JP2017513866A - パーキンソン病の運動症状変動の迅速な緩和 - Google Patents
パーキンソン病の運動症状変動の迅速な緩和 Download PDFInfo
- Publication number
- JP2017513866A JP2017513866A JP2016563193A JP2016563193A JP2017513866A JP 2017513866 A JP2017513866 A JP 2017513866A JP 2016563193 A JP2016563193 A JP 2016563193A JP 2016563193 A JP2016563193 A JP 2016563193A JP 2017513866 A JP2017513866 A JP 2017513866A
- Authority
- JP
- Japan
- Prior art keywords
- patient
- levodopa
- administration
- fpd
- administered
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 208000018737 Parkinson disease Diseases 0.000 title claims abstract description 57
- 208000024891 symptom Diseases 0.000 title claims description 31
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 claims abstract description 305
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 claims abstract description 305
- 229960004502 levodopa Drugs 0.000 claims abstract description 285
- 238000000034 method Methods 0.000 claims abstract description 98
- 230000002685 pulmonary effect Effects 0.000 claims abstract description 61
- 239000000902 placebo Substances 0.000 claims abstract description 44
- 229940068196 placebo Drugs 0.000 claims abstract description 44
- 239000002775 capsule Substances 0.000 claims description 41
- 208000012661 Dyskinesia Diseases 0.000 claims description 36
- 239000010419 fine particle Substances 0.000 claims description 15
- 229940112141 dry powder inhaler Drugs 0.000 claims description 11
- 230000002829 reductive effect Effects 0.000 claims description 9
- 229940071648 metered dose inhaler Drugs 0.000 claims description 7
- 239000002245 particle Substances 0.000 description 93
- 230000036470 plasma concentration Effects 0.000 description 50
- 229960004205 carbidopa Drugs 0.000 description 41
- TZFNLOMSOLWIDK-JTQLQIEISA-N carbidopa (anhydrous) Chemical compound NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 TZFNLOMSOLWIDK-JTQLQIEISA-N 0.000 description 41
- 239000003814 drug Substances 0.000 description 41
- 229940079593 drug Drugs 0.000 description 38
- 239000000843 powder Substances 0.000 description 29
- 238000011282 treatment Methods 0.000 description 22
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 19
- 238000012360 testing method Methods 0.000 description 18
- 210000004072 lung Anatomy 0.000 description 16
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- 239000000443 aerosol Substances 0.000 description 14
- 238000012216 screening Methods 0.000 description 12
- 230000008021 deposition Effects 0.000 description 11
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 10
- 230000007423 decrease Effects 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 239000011859 microparticle Substances 0.000 description 10
- 239000000203 mixture Substances 0.000 description 10
- 238000010521 absorption reaction Methods 0.000 description 9
- 238000004458 analytical method Methods 0.000 description 9
- 229960003638 dopamine Drugs 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- 239000000534 dopa decarboxylase inhibitor Substances 0.000 description 8
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 8
- 230000009885 systemic effect Effects 0.000 description 8
- 230000008859 change Effects 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 238000009826 distribution Methods 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 7
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 229940081615 DOPA decarboxylase inhibitor Drugs 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 6
- 210000002345 respiratory system Anatomy 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- 238000001647 drug administration Methods 0.000 description 5
- 206010044565 Tremor Diseases 0.000 description 4
- BNQDCRGUHNALGH-UHFFFAOYSA-N benserazide Chemical compound OCC(N)C(=O)NNCC1=CC=C(O)C(O)=C1O BNQDCRGUHNALGH-UHFFFAOYSA-N 0.000 description 4
- 229960000911 benserazide Drugs 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 4
- 230000007659 motor function Effects 0.000 description 4
- 229940126701 oral medication Drugs 0.000 description 4
- 150000003904 phospholipids Chemical class 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- IVTMXOXVAHXCHI-YXLMWLKOSA-N (2s)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid;(2s)-3-(3,4-dihydroxyphenyl)-2-hydrazinyl-2-methylpropanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1.NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 IVTMXOXVAHXCHI-YXLMWLKOSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 206010057249 Phagocytosis Diseases 0.000 description 3
- 230000006399 behavior Effects 0.000 description 3
- 230000036772 blood pressure Effects 0.000 description 3
- 238000011260 co-administration Methods 0.000 description 3
- 238000007405 data analysis Methods 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 210000002569 neuron Anatomy 0.000 description 3
- 230000008782 phagocytosis Effects 0.000 description 3
- 238000011002 quantification Methods 0.000 description 3
- 230000029058 respiratory gaseous exchange Effects 0.000 description 3
- 238000001694 spray drying Methods 0.000 description 3
- SLKDGVPOSSLUAI-PGUFJCEWSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine zwitterion Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OCCN)OC(=O)CCCCCCCCCCCCCCC SLKDGVPOSSLUAI-PGUFJCEWSA-N 0.000 description 2
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 2
- BIABMEZBCHDPBV-MPQUPPDSSA-N 1,2-palmitoyl-sn-glycero-3-phospho-(1'-sn-glycerol) Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@@H](O)CO)OC(=O)CCCCCCCCCCCCCCC BIABMEZBCHDPBV-MPQUPPDSSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 210000001132 alveolar macrophage Anatomy 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 229940052036 carbidopa / levodopa Drugs 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- 235000020937 fasting conditions Nutrition 0.000 description 2
- 230000030136 gastric emptying Effects 0.000 description 2
- 230000005484 gravity Effects 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000003340 mental effect Effects 0.000 description 2
- 230000004899 motility Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 description 2
- 238000013125 spirometry Methods 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 230000002618 waking effect Effects 0.000 description 2
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 1
- 208000012639 Balance disease Diseases 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- 229940123736 Decarboxylase inhibitor Drugs 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 201000006347 Intellectual Disability Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 206010024264 Lethargy Diseases 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 206010025476 Malabsorption Diseases 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 208000012287 Prolapse Diseases 0.000 description 1
- 206010071390 Resting tremor Diseases 0.000 description 1
- 206010040021 Sensory abnormalities Diseases 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 238000012387 aerosolization Methods 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- VMWNQDUVQKEIOC-CYBMUJFWSA-N apomorphine Chemical compound C([C@H]1N(C)CC2)C3=CC=C(O)C(O)=C3C3=C1C2=CC=C3 VMWNQDUVQKEIOC-CYBMUJFWSA-N 0.000 description 1
- 229960004046 apomorphine Drugs 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 210000004227 basal ganglia Anatomy 0.000 description 1
- 238000010876 biochemical test Methods 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- -1 blisters Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 230000003920 cognitive function Effects 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000010411 cooking Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000003954 decarboxylase inhibitor Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 210000005064 dopaminergic neuron Anatomy 0.000 description 1
- 230000003291 dopaminomimetic effect Effects 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000001667 episodic effect Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 230000008921 facial expression Effects 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 238000004442 gravimetric analysis Methods 0.000 description 1
- 238000012388 gravitational sedimentation Methods 0.000 description 1
- 229960003943 hypromellose Drugs 0.000 description 1
- 230000005032 impulse control Effects 0.000 description 1
- 238000002664 inhalation therapy Methods 0.000 description 1
- 231100000037 inhalation toxicity test Toxicity 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 229960001375 lactose Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960001021 lactose monohydrate Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 230000004199 lung function Effects 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 230000006996 mental state Effects 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 230000036651 mood Effects 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 210000001539 phagocyte Anatomy 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 230000009325 pulmonary function Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- 230000036387 respiratory rate Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000010079 rubber tapping Methods 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 230000036578 sleeping time Effects 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000011775 sodium fluoride Substances 0.000 description 1
- 235000013024 sodium fluoride Nutrition 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000007785 strong electrolyte Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000003746 surface roughness Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
Images









Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid, pantothenic acid
- A61K31/198—Alpha-aminoacids, e.g. alanine, edetic acids [EDTA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
- A61K9/0075—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy for inhalation via a dry powder inhaler [DPI], e.g. comprising micronized drug mixed with lactose carrier particles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Otolaryngology (AREA)
- Pulmonology (AREA)
- Psychology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
Abstract
Description
パーキンソン病(本明細書では「PD」とも呼ぶ)は、神経病理学的には大脳基底核のドパミンニューロンの変性と特徴とし、神経学的には消耗性の振戦、動作緩慢およびバランス困難を特徴とする。パーキンソン病に罹患している人は100万人を超えると推定される。大半の患者には、多くの場合、ドパ−デカルボキシラーゼ阻害剤であるカルビドパとともに、ドパミン前駆体であるレボドパ、つまり、「L−Dopa」を投与する。パーキンソン病の初期段階では、L−Dopaにより疾患の症状が適切に抑えられる。しかし、疾患の過程で、数か月〜数年にわたる期間ののちに効果が少なくなる傾向がみられる。
定義
半減期(T1/2)とは、体液または組織中の薬物の濃度(C)が濃度C/2に達する時間のことである。
と定義される。

実施例1
概要
成人健常被験者を対象に90/8/2経肺レボドパ粉末の投与後の安全性、忍容性およびレボドパ薬物動態(PK)を経口レボドパと比較して評価するため、90/8/2乾燥粉末レボドパ製剤を準備した。これらの実施例に記載する経肺レボドパ粉末は、いずれも乾燥重量で90%のレボドパ、8%のジパルミトイルホスファチジルコリンおよび2%の塩化ナトリウムの粉末からなるものであり、本明細書では90/8/2と呼ぶ。このデータは、90/8/2の単回吸入投与後のレボドパのPKおよび絶食または摂食条件で経口投与したレボドパ(LD)との比較のほか、カルビドパ(CD)による前治療を実施した場合と実施しなかった場合のPKの比較について記載するものである。これは健常な成人男性被験者および成人女性被験者を対象にした以下の2部:第A部−経口レボドパと比較した用量漸増の部;および第B部−90/8/2±カルビドパ前治療の部からなる試験であった。
・吸入90/8/2により血漿中レボドパ濃度が急速に増大した;
・CmaxおよびAUCに基づく投与後最初の10分間のレボドパへの全身曝露量は、90/8/2吸入投与の方が経口薬物投与よりもはるかに多かった;
・健常成人では微粒子用量20〜50mgの吸入後5〜10分以内に治療上関連すると思われるレボドパ血漿中濃度が得られた;
・対象間の血漿中レボドパ濃度のばらつきは、吸入後の方が経口投与よりもはるかに小さく、経肺投与で予想されるものであった;
・全身レボドパ曝露量は投与したレボドパ微粒子用量に比例していた;
・薬物動態モデリングから、吸入90/8/2の方が経口投与よりもはるかにラグタイムが短く、吸収速度が速いことがわかった;
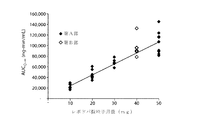
・用量で正規化した(推定微粒子用量に基づく)吸入後の曝露量は、AUCに基づくものが経口投与の1.3〜1.6倍、Cmaxに基づくものが1.6〜2.9倍であった;
・カルビドパ前治療を実施しない場合、血漿レボドパクリアランスが約4倍になり、レボドパ曝露量が減少した。
この実施例では、標準的な経口薬に対して間欠的に不十分な反応がみられるパーキンソン病患者を対象とした運動症状変動(「オフエピソード」)のエピソード治療法として90/8/2を試験した。90/8/2は、患者の既存のドパデカルボキシラーゼ阻害剤(すなわち、カルビドパまたはベンセラジド)を含むパーキンソン病投薬レジメンの補助剤として用いられ得る。この試験は、ヒトを対象に90/8/2を用いた最初の試験であり、成人健常被験者を対象に90/8/2投与後の安全性、忍容性およびレボドパ薬物動態(PK)を経口レボドパと比較して評価するようデザインされたものである。
この試験は、健常な成人男性被験者および成人女性被験者を対象にした以下の2部からなる試験である:
・第A部:経口レボドパと比較した用量漸増の部。
・第B部:90/8/2±カルビドパ前治療の部。
レジメンA:CD前治療を実施する90/8/2
レジメンB:CD前治療を実施しない90/8/2
が2種類の投与順序、A→BまたはB→Aで実施されるように無作為化しバランスをとった方法で、被験者8例にCDによる前治療を実施して、および実施せずに、単回吸入90/8/2用量(40mgのレボドパFPD)を投与した後の安全性、忍容性およびレボドパPKの評価を実施した。
非コンパートメント解析
各被験者および各治療の血漿中濃度および時間に関してデータ解析を実施した。WINNONLIN(登録商標)プロフェッショナルバージョン5.3により非コンパートメント解析を実施した。線形台形法を用いて時間0から最後の測定可能な時点までの曲線下面積(AUC0〜t)を推定した。最後の3時点以上にわたる線形回帰を用いて消失速度定数(λ)を推定し、これを用いて以下の方程式から終末相半減期(T1/2)および0〜無限大のAUC(AUC0〜∞)を推定した:
T1/2=In(2)/λ
AUC0〜∞=AUC0〜t+Ct/λ
式中、Ctは回帰直線によって予測される最後の測定可能な濃度である。血清クリアランスをバイオアベイラビリティで割った商(CL/F)および終末相の見かけの分布容積をバイオアベイラビリティで割った商(Vz/F)を以下の方程式:
CL/F=用量/AUC0〜∞
Vz/F=用量/(λ×AUC0〜∞)
から推定し、最大濃度(Cmax)およびそれが観察された時間(Tmax)をデータから直接求めた。
WINNONLTN(登録商標)プロフェッショナルバージョン5.3を用いて薬物動態モデリングを実施した。ラグタイムの有無を問わず1−コンパートメントモデルおよび2−コンパートメントモデルを含めた多数の異なるモデルを評価した。評価したモデルはいずれも一次入力とした。赤池情報量規準、残差平方和、推定パラメータの相対値およびそのそれぞれの標準誤差推定値、実測濃度と予測濃度との相関ならびに予測濃度と実測濃度との間の差の全般的傾向を含めた多数の診断基準に基づきモデルを評価した。
Ct=Ae−αt+Be−βt+Ce−k01t
によって表され、Ctは投与後の時間tにおける血漿中レボドパ濃度であり、A、BおよびCは曲線の分布相、消失相および吸収相のy軸切片であり、用量、容積および速度定数から計算される。
10〜50mgのレボドパFPDの用量で吸入により投与した90/8/2では、健常成人に20〜50mgの微粒子用量のレボドパを投与した後、用量に比例した血漿中レボドパ濃度の急速な増大がみられ、5〜10分以内に治療上関連があると思われるレベル(400〜500ng/mL)に達した。
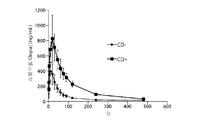
カルビドパ前治療を実施して、または実施せずに40mgのレボドパFPDの90/8/2を吸入するクロスオーバーデザインの試験第B部で得られた血漿中濃度を図7に示す。血漿中濃度および曝露量のピークはカルビドパ前治療を実施した場合の方が高かった。血漿レボドパクリアランスはCD前治療を実施しない場合の方が約4倍速かった。同じように、CD前治療を実施しない場合の方がCmaxおよびAUCが低く、TmaxおよびT1/2がいくぶん短かった(表8)。
この試験の主な知見は以下の通りであった:(i)吸入90/8/2により血漿中レボドパ濃度が急速に増大した;(ii)CmaxおよびAUCに基づく投与後最初の10分間のレボドパへの全身曝露量は、90/8/2吸入投与の方が経口薬物投与よりもはるかに多かった;(iii)健常成人では20〜50mgのレボドパ微粒子用量の90/8/2投与後5〜10分以内に治療上関連があると思われる血漿中レボドパ濃度が得られた;(iv)対象間の血漿中レボドパ濃度のばらつきは、吸入後の方が経口投与よりもはるかに小さかった;(v)全身レボドパ曝露量は投与したレボドパ微粒子用量に比例していた;(vi)薬物動態モデリングから、吸入90/8/2の方が経口投与よりもはるかにラグタイムが短く、吸収速度が速いことがわかった;vii)用量で正規化した(推定微粒子用量に基づく)吸入後の曝露量は、AUCに基づくものが経口投与の1.3〜1.6倍、Cmaxに基づくものが経口投与の1.6〜2.9倍であった;およびviii)カルビドパ前治療を実施しない場合、血漿レボドパクリアランスが約4倍になり、レボドパ曝露量が減少した。
2種類の用量の経肺レボドパ(被験薬25mgおよび50mg)を検討した第2相試験は、3群(プラセボ、25mgおよび50mg)の多施設無作為化二重盲検プラセボ対照単回投与クロスオーバーデザイン試験とし、「非盲検」経口Sinemet群を含めた。この試験で治療したPD患者24例(24)には来院毎にL−Dopa血漿中レベル、運動反応および安全性の連続評価を実施した。オフ状態の患者に被験薬を投与し、連続評価を投与前に開始し、投与後180分まで継続した。タッピング検査、統一パーキンソン病評価尺度(Unified Parkinson’s Disease Rating Scale:UPDRS)第三部(UPDRS III)ならびに「意味のある」オンおよびオフの主観的評価を用いて運動機能を測定した。モニターした安全性パラメータには、肺機能、臨床検査データ、EGCおよびバイタルサイン(血圧、心拍数および起立時血圧)を含めた。この試験は、経肺レボドパの運動機能に対する効果の時間、大きさおよび持続性を測定し、パーキンソン病患者における経肺レボドパの安全性および忍容性を評価するようデザインされたものである。
第2(b)相無作為化二重盲検プラセボ対照試験
90/8/2を用いた第2b相試験のデザインおよび方法
この試験は、運動症状変動(オフエピソード)の認められるパーキンソン病(PD)被験者を対象とした1日当たり最大3回のオフエピソードを治療する吸入(吸入レボドパ[LD]粉末)またはプラセボの無作為化二重盲検プラセボ対照多施設試験とした。被験者を無作為化により1:1の比で割り付け、吸入90/8/2(ここでは「被験薬」とも呼ぶ)またはプラセボを投与した。無作為化は被験者のヘーンとヤールの段階(2.5未満と2.5以上)によって層別化し、各グループの疾患重症度のバランスをとった。
年齢30〜80歳の男性および女性被験者で、30歳より後に特発性PDであると診断されており;UK Brain Bank基準の段階1および2を満たし;オン状態で修正版ヘーンとヤールの段階1〜3に分類され;自己報告およびPD日誌の確認により日中覚醒時当たり最低2時間の平均1日オフ時間にわたって運動症状変動が認められ(早朝のオフ時間を除く);許容可能なLD反応を示す被験者を、この試験に参加するのに適格であるとした。被験者はスクリーニング来院1回目の前に少なくとも2週間、経口LDを含む安定な治療用量/レジメンを受けていなければならず;LD/ドパミンデカルボキシラーゼ阻害剤(DDI)を含むレジメンには日中覚醒時に少なくとも4回投与する投与スケジュールが含まれていなければならない。被験者はスクリーニング来院1回目の前に少なくとも4週間、安定に他のPD投薬を受けている必要がある。被験者はスクリーニング時、オフ状態とオン状態で記録されたUPDRS第三部スコアの間に25%以上の差が認められなければならない。被験者は、試験期間中、毎日の投薬量を理解し(介護者の補助の有無を問わない)、それを変更していない者でなければならない。被験者は、ミニメンタルステート検査(MMSE)でスコアが25以上であることにより認知機能が正常であることが確認された者でなければならない。被験者はスクリーニング時、オン状態でのスパイロメトリーにより予測されるスクリーニングFEV1が60%超、FEV1/FVC比が75%以上であり、肺疾患の病歴が認められない者でなければならない。
試験の目的および変数を表9に記載する。
患者86例を試験に登録した。来院2回目の前の連日3日間、患者に覚醒時のオン/オフ状態(オフ時間、ジスキネジアが認められないオン時間、対処困難ではないジスキネジアが認められるオン時間、対処困難なジスキネジアが認められるオン時間)および睡眠時間を記録するスクリーニングPD日誌をもれなく記入させた。さらに、来院2回目の前の7日間、患者に日中覚醒時に認められる各オフエピソードに関する以下の情報:オフエピソードの開始時間、次のオン時間の開始時間および患者の標準的LD薬の使用方法を記録させるオン/オフエピソードのスクリーニングおよび投薬記録を記入させる。
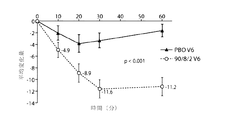
第2b相試験の主要評価項目は、被験薬とプラセボとの間で来院6回目の投与前のUPDRS第三部スコア平均値からの平均変化量(投与後10〜60分)の差を評価するためのものであった(DL2)。DL1を用いた来院4回目および来院5回目(1回目のDL2投与)におけるこれと同じ差を副次評価項目として用いた。UPDRSに従うにあたっては、UPDRS運動スコアの臨床的に重要な差(CID)は、最小が2.5ポイント、中程度が5.2ポイント、大きいCIDは10.8ポイントとする(Shulmanら,Arch Neurol,Vol.67(Jan 2010))。
本明細書に記載したデータの通り、第2b相試験では、プラセボと比較して、来院6回目の投与後10〜60分における投与前からのUPDRS第三部スコア平均値の統計学的に有意な平均変化量を示す主要評価項目が得られた。データからほかにも、UPDRSの時間曲線の形状が迅速かつ持続性のある反応を示し、用量35mg(DL1)の振幅は用量50mg(DL2)とほぼ同じであるが、効果の持続時間がわずかに短い可能性があることがわかった。このほか、UDPRS第三部スコアの最大変化量および最大変化百分率はいずれの来院および用量も統計学的に有意なものであり、プラセボの反応が減衰する前のV4(DL1)でも大きな開きがみられた。
Claims (47)
- パーキンソン病(PD)患者のオフエピソードを治療する方法であって、患者の肺系にレボドパを投与することを含み、投与後、前記患者の統一パーキンソン病評価尺度(Unified Parkinson’s Disease Rating Scale:UPDRS)第三部スコアがプラセボ対照に比して少なくとも5ポイント改善される、方法。
- 前記患者のUDPRS第三部スコアが、プラセボ対照に比して少なくとも5〜10ポイント改善される、請求項1に記載の方法。
- 前記患者の前記肺系に約30mg〜約60mgの微粒子用量(FPD)のレボドパを投与する、請求項1に記載の方法。
- 前記患者に35mgまたは50mgのFPDのレボドパを投与する、請求項1に記載の方法。
- 前記患者に前記レボドパの経肺投与前のジスキネジアのレベルに比してジスキネジアの増大が認められない、請求項1に記載の方法。
- 前記患者に1日約3〜約4回のオフエピソードが認められる、請求項1に記載の方法。
- 前記患者に1日約4〜約8時間のオフエピソードが認められる、請求項1に記載の方法。
- 前記FPDのレボドパの投与後60分以内に、前記患者のUDPRS第三部スコアがプラセボ対照に比して少なくとも5〜10ポイント改善される、請求項1に記載の方法。
- 前記FPDのレボドパを含有する少なくとも1つのカプセルの内容物を吸入により前記患者に投与する、請求項1に記載の方法。
- 前記FPDのレボドパを含む少なくとも2つのカプセルの内容物を吸入により前記患者に投与する、請求項9に記載の方法。
- 前記微粒子用量のレボドパを、吸入装置により前記少なくとも1つのカプセルから前記肺系に送達する、請求項9に記載の方法。
- 前記吸入装置が乾燥粉末吸入器(DPI)または定量吸入器(MDI)である、請求項11に記載の方法。
- 前記投与をオフ症状の出現時に実施する、請求項1に記載の方法。
- パーキンソン病患者のオフ期間を治療する方法であって、患者の肺系にレボドパを投与することを含み、前記患者のUDPRS第三部スコアが、前記FPDのレボドパを投与する前の前記患者のUPDRSスコアに比して少なくとも約5〜約12ポイント改善される、方法。
- 前記患者の前記肺系に約30mg〜約60mgの微粒子用量(FPD)のレボドパを投与する、請求項14に記載の方法。
- 前記患者に35mgまたは50mgのFPDのレボドパを投与する、請求項14に記載の方法。
- 前記患者に前記レボドパの経肺投与前のジスキネジアのレベルに比してジスキネジアの増大が認められない、請求項14に記載の方法。
- 前記患者に1日約3〜約4回のオフエピソードが認められる、請求項14に記載の方法。
- 前記患者に1日約4〜約8時間のオフエピソードが認められる、請求項14に記載の方法。
- 前記FPDのレボドパの投与後60分以内に、前記患者のUDPRS第三部スコアが少なくとも8ポイント改善される、請求項14に記載の方法。
- 前記FPDのレボドパを含有する少なくとも1つのカプセルの内容物を吸入により前記患者に投与する、請求項14に記載の方法。
- 前記FPDのレボドパを含む少なくとも2つのカプセルの内容物を吸入により前記患者に投与する、請求項21に記載の方法。
- 前記微粒子用量を、吸入装置により前記少なくとも1つのカプセルから前記肺系に送達する、請求項21に記載の方法。
- 前記吸入装置が乾燥粉末吸入器(DPI)または定量吸入器(MDI)である、請求項23に記載の方法。
- 前記投与をオフ症状の出現時に実施する、請求項14に記載の方法。
- 前記患者のUDPRS第三部スコアが、前記FPDのレボドパを投与する前の前記患者のUPDRSスコアに比して少なくとも8ポイント改善される、請求項14に記載の方法。
- パーキンソン病患者の平均1日オフ時間を短縮する方法であって、患者の肺系に少なくとも1日2回、レボドパを投与することを含み、前記患者の平均1日オフ時間が少なくとも1時間短縮される、方法。
- 前記患者の平均1日オフ時間が少なくとも3時間短縮される、請求項27に記載の方法。
- 前記患者の前記肺系に約30mg〜約60mgの微粒子用量(FPD)のレボドパを投与する、請求項27に記載の方法。
- 前記患者に35mgまたは50mgのFPDのレボドパを投与する、請求項27に記載の方法。
- 前記患者に前記レボドパの経肺投与前のジスキネジアのレベルに比してジスキネジアの増大が認められない、請求項27に記載の方法。
- 前記患者に1日約3〜約4回のオフエピソードが認められる、請求項27に記載の方法。
- 前記患者に1日約4〜約8時間のオフエピソードが認められる、請求項27に記載の方法。
- パーキンソン病患者にレボドパを送達する方法であって、患者の肺系にレボドパを投与することを含み、前記患者の統一パーキンソン病評価尺度(Unified Parkinson’s Disease Rating Scale:UPDRS)第三部スコアが、投与前の前記患者のUPDRSスコアに比して少なくとも8ポイント改善される、方法。
- 前記FPDのレボドパの投与後60分以内に、前記患者のUDPRS第三部スコアが少なくとも8ポイント改善される、請求項34に記載の方法。
- パーキンソン病患者にレボドパを送達する方法であって、患者の肺系にレボドパを投与することを含み、投与後、前記患者の統一パーキンソン病評価尺度(Unified Parkinson’s Disease Rating Scale:UPDRS)第三部スコアがプラセボ対照に比して少なくとも5ポイント改善される、方法。
- 前記FPDのレボドパの投与後60分以内に、前記患者のUDPRS第三部スコアがプラセボ対照に比して少なくとも5〜10ポイント改善される、請求項36に記載の方法。
- 前記患者の前記肺系に約30mg〜約60mgのFPDのレボドパを投与する、請求項36に記載の方法。
- 前記患者に35mgまたは50mgのFPDのレボドパを投与する、請求項36に記載の方法。
- 前記患者に前記レボドパの経肺投与前のジスキネジアのレベルに比してジスキネジアの増大が認められない、請求項36に記載の方法。
- 前記FPDのレボドパを含有する少なくとも1つのカプセルの内容物を吸入により前記患者に投与する、請求項36に記載の方法。
- 前記FPDのレボドパを含む少なくとも2つのカプセルの内容物を吸入により前記患者に投与する、請求項41に記載の方法。
- 前記微粒子用量のレボドパを、吸入装置により前記少なくとも1つのカプセルから前記肺系に送達する、請求項41に記載の方法。
- 前記吸入装置が乾燥粉末吸入器(DPI)または定量吸入器(MDI)である、請求項43に記載の方法。
- 前記投与をオフ症状の出現時に実施する、請求項36に記載の方法
- 前記患者の前記肺系に約30mg〜約60mgのFPDのレボドパを投与する、請求項34に記載の方法。
- 前記患者に35mgまたは50mgのFPDのレボドパを投与する、請求項34に記載の方法。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2014/034778 WO2015163840A1 (en) | 2014-04-21 | 2014-04-21 | Rapid relief of motor fluctuations in parkinson's disease |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019039444A Division JP2019108379A (ja) | 2019-03-05 | 2019-03-05 | パーキンソン病の運動症状変動の迅速な緩和 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017513866A true JP2017513866A (ja) | 2017-06-01 |
| JP2017513866A5 JP2017513866A5 (ja) | 2019-04-25 |
Family
ID=54332876
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016563193A Pending JP2017513866A (ja) | 2014-04-21 | 2014-04-21 | パーキンソン病の運動症状変動の迅速な緩和 |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US20170296498A1 (ja) |
| EP (2) | EP3831375A1 (ja) |
| JP (1) | JP2017513866A (ja) |
| KR (2) | KR20170008754A (ja) |
| CN (2) | CN113209055A (ja) |
| AU (2) | AU2014391721B2 (ja) |
| BR (1) | BR112016024502A8 (ja) |
| CA (1) | CA2946165C (ja) |
| IL (2) | IL309959A (ja) |
| MX (1) | MX2016013741A (ja) |
| RU (1) | RU2698330C2 (ja) |
| SG (1) | SG11201608608PA (ja) |
| WO (1) | WO2015163840A1 (ja) |
| ZA (1) | ZA201607833B (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021509676A (ja) * | 2018-01-05 | 2021-04-01 | インペル ニューロファーマ インコーポレイテッド | 精密嗅覚装置によるレボドパ粉末の鼻腔内送達 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112015010601B1 (pt) | 2012-11-09 | 2022-07-19 | Civitas Therapeutics, Inc. | Composição farmacêutica e uso da composição |
| US11517548B2 (en) | 2018-07-19 | 2022-12-06 | Impel Pharmaceuticals Inc. | Respiratory tract delivery of levodopa and DOPA decarboxylase inhibitor for treatment of Parkinson's Disease |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006507218A (ja) * | 2002-03-20 | 2006-03-02 | アドバンスト インハレーション リサーチ,インコーポレイテッド | レボドパの肺送達 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6254854B1 (en) | 1996-05-24 | 2001-07-03 | The Penn Research Foundation | Porous particles for deep lung delivery |
| US6858199B1 (en) | 2000-06-09 | 2005-02-22 | Advanced Inhalation Research, Inc. | High efficient delivery of a large therapeutic mass aerosol |
| US6613308B2 (en) * | 2000-09-19 | 2003-09-02 | Advanced Inhalation Research, Inc. | Pulmonary delivery in treating disorders of the central nervous system |
| US6514482B1 (en) | 2000-09-19 | 2003-02-04 | Advanced Inhalation Research, Inc. | Pulmonary delivery in treating disorders of the central nervous system |
| AR044007A1 (es) * | 2003-04-11 | 2005-08-24 | Newron Pharmaceuticals Inc | Metodos para el tratamiento de la enfermedad de parkinson |
| WO2008156586A2 (en) | 2007-06-12 | 2008-12-24 | Alkermes, Inc. | Inhalation device for powdered substances |
| GB2454480A (en) * | 2007-11-07 | 2009-05-13 | Vectura Group Plc | Pulmonary inhalation of levodopa containing compositions in the treatment of Parkinsons disease and other central nervous system disorders |
| AU2009265760B2 (en) * | 2008-06-30 | 2013-07-18 | Novartis Ag | Combinations comprising mGluR modulators for the treatment of parkinson's disease |
| US8399513B2 (en) * | 2008-10-20 | 2013-03-19 | Xenoport, Inc. | Levodopa prodrug mesylate hydrate |
| TW201304822A (zh) * | 2010-11-15 | 2013-02-01 | Vectura Ltd | 組成物及用途 |
| KR20150102960A (ko) * | 2012-10-22 | 2015-09-09 | 키비타스 테라퓨틱스, 인코포레이티드. | 파킨슨병의 빠른 경감을 위한 레보도파 제형 |
| US8545878B1 (en) * | 2012-11-09 | 2013-10-01 | Civitas Therapeutics, Inc. | Capsules containing high doses of levodopa for pulmonary use |
-
2014
- 2014-04-21 US US15/500,608 patent/US20170296498A1/en not_active Abandoned
- 2014-04-21 CN CN202110185196.6A patent/CN113209055A/zh active Pending
- 2014-04-21 WO PCT/US2014/034778 patent/WO2015163840A1/en active Application Filing
- 2014-04-21 RU RU2016144340A patent/RU2698330C2/ru active
- 2014-04-21 SG SG11201608608PA patent/SG11201608608PA/en unknown
- 2014-04-21 IL IL309959A patent/IL309959A/en unknown
- 2014-04-21 EP EP21150770.2A patent/EP3831375A1/en active Pending
- 2014-04-21 CN CN201480079968.6A patent/CN106659685B/zh active Active
- 2014-04-21 KR KR1020167032453A patent/KR20170008754A/ko active Application Filing
- 2014-04-21 EP EP14889874.5A patent/EP3134077A4/en not_active Ceased
- 2014-04-21 AU AU2014391721A patent/AU2014391721B2/en active Active
- 2014-04-21 CA CA2946165A patent/CA2946165C/en active Active
- 2014-04-21 KR KR1020217038097A patent/KR20210144946A/ko not_active IP Right Cessation
- 2014-04-21 JP JP2016563193A patent/JP2017513866A/ja active Pending
- 2014-04-21 BR BR112016024502A patent/BR112016024502A8/pt not_active Application Discontinuation
- 2014-04-21 MX MX2016013741A patent/MX2016013741A/es unknown
-
2016
- 2016-10-20 IL IL248445A patent/IL248445A0/en unknown
- 2016-11-14 ZA ZA2016/07833A patent/ZA201607833B/en unknown
-
2020
- 2020-09-24 AU AU2020239754A patent/AU2020239754B2/en active Active
-
2022
- 2022-01-26 US US17/584,663 patent/US20230053976A1/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006507218A (ja) * | 2002-03-20 | 2006-03-02 | アドバンスト インハレーション リサーチ,インコーポレイテッド | レボドパの肺送達 |
Non-Patent Citations (1)
| Title |
|---|
| NEUROLOGY, 2014, VOL.82, NO.10 SUPPLEMENT, S7.007, JPN6018007782, 8 April 2014 (2014-04-08), ISSN: 0004072336 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021509676A (ja) * | 2018-01-05 | 2021-04-01 | インペル ニューロファーマ インコーポレイテッド | 精密嗅覚装置によるレボドパ粉末の鼻腔内送達 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN113209055A (zh) | 2021-08-06 |
| RU2016144340A3 (ja) | 2018-05-22 |
| CN106659685A (zh) | 2017-05-10 |
| BR112016024502A2 (pt) | 2017-08-15 |
| AU2014391721B2 (en) | 2020-07-16 |
| US20170296498A1 (en) | 2017-10-19 |
| CA2946165C (en) | 2022-10-18 |
| AU2020239754A1 (en) | 2021-01-14 |
| KR20170008754A (ko) | 2017-01-24 |
| RU2016144340A (ru) | 2018-05-22 |
| EP3134077A4 (en) | 2017-12-20 |
| IL248445A0 (en) | 2016-12-29 |
| EP3134077A1 (en) | 2017-03-01 |
| EP3831375A1 (en) | 2021-06-09 |
| AU2014391721A1 (en) | 2016-11-03 |
| BR112016024502A8 (pt) | 2021-06-29 |
| CA2946165A1 (en) | 2015-10-29 |
| AU2020239754B2 (en) | 2022-06-23 |
| CN106659685B (zh) | 2021-02-05 |
| MX2016013741A (es) | 2017-04-06 |
| ZA201607833B (en) | 2018-08-29 |
| US20230053976A1 (en) | 2023-02-23 |
| IL309959A (en) | 2024-03-01 |
| KR20210144946A (ko) | 2021-11-30 |
| SG11201608608PA (en) | 2016-11-29 |
| RU2698330C2 (ru) | 2019-08-26 |
| WO2015163840A1 (en) | 2015-10-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018253538B2 (en) | Levodopa formulations for rapid relief of parkinson's disease | |
| US20230053976A1 (en) | Rapid relief of motor fluctuations in parkinson's disease | |
| WO2011020061A2 (en) | Compositions and methods of for treating bipolar disorder | |
| AU2020250280B2 (en) | Reducing Inter-Patient Variability of Levodopa Plasma Concentrations | |
| JP2019108379A (ja) | パーキンソン病の運動症状変動の迅速な緩和 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170420 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20170420 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180306 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180531 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180801 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20181106 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190305 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20190305 |
|
| C60 | Trial request (containing other claim documents, opposition documents) |
Free format text: JAPANESE INTERMEDIATE CODE: C60 Effective date: 20190305 |
|
| C11 | Written invitation by the commissioner to file amendments |
Free format text: JAPANESE INTERMEDIATE CODE: C11 Effective date: 20190319 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20190521 |
|
| C21 | Notice of transfer of a case for reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C21 Effective date: 20190528 |
|
| A912 | Re-examination (zenchi) completed and case transferred to appeal board |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20190705 |
|
| C211 | Notice of termination of reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C211 Effective date: 20190709 |
|
| C22 | Notice of designation (change) of administrative judge |
Free format text: JAPANESE INTERMEDIATE CODE: C22 Effective date: 20200107 |
|
| C22 | Notice of designation (change) of administrative judge |
Free format text: JAPANESE INTERMEDIATE CODE: C22 Effective date: 20200407 |
|
| C13 | Notice of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: C13 Effective date: 20200428 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20200715 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20200817 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20200914 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20201026 |
|
| C22 | Notice of designation (change) of administrative judge |
Free format text: JAPANESE INTERMEDIATE CODE: C22 Effective date: 20201124 |
|
| C23 | Notice of termination of proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C23 Effective date: 20210105 |
|
| C03 | Trial/appeal decision taken |
Free format text: JAPANESE INTERMEDIATE CODE: C03 Effective date: 20210209 |
|
| C30A | Notification sent |
Free format text: JAPANESE INTERMEDIATE CODE: C3012 Effective date: 20210209 |