JP2015504861A - ニコチン性受容体標的化合物および組成物 - Google Patents
ニコチン性受容体標的化合物および組成物 Download PDFInfo
- Publication number
- JP2015504861A JP2015504861A JP2014546185A JP2014546185A JP2015504861A JP 2015504861 A JP2015504861 A JP 2015504861A JP 2014546185 A JP2014546185 A JP 2014546185A JP 2014546185 A JP2014546185 A JP 2014546185A JP 2015504861 A JP2015504861 A JP 2015504861A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- alkyl
- formula
- alkoxy
- aryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 ***C(CCC1=C(*)*)NC1c1cccnc1 Chemical compound ***C(CCC1=C(*)*)NC1c1cccnc1 0.000 description 6
- YSTIUQVYRWKULO-YBFXNURJSA-N CC(CC(C)N=C1c2cccnc2)/C1=C\c(ccc(OC)c1)c1OC Chemical compound CC(CC(C)N=C1c2cccnc2)/C1=C\c(ccc(OC)c1)c1OC YSTIUQVYRWKULO-YBFXNURJSA-N 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/02—Antidotes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Abstract
Description
本願は、2011年12月12日に出願した米国仮特許出願第61/569,539号の利益を主張する。上述の出願の技術全体は、参照として本明細書に援用される。
本研究は、国立衛生研究所のNIMH承認番号第5RO1MH061412−09号および第1P50MH086383号により一部が後援されている。政府は本発明に一定の権利を有する。

式中、
R1が、発生時に独立して、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール−O−、アリール、または5〜6員環のヘテロアリールであり;または2つのR1基が、付着した結合と共に、5〜8員環を形成し;
nが、0、1、2、3、または4であり;
a1、a2、およびa3のそれぞれが、0または1であり、a1、a2、およびa3の内の2つが0であり、それ以外が1であり;
R2が、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
EおよびGが、それぞれ独立して、何もない状態、−ヘテロ(C0−C3)アルキル−、(C1−C3)アルキレン、または(C2−C3)アルケニレンであり、EおよびGが、同時に両方何もない状態とすることができず;
Aが、結合または
n´が、0、1、または2であり;
R4およびR5が、それぞれ発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Xが、アリールまたはヘテロアリールであって、このアリールおよびヘテロアリールが、5つのR3基および/または1つのRc基により任意に置換され;
R3が、それぞれ発生時に独立して、水素、ハロゲン、(C1−C3)アルキル−C(O)O−、(C1−C3)アルキル−C(O)−、(C1−C3)アルキル−C(O)N(R6)、N(R6)2−、(R6)2NC(O)−、(R6)2N(C1−C5)アルコキシ、
R6が、それぞれ発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Raが、それぞれ発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール、またはアリール−O−であり;
Rcが、水素、(C1−C5)アルコキシ、または(C1−C5)アルキルであり、この(C1−C5)アルコキシおよび(C1−C5)アルキルが、ヒドロキシル、(C1−C3)アルコキシ、ハロゲン、およびチオの群から選択される1つ以上の同一もしくは異なる置換基により任意に置換され;および
bが、0、1、2、3、または4である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはそれらの組み合わせを提供する。
式中、
R1が、発生時に独立して、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール−O−、アリール、または5〜6員環のヘテロアリールであり;または2つのR1基が、付着した結合と共に、5〜8員環を形成し;
nが、0、1、2、3、または4であり;
R2が、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
EおよびGが、それぞれ独立して、何もない状態か、−ヘテロ(C0−C3)アルキル−、(C1−C3)アルキレン、または(C2−C3)アルケニレンであり、EおよびGが、同時に何もない状態とすることができず;
Aが、結合または
n´が、0または1であり;
R4およびR5が、それぞれ発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Xが、アリールまたはヘテロアリールであって、このアリールおよびヘテロアリールが、5つのR3基および/または1つのRc基により任意に置換され;
R3が、それぞれ発生時に独立して、水素、ハロゲン、(C1−C3)アルキル−C(O)O−、(C1−C3)アルキル−C(O)N(R6)、N(R6)2−、(R6)2NC(O)−、(R6)2N(C1−C5)アルコキシ、
R6が、それぞれ発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Raが、それぞれ発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール、またはアリール−O−であり;
Rcが、水素、または、ヒドロキシル、(C1−C3)アルコキシ、ハロゲン、およびチオからなる群から選択される1つ以上の同一または異なる置換基により任意に置換され;および
bが、0、1、2、3、または4である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマーまたはそれらの組み合わせを提供する。
式中、
EおよびGのそれぞれが何もない状態か、(C1−C3)アルキレン、または(C2−C3)アルケニレンであり、かつEおよびGの両方を同時に何もない状態とすることができず、他の全ての本明細書中の変形が、式(IA)に定義されるものとする、
化合物である。
式中、
化合物である。
式中、
Wが、OまたはSであり、かつ
化合物である。
Rcが、水素、またはヒドロキシル、(C1−C3)アルコキシ、およびハロゲンの群から選択される1つ以上の同一もしくは異なる置換基により任意に置換される、(C1−C5)アルキルであり;
R1、R2、R3、Ra、bおよびnが、式(IA)に定義されるものである、
化合物である。
を含む。
式(IIb)の全ての変形は、式(IA)に定義されるものと同一である、
化合物を提供する。
式(IIb)の全ての変形は、式(IA)に定義されるものと同一である、
化合物を提供する。
式中、
式(IB)の全ての変形は、式(I)に定義されるものと同一である、
化合物に関する。
式中、
式(IC)の全ての変形は、式(I)に定義されるものと同一である、
化合物を提供する。
式中、
nが、1、2、3、4、または5であり;
n´が、1、2、または3であり;
R1が、独立して、アミノ、C1−C3アルキル、またはC1−C3アルコキシであり;
R2が、独立してC1−C3アルキルであり;かつ少なくとも1つのR2が、4、5、または6の位置に存在し;
ただし、nが2であり、かつR1が両方ともメトキシである場合、n´が2である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはそれらの組み合わせを提供する。
本発明をさらに記載する前に、本発明をより容易に理解するため、便宜上、特定の用語をまず定義しここに収集した。
本発明の新規化合物は、米国特許公開公報第2009/0215705号に開示かつ/または請求されるものを含む、先行技術の化合物を特に除外する。したがって、本発明は、米国特許公開公報第2009/0215705号に開示かつ/または請求されるものを含む、先行技術の1つ以上の化合物の内の1つを除外したものから生じる、本明細書に記載される式(I)の化合物の1つ以上の亜属(subgenuse)を含む。
式中、
R1が、発生時に独立して、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール−O−、アリール、または5〜6員環のヘテロアリールであり;または2つのR1基が、付着した結合と共に、5〜8員環を形成し;
nが、0、1、2、3、または4であり;
a1、a2、およびa3のそれぞれが、0または1であり、a1、a2、およびa3の内の2つが0であり、かつそれ以外が1であり;
R2が、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
EおよびGが、それぞれ独立して、何もない状態か、−ヘテロ(C0−C3)アルキル−、(C1−C3)アルキレン、または(C2−C3)アルケニレンであり、EおよびGが同時に何もない状態とすることができず;
Aが、結合または
n´が、0、1、または2であり;
R4およびR5が、それぞれ発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Xが、アリールまたはヘテロアリールであり、このアリールおよびヘテロアリールが、1〜5つのR3基および/または1つのRc基により任意に置換され;
R3が、発生時に独立して、水素、ハロゲン、(C1−C3)アルキル−C(O)O−、(C1−C3)アルキル−C(O)−、(C1−C3)アルキル−C(O)N(R6)、N(R6)2−、(R6)2NC(O)−、(R6)2N(C1−C5)アルコキシ、
R6が、発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Raが、発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール、またはアリール−O−であり;
Rcが、水素、(C1−C5)アルコキシ、または(C1−C5)アルキルであり、前記(C1−C5)アルコキシおよび前記(C1−C5)アルキルが、ヒドロキシル(C1−C3)アルコキシ、ハロゲン、およびチオ基から選択される1つ以上の同一または異なる置換基により任意に置換され;かつ
bが、0、1、2、3、または4である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはそれらの組み合わせを提供する。
式中、
R1が、発生時に独立して、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール−O−、アリール、または5〜6員環のヘテロアリールであり;または2つのR1基が、付着した結合と共に、5〜8員環を形成し;
nが、0、1、2、3、または4であり;
R2が、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
EおよびGが、それぞれ独立して、何もない状態か、−ヘテロ(C0−C3)アルキル−、(C1−C3)アルキレン、または(C2−C3)アルケニレンであり、EおよびGが同時に何もない状態とすることができず;
Aが、結合または
n´が、0または1であり;
R4およびR5が、発生時にそれぞれ独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Xが、アリールまたはヘテロアリールであり、このアリールおよびヘテロアリールが、1〜5つのR3基および/または1つのR3基により任意に置換され;
R3が、発生時に独立して、水素、ハロゲン、(C1−C3)アルキル−C(O)O−、(C1−C3)アルキル−C(O)N(R6)、N(R6)2−、(R6)2NC(O)−、(R6)2N(C1−C5)アルコキシ、
R6が、発生時にそれぞれ独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Raが、発生時にそれぞれ独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール、またはアリール−O−であり;
Rcが、水素、またはヒドロキシル(C1−C3)アルコキシ、ハロゲン、およびチオから選択される1つ以上の同一または異なる置換基により任意に置換される(C1−C5)アルキルであり;かつ
bが、0、1、2、3、または4である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはそれらの組み合わせである。
式中、
R1、R2、Ra、X、b、nが、式(IA)において上述されている、化合物を提供する。
式中、
式(III−A)中のSが、たとえば、1つまたは2つのR3基により任意に置換されるアリール基である、化合物を提供する。1つの実施形態において、Xは、1つまたは2つのR3基により置換されるフェニル基であり、R2は水素である。特定の実施形態において、nは0である。R3は、たとえば、アミノ、(C1−C3)アルコキシまたはヒドロキシである。
などを含む。
式中、
化合物を提供する。
を含む。
式中、
Wが、OまたはSであり、
化合物を提供する。1つの実施形態において、bは0である。別の実施形態において、R2はHである。特定の実施形態において、R3は発生時に独立して、ヒドロキシル、アルキル、アルキルアミノ、ジアルキルアミノ、またはアルコキシである。
式中、
Rcが、水素、またはヒドロキシル、(C1−C3)アルコキシ、およびハロゲンから選択される1つ以上の同一または異なる置換基により任意に置換される(C1−C5)アルキルであり;かつ
R1、R2、R3、Ra、b、およびnが、独立して、式(IA)に記載されるものである、
化合物を提供する。
式(IIb)
式中、
化合物をも含む。1つの実施形態において、Xは、任意に置換されるアリールである。別の実施形態において、bは0である。別の実施形態において、EおよびGの内の1つは、何もない状態か、または(C1−C3)アルキレンであり、もう一方が−ヘテロ(C0−C3)アルキルである。
式中、
式中、
nが、1、2、3、4、または5であり;
n´が、1、2、または3であり;
R1が、独立して、アミノ、C1−C3アルキルまたはC1−C3アルコキシであり;
R2が、独立して、C1−C3アルキルであり;かつ少なくとも1つのR2が、4、5、または6の位置で存在しており;
ただし、nが2であり、かつR1が両方ともメトキシである場合、n´が2である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはそれらの組み合わせを提供する。
などを含む。
本発明は、その必要があると同定された対象の神経系疾患または障害を治療または予防する方法をも提供する。本方法は、本発明の化合物の有効量を対象に投与することを含む。神経系疾患または障害は、たとえば、統合失調症、アルツハイマー病、パーキンソン病、薬物依存、または中毒である。
本発明は、多くの適用に有益であり、特に、α7ニコチン性受容体活性を増大させることに利点のある疾患または障害の治療に有益であると予測される。α7受容体の損失は、ADの進行において起こり、統合失調症においてはこの受容体のサブタイプの発現が不足した状態となる。α7アゴニスト様3−(2,4−ジメトキシベンジリデン)−アナバセイン(DMXBA)を慢性的に投与することにより、細胞表面に機能的なα7受容体の発現を増加させることができることが示された。したがって、α7に選択的な薬剤を慢性的に投与することは、α7の数および応答性のアップレギュレーションが起こる前よりも非常に高い効果を有し得る。応答性のアップレギュレーションは本発明の化合物単独により、または薬学的に許容可能な適切な形態での本化合物の併用により起こると予測される。アナバセイン構造に基づくこれらの新規α7アゴニストおよびアンタゴニストの潜在的な適用は、全身的に作用する抗増殖剤、抗炎症剤および創傷治癒薬剤の開発の可能性のみならず、nAChRが関与する神経変性疾患、神経発達疾患、および中毒疾患の治療処置を含む。特に、アナバセイン化合物の極性およびイオン化を変えることにより、中枢神経系に顕著に侵入することなく末梢(血液および間質液)コンパートメントへの薬剤適用および局在化が可能になることが示される。
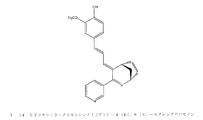
a)3−(2,4−ジメトキシベンジリデン)−4(S)−メチル−アナバセイン(“3−(DMXB)−4(S)−Me−A”)
本明細書に記載される化合物またはその医薬組成物もしくは医薬製剤は、一般的に、意図する結果を達成する有効量、たとえば治療される特定の病態を治療または予防するための有効量で使用される。本化合物またはその医薬組成物/製剤は、治療上の利益を達成するため、治療のために投与されてもよい。「治療上の利益」は、治療される基礎症状の根絶もしくは回復、および/または、未だに基礎疾患を罹患している可能性があるにも関わらず、患者が気分もしくは病態の改善を報告するような、基礎疾患に関連する1つ以上の症状の根絶または回復を意味する。また、治療上の利益は、改善が認められたかどうかにかかわらず、この病態の進行を停止または遅くさせることをも含む。
本発明は、本明細書に記載される疾患または障害の治療または予防のためのキットを提供する。1つの実施形態において、本キットは、単位剤形中に本発明の化合物の有効量を含む治療上または予防用組成物を含む。いくつかの実施形態において、本発明の化合物は、従来の治療剤と併用して提供される。他の実施形態において、本キットは、治療上または予防用組成物を含む無菌性コンテナを含む。このようなコンテナは、箱、アンプル、ビン、バイアル、チューブ、バッグ、ポーチ、ブリスター包装、または当業者に知られている他の適切なコンテナ形態であってよい。このようなコンテナは、プラスチック、ガラス、ラミネート紙、金属ホイル、または薬物の保持に適した他の物質から作製できる。
以下の実施例は、本発明のアッセイ、スクリーニング、および治療法の作製方法および使用方法の完全な開示および記載を当業者に提供するために設定されるものであり、本発明者らが本発明の範囲を限定するように意図したものではない。
本発明の化合物は、このセクションに記載される方法、実施例、および化学的な文献により合成することができる。
A)本発明の化合物の調製のスキーム
スキーム1:4,6−エチレン−アナバセインおよび3−(アリーリデン)−4,6−エチレンアナバセイン化合物の合成
薬理学的に有効でα7選択性のあるエナンチオマーの不斉合成のため、ならびにラセミ体の3−(DMXB)−4,6−エチレンアナバセインのキラルクロマトグラフィーにより得られるエナンチオマーの両方の立体配置を割り当てるために、キラル前駆体((+)−endo−2ノルカンファー)を使用した。
商業的に入手可能な化学物質をフィッシャーサイエンティフィックおよびシグマアルドリッチから購入し、さらに精製することなく使用した。全てのガラス器具は120℃で一晩乾燥させた。dl−4,6−エチレン−アナバセインの合成はアルゴン雰囲気下で実行した。7.5mlの乾燥テトラヒドロフラン(THF)を−75℃まで冷却し、5.1ml(9.2mmole)のLDA溶液(1.8M)を含むヘプタン/THF/エチルベンゼンを5分間にわたり滴下した。この溶液を再び−75℃まで冷却し、1.16g(9.3mmole)の2−aza−3−オキソ−ビシクロ[3.2.1]オクタン(Krowら、1996)を含む4.0mlのTHFを20分間にわたり添加した(この2−aza−3−オキソ−ビシクロ[3.2.1]オクタンは、真空下、55℃で2×10mlの乾燥ベンゼンを蒸溜することにより使用前に乾燥させた)。この溶液を再び−75℃まで冷却し、1.16ml(1.00g、9.2mmole)のクロロトリメチルシランを5分間にわたり滴下した。
12.93g(0.060mol)のクロロクロム酸ピリジニウムおよび乾燥した2塩化メチレン(130ml)の混合物を、氷槽中で冷却し、モレキュラーシーブ(3AO、30g)および(+)−endo−2−ノルボルネオール(3.37g、0.030mol)を添加して、氷槽中で2時間撹拌した。乾燥したエーテル(400ml)を反応混合物に室温で添加し、30分間撹拌し、デカントした。固体残留物を、乾燥したエーテル(50ml)で処置し、15分間撹拌してデカントした。この手法を3回繰り返した。組み合わせたデカント溶液を組み合わせ、シリカゲル層(グラスフィルター上で1cmの高さ)を介して濾過し、rotavaporで蒸発させて望ましい産物を得た。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 2.67 (1H, br s), 2.60 (1H, br s), 2.06 (1H, dd, J = 18.0, 2.4 Hz), 1.86 (1H, d, J = 4.2 Hz), 1.84−1.77 (2H, m), 1.73 (1H, dt, J = 10.2, 1.5 Hz), 1.59−1.49 (2H, m), 1.48−1.39 (1H, m)。 13C−NMR (75 MHz, CDCl3, delta, TMS int. standard): 218.3, 49.8, 45.2, 37.7, 35.3, 27.2, 24.2。
(1S,4R)−ノルカンファー(2.75g、0.025mole)を含む氷酢酸(150ml)溶液にヒドロキシルアミン−O−スルホン酸(4.52g、0.040mole)を添加し、125℃の油槽中で2時間撹拌した。冷却した後、この混合物を、25℃、rotavapor上で蒸発させ、残留物(11.6g)に飽和型水性炭酸水素ナトリウム溶液(60ml)を、撹拌しながらゆっくりと注意して添加し、その後、固体の炭酸水素ナトリウム(10g)をpH7.5になるまで少しずつ添加し、クロロホルム(6×10ml)で抽出した。組み合わせた抽出物を硫酸マグネシウムで乾燥させ、活性炭で脱色し、35℃で蒸発させて粗生成物(2.59g)を得た。この産物を、20%のエタノールを含むエーテルでシリカゲル(150g)上のカラムクロマトグラフィーにより精製した。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 6.77 (1H, br s), 3.55−3.53 (1H, m), 2.62−2.47 (2H, m), 2.29−2.20 (1H, m), 2.07−1.57 (6H, m)。 13C−NMR (75 MHz, CDCl3, delta, TMS int. standard): 172.1, 53.0, 41.9, 36.1, 35.3, 32.4, 32.1, 28.9。
10mlの乾燥THFを、−75℃まで冷却し、5.81ml(10.5mmole)のLDA溶液(1.8M)を含むヘプタン/THF/エチルベンゼンを5分間にわたり滴下した。これを−75℃まで冷却する際に、1.32g(10.5mmole)の(4R,6S)−4,6−エチレン−δ−バレロラクタムを含む20mlのTHF溶液を20分で添加した(このラクタムは、真空下、55℃で2×10mlの乾燥ベンゼンを蒸溜することにより使用前に乾燥させた)。この溶液を再び−75℃まで冷却する際に、1.32ml(1.14g、10.5mmole)のクロロトリメチルシランを5分間にわたり滴下した。この反応混合物を−75℃で15分間および室温で2時間撹拌した。これを再び−75℃まで冷却し、5.81ml(10.5mmole)のLDA溶液(1.8M)を含むヘプタン/THF/エチルベンゼンを、8分間にわたり滴下した。−75℃で20分間撹拌した後、1.09ml(1.21g,8.0mmole)のニコチン酸エチルを5分で滴下した。この反応混合物を−75℃で25分間および室温で一晩(16時間)撹拌した。その後1mlの水を添加し、室温で2時間撹拌し、分離した物質を濾過し、乾燥THF(2×15ml)で洗浄し、撹拌しながら10mlの氷冷した濃塩酸中にこの物質を添加し、105℃の油槽中で18時間煮沸した。
1mlの乾燥エタノール中の32mg(0.172mmol)dl−4,6−エチレンアナバセインおよび25mg(0.206mmol)4−ヒドロキシベンズアルデヒドの混合物に、濃塩酸を2滴添加し、この混合物(閉鎖フラスコ中、アルゴン下)を、一晩(18時間)83〜85℃の油槽中で撹拌した。冷却した後、この反応混合物を蒸発させ、残留物に1mlの飽和型炭酸水素ナトリウムおよび12%の水酸化ナトリウム溶液12滴をpH8.0となるまで添加し、5×1mlのジクロロメタンで抽出した。組み合わせた抽出物を、硫酸マグネシウムで乾燥させ、活性炭で脱色し蒸発させた。
1)N−BOC−4(S)−メチル−ピペリド−2−オン
4−ジメチルアミノ−ピリジン(0.61g、0.005mole)および4(S)−メチル−ピペリド−2−オン(0.65g、0.00575mole)を、ジ−tert−ブチルジカーボネート(2.18g、0.010mole)を含む乾燥ジクロロメタン(10m)トリエチルアミン(0.61g、0.005mole)溶液にアルゴン雰囲気下で添加し、室温で一晩撹拌した。この反応混合物を55℃のrotavapor上で蒸発させ、残留物(1.609g)を酢酸エチルで溶出するシリカゲル(40g)カラムクロマトグラフィーにより精製し、純粋な産物(0.673g、収率55%)を得た。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 3.84−3.75 (1H, m), 3.55−3.44 (1H, m), 2.64−2.50 (1H, m), 2.17−2.06 (1H, m), 2.05−1.87 (2H, m), 1.53 (9H, s), 1.50−1.37 (1H, m), 1.02 (3H, d, J = 6.3 Hz)。
全てのガラス製品は24時間120℃にてオーブン内で乾燥し、反応はアルゴン雰囲気下で実行した。3−ブロモ−ピリジン(0.30ml、0.49g、3.11mmol)を含む乾燥エーテル(8.5ml)の撹拌溶液を(ドライアイス/アセトンで)−78℃まで冷却し、撹拌しながらn−ブチルリチウム溶液(ヘキサン中1.6M、1.95ml、3.12mmol)を滴下し、その後、この溶液をさらに30分撹拌した。この撹拌し冷却した溶液にN−BOC−6(S)−メチル−ピペリド−2−オン(0.619g、2.90mmol、55℃のrotavapor上で10mlの乾燥ベンゼンを蒸溜することにより前もって乾燥させる)を含む乾燥テトラヒドロフラン(7ml)を1時間にわたり滴下した。この反応混合物を、−78℃で3時間撹拌し、その後、2Nの塩酸(3.1ml,6.2mmol)を、10分間の間ゆっくりと滴下し、この撹拌反応混合物を放置して室温まで温めた。
2,4−ジメトキシ−ベンズアルデヒド(0.0415g、0.25mmol)および濃塩酸(2滴)を、4(S)−メチル−アナバセイン(0.036g、0.21mmol)を含む乾燥エタノール(1.5ml)溶液に添加し、この反応混合物(pH〜1)を82℃の閉鎖系中のアルゴン雰囲気下で16時間撹拌した。2日間4℃で冷蔵庫に保存した後、これを濾過し、冷却したエタノールを用いて乾燥アルゴン雰囲気下で洗浄し、その後、乾燥器中にて5酸化リン酸で乾燥させ、非常に吸湿性の高い純粋な産物(0.011g、収率13%)を得た。1H−NMR (300 MHz, DMSO−d6, delta, TMS int. standard): 8.90 (1H, dd, J = 4.8, 1.5 Hz), 8.84 (1H, d, J = 1.5 Hz), 8.10 (1H, dt, J = 7.8, 1.8 Hz), 7.70 (1H, dd, J = 7.8, 4.8 Hz), 7.66 (1H, d, J = 8.7 Hz), 7.27 (1H, s), 6.74 (1H, dd, J = 8.7, 2.4 Hz), 6.64 (1H, d, J = 2.4 Hz), 3.86 (3H, s), 〜3.8 (2H, under the water line), 3.69 (3H, s), 3.51−3.43 (1H, m), 2.21−1.89 (2H, m), 1.35 (3H, d, J = 6.6 Hz)。
1)dl−5−メチル−アナバセイン ビスハイドロクロライド
dl−5−メチル−ピペリド−2−オン(2.00g、17.7mmol)、水性ホルムアルデヒド溶液(37%、1.7ml、21mmol)、およびジエチルアミン(2.2ml、21mmol)を含む溶液を撹拌し、115℃の油槽中で8時間還流し、その後、良好な真空下、50℃にてrotavapor上で蒸発させた。粗N−ジエチルアミノメチル−5メチル−ピペリド−2−オン残留物(2.13g、10,7mmol)に、乾燥トルエン(10ml)、ニコチン酸エチル(1.62g、10.7mmol)および(部分的に)水素化ナトリウム(鉱油中60%、0.88g、22.0mmol)を添加し、4時間撹拌しながら還流させた。さらに水素化ナトリウム分散物(0.44g、11.0mmol)を反応混合物に添加して撹拌し、さらに4時間還流させた。室温まで冷却した後、過剰な水素化ナトリウムを(注意深く破壊されるように)濾過し、この濾液を氷で一晩冷却した。沈殿物質を濾過し、ヘキサンで洗浄し、その後、濃塩酸(5ml)およびアセトン(1ml)の混合物中で一晩煮沸した。冷却した後、分離した塩化ナトリウムを濾過により除去し、イソプロパノール(40ml)を添加し、3日間5℃で冷却し、この産物の二塩酸塩(0.17g、収率4%)をゆっくりと分離した。1H−NMR (300 MHz, DMSO−d6, delta, TMS int. standard): 9.27 (1H, dd, J = 2.1, 0.6 Hz), 8.91 (1H, dd, 5.1, 1.5 Hz), 8.56 (1H, dt, J = 8.1, 2.1 Hz), 7.80 (1H, ddd, J = 8.1, 5.1, 0.6 Hz), 3.29−3.07 (2H, m), 2.89−2.73 (1H, m), 2.70−2.59 (1H, m), 1.92−1.67 (2H, m), 1.60−1.45 (1H, m), 0.98 (3H, d, J = 6.6 Hz)。
dl−5−メチル−アナバセイン ビスハイドロクロライド(0.17g、0.64mmol)、2,4−ジメトキシベンズアルデヒド(0.14g、0.83mmol)を含むエタノール(5ml)溶液ならびに濃塩酸(2滴)を75℃の油槽上で一晩煮沸した。Rotavapor上で蒸発した後、残留物(0.35g)を水(5ml)中に溶解し、炭酸水素ナトリウム(0.5g)を飽和するまで添加し、この水性溶液をジクロロメタン(3×5ml)で抽出した。組み合わせた抽出物を(硫酸マグネシウムで)乾燥し、蒸発させ、残留物(0.17g)をベンゼン−ヘキサン―ジエチルアミン(9−4−1)でシリカゲル(25g)上でのカラムクロマトグラフィーにより精製して、精製産物(0.0616g、収率30%)を得た。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.75 (1H, dd, J = 2.1, 0.9 Hz), 8.61 (1H, dd, J = 4.8, 1.8 Hz), 7.82 (1H, dt, J = 4.8, 2.1 Hz), 7.30, (1H, ddd, J = 7.8, 4.8, 0.9 Hz), 7.25 (1H, d, J = 8.4 Hz), 6.78 (1H, s), 6.51 (1H, dd, J = 8.7, 2.4 Hz), 6.43 (1H, d, J = 2.4 Hz), 4.05 (1H, dm, J = 17.4 Hz), 3.82 (3H, s), 3.72 (3H, s), 3.35 (1H, dd, J = 17.4, 9.9 Hz), 2.90 (1H, dm, J = 15.0 Hz), 2.29−2.18 (1H, m), 1.96−1.81 (1H, m), 1.02 (3H, d, J = 6.6 Hz)。
1)N−BOC−6(S)−メチル−ピペリド−2−オン
酸化ルテニウム(IV)水和物(0.8g、0.006mol)、続いてN−BOC−6(S)−メチル−ピペリジン(4.00g、0.020mol)を含む酢酸エチル(240ml)溶液を、過ヨウ素酸ナトリウム(21.36g、0.100mole)を含む水溶液(200ml)に添加し、アルゴン雰囲気下、室温で24時間激しく撹拌した。分離した後、水性相を酢酸エチル(3×100ml)で抽出した。組み合わせた有機相を硫酸マグネシウムで乾燥させ、活性炭で処置し、得られた無色の溶液を30℃にてrotavapor上で蒸発させた。残留物(3.98g)を、シクロヘキサン−酢酸エチル(1−1、v/v)でシリカゲル(125g)カラムクロマトグラフィーにより精製し、産物(2.50g、収率59%)を得た。この産物は、1H−NMRスペクトラムにより次の反応ステップのために十分な純度であることが確認された。
全てのガラス製品を、24時間、120℃で、オーブン内で乾燥させ、この反応を、アルゴン雰囲気下で実行した。3−ブロモ−ピリジン(0.70g、4.4mmol)を含む乾燥エーテル(12ml)撹拌溶液を(ドライアイス/アセトンを用いて)−78℃まで冷却し、n−ブチルリチウム溶液(ヘキサン中1.6M、2.75ml、4.4mmol)を、撹拌の間液滴下し、その後、この溶液をさらに30分間撹拌した、この冷却した溶液にN−BOC−6(S)−メチル−ピペリド−2−オン(0.935g、4.4mmol、55℃にてrotavapor上で10mlの乾燥ベンゼンを蒸溜することによりあらかじめ乾燥させた)を含む乾燥テトラヒドロフラン(7ml)を1時間で滴下した。この反応混合物を−78℃で3時間撹拌し、その後、2Nの塩酸(4.4ml、8.8mmol)を、10分間にわたりゆっくりと滴下し、この撹拌反応混合物を放置して室温まで温めた。分離した後、この水性相をエーテル(3×15ml)で抽出し、組み合わせた有機溶液を、1×5mlの飽和型炭酸水素ナトリウムおよび2×5mlの鹹水で洗浄し、(硫酸マグネシウムで)乾燥し、30℃で蒸発させた。トリフルオロ酢酸(3.5ml)を、氷冷下で残留物(0.812g)に添加し、アルゴン雰囲気下で撹拌し、その後さらに4時間室温で撹拌した。30℃にてrotavapor上での蒸発の後、水性水酸化ナトリウム(40%)を残留物に氷冷下で撹拌しながらpH12となるまで滴下し、この物質をジエチルエーテル(1×10および4×5ml)で抽出した。組み合わせた有機溶液を(硫酸マグネシウムで)乾燥し、良好な真空下、30℃にてrotavapor上で蒸発させ、粗生成物(0.192g、25%)を得た。これより分析試料を、30%のトリエチルアミンを含有するシクロヘキサンを用いたシリカゲルのカラムクロマトグラフィーにより調製した。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.95 (1H, dd, J = 2.4, 0.6 Hz), 8.61 (1H, dd, J = 4.8, 1.5 Hz), 8.12 (1H, dt, J = 7.8, 2.1 Hz), 7.31 (1H, ddd, J = 8.1, 4.8, 0.9 Hz), 3.78−3.64 (1H, m), 2.65−2.59 (1H, m), 2.57−2.44 (1H, m), 2.02−1.68 (3H, m), 1.36 (3H, d, J = 6.9 Hz), 1.32−1.1.24 (1H, m)。
2,4−ジメトキシ−ベンズアルデヒド(0.0415g、0.25mmol)および濃塩酸(2滴)を、6(S)−メチル−アナバセイン(0.037g、0.21mmol)を含む乾燥エタノール(1.5ml)溶液に添加し、この反応混合物(pH〜1)を82℃の層内の閉鎖系中、アルゴン雰囲気下で16時間撹拌した。この純粋な産物の遊離塩基(0.021g、収率26%)を、溶出に90%ヘキサン―10%イソプロパノール溶媒を用いたアイソクラティックなシリカゲル分取HPLCにより得た。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.75 (1H, dd, J = 2.1, 0.6 Hz), 8.61 (1H, dd, J = 4.8, 1.8 Hz), 7.83 (1H, dt, J = 7.8, 2.1 Hz), 7.31 (1H, ddd, J = 7.8, 4.8, 0.6 Hz), 7.27 (1H, d, J = 7.2 Hz), 6.77 (1H, s), 6.50 (1H, dd, J = 8.4, 2.4 Hz), 6.42 (1H, d, J = 2.1 Hz), 3.83 (3H, s), 3.82−3.66 (1H, m), 3.71 (3H, s), 2.86 (1H, dm, 15.6 Hz), 2.69−2.54 (1H, m), 2.10−1.88 (1H, m), 1.55−1.42 (1H, m), 1.41 (3H, d, J = 6.6 Hz)。
1)dl−2,4−ジメチルピペリジン
オガワら(1984)の方法により、2,4−ジメチルピリジン(5.00g)を使用して2,4−ジメチルピペリジン(3.53g、収率67%)を調製した。この物質は、1H−NMRスペクトルにより、純度95%であり、シスおよびトランスアイソマーを約29〜71の比率で含む(Ogawa, K., Takeuchi, Y., Suzuki, H. and Nomura, Y. (1984). Barriers to Rotation and Inversion in meso−1,1’−Bi(2−メチルピペリジン)s. Journal of the American Chemical Society 106 (4),831−841参照)。
ジ−tert−ブチル ジカーボネート(6.95g、0.032mole)およびトリエチルアミン(4.06ml、0.0291mole)をdl−2,4−ジメチルピペリジン(3.53g、0.0312mole)を含む1,4−ジオキサン(40ml)−水(40ml)に添加し、この懸濁物を室温で24時間激しく撹拌した。この反応混合物を、2Nの塩酸を用いてpH4となるまで酸性化し、エーテル(4×25ml)で抽出した。組み合わせた有機抽出物を(硫酸マグネシウムで)乾燥し、30℃にてrotavapor上で蒸発させ、シクロヘキサンを含有する10%の酢酸エチルを用いたシリカゲルのカラムクロマトグラフィーにより精製して産物を得た(5.37g、収率81%)。この産物は次のステップのために十分な純度であった(1H−NMRスペクトルによると、シスおよびトランスアイソマーは、それぞれ22%および78%のモル比で含まれる)。
過ヨウ素酸ナトリウム(26.7g、0.125mole)を含む水溶液(250ml)に、室温にてアルゴン雰囲気下で激しく撹拌しながら、酸化ルテニウム(IV)水和物(1.0g、0.0075mole)を添加し、その後dl−N−BOC−2,4−ピペリジン(5.37g、0.025mole)を含む酢酸エチル(300ml)溶液を添加し、この混合物を24時間撹拌した。相を分離した後、水性相を酢酸エチル(3×100ml)で抽出した。組み合わせた有機相を(硫酸マグネシウムで)乾燥し、活性炭で処置して無色の溶液を得た。この溶液を30℃にてrotavapor上で蒸発させた。残留物(5.80g)を、30%の酢酸エチルを含有するシクロヘキサンを用いたシリカゲル(125g)のカラムクロマトグラフィーにより精製し、純粋な産物(2.61g、46%)を得た。この産物は、1H−NMRスペクトルによると、シスおよびトランスアイソマーで構成され、次のステップのために十分な純度であった。
全てのガラス製品は24時間120℃にてオーブン内で乾燥し、反応はアルゴン雰囲気下で実行した。N−ブチルリチウム溶液(ヘキサン中1.6M、7.15ml、11.4mmol)を、−78℃まで(ドライアイス/アセトンで)冷却した3−ブロモ−ピリジン(1.80g、11.4mmol)を含む乾燥エーテルに、撹拌しながら滴下し、この溶液をさらに30分撹拌した。この冷却溶液にdl−N−BOC−4,6−ジメチルピペリド−2−one(2.59g、11.4mmol、55℃にてrotavapor上の12mlの乾燥ベンゼンを蒸溜することによりあらかじめ乾燥させた)を含む乾燥テトラヒドロフラン(20ml)を、1時間にわたり滴下した。その後、この反応混合物を、−78℃で3時間撹拌した。その後、2Nの塩酸(11.5ml、23.0mmol)を、20分間にわたりゆっくりと添加し、この撹拌混合物を放置して室温まで温めた。分離した後、水性相をエーテル(3×40ml)で抽出し、組み合わせた有機溶液を(硫酸マグネシウムで)乾燥し、30℃で蒸発させた。雰囲気下。トリフルオロ酢酸(11ml、144mmol)を氷冷中アルゴン下で撹拌しながらこの残留物(2.25g)に添加し、その後、一晩室温で撹拌した。30℃にてrotavapor上で蒸発させた後、水性水酸化ナトリウム溶液(40%)を残留物に冷却下で撹拌しながらpH10〜11となるまで滴下し、その後ジクロロメタン(10×20ml)で抽出した。組み合わせた有機溶液を(硫酸マグネシウムで)乾燥させ、良好な真空下、30℃にてrotavaporで蒸発させ、粗シス―トランスアイソマー混合物(1.34g、7.12mmol、収率62%)を得た。この産物は、低温下以外では不安定であり(−78℃で保存できる)、精製することなく次のステップに使用した。
2,4−ジメトキシ−ベンズアルデヒド(0.069g、0.345mmol)および濃塩酸(2滴)を4,6−ジメチルアナバセインアイソマー混合物(0.065g、0.345mmol)を含む乾燥エタノール溶液(2ml)に添加し、この反応混合物(pH〜1)を、82℃の槽の閉鎖系において、アルゴン雰囲気下で16時間撹拌した。この物質を、30℃にてrotavapor上で蒸発させた。その後、水(0.5ml)、炭酸水素カリウム(0.2g)、およびクロロホルム(1ml)を残留物に添加し、その後、2つの相に分離した。水性相をクロロホルム(3×1ml)で抽出した。組み合わせた有機相を、(硫酸マグネシウムで)乾燥し、30℃にてrotavapor上で蒸発させた。残留物(0.130g)を、エーテルを含む10%のトリエチルアミン収集画分2〜3mlを用いシリカゲル上でカラムクロマトグラフィーを行い、2〜3mlで分画した。
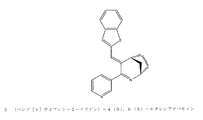
2滴の濃HClおよび4−アミノベンズアルデヒド(0.030g、0.25mmol)を含むエタノール2.0mlを6(S)−メチル−アナバセインの遊離塩基(0.021g、0.12mmol)に添加し、この溶液を70℃で一晩加熱した。その後、溶媒をrotavaporで除去し、結果として得られる残留物を、5mlの水に溶解し(得られるpHは2.5である)、その後、4×15mlのジエチルエーテルで抽出して、副産物および反応していないアルデヒドを除去した。その後、水性溶液のpHを>12まで上昇させ、次いで3×15mlのジエチルエーテルで再び抽出し、かつクロロホルムで一度抽出した。結果として得られる有機相をrotavaporで蒸発させ、シリカゲルカラムの分取HPLCにより得られる残留物から望ましい産物を得た(0.008g、収率21%)。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.72 (1H, s), 8.61 (1H, d, J = 4.5 Hz), 7.80 (1H, dm, J = 7.8 Hz), 7.31 (1H, dd, J = 7.8, 5.1 Hz), 7.15 (2H, d, J = 8.4 Hz), 6.65 (2H, d, J = 8.4 Hz), 6.50 (1H, s), 3.71−3.57 (1H, m), 2.95 (1H, dm, J = 16.5 Hz), 2.79−2.65 (1H, m), 2.02−1.92 (1H, m), 1.51−1.34 (1H, m), 1.41 (3H, d, J = 6.9 Hz). HRMS (+E SI): theoretical for [M+H]+ = 278.1652; found: 278.1654。
2滴の濃HClおよび4−アミノベンズアルデヒド(0.033g、0.27mmol)を含むエタノール2.0mlを4(R),6(S)−エチレン−アナバセインの遊離塩基(0.025g、0.134mmol)に添加し、この溶液を70℃で一晩加熱した。その後、溶媒を、rotavaporを用いて除去し、結果として得られる残留物を5mlの水に溶解し(得られるpHは2.5である)、4×15mlのジエチルエーテルで抽出して、副産物および反応していないアルデヒドを除去した。それから水性溶液のpHを、>12まで上昇させ、その後、3×15mlのジエチルエーテルで再び抽出し、クロロホルムで1度抽出した。結果として得られる有機相を、rotavaporで蒸発させ、シリカゲルカラム上の分取HPLCにより得られる残留物から望ましい産物を得た(0.0101g、収率26%)。1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.66−8.77 (2H, m), 7.71 (1H, d, J = 7.8 Hz), 7.32 (1H, dd, J = 7.5, 5.1 Hz), 7.14 (2H, d, J = 8.7 Hz), 6.65 (2H, d, J = 8.4 Hz), 6.33 (1H, s), 4.50 (1H, s), 3.61 (1H, t, J = 5.1 Hz), 2.29−2.12 (1H, m), 2.09−1.95 (2H, m), 1.93−1.80 (2H, m), 1.69−1.60 (1H, m). HRMS (+E SI): theoretical for [M+H]+ = 290.1652; found: 290.1654。
以下のアリーリデン−エチレン−アナバセインを、dl−4,6−エチレン−アナバセインの塩酸塩(第1の化合物)または4(R),6(S)−エチレン−アナバセインの塩酸塩(残存化合物)のいずれかと、わずかに過剰モル量である適切に置換されたベンズアルデヒドを含む酸性エタノールとを使用して合成した。これらの遊離塩基を、SG−クロマトグラフィーにより得た。
1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.68 (1H, dd, J = 2.4, 0.9 Hz), 8.61 (1H, dd, J = 4.8, 1.5 Hz), 7.75 (1H, dt, J = 7.5, 1.5 Hz), 7.31 (1H, ddd, J = 7.8, 5.1, 0.9), 7.24 (1H, dd, J = 8.4, 0.6 Hz), 6.55 (1H, s), 6.50 (1H, dd, J = 8.4, 2.7 Hz), 6.43 (1H, d, J = 2.4 Hz), 4.52−4.47 (1H, m), 3.47 (1H, t, J = 5.4 Hz), 2.28−2.13 (1H, m), 2.07−1.95 (2H, m), 1.92−1.80 (2H, m), 1.67−1.59 (1H, m)。 13C−NMR (75 MHz, CDCl3, delta, TMS int. standard): 165.4, 161.0, 158.7, 148.4, 149.3, 139.7, 136.2, 135.7, 130.4, 127.8, 122.8, 117.5, 104.0, 98.2, 60.2, 55.38, 55.36, 36.4, 36.3, 23.6, 31.2。
1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.65−8.60 (2H, m), 7.71 (1H, dt, J = 7.5, 1.8 Hz), 7.33 (1H, dd, J = 7.5, 4.8 Hz), 7.05−6.93 (2H, m), 6.92−6.84 (2H, m), 6.56 (1H, d, J = 15.3 Hz), 6.14 (1H, d, J = 11.4 Hz), 4.55 (1H, m), 3.92 (3H, s), 3.54 (1H, t, J = 4.8), 2.31−2.00 (2H, m), 1.98−1.81 (2H, m), 1.75−1.64 (2H, m)
1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.65−8.60 (2H, m), 7.70 (1H, dt, J = 8.1, 2.1 Hz), 7.32 (1H, ddd, J = 7.8, 4.8, 0.9 Hz), 6.44 (1H, d, J = 3.3 Hz), 6.38 (1H, d, J = 3.3 Hz), 6.15 (1H, s), 4.59−4.54 (1H, m), 3.91 (1H, t, J = 4.8 Hz), 2.25−2.12 (1H, m), 2.11−1.98 (1H, m), 1.95−1.82 (2H, m), 1.79−1.63 (2H, m)。
1H−NMR (300 MHz, CDCl3, delta, TMS int. standard): 8.69−8.64 (2H, m), 7.83−7.70 (3H, m), 7.39−7.30 (3H, m), 7.30−7.24 (1H, m), 6.64 (1H, s), 4.65−4.59 (1H, m), 3.96 (1H, t, J = 5.4 Hz), 2.36−2.21 (1H, m), 2.19−2.02 (1H, m), 2.01−1.87 (2H, m), 1.87−1.69 (2H, m)。
本発明の実施は、特に他の記載がない限り、分子生物学(組み換え技術を含む)、微生物学、細胞生物学、生化学、薬理学、および免疫学の従来技術を使用し、これらの技術は当業者に良く知られている。
オスのスプラーグドーリーラットの全脳(Pel−Freeze Biologicals、アリゾナ州ロジャース)を、結合食塩水(120mMのNaCl、5mMのKCl、2mMのCaCl2、1mMのMgCl2、50mMのTris−TrisHCl緩衝剤、pH7.4)中で、Wheaton社(場所)製の30mlのホモジナイズ用ガラス管および内筒を用いてホモジナイズした。ホモジネートを、10分間、11,000rpm(変換するまで、×G)で遠心した後、結果として得られるペレットを、新鮮な結合食塩水中に再懸濁し、ホモジナイズし再び遠心した。その後、タンパク質アッセイ(BCA、Pierce、イリノイ州ロックフォード)を実施して、ペレットに含まれるラットの脳膜のタンパク質濃度を得た。この物質を、使用まで−85℃で保存した。
Y=Bottom+(Top−Bottom)/(1+10(LogIC50−X)n)
ここで、Top=曲線の最上部での放射性リガンドの最大特異的結合であり、Bottom=高濃度の置換リガンドで観察される最小特異的結合である。放射性リガンドに対するIC50値およびKd値(125I−αBtxに対して0.32nM)は、同じnAChR含有膜を使用して前もって決定し、その後、チェン−プルソフ式(Ki=IC50/(1+(放射性リガンド)/Kd)を使用して置換リガンドの平衡解離定数(Ki)を計算するために使用した。表1に示される各化合物のα7結合選択性は、α4β2結合に対するKiをα7結合に対するKiで割ることにより推定した。3−(2,4−ジメトキシベンジリデン)−アナバセイン(DMXBA)(表1)と比べた各化合物のα7結合選択性は、α4β2結合に対するKiをα7結合に対するKiで割り、この値を測定されたDMXBAのα7選択性(Kem et al., 2004 Mol. Pharmacol. 65, page 62の表3に報告される値は1.95)で割ることにより計算した。
2つの主要なラット脳のnAChRに対する、合成化合物の結合親和性を表1に示す。アナバセイン(化合物1)は、ラットα7nAChRに対しておよそ4倍低い結合親和性(より高いKi)を示した(選択性因子=0.26)のに対し、DMXB−アナバセイン(化合物2)は、α4β2nAChRと比べα7nAChRに対してわずかに高い親和性(選択性因子=1.91)を示した。この特許出願で請求される化合物を全く含まない45 3−(ベンジリデン)‐アナバセインの試験において、α7nAChR選択性の平均値は1.8であった(Slavovら、2010)。
本明細書中で以下の書類を引用する。
Herscovici, J., Egron, M−J., and Antonakis, K. (1982). New Oxidative Systems for Alcohols: Molecular Sieves with Chromium(VI) Reagents. J. Chem. Soc. Perkin Trans. I, 1967−1973.
Kawamura, M. and Ogasawara, K. 1995). Enantio− and Stereocontrolled Syntheses of (−)−Semburin, (+)−N−Benzoylmeroquinene Aldehyde, (−)−Antirhine, and (+)−Isocorynantheol from Common (+)−Norcamphor. Tetrahedron Letters 36, 3369−3372.
Kem, W. R., Mahnir, V. M., Prokai, L., Papke, R. M., Cao, X. F., LeFrancois, S., Wildeboer, K., , Porter−Papke, J., Prokai−Tatrai, K., and Soti, F. (2004). Hydroxy metabolites of the Alzheimer’s drug candidate DMXBA (GTS−21): Their interactions with brain nicotinic receptors, and brain penetration. Mol. Pharmacol. 65, 56−67.
Krow, G. R., Cheung, O. H., Hu, Z., and Lee, Y. B. (1996). Regioselective Functionalization. 6. Migratory Preferences in Hydroxylamine−O−sulfonic Acid and Schmidt Rearrangements of 7−Substituted Norcamphors. J. Org. Chem. 61, 5574−5580.
Lattanzi, A., Iannece, P., and Scettri, A. (2004). Synthesis of a renewable hydroperoxide from (+)−norcamphor: influence of steric modifications of the bicyclic framework on asymmetric sulfoxidation. Tetrahedron: Asymmetry 15, 1779−1785.
Slavov, S.H., Radzvilovits, M., LeFrancois, S., Stoyanova−Slavova, I.B., Soti, F., Kem, W.R., Katrizky, A.R. (2010) A computational study of the binding of 3−(arylidene) anabaseines to two major brain nicotinic acetylcholine receptors and to the acetylcholine binding protein. Eur. J. Med. Chem. 45, 2433−2446.
Claims (66)
- 式(I)
式中、
R1が、発生時に独立して、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール−O−、アリール、または5〜6員のヘテロアリールであり;または2つのR1基が、付着した結合と共に、5〜8員環を形成し;
nが、0、1、2、3、または4であり;
各a1、a2、およびa3が、0または1であり、a1、a2、およびa3の内の2つが0であり、それ以外が1であり;
R2が、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
EおよびGが、それぞれ独立して、何もない状態か、またはヘテロ(C0−C3)アルキル−、(C1−C3)アルキレン、または(C2−C3)アルケニレンであり、EおよびGが、同時に何もない状態とすることができず;
Aが結合または
n´が、0、1、または2であり;
R4およびR5が、発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Xが、アリールまたはヘテロアリールであって、前記アリールおよび前記ヘテロアリールが、任意に、1〜5つのR3基および/または1つのRc基により置換され;
R3が、発生時に独立して、水素、ハロゲン、(C1−C3)アルキル−C(O)O−、(C1−C3)アルキル−C(O)−、(C1−C3)アルキル−C(O)N(R6)、N(R6)2−、(R6)2NC(O)−、(R6)2N(C1−C5)アルコキシ、
R6が、発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Raが、発生時に独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール、またはアリール−O−であり;
Rcが、水素、(C1−C5)アルコキシ、または(C1−C5)アルキルであり、前記(C1−C5)アルコキシおよび前記(C1−C5)アルキルが、ヒドロキシル、(C1−C3)アルコキシ、ハロゲン、およびチオからなる群から選択される1つ以上の同一もしくは異なる置換基により任意に置換され;かつ
bが、0、1、2、3、または4である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはその組み合わせ。 - 前記化合物が、式(IA)
式中、
R1が、発生時に独立して、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール−O−、アリール、または5〜6員のヘテロアリールであり、または2つのR1基が、付着した結合と共に、5〜8員環を形成し;
nが、0、1、2、3、または4であり;
R2が、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
EおよびGが、それぞれ独立して、何もない状態、−ヘテロ(C0−C3)アルキル−、(C1−C3)アルキレン、(C2−C3)アルケニレンであり、EおよびGが、同時に何もない状態とすることができず;
Aが、結合または
n´が、0または1であり;
R4およびR5が、発生時にそれぞれ独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Xが、アリールまたはヘテロアリールであって、前記アリールおよび前記ヘテロアリールが、任意に、1〜5つのR3基および/または1つのRc基により置換され;
R3が、発生時にそれぞれ独立して、水素、ハロゲン、(C1−C3)アルキル−C(O)O−、(C1−C3)アルキル−C(O)N(R6)、N(R6)2−、(R6)2NC(O)−、(R6)2NC1−C5)アルコキシ、
R6が、発生時にそれぞれ独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、または(C1−C3)アルコキシであり;
Raが、発生時にそれぞれ独立して、水素、(C1−C3)アルキル、ヒドロキシ(C1−C3)アルキル、(C1−C3)アルコキシ、シアノ、ハロゲン、アリール、またはアリール−O−であり;
Rcが、水素、または、ヒドロキシル、(C1−C3)アルコキシ、ハロゲン、およびチオから選択される1つ以上の同一もしくは異なる置換基により任意に置換される(C1−C5)アルキルであり;かつ
bが、0、1、2、3、または4である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはその組み合わせ。 - 各EおよびGが何もない状態か、(C1−C3)アルキレン、または(C2−C3)アルキレンであり、ただし、EおよびGが、同時に両方が何もない状態とすることができない、請求項2に記載の化合物。
- 前記化合物が、式(IIa)
- Xが、1〜5つのR3基により任意に置換されるアリール基である、請求項4に記載の化合物。
- bが0である、請求項5に記載の化合物。
- 前記化合物が、式(III−A)
- Xが、1つまたは2つのR3基により任意に置換されるアリール基である、請求項7に記載の化合物。
- R2が水素であり、かつXが、1つまたは2つのR3基により置換されるフェニル基である、請求項8に記載の化合物。
- nが0であり、かつR3が、アミノ、(C1−C3)アルコキシまたはヒドロキシルである、請求項9に記載の化合物。
- 前記化合物が、
である、請求項10に記載の化合物。 - 前記化合物が、式(III−B)
式中、
請求項6に記載の化合物。 - 前記化合物が、式(III−C)
- Xが、1つまたは2つのR3基により任意に置換されるアリール基である、請求項13に記載の化合物。
- nが0であり、R2が水素であり、かつXが、2つの(C1−C3)アルコキシ基により置換されるフェニル基である、請求項14に記載の化合物。
- 前記化合物が、
である、請求項15に記載の化合物。 - Xが、任意に置換されるヘテロアリール基である、請求項4に記載の化合物。
- 前記化合物が、式(III―D)
式中、
Wが、OまたはSであり、かつ
請求項4に記載の化合物。 - bが0であり、かつR2がHである、請求項18に記載の化合物。
- R3が、発生時にそれぞれ独立して、ヒドロキシル、アリール、アルキルアミノ、ジアルキルアミノ、またはアルコキシである、請求項19に記載の化合物。
- 前記化合物が、式(III−E)
Rcが、水素、または、ヒドロキシル、(C1−C3)アルコキシ、およびハロゲンからなる群から選択される1つ以上の同一もしくは異なる置換基により任意に置換される(C1−C5)アルキルであり;かつ
R1、R2、R3、Ra、b、およびnが、請求項2に定義されている、
請求項4に記載の化合物。 - Xが、1〜5つのR3基により置換されるアリールであり、1つのR3基が、その糖部または誘導体である、請求項3に記載の化合物。
- 前記化合物が、式(IIb)
式中、
請求項2に記載の化合物。 - 前記化合物が、式(IIc)
式中、
請求項2に記載の化合物。 - Xが、任意に置換されるアリールである、請求項24に記載の化合物。
- bが0であり、かつEおよびGの内の1つが、何もない状態、(C1−C3)アルキレン、または(C2−C3)アルキニレンであり、もう一方が、―ヘテロ(C0−C3)アルキルである、請求項25に記載の化合物。
- 前記化合物が、式(i)
- 前記化合物が、式(ii)
- 前記化合物が、式(iii)
- 前記化合物が、式(iv)
- 前記化合物が、式(IB)
式中、
請求項1に記載の化合物。 - EおよびGの内の1つが何もない状態であり、かつもう一方が、−CH2−またはヘテロ原子である、請求項31に記載の化合物。
- EおよびGの内の1つが、(C1−C3)アルキレンまたは(C2−C3)アルケニレンであり、かつもう一方が、ヘテロ(C0−C3)アルキルである、請求項31に記載の化合物。
- EおよびGの内の1つが、CH2−、−CH2−CH2−、−CH=CH−、−CH=CH−CH2−、および−CH2−CH2−CH2−からなる群から選択され、かつもう一方が、ヘテロ原子または−Het−CH2−である、請求項33に記載の化合物。
- 前記化合物が、式(v)
- 前記化合物が、式(vi)
- 前記化合物が、式(vii)
- 前記化合物が、式(viii)
- 前記化合物が、式(IC)
式中、
請求項1に記載の化合物。 - EおよびGの内の1つが何もない状態であり、かつもう一方が―CH2−またはヘテロ原子である、請求項39に記載の化合物。
- EおよびGの内の1つが、(C1−C3)アルキレンであり、かつもう一方が、ヘテロ(C0−C3)アルキルである、請求項39に記載の化合物。
- EおよびGの内の1つが、−CH2−、−CH2−CH2−、および−CH2−CH2−CH2の群から選択され、かつもう一方が、ヘテロ原子または−Het−CH2−である、請求項41に記載の化合物。
- 前記化合物が、式(ix)
- 前記化合物が、式(x)
- 前記化合物が、式(xi)
- 前記化合物が、式(xii)
- 式(IV)
式中、
nが、1、2、3、4、または5であり;
n´が、1、2、または3であり;
R1が、独立して、アミノ、C1−C3アルキル、またはC1−C3アルコキシであり;
R2が、独立して、C1−C3アルキルであり、少なくとも1つのR2が、4、5、または6の位置で存在し;
ただし、nが2であり、R1が両方ともメトキシである場合、n´が2である、
化合物、またはその薬学的に許容可能な塩、溶媒和物、水和物、クラスレート、多形体、ステレオアイソマー、エナンチオマー、またはそれらの組み合わせ。 - R1が、独立してアミノまたはメトキシであり、かつnが1または2である、請求項47に記載の化合物。
- R2がメチルであり、かつn´が1または2である、請求項47に記載の化合物。
- 前記化合物が、
である、請求項47に記載の化合物。 - 前記化合物が、α7ニコチン性アセチルコリン受容体アゴニストまたはアンタゴニストである、請求項1〜50のいずれか1項に記載の化合物。
- 前記化合物が、α4β2ニコチン性アセチルコリン受容体と比較してα7ニコチン性アセチルコリン受容体に選択的に結合する、請求項1〜50のいずれか1項に記載の化合物。
- 前記化合物が、α7ニコチン性アセチルコリン受容体と比較してα4β2ニコチン性アセチルコリン受容体に選択的に結合する、請求項1〜50のいずれか1項に記載の化合物。
- 前記化合物が、α7ニコチン性アセチルコリン受容体を選択的に活性化する、請求項1〜50のいずれか1項に記載の化合物。
- 前記化合物が、α7ニコチン性アセチルコリン受容体を選択的に阻害する、請求項1〜50のいずれか1項に記載の化合物。
- その必要のある対象の神経系疾患または障害を治療または予防する方法であって、請求項1〜50のいずれか1項に記載の化合物の有効量を前記対象に投与することを含む、方法。
- 前記神経系疾患または障害が、統合失調症、アルツハイマー病、パーキンソン病、薬物依存、および中毒から選択される、請求項56に記載の方法。
- 前記化合物が、式(III−A)または式(IV)の化合物である、請求項56に記載の方法。
- 前記化合物が、(1S,5R,E)−4−(2,4−ジメトキシベンジリデン)−3−(ピリジン−3−イル)−2−アザビシクロ[3.2.1]オクト−2−エンまたは3−(4−アミノベンジリデン)−4(R),6(S)−エチレン−アナバセインである、請求項58に記載の方法。
- その必要のある対象の疾患または障害を治療または予防する方法であって、請求項1〜50のいずれか1項に記載の化合物の有効量を前記対象に投与することを含み、前記疾患または障害が、α7ニコチン性アセチルコリン受容体活性に関連する、方法。
- 前記疾患または障害が、炎症または癌である、請求項60に記載の方法。
- 神経系疾患または障害の治療または予防のための医薬組成物であって、請求項1〜50のいずれか1項に記載の化合物の治療上有効量および薬学的に許容可能な賦形剤を含む医薬組成物。
- 前記化合物が、式(III−A)または式(IV)の化合物である、請求項62に記載の組成物。
- 神経系疾患または障害の治療または予防に使用するキットであって、請求項1〜50のいずれか1項に記載の化合物の治療上有効量およびそれらの使用のための指示書を含むキット。
- 細胞においてα7ニコチン性受容体を選択的に刺激する方法であって、請求項1〜50のいずれか1項に記載の化合物の有効量と前記細胞を接触させることを含む方法。
- 細胞中においてα7ニコチン性受容体を選択的に阻害する方法であって、請求項1〜50のいずれか1項に記載の化合物の有効量と前記細胞を接触させることを含む方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161569539P | 2011-12-12 | 2011-12-12 | |
| US61/569,539 | 2011-12-12 | ||
| PCT/US2012/068943 WO2013090260A1 (en) | 2011-12-12 | 2012-12-11 | Nicotinic receptor targeted compounds and compositions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2015504861A true JP2015504861A (ja) | 2015-02-16 |
| JP2015504861A5 JP2015504861A5 (ja) | 2016-02-04 |
Family
ID=48613106
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014546185A Pending JP2015504861A (ja) | 2011-12-12 | 2012-12-11 | ニコチン性受容体標的化合物および組成物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9150558B2 (ja) |
| EP (1) | EP2791135B8 (ja) |
| JP (1) | JP2015504861A (ja) |
| AU (1) | AU2012352510A1 (ja) |
| CA (1) | CA2858720A1 (ja) |
| WO (1) | WO2013090260A1 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130231290A1 (en) * | 2010-11-18 | 2013-09-05 | Dignity Health | Methods of diagnosing and treating neurodegenerative diseases |
| CN105985410B (zh) * | 2015-02-16 | 2019-11-29 | 海南大学 | 芋螺肽、其药物组合物及用途 |
| JP6590226B2 (ja) * | 2016-05-11 | 2019-10-16 | 株式会社シード探索研究所 | オキサアジリジン化合物およびその製造方法 |
| US11096932B2 (en) | 2016-09-29 | 2021-08-24 | The Uab Research Foundation | Methods and compositions for increasing mucus clearance |
| WO2021113311A1 (en) * | 2019-12-02 | 2021-06-10 | Bcell Solutions, Inc. | Treatment of cancer using acetylcholine modulation and immunotherapy |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992001688A1 (en) * | 1990-07-23 | 1992-02-06 | Pfizer Inc. | Quinuclidine derivatives |
| WO2001023385A2 (en) * | 1999-09-27 | 2001-04-05 | Georgetown University | Novel tropane analogs |
| WO2004019943A1 (en) * | 2002-08-30 | 2004-03-11 | Memory Pharmaceuticals Corporation | Anabaseine derivatives useful in the treatment of neurodegenerative diseases |
| WO2006133303A1 (en) * | 2005-06-07 | 2006-12-14 | University Of Florida Research Foundation, Inc. | Alpha 7 nicotinic receptor selective ligands |
| WO2008095944A1 (en) * | 2007-02-06 | 2008-08-14 | Eisai R&D Management Co. Ltd. | 7-azaindole derivatives and their use in the inhibition of c-jun n-terminal kinase |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0724570B1 (en) * | 1993-10-21 | 1999-03-03 | G.D. Searle & Co. | Amidino derivatives useful as nitric oxide synthase inhibitors |
-
2012
- 2012-12-11 JP JP2014546185A patent/JP2015504861A/ja active Pending
- 2012-12-11 CA CA2858720A patent/CA2858720A1/en not_active Abandoned
- 2012-12-11 AU AU2012352510A patent/AU2012352510A1/en not_active Abandoned
- 2012-12-11 WO PCT/US2012/068943 patent/WO2013090260A1/en active Application Filing
- 2012-12-11 EP EP12857224.5A patent/EP2791135B8/en active Active
- 2012-12-11 US US14/364,499 patent/US9150558B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992001688A1 (en) * | 1990-07-23 | 1992-02-06 | Pfizer Inc. | Quinuclidine derivatives |
| WO2001023385A2 (en) * | 1999-09-27 | 2001-04-05 | Georgetown University | Novel tropane analogs |
| WO2004019943A1 (en) * | 2002-08-30 | 2004-03-11 | Memory Pharmaceuticals Corporation | Anabaseine derivatives useful in the treatment of neurodegenerative diseases |
| WO2006133303A1 (en) * | 2005-06-07 | 2006-12-14 | University Of Florida Research Foundation, Inc. | Alpha 7 nicotinic receptor selective ligands |
| WO2008095944A1 (en) * | 2007-02-06 | 2008-08-14 | Eisai R&D Management Co. Ltd. | 7-azaindole derivatives and their use in the inhibition of c-jun n-terminal kinase |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2791135B8 (en) | 2018-07-18 |
| EP2791135A4 (en) | 2015-10-14 |
| US9150558B2 (en) | 2015-10-06 |
| CA2858720A1 (en) | 2013-06-20 |
| EP2791135B1 (en) | 2018-06-06 |
| AU2012352510A1 (en) | 2014-07-17 |
| EP2791135A1 (en) | 2014-10-22 |
| WO2013090260A1 (en) | 2013-06-20 |
| US20140343042A1 (en) | 2014-11-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6880260B2 (ja) | ムスカリン受容体アゴニスト | |
| CN107406449B (zh) | 作为毒蕈碱m1受体和/或m4受体的激动剂的螺环化合物 | |
| JP6876675B2 (ja) | ムスカリンアゴニスト | |
| JP6847093B2 (ja) | ムスカリン様アゴニスト | |
| TWI631097B (zh) | 作爲s1p調節劑及/或atx調節劑之化合物 | |
| JP6047563B2 (ja) | 新規トリフルオロメチル−オキサジアゾール誘導体および疾患の治療におけるその使用 | |
| JP2021193128A (ja) | 補体媒介障害の治療のためのアリール化合物、ヘテロアリール化合物及び複素環式化合物 | |
| JP6843114B2 (ja) | ムスカリン作動薬 | |
| TW200911254A (en) | Oxadiazole derivatives and their use as metabotropic glutamate receptor potentiators-842 | |
| JP2021511342A (ja) | 癌処置のためのbcl−2タンパク質分解剤 | |
| HUT65771A (en) | Process for producing substituted 3-amino-quinuclidine derivatives and pharmaceutical compositions containing them | |
| US20230227405A1 (en) | 5-oxopyrrolidine-3-carboxamides as nav1.8 inhibitors | |
| TWI822803B (zh) | 醫藥化合物 | |
| JPH08169884A (ja) | シクロプロパクロメンカルボン酸誘導体 | |
| TW200911255A (en) | Metabotropic glutamate receptor oxadiazole ligands and their use as potentiators-841 | |
| JPH11512443A (ja) | 新規な置換アザ環式またはアザ二環式化合物 | |
| JP2015504861A (ja) | ニコチン性受容体標的化合物および組成物 | |
| JP6837431B2 (ja) | ベータ2アドレナリンアゴニスト活性およびm3ムスカリンアンタゴニスト活性を有する新規の二環式誘導体 | |
| TR201907763T4 (tr) | Ror-gama-t modülatörleri olarak triflorometil alkoller. | |
| EP3891143B1 (en) | Quinolinone and benzoxazine derivatives as muscarinic m1 and/or m4 receptor agonists | |
| US20220380338A1 (en) | Cycloalkyl 3-oxopiperazine carboxamides and cycloheteroalkyl 3-oxopiperazine carboxamides as nav1.8 inhibitors | |
| JP6483105B2 (ja) | ピペラジン誘導体および医薬としてのその使用 | |
| JP2024517498A (ja) | 化合物 | |
| CA3102762A1 (en) | Eaat2 activators and methods of using thereof | |
| KR20150021120A (ko) | 헤테로아릴 화합물 및 이의 이용 방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20151207 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20151207 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160614 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20160616 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20170411 |