JP2021522798A - 免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 - Google Patents
免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 Download PDFInfo
- Publication number
- JP2021522798A JP2021522798A JP2020561821A JP2020561821A JP2021522798A JP 2021522798 A JP2021522798 A JP 2021522798A JP 2020561821 A JP2020561821 A JP 2020561821A JP 2020561821 A JP2020561821 A JP 2020561821A JP 2021522798 A JP2021522798 A JP 2021522798A
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- cish
- car
- antigen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 210000000822 natural killer cell Anatomy 0.000 title claims abstract description 227
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 title claims abstract description 111
- 230000005746 immune checkpoint blockade Effects 0.000 title description 2
- 102100032218 Cytokine-inducible SH2-containing protein Human genes 0.000 claims abstract description 141
- 101000943420 Homo sapiens Cytokine-inducible SH2-containing protein Proteins 0.000 claims abstract description 139
- 238000000034 method Methods 0.000 claims abstract description 105
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 45
- 201000010099 disease Diseases 0.000 claims abstract description 23
- 238000004519 manufacturing process Methods 0.000 claims abstract description 7
- 210000004027 cell Anatomy 0.000 claims description 184
- 239000000427 antigen Substances 0.000 claims description 174
- 108091007433 antigens Proteins 0.000 claims description 172
- 102000036639 antigens Human genes 0.000 claims description 172
- 206010028980 Neoplasm Diseases 0.000 claims description 139
- 102000003812 Interleukin-15 Human genes 0.000 claims description 104
- 108090000172 Interleukin-15 Proteins 0.000 claims description 104
- 108091008874 T cell receptors Proteins 0.000 claims description 88
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims description 82
- 230000014509 gene expression Effects 0.000 claims description 80
- 201000011510 cancer Diseases 0.000 claims description 54
- 108091033409 CRISPR Proteins 0.000 claims description 51
- 210000002865 immune cell Anatomy 0.000 claims description 51
- 239000013598 vector Substances 0.000 claims description 50
- 238000011282 treatment Methods 0.000 claims description 39
- -1 CA- 125 Proteins 0.000 claims description 36
- 102000004127 Cytokines Human genes 0.000 claims description 36
- 108090000695 Cytokines Proteins 0.000 claims description 36
- 230000006870 function Effects 0.000 claims description 29
- 239000003814 drug Substances 0.000 claims description 28
- 108010002350 Interleukin-2 Proteins 0.000 claims description 25
- 102000000588 Interleukin-2 Human genes 0.000 claims description 25
- 210000004700 fetal blood Anatomy 0.000 claims description 24
- 230000001404 mediated effect Effects 0.000 claims description 23
- 208000035475 disorder Diseases 0.000 claims description 22
- 230000001965 increasing effect Effects 0.000 claims description 22
- 238000010361 transduction Methods 0.000 claims description 20
- 230000026683 transduction Effects 0.000 claims description 20
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 19
- 208000024908 graft versus host disease Diseases 0.000 claims description 19
- 230000011664 signaling Effects 0.000 claims description 19
- 238000002560 therapeutic procedure Methods 0.000 claims description 18
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 claims description 15
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 claims description 15
- 239000003446 ligand Substances 0.000 claims description 15
- 201000001441 melanoma Diseases 0.000 claims description 15
- 239000000203 mixture Substances 0.000 claims description 15
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 claims description 14
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 claims description 14
- 102100030704 Interleukin-21 Human genes 0.000 claims description 13
- 238000009169 immunotherapy Methods 0.000 claims description 13
- 108010074108 interleukin-21 Proteins 0.000 claims description 13
- 108091027544 Subgenomic mRNA Proteins 0.000 claims description 12
- 238000003556 assay Methods 0.000 claims description 11
- 101100369992 Homo sapiens TNFSF10 gene Proteins 0.000 claims description 10
- 102000013462 Interleukin-12 Human genes 0.000 claims description 10
- 108010065805 Interleukin-12 Proteins 0.000 claims description 10
- 108700012411 TNFSF10 Proteins 0.000 claims description 10
- 108060008682 Tumor Necrosis Factor Proteins 0.000 claims description 10
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 claims description 10
- 230000000735 allogeneic effect Effects 0.000 claims description 10
- 208000023275 Autoimmune disease Diseases 0.000 claims description 9
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 claims description 9
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 claims description 9
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 claims description 9
- 241000713772 Human immunodeficiency virus 1 Species 0.000 claims description 9
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 claims description 9
- 238000002512 chemotherapy Methods 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- 230000001177 retroviral effect Effects 0.000 claims description 9
- 238000001356 surgical procedure Methods 0.000 claims description 9
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 claims description 8
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 claims description 8
- 101710121417 Envelope glycoprotein Proteins 0.000 claims description 8
- 108700024394 Exon Proteins 0.000 claims description 8
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 claims description 8
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 claims description 8
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 claims description 8
- 102100034256 Mucin-1 Human genes 0.000 claims description 8
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 claims description 8
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 claims description 8
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 claims description 8
- 108010087914 epidermal growth factor receptor VIII Proteins 0.000 claims description 8
- 229940124597 therapeutic agent Drugs 0.000 claims description 8
- 108010074328 Interferon-gamma Proteins 0.000 claims description 7
- 210000001185 bone marrow Anatomy 0.000 claims description 7
- 239000003937 drug carrier Substances 0.000 claims description 7
- 239000011886 peripheral blood Substances 0.000 claims description 7
- 210000005259 peripheral blood Anatomy 0.000 claims description 7
- 238000001959 radiotherapy Methods 0.000 claims description 7
- 102000016914 ras Proteins Human genes 0.000 claims description 7
- 108010014186 ras Proteins Proteins 0.000 claims description 7
- 101001005269 Arabidopsis thaliana Ceramide synthase 1 LOH3 Proteins 0.000 claims description 6
- 101001005312 Arabidopsis thaliana Ceramide synthase LOH1 Proteins 0.000 claims description 6
- 102100038080 B-cell receptor CD22 Human genes 0.000 claims description 6
- 102100033417 Glucocorticoid receptor Human genes 0.000 claims description 6
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 claims description 6
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 claims description 6
- 101001023379 Homo sapiens Lysosome-associated membrane glycoprotein 1 Proteins 0.000 claims description 6
- 101001109501 Homo sapiens NKG2-D type II integral membrane protein Proteins 0.000 claims description 6
- 101000633784 Homo sapiens SLAM family member 7 Proteins 0.000 claims description 6
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 claims description 6
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 claims description 6
- 102100035133 Lysosome-associated membrane glycoprotein 1 Human genes 0.000 claims description 6
- 102100022680 NKG2-D type II integral membrane protein Human genes 0.000 claims description 6
- 101000668858 Spinacia oleracea 30S ribosomal protein S1, chloroplastic Proteins 0.000 claims description 6
- 101000898746 Streptomyces clavuligerus Clavaminate synthase 1 Proteins 0.000 claims description 6
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 claims description 6
- 230000030279 gene silencing Effects 0.000 claims description 6
- 239000012528 membrane Substances 0.000 claims description 6
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 claims description 5
- BGFTWECWAICPDG-UHFFFAOYSA-N 2-[bis(4-chlorophenyl)methyl]-4-n-[3-[bis(4-chlorophenyl)methyl]-4-(dimethylamino)phenyl]-1-n,1-n-dimethylbenzene-1,4-diamine Chemical compound C1=C(C(C=2C=CC(Cl)=CC=2)C=2C=CC(Cl)=CC=2)C(N(C)C)=CC=C1NC(C=1)=CC=C(N(C)C)C=1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 BGFTWECWAICPDG-UHFFFAOYSA-N 0.000 claims description 5
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 claims description 5
- 102000006942 B-Cell Maturation Antigen Human genes 0.000 claims description 5
- 102100025221 CD70 antigen Human genes 0.000 claims description 5
- 102100028801 Calsyntenin-1 Human genes 0.000 claims description 5
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 claims description 5
- 101000991061 Homo sapiens MHC class I polypeptide-related sequence B Proteins 0.000 claims description 5
- 101000617830 Homo sapiens Sterol O-acyltransferase 1 Proteins 0.000 claims description 5
- 101000914496 Homo sapiens T-cell antigen CD7 Proteins 0.000 claims description 5
- 101000621309 Homo sapiens Wilms tumor protein Proteins 0.000 claims description 5
- 102100037850 Interferon gamma Human genes 0.000 claims description 5
- 102100030300 MHC class I polypeptide-related sequence B Human genes 0.000 claims description 5
- 102000003735 Mesothelin Human genes 0.000 claims description 5
- 108090000015 Mesothelin Proteins 0.000 claims description 5
- 108010008707 Mucin-1 Proteins 0.000 claims description 5
- KHGNFPUMBJSZSM-UHFFFAOYSA-N Perforine Natural products COC1=C2CCC(O)C(CCC(C)(C)O)(OC)C2=NC2=C1C=CO2 KHGNFPUMBJSZSM-UHFFFAOYSA-N 0.000 claims description 5
- 102100021993 Sterol O-acyltransferase 1 Human genes 0.000 claims description 5
- 101000697584 Streptomyces lavendulae Streptothricin acetyltransferase Proteins 0.000 claims description 5
- 102100027208 T-cell antigen CD7 Human genes 0.000 claims description 5
- 101800001690 Transmembrane protein gp41 Proteins 0.000 claims description 5
- 101710090322 Truncated surface protein Proteins 0.000 claims description 5
- 102100022748 Wilms tumor protein Human genes 0.000 claims description 5
- 239000013604 expression vector Substances 0.000 claims description 5
- 230000001605 fetal effect Effects 0.000 claims description 5
- 239000010445 mica Substances 0.000 claims description 5
- 229910052618 mica group Inorganic materials 0.000 claims description 5
- 229930192851 perforin Natural products 0.000 claims description 5
- 102100028757 Chondroitin sulfate proteoglycan 4 Human genes 0.000 claims description 4
- 108090000079 Glucocorticoid Receptors Proteins 0.000 claims description 4
- 208000002250 Hematologic Neoplasms Diseases 0.000 claims description 4
- 101000916489 Homo sapiens Chondroitin sulfate proteoglycan 4 Proteins 0.000 claims description 4
- 101001103039 Homo sapiens Inactive tyrosine-protein kinase transmembrane receptor ROR1 Proteins 0.000 claims description 4
- 101001055157 Homo sapiens Interleukin-15 Proteins 0.000 claims description 4
- 101001103036 Homo sapiens Nuclear receptor ROR-alpha Proteins 0.000 claims description 4
- 101000809875 Homo sapiens TYRO protein tyrosine kinase-binding protein Proteins 0.000 claims description 4
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 claims description 4
- 241000192019 Human endogenous retrovirus K Species 0.000 claims description 4
- 102100039615 Inactive tyrosine-protein kinase transmembrane receptor ROR1 Human genes 0.000 claims description 4
- 102100038717 TYRO protein tyrosine kinase-binding protein Human genes 0.000 claims description 4
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 claims description 4
- 102000036215 folic acid binding proteins Human genes 0.000 claims description 4
- 108091011001 folic acid binding proteins Proteins 0.000 claims description 4
- 238000012226 gene silencing method Methods 0.000 claims description 4
- 102000056003 human IL15 Human genes 0.000 claims description 4
- 230000002062 proliferating effect Effects 0.000 claims description 4
- 102100036008 CD48 antigen Human genes 0.000 claims description 3
- 229920001917 Ficoll Polymers 0.000 claims description 3
- 101000716130 Homo sapiens CD48 antigen Proteins 0.000 claims description 3
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 claims description 3
- 101000713602 Homo sapiens T-box transcription factor TBX21 Proteins 0.000 claims description 3
- 101100445364 Mus musculus Eomes gene Proteins 0.000 claims description 3
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 claims description 3
- 102100036840 T-box transcription factor TBX21 Human genes 0.000 claims description 3
- 101100445365 Xenopus laevis eomes gene Proteins 0.000 claims description 3
- 102000013529 alpha-Fetoproteins Human genes 0.000 claims description 3
- 108010026331 alpha-Fetoproteins Proteins 0.000 claims description 3
- 238000012258 culturing Methods 0.000 claims description 3
- 230000004968 inflammatory condition Effects 0.000 claims description 3
- 230000002757 inflammatory effect Effects 0.000 claims description 3
- 238000010212 intracellular staining Methods 0.000 claims description 3
- 210000005087 mononuclear cell Anatomy 0.000 claims description 3
- 230000010412 perfusion Effects 0.000 claims description 3
- 230000005909 tumor killing Effects 0.000 claims description 3
- 206010066476 Haematological malignancy Diseases 0.000 claims description 2
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 claims description 2
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 claims description 2
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 claims description 2
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 claims description 2
- 238000001815 biotherapy Methods 0.000 claims description 2
- 150000003431 steroids Chemical class 0.000 claims description 2
- 102100027723 Endogenous retrovirus group K member 6 Rec protein Human genes 0.000 claims 4
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 claims 3
- 238000010354 CRISPR gene editing Methods 0.000 claims 2
- 238000010586 diagram Methods 0.000 abstract description 29
- 108090000765 processed proteins & peptides Proteins 0.000 description 193
- 102000004196 processed proteins & peptides Human genes 0.000 description 182
- 229920001184 polypeptide Polymers 0.000 description 176
- 108090000623 proteins and genes Proteins 0.000 description 152
- 108010005327 CD19-specific chimeric antigen receptor Proteins 0.000 description 82
- 102000004169 proteins and genes Human genes 0.000 description 77
- 235000018102 proteins Nutrition 0.000 description 73
- 230000027455 binding Effects 0.000 description 55
- 230000000694 effects Effects 0.000 description 38
- 150000007523 nucleic acids Chemical class 0.000 description 34
- 230000003211 malignant effect Effects 0.000 description 33
- 241000700605 Viruses Species 0.000 description 31
- 208000009956 adenocarcinoma Diseases 0.000 description 31
- 210000001744 T-lymphocyte Anatomy 0.000 description 30
- 108020004414 DNA Proteins 0.000 description 29
- 206010025323 Lymphomas Diseases 0.000 description 24
- 102000039446 nucleic acids Human genes 0.000 description 23
- 108020004707 nucleic acids Proteins 0.000 description 23
- 210000000612 antigen-presenting cell Anatomy 0.000 description 21
- 230000008685 targeting Effects 0.000 description 20
- 102000004190 Enzymes Human genes 0.000 description 19
- 108090000790 Enzymes Proteins 0.000 description 19
- 101710163270 Nuclease Proteins 0.000 description 19
- 229940079593 drug Drugs 0.000 description 19
- 229940088598 enzyme Drugs 0.000 description 19
- 239000000047 product Substances 0.000 description 18
- 210000004881 tumor cell Anatomy 0.000 description 18
- 108060003951 Immunoglobulin Proteins 0.000 description 17
- 102000018358 immunoglobulin Human genes 0.000 description 17
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 16
- 239000003623 enhancer Substances 0.000 description 16
- 239000003550 marker Substances 0.000 description 16
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 16
- 241001529453 unidentified herpesvirus Species 0.000 description 16
- 102000006306 Antigen Receptors Human genes 0.000 description 15
- 108010083359 Antigen Receptors Proteins 0.000 description 15
- 239000012634 fragment Substances 0.000 description 15
- 208000032839 leukemia Diseases 0.000 description 15
- 230000004083 survival effect Effects 0.000 description 15
- 241000699670 Mus sp. Species 0.000 description 14
- 108091028043 Nucleic acid sequence Proteins 0.000 description 14
- 239000003795 chemical substances by application Substances 0.000 description 14
- 239000012636 effector Substances 0.000 description 14
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 13
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 13
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 13
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 13
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 13
- 230000004913 activation Effects 0.000 description 13
- 150000001413 amino acids Chemical group 0.000 description 13
- 238000003776 cleavage reaction Methods 0.000 description 13
- 238000012217 deletion Methods 0.000 description 13
- 230000037430 deletion Effects 0.000 description 13
- 230000035755 proliferation Effects 0.000 description 13
- 230000007017 scission Effects 0.000 description 13
- 238000013518 transcription Methods 0.000 description 13
- 230000035897 transcription Effects 0.000 description 13
- 108020005004 Guide RNA Proteins 0.000 description 12
- 230000003013 cytotoxicity Effects 0.000 description 12
- 231100000135 cytotoxicity Toxicity 0.000 description 12
- 238000004520 electroporation Methods 0.000 description 12
- 238000003752 polymerase chain reaction Methods 0.000 description 12
- 230000001105 regulatory effect Effects 0.000 description 12
- 230000004568 DNA-binding Effects 0.000 description 11
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 210000004698 lymphocyte Anatomy 0.000 description 11
- 102000005962 receptors Human genes 0.000 description 11
- 108020003175 receptors Proteins 0.000 description 11
- 210000001519 tissue Anatomy 0.000 description 11
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 10
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 10
- 238000002659 cell therapy Methods 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 239000003112 inhibitor Substances 0.000 description 10
- 230000003827 upregulation Effects 0.000 description 10
- 229910052725 zinc Inorganic materials 0.000 description 10
- 239000011701 zinc Substances 0.000 description 10
- 108091026890 Coding region Proteins 0.000 description 9
- 108020004705 Codon Proteins 0.000 description 9
- 241000701022 Cytomegalovirus Species 0.000 description 9
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 9
- 235000001014 amino acid Nutrition 0.000 description 9
- 238000013459 approach Methods 0.000 description 9
- 230000001939 inductive effect Effects 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 108020004999 messenger RNA Proteins 0.000 description 9
- 230000004048 modification Effects 0.000 description 9
- 238000012986 modification Methods 0.000 description 9
- 239000002773 nucleotide Substances 0.000 description 9
- 125000003729 nucleotide group Chemical group 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- 201000009030 Carcinoma Diseases 0.000 description 8
- 102000003886 Glycoproteins Human genes 0.000 description 8
- 108090000288 Glycoproteins Proteins 0.000 description 8
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 8
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 description 8
- 206010039491 Sarcoma Diseases 0.000 description 8
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 8
- 108010017070 Zinc Finger Nucleases Proteins 0.000 description 8
- 229940024606 amino acid Drugs 0.000 description 8
- 239000005557 antagonist Substances 0.000 description 8
- 239000002246 antineoplastic agent Substances 0.000 description 8
- 238000010362 genome editing Methods 0.000 description 8
- 206010025135 lupus erythematosus Diseases 0.000 description 8
- 230000006780 non-homologous end joining Effects 0.000 description 8
- 210000000056 organ Anatomy 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 208000024891 symptom Diseases 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- 238000013519 translation Methods 0.000 description 8
- 238000002054 transplantation Methods 0.000 description 8
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 7
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 7
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 7
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 7
- 241000725303 Human immunodeficiency virus Species 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- 241000193998 Streptococcus pneumoniae Species 0.000 description 7
- 108091028113 Trans-activating crRNA Proteins 0.000 description 7
- 230000001154 acute effect Effects 0.000 description 7
- 229940049595 antibody-drug conjugate Drugs 0.000 description 7
- 230000001684 chronic effect Effects 0.000 description 7
- 230000000295 complement effect Effects 0.000 description 7
- 108020001507 fusion proteins Proteins 0.000 description 7
- 102000037865 fusion proteins Human genes 0.000 description 7
- 230000002414 glycolytic effect Effects 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 238000003780 insertion Methods 0.000 description 7
- 230000037431 insertion Effects 0.000 description 7
- 230000003834 intracellular effect Effects 0.000 description 7
- 229960005386 ipilimumab Drugs 0.000 description 7
- 230000010076 replication Effects 0.000 description 7
- 238000012546 transfer Methods 0.000 description 7
- 102000007469 Actins Human genes 0.000 description 6
- 108010085238 Actins Proteins 0.000 description 6
- 101150043532 CISH gene Proteins 0.000 description 6
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 6
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 6
- 241000282412 Homo Species 0.000 description 6
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 6
- 108010002586 Interleukin-7 Proteins 0.000 description 6
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 108700026244 Open Reading Frames Proteins 0.000 description 6
- 206010035226 Plasma cell myeloma Diseases 0.000 description 6
- 102000004389 Ribonucleoproteins Human genes 0.000 description 6
- 108010081734 Ribonucleoproteins Proteins 0.000 description 6
- 108020004459 Small interfering RNA Proteins 0.000 description 6
- 101710185494 Zinc finger protein Proteins 0.000 description 6
- 102100023597 Zinc finger protein 816 Human genes 0.000 description 6
- 210000003719 b-lymphocyte Anatomy 0.000 description 6
- 230000001580 bacterial effect Effects 0.000 description 6
- 238000004422 calculation algorithm Methods 0.000 description 6
- 229960004397 cyclophosphamide Drugs 0.000 description 6
- 229940127089 cytotoxic agent Drugs 0.000 description 6
- 230000001472 cytotoxic effect Effects 0.000 description 6
- 210000003527 eukaryotic cell Anatomy 0.000 description 6
- 238000000684 flow cytometry Methods 0.000 description 6
- 238000012239 gene modification Methods 0.000 description 6
- 238000010199 gene set enrichment analysis Methods 0.000 description 6
- 239000008103 glucose Substances 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 230000028993 immune response Effects 0.000 description 6
- 210000000987 immune system Anatomy 0.000 description 6
- 210000000428 immunological synapse Anatomy 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 230000000977 initiatory effect Effects 0.000 description 6
- 238000010172 mouse model Methods 0.000 description 6
- 229960003301 nivolumab Drugs 0.000 description 6
- 229960002621 pembrolizumab Drugs 0.000 description 6
- 102000040430 polynucleotide Human genes 0.000 description 6
- 108091033319 polynucleotide Proteins 0.000 description 6
- 239000002157 polynucleotide Substances 0.000 description 6
- 230000008439 repair process Effects 0.000 description 6
- 239000004055 small Interfering RNA Substances 0.000 description 6
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 6
- 208000011580 syndromic disease Diseases 0.000 description 6
- 230000009258 tissue cross reactivity Effects 0.000 description 6
- 230000003612 virological effect Effects 0.000 description 6
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 5
- 102100031780 Endonuclease Human genes 0.000 description 5
- 108060003393 Granulin Proteins 0.000 description 5
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 description 5
- 101000914321 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 7 Proteins 0.000 description 5
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 5
- 101000617725 Homo sapiens Pregnancy-specific beta-1-glycoprotein 2 Proteins 0.000 description 5
- 101001117317 Homo sapiens Programmed cell death 1 ligand 1 Proteins 0.000 description 5
- 101001117312 Homo sapiens Programmed cell death 1 ligand 2 Proteins 0.000 description 5
- 102000015696 Interleukins Human genes 0.000 description 5
- 108010063738 Interleukins Proteins 0.000 description 5
- 102000042838 JAK family Human genes 0.000 description 5
- 102100023123 Mucin-16 Human genes 0.000 description 5
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 5
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 5
- 108020004440 Thymidine kinase Proteins 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000003242 anti bacterial agent Substances 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 239000011651 chromium Substances 0.000 description 5
- 108091008034 costimulatory receptors Proteins 0.000 description 5
- 230000005782 double-strand break Effects 0.000 description 5
- 231100000221 frame shift mutation induction Toxicity 0.000 description 5
- 230000037433 frameshift Effects 0.000 description 5
- 230000004927 fusion Effects 0.000 description 5
- 230000005017 genetic modification Effects 0.000 description 5
- 235000013617 genetically modified food Nutrition 0.000 description 5
- 230000034659 glycolysis Effects 0.000 description 5
- 239000003102 growth factor Substances 0.000 description 5
- 229940126546 immune checkpoint molecule Drugs 0.000 description 5
- 230000004068 intracellular signaling Effects 0.000 description 5
- 210000000265 leukocyte Anatomy 0.000 description 5
- 210000004185 liver Anatomy 0.000 description 5
- 230000004060 metabolic process Effects 0.000 description 5
- 210000003879 microtubule-organizing center Anatomy 0.000 description 5
- 230000002688 persistence Effects 0.000 description 5
- 210000000130 stem cell Anatomy 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 230000002103 transcriptional effect Effects 0.000 description 5
- 238000011144 upstream manufacturing Methods 0.000 description 5
- 239000013603 viral vector Substances 0.000 description 5
- 241001057184 Axion Species 0.000 description 4
- 206010004194 Bed bug infestation Diseases 0.000 description 4
- 206010006187 Breast cancer Diseases 0.000 description 4
- 238000010356 CRISPR-Cas9 genome editing Methods 0.000 description 4
- 229940045513 CTLA4 antagonist Drugs 0.000 description 4
- 108010012236 Chemokines Proteins 0.000 description 4
- 102000019034 Chemokines Human genes 0.000 description 4
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 4
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 4
- 241001414835 Cimicidae Species 0.000 description 4
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 4
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 4
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 108010042407 Endonucleases Proteins 0.000 description 4
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 4
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 4
- 101710154606 Hemagglutinin Proteins 0.000 description 4
- 208000017604 Hodgkin disease Diseases 0.000 description 4
- 101000946860 Homo sapiens T-cell surface glycoprotein CD3 epsilon chain Proteins 0.000 description 4
- 206010021143 Hypoxia Diseases 0.000 description 4
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 4
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 4
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 4
- 102100034349 Integrase Human genes 0.000 description 4
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 4
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 4
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 4
- 241000206591 Peptococcus Species 0.000 description 4
- 102000004503 Perforin Human genes 0.000 description 4
- 108010056995 Perforin Proteins 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- 101710176177 Protein A56 Proteins 0.000 description 4
- 108010017324 STAT3 Transcription Factor Proteins 0.000 description 4
- 108010029477 STAT5 Transcription Factor Proteins 0.000 description 4
- 102000001712 STAT5 Transcription Factor Human genes 0.000 description 4
- 102100024040 Signal transducer and activator of transcription 3 Human genes 0.000 description 4
- 241000191940 Staphylococcus Species 0.000 description 4
- 108091081024 Start codon Proteins 0.000 description 4
- 241000193996 Streptococcus pyogenes Species 0.000 description 4
- 102100035794 T-cell surface glycoprotein CD3 epsilon chain Human genes 0.000 description 4
- 102000006601 Thymidine Kinase Human genes 0.000 description 4
- 102000003425 Tyrosinase Human genes 0.000 description 4
- 108060008724 Tyrosinase Proteins 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 229940088710 antibiotic agent Drugs 0.000 description 4
- 239000000611 antibody drug conjugate Substances 0.000 description 4
- 238000011319 anticancer therapy Methods 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 101150038500 cas9 gene Proteins 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- 229910052804 chromium Inorganic materials 0.000 description 4
- 238000010367 cloning Methods 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 231100000433 cytotoxic Toxicity 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 229960000390 fludarabine Drugs 0.000 description 4
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 239000000185 hemagglutinin Substances 0.000 description 4
- 229940088597 hormone Drugs 0.000 description 4
- 239000005556 hormone Substances 0.000 description 4
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 4
- 229940072221 immunoglobulins Drugs 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 210000004072 lung Anatomy 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 230000037353 metabolic pathway Effects 0.000 description 4
- 210000003470 mitochondria Anatomy 0.000 description 4
- 201000000050 myeloid neoplasm Diseases 0.000 description 4
- 210000003463 organelle Anatomy 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 230000001717 pathogenic effect Effects 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 210000002307 prostate Anatomy 0.000 description 4
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 4
- 230000006798 recombination Effects 0.000 description 4
- 238000005215 recombination Methods 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 210000003491 skin Anatomy 0.000 description 4
- 208000003265 stomatitis Diseases 0.000 description 4
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 3
- 241000238876 Acari Species 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 102100038078 CD276 antigen Human genes 0.000 description 3
- 102100032937 CD40 ligand Human genes 0.000 description 3
- 201000004624 Dermatitis Diseases 0.000 description 3
- 101150029707 ERBB2 gene Proteins 0.000 description 3
- 108010003471 Fetal Proteins Proteins 0.000 description 3
- 102000004641 Fetal Proteins Human genes 0.000 description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 3
- 208000004463 Follicular Adenocarcinoma Diseases 0.000 description 3
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 3
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 3
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 3
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 3
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 3
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 108091092195 Intron Proteins 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 102000043129 MHC class I family Human genes 0.000 description 3
- 108091054437 MHC class I family Proteins 0.000 description 3
- 101710125418 Major capsid protein Proteins 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 102000008135 Mechanistic Target of Rapamycin Complex 1 Human genes 0.000 description 3
- 108010035196 Mechanistic Target of Rapamycin Complex 1 Proteins 0.000 description 3
- 108010052285 Membrane Proteins Proteins 0.000 description 3
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 3
- 101710101148 Probable 6-oxopurine nucleoside phosphorylase Proteins 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 3
- 102100038358 Prostate-specific antigen Human genes 0.000 description 3
- 102000030764 Purine-nucleoside phosphorylase Human genes 0.000 description 3
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 3
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 3
- 108091027981 Response element Proteins 0.000 description 3
- 241000606701 Rickettsia Species 0.000 description 3
- 241000191967 Staphylococcus aureus Species 0.000 description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 description 3
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 3
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 3
- 108700026226 TATA Box Proteins 0.000 description 3
- 108091005735 TGF-beta receptors Proteins 0.000 description 3
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 3
- 108020004566 Transfer RNA Proteins 0.000 description 3
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 3
- 206010047115 Vasculitis Diseases 0.000 description 3
- 238000011467 adoptive cell therapy Methods 0.000 description 3
- 230000000890 antigenic effect Effects 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 235000006708 antioxidants Nutrition 0.000 description 3
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 210000000234 capsid Anatomy 0.000 description 3
- 229960004562 carboplatin Drugs 0.000 description 3
- 230000021164 cell adhesion Effects 0.000 description 3
- 230000003915 cell function Effects 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 108700010039 chimeric receptor Proteins 0.000 description 3
- 208000006990 cholangiocarcinoma Diseases 0.000 description 3
- 238000004737 colorimetric analysis Methods 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- 238000004624 confocal microscopy Methods 0.000 description 3
- 238000010276 construction Methods 0.000 description 3
- 230000000139 costimulatory effect Effects 0.000 description 3
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 3
- 235000018417 cysteine Nutrition 0.000 description 3
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 3
- 230000016396 cytokine production Effects 0.000 description 3
- 210000005220 cytoplasmic tail Anatomy 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 210000003298 dental enamel Anatomy 0.000 description 3
- 230000004069 differentiation Effects 0.000 description 3
- 229960004679 doxorubicin Drugs 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 229960002949 fluorouracil Drugs 0.000 description 3
- 230000002538 fungal effect Effects 0.000 description 3
- 206010017758 gastric cancer Diseases 0.000 description 3
- 230000009368 gene silencing by RNA Effects 0.000 description 3
- 239000005090 green fluorescent protein Substances 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 201000005787 hematologic cancer Diseases 0.000 description 3
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 3
- 238000009396 hybridization Methods 0.000 description 3
- 230000007954 hypoxia Effects 0.000 description 3
- 208000026278 immune system disease Diseases 0.000 description 3
- 230000020287 immunological synapse formation Effects 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 244000000010 microbial pathogen Species 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- 210000004940 nucleus Anatomy 0.000 description 3
- 238000005457 optimization Methods 0.000 description 3
- 230000002611 ovarian Effects 0.000 description 3
- 210000000496 pancreas Anatomy 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 108020001580 protein domains Proteins 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 238000002271 resection Methods 0.000 description 3
- 108091008146 restriction endonucleases Proteins 0.000 description 3
- 210000004500 stellate cell Anatomy 0.000 description 3
- 230000004936 stimulating effect Effects 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 210000002784 stomach Anatomy 0.000 description 3
- 201000011549 stomach cancer Diseases 0.000 description 3
- 208000030901 thyroid gland follicular carcinoma Diseases 0.000 description 3
- 230000036962 time dependent Effects 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 229960001612 trastuzumab emtansine Drugs 0.000 description 3
- 241000701161 unidentified adenovirus Species 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 229960004528 vincristine Drugs 0.000 description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 3
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 3
- LKKMLIBUAXYLOY-UHFFFAOYSA-N 3-Amino-1-methyl-5H-pyrido[4,3-b]indole Chemical compound N1C2=CC=CC=C2C2=C1C=C(N)N=C2C LKKMLIBUAXYLOY-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- 102100023003 Ankyrin repeat domain-containing protein 30A Human genes 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 2
- 101710144268 B- and T-lymphocyte attenuator Proteins 0.000 description 2
- 208000003950 B-cell lymphoma Diseases 0.000 description 2
- 206010004593 Bile duct cancer Diseases 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- 102100040840 C-type lectin domain family 7 member A Human genes 0.000 description 2
- 102100037904 CD9 antigen Human genes 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 2
- 101710132601 Capsid protein Proteins 0.000 description 2
- 102000013392 Carboxylesterase Human genes 0.000 description 2
- 108010051152 Carboxylesterase Proteins 0.000 description 2
- 108010006303 Carboxypeptidases Proteins 0.000 description 2
- 102000005367 Carboxypeptidases Human genes 0.000 description 2
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 2
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 2
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 2
- 101710094648 Coat protein Proteins 0.000 description 2
- 108700010070 Codon Usage Proteins 0.000 description 2
- 206010009900 Colitis ulcerative Diseases 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 2
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 2
- 101710132484 Cytokine-inducible SH2-containing protein Proteins 0.000 description 2
- 108010080611 Cytosine Deaminase Proteins 0.000 description 2
- 230000005778 DNA damage Effects 0.000 description 2
- 231100000277 DNA damage Toxicity 0.000 description 2
- 230000007018 DNA scission Effects 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 101000831205 Danio rerio Dynein axonemal assembly factor 11 Proteins 0.000 description 2
- 241000702421 Dependoparvovirus Species 0.000 description 2
- 208000010772 Dog disease Diseases 0.000 description 2
- 102100024282 Dynein axonemal assembly factor 11 Human genes 0.000 description 2
- 206010014733 Endometrial cancer Diseases 0.000 description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 description 2
- 241000991587 Enterovirus C Species 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 108700039887 Essential Genes Proteins 0.000 description 2
- 102000016621 Focal Adhesion Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108010067715 Focal Adhesion Protein-Tyrosine Kinases Proteins 0.000 description 2
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 2
- 208000007465 Giant cell arteritis Diseases 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 2
- 102000009465 Growth Factor Receptors Human genes 0.000 description 2
- 108010009202 Growth Factor Receptors Proteins 0.000 description 2
- 102100028972 HLA class I histocompatibility antigen, A alpha chain Human genes 0.000 description 2
- 108010075704 HLA-A Antigens Proteins 0.000 description 2
- 108010086377 HLA-A3 Antigen Proteins 0.000 description 2
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 2
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 2
- 241001559542 Hippocampus hippocampus Species 0.000 description 2
- 101000757191 Homo sapiens Ankyrin repeat domain-containing protein 30A Proteins 0.000 description 2
- 101000884279 Homo sapiens CD276 antigen Proteins 0.000 description 2
- 101000868215 Homo sapiens CD40 ligand Proteins 0.000 description 2
- 101000831210 Homo sapiens Dynein axonemal assembly factor 11 Proteins 0.000 description 2
- 101000926939 Homo sapiens Glucocorticoid receptor Proteins 0.000 description 2
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 2
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 description 2
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 2
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 2
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 2
- 101000603882 Homo sapiens Nuclear receptor subfamily 1 group I member 3 Proteins 0.000 description 2
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 2
- 101000611183 Homo sapiens Tumor necrosis factor Proteins 0.000 description 2
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 2
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 2
- 102000002265 Human Growth Hormone Human genes 0.000 description 2
- 108010000521 Human Growth Hormone Proteins 0.000 description 2
- 239000000854 Human Growth Hormone Substances 0.000 description 2
- 108010003272 Hyaluronate lyase Proteins 0.000 description 2
- 102000001974 Hyaluronidases Human genes 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 102000008070 Interferon-gamma Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- 102000000589 Interleukin-1 Human genes 0.000 description 2
- 102000004889 Interleukin-6 Human genes 0.000 description 2
- 108090001005 Interleukin-6 Proteins 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- 102100031413 L-dopachrome tautomerase Human genes 0.000 description 2
- 101710093778 L-dopachrome tautomerase Proteins 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- 241000713666 Lentivirus Species 0.000 description 2
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 2
- 102100020862 Lymphocyte activation gene 3 protein Human genes 0.000 description 2
- 108091054438 MHC class II family Proteins 0.000 description 2
- 101710175625 Maltose/maltodextrin-binding periplasmic protein Proteins 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 206010028372 Muscular weakness Diseases 0.000 description 2
- 241000204031 Mycoplasma Species 0.000 description 2
- 108010057466 NF-kappa B Proteins 0.000 description 2
- 102000003945 NF-kappa B Human genes 0.000 description 2
- 108091061960 Naked DNA Proteins 0.000 description 2
- 206010029164 Nephrotic syndrome Diseases 0.000 description 2
- 102000005348 Neuraminidase Human genes 0.000 description 2
- 108010006232 Neuraminidase Proteins 0.000 description 2
- 102000004459 Nitroreductase Human genes 0.000 description 2
- 108010070047 Notch Receptors Proteins 0.000 description 2
- 102000005650 Notch Receptors Human genes 0.000 description 2
- 101710141454 Nucleoprotein Proteins 0.000 description 2
- 102000011931 Nucleoproteins Human genes 0.000 description 2
- 108010061100 Nucleoproteins Proteins 0.000 description 2
- 108010038807 Oligopeptides Proteins 0.000 description 2
- 102000015636 Oligopeptides Human genes 0.000 description 2
- 108091007960 PI3Ks Proteins 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 108060006580 PRAME Proteins 0.000 description 2
- 102000036673 PRAME Human genes 0.000 description 2
- 208000002193 Pain Diseases 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Natural products OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 2
- 102000004422 Phospholipase C gamma Human genes 0.000 description 2
- 108010056751 Phospholipase C gamma Proteins 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- 241000709664 Picornaviridae Species 0.000 description 2
- 101710083689 Probable capsid protein Proteins 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- 101710149951 Protein Tat Proteins 0.000 description 2
- 241000125945 Protoparvovirus Species 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 101710132082 Pyrimidine/purine nucleoside phosphorylase Proteins 0.000 description 2
- 241000233639 Pythium Species 0.000 description 2
- 238000011529 RT qPCR Methods 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- 108700008625 Reporter Genes Proteins 0.000 description 2
- 101710173694 Short transient receptor potential channel 2 Proteins 0.000 description 2
- 241000700584 Simplexvirus Species 0.000 description 2
- 241000244174 Strongyloides Species 0.000 description 2
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 2
- 102100036407 Thioredoxin Human genes 0.000 description 2
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 102100031372 Thymidine phosphorylase Human genes 0.000 description 2
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 2
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 2
- 102000004060 Transforming Growth Factor-beta Type II Receptor Human genes 0.000 description 2
- 108010082684 Transforming Growth Factor-beta Type II Receptor Proteins 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 102100040247 Tumor necrosis factor Human genes 0.000 description 2
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 201000006704 Ulcerative Colitis Diseases 0.000 description 2
- 241000571986 Uncinaria Species 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 2
- 108700005077 Viral Genes Proteins 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 238000011374 additional therapy Methods 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 238000009175 antibody therapy Methods 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 208000026900 bile duct neoplasm Diseases 0.000 description 2
- 210000000601 blood cell Anatomy 0.000 description 2
- 108091005948 blue fluorescent proteins Proteins 0.000 description 2
- 210000002798 bone marrow cell Anatomy 0.000 description 2
- 229960000455 brentuximab vedotin Drugs 0.000 description 2
- 229960002092 busulfan Drugs 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- 230000002759 chromosomal effect Effects 0.000 description 2
- 238000003501 co-culture Methods 0.000 description 2
- 230000008045 co-localization Effects 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 description 2
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 description 2
- 238000011498 curative surgery Methods 0.000 description 2
- 108010082025 cyan fluorescent protein Proteins 0.000 description 2
- YPHMISFOHDHNIV-FSZOTQKASA-N cycloheximide Chemical compound C1[C@@H](C)C[C@H](C)C(=O)[C@@H]1[C@H](O)CC1CC(=O)NC(=O)C1 YPHMISFOHDHNIV-FSZOTQKASA-N 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 108010025838 dectin 1 Proteins 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 230000003325 follicular Effects 0.000 description 2
- CHPZKNULDCNCBW-UHFFFAOYSA-N gallium nitrate Chemical compound [Ga+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CHPZKNULDCNCBW-UHFFFAOYSA-N 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 102000054766 genetic haplotypes Human genes 0.000 description 2
- 210000004907 gland Anatomy 0.000 description 2
- 239000003862 glucocorticoid Substances 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 2
- 239000000833 heterodimer Substances 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 238000002744 homologous recombination Methods 0.000 description 2
- 230000006801 homologous recombination Effects 0.000 description 2
- 229960002773 hyaluronidase Drugs 0.000 description 2
- 210000004408 hybridoma Anatomy 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000003463 hyperproliferative effect Effects 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 239000012642 immune effector Substances 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 229940121354 immunomodulator Drugs 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 108020004201 indoleamine 2,3-dioxygenase Proteins 0.000 description 2
- 102000006639 indoleamine 2,3-dioxygenase Human genes 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- 229940079322 interferon Drugs 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- 201000005249 lung adenocarcinoma Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 210000002751 lymph Anatomy 0.000 description 2
- 230000000527 lymphocytic effect Effects 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 201000010225 mixed cell type cancer Diseases 0.000 description 2
- 208000029638 mixed neoplasm Diseases 0.000 description 2
- 201000010879 mucinous adenocarcinoma Diseases 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 2
- 230000036473 myasthenia Effects 0.000 description 2
- 208000025113 myeloid leukemia Diseases 0.000 description 2
- 230000001613 neoplastic effect Effects 0.000 description 2
- 208000009928 nephrosis Diseases 0.000 description 2
- 231100001027 nephrosis Toxicity 0.000 description 2
- 108020001162 nitroreductase Proteins 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 208000004019 papillary adenocarcinoma Diseases 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- 239000013600 plasmid vector Substances 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 229950010131 puromycin Drugs 0.000 description 2
- 230000008707 rearrangement Effects 0.000 description 2
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000007480 sanger sequencing Methods 0.000 description 2
- 230000000405 serological effect Effects 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229940083542 sodium Drugs 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 210000000225 synapse Anatomy 0.000 description 2
- 229940037128 systemic glucocorticoids Drugs 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 206010043207 temporal arteritis Diseases 0.000 description 2
- 108060008226 thioredoxin Proteins 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 230000005945 translocation Effects 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 238000010200 validation analysis Methods 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 229940124676 vascular endothelial growth factor receptor Drugs 0.000 description 2
- 210000002845 virion Anatomy 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 108091005957 yellow fluorescent proteins Proteins 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- FLWWDYNPWOSLEO-HQVZTVAUSA-N (2s)-2-[[4-[1-(2-amino-4-oxo-1h-pteridin-6-yl)ethyl-methylamino]benzoyl]amino]pentanedioic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1C(C)N(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FLWWDYNPWOSLEO-HQVZTVAUSA-N 0.000 description 1
- URLVCROWVOSNPT-XOTOMLERSA-N (2s)-4-[(13r)-13-hydroxy-13-[(2r,5r)-5-[(2r,5r)-5-[(1r)-1-hydroxyundecyl]oxolan-2-yl]oxolan-2-yl]tridecyl]-2-methyl-2h-furan-5-one Chemical compound O1[C@@H]([C@H](O)CCCCCCCCCC)CC[C@@H]1[C@@H]1O[C@@H]([C@H](O)CCCCCCCCCCCCC=2C(O[C@@H](C)C=2)=O)CC1 URLVCROWVOSNPT-XOTOMLERSA-N 0.000 description 1
- TVIRNGFXQVMMGB-OFWIHYRESA-N (3s,6r,10r,13e,16s)-16-[(2r,3r,4s)-4-chloro-3-hydroxy-4-phenylbutan-2-yl]-10-[(3-chloro-4-methoxyphenyl)methyl]-6-methyl-3-(2-methylpropyl)-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetrone Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H](O)[C@@H](Cl)C=2C=CC=CC=2)C/C=C/C(=O)N1 TVIRNGFXQVMMGB-OFWIHYRESA-N 0.000 description 1
- SSOORFWOBGFTHL-OTEJMHTDSA-N (4S)-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[(2S)-2-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-5-carbamimidamido-1-[[(2S)-5-carbamimidamido-1-[[(1S)-4-carbamimidamido-1-carboxybutyl]amino]-1-oxopentan-2-yl]amino]-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxohexan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]carbamoyl]pyrrolidin-1-yl]-2-oxoethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-4-[[(2S)-2-[[(2S)-2-[[(2S)-2,6-diaminohexanoyl]amino]-3-methylbutanoyl]amino]propanoyl]amino]-5-oxopentanoic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H]1CCCN1C(=O)CNC(=O)[C@H](Cc1c[nH]c2ccccc12)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](Cc1c[nH]cn1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@@H](N)CCCCN)C(C)C)C(C)C)C(C)C)C(C)C)C(C)C)C(C)C)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O SSOORFWOBGFTHL-OTEJMHTDSA-N 0.000 description 1
- INAUWOVKEZHHDM-PEDBPRJASA-N (7s,9s)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-7-[(2r,4s,5s,6s)-5-hydroxy-6-methyl-4-morpholin-4-yloxan-2-yl]oxy-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1 INAUWOVKEZHHDM-PEDBPRJASA-N 0.000 description 1
- NOPNWHSMQOXAEI-PUCKCBAPSA-N (7s,9s)-7-[(2r,4s,5s,6s)-4-(2,3-dihydropyrrol-1-yl)-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCC=C1 NOPNWHSMQOXAEI-PUCKCBAPSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- AGNGYMCLFWQVGX-AGFFZDDWSA-N (e)-1-[(2s)-2-amino-2-carboxyethoxy]-2-diazonioethenolate Chemical compound OC(=O)[C@@H](N)CO\C([O-])=C\[N+]#N AGNGYMCLFWQVGX-AGFFZDDWSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- 150000005206 1,2-dihydroxybenzenes Chemical class 0.000 description 1
- 150000005207 1,3-dihydroxybenzenes Chemical class 0.000 description 1
- IPVFGAYTKQKGBM-BYPJNBLXSA-N 1-[(2r,3s,4r,5r)-3-fluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-iodopyrimidine-2,4-dione Chemical compound F[C@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 IPVFGAYTKQKGBM-BYPJNBLXSA-N 0.000 description 1
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 1
- BTOTXLJHDSNXMW-POYBYMJQSA-N 2,3-dideoxyuridine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(=O)NC(=O)C=C1 BTOTXLJHDSNXMW-POYBYMJQSA-N 0.000 description 1
- BOMZMNZEXMAQQW-UHFFFAOYSA-N 2,5,11-trimethyl-6h-pyrido[4,3-b]carbazol-2-ium-9-ol;acetate Chemical compound CC([O-])=O.C[N+]1=CC=C2C(C)=C(NC=3C4=CC(O)=CC=3)C4=C(C)C2=C1 BOMZMNZEXMAQQW-UHFFFAOYSA-N 0.000 description 1
- QCXJFISCRQIYID-IAEPZHFASA-N 2-amino-1-n-[(3s,6s,7r,10s,16s)-3-[(2s)-butan-2-yl]-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-10-propan-2-yl-8-oxa-1,4,11,14-tetrazabicyclo[14.3.0]nonadecan-6-yl]-4,6-dimethyl-3-oxo-9-n-[(3s,6s,7r,10s,16s)-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-3,10-di(propa Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N=C2C(C(=O)N[C@@H]3C(=O)N[C@H](C(N4CCC[C@H]4C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]3C)=O)[C@@H](C)CC)=C(N)C(=O)C(C)=C2O2)C2=C(C)C=C1 QCXJFISCRQIYID-IAEPZHFASA-N 0.000 description 1
- FDAYLTPAFBGXAB-UHFFFAOYSA-N 2-chloro-n,n-bis(2-chloroethyl)ethanamine Chemical compound ClCCN(CCCl)CCCl FDAYLTPAFBGXAB-UHFFFAOYSA-N 0.000 description 1
- PWMYMKOUNYTVQN-UHFFFAOYSA-N 3-(8,8-diethyl-2-aza-8-germaspiro[4.5]decan-2-yl)-n,n-dimethylpropan-1-amine Chemical compound C1C[Ge](CC)(CC)CCC11CN(CCCN(C)C)CC1 PWMYMKOUNYTVQN-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- 108010082808 4-1BB Ligand Proteins 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- 102100033714 40S ribosomal protein S6 Human genes 0.000 description 1
- 102100030310 5,6-dihydroxyindole-2-carboxylic acid oxidase Human genes 0.000 description 1
- 101710163881 5,6-dihydroxyindole-2-carboxylic acid oxidase Proteins 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- BSYNRYMUTXBXSQ-FOQJRBATSA-N 59096-14-9 Chemical compound CC(=O)OC1=CC=CC=C1[14C](O)=O BSYNRYMUTXBXSQ-FOQJRBATSA-N 0.000 description 1
- WYXSYVWAUAUWLD-SHUUEZRQSA-N 6-azauridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=N1 WYXSYVWAUAUWLD-SHUUEZRQSA-N 0.000 description 1
- LQNBBPPWZOXLOV-UHFFFAOYSA-N 6-methyl-7h-purine;7h-purine Chemical compound C1=NC=C2NC=NC2=N1.CC1=NC=NC2=C1NC=N2 LQNBBPPWZOXLOV-UHFFFAOYSA-N 0.000 description 1
- SYMHUEFSSMBHJA-UHFFFAOYSA-N 6-methylpurine Chemical compound CC1=NC=NC2=C1NC=N2 SYMHUEFSSMBHJA-UHFFFAOYSA-N 0.000 description 1
- ZGXJTSGNIOSYLO-UHFFFAOYSA-N 88755TAZ87 Chemical compound NCC(=O)CCC(O)=O ZGXJTSGNIOSYLO-UHFFFAOYSA-N 0.000 description 1
- 102100040079 A-kinase anchor protein 4 Human genes 0.000 description 1
- 101710109924 A-kinase anchor protein 4 Proteins 0.000 description 1
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 1
- 208000002008 AIDS-Related Lymphoma Diseases 0.000 description 1
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 description 1
- 101150019464 ARAF gene Proteins 0.000 description 1
- 241000235389 Absidia Species 0.000 description 1
- 108010009924 Aconitate hydratase Proteins 0.000 description 1
- 241001019659 Acremonium <Plectosphaerellaceae> Species 0.000 description 1
- 102100022498 Actin-like protein 8 Human genes 0.000 description 1
- 241000186046 Actinomyces Species 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 102100021305 Acyl-CoA:lysophosphatidylglycerol acyltransferase 1 Human genes 0.000 description 1
- 102000007471 Adenosine A2A receptor Human genes 0.000 description 1
- 108010085277 Adenosine A2A receptor Proteins 0.000 description 1
- 101150051188 Adora2a gene Proteins 0.000 description 1
- 208000006468 Adrenal Cortex Neoplasms Diseases 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 208000008190 Agammaglobulinemia Diseases 0.000 description 1
- 101710186708 Agglutinin Proteins 0.000 description 1
- 241000589158 Agrobacterium Species 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 206010027654 Allergic conditions Diseases 0.000 description 1
- 102100037982 Alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase A Human genes 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 241000710929 Alphavirus Species 0.000 description 1
- 241000223600 Alternaria Species 0.000 description 1
- 108091093088 Amplicon Proteins 0.000 description 1
- 244000099147 Ananas comosus Species 0.000 description 1
- 235000007119 Ananas comosus Nutrition 0.000 description 1
- 102000052587 Anaphase-Promoting Complex-Cyclosome Apc3 Subunit Human genes 0.000 description 1
- 108700004606 Anaphase-Promoting Complex-Cyclosome Apc3 Subunit Proteins 0.000 description 1
- 102100032187 Androgen receptor Human genes 0.000 description 1
- 102100022014 Angiopoietin-1 receptor Human genes 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 101710145634 Antigen 1 Proteins 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 229940088872 Apoptosis inhibitor Drugs 0.000 description 1
- 101000719121 Arabidopsis thaliana Protein MEI2-like 1 Proteins 0.000 description 1
- 241000239290 Araneae Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 102000030431 Asparaginyl endopeptidase Human genes 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 102100022717 Atypical chemokine receptor 1 Human genes 0.000 description 1
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 1
- 206010050245 Autoimmune thrombocytopenia Diseases 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 102100035526 B melanoma antigen 1 Human genes 0.000 description 1
- 208000004736 B-Cell Leukemia Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 108010045634 B7 Antigens Proteins 0.000 description 1
- 102000005738 B7 Antigens Human genes 0.000 description 1
- 241000223836 Babesia Species 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 1
- 102100027522 Baculoviral IAP repeat-containing protein 7 Human genes 0.000 description 1
- 241001235574 Balantidium Species 0.000 description 1
- 241000606660 Bartonella Species 0.000 description 1
- 208000005440 Basal Cell Neoplasms Diseases 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 208000023328 Basedow disease Diseases 0.000 description 1
- 241000235579 Basidiobolus Species 0.000 description 1
- 101000653197 Beet necrotic yellow vein virus (isolate Japan/S) Movement protein TGB3 Proteins 0.000 description 1
- 206010061692 Benign muscle neoplasm Diseases 0.000 description 1
- 241000359271 Besnoitia Species 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- 241001465178 Bipolaris Species 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- 241000335423 Blastomyces Species 0.000 description 1
- 241000588807 Bordetella Species 0.000 description 1
- 241000589968 Borrelia Species 0.000 description 1
- 241000589969 Borreliella burgdorferi Species 0.000 description 1
- 241000701822 Bovine papillomavirus Species 0.000 description 1
- 241000701922 Bovine parvovirus Species 0.000 description 1
- 244000056139 Brassica cretica Species 0.000 description 1
- 235000003351 Brassica cretica Nutrition 0.000 description 1
- 235000003343 Brassica rupestris Nutrition 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 208000007690 Brenner tumor Diseases 0.000 description 1
- 206010073258 Brenner tumour Diseases 0.000 description 1
- 208000003170 Bronchiolo-Alveolar Adenocarcinoma Diseases 0.000 description 1
- 241000589562 Brucella Species 0.000 description 1
- 241000244036 Brugia Species 0.000 description 1
- 241000931178 Bunostomum Species 0.000 description 1
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 1
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 1
- 102100024217 CAMPATH-1 antigen Human genes 0.000 description 1
- 102100027207 CD27 antigen Human genes 0.000 description 1
- 101710185679 CD276 antigen Proteins 0.000 description 1
- 108010029697 CD40 Ligand Proteins 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- 108010065524 CD52 Antigen Proteins 0.000 description 1
- 108010084313 CD58 Antigens Proteins 0.000 description 1
- 101150108242 CDC27 gene Proteins 0.000 description 1
- 108091007914 CDKs Proteins 0.000 description 1
- 201000002829 CREST Syndrome Diseases 0.000 description 1
- 101150018129 CSF2 gene Proteins 0.000 description 1
- 101150069031 CSN2 gene Proteins 0.000 description 1
- 108091058556 CTAG1B Proteins 0.000 description 1
- 102000000905 Cadherin Human genes 0.000 description 1
- 108050007957 Cadherin Proteins 0.000 description 1
- 101100005789 Caenorhabditis elegans cdk-4 gene Proteins 0.000 description 1
- 101100476671 Caenorhabditis elegans sart-3 gene Proteins 0.000 description 1
- 101710147327 Calcineurin B homologous protein 1 Proteins 0.000 description 1
- 241000282836 Camelus dromedarius Species 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- 241000589876 Campylobacter Species 0.000 description 1
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 1
- 102100039510 Cancer/testis antigen 2 Human genes 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 241000253350 Capillaria Species 0.000 description 1
- 241000190890 Capnocytophaga Species 0.000 description 1
- 108090000565 Capsid Proteins Proteins 0.000 description 1
- 101710205625 Capsid protein p24 Proteins 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- 201000000274 Carcinosarcoma Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 102100026548 Caspase-8 Human genes 0.000 description 1
- 108090000538 Caspase-8 Proteins 0.000 description 1
- 102100026550 Caspase-9 Human genes 0.000 description 1
- 108090000566 Caspase-9 Proteins 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 102100023321 Ceruloplasmin Human genes 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 241000893172 Chabertia Species 0.000 description 1
- 241000606161 Chlamydia Species 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- MKQWTWSXVILIKJ-LXGUWJNJSA-N Chlorozotocin Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC(=O)N(N=O)CCCl MKQWTWSXVILIKJ-LXGUWJNJSA-N 0.000 description 1
- 108010049048 Cholera Toxin Proteins 0.000 description 1
- 102000009016 Cholera Toxin Human genes 0.000 description 1
- 108010023736 Chondroitinases and Chondroitin Lyases Proteins 0.000 description 1
- 102100032920 Chromobox protein homolog 2 Human genes 0.000 description 1
- 208000030939 Chronic inflammatory demyelinating polyneuropathy Diseases 0.000 description 1
- 102100038449 Claudin-6 Human genes 0.000 description 1
- 241000555825 Clupeidae Species 0.000 description 1
- 241000223203 Coccidioides Species 0.000 description 1
- 102100035167 Coiled-coil domain-containing protein 54 Human genes 0.000 description 1
- 208000011038 Cold agglutinin disease Diseases 0.000 description 1
- 206010009868 Cold type haemolytic anaemia Diseases 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 241001126268 Cooperia Species 0.000 description 1
- 241000711573 Coronaviridae Species 0.000 description 1
- 241000186216 Corynebacterium Species 0.000 description 1
- 241001445332 Coxiella <snail> Species 0.000 description 1
- 241000986238 Crenosoma Species 0.000 description 1
- 208000019707 Cryoglobulinemic vasculitis Diseases 0.000 description 1
- 241001527609 Cryptococcus Species 0.000 description 1
- 229930188224 Cryptophycin Natural products 0.000 description 1
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 1
- 108010009392 Cyclin-Dependent Kinase Inhibitor p16 Proteins 0.000 description 1
- 102100024462 Cyclin-dependent kinase 4 inhibitor B Human genes 0.000 description 1
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 1
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 102000012466 Cytochrome P450 1B1 Human genes 0.000 description 1
- 108050002014 Cytochrome P450 1B1 Proteins 0.000 description 1
- 102100039868 Cytoplasmic aconitate hydratase Human genes 0.000 description 1
- 102000000311 Cytosine Deaminase Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 108010008286 DNA nucleotidylexotransferase Proteins 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 1
- 101710096438 DNA-binding protein Proteins 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 108020005199 Dehydrogenases Proteins 0.000 description 1
- 102100040606 Dermatan-sulfate epimerase Human genes 0.000 description 1
- 101710127030 Dermatan-sulfate epimerase Proteins 0.000 description 1
- 241000187831 Dermatophilus Species 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 241001147667 Dictyocaulus Species 0.000 description 1
- 101100216227 Dictyostelium discoideum anapc3 gene Proteins 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- VYZAHLCBVHPDDF-UHFFFAOYSA-N Dinitrochlorobenzene Chemical compound [O-][N+](=O)C1=CC=C(Cl)C([N+]([O-])=O)=C1 VYZAHLCBVHPDDF-UHFFFAOYSA-N 0.000 description 1
- 241000690784 Dioctophyme Species 0.000 description 1
- 102100020743 Dipeptidase 1 Human genes 0.000 description 1
- 108090000204 Dipeptidase 1 Proteins 0.000 description 1
- 241000189163 Dipetalonema Species 0.000 description 1
- 241001137876 Diphyllobothrium Species 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 102100037070 Doublecortin domain-containing protein 2 Human genes 0.000 description 1
- 101100095895 Drosophila melanogaster sle gene Proteins 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 229930193152 Dynemicin Natural products 0.000 description 1
- 108050002772 E3 ubiquitin-protein ligase Mdm2 Proteins 0.000 description 1
- 102000012199 E3 ubiquitin-protein ligase Mdm2 Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 101150049307 EEF1A2 gene Proteins 0.000 description 1
- 241001115402 Ebolavirus Species 0.000 description 1
- 241000223924 Eimeria Species 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- AFMYMMXSQGUCBK-UHFFFAOYSA-N Endynamicin A Natural products C1#CC=CC#CC2NC(C=3C(=O)C4=C(O)C=CC(O)=C4C(=O)C=3C(O)=C3)=C3C34OC32C(C)C(C(O)=O)=C(OC)C41 AFMYMMXSQGUCBK-UHFFFAOYSA-N 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- 241000224431 Entamoeba Species 0.000 description 1
- 241000498256 Enterobius Species 0.000 description 1
- 241000194033 Enterococcus Species 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- 241001480035 Epidermophyton Species 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 1
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 1
- OBMLHUPNRURLOK-XGRAFVIBSA-N Epitiostanol Chemical compound C1[C@@H]2S[C@@H]2C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 OBMLHUPNRURLOK-XGRAFVIBSA-N 0.000 description 1
- 101710122228 Epstein-Barr nuclear antigen 2 Proteins 0.000 description 1
- 101710122231 Epstein-Barr nuclear antigen 3 Proteins 0.000 description 1
- 101710122229 Epstein-Barr nuclear antigen 6 Proteins 0.000 description 1
- 229930189413 Esperamicin Natural products 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 241000223682 Exophiala Species 0.000 description 1
- 101710104359 F protein Proteins 0.000 description 1
- 208000002476 Falciparum Malaria Diseases 0.000 description 1
- 102100028043 Fibroblast growth factor 3 Human genes 0.000 description 1
- 108090000382 Fibroblast growth factor 6 Proteins 0.000 description 1
- 102100028075 Fibroblast growth factor 6 Human genes 0.000 description 1
- 208000001640 Fibromyalgia Diseases 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 241000986243 Filaroides Species 0.000 description 1
- 108010000916 Fimbriae Proteins Proteins 0.000 description 1
- 241000710831 Flavivirus Species 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 241000710198 Foot-and-mouth disease virus Species 0.000 description 1
- 241000589601 Francisella Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 241000605909 Fusobacterium Species 0.000 description 1
- 102100039717 G antigen 1 Human genes 0.000 description 1
- 101150106478 GPS1 gene Proteins 0.000 description 1
- 101001077417 Gallus gallus Potassium voltage-gated channel subfamily H member 6 Proteins 0.000 description 1
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 1
- 208000031852 Gastrointestinal stromal cancer Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 241000159512 Geotrichum Species 0.000 description 1
- 208000007569 Giant Cell Tumors Diseases 0.000 description 1
- 241000224466 Giardia Species 0.000 description 1
- 208000000779 Gingival Neoplasms Diseases 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 206010018372 Glomerulonephritis membranous Diseases 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 1
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 1
- 102000006771 Gonadotropins Human genes 0.000 description 1
- 108010086677 Gonadotropins Proteins 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 1
- 102000001398 Granzyme Human genes 0.000 description 1
- 108060005986 Granzyme Proteins 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 1
- UYTPUPDQBNUYGX-UHFFFAOYSA-N Guanine Natural products O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 102100028976 HLA class I histocompatibility antigen, B alpha chain Human genes 0.000 description 1
- 102100028971 HLA class I histocompatibility antigen, C alpha chain Human genes 0.000 description 1
- 108010058607 HLA-B Antigens Proteins 0.000 description 1
- 108010052199 HLA-C Antigens Proteins 0.000 description 1
- 102000015789 HLA-DP Antigens Human genes 0.000 description 1
- 108010010378 HLA-DP Antigens Proteins 0.000 description 1
- 108010062347 HLA-DQ Antigens Proteins 0.000 description 1
- 102000006354 HLA-DR Antigens Human genes 0.000 description 1
- 108010058597 HLA-DR Antigens Proteins 0.000 description 1
- 241000243976 Haemonchus Species 0.000 description 1
- 241000606790 Haemophilus Species 0.000 description 1
- 241000606768 Haemophilus influenzae Species 0.000 description 1
- 241000406101 Hammondia Species 0.000 description 1
- 208000001204 Hashimoto Disease Diseases 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 241000589989 Helicobacter Species 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 102100029360 Hematopoietic cell signal transducer Human genes 0.000 description 1
- 108700008783 Hepatitis C virus E1 Proteins 0.000 description 1
- 108010073141 Hepatitis C virus glycoprotein E2 Proteins 0.000 description 1
- 241001278020 Hepatozoon Species 0.000 description 1
- 101001023784 Heteractis crispa GFP-like non-fluorescent chromoprotein Proteins 0.000 description 1
- 101710121996 Hexon protein p72 Proteins 0.000 description 1
- 102100035108 High affinity nerve growth factor receptor Human genes 0.000 description 1
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 1
- 102100032742 Histone-lysine N-methyltransferase SETD2 Human genes 0.000 description 1
- 108010033040 Histones Proteins 0.000 description 1
- 241000228402 Histoplasma Species 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 102100028092 Homeobox protein Nkx-3.1 Human genes 0.000 description 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 1
- 101000779641 Homo sapiens ALK tyrosine kinase receptor Proteins 0.000 description 1
- 101000678435 Homo sapiens Actin-like protein 8 Proteins 0.000 description 1
- 101001042227 Homo sapiens Acyl-CoA:lysophosphatidylglycerol acyltransferase 1 Proteins 0.000 description 1
- 101000753291 Homo sapiens Angiopoietin-1 receptor Proteins 0.000 description 1
- 101000678879 Homo sapiens Atypical chemokine receptor 1 Proteins 0.000 description 1
- 101000874316 Homo sapiens B melanoma antigen 1 Proteins 0.000 description 1
- 101000936083 Homo sapiens Baculoviral IAP repeat-containing protein 7 Proteins 0.000 description 1
- 101000749322 Homo sapiens C-type lectin domain family 6 member A Proteins 0.000 description 1
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 1
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 description 1
- 101000889345 Homo sapiens Cancer/testis antigen 2 Proteins 0.000 description 1
- 101000797586 Homo sapiens Chromobox protein homolog 2 Proteins 0.000 description 1
- 101000882898 Homo sapiens Claudin-6 Proteins 0.000 description 1
- 101000737052 Homo sapiens Coiled-coil domain-containing protein 54 Proteins 0.000 description 1
- 101000980919 Homo sapiens Cyclin-dependent kinase 4 inhibitor B Proteins 0.000 description 1
- 101000954709 Homo sapiens Doublecortin domain-containing protein 2 Proteins 0.000 description 1
- 101000886137 Homo sapiens G antigen 1 Proteins 0.000 description 1
- 101000990188 Homo sapiens Hematopoietic cell signal transducer Proteins 0.000 description 1
- 101000985516 Homo sapiens Hermansky-Pudlak syndrome 5 protein Proteins 0.000 description 1
- 101000654725 Homo sapiens Histone-lysine N-methyltransferase SETD2 Proteins 0.000 description 1
- 101000578249 Homo sapiens Homeobox protein Nkx-3.1 Proteins 0.000 description 1
- 101000614481 Homo sapiens Kidney-associated antigen 1 Proteins 0.000 description 1
- 101001034314 Homo sapiens Lactadherin Proteins 0.000 description 1
- 101000941892 Homo sapiens Leucine-rich repeat and calponin homology domain-containing protein 4 Proteins 0.000 description 1
- 101000941871 Homo sapiens Leucine-rich repeat neuronal protein 1 Proteins 0.000 description 1
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 1
- 101001051093 Homo sapiens Low-density lipoprotein receptor Proteins 0.000 description 1
- 101001134060 Homo sapiens Melanocyte-stimulating hormone receptor Proteins 0.000 description 1
- 101001005728 Homo sapiens Melanoma-associated antigen 1 Proteins 0.000 description 1
- 101001005720 Homo sapiens Melanoma-associated antigen 4 Proteins 0.000 description 1
- 101001057131 Homo sapiens Melanoma-associated antigen D4 Proteins 0.000 description 1
- 101000628547 Homo sapiens Metalloreductase STEAP1 Proteins 0.000 description 1
- 101000615488 Homo sapiens Methyl-CpG-binding domain protein 2 Proteins 0.000 description 1
- 101000957259 Homo sapiens Mitotic spindle assembly checkpoint protein MAD2A Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101001133081 Homo sapiens Mucin-2 Proteins 0.000 description 1
- 101000721712 Homo sapiens NTF2-related export protein 1 Proteins 0.000 description 1
- 101000687346 Homo sapiens PR domain zinc finger protein 2 Proteins 0.000 description 1
- 101000613490 Homo sapiens Paired box protein Pax-3 Proteins 0.000 description 1
- 101000601724 Homo sapiens Paired box protein Pax-5 Proteins 0.000 description 1
- 101000691463 Homo sapiens Placenta-specific protein 1 Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101000874141 Homo sapiens Probable ATP-dependent RNA helicase DDX43 Proteins 0.000 description 1
- 101001136592 Homo sapiens Prostate stem cell antigen Proteins 0.000 description 1
- 101001136981 Homo sapiens Proteasome subunit beta type-9 Proteins 0.000 description 1
- 101000880770 Homo sapiens Protein SSX2 Proteins 0.000 description 1
- 101000679365 Homo sapiens Putative tyrosine-protein phosphatase TPTE Proteins 0.000 description 1
- 101001109419 Homo sapiens RNA-binding protein NOB1 Proteins 0.000 description 1
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 101000857677 Homo sapiens Runt-related transcription factor 1 Proteins 0.000 description 1
- 101000706563 Homo sapiens SUN domain-containing protein 3 Proteins 0.000 description 1
- 101000665137 Homo sapiens Scm-like with four MBT domains protein 1 Proteins 0.000 description 1
- 101001041393 Homo sapiens Serine protease HTRA1 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000825253 Homo sapiens Sperm protein associated with the nucleus on the X chromosome A Proteins 0.000 description 1
- 101000824971 Homo sapiens Sperm surface protein Sp17 Proteins 0.000 description 1
- 101000648075 Homo sapiens Trafficking protein particle complex subunit 1 Proteins 0.000 description 1
- 101000813738 Homo sapiens Transcription factor ETV6 Proteins 0.000 description 1
- 101001010792 Homo sapiens Transcriptional regulator ERG Proteins 0.000 description 1
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 1
- 101000638154 Homo sapiens Transmembrane protease serine 2 Proteins 0.000 description 1
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 description 1
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 1
- 101000814512 Homo sapiens X antigen family member 1 Proteins 0.000 description 1
- 101710146024 Horcolin Proteins 0.000 description 1
- 101800000120 Host translation inhibitor nsp1 Proteins 0.000 description 1
- 108010048209 Human Immunodeficiency Virus Proteins Proteins 0.000 description 1
- 101100048372 Human cytomegalovirus (strain AD169) H301 gene Proteins 0.000 description 1
- 101100048373 Human cytomegalovirus (strain Merlin) UL18 gene Proteins 0.000 description 1
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- 241000829111 Human polyomavirus 1 Species 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- 241000257303 Hymenoptera Species 0.000 description 1
- 206010020983 Hypogammaglobulinaemia Diseases 0.000 description 1
- MPBVHIBUJCELCL-UHFFFAOYSA-N Ibandronate Chemical compound CCCCCN(C)CCC(O)(P(O)(O)=O)P(O)(O)=O MPBVHIBUJCELCL-UHFFFAOYSA-N 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 101100028758 Influenza A virus (strain A/Swine/Wisconsin/1/1967 H1N1) PB1-F2 gene Proteins 0.000 description 1
- 108050002021 Integrator complex subunit 2 Proteins 0.000 description 1
- 102100022297 Integrin alpha-X Human genes 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000018682 Interleukin Receptor Common gamma Subunit Human genes 0.000 description 1
- 108010066719 Interleukin Receptor Common gamma Subunit Proteins 0.000 description 1
- 102000003814 Interleukin-10 Human genes 0.000 description 1
- 108090000174 Interleukin-10 Proteins 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 102000000704 Interleukin-7 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 241000567229 Isospora Species 0.000 description 1
- 241000701460 JC polyomavirus Species 0.000 description 1
- 206010023126 Jaundice Diseases 0.000 description 1
- 208000003456 Juvenile Arthritis Diseases 0.000 description 1
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 102100039648 Lactadherin Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000776461 Lagochilascaris Species 0.000 description 1
- 108010000851 Laminin Receptors Proteins 0.000 description 1
- 102000002297 Laminin Receptors Human genes 0.000 description 1
- 101800000517 Leader protein Proteins 0.000 description 1
- 101710189395 Lectin Proteins 0.000 description 1
- 241000222722 Leishmania <genus> Species 0.000 description 1
- 229920001491 Lentinan Polymers 0.000 description 1
- 241001503405 Leptosira Species 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 102100032655 Leucine-rich repeat neuronal protein 1 Human genes 0.000 description 1
- 206010024305 Leukaemia monocytic Diseases 0.000 description 1
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 1
- 241000186781 Listeria Species 0.000 description 1
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000005446 Lupus vulgaris Diseases 0.000 description 1
- 208000016604 Lyme disease Diseases 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- 206010025312 Lymphoma AIDS related Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 230000037364 MAPK/ERK pathway Effects 0.000 description 1
- 108010010995 MART-1 Antigen Proteins 0.000 description 1
- 101150047390 MCP gene Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108700012912 MYCN Proteins 0.000 description 1
- 101150022024 MYCN gene Proteins 0.000 description 1
- 241001444195 Madurella Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 101710179758 Mannose-specific lectin Proteins 0.000 description 1
- 101710150763 Mannose-specific lectin 1 Proteins 0.000 description 1
- 101710150745 Mannose-specific lectin 2 Proteins 0.000 description 1
- 241000142892 Mansonella Species 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 241001115401 Marburgvirus Species 0.000 description 1
- 101710085938 Matrix protein Proteins 0.000 description 1
- 102100025169 Max-binding protein MNT Human genes 0.000 description 1
- 241000590503 Melanitis Species 0.000 description 1
- 102100034216 Melanocyte-stimulating hormone receptor Human genes 0.000 description 1
- 108010071463 Melanoma-Specific Antigens Proteins 0.000 description 1
- 102000007557 Melanoma-Specific Antigens Human genes 0.000 description 1
- 102000000440 Melanoma-associated antigen Human genes 0.000 description 1
- 108050008953 Melanoma-associated antigen Proteins 0.000 description 1
- 102100025050 Melanoma-associated antigen 1 Human genes 0.000 description 1
- 102100025077 Melanoma-associated antigen 4 Human genes 0.000 description 1
- 101710127721 Membrane protein Proteins 0.000 description 1
- 208000027530 Meniere disease Diseases 0.000 description 1
- IVDYZAAPOLNZKG-KWHRADDSSA-N Mepitiostane Chemical compound O([C@@H]1[C@]2(CC[C@@H]3[C@@]4(C)C[C@H]5S[C@H]5C[C@@H]4CC[C@H]3[C@@H]2CC1)C)C1(OC)CCCC1 IVDYZAAPOLNZKG-KWHRADDSSA-N 0.000 description 1
- SGDBTWWWUNNDEQ-UHFFFAOYSA-N Merphalan Chemical compound OC(=O)C(N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-UHFFFAOYSA-N 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 102100026712 Metalloreductase STEAP1 Human genes 0.000 description 1
- 206010054949 Metaplasia Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 102100021299 Methyl-CpG-binding domain protein 2 Human genes 0.000 description 1
- 241001480037 Microsporum Species 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- 101710151805 Mitochondrial intermediate peptidase 1 Proteins 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 102100038792 Mitotic spindle assembly checkpoint protein MAD2A Human genes 0.000 description 1
- 208000003250 Mixed connective tissue disease Diseases 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 241000908267 Moniliella Species 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 241000235575 Mortierella Species 0.000 description 1
- 102100034263 Mucin-2 Human genes 0.000 description 1
- 241000235395 Mucor Species 0.000 description 1
- 241000986227 Muellerius Species 0.000 description 1
- 241000714177 Murine leukemia virus Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 1
- 101100219625 Mus musculus Casd1 gene Proteins 0.000 description 1
- 101001062862 Mus musculus Fatty acid-binding protein, adipocyte Proteins 0.000 description 1
- 101100346932 Mus musculus Muc1 gene Proteins 0.000 description 1
- 101100369076 Mus musculus Tdgf1 gene Proteins 0.000 description 1
- 208000007101 Muscle Cramp Diseases 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 201000004458 Myoma Diseases 0.000 description 1
- 102000003505 Myosin Human genes 0.000 description 1
- 108060008487 Myosin Proteins 0.000 description 1
- 108700026495 N-Myc Proto-Oncogene Proteins 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- 102100031688 N-acetylgalactosamine-6-sulfatase Human genes 0.000 description 1
- 102100030124 N-myc proto-oncogene protein Human genes 0.000 description 1
- 230000006051 NK cell activation Effects 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 241000498271 Necator Species 0.000 description 1
- 241000588653 Neisseria Species 0.000 description 1
- 241001137882 Nematodirus Species 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 241001147660 Neospora Species 0.000 description 1
- 241000232901 Nephroma Species 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 101100385413 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) csm-3 gene Proteins 0.000 description 1
- 241000187654 Nocardia Species 0.000 description 1
- 206010029488 Nodular melanoma Diseases 0.000 description 1
- 102220580210 Non-receptor tyrosine-protein kinase TYK2_D10A_mutation Human genes 0.000 description 1
- 101800000512 Non-structural protein 1 Proteins 0.000 description 1
- 108020004485 Nonsense Codon Proteins 0.000 description 1
- 241001126829 Nosema Species 0.000 description 1
- 108010051791 Nuclear Antigens Proteins 0.000 description 1
- 102000019040 Nuclear Antigens Human genes 0.000 description 1
- 102000007999 Nuclear Proteins Human genes 0.000 description 1
- 108010089610 Nuclear Proteins Proteins 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- 108090001074 Nucleocapsid Proteins Proteins 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 206010058461 Orchitis noninfective Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 108700006640 OspA Proteins 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 241000243795 Ostertagia Species 0.000 description 1
- 101710116435 Outer membrane protein Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 101150103639 PB1 gene Proteins 0.000 description 1
- 102100024885 PR domain zinc finger protein 2 Human genes 0.000 description 1
- 102100034640 PWWP domain-containing DNA repair factor 3A Human genes 0.000 description 1
- 108050007154 PWWP domain-containing DNA repair factor 3A Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000027868 Paget disease Diseases 0.000 description 1
- 102100040891 Paired box protein Pax-3 Human genes 0.000 description 1
- 102100037504 Paired box protein Pax-5 Human genes 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 241001631646 Papillomaviridae Species 0.000 description 1
- 206010061332 Paraganglion neoplasm Diseases 0.000 description 1
- 241001480233 Paragonimus Species 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 241000606860 Pasteurella Species 0.000 description 1
- 101000621505 Peanut clump virus (isolate 87/TGTA2) Suppressor of RNA silencing Proteins 0.000 description 1
- 108010087702 Penicillinase Proteins 0.000 description 1
- 241000228143 Penicillium Species 0.000 description 1
- 102000002508 Peptide Elongation Factors Human genes 0.000 description 1
- 108010068204 Peptide Elongation Factors Proteins 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 241000710778 Pestivirus Species 0.000 description 1
- 241000222831 Phialophora <Chaetothyriales> Species 0.000 description 1
- 101710177166 Phosphoprotein Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 241001277123 Physaloptera Species 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 102100026181 Placenta-specific protein 1 Human genes 0.000 description 1
- 102100022427 Plasmalemma vesicle-associated protein Human genes 0.000 description 1
- 101710193105 Plasmalemma vesicle-associated protein Proteins 0.000 description 1
- 241000224016 Plasmodium Species 0.000 description 1
- 241000223960 Plasmodium falciparum Species 0.000 description 1
- 206010035500 Plasmodium falciparum infection Diseases 0.000 description 1
- 201000011336 Plasmodium falciparum malaria Diseases 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 101710183389 Pneumolysin Proteins 0.000 description 1
- 208000000474 Poliomyelitis Diseases 0.000 description 1
- 206010065159 Polychondritis Diseases 0.000 description 1
- 241001505332 Polyomavirus sp. Species 0.000 description 1
- 208000037062 Polyps Diseases 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102100035724 Probable ATP-dependent RNA helicase DDX43 Human genes 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 102100036735 Prostate stem cell antigen Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102100035764 Proteasome subunit beta type-9 Human genes 0.000 description 1
- 102100037686 Protein SSX2 Human genes 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 241000588769 Proteus <enterobacteria> Species 0.000 description 1
- 241000196250 Prototheca Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 1
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 1
- 101800001295 Putative ATP-dependent helicase Proteins 0.000 description 1
- 101800001006 Putative helicase Proteins 0.000 description 1
- 102100022578 Putative tyrosine-protein phosphatase TPTE Human genes 0.000 description 1
- 101150093191 RIR1 gene Proteins 0.000 description 1
- 230000007022 RNA scission Effects 0.000 description 1
- 230000006819 RNA synthesis Effects 0.000 description 1
- 102100022491 RNA-binding protein NOB1 Human genes 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 238000003559 RNA-seq method Methods 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 101100047461 Rattus norvegicus Trpm8 gene Proteins 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 208000033464 Reiter syndrome Diseases 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 241000235527 Rhizopus Species 0.000 description 1
- 108090000221 Ribosomal protein S6 Proteins 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 241000702670 Rotavirus Species 0.000 description 1
- 102100025373 Runt-related transcription factor 1 Human genes 0.000 description 1
- 101150103019 SCP gene Proteins 0.000 description 1
- 108050003189 SH2B adapter protein 1 Proteins 0.000 description 1
- 102100031129 SUN domain-containing protein 3 Human genes 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000224003 Sarcocystis Species 0.000 description 1
- 241000242678 Schistosoma Species 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 102100038689 Scm-like with four MBT domains protein 1 Human genes 0.000 description 1
- 102000003800 Selectins Human genes 0.000 description 1
- 108090000184 Selectins Proteins 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102100021119 Serine protease HTRA1 Human genes 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108700025832 Serum Response Element Proteins 0.000 description 1
- 235000005775 Setaria Nutrition 0.000 description 1
- 241000232088 Setaria <nematode> Species 0.000 description 1
- 241000607768 Shigella Species 0.000 description 1
- 101710173693 Short transient receptor potential channel 1 Proteins 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- 101710149279 Small delta antigen Proteins 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 102100022327 Sperm protein associated with the nucleus on the X chromosome A Human genes 0.000 description 1
- 241000922629 Spirocerca Species 0.000 description 1
- 241000713880 Spleen focus-forming virus Species 0.000 description 1
- 101710192036 Sporozoite surface protein 2 Proteins 0.000 description 1
- 102100035748 Squamous cell carcinoma antigen recognized by T-cells 3 Human genes 0.000 description 1
- 101710185775 Squamous cell carcinoma antigen recognized by T-cells 3 Proteins 0.000 description 1
- 241000371621 Stemphylium Species 0.000 description 1
- 206010072148 Stiff-Person syndrome Diseases 0.000 description 1
- 241000194017 Streptococcus Species 0.000 description 1
- 241000193985 Streptococcus agalactiae Species 0.000 description 1
- 241000193990 Streptococcus sp. 'group B' Species 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 101800001271 Surface protein Proteins 0.000 description 1
- 108010002687 Survivin Proteins 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 230000024932 T cell mediated immunity Effects 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- BXFOFFBJRFZBQZ-QYWOHJEZSA-N T-2 toxin Chemical compound C([C@@]12[C@]3(C)[C@H](OC(C)=O)[C@@H](O)[C@H]1O[C@H]1[C@]3(COC(C)=O)C[C@@H](C(=C1)C)OC(=O)CC(C)C)O2 BXFOFFBJRFZBQZ-QYWOHJEZSA-N 0.000 description 1
- 108700012457 TACSTD2 Proteins 0.000 description 1
- 102000046283 TNF-Related Apoptosis-Inducing Ligand Human genes 0.000 description 1
- 101150102071 TRX1 gene Proteins 0.000 description 1
- 101150037769 TRX2 gene Proteins 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- 108010017842 Telomerase Proteins 0.000 description 1
- 241001477954 Thelazia Species 0.000 description 1
- 241001648840 Thosea asigna virus Species 0.000 description 1
- 108010046722 Thrombospondin 1 Proteins 0.000 description 1
- 102100036034 Thrombospondin-1 Human genes 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 241000244031 Toxocara Species 0.000 description 1
- 241000223996 Toxoplasma Species 0.000 description 1
- 102100025256 Trafficking protein particle complex subunit 1 Human genes 0.000 description 1
- 241000283907 Tragelaphus oryx Species 0.000 description 1
- 108700009124 Transcription Initiation Site Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102100039580 Transcription factor ETV6 Human genes 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 102000004357 Transferases Human genes 0.000 description 1
- 108090000992 Transferases Proteins 0.000 description 1
- 102000003932 Transgelin Human genes 0.000 description 1
- 102100031013 Transgelin Human genes 0.000 description 1
- 108090000333 Transgelin Proteins 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 102100023935 Transmembrane glycoprotein NMB Human genes 0.000 description 1
- 102100031989 Transmembrane protease serine 2 Human genes 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 241000589886 Treponema Species 0.000 description 1
- 241000243774 Trichinella Species 0.000 description 1
- 241000223238 Trichophyton Species 0.000 description 1
- 241000223230 Trichosporon Species 0.000 description 1
- 241000243797 Trichostrongylus Species 0.000 description 1
- 241001489151 Trichuris Species 0.000 description 1
- UMILHIMHKXVDGH-UHFFFAOYSA-N Triethylene glycol diglycidyl ether Chemical compound C1OC1COCCOCCOCCOCC1CO1 UMILHIMHKXVDGH-UHFFFAOYSA-N 0.000 description 1
- FYAMXEPQQLNQDM-UHFFFAOYSA-N Tris(1-aziridinyl)phosphine oxide Chemical compound C1CN1P(N1CC1)(=O)N1CC1 FYAMXEPQQLNQDM-UHFFFAOYSA-N 0.000 description 1
- LVTKHGUGBGNBPL-UHFFFAOYSA-N Trp-P-1 Chemical compound N1C2=CC=CC=C2C2=C1C(C)=C(N)N=C2C LVTKHGUGBGNBPL-UHFFFAOYSA-N 0.000 description 1
- 102100032101 Tumor necrosis factor ligand superfamily member 9 Human genes 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 description 1
- 102100033254 Tumor suppressor ARF Human genes 0.000 description 1
- 102100027212 Tumor-associated calcium signal transducer 2 Human genes 0.000 description 1
- 208000035896 Twin-reversed arterial perfusion sequence Diseases 0.000 description 1
- 102100027244 U4/U6.U5 tri-snRNP-associated protein 1 Human genes 0.000 description 1
- 101710155955 U4/U6.U5 tri-snRNP-associated protein 1 Proteins 0.000 description 1
- 101150109748 UL19 gene Proteins 0.000 description 1
- 101150003230 UL27 gene Proteins 0.000 description 1
- 101150023000 UL35 gene Proteins 0.000 description 1
- 101150090946 UL38 gene Proteins 0.000 description 1
- 101150048066 UL45 gene Proteins 0.000 description 1
- 101150033660 UL6 gene Proteins 0.000 description 1
- IVOMOUWHDPKRLL-UHFFFAOYSA-N UNPD107823 Natural products O1C2COP(O)(=O)OC2C(O)C1N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-UHFFFAOYSA-N 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 101710145727 Viral Fc-gamma receptor-like protein UL119 Proteins 0.000 description 1
- 108010087302 Viral Structural Proteins Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 208000019297 Well-differentiated fetal adenocarcinoma of the lung Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 241000244002 Wuchereria Species 0.000 description 1
- 102100039490 X antigen family member 1 Human genes 0.000 description 1
- 241000223673 Xylohypha Species 0.000 description 1
- 241000607734 Yersinia <bacteria> Species 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- PTFCDOFLOPIGGS-UHFFFAOYSA-N Zinc dication Chemical compound [Zn+2] PTFCDOFLOPIGGS-UHFFFAOYSA-N 0.000 description 1
- PVNFMCBFDPTNQI-UIBOPQHZSA-N [(1S,2R,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl] acetate [(1S,2R,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl] 3-methylbutanoate [(1S,2R,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl] 2-methylpropanoate [(1S,2R,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl] propanoate Chemical compound CO[C@@H]1\C=C\C=C(C)\Cc2cc(OC)c(Cl)c(c2)N(C)C(=O)C[C@H](OC(C)=O)[C@]2(C)OC2[C@H](C)[C@@H]2C[C@@]1(O)NC(=O)O2.CCC(=O)O[C@H]1CC(=O)N(C)c2cc(C\C(C)=C\C=C\[C@@H](OC)[C@@]3(O)C[C@H](OC(=O)N3)[C@@H](C)C3O[C@@]13C)cc(OC)c2Cl.CO[C@@H]1\C=C\C=C(C)\Cc2cc(OC)c(Cl)c(c2)N(C)C(=O)C[C@H](OC(=O)C(C)C)[C@]2(C)OC2[C@H](C)[C@@H]2C[C@@]1(O)NC(=O)O2.CO[C@@H]1\C=C\C=C(C)\Cc2cc(OC)c(Cl)c(c2)N(C)C(=O)C[C@H](OC(=O)CC(C)C)[C@]2(C)OC2[C@H](C)[C@@H]2C[C@@]1(O)NC(=O)O2 PVNFMCBFDPTNQI-UIBOPQHZSA-N 0.000 description 1
- XZSRRNFBEIOBDA-CFNBKWCHSA-N [2-[(2s,4s)-4-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-3,4-dihydro-1h-tetracen-2-yl]-2-oxoethyl] 2,2-diethoxyacetate Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)C(OCC)OCC)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 XZSRRNFBEIOBDA-CFNBKWCHSA-N 0.000 description 1
- 238000002679 ablation Methods 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- QUHYUSAHBDACNG-UHFFFAOYSA-N acerogenin 3 Natural products C1=CC(O)=CC=C1CCCCC(=O)CCC1=CC=C(O)C=C1 QUHYUSAHBDACNG-UHFFFAOYSA-N 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 229930183665 actinomycin Natural products 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000017488 activation-induced cell death of T cell Effects 0.000 description 1
- 230000003044 adaptive effect Effects 0.000 description 1
- 238000007792 addition Methods 0.000 description 1
- 230000002730 additional effect Effects 0.000 description 1
- 238000009098 adjuvant therapy Methods 0.000 description 1
- 201000002454 adrenal cortex cancer Diseases 0.000 description 1
- 230000001919 adrenal effect Effects 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 239000000910 agglutinin Substances 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 239000013566 allergen Substances 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 108010034034 alpha-1,6-mannosylglycoprotein beta 1,6-N-acetylglucosaminyltransferase Proteins 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229960002749 aminolevulinic acid Drugs 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- BBDAGFIXKZCXAH-CCXZUQQUSA-N ancitabine Chemical compound N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 BBDAGFIXKZCXAH-CCXZUQQUSA-N 0.000 description 1
- 229950000242 ancitabine Drugs 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 108010080146 androgen receptors Proteins 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 230000003474 anti-emetic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000001775 anti-pathogenic effect Effects 0.000 description 1
- 230000002421 anti-septic effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 229940045687 antimetabolites folic acid analogs Drugs 0.000 description 1
- 239000002257 antimetastatic agent Substances 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 238000002617 apheresis Methods 0.000 description 1
- 201000007436 apocrine adenocarcinoma Diseases 0.000 description 1
- 239000000158 apoptosis inhibitor Substances 0.000 description 1
- 150000008209 arabinosides Chemical class 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- 210000004507 artificial chromosome Anatomy 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 108010055066 asparaginylendopeptidase Proteins 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N aspartic acid group Chemical class N[C@@H](CC(=O)O)C(=O)O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 description 1
- 208000036923 autoimmune primary adrenal insufficiency Diseases 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 150000001541 aziridines Chemical class 0.000 description 1
- 201000008680 babesiosis Diseases 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 230000008236 biological pathway Effects 0.000 description 1
- 230000029918 bioluminescence Effects 0.000 description 1
- 238000005415 bioluminescence Methods 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- QKSKPIVNLNLAAV-UHFFFAOYSA-N bis(2-chloroethyl) sulfide Chemical compound ClCCSCCCl QKSKPIVNLNLAAV-UHFFFAOYSA-N 0.000 description 1
- HUTDDBSSHVOYJR-UHFFFAOYSA-H bis[(2-oxo-1,3,2$l^{5},4$l^{2}-dioxaphosphaplumbetan-2-yl)oxy]lead Chemical compound [Pb+2].[Pb+2].[Pb+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O HUTDDBSSHVOYJR-UHFFFAOYSA-H 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 201000000053 blastoma Diseases 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N butyl alcohol Substances CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108700002839 cactinomycin Proteins 0.000 description 1
- 229950009908 cactinomycin Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- XREUEWVEMYWFFA-CSKJXFQVSA-N carminomycin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XREUEWVEMYWFFA-CSKJXFQVSA-N 0.000 description 1
- 229930188550 carminomycin Natural products 0.000 description 1
- XREUEWVEMYWFFA-UHFFFAOYSA-N carminomycin I Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XREUEWVEMYWFFA-UHFFFAOYSA-N 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 229950001725 carubicin Drugs 0.000 description 1
- 101150055766 cat gene Proteins 0.000 description 1
- 210000004534 cecum Anatomy 0.000 description 1
- CZTQZXZIADLWOZ-CRAIPNDOSA-N cefaloridine Chemical compound O=C([C@@H](NC(=O)CC=1SC=CC=1)[C@H]1SC2)N1C(C(=O)[O-])=C2C[N+]1=CC=CC=C1 CZTQZXZIADLWOZ-CRAIPNDOSA-N 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 230000010307 cell transformation Effects 0.000 description 1
- 230000007248 cellular mechanism Effects 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000012627 chemopreventive agent Substances 0.000 description 1
- 229940124443 chemopreventive agent Drugs 0.000 description 1
- 239000012829 chemotherapy agent Substances 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 210000003763 chloroplast Anatomy 0.000 description 1
- 229960001480 chlorozotocin Drugs 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 201000005795 chronic inflammatory demyelinating polyneuritis Diseases 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 1
- ACSIXWWBWUQEHA-UHFFFAOYSA-N clodronic acid Chemical compound OP(O)(=O)C(Cl)(Cl)P(O)(O)=O ACSIXWWBWUQEHA-UHFFFAOYSA-N 0.000 description 1
- 229960002286 clodronic acid Drugs 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 230000000112 colonic effect Effects 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 101150055601 cops2 gene Proteins 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 201000003278 cryoglobulinemia Diseases 0.000 description 1
- 238000005138 cryopreservation Methods 0.000 description 1
- 238000002681 cryosurgery Methods 0.000 description 1
- 108010006226 cryptophycin Proteins 0.000 description 1
- 108010089438 cryptophycin 1 Proteins 0.000 description 1
- 108010090203 cryptophycin 8 Proteins 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229940095074 cyclic amp Drugs 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical class OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 208000002445 cystadenocarcinoma Diseases 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 238000004163 cytometry Methods 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 230000000779 depleting effect Effects 0.000 description 1
- URLVCROWVOSNPT-QTTMQESMSA-N desacetyluvaricin Natural products O=C1C(CCCCCCCCCCCC[C@@H](O)[C@H]2O[C@@H]([C@@H]3O[C@@H]([C@@H](O)CCCCCCCCCC)CC3)CC2)=C[C@H](C)O1 URLVCROWVOSNPT-QTTMQESMSA-N 0.000 description 1
- 229950003913 detorubicin Drugs 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- WVYXNIXAMZOZFK-UHFFFAOYSA-N diaziquone Chemical compound O=C1C(NC(=O)OCC)=C(N2CC2)C(=O)C(NC(=O)OCC)=C1N1CC1 WVYXNIXAMZOZFK-UHFFFAOYSA-N 0.000 description 1
- 229950002389 diaziquone Drugs 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 230000000447 dimerizing effect Effects 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 230000009429 distress Effects 0.000 description 1
- 150000002019 disulfides Chemical class 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000034431 double-strand break repair via homologous recombination Effects 0.000 description 1
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- AFMYMMXSQGUCBK-AKMKHHNQSA-N dynemicin a Chemical compound C1#C\C=C/C#C[C@@H]2NC(C=3C(=O)C4=C(O)C=CC(O)=C4C(=O)C=3C(O)=C3)=C3[C@@]34O[C@]32[C@@H](C)C(C(O)=O)=C(OC)[C@H]41 AFMYMMXSQGUCBK-AKMKHHNQSA-N 0.000 description 1
- 244000078703 ectoparasite Species 0.000 description 1
- FSIRXIHZBIXHKT-MHTVFEQDSA-N edatrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CC(CC)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FSIRXIHZBIXHKT-MHTVFEQDSA-N 0.000 description 1
- 229950006700 edatrexate Drugs 0.000 description 1
- 229950000549 elliptinium acetate Drugs 0.000 description 1
- 201000008184 embryoma Diseases 0.000 description 1
- 210000001671 embryonic stem cell Anatomy 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 229940125532 enzyme inhibitor Drugs 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 230000008472 epithelial growth Effects 0.000 description 1
- 208000010932 epithelial neoplasm Diseases 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 229950002973 epitiostanol Drugs 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- LJQQFQHBKUKHIS-WJHRIEJJSA-N esperamicin Chemical compound O1CC(NC(C)C)C(OC)CC1OC1C(O)C(NOC2OC(C)C(SC)C(O)C2)C(C)OC1OC1C(\C2=C/CSSSC)=C(NC(=O)OC)C(=O)C(OC3OC(C)C(O)C(OC(=O)C=4C(=CC(OC)=C(OC)C=4)NC(=O)C(=C)OC)C3)C2(O)C#C\C=C/C#C1 LJQQFQHBKUKHIS-WJHRIEJJSA-N 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- QSRLNKCNOLVZIR-KRWDZBQOSA-N ethyl (2s)-2-[[2-[4-[bis(2-chloroethyl)amino]phenyl]acetyl]amino]-4-methylsulfanylbutanoate Chemical compound CCOC(=O)[C@H](CCSC)NC(=O)CC1=CC=C(N(CCCl)CCCl)C=C1 QSRLNKCNOLVZIR-KRWDZBQOSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 229960005237 etoglucid Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 238000004299 exfoliation Methods 0.000 description 1
- 230000006539 extracellular acidification Effects 0.000 description 1
- 230000008622 extracellular signaling Effects 0.000 description 1
- 230000001815 facial effect Effects 0.000 description 1
- 125000004030 farnesyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 1
- 229960004413 flucytosine Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 108010021843 fluorescent protein 583 Proteins 0.000 description 1
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 1
- 201000005206 focal segmental glomerulosclerosis Diseases 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 150000002224 folic acids Chemical class 0.000 description 1
- 201000003444 follicular lymphoma Diseases 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 125000002446 fucosyl group Chemical group C1([C@@H](O)[C@H](O)[C@H](O)[C@@H](O1)C)* 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 230000008717 functional decline Effects 0.000 description 1
- 101150029683 gB gene Proteins 0.000 description 1
- 229940044658 gallium nitrate Drugs 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 210000003976 gap junction Anatomy 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 208000015419 gastrin-producing neuroendocrine tumor Diseases 0.000 description 1
- 201000000052 gastrinoma Diseases 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 108091008053 gene clusters Proteins 0.000 description 1
- 230000004547 gene signature Effects 0.000 description 1
- 238000010363 gene targeting Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000023266 generation of precursor metabolites and energy Effects 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 150000002338 glycosides Chemical class 0.000 description 1
- 239000002622 gonadotropin Substances 0.000 description 1
- 201000007574 granular cell carcinoma Diseases 0.000 description 1
- 210000004565 granule cell Anatomy 0.000 description 1
- 208000035474 group of disease Diseases 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 210000002768 hair cell Anatomy 0.000 description 1
- 229910000856 hastalloy Inorganic materials 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 239000003481 heat shock protein 90 inhibitor Substances 0.000 description 1
- 244000000013 helminth Species 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- 208000029824 high grade glioma Diseases 0.000 description 1
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 1
- 230000003284 homeostatic effect Effects 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 102000057744 human CLEC6A Human genes 0.000 description 1
- 102000043321 human CTLA4 Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 229940044700 hylenex Drugs 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 229940015872 ibandronate Drugs 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 238000003318 immunodepletion Methods 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 238000011532 immunohistochemical staining Methods 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 230000006057 immunotolerant effect Effects 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 208000037797 influenza A Diseases 0.000 description 1
- 208000037798 influenza B Diseases 0.000 description 1
- 208000037799 influenza C Diseases 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 108091008042 inhibitory receptors Proteins 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000008611 intercellular interaction Effects 0.000 description 1
- 230000035990 intercellular signaling Effects 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 229940076144 interleukin-10 Drugs 0.000 description 1
- 108090000237 interleukin-24 Proteins 0.000 description 1
- 102000003898 interleukin-24 Human genes 0.000 description 1
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 206010073095 invasive ductal breast carcinoma Diseases 0.000 description 1
- 201000010985 invasive ductal carcinoma Diseases 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 210000000867 larynx Anatomy 0.000 description 1
- 238000002430 laser surgery Methods 0.000 description 1
- 229940115286 lentinan Drugs 0.000 description 1
- 239000012035 limiting reagent Substances 0.000 description 1
- 125000003473 lipid group Chemical group 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 201000003866 lung sarcoma Diseases 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 208000025036 lymphosarcoma Diseases 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 150000002672 m-cresols Chemical class 0.000 description 1
- 101710130522 mRNA export factor Proteins 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 230000036244 malformation Effects 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 208000025854 malignant tumor of adrenal cortex Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 208000027202 mammary Paget disease Diseases 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- MQXVYODZCMMZEM-ZYUZMQFOSA-N mannomustine Chemical compound ClCCNC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CNCCCl MQXVYODZCMMZEM-ZYUZMQFOSA-N 0.000 description 1
- 229950008612 mannomustine Drugs 0.000 description 1
- 238000012083 mass cytometry Methods 0.000 description 1
- 208000000516 mast-cell leukemia Diseases 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 1
- 231100000855 membranous nephropathy Toxicity 0.000 description 1
- 229950009246 mepitiostane Drugs 0.000 description 1
- 201000008806 mesenchymal cell neoplasm Diseases 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Chemical class 0.000 description 1
- 229910001092 metal group alloy Inorganic materials 0.000 description 1
- 230000015689 metaplastic ossification Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Natural products OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 238000012737 microarray-based gene expression Methods 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- ZAHQPTJLOCWVPG-UHFFFAOYSA-N mitoxantrone dihydrochloride Chemical compound Cl.Cl.O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO ZAHQPTJLOCWVPG-UHFFFAOYSA-N 0.000 description 1
- 201000006894 monocytic leukemia Diseases 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 208000010492 mucinous cystadenocarcinoma Diseases 0.000 description 1
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 1
- 235000010460 mustard Nutrition 0.000 description 1
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- 210000003098 myoblast Anatomy 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 230000032965 negative regulation of cell volume Effects 0.000 description 1
- 238000009099 neoadjuvant therapy Methods 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 229910052754 neon Inorganic materials 0.000 description 1
- GKAOGPIIYCISHV-UHFFFAOYSA-N neon atom Chemical compound [Ne] GKAOGPIIYCISHV-UHFFFAOYSA-N 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 208000025351 nephroma Diseases 0.000 description 1
- 238000003012 network analysis Methods 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 201000000032 nodular malignant melanoma Diseases 0.000 description 1
- 108091027963 non-coding RNA Proteins 0.000 description 1
- 102000042567 non-coding RNA Human genes 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000001668 nucleic acid synthesis Methods 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 230000036284 oxygen consumption Effects 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 238000011499 palliative surgery Methods 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 201000010210 papillary cystadenocarcinoma Diseases 0.000 description 1
- 208000024641 papillary serous cystadenocarcinoma Diseases 0.000 description 1
- 208000007312 paraganglioma Diseases 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 238000003068 pathway analysis Methods 0.000 description 1
- 108010089193 pattern recognition receptors Proteins 0.000 description 1
- 102000007863 pattern recognition receptors Human genes 0.000 description 1
- 210000002568 pbsc Anatomy 0.000 description 1
- 229950009506 penicillinase Drugs 0.000 description 1
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical class CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 201000002628 peritoneum cancer Diseases 0.000 description 1
- 206010034674 peritonitis Diseases 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 108091005981 phosphorylated proteins Proteins 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229950001100 piposulfan Drugs 0.000 description 1
- NUKCGLDCWQXYOQ-UHFFFAOYSA-N piposulfan Chemical compound CS(=O)(=O)OCCC(=O)N1CCN(C(=O)CCOS(C)(=O)=O)CC1 NUKCGLDCWQXYOQ-UHFFFAOYSA-N 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- BLFWHYXWBKKRHI-JYBILGDPSA-N plap Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(=O)[C@@H]1CCCN1C(=O)[C@H](CO)NC(=O)[C@@H](N)CCC(O)=O BLFWHYXWBKKRHI-JYBILGDPSA-N 0.000 description 1
- 208000031223 plasma cell leukemia Diseases 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 208000005987 polymyositis Diseases 0.000 description 1
- 229920000098 polyolefin Polymers 0.000 description 1
- 208000015768 polyposis Diseases 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 1
- 230000029279 positive regulation of transcription, DNA-dependent Effects 0.000 description 1
- 229940072033 potash Drugs 0.000 description 1
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000001902 propagating effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 230000012743 protein tagging Effects 0.000 description 1
- WOLQREOUPKZMEX-UHFFFAOYSA-N pteroyltriglutamic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(=O)NC(CCC(=O)NC(CCC(O)=O)C(O)=O)C(O)=O)C(O)=O)C=C1 WOLQREOUPKZMEX-UHFFFAOYSA-N 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- JFINOWIINSTUNY-UHFFFAOYSA-N pyrrolidin-3-ylmethanesulfonamide Chemical compound NS(=O)(=O)CC1CCNC1 JFINOWIINSTUNY-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- BMKDZUISNHGIBY-UHFFFAOYSA-N razoxane Chemical compound C1C(=O)NC(=O)CN1C(C)CN1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-UHFFFAOYSA-N 0.000 description 1
- 229960000460 razoxane Drugs 0.000 description 1
- 208000002574 reactive arthritis Diseases 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 239000003488 releasing hormone Substances 0.000 description 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 description 1
- 230000008263 repair mechanism Effects 0.000 description 1
- 230000008672 reprogramming Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 230000000552 rheumatic effect Effects 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 108010038196 saccharide-binding proteins Proteins 0.000 description 1
- 235000002020 sage Nutrition 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 235000019512 sardine Nutrition 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 206010039722 scoliosis Diseases 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 201000008407 sebaceous adenocarcinoma Diseases 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- UQDJGEHQDNVPGU-UHFFFAOYSA-N serine phosphoethanolamine Chemical compound [NH3+]CCOP([O-])(=O)OCC([NH3+])C([O-])=O UQDJGEHQDNVPGU-UHFFFAOYSA-N 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 125000005630 sialyl group Chemical group 0.000 description 1
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 1
- 229910010271 silicon carbide Inorganic materials 0.000 description 1
- 230000005783 single-strand break Effects 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 231100000046 skin rash Toxicity 0.000 description 1
- 208000000649 small cell carcinoma Diseases 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 206010062261 spinal cord neoplasm Diseases 0.000 description 1
- 229950006315 spirogermanium Drugs 0.000 description 1
- 210000003046 sporozoite Anatomy 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- SFVFIFLLYFPGHH-UHFFFAOYSA-M stearalkonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 SFVFIFLLYFPGHH-UHFFFAOYSA-M 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 101150050955 stn gene Proteins 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- CCEKAJIANROZEO-UHFFFAOYSA-N sulfluramid Chemical group CCNS(=O)(=O)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F CCEKAJIANROZEO-UHFFFAOYSA-N 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000000946 synaptic effect Effects 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 229940094937 thioredoxin Drugs 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 230000001256 tonic effect Effects 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 108091006106 transcriptional activators Proteins 0.000 description 1
- 108091008023 transcriptional regulators Proteins 0.000 description 1
- 108091006107 transcriptional repressors Proteins 0.000 description 1
- 230000002463 transducing effect Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 108091007466 transmembrane glycoproteins Proteins 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229950007217 tremelimumab Drugs 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- 229960000875 trofosfamide Drugs 0.000 description 1
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 1
- 239000003744 tubulin modulator Substances 0.000 description 1
- 239000000439 tumor marker Substances 0.000 description 1
- 239000000717 tumor promoter Substances 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 238000007492 two-way ANOVA Methods 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000007484 viral process Effects 0.000 description 1
- 239000000304 virulence factor Substances 0.000 description 1
- 230000007923 virulence factor Effects 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 238000004383 yellowing Methods 0.000 description 1
- 229940055760 yervoy Drugs 0.000 description 1
Images


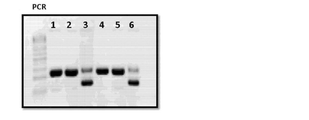






























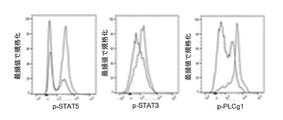






































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4702—Regulators; Modulating activity
- C07K14/4703—Inhibitors; Suppressors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/31—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/48—Blood cells, e.g. leukemia or lymphoma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4613—Natural-killer cells [NK or NK-T]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4631—Chimeric Antigen Receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4635—Cytokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4636—Immune checkpoint inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464411—Immunoglobulin superfamily
- A61K39/464412—CD19 or B4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/5434—IL-12
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/5443—IL-15
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/7051—T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/70517—CD8
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70578—NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30, CD40, CD95
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0646—Natural killers cells [NK], NKT cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/33—Fusion polypeptide fusions for targeting to specific cell types, e.g. tissue specific targeting, targeting of a bacterial subspecies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/80—Vectors containing sites for inducing double-stranded breaks, e.g. meganuclease restriction sites
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Cell Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Pharmacology & Pharmacy (AREA)
- Microbiology (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Mycology (AREA)
- Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Hematology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Transplantation (AREA)
- Hospice & Palliative Care (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Developmental Biology & Embryology (AREA)
- Virology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
【選択図】図1A
Description
2019年5月3日に作成された2KB(Microsoft Windowsで測定)の「UTFC1366WO.txt」と名付けられたファイルに含まれている配列表は、電子出願によって本明細書とともに提出され、参照により本明細書に組み込まれる。
本明細書で使用する場合、特定の成分に関して「本質的に含まない」は、特定の成分が意図的に組成物に配合されておらず、かつ/又は汚染物質として、又は微量でのみ存在することを意味するために本明細書で使用される。したがって、組成物の意図しない汚染から生じる特定の成分の総量は、0.05%をはるかに下回り、好ましくは0.01%を下回る。最も好ましいのは、特定の成分の量が標準的な分析方法で検出できない組成物である。
特定の実施形態において、NK細胞は、当技術分野で周知の方法により、ヒト末梢血単核細胞(PBMC)、非刺激白血球アフェレーシス産物(PBSC)、ヒト胚性幹細胞(hESC)、人工多能性幹細胞(iPSC)、骨髄、又は臍帯血から誘導される。特に、NK細胞は、臍帯血(CB)、末梢血(PB)、骨髄、又は幹細胞から単離することができる。特定の実施形態において、免疫細胞は、プールされたCBから単離される。CBは、2、3、4、5、6、7、8、10又はそれ以上のユニットからプールすることができる。免疫細胞は自己由来であっても、又は同種異系であってもよい。単離されたNK細胞は、細胞療法を投与される対象に対してハプロタイプが一致し得る。NK細胞は、CD3の発現がないヒトでは、CD16、CD56、CD8などの特異的表面マーカーによって検出することができる。
サイトカイン誘導SH2含有タンパク質(CIS)は、CISH遺伝子によってコードされる。これはSOCSファミリーのメンバーであり、NK細胞の細胞内チェックポイント分子である。CISHはIL−15に応答して急速に誘導され、CISHを欠失すると、NK細胞はIL−15に対して過敏になる。CISHノックアウトNK細胞は、増殖が促進され、IFNγ産生が増加し、細胞傷害活性が増強される。CISのアブレーションは、NK細胞活性にブレーキを掛ける。
インターロイキン−15(IL−15)は組織が制限されており、病理学的条件下でのみ、血清中に任意のレベルで、又は全身的に観察される。IL−15は、養子療法に望ましいいくつかの属性を保有する。IL−15は、ナチュラルキラー細胞の発生及び細胞増殖を誘導し、腫瘍常在細胞の機能抑制を緩和することで確立された腫瘍の根絶を促進し、AICDを阻害する恒常性サイトカインである。
本開示のNK細胞は、操作されたTCR及び/又はCARなどの抗原受容体を発現するように遺伝子操作することができる。例えば、NK細胞は、がん抗原に対して抗原特異性を有するTCRを発現するように改変される。異なる抗原などに対する複数のCAR及び/又はTCRをNK細胞に追加することができる。
いくつかの実施形態において、CARは、a)細胞内シグナル伝達ドメイン、b)膜貫通ドメイン、及びc)抗原結合領域を含む細胞外ドメインを含む。
いくつかの実施形態において、遺伝子操作された抗原受容体は、組換えTCR及び/又は天然に存在するT細胞からクローン化されたTCRを含む。「T細胞受容体」又は「TCR」は、可変のa及びβ鎖(それぞれ、TCRα及びTCRβとしても知られる)又は可変のγ及びδ鎖(それぞれ、TCRγ及びTCRδとしても知られる)を含む分子を指し、MHC受容体に結合した抗原ペプチドに特異的に結合することができる。いくつかの実施形態において、TCRはαβ形態である。
マクロファージ、Bリンパ球、樹状細胞を含む抗原提示細胞は、特定のMHC分子の発現によって区別される。APCは抗原を内在化し、MHC分子と一緒にその抗原の一部を細胞膜外側に再発現させる。MHCは、複数の遺伝子座を有する大きな遺伝子複合体である。MHC遺伝子座は、クラスI及びクラスII MHCと呼ばれる2つの主要なクラスのMHC膜分子をコードする。Tヘルパーリンパ球は一般にMHCクラスII分子に関連する抗原を認識し、T細胞傷害性リンパ球はMHCクラスI分子に関連する抗原を認識する。ヒトではMHCはHLA複合体と呼ばれ、マウスではH−2複合体と呼ばれる。
遺伝子操作された抗原受容体によって標的とされる抗原の中には、養子細胞療法によって標的とされる疾患、状態、又は細胞型に関連して発現されるものある。疾患及び状態の中には、増殖性、腫瘍性及び悪性の疾患及び障害があり、これには、リンパ腫、白血病及び/又は骨髄腫、例えば、B細胞性、T細胞性及び骨髄性の白血病、リンパ腫及び多発性骨髄腫などの血液がん、免疫系のがんを含むがん及び腫瘍が含まれる。いくつかの実施形態において、抗原は、正常又は非標的の細胞もしくは組織と比較して、疾患又は病態の細胞、例えば、腫瘍細胞又は病原性細胞上で選択的に発現又は過剰発現される。他の実施形態において、抗原は、正常細胞上で発現されるか、及び/又は操作された細胞上で発現される。
本開示の免疫細胞のCARは、1又は複数の自殺遺伝子を含み得る。本明細書で使用される「自殺遺伝子」という用語は、プロドラッグの投与時に、その宿主細胞を殺す化合物への遺伝子産物の転移をもたらす遺伝子として定義される。使用できる自殺遺伝子/プロドラッグの組み合わせの例は、単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)及びガンシクロビル、アシクロビル又はFIAU;オキシドレダクターゼ及びシクロヘキシミド;シトシンデアミナーゼ及び5−フルオロシトシン;チミジンキナーゼチミジン酸キナーゼ(Tdk::Tmk)及びAZT;ならびにデオキシシチジンキナーゼ及びシトシンアラビノシドである。
当業者は、本開示の抗原受容体を発現させる標準的な組換え技術(例えば、Sambrook et al.,2001及びAusubel et al.,1996を参照されたい。当該文献は双方とも参照により本明細書に両方とも組み込まれる)によってベクターを構築することを熟知していると思われる。ベクターには、限定するものではないが、プラスミド、コスミド、ウイルス(バクテリオファージ、動物ウイルス及び植物ウイルス)、人工染色体(例えば、YAC)、レトロウイルスベクター(例えば、モロニーネズミ白血病ウイルスベクター(MoMLV)、MSCV、SFFV、MPSV、SNVなどに由来)、レンチウイルスベクター(例えば、HIV−1、HIV−2、SIV、BIV、FIVなどに由来)、複製可能、複製欠損及びガットレス(gutless)型を含むアデノウイルス(Ad)ベクター、アデノ随伴ウイルス(AAV)ベクター、シミアンウイルス40(SV−40)ベクター、ウシ乳頭腫ウイルスベクター、エプスタインバーウイルスベクター、ヘルペスウイルスベクター、ワクシニアウイルスベクター、ハーベイマウス肉腫ウイルスベクター、ネズミ乳腺腫瘍ウイルスベクター、ルース肉腫ウイルスベクター、パルボウイルスベクター、ポリオウイルスベクター、水疱性口内炎ウイルスベクター、マラバウイルスベクター及びB群アデノウイルスエナデノツシレフ(enadenotucirev)ベクターが含まれる。
抗原受容体をコードするウイルスベクターを、本開示の特定の態様において提供することができる。組換えウイルスベクターの作製において、非必須遺伝子は、典型的には、異種(又は非天然)タンパク質の遺伝子又はコード配列で置き換えられる。ウイルスベクターは、ウイルス配列を利用して細胞に核酸やタンパク質を導入する一種の発現構築体である。特定のウイルスは、受容体媒介エンドサイトーシスにより細胞に感染したり、又は細胞に侵入し、宿主細胞のゲノムに組み込まれてウイルス遺伝子を安定的かつ効率的に発現したりするその能力により、外来核酸を細胞(哺乳類細胞など)に移入させる魅力的な候補となっている。本発明の特定の態様の核酸を送達するために使用され得るウイルスベクターの非限定的な例を、以下に記載する。
本開示において有用なベクターに含まれる発現カセットは、特に(5′から3′方向に)タンパク質コード配列に作動可能に連結された真核生物転写プロモーター、介在配列を含むスプライスシグナル、及び転写終結/ポリアデニル化配列を含む。真核細胞におけるタンパク質コード遺伝子の転写を制御するプロモーター及びエンハンサーは、複数の遺伝的要素で構成されている。細胞機構は、各要素によって伝達される調節情報を収集して統合することができ、異なる遺伝子が転写調節の異なる、しばしば複雑なパターンの進化を可能にする。本開示の文脈で使用されるプロモーターには、構成的、誘導性、及び組織特異的プロモーターが含まれる。
本明細書で提供される発現構築体は、抗原受容体の発現を駆動するためのプロモーターを含む。プロモーターは一般に、RNA合成の開始部位を配置するように機能する配列を含む。この最もよく知られている例はTATAボックスであるが、例えば、哺乳類のターミナルデオキシヌクレオチジルトランスフェラーゼ遺伝子のプロモーターやSV40後期遺伝子のプロモーターなど、TATAボックスのない一部のプロモーターでは、開始部位に重なる個別の要素自体が開始の場所を修正するのを助ける。追加のプロモーター要素が転写開始の頻度を調節する。多くのプロモーターが開始部位の下流にも機能要素を含んでいることが示されているが、典型的には、これらは開始部位の30110 bp上流の領域に配置されている。コード配列をプロモーターの「制御下」に持ってくるため、選択されたプロモーターの転写リーディングフレームの「下流」(すなわち、その3′)の転写開始部位の5′末端にコード配列を配置する。「上流」のプロモーターは、DNAの転写を刺激し、コードされたRNAの発現を促進する。
特定の開始シグナルはまた、コード配列の効率的な翻訳のために、本開示において提供される発現構築体において使用することができる。これらのシグナルには、ATG開始コドン又は隣接する配列が含まれる。ATG開始コドンを含む外因性翻訳制御シグナルを提供する必要がある場合がある。当業者は、これを容易に決定し、必要なシグナルを提供することができるであろう。開始コドンは、インサート全体の翻訳を確実にするために、所望のコード配列のリーディングフレームと「インフレーム」でなければならないことは周知である。外因性翻訳制御シグナル及び開始コドンは、天然であっても、又は合成であってもよい。発現の効率は、適切な転写エンハンサー要素を含めることによって向上させることができる。
(iii)複製起点
宿主細胞内でベクターを増殖させるために、ベクターは、1又は複数の複製起点部位(しばしば「ori」と呼ばれる)、例えば、上記のEBVのoriPに対応する核酸配列、又は複製が開始される特異的核酸配列である、プログラミングで同様又は高い機能を有するように遺伝子操作されたoriPを含み得る。又は、上記のような他の染色体外複製ウイルスの複製起点、又は自律複製配列(ARS)を使用することができる。
いくつかの実施形態において、本開示の構築体を含む細胞は、発現ベクター中にマーカーを含めることにより、in vitro又はin vivoで同定することができる。そのようなマーカーは、同定可能な変化を細胞に与え、発現ベクターを含む細胞の容易な同定を可能にする。一般に、選択マーカーは、選択を可能にする特性を付与するマーカーである。ポジティブ選択マーカーは、マーカーの存在により選択が可能なマーカーであり、ネガティブ選択マーカーは、マーカーの存在により選択が妨げられるマーカーである。ポジティブ選択マーカーの例は、薬剤耐性マーカーである。
抗原受容体をコードする核酸のウイルス送達に加えて、以下は、所与の宿主細胞へ組換え遺伝子を送達する、したがって、本開示において検討される追加の方法である。
いくつかの実施形態において、本開示の免疫細胞は、CISHの発現を変化させるように改変させることができる。追加の実施形態において、細胞は、グルココルチコイド受容体及び/又はTGFβ受容体(例えば、TGFβ−RII)の発現を破壊するようにさらに改変させることができる。一実施形態において、免疫細胞は、内因性TGFβを枯渇させるためのサイトカインシンクとして機能することができるドミナントネガティブTGFβ受容体II(TGFβRIIDN)を発現するように改変させることができる。
いくつかの実施形態において、DNA標的化分子は、エンドヌクレアーゼなどのエフェクタータンパク質に融合された、1又は複数のジンクフィンガータンパク質(ZFP)又は転写活性化因子様タンパク質(TAL)などのDNA結合タンパク質を含む。例としては、ZFN、TALE及びTALENが挙げられる。
いくつかの実施形態において、DNA標的化分子は、転写活性化因子様タンパク質エフェクター(TALE)タンパク質などにおいて、天然に存在する、又は操作された(天然に存在しない)転写活性化因子様タンパク質(TAL)DNA結合ドメインを含む。例えば、米国特許公開第2011/0301073号明細書を参照されたく、当該特許公開は参照によりその全体が本明細書に組み込まれる。
いくつかの実施形態において、改変は、RNA誘導型エンドヌクレアーゼ(RGEN)を介した改変など、1又は複数のDNA結合核酸を使用して実施される。例えば、改変は、クラスター化された規則的に間隔を空けた短いパリンドロームリピート(CRISPR)及びCRISPR関連(Cas)タンパク質を使用して実行することができる。一般に、「CRISPRシステム」とは、CRISPR関連(「Cas」)遺伝子の発現又は活性の誘導に関与する転写産物及び他の要素を総称して指し、Cas遺伝子をコードする配列、tracr(トランス活性化CRISPR)配列(例えば、tracrRNA又は活性な部分的tracrRNA)、tracr−メイト配列(「ダイレクトリピート」及び内因性CRISPRシステムに関連してtracrRNAによりプロセシングされた部分的ダイレクトリピートを包含する)、ガイド配列(内因性CRISPRシステムに関連して「スペーサー」とも呼ばれる)、及び/又はCRISPR遺伝子座由来の他の配列及び転写産物を含む。
いくつかの実施形態において、本開示は、有効量の本開示のNK細胞を投与することを含む免疫療法のための方法を提供する。一実施形態において、医学的疾患又は障害は、免疫応答を誘発するNK細胞集団の移入によって治療される。本開示の特定の実施形態において、がん又は感染症は、免疫応答を誘発するNK細胞集団の移入によって治療される。本明細書で提供されるのは、有効量の抗原特異的細胞療法を個体に投与することを含む、個体のがんを治療する、又はその進行を遅延させるための方法である。本発明の方法は、免疫障害、固形がん、血液がん、及びウイルス感染症の治療に適用することができる。
免疫細胞(例えば、T細胞又はNK細胞)及び薬学的に許容される担体を含む医薬組成物及び製剤も本明細書において提供される。
特定の実施形態において、本実施形態の組成物及び方法は、少なくとも1つの追加の治療と組み合わせた免疫細胞集団を含む。追加の治療法は、放射線療法、外科手術(例えば、乳腺腫瘍摘出術及び乳房切除術)、化学療法、遺伝子治療、DNA療法、ウイルス療法、RNA療法、免疫療法、骨髄移植、ナノ療法、モノクローナル抗体療法、又は前述の組み合わせであり得る。追加の治療法は、アジュバント療法又はネオアジュバント療法の形態であり得る。
A/B/A B/A/B B/B/A A/A/B A/B/B B/A/A A/B/B/B B/A/B/B
B/B/B/A B/B/A/B A/A/B/B A/B/A/B A/B/B/A B/B/A/A
B/A/B/A B/A/A/B A/A/A/B B/A/A/A A/B/A/A A/A/B/A
多種多様な化学療法剤を、本実施形態に従って使用することができる。「化学療法」という用語は、がんを治療するための薬物の使用を指す。「化学療法剤」は、がんの治療において投与される化合物又は組成物の意味を含んで使用される。これらの薬剤又は薬物は、細胞周期に影響を与えるかどうか、どの段階で影響を与えるかなど、細胞内の活性モードによって分類される。又は、薬剤は、DNAを直接架橋する、DNAに挿入する、又は核酸合成に影響を与えることによって染色体及び有糸分裂の異常を誘発するその能力に基づいて特徴付けることができる。
DNA損傷を引き起こし、広く使用されてきた他の因子には、一般に、γ線、X線、及び/又は腫瘍細胞への放射性同位元素の直接送達として公知のものが含まれる。マイクロ波、陽子線照射(米国特許第5,760,395号明細書及び第4,870,287号明細書)、及びUV照射などの他の形態のDNA損傷因子も企図されている。これらの因子はすべて、DNA、DNAの前駆体、DNAの複製と修復、及び染色体のアセンブリと維持に対する広範囲の損傷に影響を与える可能性が最も高い。X線の線量範囲は、長期間(3〜4週間)の1日線量50〜200レントゲンから単一線量2000〜6000レントゲンまでの範囲である。放射性同位元素の線量範囲は大きく異なり、放射性同位元素の半減期、放出される放射線の強度と種類、及び腫瘍細胞による取り込みに依存する。
当業者は、追加の免疫療法が、実施形態の方法と組み合わせて、又は併せて使用できることを理解すると思われる。がん治療に関連して、免疫療法は、一般に、がん細胞を標的にして破壊するために免疫エフェクター細胞及び分子の使用に依存している。リツキシマブ(RITUXAN(登録商標))はそのような例である。免疫エフェクターは、例えば、腫瘍細胞の表面上のいくつかのマーカーに特異的な抗体であり得る。抗体が単独で治療のエフェクターとして機能する場合もあれば、他の細胞を動員して実際に細胞殺滅に影響を与える場合もある。抗体はまた、薬物又は毒素(化学療法剤、放射性核種、リシンA鎖、コレラ毒素、百日咳毒素など)と結合することができ、標的化剤として役立つ。又は、エフェクターは、腫瘍細胞標的と直接的又は間接的に相互作用する表面分子を運ぶリンパ球であってもよい。さまざまなエフェクター細胞には、細胞傷害性T細胞及びNK細胞が含まれる。
がん患者の約60%は、予防的、診断的又は病期分類的、治癒的、緩和的な外科手術を含む何らかの種類の外科手術を受ける。治癒的外科手術は、がん性組織の全部又は一部が物理的に除去、切除、及び/又は破壊される摘出術を含み、本実施形態の治療、化学療法、放射線療法、ホルモン療法、遺伝子療法、免疫療法及び/又は代替療法などの他の療法と組み合わせて使用することができる。腫瘍摘出術とは、腫瘍の少なくとも一部を物理的に除去することを指す。腫瘍摘出術に加えて、外科手術による治療には、レーザー外科手術、凍結外科手術、電気外科手術、及び顕微鏡制御外科手術(モース術)が含まれる。
治療の治療有効性を改善するために、他の薬剤を本実施形態の特定の態様と組み合わせて使用可能であることが企図される。これらの追加の薬剤には、細胞表面受容体及びギャップ結合のアップレギュレーションに影響を与える薬剤、細胞増殖抑制剤及び分化剤、細胞接着の阻害剤、アポトーシス誘導物質に対する過剰増殖細胞の感受性を高める薬剤、又は他の生物学的薬剤が含まれる。ギャップ結合の数を増やすことによる細胞間シグナル伝達の増加は、隣接する過剰増殖細胞集団に対する抗過剰増殖効果を増加させる。他の実施形態において、治療の抗過剰増殖有効性を改善するために、細胞増殖抑制剤又は分化剤を、本実施形態の特定の態様と組み合わせて使用することができる。細胞接着の阻害剤は、本実施形態の有効性を改善することが企図されている。細胞接着阻害剤の例は、焦点接着キナーゼ(FAK)阻害剤及びロバスタチンである。治療有効性を改善するために、抗体c225などの、アポトーシスに対する過剰増殖性細胞の感受性を増加させる他の薬剤を、本実施形態の特定の態様と組み合わせて使用可能であることがさらに企図される。
免疫細胞を含む製品又はキットも本明細書で提供される。製品又はキットは、免疫細胞を使用して個体のがんを治療又は進行を遅延させるため、又はがんを有する個体の免疫機能を増強するための説明書を含む添付文書をさらに含むことができる。本明細書に記載の抗原特異的免疫細胞はすべて、製品又はキットに含めることができる。適切な容器としては、例えば、ボトル、バイアル、バッグ及び注射器が挙げられる。容器は、ガラス、プラスチック(ポリ塩化ビニル又はポリオレフィンなど)、又は金属合金(ステンレス鋼又はハステロイなど)などのさまざまな材料から形成することができる。いくつかの実施形態において、容器は製剤を保持し、容器上の又はそれに関連するラベルは、使用のための指示を示すことができる。製品又はキットは、他の緩衝液、希釈剤、フィルター、針、注射器、及び使用説明書を含む添付文書を含む、商業的及びユーザーの観点から望ましい他の材料をさらに含むことができる。いくつかの実施形態において、製品は、1又は複数の別の薬剤(例えば、化学療法剤及び抗腫瘍剤)をさらに含む。1又は複数の薬剤に適した容器には、例えば、ボトル、バイアル、バッグ及び注射器が含まれる。
以下の実施例は、本発明の好ましい実施形態を実証するために含まれる。以下の実施例に開示される技術は、本発明者により本発明の実施において良好に機能することが発見された技術を表し、したがって、その実施のための好ましい様式を構成すると見なし得ることが当業者により理解されるべきである。しかしながら、当業者は、本開示に照らして、開示された特定の実施形態において多くの変更を行うことができ、なおかつ本発明の精神及び範囲から逸脱することなく同様又は類似の結果を得ることができることを理解すべきである。
NK細胞を、3つの異なる臍帯血(CB)ユニットからネガティブセレクションによって単離し、増殖させた。次に、NK細胞にCAR構築体を形質導入し、CISH KOを各CBに対して異なる時点で実施した。各CBユニットに関して、NT、NT CISH KO、CAR19/IL−15、及びCAR19/IL−15 CISH KOを含む、4つの異なる細胞グループを得た。
CISH KOされたCAR NK細胞をin vivoで特性評価するために、10週齢のNSGヌルの雌マウスを研究した。マウスに20x103のRaji−FFLuc細胞を注射した。エフェクター細胞については、以下のグループに応じて、細胞#1〜細胞#3を、3x106又は107細胞/動物を注射した。
−グループ#1:Raji−FFLuc単独
−グループ#2:Raji−FFLuc+細胞#1 3x106細胞/動物
−グループ#3:Raji−FFLuc+細胞#1 107細胞/動物
−グループ#4:Raji−FFLuc+細胞#2 3x106細胞/動物
−グループ#5:Raji−FFLuc+細胞#2 107細胞/動物
−細胞#1:NKCAR19 cas9+エレクトロポレーション
−細胞#3:NKCAR19 CISH KO
CIS欠損iC9/CAR19/IL−15 NK細胞におけるin vitro機能の増強:CISの発現レベルを評価して、iC9/CAR19/IL−15 CB−NK細胞が、非改変NK細胞においてサイトカインシグナル伝達を生理学的にダウンレギュレートする同じ逆調節回路にさらされているかどうかを判断した。CB−NK細胞を、IL−2の存在下で、膜結合型IL21、4−1BBリガンド及びCD48(uAPC)を発現するように操作されたK562フィーダー細胞株とともに21日間培養した。NK細胞に、材料及び方法に記載されている、iC9/CAR19/IL−15を発現するレトロウイルスベクターを+4日目に形質導入したか、又は形質導入しなかった(NT、対照)。CISの発現は、時間の経過とともに、NT対照及びiC9/CAR19/IL−15 CB−NK細胞のインビトロ増殖中に有意に増加した(図7A)。特に、CISの発現レベルは、おそらくCIS導入に対するIL−15の追加の効果のために、NT CB−NK細胞と比較して、iC9/CAR19/IL−15においてより顕著であった(図7A)。
細胞株及び培地:Raji(バーキットリンパ腫細胞株)は、American Type Culture Collection(Manassa、VA、USA)から購入した。K562系のフィーダー細胞をレトロウイルスにより形質導入して、4−1BBL、CD48、及び膜結合型IL−21 9uAPCを共発現させた。インビボ実験に使用されたホタルルシフェラーゼ形質導入Raji(Raji−FFLUC)細胞は、Dr.Gianpietro Dotti(University of North Carolina)のご厚意により提供された。
エクソン4フォワードプライマー:CGTCTGGACTCCAACTGCTT(配列番号7)
エクソン4リバースプライマー:GTACAAAGGGCTGCACCAGT(配列番号8)
抗体コンジュゲーション:38個の金属タグ付き抗体で構成されるパネルを使用して、NK細胞の詳細な特性評価を行った。対応する金属タグ同位体を有する抗体のリスト。非標識抗体はすべて無担体の形で購入し、製造業者の指示(Fludigm)に従って、Maxpar X8ポリマーを使用して、社内で対応する金属タグと結合させた。塩化インジウム(III)(Sigma−Aldrich、St.Louis、MO)を除いて、すべての金属同位体はFludigmから入手した。Nanodrop 2000(Thermo Fisher Scientific、Waltham、MA)を使用してA280タンパク質の量を測定することにより、抗体濃度を決定した。次に、結合した抗体を、PBSベースの抗体安定化溶液又は0.05%アジドナトリウム(Sigma−Aldrich、St.Louis、MO)を添加したLowCross−Buffer(Candor Bioscience GmbH、Wangen、Germany)で最終濃度0.5mg/mlに希釈した。連続滴定実験を実施して、各抗体の最適なシグナル対ノイズ比で濃度を決定した。
NK細胞の標識化:NK細胞を、生細胞染色バッファー(abcam)と、それぞれ、ミトコンドリア、リソソーム及び核の標識化のために、最終濃度500nMのMitoTracker(商標)Deep Red FM(Invitrogen(商標))、250nMのLysoRed(abcam)、及び1μMのHoechst 33342(Sigma)を含有するRPMI(Corning)との1:1(v/v)溶液中で37℃において40分間インキュベートした。次に、細胞をハンクスの平衡塩類溶液(Cellgro)+10%HEPES(Corning)で最初に洗浄し、完全培地RPMI+10%FBS(R10)で2回目の洗浄を行った。
下記の参考文献は、それらが本明細書に記載されたものを補足する例示的な手順又は他の詳細を提供する範囲で、参照により本明細書に具体的に組み込まれる。
Austin-Ward and Villaseca, Revista Medica de Chile, 126(7):838-845, 1998.
Ausubel et al., Current Protocols in Molecular Biology, Greene Publishing Associates and John Wiley & Sons, NY, 1994.
Banerjee et al., J Immunol Methods 355(1-2): 1-13, 2010.
Bennekov et al., Mt. Sinai J. Med. 71 (2): 86-93, 2004.
Bukowski et al., Clinical Cancer Res., 4(10):2337-2347,1998.
Camacho et al. J Clin Oncology 22(145): Abstract No.2505 (antibody CP-675206), 2004.
Campbell, Curr. Top. Microbiol. Immunol., 298: 23-57, 2006.
Chothia et al., EMBO J. 7:3745, 1988.
Christodoulides et al., Microbiology, 144(Pt 11):3027-3037, 1998.
Cohen et al. J Immunol. 175:5799-5808, 2005.
Daher et al., Curr Opin Immunol 51: 146-53, 2018.
Davidson et al., J. Immunother., 21(5):389-398, 1998.
Davila et al. PLoS ONE 8(4): e61338, 2013.
Dobosy et al., BMC Biotechnol 11: 80, 2011.
Doulatov et al., Cell Stem Cell. 10:120-36, 2012.
欧州特許出願番号第EP2537416号
Fedorov et al., Sci. Transl. Medicine, 5(215), 2013.
Frolet et al., BMC Microbiol. 10:190 (2010).
Gaj et al., Trends in Biotechnology 31(7), 397-405, 2013.
Gay et al., Clin Lymphoma Myeloma Leuk 17(8): 471-8, 2017.
Hanibuchi et al., Int. J. Cancer, 78(4):480-485, 1998.
Hartmann et al., EMBO Mol Med 9(9): 1183-97, 2017.
Heemskerk et al. Hum Gene Ther. 19:496-510, 2008.
Hellstrand et al., Acta Oncologica, 37(4):347-353, 1998.
Hollander, Front. Immun., 3:3, 2012.
Hubert et al., Proc. Natl. Acad. Sci. USA 96 14523-28, 1999.
Hui and Hashimoto, Infection Immun., 66(11):5329-5336, 1998.
Hurwitz et al. Proc Natl Acad Sci USA 95(17): 10067-10071, 1998.
国際特許出願公開番号WO 00/37504 国際特許出願公開番号WO 01/14424 国際特許出願公開番号WO 2007/069666 国際特許出願公開番号WO 2007/069666
国際特許出願公開番号WO 98/42752 国際特許出願公開番号WO/2014055668 国際特許出願公開番号WO1995001994 国際特許出願公開番号WO1998042752 国際特許出願公開番号WO2000037504 国際特許出願公開番号WO200014257
国際特許出願公開番号WO2001014424 国際特許出願公開番号WO2006/121168 国際特許出願公開番号WO2007/103009 国際特許出願公開番号WO2009/101611 国際特許出願公開番号WO2009/114335 国際特許出願公開番号WO2010/027827 国際特許出願公開番号WO2011/066342 国際特許出願公開番号WO2012/129514 国際特許出願公開番号WO2013/071154 国際特許出願公開番号WO2013/123061 国際特許出願公開番号WO2013/166321 国際特許出願公開番号WO2013126726 国際特許出願公開番号WO2014/055668 国際特許出願公開番号WO2014031687 国際特許出願公開番号WO2015016718 国際特許出願公開番号WO99/40188
Janeway et al, Immunobiology: The Immune System in Health and Disease, 3rd Ed., Current Biology Publications, p. 433, 1997.
Johnson et al. Blood 114:535-46, 2009.
Jores et al., PNAS U.S.A. 87:9138, 1990.
Kim et al., Genome biology 14(4): R36, 2013.
Kim et al., Nature Biotechnology 31, 251-258, 2013.
Kirchmaier and Sugden, J. Virol., 72(6):4657-4666, 1998.
Kolesnik et al., Methods Mol Biol 967: 235-48, 2013.
Leal, M., Ann N Y Acad Sci 1321, 41-54, 2014.
Lefranc et al., Dev. Comp. Immunol. 27:55, 2003.
Li et al. Nat Biotechnol. 23:349-354, 2005.
Li et al. Proc. Natl. Acad. Sci. USA 89:4275-4279, 1992.
Linnemann, C. et al. Nat Med 21, 81-85, 2015.
Lockey et al., Front. Biosci. 13:5916-27, 2008.
Loewendorf et al., J. Intern. Med. 267(5):483-501, 2010.
Ludwig et al. Nature Biotech., (2):185-187, 2006a.
Ludwig et al. Nature Methods, 3(8):637-646, 2006b.
Magoc Bioinformatics 27(21): 2957-63, 2011.
Marschall et al., Future Microbiol. 4:731-42, 2009.
Mehta et al., Front Immunol 9: 283, 2018.
Mokyr et al. Cancer Res 58:5301-5304, 1998.
Moreno-Mateos et al., Nat Methods 12(10): 982-8, 2015.
Notta et al., Science, 218-221, 2011.
Pardoll, Nat Rev Cancer, 12(4): 252-64, 2012
Parkhurst et al. Clin Cancer Res. 15: 169-180, 2009.
Qin et al., Proc. Natl. Acad. Sci. USA, 95(24):14411-14416, 1998.
Quinlan et al., Bioinformatics 26(6): 841-2, 2010.
Rieder et al., J. Interferon Cytokine Res. (9):499-509, 2009.
Robinson et al., Bioinformatics 26(1): 139-40, 2010.
Rouce et al., Leukemia 30(4): 800-11, 2016.
Rykman, et al., J. Virol. 80(2):710-22, 2006.
Sadelain et al., Cancer Discov. 3(4): 388-398, 2013.
Sambrook et al., Molecular Cloning: A Laboratory Manual, 3rd ed., Cold Spring Harbor Press, Cold Spring Harbor, N.Y. 2001.
Shah et al., PLoS One 8(10): e76781, 2013.
Singh et al., Cancer Research, 68:2961-2971, 2008.
Singh et al., Cancer Research, 71:3516-3527, 2011.
Takahashi et al., Cell, 126(4):663-76, 2007.
Terakura et al. Blood. 1:72- 82, 2012.
Tsai et al., Nat Biotechnol 33(2): 187-97, 2015.
Tsai et al., Nat Biotechnol 34(5): 483, 2016.
Turtle et al., Curr. Opin. Immunol., 24(5): 633-39, 2012.
米国特許第4,870,287号 米国特許第5,739,169号 米国特許第5,760,395号 米国特許第5,801,005号 米国特許第5,824,311号 米国特許第5,830,880号 米国特許第5,844,905号 米国特許第5,846,945号 米国特許第5,885,796号 米国特許第5,994,136号 米国特許第6,013,516号 米国特許第6,103,470号 米国特許第6,207,156号 米国特許第6,225,042号 米国特許第6,355,479号 米国特許第6,362,001号 米国特許第6,410,319号 米国特許第6,416,998号 米国特許第6,544,518号 米国特許第6,790,662号 米国特許第7,109,304号 米国特許第7,442,548号 米国特許第7,446,190号 米国特許第7,598,364号 米国特許第7,989,425号 米国特許第8,008,449号 米国特許第8,017,114号 米国特許第8,058,065号 米国特許第8,071,369号 米国特許第8,119,129号 米国特許第8,129,187号 米国特許第8,183,038号 米国特許第8,268,620号 米国特許第8,329,867号 米国特許第8,354,509号 米国特許第8,546,140号 米国特許第8,691,574号 米国特許第8,735,553号 米国特許第8,741,648号 米国特許第8,900,871号 米国特許第9,175,268号
米国公開特許出願第2010/0210014号 米国公開特許出願第12/478,154号 米国公開特許出願第2002131960号 米国公開特許出願第2003/0211603号 米国公開特許出願第2005/0260186号 米国公開特許出願第2006/0104968号 米国公開特許出願第2009/0004142号 米国公開特許出願第2009/0017000号 米国公開特許出願第2009/0246875号 米国公開特許出願第2011/0104125号 米国公開特許出願第2011/0301073号 米国公開特許出願第20110008369号 米国公開特許出願第2012/0276636号 米国公開特許出願第2013/0315884号 米国公開特許出願第20130149337号 米国公開特許出願第2013287748号 米国公開特許出願第2014/0120622号 米国公開特許出願第2014022021号 米国公開特許出願第20140294898号
Van Gassen et al., Cytometry 87(7): 636-45, 2015.
Varela-Rohena et al. Nat Med. 14: 1390-1395, 2008.
Vera et al., Blood 108(12): 3890-7, 2006.
Wang et al. J Immunother. 35(9):689-701, 2012.
Wu et al., Adv. Cancer Res., 90: 127-56, 2003.
Wu et al., Cancer, 18(2): 160-75, 2012.
Yamanaka et al., Cell, 131(5):861-72, 2007.
Yu et al., Science, 318:1917-1920, 2007.
Zysk et al., Infect. Immun. 68(6):3740-43, 2000.
Claims (54)
- (1)キメラ抗原受容体(CAR)及び/又はT細胞受容体(TCR)と(2)ヒトIL−15(hIL−15)とを発現し、本質的にCISHを発現しないように操作された単離ナチュラルキラー(NK)細胞。
- CARを発現するように操作されている、請求項1に記載のNK細胞。
- TCRを発現するように操作されている、請求項1に記載のNK細胞。
- CAR及びTCRを発現するように操作されている、請求項1に記載のNK細胞。
- 臍帯血、末梢血、骨髄、CD34+細胞、又はiPSCに由来する、請求項1に記載のNK細胞。
- 臍帯血に由来する、請求項1に記載のNK細胞。
- 前記臍帯血が事前凍結されている、請求項6に記載のNK細胞。
- 前記CAR及び/又はTCRが、CD19、CD319/CS1、ROR1、CD20、CD5、CD7、CD22、CD70、CD30、BCMA、CD25、NKG2Dリガンド、MICA/MICB、がん胎児性抗原、アルファフェトプロテイン、CA−125、MUC−1、上皮腫瘍抗原、メラノーマ関連抗原、変異p53、変異ras、HER2/Neu、ERBB2、葉酸結合タンパク質、HIV−1エンベロープ糖タンパク質gp120、HIV−1エンベロープ糖タンパク質gp41、GD2、CD123、CD33、CD30、CD56、c−Met、メソテリン、GD3、HERV−K、IL−11Ralpha、カッパ鎖、ラムダ鎖、CSPG4、ERBB2、WT−1、EGFRvIII、TRAIL/DR4、及び/又はVEGFR2に対して抗原特異性を有する、請求項1に記載のNK細胞。
- 前記CARがCD19に対して抗原特異性を有する、請求項1に記載のNK細胞。
- 前記NK細胞が第2のサイトカインをさらに発現する、請求項1に記載の方法。
- 前記サイトカインがIL−21又はIL−12である、請求項10に記載の方法。
- 前記NK細胞が哺乳類ラパマイシン標的タンパク質(mTOR)シグナル伝達を活性化させた、請求項1〜11のいずれか一項に記載の方法。
- 前記NK細胞がJAK/STATシグナル伝達を増加させた、請求項12に記載の方法。
- 請求項1〜14のいずれか一項に記載のNK細胞を作製する方法であって、
(a)NK細胞の開始集団を取得すること、
(b)前記NK細胞の開始集団を人工提示細胞(APC)の存在下で培養すること、
(c)CAR及び/又はTCR発現ベクターを前記NK細胞に導入すること、
(d)前記NK細胞をAPCの存在下で増殖させること、それによって増殖したNK細胞を得ること、ならびに
(e)増殖したNK細胞においてCISHの発現を破壊すること、
を含む方法。 - 発現を破壊することが、CRISPR媒介遺伝子サイレンシングを使用することを含む、請求項14に記載の方法。
- CRISPR媒介遺伝子サイレンシングが、CAR NK細胞と、sgRNA及びCas9とを接触させることを含む、請求項15に記載の方法。
- 前記sgRNAがCISHのエクソン4を標的とする、請求項11に記載の方法。
- 前記sgRNAが配列番号1〜2を含む、請求項17に記載の方法。
- 前記NK細胞の開始集団が、フィコールパック密度勾配を使用して単核細胞を単離することによって得られる、請求項14に記載の方法。
- 前記APCがガンマ線照射されたAPCである、請求項14に記載の方法。
- 前記APCがユニバーサルAPC(uAPC)である、請求項14に記載の方法。
- 前記APCが41BB及びIL−21を発現するように操作されている、請求項14に記載の方法。
- 前記uAPCが、(1)CD48及び/又はCS1(CD319)、(2)膜結合インターロイキン−21(mbIL−21)、及び(3)41BBリガンド(41BBL)を発現するように操作されている、請求項21に記載の方法。
- 前記NK細胞とAPCとが1:2の比率で存在する、請求項14に記載の方法。
- ステップ(b)及び(d)のNK細胞が、IL−2の存在下でさらに増殖される、請求項14に記載の方法。
- 前記IL−2が100〜300U/mLの濃度で存在する、請求項25に記載の方法。
- 前記IL−2が200U/mLの濃度で存在する、請求項25に記載の方法。
- 導入することが形質導入を含む、請求項14に記載の方法。
- 前記CAR及び/又はTCR発現構築体が、レンチウイルスベクター又はレトロウイルスベクターである、請求項14に記載の方法。
- 前記NK細胞がGMPに準拠している、請求項14に記載の方法。
- 前記NK細胞が同種異系である、請求項14に記載の方法。
- 前記NK細胞が自己由来である、請求項14に記載の方法。
- 前記CAR及び/又はTCRが、CD19、CD319/CS1、ROR1、CD20、CD5、CD7、CD22、CD70、CD30、BCMA、CD25、NKG2Dリガンド、MICA/MICB、がん胎児性抗原、アルファフェトプロテイン、CA−125、MUC−1、上皮腫瘍抗原、メラノーマ関連抗原、変異p53、変異ras、HER2/Neu、ERBB2、葉酸結合タンパク質、HIV−1エンベロープ糖タンパク質gp120、HIV−1エンベロープ糖タンパク質gp41、GD2、CD123、CD23、CD30、CD56、c−Met、メソテリン、GD3、HERV−K、IL−11Ralpha、カッパ鎖、ラムダ鎖、CSPG4、ERBB2、WT−1、EGFRvIII、TRAIL/DR4、及び/又はVEGFR2に対して抗原特異性を有する、請求項14に記載の方法。
- 前記CARがCD19に対して抗原特異性を有する、請求項14に記載の方法。
- 前記CAR及び/又はTCR発現構築体がサイトカインをさらに発現する、請求項14に記載の方法。
- 前記サイトカインがIL−15、IL−21又はIL−12である、請求項35に記載の方法。
- 前記増殖したNK細胞の集団を凍結保存することをさらに含む、請求項14に記載の方法。
- 請求項1〜11のいずれか一項に記載のNK細胞の集団、又は請求項14〜37のいずれか一項に記載の方法により作製されたNK細胞の集団と、薬学的に許容される担体とを含む医薬組成物。
- 対象の疾患又は障害の治療に使用するための、請求項1〜11のいずれか一項に記載のNK細胞、又は請求項14〜37のいずれか一項に記載の方法によって作製されたNK細胞の有効量を含む組成物。
- 対象の免疫関連障害の治療のための、請求項1〜11のいずれか一項に記載のNK細胞、又は請求項14〜37のいずれか一項に記載の方法によって作製されたNK細胞の有効量を含む組成物の使用。
- 請求項1〜11のいずれか一項に記載のNK細胞、又は請求項14〜37のいずれか一項に記載の方法により作製されたNK細胞の有効量を対象に投与することを含む、対象の免疫関連障害を治療する方法。
- 前記免疫関連障害が、がん、自己免疫障害、移植片対宿主病、同種移植片拒絶、又は炎症状態である、請求項41に記載の方法。
- 前記免疫関連障害が炎症状態であり、前記免疫細胞が本質的にグルココルチコイド受容体の発現を有さない、請求項41に記載の方法。
- 前記対象がステロイド療法を投与されていたか、又は投与されている、請求項43に記載の方法。
- 前記NK細胞が自己由来である、請求項41に記載の方法。
- 前記NK細胞が同種異系である、請求項41に記載の方法。
- 前記免疫関連障害ががんである、請求項41に記載の方法。
- 前記がんが固形がん又は血液悪性腫瘍である、請求項47に記載の方法。
- 少なくとも第2の治療薬を投与することをさらに含む、請求項41に記載の方法。
- 前記少なくとも第2の治療薬が、化学療法、免疫療法、外科手術、放射線療法、又は生物療法を含む、請求項49に記載の方法。
- 前記NK細胞及び/又は少なくとも第2の治療薬が、静脈内、腹腔内、気管内、腫瘍内、筋肉内、内視鏡的、病変内、経皮的、皮下、局所的に、又は直接注射又は灌流によって投与される、請求項49に記載の方法。
- 前記本質的にCISHを発現しないNK細胞が、CISHを発現するNK細胞と比較して増強された機能を有する、請求項49に記載の方法。
- 前記増強された機能が、IFN−γ及びTNF−αの細胞内染色、CD107a脱顆粒、及び51Cr放出アッセイによる腫瘍殺滅によって測定される、請求項52記載の方法。
- 前記増強された機能が、グランザイム−b、パーフォリン、TRAIL、CD3z、Eomes、T−bet、DAP12、DNAM、CD25及び/又はKi67の発現の増加によって測定される、請求項52に記載の方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2024000389A JP2024045179A (ja) | 2018-05-03 | 2024-01-05 | 免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862666665P | 2018-05-03 | 2018-05-03 | |
| US62/666,665 | 2018-05-03 | ||
| US201862666965P | 2018-05-04 | 2018-05-04 | |
| US62/666,965 | 2018-05-04 | ||
| PCT/US2019/030721 WO2019213610A1 (en) | 2018-05-03 | 2019-05-03 | Natural killer cells engineered to express chimeric antigen receptors with immune checkpoint blockade |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2024000389A Division JP2024045179A (ja) | 2018-05-03 | 2024-01-05 | 免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021522798A true JP2021522798A (ja) | 2021-09-02 |
| JPWO2019213610A5 JPWO2019213610A5 (ja) | 2022-05-09 |
Family
ID=68386134
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020561821A Pending JP2021522798A (ja) | 2018-05-03 | 2019-05-03 | 免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 |
| JP2024000389A Pending JP2024045179A (ja) | 2018-05-03 | 2024-01-05 | 免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2024000389A Pending JP2024045179A (ja) | 2018-05-03 | 2024-01-05 | 免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US20210230548A1 (ja) |
| EP (1) | EP3788061A4 (ja) |
| JP (2) | JP2021522798A (ja) |
| KR (1) | KR20210005240A (ja) |
| CN (1) | CN112292390A (ja) |
| AU (1) | AU2019262218A1 (ja) |
| BR (1) | BR112020022010A2 (ja) |
| CA (1) | CA3099342A1 (ja) |
| CO (1) | CO2020015168A2 (ja) |
| MX (1) | MX2020011697A (ja) |
| SG (1) | SG11202010763VA (ja) |
| WO (1) | WO2019213610A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021523725A (ja) * | 2018-05-16 | 2021-09-09 | リサーチ インスティテュート アット ネイションワイド チルドレンズ ホスピタルResearch Institute At Nationwide Children’S Hospital | Cas9リボ核タンパク質を使用したノックアウト初代および増殖ヒトnk細胞の生成 |
Families Citing this family (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2008302276B2 (en) | 2006-09-29 | 2013-10-10 | Takeda Vaccines, Inc. | Method of conferring a protective immune response to norovirus |
| EA029470B1 (ru) | 2011-07-11 | 2018-03-30 | Такеда Вэксинс, Инк. | Способ стимулирования формирования защитного иммунитета против норовируса |
| EP3612210A4 (en) * | 2017-04-19 | 2021-01-27 | Board Of Regents, The University Of Texas System | IMMUNE CELLS EXPRESSING MODIFIED ANTIGEN RECEPTORS |
| SG11202004003YA (en) * | 2017-11-09 | 2020-05-28 | Sangamo Therapeutics Inc | Genetic modification of cytokine inducible sh2-containing protein (cish) gene |
| BR112020015512A2 (pt) | 2018-02-01 | 2021-01-26 | Nkmax Co., Ltd. | método de produção de células exterminadoras naturais e composição para tratamento de câncer |
| JP2022519935A (ja) * | 2019-03-29 | 2022-03-25 | ボード オブ リージェンツ,ザ ユニバーシティ オブ テキサス システム | Car-nk細胞の作製方法およびその使用方法 |
| WO2020247392A1 (en) * | 2019-06-04 | 2020-12-10 | Nkarta, Inc. | Combinations of engineered natural killer cells and engineered t cells for immunotherapy |
| CN114555123B (zh) | 2019-10-18 | 2024-04-02 | 四十七公司 | 用于治疗骨髓增生异常综合征和急性髓系白血病的联合疗法 |
| AU2020374947A1 (en) | 2019-10-31 | 2022-03-31 | Forty Seven, Inc. | Anti-CD47 and anti-CD20 based treatment of blood cancer |
| US20210147799A1 (en) * | 2019-11-08 | 2021-05-20 | The Broad Institute, Inc. | Engineered antigen presenting cells and uses thereof |
| CN110760007B (zh) * | 2019-11-21 | 2022-08-26 | 博生吉医药科技(苏州)有限公司 | Cd7-car-t细胞及其制备和应用 |
| EP4065140A4 (en) * | 2019-11-27 | 2024-02-21 | Board of Regents, The University of Texas System | COMBINED LARGE-SCALE CAR TRANSDUCTION AND CRISPR GENE EDITING OF NK CELLS |
| US20230045174A1 (en) * | 2019-11-27 | 2023-02-09 | Board Of Regents, The University Of Texas System | Large-scale combined car transduction and crispr gene editing of msc cells |
| US11845723B2 (en) | 2019-12-24 | 2023-12-19 | Gilead Sciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021202581A1 (en) * | 2020-03-30 | 2021-10-07 | WUGEN, Inc. | Engineered immune cells for adoptive cell therapy |
| US11591381B2 (en) | 2020-11-30 | 2023-02-28 | Crispr Therapeutics Ag | Gene-edited natural killer cells |
| US11661459B2 (en) | 2020-12-03 | 2023-05-30 | Century Therapeutics, Inc. | Artificial cell death polypeptide for chimeric antigen receptor and uses thereof |
| JP2023552895A (ja) | 2020-12-15 | 2023-12-19 | ユニフェルシテイト アントウェルペン | Cd70をターゲティングする細胞ベースの治療剤 |
| AR124414A1 (es) | 2020-12-18 | 2023-03-22 | Century Therapeutics Inc | Sistema de receptor de antígeno quimérico con especificidad de receptor adaptable |
| EP4271798A1 (en) | 2020-12-30 | 2023-11-08 | CRISPR Therapeutics AG | Compositions and methods for differentiating stem cells into nk cells |
| CN113005151A (zh) * | 2021-03-12 | 2021-06-22 | 广东药科大学 | 一种kdr-car-nk细胞的制备方法及其应用 |
| TW202302145A (zh) | 2021-04-14 | 2023-01-16 | 美商基利科學股份有限公司 | CD47/SIRPα結合及NEDD8活化酶E1調節次單元之共抑制以用於治療癌症 |
| US11976072B2 (en) | 2021-06-23 | 2024-05-07 | Gilead Sciences, Inc. | Diacylglycerol kinase modulating compounds |
| KR20240005901A (ko) | 2021-06-23 | 2024-01-12 | 길리애드 사이언시즈, 인코포레이티드 | 디아실글리세롤 키나제 조절 화합물 |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| CN113599395B (zh) * | 2021-09-22 | 2022-08-12 | 郑州源创吉因实业有限公司 | 含有nk细胞的治疗癌症的药物组合物 |
| CN118251227A (zh) * | 2021-10-01 | 2024-06-25 | 得克萨斯大学体系董事会 | 负载抗体的免疫细胞及用于癌症治疗的方法 |
| WO2023062113A1 (en) * | 2021-10-15 | 2023-04-20 | Miltenyi Biotec B.V. & Co. KG | Method for the generation of genetically modified nk cells |
| WO2023070079A1 (en) * | 2021-10-21 | 2023-04-27 | Board Of Regents, The University Of Texas System | Methods for production of therapeutic immune cells having enhanced metabolic fitness and compositions thereof |
| EP4426724A1 (en) * | 2021-11-04 | 2024-09-11 | Saliogen Therapeutics, Inc. | Immune cells with chimeric antigen receptors or chimeric autoantibody receptors |
| US20230149464A1 (en) | 2021-11-18 | 2023-05-18 | Janssen Biotech, Inc. | Feeder free cell culture methods for expanding natural killer cell preparations |
| US20230355796A1 (en) | 2022-03-24 | 2023-11-09 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| TW202345901A (zh) | 2022-04-05 | 2023-12-01 | 美商基利科學股份有限公司 | 用於治療結腸直腸癌之組合療法 |
| CN114774364B (zh) * | 2022-04-26 | 2024-04-26 | 深圳市体内生物医药科技有限公司 | 一种嵌合抗原受体t细胞及其制备方法和应用 |
| US20240091351A1 (en) | 2022-09-21 | 2024-03-21 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPa DISRUPTION ANTICANCER COMBINATION THERAPY |
| WO2024089622A1 (en) * | 2022-10-25 | 2024-05-02 | Takeda Pharmaceutical Company Limited | Adgre2 chimeric receptor nk cell compositions and methods of use |
| WO2024100203A1 (en) * | 2022-11-10 | 2024-05-16 | Onk Therapeutics Limited | Combined therapies using immunomodulating drugs |
| CN115947869B (zh) * | 2022-11-28 | 2023-12-12 | 广州佰芮慷生物科技有限公司 | 一种靶向人巨细胞病毒的嵌合抗原受体、car-nk细胞及用途 |
| CN115948344A (zh) * | 2023-02-10 | 2023-04-11 | 上海交通大学 | 嵌合抗原受体nk细胞、制备方法及其用途 |
| CN116769722B (zh) * | 2023-07-04 | 2024-07-16 | 杭州荣谷生物科技有限公司 | 功能增强型car-nk细胞、其制备方法及其在免疫疗法中的应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012136231A1 (en) * | 2010-09-08 | 2012-10-11 | Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus | Interleukin 15 as selectable marker for gene transfer in lymphocytes |
| US20140255363A1 (en) * | 2011-09-16 | 2014-09-11 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| JP2015502756A (ja) * | 2011-12-22 | 2015-01-29 | モガム バイオテクノロジー リサーチ インスティチュート | ナチュラルキラー細胞の製造方法、その方法により製造されたナチュラルキラー細胞、並びにそれを含む腫瘍及び感染性疾患治療用組成物 |
| WO2017023803A1 (en) * | 2015-07-31 | 2017-02-09 | Regents Of The University Of Minnesota | Modified cells and methods of therapy |
| WO2017100861A1 (en) * | 2015-12-16 | 2017-06-22 | The Walter And Eliza Hall Institute Of Medical Research | Inhibition of cytokine-induced sh2 protein in nk cells |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3402494B1 (en) * | 2016-01-11 | 2021-04-07 | The Board of Trustees of the Leland Stanford Junior University | Chimeric proteins and methods of immunotherapy |
| WO2017123956A1 (en) * | 2016-01-15 | 2017-07-20 | Etubics Corporation | Methods and compositions for t-cell immunotherapy |
| MX2018014602A (es) * | 2016-05-27 | 2019-06-10 | Etubics Corp | Composiciones de vacunas neoepitopos y metodos de uso de las mismas. |
| AU2017347854B2 (en) * | 2016-10-27 | 2022-12-08 | Intima Bioscience, Inc. | Viral methods of T cell therapy |
| EP3612210A4 (en) * | 2017-04-19 | 2021-01-27 | Board Of Regents, The University Of Texas System | IMMUNE CELLS EXPRESSING MODIFIED ANTIGEN RECEPTORS |
| JP2022519935A (ja) * | 2019-03-29 | 2022-03-25 | ボード オブ リージェンツ,ザ ユニバーシティ オブ テキサス システム | Car-nk細胞の作製方法およびその使用方法 |
-
2019
- 2019-05-03 WO PCT/US2019/030721 patent/WO2019213610A1/en active Application Filing
- 2019-05-03 EP EP19795874.7A patent/EP3788061A4/en active Pending
- 2019-05-03 CN CN201980037453.2A patent/CN112292390A/zh active Pending
- 2019-05-03 MX MX2020011697A patent/MX2020011697A/es unknown
- 2019-05-03 US US17/050,775 patent/US20210230548A1/en active Pending
- 2019-05-03 CA CA3099342A patent/CA3099342A1/en active Pending
- 2019-05-03 BR BR112020022010-8A patent/BR112020022010A2/pt unknown
- 2019-05-03 SG SG11202010763VA patent/SG11202010763VA/en unknown
- 2019-05-03 AU AU2019262218A patent/AU2019262218A1/en active Pending
- 2019-05-03 JP JP2020561821A patent/JP2021522798A/ja active Pending
- 2019-05-03 KR KR1020207034764A patent/KR20210005240A/ko unknown
-
2020
- 2020-12-02 CO CONC2020/0015168A patent/CO2020015168A2/es unknown
-
2024
- 2024-01-05 JP JP2024000389A patent/JP2024045179A/ja active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012136231A1 (en) * | 2010-09-08 | 2012-10-11 | Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus | Interleukin 15 as selectable marker for gene transfer in lymphocytes |
| US20140255363A1 (en) * | 2011-09-16 | 2014-09-11 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| JP2015502756A (ja) * | 2011-12-22 | 2015-01-29 | モガム バイオテクノロジー リサーチ インスティチュート | ナチュラルキラー細胞の製造方法、その方法により製造されたナチュラルキラー細胞、並びにそれを含む腫瘍及び感染性疾患治療用組成物 |
| WO2017023803A1 (en) * | 2015-07-31 | 2017-02-09 | Regents Of The University Of Minnesota | Modified cells and methods of therapy |
| WO2017100861A1 (en) * | 2015-12-16 | 2017-06-22 | The Walter And Eliza Hall Institute Of Medical Research | Inhibition of cytokine-induced sh2 protein in nk cells |
Non-Patent Citations (4)
| Title |
|---|
| C.SAHM, ET AL., CANCER IMMUNOLOGY IMMUNOTHERAPY, vol. 61, JPN6022020682, 2012, pages 1451 - 1461, ISSN: 0005147894 * |
| LEUKEMIA, vol. 32, no. 2, JPN6023017561, February 2018 (2018-02-01), pages 520 - 531, ISSN: 0005147895 * |
| NATURE IMUNOLOGY, vol. 17, no. 7, JPN6023017560, 2016, pages 816 - 824, ISSN: 0005147897 * |
| NATURE IMUNOLOGY, vol. 17, no. 9, JPN6023017562, 2016, pages 1025 - 1036, ISSN: 0005147896 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021523725A (ja) * | 2018-05-16 | 2021-09-09 | リサーチ インスティテュート アット ネイションワイド チルドレンズ ホスピタルResearch Institute At Nationwide Children’S Hospital | Cas9リボ核タンパク質を使用したノックアウト初代および増殖ヒトnk細胞の生成 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN112292390A (zh) | 2021-01-29 |
| MX2020011697A (es) | 2020-12-10 |
| JP2024045179A (ja) | 2024-04-02 |
| EP3788061A1 (en) | 2021-03-10 |
| EP3788061A4 (en) | 2022-02-23 |
| BR112020022010A2 (pt) | 2021-01-26 |
| WO2019213610A1 (en) | 2019-11-07 |
| SG11202010763VA (en) | 2020-11-27 |
| AU2019262218A1 (en) | 2020-12-10 |
| CO2020015168A2 (es) | 2021-09-09 |
| US20210230548A1 (en) | 2021-07-29 |
| KR20210005240A (ko) | 2021-01-13 |
| CA3099342A1 (en) | 2019-11-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2021522798A (ja) | 免疫チェックポイント遮断によりキメラ抗原受容体を発現するように操作されたナチュラルキラー細胞 | |
| US20220265718A1 (en) | Immune cells expressing engineered antigen receptors | |
| US20220031749A1 (en) | Multiplex genome editing of immune cells to enhance functionality and resistance to suppressive environment | |
| US20220389383A1 (en) | Large-scale combined car transduction and crispr gene editing of nk cells | |
| CN114615886A (zh) | 细胞冷冻保存培养基 | |
| JP2022519935A (ja) | Car-nk細胞の作製方法およびその使用方法 | |
| CA3162901A1 (en) | Large-scale combined car transduction and crispr gene editing of msc cells | |
| CA3162900A1 (en) | Large-scale combined car transduction and crispr gene editing of b cells | |
| US20220401479A1 (en) | Large-scale combined car transduction and crispr gene editing of t cells | |
| CN117615769A (zh) | 抑制性嵌合抗原受体防止过继细胞疗法的非肿瘤靶向效应 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220425 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220425 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20230509 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230803 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20230908 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20240105 |