JP2011001663A - 複合繊維 - Google Patents
複合繊維 Download PDFInfo
- Publication number
- JP2011001663A JP2011001663A JP2009147528A JP2009147528A JP2011001663A JP 2011001663 A JP2011001663 A JP 2011001663A JP 2009147528 A JP2009147528 A JP 2009147528A JP 2009147528 A JP2009147528 A JP 2009147528A JP 2011001663 A JP2011001663 A JP 2011001663A
- Authority
- JP
- Japan
- Prior art keywords
- dendritic polyester
- polymer
- spinning
- acid
- polyester
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


Landscapes
- Multicomponent Fibers (AREA)
Abstract
【課題】 従来とは異なり、マトリックスポリマーと分子鎖の絡み合いが抑制され、紡糸過程での伸長変形を阻害し難い樹状ポリエステルを用いることにより、紡糸温度の低温化を図るとともに、力学特性に優れた複合繊維を提供するものである。
【解決手段】 芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性であるマトリックスポリマーに0.1〜10重量%ブレンドしたポリマーが少なくとも一部を構成する複合繊維。
【選択図】 なし
【解決手段】 芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性であるマトリックスポリマーに0.1〜10重量%ブレンドしたポリマーが少なくとも一部を構成する複合繊維。
【選択図】 なし
Description
本発明は、融点や粘度等が異なる2種類以上のポリマーにより構成させる複合繊維において、樹状ポリエステルが添加されたポリマーブレンドが少なくとも一部を構成する複合繊維に関するものである。
ポリエステルやポリアミドなどの熱可塑性ポリマーを用いた繊維は力学的特性や寸法安定性に優れるため、衣料用途のみならずインテリアや車両内装、産業用途等幅広く利用されており、産業上の価値は極めて高い。しかしながら、繊維の用途が多様化する現状において、その要求特性も様々なものとなり、芯鞘複合紡糸などを活用し、2種類以上の熱可塑性ポリマーを複合繊維としたり、繊維断面を異形化する試みが検討されている。
一般に、芯鞘複合繊維では、2種類以上のポリマーを組み合わせ、芯成分を鞘成分が被覆することで、単独繊維では達成されない風合い、嵩高性などといった感性的効果、また、強度、弾性率、耐摩耗性などといった力学特性の付与が可能となる。また、溶剤に易溶出性を示すポリマーで鞘成分、難溶出成分で異形断面となるように芯成分を構成し、鞘成分を溶出することで精度の高い異形断面繊維を得ることができる。通常、ポリエステルやポリアミドなどのポリマーを溶融紡糸によって得た場合、真円形の断面を持つことが多いが、異形断面とすることで真円形の繊維では得られない特殊な風合いを付与したり、織り編みの際のこなれを良くしたり、繊維を被覆する他の樹脂との接触面積を増加させ、剥離などの問題を抑制することができる。
このように既存のポリマーを組み合わせることで機能の複合化、断面形態の制御などにより様々な特性向上が期待される複合繊維であるが、その製造においては、融点や粘度が異なるポリマーを口金内で複合化させ、同一の細孔から吐出することになる。この複合化されたポリマー流の流動性を確保するためには紡糸温度の設定が非常に重要となる。
複合繊維の溶融紡糸における温度条件は、組み合わせるポリマーのうち高融点や高粘度のものによって決まるのが一般的であり、この高融点や高粘度のポリマーが良好な良流動性を示す温度でないと実質的に紡糸はできない。このため、用途の要求特性に応じて複合繊維を構成するポリマーの組み合わせを企画しても、実際には紡糸ができなかったり、低融点や耐熱性の低いポリマーには過酷な紡糸温度で紡糸することになり、大きく熱分解を引き起こし、得られる繊維は実用に耐えないものとなってしまう場合も多く、繊維の特性や用途に制限をつくる一つの要因となっている。
例えば、地球的規模での環境に対する意識が高まり、バイオマスポリマーであるポリ乳酸は、易成形性であり、価格も低いことから多く活用方法が検討されているが、単独で繊維化した場合には耐摩耗性や耐熱性などが低いというようないくつかの欠点を有している。このため、複合繊維として、それらの欠点を克服しようとする試みがある。また、ポリ乳酸はアルカリ水溶液で簡単に溶出できるために、前記した異形断面繊維や海島複合繊維の易溶出成分(海成分)として使用することも好適である。しかしながら、ポリ乳酸は250℃以上で顕著な熱分解が発生することから紡糸温度が250℃以上であるポリエステルやポリアミドと組み合わせることはポリ乳酸の熱分解という観点から非常に困難なことであった。よって、ポリ乳酸と組み合わせるポリマーは共重合などすることにより融点を降下させることも考えられるが、この場合、ポリエステルやポリアミドが本来有する優れた特性が大きく損なわれる場合がある。
この融点を降下させる目的とは、すなわち、高融点や高粘度のポリマーの溶融時の流動性を確保することであり、これに基づけば、高融点や高粘度のポリマーの粘度を低下させるために減粘剤などを添加することで流動性を向上させることが考えられる。これが可能となれば、紡糸温度を低温化し、ポリ乳酸などの耐熱性の低いポリマーと組み合わすことができるポリマーが増え、それに応じて様々な特性の向上が可能となることが考えられる。しかしながら、一般に減粘剤と呼ばれるものは低分子量物、すなわち低融点のポリマーを添加することにより添加されたポリマー全体の粘度を低下させるものである。一般には、この減粘剤自体の耐熱性が低いものである場合が多く、溶融混練中などに揮発することにより減粘効果が大きく低減するものであったり、減粘剤の分解物が繊維の特性を大きく損なわせたりすること等から現在まで溶融紡糸に適用したもので成功した例は少ない。
一方、樹状ポリマーを熱可塑性ポリマーに添加して、ポリマーの流動性を向上することが提案されている(特許文献1)。樹状ポリマーは分岐モノマーを介して、低分子量の主鎖を連結することで超分岐構造を有し、その樹状ポリマー全体では高分子量体となるため、低粘度成分添加の減粘効果を有しつつも、耐熱性が向上することとなる。特許文献1では、熱可塑性ポリマーと非反応性の樹状ポリマーを添加することで、流動性が向上することが開示されている。しかしながら、特許文献1に用いられる樹状ポリマーの主鎖部分は脂肪族系ポリマーで構成されており、脂肪族系ポリマーはその分子構造から、溶融下においての主鎖部の柔軟性が高く、樹状ポリマーと熱可塑性ポリマーが非反応性であるとしても、樹状ポリマー主鎖部分と熱可塑性ポリマーの主鎖部分で分子鎖どうしの絡み合いが多く発生してしまう場合があった。これは、樹脂の押出加工(射出成形等)では変形量が小さく、さらに剪断変形が支配的であるため大きな問題とはならないが、紡糸などの大きな伸長変形を伴う場合には深刻な問題を引き起こしてしまう場合があった。すなわち、分子鎖の絡み合いの程度が大きくなることで、熱可塑性ポリマーの分子鎖がスムーズな伸長変形することを阻害され、紡糸性を著しく損ない、場合よっては添加された熱可塑性ポリマーの弾性的性質が強くなり過ぎ、口金直下でのポリマーが糸として繋がらない五月雨現象が発生する場合がある。特に複合紡糸の場合には2種類以上のポリマーが伸長変形することになるため、複合ポリマー流を構成するポリマーのうち1成分でも前記したような挙動を示すと、その伸長変形の制御は非常に困難なものとなり、紡糸不能に陥る場合が多い。
前記した樹状ポリマーとの絡み合いを抑制する方法としては、樹状ポリマーの主鎖に剛直成分を組み込み溶融下での柔軟性を低下させ、熱可塑性ポリマーとの主鎖との絡み合いを抑制する方法が提案されている(特許文献2)。しかしながら、特許文献2に記載される樹状ポリマーは分子末端がカルボン酸基であり、分子末端構造について考慮されていないことが課題として残る。このカルボン酸基は、溶融下においては自己触媒反応により、マトリクスポリマーである熱可塑性ポリマーの加水分解を起こし、添加した熱可塑性ポリマーの分子量低下を招く場合があった。この現象は溶融滞留時間の短い樹脂の押出加工(射出成形等)では大きな問題とならないが、複雑に入り組んだ配管やパック口金構造を有する溶融紡糸機では、滞留時間が長いものとなったり、異常滞留部ができやすかったりするため、熱可塑性ポリマーの分子量低下が顕著化し、紡糸性や複合繊維の力学物性に大きな影響を与える場合があった。
本発明は、融点や粘度等が異なる2種類以上のポリマーにより構成させる複合繊維において、樹状ポリエステルが添加されたポリマーブレンドが少なくとも一部を構成する複合繊維に関するものである。
上記目的は、以下の手段により達成される。すなわち、
(1)2種類以上のポリマーからなる複合繊維であって、芳香族オキシカルボニル単位(P)、芳香族および/または脂肪族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)から選ばれる少なくとも1種の構造単位と、3官能の有機残基(B)とを含み、かつ前記P、Q、RおよびBの含有量の合計に対してBの含有量が7.5〜50モル%であり、末端のカルボン酸基量が1×10-4当量/g以下である樹状ポリエステルを、マトリックスポリマーに0.1〜10重量%添加したポリマーブレンドを少なくとも構成の一部であることを特徴とする複合繊維、
(2)樹状ポリエステルが、カルボン酸反応性単官能化合物残基を含有することを特徴とする(1)記載の複合繊維、
(3)カルボン酸反応性単官能化合物が、オルトエステル、オキサゾリン、エポキシドから選ばれる少なくとも1種の化合物であることを特徴とする(2)に記載の複合繊維、
(4)マトリックスポリマーが、ポリエステル、ポリアミド、またはポリフェニレサルファイドであることを特徴とする(1)〜(3)いずれかに記載の複合繊維、
(5)ポリトリメチレンテレフタレートが少なくとも構成の一部であることを特徴とする(1)〜(4)のいずれかに記載の複合繊維、
(6)ポリ乳酸が少なくとも構成の一部であることを特徴とする(1)〜(5)のいずれかに記載の複合繊維、
である。
(1)2種類以上のポリマーからなる複合繊維であって、芳香族オキシカルボニル単位(P)、芳香族および/または脂肪族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)から選ばれる少なくとも1種の構造単位と、3官能の有機残基(B)とを含み、かつ前記P、Q、RおよびBの含有量の合計に対してBの含有量が7.5〜50モル%であり、末端のカルボン酸基量が1×10-4当量/g以下である樹状ポリエステルを、マトリックスポリマーに0.1〜10重量%添加したポリマーブレンドを少なくとも構成の一部であることを特徴とする複合繊維、
(2)樹状ポリエステルが、カルボン酸反応性単官能化合物残基を含有することを特徴とする(1)記載の複合繊維、
(3)カルボン酸反応性単官能化合物が、オルトエステル、オキサゾリン、エポキシドから選ばれる少なくとも1種の化合物であることを特徴とする(2)に記載の複合繊維、
(4)マトリックスポリマーが、ポリエステル、ポリアミド、またはポリフェニレサルファイドであることを特徴とする(1)〜(3)いずれかに記載の複合繊維、
(5)ポリトリメチレンテレフタレートが少なくとも構成の一部であることを特徴とする(1)〜(4)のいずれかに記載の複合繊維、
(6)ポリ乳酸が少なくとも構成の一部であることを特徴とする(1)〜(5)のいずれかに記載の複合繊維、
である。
本発明の複合繊維により、ポリマーの組み合わせに制限なく複合繊維を得ることが可能であり、更に流動性向上に伴う紡糸温度の低下が可能となり、熱分解に伴う分子量低下が抑制されるため優れた力学特性を有した複合繊維を得ることができる。
本発明の樹状ポリエステルは、芳香族オキシカルボニル単位(P)、芳香族および/または脂肪族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)から選ばれる少なくとも1種の構造単位と、3官能の有機残基(B)とを含み、かつ前記(P)、(Q)、(R)および(B)の含有量の合計に対して(B)の含有量が7.5〜50モル%の範囲にあり、末端のカルボン酸量が1×10-4当量/g以下である樹状ポリエステルである。
ここで、芳香族オキシカルボニル単位(P)、芳香族および/または脂肪族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)は、それぞれ下式で表される構造単位であることが好ましい。

ここで、R1およびR3は、それぞれ芳香族残基である。R2は、芳香族残基または脂肪族残基である。R1、R2、およびR3は、それぞれ複数の構造単位を含んでも良い。
前記の芳香族残基としては、置換または非置換のフェニレン基、ナフチレン基、ビフェニレン基などが挙げられ、脂肪族残基としてはエチレン、プロピレン、ブチレンなどが挙げられる。R1、R2およびR3は、好ましくは、それぞれ下式で表される構造単位から選ばれる少なくとも1種以上の構造単位である。
ただし、式中Yは、水素原子、ハロゲン原子およびアルキル基から選ばれる少なくとも1種である。式中nは2〜8の整数である。ここで好ましいアルキル基としては、炭素数1〜4が好ましい。
本発明の樹状ポリエステルは、3官能の有機残基(B)が、互いにエステル結合および/またはアミド結合により直接、あるいは、枝構造部分(P)、(Q)または(R)を介して結合した、3分岐の分岐構造を基本骨格としている。分岐構造は、3分岐など単一の基本骨格で形成されていてもよいし、3分岐と4分岐、3分岐と5分岐など複数の基本骨格が共存していてもよい。ポリマーの全てが該基本骨格からなる必要はなく、例えば、末端封鎖のために末端に他の構造が含まれても良い。また、樹状ポリエステル中には、Bの3つの官能基が全て反応している構造、2つだけが反応している構造、および1つだけが反応している構造が混在していてもよい。好ましくは(B)の3つの官能基が全て反応した構造が、(B)全体に対して15モル%以上であることが好ましく、より好ましくは20モル%以上であり、さらに好ましくは30モル%以上である。前記3分岐の基本骨格を模式的に示すと、下式で示される。
3官能の有機残基(B)の含有量は、前記P、Q、R、およびBの含有量の合計に対して7.5モル%以上であれば、得られたポリエステルは樹状構造に起因する効果を十分得ることができる。Bの含有量が50モル%以下であれば、剪断応答性の低下や流動性向上効果が低下することもなく、ゲル化反応の抑制が可能となる。また、この範囲内であれば、マトリクスポリマー中での樹状ポリエステル分散径を縮小できるため、マトリクスポリマーと配合して得られる熱可塑性樹ポリマーの流動性向上効果が向上することとなる。(B)の含有量は、好ましくは10〜40モル%であり、高い剪断応答性と、マトリクスポリマーに配合した際の流動性向上効果や樹状ポリエステルの分散径が小さくなるという点から15〜35モル%とすることがさらに好ましい。
ここで、(B)の含有量は樹状ポリエステルの枝構造および分岐構造を構成する構造単位に対しての値であり、末端構造を構成する残基は含まない。ここで、枝構造とは、樹状ポリエステル中での(P)、(Q)、(R)のいずれかを含有してなる直鎖ポリエステル構造を意味しており、分岐構造とは、(B)由来の構造を意味している。
本発明に用いる樹状ポリエステルは、溶融液晶性を示すことが好ましい。ここで溶融液晶性を示すとは、室温から昇温していった際に、ある温度域で液晶状態を示すことである。液晶状態とは、剪断下において光学的異方性を示す状態である。
溶融液晶性を示すために、基本骨格は、下式で示されるように、有機残基(B)が、枝構造部分(P)、(Q)または(R)により構成される構造単位(D)を介して結合していることが好ましい。
3官能の有機残基(B)としては、カルボン酸基、ヒドロキシル基、アミノ基を含有する化合物の残基であることが好ましく、例えば、フロログルシノール、トリメシン酸、トリメリット酸、無水トリメリット酸、α−レゾルシル酸、4−ヒドロキシ−1,2−ベンゼンジカルボン酸、5−ヒドロキシイソフタル酸などの残基が好ましく、さらに好ましくは、トリメシン酸、α−レゾルシル酸の残基であり、最も好ましくはトリメシン酸の残基である。
また、樹状ポリエステルの芳香族オキシカルボニル単位(P)、芳香族および/または脂肪族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)は、樹状ポリエステルの分岐間の枝構造部分を構成する単位である。p、qおよびrはそれぞれ構造単位P、QおよびRの平均含有率(モル比)であり、このp、qおよびrの値は、例えば、樹状ポリエステルをペンタフルオロフェノール50重量%:重クロロホルム50重量%の混合溶媒に溶解し、40℃でプロトン核の核磁気共鳴スペクトル分析を行い、それぞれの構造単位に由来するピーク強度比から求めることができる。各構造単位のピーク面積強度比から、平均含有率を算出し、小数点3桁は四捨五入する。
pとqの比率およびpとrの比率(p/q、p/r)は、いずれも5/95〜95/5の範囲が好ましく、より好ましくは10/90〜90/10であり、さらに好ましくは20/80〜80/20である。この範囲であれば、液晶性が発現しやすく好ましい。p/qおよびp/rの比率を95/5以下とすることで、樹状ポリエステルの融点を適当な範囲とすることができるため好ましい。また、p/qおよびp/rを5/95以上とすることで樹状ポリエステルの溶融液晶性を発現することができるため好ましい。
前記一般式の(P)構造単位において、R1は芳香族オキシカルボニル単位由来の構造単位であり、具体例としては、p−ヒドロキシ安息香酸、6−ヒドロキシ−2−ナフトエ酸から生成した構造単位などが挙げられる。好ましくはp−ヒドロキシ安息香酸由来の構造単位であり、6−ヒドロキシ−2−ナフトエ酸由来の構造単位部併用することも可能である。また本発明の効果を損なわない範囲でグリコール酸、乳酸、ヒドロキシプロピオン酸、ヒドロキシ酪酸、ヒドロキシ吉草酸、ヒドロキシカプロン酸などの脂肪族ヒドロキシカルボン酸由来の構造単位を含有しても良い。
(Q)構造単位において、R2は芳香族および/または脂肪族ジオキシ単位由来の構造単位であり、例えば、4,4’−ジヒドロキシビフェニル、ハイドロキノン、3,3’,5,5’−テトラメチル−4,4’−ジヒドロキシビフェニル、t−ブチルハイドロキノン、フェニルハイドロキノン、メチルハイドロキノン、2,6−ジヒドロキシナフタレン、2,7−ジヒドロキシナフタレン、2,2−ビス(4−ヒドロキシフェニル)プロパンおよび4,4’−ジヒドロキシジフェニルエーテル、エチレングリコール、1,3−プロピレングリコール、1,4−ブタンジオールなど由来の構造単位が挙げられる。好ましくは、4,4’−ジヒドロキシビフェニル、ハイドロキノン、およびエチレングリコール由来の構造単位であり、4,4’−ジヒドロキシビフェニルとハイドロキノンもしくは4,4’−ジヒドロキシビフェニルとエチレングリコール由来の構造単位が含まれることが液晶性の制御の点から好ましい。
(R)構造単位において、R3は芳香族ジカルボニル単位由来の構造単位であり、例えば、テレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、4,4’−ジフェニルジカルボン酸、1,2−ビス(フェノキシ)エタン−4,4’−ジカルボン酸、1,2−ビス(2−クロロフェノキシ)エタン−4,4’−ジカルボン酸および4,4’−ジフェニルエーテルジカルボン酸など由来の構造単位が挙げられる。好ましくはテレフタル酸またはイソフタル酸由来の構造単位であり、特に両者を併用した場合に融点調節がしやすく好ましい。セバシン酸やアジピン酸などの脂肪族ジカルボン酸由来の構造単位が一部含まれることもある。
本発明の樹状ポリエステルの枝構造部分は、主としてポリエステル骨格からなることが好ましいが、カーボネート構造やアミド構造、ウレタン構造などを、特性に大きな影響を与えない程度に導入することも可能である。中でもアミド構造を導入することが好ましい。このような別の結合を導入することで、多種多様なマトリクスポリマーに対する相溶性を調整することが可能であり、好ましい。アミド結合の導入の方法としては、p−アミノ安息香酸、m−アミノ安息香酸、p−アミノフェノール、m−アミノフェノール、p−フェニレンジアミン、m−フェニレンジアミン、テトラメチレンジアミンペンタメチレンジアミン、ヘキサメチレンジアミン、2−メチルペンタメチレンジアミン、ノナメチレンジアミン、ウンデカメチレンジアミン、ドデカメチレンジアミン、2,2,4−/2,4,4−トリメチルヘキサメチレンジアミン、5−メチルノナメチレンジアミン、m−キシリレンジアミン、p−キシリレンジアミン、1,3−ビス(アミノメチル)シクロヘキサン、1,4−ビス(アミノメチル)シクロヘキサン、1−アミノ−3−アミノメチル−3,5,5−トリメチルシクロヘキサン、ビス(4−アミノシクロヘキシル)メタン、ビス(3−メチル−4−アミノシクロヘキシル)メタン、2,2−ビス(4−アミノシクロヘキシル)プロパン、ビス(アミノプロピル)ピペラジン、アミノエチルピペラジンなどの脂肪族、脂環族、あるいは芳香族のアミン化合物などを共重合することが好ましい。中でもp−アミノフェノールまたはp−アミノ安息香酸の共重合が好ましい。
樹状ポリエステルの枝構造部分の具体例としては、p−ヒドロキシ安息香酸および6−ヒドロキシ−2−ナフトエ酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、6−ヒドロキシ−2−ナフトエ酸由来の構造単位、4,4’−ジヒドロキシビフェニル由来の構造単位およびテレフタル酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、4,4’−ジヒドロキシビフェニル由来の構造単位、テレフタル酸由来の構造単位およびイソフタル酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、4,4’−ジヒドロキシビフェニル由来の構造単位、ハイドロキノン由来の構造単位、テレフタル酸由来の構造単位およびイソフタル酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、エチレングリコール由来の構造単位およびテレフタル酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、エチレングリコール由来の構造単位、4,4’−ジヒドロキシビフェニル由来の構造単位およびテレフタル酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、ハイドロキノン由来の構造単位、4,4’−ジヒドロキシビフェニル由来の構造単位、テレフタル酸由来の構造単位および2,6−ナフタレンジカルボン酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、6−ヒドロキシ−2−ナフトエ酸由来の構造単位、ハイドロキノン由来の構造単位およびテレフタル酸由来の構造単位からなるものなどが挙げられる。
特に好ましいのは、枝構造部分が、下式に記載した構造単位(P−I)、(Q−II)、(Q−III)、(R−IV)および(R−V)から構成されること、
もしくは、下式に記載した構造単位(P−I)、(Q−II)、(R−VI)および(R−IV)から構成されることである。
枝構造部分が、前記構造単位(P−I)、(Q−II)、(Q−III)、(R−IV)および(R−V)から構成される場合には、構造単位(P−I)の含有量pは、各構造単位の合計p+q+rに対して30〜70モル%が好ましく、より好ましくは45〜60モル%である。
また、構造単位(Q−II)の含有量qは、構造単位(Q−II)および(Q−III)の合計含有量qに対して60〜75モル%が好ましい。また、構造単位(R−IV)の含有量rは、構造単位(R−IV)および(R−V)の合計含有量rに対して60〜92モル%が好ましい。
このような場合には、本発明の効果である、せん断応答性やマトリクスポリマーへの添加効果が顕著に発現するため好ましい。
前記のように、構造単位(Q−II)および(Q−III)の合計含有量qと(R−IV)および(R−V)の合計含有量rは実質的に等モルであることが好ましいが、いずれかの成分を過剰に加えてもよい。
枝構造部分が、前記構造単位(P−I)、(Q−II)、(R−VI)および(R−IV)から構成される場合には、前記構造単位(P−I)の含有量pは、p+q+rに対して30〜90モル%が好ましく、40〜80モル%がより好ましい。
また、有機残基(B)の含有量は、樹状ポリエステルを構成する全単量体の含有量に対して7.5モル%以上であり、10モル%以上がより好ましく、さらに好ましくは15モル%以上である。このような場合に、枝構造部分の連鎖長が、樹状ポリエステルが樹状の形態をとるのに適した長さとなるため好ましい。有機残基(B)の含有量の上限としては、50モル%以下であり、40モル%以下がより好ましい。
また本発明の樹状ポリエステルは特性に影響が出ない範囲で、部分的に架橋構造を有していてもよい。
本発明において、樹状ポリエステルの製造方法は、公知のポリエステルの重縮合法に準じて製造できる。前記R1で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、R2で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、および、3官能の多官能単量体を反応させる方法であって、該多官能単量体の添加量(モル)が、樹状ポリエステルを構成する全単量体(モル)に対して7.5モル%以上として製造する方法が好ましい。多官能単量体の添加量は、より好ましくは10モル%以上、さらに好ましくは15モル%以上である。また、添加量の上限としては、50モル%以下が好ましく、より好ましくは35モル%以下である。
また、前記反応に際して、R1、R2およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体をアシル化した後、3官能以上の多官能単量体を反応させる態様も好ましい。また、R1、R2およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、および、3官能以上の多官能単量体をアシル化した後、重合反応させる態様も好ましい。
前記構造単位(P−I)、(Q−II)、(Q−III)、(R−IV)および(R−V)とトリメシン酸残基から構成される樹状ポリエステルを製造する場合を例に挙げて、好ましい製造方法を説明する。
(1)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、ジアセトキシベンゼン、テレフタル酸およびイソフタル酸から脱酢酸縮重合反応によって液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(2)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、ジアセトキシベンゼン、テレフタル酸、イソフタル酸およびトリメシン酸から脱酢酸縮重合反応によって製造する方法。
(3)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、ハイドロキノンとテレフタル酸およびイソフタル酸に無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって液晶性ポリエステルオリゴマーを合成し、さらにトリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(4)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、ハイドロキノンとテレフタル酸、イソフタル酸およびトリメシン酸に無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって製造する方法。
(5)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、ハイドロキノン、テレフタル酸ジフェニルエステルおよびイソフタル酸ジフェニルエステルから脱フェノール重縮合反応により液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱フェノール重縮合反応によって製造する方法。
(6)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、ハイドロキノン、テレフタル酸ジフェニルエステル、イソフタル酸ジフェニルエステルおよびトリメシン酸のフェニルエステルから脱フェノール重縮合反応によって製造する方法。
(7)p−ヒドロキシ安息香酸、テレフタル酸、イソフタル酸、トリメシン酸にジフェニルカーボネートを反応させて、それぞれフェニルエステルとした後、4,4’−ジヒドロキシビフェニル、ハイドロキノンを加え、脱フェノール重縮合反応によって製造する方法。
(1)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、ジアセトキシベンゼン、テレフタル酸およびイソフタル酸から脱酢酸縮重合反応によって液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(2)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、ジアセトキシベンゼン、テレフタル酸、イソフタル酸およびトリメシン酸から脱酢酸縮重合反応によって製造する方法。
(3)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、ハイドロキノンとテレフタル酸およびイソフタル酸に無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって液晶性ポリエステルオリゴマーを合成し、さらにトリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(4)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、ハイドロキノンとテレフタル酸、イソフタル酸およびトリメシン酸に無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって製造する方法。
(5)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、ハイドロキノン、テレフタル酸ジフェニルエステルおよびイソフタル酸ジフェニルエステルから脱フェノール重縮合反応により液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱フェノール重縮合反応によって製造する方法。
(6)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、ハイドロキノン、テレフタル酸ジフェニルエステル、イソフタル酸ジフェニルエステルおよびトリメシン酸のフェニルエステルから脱フェノール重縮合反応によって製造する方法。
(7)p−ヒドロキシ安息香酸、テレフタル酸、イソフタル酸、トリメシン酸にジフェニルカーボネートを反応させて、それぞれフェニルエステルとした後、4,4’−ジヒドロキシビフェニル、ハイドロキノンを加え、脱フェノール重縮合反応によって製造する方法。
なかでも(1)〜(5)の製造方法が好ましく、(3)および(4)の方法がより好ましく、鎖長制御と立体規制の点から(4)の製造方法が最も好ましい。
分子量を上げるためには、トリメシン酸のカルボン酸量に相当する分だけ、ハイドロキノンや4,4’−ジヒドロキシビフェニルなどのジヒドロキシモノマーを、ジカルボン酸モノマーに対して過剰に加え、全単量体におけるカルボン酸と水酸基当量を合わせることが好ましい。
脱酢酸重縮合反応を行う場合には、樹状ポリエステルが溶融する温度で、場合によっては減圧下で反応させ、所定量の酢酸を留出させ、重縮合反応を完了させる溶融重合法が好ましい。例えば、所定量のp−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、ハイドロキノン、テレフタル酸、イソフタル酸、トリメシン酸および無水酢酸を、攪拌翼および留出管を備え、下部に吐出口を備えた反応容器中に仕込む。混合物を、窒素ガス雰囲気下で攪拌しながら加熱して、水酸基をアセチル化させた後、200〜350℃まで昇温して脱酢酸重縮合反応を行い、酢酸を留出させる。理論留出量の91%まで酢酸を留出させ、反応を完了させる。
アセチル化させる条件としては、実際の製造工程を考慮すると反応温度は、130〜170℃の範囲が好ましく、反応時間は、0.5〜6時間が好ましい。
重縮合させる温度は、樹状ポリエステルが溶融する温度であり、好ましくは樹状ポリエステルの融点+10℃以上の温度である。具体的には、200〜350℃の範囲が好ましい。重縮合させるときの雰囲気は、常圧窒素下でも問題ないが、減圧すると反応が早く進み、系内の残留酢酸が少なくなるため好ましい。減圧度は、0.1mmHg(13.3Pa)〜200mmHg(26600Pa)が好ましく、より好ましくは10mmHg(1330Pa)〜100mmHg(13300Pa)である。なお、アセチル化と重縮合は同一の反応容器で連続して行っても良いし、アセチル化と重縮合を異なる反応容器で行っても良い。
重縮合反応が完了した後、反応容器内を樹状ポリエステルが溶融する温度に保ち、例えば、0.01〜1.0kg/cm2(0.001〜0.1MPa)に加圧し、反応容器下部に設けられた吐出口より、樹状ポリエステルをストランド状に吐出する。吐出口には断続的に開閉する機構を設け、液滴状に吐出することも可能である。吐出した樹状ポリエステルは、空気中もしくは水中を通過して冷却された後、必要に応じて、カッティングもしくは粉砕される。
得られたペレット状、粒状または粉状の樹状ポリエステルは、さらに必要に応じて、熱乾燥や真空乾燥により水、酢酸などを除く。また、重合度の微調整、あるいは、さらに重合度を上げるために、固相重合をすることも可能である。固相重合は、例えば、前記により得られた樹状ポリエステルを、窒素気流下、または、減圧下、樹状ポリエステルの融点−5℃〜融点−50℃(例えば、200〜300℃)の温度範囲で1〜50時間加熱する方法が挙げられる。
樹状ポリエステルの重縮合反応は無触媒でも進行するが、酢酸第一錫、テトラブチルチタネート、酢酸カリウムおよび酢酸ナトリウム、三酸化アンチモン、金属マグネシウムなどの金属化合物を使用することもできる。
本発明の樹状ポリエステルは、数平均分子量は1,000〜40,000であることが好ましく、より好ましくは1,000〜20,000、さらに好ましくは1,000〜10,000であり、最も好ましくは1,000〜5,000の範囲である。なお、この数平均分子量は、樹状ポリエステルが可溶な溶媒、例えばペンタフルオロフェノール/クロロホルム(体積混合比75/25)混合溶媒を溶離液として用いたGPC−LS(ゲル浸透クロマトグラフ−光散乱)法により絶対分子量として測定した値である。
本発明では、分子量を制御するために単官能カルボン酸を重合系中に添加することができる。単官能カルボン酸を添加することにより、過剰な重合反応を抑制し、ゲル化などの副反応の発生を抑制することができる。単官能カルボン酸は、反応性、耐熱性やハンドリング性の観点から、安息香酸またはその誘導体であることが好ましい。具体的には、安息香酸、4−tert−ブチル安息香酸、3−tert−ブチル安息香酸、4−クロロ安息香酸、3−クロロ安息香酸、4−メチル安息香酸、3−メチル安息香酸、2−メチル安息香酸、3,5−ジメチル安息香酸、3,4−ジメチル安息香酸、2,3−ジメチル安息香酸、2,4−ジメチル安息香酸、2,5−ジメチル安息香酸、2,6−ジメチル安息香酸、4−エチル安息香酸などを添加することが可能である。添加方法は、樹状ポリエステルの重合反応開始前に添加する方法、重合反応途中に添加する方法のいずれかを選択できる。
樹状ポリエステルのカルボン酸末端とカルボン酸反応性単官能化合物との反応方法としては、樹状ポリエステルの重合反応途中に添加する方法、樹状ポリエステルの重合反応後に、再溶融または溶媒中に溶解せしめた樹状ポリエステルとカルボン酸反応性単官能化合物とを反応させる方法のいずれかを選択できるが、樹状ポリエステルとの反応性や安全性の観点から、樹状ポリエステルの重合反応後に、再溶融または溶媒中に溶解せしめた樹状ポリエステルとカルボン酸反応性単官能化合物とを反応させる方法を用いることが好ましい。また、マトリクスポリマーや充填剤に樹状ポリエステルを配合し、成形加工する際にカルボン酸反応性単官能化合物を同時に配合する方法を用いても良い。
こうして得られた樹状ポリエステルは優れた溶融液晶性を示し、マトリクスポリマーに配合することにより、マトリクスポリマーの流動性を改良することができるが、本発明の製造方法の要点は、用いる樹状ポリエステルの分子末端に存在するカルボン酸基の量を1×10-4当量/g以下としたことである。一般にカルボン酸基はプロトンが電離することにより自己触媒反応を起こし、加水分解を引き起こすことが知られている。また、加水分解により生成したカルボン酸基が更に自己触媒反応を起こすため、ポリエステルは大きく分子量を低下させることとなるが、本発明の製造方法に用いる樹状ポリエステルに関しても、同様である。また、他のマトリクスポリマーと溶融混練した場合には、樹状ポリエステルが元々有するカルボン酸基に加え、加水分解による発生したカルボン酸基がマトリクスポリマーの分子鎖を攻撃することとなるため、吐出されたポリマーの分子量は大きく低下してしまう。また、樹状ポリエステルに官能基が多く存在することにより、マトリクスポリマーによってはエステル交換反応などにより分子構造が変化し、特性を変化させてしまう場合がある。このような現象は滞留時間が短い押出加工(例えば射出成形など)では大きな問題とならないものの、滞留時間が長い溶融紡糸においては無視できない問題となる場合がある。参考例7に記載の通り、樹状ポリエステルは分子末端を未制御のままでは8.95×10−4当量/gと非常に多くのカルボン酸基を有する構造である。本発明者等は樹状ポリエステルの流動性向上の効果を溶融紡糸に適用すべく鋭意検討した結果、樹状ポリエステルの分子末端に存在するカルボン酸基量を1×10−4当量/g以下することでマトリクスポリマーの加水分解を抑制できることを見出し、これを満足する樹状ポリエステルであれば、流動性を向上する効果が大きく、紡糸温度を低下することにより、溶融混練時などの樹状ポリエステルおよびマトリクスポリマーの加水分解が抑制される。よって樹状ポリエステルを添加することによる悪影響は全く考慮する必要がなくなり、単純に紡糸温度を低下させた分熱分解が抑制され、吐出されるポリマーの分子量は向上することとなる。また、熱分解が抑制されることにより、分解ガスの発生が低下するため、口金汚れが大きく抑制され、長時間安定した紡糸を可能とする。
樹状ポリエステルの分子末端に存在するカルボン酸基量の定量は、中和滴定法によって行うことができる。例えば、樹状ポリエステル0.5gをo−クロロフェノールまたはo−クレゾール10mLに90℃に加熱しながら溶解させ、冷却した後、クロロホルム4mLを加える。ブロモフェノールブルー−エタノール溶液(0.2重量%)を数滴加えた後、滴定試薬(0.04M水酸化カリウム−メタノール溶液)をビュレットにて滴下し、中和点に達するまでに滴下した滴定試薬量から樹状ポリエステルの末端カルボン酸量を計算できる。
本発明の製造方法に用いる樹状ポリエステルの分子末端カルボン酸基量は、カルボン酸反応性単官能化合物を反応せしめることにより低下させることが可能であり、該単官能化合物であれば樹状ポリエステルの流動性向上の効果を損なうことなく、分子末端のカルボン酸基を低下させることができる。ここで、カルボン酸反応性単官能化合物とは、常温または加熱時にカルボン酸と反応し、エステル、アミド、ウレタン、ウレア結合を形成しうる官能基を分子内に1つ有する化合物をいう。樹状ポリエステルの分子末端に存在するカルボン酸基に、カルボン酸反応性単官能化合物を反応させ、分子末端に単官能化合物を導入することにより、樹状ポリエステルの滞留安定性や耐加水分解性を向上させ、さらに他のマトリクスポリマーと溶融混練した際には、マトリクスポリマーの分解を抑制できる。
本発明の樹状ポリエステルに用いることのできるカルボン酸反応性単官能化合物としては、オルトエステル、オキサゾリン、エポキシド、イソシアネート、カルボジイミド、ジアゾ化合物から選ばれる1種類以上の化合物である。カルボン酸との反応性およびハンドリング性の観点から、オルトエステル、オキサゾリン、エポキシド、イソシアネートが好ましく、中でも樹状ポリエステルの融点を高く維持できるという観点からオルトエステルが特に好ましい。カルボン酸反応性単官能化合物は、単独で使用または2種類以上のカルボン酸反応性単官能化合物を併用しても構わないことは言うまでもない。
前記したオルトエステル化合物としては、例えば、オルト酢酸トリメチル、オルト酢酸トリエチル、オルト酢酸トリプロピル、オルト酢酸トリブチル、オルト酢酸トリベンジル、オルト蟻酸トリメチル、オルト蟻酸トリエチル、オルト蟻酸トリプロピル、オルト蟻酸トリブチル、オルト蟻酸トリベンジル、オルトプロピオン酸トリメチル、オルトプロピオン酸トリエチル、オルトプロピオン酸トリプロピル、オルトプロピオン酸トリブチル、オルトプロピオン酸トリベンジル、オルト安息香酸トリメチル、オルト安息香酸トリエチル、オルト安息香酸トリプロピル、オルト安息香酸トリブチル、オルト安息香酸トリベンジルなどが挙げられる。このうち、樹状ポリエステルとの反応性や親和性およびハンドリング性の観点から、オルト酢酸トリメチル、オルト酢酸トリエチル、オルト蟻酸トリメチル、オルト蟻酸トリエチルが好ましく、熱可塑性繊維の特性への影響を予防するとい観点からオルト酢酸トリメチルまたはオルト酢酸トリエチルが特に好ましい。
その他のカルボン酸反応性単官能化合物として、オキサゾリン化合物としては、例えば、2−メチル−2−オキサゾリン、2−エチル−2−オキサゾリン、2−プロピル−2−オキサゾリン、2−ブチル−2−オキサゾリン、2−イソプロピル−2−オキサゾリン、2−イソブチル−2−オキサゾリン、2−sec−ブチル−2−オキサゾリン、2−tert−ブチル−2−オキサゾリン、2−フェニル−2−オキサゾリン、2−ビフェニル−2−オキサゾリンが挙げられ、エポキシ化合物としては、エチレンオキサイド、プロピレンオキサイド、ブチルグリシジルエーテル、フェニルグリシジルエーテル、安息香酸グリシジルエステルが挙げられる。
その他のカルボン酸反応性単官能化合物として、オキサゾリン化合物としては、例えば、2−メチル−2−オキサゾリン、2−エチル−2−オキサゾリン、2−プロピル−2−オキサゾリン、2−ブチル−2−オキサゾリン、2−イソプロピル−2−オキサゾリン、2−イソブチル−2−オキサゾリン、2−sec−ブチル−2−オキサゾリン、2−tert−ブチル−2−オキサゾリン、2−フェニル−2−オキサゾリン、2−ビフェニル−2−オキサゾリンが挙げられ、エポキシ化合物としては、エチレンオキサイド、プロピレンオキサイド、ブチルグリシジルエーテル、フェニルグリシジルエーテル、安息香酸グリシジルエステルが挙げられる。
理論的には、前記カルボン酸末端の封鎖に用いるカルボン酸反応性単官能化合物を、封鎖したい末端基に相当する量添加することで末端封鎖が可能である。封鎖したい末端基相当量に対して、末端封鎖に用いる有機化合物を、1.005倍当量以上用いることが好ましい。また、末端封鎖に用いる有機化合物の添加量は2.5倍当量以下であれば、樹状ポリエステルの末端封鎖が充分行われ、かつ、カルボン酸基が系中に残存して、ガスを発生したりすることもない。
本発明者らは複合繊維おいて紡糸温度を低下させ、融点や耐熱性の異なるポリマーの組み合わせを実現するため、鋭意検討した結果、請求項1記載の樹状ポリエステルをブレンドすることにより、従来の樹状ポリエステルでは問題となっていた樹状ポリエステルのカルボン酸基を起点とした加水分解を抑制しつつ、流動性を向上させることに成功し、これがブレンドされた高融点や高粘度のポリマーを複合繊維に適用することで、力学特性に優れた複合繊維を得ることに成功した。また、本発明の複合繊維では、紡糸温度を低下させることができるため、耐熱性の低いポリマーと高融点や高粘度ポリマー、あるいは耐熱性の低いポリマーどうしの組み合わせに用いることも有用であり、ポリマーの組み合わせに制限なく、用途、必要特性に合わせ、様々な複合繊維を得ることができる。また、別の観点では、複合紡糸の断面形態を制御するにあたり、組み合わせたポリマーの粘度比が非常に重要な要素となるが、この樹状ポリエステルの添加量を変更することにより、未制御、すなわち未添加の場合と比較して、易成形性等で優れた複合繊維を得ることが可能となる。ここで請求項1記載の樹状ポリエステルをブレンドするマトリックスポリマーとしては、例えば、ポリエステル、ポリアミド、ポリフェニレンスルフィド、ポリオレフィン、ポリカーボネート、ポリエステルカーボネート、ポリイミド、ポリアミドイミド、ポリエーテルケトン、ポリエーテルエーテルケトン、ポリフッ化ビニリデン、ポリビニールアルコールおよびこれらの共重合体、エチレン酢酸ビニルなどを挙げることができる。中でも、参考例7〜11に示すように紡糸温度を10〜15℃低下可能であり、汎用性が高いポリエステルやポリアミド、参考例12に示すように紡糸温度を20℃低下可能な耐熱性・薬品性に優れるポリフェニレンスルフィドが好ましい。
樹状ポリエステルは本発明の目的からすると、高融点や高粘度ポリマーにブレンドし、これらの流動性を向上することが好ましい。高融点や高粘度ポリマーに本発明の樹状ポリエステルが添加されていることにより、流動性が向上し、紡糸温度を未添加の場合と比較して、10℃以上低下させることが可能である。例えば、極限粘度が1.0dL/g以上の高分子量ポリエステルなどは、その融点はそれほど高くないものの超高粘度のため、参考例11に示すように紡糸温度を衣料用ポリエステル(通常IV0.65dL/g程度)の場合に比べ10〜20℃程度高くする場合も有る。これを複合紡糸に用いる場合には複合ポリマー流の流動性を向上させるために通常よりも紡糸温度は高く設定する必要がある。また、ポリ乳酸などの耐熱性の低いポリマーと組み合わせる場合には、熱分解が著しく進行し、複合繊維の力学的特性を低下させるだけでなく、紡糸性を低下させ紡糸自体を困難にする場合がある。本発明の複合繊維において、樹状ポリエステルは複合繊維を構成するポリマーのうち高融点ポリマーに添加することが好ましい。ここで言う融点とは、示差走査熱量測定(DSC)で観測される融解ピークのピークトップ温度を意味し、具体的な測定方法としては、以下のようにして行うことができる。すなわち、サンプルとして10mgを計量し、アルミパンに封入後、TA Instruments社製DSC2920 Modulated DSCに設置して、昇温速度16℃/分で測定を行う。そして、2nd runにおいてそのポリマーの融解ピークのピークトップ温度をそのポリマーの融点として求めたものである。
2種類のポリマーを用いる場合には、前記した測定方法によって予め各ポリマーの融点を測定し、各ポリマーの融点を比較して、高い方に樹状ポリエステルを添加すると良い。同様に3種類以上のポリマーを使用する場合には、測定された融点から最も高い融点を有するポリマーに添加することに加え、2番目以降の融点を有するポリマーに添加すると最も融点が低いポリマーに適した紡糸温度にすることができるため、好ましい。
本発明に用いるマトリクスポリマーには、熱安定性を保持するために、フェノール系およびリン系化合物の中から選ばれた1種以上の耐熱剤をあらかじめ添加することもできる。かかる耐熱剤の添加量は、耐熱改良効果の点から、0.01重量%以上が好ましく、特に0.02重量%以上であることが好ましい。得られる繊維への力学特性変化を予防するためには、添加量は、5重量%以下が好ましく、特に1重量%以下であることが好ましい。また、フェノール系およびリン系化合物を併用して使用することは、特に耐熱性、熱安定性および流動性保持効果が大きく好ましい。
フェノール系化合物としては、ヒンダードフェノール系化合物が好ましく用いられ、中でも、N、N’−ヘキサメチレンビス(3,5−ジ−t−ブチル−4−ヒドロキシ−ヒドロシンナミド)、テトラキス[メチレン−3−(3’,5’−ジ−t−ブチル−4’−ヒドロキシフェニル)プロピオネート]メタンなどが好ましく用いられる。
リン系化合物としては、ビス(2,6−ジ−t−ブチル−4−メチルフェニル)ペンタエリスリトール−ジ−ホスファイト、ビス(2,4−ジ−t−ブチルフェニル)ペンタエリスリトール−ジ−ホスファイト、ビス(2,4−ジ−クミルフェニル)ペンタエリスリトール−ジ−ホスファイト、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト、テトラキス(2,4−ジ−t−ブチルフェニル)−4,4’−ビスフェニレンホスファイト、ジ−ステアリルペンタエリスリトール−ジ−ホスファイト、トリフェニルホスファイト、3,5−ジーブチル−4−ヒドロキシベンジルホスフォネートジエチルエステルなどが挙げられる。中でも、熱可塑性繊維の製造工程において耐熱剤が揮発や分解することを少なくするために、融点が高いものが好ましく用いられる。さらに、紫外線吸収剤(たとえばレゾルシノール、サリシレート)、着色防止剤(亜リン酸塩、次亜リン酸塩など)、滑剤および離型剤(ステアリン酸、モンタン酸およびその金属塩など)、着色剤、導電剤あるいは着色剤としてのカーボンブラック、結晶核剤、可塑剤および帯電防止剤などの通常の添加剤、もしくは、マトリクスポリマー以外の重合体を配合することができる。
本発明の複合繊維では、樹状ポリエステルをマトリクスポリマーにブレンドしたポリマーブレンドの複合比率は1〜99重量%とすることが好ましいが、複合繊維を構成するポリマーのうち、複合比率が高いポリマーがその複合繊維の力学特性、特に強度、伸度あるいは弾性率を決定することとなるため、目的とする機能により複合比率を決定する必要がある。ここで言う複合比率とは、一定量の複合繊維の重量に対する樹状ポリエステルが占める重量比率のことを意味する。
本発明の複合繊維とは、2種類以上のポリマーが組み合わされた繊維のことを意味し、繊維横断面において2種類以上のポリマーが層状あるいは海島状等の形態をとって存在している繊維のことを言う。この複合繊維の断面形態としては、例えば、芯鞘、サイドバイサイドおよび海島などが挙げられる。
ここで言う芯鞘の芯成分は真円に加え、星形、三葉などの多葉断面、不定形などが製造可能である。ここで言う芯鞘複合繊維とは、図1〜3に示すように、異なる2種類以上のポリマーが繊維軸に対して垂直の断面において、芯成分を鞘成分が被覆するように構成されている繊維を意味し、樹状ポリエステルがブレンドされたポリマーブレンドはこの芯成分あるいは鞘成分を構成ことができる。また、更なる特性の向上のために芯成分と鞘成分を多層としたり、芯成分または/あるいは鞘成分が2種類以上のポリマーにより構成(例えば、ポリマーアロイ)することも好適である。芯鞘複合繊維の製造方法としては、公知の方法を用いることができ、例えば、インサート型やパイプ型などの複合口金を用いて、芯成分と鞘成分を複合流とし、細孔から吐出することで作製させる。この時、芯成分を難溶出成分で構成し、かつあらかじめ異形断面とし、複合繊維として紡糸し、巻き取り後に易溶出成分を溶出することにより、例えば星形(図2)あるいは三葉などといった多葉断面(図3)などの異形断面繊維が得られる。ここで言う異形断面とは、異形度が1.3以上のいわゆる真円でない断面形状のことを言う。
また、サイドバイサイドとは、横断面方向で見て、2種類以上のポリマーが張り合わされた形態をとり構成されているものであり、このサイドバイサイド複合繊維の製造方法としては、公知の方法を用いることができる。例えば、少なくとも吐出直後にポリマーが接触するように口金面の垂直方向に対して傾斜を付けた計量孔を2個以上有した細孔により、組み合わせるポリマーをそれぞれ計量し、吐出直前に細孔内でポリマー同士が合流し、張り合わされることにより作製される。また、別々の吐出孔を設けて、吐出直後で合流し、張り合わせる方法も採用することができる。このサイドバイサイドにいては、2種類以上のポリマーが多層に張り合わされていても良いし、3種類以上のポリマーを張り合わせることにより、3種類以上の特性を付与することも好適である。
海島型複合繊維とは、図4に示すように異なる2種類以上のポリマーが繊維方向に対し垂直な断面に海島構造を形成しており、ここでいう海島構造とは、島成分が海成分により複数に区別されている状態あるいは構造を形成しているもののことを言い、その区別された状態または島成分の断面形状に制約はなく、易溶出成分を溶出することにより、いわゆる極細繊維だけでなく、分割繊維等も得ることができる。
本発明の樹状ポリエステルはブレンドされたポリマーブレンドはこの海成分であっても島成分であってもよいが、前述した樹状ポリエステルの末端構造制御の効果を有効にするのであれば、島成分(難溶出成分)に添加するのが好ましい。カルボン酸基はポリマー中に存在することにより、自己触媒反応によって加水分解を促進させることが知られているが、従来の樹状ポリエステルにおいては多くこのカルボン酸基を有することにより、加水分解、すなわち溶出を促進させてしまう効果があった。このため、例えば、難溶出成分として高融点ポリマーを使用した場合、樹状ポリエステルを添加して、紡糸温度を低下させ、分子量低下なく、難溶出成分および易溶出成分を吐出し、繊維化したとしても、易溶出成分を溶出する工程において、難溶出成分として用いた高融点ポリマーまで加水分解による溶解が進み、得られた極細繊維や異形断面糸の力学特性を損なうことになる。
このような海島複合繊維を製造する方法としては公知のパイプ型やインサート型の海島複合口金を用いる方法、異なる2種類のポリマーをあらかじめブレンドし、アロイポリマーとして単独口金から吐出する方法、あるいはこれらの組み合わせる方法がある。
本発明の複合繊維においては、高融点ポリマーの流動性を向上させ、紡糸温度を低く設定することができ、組み合わせたいずれのポリマーの熱分解を抑制することができることで、優れた力学的特性を有した複合繊維となる。また、紡糸性の観点からもポリマーの熱分解を抑制することはポリマー流動の安定性が増すため、好ましいのである。
本発明の複合繊維は、紡糸温度を低下させることができるため、複合化のため組み合わせるポリマーをいずれも熱分解を抑制させることができ、得られる複合繊維は力学特性が大きく向上する。これは同融点であっても高分子量ポリマーを利用することにより、通常の紡糸温度よりも高く設定せざる得ない場合にも有効に作用し、例えば、実施例13と比較例7の比較からもその効果は明らかである。また、このように未延伸糸において力学特性が優れる複合繊維は図7に示すように同伸度当たりの強度が大きく向上するように優れた延伸糸となる。
更に、融点が大幅に異なるポリマー同士の組み合わせにも非常に有効であることは言うまでもなく、例えば、本発明の効果が顕著化する例としては、下記するようなポリマーの組み合わせが考えられる。
例えば、ポリ乳酸(PLA)が一部を構成する複合繊維としては、最近ではバイオ原料による合成が可能となったポリトリメチレンテレフタレート(3GT)との芯鞘構造とすることによりバイオ原料比率を向上させた繊維が着目されている。しかしながら、3GTの融点が230℃であるため、紡糸温度は3GTに合わせておのずと245℃以上に設定する必要があり、250℃が好適に用いられている。この250℃という紡糸温度は、PLAの融点が160℃であるのに対して、90℃も高く、結果的にPLAの熱分解を著しく進行させてしまうこととなる。一方、紡糸温度をできるだけ低温化しようと、紡糸温度を245℃に設定しても、紡糸は可能であるが、3GTにとっては安定紡糸限界の温度であるために、吐出が不安定になり、例えば、比較例1に示すように結果的に低い力学特性となってしまう。一方、本発明の複合繊維に関しては実施例3に示したように紡糸温度を安定紡糸限界温度かさらに10℃低下させても問題のない紡糸性が確保されるため、優れた力学特性となる。また、PLAが熱分解することにより、生産に関しては、PLAの熱分解に伴う着色や糸切れが多発するという問題、また、このように採取したPLA/3GT芯鞘複合繊維の力学特性は当然期待される値に対して低下したものとなってしまう。
また、PLAはアルカリ水溶液に易溶出性であるため、海島複合繊維における海成分として利用する場合があり、前記したいわゆる海島複合口金やポリマーアロイ技術を活用して、3GT/PLA海島複合繊維とする場合がある。この3GT/PLA海島複合繊維は海成分であるPLAを除去することにより、3GTの特性であるソフト性や染色性を生かした極細繊維を得ることができ、衣料用だけでなく、産業用途においても利用価値が高い。しかしながら、この海島複合繊維を得る際にも、前記した芯鞘複合繊維の場合と同様に紡糸温度は245℃以上にする必要があり、PLAの熱分解が著しく進行することとなる。繊維表層に存在するPLAの熱分解が進行すると、紡糸時の糸切れが多発することに加え、熱分解したPLAが紡糸機のガイドやローラとの擦過により削れ、紡糸装置を汚すこととなり、装置の清掃周期を早めるほかこの付着した汚れによって紡糸性の低下などの問題を起こす。また、このような結果、得られる3GT/PLA海島複合繊維の力学特性は低いものとなり、後加工性等が大きく低下したものとなることは言うまでもない。
一方、本発明の複合繊維においては、この組み合わせで言うと高融点である3GTに請求項1記載の樹状ポリエステルを添加することにより、3GTの流動性を向上させる効果から参考例7からわかるように紡糸温度を10℃低下させることができるため、これを複合繊維とした場合も同様に紡糸温度が低下でき、PLAの熱分解が大きく抑制されたものあとなる。このため、糸切れやPLAの削れなどの問題が大きく抑制され、生産性の向上および繊維の品位を高めるのに大きく貢献できるものとなることに加え、実施例3と比較例1との比較からわかるように優れた力学特性を有した複合繊維となる。
また、収縮特性の異なるポリマーを組み合わせることにより、独特の風合いを有した複合繊維とする場合がある。例えば、3GTとポリエチレンテレフタレート(PET)をサイドバイサイドとして、これを熱処理等することにより収縮特性を発現させる技術がある。この場合、3GTの融点が230℃であるのに対して、紡糸温度はPETに合わせることになるため、275℃以上に設定する必要があり、3GTの融点との温度差は45℃と3GTにとっては非常に過酷な温度雰囲気にさらされることとなる。ここで問題となるのは、3GTの熱分解に起因した口金汚れである。口金汚れとは、溶融ポリマーを吐出した際に分解物が吐出孔周りに付着し、これとポリマー流が接触することで糸切れの要因となる。前記したように3GT/PETのサイドバイサイド複合繊維の場合には3GTの融点と比較して、高い紡糸温度に設定する必要があり、この口金汚れのため、生産性の悪化や繊維の品位を落とすという問題があったが、本発明の複合繊維においては,PETに請求項1に記載の樹状ポリエステルを添加することにより、PETの流動性を向上させ、参考例11に示したように紡糸温度を5℃以上低下することが可能なのである。この効果により3GTの熱分解を抑制し、口金汚れを低減することが可能になることに加え、3GTの分子量が高くなることによってより優れた収縮特性を発現させることが可能となる。また、サイドバイサイドの場合は、ポリマーの吐出時の流速(吐出線速度)が異なると、ポリマーが吐出面側に屈曲してしまう糸曲がりの原因となり、紡糸性が大幅に低下することがある。紡糸温度が高いと経時的にポリマーの粘度が変化するため、この吐出線速度を合わせることが非常に困難となり、これも紡糸性や繊維の品位を落とす要因となるが、本発明の複合繊維においては、これの抑制にも大きく貢献することができる。従来方法で行った比較例11では長時間紡糸した際にこの糸曲がりが確認されたが紡糸温度を低温化した実施例18においては糸曲がりが確認されず、長時間紡糸を行った際でも良好な紡糸性が維持されていた。
前記したようにポリフェニレンサルファイド(PPS)は耐熱性や耐薬品性に優れるため、産業用途において利用価値が高い。一方、PETは熱可塑性ポリマーの中では耐酸性は比較的高い部類に入るが、耐アルカリ性は低く、この特性を補うには、PET/PPS芯鞘複合繊維とすることが考えられる。しかしながら、PPSは高融点ポリマーであるため、紡糸温度は310℃以上と非常に高く設定する必要があり、PPSを起因としたガスが発生するために口金汚れが問題となることに加え、熱媒を変更する必要があることから紡糸機を専用化しなくてはいけない場合がある。また、このPET/PPS芯鞘複合繊維を考えた場合には、この高い紡糸温度のため、PETが熱分解してしまい、繊維の力学特性を低下させる結果となる。本発明の複合繊維においては、PPSまたは/あるいはPETに請求項1記載の樹状ポリエステルを添加することにより、PPSまたは/あるいはPETの流動性を向上させることができ、参考例12に示したように紡糸温度を10℃低下させることが可能となる。この結果、PPSに起因したガス発生を抑制するとともに、紡糸温度が300℃と一般の溶融紡糸装置においても熱媒を変更することなく紡糸が可能である温度であり、産業用途における使用にも耐えうる優れた力学特性を有したPET/PPS芯鞘複合繊維を生産性良く採取することができる。
本発明の複合繊維は、優れた特性を有するため、衣料用途に用いた場合には品位に優れたものとなることが言うまでもなく、図9に示したように同伸度当たりの強度が向上するというような力学的特性が向上するために、特に産業資材用途でその特性を有効に活用することができる。
例えば、芯鞘複合繊維においては、屈曲疲労や摩耗特性も従来品よりも向上し、タイヤコードやタイヤのキャップレイヤー材などのゴム補強用途のみならず漁網や農業資材の他、スクリーン紗などにも好適に用いることができる。
本発明の複合繊維では、強度は2cN/dtex以上が好ましく、産業資材用途で必要とされる力学的特性を考えれば、5cN/dtex以上であることが好ましい。現実的な上限としては20cN/dtexである。また、伸度は延伸糸で2〜60%、特に高強度が必要とされる産業資材分野では2〜25%、衣料用では25〜60%とすることが好ましい。前記した複合繊維は、繊維巻き取りパッケージやトウ、カットファイバー、わた、ファイバーボール、コード、パイル、織編、不織布、紙、液体分散体など多用な繊維製品とすることができる。
本発明の複合繊維を得る方法、すなわち、樹状ポリエステルのマトリクスポリマーへのブレンド方法および繊維化については、公知の混練方法や複合紡糸方法を採用することで作製できる。
まず、樹状ポリエステルのブレンドについては例えば以下のような方法を用いることができる。すなわち、請求項1に記載した樹状ポリエステルとマトリックスポリマーを必要に応じ乾燥し、二軸押し出し混練機に導入する。この時、ブレンド装置としてはブレンド斑を低減するために二軸押し出し混練機とすることが好ましい。ここで、作製したポリマーブレンドをそのまま複合紡糸機に導いても、マスターペレットとして一旦ペレット化しても良い。省力化のためには混練直結紡糸が好ましく、樹状ポリエステルのブレンド率やポリエステル分子量が異なる品種をいくつかつくるなど汎用性を持たせるため場合にはマスターペレット化しておくことが好ましい。また、混練直結紡糸の場合には、二軸押し出し混練機では一軸押し出し混練機の場合とは異なり、混練機中で誘起された発泡が仕込み側に抜け難いため、発泡が繊維にまで混入し糸切れが頻発する場合がある。このため、特に高分子量ポリエステルなど高粘度ポリマーをマトリックスポリマーとする場合には、二軸押し出し混練機の吐出側でベントを行い、泡を抜く操作を行うことが好ましい。なお、マスターペレット化場合にもガット切れが頻発する時はベントを行うことが好ましい。また、本発明においては樹状ポリエステル添加による良流動化効果により、未添加の場合に比べ同一温度であればスクリュートルクが小さくなるため、混練温度の低温化が可能である。これにより、ポリマーの熱分解や熱変性、また加水分解などを抑制することができ、バージンポリマーが本来持っていた高分子量や易加工性などを利用し易くできるのである。先の樹状ポリエステルブレンドでマスターペレット化した場合には、紡糸過程でバージンポリマーにより希釈されるわけであるが、この時も二軸押し出し混練機を用いる方がブンレンドの均一性の観点から好ましい。というのは、本発明では樹状ポリエステルブレンド率で良流動化効果の程度が異なるため、ポリマーブレンド中でブレンドが不均一であるとスクリュートルクや先端圧、濾圧、口金背面圧、ひいては紡糸応力などの斑が発生し、安定した紡糸が不能となる場合があるからである。
本発明の樹状ポリエステルブレンドによる良流動化効果のため、未添加の場合に比べ混練機温度を低温化できることも有用であるが、本発明においては紡糸機、特に紡糸ヘッドの設定温度を低下させることが可能であることがその効果の特徴として挙げられる。すなわち、2つ以上の溶融押出機を具備した複合紡糸機では溶融温度についてはおのおのポリマーで独立して設定できるものの、紡糸ヘッドでポリマーらは同パック内に導かれるため、同じ温度履歴を受けることとなる。この温度を決定するのは前述したように主に高融点や高粘度ポリマーが流動性を示す温度であり、耐熱性が低いポリマーがある場合には紡糸ヘッド内で大きく熱分解を起こすこととなる。これが複合繊維の物性だけでなく、紡糸性にも悪影響を与え、紡糸自体が困難となる場合が多い。このため、組み合わせるポリマーにはおのずと制限ができてしまうのである。一方、本発明の複合繊維の場合には、樹状ポリエステル添加による流動性の向上を利用することにより、例えば、高融点や高粘度ポリマーのため通常では紡糸温度を高温化せざるを得ない場合であっても、樹状ポリエステル添加により10℃以上の低温化も可能であり、この効果は、高粘度ポリマーほど大きく発現する。このため、紡糸温度を低く設定することができ、低粘度あるいは耐熱性の低いポリマーの熱分解というような従来の問題を解消することが可能である。しかも、本発明者らは樹状ポリエステル添加による流動性向上する技術を利用した紡糸技術について鋭意検討した結果、前述したR2構造の構成単位に芳香族が含まれていることによって、紡糸といったような非常に大きい伸長変形の際にも、マトリクスポリマーへの分子鎖の絡み合いが抑制させることにより、紡糸性良く複合繊維を得ることができることを見出した。
さらに、従来の樹状ポリエステルでは、末端構造がカルボン酸基であるため、添加することで加水分解速度が高まることにより、溶融時ならびに海島複合繊維などにおいては海成分除去時に樹状ポリエステルが添加された高融点ポリマーまで加水分解が進み、分子量を大きく低下させてしまう問題があった。本発明者らは、樹状ポリエステルを活用した複合繊維において、かかる問題について鋭意検討した結果、参考例7に例示されるように樹状ポリエステルの末端基のカルボン酸基量を1×10−4当量/g以下とすることにより、前記した加水分解が大きく抑制され、分子量が維持させた様々な複合繊維を得ることが可能となった。
本発明において、紡糸温度は、可能な限り樹状ポリエステル未添加のマトリクスポリマーを単独で紡糸する場合に比較して5〜20℃低下させることが好ましい。より好ましくは、7〜15℃低下である。また、紡糸速度はマトリックスポリマーと組み合わせるポリマーの物性や複合繊維の目的によって異なるが、500〜6000m/分程度とすることができる。特に、産業資材用途で高い力学的特性が必要な場合には、高分子量ポリマーを用い、500〜2000m/分とし、その後高倍率延伸することが好ましい。
延伸に際しては、特に予熱温度を適切に設定することが好ましい。というのは本発明で用いる樹状ポリエステルはガラス転移温度などの軟化温度が70℃より高い場合があり、例えばPETの通常の予熱温度である85〜95℃程度では、樹状ポリエステルが延伸過程で異物として振る舞い結果として延伸糸のタフネスの低下を招く場合がある。この影響は、特に高倍率延伸時ほど顕著に現れる。このため、樹状ポリエステルの添加量が微量であっても予熱温度は樹状ポリエステルのガラス転移温度や軟化温度以上に設定することが好ましい。予熱温度の上限としては、予熱過程で繊維の自発伸長により糸道乱れが発生しない温度とすることが好ましい。この延伸時の予熱温度設定も糸斑低減に寄与することができる。
本発明の複合繊維から異形断面繊維を得る場合には、水酸化ナトリウム水溶液などのアルカリ水溶液にて易溶出成分を除去することにより難溶出成分からなる異形断面繊維を得ることができる。本発明の複合繊維をアルカリ水溶液にて処理する方法としては、例えば、複合繊維あるいはそれからなる繊維構造体としたあと、アルカリ水溶液に浸漬させればよい。この時、アルカリ水溶液は50℃以上に加熱すると、加水分解の進行を早めるため、好ましい。また、流体染色機などを利用し、処理すれば、一度に大量に処理をおこなうことができるため、生産性もよく、工業的な観点から好ましいことである。
以下、本発明を実施例を用いて詳細に説明する。なお、実施例中の測定方法は以下の方法を用いた。
A.樹状ポリエステルの絶対分子量
樹状ポリエステルの絶対分子量は樹状ポリエステルが可溶な溶媒であるペンタフルオロフェノールを使用して、GPC−MALLS(ゲル浸透クロマトグラフ(ShodexGPC−101)−光散乱検出器(Wyatt製DAWN HELEOS))により、試料濃度0.04%、測定温度23℃で測定した。
樹状ポリエステルの絶対分子量は樹状ポリエステルが可溶な溶媒であるペンタフルオロフェノールを使用して、GPC−MALLS(ゲル浸透クロマトグラフ(ShodexGPC−101)−光散乱検出器(Wyatt製DAWN HELEOS))により、試料濃度0.04%、測定温度23℃で測定した。
B.樹状ポリエステルの化学組成比
樹状ポリエステルの化学組成比は核磁気共鳴装置(日本電子製JNM−AL400)を用いて、ペンタフルオロフェノール/重水素化クロロホルム(50/50)混合溶媒に溶解して、40℃で1H−NMR測定を行い、ピーク強度比から各成分の化学組成比を算出した。
樹状ポリエステルの化学組成比は核磁気共鳴装置(日本電子製JNM−AL400)を用いて、ペンタフルオロフェノール/重水素化クロロホルム(50/50)混合溶媒に溶解して、40℃で1H−NMR測定を行い、ピーク強度比から各成分の化学組成比を算出した。
C.樹状ポリエステルの融点
樹状ポリエステルの融点(Tm)は、樹状ポリエステルを、示差熱量測定において、室温から20℃/分の昇温条件で測定した際に観測される吸熱ピーク温度(Tm1)の観測後、Tm1+20℃の温度で5分間保持し、20℃/分の降温条件で室温まで一旦冷却し、再度20℃/分の昇温条件で測定した際に観測される吸熱ピーク温度(Tm)とした。
樹状ポリエステルの融点(Tm)は、樹状ポリエステルを、示差熱量測定において、室温から20℃/分の昇温条件で測定した際に観測される吸熱ピーク温度(Tm1)の観測後、Tm1+20℃の温度で5分間保持し、20℃/分の降温条件で室温まで一旦冷却し、再度20℃/分の昇温条件で測定した際に観測される吸熱ピーク温度(Tm)とした。
D.樹状ポリエステルの液晶開始温度
液晶開始温度は、剪断応力加熱装置(CSS−450)により、剪断速度1.0(1/秒)、昇温速度5.0℃/分、対物レンズ60倍において測定し、視野全体が流動開始する温度とした。
液晶開始温度は、剪断応力加熱装置(CSS−450)により、剪断速度1.0(1/秒)、昇温速度5.0℃/分、対物レンズ60倍において測定し、視野全体が流動開始する温度とした。
E.樹状ポリエステルの分子末端カルボン酸基量測定
分子末端カルボン酸基量測定は、中和滴定法によって行った。樹状ポリエステル0.5gをo−クロロフェノール10mLに90℃で加熱しながら溶解させ、冷却した後、クロロホルム4mLを加えた。ブロモフェノールブルー−エタノール溶液(0.2重量%)を数滴加えた後、滴定試薬(0.04M水酸化カリウム−メタノール溶液)をビュレットを用いて滴下し、中和点に達するまでに滴下した滴定試薬量から樹状ポリエステルの末端カルボン酸量を計算した。
分子末端カルボン酸基量測定は、中和滴定法によって行った。樹状ポリエステル0.5gをo−クロロフェノール10mLに90℃で加熱しながら溶解させ、冷却した後、クロロホルム4mLを加えた。ブロモフェノールブルー−エタノール溶液(0.2重量%)を数滴加えた後、滴定試薬(0.04M水酸化カリウム−メタノール溶液)をビュレットを用いて滴下し、中和点に達するまでに滴下した滴定試薬量から樹状ポリエステルの末端カルボン酸量を計算した。
F.ポリエステルの極限粘度(IV)(マトリクスポリマーおよび吐出ポリマー)
ポリエステルの極限粘度はo−クロロフェノールに溶解してオストワルド式粘度計を用いて25℃で測定した。
ポリエステルの極限粘度はo−クロロフェノールに溶解してオストワルド式粘度計を用いて25℃で測定した。
G.ナイロンの相対粘度(ηr)(マトリクスポリマーおよび吐出ポリマー)
98%硫酸水溶液にナイロンを溶解し0.01g/mLの濃度に調整した後、オストワルド式粘度計を用いて25℃で測定した。
98%硫酸水溶液にナイロンを溶解し0.01g/mLの濃度に調整した後、オストワルド式粘度計を用いて25℃で測定した。
H.重量平均分子量
ポリ乳酸の重量平均分子量は以下のようにして求めた。試料のクロロホルム溶液にTHF(テトロヒドロフラン)を混合し測定溶液とした。これをWATERS社製GPC WATERS2690を用いて25℃で測定し、ポリスチレン換算で重量平均分子量を求めた。
ポリ乳酸の重量平均分子量は以下のようにして求めた。試料のクロロホルム溶液にTHF(テトロヒドロフラン)を混合し測定溶液とした。これをWATERS社製GPC WATERS2690を用いて25℃で測定し、ポリスチレン換算で重量平均分子量を求めた。
I.融点
TA Instruments社製DSC2920 Modulated DSCを用いて2nd runでポリマーの融解を示すピークトップ温度をポリマーの融点とした。この時の昇温速度は16℃/分、サンプル量は10mgとした。
TA Instruments社製DSC2920 Modulated DSCを用いて2nd runでポリマーの融解を示すピークトップ温度をポリマーの融点とした。この時の昇温速度は16℃/分、サンプル量は10mgとした。
J.繊維の単糸繊度
繊維を検尺機によって100mの小綛とし、その重量を100倍することにより、総繊度とした。総繊度をフィラメント数で割ることで、単糸繊度を算出した。
繊維を検尺機によって100mの小綛とし、その重量を100倍することにより、総繊度とした。総繊度をフィラメント数で割ることで、単糸繊度を算出した。
K.繊維の力学特性(強度、伸度、タフネス)
室温(25℃)で、初期試料長=200mm、引っ張り速度=200mm/分とし、JIS L1013に示される条件で荷重−伸長曲線を求めた。次に破断時の荷重値を初期の繊度で割り、それを強度とし、破断時の伸びを初期試料長で割り伸度として強伸度曲線を求めた。また、下記式に従い、強度および伸度からタフネスを算出した。弾性率は荷重−伸長曲線の初期立ち上がり部分を直線近似し、その傾きから求めた
(タフネス)=(強度)×√(伸度) (cN/dtex・%1/2)
L.複合繊維の断面形態観察
複合口金から吐出された複合ポリマー流を口金下で冷却固化後サンプリングし、得られたガット状サンプル1本を剃刀にて繊維軸に対して垂直方向に切断した。この切断面を400倍に設定した実体顕微鏡にて観察した。
室温(25℃)で、初期試料長=200mm、引っ張り速度=200mm/分とし、JIS L1013に示される条件で荷重−伸長曲線を求めた。次に破断時の荷重値を初期の繊度で割り、それを強度とし、破断時の伸びを初期試料長で割り伸度として強伸度曲線を求めた。また、下記式に従い、強度および伸度からタフネスを算出した。弾性率は荷重−伸長曲線の初期立ち上がり部分を直線近似し、その傾きから求めた
(タフネス)=(強度)×√(伸度) (cN/dtex・%1/2)
L.複合繊維の断面形態観察
複合口金から吐出された複合ポリマー流を口金下で冷却固化後サンプリングし、得られたガット状サンプル1本を剃刀にて繊維軸に対して垂直方向に切断した。この切断面を400倍に設定した実体顕微鏡にて観察した。
参考例1(樹状ポリエステルA−1の合成)
攪拌翼および留出管を備えた500mLの反応容器にp−ヒドロキシ安息香酸66.3g(0.48モル)、4,4’−ジヒドロキシビフェニル8.38g(0.045モル)、テレフタル酸7.48g(0.045モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレ−ト14.41g(0.075モル)、トリメシン酸31.52g(0.15モル)を加えておよび無水酢酸62.48g(フェノール性水酸基合計の1.00当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で2時間反応させた。その後、280℃まで昇温し、3時間攪拌し、理論留出量の91%の酢酸が留出したところで加熱および攪拌を停止し、内容物を冷水中に吐出した。
攪拌翼および留出管を備えた500mLの反応容器にp−ヒドロキシ安息香酸66.3g(0.48モル)、4,4’−ジヒドロキシビフェニル8.38g(0.045モル)、テレフタル酸7.48g(0.045モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレ−ト14.41g(0.075モル)、トリメシン酸31.52g(0.15モル)を加えておよび無水酢酸62.48g(フェノール性水酸基合計の1.00当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で2時間反応させた。その後、280℃まで昇温し、3時間攪拌し、理論留出量の91%の酢酸が留出したところで加熱および攪拌を停止し、内容物を冷水中に吐出した。
得られた樹状ポリエステルを、乾燥機を用いて110℃で5時間乾燥した後、ブレンダーを用いて粉砕し、得られた樹状ポリエステル粉末を、真空加熱乾燥機を用いて100℃で12時間加熱真空乾燥した。
乾燥後の樹状ポリエステル粉末70gと、カルボン酸反応性単官能化合物としてオルト酢酸エチル31.4g(0.19モル)を、撹拌翼を備えた500mLの反応容器に仕込み、200℃に昇温した。200℃で20分撹拌した後、内容物を冷水中に吐出した。
得られた樹状ポリエステル(A−1)について、核磁気共鳴スペクトル分析を行った結果、トリメシン酸残基に対して、p−オキシベンゾエート単位の含有量pが2.66、4,4’−ジオキシビフェニル単位とエチレンオキシド単位の含有量qが0.66、テレフタレート単位の含有量rが0.66であり、p+q+r=4であった。
得られた樹状ポリエステル(A−1)の融点Tmは172℃、液晶開始温度は131℃で、数平均分子量2100であった。
また、得られた樹状ポリエステル(A−1)の分子末端カルボン酸基量は、0.23×10−6当量/gであった。
参考例2(樹状ポリエステルA−2の合成)
カルボン酸反応性単官能化合物をオルト酢酸エチル20.9g(0.13モル)である以外は参考例1と同様にして、樹状ポリエステル(A−2)を得た。得られた樹状ポリエステルの評価結果を表1に示す。
カルボン酸反応性単官能化合物をオルト酢酸エチル20.9g(0.13モル)である以外は参考例1と同様にして、樹状ポリエステル(A−2)を得た。得られた樹状ポリエステルの評価結果を表1に示す。
参考例3(樹状ポリエステルA−3の合成)
カルボン酸反応性単官能化合物を2−フェニル−2−オキサゾリン28.5g(0.19モル)である以外は参考例1と同様にして、樹状ポリエステル(A−3)を得た。得られた樹状ポリエステルの評価結果を表1に示す。
カルボン酸反応性単官能化合物を2−フェニル−2−オキサゾリン28.5g(0.19モル)である以外は参考例1と同様にして、樹状ポリエステル(A−3)を得た。得られた樹状ポリエステルの評価結果を表1に示す。
参考例4(樹状ポリエステルA−4の合成)
カルボン酸反応性単官能化合物を安息香酸グリシジルエステル34.4g(0.19モル)である以外は参考例1と同様にして、樹状ポリエステル(A−4)を得た。得られた樹状ポリエステルの評価結果を表1に示す。
カルボン酸反応性単官能化合物を安息香酸グリシジルエステル34.4g(0.19モル)である以外は参考例1と同様にして、樹状ポリエステル(A−4)を得た。得られた樹状ポリエステルの評価結果を表1に示す。
参考例5(樹状ポリエステルB−1の合成)
攪拌翼および留出管を備えた500mLの反応容器にp−ヒドロキシ安息香酸66.3g(0.48モル)、4,4’−ジヒドロキシビフェニル8.38g(0.045モル)、テレフタル酸7.48g(0.045モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレ−ト14.41g(0.075モル)、トリメシン酸31.52g(0.15モル)を加えておよび無水酢酸62.48g(フェノール性水酸基合計の1.00当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で2時間反応させた。その後、280℃まで昇温し、3時間攪拌し、理論留出量の91%の酢酸が留出したところで加熱および攪拌を停止し、内容物を冷水中に吐出し、分子末端を封鎖していない樹状ポリエステル(B−1)を得た。樹状ポリエステルB−1の評価結果を表1に示す。
攪拌翼および留出管を備えた500mLの反応容器にp−ヒドロキシ安息香酸66.3g(0.48モル)、4,4’−ジヒドロキシビフェニル8.38g(0.045モル)、テレフタル酸7.48g(0.045モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレ−ト14.41g(0.075モル)、トリメシン酸31.52g(0.15モル)を加えておよび無水酢酸62.48g(フェノール性水酸基合計の1.00当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で2時間反応させた。その後、280℃まで昇温し、3時間攪拌し、理論留出量の91%の酢酸が留出したところで加熱および攪拌を停止し、内容物を冷水中に吐出し、分子末端を封鎖していない樹状ポリエステル(B−1)を得た。樹状ポリエステルB−1の評価結果を表1に示す。
参考例6(樹状ポリエステルB−2の合成)
カルボン酸反応性単官能化合物をオルト酢酸エチル7.85g(0.05モル)である以外は参考例1と同様にして、分子末端の封鎖を不十分とした樹状ポリエステル(B−2)を得た。樹状ポリエステルB−2の評価結果を表1に示す。
カルボン酸反応性単官能化合物をオルト酢酸エチル7.85g(0.05モル)である以外は参考例1と同様にして、分子末端の封鎖を不十分とした樹状ポリエステル(B−2)を得た。樹状ポリエステルB−2の評価結果を表1に示す。
参考例7
参考例1で合成した樹状ポリエステルA−1と、マトリックスポリマーとしてIV1.51dL/gのポリトリメチレンテレフタレート(3GT 融点230℃)を乾燥した後、別々に計量し、独立に二軸混練機(15mmφ、L/D=15)に仕込んだ。この時、樹状ポリエステルの添加量は、1重量%とした。二軸押出混練機のスクリュー回転数は100rpmとし、二軸押出混練機の吐出側でベントを行い、泡を消した。吐出量は20g/分で絶対濾過径10μの金属不織布で濾過した後、丸孔24ホール(φ=0.3mm L/D=1.5)の口金から吐出した。紡糸温度(混練機および紡糸ヘッド)を250〜235℃まで段階的に変更し、そのパック圧と吐出ポリマーのIVについて調べた。また、この比較として、紡糸温度を245℃に固定し、樹状ポリエステルを未添加の場合、樹状ポリエステルを参考例5で合成した樹状ポリエステルB−1とした場合(末端封鎖なし)および参考例6で合成した樹状ポリエステルB−2とした場合(末端封鎖が不十分)を同様に紡糸した。
参考例1で合成した樹状ポリエステルA−1と、マトリックスポリマーとしてIV1.51dL/gのポリトリメチレンテレフタレート(3GT 融点230℃)を乾燥した後、別々に計量し、独立に二軸混練機(15mmφ、L/D=15)に仕込んだ。この時、樹状ポリエステルの添加量は、1重量%とした。二軸押出混練機のスクリュー回転数は100rpmとし、二軸押出混練機の吐出側でベントを行い、泡を消した。吐出量は20g/分で絶対濾過径10μの金属不織布で濾過した後、丸孔24ホール(φ=0.3mm L/D=1.5)の口金から吐出した。紡糸温度(混練機および紡糸ヘッド)を250〜235℃まで段階的に変更し、そのパック圧と吐出ポリマーのIVについて調べた。また、この比較として、紡糸温度を245℃に固定し、樹状ポリエステルを未添加の場合、樹状ポリエステルを参考例5で合成した樹状ポリエステルB−1とした場合(末端封鎖なし)および参考例6で合成した樹状ポリエステルB−2とした場合(末端封鎖が不十分)を同様に紡糸した。
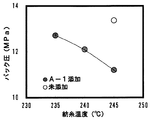
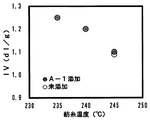
紡糸温度とパック圧の関係を図5にパック圧と吐出ポリマーIVの関係を図6に示す。
樹状ポリエステルA−1が添加された場合は、未添加の場合と比較して、パック圧合わせで紡糸温度が10℃低下できることがわかった。また、樹状ポリエステルB−1および樹状ポリエステルB−2が添加されたものについては、パック圧が低下するものの、図6からわかるように吐出ポリマーIVが樹状ポリエステルA−1添加のものと比較して低下したものであることがわかった。結果を表2に示す。
参考例8
紡糸温度を240℃とし、添加する樹状ポリエステルを参考例2で合成した樹状ポリエステルA−2、参考例3で合成した樹状ポリエステルA−3、参考例4で合成した樹状ポリエステルA−4と変更したこと以外は全て参考例7に従って紡糸した。結果、未添加の場合と比較して、紡糸温度を低下しているにも関わらずパック圧は低下しており、更に樹状ポリエステルB−1添加の場合や樹状ポリエステルB−2添加の場合と比較して、吐出ポリマーのIVは高くなることがわかった。結果を表2に示す。
紡糸温度を240℃とし、添加する樹状ポリエステルを参考例2で合成した樹状ポリエステルA−2、参考例3で合成した樹状ポリエステルA−3、参考例4で合成した樹状ポリエステルA−4と変更したこと以外は全て参考例7に従って紡糸した。結果、未添加の場合と比較して、紡糸温度を低下しているにも関わらずパック圧は低下しており、更に樹状ポリエステルB−1添加の場合や樹状ポリエステルB−2添加の場合と比較して、吐出ポリマーのIVは高くなることがわかった。結果を表2に示す。
参考例9
紡糸温度を235℃に固定し、樹状ポリエステルA−1の添加量を0.1重量%および10重量%と変更したこと以外は全て参考例7に従い実施した。
紡糸温度を235℃に固定し、樹状ポリエステルA−1の添加量を0.1重量%および10重量%と変更したこと以外は全て参考例7に従い実施した。
結果、添加量0.1重量%の場合でも良流動性の効果が得られていることがわかり、10重量%では大幅にパック圧が低下させることができることがわかった。また、吐出ポリマーのIVについても、維持されていることが確認された。結果を表2に示す。
参考例10
マトリクスポリマーをηr2.5のナイロン6(N6 融点220℃)として、表3に示す条件に示したように紡糸温度を変化させて紡糸を行い、パック圧を調べた。
マトリクスポリマーをηr2.5のナイロン6(N6 融点220℃)として、表3に示す条件に示したように紡糸温度を変化させて紡糸を行い、パック圧を調べた。
樹状ポリエステルA−1を添加した場合は未添加の場合と比較して、紡糸温度を15℃低下した場合でもパック圧が同等であり、N6に添加した場合でも効果が高く、紡糸温度低下が可能であることがわかった。結果を表3に示す。
参考例11
マトリクスポリマーをIV1.16dL/g(H−PET)およびIV0.65dL/g(R−PET)のPET(いずれも融点260℃)として、表3に示す条件に示したように紡糸温度を変化させて紡糸を行い、パック圧を調べた。
マトリクスポリマーをIV1.16dL/g(H−PET)およびIV0.65dL/g(R−PET)のPET(いずれも融点260℃)として、表3に示す条件に示したように紡糸温度を変化させて紡糸を行い、パック圧を調べた。
マトリクスポリマーをPETとした場合でも樹状ポリエステルを添加したことによる良流動性の効果から紡糸温度を15℃以上低下させることができることがわかった。またこの効果がIVに係わらず発揮されることがわかった。結果を表3に示す。
参考例12
マトリクスポリマーをPPSとして、表3に示す条件に示したように紡糸温度を変化させて紡糸を行い、パック圧を調べた。
マトリクスポリマーをPPSとして、表3に示す条件に示したように紡糸温度を変化させて紡糸を行い、パック圧を調べた。
マトリクスポリマーをPPSとした場合でも樹状ポリエステルを添加したことによる良流動性の効果から紡糸温度を18℃低下させることができことがわかった。結果を表3に示す。
実施例1〜3、比較例1
光学純度99%のポリL乳酸(PLA重量平均分子量11万 融点160℃)と参考例7で用いたIV1.51dL/gのポリトリメチレンテレフタレート(3GT 融点230℃)に樹状ポリエステルA−1を1重量%添加したポリマーブレンドをそれぞれ210℃、245℃にて溶融後、ポンプによる計量を行い、紡糸温度250℃にてPLAが芯成分、(3GT+樹状ポリエステルA−1)が鞘成分になるように図1に示すような同心円型芯鞘複合繊維になるように公知の芯鞘複合口金に流入させた。芯鞘複合比は芯成分/鞘成分=30/70となるようにし、芯鞘を合わせた単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度1500m/分で巻き取った。
光学純度99%のポリL乳酸(PLA重量平均分子量11万 融点160℃)と参考例7で用いたIV1.51dL/gのポリトリメチレンテレフタレート(3GT 融点230℃)に樹状ポリエステルA−1を1重量%添加したポリマーブレンドをそれぞれ210℃、245℃にて溶融後、ポンプによる計量を行い、紡糸温度250℃にてPLAが芯成分、(3GT+樹状ポリエステルA−1)が鞘成分になるように図1に示すような同心円型芯鞘複合繊維になるように公知の芯鞘複合口金に流入させた。芯鞘複合比は芯成分/鞘成分=30/70となるようにし、芯鞘を合わせた単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度1500m/分で巻き取った。
参考例7において3GT(鞘成分)に樹状ポリエステルがブレンドされたポリマーブレンドとすることによる流動性の向上により紡糸温度を10℃低下できることが確認されたため、紡糸温度は245℃から段階的に低下させた(実施例1〜3)。これの比較として、紡糸温度を245℃とし、樹状ポリエステル未添加とした場合も同様に紡糸を行った(比較例1)。
得られた芯鞘複合繊維の物性を比較すると、比較例1については、若干吐出が不安定になるため、力学特性が低下してしまい、結果、本発明の繊維(実施例1〜3)は、いずれも比較例1と比較して力学物性(タフネス)が向上し、品位に優れたPLA/3GT芯鞘複合繊維になっていることがわかった。また、紡糸性という観点においては、比較例1においては、8時間紡糸後の口金面を見ると、分解ガスの発生により24ホール中10ホールで吐出孔付近に白濁した分解物が付着しているのに対し、実施例3においては24ホール中1ホールに微細な付着物が見られる程度であり、本発明においては口金汚れを抑制する効果に優れたものであることがわかった。結果を表4に示す。
実施例4〜6
添加する樹状ポリエステルを参考例2で合成した樹状ポリエステルA−2(実施例4)、参考例3で合成した樹状ポリエステルA−3(実施例5)、参考例4で合成した樹状ポリエステルA−4(実施例6)としたこと以外は全て実施例2(紡糸温度240℃)に従い紡糸した。
添加する樹状ポリエステルを参考例2で合成した樹状ポリエステルA−2(実施例4)、参考例3で合成した樹状ポリエステルA−3(実施例5)、参考例4で合成した樹状ポリエステルA−4(実施例6)としたこと以外は全て実施例2(紡糸温度240℃)に従い紡糸した。
得られた芯鞘複合繊維の力学物性は比較例1と比較して向上していることがわかった。また、紡糸性についても問題なく、良好にPLA/3GT芯鞘複合繊維を採取することができた。結果を表5に示す。
比較例2,3
添加する樹状ポリエステルを参考例5で合成した樹状ポリエステルの末端が封鎖されていないB−1(比較例2)、参考例6で合成した樹状ポリエステルの末端封鎖が不十分なB−2(比較例3)としたこと以外は全て実施例3(紡糸温度240℃)に従い紡糸した。
添加する樹状ポリエステルを参考例5で合成した樹状ポリエステルの末端が封鎖されていないB−1(比較例2)、参考例6で合成した樹状ポリエステルの末端封鎖が不十分なB−2(比較例3)としたこと以外は全て実施例3(紡糸温度240℃)に従い紡糸した。
比較例1と比較すると、力学特性は向上したものであるが、本発明のPLA/3GT芯鞘複合繊維と比較すると、力学特性が低下したものであった。更に紡糸8時間後の口金面を見ると、比較例2では、24ホール中8ホールで吐出孔付近に白濁した分解物が付着しており、比較例3では、24ホール中5ホールで分解物の付着があることがわかり、参考例7の結果でも吐出ポリマーIVの低下が観られていることから、樹状ポリエステルのカルボン酸基の自己触媒反応によって加水分解が進行し、分子量が低下したものであった。結果を表3に示す。
実施例7,8、比較例4
鞘成分として、参考例10で用いたηr2.5のナイロン6(N6 融点220℃)に樹状ポリエステルA−1を2重量%添加したポリマーブレンドを用い、紡糸温度を255℃としたこと以外は全て実施例1に従い芯鞘複合繊維を得た(実施例7)。
鞘成分として、参考例10で用いたηr2.5のナイロン6(N6 融点220℃)に樹状ポリエステルA−1を2重量%添加したポリマーブレンドを用い、紡糸温度を255℃としたこと以外は全て実施例1に従い芯鞘複合繊維を得た(実施例7)。
紡糸温度の低下による力学特性向上の効果を確かめるため、紡糸温度は245℃に低下させ、同様に紡糸を行った(実施例8)。これの比較として、樹状ポリエステル未添加とした場合も実施例7に従い紡糸を行った(比較例4)。
得られた芯鞘複合繊維の物性を比較すると、実施例7および実施例8は力学物性(タフネス)が向上していることがわかった。また、紡糸性という観点においては、比較例4においては紡糸温度が高いため、分解ガスが多く発生し、紡糸ヘッドのハウジング内に分解物が多く付着しているのに対して、実施例8においては分解物の付着がほとんど見られなかった。結果を表6に示す。
実施例9,10、比較例5,6
参考例10で用いたηr2.5のN6と樹状ポリエステルA−1を2重量%となるようにドライブレンドしたものと光学純度99%のポリL乳酸(PLA重量平均分子量11万)を乾燥した後、混練比率(N6+A−1)/PLA=40/60となるように別々に計量して二軸押し出し混練機(混練温度220℃)に仕込んでアロイポリマーとして吐出し、水槽中で冷却固化することでガット化した後、ペレットとした。この(N6+A−1)/PLAアロイポリマーを単独成分の溶融紡糸機に挿入し、紡糸温度235℃、単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度3500m/分で巻き取った。採取した繊維の横断面ではPLAが海成分を形成し、N6が微細に分散して島成分をなしている図4に示すような海島複合繊維となっていた。
参考例10で用いたηr2.5のN6と樹状ポリエステルA−1を2重量%となるようにドライブレンドしたものと光学純度99%のポリL乳酸(PLA重量平均分子量11万)を乾燥した後、混練比率(N6+A−1)/PLA=40/60となるように別々に計量して二軸押し出し混練機(混練温度220℃)に仕込んでアロイポリマーとして吐出し、水槽中で冷却固化することでガット化した後、ペレットとした。この(N6+A−1)/PLAアロイポリマーを単独成分の溶融紡糸機に挿入し、紡糸温度235℃、単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度3500m/分で巻き取った。採取した繊維の横断面ではPLAが海成分を形成し、N6が微細に分散して島成分をなしている図4に示すような海島複合繊維となっていた。
また、紡糸温度低下の効果を確かめるため、紡糸温度は230℃として力学特性を調べた(実施例10)。これの比較として、樹状ポリエステル未添加として紡糸温度を235℃とした場合(比較例5)、紡糸温度を230℃とした場合(比較例6)として同様に紡糸を行った。
実施例10では紡糸温度を5℃低下させているにも係わらず、良好な紡糸性が維持されていたが、比較例6のように樹状ポリエステルを添加せず紡糸温度の低下を行った場合は、溶融吐出は可能であるものの、パック圧が増加し、吐出孔直下でポリマー流が膨れ上がる(バラス)現象が見られ、紡糸速度3500m/分では満足に巻き取ることができなかった。
更に、紡糸温度を低下させた実施例10においては比較例5と比較して力学特性に優れた海島複合繊維となり、後加工性に優れるものであった。結果を表7に示す。
更に、紡糸温度を低下させた実施例10においては比較例5と比較して力学特性に優れた海島複合繊維となり、後加工性に優れるものであった。結果を表7に示す。
実施例11〜13、比較例7
芯成分として、参考例11で用いたIV1.16dL/gの高分子量PET(H−PET 融点260℃)と参考例1で合成した樹状ポリエステルA−1を別々に計量して、樹状ポリエステルが1重量%となるように投入し、鞘成分として参考例12で用いたIV0.65dL/gのPET(R−PET 融点260℃)樹状ポリエステルを未添加のままを投入して、それぞれ押出温度300℃、280℃として、溶融し、公知の複合紡糸方法に従って、複合比率が芯成分/鞘成分=80/20となるように同心円型芯鞘複合繊維を得た。芯鞘成分を合わせた単孔吐出量は2.0g/分で丸孔10ホール(φ=0.6mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し巻き取った。紡糸速度を900m/分に固定し、紡糸温度低下に伴う、力学特性の向上効果を調べた。
芯成分として、参考例11で用いたIV1.16dL/gの高分子量PET(H−PET 融点260℃)と参考例1で合成した樹状ポリエステルA−1を別々に計量して、樹状ポリエステルが1重量%となるように投入し、鞘成分として参考例12で用いたIV0.65dL/gのPET(R−PET 融点260℃)樹状ポリエステルを未添加のままを投入して、それぞれ押出温度300℃、280℃として、溶融し、公知の複合紡糸方法に従って、複合比率が芯成分/鞘成分=80/20となるように同心円型芯鞘複合繊維を得た。芯鞘成分を合わせた単孔吐出量は2.0g/分で丸孔10ホール(φ=0.6mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し巻き取った。紡糸速度を900m/分に固定し、紡糸温度低下に伴う、力学特性の向上効果を調べた。
また、比較例7の力学物性と比較して、紡糸温度を低下させるに伴ってさらに力学特性(タフネス)が向上し、実施例13においては、比較例7と比較して力学特性(タフネス)が大きく向上したものであった。結果を表8に示す。
実施例14〜16、比較例8〜10
実施例11,13および比較例7で得られたPET芯鞘複合繊維(未延伸糸)を延伸・熱処理した。この時、延伸機としては3ホットローラー型延伸機を用い、フィードローラー(非加熱)、第1ホットローラー、第2ホットローラー、第3ホットローラー、デリバリーローラー(非加熱)と糸を通して延伸・熱処理を行った。第1ホットローラー温度(予熱温度)を90℃、第2ホットローラー温度を150℃、第3ホットローラー温度を230℃とし、第1ホットローラーと第2ホットローラー間の延伸倍率を3.4に固定し、第2ホットローラーと第3ホットローラー間の延伸倍率を変化させた。表7にはフィードローラーからデリバリーローラーまでの総延伸倍率を示した。
実施例11,13および比較例7で得られたPET芯鞘複合繊維(未延伸糸)を延伸・熱処理した。この時、延伸機としては3ホットローラー型延伸機を用い、フィードローラー(非加熱)、第1ホットローラー、第2ホットローラー、第3ホットローラー、デリバリーローラー(非加熱)と糸を通して延伸・熱処理を行った。第1ホットローラー温度(予熱温度)を90℃、第2ホットローラー温度を150℃、第3ホットローラー温度を230℃とし、第1ホットローラーと第2ホットローラー間の延伸倍率を3.4に固定し、第2ホットローラーと第3ホットローラー間の延伸倍率を変化させた。表7にはフィードローラーからデリバリーローラーまでの総延伸倍率を示した。
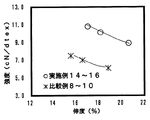
採取した延伸糸の強度と伸度の関係を図7にプロットしたが、紡糸温度を低下させた実施例13を未延伸糸とした実施例14〜16は、比較例8〜10に比べ同一伸度での強度が高いものであり、力学特性に優れた複合繊維が得られていることがわかった。また、強度と同様に実施例14〜16はいずれも比較例8〜10と比較して、同伸度当たりの弾性率が増加していることがわかった。結果を表9に示す。
実施例17,18、比較例11
A成分として参考例7で用いたIV1.51dL/gの3GT(融点230℃)とB成分として参考例12で用いたIV0.65dL/gのPET(融点260℃)に樹状ポリエステルA−1を1重量%添加したポリマーブレンドをそれぞれ250℃と275℃で溶融後、紡糸温度を275℃として、公知のサイドバイサイド複合口金においてサイドバイサイドの断面形態となるように細孔に流入させた。複合比はA成分/B成分となるようにし、A成分とB成分をあわせた単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度1500m/分で巻き取った。
A成分として参考例7で用いたIV1.51dL/gの3GT(融点230℃)とB成分として参考例12で用いたIV0.65dL/gのPET(融点260℃)に樹状ポリエステルA−1を1重量%添加したポリマーブレンドをそれぞれ250℃と275℃で溶融後、紡糸温度を275℃として、公知のサイドバイサイド複合口金においてサイドバイサイドの断面形態となるように細孔に流入させた。複合比はA成分/B成分となるようにし、A成分とB成分をあわせた単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度1500m/分で巻き取った。
参考例12においてPET(B成分)に樹状ポリエステルがブレンドされたポリマーブレンドとすることによる流動性の向上により紡糸温度を10℃以上低下できることが確認されたため、紡糸温度は275℃から低下させた(実施例18)。これの比較として、紡糸温度を275℃とし、樹状ポリエステル未添加とした場合も同様に紡糸を行った(比較例11)。
得られたサイドバイサイド複合繊維の物性を比較すると、比較例11と比較して、実施例17および実施例18は力学物性(タフネス)が向上し、品位に優れた3GT/PETサイドバイサイド複合繊維が得られていることがわかった。また、紡糸性という観点においては、比較例11においては紡糸温度が高いため、8時間紡糸後の口金面を見ると、24ホール中8ホールで糸曲がりが見られるのに対し、実施例18においては24ホール中いずれにおいても糸曲がりを確認することができなかった。本発明においては紡糸性の維持にも有効であることがわかった。結果を表10に示す。
実施例19.20、比較例12
参考例11で用いたIV1.16dL/gの高分子量PET(H−PET 融点260℃)と参考例12で用いたPPS(融点280℃)に樹状ポリエステルA−1を1重量%添加したポリマーブレンドをそれぞれ300℃、310℃にて溶融後、ポンプによる計量を行い、紡糸温度250℃にて高分子量PETが芯成分、(PPS+樹状ポリエステルA−1)が鞘成分とし、図1に示すような同心円型芯鞘複合繊維になるように公知の芯鞘複合口金に流入させた。芯鞘複合比は芯成分/鞘成分=70/30となるようにし、芯鞘を合わせた単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度1000m/分で巻き取った。
参考例11で用いたIV1.16dL/gの高分子量PET(H−PET 融点260℃)と参考例12で用いたPPS(融点280℃)に樹状ポリエステルA−1を1重量%添加したポリマーブレンドをそれぞれ300℃、310℃にて溶融後、ポンプによる計量を行い、紡糸温度250℃にて高分子量PETが芯成分、(PPS+樹状ポリエステルA−1)が鞘成分とし、図1に示すような同心円型芯鞘複合繊維になるように公知の芯鞘複合口金に流入させた。芯鞘複合比は芯成分/鞘成分=70/30となるようにし、芯鞘を合わせた単孔吐出量は0.8g/分で丸孔24ホール(φ=0.3mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し紡糸速度1000m/分で巻き取った。
参考例12においてPPS(鞘成分)に樹状ポリエステルがブレンドされたポリマーブレンドとすることによる流動性の向上により紡糸温度を10℃以上低下できることが確認されたため、紡糸温度は310℃から低下させた(実施例20)。これの比較として、紡糸温度を310℃とし、樹状ポリエステル未添加とした場合も同様に紡糸を行った(比較例12)。
得られた芯鞘複合繊維の物性を比較すると、比較例12と比較して、実施例19および実施例20と紡糸温度を低下させることにより、力学物性(タフネス)が向上し、品位に優れたH−PET/PPS芯鞘複合繊維になっていることがわかった。また、紡糸性という観点においては、比較例12においては紡糸温度が高いため、分解ガスの大量に発生し、かつ24時間巻き取りにおける糸切れ回数は5回であったのに対し、実施例20に関しては、分解ガスが大きく抑制されており、糸切れすることがなかった。本発明においては糸切れを抑制する効果に優れたものであることがわかった。結果を表11に示す。
1:芯成分
2:鞘成分
3:難溶出成分(星形)
4:易溶出成分
5:難溶出成分(三葉)
6:易溶出成分
7:島成分
8:海成分
2:鞘成分
3:難溶出成分(星形)
4:易溶出成分
5:難溶出成分(三葉)
6:易溶出成分
7:島成分
8:海成分
Claims (6)
- 2種類以上のポリマーからなる複合繊維であって、芳香族オキシカルボニル単位(P)、芳香族および/または脂肪族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)から選ばれる少なくとも1種の構造単位と、3官能の有機残基(B)とを含み、かつ前記P、Q、RおよびBの含有量の合計に対してBの含有量が7.5〜50モル%であり、末端のカルボン酸基量が1×10-4当量/g以下である樹状ポリエステルを、マトリックスポリマーに0.1〜10重量%添加したポリマーブレンドを少なくとも構成の一部であることを特徴とする複合繊維。
- 樹状ポリエステルが、カルボン酸反応性単官能化合物残基を含有することを特徴とする請求項1記載の複合繊維。
- カルボン酸反応性単官能化合物が、オルトエステル、オキサゾリン、エポキシドから選ばれる少なくとも1種の化合物であることを特徴とする請求項2に記載の複合繊維。
- マトリックスポリマーが、ポリエステル、ポリアミド、またはポリフェニレンサルファイドであることを特徴とする請求項1〜3いずれかに記載の複合繊維。
- ポリトリメチレンテレフタレートが少なくとも構成の一部であることを特徴とする請求項1〜4のいずれかに記載の複合繊維。
- ポリ乳酸が少なくとも構成の一部であることを特徴とする請求項1〜5のいずれかに記載の複合繊維。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009147528A JP2011001663A (ja) | 2009-06-22 | 2009-06-22 | 複合繊維 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009147528A JP2011001663A (ja) | 2009-06-22 | 2009-06-22 | 複合繊維 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2011001663A true JP2011001663A (ja) | 2011-01-06 |
Family
ID=43559847
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009147528A Pending JP2011001663A (ja) | 2009-06-22 | 2009-06-22 | 複合繊維 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2011001663A (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20180028393A (ko) * | 2017-10-12 | 2018-03-16 | 충남대학교산학협력단 | 반응기를 가지는 섬유 및 이의 제조방법 |
| KR101838125B1 (ko) | 2016-09-08 | 2018-04-26 | 충남대학교산학협력단 | 반응기를 가지는 섬유 및 이의 제조방법 |
| WO2018147251A1 (ja) * | 2017-02-09 | 2018-08-16 | 東レ株式会社 | 熱接着性芯鞘型複合繊維およびトリコット編み地 |
| CN115434035A (zh) * | 2022-09-20 | 2022-12-06 | 江苏中淮纺织新材料有限公司 | 一种复合纤维及其制备方法 |
-
2009
- 2009-06-22 JP JP2009147528A patent/JP2011001663A/ja active Pending
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101838125B1 (ko) | 2016-09-08 | 2018-04-26 | 충남대학교산학협력단 | 반응기를 가지는 섬유 및 이의 제조방법 |
| WO2018147251A1 (ja) * | 2017-02-09 | 2018-08-16 | 東レ株式会社 | 熱接着性芯鞘型複合繊維およびトリコット編み地 |
| KR20180028393A (ko) * | 2017-10-12 | 2018-03-16 | 충남대학교산학협력단 | 반응기를 가지는 섬유 및 이의 제조방법 |
| KR101884873B1 (ko) | 2017-10-12 | 2018-08-02 | 충남대학교산학협력단 | 반응기를 가지는 섬유 및 이의 제조방법 |
| CN115434035A (zh) * | 2022-09-20 | 2022-12-06 | 江苏中淮纺织新材料有限公司 | 一种复合纤维及其制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100567600C (zh) | 一种改性的共聚酯切片或纤维及其制备方法 | |
| JP5243410B2 (ja) | ポリ乳酸組成物およびそれよりなる繊維 | |
| CA2901965C (en) | Composite fibers, weave fabrics, knitted fabrics and composite materials | |
| KR101314878B1 (ko) | 고권축성 2성분 섬유 | |
| CN107936237B (zh) | 一种生物基可降解聚酯纤维及其制备方法 | |
| KR20150036294A (ko) | 폴리에스테르 코-포스포네이트 | |
| JP2001200034A (ja) | 伸縮自在なポリマーを製造するための組成物および方法ならびにそれらによって製造される造形物品 | |
| JP4992610B2 (ja) | 繊維 | |
| CN103789866B (zh) | 一种多羟基聚酯纤维的制备方法 | |
| JPH06508860A (ja) | 高モジュラス繊維用のコポリエステル類 | |
| JP2011001663A (ja) | 複合繊維 | |
| TWI304448B (en) | Poly (trimethylene terephthalate) bicomponent fiber process | |
| JP2010196187A (ja) | ポリフェニレンサルファイド短繊維およびその製造方法 | |
| JP5109986B2 (ja) | 溶融紡糸方法 | |
| JP4720306B2 (ja) | 液晶性樹脂繊維およびその製造方法 | |
| US8916673B2 (en) | Process for producing liquid crystalline polyester resin and apparatus for producing liquid crystalline polyester resin | |
| KR20230026439A (ko) | 액정 폴리에스테르 섬유 및 액정 폴리에스테르 섬유의 제조 방법 | |
| Choi et al. | Reinforcing properties of poly (trimethyleneterephthalate) by a thermotropic liquid crystal polymer | |
| JP3737043B2 (ja) | ポリトリメチレンテレフタレートの連続重合方法、ポリトリメチレンテレフタレート組成物の連続重合方法 | |
| WO2017022569A1 (ja) | ポリエチレンテレフタレート繊維およびポリエステル樹脂 | |
| JP2009052160A (ja) | 繊維 | |
| JP2009052161A (ja) | 複合繊維 | |
| JP2010163552A (ja) | 繊維強化複合材料およびその製造方法 | |
| JP4080221B2 (ja) | ポリエステル組成物及びそれよりなる繊維 | |
| Mao et al. | Enhancing Poly (butylene terephthalate)(PBT) fiber orientation and tenacity via controlled recycling of composition-tuned vitrimers |