JP2012201675A - カルボン酸の製造方法、及びカルボン酸の製造装置 - Google Patents
カルボン酸の製造方法、及びカルボン酸の製造装置 Download PDFInfo
- Publication number
- JP2012201675A JP2012201675A JP2011070858A JP2011070858A JP2012201675A JP 2012201675 A JP2012201675 A JP 2012201675A JP 2011070858 A JP2011070858 A JP 2011070858A JP 2011070858 A JP2011070858 A JP 2011070858A JP 2012201675 A JP2012201675 A JP 2012201675A
- Authority
- JP
- Japan
- Prior art keywords
- carboxylic acid
- catalyst
- gas
- product
- raw material
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 150000001732 carboxylic acid derivatives Chemical class 0.000 title claims abstract description 143
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 68
- 239000003054 catalyst Substances 0.000 claims abstract description 149
- 239000007789 gas Substances 0.000 claims abstract description 101
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 86
- 239000002994 raw material Substances 0.000 claims abstract description 63
- 229910052703 rhodium Inorganic materials 0.000 claims abstract description 26
- 239000010948 rhodium Substances 0.000 claims abstract description 26
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 claims abstract description 26
- 238000000034 method Methods 0.000 claims abstract description 21
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 17
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 claims abstract description 16
- 229910002091 carbon monoxide Inorganic materials 0.000 claims abstract description 16
- 239000001257 hydrogen Substances 0.000 claims abstract description 16
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 16
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 claims abstract description 15
- 229910001882 dioxygen Inorganic materials 0.000 claims abstract description 15
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 claims abstract description 15
- 239000010931 gold Substances 0.000 claims abstract description 6
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 claims abstract description 5
- 229910052737 gold Inorganic materials 0.000 claims abstract description 5
- 238000006243 chemical reaction Methods 0.000 claims description 77
- 239000007788 liquid Substances 0.000 claims description 44
- 125000004432 carbon atom Chemical group C* 0.000 claims description 37
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 28
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 20
- 239000006200 vaporizer Substances 0.000 claims description 20
- 229910052751 metal Inorganic materials 0.000 claims description 19
- 239000002184 metal Substances 0.000 claims description 19
- 230000008016 vaporization Effects 0.000 claims description 14
- 229910052783 alkali metal Inorganic materials 0.000 claims description 13
- 150000001340 alkali metals Chemical class 0.000 claims description 13
- 239000000377 silicon dioxide Substances 0.000 claims description 13
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 claims description 11
- 229910052796 boron Inorganic materials 0.000 claims description 11
- 239000011572 manganese Substances 0.000 claims description 10
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 claims description 9
- 229910052748 manganese Inorganic materials 0.000 claims description 9
- 238000000926 separation method Methods 0.000 claims description 6
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 claims description 4
- 239000002253 acid Substances 0.000 claims description 2
- ULDHMXUKGWMISQ-UHFFFAOYSA-N carvone Chemical compound CC(=C)C1CC=C(C)C(=O)C1 ULDHMXUKGWMISQ-UHFFFAOYSA-N 0.000 claims 2
- 239000005973 Carvone Substances 0.000 claims 1
- 238000006555 catalytic reaction Methods 0.000 abstract description 18
- 230000015572 biosynthetic process Effects 0.000 abstract description 12
- 238000003786 synthesis reaction Methods 0.000 abstract description 12
- 230000002194 synthesizing effect Effects 0.000 abstract description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 75
- 150000001299 aldehydes Chemical class 0.000 description 47
- 239000007864 aqueous solution Substances 0.000 description 15
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 12
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 12
- 238000009834 vaporization Methods 0.000 description 10
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 9
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 9
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 6
- 235000011054 acetic acid Nutrition 0.000 description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 6
- 235000019253 formic acid Nutrition 0.000 description 6
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 6
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 6
- 235000019260 propionic acid Nutrition 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 5
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 5
- 150000001735 carboxylic acids Chemical class 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 229910052744 lithium Inorganic materials 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 3
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 229910001873 dinitrogen Inorganic materials 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 3
- 239000011148 porous material Substances 0.000 description 3
- 239000008213 purified water Substances 0.000 description 3
- 239000010935 stainless steel Substances 0.000 description 3
- 229910001220 stainless steel Inorganic materials 0.000 description 3
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 239000002028 Biomass Substances 0.000 description 2
- 229910010413 TiO 2 Inorganic materials 0.000 description 2
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 239000004327 boric acid Substances 0.000 description 2
- 239000012018 catalyst precursor Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000005755 formation reaction Methods 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 238000005470 impregnation Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000010453 quartz Substances 0.000 description 2
- SONJTKJMTWTJCT-UHFFFAOYSA-K rhodium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Rh+3] SONJTKJMTWTJCT-UHFFFAOYSA-K 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 1
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 239000000956 alloy Substances 0.000 description 1
- 229910045601 alloy Inorganic materials 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- -1 and the like Chemical compound 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 229910002090 carbon oxide Inorganic materials 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- CETPSERCERDGAM-UHFFFAOYSA-N ceric oxide Chemical compound O=[Ce]=O CETPSERCERDGAM-UHFFFAOYSA-N 0.000 description 1
- 229910000422 cerium(IV) oxide Inorganic materials 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 150000004715 keto acids Chemical class 0.000 description 1
- 229940099607 manganese chloride Drugs 0.000 description 1
- 235000002867 manganese chloride Nutrition 0.000 description 1
- 239000011565 manganese chloride Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003345 natural gas Substances 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000003507 refrigerant Substances 0.000 description 1
- 229910052701 rubidium Inorganic materials 0.000 description 1
- IGLNJRXAVVLDKE-UHFFFAOYSA-N rubidium atom Chemical compound [Rb] IGLNJRXAVVLDKE-UHFFFAOYSA-N 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 150000004685 tetrahydrates Chemical class 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Images
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
【課題】合成ガスを原料とする触媒反応によって、高い選択率でカルボン酸を合成できるカルボン酸の製造方法、及び該製造方法に使用しうるカルボン酸の製造装置を提供する。
【解決手段】水素及び一酸化炭素を含む第一原料ガスをロジウム含有触媒αに接触させて、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む第一の生成物を得る工程Aと、前記第一の生成物から、前記アルコール及び/又は前記アルデヒドを含む第二の生成物、並びに前記カルボン酸を含む第三の生成物を得る工程Bと、気化させた前記第二の生成物と酸素ガスとを含む第二原料ガスを金含有触媒βに接触させて、炭素数1〜4のカルボン酸を含む第四の生成物を得る工程Cと、を少なくとも有することを特徴とするカルボン酸の製造方法。
【選択図】なし
【解決手段】水素及び一酸化炭素を含む第一原料ガスをロジウム含有触媒αに接触させて、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む第一の生成物を得る工程Aと、前記第一の生成物から、前記アルコール及び/又は前記アルデヒドを含む第二の生成物、並びに前記カルボン酸を含む第三の生成物を得る工程Bと、気化させた前記第二の生成物と酸素ガスとを含む第二原料ガスを金含有触媒βに接触させて、炭素数1〜4のカルボン酸を含む第四の生成物を得る工程Cと、を少なくとも有することを特徴とするカルボン酸の製造方法。
【選択図】なし
Description
本発明は、カルボン酸の製造方法、及びカルボン酸の製造装置に関する。
一酸化炭素と水素の混合ガス(合成ガス)は、C1化学における重要な原料の一つである。合成ガスは天然ガス、石炭、バイオマス等の石油以外の資源から得られるため、石油依存を脱却する化学として従来から研究が盛んである。近年では、バイオマスを有効利用するC1化学は、CO2の排出を低減し、地球温暖化を改善するとともに、持続可能な工業文明の構築にも資するものとして期待されている。
従来、ロジウムおよびアルカリ金属を含む触媒に、合成ガスを接触させることによって、酸素化物である、エタノール、酢酸、アセトアルデヒドを合成する方法が知られている(特許文献1〜2参照)。しかし、これらの方法では、酢酸以外にエタノール及びアセトアルデヒドが多く生成されるため、酢酸を従来より高い比率で製造する方法が望まれている。
本発明は上記事情に鑑みてなされたものであり、合成ガスを原料とする触媒反応によって、高い選択率でカルボン酸を合成できるカルボン酸の製造方法、及び該製造方法に使用しうるカルボン酸の製造装置の提供を課題とする。
本発明の請求項1に記載のカルボン酸の製造方法は、水素及び一酸化炭素を含む第一原料ガスを触媒αに接触させて、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む第一の生成物を得る工程Aと、前記第一の生成物から、前記アルコール及び/又は前記アルデヒドを含む第二の生成物、並びに前記カルボン酸を含む第三の生成物を得る工程Bと、気化させた前記第二の生成物と酸素ガスとを含む第二原料ガスを触媒βに接触させて、炭素数1〜4のカルボン酸を含む第四の生成物を得る工程Cと、を少なくとも有することを特徴とする。
本発明の請求項2に記載のカルボン酸の製造方法は、請求項1において、前記工程Bにおいて、前記第一の生成物に含まれる、前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを液化することによって、未反応の前記第一原料ガスから気液分離することを特徴とする。
本発明の請求項3に記載のカルボン酸の製造方法は、請求項1又は2において、前記触媒αが、ロジウム及びマンガンを含み、前記触媒α中のロジウム:マンガンのモル比が1:0.01〜1:1の範囲であることを特徴とする。
本発明の請求項4に記載のカルボン酸の製造方法は、請求項1又は2において、前記触媒αが、ロジウム、ホウ素、及びアルカリ金属を含み、前記触媒α中のロジウム:ホウ素のモル比が1:1〜1:5の範囲であり、前記触媒α中のロジウム:アルカリ金属のモル比が1:0.1〜1:1の範囲であり、前記触媒α中のホウ素:アルカリ金属のモル比が1:0.02〜1:1の範囲であることを特徴とする。
本発明の請求項5に記載のカルボン酸の製造方法は、請求項1〜4のいずれか一項において、前記触媒βが金を含むことを特徴とする。
本発明の請求項6に記載のカルボン酸の製造方法は、請求項1〜5のいずれか一項において、前記第一原料ガスを前記触媒αに接触させる際の温度が、150〜450℃の範囲であることを特徴とする。
本発明の請求項7に記載のカルボン酸の製造方法は、請求項1〜6のいずれか一項において、前記第二原料ガスを前記触媒βに接触させる際の温度が、100〜500℃の範囲であることを特徴とする。
本発明の請求項8に記載のカルボン酸の製造方法は、請求項1〜7のいずれか一項において、前記第一原料ガスを前記触媒αに接触させる際の圧力が、0.5MPa〜10MPaの範囲であることを特徴とする。
本発明の請求項9に記載のカルボン酸の製造方法は、請求項1〜8のいずれか一項において、前記第二原料ガスを前記触媒βに接触させる際の圧力が、0.1MPa〜1MPaの範囲であることを特徴とする。
本発明の請求項10に記載のカルボン酸の製造方法は、請求項1〜9のいずれか一項において、前記触媒αまたは前記触媒βが担持触媒であることを特徴とする。
本発明の請求項11に記載のカルボン酸の製造方法は、請求項10において、前記担持触媒αの担体が、シリカであることを特徴とする。
本発明の請求項12に記載のカルボン酸の製造方法は、請求項10又は11において、前記担持触媒βの担体が、チタニアであることを特徴とする。
本発明の請求項13に記載のカルボン酸の製造装置は、水素および一酸化炭素を含む第一原料ガスから、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む生成物を生成する触媒αが内部に配された第一反応管と、前記生成物中の前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを液化する気液分離器と、液化された前記アルコール及び/又は前記アルデヒドを気化する気化器と、気化された前記アルコール及び/又は前記アルデヒドと酸素ガスとを含む第二原料ガスから、炭素数1〜4のカルボン酸を含む生成物を生成する触媒βが内部に配された第二反応管と、が少なくとも備えられ、
前記第一反応管、前記第二反応管、前記気液分離器、及び前記気化器には、ガス若しくは液体の導入口及び排出口が各々設けられ、
前記第一反応管の排出口の下流に前記気液分離器の導入口が配管接続され、前記気液分離器の排出口の下流に前記気化器の導入口が配管接続され、前記気化器の排出口の下流に前記第二反応管の導入口が配管接続されたことを特徴とする。
本発明の請求項2に記載のカルボン酸の製造方法は、請求項1において、前記工程Bにおいて、前記第一の生成物に含まれる、前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを液化することによって、未反応の前記第一原料ガスから気液分離することを特徴とする。
本発明の請求項3に記載のカルボン酸の製造方法は、請求項1又は2において、前記触媒αが、ロジウム及びマンガンを含み、前記触媒α中のロジウム:マンガンのモル比が1:0.01〜1:1の範囲であることを特徴とする。
本発明の請求項4に記載のカルボン酸の製造方法は、請求項1又は2において、前記触媒αが、ロジウム、ホウ素、及びアルカリ金属を含み、前記触媒α中のロジウム:ホウ素のモル比が1:1〜1:5の範囲であり、前記触媒α中のロジウム:アルカリ金属のモル比が1:0.1〜1:1の範囲であり、前記触媒α中のホウ素:アルカリ金属のモル比が1:0.02〜1:1の範囲であることを特徴とする。
本発明の請求項5に記載のカルボン酸の製造方法は、請求項1〜4のいずれか一項において、前記触媒βが金を含むことを特徴とする。
本発明の請求項6に記載のカルボン酸の製造方法は、請求項1〜5のいずれか一項において、前記第一原料ガスを前記触媒αに接触させる際の温度が、150〜450℃の範囲であることを特徴とする。
本発明の請求項7に記載のカルボン酸の製造方法は、請求項1〜6のいずれか一項において、前記第二原料ガスを前記触媒βに接触させる際の温度が、100〜500℃の範囲であることを特徴とする。
本発明の請求項8に記載のカルボン酸の製造方法は、請求項1〜7のいずれか一項において、前記第一原料ガスを前記触媒αに接触させる際の圧力が、0.5MPa〜10MPaの範囲であることを特徴とする。
本発明の請求項9に記載のカルボン酸の製造方法は、請求項1〜8のいずれか一項において、前記第二原料ガスを前記触媒βに接触させる際の圧力が、0.1MPa〜1MPaの範囲であることを特徴とする。
本発明の請求項10に記載のカルボン酸の製造方法は、請求項1〜9のいずれか一項において、前記触媒αまたは前記触媒βが担持触媒であることを特徴とする。
本発明の請求項11に記載のカルボン酸の製造方法は、請求項10において、前記担持触媒αの担体が、シリカであることを特徴とする。
本発明の請求項12に記載のカルボン酸の製造方法は、請求項10又は11において、前記担持触媒βの担体が、チタニアであることを特徴とする。
本発明の請求項13に記載のカルボン酸の製造装置は、水素および一酸化炭素を含む第一原料ガスから、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む生成物を生成する触媒αが内部に配された第一反応管と、前記生成物中の前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを液化する気液分離器と、液化された前記アルコール及び/又は前記アルデヒドを気化する気化器と、気化された前記アルコール及び/又は前記アルデヒドと酸素ガスとを含む第二原料ガスから、炭素数1〜4のカルボン酸を含む生成物を生成する触媒βが内部に配された第二反応管と、が少なくとも備えられ、
前記第一反応管、前記第二反応管、前記気液分離器、及び前記気化器には、ガス若しくは液体の導入口及び排出口が各々設けられ、
前記第一反応管の排出口の下流に前記気液分離器の導入口が配管接続され、前記気液分離器の排出口の下流に前記気化器の導入口が配管接続され、前記気化器の排出口の下流に前記第二反応管の導入口が配管接続されたことを特徴とする。
本発明のカルボン酸の製造方法によれば、水素と一酸化炭素を含む合成ガスを原料として高い選択率でカルボン酸を合成することができる。
本発明のカルボン酸製造装置によれば、水素と一酸化炭素を含む合成ガスを原料として高い選択率でカルボン酸を合成することができる。
本発明のカルボン酸製造装置によれば、水素と一酸化炭素を含む合成ガスを原料として高い選択率でカルボン酸を合成することができる。
以下、本発明について詳しく説明する。
<<カルボン酸の製造方法>>
本発明のカルボン酸の製造方法は、水素及び一酸化炭素を含む第一原料ガスを触媒αに接触させて、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む第一の生成物を得る工程Aと、前記第一の生成物から、前記アルコール及び/又は前記アルデヒドを含む第二の生成物、並びに前記カルボン酸を含む第三の生成物を得る工程Bと、気化させた前記第二の生成物と酸素ガスとを含む第二原料ガスを触媒βに接触させて、炭素数1〜4のカルボン酸を含む第四の生成物を得る工程Cと、を少なくとも有する方法である。
本発明のカルボン酸の製造方法は、本発明の趣旨を逸脱しない限りにおいて、製造装置や製造条件に応じて、前記工程A〜C以外の工程を含んでいてもよい。
<<カルボン酸の製造方法>>
本発明のカルボン酸の製造方法は、水素及び一酸化炭素を含む第一原料ガスを触媒αに接触させて、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む第一の生成物を得る工程Aと、前記第一の生成物から、前記アルコール及び/又は前記アルデヒドを含む第二の生成物、並びに前記カルボン酸を含む第三の生成物を得る工程Bと、気化させた前記第二の生成物と酸素ガスとを含む第二原料ガスを触媒βに接触させて、炭素数1〜4のカルボン酸を含む第四の生成物を得る工程Cと、を少なくとも有する方法である。
本発明のカルボン酸の製造方法は、本発明の趣旨を逸脱しない限りにおいて、製造装置や製造条件に応じて、前記工程A〜C以外の工程を含んでいてもよい。
前記触媒αは、前記第一原料ガスに含まれる水素及び一酸化炭素を原料として、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを生成する触媒活性を有するものである。
前記触媒βは、前記第二原料ガスに含まれる炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒド、並びに酸素ガスを原料として、炭素数1〜4のカルボン酸を生成する触媒活性を有するものである。
本発明のカルボン酸の製造方法では、前記工程Bにおいて、前記第一の生成物に含まれる、前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを液化することによって、未反応の前記第一原料ガスから気液分離することが好ましい。
前記気液分離を行うことによって、前記触媒βにおける前記カルボン酸を生成する反応効率を高めて、本発明のカルボン酸の製造方法の全工程を通して最終的に得られる前記カルボン酸の選択率を一層向上させられる。
前記気液分離を行うことによって、前記触媒βにおける前記カルボン酸を生成する反応効率を高めて、本発明のカルボン酸の製造方法の全工程を通して最終的に得られる前記カルボン酸の選択率を一層向上させられる。
本発明のカルボン酸の製造方法は、工程A〜工程Cを少なくとも有する方法である。以下に、工程A〜Cの実施形態を例示するが、本発明はこの実施形態に限定されるものではない。
[工程A〜C]
工程Aにおいて、前記第一原料ガスを前記触媒αに接触させることによって、前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを含む第一の生成物を得る。
次に工程Bにおいて、前記第一の生成物に含まれる前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを冷却することによって液化し、未反応の前記第一原料ガスと後段の触媒βで用いる酸素化物とを気液分離する。この際、未反応の前記第一原料ガス及びメタン等の酸素化物以外のガス成分は系外へ適宜除去する。次いで、得られた液体に含まれる前記アルコール及び/又は前記アルデヒドを第一の気化条件で気化(蒸留)することによって、前記アルコール及び/又は前記アルデヒドを含む前記第二の生成物を得る。また、前記液体のうち、気化(蒸留)されずに残る前記カルボン酸を含む残留液を前記第三の生成物として得る。或いは、前記残留液を第二の気化条件で気化(蒸留)することによって、前記カルボン酸を含むガスを第三の生成物として得る。
次に工程Cにおいて、気化された前記第二の生成物と、別途導入する酸素ガスとを混合して前記第二原料ガスとし、前記第二原料ガスを前記触媒βに接触させることによって、前記カルボン酸を含む第四の生成物を得る。
なお、前記第二原料ガスには、窒素ガスやアルゴンガス等の不活性ガス又は別のガスを必要に応じて混合してもよい。
また、前記第三の生成物及び前記第四の生成物として得られた前記カルボン酸は、必要に応じて、蒸留等の公知の精製方法によって純度を一層高められる。
工程Aにおいて、前記第一原料ガスを前記触媒αに接触させることによって、前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを含む第一の生成物を得る。
次に工程Bにおいて、前記第一の生成物に含まれる前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを冷却することによって液化し、未反応の前記第一原料ガスと後段の触媒βで用いる酸素化物とを気液分離する。この際、未反応の前記第一原料ガス及びメタン等の酸素化物以外のガス成分は系外へ適宜除去する。次いで、得られた液体に含まれる前記アルコール及び/又は前記アルデヒドを第一の気化条件で気化(蒸留)することによって、前記アルコール及び/又は前記アルデヒドを含む前記第二の生成物を得る。また、前記液体のうち、気化(蒸留)されずに残る前記カルボン酸を含む残留液を前記第三の生成物として得る。或いは、前記残留液を第二の気化条件で気化(蒸留)することによって、前記カルボン酸を含むガスを第三の生成物として得る。
次に工程Cにおいて、気化された前記第二の生成物と、別途導入する酸素ガスとを混合して前記第二原料ガスとし、前記第二原料ガスを前記触媒βに接触させることによって、前記カルボン酸を含む第四の生成物を得る。
なお、前記第二原料ガスには、窒素ガスやアルゴンガス等の不活性ガス又は別のガスを必要に応じて混合してもよい。
また、前記第三の生成物及び前記第四の生成物として得られた前記カルボン酸は、必要に応じて、蒸留等の公知の精製方法によって純度を一層高められる。
前記第一の気化条件は前記アルコール及び/又は前記アルデヒドを気化させられる温度及び圧力であれば特に制限されない。例えば大気圧(1気圧)において、50〜150℃が好ましく、70〜90℃がより好ましい。
上記範囲の下限値以上とすることによって、前記アルコール及び/又は前記アルデヒドをより高効率で気化させることができる。上記範囲の上限値以下とすることによって、前記アルコール及び/又は前記アルデヒドを、前記カルボン酸よりも優先的に気化させることができ、前記アルコール及び/又は前記アルデヒドをより高純度で含む前記第二の生成物を得られる。
前記第一の気化条件における温度は、前記第二の気化条件における温度よりも低温であることが好ましい。
上記範囲の下限値以上とすることによって、前記アルコール及び/又は前記アルデヒドをより高効率で気化させることができる。上記範囲の上限値以下とすることによって、前記アルコール及び/又は前記アルデヒドを、前記カルボン酸よりも優先的に気化させることができ、前記アルコール及び/又は前記アルデヒドをより高純度で含む前記第二の生成物を得られる。
前記第一の気化条件における温度は、前記第二の気化条件における温度よりも低温であることが好ましい。
前記第二の気化条件は前記カルボン酸を気化させられる温度及び圧力であれば特に制限されない。例えば大気圧(1気圧)において、90〜180℃が好ましく、110〜130℃がより好ましい。
上記範囲の下限値以上とすることによって、前記カルボン酸をより高効率で気化させることができる。上記範囲の上限値以下とすることによって、前記残留液から前記カルボン酸よりも沸点の高い成分が気化することを抑制しつつ、前記カルボン酸を気化させることができ、前記カルボン酸をより高純度で含む前記第三の生成物を得られる。
前記第二の気化条件における温度は、前記第一の気化条件における温度よりも高温であることが好ましい。
上記範囲の下限値以上とすることによって、前記カルボン酸をより高効率で気化させることができる。上記範囲の上限値以下とすることによって、前記残留液から前記カルボン酸よりも沸点の高い成分が気化することを抑制しつつ、前記カルボン酸を気化させることができ、前記カルボン酸をより高純度で含む前記第三の生成物を得られる。
前記第二の気化条件における温度は、前記第一の気化条件における温度よりも高温であることが好ましい。
本発明のカルボン酸の製造方法によれば、前記触媒αで生成された前記アルコール及び/又は前記アルデヒドを、前記触媒βでカルボン酸に変換できる。つまり、前記触媒α及び前記触媒βの各触媒反応においてカルボン酸を得ることができる。この結果、原料ガスに含まれる一酸化炭素を高い選択率でカルボン酸に変換して得られる。
前記第一原料ガスにおける水素ガスと一酸化炭素との混合比(体積比)は、水素ガス:一酸化炭素ガス=5:1〜1:5の範囲が好ましく、3:1〜1:2の範囲がより好ましく、2.5:1〜1:1の範囲がさらに好ましい。
上記範囲であると、カルボン酸を高い選択率で合成でき、生成されるカルボン酸に含まれる酢酸の比率を高められる。
上記範囲であると、カルボン酸を高い選択率で合成でき、生成されるカルボン酸に含まれる酢酸の比率を高められる。
本発明において、「選択率」とは、「合成ガス中の消費されたCOのモル数のうち、特定の酸素化物へ変換されたCのモル数が占める百分率」を意味する。例えば、以下の反応式(A)によれば、酸素化物であるカルボン酸(酢酸)の選択率は100モル%である。一方、以下の反応式(B)によれば、酸素化物であるカルボン酸(酢酸)の選択率は50モル%であり、酸素化物であるアセトアルデヒドの選択率も50モル%である。
(A)2H2+2CO → CH3COOH
(B)5H2+4CO → CH3COOH+CH3CHO+H2O
(A)2H2+2CO → CH3COOH
(B)5H2+4CO → CH3COOH+CH3CHO+H2O
本発明において、前記触媒αは、前記第一原料ガスから炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを生成するものである。
前記触媒αとしては、前記第一原料ガスから炭素数1〜4のカルボン酸のうち何れか一種以上を生成し、これに加えて、炭素数1〜4のアルコール及び炭素数1〜4のアルデヒドのうち何れか一種以上を生成するものが好ましい。
前記触媒αとしては、前記第一原料ガスから炭素数1〜4のカルボン酸のうち何れか一種以上を生成し、これに加えて、炭素数1〜4のアルコール及び炭素数1〜4のアルデヒドのうち何れか一種以上を生成するものが好ましい。
前記触媒αとしては、ロジウム及びマンガンを含むものが、前記アルデヒド及び/又は前記アルコールの選択率を高められるため好ましい。
また、前記触媒αとしては、前記カルボン酸の選択率を一層高める観点から、ロジウム、並びにホウ素、アルカリ金属及びマンガンのうち、いずれか一種以上の金属を含むものが好ましく、ロジウム、ホウ素、及びアルカリ金属を含むものがより好ましい。
前記金属を含む触媒αを用いることにより、触媒αにおいて前記原料ガスから合成される酸素化物の選択率を高めて、該酸素化物に含まれる、炭素数1〜4のカルボン酸、炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドの比率(モル%)を高められる。
また、前記触媒αとしては、前記カルボン酸の選択率を一層高める観点から、ロジウム、並びにホウ素、アルカリ金属及びマンガンのうち、いずれか一種以上の金属を含むものが好ましく、ロジウム、ホウ素、及びアルカリ金属を含むものがより好ましい。
前記金属を含む触媒αを用いることにより、触媒αにおいて前記原料ガスから合成される酸素化物の選択率を高めて、該酸素化物に含まれる、炭素数1〜4のカルボン酸、炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドの比率(モル%)を高められる。
本発明において、前記触媒βは、酸素(酸素ガス)と前記アルコール及び/又は前記アルデヒドとを原料として、炭素数1〜4のカルボン酸を生成するものである。
前記触媒βとしては、前記触媒αによって生成される第一の生成物から前記アルコール及び/又はアルデヒドを分離して得られたガスと、別途導入する酸素ガスとを混合して含む前記第二原料ガスから、炭素数1〜4のカルボン酸のうち何れか一種以上を生成するものが好ましい。
前記触媒βとしては、前記触媒αによって生成される第一の生成物から前記アルコール及び/又はアルデヒドを分離して得られたガスと、別途導入する酸素ガスとを混合して含む前記第二原料ガスから、炭素数1〜4のカルボン酸のうち何れか一種以上を生成するものが好ましい。
前記触媒βとしては、金(Au)、白金(Pt)、ルテニウム(Ru)、銅(Cu)、及びマンガン(Mn)のうち、いずれか一種以上の金属を含むものが好ましく、金を含むものがより好ましい。
前記金属を含む触媒βを用いることにより、本発明のカルボン酸の製造方法の全工程を通して、前記第一原料ガスから最終的に合成される炭素数1〜4のカルボン酸の選択率(モル%)を高められる。
前記金属を含む触媒βを用いることにより、本発明のカルボン酸の製造方法の全工程を通して、前記第一原料ガスから最終的に合成される炭素数1〜4のカルボン酸の選択率(モル%)を高められる。
前記触媒αが、ロジウム及びマンガンを含むものである場合、前記触媒α中のロジウム:マンガンのモル比が1:0.01〜1:1の範囲であることが好ましく、1:0.1〜1:0.5の範囲であることがより好ましい。
上記範囲とすることによって、前記第一原料ガスから前記触媒αにおいて合成される酸素化物の選択率を高めて、該酸素化物に含まれる、前記アルコール及び/又は前記アルコールの比率を高め、前記触媒α及び触媒βによって最終的に生成される前記カルボン酸の比率(モル%)を高められる。
また、上記範囲とすることによって、前記アルコール及び/又は前記アルデヒドを、主生成物として(例えば選択率40%以上で)生成することが容易となる。
上記範囲とすることによって、前記第一原料ガスから前記触媒αにおいて合成される酸素化物の選択率を高めて、該酸素化物に含まれる、前記アルコール及び/又は前記アルコールの比率を高め、前記触媒α及び触媒βによって最終的に生成される前記カルボン酸の比率(モル%)を高められる。
また、上記範囲とすることによって、前記アルコール及び/又は前記アルデヒドを、主生成物として(例えば選択率40%以上で)生成することが容易となる。
前記触媒αが、ロジウム、ホウ素、及びアルカリ金属を含むものである場合、前記触媒α中のロジウム:ホウ素のモル比が1:1〜1:5の範囲であり、前記触媒α中のロジウム:アルカリ金属のモル比が1:0.1〜1:1の範囲であり、前記触媒α中のホウ素:アルカリ金属のモル比が1:0.02〜1:1の範囲であることが好ましい。
上記範囲とすることによって、前記第一原料ガスから前記触媒αにおいて合成される酸素化物の選択率を高めて、該酸素化物に含まれる前記カルボン酸の比率を一層高めることができ、さらに前記アルコール及び/又は前記アルデヒドも比較的高い比率(モル%)で得られる。また、前記触媒α及び触媒βによって最終的に生成されるカルボン酸の選択率(モル%)を高められる。さらに、生成されるカルボン酸のうち、酢酸の比率(モル%)を高められる。具体的には、生成される前記カルボン酸に含まれる酢酸の比率を95モル%以上とすることができる。
また、上記範囲とすることによって、前記カルボン酸を、主生成物として(例えば選択率40%以上で)生成することが容易となる。
上記範囲とすることによって、前記第一原料ガスから前記触媒αにおいて合成される酸素化物の選択率を高めて、該酸素化物に含まれる前記カルボン酸の比率を一層高めることができ、さらに前記アルコール及び/又は前記アルデヒドも比較的高い比率(モル%)で得られる。また、前記触媒α及び触媒βによって最終的に生成されるカルボン酸の選択率(モル%)を高められる。さらに、生成されるカルボン酸のうち、酢酸の比率(モル%)を高められる。具体的には、生成される前記カルボン酸に含まれる酢酸の比率を95モル%以上とすることができる。
また、上記範囲とすることによって、前記カルボン酸を、主生成物として(例えば選択率40%以上で)生成することが容易となる。
本発明において、「酸素化物」は、蟻酸、酢酸、プロパン酸、酪酸、メタノール、エタノール、プロパノール、ブタノール、ホルムアルデヒド、アセトアルデヒド、プロピオンアルデヒド、ブチルアルデヒド、蟻酸メチル、蟻酸エチル、酢酸メチル、酢酸エチルなどの炭素原子と水素原子と酸素原子からなる分子を意味する。
本発明において、最終的に生成される酸素化物は、蟻酸、酢酸、プロパン酸、及び酪酸の炭素数1〜4のカルボン酸のうち、何れか一つ以上を含むことが好ましい。
本発明において、最終的に生成される酸素化物は、蟻酸、酢酸、プロパン酸、及び酪酸の炭素数1〜4のカルボン酸のうち、何れか一つ以上を含むことが好ましい。
前記触媒αにおいて、第一の生成物として炭素数1〜4のアルコールが生成される場合、該主生成物には、メタノール、エタノール、プロパノール、及びブタノールのうち、いずれか1種以上が含まれることが好ましい。これにより、最終的に生成されるカルボン酸として、蟻酸、酢酸、プロパン酸、及び酪酸を得ることが容易になる。
前記触媒αにおいて、前記第一原料ガスから、前記アルコールが生成する触媒反応としては、以下の反応式が考えられる。
(1)2H2+CO → CH3OH
(2)4H2+2CO → CH3CH2OH+H2O
(3)6H2+3CO → CH3CH2CH2OH+2H2O
(4)8H2+4CO → CH3CH2CH2CH2OH+3H2O
前記触媒αにおいて、前記第一原料ガスから、前記アルコールが生成する触媒反応としては、以下の反応式が考えられる。
(1)2H2+CO → CH3OH
(2)4H2+2CO → CH3CH2OH+H2O
(3)6H2+3CO → CH3CH2CH2OH+2H2O
(4)8H2+4CO → CH3CH2CH2CH2OH+3H2O
前記触媒αにおいて、第一の生成物として炭素数1〜4のアルデヒドが生成される場合、該主生成物には、ホルムアルデヒド、アセトアルデヒド、プロピオンアルデヒド、及びブチルアルデヒドのうち、いずれか1種以上が含まれることが好ましい。これにより、最終的に生成されるカルボン酸として、蟻酸、酢酸、プロパン酸、及び酪酸を得ることが容易になる。
前記触媒αにおいて、前記第一原料ガスから前記アルデヒドが生成する触媒反応としては、以下の反応式が考えられる。
(1)H2+CO → HCHO
(2)3H2+2CO → CH3CHO+H2O
(3)5H2+3CO → CH3CH2CHO+2H2O
(4) 7H2+4CO → CH3CH2CH2CHO+3H2O
前記触媒αにおいて、前記第一原料ガスから前記アルデヒドが生成する触媒反応としては、以下の反応式が考えられる。
(1)H2+CO → HCHO
(2)3H2+2CO → CH3CHO+H2O
(3)5H2+3CO → CH3CH2CHO+2H2O
(4) 7H2+4CO → CH3CH2CH2CHO+3H2O
前記触媒αにおいて、第一の生成物として、炭素数1〜4のアルコール及び炭素数1〜4のアルデヒドが生成される場合、該主生成物にはメタノール、エタノール、プロパノール、及びブタノールのうち、いずれか1種以上のアルコールと、ホルムアルデヒド、アセトアルデヒド、プロピオンアルデヒド、及びブチルアルデヒドのうち、いずれか1種以上と、が含まれることが好ましい。これにより、最終的に生成されるカルボン酸として、蟻酸、酢酸、プロパン酸、及び酪酸を得ることが容易になる。これらが合成される触媒反応は、前述と同様である。
前記触媒αにおける触媒反応の温度としては、150〜450℃の範囲が好ましく、200〜400℃の範囲がより好ましく、250〜350℃の範囲がさらに好ましい。
上記範囲とすることにより、触媒αにおいて生成される酸素化物を高い選択率で得ることができ、生成される酸素化物に含まれる前記アルコール及び/又は前記アルデヒドの比率(モル%)を高められる。また、生成される前記アルコール及び/又は前記アルデヒドのうち、エタノール及び/又はアセトアルデヒドの比率(モル%)を高められる。この結果、後段の触媒βを経て最終的に生成される前記カルボン酸の選択率(モル%)を高められる。また、後段の触媒βを経て最終的に生成される前記カルボン酸のうち、酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記酸素化物の生成反応を主反応とすることができる。
上記範囲とすることにより、触媒αにおいて生成される酸素化物を高い選択率で得ることができ、生成される酸素化物に含まれる前記アルコール及び/又は前記アルデヒドの比率(モル%)を高められる。また、生成される前記アルコール及び/又は前記アルデヒドのうち、エタノール及び/又はアセトアルデヒドの比率(モル%)を高められる。この結果、後段の触媒βを経て最終的に生成される前記カルボン酸の選択率(モル%)を高められる。また、後段の触媒βを経て最終的に生成される前記カルボン酸のうち、酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記酸素化物の生成反応を主反応とすることができる。
前記触媒αにおける触媒反応の圧力としては、0.5MPa〜10MPaの範囲が好ましく、1MPa〜7.5MPaの範囲がより好ましく、2MPa〜5MPaの範囲がさらに好ましい。
上記範囲とすることにより、前記触媒αにおいて生成される酸素化物を高い選択率で得ることができ、生成される酸素化物に含まれる前記アルコール及び/又は前記アルデヒドの比率(モル%)を高められる。また、生成される前記アルコール及び/又は前記アルデヒドのうち、エタノール及び/又はアセトアルデヒドの比率(モル%)を高められる。この結果、後段の触媒βを経て最終的に生成される酸素化物を高い選択率で得ることができ、該酸素化物に含まれるカルボン酸の比率(モル%)を高められる。また、後段の触媒βを経て最終的に生成される前記カルボン酸のうち、酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記酸素化物の生成反応を主反応とすることができる。
上記範囲とすることにより、前記触媒αにおいて生成される酸素化物を高い選択率で得ることができ、生成される酸素化物に含まれる前記アルコール及び/又は前記アルデヒドの比率(モル%)を高められる。また、生成される前記アルコール及び/又は前記アルデヒドのうち、エタノール及び/又はアセトアルデヒドの比率(モル%)を高められる。この結果、後段の触媒βを経て最終的に生成される酸素化物を高い選択率で得ることができ、該酸素化物に含まれるカルボン酸の比率(モル%)を高められる。また、後段の触媒βを経て最終的に生成される前記カルボン酸のうち、酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記酸素化物の生成反応を主反応とすることができる。
前記触媒βにおいて生成される炭素数1〜4のカルボン酸としては、蟻酸、酢酸、プロパン酸、酪酸等が挙げられる。
前記触媒βにおいて、酸素ガス及び前記アルコール若しくは前記アルデヒドから、前記カルボン酸が生成する触媒反応としては、以下の反応式が考えられる。
(1)2CH3CH2OH+O2 → 2CH3CHO+2H2O
(2)2CH3CHO+O2 → 2CH3COOH
前記触媒βにおいて、酸素ガス及び前記アルコール若しくは前記アルデヒドから、前記カルボン酸が生成する触媒反応としては、以下の反応式が考えられる。
(1)2CH3CH2OH+O2 → 2CH3CHO+2H2O
(2)2CH3CHO+O2 → 2CH3COOH
前記触媒βにおける触媒反応の温度としては、100〜400℃の範囲が好ましく、150〜350℃の範囲がより好ましく、150〜250℃の範囲がさらに好ましい。
上記範囲とすることにより、前記アルコール若しくは前記アルデヒド及び酸素ガスから、前記カルボン酸を高い選択率で得ることができ、生成される酸素化物に含まれる酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記カルボン酸の生成反応を主反応とすることができる。
上記範囲とすることにより、前記アルコール若しくは前記アルデヒド及び酸素ガスから、前記カルボン酸を高い選択率で得ることができ、生成される酸素化物に含まれる酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記カルボン酸の生成反応を主反応とすることができる。
前記触媒βにおける触媒反応の圧力としては、0.1MPa〜2MPaの範囲が好ましく、0.5MPa〜1MPaの範囲がより好ましい。
上記範囲とすることにより、前記アルコール若しくは前記アルデヒド及び酸素ガスから、前記カルボン酸を高い選択率で得ることができ、生成される酸素化物に含まれる酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記カルボン酸の生成反応を主反応とすることができる。
上記範囲とすることにより、前記アルコール若しくは前記アルデヒド及び酸素ガスから、前記カルボン酸を高い選択率で得ることができ、生成される酸素化物に含まれる酢酸の比率(モル%)を高められる。
上記範囲の下限値以上とすることにより、触媒反応の速度を充分に高められる。上記範囲の上限値以下とすることにより、前記カルボン酸の生成反応を主反応とすることができる。
本発明のカルボン酸の製造方法によれば、2つの工程において前記カルボン酸を得られるので、従来よりも前記カルボン酸の選択率を高められる。すなわち、前記工程Bにおいて第三の生成物として前記カルボン酸を得ることができ、且つ前記工程Cにおいて第四の生成物として前記カルボン酸を得ることができるので、第一原料ガスに含まれる一酸化炭素を高効率で前記カルボン酸に変換できる。前記酢酸等のカルボン酸の生成比率が高められるメカニズムの一つとしては、触媒αによって高い選択率でアルデヒド及び/又はアルコールを合成し、これらを原料として触媒βにおいて高い選択率でカルボン酸を合成できるためであると考えられる。具体的には、水素及び一酸化炭素から直接カルボン酸を生成するよりも、エタノール及び/又はアセトアルデヒドから酢酸を生成する方が容易であるからだと考えられる。
ここで、前記触媒αにおいて、単位時間あたりに、前記第一原料ガスから前記アルコールと前記アルデヒドが合計1モル生成されるのに必要な触媒量を触媒αの1単位量と定義する。また、前記触媒βにおいて、単位時間あたりに、前記第二原料ガスから、前記カルボン酸が1モル生成されるのに必要な触媒量を、触媒βの1単位量と定義する。
このとき、本発明において、触媒αと触媒βの触媒量の比は、触媒α:触媒β=1:1(単位量)であることが好ましい。
触媒量が上記の比であることによって、本発明のカルボン酸の製造方法における触媒の使用効率をより高めることができる。また、前記第一原料ガスから最終的に合成される酸素化物の選択率を高めて、該酸素化物のうち、前記カルボン酸の選択率(モル%)を高められる。
触媒量が上記の比であることによって、本発明のカルボン酸の製造方法における触媒の使用効率をより高めることができる。また、前記第一原料ガスから最終的に合成される酸素化物の選択率を高めて、該酸素化物のうち、前記カルボン酸の選択率(モル%)を高められる。
前記触媒量の比を触媒α:触媒β=1:1(単位量)とする場合、下記の触媒α及び触媒βの組み合わせが好適なものとして挙げられる。
前記触媒αとしては、ロジウム及びマンガンを含むものが好ましい。より好ましくは、前記触媒αがロジウム及びマンガンを含み、且つ前記触媒α中のロジウム:マンガンのモル比が1:0.01〜1:1の範囲であることがより好ましい。
このときの触媒βは、前述した触媒βのいずれも好ましく用いられる。
このときの触媒βは、前述した触媒βのいずれも好ましく用いられる。
本発明において、前記アルカリ金属としては、リチウム(Li)、ナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)等が挙げられる。これらのなかでも、触媒反応によって生成する前記カルボン酸の選択率を高める観点から、リチウムが好ましい。
前記触媒α及び触媒βに使用しうる前記金属(元素)は、合金を形成していてもよく、形成していなくてもよい。これら金属は、前記原料ガスとの接触面積が大きくなる状態であることが好ましい。例えば、金属を粉体にしたもの、金属を多孔性の担体の表面および細孔内に担持させて担持触媒にしたもの等が挙げられる。
前記担持触媒の担体としては、金属触媒の担体として周知のものが使用できるが、比表面積が10〜1000m2/gで、多孔質性のものが好ましい。具体的には、シリカ、チタニア、アルミナ、セリア等が挙げられ、シリカ又はチタニア(TiO2)が好ましい。
前記シリカは粒子径(大きさ)の分布が狭いものが好ましい。その平均粒子径は特に制限されないが、例えば、0.5μm〜5000μmのものが好ましい。
各金属(元素)の合計の質量と、担体であるシリカ又はチタニア(TiO2)との質量比は、0.0005:1〜0.1:1の範囲が好ましい。
前記シリカは粒子径(大きさ)の分布が狭いものが好ましい。その平均粒子径は特に制限されないが、例えば、0.5μm〜5000μmのものが好ましい。
各金属(元素)の合計の質量と、担体であるシリカ又はチタニア(TiO2)との質量比は、0.0005:1〜0.1:1の範囲が好ましい。
前記触媒α及び触媒βに使用しうる前記金属(元素)をシリカ、チタニア等の担体の表面及び細孔内に担持(付着)させる方法としては、いわゆる含浸法が適用できる。より具体的には、前記金属の水溶液を調製し、これにシリカ又はチタニアを浸漬して、該水溶液をシリカの細孔中に含浸させた後、該シリカ又はチタニアを110℃で3時間加熱し、さらに450℃で3時間加熱することによって、行う方法が例示できる。前記水溶液中の各金属の濃度比(モル比)が、前記触媒中の各金属の存在比(モル比)とほぼ同じになる。
前記水溶液を調製する方法としては、例えば、各金属の塩化物やオキソ酸をそれぞれ水に溶解して調製することができる。
含浸は、各金属を同時に含浸させてもよいし、別々に水溶液を調製し、逐次含浸させてもよい。また、アルカリ金属は2種類以上を使用してもよい。
また、触媒活性、選択率を向上させるためにその他の元素を助触媒として添加しても良い。
前記水溶液を調製する方法としては、例えば、各金属の塩化物やオキソ酸をそれぞれ水に溶解して調製することができる。
含浸は、各金属を同時に含浸させてもよいし、別々に水溶液を調製し、逐次含浸させてもよい。また、アルカリ金属は2種類以上を使用してもよい。
また、触媒活性、選択率を向上させるためにその他の元素を助触媒として添加しても良い。
前記触媒α及び触媒βに使用しうる前記金属(元素)がシリカやチタニアに担持された担持触媒を、例えばステンレス製の反応管の内部に配することによって、本発明の製造装置とすることができる。製造装置については後述するが、例えば、前記反応管のガス導入口から前記原料ガスを流入することによって、該反応管のガス排出口から酸素化物を含有するガスを得ることができる。
前記触媒αを内部に配した前記反応管内に、前記第一原料ガスを流通させる空間速度(単位時間あたり原料ガス流通量÷触媒容量)は、標準状態換算で10h−1〜100000h−1が好ましく、1000〜50000h−1がより好ましく、3000〜20000h−1がさらに好ましい。上記範囲から、目的とする酸素化物に適した反応圧力、反応温度、及び第一原料ガスの組成に応じて適宜調整すればよい。
前記触媒βを内部に配した前記反応管内に、前記第二原料ガスを流通させる空間速度(単位時間あたり原料ガス流通量÷触媒容量)は、標準状態換算で10h−1〜50000h−1が好ましく、500〜20000h−1がより好ましく、1000〜10000h−1がさらに好ましい。上記範囲から、目的とする酸素化物に適した反応圧力、反応温度、及び第二原料ガスの組成に応じて適宜調整すればよい。
<<カルボン酸の製造装置>>
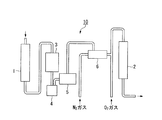
本発明のカルボン酸の製造装置の一例である製造装置10を、図1を参照して以下に説明する。製造装置10は、前記触媒αが内部に配された第一反応管1、第一の生成物に含まれる前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドと第一原料ガスとを分離する気液分離器3、液化して得られた前記アルコール及び/又は前記アルデヒドを気化する気化器6、及び前記触媒βが内部に配された第二反応管2を少なくとも備え、各反応管及び各機器には、ガス若しくは液体の導入口及び排出口が設けられており、第一反応管1の排出口が気液分離器3の導入口に配管接続され(配管によって接続され)、気液分離器3の排出口が液捕集器4の導入口に配管接続され、液捕集器4の排出口が送液ポンプ5を介して気化器6の導入口に配管接続され、気化器6の排出口が第二反応管2の導入口5に配管接続されている。
本発明のカルボン酸の製造装置の一例である製造装置10を、図1を参照して以下に説明する。製造装置10は、前記触媒αが内部に配された第一反応管1、第一の生成物に含まれる前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドと第一原料ガスとを分離する気液分離器3、液化して得られた前記アルコール及び/又は前記アルデヒドを気化する気化器6、及び前記触媒βが内部に配された第二反応管2を少なくとも備え、各反応管及び各機器には、ガス若しくは液体の導入口及び排出口が設けられており、第一反応管1の排出口が気液分離器3の導入口に配管接続され(配管によって接続され)、気液分離器3の排出口が液捕集器4の導入口に配管接続され、液捕集器4の排出口が送液ポンプ5を介して気化器6の導入口に配管接続され、気化器6の排出口が第二反応管2の導入口5に配管接続されている。
第一反応管1の導入口から第一原料ガスを導入し、前記触媒αにおける触媒反応によって前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを含む第一の生成物を合成する。第一の生成物および未反応の第一原料ガスを、第一反応管1の排出口から排出し、気液分離器3へ導入して、第一の生成物から前記アルコール及び/又は前記アルデヒドを含む第二の生成物、並びに前記カルボン酸を含む第三の生成物を液化させて、液捕集器4に溜める。この際、水素、一酸化炭素、及びメタン等を含むガス成分を装置外へ排出する。得られた第二の生成物及び第三の生成物が含まれる液を、送液ポンプ5によって気化器6に導入して、少なくとも第二の生成物に含まれる前記アルコール及び/又は前記アルデヒドを気化して、別の配管から導入する窒素ガスと混合して気化器6から排出して、さらに別の配管から導入する酸素ガスと混合して第二原料ガスを得る。第二原料ガスを第二反応管2の導入口へ導入する。第二反応管において、前記触媒βにおける触媒反応によって前記カルボン酸を含む第四の生成物を生成する。第四の生成物は、第二反応管2の排出口から回収される。
ここで、前記触媒αで生成される第一の生成物の主成分が炭素数1及び/又は2の酸素化物であれば、気化器6の温度等の条件を制御することによって、当該アルデヒド及び/又は当該アルコールを主に気化し、第二反応管2へ導入しても良い。気化器6の大きさ、流速にもよるが、例えば温度を90℃に設定することにより前記炭素数2の酸素化物のうち、アセトアルデヒド及びエタノールを気化させ、酢酸及び水は気化させずに液体のままで気化器6において回収することによって、前記気化に要するエネルギーを低減させられる。このように、本発明のカルボン酸の製造装置は、エネルギー的に有利なシステム構成とすることができる。
前記反応管としては、原料ガス及び生成物に対して不活性な材料からなるものが好ましく、100〜500℃程度の加熱、又は10Mpa程度の加圧に耐えうる形状のものが好ましい。例えば、ステンレス製の円筒型(直径1インチ、長さ15インチ)で、その両端にガス導入バルブ及びガス排出バルブが各々備えられたものが挙げられる。
前記反応管を加熱又は加圧する方法は、周知の方法が適用できる。つまり、本発明の製造装置には、電気炉等の温度制御部、マスフロー等のガスの流量を調整するガス流量制御部、圧力弁等の圧力を調整する圧力制御部、などの周知の装置構成が備えられていてもよい。
前記反応管を加熱又は加圧する方法は、周知の方法が適用できる。つまり、本発明の製造装置には、電気炉等の温度制御部、マスフロー等のガスの流量を調整するガス流量制御部、圧力弁等の圧力を調整する圧力制御部、などの周知の装置構成が備えられていてもよい。
前記反応管の内部に触媒を配する際には、前記原料ガスを流通させる空間速度(単位時間あたり原料ガス流通量×触媒容量)が、標準状態換算で10h−1〜100000h−1の範囲となるようにすることが好ましい。目的とする酸素化物に適した反応圧力、反応温度、及び原料ガスの組成に応じて、より適した空間速度となるように、上記範囲内で適宜調整すればよい。
次に、実施例により本発明をさらに詳細に説明するが、本発明はこれらの例によって限定されるものではない。
<実施例1>
[触媒αの調製]
塩化ロジウム(RhCl3・3H2O)1.00g、及び塩化マンガン(MnCl2・4H2O)0.077gを精製水22mlに溶解させて、水溶液S1を得た。この水溶液において、ロジウム:マンガン=1:0.1(モル比)である。
つぎに、比表面積270m2/gのシリカ20gに上記水溶液を少しずつ滴下し、含浸させた。水溶液S1を含浸させたシリカを110℃で3時間加熱し、さらに450℃で3時間加熱した。
得られたシリカ担持触媒前駆体を石英製反応管に充填し、常圧で水素−窒素混合ガス(体積比で、H2:N2=1:2)を300ml/分で流通させながら、350℃で3時間、水素還元を行った。放冷後、触媒αを得た。
[触媒αの調製]
塩化ロジウム(RhCl3・3H2O)1.00g、及び塩化マンガン(MnCl2・4H2O)0.077gを精製水22mlに溶解させて、水溶液S1を得た。この水溶液において、ロジウム:マンガン=1:0.1(モル比)である。
つぎに、比表面積270m2/gのシリカ20gに上記水溶液を少しずつ滴下し、含浸させた。水溶液S1を含浸させたシリカを110℃で3時間加熱し、さらに450℃で3時間加熱した。
得られたシリカ担持触媒前駆体を石英製反応管に充填し、常圧で水素−窒素混合ガス(体積比で、H2:N2=1:2)を300ml/分で流通させながら、350℃で3時間、水素還元を行った。放冷後、触媒αを得た。
[触媒βの調製]
塩化金酸四水和物1.26gを精製水7.8mlに溶解させて水溶液S2を得た。次に、比表面積50m2/gのチタニア20gに上記水溶液S2を少しずつ滴下し、含浸させた。これを110℃で3時間加熱し、さらに、さらに450℃で3時間加熱した。
得られたチタニア担持触媒前駆体を石英製反応管に充填し、常圧で水素−窒素混合ガス(体積比で、H2:N2=1:2)を300ml/分で流通させながら、350℃で3時間、水素還元を行った。放冷後、触媒βを得た。
塩化金酸四水和物1.26gを精製水7.8mlに溶解させて水溶液S2を得た。次に、比表面積50m2/gのチタニア20gに上記水溶液S2を少しずつ滴下し、含浸させた。これを110℃で3時間加熱し、さらに、さらに450℃で3時間加熱した。
得られたチタニア担持触媒前駆体を石英製反応管に充填し、常圧で水素−窒素混合ガス(体積比で、H2:N2=1:2)を300ml/分で流通させながら、350℃で3時間、水素還元を行った。放冷後、触媒βを得た。
[反応装置の構成]
直径1インチ、長さ15インチのステンレス製反応管を2本用意し、第一反応管に触媒αを10g、第二反応管に触媒βを10g充填した。
反応装置は、図1で模式的に表したように、第一反応管、気液分離器、液捕集器、送液ポンプ、気化器、第二反応管、の順に原料および生成物が流通するように構成した。なお、第一反応管の出口配管および第二反応管の出口配管には背圧弁が設けてあり、各反応管の圧力を制御できる。
また、図示しないが、各反応管及び各機器の温度を調整する機構が備わっている。
直径1インチ、長さ15インチのステンレス製反応管を2本用意し、第一反応管に触媒αを10g、第二反応管に触媒βを10g充填した。
反応装置は、図1で模式的に表したように、第一反応管、気液分離器、液捕集器、送液ポンプ、気化器、第二反応管、の順に原料および生成物が流通するように構成した。なお、第一反応管の出口配管および第二反応管の出口配管には背圧弁が設けてあり、各反応管の圧力を制御できる。
また、図示しないが、各反応管及び各機器の温度を調整する機構が備わっている。
[カルボン酸の製造]
水素と一酸化炭素を混合した第一原料ガス(体積比で、H2:CO=2:1)を第一反応管1に流通させる。このとき、第一原料ガスの流速は、空間速度4000〜15000h−1とし、温度を室温から反応温度295℃、圧力2MPaまで昇温・昇圧した。
第一反応管1での反応が安定化したら、生成物ガスを気液分離器3に通した。気液分離器3は−5℃に冷却した冷媒を流通させ、凝縮した液状成分(第二の生成物及び第三の生成物を含む液体)は、下方に落ちて、約10℃に保った液捕集器4に溜めた。溜まった液体中には、少なくともメタノール、エタノール、プロパノール、ブタノール、ホルムアルデヒド、アセトアルデヒド、酢酸エチル、酢酸メチル、及び酢酸が含まれることをガスクロマトグラフィ分析によって確認した。
溜まった液状成分を送液ポンプ5により0.18ml/minのスピードで気化器6(90℃)に導入した。気化器6には、窒素ガスを300ml/minで流通させ、気化成分(第二の生成物)と混合して排出し、さらに別の配管から導入した酸素ガスを30ml/minで混合して、第二原料ガスを得た。
次に第二原料ガスを第二反応管2に流通させ、約6時間反応を継続させ、第四の生成物を得た。この第四の生成物をガスクロマトグラフィーにより分析した。
得られたデータからCO転化率(モル%)、各成分の選択率(モル%)を計算した。この結果を表1に示す。
水素と一酸化炭素を混合した第一原料ガス(体積比で、H2:CO=2:1)を第一反応管1に流通させる。このとき、第一原料ガスの流速は、空間速度4000〜15000h−1とし、温度を室温から反応温度295℃、圧力2MPaまで昇温・昇圧した。
第一反応管1での反応が安定化したら、生成物ガスを気液分離器3に通した。気液分離器3は−5℃に冷却した冷媒を流通させ、凝縮した液状成分(第二の生成物及び第三の生成物を含む液体)は、下方に落ちて、約10℃に保った液捕集器4に溜めた。溜まった液体中には、少なくともメタノール、エタノール、プロパノール、ブタノール、ホルムアルデヒド、アセトアルデヒド、酢酸エチル、酢酸メチル、及び酢酸が含まれることをガスクロマトグラフィ分析によって確認した。
溜まった液状成分を送液ポンプ5により0.18ml/minのスピードで気化器6(90℃)に導入した。気化器6には、窒素ガスを300ml/minで流通させ、気化成分(第二の生成物)と混合して排出し、さらに別の配管から導入した酸素ガスを30ml/minで混合して、第二原料ガスを得た。
次に第二原料ガスを第二反応管2に流通させ、約6時間反応を継続させ、第四の生成物を得た。この第四の生成物をガスクロマトグラフィーにより分析した。
得られたデータからCO転化率(モル%)、各成分の選択率(モル%)を計算した。この結果を表1に示す。
ここで、「選択率」の定義は、前述のとおり、「合成ガス中の消費されたCOのモル数のうち、特定の酸素化物へ変換されたCのモル数が占める百分率」である。
「CO転化率」とは、「合成ガス中のCOのモル数のうち、消費されたCOのモル数が占める百分率」である。
「CO転化率」とは、「合成ガス中のCOのモル数のうち、消費されたCOのモル数が占める百分率」である。
[実施例2]
塩化ロジウム(RhCl3・3H2O)1.0g、ホウ酸(H3BO3)0.26g、及び塩化リチウム(LiCl・H2O)0.070gを精製水22mlに溶解させて、水溶液S3を得た。この水溶液S3において、ロジウム:ホウ酸=1:1.1、ロジウム:リチウム=1:0.31、ホウ酸:リチウム=1:0.28(モル比)である。
実施例1で用いた前記水溶液S1に代えて、実施例2で調整した前記水溶液S3を使用した以外は、実施例1と同様に反応システム(反応装置)を構築し、合成実験を行った。その結果を、表1に併記する。
塩化ロジウム(RhCl3・3H2O)1.0g、ホウ酸(H3BO3)0.26g、及び塩化リチウム(LiCl・H2O)0.070gを精製水22mlに溶解させて、水溶液S3を得た。この水溶液S3において、ロジウム:ホウ酸=1:1.1、ロジウム:リチウム=1:0.31、ホウ酸:リチウム=1:0.28(モル比)である。
実施例1で用いた前記水溶液S1に代えて、実施例2で調整した前記水溶液S3を使用した以外は、実施例1と同様に反応システム(反応装置)を構築し、合成実験を行った。その結果を、表1に併記する。
[比較例1]
実施例1において、反応管1のみのシステム(反応装置)とし、生成物を分析した。
[比較例2]
実施例2において、反応管1のみのシステム(反応装置)とし、生成物を分析した。
実施例1において、反応管1のみのシステム(反応装置)とし、生成物を分析した。
[比較例2]
実施例2において、反応管1のみのシステム(反応装置)とし、生成物を分析した。

表1の結果から、得られた炭素数2の酸素化物に含まれる酢酸の比率を求めた。結果を表2に併記する。
以上の結果から、本発明にかかる実施例1〜2のカルボン酸の製造方法は、比較例1〜
2のカルボン酸の製法よりも、酢酸の選択率が高く、生成された酸素化物に占める酢酸の比率も高かった。このことから、本発明のカルボン酸の製造方法は従来法よりも、合成ガスから高い選択率でカルボン酸を合成できることは明らかである。
2のカルボン酸の製法よりも、酢酸の選択率が高く、生成された酸素化物に占める酢酸の比率も高かった。このことから、本発明のカルボン酸の製造方法は従来法よりも、合成ガスから高い選択率でカルボン酸を合成できることは明らかである。
本発明のカルボン酸の製造方法、及びカルボン酸の製造装置は、合成ガスから工業的に有用なカルボン酸を製造するために広く利用することが可能である。
1…第一反応管、2…第二反応管、3…気液分離器、4…液捕集器、5…送液ポンプ、6…気化器、10…製造装置
Claims (13)
- 水素及び一酸化炭素を含む第一原料ガスを触媒αに接触させて、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む第一の生成物を得る工程Aと、
前記第一の生成物から、前記アルコール及び/又は前記アルデヒドを含む第二の生成物、並びに前記カルボン酸を含む第三の生成物を得る工程Bと、
気化させた前記第二の生成物と酸素ガスとを含む第二原料ガスを触媒βに接触させて、炭素数1〜4のカルボン酸を含む第四の生成物を得る工程Cと、を少なくとも有することを特徴とするカルボン酸の製造方法。 - 前記工程Bにおいて、前記第一の生成物に含まれる、前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを液化することによって、未反応の前記第一原料ガスから気液分離することを特徴とする請求項1に記載のカルボン酸の製造方法。
- 前記触媒αが、ロジウム及びマンガンを含み、前記触媒α中のロジウム:マンガンのモル比が1:0.01〜1:1の範囲であることを特徴とする請求項1又は2に記載のカルボン酸の製造方法。
- 前記触媒αが、ロジウム、ホウ素、及びアルカリ金属を含み、前記触媒α中のロジウム:ホウ素のモル比が1:1〜1:5の範囲であり、前記触媒α中のロジウム:アルカリ金属のモル比が1:0.1〜1:1の範囲であり、前記触媒α中のホウ素:アルカリ金属のモル比が1:0.02〜1:1の範囲であることを特徴とする請求項1又は2に記載のカルボン酸の製造方法。
- 前記触媒βが金を含むことを特徴とする請求項1〜4のいずれか一項に記載のカルボン酸の製造方法。
- 前記第一原料ガスを前記触媒αに接触させる際の温度が、150〜450℃の範囲であることを特徴とする請求項1〜5のいずれか一項に記載のカルボン酸の製造方法。
- 前記第二原料ガスを前記触媒βに接触させる際の温度が、100〜500℃の範囲であることを特徴とする請求項1〜6のいずれか一項に記載のカルボン酸の製造方法。
- 前記第一原料ガスを前記触媒αに接触させる際の圧力が、0.5MPa〜10MPaの範囲であることを特徴とする請求項1〜7のいずれか一項に記載のカルボン酸の製造方法。
- 前記第二原料ガスを前記触媒βに接触させる際の圧力が、0.1MPa〜1MPaの範囲であることを特徴とする請求項1〜8のいずれか一項に記載のカルボン酸の製造方法。
- 前記触媒αまたは前記触媒βが担持触媒であることを特徴とする請求項1〜9のいずれか一項に記載のカルボン酸の製造方法。
- 前記担持触媒αの担体が、シリカであることを特徴とする請求項10に記載のカルボン酸の製造方法。
- 前記担持触媒βの担体が、チタニアであることを特徴とする請求項10又は11に記載のカルボン酸の製造方法。
- 水素および一酸化炭素を含む第一原料ガスから、炭素数1〜4のカルボン酸、並びに炭素数1〜4のアルコール及び/又は炭素数1〜4のアルデヒドを含む生成物を生成する触媒αが内部に配された第一反応管と、前記生成物中の前記カルボン酸、並びに前記アルコール及び/又は前記アルデヒドを液化する気液分離器と、液化された前記アルコール及び/又は前記アルデヒドを気化する気化器と、気化された前記アルコール及び/又は前記アルデヒドに酸素ガスを混合した第二原料ガスから、炭素数1〜4のカルボン酸を含む生成物を生成する触媒βが内部に配された第二反応管と、が少なくとも備えられ、
前記第一反応管、前記第二反応管、前記気液分離器、及び前記気化器には、ガス若しくは液体の導入口及び排出口が各々設けられ、
前記第一反応管の排出口の下流に前記気液分離器の導入口が配管接続され、前記気液分離器の排出口の下流に前記気化器の導入口が配管接続され、前記気化器の排出口の下流に前記第二反応管の導入口が配管接続されたことを特徴とするカルボン酸の製造装置。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011070858A JP2012201675A (ja) | 2011-03-28 | 2011-03-28 | カルボン酸の製造方法、及びカルボン酸の製造装置 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011070858A JP2012201675A (ja) | 2011-03-28 | 2011-03-28 | カルボン酸の製造方法、及びカルボン酸の製造装置 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012201675A true JP2012201675A (ja) | 2012-10-22 |
Family
ID=47183020
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011070858A Withdrawn JP2012201675A (ja) | 2011-03-28 | 2011-03-28 | カルボン酸の製造方法、及びカルボン酸の製造装置 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2012201675A (ja) |
-
2011
- 2011-03-28 JP JP2011070858A patent/JP2012201675A/ja not_active Withdrawn
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102282117B (zh) | 乙酸氢化制造乙醇 | |
| CN101910099B (zh) | 由碳质原料生产醇的方法 | |
| Pairojpiriyakul et al. | Hydrogen production from catalytic supercritical water reforming of glycerol with cobalt-based catalysts | |
| JPH11322658A (ja) | 酢酸からのアセトアルデヒドの製造方法およびこの製造方法に用いる触媒 | |
| JP2013508423A5 (ja) | ||
| TW200906794A (en) | Process for the carbonylation of dimethyl ether | |
| CN103459020B (zh) | 用于将合成气转化为醇的催化剂 | |
| JP5376222B2 (ja) | エタノール水蒸気改質用触媒 | |
| JP6329286B2 (ja) | 酸素化物合成用の触媒の製造方法、及び酸素化物の製造方法 | |
| JP2023167854A (ja) | アセトン水素化触媒及びイソプロパノールの製造方法 | |
| JPWO2021262922A5 (ja) | ||
| JP4713895B2 (ja) | ヨウ化物の製造方法 | |
| JP2012201662A (ja) | グリセリンの水素化分解物の製造方法 | |
| EP2694206A1 (en) | Catalysts for the conversion of synthesis gas to alcohols | |
| JP2012201675A (ja) | カルボン酸の製造方法、及びカルボン酸の製造装置 | |
| JP2012131709A (ja) | アルコールの製造方法、及びアルコール製造装置 | |
| JP2017202459A (ja) | ガス分離方法、有機酸素化物の製造方法、ガス分離装置、及び有機酸素化物の製造システム | |
| JP5525420B2 (ja) | 酸素化物の合成方法、触媒、及び反応装置 | |
| US9333491B2 (en) | Catalyst for oxygenate synthesis, oxygenate production apparatus, and method of producing oxygenate | |
| JP5127516B2 (ja) | 水素生成用原料組成物および水素の製造方法 | |
| JP2009051704A (ja) | 水性ガスの製造方法 | |
| JP7756355B2 (ja) | 水素及びカルボン酸の製造方法 | |
| JP2012111693A (ja) | 酸素化物の合成方法、触媒、及び反応装置 | |
| Lino et al. | Application of Hydrogen in Mixed Higher Alcohols Production | |
| Beckmann et al. | Sustainable methyl formate generation by dehydrogenation of green methanol over Cu_SiO2/MgO |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A300 | Withdrawal of application because of no request for examination |
Free format text: JAPANESE INTERMEDIATE CODE: A300 Effective date: 20140603 |