JP2010502736A - 消化管の炎症を低減または軽減するための方法 - Google Patents
消化管の炎症を低減または軽減するための方法 Download PDFInfo
- Publication number
- JP2010502736A JP2010502736A JP2009527579A JP2009527579A JP2010502736A JP 2010502736 A JP2010502736 A JP 2010502736A JP 2009527579 A JP2009527579 A JP 2009527579A JP 2009527579 A JP2009527579 A JP 2009527579A JP 2010502736 A JP2010502736 A JP 2010502736A
- Authority
- JP
- Japan
- Prior art keywords
- use according
- ace2
- ethylamino
- carboxy
- imidazol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Plant Substances (AREA)
Abstract
消化管の炎症性疾患を有する被験者における炎症、またはそれに関連するもしくはその二次的な病理学的プロセスを低減または軽減するための方法を提供する。当該方法は、前記被験者に抗炎症有効量のACE2阻害薬を投与することを包含する。
【選択図】 図7
【選択図】 図7
Description
[0001]本出願は、2006年9月8日に提出された米国仮特許出願第60/825,075号、および2006年10月2日に提出された第60/827,807号の利益を主張し、これら各出願の全開示を本明細書において参照として援用する。
[0002]本発明は、消化管の炎症性疾患、例えば炎症性腸疾患のための薬物療法に関する。より特定すれば本発明は、消化管の炎症性疾患を有する被験者における炎症を低減または軽減するための方法に関する。
IBDの全般的背景
[0003]消化管(digestive tract、alimentary canal(nourishment canal))は腸管(gut)ともいうが、消化器系、すなわち食物を取り込み、それを消化してエネルギーおよび栄養素を抽出し、そして残りの廃棄物を排泄する、多細胞動物体内の臓器系の一部である。このプロセスを消化と呼ぶ。本明細書において定義する場合、消化管は、消化プロセスのコースにおいて食物または固形排泄物が通過する臓器を含むが、消化の手助けをする物質を保存および/または分泌する、消化管に隣接し接続する消化器系の臓器、例えば肝臓、胆嚢、および膵臓は除外する。この定義は、標準的な参考書籍、例えばDorland’s Illustrated Medical Dictionary, 第30版 (2003), Saunders, Philadelphia に提示されているものと広義では一致する。同書は、消化管は食道、胃および腸により形成される消化器系のその部分とし、本明細書では便宜上含めている口および咽頭を除外して定義している。正常な成人男性において、口から肛門までの消化管は、およそ7.5メートル長であり、以下の構成要素を持つ上部および下部の部分から成る:
・上部消化管:口(口腔または頬側口腔;唾液腺、口腔粘膜、歯、および舌を含む)、咽頭、食道および噴門、胃(洞および幽門および幽門括約筋を含む);
・下部消化管:(a)小腸、これは3つの部分:十二指腸、空腸 および回腸を有する;(b)大腸、これは3つの部分:盲腸(盲腸の憩室である虫垂を含む)、結腸(上行結腸、横行結腸、下行結腸、およびS状結腸)、および直腸を有する;ならびに(c)肛門、から成る腸または腸管。
[0003]消化管(digestive tract、alimentary canal(nourishment canal))は腸管(gut)ともいうが、消化器系、すなわち食物を取り込み、それを消化してエネルギーおよび栄養素を抽出し、そして残りの廃棄物を排泄する、多細胞動物体内の臓器系の一部である。このプロセスを消化と呼ぶ。本明細書において定義する場合、消化管は、消化プロセスのコースにおいて食物または固形排泄物が通過する臓器を含むが、消化の手助けをする物質を保存および/または分泌する、消化管に隣接し接続する消化器系の臓器、例えば肝臓、胆嚢、および膵臓は除外する。この定義は、標準的な参考書籍、例えばDorland’s Illustrated Medical Dictionary, 第30版 (2003), Saunders, Philadelphia に提示されているものと広義では一致する。同書は、消化管は食道、胃および腸により形成される消化器系のその部分とし、本明細書では便宜上含めている口および咽頭を除外して定義している。正常な成人男性において、口から肛門までの消化管は、およそ7.5メートル長であり、以下の構成要素を持つ上部および下部の部分から成る:
・上部消化管:口(口腔または頬側口腔;唾液腺、口腔粘膜、歯、および舌を含む)、咽頭、食道および噴門、胃(洞および幽門および幽門括約筋を含む);
・下部消化管:(a)小腸、これは3つの部分:十二指腸、空腸 および回腸を有する;(b)大腸、これは3つの部分:盲腸(盲腸の憩室である虫垂を含む)、結腸(上行結腸、横行結腸、下行結腸、およびS状結腸)、および直腸を有する;ならびに(c)肛門、から成る腸または腸管。
[0004]“胃腸管(gastrointestinal tract)”または“GI管”という用語は、当該技術において一般的であるように、消化管全体をいうために本明細書でも時折使用する。その厳密な意味で使用する場合は、胃および腸により形成される消化管のその部分を意味するが、そのような使用は本明細書で明確に特定されるか、または文脈により必要とされることになるだろう。
[0005]炎症性腸疾患(inflammatory bowel disease、IBD)は、自己免疫反応に関与すると考えられている消化管の特発性疾患の1つのクラスである。IBDの2つの主要なタイプ:潰瘍性大腸炎(UC)およびクローン病(CD)が認識されている。UCは、特発性直腸結腸炎としても知られており、典型的には結腸に限定される;CDは、限局性腸炎、回腸末端炎、または肉芽種性回結腸炎とも言われ、口から肛門までの消化管のあらゆる区域(1つまたは複数)を侵す可能性がある。“炎症性腸疾患”または“IBD”という用語を本明細書において使用する場合、特にCDに関しては、排他的に腸だけではなく、消化管のあらゆる部分での症状を含むものと理解されるものとする。
[0006]UCおよびCDは著しい差を示すが、双方の疾患とも腸および腸以外のいくつかの症状を共有する。ただしこれらの症状によっては、一方の疾患または他方の疾患に、より一般的に生じる傾向がある。UCおよびCDは双方とも、通例、一進一退の強度および重症度を示す。IBD患者が著しい炎症を表す症状を有する場合、その疾患は活動期にあるとみなされる;そのような患者はIBDの“フレア(flare)”を有していると言われる。炎症がより低い重症度であるかまたは存在しない場合、患者は実質的に無症状であり、疾患は寛解期にあるとみなされる。ほとんどの場合症状は、どちらかの疾患に関する炎症の存在の程度によく対応するが、例外なく当てはまる訳ではない。一部の患者は、著しい有害な副作用の潜在性を伴う薬物を投与する前に、疾患の活動性に関する客観的エビデンスを必要とすることになると思われる。
[0007]IBD、その症状、病理、および治療に関する情報は、例えば個別に以下に引用し、本明細書において参照として援用するものを含む、様々な出版物およびインターネットの情報源に見出すことができる。
[0008]Bonner (2003) “Inflammatory bowel disease: advances in medical management(炎症性腸疾患:内科的管理における進歩)”
http://www.fascrs.org/displaycommon.cfm?an=1&subarticlenbr=113。
http://www.fascrs.org/displaycommon.cfm?an=1&subarticlenbr=113。
[0009]Tung & Warner (2002) Postgraduate Medicine 112(5). “Colonic inflammatory bowel disease: medical therapies for colonic Crohn’s disease and ulcerative colitis(慢性炎症性腸疾患:クローン病および潰瘍性大腸炎に関する内科的療法)”
http://www.postgradmed.com/issues/2002/11_02/tung2.htm。
http://www.postgradmed.com/issues/2002/11_02/tung2.htm。
[0010]University of Maryland Medical Center (2002) “What are the Drug Treatments for Inflammatory Bowel Disease?(炎症性腸疾患に関する薬剤治療とは何か?)”
http://www.umm.edu/patiented/articles/what_drug_treatments_inflammatory_bowel_disease_000069_9.htm。
http://www.umm.edu/patiented/articles/what_drug_treatments_inflammatory_bowel_disease_000069_9.htm。
潰瘍性大腸炎:症状および病理
[0011]UCの患者は、最も一般的には血性下痢を呈する。より重症な場合、腹痛および痙攣、発熱および体重減少を生じる。結腸の病変の範囲が拡大するほど、患者が下痢をする可能性はより高くなる。便意ひっ迫(しぶり)は、炎症を起こした直腸に関連づけることができる。疾患が直腸に限局されていれば、患者は便を形成するかもしれない。炎症の程度が増大するにつれ、微熱、倦怠感、悪心、嘔吐、発汗、および関節痛を含む全身症状を発症する。重症のUCでは、結腸のより深い層内に進行した炎症を反映する、発熱、脱水、および腹部圧痛を発症する。
[0011]UCの患者は、最も一般的には血性下痢を呈する。より重症な場合、腹痛および痙攣、発熱および体重減少を生じる。結腸の病変の範囲が拡大するほど、患者が下痢をする可能性はより高くなる。便意ひっ迫(しぶり)は、炎症を起こした直腸に関連づけることができる。疾患が直腸に限局されていれば、患者は便を形成するかもしれない。炎症の程度が増大するにつれ、微熱、倦怠感、悪心、嘔吐、発汗、および関節痛を含む全身症状を発症する。重症のUCでは、結腸のより深い層内に進行した炎症を反映する、発熱、脱水、および腹部圧痛を発症する。
[0012]UCの診断は、内視鏡検査によりまたは放射線学的に行うことができ、典型的にな造影X線写真で正常な粘膜パターンの喪失を、より進行した疾患では結腸の膨起の喪失を示すことができる。S状結腸内視鏡検査または結腸内視鏡検査は、ほとんど場合直腸が侵されていることを明らかにする。この疾患は、患者の約25%では直腸(直腸炎)に限定されることができるが;ほとんどの患者では直腸、S状結腸、および下行結腸(左側結腸炎)に;または患者の約10%では全結腸(汎大腸炎)に及ぶ。UCは、胃腸管のその他のいかなる区域も侵すことはない。結腸切除が治療法である。
[0013]UCにおいて、明確な分界は病変粘膜および非病変粘膜の間に存在する;そして病変領域では、疾患が顕著にかつ一様に継続する。UCは最初に粘膜および粘膜下層を侵し、陰窩膿瘍および粘膜潰瘍の形成を伴う。粘膜は典型的には顆粒状で脆弱な様相を見せる。より重症な場合、浅い潰瘍を有する炎症粘膜によって囲まれた腫脹した粘膜を有する肥厚に成長した領域から成る、偽ポリープを形成する。重症のUCでは、炎症および壊死が固有層下に拡大して、粘膜下層および環状筋および縦走筋を侵し得るが、これは稀である。疾患が慢性化すると、通常の膨起(out-pouch)紋理を喪失した硬い短小化した管となり、バリウム浣腸で観察される“鉛管”の様相をきたすようになる。
[0014]UCの予後に関して、少数パーセントの患者のみが1回の発作で再発を免れるに過ぎない。しかしながら典型的には寛解と増悪がUCの特徴であり、急性の発作が数週間から数か月持続する。20パーセントの患者は、治療法である結腸切除を必要とする。長期的な罹患は、内科的療法、特に長期的なステロイドの合併症に第一に起因する。
[0015]IBDにおける最も一般的な死亡原因は、敗血症を伴う腹膜炎、悪性腫瘍、血栓塞栓性疾患、および手術の合併症である。UCの最も重篤な合併症の1つである中毒性巨大結腸症は、穿孔、敗血症、および死をもたらし得る。診断後およそ8−10年で、大腸癌のリスクが一般の集団のそれを有意に上回って上昇し始めることから、悪性腫瘍は、UCの長期的に障害された腸の最も重篤な合併症である。UCの患者の結腸狭窄は、(通常切除により)そうでないことが証明されてない限り、悪性であると推定される。
クローン病:症状および病理
[0016]CDの症状は、通常UCのそれより先行性であり、継続的な腹痛、食欲不振、下痢、体重減少および疲労感を伴う。肉眼的血便はUCの典型であるが、CDにおいてはそれほど一般的ではない。便は形成されると思われるが、結腸または回腸末端が広範に侵されていれば、主に軟便となる。CDの患者の半数は、肛門周囲の疾患(例えば瘻孔または膿瘍)を呈する。場合により虫垂炎に類似する急性の右下4分の1の腹痛および発熱が認められてもよい。一般に診断は、数年の反復性の腹痛、発熱、および下痢の後でしか確定されない。胃十二指腸の病変を伴うCDは、消化性潰瘍疾患に類似すると思われ、胃幽門部の閉塞に進行する可能性がある。
[0016]CDの症状は、通常UCのそれより先行性であり、継続的な腹痛、食欲不振、下痢、体重減少および疲労感を伴う。肉眼的血便はUCの典型であるが、CDにおいてはそれほど一般的ではない。便は形成されると思われるが、結腸または回腸末端が広範に侵されていれば、主に軟便となる。CDの患者の半数は、肛門周囲の疾患(例えば瘻孔または膿瘍)を呈する。場合により虫垂炎に類似する急性の右下4分の1の腹痛および発熱が認められてもよい。一般に診断は、数年の反復性の腹痛、発熱、および下痢の後でしか確定されない。胃十二指腸の病変を伴うCDは、消化性潰瘍疾患に類似すると思われ、胃幽門部の閉塞に進行する可能性がある。
[0017]体重減少は、小腸の疾患に関連する吸収障害のため、UCよりCDにおいてより一般的に観察される。患者は、その症状をコントロールする目的で食物摂取を減量してよい。全身症状が一般的であり、発熱、発汗、倦怠感、および関節痛を含む。微熱はフレアの最初の警告の兆候であると思われる。再発は、精神的ストレス、感染またはその他の急性の病気、妊娠、無分物な食事、下剤もしくは抗生剤の使用、または抗炎症薬もしくはステロイド薬物の中止に伴って現れると思われる。
[0018]小児は、成長遅滞、および性成熟の遅延または性成熟不全を呈すると思われる。症例の10−20%において、患者は、関節炎、ブドウ膜炎または肝疾患を含む、腸管外の症状を呈する。
[0019]CDの診断は、内視鏡検査によりまたは放射線学的に行うことができ、典型的には造影X線写真で、正常粘膜の領域と炎症粘膜の領域が交互に存在する(“スキップ病変”)敷石状パターンを粘膜に示すことができる。最も重要な病理学的特徴は、UCの特徴であるような粘膜および粘膜下層だけでなく、腸の全層の病変である。S状結腸鏡検査または結腸鏡検査は、直腸にはしばしば病変はなく、右側結腸の主病変が一般的であることを明らかにしている。CDの患者の90パーセントは、回腸末端および/または右側結腸の病変を有する。小児患者は、小腸に限定された疾患を呈する可能性がより高い(約20%)。一般的と言うにははるかに少ないが、CDは、口、舌、食道、胃、および十二指腸を含む胃腸管のより近位の部分をも侵す。
[0020]CDの患者における狭窄および閉塞はしばしば炎症性であり、往々にして内科的治療により解決される。固定化した(瘢痕化したまたは瘢痕の)狭窄は、閉塞を解放するため内視鏡的または外科的な介入を必要とすると思われる。CDの患者における瘻孔および肛門周囲の疾患は、抗生剤療法を含む積極的な内科的治療に対して難治性であると思われる。瘻孔および肛門周囲の疾患に関しては外科的介入がしばしば必要とされるが、双方とも高リスクの再発を伴う。CDの患者における癌のリスクは、全結腸が侵されている場合は、UCの患者のそれに等しいと思われ、そして小腸の悪性腫瘍のリスクはCDの患者で増加するが、この悪性腫瘍は、炎症領域と同程度に、これまで正常な領域で発生する可能性が高い。
[0021]CDの予後は、疾患の部位および範囲に依存する。周期的な寛解および増悪を常とする。患者のおよそ50%は外科的介入を必要とし;手術した患者の50%が2回目の手術を必要とし;これらの患者の内50%は3回目の手術を行っている。再発率は、内科的管理に反応した患者に関して、1年以内に25−50%である。手術を必要とする患者ではこの比率はより高くなる。
[0022]生活に質(QOL)は、一部にはCDのために行われる術後再発のため、通常UCよりCDの方が低い。栄養不良および慢性貧血が、長期化したCDで観察される。CDまたはUCの小児は、成長遅滞を示す可能性がある。
IBDの有病率および発生率
[0023]IBDは北欧および北米において最も一般的に観察される。これは先進工業国の疾患である。米国において、およそ100万人がUCまたはCDを有する。1960年以前に報告されたUCの発生率は、CDのそれより数倍高かった。より最近のデータは、現在のCDの発生率がUCそれに近づいていることを示唆しているが、この変化はCDの改善された認識および診断を反映していると思われる。
[0023]IBDは北欧および北米において最も一般的に観察される。これは先進工業国の疾患である。米国において、およそ100万人がUCまたはCDを有する。1960年以前に報告されたUCの発生率は、CDのそれより数倍高かった。より最近のデータは、現在のCDの発生率がUCそれに近づいていることを示唆しているが、この変化はCDの改善された認識および診断を反映していると思われる。
[0024]米国において、ヨーロッパの家系の人々の間のIBDの発生頻度の比率は、例えばミネソタ州オルムステッド郡において測定された。この集団において、UCの発生率は1年当たり人口100,000人当たり7.3例であり、有病率は人口100,000人当たり116例である;CDの発生率は、1年当たり人口100,000人当たり5.8例であり、有病率は人口100,000人当たり133例であると、報告されている。IBDの発生率は、アシュケナージ系ユダヤ人(すなわち北欧から移民した人々)において最も高く、通常の集団の4−5倍であり、非ユダヤ系白人の集団がこれに続くことが報告された。しかしながら最近のデータは、非ユダヤ系、アフリカ系、およびヒスパニック系の集団における発生率が増加していることを示唆する。男性−対−女性の比率は、UCおよびCD双方に関してほぼ等しい。イタリアにおける最近の研究は、UCおよびCDの発生率が米国のそれに類似することを示した。
[0025]UCおよびCDは、最も一般的には若年成人(すなわち後期青年期から30代)において診断される。新たに診断されるIBD例の年齢分布は、ベル型である;ピークの発生率は、20代前半の人々に現れ、新規診断の大多数は15−40歳の人々でなされる。第2のより小さなピークは、55−65歳の患者に現れる。しかしながら5歳より若い小児、および高齢者も時折診断される。IBDの患者の約10%のみが、18歳より若いに過ぎない。発生率は、男性より女性の方がわずかに高いと思われる。
IBDの内科的治療
[0026]IBDの患者のケアは、本質的には内科的もしくは外科的のいずれか、または一般的には双方の組み合わせであることができる。IBDのために使用される薬物は、個々の薬剤の化学的類似性、および作用機序の類似性の基づいて、いくつかのクラスに分類される。IBDの患者のための内科的アプローチは、対症的(フレアの)ケアであり、通常反応が達成されるまで、内科的投与計画の進行にそって薬物療法の段階的アプローチを踏む。このアプローチでは、最も穏やかな(または一過性の)薬剤が最初に使用される。それら薬剤で軽快を提供できない時は、より高いステップの薬剤が使用される。
[0026]IBDの患者のケアは、本質的には内科的もしくは外科的のいずれか、または一般的には双方の組み合わせであることができる。IBDのために使用される薬物は、個々の薬剤の化学的類似性、および作用機序の類似性の基づいて、いくつかのクラスに分類される。IBDの患者のための内科的アプローチは、対症的(フレアの)ケアであり、通常反応が達成されるまで、内科的投与計画の進行にそって薬物療法の段階的アプローチを踏む。このアプローチでは、最も穏やかな(または一過性の)薬剤が最初に使用される。それら薬剤で軽快を提供できない時は、より高いステップの薬剤が使用される。
[0027]アミノサリチル酸および対症薬は、このスキーム下ではステップIの薬剤である;抗生剤は、それらが使用される限定された状況を前提として、ステップIAの薬剤とみなされる。コルチコステロイドは、ステップIの薬剤で十分にIBDをコントロールできない場合に使用される、ステップIIの薬剤を構成する。免疫調節薬はステップIIIの薬剤であり、コルチコステロイドが奏功しない、または長期間必要とされる場合に使用される。腫瘍壊死因子(TNF)αに対するモノクローナル抗体であるインフリキシマブもまた、CDおよびUCの患者のいくつかの状況において使用することのできるステップIIIの薬剤である。実験薬はステップIVの薬剤であり、これまでのステップが奏功しなかった後にのみしか使用されず、その場合それら薬剤の使用に精通している医師によってしか投与されない。すべてのステップの薬剤は、付加的に使用してもよい;通常、目標はできるだけ速やかに患者からステロイドを断って、これらの薬剤による長期的な有害作用を防ぐことである。この段階的アプローチにおけるある種の薬剤の使用に関しては、種々の見解がある。
[0028]ステップI(アミノサリチル酸)。米国での使用に利用可能な経口アミノサリチル酸調製物は、スルファサラジン、メサラミン、バルサラジド、およびオルサラジンを含む。浣腸および座剤用の製剤もまた利用できる。これらはすべて5−アミノサリチル酸(5−ASA)の誘導体である;主な違いは送達機序にある。これらのいくつかはまた、このクラスの他の物質にはない固有の有害作用を有する。アミノサリチル酸はすべて、IBDのフレアを治療するため、そして寛解を維持するために有用である。アミノサリチル酸はいずれも、他のいかなる薬剤をも上回る、UCまたはCDの治療のためのより大きな有効性を有することは証明されていない。これらはすべて、CDの患者よりUCの患者において明らかにより有効である;CDでは第一の使用は慢性疾患に対してである。
[0029]ステップIA(抗生剤)。抗生剤のメトロニダゾールおよびシプロフロキサシンは、IBDにおいて最も一般的に使用される抗生剤である。UCは、抗生剤に関連する偽膜性大腸炎を発症するリスクを増大させるため、UCでは抗生剤は控えめにしか使用されない。しかしながらCDにおいては、抗生剤は多様な適用に、最も一般的には肛門周囲の疾患に対して使用される。これらはまた、瘻孔、および腹部の炎症性腫瘤に対しても使用され、回腸炎の治療にも何らかの有効性を有すると思われる。抗生剤は、悪心、食欲不振、下痢、モリニア(カンジダ)感染、そしてメトロニダゾールの場合は末梢性ニューロパチーを含む、潜在的な有害作用を有する。
[0030]ステップII(コルチコステロイド)。コルチコステロイドは、疾患の急性のフレアのみ関するIBDの治療において使用される、速効型の抗炎症薬である;コルチコステロイドには寛解の維持という役割はない。コルチコステロイドは、疾患の場所および重症度に依存して、多様な経路で投与してよい;例えば静脈内(例えばメチルプレドニゾロン、ヒドロコルチゾン)、経口(例えばプレドニゾン、プレドニゾロン、ブデソニド、デキサメタゾン)、または局所的(浣腸、座剤もしくは起泡性の調製物)にて投与してよい。
[0031]静脈内コルチコステロイドは、しばしば重症病気および入院中の患者に使用される。通常、臨床反応が一度(典型的には1−2日以内に、時折それより遅れて)観察されたら、静脈内コルチコステロイドの用量を漸減することができる。退院前に経口コルチコステロイドへの変更を行う;さらに投与量を漸減して外来患者の設定に達することができる。重ねて言うと、一度臨床反応が認められたら用量を漸減する。経口コルチコステロイドを使用するほとんどの患者は、反応が達成された後の相対的に速い漸減に耐えられることは殆どない;時には非常に長期間のステロイドの漸減が、再発を防ぐために必要となる。後者の状況が生じた場合は、代替え薬(免疫調節薬または抗TNFα療法)を使用してもよい。局所コルチコステロイドは、遠位結腸の疾患の患者に局所メサラミンと類似の様式で使用されるが、局所コルチコステロイドは寛解の維持にはたいして役に立たないため、典型的には活動期の疾患に対してのみ使用される。
[0032]コルチコステロイド使用による潜在的な合併症は多数あり、体液および電解質の異常、骨粗鬆症、骨頭無菌性壊死、消化管潰瘍、白内障、神経および内分泌の機能不全、感染性合併症、および時に精神医学的障害(精神障害)を含む。
[0033]ステップIII(免疫調節薬)。免疫調節薬であるアザチオプリンおよび6−メルカプトプリン(6−MP)は、アミノサリチル酸単独で寛解を維持することが困難なIBDの患者に使用される。免疫調節薬はリンパ球数の低減を引き起こすことにより働くが、その作用機序のため、これらの作用の開始は相対的に遅い(典型的には2−3か月)。これら薬剤は最も一般的には、不応性疾患の患者のステロイド用量減量作用のために使用される;これらはまた瘻孔の一次治療として、およびアミノサリチル酸不耐性患者の寛解維持としても使用される。
[0034]これらの薬剤の使用は、血液パラメータのモニタリングが義務付けられている;これらは、用量の低減または中止に値する著しい好中球減少または汎血球減少を引き起こし得る。免疫調節薬のその他の有害作用には、発熱、発疹、感染性合併症、肝炎、膵炎、および骨髄機能不全を含む。免疫調節薬を最初の数週間以内に中止する最も一般的な理由は、腹痛の発症である;時には生化学的に実証可能な膵炎を生じる。アザチオプリンおよび6−MPを投与している患者における悪性腫瘍の発症についての懸念も提起されている。
[0035]インフリキシマブは、異なる機序で働く付加的なステップIIIの薬剤である。インフリキシマブは、UCおよびCD双方に関して現在米国医薬品局(FDA)で承認されている抗TNFαモノクローナル抗体であるが、CDにおいてより高い有効率を確かに有するようである。インフリキシマブは通常、中程度から重症のIBDの治療のために、5mg/kgの注入として投与される。インフリキシマブは、第0、2、および6週に5mg/kgを3回、別々に注入し、しばしば引き続き8週毎に寛解維持のための用量を投与される。CDに関して、1回の用量後の反応比率は80%であり、寛解導入比率は50%である;複数回投与ではより高い比率の寛解が得られる。UCに関しては反応比率は50−70%である。インフリキシマブはまた、瘻孔を生じているCDの治療にも適用される;この適用に関して、瘻孔は、インフリキシマブで治療した患者の68%で反応(閉鎖)するが、12%は膿瘍を発症する。導入用量後に規定の投与を継続する(すなわち8週毎)ことにより、この反応を維持することができる。
[0036]インフリキシマブの治療は極めて高価であり、一般に過敏症および風邪様症状を含む有害作用伴うとも思われる。ループス様の反応、およびリンパ球増殖性悪性腫瘍という稀な例も報告されているが、悪性腫瘍が薬剤に関するのか、あるいは基底にある疾患のプロセスに関するのかは、いまだ解明されていない。
[0037]ステップIV(実験的治療)。CDに使用される実験薬は、メトトレキサート(12.5−25mg/週、経口または筋肉内)、サリドマイド(50−300mg/日、経口)およびインターロイキン11(1mg/週、皮下)を含む。UCに使用される実験薬は、シクロスポリンA、2−4mg/kg/日、静脈内(測定レベル;経口投薬には、静脈内用量の2−3倍の用量に変更する)、ニコチン、14−21mg/日、局所パッチを介して、ブチレート、浣腸(直腸当たり100ml、1日2回)、およびヘパリン(10,000U、皮下、1日2回)を含む。多数の禁忌、相互作用、および注意がこれらの薬剤に付随する。
慢性胃炎
[0038]慢性胃炎、胃粘膜の慢性的炎症は、ヘリコバクター・ピロリ(Helicobacter pylor)菌の感染により最も高頻度に引き起こされるが、非ステロイド抗炎症薬(NSAID)の使用、自己免疫、アレルギー、またはその他の因子によっても引き起こされると思われる。感染性胃炎は通例、基底にある感染を排除するための抗生剤、および炎症粘膜を治療するための1つまたはそれより多くの薬剤を包含する、多剤療法により治療される。感染性胃炎を治療するための抗生剤と共に、または他の形の胃炎を治療するために単独でのいずれかで使用される最新の薬剤は、2つの主なクラス:プロトン−ポンプ阻害薬およびH2−受容体遮断薬があり、これらの双方とも、胃酸の分泌を阻害することにより作用する。しかしながら多くの症例において、これらの方法は非効率的であり、完全に有効とは言えず、治療の新たな様式が必要とされている。
[0038]慢性胃炎、胃粘膜の慢性的炎症は、ヘリコバクター・ピロリ(Helicobacter pylor)菌の感染により最も高頻度に引き起こされるが、非ステロイド抗炎症薬(NSAID)の使用、自己免疫、アレルギー、またはその他の因子によっても引き起こされると思われる。感染性胃炎は通例、基底にある感染を排除するための抗生剤、および炎症粘膜を治療するための1つまたはそれより多くの薬剤を包含する、多剤療法により治療される。感染性胃炎を治療するための抗生剤と共に、または他の形の胃炎を治療するために単独でのいずれかで使用される最新の薬剤は、2つの主なクラス:プロトン−ポンプ阻害薬およびH2−受容体遮断薬があり、これらの双方とも、胃酸の分泌を阻害することにより作用する。しかしながら多くの症例において、これらの方法は非効率的であり、完全に有効とは言えず、治療の新たな様式が必要とされている。
[0039]IBDにおける炎症の活動性は、核内因子κB(NF−κB)の活性化を伴うことが知られている。例えばSchreiber et al. (1998) Gut 42:477-484を参照のこと。同書文献は、双方のIBDにおいて、しかし特にCDにおいて、NF−κBの増大した活性化が炎症反応の制御に関与すると思われ、したがってNF−κB活性化の阻害は、ステロイドがIBDにおいて抗炎症効果を及ぼす機序を示すものと思われる、と結論している。
[0040]さらに、IBDのための有効な治療となり得る抗TNFα抗体のインフリキシマブも、少なくともCDにおいてNF−κBの活性を低減することが報告された(Guidi et al. (2005) Int. J. Immunopathol. Pharmacol.18(1):155-164を参照のこと)。
[0041]逆にNF−κB活性の増加が、インフリキシマブに対する反応を維持することができないCD患者において、症状の再発に先行することが報告された(Nikolaus et al. (2000) Lancet 356(9240):1475-1479を参照のこと)。
[0042]NF−κBのシグナル伝達経路は広範な炎症誘発効果に関与する。例えばSchreiber et al. (1998)、上記記載、を参照のこと。レニン−アンジオテンシン系(RAS)のメンバーであり、アンジオテンシン変換酵素(ACE)の一次産物であるアンジオテンシンII(AII)は、その1型受容体および2型受容体(各々AT1およびAT2)を介して、そして多くの場合最終的には以下に示すようにNF−κBの活性化を通して、多様な組織において炎症誘発効果を及ぼすことが知られている。
[0043]血中RASにおけるAII合成の古典的経路において、AIIの前駆物質はアンジオテンシノーゲンであり、これは原則として肝臓で生成された後、レニンによって切断されてアンジオテンシンI(AI)を形成し、これがACEによりAIIに変換され、AIIが循環系を介して様々な標的細胞に運ばれる。例えばInokuchi et al. (2005) Gut 54:349-356、および同文献に引用されている出典を参照のこと。加えて、組織特異的なレニン−アンジオテンシン系が多くの臓器において同定されており、腎臓、脳、大動脈、副腎、心臓、胃および結腸を含む様々な組織が、血中RASのAIIを独立して合成する能力を有することを示唆する。
[0044]Donoghue et al. (2000) Circ. Res.87:1-9は、ヒト心不全の心室のcDNAライブラリの配列決定から、ACEに関連するカルボキシペプチダーゼの同定についてを報告した。このカルボキシダーゼ、すなわちACE2は、最初の公知のヒトACEホモログであると述べられた。著者はさらに、ACE2およびACEのメタロプロテアーゼ触媒ドメインが42%同一であること、そしてより遍在的なACEとは反対に、ACE2転写物は検討された23のヒト組織のうち、心臓、腎臓、および精巣のみに見出されることを報告した。
[0045]Actonらに対する米国特許第6,194,556号は、ACE2をコードする新規遺伝子を開示している。これらの遺伝子に基づく治療、診断、およびスクリーニングのアッセイもまた開示されている。
[0046]Harmer et al. (2002) FEBS Lett.532:107-110は、72のヒト組織におけるACE2(およびACEの2つのアイソフォーム)の転写発現プロフィールの定量的マッピングについて報告した。この研究は報告によれば、ACE2の発現は腎臓および心血管の組織において高いことを確認した。ACE2は、胃腸系、特に回腸、十二指腸、空腸、盲腸および結腸において、比較的高レベルの発現を示すこともさらに報告された。著者らは、ACE2の機能的有意性を証明する上で、胃腸の生理学および病態生理学における役割について何らかの考察がなされるべきであると提案した。
[0047]Rice et al. (2003) Bull. Br. Soc. Cardiovasc. Res. 16(2):5-11は、ACE2の潜在的な機能的役割を概説し、その発現が主として精巣、腎臓、心臓および腸管に局在することを示した。
[0048]Ferreira & Santos (2005) Braz. J. Med. Biol. Res. 38:499-507は、本明細書では図1に示したように、ACEおよびACE2の役割を含むRASの重要な経路をまとめた。
[0049]多様な炎症誘発効果における、ACEの主要産生物であるアンジオテンシンIIの意味のエビデンスとして、例えば以下を参照のこと:
・Phillips & Kagiyama (2002) Curr. Opin. Investig. Drugs 3(4):569-577、著者らは、アンジオテンシンIIが、炎症の促進において、とりわけアテローム硬化症において、核内因子κB(NF−κB)の活性化を介しての鍵となる因子であることを示す文献について概説した;
・Costanzo et al. (2003) J. Cell Physiol. 195(3):402-410、著者らは、NF−κBの活性化を通しての炎症性サイトカインを介しての、アテローム硬化症に関与する血管内簸細胞接着分子の、アンジオテンシンIIによるアップレギュレーションについて報告した;
・Sanz-Rosa et al. (2005) Am. J. Physiol. Heart Circ. Physiol. 288:H111-H115、著者らは、AT1受容体を遮断することが、本態性高血圧における血管および血中の炎症性メディエーター、例えばNF−κBおよびTNF−αのレベルを低減させる、と報告した。
・Phillips & Kagiyama (2002) Curr. Opin. Investig. Drugs 3(4):569-577、著者らは、アンジオテンシンIIが、炎症の促進において、とりわけアテローム硬化症において、核内因子κB(NF−κB)の活性化を介しての鍵となる因子であることを示す文献について概説した;
・Costanzo et al. (2003) J. Cell Physiol. 195(3):402-410、著者らは、NF−κBの活性化を通しての炎症性サイトカインを介しての、アテローム硬化症に関与する血管内簸細胞接着分子の、アンジオテンシンIIによるアップレギュレーションについて報告した;
・Sanz-Rosa et al. (2005) Am. J. Physiol. Heart Circ. Physiol. 288:H111-H115、著者らは、AT1受容体を遮断することが、本態性高血圧における血管および血中の炎症性メディエーター、例えばNF−κBおよびTNF−αのレベルを低減させる、と報告した。
・Esteban et al. (2004) J. Am. Soc. Nephrol. 15:1514-1529、著者らは閉塞腎において、アンジオテンシンIIは、AT1およびAT2を介してNF−κBを活性化し、それにより炎症を促進する、と報告した;そして
・Inokuchi et al. (2005)、上記記載、著者らは、低レベルのアンジオテンシンIIを有するアンジオテンシン遺伝子ノックアウトマウスにおいて、2,4,6−トリニトロベンゼンスルホン酸(TNBS)により誘発される炎症性結腸炎が回復すること、そしてAT1受容体の遮断もまたTNBS誘発結腸炎を回復させる、と報告した。
・Inokuchi et al. (2005)、上記記載、著者らは、低レベルのアンジオテンシンIIを有するアンジオテンシン遺伝子ノックアウトマウスにおいて、2,4,6−トリニトロベンゼンスルホン酸(TNBS)により誘発される炎症性結腸炎が回復すること、そしてAT1受容体の遮断もまたTNBS誘発結腸炎を回復させる、と報告した。
[0050]RASのアンジオテンシンが、免疫を介した炎症性腸疾患のための予防的戦略(対象)として推定された(Inokuchi et al. (2005)、上記記載)。
[0051]ACE2の破壊および/またはACE2遺伝子を欠損する突然変異体を伴う様々な研究において、ACE産生物であるアンジオテンシンIIの炎症誘発効果は、通常ACE2によって均衡されることが発見された。例えば以下を参照のこと:
・Crackower et al. (2002) Nature417(6891):822-828、著者らは、様々なラットモデルにおいてACE2遺伝子の破壊またはACE2遺伝子の欠損が、アンギオテンシンIIのレベルを上昇させる、と報告した;
・Huentelman et al. (2005) Exp. Physiol.90(5):783-790、著者らは、ACE2をコードするベクターの注入は、アンジオテンシンIIに誘発される心臓の肥大および線維化から野生型マウスを保護する、と報告した;そして
・Imai et al. (2005) Nature436(7047):112-116、著者らは、ACE遺伝子の欠損またはACE2タンパク質を与えることで、酸誘発による急性肺傷害から野生型マウスを保護する、と報告した。
・Crackower et al. (2002) Nature417(6891):822-828、著者らは、様々なラットモデルにおいてACE2遺伝子の破壊またはACE2遺伝子の欠損が、アンギオテンシンIIのレベルを上昇させる、と報告した;
・Huentelman et al. (2005) Exp. Physiol.90(5):783-790、著者らは、ACE2をコードするベクターの注入は、アンジオテンシンIIに誘発される心臓の肥大および線維化から野生型マウスを保護する、と報告した;そして
・Imai et al. (2005) Nature436(7047):112-116、著者らは、ACE遺伝子の欠損またはACE2タンパク質を与えることで、酸誘発による急性肺傷害から野生型マウスを保護する、と報告した。
[0052] その受容体(Mas)を介してのACE2の一次産生物、すなわちアンジオテンシン(1−7)は、通常ACE産生物であるアンジオテンシンIIの機能に対抗することが発見された。例えば以下を参照のこと:
・Guy et al. (2005) Biochim. Biophys. Acta 1751(1):2-8、著者らはとりわけ、ACE2は、アンジオテンシン(1−7)に相対してアンジオテンシンIIのレベルをコントロールすることにより、心臓および腎臓の機能を制御し、そしてそれ故にRAS内でのACEの効果を相殺すると思われる、と指摘する文献について概説した;
・Ferreira & Santos (2005),上記記載、著者らはとりわけ、ACE阻害薬の有益性は、ACE2産生物であるアンジオテンシン(1−7)により部分的に仲介されると思われ、アンジオテンシン(1−7)の血漿レベルはACE阻害薬の慢性的投与後に大きく増加することを示す文献について概説した。
・Guy et al. (2005) Biochim. Biophys. Acta 1751(1):2-8、著者らはとりわけ、ACE2は、アンジオテンシン(1−7)に相対してアンジオテンシンIIのレベルをコントロールすることにより、心臓および腎臓の機能を制御し、そしてそれ故にRAS内でのACEの効果を相殺すると思われる、と指摘する文献について概説した;
・Ferreira & Santos (2005),上記記載、著者らはとりわけ、ACE阻害薬の有益性は、ACE2産生物であるアンジオテンシン(1−7)により部分的に仲介されると思われ、アンジオテンシン(1−7)の血漿レベルはACE阻害薬の慢性的投与後に大きく増加することを示す文献について概説した。
・Mendes et al. (2005) Regul. Pept.125(1-3):29-34、著者らは、アンジオテンシン(1−7)の注入が心臓におけるアンジオテンシンIIレベルを低下させることを報告し、そのような低下はアンジオテンシン(1−7)の有益な効果に寄与すると思われる、と推定した;そして
・Tallant & Clark (2003) Hypertension42:574-579、著者らは、アンジオテンシン(1−7)は血管傷害後の平滑筋の増殖を低減し、そしてラット大動脈の血管平滑筋細胞において、増殖およびマイトジェン活性化タンパク質(MAP)キナーゼの活性の、アンジオテンシンIIによる刺激の逆に作用する、と報告した。
・Tallant & Clark (2003) Hypertension42:574-579、著者らは、アンジオテンシン(1−7)は血管傷害後の平滑筋の増殖を低減し、そしてラット大動脈の血管平滑筋細胞において、増殖およびマイトジェン活性化タンパク質(MAP)キナーゼの活性の、アンジオテンシンIIによる刺激の逆に作用する、と報告した。
[0053]このように低レベルのアンジオテンシンIIは、炎症性結腸炎を回復させる(Inokuchi et al. (2005),上記記載)様にであり、そしてACE2の活性は、アンジオテンシン(1−7)レベルを増加させるか、もしくはアンジオテンシンIIレベルを減少させるかのいずれか、または双方により、多様な組織においてアンジオテンシンIIの炎症作用を相殺する様である。
[0054]それ故1つのシナリオにおいて、ACE2活性の促進は、疾患、例えばIBDにおける炎症を低減するために興味深いと思われる。Huentelman et al. (2004) Hypertension 44:903-906は同様に、in vivo でのACE2の活性化は、アンジオテンシンIIの強力な血管収縮効果を相殺することにより、高血圧およびその他の心血管疾患のための保護および治療の成功をもたらし得ると提案した。
[0055]上に引用した米国特許第6,194,556号はACE2をコードする新規遺伝子を開示しており、第60段36−54行で次のように提案している:“ブラジキニンが過剰産生されていて、ブラジキニンを不活性化する能力のあるACE2アゴニストによる治療が有用であり得る、なおその他の疾患または状態として、病理学的状態、例えば敗血症および出血性のショック、アナフィラキシー、関節炎、鼻炎、喘息、炎症性腸疾患、サルコイドーシス、ならびに急性膵炎、胃摘出後のダンピング症候群、カルチノイド症候群、片頭痛、および遺伝性血管浮腫を含むある種のその他の状態を含む(参考文献省略)。
[0056]ACE2活性を促進するよりむしろ阻害する薬剤も、当該技術において述べられてきた。例えばHuentelman et al. (2004)、上記記載、は、SARS−CoV、すなわち重症急性呼吸器症候群(SARS)の原因であるコロナウイルスによる感染を阻害するACE2阻害化合物を同定するための努力について報告しており、それに関して、ACE2が機能的な受容体であることが見出された。そのように同定された化合物の中に、NAAE(N−(2−アミノエチル)−1−アジリジン−エタンアミン)があった。
[0057]Parryらに対する米国特許第6,900,033号は、ACE2タンパク質またはACE2様ポリペプチドに特異的に結合すると言われる、特定のアミノ酸配列を包含するペプチドを開示している。同文献の第53段、63−65行に、“異常に高いアンジオテンシンIIのレベルは、異常に低いACE−2活性に起因する可能性がある”、そしてその第63段21−32行で、“ACE−2に誘発されるシグナル伝達を活性化する...ACE−2結合ポリペプチドを動物に投与して、異常なACE−2発現、ACE−2機能の欠如、異常なACE−2基質の発現、またはACE−2基質の機能の欠如、に関連する疾患または障害を治療、予防、または回復させることができる。これらのACE−2結合ポリペプチドは、ACE−2を介した基質の作用の生物学的活性のすべてまたはそのサブセットのいずれかを増強または活性化すると思われる...”と、提案している。さらにその第71段、26−37行に、“本発明の”ポリペプチド(活性化または阻害のいずれかは特定していない)はとりわけ、“感染に関連する炎症(例えば敗血症性ショック、敗血症、または全身性炎症反応症候群(SIRS))、虚血再灌流障害、多発性外傷、疼痛、エンドトキシンによる死亡、関節炎(例えば変形性関節症および関節リウマチ)、合併症を介した超急性の拒絶、腎炎、サイトカインまたはケモカインに誘発される肺傷害、炎症性腸疾患、クローン病、ならびにサイトカイン(例えばTNFまたはIL−1)の過剰産生に起因するもの、を含むがこれに限定されない炎症を治療、予防、または回復するために有用である”と記されている。これとは別に、in vitroでACE2を阻害することが報告されているACE2結合ペプチドが、同文献の第127−130段の表2に同定されている。
[0058]Huang et al. (2003) J. Biol. Chem. 278(18):15532-15540は、1つのそのようなACE2阻害ペプチド、すなわちDX600が、2.8nMのACE2 Ki値を示すことを報告した。
[0059]Li et al. (2005) Am. J. Physiol. Renal Physiol. 288:F353-F362は、DX600が、ラットのネフロンセグメントにおいてアンジオテンシンIを介したアンジオテンシン(1−7)の生成を遮断することを報告した。
[0060]Actonらに対する米国特許第6,632,830号は、ACE2の活性を調節するために有用であると記された、亜鉛配位結合部分およびアミノ酸模倣部分を包含する化合物を開示している。より特定すれば、同文献に提示された一般式のACE2阻害化合物が開示されている。そのような化合物は、患者の“ACE−2に関連する状態”を治療するために有用であると記されている。“ACE−2に関連する状態”は、高血圧ならびにそれに関する疾患および障害、特に動脈性高血圧、うっ血性心不全、慢性心不全、左心室肥大、急性心不全、心筋梗塞、および心筋症;平滑細胞の増殖、特に平滑筋細胞の増殖の制御に関連する状態;腎臓の疾患および障害;その他のアドレナリンの上昇した状態;キネテンシン(kinetensin)に関連する状態(例えば失神、喘息およびアナフィラキシーショックを含む局所または全身のアレルギー反応における、異常なヒスタミン放出によって引き起こされるまたは寄与するものを含む;生殖体の成熟に関する不妊症またはその他の障害;認知障害;ブラジキニンおよびdes-Argブラジキニンに関連する障害;ならびに“SIRS...、敗血症、多発性外傷、炎症性腸疾患、急性および慢性の疼痛、関節リウマチおよび変形性関節症および歯周病における骨の破壊、月経困難症、早産、限局的損傷に続発する脳浮腫、びまん性軸索損傷、脳卒中、くも膜下出血後の再灌流傷害および脳血管痙攣、アレルギー性障害(喘息、成人呼吸促迫症候群を含む)、創傷治癒および瘢痕の形成”を含むと記されている“その他の例”(同文献の第36段、58−67行)を含むと記されている。
[0061]Dales et al. (2002) J. Am. Chem. Soc. 124:11852-11853は、一定範囲のそのような化合物のACE2 IC50値を報告した。これらのうち最も活性なのは、以下の式

を有するとして同文献で同定された化合物16であった。化合物16の4つのすべての立体異性体が調製され、最大の能力がS,S−同位体に関して報告され、報告によれば、この同位体は0.44mMのACE2に対するIC50を有した。上の化合物、2−[1−カルボキシ−2−[3−(3,5−ジクロロベンジル)−3H−イミダゾール−4−イル]−エチルアミノ]−4−メチルペンタン酸のS,S−異性体を本明細書においてGL1001と呼ぶが、従来はMLN−4760と呼ばれた。
[0062]Actonらの米国特許出願公開第2004/0082496号は、ACE2の活性を調節するために有用であると記された付加的な化合物を開示している。体重の障害を治療し、食欲を低下させ、筋肉量を増大し、体脂肪を低減し、糖尿病を治療し、そして脂質代謝異常に関連する状態を治療するための、阻害薬、および阻害薬を含有する医薬組成物を使用する方法もまた記載されている。
[0063]上に示したように、IBDおよび慢性胃炎のための現行の薬物療法は、乏しいまたは信頼性のない有効性、有害な副作用、および高いコスト、の1つまたはそれより多くを含むという弱点がある。消化管の炎症性疾患、例えばIBDおよび慢性胃炎、より特定すればUCおよびCDのいずれかまたは双方に関する付加的な薬物療法の必要性は、処方する医師、およびIBDまたは慢性胃炎の患者の利用可能な選択肢の範囲を拡大するために、依然として残されている。
[0064]今回、消化管の炎症性疾患を有する被験者における炎症、またはそれに関連するもしくはその二次的な病理学的プロセスを低減または軽減するための方法であって、前記被験者にACE2阻害薬の抗炎症有効量を投与することを包含する、前記方法を提供する。
[0065]消化管の炎症性疾患を有する被験者における粘膜の潰瘍の治癒を促進するための方法であって、前記患者に治療有効量のACE2阻害薬を投与することを包含する、前記方法をさらに提供する。
[0066]被験者における消化管の炎症性疾患の寛解を導入または維持するための方法であって、前記被験者に治療有効量のACE2阻害薬を投与することを包含する、前記方法をまださらに提供する。
[0067]上の各態様に従って、炎症性疾患は、例えば慢性胃炎であることができる。
[0068]あるいは上の各態様に従って、炎症性疾患は、例えばIBD、より特定すればUCまたはCDであることができる。
[0069]アミノサリチル酸不応性炎症性腸疾患を有する被験者における、コルチコステロイド療法を避けるための方法であって、治療有効量のACE2阻害薬を、所望によりアミノサリチル酸との併用療法において、ただしコルチコステロイドは使用せずに投与することを包含する、前記方法をまださらに提供する。
[0070]ACE2阻害薬、ならびにアミノサリチル酸、コルチコステロイド、免疫抑制薬、抗TNFα薬、およびそれらの組み合わせから成る群より選択される少なくとも1つの付加的な薬剤を包含する治療の組み合わせを、まださらに提供する。
[0071]上にまとめた態様の特定の側面を含むその他の態様は、以下の詳細な説明より明らかにする。
[0095]本明細書に様々な治療法を記載するが、それらはすべて消化管の炎症性疾患を有する被験者にACE2阻害薬を投与することを伴う。
[0096]あらゆるACE2阻害薬を使用することができる。通常、in vitroまたはin vivoのいずれで測定されるにせよ、例えばIC50またはKiにより表されるような、ACE2に対する相対的に高い活性を有するACE2阻害薬を選択することは有用であると理解されるだろう。1つの態様において、選択されるACE2阻害薬は、約1000nMより大きくない、例えば約500nMより大きくない、約250nMより大きくない、または約100nMより大きくない、in vitroのACE2 IC50および/またはACE2 Kiを示すものである。
[0097]ACE2阻害薬は、ACE2に対するそれらのアフィニティーだけでなく、ACE2への結合に関するそれらの選択性においても、より遍在的なACEとは対照的に異なることが知られている。1つの態様においてACE2阻害薬は、IC50(ACE2)に対するIC50(ACE)の比率で表した場合、少なくとも約102、少なくとも約103、または少なくとも約104の、ACE2対ACEへの選択性を示す。
[0098]ペプチドおよび非ペプチドのACE2阻害薬を使用することができる。ペプチドのACE2阻害薬の例、およびそれらを調製する方法は、例えば上に引用した米国特許第6,900,033号に見出すことができ、その全内容を本明細書において参照として援用する。相対的に強いACE2の阻害を示すペプチド化合物は、説明によれば米国特許第6,900,033号において、DX−512、DX−513、DX−524、DX−525、DX−529、DX−531、DX−599、DX−600、DX−601、およびDX−602として同定されているペプチド配列を有するものを含む。ACE2タンパク質に特異的に結合し、それによりACE2活性を阻害する抗体もまた、本発明の方法および組成物において使用することができる。
[0099]多くの目的のためには、非ペプチドまたは“低分子”のACE2阻害薬を使用することは好ましいと理解されるだろう。そのような化合物は、殊に大規模スケールまたは商業スケールで調製がより容易であり、より低コストであり、そして体内の活性部位への投与および送達においてより問題が少ない、という傾向がある。それ故様々な態様においてACE2阻害薬は、非ペプチド化合物、または医薬的に受容可能なその塩もしくはそのプロドラッグを包含する。
[0100]実例としてACE2阻害薬は、上に引用した米国特許第6,632,830号において、その調製法と並んで同文献に開示されている具体的な化合物のいずれかを含む、一般名称で開示されている1つのタイプであることができる。同文献の全内容を本明細書において参照として援用する。1つの態様において、非ペプチド化合物は、亜鉛配位結合部分およびアミノ酸模倣部分を包含する。
[0101]より具体的には非ペプチド化合物は、米国特許第6,632,830号に開示されているように、以下の式
を有することができ、式中
R6はヒドロキシルまたは保護しているプロドラッグ部分であり;
R7は水素、カルボン酸、エーテル、アルコキシ、アミド、保護しているプロドラッグ部分、ヒドロキシル、チオール、ヘテロシクリル、アルキル、またはアミンであり;
Qは、CH2、O、NH、またはNR3であり、ここでR3は、置換されたまたは置換されていないC1−5の側鎖または直鎖のアルキル、C2−5の側鎖または直鎖のアルケニル、置換されたまたは置換されていないアシル、アリール、またはC3−8環であり;
Gは、共有結合、またはCH2、エーテル、チオエーテル、アミン、またはカルボニルの連結部分であり;
Mはヘテロアリールであって、サブリンク部分である(CH2)nまたは(CH2)nO(CH2)nを通して連結される、少なくとも1つのサブアンカー部分で置換されており、ここでnは0から3の整数であり、前記サブアンカー部分は、置換されたまたは置換されていないシクロアルキル環またはアリール環を包含し;
Jは、1つの結合、または置換されたもしくは置換されていないアルキル、アルケニル、もしくはアルキニルの部分であり;そして
Dは、アルキル、アルケニル、アルキニル、アリール、またはヘテロアリールであって、所望によりGまたはMに連結して1つの環を形成する。
R6はヒドロキシルまたは保護しているプロドラッグ部分であり;
R7は水素、カルボン酸、エーテル、アルコキシ、アミド、保護しているプロドラッグ部分、ヒドロキシル、チオール、ヘテロシクリル、アルキル、またはアミンであり;
Qは、CH2、O、NH、またはNR3であり、ここでR3は、置換されたまたは置換されていないC1−5の側鎖または直鎖のアルキル、C2−5の側鎖または直鎖のアルケニル、置換されたまたは置換されていないアシル、アリール、またはC3−8環であり;
Gは、共有結合、またはCH2、エーテル、チオエーテル、アミン、またはカルボニルの連結部分であり;
Mはヘテロアリールであって、サブリンク部分である(CH2)nまたは(CH2)nO(CH2)nを通して連結される、少なくとも1つのサブアンカー部分で置換されており、ここでnは0から3の整数であり、前記サブアンカー部分は、置換されたまたは置換されていないシクロアルキル環またはアリール環を包含し;
Jは、1つの結合、または置換されたもしくは置換されていないアルキル、アルケニル、もしくはアルキニルの部分であり;そして
Dは、アルキル、アルケニル、アルキニル、アリール、またはヘテロアリールであって、所望によりGまたはMに連結して1つの環を形成する。
[0102]1つの態様において、非ペプチド化合物に関する上の式において、R6はヒドロキシでありR7はカルボン酸であり、QはNHであり、そしてGはCH2である。
[0103]1つの態様において、非ペプチド化合物に関する上の式において、Mのヘテロアリール基は、イミダゾリル、チエニル、トリアゾリル、ピラゾリル、またはチアゾリルである。ヘテロアリール基の選択とは独立して、本態様に従ってのサブアンカー部分は、C3−6シクロアルキル、フェニル、メチレンジオキシフェニル、ナフタレニル、またはハロ、C1−6アルキル、C3−6シクロアルキル、トリフルオロメチル、C1−6アルコキシ、トリフルオロメトキシ、フェニル、シアノ、ニトロ、およびカルボン酸の基より独立して選択される1から3の置換基を有するフェニルであって、(CH2)nまたは(CH2)O(CH2)のサブリンク部分を通してヘテロアリー基に連結されており、ここでnは0から3の整数である。
[0104]1つの態様において、非ペプチド化合物に関する上の式において、Jは、1つの結合またはCH2部分であり、そしてDは、C1−6アルキル、C3−6シクロアルキル、またはフェニルである。
[0105]より特定の態様において、非ペプチド化合物に関する上の式において、
R6はヒドロキシルであり;
R7は水素、カルボン酸であり;
QはNHであり;
GはCH2であり;
Mは、イミダゾリル、チエニル、トリアゾリル、ピラゾリル、またはチアゾリルであって、(CH2)nまたは(CH2)O(CH2)のサブリンク部分を通してサブアンカー部分に連結されており、ここでnは0から3の整数であり、前記サブアンカー部分は、C3−6シクロアルキル、フェニル、メチレンジオキシフェニル、ナフタレニル、またはハロ、C1−6アルキル、C3−6シクロアルキル、トリフルオロメチル、C1−6アルコキシ、トリフルオロメトキシ、フェニル、シアノ、ニトロ、およびカルボン酸の基より独立して選択される1から3の置換基を有するフェニルであり;
Jは、1つの結合、またはCH2部分であり;そして
Dは、C1−6アルキル、C3−6シクロアルキル、またはフェニルである。
R6はヒドロキシルであり;
R7は水素、カルボン酸であり;
QはNHであり;
GはCH2であり;
Mは、イミダゾリル、チエニル、トリアゾリル、ピラゾリル、またはチアゾリルであって、(CH2)nまたは(CH2)O(CH2)のサブリンク部分を通してサブアンカー部分に連結されており、ここでnは0から3の整数であり、前記サブアンカー部分は、C3−6シクロアルキル、フェニル、メチレンジオキシフェニル、ナフタレニル、またはハロ、C1−6アルキル、C3−6シクロアルキル、トリフルオロメチル、C1−6アルコキシ、トリフルオロメトキシ、フェニル、シアノ、ニトロ、およびカルボン酸の基より独立して選択される1から3の置換基を有するフェニルであり;
Jは、1つの結合、またはCH2部分であり;そして
Dは、C1−6アルキル、C3−6シクロアルキル、またはフェニルである。
[0106]上の態様のいずれかに従って化合物は、あらゆるエナンチオマーの立体配置、例えば(R,R)、(R,S)、(S,R)もしくは(S,S)、またはエナンチオマーの混合物、例えばラセミ混合物として存在することができる。しかし通常、化合物は(S,S)−立体配置で存在することが好ましいと理解されている。1つの態様において化合物は、(S,S)−立体配置にあり、実質的にエナンチオマー的に純粋である。例えば化合物は、存在する化合物のすべてのエナンチオマーの形の重量の少なくとも約90%、少なくとも約95%、または少なくとも約99%の、エナンチオマー的純度を示すことができる。
[0107]米国特許第6,632,830号に具体的に開示されている実例としての化合物は以下を含み、それら各化合物はあらゆるエナンチオマーの形、実例としては(S,S)−立体配置で存在することができる:
2−[1−カルボキシ−2−[3−(4−トリフルオロメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−ナフタレン−1−イルメチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−クロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,4−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−シアノベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−クロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,5−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,4−ジメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(3−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,5−ジメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−トリフルオロメトキシベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−イソプロピルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−t−ブチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(4−ニトロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジメトキシベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジフルオロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(3−トリフルオロメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[2−(3−ベンゾ[1,3]ジオキソール−5−イルメチル−3H−イミダゾール−4−イル)−1−カルボキシエチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2−シクロヘキシルエチル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−フェネチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−ヨードベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−フルオロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−ベンジルオキシメチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(4−ブチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[2−フェニルチアゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[1−ベンジル)−1H−ピラゾール−4−イル]エチルアミノ]−メチルペンタン酸;および
2−[1−カルボキシ−2−[3−(2−メチルビフェニル−3−イルメチル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸。
2−[1−カルボキシ−2−[3−(4−トリフルオロメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−ナフタレン−1−イルメチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−クロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,4−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−シアノベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−クロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,5−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,4−ジメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(3−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,5−ジメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−トリフルオロメトキシベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−イソプロピルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−t−ブチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(4−ニトロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジメトキシベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジフルオロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(3−トリフルオロメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[2−(3−ベンゾ[1,3]ジオキソール−5−イルメチル−3H−イミダゾール−4−イル)−1−カルボキシエチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2−シクロヘキシルエチル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−フェネチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−ヨードベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−フルオロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−ベンジルオキシメチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(4−ブチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[2−フェニルチアゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[1−ベンジル)−1H−ピラゾール−4−イル]エチルアミノ]−メチルペンタン酸;および
2−[1−カルボキシ−2−[3−(2−メチルビフェニル−3−イルメチル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸。
[0108]すべての態様におけるものとして、上の化合物のいずれも、上の式の形、または医薬的に受容可能なその塩もしくはそのプロドラッグの形で存在することができる。
[0109]本発明は、本明細書において(S,S)−2−[1−カルボキシ−2−[3−(3,5−ジクロロベンジル)−3H−イミダゾール−4−イル]−エチルアミノ]−4−メチルペンタン酸、さもなければGL1001として公知の化合物を、特に参照して説明する。この化合物は、例えばDales ら(2002)、上記記載、によりそのような化合物の調製法と合わせて開示されているように、以下の式
を有する(S,S)−エナンチオマーの化合物である。手短にはこの化合物の製法は、(S)−ヒスチジンメチルエステルをBoc2Oで処理して、完全に保護されたヒスチジン誘導体を提供することを包含する。その後N−3イミダゾールの窒素を、3,5−ジクロロベンジルアルコールのトリフレートを使用して選択的にアルキル化する。Bocの脱保護に続いて、得られたアルキル化されたヒスチジン誘導体とβ−ケトエステル間の還元的アミノ化によりジエステルアミン化合物を供給し、これを加水分解することにより、2−[1−カルボキシ−2−[3−(3,5−ジクロロベンジル)−3H−イミダゾール−4−イル]−エチルアミノ]−4−メチルペンタン酸を、ジアステレオマーの混合物として得る。ジアステレオマーは、HPLCおよび結晶化を使用して分離および精製することができる。
[0110]上で参照した米国特許第6,632,830号に記載されている製法を非限定的に含む、他の製造過程を使用してGL1001を調製することもできる。
[0111]本発明の実践において使用することのできる、ACE2阻害活性を有する付加的な化合物は、Huentelman ら(2004)、上記記載、により開示されており、NAAE(N−(2−アミノエチル)−1−アジリジンエタンアミン)を含む。
[0112]本発明の実践において使用することのできる、ACE2阻害活性を有するさらに付加的な化合物は、Rella et al. (2006) J. Chem. Inf. Model. 46(2):708-716により開示されている。この公開文献が本発明に対する先行技術を構成するという了解なく、同文献の全内容を参照として援用する。同文献は、ACE2活性の阻害効果を表示すると報告されている17化合物(その最も活性な6化合物は、62−179μMの範囲のIC50を示す)を含む新規ACE2阻害薬について、構造に基づいたファルマコフォアのデザインおよび仮想スクリーニングを開示している。
[0113]本明細書に提供した方法は、被験者の消化管の全体またはいずれか1部分もしくは複数部分の炎症性疾患を治療する上で有用である。特に本方法は、慢性胃炎、ならびにUCおよびCDを含むIBDを治療する上で有用である。
[0114]“被験者”は本明細書において、恒温動物、通常哺乳類、例えばネコ、イヌ、ウマ、ウシ、ブタ、マウス、ラット、またはヒトを含む霊長類である。1つの態様において被験者は、ヒト、例えば臨床的に診断された消化管の炎症性疾患、例えば慢性胃炎、またはUCおよびCDを含むIBDを有する患者である。ヒトの疾患に関連する実験的検討における動物モデルもまた、本明細書において“被験者”の例であり、例えばげっ歯目(例えばマウス、ラット、モルモット)、ウサギ目(例えばラビット)、食肉類(例えばネコ、イヌ)、またはヒト以外の霊長類(例えばサル、チンパンジー)を含むことができる。さらに被験者は、獣医学的にケアする動物(例えば家畜、牧畜、作業、スポーツまたは動物園の動物)であることができる。
[0115]本発明に従っての有用なある種の化合物は、酸および/または塩基の部分を有し、適切な条件下で適切な酸と塩を形成することができる。例えばGL1001は、適切な条件下で適切な塩基と塩を形成することができる2つの酸の部分、および適切な条件下で適切な酸と塩を形成することができるアミノ基を有する。内部塩を形成することもできる。化合物は、その遊離の酸/塩基の形、または内部塩、酸付加塩もしくは塩基との塩の形で使用することができる。
[0116]酸付加塩は実例として、無機酸、例えば鉱酸、例えば硫酸、リン酸またはハロゲン化水素酸(例えば塩酸もしくは臭化水素酸)と共に;有機カルボン酸、例えば(a)置換されていなくても置換されていてもよい(例えばハロで置換された)C1−4アルケンカルボン酸、例えば酢酸、(b)飽和もしくは不飽和のジカルボン酸、例えばシュウ酸、マロン酸、コハク酸、マレイン酸、フマル酸、フタル酸、もしくはテレフタル酸、(c)ヒドロキシカルボン酸、例えばアスコルビン酸、グリコール酸、乳酸、リンゴ酸、酒石酸、もしくはクエン酸、(d)アミノ酸、例えばアスパラギン酸もしくはグルタミン酸、または(e)安息香酸と共に;あるいは有機スルホン酸、例えば置換されていなくてよい(例えばハロで置換された)C1−4アルカンスルホン酸またはアリールスルホン酸、例えばメタンスルホン酸またはp−トルエンスルホン酸と共に、形成することができる。
[0117]塩基との塩は、金属塩、例えばアルカリ金属もしくはアルカリ土類金属の塩、例えばナトリウム塩、カリウム塩、もしくはマグネシウム塩;またはアンモニアもしくは有機アミン、例えばモルホリン、チオモルホリン、ピペリジン、ピロリジン、モノ−、ジ−、もしくはトリ−低級アルキルアミン、例えばエチルアミン、t−ブチルアミン、ジエチルアミン、ジイソプロピルアミン、トリエチルアミン、トリブチルアミン、もしくはジメチルプロピルアミン、もしくはモノ−、ジ−、もしくはトリ−(ヒドロキシ低級アルキル)アミン、例えばモノエタノールアミン、ジエタノールアミン、もしくはトリエタノールアミンとの塩を含む。
[0118]あるいは化合物のプロドラッグ、またはそのようなプロドラッグの塩を使用することができる。プロドラッグは、被験者の体内で活性な化合物、この場合はACE2阻害化合物に開裂、代謝、またはさもなければ変換される、典型的にはそれ自体が弱い医薬的活性を有する、または医薬的活性を有していない化合物である。プロドラッグの例は、エステル、特にアルカノイルエステル、そしてより特定すればC1−6アルカノイルエステルである。その他の例として、カルバメート、カルボネート、ケタール、アセタール、ホスフェート、ホスホネート、スルフェート、およびスルホネートを含む。GL1001の様々なプロドラッグ、およびそのようなプロドラッグを作成する方法は、例えば上で参照した米国特許第6,632,830号、および米国公開特許出願第2004/0082496号に開示されている。
[0119]ACE2阻害薬は、治療有効量、例えば抗炎症有効量で投与しなければならない。治療にまたは抗炎症の有効量を何の成分で構成するかは、特定の被験者の年齢および体重、疾患の性質、ステージおよび重症度、求められる特定の効果(例えば炎症の低減、症状の軽減、寛解の維持、など)ならびにその他の因子を含むいくつかの因子に依存するが、ほとんどの被験者に関しては、約0.5から約5000mg/日、より典型的には約5から約1000mg/日の投与量が適切であると理解されるだろう。特定の態様において使用される投与量は、約10から約800mg/日、約50から約750mg/日、または約100から約600mg/日;実例として約50、約100、約150、約200、約250、約300、約350、約400、約450、約500、約550、約600、約650、約700、または約750mg/日である。
[0120]ACE2阻害薬化合物の塩またはプロドラッグを使用する場合、投与される量は、上述のような化合物の1日の投与量を送達する量でなければならない。
[0121]したがって1つの態様において、約0.5から約5000mg/日、例えば約5から約1000mg/日、約10から約800mg/日、約50から約750mg/日、または約100から約600mg/日;実例として約50、約100、約150、約200、約250、約300、約350、約400、約450、約500、約550、約600、約650、約700、または約750mg/日の量のACE2阻害薬を、被験者に投与することを包含する、被験者における消化管の炎症性疾患を治療するための方法を提供する。
[0122]上の投与量は1日当たりを基本として示しているが、必ずしも1日1回の頻度で投与しなければならないと解釈されるべきではない。事実化合物またはその塩もしくはプロドラッグは、あらゆる適切な頻度で、例えばいくつかの因子を考慮する臨床医により従来より決定されるように、典型的には1日約4回、1日3回、1日2回、1日1回、隔日、1週間に2回、1週間に1回、1か月に2回、または1か月に1回として、投与することができる。あるいは化合物またはその塩もしくはプロドラッグは、多かれ少なかれ継続的に、例えば入院中は非経口注入により投与することができる。状況によってはただ1回の用量を投与してよいが、より典型的には投与は、治療期間にわたって反復される投与量を伴う投与計画に従う。そのような投与計画において、1日の投与量および/または投与頻度は、所望であれば治療期間のコースにわたり変更することができ、例えば当該化合物を相対的に低用量で被験者に導入し、その後上限用量に達するまで、1またはそれより多くのステップを経て用量を増加することができる。
[0123]治療期間は通常、所望の結果、例えば寛解の導入または維持、症状の軽減などを達成するために必要とされる限り継続される。状況によっては、薬剤を断続的に、例えば数日、数週関、または数か月の治療期間を、非治療期間をはさんで投与することが有用であると理解されるだろう。そのような断続的な投与は、例えば疾患のフレアに対応して調節することができる。
[0124]投与は、経口、 舌下、経鼻、眼球内、経直腸、経膣、経皮、または非経口(例えば皮内、皮下、筋肉内、静脈内、動脈内、気管内、心室内、腹腔内など)の経路を非限定的に含む、そして吸入または植込みによる経路を含む、あらゆる適切な経路によることができる。
[0125]活性な医薬成分(active pharmaceutical ingredient、API)単独として製剤化されてない化合物、またはその塩もしくはプロドラッグを投与することは可能であり得るが、通常は、APIおよび少なくとも1つの医薬的に受容可能な賦形剤を包含する医薬組成物中のAPIを投与することが好ましいと理解されるだろう。賦形剤(1つまたは複数)は集合的にAPIのためのビヒクルまたは担体を提供する。すべての可能な投与経路に適用する医薬組成物は、当該技術において周知されており、標準的な教科書およびハンドブック、例えば以下に個々に引用するものに述べられている原理および方式に従って調製することができる。
[0126]USIP 編(2005) Remington: The Science and Practice of Pharmacy, 第21版, Lippincott, Williams & Wilkins。
[0127]Allen et al. (2004) Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems, 第8版,Lippincott, Williams & Wilkins。
[0128]適切な賦形剤は、例えばKibbe,編 (2000) Handbook of Pharmaceutical Excipients, 第3版, American Pharmaceutical Association に記載されている。
[0129]本発明の実践においてAPIの送達のためのビヒクルとして使用することのできる製剤の例は、非限定的に、溶液、懸濁液、粉末、顆粒、錠剤、カプセル、ピル、ロゼンジ、咀嚼錠、クリーム、軟膏、ゲル、リポソーム調製物、ナノ粒子調製物、注射可能な調製物、浣腸、座剤、吸入可能な粉末、スプレー可能な液体、エアゾール、パッチ、デポー、および植込み錠を含む。
[0130]実例として、例えば非経口、経鼻、または経口の送達に適する液体製剤において、APIは、希釈剤、例えば水を包含する液体培地中の溶液もしくは懸濁液中に、または何か他の分散した剤形で存在することができる。そのような製剤中に存在することができる付加的な賦形剤として、浸透圧調整剤、バッファー(例えばトリス、リン酸、イミダゾール、もしくは炭酸水素のバッファー)、分散剤もしくは懸濁剤、および/または保存剤を含む。そのような製剤は、ミクロ粒子もしくはナノ粒子、ミセル、および/またはリポソームを含有することができる。非経口製剤は、注射による投与の前に液体担体、例えば水または生理食塩水の添加を必要とする、乾燥した再構築可能な剤形で調製することができる。
[0131]直腸送達用に、APIは適切な液体(例えば浣腸として)、半固体(例えばクリームまたは軟膏として)、または固体(例えば座剤として)の培地中に分散された剤形で存在することができる。
[0132]経口送達用に、APIは液体または固体の剤形で、例えば固体のユニット投与量の剤形、例えば錠剤またはカプセルとして製剤化することができる。そのような投与剤形は典型的には賦形剤として、1つまたはそれより多くの医薬的に受容可能は希釈剤、結合剤、崩壊剤、湿潤剤、および/または減摩剤(antifrictional agents)(滑剤、減粘剤(anti-adherents)および/もしくは潤沢剤)を包含する。多くの賦形剤は、医薬組成物中で2つまたはそれより多くの機能を有する。ある種の機能、例えば希釈剤、結合剤、崩壊剤などを有するものとしての特定の賦形剤の本明細書での特徴付けは、その機能に限定して読まれるべきではない。
[0133]実例として適切な希釈剤は、個別にまたは組み合わせてのいずれかで、ラクトース(無水ラクトースおよびラクトース一水和物を含む);ラクチトール;マルチトール;マンニトール;ソルビトール;キシリトール;ブドウ糖およびブドウ糖一水和物;果糖:ショ糖およびショ糖を基材とする希釈剤、例えば圧縮糖、グラニュー糖および粒状糖;マルトース;イノシトール;加水分解された穀類の固形物;デンプン類(例えばトウモロコシデンプン、小麦デンプン、米デンプン、ジャガイモデンプン、タピオカデンプンなど)、デンプン成分、例えばアミロースおよびデキストレート類、および修飾または加工されたデンプン類、例えばアルファ化デンプン;デキストリン類;セルロース類、(粉末セルロース、微結晶セルロース、ケイ化微結晶セルロース、αセルロースおよびアモルファスセルロースおよび粉末セルロースの食品グレードの原料、ならびに酢酸セルロースを含む);カルシウム塩(炭酸カルシウム、リン酸三カルシウム、リン酸二カルシウム、硫酸一カルシウム一水和物、硫酸カルシウム、および顆粒状乳酸カルシウム三水和物を含む);炭酸マグネシウム;酸化マグネシウム;ベントナイト;カオリン;塩化ナトリウム;などを含む。そのような希釈剤は、存在する場合、典型的には総計で組成物の重量の約5%から約99%、例えば約10%から約85%、または約20%から約80%を構成する。選択される1つまたは複数の希釈剤は好ましくは、適切な流動特性、そして錠剤が所望される場合は適切な圧縮率を示す。
[0134]ラクトース、微結晶セルロース、およびデンプンは、個別にまたは組み合わせてのいずれかで、特に有用な希釈剤である。
[0135]結合剤または接着剤は、特に組成物が錠剤の剤形である場合に有用な賦形剤である。そのような結合剤および接着剤は、通常の加工操作、例えばサイズ決定、潤滑剤注入、圧縮および包装を可能とするため、錠剤化するブレンドに十分な粘着性を与えるが、それでもなお錠剤が崩壊して組成物が摂取時に吸収されることを可能としなければならない。適切な結合剤および接着剤は、個別にまたは組み合わせてのいずれかで、アカシア;トラガカント;グルコース;ポリデキストロース;デンプン(アルファ化デンプンを含む);ゼラチン;修飾されたセルロース類(メチルセルロース、カルメロースナトリウム、ヒドロプロピルメチルセルロース(HPMCまたはヒプロメロース)、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、およびエチルセルロースを含む);デキストリン類(マルトデキストリンを含む);ゼイン;アルギン酸およびアルギン酸の塩、例えばアルギン酸ナトリウム;ケイ酸マグネシウムアルミニウム;ベントナイト;ポリエチレングリコール(PEG);ポリエチレンオキシド;グアーガム;多糖酸(polysaccharide acids);ポリビニルピロリジン(ポビドン)、例えばポビドンK−15、K−30およびK−29/32;ポリアクリル酸類(カーボマー類);ポリメタクリレート類;などを含む。1つまたはそれより多くの結合剤および/または接着剤は、存在する場合、典型的には総計で組成物の重量の約0.5%から約25%、例えば約0.75%から約15%、または約1%から約10%を構成する。
[0136]ポビドンは、錠剤製剤のための特に有用な結合剤であり、存在する場合、典型的には組成物の重量の約0.5%から約15%、例えば約1%から約10%、または約2%から約8%を構成する。
[0137]適切な崩壊剤は、個別にまたは組み合わせてのいずれかで、デンプン類(アルファ化デンプンおよびデンプングリコール酸ナトリウムを含む);粘土類;ケイ酸マグネシウムアルミニウム;セルロースを基材とする崩壊剤、例えば粉末セルロース、微結晶セルロース、メチルセルロース、低置換度のヒドロキシプロピルセルロース、カルメロース、カルメロースカルシウム、カルメロースナトリウム、およびクロスカルメロースナトリウム;アルギネート類;ポビドン;クロスポビドン;ポラクリリンカリウム;ガム類、例えば寒天、グアーガム、イナゴマメガム、カラヤガム、ペクチンガムおよびトラガカントガム;コロイド状二酸化ケイ素;などを含む。1つまたはそれより多くの崩壊剤は、存在する場合、典型的に総計では組成物の重量の約0.2%から約30%、例えば約0.2%から約10%、または約0.2%から約5%を構成する。
[0138]クロスカルメロースナトリウムおよびクロスポビドンは、個別にまたは組み合わせてのいずれかで、錠剤またはカプセルの製剤のためのとくに有用な崩壊剤であり、存在する場合、典型的には総計で組成物の重量の約0.2%から約10%、例えば約0.5%から約7%、または約1%から約5%を構成する。
[0139]湿潤剤は、存在する場合、水と密接に関連して、すなわち組成物のバイオアベイラビリティーを改善すると考えられる状態で、1つまたは複数の薬剤を維持するために通常は選択される。湿潤剤として使用することができる界面活性剤の非限定的例は、個別にまたは組み合わせてのいずれかで、第四級アンモニウム化合物、例えば塩化ベンザルコニウム、塩化ベンゼトニウム、および塩化セチルピリジニウム;ジオクチルナトリウムスルホスクシネート;ポリオキシエチレンアルキルフェニルエーテル類、例えばノンオキシノール9、ノンオキシノール10、およびオクトオキシノール9;ポロキサマー類(ポリオキシエチレンおよびポリオキシプロピレンのブロック共重合体);ポリオキシエチレン脂肪酸グリセリドおよび油類、例えばポリオキシエチレン(8)カプリル酸/カプリン酸のモノおよびジグリセリド、ポリオキシエチレン(35)カスター油およびポリオキシエチレン(40)水素化カスター油;ポリオキシエチレンアルキルエーテル類、例えばセテス−10、ラウレス−4、ラウレス−23、オレス−2、オレス−10、オレス−20、ステアレス−2、ステアレス−10、ステアレス−20、ステアレス−100、およびポリオキシエチレン(20)セトステアリルエーテル;ポリオキシエチレン脂肪酸エステル類、例えばポリオキシエチレン(20)ステアレート、ポリオキシエチレン(40)ステアレート、およびポリオキシエチレン(100)ステアレート;ソルビタンエステル類;ポリオキシエチレンソルビタンエステル類、例えばポリソルベート20およびポリソルベート80;ポリエチレングリコール脂肪酸エステル類、例えばラウリン酸プロピレングリコール;ラウリル硫酸ナトリウム;脂肪酸およびその塩、例えばオレイン酸、オレイン酸ナトリウム、およびオレイン酸トリエタノールアミン;グリセリル脂肪酸エステル、例えばモノオレイン酸グリセリン、モノステアリン酸グリセリン、およびパルミトステアリン酸グリセリン;ソルビタンエステル類、例えばモノラウリン酸ソルビタン、モノオレイン酸ソルビタン、モノパルミチン酸ソルビタン、およびモノステアリン酸ソルビタン;チロキサポール;などを含む。1つまたはそれより多くの湿潤剤は、存在する場合、典型的には総計で組成物の重量の約0.25%から約15%、好ましくは約0.4%から約10%、そしてより好ましくは約0.5%から約5%を構成する。
[0140]アニオンの界面活性剤である湿潤剤がとくに有用である。実例として、ラウリル硫酸ナトリウムは、存在する場合、典型的には組成物の重量の約0.25%から約7%、例えば約0.4%から約4%、または約0.5%から約2%を構成する。
[0141]滑剤は、錠剤製剤の圧縮時に錠剤混合物と錠剤成型装置間の摩擦を低減する。適切な滑剤として、個別にまたは組み合わせてのいずれかで、ベヘン酸グリセリン、ステアリン酸およびその塩(ステアリン酸マグネシウム、ステアリン酸カルシウム、およびステアリン酸ナトリウムを含む);水素化した植物油類;パルミトステアリン酸グリセリン;タルク;ワックス類;安息香酸ナトリウム;酢酸ナトリウム;フマル酸ナトリウム;フマル酸ステアリルナトリウム;PEG類(例えばPEG4000およびPEG6000);ポロキサマー類;ポリビニルアルコール;オレイン酸ナトリウム;ラウリル硫酸ナトリウム;ラウリル硫酸マグネシウム;などを含む。1つまたはそれより多くの滑剤は、存在する場合、典型的には総計で組成物の重量の約0.05%から約10%、例えば約0.1%から約8%、または約0.2%から約5%を構成する。ステアリン酸マグネシウムは、特に有用な滑剤である。
[0142]減粘剤は、成型装置表面への錠剤製剤の粘着を低減する。適切な減粘剤として、個別にまたは組み合わせてのいずれかで、タルク、コロイド状二酸化ケイ素、デンプン、DL−ロイシン、ラウリル硫酸ナトリウム、およびステアリン酸の金属塩を含む。1つまたはそれより多くの減粘剤は、存在する場合、典型的には総計で組成物の重量の約0.1%から約10%、例えば約0.1%から約5%、または約0.1%から約2%を構成する。
[0143]流動促進剤は、錠剤混合物中の流動特性を改善し、静電気を低減する。適切な流動促進剤として、個別にまたは組み合わせてのいずれかで、コロイド状二酸化ケイ素、デンプン、粉末セルロース、ラウリル硫酸ナトリウム、三ケイ酸マグネシウム、およびステアリン酸の金属塩を含む。1つまたはそれより多くの流動促進剤は、存在する場合、典型的には総計で組成物の重量の約0.1%から約10%、例えば約0.1%から約5%、または約0.1%から約2%を構成する。
[0144]タルクおよびコロイド状二酸化ケイ素は、個別にまたは組み合わせてのいずれかで、特に有用な減粘剤および流動促進剤である。
[0145]その他の賦形剤、例えばバッファー剤、安定剤、抗酸化剤、抗菌剤、着色剤、芳香剤、甘味剤も、医薬技術において公知であり、使用することができる。錠剤は、コーティングしないことも、あるいは例えば機能を持たないフィルム、または放出を修飾するもしくは腸溶性のコーティングでコーティングされたコアを包含することもできる。カプセルは、例えばゼラチンおよび/またはHPLCを、所望により1つまたはそれより多くの可塑剤と共に包含する、ハードまたはソフトの外殻を有することができる。
[0146]本明細書において有用な医薬組成物は、典型的には当該化合物またはその塩もしくはプロドラッグを、組成物の重量の約1%から約99%、より典型的には約5%から約90%、または約10%から約60%の量で含有する。ユニット投与剤形、例えば錠剤またはカプセルは、便利なように1回の用量を提供する量の化合物を含有することができるが、必要とされる用量が多い場合は、1回の用量として複数個の投与剤形を投与することが必要または望ましいと思われる。実例としてユニット投与剤形は化合物を、約10から約800mg、例えば約50から約750mgもしくは約100から約600mg;または特に具体的な例として、約50、約100、約150、約200、約250、約300、約350、約400、約450、約500、約550、約600、約650、約700または、約750mgの量で包含することができる。
[0147]本発明の1つの態様において、消化管の炎症性疾患、例えばIBDを有する被験者における炎症、またはそれに関連するもしくはその二次的な病理学的プロセスを低減または軽減するための方法を提供する。
[0148]もう1つの態様において、消化管の炎症性疾患、例えばIBDを有する被験者における粘膜の潰瘍の治癒を促進するための方法を提供する。
[0149]なおもう1つの態様において、被験者における消化管の炎症性疾患、例えばIBDの寛解を導入または維持するための方法を提供する。
[0150]これらの各態様に従って当該方法は、より完全には上に記載したように、治療有効量のACE2阻害薬を被験者に投与することを包含する。
[0151]なおもう1つの態様において、より完全には上に記載したように、ACE2阻害薬を約0.5から約5000mg/日の量で被験者に投与することを包含する、被験者における消化管の炎症性疾患、例えばIBDを治療するための方法を提供する。
[0152]文脈において別段の要求がない限り、“治療する”“治療すること”または“治療”という用語は本明細書において、消化管の炎症性疾患のリスクのある、または同疾患を含む予後を有する被験者における、薬剤、例えばACE2阻害薬の予防的または予後的使用を含み、同様に疾患もしくはその基底にある原因の1つまたはそれより多くの症状を軽減する、軽快する、強度を低減する、または排除するための治療法として、既にそのような疾患を経験している被験者におけるそのような薬剤の使用を含む。したがって治療は、(a)状態または疾患の素因があると思われるが、その状態または疾患がまだ診断されていない被験者に状態または疾患が起こらないように予防すること;(b)その発症を阻止することを含む、状態または疾患を阻害すること;および/または(c)疾患の寛解を促進、導入または維持することを含む、状態もしくは疾患、または一次的もしくは二次的なその兆候および症状を軽快、軽減または回復させること、を含む。
[0153]本発明の方法に従って、ACE2阻害薬、すなわちGL1001が、リコンビナントHeLaレポーター細胞においてTNFα誘発によるNF−κBの活性化を阻害することが、驚くことに発見された。この発見は、以下の実施例2にさらに詳細に報告する。レニン−アンジオテンシン系(RAS)におけるACE2阻害薬の効果は、上に指摘したように多様な炎症誘発効果において結びつけられている、アンジオテンシンIIのレベルの増加に関与する(図1参照のこと)と予測されると思われる。本発明者らはそのような予測に反して、ACE2阻害薬によって、炎症誘発サイトカインの合成の鍵となるメディエーターであるNF−κBの活性化が促進されないのではなく、阻害されることを発見した。
[0154]さらに驚くことにACE2阻害薬GL1001は、リコンビナントレポーターマウスにおいて、in vivoの基礎状態のNF−κB依存性転写を阻害することが発見された。この発見は以下の実施例3にさらに詳細に報告するが、RASにおけるACE2の役割に関する現在の理解に基づいた予測に反する、ACE阻害薬の抗炎症効果をさらに支持するようにみえる。
[0155]まださらに驚くことに、IBDに関するマウスモデル(デキストラン硫酸ナトリウム(DSS)マウスモデル)において、ACE阻害薬GL1001の投与は疾患の進行を遅らせることが発見された。この発見は、ヒトIBDにおけるACE2阻害薬の治療有効性を示唆する強力なエビデンスである。
[0156]まださらに驚くことに、消化管の組織におけるACE2 mRNAの発現が、慢性胃炎において特に強く上昇することが発見された。したがって、慢性胃炎におけるACE2の上昇は、この疾患の潜在的な病理学的因子であり、ACE2阻害薬、例えばGL1001の投与は、慢性胃炎の治療に有益であると考察される。
[0157]一部の態様において、被験者はクローン病(CD)を有する。CDは、活動期または寛解期であることができる。CDの活動性の程度は、あらゆる適切なスコアまたは指数を使用して定量化することができる。様々な指標について、例えば Naber & de Jong (2003) Neth. J. Med. 61(4):105-110により概説されている。
[0158]“活動指数”は本明細書において使用する場合、Best et al. (1976) Gastroenterology 70(3):439-444により開発されたクローン病活動指数(CDAI)として定義する。通常少なくとも約220の活動指数が活動期CDに関連する。
[0159]活動期CDを有する患者に対して、ACE2阻害薬は、活動指数の臨床的に有意義な低下を達成するために有効な、用量、頻度、および治療期間を含む投与計画に従って投与することができる。様々な態様において、活動指数の少なくとも約30ポイント、少なくとも約50ポイント、少なくとも約70ポイント、または少なくとも約90ポイントの低下が得られる。いくつかの態様に従ってこの低下は、活動指数を約220以下にする、またはCDの臨床的寛解を達成するために十分である。
[0160]いくつかの態様において、CDを有する被験者は瘻孔を生じるCDを有する可能性がある。そのような場合ACE2阻害薬を、例えば漏出している瘻孔数の減少を達成するために、または瘻孔の閉鎖を維持するために有効な、用量、頻度、および治療期間を含む投与計画に従って投与することができる。
[0161]いくつかの態様において、CDを有する被験者は小児患者である。
[0162]いくつかの態様において、被験者は潰瘍性大腸炎(UC)を有する。UCは、活動期または寛解期にあることができる。UCの活動性の程度は、Naber & de Jong (2003),上記記載、により参照されているようなメイヨー(Mayo)スコアを含む、この疾患に利用できるいずれかの指標を使用して定量化することができる。
[0163]本発明の方法は、例えば中程度から重症の活動期UCを有する被験者、典型的には少なくとも約6のメイヨースコアを示す被験者において、有用であることができる。そのような被験者に対して、ACE2阻害薬は、メイヨースコアの臨床的に有意義な低下を達成するために有効な、用量、頻度、および治療期間を含む投与計画に従って投与することができる。様々な態様において、メイヨースコアにおける少なくとも2ポイントの低下、少なくとも3ポイント、少なくとも4ポイント、もしくは少なくとも5ポイントの低下;または少なくとも20%、少なくとも30%、少なくとも40%、または少なくとも50%の低下が得られる。1つの態様において、少なくとも約30%および少なくとも約3ポイントの低下が得られる。いくつかの態様に従ってのこの低下は、メイヨースコアを約6以下にする、またはUCの臨床的寛解を達成するために十分である。
[0164]UCを有する被験者は、潰瘍性直腸炎、左側結腸炎、汎大腸炎、および劇症型大腸炎を含む、UCの公知のバリアントまたはタイプのいずれかを有し得る。劇症型大腸炎の患者において、本発明の方法に従っての治療は、重症の合併症、例えば大腸破裂および中毒性巨大結腸症のリスクを低減することができる。
[0165]本発明の方法はまた、非活動期または寛解期にあるIBD(CDまたはUCのいずれか)を有する被験者においても有用である。そのような被験者に対して、ACE2阻害薬は、非活動期または寛解期の延長を達成するために有効な、用量、頻度、および治療期間を含む投与計画に従って投与することができる。
[0166]いくつかの態様においてACE2阻害薬の投与は、IBDの少なくとも1つの兆候または症状の軽減に関連する、または帰着する。軽減することのできる兆候または症状の例として非限定的に、下痢(脱水およびショックさえきたすに十分なほど重症となり得る)、軟便、腹痛(中程度から重症である可能性があり、そして悪心および/または嘔吐を伴い得る)、腹部痙攣、直腸痛、しぶり、直腸からの出血、血便(より重症度の低いケースでは潜血を含む)、食欲低下、体重減少、およびそれらの組み合わせを含む。二次的症状もまた軽減することができ、これには、発熱、寝汗、疲労感、および消化管を超えて、例えば関節および/または皮膚にまで拡大する炎症を含む。
[0167]より特定の態様において、下痢、直腸からの出血、体重減少、およびそれらの組み合わせより選択される、少なくとも1つの兆候または症状が軽減される。
[0168]様々な態様において被験者は、アミノサリチル酸、コルチコステロイド、免疫抑制薬、抗生剤、およびそれらの組み合わせから成る群より選択される、少なくとも1つの基本的な薬剤の上限用量の投与を包含する基本的な療法に対して不応性である、または不応性になったIBD(CDまたはUCのいずれか)を有する。被験者が不応性である基本的療法は、第一選択療法または第二選択療法を包含することが出来る。
[0169]理論に拘束されるものではないが、ACE阻害薬は基本的な薬剤の作用機序とは異なるIBDにおける作用機序を有すると考えられる。不応性IBDの治療におけるACE2阻害薬の有用性は、この異なる作用機序をある程度反映すると思われるが、それに関する予測は行っていない。
[0170]不応性IBDの被験者において、ACE2阻害薬は単独療法で、または基本的な療法もしくはその一部分と併用して投与することができる。1つの態様において、例えばACE2阻害薬は、少なくとも最初は基本的療法と併用して投与する。もう1つの態様においてACE2阻害薬は、少なくとも上限より少ない用量で投与されている少なくとも1つの基本的な薬剤と併用して投与する。なおもう1つの態様においてACE2阻害薬は、用量、頻度、および治療期間を含む投与計画に従って、少なくとも1つの基本的な薬剤と併用して投与し、その場合IBDの臨床的寛解に達した時点で、少なくとも1つの基本的な薬剤を中止する。少なくとも1つの基本的な薬剤の中止は一度に実行することもできるが、より典型的には、漸減によるまたは段階的な用量低減により一定期間にわたって実行する。
[0171]少なくとも1つの基本的な薬剤がコルチコステロイドを包含する場合、そのような薬剤の長期的な使用に伴い得る有害な副作用のため、例えば漸減しての用量低減による中止が、しばしば殊に望ましい。
[0172]もう1つの態様においてACE2阻害薬は、アミノサリチル酸を包含する第一選択療法に対して不応性である、または不応性になった被験者に投与する、したがってそのような投与はコルチコステロイドに代わるものである。したがって治療有効量のACE2阻害薬を、所望によりアミノサリチル酸との併用療法において、ただしコルチコステロイドは使用せずに、投与することを包含する、アミノサリチル酸不応性IBDを有する被験者における、コルチコステロイド療法を回避するための方法を提供する。コルチコステロイドの回避は、コルチコステロイドに対する有害な反応の病歴のある被験者、またはそのような有害な反応の素因となるリスク因子を有する被験者において、特に重要である。
[0173]疾患が他の薬剤に対して不応性であるかどうかにかかわりなく、ACE阻害薬は、1つまたはそれより多くの付加的な薬剤、例えばIBDに関連する兆候、症状、基底にある原因、寄与因子、または二次的状態に対応する薬剤、との共同療法にて投与することができる。
[0174]“治療の組み合わせ”という用語は、本明細書において、一緒にまたは別々に被験者に投与した時に、被験者に治療の有益性をもたらす上で共同活性的な複数の薬剤をいう。そのような投与を、“組み合わせ療法”、“共同療法”“併用療法”、または“付加的療法”という。例えば1つの薬剤はもう1つの薬剤の治療効果を強化もしくは増強する、またはもう1つの薬剤の有害な副作用を低減することができる、あるいは1つもしくはそれより多くの薬剤は、単独で使用する場合より低用量で効果的に投与することができる、または単独で使用する場合より大きな治療の有益性を提供することができる、または疾患もしくは状態の異なる側面、症状もしくは病原学的因子に相補的に対応することができる。
[0175]例えばACE2阻害薬は、アミノサリチル酸、コルチコステロイド、免疫抑制薬、抗TNFα薬、およびそれらの組み合わせより選択される少なくとも1つの付加的な薬剤、との組み合わせ療法または併用療法にて投与することができる。
[0176]アミノサリチル酸の非限定的例は、バルサラジド、メサラミン、オルサラジン、スルファサラジン、医薬的に受容可能なそれらの塩、およびそれらの組み合わせを含む。
[0177]コルチコステロイドの非限定的例は、べクロメタゾン、べクロメタゾンジプロピオネート、ブデソニド、デキサメタゾン、フルチカゾン、ヒドロコルチゾン、メチルプレドニゾロン、プレドニゾン、プレドニゾロン、プレドニゾロン−21−メタスルホベンゾエート、チキソコルトール、医薬的に受容可能なそれらの塩、およびそれらの組み合わせを含む。
[0178]免疫抑制薬の非限定的例は、アザチオプリン、シクロスポリン(例えばシクロスポリンA)、メルカプトプリン、メトトレキセート、タクロリムス、医薬的に受容可能なそれらの塩、およびそれらの組み合わせを含む。
[0179]1つの態様においてACE2阻害薬は、抗TNFα薬、例えばインフリキシマブとの組み合わせ療法または併用療法にて投与する。
[0180]組み合わせ療法または併用療法において投与される2つまたはそれより多くの活性な薬剤は、被験者に同時に投与するための1つの医薬的調製物(1つの投与剤形)中に、または被験者に同時にもしくは異なる時間に、例えば逐次的に投与するための、2つもしくはそれより多くの別個の調製物(別々の投与剤形)中に製剤化することができる。2つの別個の調製物は、同じ投与経路または異なる投与経路による投与用に製剤化することができる。
[0181]別々の投与剤形として、所望により、例えば1つの外装内の1つの容器もしくは複数の容器の中に合わせて包装する、また別々の包装で合わせて提示する(“一般的な提示”)ことができる。合わせての包装、または一般的な提示の一例として、第一の容器にACE2阻害薬、そして第二の容器に付加的な薬剤、例えば上述の薬剤のいずれか、を包含するキットを意図する。もう1つの例においてACE2阻害薬および付加的な薬剤は別々に包装し、互いに独立した販売を利用することができる、ただし本発明に従っての使用のために合わせて市販するか、または合わせて販売促進する。別々の投与剤形をまた、本発明に従っての使用のために、別々にそして独立して被験者に提示してもよい。
[0182]投与剤形は同一でも異なっていてもよく、例えば速放性の投与剤形、コントロールされた放出の投与剤形、またはデポー剤形といったその投与剤形に依存して、ACE2阻害薬および付加的な薬剤は、同一のまたは異なるスケジュールで、例えば毎日、毎週、または毎月を基準として投与してよい。
[0183]1つの態様において本発明は、ACE2阻害薬、ならびにアミノサリチル酸、コルチコステロイド、免疫抑制薬、抗TNFα薬、およびそれらの組み合わせより選択される少なくとも1つの付加的な薬剤を包含する治療の組み合わせを提供する。そのような付加的な薬剤の具体的な例は、実例として上に列記している。
実施例1:健常な状態および疾患の状態におけるACE2 mRNAの発現。
[0184] Donoghue et al. (2000),上記記載、は、検討した23名の正常なヒト組織から、ACE2転写物を主として心臓、腎臓および精巣中に、そしてACE2タンパク質を(免疫組織化学により)主に冠動脈および腎臓内血管の内皮、および腎臓の尿細管上皮中に発見したことを報告した。
[0185]さらにTipnis et al. (2000) J. Biol. Chem. 275(43):33238-33243 は、ノーザンブロット分析で、ACE2 mRNA転写物が精巣、腎臓、および心臓において最も高度に発現されていることを示した、と報告した。
[0186] Komatsu et al. (2002) DNA Seq.13:217-220 は、マウスのアンジオテンシン変換酵素関連カルボキシペプチダーゼ(mACE2)の分子のクローニングが、ヒトACE2との83%の同一性を示し、ノーザンブロット分析で、転写物が主として腎臓および肺で発現されていることを示した、と報告した。
[0187]より最近Gembardt et al. (2005) Peptides 26:1270-1277は、マウスおよびラットの様々な正常組織におけるACE2 mRNAおよびACE2タンパク質の発現を分析し、双方の種の検査したすべての臓器(心室、腎臓、肺、肝臓、精巣、胆嚢、前脳、脾臓、胸腺、胃、回腸、結腸、脳幹、心房、および脂肪組織)における、少なくとも検出可能なレベルのACE2 mRNAを報告した。双方の種において回腸組織がACE2 mRNAの最も高い発現を示したが、マウスは、この臓器ならびに腎臓および結腸でもACE2 mRNAの発現においてラットを上回っていた。
[0188] Burrel et al. (2005) Eur. Heart J. 26:369-375は最近、心筋梗塞は、ラットおよびヒトの心臓においてACE2の発現を増大する、と報告した。
[0189]今日ACE2 mRNAの発現は、Gene Logic Inc.のBioExpress(登録商標)システムを使用して、健常な被験者および病気の被験者に由来する様々なヒト組織において検討されている。このシステムは、正常なサンプル、および約435の疾患状態由来の疾患サンプルの双方を包含する、約18,000のサンプル(そのうち約90%はヒト組織由来である)由来のmRNA発現データを含む。手短に言えば、手術の生検または死亡後摘出のいずれかに由来するヒト組織サンプルが、Affymetrix GeneChips(登録商標)を使用してmRNA発現プロフィール分析用に処理された。各組織サンプルは有資格の病理学者により検討されて、病理学的診断が確認された。RNAの単離、cDNAの合成、cRNAの増幅および標識化、ハイブリダイゼーション、ならびにシグナルの標準化は、標準的なAffymetrixプロトコルを使用して行われた。コンピューター分析は、Genesis Enterprise System(登録商標)ソフトウェア、およびAscenta(登録商標)ソフトウェアシステム (Gene Logic Inc)を使用して施行された。
[0190]Donoghue et al. (2000),上記記載、および Tipnis et al. (2000), 上記記載、と一致して、本研究は、正常なヒトの心臓、腎臓および精巣において、相対的に高いレベルのACE2転写物を示した(データは示していない)。しかしこれら3つの正常な組織を除き、検討した70の付加的な正常なヒト組織の中で8つの最も高い発現レベルのACE2 mRNAを、以下の表1に平均発現レベルの高い方から順に列記する(“平均相対レベル”、すなわち任意の単位でシグナルレベルを設定し、検査したすべてのサンプルのうちで最も低いシグナルレベルに対して標準化し、2つの異なるプローブのフラグメントについて平均して得た)。
[0191]表1のこれらの最も高い8つの正常組織(ならびに心臓、腎臓および精巣も同様に)は、4.0より高いACE2 mRNA発現の平均相対レベルを示し、一方検討した残りの62の正常組織は、4.0より低い平均相対レベルを示した。
[0192]表1はまた、正常ヒト組織の中で5つの最も高い発現レベルのACE2 mRNAの内4つ(心臓、腎臓および精巣以外)は、胃腸管の構成要素、すなわち(発現レベルの高い方から順に):十二指腸、小腸、結腸および胃であったことを示す。
[0193]BioExpress(登録商標)システムにより包括的に含まれる疾患の状態におけるACE2 mRNA発現の検討では、いくつかの状態のみ、主として胃腸管の構成要素の炎症状態において、ACE2 mRNAの増加を示した。したがって表2は、ACE2 mRNA発現が、(正常値に対する変化の倍数表示の平均値の高い方から順に)炎症状態の胃(慢性胃炎)、大唾液腺(耳下腺を除く)(慢性唾液腺炎)、および結腸(クローン病、活動期(慢性または急性の炎症))において上昇していたことを示す。反対に、活動期潰瘍性大腸炎(慢性または急性の炎症)、および活動期クローン病(慢性または急性の炎症)の小腸におけるACE2 mRNAのレベルは、表1に示した対応する正常組織でのすでに有意なレベルからは実質的に変化していなかった。
[0194]上の知見を合わせると、正常ヒト組織で見出された11の最も高い発現レベルのACE2 mRNAの内4つが消化管の構成成分であり、増加したACE2 mRNAの発現に関与する、検討した疾患の状態の大半が、炎症状態の消化管であることを示す。故にこれらの知見は、高レベルのACE2 mRNA発現は病理学的因子である可能性があり、そしてそれ故、ACE2活性の低減は、少なくともいくつかの炎症状態の消化管、特に胃(慢性胃炎)、大唾液腺(慢性唾液腺炎)、および結腸(慢性または急性の炎症を伴うクローン病)において、治療の有益性を提供し得ることを示唆する。さらにACE2 mRNAレベルは、潰瘍性大腸炎の結腸、またはクローン病の小腸では上昇していないが、正常な結腸および小腸でのそのようなmRNAのすでに実質的なレベルが、少なくともACE2活性が存在しており、そしてそれ故これら2つの疾患組織においてもなお病理学的因子を構成し得ることを示唆する。
実施例2:リコンビナントHeLaレポーター細胞におけるTNFα誘発によるNF−κB活性化の、GL1001による阻害
[0195]ヒトIBDおよびIBDのげっ歯類モデルの双方において、炎症は少なくとも部分的には、NF-κBファミリーメンバーの活性化および核移行に依存する可能性が高い。例えばFichtner-Feigl et al. (2005) J. Clin. Invest. 115:3057-3071、および同文献に引用された出典を参照のこと。したがってIL−12および/またはIL−23に依存するTh1を介した炎症において、これらのサイトカインの合成はNF-κB転写因子によって制御される。IL−4またはIL−13に依存するTh2を介した炎症においては、IL−12およびIL−23のそれより間接的ではあるが、これらのサイトカインの合成もまたNF-κB転写因子に依存する。したがってIBDの炎症を治療する1つの方法は、NF-κB活性を阻害する薬剤を投与することである可能性があり、そして事実Fichtner-Feigl et al. (2005)、上記記載、は、NF-κBによる遺伝子発現の活性化を防ぐNF-κBデコイのオリゴデオキシヌクレオチド(ODN)が、急性トリニトロベンゼンスルホン酸(TNBS)誘発結腸炎(臨床コースおよびTh1サイトカインの産生における効果により評価されるように);慢性TNBS誘発結腸炎(IL−23/IL-17の産生および線維症の発症を阻害する);およびオキサゾロン誘発結腸炎(Th2を介した炎症プロセス)を含む、マウスにおけるTh1およびTh2を介したIBDの様々なモデルを治療および予防する上で有効であることを示した。
[0195]ヒトIBDおよびIBDのげっ歯類モデルの双方において、炎症は少なくとも部分的には、NF-κBファミリーメンバーの活性化および核移行に依存する可能性が高い。例えばFichtner-Feigl et al. (2005) J. Clin. Invest. 115:3057-3071、および同文献に引用された出典を参照のこと。したがってIL−12および/またはIL−23に依存するTh1を介した炎症において、これらのサイトカインの合成はNF-κB転写因子によって制御される。IL−4またはIL−13に依存するTh2を介した炎症においては、IL−12およびIL−23のそれより間接的ではあるが、これらのサイトカインの合成もまたNF-κB転写因子に依存する。したがってIBDの炎症を治療する1つの方法は、NF-κB活性を阻害する薬剤を投与することである可能性があり、そして事実Fichtner-Feigl et al. (2005)、上記記載、は、NF-κBによる遺伝子発現の活性化を防ぐNF-κBデコイのオリゴデオキシヌクレオチド(ODN)が、急性トリニトロベンゼンスルホン酸(TNBS)誘発結腸炎(臨床コースおよびTh1サイトカインの産生における効果により評価されるように);慢性TNBS誘発結腸炎(IL−23/IL-17の産生および線維症の発症を阻害する);およびオキサゾロン誘発結腸炎(Th2を介した炎症プロセス)を含む、マウスにおけるTh1およびTh2を介したIBDの様々なモデルを治療および予防する上で有効であることを示した。
[0196]IBDに関する抗炎症活性についてACE2阻害薬GL1001を検査するため、TNF-αによるNF-κB依存性転写の活性化における化合物の効果を、NF-κBに依存する制御配列のコントロール下で、ルシフェラーゼレポーター遺伝子を含むコンストラクトを含有するリコンビナントレポーター細胞において検討し、それにより、生成される光の検出に基づいた従来のルシフェラーゼ活性アッセイを使用してレポーター酵素を測定することにより、NF-κB依存性転写の検出を可能にした。
[0197]特にHeLa細胞(American Type Culture Collection)は、以下のように(別に指摘していなければすべてのインキュベーションステップは37℃にて)、10%ウシ胎児血清を補充したダルベッコ変法イーグル培地(DMEM)中で成長させ、そして一過的にNF-κB−lucコンストラクト(Stratagene, Inc.)を形質移入した。細胞は10cm細胞培養ディシュ内に播種し、約70%の培養密度まで成長させた。プラスミドDNA(10μg)を、試験管内の1mlの血清不含DMEM培地に加えた。次に図6のトランスフェクション試薬(30μl)(Roche)を試験管内にゆっくりピペットで加え、内容物を反転させて緩やかに混合した。混合物を室温で15分間インキュベーションした後、10cmディッシュ内の細胞に滴下させながら加えた。24時間のインキュベーション後、トリプシン−EDTA(Gibco-BRL)を用いて細胞をプレートからはがし、1ウェル当たり100μlの血清不含DMEMを含む、透明底面の白色96ウェル検査プレート(Fisher)のウェルに、1ウェル当たり3×104細胞の密度で移し、一晩放置して細胞を付着させた。次に化合物(GL1001)を、およそ0、0.008、0.04、0.2、1.0または5.0μMの濃度でウェルに加え、続いて直ちにTNFα(R&D)を最終濃度20ng/mlにて加えた。6時間のインキュベーション後、100μlのBright-Glo ルシフェラーゼバッファ-(Promega, Cat# E2610)を加え、室温で、プレートを軽く振盪させながら、10分間インキュベーションした。その後生物発光を、Veritas ルミノメーター(Turner BioSystems)を使用して測定した。プロットした各データの値は、4つの独立したウェルの生物発光の平均値を表す。
[0198]図2に示したように、GL1001は、TNF-α誘発によるNF-κB依存性転写の活性化を、検査したすべての濃度で有意に阻害し、8nMで80%を超える阻害、そして0.2μMで95%を超える最大の阻害を示した。これらの結果は、ACE2阻害薬GL1001がIBDに関連する強力な抗炎症活性を有すること、すなわち炎症性サイトカインTNF-αによるNF-κBシグナル伝達経路の活性化を阻害することを示す。本発明者らは、あらゆるACE2阻害薬に関する、そのような抗炎症活性のいかなる報告もこれまで耳にしていない。
実施例3:リコンビナントレポーターマウスにおけるin vivo の基礎状態のNF-κB依存性転写の、GL1001による阻害
[0199]ルシフェラーゼ遺伝子に連結したNF-κBエンハンサーを含有するコンストラクトを用いて、このNF-κBレポーターコンストラクトがマウスの全細胞に存在するように、生殖系列に遺伝子操作を行ったマウス(すなわち、NF-κB::Lucマウス)において、NF-κB依存性転写の基礎状態レベルにおけるGL1001の効果を検討することにより、in vivo の抗炎症活性について、GL1001をさらに検査した。
[0199]ルシフェラーゼ遺伝子に連結したNF-κBエンハンサーを含有するコンストラクトを用いて、このNF-κBレポーターコンストラクトがマウスの全細胞に存在するように、生殖系列に遺伝子操作を行ったマウス(すなわち、NF-κB::Lucマウス)において、NF-κB依存性転写の基礎状態レベルにおけるGL1001の効果を検討することにより、in vivo の抗炎症活性について、GL1001をさらに検査した。
[0200]より特定すれば、遺伝子導入NF-κB::Lucマウスは、Carlsen et al. (2002) J. Immunol. 168:1441-1446に記載されているように、ホタルルシフェラーゼ遺伝子に融合させたIgκ軽鎖プロモーター由来の3つのNF-κB応答エレメントを使用して作成した。精製したコンストラクトDNAの前核へのマイクロインジェクションを使用して、C57BL/6 XCBA/Jバックグラウンドの遺伝子導入創始動物を作成した。創始動物は引き続きC57BL/6白色種バックグラウンドに戻し交配を行った。すべての実験プロトコルは、研究機関の動物管理使用委員会(Institutional Animal Care and Use Committee)による承認を得ており、実験動物の管理および使用に関するILAR指針に従うものである。in vivoイメージングのため、NF-κB::Lucマウスはイメージングの10分前にルシフェリン(150mg/kg)を腹腔内に注射し、麻酔(1−3%イソフルラン使用)し、遮光性のカメラボックス内に置いた。マウスは、20cmの視野角の高解像度の設定で、背側または腹側から2分間までイメージングを行った。導入遺伝子から放射される光を、IVIS(登録商標)イメージングシステム200シリーズ (Xenogen Corporation, Alameda, CA)により検出し、デジタル化し、モニターにディスプレイした。Living Image(登録商標)ソフトウェア(Xenogen Corporation, Alameda, CA; see Rice et al. (2002) J. Biomed. Opt. 6:432-440)は、カメラからのデータを、シグナル強度の変動を色で表す疑似カラースケールを使用してディスプレイする。シグナルデータはまた、Living Image(登録商標)ソフトウェアを使用して定量化し記録した。光の光子は、以下にさらに記載するように、方法に依存してサイズを変化させる楕円形の関心領域(ROI)を使用して定量した。
[0201]ルシフェラーゼアッセイ用に、臓器を抽出し、液体窒素内で急速冷凍した。すべての組織サンプルは、阻害剤を含む溶解バッファー(Passive Lysis Buffer (Promega) およびComplete Mini Protease Inhibitor Cocktail (Roche, Indianapolis, IN))中に入れ、組織用ホモジェナイザー(Handishear, Hand-held homogenizer, VirTis, Gardiner, NY)を使用して破砕した。組織ホモジェネートを遠心し、濁りをとった溶菌液を、ルミノメーターアッセイおよびウェスタンブロットに使用した。ルミノメーターアッセイ用には、ルシフェラーゼアッセイ基質(Luciferase Assay Substrate 、Luciferase Assay System, Promega)を、製造元の指示どおりに調製し、ディスポーザブルのキュベット内に入れた。組織ホモジェネート(20μl)および基質(100μl)を混合し、Veritas Microplateルミノメーター (Turner Designs, Sunnyvale, CA)にて、パラメータをdelay(遅延時間)2秒の10秒として測定した。バックグラウンドの発光の読み値を得、発光データからバックグラウンドの読み値を差し引いた。タンパク質濃度は、製造元のプロトコルに従って、BCA タンパク質アッセイキット(BCA Protein Assay Kit 、Pierce, Rockford, IL)を使用して決定し、VERSAmax可変波長マイクロプレートリーダー、およびそれに連結したSoftmax Pro バージョン3.1.2 ソフトウェア(Molecular Devices, Sunnyvale CA)を使用して分析した。各タンパク質溶解液の発光値は、タンパク質1マイクログラム当たりの光の任意の単位として算出した。統計分析は、治療群間のMEAN、SEMおよびANOVAおよびスチューデントt−検定を含む。
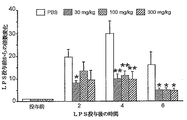
[0202]NF−κB依存性転写の基礎状態レベルにおけるGL1001のin vivoの効果を検査するため、オスNF−κB::Lucマウスに、生理食塩水中0、3、30、または100mg/kg GL1001の皮下投与直前、ならびに投与後2、4および6時間に、上に記載したように(2.76×3.7cmの固定されたROIを使用して)腹部領域の定量的in vivoイメージングを行った。全身のイメージングは、GL1001が主として腹部領域において、ルシフェラーゼレポーター遺伝子のNF−κB依存性転写の基礎状態のin vivoレベルを、有意に阻害することを示した。図3の定量的イメージングデータにより示したように、LPS投与後4時間にGL1001は、選択された腹部ROIにおけるNF−κB依存性転写の基礎状態のin vivoレベルを、300mg/kgで40%を超えてまで阻害し(p<0.01、ANOVAおよびスチューデントt−検定による)、2つのより低用量ではより低いがまだ有意な程度に阻害した。
[0203]NF−κB::Lucマウスで観察された結果とは反対に、本研究のNF−κB::Lucマウスと同様にコンストラクトしたAP−1::Lucマウスでは、レポータールシフェラーゼ発現の基礎状態のin vivoレベルにおいて、GL1001の有意な効果は観察されなかった(データは示していない)。AP−1::Lucマウスではレポーターの転写は、細胞増殖および腫瘍促進に関与すると考えられている公知の癌原遺伝子である、アクチベータータンパク質1(AP−1)に応答するエンハンサーエレメントにより推し進められる。
実施例4:GL1001は、リコンビナントレポーターマウスにおけるin vivoのLPS誘発によるNF−κB依存性転写を阻害する。
[0204]グラム陰性菌の細胞壁の主要成分である細菌リポ多糖(LPS)は、マクロファージのTNFα産生および放出を刺激する、非常に生物学的に活性な分子である。例えばJersmann et al. (2001) Infection and Immunity 69(3):1273-1279、および同文献に引用されている出典を参照のこと。細菌感染と心血管イベントとの認識されている関連性の1つは、内皮の活性化および接着分子のアップレギュレーションである。心血管イベントの因果関係に結びつけられる2つの主要な炎症誘発メディエーターである、細菌LPSおよびTNFαは、NF−κBおよびp38マイトジェン活性化タンパク質キナーゼのシグナル伝達経路の活性化を通して、ヒト内皮接着分子の発現を相乗的に増大させることにより、内皮細胞の接着特性を共同して上昇させることが発見された。
[0205]GL1001はさらに、NF−κB::Lucマウスにおいて、細菌LPS誘発によるNF−κB依存性転写におけるその効果を検討することにより、in vivoの抗炎症活性について検査した。特に炎症は、これらの6−10週齢マウスにおいて、GL1001投与後1時間での0.5mg/kg(静注)の可溶性LPS(sLPS; Sigma)の投与により誘発した。マウスには、上に記載したようにLPS投与後2、4および6時間に、定量的な腹部イメージングを行った。検証実験では、ルシフェラーゼシグナルの最大の変動を示したタイムポイントで、動物を安楽死させ、組織を採集し、さらなる分析のため保存した。ルシフェラーゼシグナルは、興味あるいくつかの領域から定量した。統計分析は、治療群間のMEAN、SEMおよびANOVAおよびスチューデントt−検定を含む。
[0206]全身のイメージングは、これもまた第一に腹部領域において、LPS誘発によるin vivoでのルシフェラーゼレポーター遺伝子のNF−κB依存性転写レベルを、有意に阻害することを示した。図4の定量的イメージングデータにより示したように、LPSは、コントロールマウスにおいて強いNF−κB依存性ルシフェラーゼシグナルを誘発し、予想された様に強いNF−κBシグナル伝達応答を示した。反対にGL1001で前処置したマウスは、腹部領域において定量的に測定することのできた、有意に低減したLPS誘発によるNF−κBシグナル伝達応答を示した。NF−κB依存性ルシフェラーゼ活性の阻害が、本実験ではGL1001の全用量範囲(30mg/kg、100mg/kg、300mg/kg)で観察されたため、わずかにより低用量の範囲(3−100mg/kg)を使用して、この実験を繰り返し行った。図5に示したように、このより低用量の範囲ではGL1001は、30mg/kgおよび100mg/kgで、LPS誘発によるNF−κBシグナル伝達を有意に低減した。これらの結果は、ACE2阻害薬GL1001の全身(皮下)投与が、主として腹部領域において、細菌LPS誘発によるNF−κB依存性転写、ならびに基礎状態のNF−κB依存性転写に対して有意なin vivoの抗炎症活性を示したことを示す。
[0207]0.5mg/kgのLPSおよび30mg/kgのGL1001、または0.5mg/kgのLPS単独で処置したNF−κB::Lucマウスから抽出された選択された臓器の検討(図6)は、LPS単独で処置したマウスと比較して、GL1001処置マウスの胃においてLPS誘発によるNF−κB依存性転写の有意な(約37倍)低減を示したが、膵臓および子宮、または分析を行ったその他のいかなる臓器または臓器部分でも、すなわち肝臓、腎臓、脾臓、小腸、大腸(結腸)、腸間リンパ節、盲腸(小腸の後の回腸の最初の部分)、卵巣、子宮、顎下リンパ節、脳、心臓、および肺においては、LPS誘発によるNF−κBシグナル伝達の統計的に有意な低下は認めらなかった(データは示してない)。
[0208]マウス胃におけるLPS誘発によるNF−κB活性のGL1001による阻害は、ヒト被験者の正常な胃組織におけるACE2 mRNA発現に関する本発明の観察(上記)、およびGembardt et al. (2005)、上記記載、によるマウス胃におけるACE2 mRNA発現の報告と一致する。高レベルのACE2 mRNAを発現することがこれまでに報告されているその他のげっ歯類の組織(例えば腎臓、小腸または結腸;Gembardt et al. (2005)、上記記載、を参照のこと)においては、LPS誘発によるNF−κB活性の阻害が観察されなかったという事実は、GL1001の全身(皮下)投与に続発する主に腹部領域におけるLPS誘発によるNF−κBシグナル伝達への阻害効果が、胃におけるこのACE2阻害薬の何らかの活性に、何もよりもまずよるものであることを示す。
実施例5:GL1001は、デキストラン硫酸ナトリウム(DSS)によりマウスにおいて誘発されるIBDを阻害する。
[0209]GL1001はさらに、マウスのデキストラン硫酸ナトリウム(DSS)誘発結腸炎におけるその効果を検討することにより、in vivoの抗炎症活性について検査した。このIBDモデルは、潰瘍性大腸炎の患者に認められるものと非常に類似する、再現性ある形態的変化を示す。例えばHollenbach et al. (2004) FASEB J. 18(13):1550-1552を参照のこと。またBryne et al. (2006), Current Opinion in Drug Discovery & Development 8(2):207-217、および同文献に引用されている出典も参照のこと。これらの病理には、双方の生物学的系における、主に左側結腸の炎症、結腸癌をきたす異形成を伴う結腸粘膜細胞の顕著な再生、大腸の短小化、限局的な陰窩の損傷、および高頻度のリンパ系の過形成を含む。さらに、Hollenbach et al. (2004)、上記記載、によれば、マウスのDSS誘発結腸炎は、結腸炎の治療に一般的に使用される治療薬の有効性を評価する上で高い価値を有する。というのはヒトIBDにおいて治療上有益な物質はすべて、このマウスモデルの疾患の活動もまた低減することが示されたからである。
[0210]本研究は、3群:コントロール(マウス5頭)、2.5%DSS単独(マウス10頭)、およびGL1001処置(1日当たり100mg/kg、皮下)を伴う2.5%DSS(マウス10頭)にてデザインした。NF−κB::Lucマウスを使用して、上に記載したように炎症活性の指標としてNF−κB活性化を測定した。体重、液体摂取、便潜血、臓器の重量、および好中球浸潤(WPOアッセイ)に加えて、特に臓器特異的なルシフェラーゼ活性を測定した。6−8週齢のNF−κB::luc BL/6白色種バックグラウンドのマウスに、飲用水中の2.5%デキストラン硫酸ナトリウム(DSS、MW 40,000;MP Biomedicals)を与えた。マウスは体重測定およびイメージングを行い、GL1001を毎日投与した。便サンプルは、各治療群のケージの底から採集し、便性および潜血を、製造元(Fisher Scientific)に指示どおりにHemocult テープを使用して検査し、液体消費量を測定した。本研究の最後に、GI管を摘出し、様々な切片を清浄にして重量測定し、組織サンプルを、生物発光アッセイ、および好中球浸潤を見るためのミエロペルオキシダーゼ(Myeloperoxidaseアッセイキット、 Cytostore)用に調製した。
[0211]マウスは、毎日のGL1001またはビヒクルコントロールの投与時に、体重測定およびイメージングを行った。マウスのバイオフォトニック画像を、上に記載したように毎日得、図7に示した定量的な腹部イメージングの結果を得た。この実験において、DSS処置を受けた双方の群でNF−κBに推し進められるルシフェラーゼ発現に最初の減少が見られたが、実験開始から研究第6日までを通して、DSS単独群、およびDSS+100mg/kg GL1001群との間に維持されたルシフェラーゼの発現において、統計学的な有意差はなかった。水の消費はすべての動物についてモニタリングし、類似の消費率から、DSS処置マウスはすべて類似する量のDSSを受容していたことを示す(図8)。
[0212]炎症性腸疾患の進行は、3項目に分けた体重減少のパーセント、便性、および便潜血の合計から成る炎症性腸疾患の活動指数を使用してモニタリングした。表3は、測定した各パラメータのランキングシステムを示す。
[0213]図9にプロットした炎症性腸疾患の活動指数の結果として示したように、本研究の第3日から8日の間に、GL1001群において疾患の活動性のわずかな遅延が認められた。体重減少は、DSSのみの処置群に比してGL1001投与群において、第4日から9日の間に有意に遅延された(図10)。
[0214]本研究の最後に、胃腸管の選択された臓器を摘出、清浄、および重量測定し、最終体重に対する臓器の重量の比率を決定した。図11に示したように、DSS誘発による臓器の有意な重量増加が観察され、そして盲腸および大腸(結腸)の双方ではGL1001により重量増加を完全に防ぐことができたが、胃または小腸では防ぐことはできなかった。
[0215]加えて、胃腸管の切片、同様にコントロールとして肝臓および腎臓の切片をホモジェナイズし、図12において、ルシフェラーゼ発現をタンパク質1μg当たりの光の単位として記録した。DSS単独群では、ルシフェラーゼ発現の増加を示す臓器は、盲腸と大腸であった。GL1001処置群は、水のみを与えたコントロール群のレベルに類似するルシフェラーゼ発現レベルを示した。
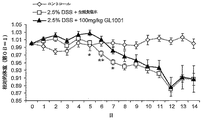
[0216]まとめると、疾患に関するパラメータのすべてのアッセイが、DSS群およびDSS+GL1001処置群間の有意差または対応する傾向のいずれかを示したことから、ACE2阻害薬GL1001は、マウスにおけるデキストラン硫酸ナトリウム(DSS)誘発結腸炎において、in vivoの抗炎症活性を示すことが示された。GL1001の全身(皮下)投与が、盲腸および大腸の残り部分(結腸)において、臓器の重量を低減させ、DSS誘発によるNF−κBシグナル伝達を低減したという事実は、このACE2阻害薬が、ヒトIBDの双方の形、すなわちUCおよびCDに関連する胃腸管の部分において抗炎症活性を有すること、そしてそのような活性に加えて、胃における基礎状態、およびLPS誘発によるNF−κBシグナル伝達に対しても活性を有することを示す。
[0217]加えてGL1001は、例えば炎症性腸疾患の活動指数の減少により示されたように、本研究の第1週に疾患の進行を有意に遅延させた。この活動指数は、3つのIBDの症状、すなわち体重減少、便性(すなわち下痢)、および便潜血(すなわち血便)の複合的評価を表す。上述のように、UCの患者のほとんどは一般に血性下痢を呈し、より重症なケースでは体重減少も生じる。同様にCDの患者は通常、継続的な下痢および体重減少を有し、血便もまた有すると思われる。
[0218]従って本研究は、報告によれば、ヒトIBDにおいて治療上有益な物質すべてが、このマウスモデルの疾患の活動性もまた低減することが示されたことから、結腸炎の治療に一般的に使用される治療薬の効能を評価する上で高い価値を有するという動物モデルにおいて、GL1001がヒトIBDの一般的な症状を効果的に治療することを示す。例えばHollenbach et al. (2004)、上記記載、を参照のこと。
[0219]NF−κB::Lucマウスにおける引き続いての研究において、2.5%DSS処置は、ルシフェラーゼ発現により測定した場合、遠位結腸および腸管膜リンパ組織においてNF−κBシグナル伝達を増大する傾向を示した。ただしこれらの増大は一般に統計的に有意ではない。GL1001の300mg/kg/日、経口経管による1日2回投与は、本研究ではDSS誘発NF-κBシグナル伝達を低下させなかった。本研究においてGL1001の効果が見られなかった理由は、現在完全には解明されていない。
実施例6:GL1001は、Balb/cマウスの結腸におけるDSS誘発による組織学的効果を低減する。
[0220]研究は、Balb/cマウスの5群:未処置コントロール、DSS後ビヒクル、DSS後デキサメタゾン 3mg/kg/日、DSS後GL1001 10mg/kg/日、およびDSS後GL1001 100mg/kg/日、にてデザインした。DSSの投与は飲用水(水中DSS 5%)を介して、研究の第0日から7日とした。その後飲用水にはDSSを含めなかった。ビヒクル、デキサメタゾンおよびGL1001の投与は皮下にて、1日1回、第7日から研究終了(第14日)までとした。
[0221]研究終了時に、各動物の結腸の近位、横行、および遠位の部分由来のサンプルを、以下を含む組織学的分析:
(a)炎症スコア(0−5)、粘膜、粘膜下層、陰窩膿瘍、および浮腫におけるリンパ球浸潤に基づく;
(b)腺スコア(0−5)、陰窩の破壊(ムチンを産生し、上皮を生成する陰窩の機能の破壊)に基づく;および
(c)びらんスコア(0−5)、上皮の完全性、またはその潰瘍化の程度に基づく
のために採集した。総合組織病理学スコアは、上の(a)、(b)および(c)のスコアを加算して得た。
(a)炎症スコア(0−5)、粘膜、粘膜下層、陰窩膿瘍、および浮腫におけるリンパ球浸潤に基づく;
(b)腺スコア(0−5)、陰窩の破壊(ムチンを産生し、上皮を生成する陰窩の機能の破壊)に基づく;および
(c)びらんスコア(0−5)、上皮の完全性、またはその潰瘍化の程度に基づく
のために採集した。総合組織病理学スコアは、上の(a)、(b)および(c)のスコアを加算して得た。
[0222](便サンプルに基づいた)疾患の活動性における有意な改善は、本研究においてデキサメタゾンまたはGL1001の治療からは観察されなかった。しかし図13に示したように、GL1001 100mg/kg/日は、遠位結腸サンプルにおける炎症、腺、びらん、および総合組織病理学スコアを有意に低減した。10mg/kg/日のデキサメタゾンまたはGL1001では、有意な組織学的効果は認められなかった。
[0223]遠位結腸切片におけるGL1001の組織学的効果(炎症の低減、および腺の喪失、およびびらんの存否)は、結腸の遠位切片から採集した組織学的切片の顕微鏡写真(50×)の比較を表す図14に、明確に認められる。これらの顕微鏡写真において、Mはより重症の病変粘膜を示し、Eは浮腫を示す。
[0224]図14の上の顕微鏡写真は、DSS後1日1回のビヒクルの皮下投与にて処置した動物に由来する。重症の炎症、腺の喪失、およびびらんが認められる。
[0225]図14の下の顕微鏡写真は、DSS後GL1001 100mg/kgの1日1回皮下投与にて処置した動物に由来する。炎症および腺の喪失は軽度であり、びらんは認められない。矢印は、より重症度の低い病変粘膜を示す。
実施例7:GL1001は、Balb/cマウスにおけるDSS誘発結腸炎を阻害する。
[0226]研究は、Balb/cマウスの6群:未処置コントロール、DSS後ビヒクル、DSS後GL1001 30mg/kg/日、DSS後GL1001 100mg/kg/日、DSS後GL1001 300mg/kg/日、およびDSS後スルファサラジン 150mg/kg/日にてデザインした。DSSの投与は飲用水を介して、研究の第0日から6日とした。その後飲用水にはDSSを含めなかった。ビヒクル、GL1001、およびスルファサラジンの投与は皮下にて、1日2回、第6日から研究終了(第16日)までとした。
[0227]各動物の体重は、第1、3および5日(GL1001またはスルファサラジンの処置開始前)および第7,9,11および13日(GL1001またはスルファサラジンの処置開始後)に測定した。これらと同じ日に、以下:
(a)直腸脱(0=直腸脱なし、1=部分的直腸脱、2=中程度の直腸脱、3=完全な直腸脱)
(b)便性(0=固体のペレット、1=半固体、2=軟便、3=下痢);および
(c)便潜血(0=血液なし、1=潜血、2=肉眼的血便)
を含む、疾患の活動性の測定を記録した。
(a)直腸脱(0=直腸脱なし、1=部分的直腸脱、2=中程度の直腸脱、3=完全な直腸脱)
(b)便性(0=固体のペレット、1=半固体、2=軟便、3=下痢);および
(c)便潜血(0=血液なし、1=潜血、2=肉眼的血便)
を含む、疾患の活動性の測定を記録した。
[0228]研究終了時に、結腸の長さを決定し、組織学的分析を実施例6のように行った。
[0229]体重、疾患の活動性、結腸の長さ、および組織学における改善が、GL1001の少なくとも300mg/kg/日の用量で得られた。通常これらの改善は、スルファサラジン処置で得られたものに匹敵し、ケースによってはスルファサラジンより明らかに大きかった。
[0230]DSS投与中および投与後の体重減少は、第9日に最大に達した。第9日の体重減少における様々な治療の効果を図15に示す。
[0231]直腸脱のスコアは、第9日に最大に達した。第9日の直腸脱における様々な治療の効果を図16に示す。便性および便潜血のスコアは、第7日に最大に達した。第7日の便性および便潜血における様々な治療の効果を、各々図17および18に示す。
[0232]結腸の長さにおける様々な治療の効果を図19に示す。炎症、陰窩(腺)、びらん、および総合組織病理学スコアにおける様々な治療の効果を、各々図20、21、22、および23に示す。
[0233]本明細書に引用したすべての特許および公開文献は、それらの全内容において本明細書にて参照として援用する。
[0234]“包含する”および“包含すること”という用語は、排他的というよりむしろ包括的なものとして解釈されるものとする。
Claims (70)
- 消化管の炎症性疾患を有する被験者における、炎症、またはそれに関連するもしくはその二次的な病理学的プロセスを低減または軽減するため、前記被験者に抗炎症有効量で投与されるための、医薬剤の調製のためのACE2阻害薬の使用。
- 疾患が慢性胃炎である、請求項1に記載の使用。
- 疾患が炎症性腸疾患である、請求項1に記載の使用。
- 炎症性腸疾患がクローン病である、請求項3に記載の使用。
- クローン病が活動期にあり、活動指数が少なくとも220を示す、請求項4に記載の使用。
- 医薬剤が、活動指数の少なくとも約50ポイントの減少を達成するために有効な投与計画に従って投与されるために調製される、請求項5に記載の使用。
- 医薬剤が、活動指数の少なくとも約70ポイントの減少を達成するために有効な投与計画に従って投与されるために調製される、請求項5に記載の使用。
- 医薬剤が、クローン病の臨床的寛解を達成するために有効な投与計画に従って投与されるために調製される、請求項5に記載の使用。
- クローン病が、瘻孔を生じているクローン病である、請求項4に記載の使用。
- 医薬剤が、漏出している瘻孔数の減少を達成するために有効な投与計画に従って投与されるために調製される、請求項9に記載の使用。
- 医薬剤が、瘻孔の閉鎖を維持するために有効な投与計画に従って投与されるために調製される、請求項9に記載の使用。
- 被験者が小児患者である、請求項4に記載の使用。
- 炎症性腸疾患が潰瘍性大腸炎である、請求項3に記載の使用。
- 潰瘍性大腸炎が中程度から重症の活動期であり、メイヨースコアが少なくとも約6を示す、請求項13に記載の使用。
- 医薬剤が、メイヨースコアの少なくとも約3ポイントの減少を達成するために有効な投与計画に従って投与されるために調製される、請求項14に記載の使用。
- 医薬剤が、メイヨースコアの少なくとも約30%、および少なくとも約3ポイントの減少を達成するために有効な投与計画に従って投与されるために調製される、請求項14に記載の使用。
- 医薬剤が、潰瘍性大腸炎の臨床的寛解を達成するために有効な投与計画に従って投与されるために調製される、請求項13に記載の使用。
- 炎症性腸疾患が、非活動期または寛解期にある、請求項3に記載の使用。
- 医薬剤が、非活動期または寛解期の延長を達成するために有効な投与計画に従って投与されるために調製される、請求項18に記載の使用。
- 炎症性腸疾患が、アミノサリチル酸、コルチコステロイド、免疫抑制薬、抗生剤、およびそれらの組み合わせから成る群より選択される、少なくとも1つの基本的な薬剤の上限用量の投与を包含する基本的療法に対して不応性である、請求項3に記載の使用。
- 医薬剤が、少なくとも最初は、前記基本的療法と併用して投与されるために調製される、請求項20に記載の使用。
- 医薬剤が、少なくとも最初は、上限より少ない用量で投与される少なくとも1つの基本的な薬剤と併用して投与されるために調製される、請求項20に記載の使用。
- 医薬剤が、投与計画に従って少なくとも1つの基本的な薬剤と併用して投与されるために調製され、その場合炎症性腸疾患の臨床的寛解を達成した時点で、少なくとも1つの基本的な薬剤が中止される、請求項20に記載の使用。
- 少なくとも1つの基本的な薬剤の中止が、漸減による用量の低減により実行される、請求項23に記載の使用。
- 少なくとも1つの基本的な薬剤がコルチコステロイドを包含する、請求項23に記載の使用。
- 炎症性腸疾患の少なくとも1つの兆候または症状が、医薬剤の投与により軽減される、請求項3に記載の使用。
- 少なくとも1つの兆候または症状が、下痢、直腸からの出血、体重減少、およびそれらの組み合わせから成る群より選択される、請求項26に記載の使用。
- 医薬剤が、アミノサリチル酸、コルチコステロイド、免疫抑制薬、抗TNF-α薬、およびそれらの組み合わせから成る群より選択される、少なくとも1つの付加的な薬剤との併用療法において被験者に投与されるために調製される、請求項3に記載の使用。
- 少なくとも1つの付加的な薬剤が、バルサラジド、メサラミン、オルサラジン、スルファサラジン、医薬的に受容可能なそれらの塩、およびそれらの組み合わせ、から成る群より選択されるアミノサリチル酸を包含する、請求項28に記載の使用。
- 少なくとも1つの付加的な薬剤が、べクロメタゾン、べクロメタゾンジプロピオネート、ブデソニド、デキサメタゾン、フルチカゾン、ヒドロコルチゾン、メチルプレドニゾロン、プレドニゾン、プレドニゾロン、プレドニゾロン−21−メタスルホベンゾエート、チキソコルトール、医薬的に受容可能なそれらの塩、およびそれらの組み合わせ、から成る群より選択されるコルチコステロイドを包含する、請求項28に記載の使用。
- 少なくとも1つの付加的な薬剤が、アザチオプリン、シクロスポリン、メルカプトプリン、メトトレキセート、タクロリムス、医薬的に受容可能なそれらの塩、およびそれらの組み合わせ、から成る群より選択される免疫抑制薬を包含する、請求項28に記載の使用。
- 少なくとも1つの付加的な薬剤が、抗TNF-α薬を包含する、請求項28に記載の使用。
- 抗TNF-α薬がインフリキシマブを包含する、請求項32に記載の使用。
- 前記抗炎症有効量が、約0.5から約5000mg/日のACE2阻害薬の投与量を包含する、請求項1に記載の使用。
- 前記抗炎症有効量が、約5から約1000mg/日のACE2阻害薬の投与量を包含する、請求項1に記載の使用。
- 医薬剤が、経口、頬側、舌下、経粘膜、経鼻、眼球内、経直腸、経膣、経皮、非経口、吸入、または植込みによる投与のために調製される、請求項1に記載の使用。
- 医薬剤が、当該化合物および少なくとも1つの医薬的に受容可能な賦形剤を包含する医薬組成物を包含する、請求項1に記載の使用。
- ACE2阻害薬が、約1000nMより大きくないin vitroのACE2 IC50および/またはACE2 Kiを示す、請求項1に記載の使用。
- ACE2阻害薬が、約100nMより大きくないin vitroのACE2 IC50および/またはACE2 Kiを示す、請求項1に記載の使用。
- ACE2阻害薬が、IC50(ACE2)に対するIC50(ACE)の比率で表した場合、少なくとも約103のACE2対ACEへの選択性を示す、請求項1に記載の使用。
- ACE2阻害薬が、IC50(ACE2)に対するIC50(ACE)の比率で表した場合、少なくとも約104のACE2対ACEへの選択性を示す、請求項1に記載の使用。
- ACE2阻害薬がペプチド化合物を包含する、請求項1に記載の使用。
- ACE2阻害薬が非ペプチド化合物、または医薬的に受容可能なその塩もしくはそのプロドラッグを包含する、請求項1に記載の使用。
- 非ペプチド化合物が、亜鉛配位結合部分およびアミノ酸模倣部分を包含する、請求項43に記載の使用。
- 非ペプチド化合物が以下の式
R6はヒドロキシルまたは保護しているプロドラッグ部分であり;
R7は水素、カルボン酸、エーテル、アルコキシ、アミド、保護しているプロドラッグ部分、ヒドロキシル、チオール、ヘテロシクリル、アルキル、またはアミンであり;
Qは、CH2、O、NH、またはNR3であり、ここでR3は、置換されたまたは置換されていないC1−5の側鎖または直鎖のアルキル、C2−5の側鎖または直鎖のアルケニル、置換されたまたは置換されていないアシル、アリール、またはC3−8環であり;
Gは、共有結合、またはCH2、エーテル、チオエーテル、アミン、またはカルボニルの連結部分であり;
Mはヘテロアリールであって、サブリンク部分である(CH2)nまたは(CH2)nO(CH2)nを通して連結される、少なくとも1つのサブアンカー部分で置換されており、ここでnは0から3の整数であり、前記サブアンカー部分は、置換されたまたは置換されていないシクロアルキル環またはアリール環を包含し;
Jは、1つの結合、または置換されたもしくは置換されていないアルキル、アルケニル、もしくはアルキニルの部分であり;そして
Dは、アルキル、アルケニル、アルキニル、アリール、またはヘテロアリールであって、所望によりGまたはMに連結して1つの環を形成する]
を有する、請求項43に記載の使用。 - 非ペプチド化合物に関する式において、R6がヒドロキシであり、R7がカルボン酸であり、QがNHであり、そしてGがCH2である、請求項45に記載の使用。
- 非ペプチド化合物に関する式において、Mのヘテロアリール基が、イミダゾリル、チエニル、トリアゾリル、ピラゾリル、またはチアゾリルである、請求項45に記載の使用。
- サブアンカー部分が、C3−6シクロアルキル、フェニル、メチレンジオキシフェニル、ナフタレニル、またはハロ、C1−6アルキル、C3−6シクロアルキル、トリフルオロメチル、C1−6アルコキシ、トリフルオロメトキシ、フェニル、シアノ、ニトロ、およびカルボン酸の基より独立して選択される1から3の置換基を有するフェニルであって、(CH2)nまたは(CH2)O(CH2)のサブリンク部分を通してヘテロアリー基に連結されており、ここでnは0から3の整数である、請求項47に記載の使用。
- 非ペプチド化合物に関する式において、Jが1つの結合またはCH2部分であり、そしてDがC1−6アルキル、C3−6シクロアルキル、またはフェニルである、請求項45に記載の使用。
- 非ペプチド化合物に関する式において:
R6がヒドロキシルであり;
R7がカルボン酸であり;
QがNHであり;
GがCH2であり;
Mが、イミダゾリル、チエニル、トリアゾリル、ピラゾリル、またはチアゾリルであって、(CH2)nまたは(CH2)O(CH2)のサブリンク部分を通してサブアンカー部分に連結されており、ここでnは0から3の整数であり、前記サブアンカー部分が、C3−6シクロアルキル、フェニル、メチレンジオキシフェニル、ナフタレニル、またはハロ、C1−6アルキル、C3−6シクロアルキル、トリフルオロメチル、C1−6アルコキシ、トリフルオロメトキシ、フェニル、シアノ、ニトロ、およびカルボン酸の基より独立して選択される1から3の置換基を有するフェニルであり;
Jが、1つの結合、またはCH2部分であり;そして
Dが、C1−6アルキル、C3−6シクロアルキル、またはフェニルである、
請求項45に記載の使用。 - 当該化合物が(S,S)−立体配置で存在する、請求項45に記載の使用。
- 当該化合物が実質的にエナンチオマー的に純粋である、請求項51に記載の使用。
- ACE2阻害薬が、以下から成る群
2−[1−カルボキシ−2−[3−(4−トリフルオロメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−ナフタレン−1−イルメチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−クロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,4−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−シアノベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−クロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,5−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,4−ジメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(3−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3,5−ジメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−トリフルオロメトキシベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−イソプロピルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(4−t−ブチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(4−ニトロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジメトキシベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジフルオロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(2,3−ジクロロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−(3−トリフルオロメチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[2−(3−ベンゾ[1,3]ジオキソール−5−イルメチル−3H−イミダゾール−4−イル)−1−カルボキシエチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2−シクロヘキシルエチル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチル−ペンタン酸;
2−[1−カルボキシ−2−[3−フェネチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−ヨードベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(3−フルオロベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−ベンジルオキシメチル−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(4−ブチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[3−(2−メチルベンジル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[2−フェニルチアゾール−4−イル]エチルアミノ]−4−メチルペンタン酸;
2−[1−カルボキシ−2−[1−ベンジル)−1H−ピラゾール−4−イル]エチルアミノ]−メチルペンタン酸;および
2−[1−カルボキシ−2−[3−(2−メチルビフェニル−3−イルメチル)−3H−イミダゾール−4−イル]エチルアミノ]−4−メチルペンタン酸、
より選択される(S,S)−立体配置の化合物を包含する、請求項43に記載の使用。 - 被験者における消化管の炎症性疾患を治療するため、前記被験者に約0.5から約5000mg/日の量で投与されるための、医薬剤の調製のためのACE2阻害薬の使用。
- 医薬剤が、約5から約1000mg/日の量で投与されるために調製される、請求項54に記載の使用。
- 消化管の炎症性疾患を有する被験者における粘膜の潰瘍の治癒を促進するための治療有効量で投与されるための、医薬剤の調製のためのACE2阻害薬の使用。
- 疾患が慢性胃炎である、請求項56に記載の使用。
- 疾患が、クローン病および潰瘍性大腸炎より選択される炎症性腸疾患である、請求項56に記載の使用。
- 被験者における消化管の炎症性疾患の寛解を導入または維持するための治療有効量で前記被験者に投与されるための、医薬剤の調製のためのACE2阻害薬の使用。
- 疾患が慢性胃炎である、請求項59に記載の使用。
- 疾患が、クローン病および潰瘍性大腸炎より選択される炎症性腸疾患である、請求項59に記載の使用。
- アミノサリチル酸不応性炎症性腸疾患を、所望によりアミノサリチル酸との併用療法において、ただしコルチコステロイドは使用せずに治療するための治療有効量で前記被験者に投与されるための、医薬剤の調製のためのACE2阻害薬の使用。
- ACE2阻害薬、ならびにアミノサリチル酸、コルチコステロイド、免疫抑制薬、抗TNFα薬、およびそれらの組み合わせから成る群より選択される少なくとも1つの付加的な薬剤を包含する治療の組み合わせ。
- 少なくとも1つの付加的な薬剤が、バルサラジド、メサラミン、オルサラジン、スルファサラジン、医薬的に受容可能なそれらの塩、およびそれらの組み合わせ、から成る群より選択されるアミノサリチル酸を包含する、請求項63に記載の組み合わせ。
- 少なくとも1つの付加的な薬剤が、べクロメタゾン、べクロメタゾンジプロピオネート、ブデソニド、フルチカゾン、ヒドロコルチゾン、メチルプレドニゾロン、プレドニゾン、プレドニゾロン、プレドニゾロン−21−メタスルホベンゾエート、チキソコルトール、医薬的に受容可能なそれらの塩、およびそれらの組み合わせ、から成る群より選択されるコルチコステロイドを包含する、請求項63に記載の組み合わせ。
- 少なくとも1つの付加的な薬剤が、アザチオプリン、シクロスポリン、メルカプトプリン、メトトレキセート、タクロリムス、医薬的に受容可能なそれらの塩、およびそれらの組み合わせ、から成る群より選択される免疫抑制薬を包含する、請求項63に記載の組み合わせ。
- 少なくとも1つの付加的な薬剤が、抗TNF-α薬を包含する、請求項63に記載の組み合わせ。
- 抗TNF-α薬がインフリキシマブを包含する、請求項67に記載の組み合わせ。
- 当該化合物および少なくとも1つの付加的な薬剤が、同時にまたは異なる時間に投与されるために別々に製剤化される、請求項63に記載の組み合わせ。
- 当該化合物および少なくとも1つの付加的な薬剤が、1つの投与剤形中に合剤化される、請求項63に記載の組み合わせ。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82507506P | 2006-09-08 | 2006-09-08 | |
| US82780706P | 2006-10-02 | 2006-10-02 | |
| PCT/US2007/077857 WO2008031014A1 (en) | 2006-09-08 | 2007-09-07 | Method for reducing or alleviating inflammation in the digestive tract |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010502736A true JP2010502736A (ja) | 2010-01-28 |
| JP2010502736A5 JP2010502736A5 (ja) | 2010-10-14 |
Family
ID=38670723
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009527579A Pending JP2010502736A (ja) | 2006-09-08 | 2007-09-07 | 消化管の炎症を低減または軽減するための方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20080234345A1 (ja) |
| EP (1) | EP2059234B1 (ja) |
| JP (1) | JP2010502736A (ja) |
| CN (1) | CN101568331A (ja) |
| AT (1) | ATE531362T1 (ja) |
| AU (1) | AU2007292247A1 (ja) |
| CA (1) | CA2662535A1 (ja) |
| DK (1) | DK2059234T3 (ja) |
| ES (1) | ES2376493T3 (ja) |
| IL (1) | IL197362A0 (ja) |
| WO (1) | WO2008031014A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018502107A (ja) * | 2014-12-26 | 2018-01-25 | セルジーン アルパイン インベストメント カンパニー Ii, エルエルシー | Smad7アンチセンスオリゴヌクレオチドの使用方法 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2095819A1 (en) * | 2008-02-28 | 2009-09-02 | Maastricht University | N-benzyl imidazole derivatives and their use as aldosterone synthase inhibitors |
| WO2009114516A1 (en) * | 2008-03-10 | 2009-09-17 | Ore Pharmaceuticals Inc. | Therapy for disorders of the proximal digestive tract |
| US20100204286A1 (en) * | 2009-02-12 | 2010-08-12 | Donahue Stephen R | Method for reducing gastrointestinal adverse effects of cytotoxic agents |
| JP5845171B2 (ja) * | 2010-03-04 | 2016-01-20 | 大日本住友製薬株式会社 | 炎症性腸疾患用薬剤 |
| KR101713453B1 (ko) | 2010-03-12 | 2017-03-07 | 오메로스 코포레이션 | Pde10 억제제 및 관련 조성물 및 방법 |
| NZ630810A (en) | 2014-04-28 | 2016-03-31 | Omeros Corp | Processes and intermediates for the preparation of a pde10 inhibitor |
| NZ716462A (en) | 2014-04-28 | 2017-11-24 | Omeros Corp | Optically active pde10 inhibitor |
| AU2016250843A1 (en) | 2015-04-24 | 2017-10-12 | Omeros Corporation | PDE10 inhibitors and related compositions and methods |
| JP2018535969A (ja) | 2015-11-04 | 2018-12-06 | オメロス コーポレーション | Pde10阻害剤の固体状態形態 |
| GB202116266D0 (en) * | 2021-11-11 | 2021-12-29 | Bicycletx Ltd | Novel use |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002098448A1 (en) * | 2001-06-04 | 2002-12-12 | Human Genome Sciences, Inc. | Methods and compositions for modulating ace-2 activity |
| JP2002543120A (ja) * | 1999-04-30 | 2002-12-17 | ミレニアム ファーマスーティカルズ インク | Ace−2阻害化合物及びその使用方法 |
| JP2006503905A (ja) * | 2002-09-24 | 2006-02-02 | コンビナトアールエックス インコーポレーティッド | 炎症性サイトカインのレベルの増大と関連する病気および障害の治療のための方法および試薬 |
| JP2006517969A (ja) * | 2003-02-14 | 2006-08-03 | コンビナトアールエックス インコーポレーティッド | 免疫炎症性障害の治療のための組み合わせ療法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6194556B1 (en) * | 1997-12-11 | 2001-02-27 | Millennium Pharmaceuticals, Inc. | Angiotensin converting enzyme homolog and therapeutic and diagnostic uses therfor |
| US7045532B2 (en) * | 1999-04-30 | 2006-05-16 | Millennium Pharmaceuticals, Inc. | ACE-2 modulating compounds and methods of use thereof |
| GB0203061D0 (en) | 2002-02-08 | 2002-03-27 | Novartis Ag | Organic compounds |
| US20050203168A1 (en) * | 2004-03-11 | 2005-09-15 | The Regents Of The University Of Michigan | Angiotensin converting enzyme inhibitor use for treatment and prevention of gastrointestinal disorders |
| US7842709B2 (en) * | 2006-09-08 | 2010-11-30 | Ore Pharmaceuticals Inc. | Method for treating inflammatory diseases of the digestive tract |
-
2007
- 2007-09-07 ES ES07814746T patent/ES2376493T3/es active Active
- 2007-09-07 AT AT07814746T patent/ATE531362T1/de active
- 2007-09-07 JP JP2009527579A patent/JP2010502736A/ja active Pending
- 2007-09-07 AU AU2007292247A patent/AU2007292247A1/en not_active Abandoned
- 2007-09-07 WO PCT/US2007/077857 patent/WO2008031014A1/en active Application Filing
- 2007-09-07 US US11/851,694 patent/US20080234345A1/en not_active Abandoned
- 2007-09-07 CN CNA2007800394181A patent/CN101568331A/zh active Pending
- 2007-09-07 CA CA002662535A patent/CA2662535A1/en not_active Abandoned
- 2007-09-07 DK DK07814746.9T patent/DK2059234T3/da active
- 2007-09-07 EP EP07814746A patent/EP2059234B1/en not_active Not-in-force
-
2009
- 2009-03-03 IL IL197362A patent/IL197362A0/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002543120A (ja) * | 1999-04-30 | 2002-12-17 | ミレニアム ファーマスーティカルズ インク | Ace−2阻害化合物及びその使用方法 |
| US6632830B1 (en) * | 1999-04-30 | 2003-10-14 | Millennium Pharmaceuticals, Inc. | ACE-2 inhibiting compounds and methods of use thereof |
| WO2002098448A1 (en) * | 2001-06-04 | 2002-12-12 | Human Genome Sciences, Inc. | Methods and compositions for modulating ace-2 activity |
| JP2006503905A (ja) * | 2002-09-24 | 2006-02-02 | コンビナトアールエックス インコーポレーティッド | 炎症性サイトカインのレベルの増大と関連する病気および障害の治療のための方法および試薬 |
| JP2006517969A (ja) * | 2003-02-14 | 2006-08-03 | コンビナトアールエックス インコーポレーティッド | 免疫炎症性障害の治療のための組み合わせ療法 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018502107A (ja) * | 2014-12-26 | 2018-01-25 | セルジーン アルパイン インベストメント カンパニー Ii, エルエルシー | Smad7アンチセンスオリゴヌクレオチドの使用方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2007292247A1 (en) | 2008-03-13 |
| IL197362A0 (en) | 2009-12-24 |
| EP2059234A1 (en) | 2009-05-20 |
| AU2007292247A2 (en) | 2009-03-26 |
| DK2059234T3 (da) | 2011-11-28 |
| EP2059234B1 (en) | 2011-11-02 |
| ES2376493T3 (es) | 2012-03-14 |
| US20080234345A1 (en) | 2008-09-25 |
| WO2008031014A1 (en) | 2008-03-13 |
| ATE531362T1 (de) | 2011-11-15 |
| CN101568331A (zh) | 2009-10-28 |
| CA2662535A1 (en) | 2008-03-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7842709B2 (en) | Method for treating inflammatory diseases of the digestive tract | |
| JP2010502736A (ja) | 消化管の炎症を低減または軽減するための方法 | |
| US20120009183A1 (en) | Method for treatment of pancreatitis | |
| JP6178657B2 (ja) | 胃内でアセトアルデヒドを結合するための組成物および方法 | |
| JP2012505160A (ja) | 炎症を処置する方法 | |
| JP2007531769A (ja) | 成長ホルモン分泌促進薬を使用したc−反応性蛋白質の低減方法 | |
| JP2016106120A (ja) | 癌治療のためのチオキサントンをベースとしたオートファジー阻害剤療法 | |
| US20190167615A1 (en) | Metabolites for treatment and prevention of autoimmune disease | |
| KR20210100594A (ko) | 염증성 장애의 치료에 사용하기 위한 벤조이미다졸 유도체 | |
| US9539236B2 (en) | Pharmaceutical composition for preventing or treating cancer | |
| US20100035848A1 (en) | Therapy for disorders of the proximal digestive tract | |
| US12016847B2 (en) | Methods of treating prostate cancer | |
| US20100204286A1 (en) | Method for reducing gastrointestinal adverse effects of cytotoxic agents | |
| WO2006046746A1 (ja) | 内臓痛予防・治療剤 | |
| FR3109089A1 (fr) | Composition pharmaceutique pour le traitement des maladies inflammatoires intestinales | |
| FR3090317A1 (fr) | Utilisation d’un antagoniste de par-1 pour le traitement d’une maladie inflammatoire chronique intestinale | |
| Larauche et al. | Effect of dietary nitric oxide on gastric mucosal mast cells in absence or presence of an experimental gastritis in rats | |
| US20240009160A1 (en) | Use of (s)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid in the treatment of cancer | |
| EA030590B1 (ru) | Применение тиазолопиримидинона для лечения воспалительного заболевания кишечника | |
| JP2023522756A (ja) | セリアック病のためのピリジノン誘導体の全身性製剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100830 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100830 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120803 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20130206 |