JP2019501190A - Dglaを含む薬学的組成物及びその使用 - Google Patents
Dglaを含む薬学的組成物及びその使用 Download PDFInfo
- Publication number
- JP2019501190A JP2019501190A JP2018535278A JP2018535278A JP2019501190A JP 2019501190 A JP2019501190 A JP 2019501190A JP 2018535278 A JP2018535278 A JP 2018535278A JP 2018535278 A JP2018535278 A JP 2018535278A JP 2019501190 A JP2019501190 A JP 2019501190A
- Authority
- JP
- Japan
- Prior art keywords
- dgla
- subject
- disease
- baseline
- dose
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/20—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids
- A61K31/202—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids having three or more double bonds, e.g. linolenic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4816—Wall or shell material
- A61K9/4825—Proteins, e.g. gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
Abstract
本開示は、様々な状態及び障害を治療するためのDGLAを含む経口送達可能な薬学的組成物、ならびにその使用方法を提供する。【選択図】図1
Description
本出願は、概して、DGLAを含む薬学的組成物及びその使用方法に関する。
ジホモガンマリノレン酸(DGLA)は、ガンマリノレン酸(GLA)の伸長生成物として体内に自然に見出される必須脂肪酸である。同様に、GLAは、リノール酸の脱飽和生成物である。DGLAの軟ゼラチンカプセル封入は、それが酸化してアルデヒドになりやすく、カプセルシェル内でゼラチンポリマー中のアミノ基と相互作用する可能性があるため、困難である。これは、ゼラチンポリマーの架橋に起因して薬物放出の減速をもたらし得る。
本開示の多くの態様は、以下の図面を参照することでよりよく理解することができる。図面内の構成要素は必ずしも縮尺通りではない。代わりに、本開示の原理を明確に例示することに重点が置かれている。
本開示は、様々な状態及び障害を治療するためのDGLAを含む経口送達可能な薬学的組成物、ならびにその使用方法を提供する。
一態様において、本開示は、必要とする対象において治験責任医師による包括的評価レベルを減少させる方法を提供し、該方法は、1日当たり最大4gのDGLAまたはその誘導体を対象に経口投与することを含む。
別の態様において、本開示は、必要とする対象において湿疹面積及び重症度インデックス(EASI)スコアを減少させる方法を提供し、該方法は、1日当たり最大4gのDGLAまたはその誘導体を対象に経口投与することを含む。
別の態様において、本開示は、必要とする対象においてアトピー性皮膚炎に冒された解剖学的部位の面積のパーセンテージを減少させる方法を提供し、該方法は、1日当たり最大4gのDGLAまたはその誘導体を対象に経口投与することを含む。
別の態様において、本開示は、必要とする対象においてアトピー性皮膚炎スコアリング(SCORAD)を減少させる方法を提供し、該方法は、1日当たり最大4gのDGLAまたはその誘導体を対象に経口投与することを含む。
別の態様において、本開示は、必要とする対象においてアトピー性皮膚炎に冒された体表面積を減少させる方法を提供し、該方法は、1日当たり最大4gのDGLAまたはその誘導体を対象に経口投与することを含む。
別の態様において、本開示は、必要とする対象において視覚的アナログ尺度(VAS)掻痒スコアを減少させる方法を提供し、該方法は、1日当たり最大4gのDGLAまたはその誘導体を対象に経口投与することを含む。
別の態様において、本開示は、必要とする対象において疾患または障害を治療する方法を提供し、該方法は、DGLAまたはその誘導体を含む薬学的組成物を対象に経口投与することを含む。
いくつかの実施形態において、1日当たり約0.2〜約3gのDGLAまたはその誘導体が対象に経口投与される。他の実施形態において、1日当たり約0.5g、約1g、または約2gのDGLAまたはその誘導体が対象に経口投与される。さらに他の実施形態において、1日当たり1g未満のDGLAまたはその誘導体が対象に経口投与される。さらに他の実施形態において、組成物は、1日当たり約1g〜約4gのDGLAまたはその誘導体を提供するのに十分な量で対象に投与される。
いくつかの実施形態において、対象は小児対象である。
いくつかの実施形態において、DGLAまたはその誘導体は、少なくとも約2週間、少なくとも約4週間、または少なくとも約8週間の期間対象に投与される。
いくつかの実施形態において、薬学的組成物は、液体または半液体形態のDGLAまたはその誘導体を含む。
いくつかの実施形態において、対象は、例えば、基準レベルに基づいてまたは基準レベルと比較して決定される、低い好酸球細胞数を有する。
いくつかの実施形態において、疾患または障害は以下から選択される:尋常性ざ瘡、酒さ性ざ瘡、軽度、中程度、もしくは重度のアトピー性皮膚炎、乾癬、掻痒/かゆみ、放射線防護、皮膚乾燥、滑らかな皮膚、健康な皮膚、抗老化、及び光防護を含む皮膚障害及び皮膚疾患;膀胱癌、膀胱瘤、血尿、間質性膀胱炎、神経因性膀胱、ペロニー病、前立腺疾患、失禁、尿路感染症、及び膀胱尿管逆流を含む泌尿器障害及び泌尿器疾患;腎不全、急性腎傷害、慢性腎疾患、及び多発性嚢胞腎を含む腎疾患及び腎障害;強直性脊椎炎、線維筋痛症、痛風、感染性関節炎、狼瘡、変形性関節症、リウマチ性多発筋痛症、乾癬性関節炎、反応性関節炎、関節リウマチ、強皮症を含むリウマチ性疾患;炎症性肺疾患、気道感染症、胸膜腔疾患、肺血管疾患、肺炎、肺塞栓症、及び肺癌を含む呼吸器疾患;ならびに急性心虚血事象、急性心筋梗塞、狭心症、不整脈、心房細動、粥状動脈硬化、動脈細動、心不全、心血管疾患、慢性心不全、慢性安定狭心症、うっ血性心不全、冠動脈疾患、冠動脈性心疾患、深部静脈血栓症、糖尿病、真性糖尿病、糖尿病性神経障害、真性糖尿病を有する対象における拡張障害、浮腫、本態性高血圧、最終的な肺塞栓症、脂肪性肝疾患、心疾患、心不全、ホモ接合性家族性高コレステロール血症(HoFH)、ホモ接合性家族性シトステロール血症、高コレステロール血症、高脂血症、高血圧症、高トリグリセリド血症、代謝症候群、混合型脂質異常症、中程度から軽度の心不全、心筋梗塞、肥満管理、発作性心房細動/動脈細動/細動/粗動、発作性上室性頻拍(PSVT)、特に重度のまたは急激に発症する浮腫、血小板凝集、原発性高コレステロール血症、原発性高脂血症、肺動脈性高血圧症、肺高血圧症、再発性の血行動態的に不安定な心室性頻脈(VT)、再発性心室性不整脈、再発心室細動(VF)、破裂動脈瘤、シトステロール血症、脳卒中、上室性頻拍、症候性心房細動/粗動、頻拍、II型糖尿病、血管疾患、静脈血栓塞栓症、ならびに心室性不整脈を含む心血管障害。
いくつかの実施形態において、薬学的組成物は、カプセルシェルに封入されたDGLAまたはその誘導体を含む。
いくつかの実施形態において、薬学的組成物は、液体または半液体形態のDGLAまたはその誘導体を含む。
いくつかの実施形態において、組成物は、1日当たり1〜8カプセル投与される。
いくつかの実施形態において、各カプセルは、約200mg〜約1gのDGLAまたはその誘導体を含む。
いくつかの実施形態において、DGLAまたはその誘導体は、少なくとも約2週間、少なくとも約4週間、または少なくとも約8週間の期間対象に投与される。
一実施形態において、本開示は、DGLAを含む薬学的組成物を提供する。一実施形態において、組成物はカプセルシェルに封入される。一実施形態において、本開示は、ゼラチン、D−ソルビトール、及び1,4−ソルビタン糖アルコールを含むカプセルシェルに封入されたDGLAを含む薬学的組成物を提供する。一実施形態において、約500mg〜約1gのDGLAまたはその誘導体がカプセルシェルに封入される。
一実施形態において、本開示は、必要とする対象において皮膚疾患または皮膚障害を治療する方法を提供し、該方法は、DGLAを含む薬学的組成物を対象に経口投与することを含む。任意選択的に、薬学的組成物は、ゼラチン、D−ソルビトール、及び1,4−ソルビタン糖アルコールを含むカプセルシェルに封入されたDGLAを含む。任意選択的に、組成物は、1日当たり約1g〜約4gのDGLAを提供するのに十分な量で対象に投与される。一実施形態において、ゼラチンは、約9500〜約11000、例えば、約9775または約10,500のゲル質量粘度を有する。別の実施形態において、ゼラチンは、約165〜約190、例えば、約170〜約185のブルームを有する。別の実施形態において、ゼラチンは、約0.33より大きい灰分パーセンテージを有する。
一実施形態において、本開示は、必要とする対象において過活動膀胱を治療する方法を提供する。該方法は、DGLAを含む薬学的組成物を対象に経口投与することを含む。任意選択的に、薬学的組成物は、ゼラチン、D−ソルビトール、及び1,4−ソルビタン糖アルコールを含むカプセルシェルに封入されたDGLAを含む。任意選択的に、組成物は、1日当たり約1g〜約4gのDGLAを提供するのに十分な量で対象に投与される。一実施形態において、ゼラチンは、約9500〜約11000、例えば、約9775または約10,500のゲル質量粘度を有する。別の実施形態において、ゼラチンは、約165〜約190、例えば、約170〜約185のブルームを有する。別の実施形態において、ゼラチンは、約0.33より大きい灰分パーセンテージを有する。本発明のこれら及び他の実施形態を、以下により詳細に記載する。
本発明は種々の形態において具現化することが可能であるが、いくつかの実施形態の以下の説明は、本開示が本発明の例示として見なされるべきであり、本発明が示される特定の実施形態に限定されることを意図するものではないという理解によって行われる。見出しは、便宜的に提供されているに過ぎず、本発明をいかようにも限定すると解釈されるべきではない。任意の見出しの下に示される実施形態は、他のいずれかの見出しの下に示される実施形態と組み合わされてもよい。
本出願において特定される種々の定量値における数値の使用は、明示的な別段の規定がない限り、特定された範囲内の最小値及び最大値の両方に「約」という単語が前に付されているかのように近似値として記載される。このように、記載される値からのわずかな変動を使用して、記載される値と実質的に同じ結果を達成することができる。また、範囲の開示は、列挙される最小値と最大値との間のあらゆる値を含む連続的な範囲、及びそのような値によって形成され得る任意の範囲として意図される。また、本明細書で、列挙される数値を任意の他の列挙される数値に分割することによって形成され得るあらゆる全ての比(及び任意のそのような比の範囲)も開示される。したがって、当業者は、多くのそのような比、範囲、及び比の範囲が、本明細書に提示される数値から明確に得ることができ、全ての場合において、そのような比、範囲、及び比の範囲が、本発明の種々の実施形態を表すことを理解するであろう。
組成物
種々の実施形態において、本開示は、DGLAまたはその誘導体を含む経口送達可能な薬学的組成物を提供する。本明細書におけるDGLAという用語は、遊離酸形態のDGLAを指す。本発明の組成物はまた、DGLAに加えてまたはDGLAの代わりにDGLA誘導体を含んでもよい。そのような誘導体は、アルキルエステル、メチルエステルもしくはDGLAエチルエステル等の低級アルキルエステル、またはトリグリセリド形態のDGLAを含む。一実施形態において、本開示は、カプセルシェルに封入されたDGLAまたはその誘導体を含む薬学的組成物を提供する。一実施形態において、約500mg〜約1gのDGLAまたはその誘導体がカプセルシェルに封入される。
種々の実施形態において、本開示は、DGLAまたはその誘導体を含む経口送達可能な薬学的組成物を提供する。本明細書におけるDGLAという用語は、遊離酸形態のDGLAを指す。本発明の組成物はまた、DGLAに加えてまたはDGLAの代わりにDGLA誘導体を含んでもよい。そのような誘導体は、アルキルエステル、メチルエステルもしくはDGLAエチルエステル等の低級アルキルエステル、またはトリグリセリド形態のDGLAを含む。一実施形態において、本開示は、カプセルシェルに封入されたDGLAまたはその誘導体を含む薬学的組成物を提供する。一実施形態において、約500mg〜約1gのDGLAまたはその誘導体がカプセルシェルに封入される。
一実施形態において、カプセルシェルは、ゼラチン、例えば、より分子量の低いゼラチンRXLまたは石灰骨ゼラチンを含む。別の実施形態において、カプセルシェルは、ゼラチンパターンを切断してその分子量を効果的に減少させるために、タンパク質分解酵素によって処理されたゼラチンRXLを含む。別の実施形態において、薬学的組成物は、D−ソルビトール及び1,4−ソルビタンのDGLAエステルを含む。一実施形態において、カプセルシェルは、(a)ゼラチンと、(b)d−ソルビトール及び1,4−ソルビタンのうちの1つ以上から選択される可塑剤とを含む。一実施形態において、ゼラチンは、米国特許第7,485,323号(参照によりその全体が本明細書に組み込まれる)に記載されるようなものである。
一実施形態において、可塑剤は、20%〜30%、例えば、約24%及び28%(無水ベースで)の量の1−4ソルビタンと、約30%〜50%、例えば、約35%〜45%(無水ベースで)のD−ソルビトール含有量とを含む。
いくつかの実施形態において、カプセルシェルは、グリセロール、精製水、二酸化チタン、中鎖トリグリセリド、及びレシチンをさらに含む。
種々の実施形態において、DGLAまたは誘導体は、本発明の組成物中に約50mg〜約5000mg、約75mg〜約2500mg、または約100mg〜約1000mg、例えば、約75mg、約100mg、約125mg、約150mg、約175mg、約200mg、約225mg、約250mg、約275mg、約300mg、約325mg、約350mg、約375mg、約400mg、約425mg、約450mg、約475mg、約500mg、約525mg、約550mg、約575mg、約600mg、約625mg、約650mg、約675mg、約700mg、約725mg、約750mg、約775mg、約800mg、約825mg、約850mg、約875mg、約900mg、約925mg、約950mg、約975mg、約1000mg、約1025mg、約1050mg、約1075mg、約1100mg、約1025mg、約1050mg、約1075mg、約1200mg、約1225mg、約1250mg、約1275mg、約1300mg、約1325mg、約1350mg、約1375mg、約1400mg、約1425mg、約1450mg、約1475mg、約1500mg、約1525mg、約1550mg、約1575mg、約1600mg、約1625mg、約1650mg、約1675mg、約1700mg、約1725mg、約1750mg、約1775mg、約1800mg、約1825mg、約1850mg、約1875mg、約1900mg、約1925mg、約1950mg、約1975mg、約2000mg、約2025mg、約2050mg、約2075mg、約2100mg、約2125mg、約2150mg、約2175mg、約2200mg、約2225mg、約2250mg、約2275mg、約2300mg、約2325mg、約2350mg、約2375mg、約2400mg、約2425mg、約2450mg、約2475mg、または約2500mgの量で存在する。任意のそのような実施形態において、組成物は、D−ソルビトール及び1,4−ソルビタンのDGLAエステルをさらに含むことができる。
一実施形態において、本発明の組成物は、全脂肪酸の約10重量%以下、約9重量%以下、約8重量%以下、約7重量%以下、約6重量%以下、約5重量%以下、約4重量%以下、約3重量%以下、約2重量%以下、約1重量%、または約0.5重量%以下のDGLA以外の脂肪酸を含有する。
別の実施形態において、DGLAまたはその誘導体は、本発明の組成物中に存在する全ての脂肪酸の少なくとも約30重量%、約40重量%、約50重量%、少なくとも約60重量%、少なくとも約70重量%、少なくとも約80重量%、少なくとも約90重量%、少なくとも約95重量%、少なくとも約97重量%、少なくとも約98重量%、少なくとも約99重量%、または100重量%に相当する。
一実施形態において、本発明の組成物は、例えば、媒体として水を用いて、USP2040(Disintegration and Dissolution of Dietary Supplements)に規定されるような標準的な崩壊試験を行った場合、40℃/75%相対湿度で約1ヶ月、約2ヶ月、または約3ヶ月保存した後、約60分未満、約50分未満、約40分未満、約30分未満、または20分未満のDGLA放出速度を有する。
一実施形態において、40℃/75%相対湿度で約1ヶ月、約2ヶ月、約3ヶ月または約6ヶ月保存した後、本発明の組成物は、全ての脂肪酸の約5重量%未満のDGLAエステル、全ての脂肪酸の約4重量%未満のDGLAエステル、全ての脂肪酸の約3重量%未満のDGLAエステル、全ての脂肪酸の約2重量%未満のDGLAエステル、または全ての脂肪酸の約1重量%未満のDGLAエステルを含む。
方法
本明細書において上述した組成物または本開示の種々の実施形態を組み合わせることによって配合することができる組成物を含む本発明の任意の組成物は、尋常性ざ瘡、酒さ性ざ瘡、アトピー性皮膚炎、乾癬、掻痒/かゆみ、放射線防護、皮膚乾燥、滑らかな皮膚、健康な皮膚、抗老化、及び光防護を含む皮膚障害及び皮膚疾患;膀胱癌、膀胱瘤、血尿、間質性膀胱炎、神経因性膀胱、ペロニー病、前立腺疾患、失禁、尿路感染症、及び膀胱尿管逆流を含む泌尿器障害及び泌尿器疾患;腎不全、急性腎傷害、慢性腎疾患、及び多発性嚢胞腎を含む腎疾患及び腎障害;強直性脊椎炎、線維筋痛症、痛風、感染性関節炎、狼瘡、変形性関節症、リウマチ性多発筋痛症、乾癬性関節炎、反応性関節炎、関節リウマチ、強皮症を含むリウマチ性疾患;炎症性肺疾患、気道感染症、胸膜腔疾患、肺血管疾患、肺炎、肺塞栓症、及び肺癌を含む呼吸器疾患;ならびに急性心虚血事象、急性心筋梗塞、狭心症、不整脈、心房細動、粥状動脈硬化、動脈細動、心不全、心血管疾患、慢性心不全、慢性安定狭心症、うっ血性心不全、冠動脈疾患、冠動脈性心疾患、深部静脈血栓症、糖尿病、真性糖尿病、糖尿病性神経障害、真性糖尿病を有する対象における拡張障害、浮腫、本態性高血圧、最終的な肺塞栓症、脂肪性肝疾患、心疾患、心不全、ホモ接合性家族性高コレステロール血症(HoFH)、ホモ接合性家族性シトステロール血症、高コレステロール血症、高脂血症、高血圧症、高トリグリセリド血症、代謝症候群、混合型脂質異常症、中程度から軽度の心不全、心筋梗塞、肥満管理、発作性心房細動/動脈細動/細動/粗動、発作性上室性頻拍(PSVT)、特に重度のまたは急激に発症する浮腫、血小板凝集、原発性高コレステロール血症、原発性高脂血症、肺動脈性高血圧症、肺高血圧症、再発性の血行動態的に不安定な心室性頻脈(VT)、再発性心室性不整脈、再発心室細動(VF)、破裂動脈瘤、シトステロール血症、脳卒中、上室性頻拍、症候性心房細動/粗動、頻拍、II型糖尿病、血管疾患、静脈血栓塞栓症、心室性不整脈、ならびに他の心血管イベントを含む心血管障害の治療または予防において使用することができる。
本明細書において上述した組成物または本開示の種々の実施形態を組み合わせることによって配合することができる組成物を含む本発明の任意の組成物は、尋常性ざ瘡、酒さ性ざ瘡、アトピー性皮膚炎、乾癬、掻痒/かゆみ、放射線防護、皮膚乾燥、滑らかな皮膚、健康な皮膚、抗老化、及び光防護を含む皮膚障害及び皮膚疾患;膀胱癌、膀胱瘤、血尿、間質性膀胱炎、神経因性膀胱、ペロニー病、前立腺疾患、失禁、尿路感染症、及び膀胱尿管逆流を含む泌尿器障害及び泌尿器疾患;腎不全、急性腎傷害、慢性腎疾患、及び多発性嚢胞腎を含む腎疾患及び腎障害;強直性脊椎炎、線維筋痛症、痛風、感染性関節炎、狼瘡、変形性関節症、リウマチ性多発筋痛症、乾癬性関節炎、反応性関節炎、関節リウマチ、強皮症を含むリウマチ性疾患;炎症性肺疾患、気道感染症、胸膜腔疾患、肺血管疾患、肺炎、肺塞栓症、及び肺癌を含む呼吸器疾患;ならびに急性心虚血事象、急性心筋梗塞、狭心症、不整脈、心房細動、粥状動脈硬化、動脈細動、心不全、心血管疾患、慢性心不全、慢性安定狭心症、うっ血性心不全、冠動脈疾患、冠動脈性心疾患、深部静脈血栓症、糖尿病、真性糖尿病、糖尿病性神経障害、真性糖尿病を有する対象における拡張障害、浮腫、本態性高血圧、最終的な肺塞栓症、脂肪性肝疾患、心疾患、心不全、ホモ接合性家族性高コレステロール血症(HoFH)、ホモ接合性家族性シトステロール血症、高コレステロール血症、高脂血症、高血圧症、高トリグリセリド血症、代謝症候群、混合型脂質異常症、中程度から軽度の心不全、心筋梗塞、肥満管理、発作性心房細動/動脈細動/細動/粗動、発作性上室性頻拍(PSVT)、特に重度のまたは急激に発症する浮腫、血小板凝集、原発性高コレステロール血症、原発性高脂血症、肺動脈性高血圧症、肺高血圧症、再発性の血行動態的に不安定な心室性頻脈(VT)、再発性心室性不整脈、再発心室細動(VF)、破裂動脈瘤、シトステロール血症、脳卒中、上室性頻拍、症候性心房細動/粗動、頻拍、II型糖尿病、血管疾患、静脈血栓塞栓症、心室性不整脈、ならびに他の心血管イベントを含む心血管障害の治療または予防において使用することができる。
所与の疾患または障害に関連する「治療」という用語は、限定されないが、疾患もしくは障害を抑制すること、例えば、疾患もしくは障害の進行を停止すること;疾患もしくは障害を緩和すること、例えば、疾患もしくは障害の退行をもたらすこと;疾患もしくは障害によって引き起こされるまたは疾患もしくは障害に起因する状態を緩和すること、例えば、疾患もしくは障害の症状を緩和、予防、もしくは治療することを含む。所与の疾患または障害に関連する「予防」という用語は、何も起こっていない場合は疾患進行の開始を予防すること、障害もしくは疾患に罹患しやすい素因を有し得るが、まだ障害もしくは疾患を有すると診断されていない対象において疾患もしくは障害が起こるのを予防すること、及び/または、既に存在する場合、さらなる疾患/障害の進行を予防することを意味する。
一実施形態において、対象は、基準レベルと比較して少ないベースラインの好酸球数を有すると決定される。一実施形態において、対象は、DGLAの投与前に少ないベースラインの好酸球数を有すると決定される。
「基準レベル」という用語は、限定されないが、健康な患者から採取された試料からのレベルを含む。基準レベルはまた、健康な患者の集団から採取された複数の試料から決定されてもよい。一例として、少ない好酸球細胞数は、健康な患者の集団または健康な患者のサブセット、例えば、特定の民族性の健康な患者から決定された好酸球細胞数に基づいて決定することができる。他の実施形態において、基準レベルは、治療を受けている同じ患者から、より初期の時点(例えば、1日、3日、1週、1ヶ月、3ヶ月、6ヶ月、12ヶ月、またはそれ以上)で採取された試料から決定される値である。いくつかの実施形態において、基準レベルは、当業者に既知の値に基づいていてもよいか、または医療機関によって開発されてもよい。
種々の実施形態において、本発明の組成物は、1日当たり約50mg〜約10000mg、約100mg〜約7500mg、または約100mg〜約5000mg、例えば、約200mg、約300mg、約400mg、約500mg、約600mg、約700mg、約800mg、約900mg、約1000mg、約1100mg、約1200mg、約1300mg、約1400mg、約1500mg、約1600mg、約1700mg、約1800mg、約1900mg、約2000mg、約2100mg、約2200mg、約2300mg、約2400mg、約2500mg、訳2600mg、約2700mg、約2800mg、約2900mg、約3000mg、約3100mg、約3200mg、約3300mg、約3400mg、約3500mg、3600mg、約3700mg、約3800mg、約3900mg、約4000mg、約4100mg、約4200mg、約4300mg、約4400mg、約4500mg、4600mg、約4700mg、約4800mg、約4900mg、約5000mg、約5100mg、約5200mg、約5300mg、約5400mg、約5500mgのDGLAの、1日DGLA用量を提供するのに十分な量で投与される。
一実施形態において、本発明は、アトピー性皮膚炎、例えば、軽度から中程度のアトピー性皮膚炎を治療する方法を提供する。一実施形態において、該方法は、そのような治療を必要とする対象に、1日当たり約500mg〜約3g、1日当たり約1g〜約2.5g、1日当たり約1g、または1日当たり約2gの量のDGLAを投与することを含む。一実施形態において、DGLAは、少なくとも約2週間、少なくとも約4週間、または少なくとも約8週間の期間、毎日対象に投与される。関連する実施形態において、例えば、約1〜約12週間、約1〜約8週間、または約1〜約4週間の期間にわたって、本発明に従って治療を行うと、対象または対象群は、以下の結果のうちの1つ以上を呈する:
(a)ベースラインもしくはプラセボ対照に対する湿疹面積及び重症度インデックス(EASI)スコアの減少、
(b)ベースラインもしくは対照に対するアトピー性皮膚炎に冒された解剖学的部位の面積のパーセンテージの減少、
(c)ベースラインもしくはプラセボ対照に対する治験責任者の包括的評価スコアにおける減少、
(d)ベースラインもしくはプラセボ対照に対する紅斑、浮腫/集団、滲出/痂皮、表皮剥離、苔癬化、及び/もしくは乾燥の強度の減少、
(e)ベースラインもしくはプラセボ対照に対する紅斑、浮腫/集団、滲出/痂皮、表皮剥離、苔癬化、及び/もしくは乾燥の減少、
(f)ベースラインもしくはプラセボ対照に対するアトピー性皮膚炎に冒された体表面積(BSA)の減少、
(g)ベースラインもしくはプラセボ対照に対する不眠の減少、
(h)ベースラインもしくはプラセボ対照に対する掻痒症(痒み)の発生の減少、
(i)視覚的アナログ尺度での過去3日及び/もしくは3夜の平均としての掻痒症の重症度の減少、
(j)ベースラインもしくはプラセボ対照に対するSCORADスコアの減少、
(k)ベースラインもしくはプラセボ対照と比較した患者自身による湿疹評価(POEM)の改善、
(l)前週に対象が湿疹に起因して皮膚が痒いと報告した日数の減少、
(m)前週に対象が自身の湿疹に起因して自身の睡眠が妨げられたと報告した日数の減少、
(n)前週に対象が皮膚の出血を経験した日数の減少、
(o)前週に対象が皮膚からの透明な体液の浸出もしくは滲出を経験した日数の減少、
(p)前週に対象の皮膚がひび割れた日数の減少、
(q)前週に対象の皮膚が剥がれ落ちた日数の減少、
(r)前週に対象が皮膚の乾燥を経験した日数の減少、
(s)ベースラインもしくはプラセボ対照と比較した経皮水分損失の増加、
(t)ベースラインと比較した血漿中総DGLA及び遊離DGLAの増加、
(u)ベースラインもしくはプラセボ対照と比較したDGLA:AA比の増加、ならびに/または
(v)ベースラインもしくはプラセボ対照と比較した動脈圧の減少。
一実施形態において、本発明の方法は、対象または対象群に投薬する前に、(a)〜(v)に記載される1つ以上のマーカーまたはパラメーターのベースラインレベルを測定することを含む。別の実施形態において、該方法は、(a)〜(v)に記載される1つ以上のマーカーまたはパラメーターのベースラインレベルが決定された後に、本明細書に開示される組成物を対象に投与することと、続いて前記1つ以上のマーカーのさらなる測定を行うこととを含む。
別の実施形態において、例えば、約1〜約12週間、約1〜約8週間、または約1〜約4週間の期間にわたって、本発明の組成物を用いて治療を行うと、対象または対象群は、すぐ上に記載される転帰(a)〜(v)のうちのいずれか2個以上、いずれか3個以上、いずれか4個以上、いずれか5個以上、いずれか6個以上、いずれか7個以上、いずれか8個以上、いずれか9個以上、いずれか10個以上、いずれか11個以上、いずれか12個以上、いずれか13個以上、いずれか14個以上、いずれか15個以上、いずれか16個以上、いずれか17個以上、いずれか18個以上、いずれか19個以上、いずれか20個以上、いずれか21個以上、または22個全てを呈する。
別の実施形態において、本発明の組成物を用いて治療を行うと、対象または対象群は、以下の結果のうちの1つ以上を呈する:
(a)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対する湿疹面積及び重症度インデックス(EASI)スコアの減少、
(b)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくは対照に対するアトピー性皮膚炎に冒された解剖学的部位の面積のパーセンテージの減少、
(c)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対する治験責任者の包括的評価スコアにおける減少、
(d)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対する紅斑、浮腫/集団、滲出/痂皮、表皮剥離、苔癬化、及び/もしくは乾燥の強度の減少、
(e)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対する紅斑、浮腫/集団、滲出/痂皮、表皮剥離、苔癬化、及び/もしくは乾燥の減少、
(f)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対するアトピー性皮膚炎に冒された体表面積(BSA)の減少、
(g)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対する不眠の減少、
(h)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対する掻痒症(痒み)の発生の減少、
(i)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、視覚的アナログ尺度での過去3日及び/もしくは3夜の平均としての掻痒症の重症度の減少、
(j)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照に対するSCORADスコアの減少、
(k)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照と比較した患者自身による湿疹評価(POEM)の改善、
(l)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、前週に対象が湿疹のために皮膚が痒いと報告した日数の減少、
(m)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、前週に対象が自身の湿疹に起因して自身の睡眠が妨げられたと報告した日数の減少、
(n)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、前週に対象が皮膚の出血を経験した日数の減少、
(o)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、前週に対象が皮膚からの透明な体液の浸出もしくは滲出を経験した日数の減少、
(p)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、前週に対象の皮膚がひび割れた日数の減少、
(q)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、前週に対象の皮膚が剥がれ落ちた日数の減少、
(r)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、前週に対象が皮膚の乾燥を経験した日数の減少、
(s)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照と比較した経皮水分損失の増加、
(t)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%のベースラインと比較した血漿中総DGLA及び遊離DGLAの増加、ならびに/または
(u)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、ベースラインもしくはプラセボ対照と比較したDGLA:AA比の増加、ならびに/または
(v)少なくとも約5%、少なくとも約10%、少なくとも約15%、少なくとも約20%、少なくとも約25%、少なくとも約30%、少なくとも約35%、少なくとも約40%、少なくとも約45%、少なくとも約50%、少なくとも約55%、少なくとも約60%、少なくとも約65%、少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、もしくは少なくとも約95%の、平均動脈圧の減少。
別の実施形態において、例えば、約1〜約12週間、約1〜約8週間、または約1〜約4週間の期間にわたって、単一用量投与または複数用量投与後に本発明の組成物を用いて治療を行うと、対象または対象群は、すぐ上に記載される転帰(a)〜(v)のうちのいずれか2個以上、いずれか3個以上、いずれか4個以上、いずれか5個以上、いずれか6個以上、いずれか7個以上、いずれか8個以上、いずれか9個以上、いずれか10個以上、いずれか11個以上、いずれか12個以上、いずれか13個以上、いずれか14個以上、いずれか15個以上、いずれか16個以上、いずれか17個以上、いずれか18個以上、いずれか19個以上、いずれか20個以上、いずれか21個以上、または22個全てを呈する。
別の実施形態において、約200mgのDGLA〜約8000mgのDGLA(1つ以上の投薬単位として、例えば、約500mg、約1000mg、約2000mg、約3000mg、約4000mg、約5000mg、約6000mg、約7000mg、または約8000mgの合計1日DGLA用量に等しい500mgまたは1gの投薬単位として)を含む組成物を用いて対象または対象群(摂食または絶食)の治療を行うと、単一用量投与後または複数用量投与後に、対象または対象群は、以下の結果のうちの1つ以上を呈する:
(a)約400ng/ml〜約4500ng/ml、約500ng/ml〜約3400ng/ml、約600ng/ml〜約3300ng/ml、約700ng/ml〜約3200ng/ml、例えば、約900ng/ml、約1000ng/ml、約1100ng/ml、約1200ng/ml、約1300ng/ml、約1400ng/ml、約1500ng/ml、約1600ng/ml、約1700ng/ml、約1800ng/ml、約1900ng/ml、約2000ng/ml、約2100ng/ml、約2200ng/ml、約2300ng/ml、約2400ng/ml、約2500ng/ml、約2600ng/ml、約2700ng/ml、約2800ng/ml、約2900ng/ml、約3000ng/ml、約3100ng/ml、約3200ng/ml、約3300ng/ml、約3400ng/ml、約3500ng/ml、約3600ng/ml、約3700ng/ml、約3800ng/ml、約3900ng/ml、約4000ng/ml、約4100ng/ml、約4200ng/ml、約4300ng/ml、約4400ng/ml、または約4500ng/mlの遊離DGLA Cmax(または平均CmaxもしくはCmax中央値)、
(b)約0.5(1/kL)〜約3(1/kL)、約0.6(1/kL)〜約2.5(1/kL)または約0.7(1/kL)〜約2(1/kL)、例えば、約0.7(1/kL)、約0.8(1/kL)、約0.9(1/kL)、約1(1/kL)、約1.5(1/kL)、約1.6(1/kL)、約1.7(1/kL)または約1.8(1/kL)の遊離DGLA Cmax/用量(または平均Cmax/用量もしくはCmax中央値/用量)、
(c)約1500ng・時間/ml〜約12000ng・時間/ml、約2000ng・時間/ml〜約11000ng・時間/ml、または約2500ng・時間/ml〜約10000ng・時間/ml、例えば、約1000ng・時間/ml、約1500ng・時間/ml、約2000ng・時間/ml、約2500ng・時間/ml、約3000ng・時間/ml、約3500ng・時間/ml、約4000ng・時間/ml、約4500ng・時間/ml、約5000ng・時間/ml、約5500ng・時間/ml、約6000ng・時間/ml、約6500ng・時間/ml、約7000ng・時間/ml、約7500ng・時間/ml、約8000ng・時間/ml、約8500ng・時間/ml、約9000ng・時間/ml、約9500ng・時間/ml、約10000ng・時間/ml、約10500ng・時間/ml、約11000ng・時間/ml、約11500ng・時間/ml、または約12000ng・時間/mlの遊離DGLA AUC0−24(または平均AUC0−24もしくはAUC0−24中央値)、
(d)約1.5〜約10時間/kL、約1.7〜約8時間/kL、または約2〜約6時間/kL、例えば、約2時間/kL、約2.5時間/kL、約3時間/kL、約3.5時間/kL、約4時間/kL、約4.5時間/kL、約5時間/kL、または約5.5時間/kLの遊離DGLA AUC0−24/用量(または平均AUC0−24/用量もしくはAUC0−24中央値/用量)、
(e)約2〜約10時間、約3〜約8時間、例えば約3時間、約4時間、約5時間、約6時間、約7時間、または約8時間の遊離DGLA tmax(h)、
(f)約4000ng/ml〜約45000ng/ml、約5000ng/ml〜約34000ng/ml、約6000ng/ml〜約33000ng/ml、または約7000ng/ml〜約32000ng/ml、例えば、約7000ng/ml、約7200ng/ml、約7500ng/ml、約8000ng/ml、約8500ng/ml、約9000ng/ml、約9500ng/ml、約10000ng/ml、約11000ng/ml、約12000ng/ml、約13000ng/ml、約14000ng/ml、約15000ng/ml、約16000ng/ml、約17000ng/ml、約18000ng/ml、約19000ng/ml、約20000ng/ml、約21000ng/ml、約22000ng/ml、約23000ng/ml、約24000ng/ml、約25000ng/ml、約26000ng/ml、約27000ng/ml、約28000ng/ml、約29000ng/ml、約30000ng/ml、約31000ng/ml、約32000ng/ml、約33000ng/ml、約34000ng/ml、または約35000ng/mlの総DGLA Cmax(または平均総DGLA Cmaxまたは総DGLA Cmax中央値)、
(g)約2(1/kL)〜約25(1/kl)、約4(1/kl)〜約20(1/kl)、または約5(1/kl)〜約17(1/kl)、例えば、約6(1/kl)、約9(1/kl)、約14(1/kl)、または約16(1/kl)の総DGLA Cmax/用量(または平均総DGLA Cmax/用量もしくは総DGLA Cmax/用量中央値)、
(h)約15000ng・時間/ml〜約900,000ng・時間/ml、約20,000ng・時間/ml〜約250,000ng・時間/ml、または約25,000ng・時間/ml〜約225,000ng・時間/ml、例えば、約40,000ng・時間/ml、約210,000ng・時間/ml、約215,000ng・時間/ml、または約435,000ng・時間/mlの総DGLA AUC0−24(または平均DGLA AUC0−24もしくはDGLA AUC0−24中央値)、
(i)50〜約400時間/kL、約60〜約250時間/kL、または約70〜約225時間/kL、例えば、約80時間/kL、約100時間/kL、約110時間/kL、または約215時間/kLの総DGLA AUC0−24/用量(または平均総DGLA AUC0−24/用量もしくは総DGLA AUC0−24/用量中央値)、
(j)約2〜約25時間、または約3〜約20時間、例えば、約8時間、約10時間、または約18時間の総DGLA tmax(h)、
(k)約5:1〜約12:1、約6:1〜約10:1、または約7:1〜約9:1、例えば、7.7:1、約8.6:1、約8.8:1、または約9.8:1の総DGLA Cmax対遊離DGLA Cmaxの比、
(l)連続1〜約30日、1〜約28日、1〜約14日、または1〜約10日間の連日投与後の、最大約2000ng/ml、最大約750ng/ml、または最大約700ng/ml、例えば、約385ng/ml、または約675ng/mlの定常状態遊離DGLA血漿レベル(Cavg)または平均定常状態遊離DGLA血漿レベル(Cavg)もしくは定常状態遊離DGLA血漿レベル(Cavg)中央値、
(m)連続1〜約30日、1〜約28日、1〜約14日、または1〜約10日間の連日投与後の、最大250,000ng/ml、最大180,000ng/ml、最大150,000ng/ml、最大125,000ng/ml、または最大100,000ng/mlの定常状態総DGLA血漿レベル(Cavg)または平均定常状態総DGLA血漿レベル(Cavg)もしくは定常状態総DGLA血漿レベル(Cavg)中央値、及び/あるいは
(n)約0.2:1〜約5:1、約0.5:1〜約2.5:1、または約0.6:1〜約1.5:1の遊離DGLA血漿対DGLA皮膚(例えば、皮膚の水疱において測定されるような)の比。別の実施形態において、例えば、約1〜約12週間、約1〜約8週間、または約1〜約4週間の期間にわたって、本発明の組成物を用いて治療を行うと、対象または対象群は、すぐ上に記載される転帰(a)〜(n)のうちのいずれか2個以上、いずれか3個以上、いずれか4個以上、いずれか5個以上、いずれか6個以上、いずれか7個以上、いずれか8個以上、いずれか9個以上、いずれか10個以上、いずれか11個以上、いずれか12個以上、いずれか13個以上、いずれか14個以上、いずれか15個以上、いずれか16個以上、いずれか17個以上、いずれか18個以上、いずれか19個以上、いずれか20個以上、いずれか21個以上、または22個全てを呈する。
別の実施形態において、約200mgのDGLA〜約8000mgのDGLA(1つ以上の投薬単位として、例えば、約500mg、約1000mg、約2000mg、約3000mg、約4000mg、約5000mg、約6000mg、約7000mg、または約8000mgの合計1日DGLA用量に等しい500mgまたは1gの投薬単位として)を含む組成物を用いて絶食及び摂食対象または絶食及び摂食対象群の治療を行うと、単一用量投与後または複数用量投与後に、対象または対象群は、以下の結果のうちの1つ以上を呈する:
(a)約1:1〜約5:1、例えば、約2.5:1、約3:1、もしくは約3.5:1の遊離DGLA Cmax(絶食):遊離DGLA Cmax(摂食)の比、
(b)約1:1〜約5:1、例えば、約1.5:1、約2:1、もしくは約2.5:1の遊離DGLA AUC0−24(絶食):遊離DGLA AUC0−24(摂食)の比、
(c)約1:1〜約5:1、例えば、約1:1、約1.5:1、もしくは約2:1の総DGLA Cmax(絶食):総DGLA Cmax(摂食)の比、及び/または
(d)約1:1〜約5:1、例えば、約1.5:1、約2:1、もしくは約2.5:1の総DGLA AUC0−24(絶食):総DGLA AUC0−24(摂食)の比。
一実施形態において、本発明のDGLA含有組成物は、以下の脂肪酸フィンガープリントを含む:

一実施形態において、本発明のDGLA含有組成物は、以下の脂肪酸フィンガープリントを含む:
例示的な本発明のDGLA含有組成物は、以下の脂肪酸フィンガープリントを含む:
一実施形態において、本発明のDGLA含有組成物は、以下の脂肪酸フィンガープリントを含む:
一実施形態において、本発明のDGLA含有組成物は、以下の脂肪酸フィンガープリントを含む:
実施例1
ゼラチンカプセルに充填された(2000pm dl−αトコフェロールとともに)DGLAを含む薬学的組成物の3つのバッチを表1に示すように調製した。
ゼラチンカプセルに充填された(2000pm dl−αトコフェロールとともに)DGLAを含む薬学的組成物の3つのバッチを表1に示すように調製した。
カプセルシェルは、以下の賦形剤を含んでいた:ゼラチン、精製水、グリセロール、二酸化チタン、ならびに加工助剤レシチン及び中鎖トリグリセリド。
表2に示すように、ゼラチン、Polysorb、またはグリセロール/Polysorbの混合物、精製水、二酸化チタン、ならびに加工助剤レシチン、及び中鎖トリグリセリドを含有するカプセル中にDGLA FFA(微量の2000ppm dl−αトコフェロールで安定化した)を含むDGLAカプセルの追加のバッチも調製した。
各バッチのカプセルシェル組成物を、下の表3及び4に示す。
上記カプセルの安定性試験を行った。各バッチからのカプセルを最長6ヶ月間維持し、定性的または定量的USP2040の溶解及び溶出試験プロトコルを使用して評価した。結果を表5〜7に示す。
上に見られるように、グリセロール及び標準酸ウシゼラチン(E09726/01及びE09727)を用いて配合されたカプセルには、経時的に水中での溶出速度の減速が見られた。模擬胃液(pH1.2、ペプシン)中、40℃/75%の相対湿度で6ヶ月後には、DGLA放出速度は30分超であった。
40℃/75%の相対湿度で6ヶ月後に見られた30分未満のDGLA放出速度は、より低い分子量(Mw)を有する石灰骨ゼラチンを含有するカプセルを用いて模擬胃液(pH1.2、ペプシン)中でのみ達成された(E09777/02)。
グリセロールを含有するDGLAカプセルシェル中で、経時的にDGLAグリセリド形成における著しい増加が見られた(表4)。これは、40℃/75%の相対湿度で形成されたDGLAの最も高い濃度で温度依存性であった。
Polysorbは、カプセル充填媒体とシェルとの間の交換を制限するための親水性可塑剤として一般的に使用されている。D−ソルビトール及び1,4−ソルビタンは、グリセロールよりも高いMWを有し、ゼラチンシェルを通るグリセロールの可動性を制限する。それにもかかわらず、バッチE09777 1/2及び3において、DGLA FFAエステルを形成するD−ソルビトールと1,4−ソルビタンとの相互作用が依然として存在していた。
D−ソルビトール及び1,4−ソルビタンを用いて配合したバッチ(E09777 1/02/3)にはDGLAの酸価における減少が見られなかったのに対し、グリセロールを用いて配合したE09778には酸価の減少が見られた。
D−ソルビトール及び1,4−ソルビタンを用いて配合したカプセルには、経時的に水中での溶出速度の減速は見られなかった(E09777/03)。40℃/75%の相対湿度で3ヶ月後、DGLA放出速度は水中で30分未満であった。
実施例2
中程度から重度のアトピー性皮膚炎を有する患者に経口投与されたDS107G(DGLA)の有効性及び安全性を評価するために、ランダム化、二重盲検、プラセボ対照、第II相試験を行った。この試験の主目的は、中程度から重度のアトピー性皮膚炎を有する成人患者の治療において、経口投与されたDS107Gカプセルとプラセボの有効性を比較することであった。副次的目的は、中程度から重度のアトピー性皮膚炎を有する成人患者において、経口投与されたDS107Gカプセルとプラセボの安全性を評価することであった。試験の主要エンドポイントは、8週目に0(消失)または1(ほぼ消失)のIGA(治験責任医師による包括的評価)、及び少なくとも2ポイントのIGAの減少を達成する患者の割合であった。試験の副次エンドポイントは以下を含んでいた:
中程度から重度のアトピー性皮膚炎を有する患者に経口投与されたDS107G(DGLA)の有効性及び安全性を評価するために、ランダム化、二重盲検、プラセボ対照、第II相試験を行った。この試験の主目的は、中程度から重度のアトピー性皮膚炎を有する成人患者の治療において、経口投与されたDS107Gカプセルとプラセボの有効性を比較することであった。副次的目的は、中程度から重度のアトピー性皮膚炎を有する成人患者において、経口投与されたDS107Gカプセルとプラセボの安全性を評価することであった。試験の主要エンドポイントは、8週目に0(消失)または1(ほぼ消失)のIGA(治験責任医師による包括的評価)、及び少なくとも2ポイントのIGAの減少を達成する患者の割合であった。試験の副次エンドポイントは以下を含んでいた:
2、4、及び8週目のIGAにおけるベースラインからの変化、
2、4、及び8週目のEASI(湿疹面積及び重症度インデックス)におけるベースラインからの変化、
8週目のIGAにおいて少なくとも1ポイントの減少を達成する患者の割合、
2、4、及び8週目の患者自身による湿疹評価(POEM)におけるベースラインからの変化、
2、4、及び8週目の皮膚疾患特異的QOL尺度(DLQI)スコアにおけるベースラインからの変化、
2、4、及び8週目のSCORADにおけるベースラインからの変化、
2、4、及び8週目の患者の視覚的アナログ尺度(VAS)掻痒スコアにおけるベースラインからの変化、
2、4、及び8週目の体表面積(BSA)におけるベースラインからの変化、
各治療群における治療中に発生した有害事象(TEAE)の数。
探索的エンドポイントは以下を含む:
2、4、及び8週目の経皮水分損失(TEWL)におけるベースラインからの変化(選択された部位のみ)、
ベースライン時の血漿中総DGLA及び遊離DGLA濃度、4週目及び8週目、
ベースライン時の血漿中総脂肪酸プロファイル、4週目及び8週目(試料を保持し、後日分析)、
ベースライン時のインターロイキンプロファイル、4週目及び8週目(試料を保持し、後日分析)。
中程度から重度のアトピー性皮膚炎を有する約100人の患者が、この多施設、二重盲検、プラセボ対照、第IIa相試験に組み入れられた。全ての対象は、インフォームドコンセントに署名し、試験適格性についてスクリーニングを受けた。対象は、絶食状態で8週間1日1回、経口DS107G(2g)またはプラセボのいずれかを服薬するようにベースライン来院時に無作為化された(1:1)。
対象は、スクリーニング、ベースライン、2週目、4週目、8週目(治療の終了/早期中止)、及び10週目(経過観察)の6回クリニックに来院した。全ての対象は、10週目の来院で試験を終了した。一次有効性変数は、8週目に0(消失)または1(ほぼ消失)のIGA、及びIGAにおける少なくとも2点の減少を達成した患者の割合であった。二次有効性変数は、他の来院時のIGA、掻痒(SCORAD視覚的アナログ尺度から得られた)、EASI、BSA、POEM、DLQI、SCORAD、及びTEWL(選択された部位のみ)を含んでいた。安全性は、有害事象、身体検査、バイタルサイン、及び安全性臨床検査(妊娠の可能性がある女性の妊娠検査を含む)によって評価された。総DGLA及び遊離DGLAの血漿中トラフレベルを測定するために、ベースライン時(0日目)、4週目、及び8週目の来院時に薬物動態試料を得た。総脂肪酸プロファイル及びインターロイキンプロファイルを後に分析するために別の血漿試料を保持した。
EASIは、4つの身体領域の各々について紅斑、硬結/丘疹形成、表皮剥離、及び苔癬化(各々が0〜3で別個にスコア化される)の程度を考慮に入れ、各身体領域の関与するBSAのパーセント、及び全身に対する身体領域の割合を調整した、0〜72に及ぶ複合スコアである。EASIスコアの計算の詳細な手順を以下に提供する。
頭部、上肢、胴体、及び下肢の4つの解剖学的部位を、検査日に見られる紅斑、硬結(丘疹)、表皮剥離、及び苔癬化について評価する。各徴候の重症度は、4点スケールを用いて評価される。
0=症状なし
1=軽微
2=中程度
3=高度
0=症状なし
1=軽微
2=中程度
3=高度
所与の解剖学的部位内でアトピー性皮膚炎に冒された領域を、解剖学的部位の総面積のパーセンテージとして推定し、以下の通りにアトピー性皮膚炎の関与の程度に従って数値を割り当てる:
0=罹患なし
1=<10%
2=10〜<30%
3=30〜<50%
4=50〜<70%
5=70〜<90%
6=90〜100%
0=罹患なし
1=<10%
2=10〜<30%
3=30〜<50%
4=50〜<70%
5=70〜<90%
6=90〜100%
EASIスコアは、以下の式を使用することによって得る。
EASI=0.1(Eh+Ih+Exh+Lh)Ah+0.2(Eu+Iu+Exu+Exu)Au+0.3(Et+It+Ext+Ext)At+0.4(El+Il+Exl+Exl)Al
式中、E、I、Ex、L、及びAは、紅斑、硬結、表皮剥離、苔癬化、及び面積をそれぞれ示し、h、u、t、及びlは、それぞれ、頭部、上肢、胴体、及び下肢を示す。
SCORADは、以下の通りに計算される。ADの重症度を評価するために6つの項目(紅斑、浮腫/丘疹形成、滲出/痂皮、表皮剥離、苔癬化、及び乾燥)が選択される。各項目の強度は、4点スケールを用いて格付けされる。
0=症状なし
1=軽度
2=中程度
3=重度
0=症状なし
1=軽度
2=中程度
3=重度
格付けのために選択される領域は、各項目の代表(平均強度)でなければならない。次いで、各項目の個々の強度評点を加算し(0〜18に及ぶ)、3.5を乗じ、63の最大スコアを得る。
ADに冒されたBSA全体を評価し(0〜100%)、5で除す。1人の患者の手のひらは、該患者の総BSAの1%を意味する。最大は20である。
客観的項目は、不眠及び掻痒の発生である。これらは、評価記入用紙の10cmスケール(0〜10)上で、過去3日/3夜の平均値に対応する点を記入するように患者に依頼することにより評価される。これら2つを合わせた最大スコアは20である。
上記スケールの合計は、0〜103で変化し得るSCORADを意味する。掻痒及び不眠の主観的スコアが除外される場合、そのSCORADは客観的SCORAD(スコア範囲0〜83)になる。
治験責任医師による包括的評価は、次の通りに決定される。
体表面積(BSA)ADに冒されたBSA全体を各来院時に評価する(0〜100%)。1人の患者の手のひらは、該患者の総BSAの1%を意味する。スクリーニング時を除く全ての治験来院で、病変のある皮膚のBSAをSCORAD測定により測定し、別のエンドポイントとして評価する。
患者自身による湿疹評価(POEM)は、以下の質問票を使用して決定される。
皮膚疾患特異的QOL尺度(DLQI)は、以下の質問票を使用して決定される。
組み入れ基準は次の通りであった:
1.インフォームドコンセント用紙(ICF)に署名する日に18歳以上の男性または女性の対象。
2.ハニフィンとライカの基準(付録G)による活動性アトピー性皮膚炎の臨床的確定診断。
3.ベースライン来院時に最低3のIGAによって定義される、ベースライン時の中程度から重度のアトピー性皮膚炎。
4.ベースライン時に体表面積の最低10%を覆うアトピー性皮膚炎。
5.肥満度指数(BMI)が18〜35kg/m2(両端の値を含む)である。
6.妊娠の可能性がある女性患者は、試験期間中、適切な避妊法を用いるか、またはパートナーに不妊化してもらうか(全身性ホルモン避妊薬、子宮内避妊器具、もしくは殺精子薬と併せたバリアー避妊法)、または性的禁欲に合意してもらわなければならない。ホルモン避妊薬は、ベースラインの前に少なくとも1ヶ月間安定した用量を服用しなければならない。注:妊娠の可能性がない女性とは、
−外科的不妊化(子宮摘出術または両側卵巣摘出術または卵管結紮)を経験している女性、
−60歳超の女性、
少なくとも12ヶ月間生理が停止しており、卵胞刺激ホルモン(FSH)検査によって妊娠の可能性がないこと裏付けられているか(FSH≧40mIU/mL)、またはFSHレベルによる裏付けはないが少なくとも24ヶ月間生理が停止している、年齢が40歳超〜60歳未満の女性である。
7.試験の間中(許可された皮膚軟化剤を除いて)アトピー性皮膚炎の治療を中止することができ、かつそうする意思のある患者。
8.署名済みのインフォームドコンセントを提出する能力があり、かつそうする意思がある(コンセントは、いずれの試験関連手順の前に得られなければならない)。
1.インフォームドコンセント用紙(ICF)に署名する日に18歳以上の男性または女性の対象。
2.ハニフィンとライカの基準(付録G)による活動性アトピー性皮膚炎の臨床的確定診断。
3.ベースライン来院時に最低3のIGAによって定義される、ベースライン時の中程度から重度のアトピー性皮膚炎。
4.ベースライン時に体表面積の最低10%を覆うアトピー性皮膚炎。
5.肥満度指数(BMI)が18〜35kg/m2(両端の値を含む)である。
6.妊娠の可能性がある女性患者は、試験期間中、適切な避妊法を用いるか、またはパートナーに不妊化してもらうか(全身性ホルモン避妊薬、子宮内避妊器具、もしくは殺精子薬と併せたバリアー避妊法)、または性的禁欲に合意してもらわなければならない。ホルモン避妊薬は、ベースラインの前に少なくとも1ヶ月間安定した用量を服用しなければならない。注:妊娠の可能性がない女性とは、
−外科的不妊化(子宮摘出術または両側卵巣摘出術または卵管結紮)を経験している女性、
−60歳超の女性、
少なくとも12ヶ月間生理が停止しており、卵胞刺激ホルモン(FSH)検査によって妊娠の可能性がないこと裏付けられているか(FSH≧40mIU/mL)、またはFSHレベルによる裏付けはないが少なくとも24ヶ月間生理が停止している、年齢が40歳超〜60歳未満の女性である。
7.試験の間中(許可された皮膚軟化剤を除いて)アトピー性皮膚炎の治療を中止することができ、かつそうする意思のある患者。
8.署名済みのインフォームドコンセントを提出する能力があり、かつそうする意思がある(コンセントは、いずれの試験関連手順の前に得られなければならない)。
除外基準は次の通りである:
1.スクリーニング時もしくは0日目の来院時(ベースライン)に妊娠検査が陽性の女性患者、または授乳中の女性。
2.患者を過度の危険にさらすかまたは試験結果の解釈を妨げるであろう、治験責任医師の判断による、あらゆる臨床的に重大な制御されたまたは制御不能の医学的状態または検査所見の異常。
3.腎機能または肝機能の臨床的に重大な障害。
4.アトピー性皮膚炎の診断及び/または評価を妨げる可能性がある他の皮膚疾患(乾癬または流行しているウイルス性、細菌性、及び真菌性の皮膚感染症等)。
5.DS107Gまたはプラセボカプセル中のあらゆる物質に対する過敏症の病歴。
6.治療開始/0日目の来院(ベースライン)前の3ヶ月または5半減期(いずれか長い方)の生物製剤の使用。
7.ベースライン来院(0日目)前の4週間以内のアトピー性皮膚炎に影響を及ぼし得る全身的治療(生物製剤以外)、例えば、レチノイド、カルシニューリン阻害剤、メトトレキサート、シクロスポリン、ヒドロキシカルバミド(ヒドロキシ尿素)、アザチオプリン、及び経口/注入可能なコルチコステロイドの使用:安定した医学的状態ための鼻腔内コルチコステロイド及び吸入コルチコステロイドは許容される)。
8.0日目の来院(ベースライン)前の30日以内または5半減期(いずれか長い方)の実験薬を用いた治療。
9.0日目の来院(ベースライン)前の4週間の過度の日光曝露、日焼け室もしくは他の紫外線(UV)光源の使用、及び/またはスクリーニングと経過観察来院との間に、日射量の多い地域への旅行、または日焼け室もしくは他のUV源を使用することを予定している。
10.治療開始/0日目の来院(ベースライン)前の2週間の、限定されないが、局所コルチコステロイド、カルシニューリン阻害剤、タール、漂白剤、抗菌薬、及び漂白剤浴を含む、アトピー性皮膚炎のためのあらゆる局所的薬物治療。
11.0日目前の2週間の尿素、セラミド、またはヒアルロン酸を含有する局所製品の使用。
12.ベースラインの2週間以内のアトピー性皮膚炎のための抗ヒスタミン剤の使用。
13.重大な制御不能の心血管疾患(治験責任医師の判断で臨床的に重大なECG異常の病歴)、神経疾患、悪性疾患、精神疾患、呼吸器疾患、または高血圧疾患、ならびに糖尿病及び関節炎。
14.慢性伝染性疾患(例えば、B型肝炎、C型肝炎、またはヒト免疫不全ウイルスへの感染)の治療歴。
15.0日目(ベースライン)前の臨床的に重大な薬物またはアルコール乱用の病歴。
1.スクリーニング時もしくは0日目の来院時(ベースライン)に妊娠検査が陽性の女性患者、または授乳中の女性。
2.患者を過度の危険にさらすかまたは試験結果の解釈を妨げるであろう、治験責任医師の判断による、あらゆる臨床的に重大な制御されたまたは制御不能の医学的状態または検査所見の異常。
3.腎機能または肝機能の臨床的に重大な障害。
4.アトピー性皮膚炎の診断及び/または評価を妨げる可能性がある他の皮膚疾患(乾癬または流行しているウイルス性、細菌性、及び真菌性の皮膚感染症等)。
5.DS107Gまたはプラセボカプセル中のあらゆる物質に対する過敏症の病歴。
6.治療開始/0日目の来院(ベースライン)前の3ヶ月または5半減期(いずれか長い方)の生物製剤の使用。
7.ベースライン来院(0日目)前の4週間以内のアトピー性皮膚炎に影響を及ぼし得る全身的治療(生物製剤以外)、例えば、レチノイド、カルシニューリン阻害剤、メトトレキサート、シクロスポリン、ヒドロキシカルバミド(ヒドロキシ尿素)、アザチオプリン、及び経口/注入可能なコルチコステロイドの使用:安定した医学的状態ための鼻腔内コルチコステロイド及び吸入コルチコステロイドは許容される)。
8.0日目の来院(ベースライン)前の30日以内または5半減期(いずれか長い方)の実験薬を用いた治療。
9.0日目の来院(ベースライン)前の4週間の過度の日光曝露、日焼け室もしくは他の紫外線(UV)光源の使用、及び/またはスクリーニングと経過観察来院との間に、日射量の多い地域への旅行、または日焼け室もしくは他のUV源を使用することを予定している。
10.治療開始/0日目の来院(ベースライン)前の2週間の、限定されないが、局所コルチコステロイド、カルシニューリン阻害剤、タール、漂白剤、抗菌薬、及び漂白剤浴を含む、アトピー性皮膚炎のためのあらゆる局所的薬物治療。
11.0日目前の2週間の尿素、セラミド、またはヒアルロン酸を含有する局所製品の使用。
12.ベースラインの2週間以内のアトピー性皮膚炎のための抗ヒスタミン剤の使用。
13.重大な制御不能の心血管疾患(治験責任医師の判断で臨床的に重大なECG異常の病歴)、神経疾患、悪性疾患、精神疾患、呼吸器疾患、または高血圧疾患、ならびに糖尿病及び関節炎。
14.慢性伝染性疾患(例えば、B型肝炎、C型肝炎、またはヒト免疫不全ウイルスへの感染)の治療歴。
15.0日目(ベースライン)前の臨床的に重大な薬物またはアルコール乱用の病歴。
連続変数を表にまとめ、それには対象の数、平均値、標準偏差、中央値、最小、最大、及び四分位範囲が含まれた。カテゴリー変数を頻度及びパーセンテージとして表に提示する。全ての統計的検定は両側検定であり、別途指定のない限り、0.05の有意レベルで行われた。
各対象に、薬物の投与前に、以下に列挙する特定の事項について質問した。対象がこれらの制約のノンコンプライアンスを認めた場合、治験責任医師(もしくは被指名者)及び/または治験依頼者は、その対象が試験に留まることを許可されるかどうかを決定した。これらの制約のノンコンプライアンスを記録した。
対象に、スクリーニングと経過観察来院との間は、日射量の多い地域への旅行、または日焼け用機器の使用を控えるように指示した。
対象に、試験の間中、アトピー性皮膚炎に影響を及ぼし得るあらゆる薬物/治療の使用を控えるように指示した(除外基準及び禁止されている治療方または手技の項を参照されたい)。
対象は、起床してから、薬物投与前に少なくとも8時間の絶食を開始する必要があった。絶食は、薬物投与後少なくとも60分間継続し、その後、対象は朝食を食べることができた。水は、絶食期間中いつでも許可されていたが、他の液体は許可されていなかった。本試験において許可されている他の状態のための薬(複数可)は、通常通り服用することができた。
ベースライン(0日目)、4週目、及び8週目の来院時に、PK分析のために採血が行われた。PK試料は投与前に採取され、したがって、治験薬の投与は、0日目及び4週目の来院の来院中に行われた。投薬は、0日目及び4週目にクリニックで行われたため、対象は治験薬投与の少なくとも8時間前に絶食する必要があり、治験薬投与の60分後に食事をとることが許可された。
対象は、理由の如何を問わず、ペナルティなしに、いつでも試験から離脱する権利を有していた。治験責任医師も、対象の最善の利益になるように、または対象が非協力的であるかもしくはノンコンプライアンスである場合、対象を試験から離脱させる権利を有していた。治験責任医師または職員のうちの1人は、離脱の理由をできるだけ完全に究明して、対象の原資料及びCRFにその理由を記録するために、電話によりまたは個人的な訪問を通してのいずれかで対象に連絡を取った。対象の離脱時の完全な最終早期中止(8週目)評価は、対象がなぜ試験から離脱したのかの説明とともに行われた。対象の脱落の理由が有害事象または異常な臨床検査結果である場合、主要な特定の事象または検査を記録した。8週目の来院より前に試験を中止した対象には、できるだけ早く早期中止来院のために来て、8週目に記載される評価を行うよう依頼した。また、彼らには、10週目に記載される安全性評価のために2週間後に再来院するよう依頼した。
全ての組み入れ条件を満たし、かつ除外基準のいずれも満たさなかった対象を試験に参加させた。各対象は、あらゆるスクリーニング手順が行われる前に、インフォームドコンセント用紙を読んで署名した。本試験は、8週間の全期間中、起床時に1日1回経口投与されるDS107G(DGLA)とプラセボとの比較を含んでいた。対象は、1:1の比で2つの治療群のうちの1つに無作為化される:
治療群A:2グラムのDS107G(4カプセル)
治療群B:2グラムのプラセボカプセル(4カプセル)
対象は、起床してから、薬物投与前に少なくとも8時間の絶食を開始する必要があった。絶食は、薬物投与後少なくとも60分間継続し、その後、対象は朝食を食べることを許可された。水は、絶食期間中いつでも許可されていたが、他の液体は許可されていなかった。本試験において許可されている他の状態のための薬(複数可)は、通常通り服用することができた。
DS107Gカプセルは、500mgのDGLA遊離脂肪酸(FFA)を含有する、不透明な楕円形の軟ゼラチンカプセルとしてDignity Sciencesによって提供された。プラセボカプセルも、500mgの液体パラフィンを含有する不透明な楕円形の軟ゼラチンカプセルとしてDignity Sciencesによって提供された。DS107Gカプセルは、28単位のアルミホイルブリスターに包装された製造形態で供給された(盲検)。プラセボは、同一のブリスター及びパック内に提示され、DS107Gカプセルと同じように保存/包装された。治験薬は、米国及びカナダの法令に従ってラベリングされた。
治験薬は、治験依頼者によって治験責任医師に提供され、施設内で、アクセスの制限された鍵のかかる部屋またはキャビネット内に保持された。DS107G及びプラセボカプセルは、15〜30℃の管理された室温で保存し、治験責任医師の管理下でのみ治験の対象に供給された。治験薬は、各治験来院時に試験施設によって対象に分配された。対象は、(使用済み及び未使用の)全ての治験薬ブリスターパックを試験施設に返却するようになっていた。ブリスターパック内のカプセルを分配の前及び返却の際に数え、その数を原資料及びeCRFに記録した。各対象は、次の治験来院時に治験薬を返却することの重要性について指導を受けた。対象が治験薬を返却しなかった場合、その対象は、できるだけ早く返却するよう指導を受けた。
スクリーニング前4週間以内及び試験の間中に服用した全ての薬物(店頭薬、ビタミン、及び制酸剤を含む)を記録した。スクリーニングの2ヶ月前にアトピー性皮膚炎のために服用した全ての薬物を記録した。対象は、自身が選択した無刺激性の皮膚軟化剤を、AD病変を含む自身の皮膚に適用することを許可されたが、但し、皮膚軟化剤の使用は、0日目の少なくとも2週間前までに開始し、試験の間中同じ頻度で同じ皮膚領域に継続されるものとした。以下の成分を含む、アトピー性皮膚炎に影響を及ぼすまたは及ぼし得るあらゆる活性成分を含有する皮膚軟化薬の使用を避けるよう対象に依頼した:尿素、セラミド、またはヒアルロン酸。
非鎮静性抗ヒスタミン剤(例えば、ロラタジン、フェキソフェナジン)は、アトピー性皮膚炎以外の医学的状態を治療するために使用される場合にのみ、試験中許可された。そのような薬物は、対象が0日目の少なくとも2週間前に安定した用量を服用していた場合にのみ試験中許可され、試験の間中毎日同じ薬剤を使用し続けた。安定な医学的状態のための吸入コルチコステロイド及び鼻腔内コルチコステロイドは許可された。
以下の局所療法または手順は、試験期間中、全ての対象に対して禁じられていた:
限定されないが、局所コルチコステロイド、カルシニューリン阻害剤、タール、漂白剤、抗菌薬、及び漂白剤浴を含む、アトピー性皮膚炎に影響を及ぼし得る局所的薬物治療、
尿素、セラミド、またはヒアルロン酸を含有するあらゆる局所製品、
アトピー性皮膚炎に影響を及ぼし得る全身療法、例えば、レチノイド、カルシニューリン阻害剤、メトトレキサート、シクロスポリン、ヒドロキシカルバミド(ヒドロキシ尿素)、アザチオプリン、及び経口/注入可能なコルチコステロイド、
抗ヒスタミン剤(非鎮静性抗ヒスタミン剤を除く)、
あらゆる生物学的製剤、
UVAまたはUVB光線療法、
ソラレン長波長紫外線治療(PUVA)、
過度の日光暴露または日焼け室の使用、
あらゆる治験薬。
治療コンプライアンスは、直接的な質問、対象のコンプライアンスログの審査、及びカプセル数によって各来院時に評価され、後者に基づいていた。対象は、各来院時に治験薬とともに紙の日記を支給された。対象は、あらゆる服用しそこねた用量、ならびに治験薬投与より前の最後の食物摂取及び治験薬投与後の食物摂取のタイミングを日記に記した。対象は、全てのカプセル及びブリスターパック(使用済み及び未使用)、ならびにコンプライアンスログを、次の治験来院時に持ってくるよう指示された。処方された投与レジメンからのあらゆる逸脱を原資料及びeCRFに記録した。大幅にノンコンプライアンスな対象にはカウンセリングが行われた。
アトピー性皮膚炎の臨床評価は、経験豊富な、(委員会認定かまたは同等の)資格のある皮膚科医によって行われた。一貫性を保証し、かつ変動を低減するために、可能な限り同じ評価者が所与の対象について全ての評価を行った。
疾患重症度の治験責任医師による包括的評価(IGA)を、各来院時に評価した。IGAは、疾患の現在の状態の包括的評価である。これは6点からなる全体的な疾患重症度の形態学的評価であり、以下の定義に従って決定される:0(消失)、1(ほぼ消失)、2(軽度)、3(中程度)、4(重度)、及び5(極めて重度)。適格であるために、対象は、ベースライン来院時(0日目)に3以上のIGAスコアを有していなければならない。
湿疹面積及び重症度インデックス(EASI)を、スクリーニング来院を除く各来院時に評価した。これは、病変重症度及び冒されたBSAのパーセントの両方に基づいて、対象のアトピー性皮膚炎の重症度を定量化する。EASIは、4つの身体領域の各々について紅斑、硬結/丘疹形成、表皮剥離、及び苔癬化(各々が0〜3で別個にスコア化される)の程度を考慮に入れ、各身体領域の関与するBSAのパーセント、及び全身に対する身体領域の割合を調整した、0〜72に及ぶ複合スコアである。
ADに冒されたBSA全体を各来院時に評価した(0〜100%)。1人の患者の手のひらは、該患者の総BSAの1%を意味する。スクリーニング時を除く全ての治験来院で、関与する皮膚のBSAをSCORAD測定(説明については以下を参照)により測定し、別のエンドポイントとして評価する。適格であるために、対象は、ベースライン来院時(0日目)に少なくとも10%のBSAを有していなければならない。
SCORADを、スクリーニング来院を除く各来院時に測定した。SCORAD格付けシステムは、European Task Force on Atopic Dermatitis(1993)によって開発され、欧州では臨床試験においてADの重症度を評価するための標準的なツールとなっている。ADの重症度を評価するために6つの項目(紅斑、浮腫/丘疹形成、滲出/痂皮、表皮剥離、苔癬化、及び乾燥)が選択された。ADに冒されたBSA全体を評価し(0〜100%)、SCORADスコアに含めた。不眠及び掻痒は、視覚的アナログ尺度(0〜10)で患者によって評価された。これらの測定値の合計は、0〜103で変化し得るSCORADを意味する。
スクリーニングを除く全ての治験来院で、SCORAD測定により掻痒重症度スコアを記録し、別のエンドポイントとして評価した。これは、評価記入用紙の10cmスケール(0〜10)上で、過去3日/3夜の平均値に対応する点を記入するように対象に依頼することにより評価した。
TEWL.ADの臨床的重症度、及び皮膚バリア機能への関連する影響を、スクリーニングを除く全ての来院で評価する。この評価は、この装置を用いて以前の経験を実証したことのある選択された部位で行われる。
ベースライン時(0日目)に、治験責任医師は、各対象の活動性ADの3つの代表的な領域を選択し、これらの部位の位置を記録する。その後の来院時に、標準的な室内の環境条件(22〜25℃、相対湿度40〜60%)でADの各領域についてTEWLを読み取り、TEWL測定の平均値を分析に使用する。
患者自身による湿疹評価(POEM)を、スクリーニング来院を除く各来院で評価した。POEMは、患者による疾患重症度の自己評価である。POEMは、以下のスケールに従ってスコア化される7つの質問に対する患者の回答に基づいて28の最大値を有する:
−全くなかった=0
−1〜2日=1
−3〜4日=2
−5〜6日=3
−毎日=4
−全くなかった=0
−1〜2日=1
−3〜4日=2
−5〜6日=3
−毎日=4
DLQIは、単純な10個の質問からなる有効な質問票であり、スクリーニングを除く各来院時に完了された。
ADの臨床的重症度、及び皮膚バリア機能への関連する影響を、スクリーニングを除く全ての来院で評価した。この評価は、この装置を用いて以前の経験を実証したことのある選択された部位で行われた。
ベースライン時(0日目)に、治験責任医師は、各対象の活動性ADの3つの代表的な領域を選択し、これらの部位の位置を記録した。その後の来院時に、標準的な室内の環境条件(22−25℃、相対湿度40〜60%)でADの各領域についてTEWLを読み取り、TEWL測定の平均値を分析に使用した。
臨床検査を、スクリーニング時、0日目、4週目、及び8週目に行った。8週目の結果がベースラインから臨床的に有意な変化を示した場合、10週目にも臨床検査を行った。検査は、検尿、差分及び凝固検査を用いた血液学的検査、標準的な化学パネル(化学は肝機能検査及びコレステロールを含む)、凝固検査、妊娠の可能性がある女性(WOCBP)の場合は血清妊娠検査(スクリーニング及び8週目/早期中止来院)を含んでいた。ベースライン(0日目)、2週目、4週目、及び10週目の来院時に、妊娠の可能性がある女性に尿妊娠検査を行った(治験責任医師のいる施設で実施される)。スクリーニング来院時に、少なくとも12ヶ月〜24ヶ月間未満生理が停止している40歳超〜60歳未満の女性のFSHレベルを検査した。
ベースライン、4週目、及び8週目の来院時に治験薬投与前に採血を行った(8週目の来院時には治験薬投与はなし)。対象が1日用量の治験薬を服用した後でクリニックを訪れた場合、この対象には、翌日PK採血のために再来院するよう依頼した。総DGLA及び遊離DGLAのトラフ血漿レベルを測定した。血漿中の総脂肪酸プロファイルを後に評価するために、2回目の採血を行った。血清化学分析のための採血を、化学分析と、後のインターロイキンの評価のために2つのアリコートに分割した。
主要エンドポイントはレスポンダー分析として解釈することができ、対象は、8週目に0(消失)または1(ほぼ消失)のIGAスコアを達成した場合に、ベースラインからの2点の減少を考慮して、レスポンダーとして分類される。45人の対象の試料サイズは、カイ二乗検定及び0.05のαに基づいて、治療群及びプラセボ群からのレスポンダー間の25%の統計的有意差を検出するための80%の検出力を有する。文献レビューに基づいて、プラセボは最大7重量%に達することができると予想されたため、治療群で予想された最小の割合は少なくとも32%であった。10%の脱落者を考慮に入れると、全部で100人の対象が試験に登録することが予定された。
ITT集団に基づいて有効性を評価し、受けた治療ではなく、無作為化治療に基づいて分析を行った。パー・プロトコル(PP)集団は、重大なプロトコル逸脱のなかった、無作為化された全ての対象を含んでいた。安全性集団は、少なくとも1用量の薬物を投与された全ての対象として定義された。分析は、対象が受けた実際の治療に従って行われる。
主要エンドポイントはレスポンダー分析として解釈することができ、対象は、8週目に0(消失)または1(ほぼ消失)のIGAスコアを達成した場合に、ベースラインからの2点の減少を考慮して、レスポンダーとして分類される。主要エンドポイントの群間比較は、層別化因子として施設を含めたコクラン・マンテル・ヘンツェル検定を使用して行った。補助的分析は、フィッシャーの直接確率検定を用いて行った。主要有効性分析は、観察された値を用いて行い、補助的分析は、最終観察繰越(LOCF)手法を用いて実施した。分析は、ITT集団を用いて行われ、一次分析としての役割を果たす一方で、PP集団を用いた主要エンドポイントの分析は、感度分析として用いられた。
ベースラインからの変化に関する副次エンドポイントは、ベースラインからの変化を従属変数として、施設群及び治療群及び施設を固定因子として、ベースライン値を共変量として含む、共分散分析(ANCOVA)を用いて分析した。最小二乗平均及び95%CIを、治療の比較からの対応するp値とともに提示した。割合に関する副次エンドポイントは、施設によって層別化したコクラン・マンテル・ヘンツェル検定を用いて分析し、p値を提示した。副次エンドポイントの分析は観察されたデータを用いて行われ、欠測値にインピューテーションは使用されない。
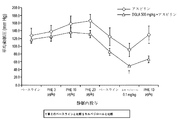
試験の結果を表8〜11及び図1〜6に示す。アトピー性皮膚炎臨床試験に関するFDA基準の主要有効性エンドポイント(治験責任医師による包括的評価(IGA)の2点減少、及び0(消失)または1(ほぼ消失)の治療終了時IGA)に基づいて、Intent−to−Treat(ITT)分析からの全体的な結果は、プラセボよりもDS107に有利な臨床的に有意な傾向(p=0.057)を示した。特に、この分析は、中程度の患者サブグループ(n=67)においてプラセボよりも有利な統計的に有意な改善(p=0.036)を示した。
評価した副次エンドポイントの中でも、掻痒スコア[視覚的アナログ尺度(VAS)]は、DS107で処置した群において4週間以内の投薬で統計的に有意な痒みの減少を示した(p=0.015)。
安全性データは、経口DS107が、幅広い安全性路ファイルを有し、非常に良好な忍容性を示したことを示唆した。薬物関連の重篤な有害事象は、いずれの患者群にも見られなかった。経験された有害事象のほとんどは軽度であり、いずれの介入もなしに解決され、群間で差があるようには見えなかった。
表8は、DGLA及びプラセボのベースラインからの湿疹面積及び重症度インデックスにおける変化を示す。
表9A/Bは、DGLA及びプラセボのベースラインからの掻痒の視覚的アナログ尺度における変化を示す。
表10は、DGLA及びプラセボのベースラインからのアトピー性皮膚炎のスコアにおける変化を示す。
表11は、DGLA及びプラセボのアトピー性皮膚炎に冒された体表面積における変化を示す。
データをさらに調べたところ、驚くべき発見があった。具体的には、少ないベースライン好酸球数を有する対象は、DS107による処置に対してより高い反応率を示したことが分かった。
好酸球は、アレルギー反応、喘息、及び寄生虫感染に対する体の反応において重要な役割を果たす、白血球の一種である。好酸球は、通常、循環白血球の7%未満を占める(1マイクロリットルの血液当たり100〜500個の好酸球)。これらの細胞は、特定の寄生虫に対する防御免疫において役割を果たすが、アレルギー性疾患において発生する炎症の原因でもある。
分析の結果を図30〜35に示す。このデータは、測定した各時点で、DS107で処置した白人患者と比較してアフリカ系アメリカ人の患者における好酸球濃度の統計的に有意な減少を示している。図30A〜B。具体的には、DS107で処置したアフリカ系アメリカ人患者の56%がレスポンダーとして分類されたのに対し、白人患者はわずか7%のみであった。図31A〜B。上で詳述したように、対象は、8週目に0(消失)または1(ほぼ消失)のIGAスコアを達成した場合に、ベースラインからの2点の減少を考慮して、レスポンダーとして分類される。図32は、全集団(図32A)、及びDS107で処置した患者(図32B)、及びプラセボで処置した患者(図32C)における全てのレスポンダーの好酸球濃度に関する同じデータを示している。さらに、データは、全集団(レスポンダー及びノンレスポンダー)で測定した全ての時点で、好酸球濃度に統計的に有意な増加を示した。図33A〜C。最後に、このデータから、プラセボ対照に対するノンレスポンダーと比較して、DS107で処置したレスポンダーでは全ての時点で好酸球濃度における統計的に有意な減少も明らかになった。図34A〜C及び図35A〜C。
総合すると、これらのデータは、低い好酸球レベルを有するように予め決められた患者が、DGLA、特に、DS107等の経口DGLAを用いた治療の有力候補となることを意味すると示唆している。そのような知見は、DGLA療法患者の有効性を予測する上で有用である。
実施例3
自然発症高血圧ラットにおけるDGLA及びアスピリンの同時投与後の降圧効果に関するin vivo調査を行った。この実験により、アスピリンとともに長期同時投与した場合に、DGLAが自然発症高血圧ラットにおいてフェニレフリンによって急激に誘導された高血圧反応を低減する有効性を調査した。Charles River Laboratoriesによって飼育された自然発症高血圧成獣雄ラット(250−300g)をこのアッセイに用いた。動物は、到着時にCCPAガイドラインに従って同定された。手術前の経口経管栄養の期間中、動物を群ごと(低用量+アスピリン、高用量+アスピリン、またはアスピリン単独)に2匹ずつ収容した。全ての動物の世話及び飼育施設の維持管理について記録し、文書を試験施設で保管した。
自然発症高血圧ラットにおけるDGLA及びアスピリンの同時投与後の降圧効果に関するin vivo調査を行った。この実験により、アスピリンとともに長期同時投与した場合に、DGLAが自然発症高血圧ラットにおいてフェニレフリンによって急激に誘導された高血圧反応を低減する有効性を調査した。Charles River Laboratoriesによって飼育された自然発症高血圧成獣雄ラット(250−300g)をこのアッセイに用いた。動物は、到着時にCCPAガイドラインに従って同定された。手術前の経口経管栄養の期間中、動物を群ごと(低用量+アスピリン、高用量+アスピリン、またはアスピリン単独)に2匹ずつ収容した。全ての動物の世話及び飼育施設の維持管理について記録し、文書を試験施設で保管した。
ラットは、血管緊張作用の判定、及び被験物質への長期曝露後の有効性の評価のために、よく特徴付けられた高感度モデルである。この試験設計は、最新のInternational Conference on Harmonization(ICH)Harmonized Tripartite Guidelines[ICH S7A]に基づいており、薬学的化合物の試験のために一般に許容される手順である。この非臨床試験は、GLPに準拠しない試験として設計され、QAを必要としなかった。
試験されるDGLAの濃度(50及び500mg/kg)及びアスピリンの濃度(10mg/kg)は、本試験の設計時に入手可能な情報に基づいて治験依頼者によって選択された。それらは、被験物質の作用機序の同定に有用な濃度範囲を網羅するように選択された。
陽性対照の濃度(カルベジロール0.1mg/kg)及び昇圧薬の濃度(フェニレフリン3、10、及び20μg/kg)は、参考文献に基づいて選択された。全ての溶液の調製をSolution Contents Formに記述し、ラベリング情報、ならびに試薬に関する全ての関連情報(バッチ番号、保存条件、含有量、及び調製日/使用期限)を定義した。
被験物質は、オリーブオイルをビヒクルとして使用して配合された。次いで、この原液を、経口経管栄養により連続7日間、1日1回投与した。低用量及び高用量(50mg/kg及び500mg/kg)を試験した。
適量のアスピリンを100%DMSOに溶解し、次いでそれを水に希釈することにより、アスピリンの5mg/ml原液を調製した。DMSOの濃度は1%未満であった。適切な体積を、経口経管栄養により連続7日間、1日1回各動物に投与した。原液は、室温で保存され、経管栄養の期間中安定であると考えられた。
カルベジロールの0.4mg/ml原液をPBS(pH=4.00±0.05)中に調製した。手術日の実験終了時に、適切な体積を静脈内注射により各動物に投与した。原液は4℃で保存され、実験期間中安定であると考えられた。
1mg/mlのフェニレフリンの原液をPBS(pH=7.4±0.05)中に調製した。原液は4℃で保存した。使用期限は、調製後14日に設定した。
試験終了後に、あらゆる残りの被験物質を破棄する。
体重約300gの雄の自然発症高血圧ラットは、経口経管栄養を開始する少なくとも2日前に施設に到着した。動物を、3つの群のうちの1つ(低用量+アスピリン、高用量+アスピリン、及びアスピリン)に割り当て、順化期間の間2匹ずつ収容した。各群(n=4)に、16G供給針を使用して経口経管栄養により適切な化合物を投与した。原液を連続7日間、1日1回投与した。投与7日目に、動物は手術及び高血圧モニタリングを受けた。
誘導箱内で、2.5〜3.0%イソフルラン−O2混合物を用いてラットを麻酔した。動物を誘導箱から移動させたとき、動物は、気管切開中に麻酔を維持するためにノーズコーンに連結されていた。自発呼吸を促進し、血行動態を安定させ、イソフルラン−O2混合物でラットを麻酔下に維持するために、気管内チューブ(コネクタから先端部まで7cm、BD社製のPE205管及び16G針でできている)を用いて動物の気管切開を行った。2本のカテーテルを挿入した:全身動脈圧(SAP)測定のために右大腿動脈に1本、昇圧薬及び陽性対照の送達のために右大腿静脈に1本。麻酔下の動物にリード1構成で配置されたECG接触部を用いてECGシグナルを取得し、手術中にラットの全身状態を継続してモニタリングできるように、パルスオキシメーターを動物の後肢に取り付けた。ベースライン測定後、3つの用量のフェニレフリン(3、10、及び20μg/kg)を、投与間隔5分でIVボーラス注射により送達した。最高濃度のフェニレフリンに続いて、1用量の陽性対照カルベジロール(0.1mg/kg)を投与した。陽性対照の後に、10μg/kgのフェニレフリンを再び投与した。血圧、全身動脈圧(SAP)、心拍数を、合計で30分間、継続的にモニタリングした。高血圧症は、拡張期血圧、収縮期血圧、または平均血圧における上昇として検出された。実験終了時に、動物を失血により安楽死させた。
使用した分析ソフトウェアは、Axon InstrumentsのClampfit 10.2.0.14であり、Microsoft Windowsを実行しているネットワーク化されたパーソナルコンピュータに実装した。Clampfit 10.2.0.14は、使用される接続コンテキストにおいて十分に確認されている。図解用のグラフィックソフトウェアは、Microsoft Windowsを実行しているネットワーク化されたパーソナルコンピュータに実装されたMicrosoft Office Excel 2007である。継続的に記録した全身動脈圧(SAP)を用いて、各条件ごとに平均全身動脈圧(mSAP)を計算した。麻酔が導入されたときから心拍数を継続的にモニタリングした。実験群にわたって曝露前と曝露後のパラメータを比較する一方向ANOVAを分析した。有意差はp≦0.05で確認した。
表14.フェニレフリンの静脈内投与前の、7日間の経管栄養後の動脈圧及び平均全身動脈圧(mmHg)。
表15は、10mg/kg/日のアスピリンを用いた連続7日間の経管栄養後の、静脈内フェニレフリン投与による平均全身動脈圧における変化を示す。
図7は、フェニレフリン投与後の平均動脈圧(mmHg)における変化を示す。5匹のラットに、10mg/kg/日のアスピリンを用いた連続7日間の経管栄養後にフェニレフリンを静脈内投与した。フェニレフリンの3つ各々の用量における平均全身動脈圧を第1のベースラインと統計的に比較した一方で、カルベジロール条件を投与の直前にベースラインと統計的に比較し、最終用量のフェニレフリン(10μg/kg)を直前に陽性対照条件と比較した。
これらの結果は、フェニレフリンの用量増加に伴う平均全身動脈圧の上昇を実証している。得られた全てのデータはベースラインとは統計的に異なっており、アスピリンを与えた自然発症高血圧ラットモデルにおけるフェニレフリンの昇圧作用を裏付けるものであった。フェニレフリンの効果は完全に可逆的であった:第2のベースライン(20μg/kg後)の平均動脈圧は、実際に元のベースラインと非常に類似していた。
カルベジロールは、フェニレフリンの直後に加えられたときに有意な低下を引き起こし、高い血圧を下げるという該薬物のよく知られた能力を裏付けた(カルベジロールは、非特異的なβ遮断薬/α1遮断薬であり、フェニレフリンのα1アドレナリン受容体への結合を遮断する)。カルベジロールに続いてフェニレフリンを最後に加えることにより、平均全身動脈圧において限られた上昇を引き起こした。
図8は、50mg/kgのDGLA+10mg/kgのアスピリンを用いた連続7日間の経管栄養後の、フェニレフリンの静脈内投与による平均動脈圧(mmHg)における変化を示す。
図16は、50mg/kgのDGLA+10mg/kgのアスピリンを用いた連続7日間の経管栄養後の、静脈内フェニレフリン投与による平均動脈圧におけるベースラインからの変化を示す。
図8に提示される結果は、50mg/kgのDGLA+10mg/kgのアスピリンを用いた連続7日間の経口投与後の、5匹の自然発症高血圧ラットにおけるフェニレフリンの静脈内投与後の平均動脈圧(mmHg)における変化を示している。被験物質の結果をアスピリン単独群(10mg/kg)で得られたデータと比較した。カルベジロールを第2のベースラインと比較し、最終用量のフェニレフリンをカルベジロールと統計的に比較した。
フェニレフリン濃度を増加させると、動物は用量依存性の血圧上昇を示した。DGLA50mg/kg及びアスピリンを投与されたラットは、所与の用量のフェニレフリンで動脈圧の上昇を示したが、それは10日にわたってアスピリンを投与されたラットにおいて測定されたのと実質的に同じであった。10mg/kgのアスピリンに加えて50mg/kgのDGLAを与えられたラットの場合、いずれのフェニレフリン濃度においてもアスピリン群と比較して平均動脈圧に有意差は見られなかった。したがって、連日のアスピリンは、1日用量50mg/kgのDGLAから得られる利益を妨げると考えられる。
カルベジロールは、DGLA+アスピリンを与えた動物において有意な血圧低下を引き起こした。麻酔による問題のために、ラット番号3には最終用量のフェニレフリンを与えなかった(この条件のためにn=4)。
表17は、10mg/kgのアスピリンと同時投与した500mg/kgのDGLAを用いた連続7日間の経管栄養後の、静脈内フェニレフリン投与による平均動脈圧における変化を示す。
図9は、4匹の自然発症高血圧ラットにおける10mg/kgのアスピリンと同時投与した500mg/kgのDGLAを用いた連続7日間の経管栄養後の、フェニレフリンの静脈内投与後の平均動脈圧における変化を示す。より低い用量のDGLA+アスピリンと同様に、フェニレフリン条件をアスピリン単独群の条件と比較した。カルベジロールを投与直前のベースラインと統計的に比較し、最終用量のフェニレフリン(10μg/kg)を陽性対照と比較した。
より低い平均動脈圧が観察されたにもかかわらず、DGLA及びアスピリンの両方を与えられたラットの平均全身動脈圧と、アスピリンを単独で与えられたラットの平均全身動脈圧との間に有意な統計差は見られなかった。したがって、この場合も同様に、アスピリンが500mg/kgのDGLAの連日投与の利益を妨げたと考えられる。陽性対照は、他の群において観察されたように、動物の動脈圧を統計的に低下させることができた。フェニレフリンの最終用量によって引き起こされた上昇は、有意であるとは見なされなかった。
表18は、7日間連続して6つの異なる経管栄養群を用いた後の、ベースライン時の平均動脈圧を示す。
図10は、本試験の第2部の間に摂食させた6つ群のベースライン時の平均動脈圧を表す。気管切開は血行動態を安定させたがラットの動脈圧に変化を引き起こした。アスピリンを投与されなかったラット(ビヒクル、DGLA50mg/kg、及びDGLA500mg/kg)の動脈圧は、本試験の第1段階(試験番号20131022−1)の間に得られ、他の群の動脈圧は、本試験(Charles Riverからの動物の異なるバッチであるが、同じ系統及びサイズのラット)中に記録された。50または500mg/kgのDGLAを投与された群と、10mg/kgのアスピリンを加えたそれらの均等物との間で統計的t検定を行った。アスピリン10mg/kgを含むDGLA500mg/kgと比較して、50mg/kgのDGLA単独と、10mg/kgのアスピリンと同時投与した50mg/kgのDGLAとの間、及びDGLAの500mg/kg群との間で、ベースライン時の平均動脈圧に統計的に有意な上昇が見られた(有意な上昇は*で印が付けられている)。したがって、DGLAは、自然発症高血圧ラットにおいて動脈圧を低下させること(アスピリンによって排除される効果)ができると考えられる。
表19は、7日間連続して6つの異なる経管栄養群を用いた後の、20μk/kgのフェニレフリンの静脈内投与による平均動脈圧を示す。
図11は、DGLAについて調査する2つの試験にわたって分析された全ての群の平均動脈圧を提示する。これらの動脈圧は、20μg/kgのフェニレフリンの静脈内投与後に得られた。ベースラインにおいて取得された結果と同様に、各用量のDGLAを、アスピリン単独群におけるその均等な条件と統計的に比較した。両方の対応する群間に、平均動脈圧の有意な増大が認められた(*で印が付けられている)。
この試験は、DGLAが単独で及びアスピリンと長期同時投与された場合に、高血圧ラットにおいて測定される動脈圧を低下させる有効性、ならびに同じ系統の自然発症高血圧ラットにおいてフェニレフリンによって急激に誘導された高血圧反応の低減の有効性を調査するために設計された。
自然発症高血圧ラット(SHR)は、各コロニー内で、心血管系に対する直接的及び間接的なエフェクターに応答して均一な変化をもたらすため、高血圧症の研究において一般的に使用されるモデルである。ラットは、選択され、何世代にもわたって同種交配されており、1ヶ月超持続する150mmHgを上回る収縮期血圧によって高血圧であると定義される。
この試験により、DGLA及びアスピリンの長期同時投与が、DGLAで処置した自然発症高血圧ラットにおいて以前に観察されたようなDGLAによる平均全身動脈血圧の低下(最大25%)を妨げたことが明らかになった。
10mg/kgのアスピリンと組み合わせて試験したDGLAの用量(50及び500mg/kg)は、アスピリンを単独で与えられた動物と比較して、フェニレフリンの注射後に平均動脈圧の用量依存性の上昇を有意に低下させなかった。2つの用量のDGLAが同様の効果を引き起こしたことから、アスピリンが同時投与される場合、より高い用量のDGLAの利益は最小限であり得ることが示唆される。両方の用量のDGLAをそれらの均等なアスピリン単独条件と比較した場合、アスピリンは、ベースライン条件において、またラットが20μg/kgのフェニレフリンの静脈内投与により負荷をかけられた場合に、平均動脈圧の明らかな上昇を引き起こした。
この試験では、陽性対照としてカルベジロールを用いた。これは、心不全及び高血圧を治療するために用いられるβ遮断薬である。カルベジロールは、血管を弛緩させて心拍数を下げることにより作用し、したがって心室の再充満、血流を改善し、また血圧を低下させる。自然発症高血圧ラットに静脈内注射されたときのカルベジロールの効果は、フェニレフリンの3つの用量の各々の後に血圧を下げる能力を裏付け、試験系の感度を証明した。
実施例4
18〜45歳(両端の値を含む)の健康な男性及び女性対象において、単施設、二重盲検、無作為化、プラセボ対照、第2部構成、第I相試験を行った。この試験の主目的は、健康な対象において(1日1回、28日間)経口投与される単一用量、及び経口投与される複数用量のDS107Gカプセルの安全性及び血漿中薬物動態(PK)を評価することであった。
18〜45歳(両端の値を含む)の健康な男性及び女性対象において、単施設、二重盲検、無作為化、プラセボ対照、第2部構成、第I相試験を行った。この試験の主目的は、健康な対象において(1日1回、28日間)経口投与される単一用量、及び経口投与される複数用量のDS107Gカプセルの安全性及び血漿中薬物動態(PK)を評価することであった。
この試験の副次的目的は、以下を評価することであった:
健康な対象に経口投与される単一用量のDS107GカプセルのPKに食物が与える影響。
健康な対象に1日1回、28日間与えられる複数用量のDS107Gカプセル後のヒト皮膚におけるDGLAのPK。
全部で48人の対象が登録された(単一用量試験の32人及び複数用量試験の16人を含む)。単一及び複数用量試験は、コホート当たり8人の対象から構成される(理想的には男性4人及び女性4人であるが、性別当たり少なくとも3人とする)。
ブロック無作為化により、対象を3:1の比でDG107G対プラセボに無作為に割り当てた。単一用量を用いる本試験の第1部は、それぞれが8人の対象からなる3つのコホートから構成され、Safety Monitoring Committee(SMC)によるコホート3の安全性データの評価に基づいてコホート4が追加された。単一経口用量のDS107Gまたは対応するプラセボを、コホート1〜3(それぞれ、500、1000、及び2000mg)の絶食対象に並行して投与した:コホート4の対象には、コホート3の終了後に4000mgの用量を投与した。コホート2の対象には、1回目の投与後14日以内に摂食状態で1000mgまたは対応するプラセボを投与した。
複数用量を用いる本試験の第2部は、それぞれが8人の対象からなる2つのコホート(コホート5及び6)から構成され、絶食状態で28日間、複数用量の治験薬を投与された。コホート5及び6の対象には、それぞれ、DS107Gを2000mgもしくは4000mg、または対応するプラセボを1日1回28日間投与した。コホート6は、SMCによってコホート5の安全性データが評価された後に開始された。中間解析は予定されていなかった。
本試験の第1部において、試験の1日目、5日目、8日目、及び14日目、ならびに経過観察来院日に、投与後最長312時間、コホート1〜4で絶食対象のみにジホモガンマリノレン酸(DGLA)及び15−ヒドロキシエイコサテトラエン酸(15−HETrE)の血漿PK分析を行った。第2部では、試験の1日目、2日目、3日目、5日目、及び8日目に、投与後最長168時間、コホート5及び6についてDGLAの血漿PK分析を行った。安全性評価は、試験の間中モニタリングされた。
無作為化、プラセボ対照、二重盲検設計を用いて、安全性及び忍容性評価の間のバイアスを最小化した。
本試験の第1部の後には、4000mgまで用量漸増した単一用量漸増設計が続いた。4000mg用量を除いて、本試験の第1部の全ての用量は、以前にヒトで試験を行った。治療関連有害事象(TEAE)は、150mg/日を28日間、450mg/日を28日間、1000mg/日を14日間、及び2000mg/日を10日間等の以前に試験した複数用量投与計画において観察されなかった。用量の選択は、以前の臨床試験で試験した経口DGLAの用量に基づいており、PKの特徴付けならびに以前に試験した用量及びより高い用量の安全性は、主目的として観察された。
食物がDGLAの経口バイオアベイラビリティに与える影響も評価した。
DGLAはステロイド代謝に関与するため、バイオマーカーであるジヒドロテストステロンに対する潜在的な性別要因を評価するために、健康な男性対象が本試験に含まれた。
本試験の第1部に各対象が参加していた全期間は、スクリーニング期間を除いて約14日であった。本試験の第2部では、期間は約42日であった。
スクリーニング手順は、−1日目(ベースライン)の試験開始前に、安全性評価を評価することにより第1部及び第2部の両方に対して実施した。安全性評価は、有害事象(AE)、臨床検査(血液学、生化学、ウイルス学[B型肝炎表面抗原、ヒト免疫不全ウイルス(HIV)抗体、C型肝炎抗体]、及び尿検査)、依存性薬物(DOA)試験結果、及び女性対象の場合は妊娠検査(尿中ヒト絨毛性ゴナドトロピンβ[βHCG])、バイタルサイン(血圧[BP]、脈拍、体温)、12リード心電図(ECG)、身体検査、ならびに併用薬の評価(本試験の第2部のみ)を含んだ。
全ての対象は、以下に記載する組み入れ基準及び除外基準を満たす必要があった。しかしながら、臨床的に重大ではなく、安全性に関する懸念を引き起こさないわずかな逸脱は、プロトコルに従って治験責任医師及び治験依頼者により許容可能であると見なされた。
試験参加が考慮される全ての対象は、以下の組み入れ基準を満たしていることが必要であった:
1.対象は、男性または女性であった。
2.女性対象、及び男性対象の女性パートナーは、
●妊娠していないこと(女性対象は、試験に参加する前に尿妊娠検査陰性でなければならない)。
●授乳していないこと。
●試験期間中または試験後3ヶ月以内に妊娠する計画がないこと。
●試験に参加する前、及び経過観察来院後さらに3ヶ月間、適切な形態の避妊を遵守していること。
3.対象は、18〜45歳であった(両端の値を含む)。
4.対象は、インフォームドコンセント用紙に署名した。
5.対象の肥満度指数(BMI)は、18.0〜30.0kg/m2であった(両端の値を含む)。
6.対象は、安全性評価を評価することにより、治験責任医師の判断で良好な健康状態にあると見なされた。
7.対象は、治験責任医師と十分にコミュニケーションを取れていること、試験の要件を理解して従うこと、そして書面によるICFに署名していること。以下の基準のうちのいずれかの証拠があった場合、対象は試験から除外された:スクリーニング前の4週間以内に臨床的に重大な疾病を有していた。
8.試験前の3ヶ月間、安全な避妊方法を使用しておらず、試験の期間中に安全な避妊方法を使用する意思のない、妊娠の可能性がある女性、及び男性対象の女性パートナー:安全な避妊方法の例として、子宮内避妊器具もしくは経口避妊薬、ダイアフラム、または殺精子薬と併用する場合はコンドームが挙げられる。
9.投薬前の2週間以内に処方薬を使用したか、または投薬前の1週間に店頭医薬品(ビタミン及びサプリメントを含む)(投薬の48時間前まで許可されているパラセタモールを例外とする)及びホルモン避妊薬を使用した対象。
10.対象がω3またはω6脂肪酸が豊富な栄養補助食品を使用した。
11.対象に薬物/溶剤乱用の有意な病歴があるか、またはスクリーニング時にDOAに陽性の検査結果を有していた。
12.治験責任医師の判断によると、対象にはアルコール乱用の病歴があり、スクリーニングの時点で1週間当たり28単位(男性)または1週間当たり21単位(女性)の飲酒をしていた。
13.対象は、治験責任医師の判断によると、試験に参加するのに適していなかった。
14.対象は、治験薬の投与初日前3ヶ月以内に、治験薬/デバイスを用いた別の臨床試験に参加していた。
15.対象は、スクリーニング時に、HIV抗体、B型肝炎表面抗原、またはC型肝炎抗体に陽性の試験結果を有していた。
16.対象は、いずれかの薬物に対して重度の有害反応または著しい過敏性を示した。
17.対象は、スクリーニング前の3ヶ月以内に血液または血液製剤を献血していた。
18.対象は、治験薬のいずれかの成分に対して既知の過敏性を有していた。
1.対象は、男性または女性であった。
2.女性対象、及び男性対象の女性パートナーは、
●妊娠していないこと(女性対象は、試験に参加する前に尿妊娠検査陰性でなければならない)。
●授乳していないこと。
●試験期間中または試験後3ヶ月以内に妊娠する計画がないこと。
●試験に参加する前、及び経過観察来院後さらに3ヶ月間、適切な形態の避妊を遵守していること。
3.対象は、18〜45歳であった(両端の値を含む)。
4.対象は、インフォームドコンセント用紙に署名した。
5.対象の肥満度指数(BMI)は、18.0〜30.0kg/m2であった(両端の値を含む)。
6.対象は、安全性評価を評価することにより、治験責任医師の判断で良好な健康状態にあると見なされた。
7.対象は、治験責任医師と十分にコミュニケーションを取れていること、試験の要件を理解して従うこと、そして書面によるICFに署名していること。以下の基準のうちのいずれかの証拠があった場合、対象は試験から除外された:スクリーニング前の4週間以内に臨床的に重大な疾病を有していた。
8.試験前の3ヶ月間、安全な避妊方法を使用しておらず、試験の期間中に安全な避妊方法を使用する意思のない、妊娠の可能性がある女性、及び男性対象の女性パートナー:安全な避妊方法の例として、子宮内避妊器具もしくは経口避妊薬、ダイアフラム、または殺精子薬と併用する場合はコンドームが挙げられる。
9.投薬前の2週間以内に処方薬を使用したか、または投薬前の1週間に店頭医薬品(ビタミン及びサプリメントを含む)(投薬の48時間前まで許可されているパラセタモールを例外とする)及びホルモン避妊薬を使用した対象。
10.対象がω3またはω6脂肪酸が豊富な栄養補助食品を使用した。
11.対象に薬物/溶剤乱用の有意な病歴があるか、またはスクリーニング時にDOAに陽性の検査結果を有していた。
12.治験責任医師の判断によると、対象にはアルコール乱用の病歴があり、スクリーニングの時点で1週間当たり28単位(男性)または1週間当たり21単位(女性)の飲酒をしていた。
13.対象は、治験責任医師の判断によると、試験に参加するのに適していなかった。
14.対象は、治験薬の投与初日前3ヶ月以内に、治験薬/デバイスを用いた別の臨床試験に参加していた。
15.対象は、スクリーニング時に、HIV抗体、B型肝炎表面抗原、またはC型肝炎抗体に陽性の試験結果を有していた。
16.対象は、いずれかの薬物に対して重度の有害反応または著しい過敏性を示した。
17.対象は、スクリーニング前の3ヶ月以内に血液または血液製剤を献血していた。
18.対象は、治験薬のいずれかの成分に対して既知の過敏性を有していた。
対象は、理由の如何を問わず、いつでも自由に試験への同意を撤回することができた。また、治験責任医師は、治験責任医師の判断で、それが対象の最善の利益になる場合、治験参加から対象を離脱させることができた。対象は、以下の理由のいずれかのために試験から離脱した:
●任意の時点での同意の撤回
●プロトコルからの逸脱
●偶発的な疾病
●AE(有害作用)
●任意の時点での同意の撤回
●プロトコルからの逸脱
●偶発的な疾病
●AE(有害作用)
対象には早期離脱の理由を説明する義務はなかったが、治験責任医師は、対象の権利を十分に尊重する一方で、その理由を明らかにするための妥当な努力を行うことになっていた。同意の撤回に医療上の理由がある場合、対象が満足できる健康状態になるまで、その対象は治験責任医師の監視下に留まった:治験責任医師は経過観察評価を実施した。
治験責任医師が、対象の福利を最大限にすると考えられると見なした場合、治験責任医師は、対象のかかりつけ医に、対象が試験から離脱した医療上の理由を通知することになっていた。治験責任医師は、対象が満足できる健康状態になることを確実にするために、経過観察のために施設に戻ってこなかった対象に連絡を取るようあらゆる努力を行うことになっていた。
本試験の第1部において、表20に従って、少なくとも8時間の絶食後、試験1日目にコホート1〜3の対象に単一用量の治験薬(500mgのDS107Gカプセルとしてまたは対応するプラセボカプセルとして、500mg、1000mg、または2000mgのいずれか)を並行して投与した。コホート4は、SMCによるコホート3の安全性データの検討後に開始された。
1回目の投与の少なくとも14日後に、コホート2において食物の影響を評価し、その時点で第2の単一用量を投与した。10時間の絶食期間後、かつ食事の摂取を開始してから30分後に、240mLの水とともに1000mg用量の治験薬を対象に投与した。次いで、対象は、投薬後少なくとも4時間は食物摂取を控えた。標準高脂肪食(800〜1000kcal:脂肪から500〜600kcal及び炭水化物から250kcal)を用いて、投薬後少なくとも12時間は食物摂取を標準化した。典型的な標準試験食は、バターで焼いた卵2個、ベーコン2本、バター付きトースト2枚、ハッシュブラウンポテト120mL、及び全乳240mLから構成されていた。
本試験の第2部(コホート5及び6)において、対象に、絶食状態で28日間1日1回、治験薬(500mgのDS107Gカプセルまたは対応するプラセボカプセル)を投与した。コホート5の対象に最初に治験薬を投与し、最初の14日間、彼らが2000mgの1日用量に忍容性を示した場合、コホート6の対象に28日間4000mgの1日用量を開始した。
用量レベルにおける任意の漸増、またはその後のコホート開始は、治験責任医師によって決定された。コホート4(4000mgの用量)を開始する前に、コホート3からの評価可能な安全性データ(2000mgの用量)を有する最低でも5人の対象が必要であった。第2部では、コホート6(4000mg/日)を開始する前に、コホート5からの14日分の評価可能な安全性データ(2000mg/日)を有する最低でも5人の対象が必要であった。
500mgのDGLA遊離脂肪酸(FFA)を含有する、ある強度のDS107G DGLAカプセルが開発された。カプセルは以下の賦形剤を含んでいた:DGLA FFA(微量の2000ppm dl−αトコフェロールで安定化した)。カプセルシェル内の全ての賦形剤は、軟ゼラチン製品に一般的に使用されており、以下の成分を含有する伝達性海綿状脳症(TSE)認定ゼラチンシェルを含む:精製水、可塑剤グリセロール、着色剤の二酸化チタン、及び加工助剤レシチン及び中鎖トリグリセリド。臨床試験用のプラセボカプセル(DS107Gプラセボカプセル)は、軟ゼラチンシェルに封入された液体パラフィンから構成され、DGLAカプセルと全く同じ外観であった。
適格性基準を満たす対象を、無作為化スケジュールを用いて、DGLA(500、1000、2000、4000mgの用量)または対応するプラセボカプセルを投与するように無作為に割り当てた。無作為化は、3:1の積極的治療対プラセボの比を用いたブロック無作為化であった。無作為化スケジュールは、SAS(登録商標)9.1.3 SP4を使用してPlanimeterにより作成した。
対象は、投薬前の2週間処方薬を使用すること、または投薬前の1週間、店頭医薬品(ビタミン及びサプリメントを含む)及びホルモン避妊薬を使用することを許可されなかったが、投薬の48時間前まで許可されているパラセタモールは例外であった。また、対象は、ω3またはω6脂肪酸が豊富な栄養補助食品を使用することを許可されなかった。対象は、1週間当たり28単位(男性対象)または1週間当たり21単位(女性対象)を超えるアルコールを摂取することを許可されなかった。
対象は、スクリーニング前の4週間及び試験中は、ω3またはω6脂肪酸が豊富な栄養補助食品(例えば、タラ肝油カプセル)の摂取を避けるように助言された。
対象は、DOAを検査するための尿試料採取の少なくとも24時間は、ポピーシード及びポピーシードを含有する食物を食べることを避けるよう助言された(ポピーシードは、時に陽性の試験結果をもたらす可能性があるため)。
食物が経口1000mgのDGLAカプセルに与える影響の評価を受けるコホート2の対象は、投与後少なくとも4時間は食物摂取を控えた。標準高脂肪食を用いて、投与後少なくとも12時間は食物摂取を標準化した。
対象は、ホルモン避妊薬を例外として、いずれの全身薬及び店頭薬(ビタミン及びサプリメントを含む)も服用することを控えなければならなかった。パラセタモール(最大4g/日の用量)は、投薬の48時間前まで許可されていた。対象はまた、最初の治験薬投与(1日目)の48時間前から経過観察来院までアルコール摂取を控えなければならなかった。
試験前または試験中に、カフェイン摂取またはたばこの使用に関する制限はなかった。対象は、臨床検査のために血液が採取される前の少なくとも3〜4時間は、運動及び激しい身体活動を避ける必要があった。
対象から得た血液試料中の(遊離及び総)DGLA及び遊離15−HETrEの血漿中濃度の分析を行った。さらに、遊離及び総DGLAの分析のために、1、7、14、及び28日目に皮膚水疱液を得た。
ジヒドロオキシテストステロン(DHT)の血漿中濃度は、バイオマーカーまたは探索的有効性エンドポイントとして第2部において評価した。
全血漿及び皮膚水疱液試料の分析は、妥当性のある方法を用いて行った。遊離及び総DGLAの濃度は、液体クロマトグラフィー/タンデム質量分析法(LC/MS/MS)により血漿及び皮膚水疱液中で測定した:定量範囲は、遊離DGLAで100〜10,000ng/mL、総DGLAで5000〜500,000ng/mLであった。遊離15−HETrEの血漿中濃度は、100〜10,000ng/mLの定量範囲でLC/MS/MSにより測定した。DHTの血漿中濃度は、0.02〜1.5ng/mLの定量範囲でLC/MSにより測定した。
対象は、治験薬及び/または手順に対する有害反応について、第1相の拘束期間を通してモニタリングされた。
本試験で用いられた薬物動態評価は、可能性のある治療薬の評価のための標準であった。安全性評価は、第1相臨床試験の標準であると考えられる方法を含んでいた。
この試験は探索的であり、正式な検出力の計算は行われなかった。各コホートに予定される対象の数(8人の対象)は、DS107Gカプセルの安全性及び全身曝露の評価を可能にするのに十分であると考えられた。
集団の分析は以下を含んでいた:
●Intention−to−Treat(ITT)集団は、少なくとも1用量の治験薬を投与された全ての無作為化対照から構成される。
●パー・プロトコル(PP)集団は、SAPに定義されるように、重大なプロトコル逸脱のなかったITT集団の全ての対象から構成される。
●PK集団は、血漿から得られた評価可能なPKデータを有していたPP集団に含まれる全ての対象を含む。以下のデータが入手可能である場合、血漿中濃度の観察は妥当な評価可能な測定値であると考えられた:試験識別番号、無作為化番号、試料採取の日時、用量及び濃度。各プロトコルに規定された濃度が評価可能であった場合、同じ試料からの一連のそのような測定値は完全であると考えられた。データが完全な一連の観察を含んでいた場合、血漿PKデータは定義により評価可能であった。いずれかの血漿PKの欠測は不完全なPKデータをもたらすため、いずれかの血漿PKの欠測がある対象は、PK集団から除外された。プラセボに無作為化された対象も、PK集団から除外された。
●Intention−to−Treat(ITT)集団は、少なくとも1用量の治験薬を投与された全ての無作為化対照から構成される。
●パー・プロトコル(PP)集団は、SAPに定義されるように、重大なプロトコル逸脱のなかったITT集団の全ての対象から構成される。
●PK集団は、血漿から得られた評価可能なPKデータを有していたPP集団に含まれる全ての対象を含む。以下のデータが入手可能である場合、血漿中濃度の観察は妥当な評価可能な測定値であると考えられた:試験識別番号、無作為化番号、試料採取の日時、用量及び濃度。各プロトコルに規定された濃度が評価可能であった場合、同じ試料からの一連のそのような測定値は完全であると考えられた。データが完全な一連の観察を含んでいた場合、血漿PKデータは定義により評価可能であった。いずれかの血漿PKの欠測は不完全なPKデータをもたらすため、いずれかの血漿PKの欠測がある対象は、PK集団から除外された。プラセボに無作為化された対象も、PK集団から除外された。
上に定義した集団は、試験の第1部及び第2部のデータのために別個に作製された。安全性分析は、ITT集団に対して行われた。
安全性分析は、治療アーム、コホート、及び来院ごとに表にした安全性集団に対して行われた。全ての安全性データは、記述統計ツールによって特徴付けられた。試験プロトコルにおいて調査するための仮説は立てなかった。安全性評価の評価は、記述的に行われた。連続変数は、それらの平均値、標準偏差(SD)、中央値、最小値、及び最大値によって特徴付けられた;離散変数は、それらの絶対値(度数)及び相対(パーセンテージ[%])分布によって特徴付けられた。治療群はまた
主要エンドポイント(DS107Gカプセルの単一及び複数経口用量から得られたPK特性)は、非コンパートメントPKモデルの助けを借りて得た。副次エンドポイントは、食物が単一経口用量のDS107GカプセルのPKに与える影響を特徴付けすること、及びDS107Gカプセルの複数経口用量後にヒトの皮膚におけるDGLAのPKを特徴付けることから構成されていた。
副次エンドポイントは、以下の記述分析ツールを用いて報告された:治療アーム、来院、及びコホートごとにグループ化した連続パラメータから得た、妥当な観察の数、平均値、標準偏差(SD)、中央値、最小値及び最大値。
試験データを評価する間に正式な仮説検証は行われなかった。第1部(単一漸増用量)では、曝露の程度は定義により1日であった(治験薬の投与が成功した場合)。第2部(複数漸増用量)では、曝露の程度は、最後の治験薬摂取日から最初の治験薬摂取日を差し引いたものに1を足したものとして算出した(治験薬投与の中断が報告されなかった場合)。中断(複数可)があった場合、上記式の結果は、中断(複数可)の数とともに減少した。
PKデータの統計分析は、SASソフトウェア(バージョン9.1.3)を使用して行われた。薬力学的分析は行われなかった。
DGLA及び15−HETrEの血漿PKパラメータは、モデル非依存的方法(非コンパートメント分析)を用いて推定し、また以下を含んでいた:第1部及び第2部は、Cmax、tmax、Clast、Tlast、AUC0−24、AUC0−inf、AUClast、λz、CL、V、及びt1/2、ならびに第2部(定常状態)のみのデータは、tmin、cmin、CLss、Vss、Cavg、及び%PTF。
全てのPKパラメータを記述的に要約し、本試験の探索的な性質のために、正式な統計的検定は行われなかった。
単一用量設定(第1部)において、治験薬の中止はコホート1、3、及び4における潜在的な結果ではなかった:したがって、これらのコホートについて記録活動は必要なかった。コホート2の場合、登録された対象に治験薬を2回投与した:絶食状態で投与された単一用量、及び摂食状態で投与された第2の単一用量。
また、プロトコルに以下の修正がなされた。
●経口DGLAを用いた以前の臨床試験及びそれらの安全性の結論を明確にするために、臨床試験の表を追加した。
●治験責任医師及び倫理委員会のために提案された治験の評価されるリスク及び便益を明確にするために、リスク/便益評価を追加した。
●本試験の第1部(単一用量コホート)に登録された対象を呼び戻して第2部(複数用量コホート)に再登録することができるように登録基準が変更された:但し、対象が治験薬に関連するAEを示さず、複数用量の投与計画を開始する前に少なくとも14日間の休薬期間を設けることとした。この変更の理論的根拠は、ボランティアの安全性を犠牲にすることなく募集を支援することであった。
●本試験の第2部(複数用量コホート5及び6)における全ての臨床検査評価時期の評価として、凝固に関する臨床検査(プロトロンビン時間及び活性化部分プロトロンビン時間[APTT])を追加した。これらの評価は、将来の試験のための探索的マーカーとして凝固因子におけるあらゆる潜在的な変化をモニタリングするために追加された。
●単一のECG記録が得られるように曖昧な表現は明確なものに置き換えた。
●第1部のコホートからの予備PKデータにより、15−HETrEの全ての試料が定量限界未満(BLQ)であったことが明らかになったため、15−HETrEを第2部(複数用量コホート)における試験のための分析物として除去した。
●「総」DGLAの血漿PKプロファイルは「遊離」DGLAと異なり得るため、遊離(非エステル化)DGLAに加えて、全ての分析に「総」DGLAの定量化を追加した。
●また、追加の分析及び統計解析アウトプットを提供するために、予定されていた分析に対する変更も含められた。
●経口DGLAを用いた以前の臨床試験及びそれらの安全性の結論を明確にするために、臨床試験の表を追加した。
●治験責任医師及び倫理委員会のために提案された治験の評価されるリスク及び便益を明確にするために、リスク/便益評価を追加した。
●本試験の第1部(単一用量コホート)に登録された対象を呼び戻して第2部(複数用量コホート)に再登録することができるように登録基準が変更された:但し、対象が治験薬に関連するAEを示さず、複数用量の投与計画を開始する前に少なくとも14日間の休薬期間を設けることとした。この変更の理論的根拠は、ボランティアの安全性を犠牲にすることなく募集を支援することであった。
●本試験の第2部(複数用量コホート5及び6)における全ての臨床検査評価時期の評価として、凝固に関する臨床検査(プロトロンビン時間及び活性化部分プロトロンビン時間[APTT])を追加した。これらの評価は、将来の試験のための探索的マーカーとして凝固因子におけるあらゆる潜在的な変化をモニタリングするために追加された。
●単一のECG記録が得られるように曖昧な表現は明確なものに置き換えた。
●第1部のコホートからの予備PKデータにより、15−HETrEの全ての試料が定量限界未満(BLQ)であったことが明らかになったため、15−HETrEを第2部(複数用量コホート)における試験のための分析物として除去した。
●「総」DGLAの血漿PKプロファイルは「遊離」DGLAと異なり得るため、遊離(非エステル化)DGLAに加えて、全ての分析に「総」DGLAの定量化を追加した。
●また、追加の分析及び統計解析アウトプットを提供するために、予定されていた分析に対する変更も含められた。
40人の対象をスクリーニングした:それら40人のうち、4人の対象が組み入れ/除外基準を満たしていないために除外され、4人の対象が同意を撤回した。治験薬に無作為化された32人の対象の内訳を表21に提示する。
DS107Gの投与前の、ベースライン時の遊離DGLA及び総DGLAの血漿中濃度を表22にまとめた。これらの濃度は、概して変動しやすかった。
単一用量のDS107G後の平均血漿中濃度を用量コホートごとにグラフで示す。遊離DGLA:図12(線形プロット)及び図13(対数線形プロット)、ならびに総DGLA:図14(線形)及び図15(対数線形プロット)。
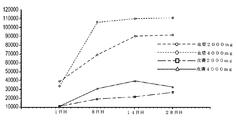
DS107Gカプセルの単一用量(500、1000、2000、及び4000mg)後、遊離DGLA及び総DGLAの対象間の変動(SDによって測定される)は、血漿中濃度及びベースライン補正したPKパラメータの両方で高かった。絶食条件下では、遊離DGLA及び総DGLAの両方のベースライン補正した平均Cmax及びAUC0−24が線形に増加した(表23、表24)。遊離DGLA(表23)の場合、最大濃度(Tmax)の期間中央値は4であり、用量にわたって一貫性がなく、2段階低い用量では8時間の値であり、2段階高い用量では18時間の値であった(表24)。ベースライン補正した排出PKパラメータは、いくつかのコホートでは対象の半数未満について決定することができたが、遊離DGLAまたは総DGLAのいずれについても排出半減期またはクリアランスの非線形薬物動態の証拠は見られなかった(表24及び表25)。統計的に評価したわけではないが、絶食条件下での単一1000mg用量のDS107Gの投与は、ベースライン補正した平均Cmax及びAUC0−24に基づいて約50%高い総DGLAの吸収速度及び範囲をもたらした(表22)。
1000mg用量(コホート2)を用いて単一用量でベースライン補正した遊離DGLA及び総DGLAのPKに食物が与える影響を評価し、表26に報告する。端的に述べると、ベースライン補正した遊離DGLAの平均Cmaxは、絶食条件下では約3倍高く、摂食条件下よりも1時間(中央値)早く生じた(表26)。絶食条件下でベースライン補正した遊離DGLAの平均AUC0〜24は、摂食条件下よりも約2倍高かった。このようにして、絶食条件下でDGLAの吸収速度及び範囲の増加が観察された。遊離DGLAの排出において、絶食条件と摂食条件との間に明確な差は見られなかった(表25)。
総DGLAの場合、ベースライン補正した平均Cmaxは、摂食条件下よりも絶食条件下で約1.5倍高く、tmaxは約50%早く生じた(中央値8対15時間)(表25)。絶食条件下でベースライン補正した平均AUC0−24は、摂食条件下で約1.8倍高かった。半数弱の対象(絶食2/6人、摂食3/6人)が、総DGLAのλz、t1/2、クリアランス、及び分布容積を推定するのに十分なデータを有していた。これらのデータは、絶食条件下でのDGLAの吸収速度及び範囲の増加を示唆するものである。データ集団が小さいことに起因して、総DGLAの排出または容積分布に関する信頼できる結論は出なかった。
表25に示すPKパラメータに基づいて、ベースライン補正した平均Cmaxは、遊離DGLAよりも総DGLAで約10倍(絶食)及び約20倍(摂食)高かった。ベースライン補正した平均AUC0−24は、遊離DGLAよりも総DGLAで約54倍(絶食)及び約56倍(摂食)高かった。
DSI07Gは、健康なボランティアに対する500、1000、2000、または4000mgの量の単一用量として良好な忍容性を示した。積極的治療の対象及びプラセボ対照の対象の同様のパーセンテージの対象から報告された最も一般的なTEAEは、軽度から中程度の下痢(報告された用語:軟便)であった(積極的に治療した対象の発生率:5/24[20.8%]、プラセボ対照の対象の発生率:2/8[25.0%])。下痢事象は、比較的短期間であり、(プラセボ対照の対象に起こったものを含めて)全てが治験責任医師によって治験薬に関連する可能性があると見なされた。注目すべきは、絶食状態で第2の単一用量のDS107Gを投与され、下痢のTEAEを示した対象には、下痢の再発が見られなかった。軽度の感染症、中咽頭痛、及び咽頭炎、ならびに摂食状態で投薬した後の中程度の重症度の発熱及び尿路感染症を含む他の全てのTEAEは、各1人の対象にのみ、かつ積極的治療群においてのみ起こった。
複数用量の結果−試験第2部
第2部の定常状態での平均血漿中濃度及び平均濃度を用量コホートごとにグラフで示す。遊離DGLA:図16(線形プロット)及び図17(対数線形プロット)、ならびに総DGLA:図18(線形プロット)及び図19(対数線形プロット)。1日目及び28日目に、遊離DGLAの平均濃度が投薬後約4時間でピークを迎えたのに対し、総DGLAでは平均ピーク濃度は明らかではなかった。遊離DGLA及び総DGLAの両方の平均濃度が反復投与により増加したが、総DGLAの増加がより顕著であった。平均濃度プロットの目視検査によれば、血漿中濃度は、両方の用量(1日2000及び4000mg)及び分析物(遊離DGLA及び総DGLA)で、14日目辺りに定常状態に達したと考えられた。1日用量を2000mgから4000mgへと2倍にすると、定常状態での平均濃度が遊離DGLAでは1.6倍増加したが、総DGLAでは1.2倍増加したのみであったことから、より高い用量では1つ以上の飽和プロセスがあることが示唆される。
第2部の定常状態での平均血漿中濃度及び平均濃度を用量コホートごとにグラフで示す。遊離DGLA:図16(線形プロット)及び図17(対数線形プロット)、ならびに総DGLA:図18(線形プロット)及び図19(対数線形プロット)。1日目及び28日目に、遊離DGLAの平均濃度が投薬後約4時間でピークを迎えたのに対し、総DGLAでは平均ピーク濃度は明らかではなかった。遊離DGLA及び総DGLAの両方の平均濃度が反復投与により増加したが、総DGLAの増加がより顕著であった。平均濃度プロットの目視検査によれば、血漿中濃度は、両方の用量(1日2000及び4000mg)及び分析物(遊離DGLA及び総DGLA)で、14日目辺りに定常状態に達したと考えられた。1日用量を2000mgから4000mgへと2倍にすると、定常状態での平均濃度が遊離DGLAでは1.6倍増加したが、総DGLAでは1.2倍増加したのみであったことから、より高い用量では1つ以上の飽和プロセスがあることが示唆される。
PKパラメータは、ベースラインDGLA濃度を用いて投与したDGLA濃度を補正した後に算出した。
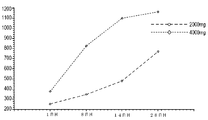
遊離DGLAのベースライン補正した血漿薬物動態を表26に報告する。端的に、遊離DGLAのベースライン補正した平均Cmax及びAUCは、評価した両方の日ともより高いDS107G用量コホートにおいてより高かった。4000mg用量のベースライン補正した平均Cmaxは、1日目には2000mg用量の場合よりも約3倍高かったが、28日目には約1.4倍高いのみであった。4000mg用量のベースライン補正した平均AUC0−24は、1日目には2000mg用量の場合よりも約2.5倍高く、28日目には約1.7倍高いのみであった。用量による変化は、1日目にはベースライン補正したCmax及びAUC0−24で線形であったが、28日目にはベースライン補正したAUC0−24のみで線形であった。大きな対象間変動がこの矛盾を引き起こした可能性がある。中央値tmaxは、1日目及び28日目の両方の用量コホートで同様であり、その値は4または5時間であった。排出半減期t1/2は、28日目のほうが1日目よりも長く、28日目の値は評価した時間間隔に依存していた。複数用量により、平均クリアランスは減少し、平均分布容積は増加した。
遊離DGLAのベースライン補正した定常状態での血漿薬物動態を表27に報告する。端的に述べると、反復投与によって遊離DGLA及び総DGLAの血漿中濃度が増加し、約14日目に定常状態を達成した。定常状態で、28日目(0−24時間)に評価したところ、ピーク値トラフ値変動(PTF)が両方の用量コホートで極めて高かった(平均値約430%及び約490%)。平均蓄積比(AR)は、Cmax及びAUCの両方とも4000mg用量コホートよりも2000mg用量コホートの方が高かった(2000mgでは約2.8及び約3.3のAR、4000mgでは約1.4及び約1.6のAR)。このデータは、遊離DGLAの反復投与による飽和動態及び/または分布容積の変化の存在を示唆している。
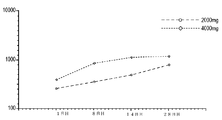
総DGLAのベースライン補正した血漿薬物動態を表28に報告する。端的に、総DGLAのベースライン補正した平均Cmax及びAUC0−24は、予想されたように、評価した両方の日ともより高いDS107G用量コホートにおいてより高かった。4000mg用量のベースライン補正した平均Cmax及びAUC0−24は、1日目には2000mg用量の場合よりもそれぞれ約1.5倍及び約1.5倍高かったが、28日目には、2000mg用量の場合よりも約1.2倍及び約1.4倍高いのみであった。
ベースライン補正したCmax及びAUC4における用量による変化は、評価したいずれの日も総DGLAの場合は線形ではなかった。大きな対象間変動がこの矛盾を引き起こした可能性がある。中央値Tmaxは、両方のコホートにおいて、単一用量を用いたとき(10〜18時間)よりも複数用量を用いたとき(8〜10時間)により迅速に生じた。2000mg用量コホートの場合、総DGLAの排出半減期t1/2は、24時間にわたって(それぞれ1日目及び28日目)評価したときには34.4〜44.0時間であり、28日目に0〜168時間にわたって評価したときには62.6時間であった。複数用量により、平均クリアランス及び分布容積は減少した。
遊離DGLAのベースライン補正した定常状態での血漿薬物動態を表29に報告する。端的に述べると、定常状態で、28日目(0〜24時間)に評価したところ、ピーク値トラフ値変動(PTF)が両方の用量コホートで極めて高かった(平均値62.5%及び44.9%)。平均ARは、Cmax及びAUCの両方とも4000mg用量コホートよりも2000mg用量コホートの方が高かった。このデータは、総DGLAの反復投与による飽和動態及び/または分布容積の変化の存在を示唆している。
皮膚水疱液中の平均遊離DGLA濃度を用量コホートごとに図20(線形プロット)及び図21(対数線形プロット)に示す。用量を2倍にすると平均濃度はほぼ2倍になり(1日目、8日目、14日目、及び28日目の濃度に基づいて)、両方の投与計画において反復投与により蓄積した。28日目の平均遊離DGLA濃度は、1日用量2000mg及び4000mgの両方とも1日目の濃度よりも約3倍高かった。
用量コホート(複数用量、PK集団)ごとの総DGLAの平均皮膚水疱液中濃度(ng/mL、線形プロット)を図22に示す。用量コホート(複数用量、PK集団)ごとの総DGLAの平均皮膚水疱液中濃度(ng/mL、対数線形プロット)を図23に示す。
皮膚水疱液中の平均総DGLA濃度を用量コホートごとに図24(線形プロット)及び図25(対数線形プロット)に示す。用量を2倍にすると総DGLAの平均濃度は約1.4倍増加した(1日目、8日目、14日目、及び28日目の濃度に基づいて)。28日目の平均総DGLA濃度は、1日用量2000mg及び4000mgの両方とも1日目の濃度よりもそれぞれ2.5倍及び3倍高かった。
血漿中及び皮膚水疱液中の濃度プロファイルを重ね合わせたところ、8日目及び14日目の同じDS107G用量では、遊離DGLAの平均濃度が血漿中及び皮膚水疱液中である程度類似していた(28日目はそうではなかった)。図26[線形プロット]及び図27[対数線形プロット])は、DGLAが血漿及び皮膚に同様に分布することを示唆している。総DGLAの場合、1日目後の同じDS107G用量では、平均濃度が皮膚水疱液中よりも血漿中ではるかに高かったことから(図26[線形プロット]及び図27[対数線形プロット])、総DGLAは皮膚内よりも血漿中でより容易に見られることが示唆される。総DGLAの皮膚内への限定された分布の機序は、血漿と比較して皮膚中の脂質の量が少ないことに関連している可能性が高い。
血漿ジヒドロテストステロン(DHT)濃度を、探索的有効性エンドポイントまたはバイオマーカーとして定量化した。ほとんどの時点で、SDに基づく濃度データにおける対象間の変動が高かった。DHTの平均血漿中濃度を用量コホートごとに図28(線形)及び図29(対数線形)に示す。
いずれの試料も、遊離15−HETrE濃度は測定できるほどではなく、全ての濃度はLLOQ(100ng/mL)未満であった。
複数用量試験に死亡は見られなかった。連続28日間、1日1回、2000mgまたは4000mgの用量として健康なボランティアに投与された場合、DSI07Gは非常に良好な忍容性を示し、最悪のTEAEは、比較的短期間の軽度から中程度の下痢(報告された用語:軟便)であった。下痢事象の大半(積極的に治療した対象の発生率:7/12[43.8%]、プラセボ対照の対象の発生率:0/4[0.0%])は、治験責任医師によって治験薬に関連する可能性があると見なされた。より高い割合の対象が、2000mg群(3/6[50%])よりも4000mg群(4/6[66.7%])で下痢を報告した。積極的に治療した対象の間でのTEAEの発生率は、プラセボ対照の対象の発生率よりもはるかに高かった(それぞれ、11/12[91.73%]の対象が全部で52件のTEAEを報告したのに対し、1/4[25.0%]の対象が全部で1件のTEAEを報告している)。重度のTEAEは見られず、下痢のTEAE以外は、全ての事象が治験薬と関連していないか、または関連している可能性が低いと見なされた。悪心が次に最もよく報告されたTEAEであった(4000mg治療群において4/6[66.7%]の対象の間で10の事象):悪心事象の9/10が軽度であり、他は中程度の重症度であった。下痢以外では、他の残りのTEAEは各2人の対象(気管支炎及び上咽頭炎)または各1人の対象(腹痛、無力症、発熱、血中CPKの増加、CRPの増加、WBC数の減少、めまい、頭痛、咳、及び血腫)、それらの大半は、治験責任医師によって治験薬と関連している可能性が低いか、または関連していないと見なされた。治験薬と関連している可能性があると考えられる他のTEAEは、腹痛及び無力症(報告された用語「脱力感」)であり、それらはどちらも軟便事象と時間的に関連していた。
複数用量試験において任意の患者に観察されたバイタルサインまたはECGには、臨床的に重大な異常は見られなかった。
Claims (21)
- 必要とする対象において、治験責任医師による包括的評価レベル、湿疹面積及び重症度インデックス(EASI)スコア、アトピー性皮膚炎に冒された解剖学的部位の面積のパーセンテージ、アトピー性皮膚炎スコアリング(SCORAD)、アトピー性皮膚炎に冒された体表面積、または視覚的アナログ尺度(VAS)掻痒スコアのうちの少なくとも1つを減少させる方法であって、1日当たり最大4gのDGLAまたはその誘導体を対象に経口投与することを含む、方法。
- 1日当たり約0.2〜約3gのDGLAまたはその誘導体が前記対象に経口投与される、請求項1に記載の方法。
- 1日当たり約0.5g、約1g、または約2gのDGLAまたはその誘導体が前記対象に経口投与される、請求項1に記載の方法。
- 1日当たり1g未満のDGLAまたはその誘導体が前記対象に経口投与される、請求項1に記載の方法。
- 前記対象は小児対象である、請求項1〜4のいずれか一項に記載の方法。
- 前記DGLAまたはその誘導体は、少なくとも約2週間、少なくとも約4週間、または少なくとも約8週間の期間、前記対象に投与される、請求項1〜5のいずれか一項に記載の方法。
- 薬学的組成物は、液体または半液体形態のDGLAまたはその誘導体を含む、請求項1〜6のいずれか一項に記載の方法。
- 前記対象は、基準レベルと比較して少ないベースライン好酸球数を有すると決定される、請求項1〜7のいずれか一項に記載の方法。
- 必要とする対象において疾患または障害を治療する方法であって、DGLAまたはその誘導体を含む薬学的組成物を前記対象に経口投与することを含む、方法。
- 前記疾患または障害は、尋常性ざ瘡、酒さ性ざ瘡、軽度、中程度、もしくは重度のアトピー性皮膚炎、乾癬、掻痒/かゆみ、放射線防護、皮膚乾燥、滑らかな皮膚、健康な皮膚、抗老化、及び光防護を含む皮膚障害及び皮膚疾患;膀胱癌、膀胱瘤、血尿、間質性膀胱炎、神経因性膀胱、ペロニー病、前立腺疾患、失禁、尿路感染症、及び膀胱尿管逆流を含む泌尿器障害及び泌尿器疾患;腎不全、急性腎傷害、慢性腎疾患、及び多発性嚢胞腎を含む腎疾患及び腎障害;強直性脊椎炎、線維筋痛症、痛風、感染性関節炎、狼瘡、変形性関節症、リウマチ性多発筋痛症、乾癬性関節炎、反応性関節炎、関節リウマチ、強皮症を含むリウマチ性疾患;炎症性肺疾患、気道感染症、胸膜腔疾患、肺血管疾患、肺炎、肺塞栓症、及び肺癌を含む呼吸器疾患;ならびに急性心虚血事象、急性心筋梗塞、狭心症、不整脈、心房細動、粥状動脈硬化、動脈細動、心不全、心血管疾患、慢性心不全、慢性安定狭心症、うっ血性心不全、冠動脈疾患、冠動脈性心疾患、深部静脈血栓症、糖尿病、真性糖尿病、糖尿病性神経障害、真性糖尿病を有する対象における拡張障害、浮腫、本態性高血圧、最終的な肺塞栓症、脂肪性肝疾患、心疾患、心不全、ホモ接合性家族性高コレステロール血症(HoFH)、ホモ接合性家族性シトステロール血症、高コレステロール血症、高脂血症、高血圧症、高トリグリセリド血症、代謝症候群、混合型脂質異常症、中程度から軽度の心不全、心筋梗塞、肥満管理、発作性心房細動/動脈細動/細動/粗動、発作性上室性頻拍(PSVT)、特に重度のまたは急激に発症する浮腫、血小板凝集、原発性高コレステロール血症、原発性高脂血症、肺動脈性高血圧症、肺高血圧症、再発性の血行動態的に不安定な心室性頻脈(VT)、再発性心室性不整脈、再発心室細動(VF)、破裂動脈瘤、シトステロール血症、脳卒中、上室性頻拍、症候性心房細動/粗動、頻拍、II型糖尿病、血管疾患、静脈血栓塞栓症、ならびに心室性不整脈を含む心血管障害から選択される、請求項9に記載の方法。
- 前記薬学的組成物は、カプセルシェルに封入されたDGLAまたはその誘導体を含む、請求項9に記載の方法。
- 前記薬学的組成物は、液体または半液体形態のDGLAまたはその誘導体を含む、請求項9〜11のいずれか一項に記載の方法。
- 前記組成物は、1日当たり最大約1gのDGLAまたはその誘導体を提供するのに十分な量で前記対象に投与される、請求項9〜12のいずれか一項に記載の方法。
- 前記組成物は、1日当たり約0.2g〜約8gのDGLAまたはその誘導体を提供するのに十分な量で前記対象に投与される、請求項9〜13のいずれか一項に記載の方法。
- 前記組成物は、1日当たり約1g〜約4gのDGLAまたはその誘導体を提供するのに十分な量で前記対象に投与される、請求項9〜11のいずれか一項に記載の方法。
- 前記組成物は、1日当たり1〜8カプセル投与される、請求項9〜15のいずれか一項に記載の方法。
- 各カプセルは、約200mg〜約1gのDGLAまたはその誘導体を含む、請求項9〜16のいずれか一項に記載の方法。
- 前記DGLAまたはその誘導体は、少なくとも約2週間、少なくとも約4週間、または少なくとも約8週間の期間、前記対象に投与される、請求項9〜17のいずれか一項に記載の方法。
- 前記対象は小児対象である、請求項9〜18のいずれか一項に記載の方法。
- 前記対象は、少ない好酸球細胞数を有する、請求項9〜19のいずれか一項に記載の方法。
- 前記少ない好酸球細胞数は、基準レベルに基づいて決定される、請求項20のいずれか一項に記載の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662276019P | 2016-01-07 | 2016-01-07 | |
| US62/276,019 | 2016-01-07 | ||
| US201662357000P | 2016-06-30 | 2016-06-30 | |
| US62/357,000 | 2016-06-30 | ||
| PCT/IB2017/000066 WO2017118911A1 (en) | 2016-01-07 | 2017-01-06 | Pharmaceutical compositions comprising dgla and use of same |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2019501190A true JP2019501190A (ja) | 2019-01-17 |
| JP2019501190A5 JP2019501190A5 (ja) | 2020-02-13 |
Family
ID=58213263
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018535278A Pending JP2019501190A (ja) | 2016-01-07 | 2017-01-06 | Dglaを含む薬学的組成物及びその使用 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US20170196825A1 (ja) |
| EP (1) | EP3399971A1 (ja) |
| JP (1) | JP2019501190A (ja) |
| CN (1) | CN108778266A (ja) |
| CA (1) | CA3027867A1 (ja) |
| WO (1) | WO2017118911A1 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0907413D0 (en) | 2009-04-29 | 2009-06-10 | Equateq Ltd | Novel methods |
| ES2727387T3 (es) | 2013-11-15 | 2019-10-15 | Ds Biopharma Ltd | Sal de lisina de ácido 15-hidroxi-8(Z),11(Z),13(E)-eicosatrienoico |
| RU2020101477A (ru) | 2014-06-04 | 2020-06-19 | Дигнити Сайенсиз Лимитед | Фармацевтические композиции, содержащие дглк, и их применение |
| JP2020520938A (ja) * | 2017-05-19 | 2020-07-16 | ディーエス バイオファーマ リミテッド | Dglaを含む薬学的組成物およびその使用 |
| US10639313B2 (en) | 2017-09-01 | 2020-05-05 | Ndsu Research Foundation | Compound for inhibition of delta-5-desaturase (D5D) and treatment of cancer and inflammation |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59152324A (ja) * | 1983-02-01 | 1984-08-31 | エフア−モル・リミテツド | 医薬または健康食品組成物 |
| JP2006219454A (ja) * | 2005-02-14 | 2006-08-24 | Suntory Ltd | 皮膚疾患経口治療または予防剤 |
| WO2015185698A1 (en) * | 2014-06-04 | 2015-12-10 | Dignity Sciences Limited | Pharmaceutical compositions comprising dgla and use of same |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006085687A1 (ja) * | 2005-02-14 | 2006-08-17 | Suntory Limited | ジホモ−γ−リノレン酸(DGLA)を有効成分として含んで成る組成物 |
| US7485323B2 (en) | 2005-05-31 | 2009-02-03 | Gelita Ag | Process for making a low molecular weight gelatine hydrolysate and gelatine hydrolysate compositions |
-
2017
- 2017-01-06 JP JP2018535278A patent/JP2019501190A/ja active Pending
- 2017-01-06 EP EP17708574.3A patent/EP3399971A1/en not_active Withdrawn
- 2017-01-06 CA CA3027867A patent/CA3027867A1/en not_active Abandoned
- 2017-01-06 WO PCT/IB2017/000066 patent/WO2017118911A1/en active Application Filing
- 2017-01-06 US US15/400,132 patent/US20170196825A1/en not_active Abandoned
- 2017-01-06 CN CN201780015787.0A patent/CN108778266A/zh active Pending
-
2018
- 2018-10-11 US US16/158,085 patent/US20190175534A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59152324A (ja) * | 1983-02-01 | 1984-08-31 | エフア−モル・リミテツド | 医薬または健康食品組成物 |
| JP2006219454A (ja) * | 2005-02-14 | 2006-08-24 | Suntory Ltd | 皮膚疾患経口治療または予防剤 |
| WO2015185698A1 (en) * | 2014-06-04 | 2015-12-10 | Dignity Sciences Limited | Pharmaceutical compositions comprising dgla and use of same |
Also Published As
| Publication number | Publication date |
|---|---|
| US20190175534A1 (en) | 2019-06-13 |
| WO2017118911A1 (en) | 2017-07-13 |
| CN108778266A (zh) | 2018-11-09 |
| CA3027867A1 (en) | 2017-07-13 |
| EP3399971A1 (en) | 2018-11-14 |
| US20170196825A1 (en) | 2017-07-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7020756B2 (ja) | 対象の心血管イベントのリスクを低減する方法 | |
| Davidson et al. | The safety and immunogenicity of a CETP vaccine in healthy adults | |
| JP2019501190A (ja) | Dglaを含む薬学的組成物及びその使用 | |
| US10849870B2 (en) | Pharmaceutical compositions comprising DGLA and use of same | |
| JP2018515460A (ja) | 炎症または神経障害性疼痛を処置する方法 | |
| TW202142229A (zh) | 治療雷葛氏症候群之病患的方法 | |
| EP4342475A1 (en) | Composition for treatment of covid-19 comprising taurodeoxycholic acid or pharmaceutically acceptable salt thereof as active ingredient | |
| JP2020520938A (ja) | Dglaを含む薬学的組成物およびその使用 | |
| CN113164377A (zh) | 用于治疗炎性、纤维化和增生性病状的dgla和/或15-hetre | |
| TW202131910A (zh) | 使用mtorc1調節劑的治療方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20191218 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20191218 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20201013 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20210511 |