JP4166846B2 - ヘパリン結合担体及びフルクトース−1,6−ビスリン酸アルドラーゼの分離方法 - Google Patents
ヘパリン結合担体及びフルクトース−1,6−ビスリン酸アルドラーゼの分離方法 Download PDFInfo
- Publication number
- JP4166846B2 JP4166846B2 JP21271497A JP21271497A JP4166846B2 JP 4166846 B2 JP4166846 B2 JP 4166846B2 JP 21271497 A JP21271497 A JP 21271497A JP 21271497 A JP21271497 A JP 21271497A JP 4166846 B2 JP4166846 B2 JP 4166846B2
- Authority
- JP
- Japan
- Prior art keywords
- heparin
- δdihs
- fructose
- carrier
- bisphosphate aldolase
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images





Landscapes
- Enzymes And Modification Thereof (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
Description
【発明の属する技術分野】
本発明は、ヘパリンが結合した担体及び該ヘパリン結合担体を使用したアフィニティークロマトグラフィーにより、フルクトース−1,6−ビスリン酸アルドラーゼの含有物から当該酵素を分離する方法に関するものであり、さらに、当該酵素に特異的な親和性を有するヘパリンに関するものである。
【0002】
【従来の技術】
ヘパリンは、ウロン酸残基とグルコサミン残基から成る二糖単位の連なったヘパリン骨格を有し、そのウロン酸残基の2位水酸基並びにグルコサミン残基の2位アミノ基及び6位水酸基が様々な程度に硫酸化されているグリコサミノグリカンの一種である。この物質は一般にアンチトロンビンIII結合部位を有し(FEBS Lett.(1980)117, 203-206)、ATIIIとの結合の結果としてトロンビン活性を阻害して抗凝固活性を発現するため、古くから透析治療薬等の成果を向上させるための医薬品として盛んに用いられてきた。また、ヘパリンがリポプロテインリパーゼと相互作用すること(J. Biol. Chem.(1981)256, 12893-12898)、塩基性繊維芽細胞増殖因子と親和性を有すること(J. Cell Biol.(1990)111, 1651-1659)など、様々な生理活性物質と相互作用することが明らかになってきた。
【0003】
上記のようにヘパリンは様々な物質と親和性を有することから、ヘパリンと生理活性物質の結合に関与する構造の解明が着目されつつあり、またATIII結合部位に由来する抗凝固作用を低減させた上で、生理活性物質との相互作用を増強させることを目的として、ヘパリンポリマーの脱硫酸化を中心とした化学修飾が盛んに行われている(J. Carbohydr.Chem.(1993)12, 507-521;Carbohydr. Res.,193, 165-172(1989);Carbohydr. Res.46,87-95(1976);Can. J. Chem.,67, 1449(1989);米国特許第5,296,471号)。また、ヘパリンを硫酸化及び脱硫酸化等の手法を用いて修飾することによりヘパリン誘導体が創作されているが、そのヘパリン誘導体の中には生理活性物質と親和性を有するものがあるので、生理活性物質と親和性を有したり、あるいは生理活性物質の活性を制御し得るヘパリン誘導体の探索が行われている。
【0004】
一方、進行性筋ジストロフィー、急性肝炎、心筋梗塞、悪性腫瘍の診断上有用な酵素であるフルクトース−1,6−ビスリン酸アルドラーゼ(EC 4.1.2.13)は、これまで筋肉、肝臓、心臓、腎臓、および脳などから得られた粗抽出物から、硫安沈殿、リン酸セルロースカラムクロマトグラフィー、DEAEセルロースカラムクロマトグラフィーさらには結晶化法等の手法を複雑に組み合わせることにより単離・精製されたもの(Methods Enzymol.(1975)42, 240-249)が供給されてきた。
【0005】
【発明が解決しようとする課題】
フルクトース−1,6−ビスリン酸アルドラーゼは上述の各種疾患の診断上有用であり、単離された状態で必要とされているにもかかわらず、当該酵素を得る手段は複雑なカラムワークが必須であった。しかし、前記カラムワークによって得られる酵素は非常に微量であり、大量の当該酵素を得るためには簡略化された効率のよい精製方法が必要とされていた。
本発明は、特定の担体を用いるアフィニティークロマトグラフィーによる極めて簡便な手法で当該酵素を分離・精製する方法を提供し、更にその為の該酵素にたいする親和性を有する特定構造のヘパリン及びヘパリン結合担体を提供するものである。
【0006】
【課題を解決するための手段】
本発明者らは、ヘパリン骨格(ウロン酸とグルコサミンの繰り返し構造)を有するグリコサミノグリカンが様々な生理活性物質と親和性を有することに注目し、フルクトース−1,6−ビスリン酸アルドラーゼが特定の構造を有するヘパリンに親和性が高く、当該ヘパリンが結合した担体を使用するアフィニティクロマトグラフィーにより、上記酵素の含有物から該酵素のみを高純度に分離・精製することができることを見出し本発明を完成した。すなわち、50%以上のグルコサミンの6位の水酸基が硫酸化されたヘパリンは上記酵素との高い親和性を有しているので、このようなヘパリンを結合したヘパリン結合担体を用いて、上記酵素の含有物から酵素を単離、精製する過程でアフィニティーカラムクロマトグラフィーを行えば前記酵素を効率的に、且つ高純度で分離・精製し得るのである。そして、上記アフィニティ担体に結合するヘパリンとしては、6位の水酸基が硫酸化されたグルコサミンが50%以上で、しかも2位のアミノ基に硫酸基が結合したグルコサミン残基が多くても10%であり、更にヘパリン中のウロン酸残基の50%以上が2−O−硫酸基を有しないヘパリンが最も効率よく前記酵素を分離・精製することを可能にするのである。
【0007】
本発明の第1の要旨は、グリコサミノグリカン分解酵素による分解と高速液体クロマトグラフィーによる分析を組み合わせた二糖分析により得られる下記構造式で表される二糖体組成において(a)の規定を充たし、且つ(b)に規定する物性を有するヘパリンがアフィニティークロマトグラフィー用担体に結合していることを特徴とするヘパリン結合担体であり、
(a)ΔDiHS−tri(U,6,N)S、ΔDiHS−di(6,N)S及びΔDiHS−di(U,N)Sのそれぞれのモル%が全て3%以下であり、ΔDiHS−6Sのモル%が50%以上である。(但し、ΔDiHS−tri(U,6,N)Sは、式中においてR 1 、R 2 、R 3 がSO 3 - であることを意味し、ΔDiHS−di(6,N)Sは、式中においてR 1 及びR 2 がSO 3 - であり、R 3 がHであることを意味し、ΔDiHS−di(U,N)Sは、式中においてR 1 がHであり、R 2 及びR 3 がSO 3 - であることを意味し、更にΔDiHS−6Sは、式中においてR 1 がSO 3 - であり、R 2 及びR 3 がHであることを意味する。)
(b)平均分子量が9,000〜11,000Daである。
【化1】

【0008】
【発明の実施の形態】
以下に本発明の実施の形態を詳説する。
本発明のヘパリン結合担体は50%以上のグルコサミン残基の6位が硫酸化しているヘパリン、特に、50%以上のグルコサミン残基の6位が硫酸化し、さらに2位にN−硫酸基を有するグルコサミン残基は多くても10%であるヘパリンが担体に結合しているものである。
本発明の結合担体における担体としては、特に限定はされず、不溶性担体、水溶性高分子担体が使用される。不溶性担体としては、クロマトグラフィー用担体として使用されている不溶性天然高分子及びその活性化物のような担体が好ましい。例えばアミノアガロース、アミノヘキシルアガロース及び臭化シアン活性化アガロース等が挙げられ、アミノアガロースが最も好ましいが特に限定はされない。これらの担体としては、セファロース、バイオゲル等の商品名で市販されているものから適宜選ぶこともできる。水溶性高分子担体としては、ポリエチレングリコール等が用いられる。
【0009】
本発明において上記担体に結合するヘパリンとしては、その平均分子量は特に限定されないが、通常8,000〜13,000Da、好ましくは9,000〜11,000Daである。又、その好ましい構造としては、ヘパリン骨格中50%以上のグルコサミン残基の6位の水酸基が硫酸化されており、しかも2位にN−硫酸基を有するグルコサミン残基は多くても10%であることを特徴とする。このようなヘパリンとしては、グリコサミノグリカン分解酵素による分解と高速液体クロマトグラフィーによる分析を組み合わせた二糖分析により得られる下記構造式で表される二糖体組成において(a)の規定を充たし、且つ(b)に規定する物性を有することを特徴とするヘパリンが最も好ましい。
(a)ΔDiHS−tri(U,6,N)S、ΔDiHS−di(6,N)S及びΔDiHS−di(U,N)Sのそれぞれのモル%が全て3%以下であり、ΔDiHS−6Sのモル%が50%以上である。
(但し、ΔDiHS−tri(U,6,N)Sは、式中においてR1、R2、R3がSO3 -であることを意味し、ΔDiHS−di(6,N)Sは、式中においてR1及びR2がSO3 -であり、R3がHであることを意味し、ΔDiHS−di(U,N)Sは、式中においてR1がHであり、R2及びR3がSO3 -であることを意味し、更にΔDiHS−6Sは、式中においてR1がSO3 -であり、R2及びR3がHであることを意味する。)
(b)平均分子量が9,000〜11,000Daである。
【0010】
【化1】
更に好ましいヘパリンは、上記(a)及び(b)の規定に加えて、二糖体組成においてΔDiHS−NSのモル%が3%以下であることを満たすものである(但し、ΔDiHS−NSは、上記式中においてR1及びR3がHでありR2がSO3 -であることを意味する。)。
本発明のヘパリン結合担体としては、これらの最適ヘパリンをアミノアガロースに結合させたものが最も好ましい。
本発明の上記特定の構造を有するヘパリンは、フルクトース−1,6−ビスリン酸アルドラーゼに対し親和性を有するものである。ここで、親和性とは、後述の試験例4に示すフルクトース−1,6−ビスリン酸アルドラーゼの活性測定法により測定し、その比活性が少なくとも1.0であることを意味する。
【0012】
本発明における上記ヘパリンの二糖体組成は、後述する実施例に記載の二糖分析法による測定値から算出したものであり、また分子量は実施例に記載の分子量測定法による測定値である。二糖体組成は試験法1に記載の酵素消化による二糖分析により得られる特定が可能な構造をもつ不飽和二糖の量[ΔDiHS−0S、ΔDiHS−6S、ΔDiHS−NS、ΔDiHS−US、ΔDiHS−di(6,N)S、ΔDiHS−di(U,N)S、ΔDiHS−di(U,6)S及びΔDiHS−tri(U,6,N)Sのモル%の合計]を100%として、上記特定の構造をもつ二糖の割合を示したものであり、当該数値は酵素消化前のヘパリンの構成糖における硫酸化の位置を反映するものである。
なお、本願明細書において、二糖体組成の表記は、下記を意味する。
【0013】
【表1】
本発明ヘパリン結合担体に結合するヘパリンの製造方法としては、市販のヘパリン中のグルコサミンの6位の水酸基を硫酸化することによって得ることが可能であるが、上述の最も好ましいヘパリンは、市販のヘパリンを公知の完全脱硫酸化法により脱硫酸化した後にグルコサミンのアミノ基をアセチル化した後、特公平6−99485に記載の方法等によりグルコサミンの6位の水酸基を選択的に硫酸化することによって得ることが可能である。グルコサミンの6位の水酸基が選択的に硫酸化されたヘパリンを、以下選択的6−O−硫酸化ヘパリンと記載する。
【0016】
最適ヘパリンの具体的製造法としては、例えば以下の方法が挙げられる。500mgのヘパリン(ブタ小腸由来:SPL社製)を10mlの蒸留水に溶解後、蒸留水で平衡化したアンバーライトIR−120Bカラム(φ2 x 15cm)に吸着させ、溶出液の酸性画分を回収し、ピリジンを添加してpHを5.0〜7.5、好ましくは6.0〜7.0に調整する。この溶液を凍結乾燥することにより、ヘパリンピリジニウム塩を得る。得られたヘパリンピリジニウム塩500mgに対し、例えば5mlのメタノール、30mlのジメチルスルホキシド(DMSO)及び15mlの1,4−ジオキサンを添加してヘパリンピリジニウム塩を完全に溶解し、この溶液を、Ogamoらの方法(Carbohydr. Res.,193, 165-172(1989))に従って、80〜95℃にて20時間以上、好ましくは40時間以上、より好ましくは60〜75時間加熱することによりヘパリンの完全脱硫酸化を行う。脱硫酸反応終了後、100〜200mlの氷冷蒸留水を添加し、反応混液のpHをNaOHを添加することにより8〜11、好ましくは9〜10に調整する。次いで、反応混液を蒸留水に対して20時間以上透析した後、透析内液を凍結乾燥処理し、完全脱硫酸化ヘパリンを黄色パウダーとして回収できる。
【0017】
次いで、Danishefskyらの方法(Arch. Biochem. Biophys.,90, 114-121(1960))に従い、完全脱硫酸化ヘパリンのグルコサミンのN−アセチル化反応を行う。例えば、400mgの完全脱硫酸化ヘパリンを60mlの50mMの炭酸ナトリウムを含む10%メタノールに溶解した後、酢酸を適量添加して溶液のpHを6以上、好ましくは7〜8の間に調整する。次いで、反応混液を氷冷しながら、よく撹拌する。次の1時間の間に300〜500μlの無水酢酸を5〜8回添加し、そのたびごとに低下した反応混液のpHを、飽和炭酸ナトリウムを含む10%メタノールを適量添加することにより、7〜8の間に調整する。反応終了後、100〜150mlの蒸留水を添加し、蒸留水に対して48時間以上、好ましくは72時間程度透析する。透析内液を凍結乾燥することにより 完全脱硫酸化/N−アセチル化ヘパリンを得ることができる。
【0018】
次いで、400mgの完全脱硫酸化/N−アセチル化ヘパリンを10ml程度の蒸留水に溶解した後、蒸留水で平衡化したアンバーライトIR−120Bカラム(φ2×15cm)に通塔し、溶出液の酸性画分を回収し、10%トリブチルアミンエタノール溶液を添加してpHを4.5〜7.0、好ましくは5.5〜6.5に調整する。この溶液を凍結乾燥することにより得られる完全脱硫酸化/N−アセチル化ヘパリントリブチルアンモニウム塩を、Takanoらの方法(Biosci. Biotech. Biochem.,56, 1413-1416(1992))に従い、5〜10mlのジメチルホルムアミド(DMF)に溶解して氷冷した後、10〜15mlの4.6mlジクロロヘキシルカルボジイミド(DCC)を含むDMFを添加する。さらに撹拌した後、4〜5mlの1.2mmol硫酸を含むDMFを添加し、氷冷条件下にて10〜20分間撹拌を続けて反応を進行させる。反応終了後、反応混液全量を50〜100gの氷中に投入し、直ちに炭酸水素ナトリウムで中和してpHを6.5〜7.5に調整する。次いで、この反応混液を蒸留水に対して透析した後、ガラス繊維濾紙、メンブレンフィルター(4.5μm)の順に通して、ジシクロヘキシル尿素の塩を除去した後、凍結乾燥処理を施すことにより選択的6−O−硫酸化ヘパリンを合成することができる。
【0019】
本発明においてヘパリンの担体への結合方法は特に限定はされないが、共有結合が好ましく、ヘパリンの還元末端部に存在しうるアルデヒド基と担体が共有結合していることが結合力が強く、アフィニティー担体として最も好ましい。
上記のようにして得た選択的6−O−硫酸化ヘパリンを担体に結合することにより本発明ヘパリン結合担体とすることができる。選択的6−O−硫酸化ヘパリンの種々の官能基と活性化クロマトグラフィー用坦体(アミノ基等の官能基を導入した誘導体)の官能基とのカップリング反応を利用して選択的6−O−硫酸化ヘパリンの坦体への固定化を行うことができる。一般には、選択的6−O−硫酸化ヘパリン分子中の還元末端部に存在しうるアルデヒド基を担体とのカップリング反応に使用することが好ましく、この場合は、担体としてアミノ基を有する不溶性担体(例えば、アミノヘキシルアガロース又はアミノアガロース)を使用する。又、選択的6−O−硫酸化ヘパリン分子中のカルボキシル基を担体とのカップリング反応に使用する場合は、担体として上記と同様のアミノ基を有する担体を使用し、選択的6−O−硫酸化ヘパリン分子中の遊離のアミノ基を担体とのカップリング反応に使用する場合は臭化シアン活性化アガロースの担体を使用する。アミノアガロースの担体がヘパリンと最も結合し易いため好ましい。
【0020】
選択的6−O−硫酸化ヘパリンと担体とを結合させる方法としては以下の方法が挙げられる。選択的6−O−硫酸化ヘパリン中の還元末端部に存在しうるアルデヒド基とアミノアガロースゲルのアミノ基を結合する方法としては、シッフ塩基を形成した後、NaB(CN)H 3 等の還元剤を作用させ、アルキルアミノ架橋を生成させる。この方法としては、例えば公知の一般的な方法で行うことが可能であり、Sakaiらの方法(J. Chromatogr.,400, 123(1987))等が挙げられる。
また、選択的6−O−硫酸化ヘパリン中ウロン酸残基中のカルボキシル基とアミノアガロースゲルのアミノ基を結合する方法としては、該カルボキシル基を1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(EDC)等のカルボジイミド試薬により活性化して酸アミド結合(-CONH-)の架橋を生成させる方法である。しかし、選択的6−O−硫酸化ヘパリンの立体構造上、担体としてより結合力が強いと考えられる前者の結合方法が好ましい。
【0021】
選択的6−O−硫酸化ヘパリンが結合したヘパリン結合担体中に未反応(遊離)のアミノ基が残存すると、非特異的な相互作用を引き起こし、特にアフィニティークロマトグラフィーの担体として使用した際に特異的な結合が起こらず、不特定物質を結合するため好ましくない。従って、ヘパリン結合担体中の未反応の残存アミノ基は以下の方法によりアセチル化して不活化することが好ましい。すなわち例えば、上記で調製したヘパリン結合担体を5mlの0.2M 酢酸ナトリウム溶液に懸濁させ、2.5mlの無水酢酸を添加して氷冷条件下にて20〜40分間、次いで室温にて20〜40分間反応させることによってアミノ基をアセチル化し不活化することが可能である。
ヘパリンの固定化(場合により、更にアセチル化反応後)したヘパリン結合担体は、水洗して完全に脱塩することが好ましい。
また、Bitter&Muirの方法(Anal. Biochem.,4, 330-334(1962))に従ってカルボキシル基の定量を行い、単位重量あたりのアガロースゲルに固定化された選択的6−O−硫酸化ヘパリンの重量を見積ることにより、ヘパリンの固定化量を確認することが可能である。
【0022】
本発明の不溶性担体にヘパリンが結合した結合担体は、カラムに充填して酵素含有物と接触させ、非吸着画分と分離するのに使用出来、またバッチ法で酵素含有物と接触後、濾過、遠心分離等により分離することもできる。また、水溶性高分子担体とヘパリンとの結合担体は、酵素含有物と接触させた後、分子ふるい等の方法で分離することができる。
【0023】
本発明のフルクトース−1,6−ビスリン酸アルドラーゼを分離・精製する方法は、上記本発明ヘパリン結合担体を用いたアフィニティークロマトグラフィーにより、フルクトース−1,6−ビスリン酸アルドラーゼの含有物から当該酵素を分離・精製する方法である。
本発明方法においては、本発明ヘパリン結合担体を使用したアフィニティークロマトグラフィーを用いる方法であれば、フルクトース−1,6−ビスリン酸アルドラーゼを分離・精製する為に、常法に従い付随的に行われる予備処理等の他の処理操作は特に制限されない。本発明の好ましい態様として、例えば、当該酵素の素材原料をホモジナイズして得られた液状物をコンドロイチンポリ硫酸固定化アフィニティークロマトグラフィーを行って分画し、その吸着画分の流出物を本発明ヘパリン結合担体を用いるアフィニティークロマトグラフィーにより分画し、その吸着画分から上記酵素を分離・取得する方法が挙げられる。
【0024】
本発明方法におけるアフィニティークロマトグラフィーは、本発明のヘパリン結合担体を充填したカラムを使用し、それ以外の操作は常法に従っておこなわれ、その特異的吸着画分を溶離剤(例えば、NaCl、KClなどのアルカリ金属塩水溶液)で処理することにより、酵素を含有した溶出物を得ることが出来る。更に、この溶出物をゲル濾過クロマトグラフィー、限外濾過等の分子ふるいを行って、より高純度の酵素を得ることができる。
【0025】
本発明方法においてフルクトース−1,6−ビスリン酸アルドラーゼを分離・精製するための素材原料は、上記酵素が発現している組織であれば限定はされないが、哺乳動物類由来の組織が好ましい。組織としては、好ましくは筋肉、肝臓、心臓、腎臓及び脳などが挙げられ、その中でも脳が好ましく、特に大脳が好ましい。これらの組織を、通常、緩衝液中でホモジナイズし、遠心分離したものを酵素源として用いる。
【0026】
【実施例】
以下、本発明を実施例により更に詳細に説明するが、本発明はその要旨を超えない限り本実施例により限定されることはない。
なお、実施例における各試験法は、以下の通りである。
【0027】
試験法1
[酵素消化による二糖分析]
ヘパリンの硫酸基の位置の分析は、次のようにして行った。すなわち、それぞれの硫酸化多糖を酵素消化し、生成した不飽和二糖(前記構造式:化1)を高速液体クロマトグラフィー(HPLC)で分析した(新生化学実験講座3、糖質II(東京化学同人刊、1991)、49−62頁に記載の「2・8グリコサミノグリカン分解酵素とHPLCを組み合わせた構造解析」参照)。各不飽和二糖のピーク面積を計算して、全面積に対するピーク面積をパーセントとして表した。
【0028】
(1)ヘパリンの完全脱硫酸化6−O−硫酸化処理によって生成したヘパリンの分解酵素による消化
新生化学実験講座3、糖質II 54−59頁に記載の方法により、ヘパリン1.0mgを2mM酢酸カルシウムを含む20mM酢酸ナトリウム(pH7.0)溶液220μlに溶解して、20mUのヘパリチナーゼ、20mUのヘパリチナーゼI及びIIを加えて、37℃、2時間反応させた。
【0029】
(2)HPLCによる分析
ヘパリンの分解酵素による消化を行った後の溶液20μlを、HPLC(医理化、モデル852型)を用いて分析した。イオン交換カラム(Dionex社、CarboPac PA−1カラム φ4.0mm×250mm)を使用し、232nmでの吸光度を測定した。流速1ml/分で、塩化リチウムを用いたグラジエント系(50mM→2.5M)を用いる方法に準拠した(Kariya, et al.,Comp.Biochem.Physiol.,103B,473,(1992))。
【0030】
試験法2
[分子量測定]
ヘパリンの3%溶液10μlをHPLCによるゲルろ過で分析した。カラムはTSKgel−(G4000+G3000+G2500)PW XL (東ソー、7.8mm×30cm)を用い、溶離液に0.2M塩化ナトリウムを使用して、1.0ml/分の流速で展開した。ヘパリンの検出には、示差屈折計(島津製作所、AID−2A)を用いた。平均分子量はヘパリンの分子量標準品を対照にして求めた(Kaneda et al.,Biochem. Biophys. Res. Comm.,220 ,108-112(1996))。標準ヘパリンの分子量の測定は光散乱法を用いて行った(Nagasawa et al., J. Biochem.,81 ,989-993(1977))。
【0031】
試験例3
[NMR分析]
13C−NMRは、GE社製QE300型NMRスペクトロメーターを用い、100%D2O置換を行った試料につき10%濃度のD2O溶液を調製したうえ、TSPを0ppmとした時のケミカルシフトが51.66ppmのメタノールを内部標準とし、測定温度80℃、パルス幅60°、積算回数90,000回の条件にて実施した。
【0032】
試験例4
[フルクトース−1,6−ビスリン酸アルドラーゼ活性測定]
Herbert G. Lebherz and William J. Rutterらの方法( Methods Enzymol.,42, 249 - 258(1975))に準じ、下記の如く活性測定を行った。すなわち、試薬として次の5種類のものを調製した。
▲1▼0.1M グリシルグリシン緩衝液(pH 7.5)、
▲2▼50mM フルクトース−1,6−ビスリン酸ナトリウム溶液、
▲3▼0.1M フルクトース−1−リン酸ジモノシクロヘキシルアンモニウム溶液、 ▲4▼10mg/ml濃度のグリセロール−3−リン酸デヒドロゲナーゼ/トリオースリン酸イソメラーゼ複合体溶液、
▲5▼NADH。
【0033】
4mgのNADH、50μlのグリセロール−3−リン酸デヒドロゲナーゼ/トリオースリン酸イソメラーゼ複合体溶液、1mlのフルクトース1,6−ビスリン酸ナトリウム溶液および1mlのフルクトース−1−リン酸ジモノシクロヘキシルアンモニウム溶液を含む0.1Mグリシルグリシン緩衝液(pH 7.5)の20mlに対し、5〜50μlのフルクトース−1、6−ビスリン酸アルドラーゼを含む測定検体を添加することにより反応を開始させた。単位時間あたりの340nmの吸光度の減少量につき、25℃の温度条件下で測定した。340nmにおけるNADHの分子吸光係数が6.22×106cm2・mol なので、吸光度の変化が12.44から6.22への減少であった場合、それぞれ1μmolのフルクトース−1、6−ビスリン酸あるいは1μmolのフルクトース−1−リン酸が開裂したとし、これに基づき、様々な試料における開裂量を算出した。フルクトース−1、6−ビスリン酸アルドラーゼの単位(活性;unit)は、上記の反応系において25℃にて1μmolの基質を1分間で開裂する酵素量を1unitと定義した。比活性は、1mgのタンパク質あたりのunit数として定義した。
【0034】
実施例1 選択的6−O−硫酸化ヘパリンの合成
500mgのヘパリン(ブタ小腸由来;SPL社製)を10mlの蒸留水に溶解した後、蒸留水で平衡化したアンバーライトIR−120Bカラム(φ2×15cm)に通塔し、溶出液を3mlごとに分画した。最終的に得られた50本の各画分のpHを測定した後、酸性画分を集め ピリジンを添加してpHを6.5に調整した。この溶液を凍結乾燥することによりヘパリンピリジニウム塩を約510mg得た。完全脱硫酸化反応を行うために、このうちの500mgに対し、5mlのメタノール、30mlのジメチルスルホキシド(DMSO)、および15mlの1,4-ジオキサンを添加してヘパリンピリジニウム塩を完全に溶解した後、Ogamoらの方法(Carbohydr. Res.,193, 165-172(1989))に従って、この反応混液を90℃にて72時間加熱した。脱硫酸化反応終了後、150mlの氷冷蒸留水を添加し、反応混液のpHを、1Nの水酸化ナトリウムを添加することにより9.5に調整した。次いで、反応混液を蒸留水に対して24時間透析した後、透析内液を凍結乾燥処理に付した。その結果、430mgの完全脱硫酸化ヘパリンを黄色パウダーとして回収した。
【0035】
次いで、Danishefskyらの方法(Arch. Biochem. Biophys.,90, 114-121(1960))に従い、N−アセチル化反応を行った。すなわち、400mgの完全脱硫酸化ヘパリンを、60mlの50mM 炭酸ナトリウムを含む10% メタノールに溶解した後、酢酸を適量添加して溶液のpHを7〜8の間に調整した。次いで、反応混液を氷冷水槽中にて冷却しながら、よく撹拌を続けた。次の1時間の間に400μlの無水酢酸を6〜7回添加し、そのたびごとに低下した反応混液のpHを、飽和炭酸ナトリウムを含む10% メタノールを適量添加することにより、7〜8の間に調整した。反応終了後、120mlの蒸留水を添加し、蒸留水に対して72時間透析した。透析内液を凍結乾燥することにより410mgの完全脱硫酸化/N−アセチル化ヘパリンを得た。
【0036】
次いで、400mgの完全脱硫酸化/N−アセチル化ヘパリンを10mlの蒸留水に溶解した後、蒸留水で平衡化したアンバーライトIR−120Bカラム(φ2×15cm)に通塔し、溶出液を3mlごとに分画した。最終的に得られた50本の各画分のpHを測定した後、酸性画分を集め10%トリブチルアミンエタノール溶液を添加してpHを5.5に調整した。この溶液を凍結乾燥することにより完全脱硫酸化/N−アセチル化ヘパリントリブチルアンモニウム塩を430mg得た。このものを Takanoらの方法(Biosci. Biotech. Biochem.,56, 1413-1416(1992))に従い、8mlのジメチルホルムアミド(DMF)に溶解して0℃に冷却した後、4.6mlのジクロロヘキシルカルボジイミド(DCC)を含む12mlのDMFを添加した。さらによく撹拌した後、4.4mlの1.2mmol硫酸を含むDMFを添加し、0℃にて15分間撹拌を続けて反応を進行させた。
【0037】
反応終了後、反応混液全体を80gの氷中に投入し、直ちに炭酸水素ナトリウムで中和してpHを7.0に調整した。次いで、この反応混液を蒸留水に対して透析した後、ガラス繊維濾紙、メンブレンフィルター(4.5μm)の順に通して、ジシクロヘキシル尿素の塩を除去した。最終的に、凍結乾燥処理を施し、選択的6−O−硫酸化ヘパリンを450mg合成した。この選択的6−O−硫酸化ヘパリンを光散乱法により分子量を測定すると、未処理のヘパリンの1.37×104Daに対し、選択的6−O−硫酸化ヘパリンは0.97×104Daであった。これらのゲルろ過HPLCによる溶出パターンは(図1)の通りであり、未処理ヘパリンの溶出時間は33.5分(a)、選択的6−O−硫酸化ヘパリンの溶出時間は35.1分(b)であった。また、これらの物質の二糖分析を行った(図2)。この溶出パターンから二糖体組成を算出した(表−2)。
【0038】
【表2】
また、選択的6−O−硫酸化ヘパリンを13C−NMRにより分析した(図3)。その結果、二糖体組成同様、選択的6−O−硫酸化ヘパリンは従来のヘパリンでは得られなかった、グルコサミン残基にN−硫酸基が存在せず、6−O−硫酸を多く有するヘパリンであることが明らかになった。また、当該ヘパリンをアフィニティークロマトグラフィーの担体に結合する場合、特異的な結合を有するヘパリンはΔDiHS−tri(U,6,N)S、ΔDiHS−di(U,N)S及びΔDiHS−di(6,N)Sのモル%はそれぞれ全て3%以下であることが好ましい。
【0040】
実施例2 選択的6-O-硫酸化ヘパリンをリガンドとするヘパリン結合坦体の調製 Sakaiらの方法(J. Chromatogr.,400, 123(1987))に従い、選択的6−O−硫酸化ヘパリン200mgを5mlの2Mリン酸緩衝液(pH 7.2)に溶解した。次いで、5gのアミノアガロースゲル粉末を懸濁し、15mgのNaB(CN)H3を添加して十分撹拌しながら室温にて24時間反応を進行させた。反応終了後、カップリング産物を含有したゲルを濾過した。さらに、水洗処理を2〜3回施すことにより、完全に脱塩した。
【0041】
未反応(遊離)のアミノ基が残存すると、非特異的な相互作用を引き起こす可能性があるので、残存アミノ基はアセチル化して不活化することにした。すなわち、調製した選択的6−O−硫酸化ヘパリン固定化アガロースゲルを 5mlの0.2M 酢酸ナトリウム溶液に懸濁させ、2.5mlの無水酢酸を添加して0℃にて30分、次いで室温にて30分間反応させた。反応終了後、生成したゲルを水洗して完全に脱塩した。Bitter & Muirの方法(Anal. Biochem.,4, 330-334(1962))に従い、カルボキシル基の定量を行うことにより、単位重量あたりのアガロースゲルに固定化された選択的6−O−硫酸化ヘパリンの重量を見積もった。その結果、従来報告されている最大結合量に匹敵する、ゲル湿重量1gあたり、約10mgの選択的6−O−硫酸化ヘパリンの固定化が確認されたため、遊離のアミノ基は存在しないことが確認された。
【0042】
実施例3 選択的6−O−硫酸化ヘパリン結合坦体を用いたフルクトース−1,6ービスリン酸アルドラーゼの精製
9gのウシ大脳に対し、0℃に冷却した80mlの1mM EDTAを含む10mM Tris−HCl(pH 7.4緩衝液)を添加し、大脳組織を冷却した。次いで、大脳組織を取り出し、剃刀を用いて細片化した後、再度0℃に冷却した上記緩衝液を2倍量(V/W)添加し、グラスホモジナイザー中でホモジナイズした。このように調製したホモジネイトを適量の遠沈管に入れ、0℃にて100,000×gの加速度の下、60分間の遠心分離を行った。遠心終了後、上澄液(粗抽出液)を用いさらに精製を進めた。
【0043】
上記粗抽出液をコンドロイチンポリ硫酸固定化アガロースアフィニティーカラム(φ2×20cm)を1mM EDTAを含む10mM Tris−HCl(pH 7.4)緩衝液で平衡化した。このアフィニティーカラムに粗抽出液を全量をアプライした後、60mlの 1mM EDTAを含む10mM Tris−HCl(pH 7.4)緩衝液で洗浄し、1mM EDTA、0.5mM メルカプトエタノール及び10%グリセロールを含む10mM Tris−HCl(pH 7.4)緩衝液100mlにより吸着画分を溶出させた。この吸着画分はSDS−PAGE(Laemmli, U. K., J. Biol. Chem., 252, 1102-1106(1977))により多数のバンドを示した。
【0044】
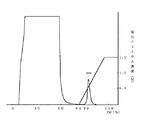
次いで、上記実施例2で調製したヘパリン結合担体を充填したカラム(φ1×10cm)を1mM EDTA、0.5mM メルカプトエタノール、10%グリセロールを含む10mM Tris−HCl(pH 7.4)緩衝液で平衡化した。このアフィニティーカラムに、コンドロイチンポリ硫酸固定化アガロースアフィニティーカラムクロマトグラフィー吸着画分のうち40mlをアプライした後、1mM EDTA、0.5mM メルカプトエタノール及び10%グリセロールを含む10mM Tris−HCl(pH 7.4)緩衝液80mlで洗浄した。特異的吸着画分につき、1mM EDTA、0.5mMメルカプトエタノール及び10%グリセロールを含む10mM Tris−HCl(pH 7.4)緩衝液15mlおよび1.5M 塩化ナトリウム、1mM EDTA、0.5mM メルカプトエタノール及び10%グリセロールを含む10mM Tris−HCl(pH 7.4)緩衝液15mlの2種溶出液による塩化ナトリウムの直線的濃度勾配(0〜1.5M)を用いて溶出した。なお、検出は220nmの吸光度により行い、その溶出パターンを図4に示した。塩化ナトリウムの直線的濃度勾配においてシングルピークが出現したので、この画分を分取してSDS−PAGEに付したところ、分子量約40kDaのシングルバンドが観察された。また、この画分はフルクトース−1、6−ビスリン酸アルドラーゼ活性を有していた。
【0045】
実施例4 フルクトース−1、6−ビスリン酸アルドラーゼのアイソフォーム
次いで、1mM EDTAを含む10mM Tris−HCl(pH 7.4)緩衝液で十分平衡化したMonoQカラム(商品名:ファルマシア社製)を装着したHPLCに、選択的6−O−硫酸化ヘパリンアフィニティークロマトグラフィーの特異的吸着画分をアプライし、15mlの1mM EDTAを含む10mM Tris−HCl(pH 7.4)緩衝液で洗浄した。吸着画分につき、7.5mlの1mM EDTAを含む10mM Tris−HCl(pH 7.4)緩衝液並びに7.5mlの0.5M 塩化ナトリウム及び1mM EDTAを含む10mM Tris−HCl(pH 7.4)緩衝液の2種溶出液による塩化ナトリウムの直線的濃度勾配(0〜0.5M)を用いて溶出を試みた。なお、検出は220nmの吸光度により行い、その溶出パターンを図5に示す。
【0046】
塩化ナトリウムの直線的濃度勾配の開始直前にブロードなピークが1本(Fr.1)、塩化ナトリウムの直線的濃度勾配においてシャープなピークが4本(Fr.2〜Fr.5)出現した。得られた5種画分につき、Superdex G-200を装着したHPLCによりゲル濾過分析と分取を行い純度の検定ならびに最終精製を行ったところ、図6に示すようにいずれの画分も約13分の保持時間にシャープなピークが検出され(220nmの吸光度による検出)、すべての画分ともきわめて高純度に精製されていることが判明した。Fr.1〜Fr.5の収量は、それぞれ 2.7mg、16.5mg、7.1mg、5.4mg、7.7mgであった。なお、実施例5に示すように、これら5種画分のフルクトース−1、6−ビスリン酸アルドラーゼ活性を測定したところ、全ての画分が高い活性を有した。
【0047】
これら5種画分につき、さらに Davidの方法(Ann. N. Y. Acad. Sci.,121, 404(1964))に従ってNative−PAGEを行った。その結果、Fr.1、Fr.2、Fr.3、Fr.4 および Fr.5 の順に移動度が大きくなることから、マイナスの帯電はFr.1<Fr.2<Fr.3<Fr.4<Fr.5 の順に大きいことが判明した。したがって、Fr.1〜Fr.5 はフルクトース−1、6−ビスリン酸アルドラーゼのアイソフォームであると考えられる。脳に存在するフルクトース−1、6−ビスリン酸アルドラーゼは等電点の高いAおよび等電点の低いCの2種サブユニットの4量体であり、A4、A3C、A2C2、AC3、C4 の5種アイソフォームが存在することが報告されている(Methods Enzymol.,42, 240-249(1975))。AとCは分子量はほとんど変わらない。Fr.1、2、3、4、および 5 は、それぞれA4、A3C、A2C2、AC3、C4に相当すると考えられる。
【0048】
実施例5 フルクトース−1、6−ビスリン酸アルドラーゼの活性測定
実施例3で得た選択的6−O−硫酸化ヘパリンを結合したヘパリン結合坦体への特異的吸着画分、実施例4においてMonoQカラムによるイオン交換で分画されたFr.1〜5の各画分及びSuperdex G-200 により精製後の分画Fr.1〜5のフルクトース−1、6−ビスリン酸アルドラーゼの活性測定を行った。その結果、選択的6−O−硫酸化ヘパリン結合アフィニティーカラムの特異的吸着画分の全活性および比活性は 142.65および1.48であった。又、MonoQカラムからの画分Fr.1の活性および比活性は2.92および2.17、Fr.2の活性および比活性は32.42および5.0、Fr.3の活性および比活性は40.98および13.5、Fr.4の活性および比活性は25.19および8.8、Fr.5の活性および比活性は16.11および4.6であった(表−3)。尚、表−3中のMonoQカラム及びSuperdex columnの全活性及び比活性はそれぞれFr.1〜5の活性の合計及び各Fr.の活性の平均を表す。
【0049】
【表3】
【発明の効果】
本発明により、進行性筋ジストロフィー等の各種疾患の診断に有用なフルクトース−1,6−ビスリン酸アルドラーゼに対して親和性を有する特異的構造を有するヘパリン及びこのヘパリンが結合した新規ヘパリン結合担体が提供される。また、このヘパリン結合担体を用いたアフィニティーカラムクロマトグラフィーを、フルクトース−1,6−ビスリン酸アルドラーゼ含有物から該酵素の分離・精製に使用することにより、単純な操作で、高度な精製を可能とする方法が提供される。
【図面の簡単な説明】
【図1】 (a)未処理ヘパリン及び(b)選択的6−O−硫酸化ヘパリンのゲルろ過HPLC溶出パターンを示す図。
【図2】 (a)不飽和二糖標品、(b)未処理ヘパリン、(c)選択的6−O−硫酸化ヘパリンの酵素消化物のイオン交換HPLC溶出パターンを示す図。
【図3】 選択的6−O−硫酸化ヘパリンの13C−NMRスペクトルを示す図。
【図4】 選択的6−O−硫酸化ヘパリン結合アフィニティー担体を用いたアフィニティークロマトグラフィーの溶出パターンを示す図。
【図5】 MonoQカラムを装着したHPLCにおけるイオン交換クロマトグラフィーの溶出パターンを示す図。
【図6】 Superdex カラムを装着したHPLCによるゲル濾過クロマトグラフィーの溶出パターンを示す図。
Claims (8)
- グリコサミノグリカン分解酵素による分解と高速液体クロマトグラフィーによる分析を組み合わせた二糖分析により得られる下記構造式で表される二糖体組成において(a)の規定を充たし、且つ(b)に規定する物性を有するヘパリンがアフィニティークロマトグラフィー用担体に結合していることを特徴とするヘパリン結合担体。
(a)ΔDiHS−tri(U,6,N)S、ΔDiHS−di(6,N)S及びΔDiHS−di(U,N)Sのそれぞれのモル%が全て3%以下であり、ΔDiHS−6Sのモル%が50%以上である。(但し、ΔDiHS−tri(U,6,N)Sは、式中においてR1、R2、R3がSO3 -であることを意味し、ΔDiHS−di(6,N)Sは、式中においてR1及びR2がSO3 -であり、R3がHであることを意味し、ΔDiHS−di(U,N)Sは、式中においてR1がHであり、R2及びR3がSO3 -であることを意味し、更にΔDiHS−6Sは、式中においてR1がSO3 -であり、R2及びR3がHであることを意味する。)
(b)平均分子量が9,000〜11,000Daである。
- ヘパリンがその還元末端において共有結合を介して該担体に結合していることを特徴とする請求項1記載のヘパリン結合担体。
- 該担体が、アミノアガロースであることを特徴とする請求項1又は2に記載のヘパリン結合担体。
- フルクトース−1,6−ビスリン酸アルドラーゼの含有物から、請求項1〜3のいずれか一項記載のヘパリン結合担体を用いたアフィニティークロマトグラフィーにより当該酵素を分離することを特徴とするフルクトース−1,6−ビスリン酸アルドラーゼの分離方法。
- 下記の工程(a)及び(b)を包含することを特徴とする請求項4記載のフルクトース−1,6−ビスリン酸アルドラーゼの分離方法。
(a) フルクトース−1,6−ビスリン酸アルドラーゼの含有物を、コンドロイチンポリ硫酸固定化アフィニティークロマトグラフィーを行い分画すること及び(b) 工程(a)で得られる吸着画分を、請求項1〜3のいずれか一項に記載のヘパリン結合担体を用いるアフィニティークロマトグラフィーにより、該酵素を選択的に吸着させ、溶出すること。 - 請求項5記載の分離方法において、更に工程(b)で得られる溶出物を分子ふるいにより精製することを特徴とするフルクトース−1,6−ビスリン酸アルドラーゼの分離方法。
- フルクトース−1,6−ビスリン酸アルドラーゼの含有物は哺乳動物の組織から得られることを特徴とする請求項4〜6のいずれか一項記載のフルクトース−1,6−ビスリン酸アルドラーゼの分離方法。
- 請求項7記載の分離方法において、哺乳動物の組織が筋肉、肝臓、心臓、腎臓及び脳からなる群から選択される1つ以上の組織であることを特徴とするフルクトース−1,6−ビスリン酸アルドラーゼの分離方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP21271497A JP4166846B2 (ja) | 1997-07-24 | 1997-07-24 | ヘパリン結合担体及びフルクトース−1,6−ビスリン酸アルドラーゼの分離方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP21271497A JP4166846B2 (ja) | 1997-07-24 | 1997-07-24 | ヘパリン結合担体及びフルクトース−1,6−ビスリン酸アルドラーゼの分離方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH1135603A JPH1135603A (ja) | 1999-02-09 |
| JP4166846B2 true JP4166846B2 (ja) | 2008-10-15 |
Family
ID=16627219
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP21271497A Expired - Fee Related JP4166846B2 (ja) | 1997-07-24 | 1997-07-24 | ヘパリン結合担体及びフルクトース−1,6−ビスリン酸アルドラーゼの分離方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4166846B2 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4633223B2 (ja) * | 1999-03-31 | 2011-02-16 | 生化学工業株式会社 | 血管内皮細胞増殖因子依存性血管内皮細胞増殖の抑制剤 |
| JP4633233B2 (ja) * | 2000-06-29 | 2011-02-16 | 生化学工業株式会社 | クラミジア感染症処置剤 |
| US7687609B2 (en) * | 2006-05-19 | 2010-03-30 | National Institute Of Advanced Industrial Science And Technology | Galectin-glycosaminoglycan complex and method for controlling galectin activity |
| CN103157452B (zh) * | 2013-03-30 | 2014-12-31 | 福州大学 | 脱硫酸化肝素衍生物亲和色谱材料的制备方法 |
-
1997
- 1997-07-24 JP JP21271497A patent/JP4166846B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JPH1135603A (ja) | 1999-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5013724A (en) | Process for the sulfation of glycosaminoglycans, the sulfated glycosaminoglycans and their biological applications | |
| Hook et al. | Anticoagulant activity of heparin: separation of high-activity and low-activity heparin species by affinity chromatography on immobilized antithrombin | |
| US4401758A (en) | Process for making oligosaccharides having anti-Xa activity and the resulting oligosaccharides | |
| Linhardt et al. | Structure and activity of a unique heparin-derived hexasaccharide. | |
| US4826827A (en) | Short chained oligosaccharides having biological properties, a process for making the same and the use thereof as drugs | |
| US5384398A (en) | High molecular mass N,O-sulphated heparosans, process for their preparation and the pharmaceutical compositions which contain them | |
| BG60479B1 (bg) | N,о-сулфатирани хепарозани, метод за получаването им и фармацевтичните смеси, които ги съдържат | |
| Danishefsky et al. | Synthesis of heparin-sepharoses and their binding with thrombin and antithrombin-heparin cofactor | |
| WO1990001501A1 (en) | Oligosaccharide heparin fragments as inhibitors of complement cascade | |
| JPH07501684A (ja) | 抗凝固剤およびその調製方法 | |
| CA1334653C (en) | Purification of glycosaminoglycan degrading enzymes | |
| Linhardt et al. | CS lyases: structure, activity, and applications in analysis and the treatment of diseases | |
| JP2007501305A (ja) | 血漿中で高度の抗血栓活性を有する多糖類誘導体 | |
| EP0064452B1 (fr) | Oligosaccharides à chaînes courtes possédant des propriétés biologiques, leur préparation et leurs applications en tant que médicaments | |
| US20080207895A1 (en) | Methods for synthesizing polysaccharides | |
| JP4166846B2 (ja) | ヘパリン結合担体及びフルクトース−1,6−ビスリン酸アルドラーゼの分離方法 | |
| RU2283319C2 (ru) | Гликозаминогликаны, производные к5-полисахарида, обладающие высокой антикоагулянтной и антитромботической активностью, и способ их получения | |
| JPWO2017115675A1 (ja) | グルコサミン残基の3−o−硫酸化率が高いヘパラン硫酸 | |
| Rosenfeld et al. | Location of specific oligosaccharides in heparin in terms of their distance from the protein linkage region in the native proteoglycan. | |
| JP3522775B2 (ja) | ヘパリン不飽和4糖およびその製造法 | |
| JP4768936B2 (ja) | 硫酸基を有するオリゴ糖 | |
| JP3497209B2 (ja) | 硫酸化糖の脱硫酸化方法 | |
| AU2013237670B2 (en) | Targeted glycosaminoglycan polymers by polymer grafting and methods of making and using same | |
| JP3041657B2 (ja) | 新規なβ1→3−N−アセチルグルコサミニル転移酵素、その製造法及びN−アセチルグルコサミニル転移生成物の製造方法 | |
| JP4469442B2 (ja) | フルクトース1,6−ビスリン酸アルドラーゼ阻害剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040714 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20040714 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080507 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080702 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20080702 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080729 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080731 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110808 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110808 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120808 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120808 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130808 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |