JP2016500403A - 官能化されたポリオキシメチレンブロックコポリマー - Google Patents
官能化されたポリオキシメチレンブロックコポリマー Download PDFInfo
- Publication number
- JP2016500403A JP2016500403A JP2015548472A JP2015548472A JP2016500403A JP 2016500403 A JP2016500403 A JP 2016500403A JP 2015548472 A JP2015548472 A JP 2015548472A JP 2015548472 A JP2015548472 A JP 2015548472A JP 2016500403 A JP2016500403 A JP 2016500403A
- Authority
- JP
- Japan
- Prior art keywords
- formaldehyde
- block copolymer
- mol
- polyoxymethylene block
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 C*C(O1)=C(*)*(*)C(C*)C1=O Chemical compound C*C(O1)=C(*)*(*)C(C*)C1=O 0.000 description 3
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4825—Polyethers containing two hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4829—Polyethers containing at least three hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4887—Polyethers containing carboxylic ester groups derived from carboxylic acids other than acids of higher fatty oils or other than resin acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2/00—Addition polymers of aldehydes or cyclic oligomers thereof or of ketones; Addition copolymers thereof with less than 50 molar percent of other substances
- C08G2/38—Block or graft polymers prepared by polymerisation of aldehydes or ketones on to macromolecular compounds
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
- Polyesters Or Polycarbonates (AREA)
- General Chemical & Material Sciences (AREA)
- Polyurethanes Or Polyureas (AREA)
Abstract
Description
nエステル、最小=(mスターター/M.W.スターター)×Fスターター (等式1)
式中、mスターターは、使用するスターター化合物の質量であり、M.W.スターターは、その数平均分子量であり、Fスターターは、その平均官能性、即ちスターター分子1個あたりの官能基の平均数である。

R1、R2、R3およびR4は、互いに独立して、水素、直鎖状または分岐状の、任意にヘテロ原子を含むC1〜C22のアルキルラジカル、直鎖状または分岐状の、モノまたはポリ不飽和の、任意にヘテロ原子を含むC1〜C22のアルケニルラジカル、または任意にモノまたはポリ置換された、任意にヘテロ原子を含むC6〜C18のアリールラジカルであるか、または飽和または不飽和の4〜7員環、または任意にヘテロ原子を含む多環系の構成員でよく、
式Iの化合物は、塩素、臭素、ニトロ基またはアルコキシ基により置換されていてよく、
nは、1以上の整数、好ましくは1、2、3または4であり、
反復単位(n>1)中のR3およびR4はそれぞれ異なっていてよい。
β−プロピオラクトン、β−ブチロラクトン、β−イソバレロラクトン、β−カプロラクトン、β−イソカプロラクトン、β−メチル−β−バレロラクトンなどの4員環ラクトン、
γ−ブチロラクトン、γ−バレロラクトン、5−メチルフラン−2(3H)−オン、5−メチリデンジヒドロフラン−2(3H)−オン、5−ヒドロキシフラン−2(5H)−オン、2−ベンゾフラン−1(3H)−オンおよび6−メチル−2−ベンゾフラン−1(3H)−オンなどの5員環ラクトン、
δ−バレロラクトン、1,4−ジオキサン−2−オン、ジヒドロクマリン、1H−イソクロメン−1−オン、8H−ピラノ[3,4−b]ピリジン−8−オン、1,4−ジヒドロ−3H−イソクロメン−3−オン、7,8−ジヒドロ−5H−ピラノ[4,3−b]ピリジン-5−オン、4−メチル−3,4−ジヒドロ−1H-ピラノ[3,4−b]ピリジン−1−オン、6−ヒドロキシ−3,4−ジヒドロ−1H−イソクロメン−1−オン、7−ヒドロキシ−3,4−ジヒドロ−2H−クロメン−2−オン、3−エチル−1H−イソクロメン−1−オン、3−(ヒドロキシメチル)−1H−イソクロメン−1−オン、9−ヒドロキシ−1H,3H−ベンゾ[デ]−イソクロメン−1−オン、6,7−ジメトキシ−1,4−ジヒドロ−3H−イソクロメン−3−オンおよび3−フェニル−3,4−ジヒドロ−1H−イソクロメン−1−オンなどの6員環ラクトン、
ε−カプロラクトン、p−ジオキサノンおよび1,5−ジオキセパン−2−オン、5−メチルオキセパン−2−オン、オキセパン−2,7−ジオン、チエパン−2−オン、5−クロロオキセパン−2−オン、(4S)−4−(プロパン−2−イル)オキセパン−2−オン、7−ブチルオキセパン−2−オン、5−(4−アミノブチル)オキセパン−2−オン、5−フェニルオキセパン−2−オン、7−ヘキシルオキセパン−2−オン、(5S,7S)−5−メチル−7−(プロパン−2−イル)オキセパン−2−オン、4−メチル−7−(プロパン−2−イル)オキセパン−2−オンなどの7員環ラクト、
(7E)−オキサシクロヘプタデカ−7−エン−2−オンなどの、より高い構成員を有する環状ラクトンである。
式中、R1、R2、R3およびR4は、それぞれ上に定義した通りであり、
mおよびnは、それぞれ独立して1以上の整数、好ましくは1、2、3または4であり、
反復単位(m>1)中のR1およびR2ならびに反復単位(n>1)中のR3およびR4はそれぞれ異なっていてよい。
グリコリド(1,4−ジオキサン−2,5−ジオン)、L−ラクチド(L−3,6−ジメチル−1,4−ジオキサン−2,5−ジオン)、D−ラクチド、DL−ラクチド、メソラクチドおよび3−メチル−1,4−ジオキサン−2,5−ジオン、3−メチル−6−(プロパ−2−エン−1−イル)−1,4−ジオキサン−2,5−ジオン、3−ヘキシル−6−メチル−1,4−ジオキサン−2,5−ジオン、3,6−ジ(ブタ−3−エン−1−イル)−1,4−ジオキサン−2,5−ジオンである(それぞれの場合に光学活性形態を含む)。
R5、R6、R7、R8、R9およびR10は、それぞれ独立して、水素、直鎖状または分岐状の、任意にヘテロ原子を含むC1〜C22のアルキルラジカル、直鎖状または分岐状の、モノまたはポリ不飽和の、任意にヘテロ原子を含むC1〜C22のアルケニルラジカル、または任意にモノまたはポリ置換された、任意にヘテロ原子を含むC6〜C18のアリールラジカルであるか、または、飽和もしくは不飽和の4〜7員環または任意にヘテロ原子を含む多環系の構成員であり、
nは、0以上の整数、好ましくは0、1、2または3であり、
および反復単位(n>1)中のR9およびR10はそれぞれ異なっていてよく、
式(V)の化合物は、塩素、臭素、ニトロ基またはアルコキシ基により置換されていてよい。
R11およびR12は、水素、ハロゲン、直鎖状または分岐状の、任意にヘテロ原子を含むC1〜C22のアルキル置換基、直鎖状または分岐状の、モノまたはポリ不飽和の、任意にヘテロ原子を含むC1〜C22のアルケニル置換基、または任意にモノまたはポリ置換された、任意にヘテロ原子を含むC6〜C18のアリール置換基であるか、またはR11およびR12は、飽和または不飽和の4〜7員環または任意にヘテロ原子を含む多環系の構成員であってよく、R11およびR12はともに、好ましくはベンゼン環を形成し、任意に追加の置換基を有し、
R13、R14、R15およびR16は、水素、直鎖状または分岐状の、任意にヘテロ原子を含むC1〜C22のアルキル置換基、直鎖状または分岐状の、モノまたはポリ不飽和の、任意にヘテロ原子を含むC1〜C22のアルケニル置換基、または任意にモノまたはポリ置換された、任意にヘテロ原子を含むC6〜C18のアリール置換基であるか、または飽和または不飽和の4〜7員環または任意にヘテロ原子を含む多環系の構成員であってよく、
R17、R18、R19、R20、R21およびR22は、水素、直鎖状または分岐状の、任意にヘテロ原子を含むC1〜C22のアルキル置換基、直鎖状または分岐状の、モノまたはポリ不飽和の、任意にヘテロ原子を含むC1〜C22のアルケニル置換基、または任意にモノまたはポリ置換された、任意にヘテロ原子を含むC6〜C18のアリール置換基であるか、または飽和または不飽和の4〜7員環または任意にヘテロ原子を含む多環系の構成員であってよく、
式(VI)および(VII)および(VIII)の化合物は、塩素、臭素、ニトロ基またはアルコキシ基により置換されていてよい。
実施例
PET−1 名目分子量M.W.=600g/molおよび平均実験式HO(CH2CH2O)13.23Hの二官能性ポリ(オキシエチレン)ポリオール。OH価は187.15mgKOH/g、数平均分子量Mn=658g/molおよび多分散性インデックスPDI=1.087(クロロホルム中で、ポリプロピレングリコール標準に対してGPCにより測定)を測定した。
使用したイソシアネート
平均官能性2.6およびNCO値31.1〜31.1%であり、42.4%の4,4’−MDI、12.6%の2,4’−MDI、2.2%の2,2’−MDI(Bayerから市販のDesmodur VP PU 0325)を含むイソシアネート1。
使用したホルムアルデヒド源は、Aldrichから市販のトリオキサン(CAS[110−88−3])(カタログ番号T81108)。
モル質量分布は、ゲル透過クロマトグラフィー(GPC)により測定した。
ゲル透過クロマトグラフィー(GPC):測定は、Agilent 1200 Series計器(G1310A Iso Pump、G1329A ALS、G1316A TCC、G1362A RID、G1365D NWD)で、RIDにより検出;溶離液:クロロホルム(GPC等級)、流量1.0ml/min;カラム組合せ:PSS SDV予備カラム8x50mm(5μm)、2×PSS SDV直線S 8×300mL(5μm)で行った。PSS Polymer Standards Serviceから市販のモル質量が既知であるポリプロピレングリコール試料を校正用に使用した。使用した測定記録および評価ソフトウエアは、ソフトウエアパッケージ「PSS WinGPC Unity」であった。GPCクロマトグラムは、溶離液としてTHFの代わりにクロロホルムを使用した以外は、DIN 55672-1により記録した。
OH価[mgKOH/g]=56100[mgKOH/mol]/等価モル質量[g/mol]
等価モル質量は、活性水素原子を含む材料の数平均総モル質量を、活性水素原子の数(官能性)で割った値であると理解される。
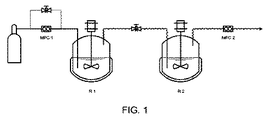
反応器1に最初に、1,3,5-トリオキサン30.18g(0.335mol)および4-ドデシルベンゼンスルホン酸0.62g(1.92mmol)をウンデカン30mlに入れた懸濁液を仕込んだ。反応器2は、ジブチルスズジラウレート(DBTL)430mg(0.68mmol)および炭酸セシウム1.09g(3.34mmol)をPET−1 20.11g(33.5mmol)に入れた溶液を含んでいた。MFC1に接続したバイパスラインを経由して、ブリッジを開いた状態で全システムを、CO2で20barに加圧した。次いで、ブリッジを閉じ、反応器1の圧力を、ガス排出口バルブにより5barに下げた。ブリッジを閉じ、反応器1内の反応混合物を65℃に加熱し、この温度で1.5時間維持した。続いて、反応器1内の温度を110℃に上昇させた。反応器2内の反応混合物を、撹拌しながら60℃に加熱した。ブリッジ温度を170℃に調節した。反応温度に達した時、反応器2内の圧力を17〜19barの値に調節した。反応器1内の圧力を、バイパスを経由してCO2で20barに調節した。バイパスラインを閉じ、マスフロー調整器を使用して、一定CO2流量Vin=160ml/minとした。その直後、ブリッジを開き、MFC2により、システム内の総圧を20barに調節した。3.2時間後、ブリッジを閉じ、システムを室温に冷却し、圧力を反応器1および反応器2で別々に解除した。続いて、内部温度40℃で撹拌しながら、HPLCポンプを使用して、無水グルタル酸7.53g(65.9mmol)をCH2Cl2 20mLに入れた溶液を反応器2中に流量5mL/minで導入した。添加が終わった後、反応器2内の反応混合物を100℃でさらに16時間撹拌した。生成物を除去する前の反応器2の秤量は、反応により3.39gの重量増加を示し(供給した無水グルタル酸の質量を差し引いた質量の差)、これは、気体状ホルムアルデヒドの移動3.39g(113mmol)に相当する。粘性の無色オイル30.92gを反応器2から除去した。ホルムアルデヒド3.39g(113mmol)が移動した結果、オリゴマーとして使用したPET−1は、分子1個あたり平均3.4ホルムアルデヒド単位、または鎖末端あたり1.7ホルムアルデヒド単位に広がった。
粘度:1600mPa・s
酸価:126.7mgKOH/g
1H NMR分光法 (400MHz, CDCl3):δ=0.47-0.62 (m, 0.045H), 0.92 (bs, 0.14H), 1.49-1.65 (m, 0.47H, OC(O)CH2CH 2CH2C(O)O), 1.96-2.18 (m, 1H, OC(O)CH 2CH2CH 2C(O)O), 3.06-3.87 (m, 6.58H, PET-1-CH2), 4.41-4.59 (m, 0.28H, OCH2O), 4.79 (s, 0.26H, OCH2O), 4.95-5.08 (m, 0.32H, OCH2O), 5.39 (s, 0.03H) ppm.
13C APT NMR分光法 (400 MHz, CDCl3): δ=18.8 (+, OC(O)CH2 CH2), 18.9 (+, OC(O)CH2 CH2), 19.0 (+, OC(O)CH2 CH2), 19.0 (+, OC(O)CH2 CH2), 19.2 (+, OC(O)CH2 CH2), 21.8 (+), 28.4 (+), 28.7 (+), 17.3 (-), 31.0 (+), 32.0 (+, OC(O)CH2CH2), 32.0 (+, OC(O)CH2CH2), 32.1 (+, OC(O)CH2CH2), 32.2 (+, OC(O)CH2CH2), 32.3 (+, OC(O)CH2CH2), 60.3 (+, PET-1-CH2), 62.6 (+, PET-1-CH2), 66.6 (+, PET-1-CH2), 66.8 (+, PET-1-CH2), 68.1 (+, PET-1-CH2), 68.6 (+, PET-1-CH2), 69.2 (+, PET-1-CH2), 69.5 (+, PET-1-CH2), 71.6 (+, PET-1-CH2), 84.3 (+, O-CH2-O), 84.7 (+, O-CH2-O), 85.7 (+, O-CH2-O), 87.8 (+, O-CH2-O), 88.3 (+, O-CH2-O), 88.3 (+, O-CH2-O), 89.0 (+, O-CH2-O), 89.9 (+, O-CH2-O), 91.4 (+, O-CH2-O), 91.5 (+, O-CH2-O), 92.6 (+, O-CH2-O), 93.1 (+, O-CH2-O), 171.5 (+, C(O)OCH2), 171.6 (+, C(O)OCH2), 172.0 (+, C(O)OCH2), 174.8 (+, C(O)OH), 175.1 (+, C(O)OH) ppm.
ESI質量スペクトルで、下記の信号系列が確認され、これは下記の一般実験式に帰せられる。
[HOC(O)(CH2)3C(O)O−(CH2O)a−(C3H6O)m−(CH2O)b−C(O)(CH2)3C(O)OH−H]+
系列1 (a + b = 8): m/z (%) [m] = 839 (1) [8], 883 (5) [9], 927 (14) [10], 971 (28) [11], 1015 (42) [12], 1059 (48) [13], 1103 (46) [14], 1147 (37) [15], 1191 (27) [16], 1235 (19) [17], 1279 (16) [18], 1323 (15) [19], 1367 (17) [20]。
系列2 (a + b = 7): m/z (%) [m] = 809 (1) [8], 853 (5) [9], 897 (16) [10], 941 (35) [11], 985 (60) [12], 1029 (75) [13], 1073 (76) [14], 1117 (64) [15], 1161 (47) [16], 1205 (32) [17], 1249 (22) [18], 1293 (18) [19], 1337 (18) [20]。
系列3 (a + b = 6): m/z (%) [m] = 823 (5) [9], 867 (14) [10], 911 (38) [11], 955 (69) [12], 999 (93) [13], 1043 (100) [14], 1087 (87) [15], 1131 (64) [16], 1175 (42) [17], 1219 (27) [18], 1263 (18) [19], 1307 (13) [20]。
系列4 (a + b = 5): m/z (%) [m] = 793 (1) [9], 837 (5) [10], 881 (12) [11], 925 (26) [12], 969 (40) [13], 1013 (48) [14], 1057 (44) [15], 1101 (33) [16], 1145 (22) [17], 1189 (18) [18], 1233 (7) [19], 1277 (2) [20]。
系列5 (a + b = 4): m/z (%) [m] = 939 (1) [13], 983 (5) [14], 1027 (10) [15], 1071 (17) [16], 1115 (23) [17], 1159 (26) [18], 1203 (7) [25], 1247 (21) [20]。
系列6 (a + b = 3): m/z (%) [m] = 909 (2) [13], 953 (5) [14], 997 (14) [15], 1041 (26) [16], 1085 (38) [17], 1129 (45) [18], 1173 (21) [25], 1217 (37) [20]。
系列7 (a + b = 2): m/z (%) [m] = 879 (1) [13], 923 (6) [14], 967 (15) [15], 1011 (33) [16], 1055 (51) [17], 1099 (61) [18], 1143 (62) [25], 1187 (52) [20]。
系列8 (a + b = 1): m/z (%) [m] = 849 (1) [13], 893 (3) [14], 937 (8) [15], 981 (19) [16], 1025 (33) [17], 1069 (44) [18], 1113 (46) [25], 1157 (40) [20]。
[HOC(O)(CH2)3C(O)O−(CH2O)a−(C3H6O)m−(CH2O)b−C(O)(CH2)3C(O)O−(CH2O)c−(C3H6O)n−(CH2O)d−C(O)(CH2)3C(O)O−H]+
系列9 (a + b + c + d = 3): m/z (%) [m+n] = 909 (1) [10], 953 (5) [11], 997 (14) [12], 1041 (27) [13], 1085 (38) [14], 1129 (45) [15], 1173 (44) [16], 1217 (37) [17], 1261 (29) [18], 1305 (21) [19], 1349 (19) [20], 1393 (20) [21], 1437 (26) [22], 1481 (32) [23], 1525 (39) [24], 1569 (45) [25], 1613 (49) [26], 1657 (49) [27], 1701 (45) [28], 1745 (39) [29], 1789 (32) [30], 1833 (25) [31], 1877 (17) [32], 1921 (12) [33], 1965 (8) [34], 2009 (5) [35], 2053 (5) [36], 2097 (2) [37]。
系列10 (a + b + c + d = 2): m/z (%) [m+n] = 879 (1) [10], 923 (6) [11], 967 (15) [12], 1011 (33) [13], 1055 (51) [14], 1099 (61) [15], 1143 (62) [16], 1187 (52) [17], 1231 (41) [18], 1275 (28) [19], 1319 (22) [20], 1363 (20) [21], 1407 (22) [22], 1451 (26) [23], 1495 (31) [24], 1539 (35) [25], 1583 (38) [26], 1627 (38) [27], 1671 (35) [28], 1715 (30) [29], 1759 (25) [30], 1803 (18) [31], 1847 (13) [32], 1891 (9) [33], 1935 (6) [34], 1979 (4) [35], 2023 (2) [36], 2067 (1) [37]。
系列11 (a + b + c + d = 1): m/z (%) [m+n] = 849 (1) [10], 893 (3) [11], 937 (9) [12], 981 (19) [13], 1025 (38) [14], 1069 (44) [15], 1113 (46) [16], 1157 (40) [17], 1201 (17) [18], 1245 (22) [19], 1289 (16) [20], 1333 (12) [21], 1377 (10) [22], 1421 (10) [23], 1465 (12) [24], 1509 (14) [25], 1553 (15) [26], 1597 (14) [27], 1641 (14) [28], 1685 (12) [29], 1729 (10) [30], 1773 (8) [31], 1817 (6) [32], 1861 (4) [33], 1905 (3) [34], 1979 (4) [35]。
反応器1に最初に、1,3,5-トリオキサン29.83g(0.331mol)および4-ドデシルベンゼンスルホン酸0.52g(1.6mmol)をウンデカン30mlに入れた懸濁液を仕込んだ。反応器2は、ジブチルスズジラウラート(DBTL)460mg(0.73mmol)、炭酸セシウム22.6g(69.5mmol)およびPET−1 20.5g(34.2mmol)の混合物を含んでいた。MFC1に接続したバイパスラインを経由して、ブリッジを開いた状態で全システムを、CO2で20barに加圧した。次いで、ブリッジを閉じ、反応器1の圧力を、ガス排出口バルブにより5barに下げた。ブリッジを閉じ、反応器1内の反応混合物を65℃に加熱し、この温度で1.5時間維持した。反応器2内の反応混合物を、撹拌しながら、60℃に加熱した。ブリッジ温度を170℃に調節した。反応器1内の反応混合物を65℃で1.5時間加熱した後、圧力を、バイパスを経由してCO2で20barに調節した。反応器2内の圧力を17〜19barの値に調節した。バイパスラインを閉じ、マスフロー調整器を使用して、一定CO2流量Vin=160ml/minとした。実験の残りの部分で、MFC2により、システム内の総圧を20barで一定に維持した。次いで、一定CO2流量下でブリッジを開き、反応器1内の温度を110℃に上昇した。6時間後、ブリッジを閉じ、反応器2を40℃に冷却した。続いて、内部温度40℃で撹拌しながら、HPLCポンプを使用して、無水グルタル酸7.53g(65.9mmol)を1,4-ジオキサン20mLに入れた溶液を反応器2中に流量5mL/minで導入した。添加が終わった後、反応器2内の反応混合物を100℃でさらに16時間撹拌した。続いて、システムを室温に冷却し、反応器1および2内の圧力を別々に解除した。反応器2から粘性の無色オイルを除去した。
粘度:生成物は、せん断速度範囲10〜162s−1でせん断-粘度上昇挙動を示し、せん断速度範囲162〜1000s−1でせん断-粘度低下挙動を示した。
せん断速度10s−1における粘度:3056mPa・s
せん断速度162s−1における粘度:3173mPa・s
せん断速度1000s−1における粘度:2986mPa・s
1H NMR分光法(400 MHz, CDCl3): δ=0.75-0.88 (m, 0.359H), 1.12-1.27 (m, 1.082H), 1.78-1.96 (m, 2.988H, -C(O)CH2CH 2CH2C(O)OH), 2.20-2.43 (m, 6.099H, -C(O)CH 2CH2CH 2C(O)OH), 3.36-3.44 (m, 0.311H, PET-1-CH2), 3.44-3.69 (m, 45.87H, PET-1-CH2), 3.69-3.80 (m, 2.148H, PET-1-CH2), 4.10-4.20 (m, 0.656H), 4.70-4.80 (m, 1.595H, OCH2O), 4.80-4.90 (m, 1.602H, OCH2O), 5.09 (s, 0.433H, OCH2O), 5.19-5.26 (m, 1.112H, OCH2O), 5.26-5.31 (m, 0.999H, OCH2O), 9.17 (bs, 1.859H) ppm.
最初にビーカーに、実施例2で得たカルボン酸末端を有するポリエチレンオキシド−ポリオキシメチレンブロックコポリマー5.02g、水0.25mLおよびジブチルスズジラウラート(DBTL)10.3mg(0.016mmol)を入れ、混合物を60℃に加熱した。続いて、撹拌しながら0.62gのイソシアネート1を加え、混合物を強く撹拌した。3時間後、著しい発泡が観察され、20秒後に減少した。黄色のゲルが得られた。
粘度:生成物は、せん断−粘度低下挙動を示した。
せん断速度0.01s−1における粘度:10500mPa・s
せん断速度927s−1における粘度:8684mPa・s
実施例2で得たカルボン酸末端を有するポリエチレンオキシド−ポリオキシメチレンブロックコポリマー5.04gを、トリフェニルホスフィン26.7mg(0.102mmol)およびフェニルグリシジルエーテル(PGE)1.41g(9.39mmol)と共に、ガラス製フラスコに秤量して取り、混合物を80℃で18時間環流下で撹拌し、その間に混合物は、黄色から暗赤色に変色した。生成物を得られた状態でさらに使用した。
粘度:生成物は、せん断−粘度低下挙動を示した。
せん断速度0.01s−1における粘度:3890mPa・s
せん断速度589s−1における粘度:3658mPa・s
せん断速度1000s−1における粘度:3574mPa・s
Tg = -42.59℃
1H NMR分光法(400 MHz, CDCl3): δ=0.79-0.88 (m, 0.135H), 1.15-1.30 (m, 0.465H), 1.81-2.01 (m, 1.971H, -C(O)CH2CH 2CH2C(O)-), 2.17-2.49 (m, 3.892H, -C(O)CH 2CH 2CH 2C(O)-), 3.38-3.45 (m, 0.181H), 3.45-3.72 (m, 29.28H, PET-1-CH2), 3.72-3.88 (m, 1.301H, PET-CH2), 3.91-4.02 (m, 1.622H), 4.02-4.36 (m, 5.773H, PGE-CH/PGE/CH2), 4.36-4.50 (0.257H), 4.74-4.82 (m, 0.256H, OCH2O), 4.82-4.89 (m, 0.243H, OCH2O), 5.12 (s, 0.187H, OCH2O), 5.15-5.39 (m, 0.956H), OCH2O), 6.80-6.97 (m, 3.000H, PGE-CHar), 7.17-7.39 (m, 2.160H, PGE-CHar/CHCl3), 7.39-7.48 (m, 0.105H, PPh3), 7.48-7.55 (m, 0.0458H, PPh3), 7.57-7.67 (m, 0.0896H, PPh3) ppm。
最初にビーカーに、実施例4で得たヒドロキシ官能化されたポリエチレンオキシド−ポリオキシメチレンブロックコポリマー4.13g、水0.25mLおよびDBTL8.0mg(0.013mmol)を入れ、混合物を60℃に加熱した。続いて、撹拌しながら0.62gのイソシアネート1を加え、混合物を強く撹拌した。4時間後、著しい発泡が観察され、20秒後に減少した。茶色のゲルが得られた。
粘度:生成物は、せん断-粘度低下挙動を示した。
せん断速度3.35s−1における粘度:1447000mPa・s
せん断速度1000s−1における粘度:31950mPa・s
Tg=-29.41℃
Claims (12)
- 反応容器中で触媒の存在下でホルムアルデヒドを重合させる工程を含んでなる、官能化されたポリオキシメチレンブロックコポリマーの製造方法であって、
ホルムアルデヒドの重合を、少なくとも2個のZerewitinoff活性水素原子を有するスターター化合物の存在下でさらに行い、数平均分子量<4500g/molである中間体を得、
得られた中間体を環状カルボン酸または炭酸エステルと反応させ、官能化されたポリオキシメチレンブロックコポリマーを得る、方法。 - 前記触媒が、塩基性触媒および/またはルイス酸触媒の群から選択される、請求項1に記載の方法。
- 前記中間体と前記環状カルボン酸または炭酸エステルとの反応が、先行するホルムアルデヒドの重合で使用したのと同一の触媒の存在下で行われる、請求項1または2に記載の方法。
- 前記スターター分子の数平均分子量が、≧62g/mol〜≦4470g/mol、特に≧100g/mol〜≦3000g/molである、請求項1〜3のいずれか一項に記載の方法。
- 前記スターター分子が、ポリエーテルポリオール、ポリエステルポリオール、ポリエーテルエステルポリオール、ポリカーボネートポリオールおよび/またはポリアクリレートポリオールの群から選択される、請求項1〜4のいずれか一項に記載の方法。
- 前記環状カルボン酸または炭酸エステルが、脂肪族または芳香族のラクトン、ラクチド、環状カーボネートおよび/または環状酸無水物の群から選択される、請求項1〜5のいずれか一項に記載の方法。
- 前記ホルムアルデヒドが、気体状ホルムアルデヒドとして前記反応容器中に導入される、請求項1〜6のいずれか一項に記載の方法。
- 前記重合が、コモノマーの存在下でさらに行われる、請求項1〜7のいずれか一項に記載の方法。
- 請求項1〜8のいずれか一項に記載の方法により得られる、官能化されたポリオキシメチレンブロックコポリマー。
- 数平均分子量が≦12000g/molである、請求項9項に記載の官能化されたポリオキシメチレンブロックコポリマー。
- 20℃における粘度が≦100000mPasである、請求項9または10に記載の官能化されたポリオキシメチレンブロックコポリマー。
- 請求項9〜11のいずれか一項に記載の官能化されたポリオキシメチレンブロックコポリマーの、ポリアミド、ポリウレタン、洗浄およびクリーニング組成物処方物、掘削流体、燃料添加剤、イオン系および非イオン系界面活性剤、潤滑剤、製紙もしくは織物製造用の処理化学薬品、または化粧品処方物の製造への使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12199053 | 2012-12-21 | ||
| EP12199053.5 | 2012-12-21 | ||
| PCT/EP2013/077049 WO2014095971A2 (de) | 2012-12-21 | 2013-12-18 | Funktionalisierte polyoxymethylen-block-copolymere |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016500403A true JP2016500403A (ja) | 2016-01-12 |
| JP2016500403A5 JP2016500403A5 (ja) | 2016-12-28 |
Family
ID=47603106
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015548472A Pending JP2016500403A (ja) | 2012-12-21 | 2013-12-18 | 官能化されたポリオキシメチレンブロックコポリマー |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9688814B2 (ja) |
| EP (1) | EP2935381B1 (ja) |
| JP (1) | JP2016500403A (ja) |
| KR (1) | KR20150097717A (ja) |
| CN (1) | CN104937001B (ja) |
| CA (1) | CA2896628A1 (ja) |
| SG (1) | SG11201504408QA (ja) |
| WO (1) | WO2014095971A2 (ja) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104937001B (zh) | 2012-12-21 | 2017-03-08 | 科思创德国股份有限公司 | 官能化的聚甲醛嵌段共聚物 |
| PL2935387T3 (pl) | 2012-12-21 | 2017-08-31 | Covestro Deutschland Ag | Zmodyfikowane NCO polioksymetylenowe kopolimery blokowe |
| CA2895583A1 (en) * | 2012-12-21 | 2014-06-26 | Bayer Materialscience Ag | Method for producing formaldehyde/co2 copolymers |
| KR102529691B1 (ko) | 2015-01-08 | 2023-05-09 | 다우 글로벌 테크놀로지스 엘엘씨 | 폴리에테르-아세탈 폴리올 조성물 |
| CN109563230A (zh) * | 2016-08-25 | 2019-04-02 | 科思创德国股份有限公司 | 制备具有降低的热值的聚氨酯聚合物的方法 |
| EP3339340A1 (de) * | 2016-12-20 | 2018-06-27 | Covestro Deutschland AG | Verfahren zur herstellung eines polyoxymethylen-block-copolymers mit erhöhtem anteil an eingebautem formaldehyd |
| AU2018203941B2 (en) * | 2017-06-27 | 2024-04-18 | Dow Global Technologies Llc | Glycidyl ether alkoxylate block copolymers |
| EP3656797A1 (de) * | 2018-11-22 | 2020-05-27 | Covestro Deutschland AG | Verfahren zur herstellung von polyoxymethylen-polyalkylenoxid-blockcopolymeren |
| EP3656796A1 (de) * | 2018-11-22 | 2020-05-27 | Covestro Deutschland AG | Verfahren zur herstellung von polyoxymethylen-polymeren mit mittlerer kettenlänge |
| WO2020173568A1 (de) * | 2019-02-28 | 2020-09-03 | Covestro Intellectual Property Gmbh & Co. Kg | Isocyanat-terminierte prepolymere für die herstellung von polyurethan-integral-schaumstoffen |
| EP4347676A1 (de) | 2021-05-31 | 2024-04-10 | Wacker Chemie AG | Flüssige monofunktionelle 1,3-dioxolan copolymere |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5098536A (ja) * | 1973-12-28 | 1975-08-05 | ||
| JPS5531888A (en) * | 1978-08-28 | 1980-03-06 | Bayer Ag | Manufacture of diphenol carbonate end group contained polymer |
| JP2007211082A (ja) * | 2006-02-08 | 2007-08-23 | Asahi Glass Co Ltd | ポリオキシメチレン−ポリオキシアルキレンブロック共重合体の製造方法 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL109080C (ja) | 1955-11-30 | |||
| US3218295A (en) * | 1955-11-30 | 1965-11-16 | Du Pont | Copolymers of formaldehyde with active hydrogen-containing polymers |
| NL255237A (ja) * | 1959-08-29 | 1900-01-01 | ||
| NL133340C (ja) * | 1962-07-27 | |||
| US3754053A (en) | 1964-05-15 | 1973-08-21 | Celanese Corp | Polyoxymethylene-oxyalkylene block polymers |
| GB1164997A (en) | 1965-11-25 | 1969-09-24 | B P Chemicals U K Ltd | Improvements in Polyether Polyols and Polyurethanes Derived therefrom |
| US3575930A (en) | 1968-12-12 | 1971-04-20 | Goodrich Co B F | Polyoxymethylene glycol polymers |
| IT1014528B (it) * | 1973-12-28 | 1977-04-30 | Sir Soc Italiana Resine Spa | Copolimeri a blocchi |
| US4352914A (en) | 1980-10-06 | 1982-10-05 | Mitsubishi Petrochemical Company Limited | Binder composition for foundry sand molds and cores |
| US4535127A (en) | 1983-03-23 | 1985-08-13 | Asahi Kasei Kogyo Kabushiki Kaisha | Polyacetal copolymers and process for production thereof |
| JP3110788B2 (ja) | 1991-04-03 | 2000-11-20 | 旭化成工業株式会社 | 潤滑特性に優れたポリアセタールブロック共重合体及びその製法 |
| WO1996006118A1 (en) | 1994-08-18 | 1996-02-29 | The University Of North Carolina At Chapel Hill | Cationic polymerization in carbon dioxide |
| US5432207A (en) | 1994-10-25 | 1995-07-11 | Jiffy Foam, Inc. | Phenolic foam composition and use thereof for "in place" foaming |
| JP4560261B2 (ja) | 1999-07-30 | 2010-10-13 | 旭化成ケミカルズ株式会社 | ポリアセタールブロックコポリマー |
| DE10251332B4 (de) | 2002-11-05 | 2006-07-27 | Ticona Gmbh | Polyoxymethylen-Copolymere, deren Herstellung und Verwendung |
| US7928184B2 (en) * | 2005-03-15 | 2011-04-19 | Polyplastics Co., Ltd. | Unstable terminal group decomposer, and stabilized polyacetal resin, manufacturing method, composition and molded article using the same |
| JP2009215340A (ja) * | 2008-03-07 | 2009-09-24 | Polyplastics Co | 安定化ポリアセタール共重合体の製造方法 |
| PL2935387T3 (pl) | 2012-12-21 | 2017-08-31 | Covestro Deutschland Ag | Zmodyfikowane NCO polioksymetylenowe kopolimery blokowe |
| CN104937001B (zh) | 2012-12-21 | 2017-03-08 | 科思创德国股份有限公司 | 官能化的聚甲醛嵌段共聚物 |
-
2013
- 2013-12-18 CN CN201380071497.XA patent/CN104937001B/zh active Active
- 2013-12-18 EP EP13814099.1A patent/EP2935381B1/de not_active Not-in-force
- 2013-12-18 SG SG11201504408QA patent/SG11201504408QA/en unknown
- 2013-12-18 US US14/654,725 patent/US9688814B2/en active Active
- 2013-12-18 KR KR1020157019349A patent/KR20150097717A/ko not_active Application Discontinuation
- 2013-12-18 JP JP2015548472A patent/JP2016500403A/ja active Pending
- 2013-12-18 CA CA2896628A patent/CA2896628A1/en not_active Abandoned
- 2013-12-18 WO PCT/EP2013/077049 patent/WO2014095971A2/de active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5098536A (ja) * | 1973-12-28 | 1975-08-05 | ||
| JPS5531888A (en) * | 1978-08-28 | 1980-03-06 | Bayer Ag | Manufacture of diphenol carbonate end group contained polymer |
| JP2007211082A (ja) * | 2006-02-08 | 2007-08-23 | Asahi Glass Co Ltd | ポリオキシメチレン−ポリオキシアルキレンブロック共重合体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2014095971A2 (de) | 2014-06-26 |
| US20150368396A1 (en) | 2015-12-24 |
| WO2014095971A3 (de) | 2014-09-18 |
| CA2896628A1 (en) | 2014-06-26 |
| SG11201504408QA (en) | 2015-07-30 |
| CN104937001B (zh) | 2017-03-08 |
| KR20150097717A (ko) | 2015-08-26 |
| US9688814B2 (en) | 2017-06-27 |
| EP2935381B1 (de) | 2018-03-21 |
| CN104937001A (zh) | 2015-09-23 |
| EP2935381A2 (de) | 2015-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2016500403A (ja) | 官能化されたポリオキシメチレンブロックコポリマー | |
| US9790328B2 (en) | Polyethercarbonate-polyoxymethylene block copolymers | |
| KR102215796B1 (ko) | 전자-부족 및 전자-풍부 이중 결합을 갖는 폴리에테르 카르보네이트 폴리올의 라디칼 가교 | |
| US10759902B2 (en) | Process for the production of polyoxymethylene block copolymers | |
| US9534090B2 (en) | NCO-modified polyoxymethylene block copolymers | |
| JP6339078B2 (ja) | ポリエーテルカーボネートポリオールの製造方法 | |
| CN105899572B (zh) | 支化的聚醚碳酸酯多元醇及其制备方法 | |
| US11999821B2 (en) | Compound, a reaction product of said compound and production methods thereof | |
| CN109790275B (zh) | 制备含多重键的预聚物作为弹性体前体的方法 | |
| EP3497148B1 (de) | Verfahren zur herstellung von polymeren ringöffnungsprodukten | |
| JP2019515119A (ja) | ポリオキシアルキレンポリオールの製造方法 | |
| US11091589B1 (en) | Method for producing a polymer which contains double bonds as an elastomer precursor | |
| ES2884027T3 (es) | Procedimiento para la preparación de polioles de polioxialquileno funcionalizados | |
| Báez et al. | Degradable poly (ester-ether urethane) s derived of AB2 miktoarm star copolymer poly (ethylene glycol-(ε-caprolactone) 2) diol: Synthesis, characterization and degradation | |
| Nishiwaki et al. | High‐molecular‐weight poly (1, 2‐propylene succinate): A soft biobased polyester applicable as an effective modifier of poly (l‐Lactide) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20161110 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20161110 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20161118 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170809 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170815 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20171115 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20180302 |