYSA-N 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 229960003964 deoxycholic acid Drugs 0.000 description 1
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 125000004914 dipropylamino group Chemical group C(CC)N(CCC)* 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000010408 film Substances 0.000 description 1
- 230000022244 formylation Effects 0.000 description 1
- 238000006170 formylation reaction Methods 0.000 description 1
- 239000002803 fossil fuel Substances 0.000 description 1
- 239000011245 gel electrolyte Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229940091173 hydantoin Drugs 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-M iodide Chemical compound [I-] XMBWDFGMSWQBCA-UHFFFAOYSA-M 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- QDLAGTHXVHQKRE-UHFFFAOYSA-N lichenxanthone Natural products COC1=CC(O)=C2C(=O)C3=C(C)C=C(OC)C=C3OC2=C1 QDLAGTHXVHQKRE-UHFFFAOYSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910001507 metal halide Inorganic materials 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000004767 nitrides Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- BHAAPTBBJKJZER-UHFFFAOYSA-N p-anisidine Chemical compound COC1=CC=C(N)C=C1 BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 1
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 230000002165 photosensitisation Effects 0.000 description 1
- 239000003504 photosensitizing agent Substances 0.000 description 1
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical compound N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001197 polyacetylene Polymers 0.000 description 1
- 229920000767 polyaniline Polymers 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920006254 polymer film Polymers 0.000 description 1
- 239000002861 polymer material Substances 0.000 description 1
- 229920000123 polythiophene Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000011164 primary particle Substances 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- USPWKWBDZOARPV-UHFFFAOYSA-N pyrazolidine Chemical compound C1CNNC1 USPWKWBDZOARPV-UHFFFAOYSA-N 0.000 description 1
- BJDYCCHRZIFCGN-UHFFFAOYSA-N pyridin-1-ium;iodide Chemical compound I.C1=CC=NC=C1 BJDYCCHRZIFCGN-UHFFFAOYSA-N 0.000 description 1
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical group OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 150000003303 ruthenium Chemical class 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 238000005245 sintering Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- NRHMKIHPTBHXPF-TUJRSCDTSA-M sodium cholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 NRHMKIHPTBHXPF-TUJRSCDTSA-M 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- GKXDJYKZFZVASJ-UHFFFAOYSA-M tetrapropylazanium;iodide Chemical compound [I-].CCC[N+](CCC)(CCC)CCC GKXDJYKZFZVASJ-UHFFFAOYSA-M 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 1
- 229910001887 tin oxide Inorganic materials 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- JMXKSZRRTHPKDL-UHFFFAOYSA-N titanium ethoxide Chemical compound [Ti+4].CC[O-].CC[O-].CC[O-].CC[O-] JMXKSZRRTHPKDL-UHFFFAOYSA-N 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- CURCMGVZNYCRNY-UHFFFAOYSA-N trimethylazanium;iodide Chemical compound I.CN(C)C CURCMGVZNYCRNY-UHFFFAOYSA-N 0.000 description 1
- AAAQKTZKLRYKHR-UHFFFAOYSA-N triphenylmethane Chemical compound C1=CC=CC=C1C(C=1C=CC=CC=1)C1=CC=CC=C1 AAAQKTZKLRYKHR-UHFFFAOYSA-N 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/542—Dye sensitized solar cells
Landscapes
- Hybrid Cells (AREA)
- Photovoltaic Devices (AREA)
Description
【0001】
【本発明の属する技術分野】
本発明は有機色素で増感された半導体微粒子、光電変換素子及び太陽電池に関し、詳しくはチエニルピロール系の色素によって増感された酸化物半導体微粒子、及びそれを用いることを特徴とする光電変換素子及びそれを利用した太陽電池に関する。
【0002】
【従来の技術】
石油、石炭等の化石燃料に代わるエネルギー資源として太陽光を利用する太陽電池が注目されている。現在、結晶又はアモルファスのシリコンを用いたシリコン太陽電池、あるいはガリウム、ヒ素等を用いた化合物半導体太陽電池等について盛んに高効率化など、開発検討がなされている。しかしそれらは製造に要するエネルギー及びコストが高いため、汎用的に使用するのが困難であるという問題点がある。また色素で増感した半導体微粒子を用いた光電変換素子、あるいはこれを用いた太陽電池も知られ、これを作成する材料、製造技術が開示されている。(特許文献1、非特許文献1、非特許文献2を参照) この光電変換素子は酸化チタン等の比較的安価な酸化物半導体を用いて製造され、従来のシリコン等を用いた太陽電池に比べコストの安い光電変換素子が得られる可能性があり、またカラフルな太陽電池が得られることなどより注目を集めている。しかし変換効率の高い素子を得るために増感色素としてルテニウム系の錯体を使用されており、色素自体のコストが高く、またその供給にも問題が残っている。また増感色素として有機色素を用いる試みも既に行われているが、変換効率、安定性、耐久性が低いなどまだ実用化には至っていないというのが現状にある。
【特許文献1】
特許第2664194号
【特許文献2】
特開平11-273754号公報
【特許文献3】
特開2000−26487号公報
【特許文献4】
WO2002011213号公報
【非特許文献1】
B.O'Regan and M.Graetzel Nature, 第353巻, 737頁 (1991年)
【非特許文献2】
M.K.Nazeeruddin, A.Kay, I.Rodicio, R.Humphry-Baker, E.Muller, P.Liska, N.Vlachopoulos, M.Graetzel, J.Am.Chem.Soc., 第115巻, 6382頁 (1993年)
【非特許文献3】
W.Kubo, K.Murakoshi, T.Kitamura, K.Hanabusa, H.Shirai, and S.Yanagida, Chem.Lett., 1241頁(1998年)
【非特許文献4】
Tetrahedron Lett. 第40巻、50号 8887-8891頁(1990年)
【非特許文献5】
Chem.Lett., 238-239頁(2000年)
【0003】
【発明が解決しようとする課題】
有機色素増感半導体を用いた光電変換素子において、安価な有機色素を用い、変換効率の高い実用性の高い光電変換素子の開発が求められている。
【0004】
【課題を解決するための手段】
本発明者等は上記の課題を解決するために鋭意努力した結果、特定の構造を有する色素を用いて半導体微粒子を増感し、光電変換素子を作成する事により変換効率の高い光電変換素子が得られることを見出し、本発明を完成させるに至った。すなわち本発明は、
【0005】
(1)一般式(1)の構造を有する色素により増感された酸化物半導体微粒子を用いることを特徴とする光電変換素子、
【0006】
【化5】
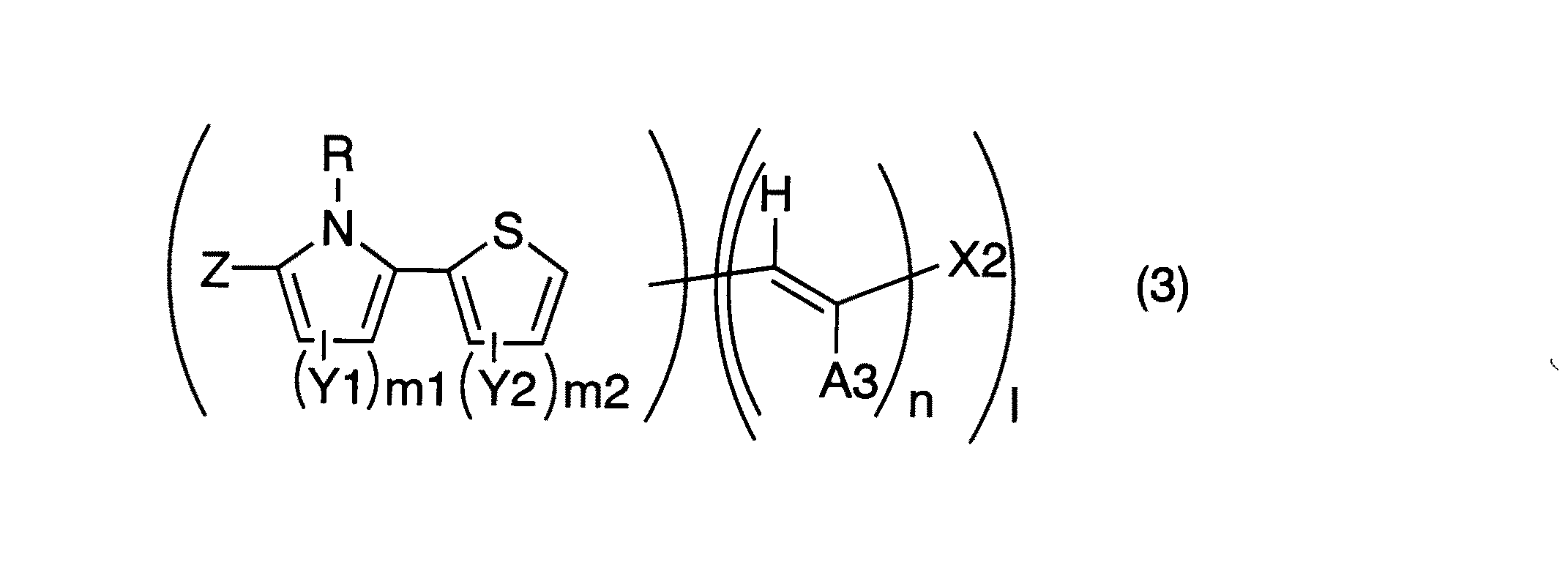
(一般式(1)中のR及びZはそれぞれ独立に水素原子、置換基を有してもよいアルキル基、置換基を有してもよい芳香族炭化水素残基又は置換基を有してもよい複素環残基を表す。Y1及びY2はそれぞれ独立に置換基を表し、環を形成してもよい。m1及びm2はそれぞれ独立に0〜3の整数を示す。)
(2)一般式(2)で示される色素により増感された酸化物半導体微粒子を用いることを特徴とする光電変換素子、
【0008】
【化6】
(式中、A1及びA2はそれぞれ独立に、置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基、ヒドロキシル基、シアノ基、水素原子、ハロゲン原子若しくは置換基を有してもよいアルキル基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基及びアシル基などのカルボニル基を有する基をあらわす。X1は置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基、シアノ基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基及びアシル基等の置換カルボニル基を有する基をあらわす。Y1及びY2はそれぞれ独立に置換基を表し、環を形成してもよい。R及びZはそれぞれ独立に水素原子、置換基を有してもよいアルキル基、置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基を表す。nは1〜4の整数を示す。m1及びm2はそれぞれ独立に0〜3の整数を示す。lは1〜3の整数を示す。またnが2以上でA1及びA2が複数存在する場合、それぞれのA1及びそれぞれのA2は互いにに同じ又は異なってもよい前記の基を示す。またA1若しくはA1が複数存在する場合にはそれぞれのA1、A2若しくはA2が複数存在する場合にはそれぞれのA2及びX1の中の複数の基により置換基を有してもよい環を形成してもよい。)
(3)一般式(2)のX1がカルボキシル基である(2)記載の光電変換素子、
(4)一般式(2)のX1がカルボキシル基でかつX1に最も近いA2がシアノ基又はカルボキシル基である(2)乃至(3)記載の光電変換素子、
(5)一般式(2)のX1又はX1とX1に最も近いA2が形成する環が置換基を有しても良い複素環残基である(2)記載の光電変換素子、
(6)(5)記載の複素環残基がチアゾール、オキサゾール、イミダゾール、セレナゾール、インドール、ピリジン及びそれらのベンゼン又はナフタレン増環体であるか、ロダニン、オキサゾリドン、チオオキサゾリドン、ヒダントイン、チオヒダントイン、チアナフテン、ピラゾロン、バルビツール酸、チオバルビツール酸の残基であることを特徴とする光電変換素子、
(7)一般式(1)及び(2)のRが置換基を有しても良い芳香族炭化水素基である(1)乃至(6)記載の光電変換素子、
(8)一般式(2)のnが1又は2である(2)乃至(7)記載の光電変換素子、
(9)(1)〜(8)記載の色素を少なくとも1つ含み、かつ他の金属錯体色素及び他の構造を有する有機色素によりなる群から選ばれた色素のうち、2種以上の色素の併用することにより増感された酸化物半導体微粒子を用いることを特徴とする光電変換素子
(10)酸化物半導体微粒子が二酸化チタンを必須成分として含有する(1)乃至(9)のいずれか1項に記載の光電変換素子
(11)酸化物半導体微粒子に包摂化合物の存在下、色素を担持させた(1)乃至(10)のいずれか1項に記載の光電変換素子
(12)(1)乃至(11)のいずれか1項に記載の光電変換素子を用いる事を特徴とする太陽電池
(13)一般式(1)又は(2)で表される色素により増感された(1)乃至(11)の何れか1項に記載の酸化物半導体微粒子
(14)一般式(3)で示される化合物
【0010】
【化7】
(式中、A3は水素原子、シアノ基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基若しくはアシル基などのカルボニル基を有する基をあらわす。またnが2以上でA3が複数存在する場合は互いに独立に同じ又は異なってもよい。X2は置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基及びアシル基等の置換カルボニル基を有する基をあらわす。Y1及びY2はそれぞれ独立に置換基を表し、環を形成してもよい。R及びZはそれぞれ独立に水素原子、置換基を有してもよいアルキル基、置換基を有してもよい芳香族炭化水素残基又は置換基を有してもよい複素環残基を表す。nは1〜4の整数を示す。m1及びm2はそれぞれ独立に0〜3の整数を示す。lは1〜3の整数を示す。またA3とXの置換基を用いて置換基を有してもよい環を形成してもよい。)
(15)一般式(4)又は(5)で示される化合物。
【0012】
【化8】
(式中、A3、X2、R、Z及びnは式(3)と同様である。)、
に関する。
【0014】
【発明の実施の形態】
以下に本発明を詳細に説明する。本発明の光電変換素子は、更に置換基を有しても良い一般式(1)の構造を有する色素によって増感された酸化物半導体を用いることを特徴とする。
【0015】
【化9】
一般式(1)においてR及びZは水素原子、置換基を有してもよいアルキル基、置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基を表す。芳香族炭化水素残基とは、芳香族炭化水素から水素原子を1つ除いた基を意味する。芳香族炭化水素残基としては例えばベンゼン、ナフタレン、アントラセン、フェナンスレン、ピレン、インデン、アズレン、フルオレン、ペリレン等が挙げられる。これらは通常炭素数6〜16の芳香環(芳香環及び芳香環を含む縮合環等)を有する芳香族炭化水素残基である。好ましくはベンゼン、ナフタレン、ピレンなどが挙げられる。また複素環残基とは、複素環化合物から水素原子を1つ除いた基を意味し、複素環化合物としては例えば、ピリジン、ピラジン、ピリミジン、ピラゾール、ピラゾリジン、チアゾリジン、オキサゾリジン、ピラン、クロメン、ピロール、ベンゾイミダゾール、イミダゾリン、イミダゾリジン、イミダゾール、ピラゾール、トリアゾール、トリアジン、ジアゾール、モルホリン、インドリン、チオフェン、フラン、オキサゾール、チアジン、チアゾール、インドール、ベンゾチオフェン、ベンゾチアゾール、ナフトチアゾール、ベンゾオキサゾール、ナフトオキサゾール、セレナゾール、ベンゾセレナゾール、インドレニン、ベンゾインドレニン、ピラジン、キノリン、キナゾリン、カルバゾール等が挙げられる。好ましくはチオフェン、ベンゾチオフェン、ピリジン、キノリン、チアゾール、インドールなどが挙げられる。さらに好ましくはチオフェン、ベンゾチオフェンが挙げられる。
Y1及びY2はそれぞれ独立に置換基を表し、環を形成してもよい。置換基としては以下に示す置換基を有しても良い芳香族炭化水素残基が有しても良い置換基と同じでよく、環を形成する場合はベンゼン環及びナフタレン環などの芳香族炭化水素が増環した形をとるものが好ましく、ベンゾチオフェン環、ベンゾピロール環などが具体的に挙げられる。m1及びm2はそれぞれ独立に0〜3の整数を示す。
【0017】
置換基を有してもよい芳香族炭化水素残基及び置換基を有してもよい複素環残基における置換基としては、特に制限はないが、アルキル基、アリール基、シアノ基、イソシアノ基、チオシアナト基、イソチオシアナト基、ニトロ基、ニトロシル基、ハロゲン原子、ヒドロキシル基、スルホ基、リン酸基、リン酸エステル基、置換もしくは非置換メルカプト基、置換もしくは非置換アミノ基、置換もしくは非置換アミド基、アルコキシル基又はアルコキシカルボニル基、カルボキシル基、カルボンアミド基、アシル基等の置換カルボニル基等が挙げられ、またさらに置換基を有してもよい芳香族炭化水素残基及び置換基を有してもよい複素環残基が置換されていても良い。アルキル基としては置換基を有してもよい飽和及び不飽和の直鎖、分岐及び環状のアルキル基が挙げられ、炭素数は1から36が好ましく、さらに好ましくは置換基を有しても良い飽和の直鎖アルキル基で、炭素数は1から20であるものが挙げられる。環状のものとして例えば炭素数3乃至8のシクロアルキルなどが挙げられる。これらのアルキル基は上記の置換基(アルキル基を除く)で更に置換されていてもよい。
【0018】
アリール基としては、後記する芳香族炭化水素残基の項で挙げられる芳香環化合物から水素原子を除いた基等が挙げられる。アリール基は更に上記の基などで置換されていてもよい。
アシル基としては例えば炭素数1乃至10のアルキルカルボニル基、アリールカルボニル基等が挙げられ、好ましくは炭素数1乃至4のアルキルカルボニル基、具体的にはアセチル基、プロピオニル基等が挙げられる。ハロゲン原子としてはフッ素、塩素、臭素、ヨウ素等の原子が挙げられ、塩素、臭素、ヨウ素が好ましい。リン酸エステルとしてはリン酸(炭素数1ないし4の)アルキルエステル基等が挙げられる。置換もしくは非置換メルカプト基としてはメルカプト基、アルキルメルカプト基等が挙げられる。置換もしくは非置換アミノ基としては、アミノ基、モノ又はジメチルアミノ基、モノ又はジエチルアミノ基、モノ又はジプロピルアミノ基等のアルキル置換アミノ基、モノ又はジフェニルアミノ基、モノ又はジナフチルアミノ基等の芳香族置換アミノ基、又はベンジルアミノ基等が挙げられる。置換もしくは非置換アミド基としては、アミド基、アルキルアミド基、芳香族アミド基等が挙げられる。アルコキシル基としては、例えば炭素数1ないし10のアルコキシル基等が挙げられる。アルコキシカルボニル基としては例えば炭素数1ないし10のアルコキシカルボニル基等が挙げられる。
また、カルボキシル基、スルホ基及びリン酸基等の酸性基及びヒドロキシル基は、塩を形成してもよく、塩としては例えばリチウム、ナトリウム、カリウム、マグネシウム、カルシウムなどのアルカリ金属又はアルカリ土類金属などとの塩、又は有機塩基、例えばテトラメチルアンモニウム、テトラブチルアンモニウム、ピリジニウム、イミダゾリウムなどの4級アンモニウム塩のような塩を挙げることができる。
【0019】
更に好適なものは一般式(2)で示される様なメチン基が導入された色素により増感された酸化物半導体微粒子を用いる光電変換素子である。
【0020】
【化10】
式中、A1及びA2はそれぞれ独立に、置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基、ヒドロキシル基、シアノ基、水素原子、ハロゲン原子若しくは置換基を有してもよいアルキル基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基及びアシル基などのカルボニル基を有する基を表す。芳香族炭化水素残基、複素環残基とは前述と同じ意味を示し、有しても良い置換基としても前記と同様である。nが2以上でA1及びA2が複数存在する場合、それぞれのA1及びそれぞれのA2は互いに独立に同じ又は異なってもよい。好ましくはA1及びA2が独立に水素原子、シアノ基、アルキル基、ハロゲン原子及びカルボキシル基である物が挙げられる。組み合わせとして好ましくはnが1の場合、A1,A2が共にシアノ基であるもの、A1が水素原子でA2が水素原子又はシアノ基であるもの。またnが2以上の場合、A1,A2が全てシアノ基であるもの、A1が全て水素原子でXに最も近いA2がシアノ基でその他のA2が水素原子あるものが好ましい。
またA1若しくはA1が複数存在する場合にはそれぞれのA1、A2若しくはA2が複数存在する場合にはそれぞれのA2及びX1の中の複数の基により置換基を有してもよい環を形成してもよい。
形成する環としては不飽和炭化水素環又は複素環が挙げられる。不飽和炭化水素環としてはベンゼン環、ナフタレン環、アントラセン環、フェナンスレン環、ピレン環、インデン環、アズレン環、フルオレン環、シクロブテン環、シクロヘキセン環、シクロペンテン環、シクロヘキサジエン環、シクロペンタジエン環等が挙げられ、複素環基としては、ピリジン環、ピラジン環、ピペリジン環、インドリン環、チオフェン環、フラン環、ピラン環、オキサゾール環、チアゾール環、インドール環、ベンゾチアゾール環、ベンゾオキサゾール環、キノリン環、カルバゾール環、ベンゾピラン環等が挙げられる。またこれらのうちの好ましいものはシクロブテン環、シクロペンテン環、シクロヘキセン環、ピラン環などが挙げられる。また、カルボニル基、チオカルボニル基等を有する場合には環状ケトン又は環状チオケトンなどを形成しても良い。
【0022】
X1は置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基、シアノ基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基及びアシル基等の置換カルボニル基を有する基を表す。芳香族炭化水素残基、複素環残基とは前記と同じ意味を示し、有しても良い置換基としても前記と同様である。好ましくは置換基を有してもよい複素環残基、シアノ基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基及びアシル基等の置換カルボニル基を有する基でさらに好ましくは置換基を有しても良い複素環残基及びカルボキシル基である。ここで示す複素環残基としては、好ましくはピリジン、ピラゾール、ピラン、クロメン、ベンゾイミダゾール、イミダゾール、ピラゾール、チアゾール、オキサゾール、インドール、ベンゾチアゾール、ナフトチアゾール、ベンゾオキサゾール、ナフトオキサゾール、セレナゾール、ベンゾセレナゾール、インドレニン、ベンゾインドレニン、キノリン等の残基が挙げられる。
また前記したようにX1とA1又はA2は環を形成することが出来る。特にX1とX1に最も近いA2が形成することが好ましく、その時の環は置換基を有しても良い複素環残基であることが更に好ましい。具体的にはピリジン、キノリン、ピラン、クロメン、ピリミジン、ピロール、チアゾール、ベンゾチアゾール、オキサゾール、ベンゾオキサゾール、セレナゾール、ベンゾセレナゾール、イミダゾール、ベンゾイミダゾール、ピラゾール、チオフェン等の残基が挙げられ、それぞれの複素環残基は増環や水素化されていても良くまた、これらは前記するように置換基を有しても良い。
これら置換基を合わせて環式炭化水素、複素環としてロダニン環、オキサゾリドン環、チオオキサゾリドン環、ヒダントイン環、チオヒダントイン環、インダンジオン環、チアナフテン環、ピラゾロン環、バルビツール環、チオバルビツール環、ピリドン環などを形成する構造が好ましい。さらにX又はXとAで形成する複素環は4級化されていても良く、その時に対イオンを有しても良い。具体的には特に限定はされないが、一般的なアニオンで良い。具体例としてはF-, Cl-, Br-, I-, ClO4 -, BF4 -, PF6 -, OH-, SO4 2-, CH3SO4 -, トルエンスルホン酸イオンなどが挙げられ、Br-, I-, ClO4 -, BF4 -, PF6 -, CH3SO4 -, トルエンスルホン酸イオンなどが好ましい。また対イオンではなく分子内又は分子間のカルボキシル基などの酸性基により中和されていても良い。
【0023】
Y1及びY2はそれぞれ独立に置換基を表し、環を形成してもよい。置換基としては前記の一般式(1)におけるものと同じでよく、具体的には水素原子、アルキル基又はアリール基が好ましい。環を形成する場合はベンゼン環及びナフタレン環などの芳香族炭化水素が増環した形をとるものが好ましく、ベンゾチオフェン環、ベンゾピロール環などが具体的に挙げられる。m1及びm2はそれぞれ独立に0〜3の整数を示す。
【0024】
R及びZは前記と同様の置換基が挙げられる。Rとして好ましくは置換基を有してもよい芳香族炭化水素残基で、具体的には置換基を有しても良いベンゼン環、ナフタレン環などが挙げられる。好ましい置換基としてはアルキル基、シアノ基、イソシアノ基、チオシアナト基、イソチオシアナト基、ハロゲン原子、ヒドロキシル基、置換もしくは非置換アミノ基、アルコキシル基又はアルコキシカルボニル基、カルボキシル基、カルボンアミド基、アシル基等の置換カルボニル基等が挙げられる。さらに好ましくはアルキル基、アルコキシル基、フェノキシ基、フッ素、塩素、臭素、ヨウ素などのハロゲン原子、ジアルキルアミノ基、モノアルキルアミノ基、ジフェニルアミノ基、モノフェニルアミノ基などが挙げられる。Zとして好ましくは水素原子、置換基を有してもよい芳香族炭化水素残基及び置換基を有してもよい複素環残基で、具体的には置換基を有しても良いベンゼン環、ナフタレン環、チオフェン環、ベンズチオフェン環などが挙げられ、更に好ましくはチオフェン環である。好ましい置換基としては前記と同様である。
【0025】
また一般式(2)のメチン基が導入される位置としては以下の一般式(6)における置換位置a〜eが挙げられるが、a,d又はeが好ましい。これらの置換位置は以下に述べる化合物(7)を製造する時のアルデヒド系置換基の導入位置による。導入位置はRやZなどが有する置換基の性質や製造の反応条件によりコントロールする。
【0026】
【化11】
nは1〜4の整数を示す。好ましくは1〜3の整数で更に好ましくは1〜2の整数である。
lは1〜3の整数を示す。好ましくは1〜2の整数で更に好ましくは1である。一般式(2)で示される化合物はシス体、トランス体などの構造異性体をとり得るが、特に限定されず、いずれも光増感用色素として良好に使用しうるものである。
いずれの場合も、分子内に少なくとも一つのカルボン酸基、カルボンアミド基、エステル基、アシル基などのカルボニル基を有することが酸化物半導体との吸着結合にとって好ましい。
【0028】
【化12】
更に構造を特定すると、上記の一般式(3)に挙げられる構造が挙げられる。このときのR、Z、Y1、Y2、m1、m2、n、lは前記と同様である。
A3は水素原子、シアノ基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基若しくはアシル基などのカルボニル基を有する基を表す。またnが2以上でA3が複数存在する場合は互いに独立に同じ又は異なってもよいが、好ましくは水素原子が挙げられ、X2に最も近いA3としてはシアノ基、カルボキシル基、アルコキシカルボニル基が挙げられる。
X2は置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基又はカルボキシル基、カルボンアミド基、アルコキシカルボニル基及びアシル基等の置換カルボニル基を有する基を表す。好ましくは置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基又はカルボキシル基及びアルコキシカルボニル基が挙げられる。置換基を有してもよい芳香族炭化水素残基、置換基を有してもよい複素環残基の例としては前記のX1で挙げた例と同様である。
またX2とA3の置換基を用いて置換基を有してもよい環を形成してもよい。これはX1とX1に最も近いA2が形成する環と同様である。
【0030】
【化13】
更に色素の構造を特定すると上記の一般式(4)及び(5)が挙げられる。このときのR、Z、A3、X2、nは前記と同様である。
【0032】
一般式(1)の化合物は以下に示す反応式の様に一般的に小倉らの方法によって製造できる。(例えば非特許文献4)これをホルミル化(例えば非特許文献5)(1)をブチルリチウムなどの塩基を用いて金属化した後、ジメチルホルムアミドなどのアミド誘導体を作用させる方法や、ジメチルホルムアミドなどに塩化ホスホリルなどを作用させビルスマイヤー試薬とし、これを(1)に作用させることで化合物(2)の前駆体である化合物(7)が得られる。ホルミル基の置換位置は(1)の有する置換基や製造方法により制御する。nが2以上の場合はさらにアセトアルデヒドなどをクライゼン縮合する方法や、ジメチルアミノアクロレインなどの試薬を用いることにより得ることができる。またlの数は温度や試薬の量により制御する。さらに化合物(7)と活性メチレンを有する化合物を必要であればナトリウムエトキシド、ピペリジン、ピペラジンなどの塩基性触媒の存在下、メタノール、エタノール、イソプロパノールなどのアルコールやジメチルホルムアミドなどの非プロトン性極性溶媒や無水酢酸などの溶媒中、20℃〜120℃好ましくは50℃〜80℃程度で縮合することにより化合物(2)が得られる。
また一般式(3)の化合物は一般式(2)と同様な方法で製造できる。即ち、一般式(4)の化合物は通常、一般式(1)をブチルリチウムなどの塩基を用いて金属化した後、ジメチルホルムアミド、ジメチルアミノアクロレイン、ジメチルアミノビニルアクロレインなどのアミド誘導体を作用させる方法で式(6)に示した置換位置aにホルミル基、プロペナール基、ペンタジエナール基などを有する一般式(4)の前駆体を得、これに活性メチレンを有する化合物を、必要であれば、ナトリウムエトキシド、ピペリジン、ピペラジンなどの塩基性触媒の存在下、メタノール、エタノール、ブタノールなどのアルコールやジメチルホルムアミドなどの非プロトン性極性溶媒や無水酢酸などの溶媒中、20〜120℃、好ましくは50〜80℃で縮合することにより一般式(4)の化合物は得られる。一般式(5)の化合物は通常、一般式(1)をジメチルホルムアミドやジメチルアミノアクロレイン、ジメチルアミノビニルアクロレインなどに塩化ホスホリルなどを作用させて出来たビルスマイヤー試薬と反応させる方法で式(6)に示した置換位置dにホルミル基、プロペナール基、ペンタジエナール基などを有する(5)の前駆体を得、これに活性メチレンを有する化合物を、必要であれば、ナトリウムエトキシド、ピペリジン、ピペラジンなどの塩基性触媒の存在下、メタノール、エタノール、ブタノールなどのアルコールやジメチルホルムアミドなどの非プロトン性極性溶媒や無水酢酸などの溶媒中、20〜120℃好ましくは50〜80℃で縮合することにより一般式(5)の化合物は得られる。
【0033】
【化14】
(上記式中のZ、R、Y1、Y2、A1、X1、A2、n、l、m1、m2は前記と同じものを表す。)
以下に化合物例を列記する。まず一般式(2)のZがチオフェン環でRが置換基を有しても良いベンゼン環(図中にフェニルの置換基をR1〜R3として記載した)でnが1である一般式(8)の化合物例を表1に表す。フェニル基をPh、ジエチルアミノ基をDEA、ジフェニルアミノ基をDPAと略する。またX1及びXとA2で環を形成した場合の環(環B)を以下に記載する。
【0035】
【化15】
【表1】
(環Bの例)
【0041】
【化16】
【化17】
その他の化合物の例を以下に示す。
【化18】
【化19】
【化20】
【化21】
【化22】
【化23】
【化24】
【化25】
【化26】
【化27】
【化28】
【化29】
【化30】
本発明の色素増感光電変換素子は、例えば、酸化物半導体微粒子を用いて基板上に酸化物半導体の薄膜を製造し、次いでこの薄膜に色素を担持させたものである。
本発明で酸化物半導体の薄膜を設ける基板としてはその表面が導電性であるものが好ましいが、そのような基板は市場にて容易に入手可能である。具体的には、例えば、ガラスの表面又はポリエチレンテレフタレート若しくはポリエーテルスルフォン等の透明性のある高分子材料の表面にインジウム、フッ素、アンチモンをドープした酸化スズなどの導電性金属酸化物や銅、銀、金等の金属の薄膜を設けたものを用いることが出来る。その導電性としては通常1000Ω以下であれば良く、特に100Ω以下のものが好ましい。
また、酸化物半導体の微粒子としては金属酸化物が好ましく、その具体例としてはチタン、スズ、亜鉛、タングステン、ジルコニウム、ガリウム、インジウム、イットリウム、ニオブ、タンタル、バナジウムなどの酸化物が挙げられる。これらのうちチタン、スズ、亜鉛、ニオブ、インジウム等の酸化物が好ましく、これらのうち酸化チタンが最も好ましい。これらの酸化物半導体は単一で使用することも出来るが、混合したり、半導体の表面にコーティングさせて使用することも出来る。また酸化物半導体の微粒子の粒径は平均粒径として、通常1〜500nmで、好ましくは1〜100nmである。またこの酸化物半導体の微粒子は大きな粒径のものと小さな粒径のものを混合したり、相重ねて用いることも出来る。
【0057】
酸化物半導体薄膜は酸化物半導体微粒子をスプレイ噴霧などで直接基板上に薄膜として形成する方法、基板を電極として電気的に半導体微粒子薄膜を析出させる方法、半導体微粒子のスラリー又は半導体アルコキサイド等の半導体微粒子の前駆体を加水分解することにより得られた微粒子を含有するペーストを基板上に塗布した後、乾燥、硬化もしくは焼成する等によって製造することが出来る。酸化物半導体電極の性能上、スラリーを用いる方法等が好ましい。この方法の場合、スラリーは2次凝集している酸化物半導体微粒子を常法により分散媒中に平均1次粒子径が1〜200nmになるように分散させることにより得られる。
【0058】
スラリーを分散させる分散媒としては半導体微粒子を分散させ得るものであれば何でも良く、水あるいはエタノール等のアルコール、アセトン、アセチルアセトン等のケトンもしくはヘキサン等の炭化水素等の有機溶媒が用いられ、これらは混合して用いても良く、また水を用いることはスラリーの粘度変化を少なくするという点で好ましい。また酸化物半導体微粒子の分散状態を安定化させる目的で分散安定剤を用いることが出来る。用いうる分散安定剤の例としては例えば酢酸、塩酸、硝酸などの酸、又はアセチルアセトン、アクリル酸、ポリエチレングリコール、ポリビニルアルコールなどが挙げられる。
【0059】
スラリーを塗布した基板は焼成してもよく、その焼成温度は通常100℃以上、好ましくは200℃以上で、かつ上限はおおむね基材の融点(軟化点)以下であり、通常上限は900℃であり、好ましくは600℃以下である。また焼成時間には特に限定はないがおおむね4時間以内が好ましい。基板上の薄膜の厚みは通常1〜200μmで好ましくは1〜50μmである。
【0060】
酸化物半導体薄膜に2次処理を施してもよい。すなわち例えば半導体と同一の金属のアルコキサイド、塩化物、硝化物、硫化物等の溶液に直接、基板ごと薄膜を浸積させて乾燥もしくは再焼成することにより半導体薄膜の性能を向上させることもできる。金属アルコキサイドとしてはチタンエトキサイド、チタンイソプロポキサイド、チタンtーブトキサイド、n−ジブチルージアセチルスズ等が挙げられ、そのアルコール溶液が用いられる。塩化物としては例えば四塩化チタン、四塩化スズ、塩化亜鉛等が挙げられ、その水溶液が用いられる。このようにして得られた酸化物半導体薄膜は酸化物半導体の微粒子から成っている。
【0061】
次に酸化物半導体薄膜に色素を担持させる方法について説明する。前記一般式(1)色素を担持させる方法としては、該色素を溶解しうる溶媒にて色素を溶解して得た溶液、又は溶解性の低い色素にあっては色素を分散せしめて得た分散液に上記酸化物半導体薄膜の設けられた基板を浸漬する方法が挙げられる。溶液又は分散液中の濃度は色素によって適宜決める。その溶液中に基板上に作成した半導体薄膜を浸す。浸積時間はおおむね常温から溶媒の沸点までであり、また浸積時間は1時間から48時間程度である。色素を溶解させるのに使用しうる溶媒の具体例として、例えば、メタノール、エタノール、アセトニトリル、ジメチルスルホキサイド、ジメチルホルムアミド、アセトン、t -ブタノール等が挙げられる。溶液の色素濃度は通常1×10-6M〜1Mが良く、好ましくは1×10-5 M〜1×10-1Mである。この様にして色素で増感した酸化物半導体微粒子薄膜の光電変換素子が得られる。
【0062】
担持する前記一般式(1)色素は1種類でも良いし、数種類混合しても良い。又、混合する場合は本発明の色素同志でも良いし、他の色素や金属錯体色素を混合しても良い。特に吸収波長の異なる色素同志を混合することにより、幅広い吸収波長を利用することが出来、変換効率の高い太陽電池が得られる。混合しうる金属錯体色素の例としては特に制限は無いが非特許文献2や特許文献2に示されているルテニウム錯体やその4級塩化合物、フタロシアニン、ポルフィリンなどが好ましく、混合利用する有機色素としては無金属のフタロシアニン、ポルフィリンやシアニン、メロシアニン、オキソノール、トリフェニルメタン系、特許文献4に示されるアクリル酸系色素などのメチン系色素や、キサンテン系、アゾ系、アンスラキノン系、ペリレン系等の色素が挙げられる。好ましくはルテニウム錯体やメロシアニン、アクリル酸系等のメチン系色素が挙げられる。色素を2種以上用いる場合は色素を半導体薄膜に順次吸着させても、混合溶解して吸着させても良い。
【0063】
混合する色素の比率は特に限定は無く、それぞれの色素より最適化選択されるが、一般的に等モルずつの混合から、1つの色素につき、10%モル程度以上使用するのが好ましい。混合色素を混合溶解もしくは分散した溶液を用いて、酸化物半導体微粒子薄膜に色素を吸着する場合、溶液中の色素合計の濃度は1種類のみ担持する場合と同様でよい。色素を混合して使用する場合の溶媒としては前記したような溶媒が使用可能であり、使用する各色素用の溶媒は同一でも異なっていてもよい。
【0064】
酸化物半導体微粒子の薄膜に色素を担持する際、色素同士の会合を防ぐために包摂化合物の共存下、色素を担持することが効果的である。ここで包摂化合物としてはコール酸等のステロイド系化合物、クラウンエーテル、シクロデキストリン、カリックスアレン、ポリエチレンオキサイドなどが挙げられるが、好ましいものとしてはデオキシコール酸、デヒドロデオキシコール酸、ケノデオキシコール酸、コール酸メチルエステル、コール酸ナトリウム等のコール酸類、ポリエチレンオキサイド等が挙げられる。又、色素を担持させた後、4ーt−ブチルピリジン等のアミン化合物で半導体電極表面を処理しても良い。処理の方法は例えばアミンのエタノール溶液に色素を担持した半導体微粒子薄膜の設けられた基板を浸す方法等が採られる。
【0065】
本発明の太陽電池は上記酸化物半導体薄膜に色素を担持させた光電変換素子電極と対極としてレドックス電解質又は正孔輸送材料又はp型半導体等から構成される。レドックス電解質、正孔輸送材料、p型半導体等の形態としては、液体、擬固体(ゲル及びゲル状)、固体などが挙げられる。液状のものとしてはレドックス電解質、溶融塩、正孔輸送材料、p型半導体等をそれぞれ溶媒に溶解させたものや常温溶融塩などが、擬固体(ゲル及びゲル状)の場合は、これらをポリマーマトリックスや低分子ゲル化剤等に含ませたもの等がそれぞれ挙げられる。固体のものとしてはレドックス電解質、溶融塩、正孔輸送材料、p型半導体等を用いることができる。正孔輸送材料としてはアミン誘導体やポリアセチレン、ポリアニリン、ポリチオフェンなどの導電性高分子、ポリフェニレンなどのディスコティック液晶相を用いる物などが挙げられる。又、p型半導体としてはCuI、CuSCN等が挙げられる。対極としては導電性を持っており、レドックス電解質の還元反応を触媒的に作用するものが好ましい。例えばガラス、もしくは高分子フィルムに白金、カーボン、ロジウム、ルテニウム等を蒸着したり、導電性微粒子を塗り付けたものが用いうる。
【0066】
本発明の太陽電池に用いるレドックス電解質としてはハロゲンイオンを対イオンとするハロゲン化合物及びハロゲン分子からなるハロゲン酸化還元系電解質、フェロシアン酸塩−フェリシアン酸塩やフェロセン−フェリシニウムイオン、コバルト錯体などの金属錯体等の金属酸化還元系電解質、アルキルチオール−アルキルジスルフィド、ビオロゲン色素、ヒドロキノン−キノン等の有機酸化還元系電解質などをあげることができるが、ハロゲン酸化還元系電解質が好ましい。ハロゲン化合物−ハロゲン分子からなるハロゲン酸化還元系電解質におけるハロゲン分子としては、例えばヨウ素分子や臭素分子等があげられ、ヨウ素分子が好ましい。又、ハロゲンイオンを対イオンとするハロゲン化合物としては、例えばLiI、NaI、KI、CsI、CaI2、CuI等のハロゲン化金属塩あるいはテトラアルキルアンモニウムヨーダイド、イミダゾリウムヨーダイド、ピリジニウムヨーダイドなどのハロゲンの有機4級アンモニウム塩等があげられるが、ヨウ素イオンを対イオンとする塩類化合物が好ましい。ヨウ素イオンを対イオンとする塩類化合物としては、例えばヨウ化リチウム、ヨウ化ナトリウム、ヨウ化トリメチルアンモニウム塩等があげられる。
【0067】
又、レドックス電解質はそれを含む溶液の形で構成されている場合、その溶媒には電気化学的に不活性なものが用いられる。例えばアセトニトリル、プロピレンカーボネート、エチレンカーボネート、3−メトキシプロピオニトリル、メトキシアセトニトリル、エチレングリコール、プロピレングリコール、ジエチレングリコール、トリエチレングリコール、γ−ブチロラクトン、ジメトキシエタン、ジエチルカーボネート、ジエチルエーテル、ジエチルカーボネート、ジメチルカーボネート、1、2−ジメトキシエタン、ジメチルホルムアミド、ジメチルスルホキサイド、1、3−ジオキソラン、メチルフォルメート、2ーメチルテトラヒドロフラン、3−メトキシーオキサジリジン−2−オン、γ−ブチロラクトン、スルフォラン、テトラヒドロフラン、水等が挙げられ、これらの中でも、特に、アセトニトリル、プロピレンカーボネート、エチレンカーボネート、3−メトキシプロピオニトリル、メトキシアセトニトリル、エチレングリコール、3−メトキシオキサジリジン−2−オン、γ−ブチロラクトン等が好ましい。これらは単独もしくは2種以上組み合わせて用いても良い。ゲル状電解質の場合は、オリゴマ−及びポリマー等のマトリックスに電解質あるいは電解質溶液を含有させたものや、非特許文献3に記載の低分子ゲル化剤等に同じく電解質あるいは電解質溶液を含有させたもの等が挙げられる。レドックス電解質の濃度は通常0.01〜99重量%で好ましくは0.1〜90重量%程度である。
【0068】
本発明の太陽電池は、基板上の酸化物半導体薄膜に色素を担持した光電変換素子の電極に、それを挟むように対極を配置する。その間にレドックス電解質を含んだ溶液を充填することにより本発明の太陽電池が得られる。
【0069】
【実施例】
以下に実施例に基づき、本発明を更に具体的に説明するが、本発明がこれらの実施例に限定されるものではない。実施例中、部は特に指定しない限り質量部を、また%は質量%をそれぞれ表す。
【0070】
合成例1
(化合物(57)の合成)
下記の反応式に従い得られた化合物(157)を用いて化合物(57)を合成した。
1,4―ジ(2−チエニル)−1,4−ブタンジオン(154)4部とN,N−ジエチル−1,4−フェニレンジアミン(155)5部、プロピオン酸8部及びトルエン25部を還流下で4時間反応する。冷却後、水100部を加え、分液、トルエン層を抽出し、さらにヘキサン:酢酸エチル混合溶液でカラムクロマトグラフィー、エタノール:ヘキサン混合溶媒で再結晶し、化合物(156)を白色板状結晶として得た。(融点:79〜81℃、吸収極大(EtOH):272,342nm)
ジメチルホルムアミド5部に塩化ホスホリル0.6部を5℃以下で滴下し、10℃以下で1時間撹拌する。ここに化合物(156)1部をジメチルホルムアミド5部に溶解した液を10℃以下に保ったまま滴下しそのまま3時間反応を続ける。反応終了後、苛性ソーダ水溶液にて中和し、析出した沈殿を濾過、水洗し、化合物(157)を淡黄色粉末として得た。(融点:154〜158℃、吸収極大(EtOH):270,308,430(Sh)nm)
【0071】
【化31】
エタノール20部に化合物(157)1部とシアノ酢酸メチル0.3部、ピペラジン0.2部を加え2時間還流反応を行なう。反応終了後、析出した沈殿をろ過し、エタノールで再結晶し化合物(56)を褐色結晶として得た。(融点:178〜180℃、吸収極大(EtOH:276,308,396nm)
化合物(56)0.2部と水酸化カリウム0.5部をエタノール10部中で2時間還流反応する。反応溶液に水50部を添加し、さらに塩酸で中和し、析出した黄色結晶をろ過、水洗し、さらにエタノールで再結晶することで化合物(57)を褐色結晶として得た。(融点:233〜235℃、吸収極大(EtOH:274, 308, 390nm)、発光極大(EtOH:434nm))
1H-NMR(PPM:d6-DMSO):1.09(t.CH3.6H), 3.30(m.CH2.4H), 6.64(d.arom.2H), 6.83(d.thio.1H), 6.98(dd.thio.1H), 7.07(d.arom.2H), 7.10(d.thio.1H), 7.12(d.thio.1H), 7.44(s.pyrr.1H), 7.47(dd.thio.1H), 7.73(dd.thio.1H), 8.00(s.-CH=.1H)
【0073】
合成例2
(化合物(58)の合成)
エタノール20部に化合物(157)0.5部と硫酸メチル=1,2,3,3−トリメチルー5−カルボキシインドレニウム0.5部、ピペラジン0.1部を加え1時間還流下に反応を行なう。反応終了後、冷却しヨウ化水素酸1部を加え、析出した沈殿をろ過し、エタノールで再結晶し化合物(58)をこげ茶色粉末として得た。(融点:174〜176℃、吸収極大(EtOH:292, 364, 528nm)、発光極大(EtOH:569nm))
【0074】
合成例3
(化合物(62)の合成)
エタノール10部に化合物(157)0.5部とロダニン−3−酢酸0.3部、ピペラジン0.1部を加え2時間還流反応を行なう。反応終了後、析出した沈殿をろ過し、エタノールで再結晶し化合物(62)を茶色粉末として得た。(融点:234〜237℃、吸収極大(EtOH:272, 442nm)、発光極大(EtOH:625nm))
1H-NMR(PPM:d6-DMSO):1.09(t.CH3.6H), 3.30(m.CH2.4H), 4.47(s.CH2.2H)
6.65(d.arom.2H), 6.84(s.pyrr.1H), 6.98(dd.thio.1H), 7.09(d.arom.2H),
7.10(dd.thio.1H), 7.12(dd.thio.1H), 7.13(dd.thio.1H), 7.45(dd.thio.1H),
7.55(s.-CH=.1H), 7.71(dd.thio.1H),
【0075】
合成例4
(化合物(61)の合成)
合成例3においてロダニン−3−酢酸の代わりにバルビツール酸を用いて同様な操作を行い、化合物(61)を茶色粉末として得た。(融点:288℃分解(TG−DTA使用)、吸収極大(EtOH:264, 314, 436nm)、発光極大(EtOH:649nm))
【0076】
合成例5
(化合物(63)の合成)
合成例3においてロダニン−3−酢酸の代わりに1−フェニルー3−カルボキシ−5−ピラゾロンを用いて同様な操作を行い、化合物(63)を茶色粉末として得た。(融点:223〜225℃、吸収極大(EtOH:256, 506nm)、発光極大(EtOH:658nm))
【0077】
合成例6
(化合物(80)の合成)
合成例3においてロダニン−3−酢酸の代わりにマロン酸ジエチルを用いて同様な操作を行い、化合物(80)を黄色粉末として得た。(融点:158〜160℃、吸収極大(EtOH:206, 276nm)、発光極大(EtOH:417nm))
【0078】
合成例7
(化合物(11)の合成)
下記の反応式に従い得られた化合物(160)を用いて化合物(11)を合成した。
1,4―ジ(2−チエニル)−1,4−ブタンジオン(154)4部とp−アニシジン(158)4部、プロピオン酸9部及びトルエン30部を還流で8時間反応する。冷却後、析出した結晶を濾過し酢酸10部で洗浄し、さらにヘキサン:酢酸エチル混合溶液でカラムクロマトグラフィー、エタノール:ヘキサン混合溶媒で再結晶し、化合物(159)を白色板状結晶として得た。
化合物(159)0.88部に無水テトラヒドロフラン53部を加え、窒素雰囲気下に−70℃に冷却し攪拌した。ここにn-ブチルリチウム(1.58mol/l,n−へキサン溶液)1.1部を加え、同温にて40分攪拌した。その後N,N−ジメチルホルムアミド1部を加え、更に30分攪拌し、室温にて更に3時間攪拌した。塩化アンモニウム溶液40部を加え、トルエン175部を用い分液、トルエン層を抽出し、さらにヘキサン:酢酸エチル混合溶液でカラムクロマトグラフィー、エタノール:ヘキサン混合溶媒で再結晶し、化合物(160)を黄色粉末として得た。
1H-NMR(PPM:d6-DMSO):3.90(s.CH3.3H), 6.59(d.pyrr.1H), 6.65(d.thio.1H), 6.74(d.thio.1H), 6.78(d.pyrr.1H), 6.85(dd.thio.1H), 7.00(d.arom.2H), 7.09(d.thio.1H), 7.27(d.arom.2H), 7.48(d.thio.1H), 9.70(s.CHO.1H),
【0079】
【化32】
エタノール20部に化合物(160)1.5部とシアノ酢酸0.5部、ピペリジン0.1部を加え2時間還流下に反応を行なう。反応終了後、析出した沈殿をろ過し、エタノールで再結晶し化合物(11)を橙色粉末として得た。
(吸収極大(EtOH:444nm)、発光極大(EtOH:587nm))
1H-NMR(PPM:d6-DMSO):3.84(s.CH3.3H), 6.67(d.pyrr.1H), 6.83(m.thio.3H), 6.92(dd.thio.1H), 7.08(d.arom.2H), 7.33(d.pyrr.1H), 7.35(d.arom.2H), 7.47(d.thio.1H), 7.92(s.-CH=.1H)
【0081】
合成例8
(化合物(110)の合成)
下記の反応式に従い得られた化合物(161)を用いて化合物(110)を合成した。
【0082】
【化33】
n-ブチルリチウム(1.58mol/l,n−へキサン溶液)1.1部を4.5部とすること以外、上記化合物(160)の合成におけるのと同様の操作を行い、化合物(161)を橙色粉末で得た。
1H-NMR(PPM:d6-DMSO):3.90(s.CH3.3H), 7.11(s.pyrr.2H), 7.20(d.arom.2H), 7.26(d.thio.2H), 7.46(d.arom.2H), 7.84(d.thio.2H), 9.72(s.CHO.2H)
【0084】
エタノール20部に化合物(161)1部とシアノ酢酸メチル0.8部を加え2時間還流反応を行なう。反応終了後、析出した沈殿をろ過し、エタノールで再結晶し化合物(109)を赤色粉末で得た。
化合物(109)0.5部と水酸化カリウム1部をエタノール20部中で2時間還流反応させる。反応溶液に水40部を添加し、さらに塩酸で中和し、析出した赤色結晶を濾過、水洗し、更にエタノールで再結晶することで化合物(110)を赤色結晶として得た。
(吸収極大(EtOH:484nm)、発光極大(EtOH:610nm))
1H-NMR(PPM:d6-DMSO):3.84(s.CH3.3H), 7.14(d.arom.2H), 7.17(s.pyrr.2H), 7.32(d.thio.2H), 7.45(d.arom.2H), 7.80(d.thio.2H), 8.29(s.-CH=.2H)
【0085】
合成例9
(化合物(113)の合成)
化合物(161)2.8部とバルビツール酸1部をエタノール50部中で3時間還流反応を行なう。反応終了後、析出した沈殿をろ過し、さらにヘキサン:酢酸エチル混合溶液でカラムクロマトグラフィー、エタノール:ヘキサン混合溶媒で再結晶し、化合物(113)を赤紫結晶として得た。
(吸収極大(EtOH:534nm)、発光極大(EtOH:636nm))
1H-NMR(PPM:d6-DMSO):3.92(s.CH3.3H), 6.99(d.thio.1H), 7.13(m.pyrr.2H), 7.18(d.arom.2H), 7.32(d.thio.1H), 7.46(d.arom.2H), 7.85(d.thio.1H), 7.95(d.thio.1H), 8.31(s.-CH=.1H), 9.74(s.CHO.1H)
【0086】
実施例
色素を3.2×10-4MになるようにEtOHに溶解した。この溶液中に多孔質基板(透明導電性ガラス電極上に多孔質酸化チタンを450℃にて30分焼結した半導体薄膜電極)を室温で3時間から一晩浸漬し色素を担持せしめ、溶剤で洗浄し、乾燥させ、色素増感された半導体微粒子からなる薄膜を有する光電変換素子を得た。実施例17〜20については2種類の色素をそれぞれ1.6×10-4MになるようにEtOH溶液を調製し、2種類の色素を担持することで同様に光電変換素子を得た。また実施例4、5、8、10及び13においては半導体薄膜電極の酸化チタン薄膜部分に0.2M四塩化チタン水溶液を滴下し、室温にて24時間静置後、水洗して、再度450℃にて30分焼成して得た、四塩化チタン処理半導体薄膜電極を用いて色素を同様に担持した。さらに実施例3及び12については色素の担持時に包摂化合物としてコール酸を3×10-2Mとなるように加えて先の色素溶液を調製し、半導体薄膜に担持して、コール酸処理色素増感半導体薄膜を得た。これと挟むように表面を白金でスパッタされた導電性ガラスを固定してその空隙に電解質を含む溶液を注入した。実施例1、3〜6、8、15、18、19及び比較例1については、3ーメトキシプロピオニトリルにヨウ素/ヨウ化リチウム/1、2ージメチルー3ーn−プロピルイミダゾリウムアイオダイド/t−ブチルピリジンをそれぞれ0.1M/0.1M/0.6M/1Mになるように溶解した電解液Aを、実施例2、7、9〜14、16、17、20及び比較例2については、エチレンカーボネートとアセトニトリルの6対4の溶液にヨウ素/テトラ−n−プロピルアンモニウムアイオーダイドを0.02M/0.5Mになるように溶解した電解液Bをそれぞれ使用した。
測定する電池の大きさは実行部分を0.25cm2とした。光源は500Wキセノンランプを用いて、AM(エアマス)1.5フィルターを通して100mW/cmとした。短絡電流、解放電圧、変換効率はポテンシオ・ガルバノスタットを用いて測定した。
【0087】
【化34】
【表2】
【発明の効果】
更に置換されていてもよい一般式(1)で表される色素の1種又は2種以上を用いることにより、変換効率の高い色素増感光電変換素子が得られた。[0001]
[Technical field to which the present invention pertains]
The present invention relates to a semiconductor fine particle sensitized with an organic dye, a photoelectric conversion element, and a solar cell, and more specifically, an oxide semiconductor fine particle sensitized with a thienylpyrrole dye, and a photoelectric conversion element using the same. And a solar cell using the same.
[0002]
[Prior art]
Solar cells that use sunlight as an energy resource to replace fossil fuels such as oil and coal are drawing attention. Currently, development studies such as high efficiency are being actively conducted on silicon solar cells using crystalline or amorphous silicon, or compound semiconductor solar cells using gallium, arsenic, or the like. However, there is a problem that they are difficult to use for general purposes because of the high energy and cost required for production. A photoelectric conversion element using semiconductor fine particles sensitized with a dye or a solar cell using the same is also known, and a material and a manufacturing technique for producing the photoelectric conversion element are disclosed. (Refer to Patent Document 1, Non-Patent Document 1, and Non-Patent Document 2) This photoelectric conversion element is manufactured using a relatively inexpensive oxide semiconductor such as titanium oxide, and compared with a conventional solar cell using silicon or the like. There is a possibility that a low-cost photoelectric conversion element can be obtained, and a colorful solar cell can be obtained. However, in order to obtain an element with high conversion efficiency, a ruthenium-based complex is used as a sensitizing dye, and the cost of the dye itself is high, and a problem still remains in its supply. Attempts have also been made to use organic dyes as sensitizing dyes, but there are currently no practical applications such as low conversion efficiency, stability and durability.
[Patent Document 1]
Japanese Patent No. 2664194
[Patent Document 2]
Japanese Patent Laid-Open No. 11-273754
[Patent Document 3]
JP 2000-26487 A
[Patent Document 4]
WO2002011213 Publication
[Non-Patent Document 1]
B.O'Regan and M.Graetzel Nature, 353, 737 (1991)
[Non-Patent Document 2]
MKNazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Muller, P. Liska, N. Vlachopoulos, M. Graetzel, J. Am. Chem. Soc., 115, 6382 (1993 Year)
[Non-Patent Document 3]
W. Kubo, K. Murakoshi, T. Kitamura, K. Hanabusa, H. Shirai, and S. Yanagida, Chem. Lett., P. 1241 (1998)
[Non-Patent Document 4]
Tetrahedron Lett. 40, 50, 8887-8891 (1990)
[Non-Patent Document 5]
Chem. Lett., 238-239 (2000)
[0003]
[Problems to be solved by the invention]
In photoelectric conversion elements using organic dye-sensitized semiconductors, development of highly practical photoelectric conversion elements with high conversion efficiency using inexpensive organic dyes is required.
[0004]
[Means for Solving the Problems]
As a result of diligent efforts to solve the above-mentioned problems, the present inventors have sensitized semiconductor fine particles using a dye having a specific structure, and by creating a photoelectric conversion element, a photoelectric conversion element with high conversion efficiency can be obtained. The inventors have found that the present invention can be obtained and have completed the present invention. That is, the present invention
[0005]
(1) A photoelectric conversion element characterized by using oxide semiconductor fine particles sensitized with a dye having a structure of the general formula (1),
[0006]
[Chemical formula 5]
(R and Z in the general formula (1) each independently have a hydrogen atom, an alkyl group which may have a substituent, an aromatic hydrocarbon residue which may have a substituent or a substituent. Y1 and Y2 each independently represent a substituent and may form a ring, and m1 and m2 each independently represents an integer of 0 to 3.)
(2) A photoelectric conversion element characterized by using oxide semiconductor fine particles sensitized with a dye represented by the general formula (2),
[0008]
[Chemical 6]
(In the formula, A1 and A2 are each independently an aromatic hydrocarbon residue which may have a substituent, a heterocyclic residue which may have a substituent, a hydroxyl group, a cyano group, a hydrogen atom, a halogen, An alkyl group which may have an atom or a substituent, or a group having a carbonyl group such as a carboxyl group, a carbonamido group, an alkoxycarbonyl group and an acyl group, where X1 is an aromatic hydrocarbon which may have a substituent. And a group having a substituted carbonyl group such as a residue, a heterocyclic residue which may have a substituent, a cyano group or a carboxyl group, a carbonamido group, an alkoxycarbonyl group, an acyl group, etc. Y1 and Y2 are each independently R and Z each independently represent a hydrogen atom, an alkyl group that may have a substituent, or an aromatic carbon that may have a substituent And n represents an integer of 1 to 4. m1 and m2 each independently represents an integer of 0 to 3. l represents 1 to 3. When n is 2 or more and A1 and A2 are present in plural, each A1 and each A2 are the same or different from each other, and when A1 or A1 is present in plural In the case where a plurality of each A1, A2 or A2 are present, a plurality of groups in each A2 and X1 may form a ring which may have a substituent.)
(3) The photoelectric conversion element according to (2), wherein X1 in the general formula (2) is a carboxyl group,
(4) The photoelectric conversion element according to (2) to (3), wherein X1 in the general formula (2) is a carboxyl group, and A2 closest to X1 is a cyano group or a carboxyl group,
(5) The photoelectric conversion device according to (2), wherein the ring formed by X1 in the general formula (2) or A2 closest to X1 and X1 is a heterocyclic residue which may have a substituent,
(6) The heterocyclic residue described in (5) is thiazole, oxazole, imidazole, selenazole, indole, pyridine and their benzene or naphthalene ring-increased substances, rhodanine, oxazolidone, thiooxazolidone, hydantoin, thiohydantoin, thianaphthene , A photoelectric conversion element characterized by being a residue of pyrazolone, barbituric acid, thiobarbituric acid,
(7) The photoelectric conversion element according to any one of (1) to (6), wherein R in the general formulas (1) and (2) is an aromatic hydrocarbon group which may have a substituent.
(8) The photoelectric conversion element according to any one of (2) to (7), wherein n in the general formula (2) is 1 or 2.
(9) Of at least one dye selected from the group consisting of other metal complex dyes and organic dyes having other structures, including at least one dye described in (1) to (8), Photoelectric conversion element using oxide semiconductor fine particles sensitized by combined use
(10) The photoelectric conversion element according to any one of (1) to (9), wherein the oxide semiconductor fine particles contain titanium dioxide as an essential component.
(11) The photoelectric conversion element according to any one of (1) to (10), wherein a dye is supported on an oxide semiconductor fine particle in the presence of an inclusion compound.
(12) A solar cell using the photoelectric conversion element according to any one of (1) to (11)
(13) The oxide semiconductor fine particles according to any one of (1) to (11) sensitized by the dye represented by the general formula (1) or (2)
(14) Compound represented by general formula (3)
[0010]
[Chemical 7]
(In the formula, A3 represents a hydrogen atom, a cyano group or a group having a carbonyl group such as a carboxyl group, a carbonamido group, an alkoxycarbonyl group or an acyl group. Also, when n is 2 or more and a plurality of A3 are present, they are independent of each other. X2 may be an aromatic hydrocarbon residue which may have a substituent, a heterocyclic residue which may have a substituent or a carboxyl group, a carbonamido group, an alkoxycarbonyl group and an acyl. Represents a group having a substituted carbonyl group such as Y. Y1 and Y2 each independently represent a substituent and may form a ring, and R and Z may each independently have a hydrogen atom or a substituent. An alkyl group, an aromatic hydrocarbon residue which may have a substituent, or a heterocyclic residue which may have a substituent, n represents an integer of 1 to 4, and m1 and m2 each represent .l represents an integer of independently 0-3 may form a ring which may have a substituent with 1 to 3 of an integer. The A3 and X substituents.)
(15) A compound represented by the general formula (4) or (5).
[0012]
[Chemical 8]
(Wherein A3, X2, R, Z and n are the same as those in formula (3)),
About.
[0014]
DETAILED DESCRIPTION OF THE INVENTION
The present invention is described in detail below. The photoelectric conversion element of the present invention is characterized by using an oxide semiconductor sensitized by a dye having a structure of the general formula (1) which may further have a substituent.
[0015]
[Chemical 9]
In the general formula (1), R and Z are a hydrogen atom, an alkyl group which may have a substituent, an aromatic hydrocarbon residue which may have a substituent, a heterocyclic residue which may have a substituent. Represents a group. The aromatic hydrocarbon residue means a group obtained by removing one hydrogen atom from an aromatic hydrocarbon. Examples of the aromatic hydrocarbon residue include benzene, naphthalene, anthracene, phenanthrene, pyrene, indene, azulene, fluorene, and perylene. These are usually aromatic hydrocarbon residues having an aromatic ring having 6 to 16 carbon atoms (such as an aromatic ring and a condensed ring containing an aromatic ring). Preferably, benzene, naphthalene, pyrene, etc. are mentioned. The heterocyclic residue means a group obtained by removing one hydrogen atom from a heterocyclic compound. Examples of the heterocyclic compound include pyridine, pyrazine, pyrimidine, pyrazole, pyrazolidine, thiazolidine, oxazolidine, pyran, chromene, and pyrrole. , Benzimidazole, imidazoline, imidazolidine, imidazole, pyrazole, triazole, triazine, diazole, morpholine, indoline, thiophene, furan, oxazole, thiazine, thiazole, indole, benzothiophene, benzothiazole, naphthothiazole, benzoxazole, naphthoxazole, Examples include selenazole, benzoselenazole, indolenine, benzoindolenine, pyrazine, quinoline, quinazoline, and carbazole. Preferred examples include thiophene, benzothiophene, pyridine, quinoline, thiazole, indole and the like. More preferred are thiophene and benzothiophene.
Y1 and Y2 each independently represent a substituent, and may form a ring. The substituent may be the same as the substituent which the aromatic hydrocarbon residue which may have the following substituents may have, and when forming a ring, aromatic carbon such as benzene ring and naphthalene ring Those having a hydrogen-increased form are preferred, and specific examples include a benzothiophene ring and a benzopyrrole ring. m1 and m2 each independently represent an integer of 0 to 3.
[0017]
The substituent in the aromatic hydrocarbon residue which may have a substituent and the heterocyclic residue which may have a substituent is not particularly limited, but is an alkyl group, an aryl group, a cyano group, an isocyano group. , Thiocyanato group, isothiocyanato group, nitro group, nitrosyl group, halogen atom, hydroxyl group, sulfo group, phosphate group, phosphate ester group, substituted or unsubstituted mercapto group, substituted or unsubstituted amino group, substituted or unsubstituted amide A substituted carbonyl group such as a group, an alkoxyl group or an alkoxycarbonyl group, a carboxyl group, a carbonamido group, an acyl group, etc., and further having an aromatic hydrocarbon residue and a substituent which may have a substituent. An optional heterocyclic residue may be substituted. Examples of the alkyl group include saturated and unsaturated linear, branched and cyclic alkyl groups which may have a substituent. The number of carbon atoms is preferably 1 to 36, more preferably a substituent. A saturated linear alkyl group having 1 to 20 carbon atoms can be mentioned. Examples of cyclic compounds include cycloalkyl having 3 to 8 carbon atoms. These alkyl groups may be further substituted with the above substituents (excluding alkyl groups).
[0018]
Examples of the aryl group include a group obtained by removing a hydrogen atom from an aromatic ring compound mentioned in the section of the aromatic hydrocarbon residue described later. The aryl group may be further substituted with the above groups.
Examples of the acyl group include an alkylcarbonyl group having 1 to 10 carbon atoms, an arylcarbonyl group, and the like, preferably an alkylcarbonyl group having 1 to 4 carbon atoms, specifically an acetyl group, a propionyl group, and the like. The halogen atom includes atoms such as fluorine, chlorine, bromine and iodine, and chlorine, bromine and iodine are preferable. Examples of phosphoric acid esters include phosphoric acid (having 1 to 4 carbon atoms) alkyl ester groups. Examples of the substituted or unsubstituted mercapto group include a mercapto group and an alkyl mercapto group. Examples of substituted or unsubstituted amino groups include amino groups, mono- or dimethylamino groups, mono- or diethylamino groups, alkyl-substituted amino groups such as mono- or dipropylamino groups, mono- or diphenylamino groups, mono- or dinaphthylamino groups, etc. An aromatic substituted amino group, a benzylamino group, etc. are mentioned. Examples of the substituted or unsubstituted amide group include an amide group, an alkylamide group, and an aromatic amide group. Examples of the alkoxyl group include an alkoxyl group having 1 to 10 carbon atoms. Examples of the alkoxycarbonyl group include an alkoxycarbonyl group having 1 to 10 carbon atoms.
In addition, an acidic group such as a carboxyl group, a sulfo group, and a phosphoric acid group and a hydroxyl group may form a salt. Examples of the salt include alkali metals or alkaline earth metals such as lithium, sodium, potassium, magnesium, and calcium. Or organic bases such as quaternary ammonium salts such as tetramethylammonium, tetrabutylammonium, pyridinium, imidazolium and the like.
[0019]
More preferred is a photoelectric conversion element using oxide semiconductor fine particles sensitized by a dye having a methine group introduced as represented by the general formula (2).
[0020]
[Chemical Formula 10]
In the formula, A1 and A2 are each independently an aromatic hydrocarbon residue which may have a substituent, a heterocyclic residue which may have a substituent, a hydroxyl group, a cyano group, a hydrogen atom, a halogen atom. Alternatively, it represents an alkyl group which may have a substituent or a group having a carbonyl group such as a carboxyl group, a carbonamido group, an alkoxycarbonyl group and an acyl group. The aromatic hydrocarbon residue and heterocyclic residue have the same meaning as described above, and the substituents that may be present are the same as described above. When n is 2 or more and a plurality of A1 and A2 are present, each A1 and each A2 may be the same or different independently of each other. Preferably, A1 and A2 are independently a hydrogen atom, a cyano group, an alkyl group, a halogen atom and a carboxyl group. As a combination, when n is 1, A1 and A2 are both cyano groups, A1 is a hydrogen atom, and A2 is a hydrogen atom or a cyano group. When n is 2 or more, it is preferable that A1 and A2 are all cyano groups, A1 is all hydrogen atoms, A2 closest to X is cyano group, and other A2 is hydrogen atom.
In addition, when there are a plurality of A1 or A1, when there are a plurality of each of A1, A2 or A2, a ring which may have a substituent is formed by a plurality of groups in each of A2 and X1. Also good.
Examples of the ring to be formed include an unsaturated hydrocarbon ring or a heterocyclic ring. Examples of unsaturated hydrocarbon rings include benzene, naphthalene, anthracene, phenanthrene, pyrene, indene, azulene, fluorene, cyclobutene, cyclohexene, cyclopentene, cyclohexadiene, cyclopentadiene, and the like. And heterocyclic groups include pyridine ring, pyrazine ring, piperidine ring, indoline ring, thiophene ring, furan ring, pyran ring, oxazole ring, thiazole ring, indole ring, benzothiazole ring, benzoxazole ring, quinoline ring, carbazole Ring, benzopyran ring and the like. Of these, preferred are a cyclobutene ring, a cyclopentene ring, a cyclohexene ring, and a pyran ring. Moreover, when it has a carbonyl group, a thiocarbonyl group, etc., you may form cyclic ketone or cyclic thioketone.
[0022]
X1 represents an aromatic hydrocarbon residue which may have a substituent, a heterocyclic residue which may have a substituent, a substituted carbonyl such as a cyano group or a carboxyl group, a carbonamido group, an alkoxycarbonyl group and an acyl group. A group having a group is represented. The aromatic hydrocarbon residue and heterocyclic residue have the same meaning as described above, and the substituents that may be present are the same as described above. A group having a substituted carbonyl group such as a heterocyclic residue which may have a substituent, a cyano group or a carboxyl group, a carbonamido group, an alkoxycarbonyl group and an acyl group, and more preferably a substituent. Good heterocyclic residue and carboxyl group. The heterocyclic residues shown here are preferably pyridine, pyrazole, pyran, chromene, benzimidazole, imidazole, pyrazole, thiazole, oxazole, indole, benzothiazole, naphthothiazole, benzoxazole, naphthoxazole, selenazole, benzoselenazole , Residues such as indolenine, benzoindolenine, and quinoline.
As described above, X1 and A1 or A2 can form a ring. In particular, X1 and A2 closest to X1 are preferably formed, and the ring at that time is more preferably a heterocyclic residue which may have a substituent. Specific examples include pyridine, quinoline, pyran, chromene, pyrimidine, pyrrole, thiazole, benzothiazole, oxazole, benzoxazole, selenazole, benzoselenazole, imidazole, benzimidazole, pyrazole, thiophene, and the like. The heterocyclic residues may be ring-increased or hydrogenated, and these may have a substituent as described above.
Combined with these substituents, cyclic hydrocarbons, rhodanine rings as heterocycles, oxazolidone rings, thiooxazolidone rings, hydantoin rings, thiohydantoin rings, indandione rings, thianaphthene rings, pyrazolone rings, barbiturates rings, thiobarbitur rings, A structure forming a pyridone ring or the like is preferable. Further, the heterocyclic ring formed by X or X and A may be quaternized and may have a counter ion at that time. Specifically, although not particularly limited, a general anion may be used. A specific example is F-, Cl-, Br-, I-, ClOFour -, BFFour -, PF6 -, OH-, SOFour 2-, CHThreeSOFour -, Toluenesulfonic acid ions, etc., Br-, I-, ClOFour -, BFFour -, PF6 -, CHThreeSOFour -Toluene sulfonate ions are preferred. Moreover, you may neutralize by acidic groups, such as a carboxyl group in a molecule | numerator or between molecules instead of a counter ion.
[0023]
Y1 and Y2 each independently represent a substituent, and may form a ring. The substituent may be the same as that in the general formula (1), and specifically, a hydrogen atom, an alkyl group or an aryl group is preferable. In the case of forming a ring, a ring in which aromatic hydrocarbons such as a benzene ring and a naphthalene ring are increased is preferable, and specific examples include a benzothiophene ring and a benzopyrrole ring. m1 and m2 each independently represent an integer of 0 to 3.
[0024]
Examples of R and Z include the same substituents as described above. R is preferably an aromatic hydrocarbon residue which may have a substituent, and specific examples include a benzene ring and a naphthalene ring which may have a substituent. Preferred substituents include alkyl groups, cyano groups, isocyano groups, thiocyanato groups, isothiocyanato groups, halogen atoms, hydroxyl groups, substituted or unsubstituted amino groups, alkoxyl groups or alkoxycarbonyl groups, carboxyl groups, carbonamido groups, acyl groups, etc. And a substituted carbonyl group. More preferable examples include alkyl groups, alkoxyl groups, phenoxy groups, halogen atoms such as fluorine, chlorine, bromine and iodine, dialkylamino groups, monoalkylamino groups, diphenylamino groups and monophenylamino groups. Z is preferably a hydrogen atom, an aromatic hydrocarbon residue which may have a substituent, and a heterocyclic residue which may have a substituent, specifically a benzene ring which may have a substituent , A naphthalene ring, a thiophene ring, a benzthiophene ring, and the like, more preferably a thiophene ring. Preferred substituents are the same as described above.
[0025]
Examples of the position at which the methine group of the general formula (2) is introduced include substitution positions a to e in the following general formula (6), and a, d, or e is preferable. These substitution positions depend on the introduction position of the aldehyde substituent when the compound (7) described below is produced. The introduction position is controlled by the nature of the substituents such as R and Z and the reaction conditions for the production.
[0026]
Embedded image
n shows the integer of 1-4. Preferably it is an integer of 1-3, More preferably, it is an integer of 1-2.
l shows the integer of 1-3. Preferably it is an integer of 1-2, and more preferably 1. The compound represented by the general formula (2) may take a structural isomer such as a cis isomer or a trans isomer, but is not particularly limited, and any of them can be used favorably as a photosensitizing dye.
In any case, it is preferable for an adsorptive bond with an oxide semiconductor to have at least one carbonyl group such as a carboxylic acid group, a carbonamide group, an ester group, or an acyl group in the molecule.
[0028]
Embedded image
Further, when the structure is specified, the structure listed in the general formula (3) can be mentioned. At this time, R, Z, Y1, Y2, m1, m2, n, and l are the same as described above.
A3 represents a group having a carbonyl group such as a hydrogen atom, a cyano group or a carboxyl group, a carbonamido group, an alkoxycarbonyl group or an acyl group. In the case where n is 2 or more and a plurality of A3 are present, they may be the same or different from each other, preferably a hydrogen atom, and A3 closest to X2 includes a cyano group, a carboxyl group, and an alkoxycarbonyl group. It is done.
X2 has an aromatic hydrocarbon residue which may have a substituent, a heterocyclic residue which may have a substituent, or a substituted carbonyl group such as a carboxyl group, a carbonamido group, an alkoxycarbonyl group and an acyl group. Represents a group. Preferred examples include an aromatic hydrocarbon residue which may have a substituent, a heterocyclic residue which may have a substituent, a carboxyl group, and an alkoxycarbonyl group. Examples of the aromatic hydrocarbon residue which may have a substituent and the heterocyclic residue which may have a substituent are the same as the examples given for X1 above.
Moreover, you may form the ring which may have a substituent using the substituent of X2 and A3. This is the same as the ring formed by X1 and A2 closest to X1.
[0030]
Embedded image
Further, when the structure of the dye is specified, the above general formulas (4) and (5) can be mentioned. In this case, R, Z, A3, X2, and n are the same as described above.
[0032]
The compound of the general formula (1) can be generally produced by the method of Ogura et al. (For example, Non-Patent Document 4) Formylation (for example, Non-Patent Document 5) (1) After metallizing (1) with a base such as butyllithium, an amide derivative such as dimethylformamide is allowed to act, dimethylformamide or the like Compound (7), which is a precursor of compound (2), is obtained by allowing phosphoryl chloride or the like to act on a bismuth reagent and causing this to act on (1). The substitution position of the formyl group is controlled by the substituent (1) and the production method. When n is 2 or more, it can be obtained by a method of further Claisen condensation of acetaldehyde or a reagent such as dimethylaminoacrolein. The number of l is controlled by the temperature and the amount of reagent. Further, if necessary, a compound having active methylene and the compound (7) in the presence of a basic catalyst such as sodium ethoxide, piperidine or piperazine, an alcohol such as methanol, ethanol or isopropanol, or an aprotic polar solvent such as dimethylformamide Compound (2) can be obtained by condensation at 20 ° C. to 120 ° C., preferably about 50 ° C. to 80 ° C., in a solvent such as water or acetic anhydride.
Moreover, the compound of General formula (3) can be manufactured by the method similar to General formula (2). That is, the compound of the general formula (4) is usually a method in which the general formula (1) is metallized with a base such as butyl lithium and then an amide derivative such as dimethylformamide, dimethylaminoacrolein, dimethylaminovinylacrolein or the like is allowed to act. To obtain a precursor of the general formula (4) having a formyl group, a propenal group, a pentadienal group, etc. at the substitution position a shown in the formula (6), and if necessary, a compound having active methylene, In the presence of a basic catalyst such as sodium ethoxide, piperidine, piperazine, in an aprotic polar solvent such as methanol, ethanol, butanol or the like, dimethylformamide or a solvent such as acetic anhydride, 20 to 120 ° C., preferably 50 A compound of the general formula (4) is obtained by condensation at -80 ° C. The compound of the general formula (5) is usually obtained by reacting the general formula (1) with a Vilsmeier reagent obtained by reacting dimethylformamide, dimethylaminoacrolein, dimethylaminovinylacrolein or the like with phosphoryl chloride or the like. The precursor of (5) having a formyl group, propenal group, pentadienal group and the like at the substitution position d shown in Fig. 5 is obtained, and if necessary, a compound having active methylene is added to sodium ethoxide, piperidine, piperazine if necessary. By condensation at 20 to 120 ° C., preferably 50 to 80 ° C. in an aprotic polar solvent such as methanol, ethanol and butanol, an aprotic polar solvent such as dimethylformamide, and a solvent such as acetic anhydride. A compound of general formula (5) is obtained.
[0033]
Embedded image
(Z, R, Y1, Y2, A1, X1, A2, n, l, m1, and m2 in the above formula are the same as described above.)
Examples of compounds are listed below. First, in the general formula (2), Z is a thiophene ring and R may have a substituent (in the figure, phenyl substituents are described as R1 to R3) and n is 1. Table 1 shows compound examples. The phenyl group is abbreviated as Ph, the diethylamino group as DEA, and the diphenylamino group as DPA. In addition, the ring (ring B) when X1 and X and A2 form a ring is described below.
[0035]
Embedded image
[Table 1]
(Example of ring B)
[0041]
Embedded image
Embedded image
Examples of other compounds are shown below.
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
Embedded image
In the dye-sensitized photoelectric conversion element of the present invention, for example, an oxide semiconductor thin film is produced on a substrate using oxide semiconductor fine particles, and then the dye is supported on the thin film.
In the present invention, the substrate on which the oxide semiconductor thin film is provided is preferably one having a conductive surface, but such a substrate is readily available in the market. Specifically, for example, conductive metal oxide such as tin oxide doped with indium, fluorine or antimony on the surface of glass or the surface of a transparent polymer material such as polyethylene terephthalate or polyether sulfone, copper, silver, etc. A film provided with a metal thin film such as gold can be used. The conductivity is usually 1000Ω or less, particularly preferably 100Ω or less.
The oxide semiconductor fine particles are preferably metal oxides, and specific examples thereof include oxides of titanium, tin, zinc, tungsten, zirconium, gallium, indium, yttrium, niobium, tantalum, vanadium, and the like. Of these, oxides such as titanium, tin, zinc, niobium, and indium are preferable, and among these, titanium oxide is most preferable. These oxide semiconductors can be used alone, but can also be used by mixing or coating the surface of the semiconductor. The average particle diameter of the oxide semiconductor fine particles is usually 1 to 500 nm, preferably 1 to 100 nm. The oxide semiconductor fine particles having a large particle size and a small particle size can be mixed or used in combination.
[0057]
An oxide semiconductor thin film is a method in which oxide semiconductor fine particles are directly formed on a substrate by spray spraying, a method in which a semiconductor fine particle thin film is electrically deposited using a substrate as an electrode, a semiconductor fine particle such as a slurry of semiconductor fine particles or a semiconductor alkoxide. After the paste containing fine particles obtained by hydrolyzing the precursor is applied on a substrate, it can be produced by drying, curing or baking. In view of the performance of the oxide semiconductor electrode, a method using slurry is preferable. In the case of this method, the slurry is obtained by dispersing the oxide semiconductor fine particles, which are secondarily aggregated, in a dispersion medium so as to have an average primary particle diameter of 1 to 200 nm by a conventional method.
[0058]
The dispersion medium for dispersing the slurry may be anything as long as it can disperse the semiconductor fine particles. Water or alcohol such as ethanol, ketone such as acetone, acetylacetone or organic solvent such as hydrocarbon such as hexane is used. A mixture may be used, and the use of water is preferable in that the viscosity change of the slurry is reduced. A dispersion stabilizer can be used for the purpose of stabilizing the dispersion state of the oxide semiconductor fine particles. Examples of the dispersion stabilizer that can be used include acids such as acetic acid, hydrochloric acid, and nitric acid, or acetylacetone, acrylic acid, polyethylene glycol, polyvinyl alcohol, and the like.
[0059]
The substrate coated with the slurry may be fired, and the firing temperature is usually 100 ° C. or higher, preferably 200 ° C. or higher, and the upper limit is generally lower than the melting point (softening point) of the base material. Yes, preferably 600 ° C. or lower. The firing time is not particularly limited but is preferably within 4 hours. The thickness of the thin film on a board | substrate is 1-200 micrometers normally, Preferably it is 1-50 micrometers.
[0060]
A secondary treatment may be performed on the oxide semiconductor thin film. That is, for example, the performance of the semiconductor thin film can be improved by immersing the thin film together with the substrate directly in a solution of the same metal alkoxide, chloride, nitride, sulfide, etc. as the semiconductor and drying or refiring. Examples of the metal alkoxide include titanium ethoxide, titanium isopropoxide, titanium tert-butoxide, n-dibutyl-diacetyltin, and the alcohol solution thereof is used. Examples of the chloride include titanium tetrachloride, tin tetrachloride, zinc chloride and the like, and an aqueous solution thereof is used. The oxide semiconductor thin film thus obtained is composed of fine particles of an oxide semiconductor.
[0061]
Next, a method for supporting a dye on an oxide semiconductor thin film will be described. As a method for supporting the dye of the general formula (1), a solution obtained by dissolving the dye in a solvent capable of dissolving the dye, or a dispersion obtained by dispersing the dye in the case of a dye having low solubility A method of immersing the substrate provided with the oxide semiconductor thin film in a liquid is given. The concentration in the solution or dispersion is appropriately determined depending on the dye. The semiconductor thin film prepared on the substrate is immersed in the solution. The immersion time is generally from room temperature to the boiling point of the solvent, and the immersion time is about 1 to 48 hours. Specific examples of the solvent that can be used for dissolving the dye include methanol, ethanol, acetonitrile, dimethyl sulfoxide, dimethylformamide, acetone, t-butanol and the like. The dye concentration of the solution is usually 1 × 10-6M to 1M is good, preferably 1 × 10-Five M ~ 1x10-1M. In this way, a photoelectric conversion element of an oxide semiconductor fine particle thin film sensitized with a dye is obtained.
[0062]
The dye of the general formula (1) to be carried may be one kind or a mixture of several kinds. Further, when mixing, the dyes of the present invention may be mixed, or other dyes or metal complex dyes may be mixed. In particular, by mixing pigments having different absorption wavelengths, a wide absorption wavelength can be used, and a solar cell with high conversion efficiency can be obtained. Examples of metal complex dyes that can be mixed are not particularly limited, but ruthenium complexes and their quaternary salt compounds, phthalocyanines, porphyrins, and the like shown in Non-Patent Document 2 and Patent Document 2 are preferable. Is a metal-free phthalocyanine, porphyrin, cyanine, merocyanine, oxonol, triphenylmethane, methine dyes such as acrylic acid dyes shown in Patent Document 4, xanthene, azo, anthraquinone, perylene, etc. Pigments. Preferably, a ruthenium complex, a merocyanine, an acrylic acid-based methine dye, or the like is used. When two or more dyes are used, the dyes may be sequentially adsorbed on the semiconductor thin film, or may be admixed and dissolved.
[0063]
The ratio of the dye to be mixed is not particularly limited, and is selected and optimized from the respective dyes. Generally, it is preferable to use about 10% mol or more per one dye from a mixture of equimolar amounts. When the dye is adsorbed to the oxide semiconductor fine particle thin film using a solution in which the mixed dye is mixed and dissolved or dispersed, the total concentration of the dye in the solution may be the same as when only one kind is supported. As the solvent in the case of using a mixture of dyes, the above-mentioned solvents can be used, and the solvents for the respective dyes to be used may be the same or different.
[0064]
When the dye is supported on the thin film of oxide semiconductor fine particles, it is effective to support the dye in the coexistence of the inclusion compound in order to prevent association between the dyes. Examples of inclusion compounds include steroidal compounds such as cholic acid, crown ether, cyclodextrin, calixarene, polyethylene oxide, and the like. Deoxycholic acid, dehydrodeoxycholic acid, chenodeoxycholic acid, methyl cholate are preferable. Examples include esters, cholic acids such as sodium cholate, polyethylene oxide, and the like. Alternatively, after the dye is supported, the surface of the semiconductor electrode may be treated with an amine compound such as 4-t-butylpyridine. As a treatment method, for example, a method in which a substrate provided with a semiconductor fine particle thin film carrying a dye in an ethanol solution of amine is immersed.
[0065]
The solar cell of the present invention is composed of a redox electrolyte, a hole transport material, a p-type semiconductor, or the like as a counter electrode with a photoelectric conversion element electrode having a dye supported on the oxide semiconductor thin film. Examples of the form of the redox electrolyte, the hole transport material, the p-type semiconductor, and the like include liquid, pseudo-solid (gel and gel), and solid. Liquids such as redox electrolytes, molten salts, hole transport materials, p-type semiconductors, etc. dissolved in solvents or room temperature molten salts are quasi-solids (gels and gels). Examples include a matrix and a low molecular gelling agent. As a solid material, a redox electrolyte, a molten salt, a hole transport material, a p-type semiconductor, or the like can be used. Examples of the hole transport material include amine derivatives, conductive polymers such as polyacetylene, polyaniline, and polythiophene, and materials using a discotic liquid crystal phase such as polyphenylene. Examples of the p-type semiconductor include CuI and CuSCN. The counter electrode is preferably conductive and has a catalytic action on the reduction reaction of the redox electrolyte. For example, glass, a polymer film deposited with platinum, carbon, rhodium, ruthenium or the like, or coated with conductive fine particles can be used.
[0066]
The redox electrolyte used in the solar cell of the present invention is a halogen redox electrolyte composed of a halogen compound and a halogen molecule as a counter ion, ferrocyanate-ferricyanate, ferrocene-ferricinium ion, cobalt complex, etc. Metal redox electrolytes such as metal complexes, and organic redox electrolytes such as alkylthiol-alkyldisulfides, viologen dyes, hydroquinone-quinones, and the like, and halogen redox electrolytes are preferred. Examples of the halogen molecule in the halogen redox electrolyte comprising a halogen compound-halogen molecule include iodine molecule and bromine molecule, and iodine molecule is preferable. Examples of halogen compounds having halogen ions as counter ions include LiI, NaI, KI, CsI, and CaI.2Metal halides such as CuI, or organic quaternary ammonium salts of halogens such as tetraalkylammonium iodide, imidazolium iodide, pyridinium iodide, and the like, and salt compounds having iodine ions as counter ions are preferred. Examples of the salt compound having iodine ion as a counter ion include lithium iodide, sodium iodide, trimethylammonium iodide salt and the like.
[0067]
When the redox electrolyte is formed in the form of a solution containing the redox electrolyte, an electrochemically inert solvent is used as the solvent. For example, acetonitrile, propylene carbonate, ethylene carbonate, 3-methoxypropionitrile, methoxyacetonitrile, ethylene glycol, propylene glycol, diethylene glycol, triethylene glycol, γ-butyrolactone, dimethoxyethane, diethyl carbonate, diethyl ether, diethyl carbonate, dimethyl carbonate, 1,2-dimethoxyethane, dimethylformamide, dimethyl sulfoxide, 1,3-dioxolane, methyl formate, 2-methyltetrahydrofuran, 3-methoxyoxaziridin-2-one, γ-butyrolactone, sulfolane, tetrahydrofuran, Water, etc., among them, acetonitrile, propylene carbonate, ethylene carbonate, among others 3-methoxypropionitrile, methoxyacetonitrile, ethylene glycol, 3-methoxyoxaziridin-2-one, γ-butyrolactone and the like are preferable. These may be used alone or in combination of two or more. In the case of a gel electrolyte, an electrolyte or electrolyte solution contained in a matrix such as an oligomer or polymer, or a low molecular gelling agent described in Non-Patent Document 3 that also contains an electrolyte or electrolyte solution Etc. The concentration of the redox electrolyte is usually 0.01 to 99% by weight, preferably about 0.1 to 90% by weight.
[0068]
In the solar cell of the present invention, a counter electrode is disposed between electrodes of a photoelectric conversion element in which a dye is supported on an oxide semiconductor thin film on a substrate so as to sandwich the electrode. In the meantime, the solar cell of the present invention is obtained by filling a solution containing the redox electrolyte.
[0069]
【Example】
Hereinafter, the present invention will be described in more detail based on examples, but the present invention is not limited to these examples. In the examples, unless otherwise specified, parts represent parts by mass, and% represents mass%.
[0070]
Synthesis example 1
(Synthesis of Compound (57))
Compound (57) was synthesized using compound (157) obtained according to the following reaction formula.
Reflux 4 parts of 1,4-di (2-thienyl) -1,4-butanedione (154), 5 parts of N, N-diethyl-1,4-phenylenediamine (155), 8 parts of propionic acid and 25 parts of toluene. React for 4 hours under. After cooling, 100 parts of water is added, and the liquid separation and toluene layer are extracted. Further, column chromatography is performed with a mixed solution of hexane: ethyl acetate, and recrystallization is performed with a mixed solvent of ethanol: hexane, and the compound (156) is converted into white plate crystals. Obtained. (Melting point: 79-81 ° C., absorption maximum (EtOH): 272,342 nm)
To 5 parts of dimethylformamide, 0.6 part of phosphoryl chloride is added dropwise at 5 ° C. or lower and stirred at 10 ° C. or lower for 1 hour. A solution prepared by dissolving 1 part of Compound (156) in 5 parts of dimethylformamide was added dropwise while maintaining the temperature at 10 ° C. or lower, and the reaction was continued for 3 hours. After completion of the reaction, the mixture was neutralized with an aqueous sodium hydroxide solution, and the deposited precipitate was filtered and washed with water to obtain the compound (157) as a pale yellow powder. (Melting point: 154-158 ° C, absorption maximum (EtOH): 270, 308, 430 (Sh) nm)
[0071]
Embedded image
1 part of compound (157), 0.3 part of methyl cyanoacetate and 0.2 part of piperazine are added to 20 parts of ethanol and refluxed for 2 hours. After completion of the reaction, the deposited precipitate was filtered and recrystallized with ethanol to obtain Compound (56) as brown crystals. (Melting point: 178-180 ° C, absorption maximum (EtOH: 276,308,396nm)
0.2 part of compound (56) and 0.5 part of potassium hydroxide are refluxed in 10 parts of ethanol for 2 hours. 50 parts of water was added to the reaction solution, neutralized with hydrochloric acid, and the precipitated yellow crystals were filtered, washed with water, and recrystallized with ethanol to obtain Compound (57) as brown crystals. (Melting point: 233-235 ° C., absorption maximum (EtOH: 274, 308, 390 nm), emission maximum (EtOH: 434 nm))
1H-NMR (PPM: d6-DMSO): 1.09 (t.CH3.6H), 3.30 (m.CH2.4H), 6.64 (d.arom.2H), 6.83 (d.thio.1H), 6.98 (dd .thio.1H), 7.07 (d.arom.2H), 7.10 (d.thio.1H), 7.12 (d.thio.1H), 7.44 (s.pyrr.1H), 7.47 (dd.thio.1H) , 7.73 (dd.thio.1H), 8.00 (s.-CH = .1H)
[0073]
Synthesis example 2
(Synthesis of Compound (58))
To 20 parts of ethanol, 0.5 part of compound (157), 0.5 parts of methyl sulfate = 1,2,3,3-trimethyl-5-carboxyindolenium and 0.1 part of piperazine are added, and the reaction is carried out under reflux for 1 hour. . After completion of the reaction, the reaction mixture was cooled, 1 part of hydroiodic acid was added, and the deposited precipitate was filtered and recrystallized with ethanol to obtain Compound (58) as a dark brown powder. (Melting point: 174 to 176 ° C, absorption maximum (EtOH: 292, 364, 528nm), emission maximum (EtOH: 569nm))
[0074]
Synthesis example 3
(Synthesis of Compound (62))
To 10 parts of ethanol, 0.5 part of the compound (157), 0.3 part of rhodanine-3-acetic acid and 0.1 part of piperazine are added and refluxed for 2 hours. After completion of the reaction, the deposited precipitate was filtered and recrystallized with ethanol to obtain Compound (62) as a brown powder. (Melting point: 234-237 ° C., absorption maximum (EtOH: 272, 442 nm), emission maximum (EtOH: 625 nm))
1H-NMR (PPM: d6-DMSO): 1.09 (t.CH3.6H), 3.30 (m.CH2.4H), 4.47 (s.CH2.2H)
6.65 (d.arom.2H), 6.84 (s.pyrr.1H), 6.98 (dd.thio.1H), 7.09 (d.arom.2H),
7.10 (dd.thio.1H), 7.12 (dd.thio.1H), 7.13 (dd.thio.1H), 7.45 (dd.thio.1H),
7.55 (s.-CH = .1H), 7.71 (dd.thio.1H),
[0075]
Synthesis example 4
(Synthesis of Compound (61))
The same operation was carried out using barbituric acid instead of rhodanine-3-acetic acid in Synthesis Example 3 to obtain compound (61) as a brown powder. (Melting point: 288 ° C. decomposition (using TG-DTA), absorption maximum (EtOH: 264, 314, 436 nm), emission maximum (EtOH: 649 nm))
[0076]
Synthesis example 5
(Synthesis of Compound (63))
The same operation was performed using 1-phenyl-3-carboxy-5-pyrazolone instead of rhodanine-3-acetic acid in Synthesis Example 3 to obtain compound (63) as a brown powder. (Melting point: 223-225 ° C, absorption maximum (EtOH: 256, 506 nm), emission maximum (EtOH: 658 nm))
[0077]
Synthesis Example 6
(Synthesis of Compound (80))
The same operation was performed in Synthesis Example 3 using diethyl malonate instead of rhodanine-3-acetic acid to obtain compound (80) as a yellow powder. (Melting point: 158 to 160 ° C., absorption maximum (EtOH: 206, 276 nm), emission maximum (EtOH: 417 nm))
[0078]
Synthesis example 7
(Synthesis of Compound (11))
Compound (11) was synthesized using compound (160) obtained according to the following reaction formula.
4 parts of 1,4-di (2-thienyl) -1,4-butanedione (154), 4 parts of p-anisidine (158), 9 parts of propionic acid and 30 parts of toluene are reacted at reflux for 8 hours. After cooling, the precipitated crystals were filtered and washed with 10 parts of acetic acid, and further recrystallized with column chromatography using a mixed solution of hexane: ethyl acetate and a mixed solvent of ethanol: hexane to obtain Compound (159) as white plate crystals. .
53 parts of anhydrous tetrahydrofuran was added to 0.88 part of the compound (159), and the mixture was cooled to -70 ° C. and stirred under a nitrogen atmosphere. To this was added 1.1 parts of n-butyllithium (1.58 mol / l, n-hexane solution), and the mixture was stirred at the same temperature for 40 minutes. Thereafter, 1 part of N, N-dimethylformamide was added, and the mixture was further stirred for 30 minutes and further stirred at room temperature for 3 hours. 40 parts of ammonium chloride solution was added, liquid separation was performed using 175 parts of toluene, and the toluene layer was extracted, followed by column chromatography with a mixed solution of hexane: ethyl acetate, and recrystallization with a mixed solvent of ethanol: hexane to give Compound (160) yellow Obtained as a powder.
1H-NMR (PPM: d6-DMSO): 3.90 (s.CH3.3H), 6.59 (d.pyrr.1H), 6.65 (d.thio.1H), 6.74 (d.thio.1H), 6.78 (d .pyrr.1H), 6.85 (dd.thio.1H), 7.00 (d.arom.2H), 7.09 (d.thio.1H), 7.27 (d.arom.2H), 7.48 (d.thio.1H) , 9.70 (s.CHO.1H),
[0079]
Embedded image
To 20 parts of ethanol, 1.5 parts of compound (160), 0.5 part of cyanoacetic acid and 0.1 part of piperidine are added and the reaction is carried out under reflux for 2 hours. After completion of the reaction, the deposited precipitate was filtered and recrystallized with ethanol to obtain Compound (11) as an orange powder.
(Absorption maximum (EtOH: 444 nm), emission maximum (EtOH: 587 nm))
1H-NMR (PPM: d6-DMSO): 3.84 (s.CH3.3H), 6.67 (d.pyrr.1H), 6.83 (m.thio.3H), 6.92 (dd.thio.1H), 7.08 (d .arom.2H), 7.33 (d.pyrr.1H), 7.35 (d.arom.2H), 7.47 (d.thio.1H), 7.92 (s.-CH = .1H)
[0081]
Synthesis Example 8
(Synthesis of Compound (110))
Compound (110) was synthesized using compound (161) obtained according to the following reaction formula.
[0082]
Embedded image
Compound (161) was prepared in the same manner as in the synthesis of compound (160) except that 1.1 part of n-butyllithium (1.58 mol / l, n-hexane solution) was changed to 4.5 parts. ) Was obtained as an orange powder.
1H-NMR (PPM: d6-DMSO): 3.90 (s.CH3.3H), 7.11 (s.pyrr.2H), 7.20 (d.arom.2H), 7.26 (d.thio.2H), 7.46 (d .arom.2H), 7.84 (d.thio.2H), 9.72 (s.CHO.2H)
[0084]
1 part of compound (161) and 0.8 part of methyl cyanoacetate are added to 20 parts of ethanol and refluxed for 2 hours. After completion of the reaction, the deposited precipitate was filtered and recrystallized with ethanol to obtain Compound (109) as a red powder.
0.5 part of compound (109) and 1 part of potassium hydroxide are refluxed in 20 parts of ethanol for 2 hours. 40 parts of water was added to the reaction solution, further neutralized with hydrochloric acid, the precipitated red crystals were filtered, washed with water, and recrystallized with ethanol to obtain compound (110) as red crystals.
(Absorption maximum (EtOH: 484 nm), emission maximum (EtOH: 610 nm))
1H-NMR (PPM: d6-DMSO): 3.84 (s.CH3.3H), 7.14 (d.arom.2H), 7.17 (s.pyrr.2H), 7.32 (d.thio.2H), 7.45 (d .arom.2H), 7.80 (d.thio.2H), 8.29 (s.-CH = .2H)
[0085]
Synthesis Example 9
(Synthesis of Compound (113))
2.8 parts of compound (161) and 1 part of barbituric acid are refluxed in 50 parts of ethanol for 3 hours. After completion of the reaction, the deposited precipitate was filtered, and further recrystallized with column chromatography using a mixed solution of hexane: ethyl acetate and a mixed solvent of ethanol: hexane to obtain Compound (113) as reddish purple crystals.
(Absorption maximum (EtOH: 534 nm), emission maximum (EtOH: 636 nm))
1H-NMR (PPM: d6-DMSO): 3.92 (s.CH3.3H), 6.99 (d.thio.1H), 7.13 (m.pyrr.2H), 7.18 (d.arom.2H), 7.32 (d thio.1H), 7.46 (d.arom.2H), 7.85 (d.thio.1H), 7.95 (d.thio.1H), 8.31 (s.-CH = .1H), 9.74 (s.CHO. 1H)
[0086]
Example
3.2 × 10 dye-FourDissolved in EtOH to M. In this solution, a porous substrate (semiconductor thin film electrode obtained by sintering porous titanium oxide on a transparent conductive glass electrode for 30 minutes at 450 ° C. for 30 minutes) is immersed at room temperature for 3 hours to overnight to support the dye, The photoelectric conversion element which has the thin film which consists of a semiconductor fine particle wash | cleaned and dried and dye-sensitized was obtained. For Examples 17 to 20, the two types of dyes were each 1.6 × 10-FourEtOH solution was prepared so that it might become M, and the photoelectric conversion element was similarly obtained by carrying | supporting 2 types of pigment | dyes. In Examples 4, 5, 8, 10, and 13, a 0.2M titanium tetrachloride aqueous solution was dropped on the titanium oxide thin film portion of the semiconductor thin film electrode, allowed to stand at room temperature for 24 hours, washed with water, and again 450 ° C. The dye was similarly supported using a titanium tetrachloride-treated semiconductor thin-film electrode obtained by baking for 30 minutes. Further, in Examples 3 and 12, cholic acid was used as an inclusion compound at the time of supporting the dye at 3 × 10-2In addition to the M, the above dye solution was prepared and supported on the semiconductor thin film to obtain a cholic acid-treated dye-sensitized semiconductor thin film. A conductive glass whose surface was sputtered with platinum was fixed so as to sandwich it, and a solution containing an electrolyte was injected into the gap. For Examples 1, 3-6, 8, 15, 18, 19 and Comparative Example 1, 3-methoxypropionitrile and iodine / lithium iodide / 1,2-dimethyl-3-n-propylimidazolium iodide / t -About the electrolyte solution A which melt | dissolved butylpyridine so that it might become 0.1M / 0.1M / 0.6M / 1M, respectively, about Example 2, 7, 9-14, 16, 17, 20, and the comparative example 2, Electrolytic solutions B in which iodine / tetra-n-propylammonium iodide was dissolved in a 6 to 4 solution of ethylene carbonate and acetonitrile to a concentration of 0.02 M / 0.5 M were used.
The size of the battery to be measured is 0.25 cm in the effective part.2It was. The light source used was a 500 W xenon lamp, and was set to 100 mW / cm through an AM (air mass) 1.5 filter. Short-circuit current, release voltage, and conversion efficiency were measured using a potentio galvanostat.
[0087]
Embedded image
[Table 2]
【The invention's effect】
Furthermore, the dye-sensitized photoelectric conversion element with high conversion efficiency was obtained by using 1 type, or 2 or more types of the pigment | dye represented by General formula (1) which may be substituted.
Claims (10)
をあらわす。Y1及びY2は水素原子を表す。Rは、炭素数1ないし4のアルコキシル基、炭素数1ないし4のアルキル基を有するジアルキルアミノ基又は炭素数1ないし4のアルキル基を有するモノアルキルアミノ基を置換基として有するベンゼン環若しくはナフタレン環を表す。Zは、下記式(11)〜(13)
のいずれかで表される環を形成してもよい。)A photoelectric conversion element using oxide semiconductor fine particles sensitized with a dye represented by the general formula (2) .
Is expressed. Y1 and Y2 represent a hydrogen atom . R is a benzene ring or naphthalene ring having a substituent having a C1-C4 alkoxyl group, a dialkylamino group having a C1-C4 alkyl group, or a monoalkylamino group having a C1-C4 alkyl group. Represents. Z represents the following formulas (11) to (13)
A ring represented by any of the above may be formed. )
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003007360A JP4230228B2 (en) | 2002-01-16 | 2003-01-15 | Dye-sensitized photoelectric conversion element |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002-7062 | 2002-01-16 | ||
| JP2002007062 | 2002-01-16 | ||
| JP2003007360A JP4230228B2 (en) | 2002-01-16 | 2003-01-15 | Dye-sensitized photoelectric conversion element |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2003282165A JP2003282165A (en) | 2003-10-03 |
| JP4230228B2 true JP4230228B2 (en) | 2009-02-25 |
Family
ID=29252914
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003007360A Expired - Fee Related JP4230228B2 (en) | 2002-01-16 | 2003-01-15 | Dye-sensitized photoelectric conversion element |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4230228B2 (en) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090114272A1 (en) | 2005-07-07 | 2009-05-07 | Nippon Kayaku Kabushiki Kaisha | Sealing Agent for Photoelectric Converter and Photoelectric Converter Using Same |
| JP2007234580A (en) * | 2006-02-02 | 2007-09-13 | Sony Corp | Dye sensitized photoelectric conversion device |
| CN101416345B (en) * | 2006-02-02 | 2012-09-26 | 索尼株式会社 | Dye sensitization photoelectric converter |
| US20090242027A1 (en) | 2006-07-05 | 2009-10-01 | Teruhisa Inoue | Dye-Sensitized Solar Cell |
| JP2008186752A (en) * | 2007-01-31 | 2008-08-14 | Konica Minolta Business Technologies Inc | Photoelectric conversion element and solar cell |
| TWI491677B (en) * | 2007-10-15 | 2015-07-11 | Dongjin Semichem Co Ltd | Novel thiophene-based dye and preparation thereof |
| TWI384034B (en) * | 2008-04-07 | 2013-02-01 | Everlight Chem Ind Corp | Dye compound for dye-sensitized solar cell |
| US20100132796A1 (en) * | 2008-11-28 | 2010-06-03 | Samsung Electro-Mechanics Co., Ltd. | Dye compound for dye-sensitized solar cells, dye-sensitized photoelectric converter and dye-sensitized solar cells |
| EP2573862A4 (en) | 2010-05-17 | 2015-03-04 | Nippon Kayaku Kk | Photoelectric conversion element using thermosetting sealing agent for photoelectric conversion element |
| JPWO2012081614A1 (en) * | 2010-12-14 | 2014-05-22 | 日本電気株式会社 | Pyrrole compounds and uses thereof |
| JPWO2013089217A1 (en) * | 2011-12-15 | 2015-04-27 | 国立大学法人九州大学 | Organic electroluminescence device |
| CN104823254A (en) | 2012-11-30 | 2015-08-05 | 日本化药株式会社 | Dye-sensitized solar cell |
| JP2016193954A (en) * | 2013-09-12 | 2016-11-17 | 日本化薬株式会社 | Methine dye and dye-sensitized photoelectric conversion element using the same |
-
2003
- 2003-01-15 JP JP2003007360A patent/JP4230228B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2003282165A (en) | 2003-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4841248B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4963343B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP5054269B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4986205B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP5138371B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP5145037B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4287655B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP5051810B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4230228B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP2004227825A (en) | Dye-sensitized photoelectric conversion element | |
| JP4274306B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4450573B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP2006294360A (en) | Dye-sensitized photoelectric transducer | |
| JP4822383B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP2002334729A (en) | Dye-sensitized photoelectric conversion element | |
| JP4266573B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP2006134649A (en) | Photoelectric conversion element | |
| JP4334185B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4230182B2 (en) | Dye-sensitized photoelectric conversion element and solar cell using the same | |
| JP4230185B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4005330B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP4278023B2 (en) | Dye-sensitized photoelectric conversion element | |
| JP2005019130A (en) | Dye-sensitized photoelectric conversion device | |
| JP2004022387A (en) | Dye-sensitizing photoelectric converter element |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050721 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080724 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080905 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20081126 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20081203 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20111212 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20141212 Year of fee payment: 6 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| LAPS | Cancellation because of no payment of annual fees |