JP2020037592A - トリアルキルガリウム化合物の調製及びその使用 - Google Patents
トリアルキルガリウム化合物の調製及びその使用 Download PDFInfo
- Publication number
- JP2020037592A JP2020037592A JP2019220438A JP2019220438A JP2020037592A JP 2020037592 A JP2020037592 A JP 2020037592A JP 2019220438 A JP2019220438 A JP 2019220438A JP 2019220438 A JP2019220438 A JP 2019220438A JP 2020037592 A JP2020037592 A JP 2020037592A
- Authority
- JP
- Japan
- Prior art keywords
- mixture
- rgacl
- compound
- gacl
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C16/00—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes
- C23C16/06—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the deposition of metallic material
- C23C16/18—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the deposition of metallic material from metallo-organic compounds
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10P—GENERIC PROCESSES OR APPARATUS FOR THE MANUFACTURE OR TREATMENT OF DEVICES COVERED BY CLASS H10
- H10P14/00—Formation of materials, e.g. in the shape of layers or pillars
- H10P14/20—Formation of materials, e.g. in the shape of layers or pillars of semiconductor materials
- H10P14/24—Formation of materials, e.g. in the shape of layers or pillars of semiconductor materials using chemical vapour deposition [CVD]
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Mechanical Engineering (AREA)
- Metallurgy (AREA)
- Chemical Vapour Deposition (AREA)
Abstract
【解決手段】本発明は、一般式:R3Gaのトリアルキルガリウム化合物を、高い収率及び選択性で安価かつ環境に無害であるように調製するための改善されたプロセスに関し、式中、Rは、1〜4個の炭素原子のアルキルである。トリアルキルガリウムは、本発明に従って、中間段階であるアルキルガリウムジクロリド(RGaCl2)又はジアルキルガリウムクロリド/アルキルガリウムジクロリド混合物(R2GaCl/RGaCl2)を介して調製される。得られるRGaCl2又はR2GaCl/RGaCl2混合物も、本発明の主題の一部を形成する。
本発明の新規のプロセスは、改善されたプロセス管理のため注目に値する。本プロセスは、環境への影響が低い安価な出発材料及び試薬の実質的な使用を意図的に行い、そのため工業規模で有用でもある。
得られるトリアルキルガリウム化合物は非常に純粋であり、そのため、半導体及びマイクロシステム技術において有機金属化学蒸着法(MOCVD)又は有機金属気相エピタキシー法(MOVPE)のための有機金属前駆体として特に有用である。
【選択図】なし
Description
R3Ga
のトリアルキルガリウム化合物を調製するための改善されたプロセスによって達成され、式中、Rは、1〜4個の炭素原子のアルキルである。前記アルキルは、分岐状又は非分岐状、好ましくは非分岐状であり得る。本発明のプロセスは、トリエチルガリウム及びトリメチルガリウムの調製に特に有用であり、トリメチルガリウムの調製に格別に有用である。したがってRは、好ましくはエチル及びメチルから選択され、最も好ましくは、以下Meとも略されるメチルである。
RGaCl2、又は
R2GaClとRGaCl2との混合物
のいずれかである、化合物(A)を調製するためのプロセスであって、
a1)ガリウムとアルキル供与体とを活性化剤の存在下で反応させて化合物(A)を形成する反応工程であって、アルキル供与体が塩化アルキルであり、活性化剤がガリウム成分である、反応工程と、
a2)任意に、前記化合物(A)を反応混合物から単離させる反応工程と、を含み、
式中、Rが、1〜4個の炭素原子の分岐状又は非分岐状アルキルであり、好ましくはR2GaCl対RGaCl2の比が、モル量に基づいて10:90〜90:10、特に10:90〜50:50、特に20:80〜40:60の範囲内である、プロセス。
2.アルキル供与体が、一般式:
RCl
を有し、式中、Rが、上記の定義の通りであり、アルキル供与体が、好ましくは気体形態にある、項目1に記載のプロセス。
3.活性化剤が、次の一般式:
RaGabClc
を有する化合物又は化合物の混合物であり、式中、aが、0、1、2、及び3から選択され、bが、1及び2から選択され、cが、0、1、2、及び3から選択されるが、但し、a及びcの両方が0になることはなく、a+b+cが4と等しいか又は4の倍数であることを条件とし、Rが、上記の定義の通りであり、bが1であるとき、a及びcの合計は3であるか、又はbが2であるとき、a及びcの合計は6である、項目1又は2に記載のプロセス。
4.活性化剤が、GaCl3、R2GaCl、R3Ga2Cl3、RGaCl2、及びこれらの混合物から選択されるか、又は、好ましくは、反応生成物である化合物(A)自体が活性化剤として使用される、任意の先行項目に記載のプロセス。
5.活性化剤が、RGaCl2、GaCl3、R3Ga2Cl3、及びこれらの混合物から選択される、任意の先行項目に記載のプロセス。
6.Rがメチル又はエチル、好ましくはメチルである、任意の先行項目に記載のプロセス。
7.アルキル供与体対ガリウムのモル比が少なくとも1.4:1である、任意の先行項目に記載のプロセス。
8.ガリウムと活性化剤とのプレミックスが反応工程a1)において反応容器に最初に投入され、アルキル供与体が後に添加される、先行項目のいずれか一項に記載のプロセス。
9.反応体混合物が、120℃〜200℃の温度に加熱される、項目8に記載のプロセス。
10.該温度が、少なくとも30分間、最大でも50時間にわたって維持される、項目9に記載のプロセス。
11.反応工程a1)が有機溶剤の非存在下で実施される、任意の先行項目に記載のプロセス。
12.工程a)が、前記化合物(A)を反応混合物から単離させることを反応工程a2)として含み、前記化合物(A)を反応混合物から単離させる前記工程が、未変換の反応体を反応混合物から分離させることを含む、任意の先行項目に記載のプロセス。
13.化合物(A)の収率が、使用されるガリウム金属に基づいて90%超である、任意の先行項目に記載のプロセス。
14.化合物(A)の純度が99%超である、任意の先行項目に記載のプロセス。
15.一般式:
R3Ga
の化合物(B)を調製するためのプロセスであって、先行項目のいずれか一項に記載の化合物(A)を提供することと、
b)前記化合物(A)を金属アルキル成分と反応させて、一般式:
R3Ga
の化合物を得ることと、を含み、式中、Rが、任意の先行項目に定義された通りである、プロセス。
16.金属アルキル成分が、一般式:
RdMeXf
を有し、式中、dが、1、2、及び3から選択され、eが、1及び2から選択され、fが、0、1、2、及び3から選択されるが、但し、d及びfの両方が0になることはないことを条件とし、Rが、上記の定義の通りであり、Mが、アルミニウム、リチウム、及びマグネシウムから選択され、Xが、Cl、Br、及びIから選択される、項目15に記載のプロセス。
17.Mがアルミニウムであり、eが1と等しいか又は2であり、d、e、及びfの合計が4と等しいか又は8であり、dが≠0であり、XがClである、項目16に記載のプロセス。
18.金属アルキル成分が、RMgCl、R2AlCl、R3Al、R3Al2Cl3、及びRLiから選択される、項目15又は16に記載のプロセス。
19.反応工程b)において補助塩基も添加され、補助塩基が、塩化ナトリウム、塩化カリウム、塩化アルミニウム、及びこれらの混合物から選択される、項目15〜18のいずれか一項に記載のプロセス。
20.化合物(B)の収率が90%超である、項目15〜19のいずれか一項に記載のプロセス。
21.化合物(B)の純度が少なくとも99%である、項目15〜20のいずれか一項に記載のプロセス。
22.項目15〜21のいずれか一項に記載のプロセスによって得られた化合物(B)の、有機金属化学蒸着法(MOCVD)又は有機金属気相エピタキシー法(MOVPE)のための前駆体としての使用。
23.項目15に記載の化合物(B)を生成するための、項目1に記載の化合物(A)の使用。
24.項目1〜14のいずれか一項に記載のプロセスによって得られる化合物(A)。
25.項目15〜21のいずれか一項に記載のプロセスによって得られる化合物(B)。
RGaCl2、又は、それぞれ、R2GaCl/RGaCl2
であり得る化合物(A)の中間段階を介して調製され、式中、Rは、1〜4個の炭素原子のアルキルである。R2GaClとRGaCl2との間の比は、モル量に基づいて10:90〜90:10、特に10:90〜50:50、特に20:80〜40:60の範囲内である。アルキル部分は、分岐状又は非分岐状、好ましくは非分岐状であり得る。Rは、好ましくはエチル及びメチルから選択され、最も好ましくは、Rはメチルである。高速様式の本発明のプロセスにより高い収率及び純度で取得可能なRGaCl2又はR2GaCl/RGaCl2は、本発明のトリアルキルガリウム化合物を調製するために後に使用することができる。
a)アルキルガリウムジクロリド又はR2GaCl/RGaCl2混合物を調製する工程を含む。
b)トリアルキルガリウム化合物を、アルキルガリウムジクロリド又はR2GaCl/RGaCl2混合物から調製する、更なる工程が続き得る。
RGaCl2又はR2GaCl/RGaCl2混合物の調製は、次の反応工程を含む:
a1)ガリウム元素とアルキル供与体とを活性化剤の存在下で反応させてRGaCl2又はR2GaCl/RGaCl2混合物を形成する反応工程、及び
a2)任意に、RGaCl2又はR2GaCl/RGaCl2混合物を反応混合物から単離させる反応工程。
本発明においてアルキル供与体は塩化アルキル、すなわち少なくとも1個の塩素原子並びにアルキル基を含む化合物であることから、本発明におけるアルキル供与体は、アルキル基を含む任意の化合物である。アルキル供与体は、好ましくは、次の一般式:
RCl
を有し、式中、Rは、上記の定義の通りである。この種類のアルキル供与体は、例えばアルキルリチウム化合物と比較して、低コストで利用可能である。Rは、より好ましくはメチル及びエチルから選択され、更に好ましくはメチルである。このように、アルキル供与体が塩化メチルであることが特に好ましい。
RaGabClc
を有する化合物又は化合物の混合物であり、式中、aは、0、1、2、及び3から選択され、bは、1及び2から選択され、cは、0、1、2、及び3から選択されるが、但し、a及びcの両方が0になることはなく、a+b+cが4と等しいか又は4の倍数であること、より好ましくは、a、b、及びcの合計が4と等しいか又は8であることを条件とし、Rは、上記の定義の通りであり、bが1であるとき、a及びcの合計は3であるか、又はbが2であるとき、a及びcの合計は6である。
本発明では反応工程a2)と称される、化合物(A)すなわちRGaCl2又はR2GaCl/RGaCl2混合物の任意の単離は、好ましくは、いかなる未変換の反応体をも反応混合物から分離させること、及び/又はRGaCl2若しくはR2GaCl/RGaCl2混合物を反応容器から除去することを含む。これは、例えば、RGaCl2若しくはR2GaCl/RGaCl2混合物を機械的に除去すること、又は反応容器からRGaCl2若しくはR2GaCl/RGaCl2混合物を昇華させることによって達成され得る。
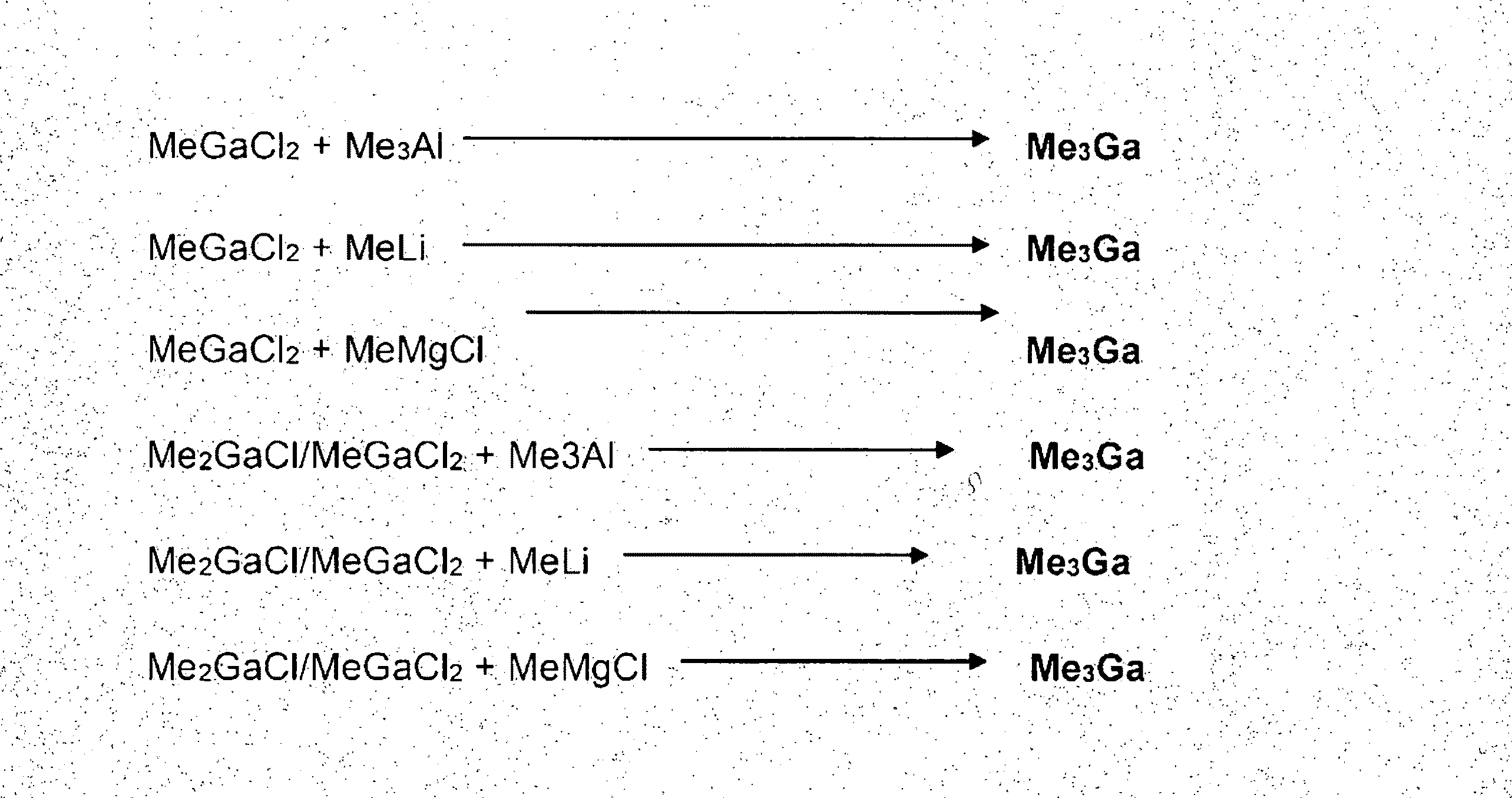
トリアルキルガリウム化合物は、RGaCl2又はR2GaCl/RGaCl2混合物と金属アルキル成分との反応によって、本発明の様式で、RGaCl2又はR2GaCl/RGaCl2混合物から調製される。
RdMeXf
を有し、式中、dは、1、2、及び3から選択され、eは、1及び2から選択され、fは、0、1、2、及び3から選択されるが、但し、d及びfの両方が0になることはないことを条件とする。Rは上記の定義の通りである。Mは、アルミニウム、リチウム、及びマグネシウムから選択され、特に好ましくはアルミニウム又はリチウムである。Xは、Cl、Br、及びIから選択され、好ましくはClである。
トリメチルガリウムを、メチルガリウムジクロリドの中間段階を介して、本発明のプロセスによって調製した。
1.1.活性化剤としてのMe3Ga2Cl3(原位置で形成、ガリウム対活性化剤のモル比:約8.8:1)の存在下でのガリウムと塩化メチルとの反応
1.17gのガリウム(16.8mmol)、0.21gのMe3Ga(1.9mmol)、及び0.32gのGaCl3(1.8mmol)を、磁気撹拌棒と一緒に250mlのParrボンベ(bomb)に量り入れた。この容器を0℃まで冷却し、脱気した。次いで、3.06gの塩化メチル(60.7mmol)を−196℃でParrボンベ内に凝縮させ、室温まで加温した後、Parrボンベを160℃まで加熱した。160℃で24時間後、ガリウムは完全に溶解しており、次いでParrボンベを室温まで冷却し、室温に達したらParrボンベを0℃で脱気して、塩化メチルの残渣を除去し、質量の増加を判定した。その後、このParrボンベをグローブボックス内で開放し、MeGaCl2を機械的に単離させた。質量増加によるMeGaCl2の収量は3.18g(20.4mmol、99%)であった。そのうち、2.57g(16.5mmol、80%)が機械的に単離された。同定はNMR及びIRによるものであった。
1.14gのガリウム(16.4mmol)及び1.46gのMeGaCl2(9.4mmol)を、磁気撹拌棒と一緒に250mlのParrボンベに量り入れた。この容器を0℃まで冷却し、脱気した。次いで、2.99gの塩化メチル(59.2mmol)を−196℃でParrボンベ内に凝縮させ、室温まで加温した後、Parrボンベを160℃まで加熱した。160℃で21時間後、ガリウムは完全に溶解しており、次いでParrボンベを室温まで冷却し、室温に達したらParrボンベを0℃で脱気して、塩化メチルの残渣を除去し、質量の増加を判定した。その後、このParrボンベをグローブボックス内で開放し、MeGaCl2を単離させた。質量増加によるMeGaCl2の収量は4.03g(25.9mmol、99%)であった。そのうち、3.87g(24.9mmol、96%)が機械的に単離された。同定はNMR及びIRによるものであった。
0.70gのガリウム(10.0mmol)及び0.89gのGaCl3(5.0mmol)を、磁気撹拌棒と一緒に125mlのParrボンベに量り入れた。この容器を0℃まで冷却し、脱気した。次いで、1.03gの塩化メチル(20.4mmol)を−196℃でParrボンベ内に凝縮させ、室温まで加温した後、Parrボンベを160℃まで加熱した。160℃で90分後、ガリウムは完全に溶解しており、次いでParrボンベを室温まで冷却し、室温に達したらParrボンベを0℃で脱気して、塩化メチルの残渣を除去し、質量の増加を判定した。その後、このParrボンベをグローブボックス内で開放し、MeGaCl2を単離させた。MeGaCl2の残渣がParrボンベ内に残り、機械的に除去され得なかった。質量増加によるMeGaCl2の収量は2.28g(14.6mmol、98%)であった。そのうち、1.94g(12.5mmol、83%)が機械的に単離された。同定はNMR及びIRによるものであった。
先行する実施例1.3からの125mlのParrボンベを使用した。Parrボンベも磁気撹拌棒も洗浄しなかった。MeGaCl2の残渣がParrボンベ内に残った。0.70gのガリウム(10.0mmol)及び0.89gのGaCl3(5.0mmol)を、磁気撹拌棒と一緒にParrボンベに投入した。この容器を0℃まで冷却し、脱気した。次いで、1.18gの塩化メチル(23.4mmol)を−196℃でParrボンベ内に凝縮させ、室温まで加温した後、この容器を160℃まで加熱した。反応中、溶融物は暗色であった。160℃で60分後、Parrボンベを室温まで冷却し、次いで0℃で脱気して、塩化メチルの残渣を除去し、質量の増加を判定した。その後、このParrボンベをグローブボックス内で開放し、MeGaCl2を単離させた。MeGaCl2の残渣がParrボンベ内に残り、機械的に除去され得なかった。先行するバッチと比較して、単離されたMeGaCl2は茶色を有した。質量増加によるMeGaCl2の収量は2.24gであった(15.4mmol、定量的、使用したGa及びGaCl3に基づく)。そのうち、2.54g(16.3mmol)が機械的に単離された。同定はNMR及びIRによるものであった。
1Lの圧力反応器に、86.63g(1.24mol)のガリウムを最初に投入した。この反応器を三重の脱気/アルゴンの充満によって不活性化(inertize)し、最後の脱気後、クロロメタンを使用して、反応器をアルゴンの代わりに大気圧に戻した。90℃に加熱した滴下漏斗を使用して、198.77gのMe2GaCl/MeGaCl2混合物(前の投入から)を液体形態で反応器内に排出した。反応を開始させるために、反応混合物を撹拌しながら150℃まで加熱し、その後、5バールのクロロメタン(絶対、4バールの過剰圧力に相当)を反応器内に注入した。反応の開始は、気体吸収の発生及び発熱から明らかであった。反応器内へのMeCl供給を、反応圧力を一定の5バールに維持するように制御した。反応器を冷却することによって、反応温度を150℃に維持した。約4時間後、溶融物中にいくらかのガリウムが依然として存在したにもかかわらず、反応の気体吸収は100mL/分未満に低下しており、これが、反応器の過剰圧力を解放し、次いで更に5バールのMeClを注入し、反応を継続させた理由である。更に30分後、ガリウムは反応器内で目に見えなくなっており、反応は気体を消費しなくなった。気体供給を停止させ、反応混合物を室温まで冷却した。反応器内に残っていた過剰圧力を解放し、次いで反応器を脱気して、残りのクロロメタン及び揮発性副生成物を除去し、アルゴンを用いて標準圧力に戻した。
不活性な1Lの圧力反応器に、105.2g(1.51mol)のガリウム及び127.9g(0.73mol)のGaCl3を最初に投入した。反応器を脱気し、クロロメタンを注入した。この反応混合物を撹拌しながら150℃まで加熱し、全てのガリウムが消費され、かつクロロメタンが消費されなくなるまで、4.5絶対バール(bara)の標的過剰圧力で、最大1000mL/分で1.5時間かけて、クロロメタンを反応器内に150℃で導入した。クロロメタンの総消費量は58Lであった。反応器内の過剰圧力を解放し、反応器を冷却し、反応生成物の約1/3を液体形態で反応器からシュレンクフラスコ内に排出した。反応生成物の残部を室温まで冷却し、後の実験で使用するために反応器内で固形物の状態に留めた。
前の実施例からのMe2GaCl/MeGaCl2混合物を含む反応器に、101.6g(1.46mol)のガリウムを投入した。次いで、この反応混合物を再び撹拌しながら150℃まで加熱し、全てのガリウムが消費され、かつクロロメタンが消費されなくなるまで、4.5絶対バールの標的過剰圧力で、最大1000mL/分で2時間かけて、クロロメタンを反応器内に150℃で導入した。クロロメタンの総消費量は55.5Lであった。反応器内の過剰圧力を解放し、反応器を冷却し、100℃において、反応生成物の約1/3を液体形態で反応器からシュレンクフラスコ内に排出した。反応生成物の残部を室温まで冷却し、後の実験で使用するために反応器内で固形物の状態に留めた。単離された生成物を、減圧下の昇華によって精製した。
前の実施例からのMe2GaCl/MeGaCl2混合物を含む反応器に、105.6g(1.51mol)のガリウムを投入した。次いで、この反応混合物を再び撹拌しながら150℃まで加熱し、4.5絶対バールの標的過剰圧力で、クロロメタンを反応器内に150℃で導入した。4時間後、気体消費量は実質的に減少し、この時点で反応器内の圧力を解放し、新しいクロロメタンを使用して、標的過剰圧力を再確立した。全てのガリウムが消費されるまで、更に1.5時間かけてクロロメタンを導入した。クロロメタンの総消費量は55.5Lであった。反応器内の過剰圧力を解放し、反応器を冷却し、100℃において、反応生成物の約1/3を液体形態で反応器からシュレンクフラスコ内に排出した。反応生成物の残部を室温まで冷却し、後の実験で使用するために反応器内で固形物の状態に留めた。単離された生成物は、依然として少量の短鎖炭化水素を不純物として含有し、減圧下の昇華によって精製された。
2.1メチルガリウムジクロリドからトリメチルガリウムへの変換
撹拌器及び70℃に加熱した分離子を備えた500mLフラスコに、保護ガス下で、実施例1.1、1.2、又は1.3からの1.94g(12.5mmol)のMeGaCl2、1.02g(17.5mmol)の乾燥NaCl、及び0.56g(7.5mmol)の乾燥KClを最初に投入した。撹拌しながら、2.56g(2.2ml、12.5mmol)のMe3Al2Cl3を、反応混合物中の温度が130℃超に上昇しないように混加した。その後の加熱中、Me3Gaが、約150℃超で、1.25g(10.9mmol、使用したMeGaCl2に基づいて87.6%の直接収率)の量で単離された。反応温度が200℃超に上昇したらすぐに分離子による生成物の単離を終了し、その後、残りのガリウム含有化合物を、高真空において第2の出口を介して反応混合物から除去した(0.095g、Me3GaとMe2GaClとの混合物)。全収率:94.8%のMe3Ga、5.0%のMe2GaCl、全体的なガリウム変換率:99.8%。
1.39gのガリウム(19.9mmol)及び1.76gのGaCl3(10.0mmol)を、125mLのParrボンベに量り入れ、脱気後、1.87gの塩化メチル(37.0mmol)をParrボンベ内に凝縮させた。Parrボンベを160℃で1時間加熱した。この期間中、ガリウムは完全に溶解し、無色の溶融物が観察された。過剰な塩化メチルをポンプで排出した後、1.60gの質量増加を判定した。これは、Ga/GaCl3混合物からMeGaCl2への定量的な変換に相当する。その後、1.63gのNaCl(27.9mmol)、0.89gのKCl(11.9mmol)、及び4.06gのメチルアルミニウムセスキクロライド(19.7mmol)を、Parrボンベに量り入れた。Parrボンベを一晩130〜140℃に加熱した。室温に冷却した後、揮発性構成物質を、減圧下で−196℃に冷却した冷トラップ内に凝縮させた。この間、Parrボンベを減圧下で160℃まで加熱した。Me2GaClとGaMe3との混合物が冷トラップ内で得られた。より揮発性のGaMe3を、−196℃に冷却した更なる冷トラップ内に、大気圧で凝縮させた。3.46gのMe2GaCl(25.6mmol、86%)及び0.18gのGaMe3(1.6mmol、5%)が単離された。
Claims (14)
- 一般式
RGaCl2、又はRGaCl2とR2GaClとの混合物
のいずれかである、化合物又は混合物(A)を調製するためのプロセスであって、
a1)ガリウムと、凝縮されたアルキル供与体と、活性化剤とを反応容器に投入し、ガリウムと前記アルキル供与体とを活性化剤の存在下で反応させて化合物又は混合物(A)を形成する反応工程と、
a2)任意に、前記化合物又は混合物(A)を前記反応混合物から単離させる反応工程と、を含み、
前記アルキル供与体が、一般式:
RCl
を有し、
前記活性化剤が、次の一般式:
RaGabClc
を有する化合物又は化合物の混合物であり、
式中、Rが、1〜4個の炭素原子の分岐状又は非分岐状アルキルであり、aが、1、2、及び3から選択され、bが、1及び2から選択され、cが、0、1、2、及び3から選択されるが、但し、a+b+cが4と等しいか又は4の倍数であることを条件とし、bが1であるとき、a及びcの合計は3であるか、又はbが2であるとき、a及びcの合計は6である、プロセス。 - 前記活性化剤が、R2GaCl、R3Ga2Cl3、RGaCl2、及びこれらの混合物から選択されるか、又は、R2GaClとRGaCl2との混合物であるか、あるいは、前記反応生成物である化合物又は混合物(A)自体が活性化剤として使用され、Rは請求項1に定義された通りである、請求項1に記載のプロセス。
- アルキル供与体対ガリウムのモル比が少なくとも1.4:1である、請求項1又は2に記載のプロセス。
- ガリウムと活性化剤とのプレミックスが反応工程a1)において反応容器に最初に投入され、前記アルキル供与体が後に添加される、請求項1〜3のいずれか一項に記載のプロセス。
- 反応工程a1)が有機溶剤の非存在下で実施される、請求項1〜4のいずれか一項に記載のプロセス。
- 一般式:
R3Ga
の化合物(B)を調製するためのプロセスであって、請求項1〜5のいずれか一項に記載のプロセスによる化合物又は混合物(A)を提供することと、
b)前記化合物又は混合物(A)を金属アルキル成分と反応させて、一般式:
R3Ga
の化合物(B)を得ることと、を含み、式中、Rは請求項1に定義された通りである、プロセス。 - 前記金属アルキル成分が、一般式:
RdMeXf
を有し、式中、dが、1、2、及び3から選択され、eが、1及び2から選択され、fが、0、1、2、及び3から選択されるが、但し、d及びfの両方が0になることはないことを条件とし、Rは請求項1に定義された通りであり、Mが、アルミニウム、リチウム、及びマグネシウムから選択され、Xが、Cl、Br、及びIから選択される、請求項6に記載のプロセス。 - Mがアルミニウムであり、eが1と等しいか又は2であり、d、e、及びfの合計が4と等しいか又は8であり、dが≠0であり、XがClである、請求項7に記載のプロセス。
- 前記金属アルキル成分が、RMgCl、R2AlCl、R3Al、R3Al2Cl3、及びRLiから選択され、Rは請求項1に定義された通りである、請求項6又は7に記載のプロセス。
- 反応工程b)において補助塩基も添加され、前記補助塩基が、塩化ナトリウム、塩化カリウム、塩化アルミニウム、及びこれらの混合物から選択される、請求項6〜9のいずれか一項に記載のプロセス。
- 化合物(B)の、有機金属化学蒸着法(MOCVD)又は有機金属気相エピタキシー法(MOVPE)のための前駆体としての使用であって、前記使用は、請求項6〜10のいずれか一項に記載のプロセスによって前記化合物(B)を調製することを含む、使用。
- 前記化合物又は混合物(A)が、前記化合物R2GaClとRGaCl2との混合物であり、式中、Rは請求項1に定義された通りであり、R2GaCl対RGaCl2の比が、モル量に基づいて10:90〜90:10である、請求項1〜10のいずれか一項に記載のプロセス。
- Rがメチル又はエチルである、請求項1〜10及び12のいずれか一項に記載のプロセス。
- Rがメチル又はエチルである、請求項11に記載の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14159973.8 | 2014-03-14 | ||
| EP14159973 | 2014-03-14 | ||
| JP2016574486A JP6742921B2 (ja) | 2014-03-14 | 2015-03-12 | トリアルキルガリウム化合物の調製及びその使用 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016574486A Division JP6742921B2 (ja) | 2014-03-14 | 2015-03-12 | トリアルキルガリウム化合物の調製及びその使用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2020037592A true JP2020037592A (ja) | 2020-03-12 |
| JP6890649B2 JP6890649B2 (ja) | 2021-06-18 |
Family
ID=50277091
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016574486A Expired - Fee Related JP6742921B2 (ja) | 2014-03-14 | 2015-03-12 | トリアルキルガリウム化合物の調製及びその使用 |
| JP2019220438A Expired - Fee Related JP6890649B2 (ja) | 2014-03-14 | 2019-12-05 | トリアルキルガリウム化合物の調製及びその使用 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016574486A Expired - Fee Related JP6742921B2 (ja) | 2014-03-14 | 2015-03-12 | トリアルキルガリウム化合物の調製及びその使用 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US10428089B2 (ja) |
| EP (2) | EP3116883B1 (ja) |
| JP (2) | JP6742921B2 (ja) |
| KR (1) | KR102383917B1 (ja) |
| CN (1) | CN106103454B (ja) |
| RU (1) | RU2016140388A (ja) |
| TW (2) | TWI672309B (ja) |
| WO (1) | WO2015136049A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102228897B1 (ko) * | 2017-10-31 | 2021-03-17 | 누리온 케미칼즈 인터내셔널 비.브이. | 갈륨 클로라이드의 저장 및/또는 수송 방법 |
| CN111116618B (zh) * | 2019-12-20 | 2022-06-21 | 南京奥格美化学研究所有限公司 | 制备烷基金属化合物的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02295991A (ja) * | 1989-04-28 | 1990-12-06 | Messer Griesheim Gmbh | ガリウムアルキル化合物の製法 |
| WO2012150229A1 (en) * | 2011-05-03 | 2012-11-08 | Akzo Nobel Chemicals International B.V. | Process for the preparation of trialkyl gallium |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3913165C2 (de) | 1989-04-21 | 2000-05-18 | Merck Patent Gmbh | Metallorganische Verbindungen sowie deren Verwendung zur Herstellung dünner Schichten |
| US5043462A (en) * | 1989-04-28 | 1991-08-27 | Messer Greisheim | Process for the production of gallium-alkyl compounds |
| US5663390A (en) | 1996-08-29 | 1997-09-02 | Libbey-Owens-Ford Co. | Method of producing organo indium chlorides |
| CN1206418A (zh) * | 1996-08-29 | 1999-01-27 | 利比-欧文斯-福特公司 | 生产有机氯化铟的方法 |
| JP4054997B2 (ja) * | 2003-06-19 | 2008-03-05 | 信越化学工業株式会社 | 高純度アルキルガリウムの製造方法 |
| TWI632149B (zh) | 2011-11-28 | 2018-08-11 | 烏明克股份有限兩合公司 | 第iii a族金屬的三烷基化合物之製法 |
| WO2014093419A1 (en) | 2012-12-12 | 2014-06-19 | Dow Global Technologies Llc | Production of tri-alkyl compounds of group 3a metals |
-
2015
- 2015-03-12 EP EP15709677.7A patent/EP3116883B1/de active Active
- 2015-03-12 JP JP2016574486A patent/JP6742921B2/ja not_active Expired - Fee Related
- 2015-03-12 US US15/126,043 patent/US10428089B2/en active Active
- 2015-03-12 RU RU2016140388A patent/RU2016140388A/ru not_active Application Discontinuation
- 2015-03-12 CN CN201580013576.4A patent/CN106103454B/zh not_active Expired - Fee Related
- 2015-03-12 KR KR1020167027983A patent/KR102383917B1/ko active Active
- 2015-03-12 EP EP19190564.5A patent/EP3613748B1/de not_active Not-in-force
- 2015-03-12 WO PCT/EP2015/055211 patent/WO2015136049A1/de not_active Ceased
- 2015-03-13 TW TW108102056A patent/TWI672309B/zh active
- 2015-03-13 TW TW104108161A patent/TWI662044B/zh active
-
2019
- 2019-12-05 JP JP2019220438A patent/JP6890649B2/ja not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02295991A (ja) * | 1989-04-28 | 1990-12-06 | Messer Griesheim Gmbh | ガリウムアルキル化合物の製法 |
| WO2012150229A1 (en) * | 2011-05-03 | 2012-11-08 | Akzo Nobel Chemicals International B.V. | Process for the preparation of trialkyl gallium |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102383917B1 (ko) | 2022-04-08 |
| JP2017512827A (ja) | 2017-05-25 |
| TW201920206A (zh) | 2019-06-01 |
| JP6890649B2 (ja) | 2021-06-18 |
| RU2016140388A (ru) | 2018-04-17 |
| US20170081344A1 (en) | 2017-03-23 |
| US10428089B2 (en) | 2019-10-01 |
| CN106103454B (zh) | 2019-06-14 |
| EP3116883B1 (de) | 2019-08-14 |
| WO2015136049A1 (de) | 2015-09-17 |
| KR20160135244A (ko) | 2016-11-25 |
| CN106103454A (zh) | 2016-11-09 |
| EP3613748A1 (de) | 2020-02-26 |
| EP3116883A1 (de) | 2017-01-18 |
| TWI672309B (zh) | 2019-09-21 |
| TW201542572A (zh) | 2015-11-16 |
| RU2016140388A3 (ja) | 2018-09-19 |
| JP6742921B2 (ja) | 2020-08-19 |
| TWI662044B (zh) | 2019-06-11 |
| EP3613748B1 (de) | 2021-11-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10745421B2 (en) | Method for producing alkyl-indium compounds and the use thereof | |
| KR102581618B1 (ko) | 알킬-인듐 화합물의 제조 방법 및 이의 용도 | |
| KR20140099863A (ko) | Iiia족 금속의 트리알킬 화합물의 제조 방법 | |
| JP6890649B2 (ja) | トリアルキルガリウム化合物の調製及びその使用 | |
| CN105473599B (zh) | 用于生产烷基铟化合物的方法及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20191212 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200928 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200925 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20201203 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20210426 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20210525 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6890649 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |