JP2012515222A - 組換えヒトアルブミン−ヒト顆粒球コロニー刺激因子融合タンパク質の安定な製剤 - Google Patents
組換えヒトアルブミン−ヒト顆粒球コロニー刺激因子融合タンパク質の安定な製剤 Download PDFInfo
- Publication number
- JP2012515222A JP2012515222A JP2011546399A JP2011546399A JP2012515222A JP 2012515222 A JP2012515222 A JP 2012515222A JP 2011546399 A JP2011546399 A JP 2011546399A JP 2011546399 A JP2011546399 A JP 2011546399A JP 2012515222 A JP2012515222 A JP 2012515222A
- Authority
- JP
- Japan
- Prior art keywords
- neug
- neutropenia
- subject
- dose
- albumin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/38—Albumins
- A61K38/385—Serum albumin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/193—Colony stimulating factors [CSF]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/38—Albumins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/643—Albumins, e.g. HSA, BSA, ovalbumin or a Keyhole Limpet Hemocyanin [KHL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
Abstract
Description
白血球減少症は、循環する白血球(WBC)の減少であり、多くの場合、WBC数4000/mL未満と規定される。白血球減少症に関与する主な細胞は、好中球である。しかしながら、リンパ球、単球、好酸球、または好塩基球の数の減少も総白血球数の低下に寄与する場合がある(メルクマニュアル17版(非特許文献1))。
正常より低い白血球数を特徴とする症状の治療、改善、および予防に有用な方法および組成物を本明細書に記載する。このような症状としては、白血球減少症および好中球減少症が挙げられるが、これらに限定されるわけではない。
本明細書には、白血球数の低下を特徴とする症状および病気を治療、予防、および改善する組成物および方法が開示される。本明細書に記載の方法および組成物は、ヒト血清アルブミンタンパク質(「HSA」)およびヒト顆粒球コロニー刺激因子(「G-CSF」)から形成された融合ポリペプチドを含む。好ましい一態様では、融合ポリペプチドはアミノ酸759個の長さがあり、当該融合物のアミノ酸1〜585は、成熟型HSAからのアミノ酸に対応し、当該融合物のアミノ酸586〜759は、成熟型ヒトG-CSFのアミノ酸に対応する。融合タンパク質のアミノ酸配列を図1に示す。
本明細書では、「ポリヌクレオチド」は、少なくとも1個の顆粒球コロニー刺激因子(G-CSF)分子(またはその断片もしくは変種)にインフレームで結合した少なくとも1個のアルブミン分子(またはその断片もしくは変種)を含むかあるいはそれらからなる融合タンパク質をコードするヌクレオチド配列を有する核酸分子を指す。
顆粒球コロニー刺激因子(G-CSF)は、好中球の産生を促す造血成長因子である。刺激される生存前駆細胞がある場合、G-CSFの投与は、好中球の増加を迅速に誘導する。G-CSFの他の重要なインビボ活性は、末梢血中への造血前駆細胞の動員である(Duhrsen et al., 1988; Molineux et al, 1999; Roberts et al, 1994)。この効果は、好中球系統だけを含むものではなく、他の単一系統および多系統前駆細胞ならびに造血多能性幹細胞にまで及ぶ(Molineux et al, 1999)。G-CSFはまた、好中球をプライミングすることによって感染に対する防衛機構の一部である細胞事象を高め、これによって、オプソニン化黄色ブドウ球菌に対する食作用および抗菌作用の両方が高まる。G-CSFはまた、好中球および単球の走化性および好中球の接着性を誘導する(Yuo et al., 1989; Wang et al., 1988)。
ヒト血清アルブミン(HSAまたはHA)は、ヒト循環系で最も一般的な天然の血液タンパク質であり、1リットル当たり約40グラムのアルブミンが測定され、20日以上の間循環系に存続する。アルブミンは、生理的濃度で最小限の活性を有する担体タンパク質である。HSAは、酵素的または免疫的な機能を有さないが、生体内に広く分布し、血液中で様々な物質(例えば、ホルモン、脂肪酸、非抱合ビリルビンなど)の担体となることが知られている(Yeh et al., 1992)。HSAおよび組換えHA(rHA)はいずれも、ヒトにおいて同様の長い血中半減期を有する。
A.断片
本発明はさらに、G-CSFタンパク質の、アルブミンタンパク質の、および/または本発明のアルブミン融合タンパク質の断片に関する。本発明はまた、G-CSFタンパク質の、アルブミンタンパク質の、および/または本発明のアルブミン融合タンパク質の断片をコードするポリヌクレオチドに関する。タンパク質のN末端から1個以上のアミノ酸が欠失して、G-CSFタンパク質、アルブミンタンパク質、および/または本発明のアルブミン融合タンパク質の1つ以上の生物学的機能が修飾または喪失しても、他の治療的活性および/または機能的活性(例えば、生物学的活性、多量体化能、リガンド結合能)はなお保持され得る。例えば、完全型または成熟型のポリペプチドを認識する抗体を誘導しかつ/またはそれに結合するN末端欠失を有するポリペプチドの能力は、N末端から除去される完全型ポリペプチドの残基が過半数未満であれば、一般に保持される。完全型のポリペプチドのN末端が欠失した特定のポリペプチドが、そのような免疫学的活性を保持するかどうかは、本明細書に記載されるかまたは当技術分野で公知である常法によって容易に決定することができる。N末端アミノ酸残基が多数欠失した変異タンパク質が、一定の生物学的または免疫原的活性を保持している可能性がある。事実、わずか6個のアミノ酸残基からなるペプチドがしばしば免疫反応を誘発する場合がある。
「変種」は、参照核酸またはポリペプチドとは異なるがそれらの本質的な特性を保持するポリヌクレオチドまたは核酸を指す。一般的に、変種は、全体として密接に類似し、多くの領域で参照核酸またはポリペプチドと同一である。
「機能的活性を有するポリペプチド」は、完全長、前駆タンパク質、および/または成熟型G-CSFタンパク質に関連する1つ以上の公知の機能的活性を示すことができるポリペプチドを指す。このような機能的活性としては、生物学的活性、抗原性[抗ポリペプチド抗体に結合する(または結合をめぐってポリペプチドと競合する)能力])、免疫原性(本発明の特定のポリペプチドに結合する抗体を生じる能力)、本発明のポリペプチドで多量体を形成する能力、およびポリペプチドの受容体またはリガンドに結合する能力が挙げられるが、これらに限定されるわけではない。
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子(rHA-G-CSF)はG-CSFアナログである。rHA-G-CSFの例は、米国特許第5,665,863号および米国特許第7,041,478号に記載されており、これらはいずれも参照により本明細書に組み入れられる。
rHA-G-CSFの合成方法の例は、米国特許出願番号第11/929,828号に記載されており、これはその全体において参照により本明細書に組み入れられる。いくつかの態様では、NEUGは、NEUG融合タンパク質を発現するように遺伝子操作された酵母宿主系(例えば、サッカロミセス・セレビジエ(Saccharomyces cerevisiae))を用いて産生される。NEUGは、酵母培養物の発酵培地から集菌され、当技術分野で周知の方法を用いて精製される(例えば、一連のクロマトグラフィーおよびろ過工程、例えば、アフィニティークロマトグラフィーおよびイオン交換クロマトグラフィーなど)。
上に記載のように、白血球減少症は循環する白血球(WBC)数の減少であり、好中球減少症は血中好中球数の減少を特徴とし、しばしば細菌感染や真菌感染に対する感受性の増加につながる。以下は、白血球減少症または好中球減少症を発症するリスクにヒト被験者をさらす可能性のある因子の非包括的なリストである:薬剤(例えば、フェニトイン、クロラムフェニコール、サルファ剤、および化学療法剤);ビタミンB12または葉酸欠乏症;過剰な飲酒;骨髄に関与する癌またはその他の病気(例えば、再生不良性貧血、異常ガンマグロブリン血症、発作性夜間ヘモグロビン尿症、ヘモグロビン血症、脊髄形成異常、骨髄異形成症候群、骨髄繊維症、白血病、骨髄腫、リンパ腫、または骨髄を浸潤し置換する転移性固形腫瘍);ウイルス感染症(例えば、インフルエンザ、HIV、早期伝染性単核球症、幼年期ウイルス性疾患);細菌感染症(例えば、結核);放射線;毒素(例えば、ベンゼンおよび殺虫剤);骨髄機能不全(例えば、シュワヒマン-ダイアモンド症候群、軟骨毛髪形成不全症、先天性角化異常症、IB型糖原貯蔵障害);脾臓障害、何らかの原因による脾腫;骨髄細胞またはその前駆体の内在性異常;アレルギー性疾患;自己免疫疾患;T-γリンパ増殖性疾患(T-γLPD);血液透析または移植;毒素。

以下の実施例は本発明を例証するものである。しかしながら、本発明は、当該実施例に記載の特定の条件または詳細に限定されるものではないことを理解されたい。限定されるものではないが米国特許を含む本明細書で参照する公開されている文献はいずれも参照により本明細書に組み入れられる。
NEUGのインビトロおよびインビボ試験の結果を次のようにまとめ、下記実験例で詳述する。
1.NEUGは、用量依存的にNFS-60細胞の増殖を誘導する。
2.NEUGは、インビトロで、Neupogen(登録商標)(フィルグラスチム)の約1/3の効能である。
3.NEUGは、モル基準で評価するとNeulasta(登録商標)(ペグフィルグラスチム)と効力が等しい(Neulasta(登録商標)(ペグフィルグラスチム)1mgは、NEUG4.5mgと効果が等しい)。
1.NEUGは、マウスおよびカニクイザルにおいて耐容性が良好であった。
2.マウスでは、NEUGの単回投与は、末梢血中の好中球および造血前駆細胞の用量依存的で迅速かつ持続的な増加を誘導した。現在の市販されているG-CSF製品と比較すると、好中球数および前駆細胞数の増加は、等モル量のNeupogen(登録商標)(フィルグラスチム)により達成されるものよりも持続期間が長く、等モル量のNeulasta(登録商標)(ペグフィルグラスチム)での持続期間および程度に類似していた。
3.マウスでは、等価量およびAUCANCは、Neulasta(登録商標)(ペグフィルグラスチム)よりも7.7倍高いミリグラム用量のNEUGで達成された。
4.等モル量のNEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)の単回注射投与は、5-FU誘発性好中球減少症のマウスでは、類似の反応速度および効果の程度で、効果的に好中球の回復を促進した。
5.NEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)の単回および反復(毎週1回)投与は、サルにおける末梢好中球数の類似の増加を誘導する。等モル量では、サルにおける好中球増加の程度および持続期間のいずれにおいても、Neulasta(登録商標)(ペグフィルグラスチム)で治療された動物とNEUGで治療された動物とで類似した。
6.マウスおよびサルのいずれにおいても、IVまたはSC投与されると、NEUGは、Neupogen(登録商標)(フィルグラスチム)よりも、クリアランスが遅く、終末相半減期が長く、かつ平均滞留時間が長い。
7.カニクイザルでは、SC注射後、NEUGの終末相半減期(12.6時間)は、Neulasta(登録商標)(ペグフィルグラスチム)のもの(9.49時間)よりも約33%長く、NEUGのクリアランス対バイオアベイラビリティー(「CL/F」)は、Neulasta(登録商標)(ペグフィルグラスチム)の約半分である。
8.NEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)両方のクリアランスは、正常なマウスよりも5-FU誘発性血球減少症を経たマウスで遅く、これは両方のタンパク質の受容体仲介性クリアランスがそれらのクリアランスに寄与することを示唆している。
9.腎排泄(ラットで評価)は、Neupogen(登録商標)(フィルグラスチム)のクリアランスに有意に寄与し、Neulasta(登録商標)(ペグフィルグラスチム)に若干影響し、NEUGのクリアランスに実質的に寄与しない。
10.NEUG(ヒト血清アルブミンおよびヒトコロニー刺激因子からなるヒトタンパク質)は、マウスおよびカニクイザルにおいて免疫原性がある。Neulasta(登録商標)(ペグフィルグラスチム)は、サルにおいて類似の発生率および抗体力価を誘導した。NEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)で処理された数匹のサルからの抗体は、インビトロで中和性であり、NEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)のいずれも、繰り返し曝露されるにつれて好中球反応は減少したが、抗体陽性動物は、回復の際に、抗体陰性動物と類似の好中球の基礎レベルを有していた。
NSF-60は、G-CSF活性の測定のためにバイオアッセイ中で慣例的に使用される細胞株である。この細胞株は、G-CSFに反応して増殖速度が増加する。組換えC-CSF(Neupogen(登録商標)、フィルグラスチム)およびNEUGの相対効力を比較した。
この試験の目的は、BDF-1マウスにおける末梢血好中球および造血幹細胞に対するNEUGの単回皮下投与の効果を評価することであった。BDF-1マウスは、NEUGを3種類の用量レベル(0.25mg/kg、1.25mg/kg、もしくは5.0mg/kg)で、またはNeupogen(登録商標)(フィルグラスチム)を2種類の用量レベル(0.25mg/kgおよび1.25mg/kg)で、単回投与で皮下注射(「SC」)した。末梢顆粒球(Gr.1+)および造血前駆細胞(c-kit+)は、1日目から5日目まで毎日フローサイトメトリーにより定量し、ビヒクルで処置した動物で得られたレベルと比較した。
この試験の目的は、BDF-1マウスの末梢血中の末梢血好中球および造血前駆細胞に対するNEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)の単回皮下(SC)注射の効果を比較することであった。これは、NEUGを5mg/kgまたは10mg/kgの1回量でBDF-1マウス(n=5)に注射することにより評価した。NEUGの効果を、単回投与として投薬した等モル量のペグフィルグラスチム(Neulasta(登録商標))(1.12mg/kgおよび2.24mg/kg)の効果と比較した。Neulasta(登録商標)(ペグフィルグラスチム)およびNEUGのこれらの2種類の用量はほぼ等モルである。
G-CSF製品は、骨髄抑制性化学療法後に好中球の回復を促進するために臨床使用される。この試験の目的は、致死未満量の5-FU(150mg/kg)により好中球減少症が誘導されたモデルにおける好中球の回復に対するNEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)の単回皮下(SC)注射の効果を比較することであった。BDF1マウスに、NEUGを5mg/kgまたは10mg/kg単回投与した。NEUGの効果は、Neulasta(1.12mg/kg、NEUG5mg/kgに対する等モル量)の単回投与の効果と比較した。薬剤はいずれも5-FU単回投与の1日後に投与した。末梢血好中球数は、6日目から10日目まで毎日測定した。この期間では、5-FUが投与されたマウスは、好中球の最下点に続く遅い回復期によって特徴付けられる。実験は、好中球回復の時間および程度に対するNEUGの効果を測定するように設計された。
NEUGの反復投与の効果を測定するためにカニクイザルを選択した。NEUGの反復投与後に血液学的な連続評価を行い、サルでの2種類の試験、すなわちNEUGおよびNeupogen(登録商標)(フィルグラスチム)の皮下投与を比較する2週間の薬理試験、ならびに皮下および静脈内NEUGの効果と皮下Neulasta(登録商標)(フィルグラスチム)の効果とを比較する、より長い(5か月)免疫原性試験を行った。試験はいずれも、NEUGが、Neulasta(登録商標)(ペグフィルグラスチム)と類似する効力および薬力学的プロファイルで、サルにおいて末梢血好中球の持続的な増加をもたらすことを示す。
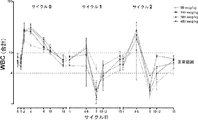
サルにおけるNEUGの薬力を評価するために、効能評価項目として血液学的パラメータを用いて2週間にわたる反復用量試験を行った。実験的に未処理(experimentally naive)のカニクイザルのオスおよびメス20匹を、無作為に各群オス2匹およびメス2匹の5治療群へ分けた。サルは、肩甲部の中央に、ビヒクル、NEUG、またはNeupogen(登録商標)(フィルグラスチム)を皮下(「SC」)注射した。試験の14日間の治療期間の間、ビヒクルは4日毎(Q4D)に投与され、NEUGは4日毎(Q4D)に25μg/kgまたは4日もしくは7日毎(それぞれQ4DもしくはQ7D)に100μg/kg投与され、Neupogen(登録商標)(フィルグラスチム)は毎日5μg/kg投与された。
単核細胞数はG-CSFに反応して末梢にて増加することが報告されているが、好中球で観察されるよりも程度は劣る。この試験では、4日毎に投与された100μg/kgのNEUGだけが、単球の絶対数の増加を誘導した。末梢血リンパ球の絶対数は、NEUGまたはNeupogen(登録商標)(フィルグラスチム)のどちらで治療しても影響されなかった。
カニクイザルにおけるNEUGの非GLP反復用量投与試験は、主に免疫原性を評価する目的で行った(Covance Study No. 6962-129)。血液学的パラメータは、試験評価項目として評価し、この試験はまた、等モル量のNEUGおよびNeulasta(登録商標)(ペグフィルグラスチム)を比較するのに有用な薬理情報を提供する。NEUGおよびNeulastaはいずれも、毎週1回3週間投与された。図18は、NEUGまたはNeulasta(登録商標)(ペグフィルグラスチム)の最初の3回の投与後のANCを示す。
NEUGの薬物動態は、正常BDF-1マウスにおいて単回IVまたはSC注射後に、そして5-FUで治療された好中球減少症BDF-1マウスにおいてSC注射後に、そして腎摘出を受けたラットにおいてIV注射後に、そしてカニクイザルにおいて単回および複数回IVおよびSC注射後に評価した。さらに、rhG-CSF(Neupogen(登録商標))のPKおよびペグ化rhG-CSF(Neulasta(登録商標))のPKを比較した。これらの試験を下記にまとめる。
NEUGは、マウスおよびサルにおいて良好な耐容性があった。4週間毎週1回100、500、または1000μg/kg/投与でNEUGを皮下投与されたサルでは有害な所見はなかった。NEUG治療に対する薬力学的反応は、カニクイザルにおいて複数回のSCまたはIV投与後に観察され、以前に報告されたG-CSFの効果と一致していた。NEUGは、顕著で用量依存的な白血球増多症および好中球増加症を一貫して誘導し、単球、好酸球、および好塩基球の増加は顕著でなく、リンパ球の増加は一貫性がなかった。サルにおけるNEUGの無作用量(NOEL)は、GLPまたは非GLP試験では確認されなかったので、皮下投与については25μg/kg/投与未満であると考えられる。副作用はSCでNEUG処理されたサルでは観察されなかったので、サルにおけるNEUGの皮下投与に関する無毒性量(NOAEL)は、1000μg/kg/投与よりも大きい。NEUGの薬理作用と一致するさらなる所見としては、脾臓重量の増加、骨髄増生の顕微鏡的証拠、および白血球増多症が挙げられる。
NEUGは、患者において、G-CSFに対して中和性である免疫反応を(理論的には)誘導し得た。HSAに対する抗体も可能性はあるが、血液(40mg/mL)中でHSAは非常に高濃度であるので当該抗体の臨床有意性は不明である。NEUGの全成分に対する抗体を検出することができる一連の非常に高感度なアッセイを使用して、ヒトでの免疫原性を評価した。
次の実施例は、「第I相」および「第II相」と題した2つのメインセクションにおいて記載する。各相は、2つの部分、即ちA部およびB部を含む。第I相および第II相の実施例は、下記表2にまとめる。
1.目的
第I相A/B、第II相A/B試験は、骨髄抑制性化学療法剤(ドキソルビシン/ドセタキセル)が投与された被験者における、皮下投与したNeugranin(商標)(「NEUG」)(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子)の安全性、耐容性、免疫原性、薬物動態、および薬力を評価するために行った。
第I相では、患者は、下記の特徴またはパラメータに基づいてスクリーニングされた。
1.ドキソルビシンおよびドセタキセルを投与される予定の組織学的に確認された乳癌患者。
2.18歳以上。
3.十分な血液学的機能。
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.十分な肝機能および腎機能:
7.血清クレアチニン<2.0x正常上限値
8.検査室(local laboratory)での正常範囲内(WNL)の総ビリルビン
9.血清トランスアミナーゼ(SGOT/SGPT)<1.5x正常上限値
10.アルカリフォスファターゼ<2.5x正常上限値
11.ECOG一般状態0または1
12.正常範囲内の左心室駆出分画(LVEF)に基づいたドキソルビシン投与を受ける資格。
13.試験条件を理解する能力を有すること、書面によるインフォームドコンセント(研究に関連する健康情報の使用および開示に関する同意書を含む)を提供すること、ならびに試験プロトコル手順を順守すること。
1.過去に1を超える化学療法レジメン(過去12か月以内に行われたのであればアジュバント療法も含む);試験開始に先立つ4週間以内の任意の化学療法/免疫療法;この試験におけるドキソルビシンの2度の全量サイクルを除く累積的なアントラサイクリン投与。
2.試験化学療法の6週間以内の事前のニトロソウレア(BCNU、CCNU)またはマイトマイシンCのいずれかの使用。
3.アントラサイクリンに基づいた化学療法レジメンの使用を除く、研究者の見解における心臓の病歴、兆候、または症状。
4.試験化学療法の2週間以内の事前の手術または放射線治療。
5.骨盤に対するまたは骨髄支持面積の20%を超える面積に対する、または骨髄障害に対する事前の広範囲照射。
6.造血幹細胞移植による事前の高用量化学療法。
7.試験化学療法の4週間以内の骨髄性(G-CSFまたはGM-CSF)成長因子の事前使用。
8.試験化学療法の4週間以内のエリスロポエチンの事前使用。
9.骨髄性の悪性腫瘍または脊髄形成異常の病歴。
10.既知の脳転移(十分に治療(手術または放射線療法)され、最低3週間の観察で進行が見られず、抗痙攣薬およびステロイドを使用しないで神経学的に安定している場合を除く)。
11.既知の鎌状赤血球症。
12.成人型呼吸窮迫症候群(ARDS)の診断。
13.静脈内または経口抗生物質を必要とする現行の感染症。
14.酵母由来製品に対するアレルギーの既知の履歴。
15.大腸菌由来タンパク質、ペグフィルグラスチム、フィルグラスチム、またはペグフィルグラスチムの任意の他の成分(第2相のみ)に対する既知の過敏症。
16.妊娠女性または授乳中の女性(試験期間中は、女性は全員、確実性が90%を超える避妊法を実施するか、不妊または閉経後でなければならない)。
17.HIV陽性または活動性肝炎が既知(未知の状態の患者は試験されない)。
18.試験期間中および試験薬剤の最後の投与後30日間効果的な避妊を使用することに合意しない男性。
1.疾患進行
2.最善の治療にもかかわらず容認できない毒性
3.研究者の裁量による併発性の病気
4.ドキソルビシンレジメン-寿命中で許容される最大蓄積量に到達(適格基準参照)
5.同意の撤回
6.経過観察への非遵守/喪失
7.妊娠
NEUG(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、rHSA-G-CSF)は、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合タンパク質である。NEUGの治療有効部分は、組換えヒトDNA由来G-CSFである。
a.試験スケジュールおよび継続期間
この試験は、ドキソルビシン/ドセタキセルを投与される予定である乳癌患者62人において行われた、ヒト初回投与、多施設、非盲検、非対照、逐次用量漸進試験であり、続いて対照無作為化試験を行った。この試験は、二部構成であった。A部は、13人の被験者、4つの用量コホート(50、150、300、または450μg/kg)における逐次用量漸進試験であり、50、150、および450μg/kgのコホートのそれぞれには3人の被験者で、300μg/kgのコホートには4人の被験者で、無作為化の実験B部を行う前に安全性を評価した。
化学療法
この実験の化学療法レジメンは、最大2回までの21日サイクルを、治療の1日目に静脈注射により逐次投与するドキソルビシン50mg/m2およびドセタキセル75mg/m2からなった。
薬理学的データ
塩酸ドキソルビシンは、DNAおよびDNA依存RNA合成ならびにタンパク質合成を阻害するストレプトミセス・ペウセティウス var カエシウス(Streptomyces peucetius var caesius)から得られるアントラサイクリン系抗生物質である。ドキソルビシンは、細胞周期のどの期においても活性があるが、S期において最も細胞毒性がある。当該薬物の排出は、主に肝臓によるものであり、腎クリアランスは小さい。
前記薬物は、10、20、50、100、または200mgバイアル中で市販されている。凍結乾燥製剤は、注射用滅菌水、5%デキストロース溶液、または0.9%注射用生理食塩水で再構成されてもよい。
約10〜14日間に最下点を有する骨髄抑制(主に白血球減少症)、ならびにまれな急性心嚢炎心筋炎症候群および累積的な容量に伴う後発性の心筋症を含む心毒性が、ドキソルビシンの用量制限毒性である。著しい脱毛症および中程度の悪心/嘔吐は、予想される毒性である。偶発的な溢出の部位での局所的な皮膚および組織損傷を生ずる溢出反応、口内炎、皮膚(特に爪床)の色素過剰症、ならびに過去に照射した部位での「メモリ」現象が報告されている。
薬理学的データ
ドセタキセルは、遊離チューブリンに結合する半合成タキソイドであり、安定した微小管の構築を促進し、有糸分裂および細胞複製(M期に特異的な細胞周期)を妨害する。ドセタキセルは、タンパク質に広く結合し、肝臓において多くは代謝され、7日以内に用量の約75%が糞便中に排泄される。
ドセタキセル(Taxotere(商標)、Sanofi Aventis)は、80mg/2mLまたは20mg/0.5mlの単回投与バイアル中に提供され、希釈剤(注射用水中13%エタノール)バイアルが付帯される。Taxotereは1ml毎にドセタキセル(無水)を40mgおよびポリソルベート80を1080mg含む。
ドセタキセルは、ドセタキセルまたはエトポシドおよびビタミンEなどのポリソルベート80と調製された他の薬剤に対して激しい過敏反応の既往歴のある患者には投薬すべきではない。
各治療サイクルの1日目に、化学療法剤(ドキソルビシンに続いてドセタキセル)を投与した。
経口(必要であればIV)コルチコステロイド(例えば、デキサメタゾン8mgBID)を、体液鬱滞および過敏反応の発生および重篤さを低下させるため、ドセタキセル投与の1日前に開始して3日間投与した。
被験者は、この試験中および下に規定するさらなる期間中は、次の薬剤および/または処置のいずれも受けないのがよい。
1.試験薬剤の開始の30日以内および実験期間中における他の治験薬。
2.後続の化学療法サイクルは、NEUGの投与後14日まで開始すべきではない。
3.持続性または発熱性の好中球減少症が起こらない限り、実験期間中におけるサイトカイン、他の造血成長因子、および予防用抗生物質。被験者が、スクリーニング期間から0日目の間の任意の時点でG-CSFで治療された場合、NEUGの投与に適格ではなく、試験を中断した。
被験者にベースライン薬剤を継続させた。可能であれば、試験全体にわたって各薬剤の一日量を維持した。何らかの理由で研究者によって必要であると認められた場合、さらなる薬剤、または用量、薬剤、投与経路の変更を必要とした被験者、および薬剤が投与された適応症は、CRFの適切なページに記録した。
被験者は全員、感染の可能性を減らすため、化学療法の各コースに続いて予防用経口抗生物質(例えば、シプロフロキサシン)を投与された。発熱性好中球減少症または持続性の重度の好中球減少症(ANC<0.5x109/L、≧5日)が起こった場合、被験者は、治療不成功と考えられ、試験から除外され、経過観察を完了し、研究者の裁量による成長因子支持を含む標準的な支持療法すべて受けた。
NEUGの安全性は、有害事象(「AE」)の種類、頻度、および重度の評価、臨床検査(血液学および臨床化学)の変化、免疫原性、身体検査、ならびに継時的なバイタルサインのモニタリングにより評価した。AEおよび実験室毒性はすべて、国立癌研究所有害事象共通用語基準に基づいて等級分けした(NCI-CTCAEバージョン3.0、2003年12月12日)。有害事象(重度有害事象「SAE」を含む)は、試験薬剤の投与開始から任意の試験薬剤の最後の投与後30日まで記録した。実験的評価は、評価スケジュール(Schedule of Assessments)で概説されているように得た。グレード4好中球減少症毒性の場合には、ANCが>500になるまで毎日実験的評価を得た。被験者の次の治療サイクルが遅延した場合(そして最後の処置サイクルの後)、ANCが>1500になるまで、鑑別を伴う全血球計算(CBC)を毎週少なくとも2回行った。
a.概論
統計的方法:
安全性、薬物動態(PK)、薬力(PD)、および免疫原性パラメータに関連するデータは、記述統計法を用いて分析した。
合計13人の被験者が、A部(実験において用量が逐次的に漸進する部分)に参加した。合計51人の被験者がB部に参加し、無作為にNEUG300μg/kg(n=20)、NEUG450μg/kg(N=21)、またはペグフィルグラスチム6mg(n=10)に分けられた。
最初の用量設定試験では、化学療法剤の非存在下で、NEUGは、良好な耐容性を示し、予想されたようにANCが上昇し、これは2日目と4日目の間にピークに達し、14日目までに正常に戻った(図22)。
NEUGが投与される被験者において、NEUGに対する抗体の血清試料は、毎NEUGサイクルの1日目での投与前および治療訪問の終わり(最後の投与の少なくとも15日後)に得た。被験者が試験中の任意の時点で陽性の抗NEUG抗体反応を生じた場合、最後のNEUG投与の約6か月後に反復試料を得た。
A部では、用量制限毒性(DLT)は、グレード2髄骨痛を除いて、試験薬剤と、場合によっては関連する、恐らく関連する、または明確に関連すると考えられるグレード2以上の臨床的に有意な有害事象として規定された。
2非関連=恐らく関連していないか、関連していないと考えられる
NEUGが投与される被験者は全員、試験期間中に渡って血清NEUG濃度をサンプリングされた。薬剤は、NEUG特異的なサンドイッチ酵素結合免疫吸着測定(ELISA)を用いて検出した。血清薬剤濃度時間データは、ノンコンパートメントまたはモデルベース分析を用い、WinNonlin Enterprise Edition(バージョン4.1以上)を用いてPK分析に供した。
試験の第I相A部からのデータの分析によって次の所見が得られた:
1.NEUGは、サイクル0(化学療法前)においてWBCおよびANCの用量依存的な上昇を誘導する(図7におけるサイクル0データ参照)。
2.サイクル0におけるANCの増加は、等モル量のペグフィルグラスチムの病歴データと同程度であった
3.予想されたように、化学療法後にWBCおよびANCは低下する
4.最下点ANCからの回復は、用量依存依存的であると考えられる
5.ANCおよびWBCは、15日目までに正常値まで回復した
乳癌治療のサイクル1におけるドキソルビシン/ドセタキセル投与の1日後にNEUGが450μg/kg投与された患者からのPK/PDプロファイルを図29に示す。NEUGのCmaxは、投与の1日以内に達成され、10日目までに検出できないレベルにまで徐々に低下した。NEUGの投与後に、ANCは4日目までにピークにまで上昇し、ANCは、ドキソルビシン/ドセタキセルおよびG-CSF治療を受ける患者において予想される通りに、8日目に最下点まで低下し、10日目に正常値にまで戻る。12日目までには、ANC値は正常範囲にあり、NEUGは検出できない。なお、予防的なG-CSF治療を受けない患者では、最下点ANCの期間およびANCが回復するまでにかかる時間はずっと長い(例えば、5〜7日)。450μg/kg投与後、NEUG排出半減期のメジアンは約30時間であり、これに対し、ペグフィルグラスチムの標準用量では15〜80時間と報告されている。
好中球減少症からの回復を促進させる効果における試験用量でのNEUGとペグフィルグラスチムとの間のより詳細な差異は、治療のサイクル1における個々のANCプロファイルを比較すれば明らかである。すべての群のピークANCは非常に類似しており、NEUGが300μg/kg投与された被験者の最下点ANCは、NEUGが450μg/kg投与された被験者より低く、ペグフィルグラスチムが投与された被験者のANC最下点は平均して最も高かった。最下点ANCからベースラインまでの回復がすべての治療群で14日目までに起きたが、NEUGが450μg/kg投与された被験者よりもNEUGが300μg/kg投与された被験者では遅く、ペグフィルグラスチムが投与された被験者で最も速かった。
1.NEUG用量が150μg/kgのEmax(最大観測ANC)は、サイクル0におけるペグフィルグラスチム用量30μg/kg(確認された有効なペグフィルグラスチム用量である100μg/kgに対して効能で劣ることが後に示された用量)と一致する。
2.Neugranin用量が300および450μg/kgのEmaxは、ペグフィルグラスチム用量が100μg/kgのサイクル0レベルとより一致していた。
3.NEUG300μg/kgおよび450μg/kgのCmaxのメジアンおよびEmaxのメジアンはほぼ同じであり、したがって、Cmaxによって引き続きEmaxが予測される。
4.ANC増加は、等モル量のペグフィルグラスチムの出版済みデータと同程度であった。
第I相薬物動態評価の結果は次のとおりである。
NEUGは、サイクル0およびサイクル1で、150μg/kg、300μg/kg、および450μg/kgの用量にてNEUGで処理されたすべての被験者からの血清試料において検出された。
サイクル1では、NEUGは、150mg/kg、300μg/kg、および450μg/kg用量群において、ほとんどの被験者(45/50でサンプリングされた)で最大144時間まで検出された。実質的に、サイクル間での薬剤蓄積は観察されなかった。
薬物暴露は、各用量群でサイクル1およびサイクル0(前化学療法)においてより高かった。サイクル1におけるNEUGへの暴露の増加は、受容体が仲介するG-CSFクリアランスにおいて役割を果たす好中球の数の減少による可能性がある。
サイクル1におけるNEUGの排出半減期のメジアンは、300μg/kg用量群で約36時間であり、450μg/kg用量群で30時間であった。排出半減期は、フィルグラスチムで3〜4時間であり、ペグフィルグラスチムで、用量に応じて42〜67.5時間であると報告されている。
用量間の統計的有意差は、最大血清濃度(tmax)までの時間および吸収半減期(t1/2、abs)に見られた。これらのパラメータはいずれも、NEUG用量の増加と共に増加した。他の用量正規化PKパラメータは、用量間の統計的有意差を示さなかった。
試験の第II相は、ドキソルビシン/ドセタキセルを最大4回まで投与した乳癌を有する被験者334人で行われた対照無作為化試験であった。試験は、臨床現場約45カ所で行われ、皮下投与したNEUGとペグフィルグラスチムとの安全性および効果を評価するための二元配置無作為化パイロットフェーズ、その後の、被験者がパイロットフェーズに基づいて選ばれるペグフィルグラスチムおよび2つの良好な耐性用量のNEUGに無作為に分けられる(1:1:1)メインフェーズからなった。主要評価項目である化学療法サイクル1中の重度(グレード4)の好中球減少(DSN)の持続期間に関してペグフィルグラスチムに対してNEUGが劣っていないことを実証するため、メインフェーズのサンプルサイズを検出した。試験の設計を下に概略的に示す。
第II相の第一の目的は、ペグフィルグラスチムに対して同等の効果を示すNEUG用量を選択すること、およびNEUGでの治療後の化学療法のサイクル1において重度の好中球減少(DSN)の持続期間を評価することであった。第二の目的は、サイクル2〜4におけるDSNを評価すること、好中球絶対数が回復するまでの時間およびサイクル1〜4における発熱性好中球減少症の割合を評価すること;ならびにNEUGの安全性、耐容性、薬物動態(サイクル1において)、および免疫原性を評価することであった。
第II相に関連して、患者は、下記の特徴またはパラメータに基づいてスクリーニングされた。
1.ドキソルビシンを60mg/m2およびドセタキセルを75mg/m2投与される予定の組織学的に確認された乳癌患者。
2.18歳以上
3.十分な血液学的機能:
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.十分な肝機能および腎機能:
7.血清クレアチニン<1.5x正常上限値
8.検査室(local laboratory)での正常範囲内(WNL)の総ビリルビン
9.血清トランスアミナーゼ(SGOT/SGPT)<1.5x正常上限値
10.アルカリフォスファターゼ<2.5x正常上限値
11.米国東海岸癌臨床試験グループ(「ECOG」)一般状態0〜2
12.正常範囲内の左心室駆出分画(LVEF)に基づいたドキソルビシン投与を受ける資格
13.試験条件を理解する能力を有すること、書面によるインフォームドコンセント(研究に関連する健康情報の使用および開示に関する同意書を含む)を提供すること、ならびに試験プロトコル手順を順守すること。
1.過去に1を超える化学療法レジメン(過去12か月以内に行われたのであればアジュバント療法も含む)
2.この試験におけるドキソルビシンの4度の全量サイクルを除く累積的なアントラサイクリン投与
3.試験化学療法剤の30日以内の事前の化学療法/免疫療法(ニトロソウレア(BCNU、CCNU)またはマイトマイシンCの試験化学療法剤の6週間以内)
4.トラスツズマブ(ハーセプチン)併用療法
5.過去30日での治験薬の投与
6.アントラサイクリンに基づいた化学療法レジメンの使用を除く、研究者の見解における心臓の病歴、兆候、または症状
7.試験化学療法の2週間以内の事前手術
8.試験化学療法の4週間以内の事前手術(骨転移のためのスポット照射を除く)
9.造血幹細胞移植による事前の高用量化学療法
10.試験化学療法の4週間以内のG-CSF、GM-CSF、またはエリスロポエチンの事前使用
11.試験化学療法の72時間以内の抗生物質の全身投与
12.骨髄性の悪性腫瘍または脊髄形成異常の病歴
13.既知の脳転移(十分に治療(手術または放射線療法)され、最低3週間の観察で進行が見られず、抗痙攣薬およびステロイドを使用しないで神経学的に安定している場合を除く)。
14.既知の鎌状赤血球症
15.成人型呼吸窮迫症候群(ARDS)の診断
16.酵母由来製品に対するアレルギーの既知の履歴
17.大腸菌由来タンパク質、ペグフィルグラスチム、フィルグラスチム、またはペグフィルグラスチムの任意の他の成分に対する既知の過敏症
18.妊娠女性または授乳中の女性。(子宮が無傷である女性は全員スクリーニングされて血清妊娠テストが陰性でなければならない。不妊でないかまたは非閉経前の女性は全員、試験期間中および試験薬剤の最後の投与後の30日間医学的に認められた避妊法を実施しなければならない。)
19.試験期間中および試験薬剤の最後の投与後30日間効果的な避妊を使用することに合意しない男性
20.HIV陽性または活動性肝炎が既知(未知の状態の患者は試験されない)
1.疾患進行
2.最善の治療にもかかわらず容認できない毒性
3.研究者の裁量による併発性の病気
4.ドキソルビシンレジメン-寿命中で許容される最大蓄積量に到達(適格基準参照)
5.同意の撤回
6.経過観察への非遵守/喪失
7.妊娠
NEUG(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、rHSA-GCSF)は、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合タンパク質である。NEUGの治療有効部分は、組換えヒトDNA由来G-CSFである。
第I相からのデータは、300および450μg/kgのNEUG用量が安全であり良好な耐容性があることを示した。さらに、ペグフィルグラスチムの承認された固定用量と比較して、NEUGのいずれの用量も、細胞毒性を有する化学療法を受ける乳癌患者におけるANCプロファイルに対して同様の効果をもたらした。ANCプロファイルのAUCは、効果の一点計測値(single-point measure of effect)としての役割を果たす。AUCANCについてはこれらの治療群間で統計的有意差はなかったが、450μg/kg群のAUCは、300μg/kg群で観察されたものよりもわずかに高く、ペグフィルグラスチム群で観察されたものとほとんど同じである(図23)。入手可能なデータに基づくと、NEUGは、300μg/kgではペグフィルグラスチムより効果が低く、450μg/kgでペグフィルグラスチムと同様の効果を与えるためのおおよその最小必要用量であると推定された。
a.試験スケジュールおよび継続時間
この試験は、ドキソルビシン/ドセタキセルが最大4回まで投与される予定の乳癌患者被験者約330人で行われた対照無作為化試験であった。45カ所の臨床現場で行ったこの試験は、パイロットフェーズおよびメインフェーズの2相からなった。
NEUGの安全性は、AEの種類、頻度、および重症度の評価、臨床検査(血液学および臨床化学)の変化、免疫原性、身体検査、ならびに継時的なバイタルサインのモニタリングにより評価した。AEおよび実験室毒性はすべて、国立癌研究所有害事象共通用語基準(NCI-CTCAEバージョン3.0、2003年12月12日)に基づいて等級分けした。
化学療法
この実験の化学療法レジメンは、最大4回の21日サイクルを、治療の1日目に静脈注射により逐次的に投与するドキソルビシン60mg/m2およびドセタキセル75mg/m2からなった。
薬理学的データ
塩酸ドキソルビシンは、DNA塩基対に直接結合し(インターカレートし)、DNA合成およびDNA依存RNA合成ならびにタンパク質合成を阻害するストレプトミセス・ペウセティウス var カエシウスから得られるアントラサイクリン系抗生物質である。ドキソルビシンは、細胞周期のどの期においても活性があるが、S期において最も細胞毒性がある。当該薬物の排出は、主に肝臓によるものであり、腎クリアランスは小さい。
前記薬物は、10、20、50、100、または200mgバイアル中で市販されている。凍結乾燥製剤は、注射用滅菌水、5%デキストロース溶液、または0.9%注射用生理食塩水で再構成されてもよい。
約10〜14日間に最下点を有する骨髄抑制(主に白血球減少症)、ならびにまれな急性心嚢炎心筋炎症候群および累積的な容量に伴う後発性の心筋症を含む心毒性が、ドキソルビシンの用量制限毒性である。
薬理学的データ
ドセタキセルは、遊離チューブリンに結合する半合成タキソイドであり、安定した微小管の構築を促進し、有糸分裂および細胞複製(M期に特異的な細胞周期)を妨害する。ドセタキセルは、タンパク質に広く結合し、肝臓において多くは代謝され、7日以内に用量の約75%が糞便中に排泄される。
ドセタキセル(Taxotere(商標)、Sanofi Aventis)は、80mg/2mLまたは20mg/0.5mlの単回投与バイアル中に提供され、希釈剤(注射用水中13%エタノール)バイアルが付帯される。Taxotereは1ml毎にドセタキセル(無水)を40mgおよびポリソルベート80を1080mg含む。
ドセタキセルは、ドセタキセルまたはエトポシドおよびビタミンEなどのポリソルベート80と調製された他の薬剤に対して激しい過敏反応の既往歴のある患者には投薬すべきではない。
各治療サイクルの1日目に、化学療法剤(ドキソルビシンに続いてドセタキセル)を投与した。
経口(必要であればIV)コルチコステロイド(例えば、デキサメタゾン8mgBID)を、体液鬱滞および過敏反応の発生および重篤さを低下させるため、ドセタキセル投与の1日前に開始して3日間投与した。
被験者は、この試験中および下に規定するさらなる期間中は、次の薬剤および/または処置のいずれも受けてはならない。
1.サイクル1化学療法の72時間以内の抗生物質の全身投与。
2.試験薬剤の開始の30日以内および実験期間中における他の治験薬。
3.後続の化学療法サイクルは、NEUGの投与後14日まで開始すべきではない。
4.持続性または発熱性の好中球減少症が起こらない限り、実験期間中におけるサイトカイン、他の造血成長因子、および予防用抗生物質。被験者が、スクリーニング期間から0日目の間の任意の時点でG-CSFで治療される場合、NEUGの投与に適格ではなく、試験を中断する。
被験者にベースライン薬剤を継続させた。可能であれば、試験全体にわたって各薬剤の一日量を維持した。何らかの理由で研究者によって必要であると認められた場合、さらなる薬剤、または用量、薬剤、投与経路の変更を必要とした被験者、および薬剤が投与された適応症、を記録した。
NEUGが投与される被験者は全員、サイクル1中に血清NEUG濃度をサンプリングされた。薬剤は、NEUG特異的なサンドイッチ酵素結合免疫吸着測定(ELISA)を用いて検出した。血清薬剤濃度時間データは、ノンコンパートメントまたはモデルベース分析を用い、WinNonlin Enterprise Edition(バージョン5.0以上)を用いてPK分析に供した。次のPKパラメータを測定した:曲線下面積(AUC0-∞)、クリアランス(CL/F)、分布容積(Vz/F)、最大濃度(Cmax)、吸収半減期(t1/2、abs)、排出半減期(t1/2、elim)、および平均滞留時間(MRT)。
NEUGが投与される被験者において、NEUGに対する抗体の血清試料は、毎NEUGサイクルの1日目における投与前および治療訪問の終わり(最後の投与の約30日後)に得た。被験者が試験中の任意の時点で陽性の抗NEUG抗体反応を生じた場合、最後のNEUG投与の約6か月後に反復試料を得、この試料が陽性であった場合、12か月目に試料を得た。プロトコルは、被験者全員からの6か月および12か月免疫原性試料を必要とするようにその後修正された。
a.概論
統計的方法
サイクル1における重度の好中球減少(DSN)の平均持続期間の主要評価項目に関連してペグフィルグラスチムに対してNEUGが劣っていないことを実証するため、この実験のメインフェーズ(B部)における1治療群当たり被験者約85人のサンプルサイズは、非劣性マージンが1日で、複数回の試験(Hochberg法による)のために調整した片側有意水準が全体で0.025で、91%の検出を提供するように選択した。サンプルサイズは、2つの独立群に対する正規近似、サイクル1 DSNの治療内標準偏差として推定1.6日、およびサイクル1 DSNの主要評価項目に関して評価できない最高率20%に基づいて計算した。
全血球計算値(「CBC」)は、1日目、3日目、そして5日目以降、最下点後にANCが>2.0x109Lになるまで、その後、週2回、および治療の終わりに得た。
試験のパイロットフェーズに参加した被験者78人のうち、被験者13人が試験を完了せず、3人(27.3%)がNEUG30mgで治療され、3人(14.3%)がNEUG40mgで治療され、3人(15.0%)がNEUG50mgで治療され、4人(15.4%)がペグフィルグラスチムで治療された。早期中断の最も多い理由は、同意の撤回(被験者7人)および研究者の判断によるもの(被験者3人)であった。NEUGを30mg投与した被験者の1人は、有害事象(糖尿病性足病変)によるとりやめであった。
試験のメインフェーズに参加した被験者256人のうち、被験者18人が試験を完了せず、10人(11.6%)がNEUG40mgで治療され、5人(6.0%)がNEUG50mgで治療され、3人(3.5%)がペグフィルグラスチムで治療された。早期中断の最も多い理由は、同意の撤回(被験者7人)および2人の死者を含むAE(被験者4人)であった。研究者は、AEはすべて、試験薬剤または化学療法と関連しないと見なした。メインフェーズでは、NEUG40mgが投与された被験者の1人(1.2%)が、試験薬剤で治療される前に除かれた。
血清Neugranin濃度は、6.312ng/mLの定量範囲の下限値(LLQ)で、有効なサンドイッチELISAを用いて測定した。薬物動態パラメータは、吸収半減期以外はノンコンパートメントモデル法を用いて計算し、吸収半減期は、一次吸収、一次排出1コンパートメントモデルを用いて測定した。モデリングは、WinNonlin Professional(バージョン5.0.1)を用いて行った。血清NEUG濃度は、第II相においてNEUGで治療されたすべての被験者における化学療法サイクル1で測定した。第II相A部では、NEUGの排出半減期の中間値は、30mg投与群で33時間であり、40mg投与群で46時間であり、50mg投与群で18時間であった(表16)。B部では、NEUGの排出半減期の中間値は、40mg投与群で40時間であり、50mg投与群で39時間であった(表17)。A部中は、PKサンプリングは、B部と比較して(投与前、3日目、5〜8日目)、より頻繁に行われた(投与前、3時間、6時間、12時間、24時間、3日目、5〜9日目、11日目)。
試験参加者間で、Neugraninで治療された被験者において、確認された抗G-CSF/新エピトープ抗体反応1つおよびペグフィルグラスチム治療群において抗G-CSF反応が1つ、すなわちそれぞれ0.5%および0.9%があった(表18)。どちらの場合も、被験者は、前投与試料中で非特異的結合の上昇があった。
第II相B部では、各治療群中の被験者の>90%が、治療により発現した有害事象(TEAE)を少なくとも1つ経験し、試験薬剤と関連する少なくとも1つのTEAEを有する被験者のパーセントは、ペグフィルグラスチム群で23.1%からNeugranin50mg群で35.0%の範囲であった。少なくとも1つのSAEを有する被験者のパーセントは、NEUG30mg群で最も高かった(30%)が、他の3つの治療群では約15%であった。SAEはどれも試験薬剤と関連していなかった。患者1人(NEUG30mg)は、糖尿病性足病変により試験から除かれ、これは試験薬剤とは関連しないと考えられた。B部では、被験者8人(NEUG40mg2人、NEUG50mg3人、ペグフィルグラスチム3人)以外はみな少なくとも1つのTEAEを有した。試験薬剤と関連する少なくとも1つのTEAEを有する被験者のパーセントは、NEUG50mg群で20.2%、NEUG 40mg群で22.4%、ペグフィルグラスチムが投与される被験者で22.1%であった。被験者2人(NEUG 40mg 1人、ペグフィルグラスチム1人)が試験中に死亡し、各治療群の被験者6〜8人がSAEを少なくとも1つ経験した。死亡やSAEは試験薬剤と関連しないと考えられた。
第II相の結果は、NEUGの40および50mg固定用量がいずれも、骨髄毒性の化学療法で治療される乳癌被験者においてペグフィルグラスチム6mgと同等の安全性および効能をもたらすことを実証した。40mg治療群の平均DSNは、50mg群の平均DSNよりわずかに低かったが、これらの差異は統計的に有意ではなかった。用量応答は、体重により調整した用量を考慮した場合と固定用量コホートに対してとの両方でAUCANCについて観察した(サイクル1における0〜15日目)(図24)。30mgコホートのAUCANCは、ペグフィルグラスチムのそれよりわずかに低く、これは、30mg固定用量がこの研究では効果が劣るが、40mgおよび50mgコホートのAUCANCは用量相関性があり、ペグフィルグラスチム治療被験者のAUCANCよりも高い(有意ではないが)ことを示している。上の分析から、NEUGが体重調節基準(mg/kg)で投与されると、用量反応が明らかである。しかしながら、第II相B部のサイクル1におけるDSNの比較によって、治療群(40および50mgNEUGおよびペグフィルグラスチム)において、そして体重調節用量(mg/kg)で、DSNが有意に異ならなかったので、体重で四分した患者はすべて十分にサポートされることが示唆された。さらに、関連する有害事象(特に骨痛;データ示さず)の発生率および重症度が、体重1キログラム当たりに投与される用量と相関せず、またペグフィルグラスチムでの有害事象と差異はなかったので、固定用量が、より低体重の患者において安全性プロファイルに変更をもたらす可能性があるという証拠はなかった。
Claims (22)
- 好中球減少症を示しているかまたは好中球減少症を発症する危険性のあるヒト被験者に、被験者を治療するのに有効な量で組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を投与する段階を含む、ヒト被験者における好中球減少症を治療するまたは予防する方法。
- 白血球減少症を示しているかまたは白血球減少症を発症する危険性のあるヒト被験者に、被験者を治療するのに有効な量で組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を投与する段階を含む、ヒト被験者における白血球減少症を治療するまたは予防する方法。
- ヒト被験者が、非脊髄性の悪性腫瘍を患っており、臨床的に有意な発生率の発熱性好中球減少症を伴う少なくとも一種類の骨髄抑制性抗癌剤が投与されている、請求項1または2記載の方法。
- 非脊髄性の悪性腫瘍を患っており、臨床的に有意な発生率の発熱性好中球減少症を伴う少なくとも一種類の骨髄抑制性抗癌剤が投与されているヒト被験者における、発熱性好中球減少症により顕在化する感染症の発生率を減少させる方法であって、被験者を治療するのに有効な量で組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を被験者に投与する段階を含む、方法。
- (a)被験者におけるグレード4好中球減少症がなくなる;
(b)被験者におけるグレード4好中球減少症が減少する;
(c)被験者において重度の好中球減少症の持続期間が短縮される;
(d)被験者におけるグレード4好中球減少症の持続期間が5日未満になる;
(e)被験者におけるグレード3好中球減少症の持続期間がなくなる;
(f)被験者におけるグレード3好中球減少症の持続期間が短縮される;または
(g)これらの任意の組み合わせである、
請求項1〜4のいずれか一項記載の方法。 - 組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の投与が、白血球(WBC)の増加をもたらす、請求項1〜5のいずれか一項記載の方法。
- (a)被験者において好中球数が増加する;
(b)被験者において好中球数の減少が抑制される;
(c)被験者において最下点の絶対好中球数(ANC)が増加する;
(d)被験者において回復ANCが増加する;
(e)被験者においてANC回復までの時間が短縮される;または
(f)これらの任意の組み合わせである、
請求項1〜6のいずれか一項記載の方法。 - 被験者に投与される組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の量が、
(a)約50μg/kgから約450μg/kgまで;
(b)約50μg/kg;
(c)約150μg/kg;
(d)約300μg/kg;
(e)約450μg/kg;
(f)約30mgから約60mgまで;
(g)約30mg;
(h)約40mg;
(i)約50mg;
(j)約60mg;または
(k)これらの任意の組み合わせ
からなる群より選択される、請求項1〜7のいずれか一項記載の方法。 - 好中球減少症が、原発性好中球減少症、急性好中球減少症、重度の慢性好中球減少症(SCN)、重度の先天性好中球減少症(コストマン症候群)、重度の小児遺伝性無顆粒球症、良性好中球減少症、周期性好中球減少症、慢性特発性好中球減少症、続発性好中球減少症、好中球減少症関連症候群、および免疫媒介好中球減少症からなる群より選択される、請求項1〜8のいずれか一項記載の方法。
- 好中球減少症が、放射線、アルコール依存症、薬物、アレルギー性疾患、再生不良性貧血、自己免疫疾患、T-γリンパ増殖性疾患(T-γLPD)、脊髄形成異常、骨髄繊維症、異常ガンマグロブリン血症、発作性夜間血色素尿症、癌、ビタミンB12欠乏症、葉酸欠乏症、ウイルス感染症、細菌感染症、脾臓障害、血液透析もしくは移植、白血病、骨髄腫、リンパ腫、骨髄を浸潤し置換する転移性固形腫瘍、毒素、骨髄機能不全、シュワヒマン-ダイアモンド症候群、軟骨毛髪形成不全症、先天性角化異常症、IB型糖原貯蔵障害、任意の原因の脾腫、骨髄細胞もしくはその前駆体の内在性異常を原因とするかまたはそれらに関連するものである、請求項1〜9のいずれか一項記載の方法。
- 組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子が、
(a)骨髄抑制性抗癌剤投与の少なくとも12時間後;
(b)骨髄抑制性抗癌剤投与の少なくとも18時間後;
(c)骨髄抑制性抗癌剤投与の少なくとも24時間後
からなる群より選択される時期に投与される、請求項3に記載の方法。 - 骨髄抑制性抗癌薬の前に組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を投与することがWBCの増加をもたらす、請求項11記載の方法。
- 化学療法前に組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を投与することがANCの上昇をもたらす、請求項11または12のいずれか一項記載の方法。
- 非脊髄性の悪性腫瘍が乳癌を含む、請求項3または11〜13のいずれか一項記載の方法。
- 骨髄抑制性抗癌剤がドキソルビシンおよびドセタキセルを含む、請求項3または11〜14のいずれか一項記載の方法。
- ドキソルビシン約50mg/m2およびドセタキセル約75mg/m2が、少なくとも一つの治療サイクルの同じ日に静脈点滴により順次投与される、請求項15記載の方法。
- ドキソルビシン約60mg/m2およびドセタキセル約75mg/m2が、少なくとも一つの治療サイクルの同じ日に静脈点滴により順次投与される、請求項15記載の方法。
- ANCおよびWBCが、
(a)化学療法後10日目;
(b)化学療法後11日目;
(c)化学療法後12日目;
(d)化学療法後13日目;
(e)化学療法後14日目;または
(f)化学療法後15日目
からなる群より選択される時期までに正常に戻る、請求項3または11〜17のいずれか一項記載の方法。 - 化学療法剤の投与後14日目において、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子で治療された患者のANCの上昇が、等価量のペグフィルグラスチムで治療された患者のANCの上昇よりも低い、請求項3または11〜18のいずれか一項記載の方法。
- 組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の投与が、リンパ球、単球、好酸球、好塩基球、またはこれらの任意の組み合わせの増加をもたらす、請求項1〜19のいずれか一項記載の方法。
- 被験者において、リンパ球数、単球数、好酸球数、好塩基球数、またはこれらの任意の組み合わせが増加する、請求項1〜20のいずれか一項記載の方法。
- 被験者において、リンパ球数、単球数、好酸球数、または好塩基球数の減少が抑制される、請求項1〜21のいずれか一項記載の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14544009P | 2009-01-16 | 2009-01-16 | |
| US14543609P | 2009-01-16 | 2009-01-16 | |
| US61/145,440 | 2009-01-16 | ||
| US61/145,436 | 2009-01-16 | ||
| PCT/US2010/021241 WO2010083439A2 (en) | 2009-01-16 | 2010-01-15 | Recombinant human albumin-human granulocyte colony stimulating factor for the prevention of neutropenia |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012515222A true JP2012515222A (ja) | 2012-07-05 |
| JP2012515222A5 JP2012515222A5 (ja) | 2013-02-28 |
Family
ID=42260335
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011546399A Pending JP2012515222A (ja) | 2009-01-16 | 2010-01-15 | 組換えヒトアルブミン−ヒト顆粒球コロニー刺激因子融合タンパク質の安定な製剤 |
| JP2011546396A Expired - Fee Related JP5753095B2 (ja) | 2009-01-16 | 2010-01-15 | 組換えヒトアルブミン−ヒト顆粒球コロニー刺激因子融合タンパク質の安定な製剤 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011546396A Expired - Fee Related JP5753095B2 (ja) | 2009-01-16 | 2010-01-15 | 組換えヒトアルブミン−ヒト顆粒球コロニー刺激因子融合タンパク質の安定な製剤 |
Country Status (16)
| Country | Link |
|---|---|
| US (4) | US20100227818A1 (ja) |
| EP (2) | EP2387420A2 (ja) |
| JP (2) | JP2012515222A (ja) |
| KR (2) | KR20110132327A (ja) |
| CN (2) | CN102378635A (ja) |
| AU (2) | AU2010204552B2 (ja) |
| BR (2) | BRPI1004940A2 (ja) |
| CA (2) | CA2749802C (ja) |
| EA (2) | EA201190079A1 (ja) |
| IL (2) | IL214051A0 (ja) |
| MX (2) | MX2011007583A (ja) |
| NZ (2) | NZ594055A (ja) |
| SG (3) | SG196821A1 (ja) |
| UA (2) | UA105201C2 (ja) |
| WO (2) | WO2010083439A2 (ja) |
| ZA (1) | ZA201105171B (ja) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102378635A (zh) * | 2009-01-16 | 2012-03-14 | 特瓦制药工业有限公司 | 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 |
| EP2384759A1 (en) | 2010-05-06 | 2011-11-09 | Suomen Punainen Risti Veripalvelu | Sulphated hyaluronic acid in combination with G-CSF for use in mobilising blood stem cells |
| HRP20150963T4 (hr) * | 2010-06-07 | 2023-12-08 | Amgen Inc. | Uređaj za primjenu lijeka |
| CN102628869B (zh) * | 2012-04-19 | 2014-04-02 | 上海蓝怡科技有限公司 | 提高甲胎蛋白抗体冻干稳定性的制剂 |
| AR094481A1 (es) | 2013-01-15 | 2015-08-05 | Teva Pharma | Formulaciones de albu-bche (albúmina-butirilcolinesterasa), preparación y sus usos |
| US20140271538A1 (en) | 2013-03-15 | 2014-09-18 | Teva Pharmaceutical Industries Ltd. | Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor for the Prevention of Neutropenia in Pediatric Patients |
| MA40904A (fr) * | 2014-11-03 | 2017-09-12 | Hygeia Tech Inc | Compositions d'ester de phorbol et procédés pour traiter ou réduire la durée d'une cytopénie |
| MX2017011374A (es) | 2015-03-06 | 2018-01-23 | Beyondspring Pharmaceuticals Inc | Método de tratamiento de cáncer asociado con una mutación de ras. |
| BE1023343B1 (nl) * | 2015-05-20 | 2017-02-09 | Mycartis Nv | Opslagbuffer |
| EP3328415B1 (en) * | 2015-07-30 | 2020-11-25 | Endor Technologies, S.L. | Colony stimulating factor for use in pancreatic or colon cancer treatment |
| WO2017139231A1 (en) | 2016-02-08 | 2017-08-17 | Beyondspring Pharmaceuticals, Inc. | Compositions containing tucaresol or its analogs |
| CN106222221A (zh) * | 2016-08-05 | 2016-12-14 | 山东科兴生物制品有限公司 | 制备重组人粒细胞刺激因子原液的纯化方法 |
| WO2018096534A1 (en) | 2016-11-22 | 2018-05-31 | Sorrel Medical Ltd. | Apparatus for delivering a therapeutic substance |
| CN110431135A (zh) | 2017-01-06 | 2019-11-08 | 大连万春布林医药有限公司 | 微管蛋白结合化合物及其治疗用途 |
| CA3052190A1 (en) | 2017-02-01 | 2018-08-09 | Beyondspring Pharmaceuticals, Inc. | Method of reducing neutropenia |
| US20210088505A1 (en) * | 2017-04-07 | 2021-03-25 | La Jolla Institute For Allergy And Immunology | Unipotent Neutrophil Progenitor Cells, Methods of Preparation, and Uses Thereof |
| CN109420159A (zh) * | 2017-08-23 | 2019-03-05 | 江苏泰康生物医药有限公司 | 一种重组蛋白药物的新型稳定制剂 |
| US11786523B2 (en) | 2018-01-24 | 2023-10-17 | Beyondspring Pharmaceuticals, Inc. | Composition and method for reducing thrombocytopenia |
| SG11202006990TA (en) * | 2018-02-01 | 2020-08-28 | Beyondspring Pharmaceuticals Inc | Composition and method for reducing chemotherapy-induced neutropenia via the administration of plinabulin and a g-csf agent |
| US11583633B2 (en) | 2018-04-03 | 2023-02-21 | Amgen Inc. | Systems and methods for delayed drug delivery |
| US11357909B2 (en) | 2018-10-05 | 2022-06-14 | Eitan Medical Ltd. | Triggering sequence |
| CN111383745A (zh) * | 2018-12-29 | 2020-07-07 | 医渡云(北京)技术有限公司 | 计算机数据处理方法、装置、存储介质及设备 |
| US11684655B2 (en) | 2019-05-31 | 2023-06-27 | Spectrum Pharmaceuticals, Inc. | Methods of treating neutorpenia using G-CSF protein complex |
| US11267858B2 (en) | 2019-05-31 | 2022-03-08 | Spectrum Pharmaceuticals, Inc. | Methods of treatment using G-CSF protein complex |
| CN112316120A (zh) * | 2019-08-05 | 2021-02-05 | 天津派格生物技术有限公司 | 使用低动员型g-csf有效且安全治疗粒细胞减少症的方法 |
| KR102485892B1 (ko) * | 2020-04-09 | 2023-01-09 | 주식회사 에이프릴바이오 | 고양이 과립구 집락 자극인자 및 혈청 알부민에 대한 항원 결합 단편을 포함하는 융합 단백질 및 이의 용도 |
| CN111751552B (zh) * | 2020-06-18 | 2022-09-16 | 广州市伊川生物科技有限公司 | 一种缺血修饰白蛋白测定试剂盒及其使用方法 |
| US11723955B1 (en) * | 2022-05-13 | 2023-08-15 | Allgenesis Biotherapeutics Inc. | VEGFR fusion protein pharmaceutical composition |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005514060A (ja) * | 2001-12-21 | 2005-05-19 | ヒューマン ジノーム サイエンシーズ, インコーポレイテッド | アルブミン融合タンパク質 |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3888381T2 (de) | 1987-04-09 | 1994-07-28 | Delta Biotechnology Ltd | Hefevektor. |
| JPH01215289A (ja) | 1988-02-22 | 1989-08-29 | Toa Nenryo Kogyo Kk | 遺伝子組換えによる正常ヒト血清アルブミンaの製造方法 |
| FR2649991B2 (fr) | 1988-08-05 | 1994-03-04 | Rhone Poulenc Sante | Utilisation de derives stables du plasmide pkd1 pour l'expression et la secretion de proteines heterologues dans les levures du genre kluyveromyces |
| FR2686899B1 (fr) * | 1992-01-31 | 1995-09-01 | Rhone Poulenc Rorer Sa | Nouveaux polypeptides biologiquement actifs, leur preparation et compositions pharmaceutiques les contenant. |
| FR2686900B1 (fr) * | 1992-01-31 | 1995-07-21 | Rhone Poulenc Rorer Sa | Nouveaux polypeptides ayant une activite de stimulation des colonies de granulocytes, leur preparation et compositions pharmaceutiques les contenant. |
| DE4242863A1 (de) * | 1992-12-18 | 1994-06-23 | Boehringer Mannheim Gmbh | Stabile lyophilisierte pharmazeutische Zubereitungen von G-CSF |
| DE19549232C2 (de) * | 1995-12-20 | 1998-05-20 | Boehringer Mannheim Gmbh | Verwendung von G-CSF in Kombination mit einem Chemotherapeutikum bei der Behandlung von Erkrankungen, die eine periphere Stammzelltransplantation erfordern |
| US7321023B2 (en) * | 1997-11-07 | 2008-01-22 | Incyte Corporation | SP16 protein |
| JP3895109B2 (ja) * | 1998-03-06 | 2007-03-22 | 中外製薬株式会社 | 蛋白非添加製剤 |
| US6831158B2 (en) * | 2000-01-10 | 2004-12-14 | Maxygen Holdings Ltd. | G-CSF conjugates |
| US6555660B2 (en) * | 2000-01-10 | 2003-04-29 | Maxygen Holdings Ltd. | G-CSF conjugates |
| JP2003530839A (ja) | 2000-04-12 | 2003-10-21 | プリンシピア ファーマスーティカル コーポレイション | アルブミン融合タンパク質 |
| CA2449593A1 (en) * | 2001-06-08 | 2002-12-19 | Powderject Vaccines, Inc. | Spray freeze-dried compositions |
| WO2005003296A2 (en) | 2003-01-22 | 2005-01-13 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| EP1572936A2 (en) * | 2002-03-05 | 2005-09-14 | Eli Lilly And Company | Heterologous g-csf fusion proteins |
| CN1241946C (zh) * | 2002-07-01 | 2006-02-15 | 美国福源集团 | 对多种细胞具刺激增生作用的人血清白蛋白重组融合蛋白 |
| US20070041987A1 (en) * | 2003-03-19 | 2007-02-22 | Daniel Carter | Fragments or polymers of albumin with tunable vascular residence time for use in therapeutic delivery and vaccine development |
| US6972332B1 (en) * | 2004-05-20 | 2005-12-06 | Acura Pharmaceuticals, Inc. | Process for the production of opiates |
| WO2007021494A2 (en) | 2005-08-12 | 2007-02-22 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| MX2007004529A (es) * | 2006-04-13 | 2008-12-01 | Wix Filtration Corp Llc | Elemento de filtro. |
| JP2008146587A (ja) * | 2006-12-13 | 2008-06-26 | Sony Corp | 表示装置、表示プログラム、表示方法、画像提供装置、画像提供プログラム、画像提供方法及び記録媒体 |
| CN102378635A (zh) | 2009-01-16 | 2012-03-14 | 特瓦制药工业有限公司 | 重组人白蛋白-人粒细胞集落刺激因子融合蛋白的新稳定制剂 |
-
2010
- 2010-01-15 CN CN2010800127233A patent/CN102378635A/zh active Pending
- 2010-01-15 EP EP10701082A patent/EP2387420A2/en not_active Withdrawn
- 2010-01-15 UA UAA201109961A patent/UA105201C2/uk unknown
- 2010-01-15 SG SG2014003073A patent/SG196821A1/en unknown
- 2010-01-15 JP JP2011546399A patent/JP2012515222A/ja active Pending
- 2010-01-15 SG SG2011050358A patent/SG172940A1/en unknown
- 2010-01-15 MX MX2011007583A patent/MX2011007583A/es active IP Right Grant
- 2010-01-15 SG SG2011050366A patent/SG172941A1/en unknown
- 2010-01-15 CA CA2749802A patent/CA2749802C/en not_active Expired - Fee Related
- 2010-01-15 CA CA2749786A patent/CA2749786A1/en not_active Abandoned
- 2010-01-15 EP EP10700685A patent/EP2387419A2/en not_active Withdrawn
- 2010-01-15 AU AU2010204552A patent/AU2010204552B2/en not_active Ceased
- 2010-01-15 US US12/688,754 patent/US20100227818A1/en not_active Abandoned
- 2010-01-15 US US12/688,655 patent/US8323634B2/en not_active Expired - Fee Related
- 2010-01-15 JP JP2011546396A patent/JP5753095B2/ja not_active Expired - Fee Related
- 2010-01-15 WO PCT/US2010/021241 patent/WO2010083439A2/en active Application Filing
- 2010-01-15 UA UAA201109963A patent/UA103221C2/uk unknown
- 2010-01-15 EA EA201190079A patent/EA201190079A1/ru unknown
- 2010-01-15 MX MX2011007582A patent/MX2011007582A/es active IP Right Grant
- 2010-01-15 EA EA201190080A patent/EA023344B1/ru not_active IP Right Cessation
- 2010-01-15 KR KR1020117018162A patent/KR20110132327A/ko active Search and Examination
- 2010-01-15 CN CN201080012749.8A patent/CN102395379B/zh not_active Expired - Fee Related
- 2010-01-15 BR BRPI1004940A patent/BRPI1004940A2/pt not_active IP Right Cessation
- 2010-01-15 WO PCT/US2010/021235 patent/WO2010083434A2/en active Application Filing
- 2010-01-15 NZ NZ594055A patent/NZ594055A/xx not_active IP Right Cessation
- 2010-01-15 BR BRPI1005159A patent/BRPI1005159A2/pt not_active IP Right Cessation
- 2010-01-15 NZ NZ594056A patent/NZ594056A/xx not_active IP Right Cessation
- 2010-01-15 KR KR1020117018153A patent/KR20110132326A/ko not_active Application Discontinuation
- 2010-01-15 AU AU2010204547A patent/AU2010204547B2/en not_active Ceased
-
2011
- 2011-07-12 IL IL214051A patent/IL214051A0/en unknown
- 2011-07-12 IL IL214052A patent/IL214052A0/en unknown
- 2011-07-13 ZA ZA2011/05171A patent/ZA201105171B/en unknown
-
2012
- 2012-10-25 US US13/660,915 patent/US8993519B2/en not_active Expired - Fee Related
-
2014
- 2014-12-18 US US14/574,553 patent/US20150202268A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005514060A (ja) * | 2001-12-21 | 2005-05-19 | ヒューマン ジノーム サイエンシーズ, インコーポレイテッド | アルブミン融合タンパク質 |
Non-Patent Citations (2)
| Title |
|---|
| JOURNAL OF CLINICAL ONCOLOGY,2000,VOL.18,NO.12,P.2369-2377, JPN6014008112, ISSN: 0002754759 * |
| PHARMACEUTICAL RESEARCH,2002,VOL.19,NO.11,P.1720-1729, JPN6014008109, ISSN: 0002754758 * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2010204552B2 (en) | New stable formulations of recombinant human albumin-human granulocyte colony stimulating factor fusion protein | |
| US10653751B2 (en) | Methods of treating cancer metastasis by using interleukin-10 | |
| US9943568B2 (en) | Methods of using pegylated interleukin-10 for treating cancer | |
| KR20150121715A (ko) | Csf1 치료제 | |
| JP2018530525A (ja) | 疾患及び障害を治療するためのインターロイキン10の使用方法 | |
| AU2014208224A1 (en) | New Stable Formulations of Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor Fusion Protein | |
| US20140271538A1 (en) | Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor for the Prevention of Neutropenia in Pediatric Patients | |
| AU2014202190B2 (en) | New Stable Formulations of Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor Fusion Protein | |
| CN117683140A (zh) | 肿瘤靶向的以白介素2为活性成分的融合蛋白型药物前体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130111 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130111 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140224 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140523 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140530 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20141127 |