JP2010189633A - 樹脂分散体の製造方法及び樹脂粒子 - Google Patents
樹脂分散体の製造方法及び樹脂粒子 Download PDFInfo
- Publication number
- JP2010189633A JP2010189633A JP2010010887A JP2010010887A JP2010189633A JP 2010189633 A JP2010189633 A JP 2010189633A JP 2010010887 A JP2010010887 A JP 2010010887A JP 2010010887 A JP2010010887 A JP 2010010887A JP 2010189633 A JP2010189633 A JP 2010189633A
- Authority
- JP
- Japan
- Prior art keywords
- resin
- acid
- group
- resin particles
- parts
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Abstract
【課題】 低温溶融性と耐ブロッキング性を両立し、粒径が均一な樹脂粒子を含有する樹脂分散体及び樹脂粒子の安定的な製造法の提供。
【解決手段】 樹脂(a)を含有する樹脂粒子(A)の水性分散液(W)と、樹脂(b)及び/又はその前駆体(b0)並びに必要により有機溶剤(u)を含有する油性液(OL)とを混合し、(W)中に(OL)を分散させ、前駆体(b0)を使用する場合は更に(W)中で(b0)を反応させて、(b)を含有する樹脂粒子(B)を形成させることにより、(B)の表面に(A)が付着された構造の樹脂粒子(C)の水性分散体(X1)を得る工程を含み、樹脂(b)が、特定のポリオール成分(x)と特定ポリカルボン酸成分(y)とが重縮合されてなるポリエステル樹脂(p1)及び/又は(p1)を構成単位として有する樹脂(p2)を含有することを特徴とする水性分散体(X1)の製造方法。
【選択図】 なし
【解決手段】 樹脂(a)を含有する樹脂粒子(A)の水性分散液(W)と、樹脂(b)及び/又はその前駆体(b0)並びに必要により有機溶剤(u)を含有する油性液(OL)とを混合し、(W)中に(OL)を分散させ、前駆体(b0)を使用する場合は更に(W)中で(b0)を反応させて、(b)を含有する樹脂粒子(B)を形成させることにより、(B)の表面に(A)が付着された構造の樹脂粒子(C)の水性分散体(X1)を得る工程を含み、樹脂(b)が、特定のポリオール成分(x)と特定ポリカルボン酸成分(y)とが重縮合されてなるポリエステル樹脂(p1)及び/又は(p1)を構成単位として有する樹脂(p2)を含有することを特徴とする水性分散体(X1)の製造方法。
【選択図】 なし
Description
本発明は樹脂分散体及び樹脂粒子の製造方法並びに樹脂粒子に関する。更に詳しくは、各種用途に有用な樹脂粒子とその水性分散体の製造方法並びに樹脂粒子に関する。
粒径及び形状が均一で、かつ、電気的特性、熱的特性、化学的安定性等に優れた粒子として、樹脂と有機溶媒を含有する混合液と、水系媒体との懸濁液から有機溶媒を除去することによって得られる樹脂粒子が知られている(例えば、特許文献1参照)。
しかしながら、熱定着方式・熱加工方式に用いられる樹脂粒子では、低温溶融性と耐ブロッキング性、特に高温高湿環境下での保存安定性に関係する耐ブロッキング性、の両立のさらなる向上が求められており、特にスラッシュ成型用樹脂、粉体塗料、電子写真トナー、静電記録トナー、静電印刷トナー又はホットメルト接着剤としては、必ずしも十分ではなかった。
本発明者は、これらの問題点を解決するべく鋭意検討した結果、本発明に到達した。即ち本発明は、樹脂(a)を含有する樹脂粒子(A)の水性分散液(W)と、樹脂(b)及び/又はその前駆体(b0)並びに必要により有機溶剤(u)を含有する油性液(OL)とを混合し、(W)中に(OL)を分散させ、前駆体(b0)を使用する場合は更に(W)中で(b0)を反応させて、(b)を含有する樹脂粒子(B)を形成させることにより、(B)の表面に(A)が付着された構造の樹脂粒子(C)の水性分散体(X1)を得る工程を含み、樹脂(b)が、ポリオール成分(x)とポリカルボン酸成分(y)とが重縮合されてなるポリエステル樹脂(p1)であって、(y)中にその70モル%以上の、テレフタル酸、イソフタル酸及び/又はそれらの低級アルキルエステル(アルキル基の炭素数:1〜4)を含有し、かつ、(x)から重縮合反応中に系外に留去されるポリオールを除いた(p1)を構成するポリオールの80〜99.99モル%が1,2−プロピレングリコールであり、0.01〜11モル%がネオペンチルグリコールであるポリエステル樹脂(p1)、及び/又は、(p1)を構成単位として有する樹脂(p2)を含有することを特徴とする水性分散体(X1)の製造方法;該製造方法により得られた水性分散体(X1)から水性溶剤を除去して樹脂粒子(C)を得る工程を含む樹脂粒子の製造方法;並びに該製造方法により得られた樹脂粒子である。
本発明は以下の効果を有する。
1.耐ブロッキング性、及び低温溶融性のいずれにも優れた樹脂粒子を得ることができる。したがって、たとえば、本発明の樹脂粒子をトナーの母体粒子として用いた場合、高温高湿時の保存性、低温定着性に優れる。
2.粒径が均一な樹脂粒子分散体及び樹脂粒子を安定的に製造できる。
3.水性分散液中で樹脂粒子が得られるため、安全かつ低コストで樹脂粒子を製造できる。
1.耐ブロッキング性、及び低温溶融性のいずれにも優れた樹脂粒子を得ることができる。したがって、たとえば、本発明の樹脂粒子をトナーの母体粒子として用いた場合、高温高湿時の保存性、低温定着性に優れる。
2.粒径が均一な樹脂粒子分散体及び樹脂粒子を安定的に製造できる。
3.水性分散液中で樹脂粒子が得られるため、安全かつ低コストで樹脂粒子を製造できる。
本発明において、樹脂粒子(B)中の樹脂(b)に含有されるポリエステル樹脂(p1)は、ポリオール成分(x)とポリカルボン酸成分(y)とが重縮合されて得られたものである。
ポリオール成分(x)中には、1,2−プロピレングリコールとネオペンチルグリコールを必須成分として含有する。尚、後述のように、テレフタル酸及び/又はイソフタル酸の1,2−プロピレングリコールジエステルを用いる場合は、必ずしも1,2−プロピレングリコールを用いる必要はない。
得られるポリエステル樹脂(p1)を構成するポリオール中の1,2−プロピレングリコールの含有量は、通常80〜99.99モル%、好ましくは85〜99.98モル%である。また、ネオペンチルグリコールの含有量は、通常0.01〜11モル%、好ましくは0.02〜10モル%、更に好ましくは0.02〜3モル%である。
ポリオール成分(x)中には、1,2−プロピレングリコールとネオペンチルグリコールを必須成分として含有する。尚、後述のように、テレフタル酸及び/又はイソフタル酸の1,2−プロピレングリコールジエステルを用いる場合は、必ずしも1,2−プロピレングリコールを用いる必要はない。
得られるポリエステル樹脂(p1)を構成するポリオール中の1,2−プロピレングリコールの含有量は、通常80〜99.99モル%、好ましくは85〜99.98モル%である。また、ネオペンチルグリコールの含有量は、通常0.01〜11モル%、好ましくは0.02〜10モル%、更に好ましくは0.02〜3モル%である。
1,2−プロピレングリコールの含有量が80モル%未満であると、樹脂強度が低下し、低温溶融性も低下する。1,2−プロピレングリコールの含有量が99.99モル%を越えるか、あるいはネオペンチルグリコールの含有量が0.01モル%未満の場合、高温高湿環境下での耐ブロッキング性が低下する。また、ネオペンチルグリコールの含有量が11モル%を越えると低温溶融性が低下する。1,2−プロピレングリコールの含有量が85〜99.98モル%で、かつネオペンチルグリコールの含有量が0.02〜3モル%であると、耐ブロッキング性と低温溶融性のバランスが特に良好で
ある。
尚、上記の含有量は、ポリエステル樹脂(p1)を構成するポリオール中の含有量を意味し、ポリオール成分(x)のうちで重縮合反応中に系外に留去されるものは除き、かつテレフタル酸及び/又はイソフタル酸の1,2−プロピレングリコールジエステルに由来する1,2−プロピレングリコールも含めた量である。
尚、上記の含有量は、ポリエステル樹脂(p1)を構成するポリオール中の含有量を意味し、ポリオール成分(x)のうちで重縮合反応中に系外に留去されるものは除き、かつテレフタル酸及び/又はイソフタル酸の1,2−プロピレングリコールジエステルに由来する1,2−プロピレングリコールも含めた量である。
ポリオール成分(x)のうちで、重縮合反応中に系外に留去されるものが無い場合は、(p1)を構成するポリオール成分中の各ポリオールの含有量(モル%)と(x)中に含有するポリオール成分中の各ポリオールの含有量(モル%)は等しくなる。重縮合反応中に系外に留去されるものがある場合は、そのポリオールについては、(p1)を構成するポリオールよりも、留去される分だけ過剰量用いる。留去されるポリオールが1,2−プロピレングリコールの場合は、(x)中に(p1)中の量に対して、例えば、120〜500モル%用いる。
ポリオール成分(x)中には、必要により、1,2−プロピレングリコールとネオペンチルグリコール以外の1種以上の他のポリオールを含有してもよい。他のポリオールとしては、ジオール(1)及び3〜8価若しくはそれ以上のポリオール(2)が挙げられる。
ジオール(1)としては、1,2−プロピレングリコールとネオペンチルグリコール以外の炭素数2〜36のアルキレングリコール(エチレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、及び1,6−ヘキサンジオール等);炭素数4〜36のアルキレンエーテルグリコール(ジエチレングリコール、トリエチレングリコール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、及びポリテトラメチレンエーテルグリコール等);炭素数6〜36の脂環式ジオール(1,4−シクロヘキサンジメタノール、及び水素添加ビスフェノールA等);上記脂環式ジオールの(ポリ)オキシアルキレン(アルキレン基の炭素数2〜4、以下のポリオキシアルキレン基も同じ)エーテル〔オキシアルキレン単位(以下AO単位と略記)の数1〜30〕;及び2価フェノール〔単環2価フェノール(例えばハイドロキノン)、及びビスフェノール類(ビスフェノールA、ビスフェノールF及びビスフェノールS等)〕のポリオキシアルキレンエーテル(AO単位の数2〜30)等が挙げられる。
3価〜8価若しくはそれ以上のポリオール(2)としては、炭素数3〜36の3価〜8価若しくはそれ以上の脂肪族多価アルコール(アルカンポリオール及びその分子内若しくは分子間脱水物、例えばグリセリン、トリメチロールエタン、トリメチロールプロパン、ペンタエリスリトール、ソルビトール、ソルビタン、ポリグリセリン、及びジペンタエリスリトール;糖類及びその誘導体、例えばショ糖及びメチルグルコシド);上記脂肪族多価アルコールの(ポリ)オキシアルキレンエーテル(AO単位の数1〜30);トリスフェノール類(トリスフェノールPA等)のポリオキシアルキレンエーテル(AO単位の数2〜30);ノボラック樹脂(フェノールノボラック及びクレゾールノボラック等、平均重合度3〜60)のポリオキシアルキレンエーテル(AO単位の数2〜30)等が挙げられる。
ジオールで好ましいものは、炭素数2〜12のアルキレングリコール、ビスフェノール類のポリオキシアルキレンエーテル(AO単位の数2〜30)及びこれらの併用であり、更に好ましいものは、ビスフェノールAのポリオキシアルキレンエーテル(アルキレン基の炭素数2又は3、AO単位の数2〜8)、炭素数2〜12のアルキレングリコール(特にエチレングリコール)、及びこれらの併用である。
3価〜8価若しくはそれ以上のポリオールで好ましいものは、炭素数3〜36の3〜8価若しくはそれ以上の脂肪族多価アルコール、及びノボラック樹脂のポリオキシアルキレンエーテル(AO単位の数2〜30)であり、更に好ましいものはノボラック樹脂のポリオキシアルキレンエーテル(AO単位の数2〜30)である。
3価〜8価若しくはそれ以上のポリオールで好ましいものは、炭素数3〜36の3〜8価若しくはそれ以上の脂肪族多価アルコール、及びノボラック樹脂のポリオキシアルキレンエーテル(AO単位の数2〜30)であり、更に好ましいものはノボラック樹脂のポリオキシアルキレンエーテル(AO単位の数2〜30)である。
これらのうち、他のポリオールとして、特に好ましいものは、ノボラック樹脂のポリオキシアルキレンエーテル(アルキレン基の炭素数2及び/又は3、AO単位の数2〜30)である。
これらの1,2−プロピレングリコールとネオペンチルグリコール以外のポリオールのポリエステル樹脂(p1)中の含有量は、好ましくは18モル%以下、更に好ましくは0.01〜15モル%である。
これらの1,2−プロピレングリコールとネオペンチルグリコール以外のポリオールのポリエステル樹脂(p1)中の含有量は、好ましくは18モル%以下、更に好ましくは0.01〜15モル%である。
本発明においては、ポリエステル樹脂(p1)の原料のポリカルボン酸成分(y)中に、テレフタル酸、イソフタル酸、及び/又はそれらの低級アルキルエステル(アルキル基の炭素数:1〜4)(y1)を必須成分として含有する。尚、低級アルキルエステルは、ヒドロキシアルキルエステルを含む意味で用いる。
低級アルキルエステルの具体例としては、テレフタル酸ジメチル、イソフタル酸ジメチル、テレフタル酸ジエチル、テレフタル酸ジブチル、テレフタル酸1,2−プロピレングリコールジエステル等が挙げられる。これらの中では、反応速度及びコストの点で、テレフタル酸、イソフタル酸、テレフタル酸ジメチル、テレフタル酸1,2−プロピレングリコールジエステル、及びこれらの2種以上の併用が好ましい。
低級アルキルエステルの具体例としては、テレフタル酸ジメチル、イソフタル酸ジメチル、テレフタル酸ジエチル、テレフタル酸ジブチル、テレフタル酸1,2−プロピレングリコールジエステル等が挙げられる。これらの中では、反応速度及びコストの点で、テレフタル酸、イソフタル酸、テレフタル酸ジメチル、テレフタル酸1,2−プロピレングリコールジエステル、及びこれらの2種以上の併用が好ましい。
(y1)中の、テレフタル酸、その低級アルキルエステル、及びそのヒドロキシアルキルエステルから選ばれる1種以上のテレフタル酸又はその誘導体〔以下、テレフタル酸(誘導体)〕と、イソフタル酸、その低級アルキルエステル、及びそのヒドロキシアルキルエステルから選ばれる1種以上のイソフタル酸又はその誘導体〔以下、イソフタル酸(誘導体)〕とのモル比は、〔テレフタル酸(誘導体)〕:〔イソフタル酸(誘導体)〕が、好ましくは(1〜100):(99〜0)、更に好ましくは(50〜99.99):(50〜0.01)、特に好ましくは(90〜99.98):(10〜0.02)である。
(y1)は、得られるポリエステル樹脂のガラス転移温度(Tg)を上げて、トナーの耐ブロッキング性を向上させる効果があるため、ポリカルボン酸成分(y)に対し、70モル%以上含有する必要があり、74〜95モル%の範囲で含有するのが好ましい。
(y1)は、得られるポリエステル樹脂のガラス転移温度(Tg)を上げて、トナーの耐ブロッキング性を向上させる効果があるため、ポリカルボン酸成分(y)に対し、70モル%以上含有する必要があり、74〜95モル%の範囲で含有するのが好ましい。
ポリカルボン酸成分(y)中には、必要により、(y1)以外の1種以上のポリカルボン酸を含有してもよい。ポリカルボン酸としては、(y1)以外のジカルボン酸(3)、及び3〜6価若しくはそれ以上のポリカルボン酸(4)が挙げられる。
(y1)以外のジカルボン酸(3)としては、炭素数2〜50のアルカンジカルボン酸(シュウ酸、マロン酸、コハク酸、アジピン酸、レパルギン酸、及びセバシン酸等);炭素数4〜50のアルケンジカルボン酸(ドデセニルコハク酸等のアルケニルコハク酸、(無水)マレイン酸、フマル酸、シトラコン酸、メサコン酸、イタコン酸、及びグルタコン酸等);(y1)以外の炭素数8〜36の芳香族ジカルボン酸(ナフタレンジカルボン酸等);及びこれらの無水物又は低級アルキル(炭素数1〜4)エステル〔(無水)フタル酸等〕等が挙げられる。これらを単独で又は2種以上を併用して用いることができる。
尚、上記において、(無水)マレイン酸とは、マレイン酸及び/又は無水マレイン酸を意味し、以下同様の記載法を用いる。
(y1)以外のジカルボン酸(3)としては、炭素数2〜50のアルカンジカルボン酸(シュウ酸、マロン酸、コハク酸、アジピン酸、レパルギン酸、及びセバシン酸等);炭素数4〜50のアルケンジカルボン酸(ドデセニルコハク酸等のアルケニルコハク酸、(無水)マレイン酸、フマル酸、シトラコン酸、メサコン酸、イタコン酸、及びグルタコン酸等);(y1)以外の炭素数8〜36の芳香族ジカルボン酸(ナフタレンジカルボン酸等);及びこれらの無水物又は低級アルキル(炭素数1〜4)エステル〔(無水)フタル酸等〕等が挙げられる。これらを単独で又は2種以上を併用して用いることができる。
尚、上記において、(無水)マレイン酸とは、マレイン酸及び/又は無水マレイン酸を意味し、以下同様の記載法を用いる。
3〜6価若しくはそれ以上のポリカルボン酸(4)としては、炭素数9〜20の芳香族ポリカルボン酸(トリメリット酸、及びピロメリット酸等);炭素数6〜36の脂肪族(脂環式を含む)ポリカルボン酸(ヘキサントリカルボン酸等);及びこれらの無水物又は低級アルキル(炭素数1〜4)エステル〔(無水)トリメリット酸、無水ピロメリット酸等〕;(メタ)アクリル酸と必要により他のビニルモノマーとの(メタ)アクリル酸(共)重合体等のポリカルボン酸重合体;等が挙げられる。
これらの(y1)以外のポリカルボン酸のうち好ましいものは、炭素数2〜50のアルカンジカルボン酸、炭素数4〜50のアルケンジカルボン酸、炭素数9〜20の芳香族ポリカルボン酸、及びそれらの無水物又は低級アルキルエステルである。
更に好ましいものは、炭素数4〜18のアルカンジカルボン酸(特にアジピン酸)、及び/又は、3〜6価の炭素数9〜20の芳香族ポリカルボン酸(無水物)(特に、トリメリット酸、及びその無水物)〔好ましくは各々(y)中30モル%以下、更に好ましくは各々0.1〜28モル%、特に好ましくは各々5〜25モル%〕である。
更に好ましいものは、炭素数4〜18のアルカンジカルボン酸(特にアジピン酸)、及び/又は、3〜6価の炭素数9〜20の芳香族ポリカルボン酸(無水物)(特に、トリメリット酸、及びその無水物)〔好ましくは各々(y)中30モル%以下、更に好ましくは各々0.1〜28モル%、特に好ましくは各々5〜25モル%〕である。
また、本発明においては、ポリエステル樹脂の特性を損なわない限り、ポリオール成分(x)及びポリカルボン酸成分(y)の合計に対して、10モル%以下の範囲で、上記以外の他のモノマー、例えば、安息香酸、p−置換安息香酸、o−置換安息香酸、酢酸、プロピオン酸、酪酸等及びこれらのメチル、エチルエステル等及びこれらの酸無水物等のモノカルボン酸;ベンジルアルコール、p−置換ベンジルアルコール、o−置換ベンジルアルコール、ラウリルアルコール、ミリスチルアルコール、ステアリルアルコール等のモノオール類、ε−カプロラクトン、メチルバレロラクトン等及びその開環重合物等のヒドロキシカルボン酸誘導体類等を使用することもできる。
ポリエステル樹脂(p1)、又は(p1)を構成単位として有する樹脂(p2)の数平均分子量(ゲルパーミエーションクロマトグラフィーにて測定、以下Mnと略記)は、好ましくは1,000〜50万、更に好ましくは1,500〜20万である。
(p1)及び(p2)の融点(DSCにて測定される。以下同じである。)は、好ましくは0℃〜200℃、更に好ましくは、35℃〜150℃である。
(p1)及び(p2)の融点(DSCにて測定される。以下同じである。)は、好ましくは0℃〜200℃、更に好ましくは、35℃〜150℃である。
(p1)及び(p2)のガラス転移温度(Tg)(DSCにて測定される。以下同じである。)は、好ましくは−60℃〜100℃、更に好ましくは−30℃〜60℃である。
ポリエステル樹脂(p1)及び樹脂(p2)のSP値(SP値はPolymer Engineering and Science,Feburuary,1974,Vol.14,No.2 P.147〜154に記載された方法によって計算される。)は、好ましくは7〜18、更に好ましくは8〜14である。
ポリエステル樹脂(p1)及び樹脂(p2)のSP値(SP値はPolymer Engineering and Science,Feburuary,1974,Vol.14,No.2 P.147〜154に記載された方法によって計算される。)は、好ましくは7〜18、更に好ましくは8〜14である。
本発明においてポリエステル樹脂(p1)は、通常のポリエステル製造法と同様にして製造することができる。例えば、不活性ガス(窒素ガス等)雰囲気中で、反応温度が好ましくは150〜280℃、更に好ましくは160〜260℃、特に好ましくは170〜240℃で反応させることにより行うことができる。また反応時間は、重縮合反応を確実に行う観点から、好ましくは30分以上、特に2〜40時間である。
また、脂肪族ジオール成分の一部、又はポリカルボン酸の低級アルキルエステルに由来する炭素数1〜4のアルコールを系外に留出除去させながら重縮合を行ってもよい。
更に反応末期の反応速度を向上させるために減圧することも有効である。
また、脂肪族ジオール成分の一部、又はポリカルボン酸の低級アルキルエステルに由来する炭素数1〜4のアルコールを系外に留出除去させながら重縮合を行ってもよい。
更に反応末期の反応速度を向上させるために減圧することも有効である。
重縮合反応の反応速度を向上させるためには、エステル化触媒を使用するのが好ましい。エステル化触媒の例には、スズ含有触媒(例えばジブチルスズオキシド、ジオクチルスズオキシド、ジオクチルスズジラウレート)、アンチモン含有触媒(例えば三酸化アンチモン)、チタン含有触媒(t)(後述)、ジルコニウム含有触媒(例えば酢酸ジルコニル)、ニッケル含有触媒(例えばニッケルアセチルアセトナート)、アルミニウム含有触媒(例えば水酸化アルミニウム、アルミニウムトリイソプロポキシド)、及び酢酸亜鉛、酢酸マンガン等が挙げられる。これらの中では、反応性と環境衛生の点から、チタン、アンチモン、ジルコニウム、ニッケル、及びアルミニウムから選ばれる1種以上の金属を含有する触媒が好ましく、ポリエステル樹脂中の低分子量成分の量が少なくなり、且つ現像性が良好であることから、チタン含有触媒(t)が更に好ましい。
触媒の添加量は、反応速度が最大になるように適宜決定することが望ましい。添加量としては、全原料に対し、好ましくは10ppm〜1.9%、更に好ましくは100ppm〜1.7%である。添加量を10ppm以上とすることで反応速度が大きくなる点で好ましい。
上記及び以下において、%は、特に断りのない場合は、重量%を意味する。
上記及び以下において、%は、特に断りのない場合は、重量%を意味する。
以下にチタン含有触媒(t)について詳述する。
チタン含有触媒(t)としては、チタンテトラブトキシド等のチタンアルコキシドでもよいが、トナー化したときの帯電特性と耐ブロッキング性の観点から、ハロゲン化チタン(t1)、チタンジケトンエノレート(t2)、カルボン酸チタン(t3)、カルボン酸チタニル(t4)、カルボン酸チタニル塩(t5)、下記一般式(I)又は(II)で表されるチタン含有化合物(t6)、及び下記一般式(III)で表されるチタン含有化合物(t7)からなる群より選ばれる少なくとも1種のチタン含有触媒(t)を用いるのが特に好ましい。
Ti(−X)m(−OH)n (I)
O=Ti(−X)p(−OR1)q (II)
Ti(−Z)r(−OR2)s (III)
チタン含有触媒(t)としては、チタンテトラブトキシド等のチタンアルコキシドでもよいが、トナー化したときの帯電特性と耐ブロッキング性の観点から、ハロゲン化チタン(t1)、チタンジケトンエノレート(t2)、カルボン酸チタン(t3)、カルボン酸チタニル(t4)、カルボン酸チタニル塩(t5)、下記一般式(I)又は(II)で表されるチタン含有化合物(t6)、及び下記一般式(III)で表されるチタン含有化合物(t7)からなる群より選ばれる少なくとも1種のチタン含有触媒(t)を用いるのが特に好ましい。
Ti(−X)m(−OH)n (I)
O=Ti(−X)p(−OR1)q (II)
Ti(−Z)r(−OR2)s (III)
式(I)及び(II)中、Xは炭素数2〜12のモノ若しくはポリアルカノールアミンから1個のOH基のHを除いた残基であり、ポリアルカノールアミンの場合、他のOH基が同一のTi原子に直接結合したOH基と分子内で重縮合し環構造を形成していてもよく、他のTi原子に直接結合したOH基と分子間で重縮合し繰り返し構造を形成していてもよい。繰り返し構造を形成する場合の重合度は2〜5である。R1はH、又は1〜3個のエーテル結合を含んでいてもよい炭素数1〜8のアルキル基である。mは1〜4の整数、nは0〜3の整数、mとnの和は4である。pは1又は2の整数、qは0〜1の整数、pとqの和は2である。m又はpが2以上の場合、一般式(I)中又は(II)中、それぞれのXは同一であっても異なっていてもよい。
式(III)中、R2はH、又は1〜3個のエーテル結合及び/若しくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。Zは芳香族モノ若しくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、他のカルボキシル基が同一分子内のOR基と分子内で重縮合し環構造を形成していてもよく、又は、別の分子のOR基と分子間で重縮合し2〜5個のTi原子を含む構造を形成していてもよい。r及びsはそれぞれ1〜3の整数であり、rとsの和は4である。
(t)の使用量としては特に限定されないが、ポリエステル樹脂(p1)を得るのに用いるポリオール成分(x)とポリカルボン酸成分(y)の合計重量を基準として、下限は0.01%が好ましく、0.02%が更に好ましく、0.03%が特に好ましく、0.05%が最も好ましい。上限は5%が好ましく、2%が更に好ましく、1.5%が特に好ましく、0.8%が最も好ましい。0.01%以上では重縮合触媒としての作用が十分得られ、5%以下であると、触媒量に応じて高い触媒作用が得られる。また上記触媒量の範囲内であれば、得られるポリエステル樹脂をトナーバインダーとして用いた場合のトナーの、必要な諸特性、特に低温低湿度条件下での感光体の画質がより良好となる。
(t)のうち、ハロゲン化チタン(t1)としては、例えば、ジクロロチタン、トリクロロチタン、テトラクロロチタン、トリフルオロチタン、テトラフルオロチタン、テトラブロモチタン等が挙げられる。
チタンジケトンエノレート(t2)としては、例えば、チタンアセチルアセトナート、チタンジイソプロポキシドビスアセチルアセトナート、チタニルアセチルアセトナート等が挙げられる。これら(t2)の中ではチタンアセチルアセトナートが好ましい。
カルボン酸チタン(t3)としては、例えば、炭素数1〜32の脂肪族カルボン酸チタン(t3−1)、炭素数7〜38の芳香族カルボン酸チタン(t3−2)等が挙げられる。2価以上のポリカルボン酸チタンの場合、チタンに配位するカルボキシル基は、1個でも2個以上でもよく、チタンに配位せず遊離のカルボキシル基が存在していてもよい。
(t3−1)としては、例えば、脂肪族モノカルボン酸チタン(t3−1a)、脂肪族ジカルボン酸チタン(t3−1b)、脂肪族トリカルボン酸チタン(t3−1c)及び4〜8価又はそれ以上の脂肪族ポリカルボン酸チタン(t3−1d)等が挙げられる。
(t3−1a)としては、例えば、ぎ酸チタン、酢酸チタン、プロピオン酸チタン、オクタン酸チタン等が挙げられる。(t3−1b)としては、例えば、シュウ酸チタン、コハク酸チタン、マレイン酸チタン、アジピン酸チタン、セバシン酸チタン等が挙げられる。(t3−1c)としては、例えば、ヘキサントリカルボン酸チタン、イソオクタントリカルボン酸チタン等が挙げられる。(t3−1d)としては、例えば、オクタンテトラカルボン酸チタン、デカンテトラカルボン酸チタン等が挙げられる。
(t3−2)としては、例えば、芳香族モノカルボン酸チタン(t3−2a)、芳香族ジカルボン酸チタン(t3−2b)、芳香族トリカルボン酸チタン(t3−2c)及び4〜8価又はそれ以上の芳香族ポリカルボン酸チタン(t3−2d)等が挙げられる。
(t3−2a)としては、例えば、安息香酸チタン等が挙げられる。(t3−2b)としては、例えば、フタル酸チタン、テレフタル酸チタン、イソフタル酸チタン、1,3−ナフタレンジカルボン酸チタン、4,4−ビフェニルジカルボン酸チタン、2,5−トルエンジカルボン酸チタン、アントラセンジカルボン酸チタン等が挙げられる。(t3−2c)としては、例えば、トリメリット酸チタン、2,4,6−ナフタレントリカルボン酸チタン等が挙げられる。(t3−2d)としては、例えば、ピロメリット酸チタン、2,3,4,6−ナフタレンテトラカルボン酸チタン等が挙げられる。
これら(t3)の中では(t3−2)が好ましく、(t3−2b)が更に好ましい。
(t3−2a)としては、例えば、安息香酸チタン等が挙げられる。(t3−2b)としては、例えば、フタル酸チタン、テレフタル酸チタン、イソフタル酸チタン、1,3−ナフタレンジカルボン酸チタン、4,4−ビフェニルジカルボン酸チタン、2,5−トルエンジカルボン酸チタン、アントラセンジカルボン酸チタン等が挙げられる。(t3−2c)としては、例えば、トリメリット酸チタン、2,4,6−ナフタレントリカルボン酸チタン等が挙げられる。(t3−2d)としては、例えば、ピロメリット酸チタン、2,3,4,6−ナフタレンテトラカルボン酸チタン等が挙げられる。
これら(t3)の中では(t3−2)が好ましく、(t3−2b)が更に好ましい。
カルボン酸チタニル(t4)としては、例えば、炭素数1〜32の脂肪族カルボン酸チタニル(t4−1)、炭素数7〜38の芳香族カルボン酸チタニル(t4−2)等が挙げられる。2価以上のポリカルボン酸チタニルの場合、チタンに配位するカルボキシル基は、1個でも2個以上でもよく、チタンに配位せず遊離のカルボキシル基が存在していてもよい。
(t4−1)としては、例えば、脂肪族モノカルボン酸チタニル(t4−1a)、脂肪族ジカルボン酸チタニル(t4−1b)、脂肪族トリカルボン酸チタニル(t4−1c)及び4〜8価又はそれ以上の脂肪族ポリカルボン酸チタニル(t4−1d)等が挙げられる。
(t4−1a)としては、例えば、ぎ酸チタニル、酢酸チタニル、プロピオン酸チタニル、オクタン酸チタニル等が挙げられる。(t4−1b)としては、例えば、シュウ酸チタニル、コハク酸チタニル、マレイン酸チタニル、アジピン酸チタニル、セバシン酸チタニル等が挙げられる。(t4−1c)としては、例えば、ヘキサントリカルボン酸チタニル、イソオクタントリカルボン酸チタニル等が挙げられる。(t4−1d)としては、例えば、オクタンテトラカルボン酸チタニル、デカンテトラカルボン酸チタニル等が挙げられる。
(t4−1a)としては、例えば、ぎ酸チタニル、酢酸チタニル、プロピオン酸チタニル、オクタン酸チタニル等が挙げられる。(t4−1b)としては、例えば、シュウ酸チタニル、コハク酸チタニル、マレイン酸チタニル、アジピン酸チタニル、セバシン酸チタニル等が挙げられる。(t4−1c)としては、例えば、ヘキサントリカルボン酸チタニル、イソオクタントリカルボン酸チタニル等が挙げられる。(t4−1d)としては、例えば、オクタンテトラカルボン酸チタニル、デカンテトラカルボン酸チタニル等が挙げられる。
(t4−2)としては、例えば、芳香族モノカルボン酸チタニル(t4−2a)、芳香族ジカルボン酸チタニル(t4−2b)、芳香族トリカルボン酸チタニル(t4−2c)及び4〜8価又はそれ以上の芳香族ポリカルボン酸チタニル(t4−2d)等が挙げられる。
(t4−2a)としては、例えば、安息香酸チタニル等が挙げられる。(t4−2b)としては、例えば、フタル酸チタニル、テレフタル酸チタニル、イソフタル酸チタニル、1,3−ナフタレンジカルボン酸チタニル、4,4−ビフェニルジカルボン酸チタニル、2,5−トルエンジカルボン酸チタニル、アントラセンジカルボン酸チタニル等が挙げられる。(t4−2c)としては、例えば、トリメリット酸チタニル、2,4,6−ナフタレントリカルボン酸チタニル等が挙げられる。(t4−2d)としては、例えば、ピロメリット酸チタニル、2,3,4,6−ナフタレンテトラカルボン酸チタニル等が挙げられる。
(t4−2a)としては、例えば、安息香酸チタニル等が挙げられる。(t4−2b)としては、例えば、フタル酸チタニル、テレフタル酸チタニル、イソフタル酸チタニル、1,3−ナフタレンジカルボン酸チタニル、4,4−ビフェニルジカルボン酸チタニル、2,5−トルエンジカルボン酸チタニル、アントラセンジカルボン酸チタニル等が挙げられる。(t4−2c)としては、例えば、トリメリット酸チタニル、2,4,6−ナフタレントリカルボン酸チタニル等が挙げられる。(t4−2d)としては、例えば、ピロメリット酸チタニル、2,3,4,6−ナフタレンテトラカルボン酸チタニル等が挙げられる。
カルボン酸チタニル塩(t5)としては、例えば、(t4−1b)、(t4−1c)、(t4−1d)、(t4−2b)、(t4−2c)、又は(t4−2d)に挙げたカルボン酸チタニルの、アルカリ金属(リチウム、ナトリウム、カリウム等)塩若しくはアルカリ土類金属(マグネシウム、カルシウム、バリウム等)塩〔(t5−1b)、(t5−1c)、(t5−1d)、(t5−2b)、(t5−2c)、及び(t5−2d)〕等が挙げられる。これら(t5)の中では、マレイン酸チタニル塩及びシュウ酸チタニル塩が好ましい。
前記一般式(I)又は(II)で表される触媒(t6)において、Xは炭素数2〜12のモノ若しくはポリアルカノールアミンから1個のOH基のH原子を除いた残基であり、窒素原子の数、即ち、1級、2級、及び3級アミノ基の合計数は、通常1〜2個、好ましくは1個である。
上記モノアルカノールアミンとしては、エタノールアミン、及びプロパノールアミン等が挙げられる。ポリアルカノールアミンとしては、ジアルカノールアミン(ジエタノールアミン、N−メチルジエタノールアミン、及びN−ブチルジエタノールアミン等)、トリアルカノールアミン(トリエタノールアミン、及びトリプロパノールアミン等)、及びテトラアルカノールアミン(N,N,N’,N’−テトラヒドロキシエチルエチレンジアミン等)が挙げられる。
ポリアルカノールアミンの場合、Ti原子とTi−O−C結合を形成するのに用いられるHを除いた残基となるOH基以外にOH基が1個以上存在し、それが同一のTi原子に直接結合したOH基と分子内で重縮合し環構造を形成していてもよく、他のTi原子に直接結合したOH基と分子間で重縮合し繰り返し構造を形成していてもよい。繰り返し構造を形成する場合の重合度は2〜5である。重合度が6以上の場合、触媒活性が低下するためオリゴマー成分が増え、樹脂粒子のブロッキング性悪化の原因になる。
Xとして好ましいものは、モノアルカノールアミン(特にエタノールアミン)の残基、ジアルカノールアミン(特にジエタノールアミン)の残基、及びトリアルカノールアミン(特にトリエタノールアミン)の残基であり、特に好ましいものはトリエタノールアミンの残基である。
R1はH、又は1〜3個のエーテル結合を含んでいてもよい炭素数1〜8のアルキル基である。炭素数1〜8のアルキル基の具体例としては、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、n−ヘキシル基、n−オクチル基、β−メトキシエチル基、及びβ−エトキシエチル基等が挙げられる。これらR1のうち好ましくは、H、及びエーテル結合を含まない炭素数1〜4のアルキル基であり、更に好ましくは、H、エチル基、及びイソプロピル基である。
式(I)中、mは1〜4の整数であり、好ましくは2〜4の整数である。nは0〜3の整数であり、好ましくは0〜2の整数である。mとnの和は4である。
式(II)中、pは1又は2、qは0又は1であり、pとqの和は2である。m又はpが2以上の場合、複数存在するXは同一であっても異なっていてもよいが、すべて同一である方が好ましい。
式(II)中、pは1又は2、qは0又は1であり、pとqの和は2である。m又はpが2以上の場合、複数存在するXは同一であっても異なっていてもよいが、すべて同一である方が好ましい。
チタン含有触媒(t6)のうち、一般式(I)で表されるものの具体例としては、チタニウムテトラキス(モノエタノールアミネート)、チタニウムモノヒドロキシトリス(トリエタノールアミネート)、チタニウムジヒドロキシビス(トリエタノールアミネート)、チタニウムトリヒドロキシトリエタノールアミネート、チタニウムジヒドロキシビス(ジエタノールアミネート)、チタニウムジヒドロキシビス(モノエタノールアミネート)、チタニウムジヒドロキシビス(モノプロパノールアミネート)、チタニウムジヒドロキシビス(N−メチルジエタノールアミネート)、チタニウムジヒドロキシビス(N−ブチルジエタノールアミネート)、テトラヒドロキシチタンとN,N,N’,N’−テトラヒドロキシエチルエチレンジアミンとの反応生成物、及びこれらの分子内又は分子間重縮合物が挙げられる。
分子内又は分子間重縮合物の例としては、下記一般式(I−1)、(I−2)、又は(I−3)で表される化合物等が挙げられる。
分子内又は分子間重縮合物の例としては、下記一般式(I−1)、(I−2)、又は(I−3)で表される化合物等が挙げられる。
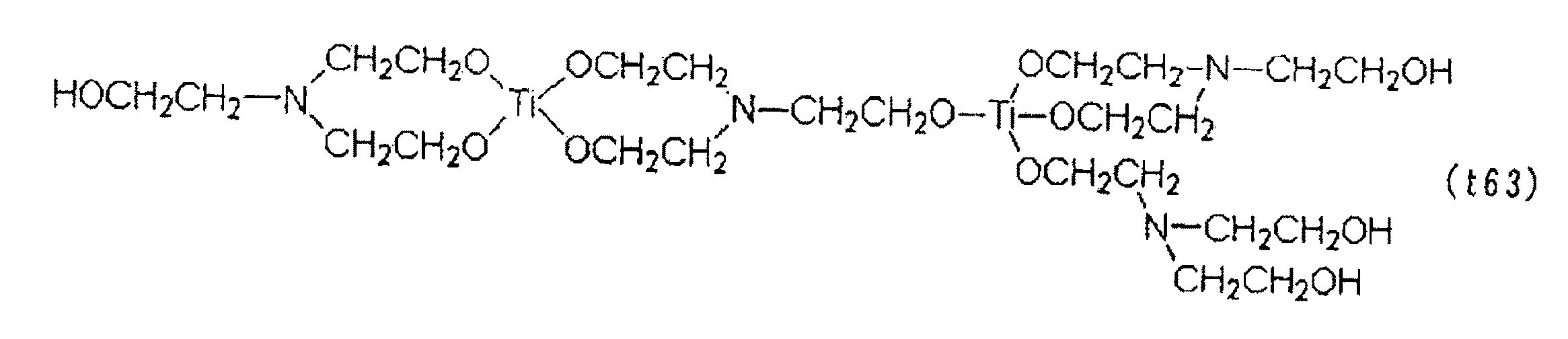
[式中、Q1及びQ6はH、又は炭素数1〜4のアルキル基若しくはヒドロキシアルキル基である。Q2〜Q5及びQ7〜Q9は炭素数1〜6のアルキレン基である。Xは炭素数2〜12のモノ若しくはポリアルカノールアミンから1個のOH基のHを除いた残基である。]
一般式(II)で表されるものの具体例としては、チタニルビス(トリエタノールアミネート)、チタニルビス(ジエタノールアミネート)、チタニルビス(モノエタノールアミネート)、チタニルヒドロキシエタノールアミネート、チタニルヒドロキシトリエタノールアミネート、チタニルエトキシトリエタノールアミネート、チタニルイソプロポキシトリエタノールアミネート、及びこれらの分子内又は分子間重縮合物が挙げられる。分子内又は分子間重縮合物の例としては、下記一般式(II−1)又は(II−2)で表される化合物等が挙げられる。
[式中、Q1及びQ6はH、又は炭素数1〜4のアルキル基若しくはヒドロキシアルキル基である。Q2〜Q5は炭素数1〜6のアルキレン基である。]
これら(t6)のうちで好ましいものは、チタニウムジヒドロキシビス(トリエタノールアミネート)、チタニウムジヒドロキシビス(ジエタノールアミネート)、チタニウムモノヒドロキシトリス(トリエタノールアミネート)、チタニウムテトラキス(エタノールアミネート)、チタニルヒドロキシトリエタノールアミネート、チタニルビス(トリエタノールアミネート)、チタニウムジヒドロキシビス(トリエタノールアミネート)の分子内重縮合物〔下記(t61)〕若しくは分子間重縮合物〔下記(t63)〕、チタニウムモノヒドロキシトリス(トリエタノールアミネート)の分子内重縮合物〔下記(t62)〕、及びこれらの併用であり、更に好ましくは、チタニウムジヒドロキシビス(トリエタノールアミネート)、チタニウムモノヒドロキシトリス(トリエタノールアミネート)、それらの分子内重縮合物〔(t61)及び(t62)〕、特に(t61)である。
これらのチタン含有触媒(t6)は、例えば市販されているチタニウムジアルコキシビス(アルコールアミネート)(Dupont製等)を、水存在下で70〜90℃にて反応させること、あるいは、市販されているチタニウムアルコキシド(日本曹達株式会社製チタニウムテトライソプロポキシド等)をアルコキシアミンと水存在下で20〜90℃にて反応させること、で安定的に得ることができる。また、重縮合物は、更に100℃にて縮合水を減圧留去することで得ることができる。
前記一般式(III)で表される触媒(t7)において、R2はH、又は1〜3個のエーテル結合及び/若しくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。炭化水素基の炭素数は、好ましくは1〜6、更に好ましくは1〜4である。
1〜3個のエーテル結合及び/若しくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基の具体例としては、脂肪族炭化水素基並びにエーテル結合及び/若しくは水酸基を含む脂肪族炭化水素基(メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、n−ヘキシル基、n−オクチル基、β−メトキシエチル基、β−エトキシエチル基、及びβ−ヒドロキシエチル基等)、芳香族炭化水素基並びにエーテル結合及び/若しくは水酸基を含む芳香族炭化水素基[フェニル基;ヒドロキシフェニル基;ビスフェノールA、ビスフェノールF及びビスフェノールS等の炭素数2〜4のエチレンオキサイド(以下、EOと記載)、プロピレンオキサイド(以下、POと記載)、及びブチレンキサイド等〕付加物(付加モル数1〜3)から1個のOHを除いた残基等]が挙げられる。
これらR2のうち好ましくは、炭素数1〜6の炭化水素基であり、更に好ましくは、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、及びn−ヘキシル基であり、特に好ましくは、n−プロピル基、イソプロピル基、及びn−ブチル基である。
これらR2のうち好ましくは、炭素数1〜6の炭化水素基であり、更に好ましくは、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、及びn−ヘキシル基であり、特に好ましくは、n−プロピル基、イソプロピル基、及びn−ブチル基である。
Zは芳香族モノ若しくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、Ti原子に結合し残基を形成するのと別のカルボキシル基が、同一分子内のOR2基{Ti原子に直接結合した水酸基(R2がHの場合)、アルコキシ基(R2が炭化水素基の場合)、又はR2が1〜2個の水酸基を含む炭化水素基の場合の該水酸基}と分子内で重縮合し環構造を形成していてもよく、チタン含有触媒(t7)の別の分子のOR2基(上記と同様)と分子間で重縮合し、複数のTi原子を含む繰り返し構造を形成していてもよい。
上記芳香族カルボン酸としては、炭素数7〜50のものが好ましく、安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸等)、ナフタレンモノカルボン酸等の芳香族モノカルボン酸;フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸等)、ナフタレンジカルボン酸、トリメリット酸、及びピロメリット酸等の2〜6価又はそれ以上の芳香族ポリカルボン酸;が挙げられる。
上記芳香族カルボン酸としては、炭素数7〜50のものが好ましく、安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸等)、ナフタレンモノカルボン酸等の芳香族モノカルボン酸;フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸等)、ナフタレンジカルボン酸、トリメリット酸、及びピロメリット酸等の2〜6価又はそれ以上の芳香族ポリカルボン酸;が挙げられる。
芳香族ポリカルボン酸の場合、前述のようにその複数のカルボキシル基により、複数のTi原子を含む繰り返し構造を形成していてもよいが、この場合の1分子内のTi原子数は2〜5である。1分子内のTi原子数が6以上の場合、触媒活性が低下し好ましくない。
Zとして好ましいものは、フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸等)の残基、及び安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸等)の残基であり、特に好ましいものはテレフタル酸、イソフタル酸、及びオルトフタル酸の残基である。
Zとして好ましいものは、フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸等)の残基、及び安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸等)の残基であり、特に好ましいものはテレフタル酸、イソフタル酸、及びオルトフタル酸の残基である。
式(III)中、r及びsはそれぞれ1〜3の整数であり、rとsの和、即ちTi原子の結合価数は4である。好ましくは、rは1又は2、sは2又は3である。rが3を超えると触媒活性が低下し、sが3を超えると耐加水分解性が低下し、いずれもポリエステル製造上好ましくない。rが1又は2の場合、触媒活性が特に高く好ましい。Ti原子の結合価数が4以外の場合は、式(III)と類似の構造でも触媒活性が劣るか副反応が起き好ましくない。
一般式(III)で表される化合物の具体例としては、チタントリイソプロポキシベンゼンカルボキシレート、チタントリブトキシベンゼンカルボキシレート、チタントリイソプロポキシテレフタレート、チタントリブトキシテレフタレート、チタントリイソプロポキシイソフタレート、チタントリイソプロポキシフタレート、チタンジイソプロポキシジベンゼンカルボキシレート、チタンジブトキシジベンゼンカルボキシレート、チタンジイソプロポキシジテレフタレート、チタンジブトキシジテレフタレート、チタンジイソプロポキシジイソフタレート、チタンジイソプロポキシジフタレート、チタンジヒドロキシジベンゼンカルボキシレート、チタンジヒドロキシジテレフタレート、チタンジヒドロキシジイソフタレート、チタンジヒドロキシジフタレート、及びこれらの分子内又は分子間重縮合物等が挙げられる。
本発明に用いるチタン含有触媒(t7)は、ポリエステル重合時の触媒活性の観点から、30℃の水への溶解度が5g/100ml以下であることが好ましく、2g/100ml以下であることが更に好ましく、1g/100ml以下であることが特に好ましい。溶解度が5g/100ml以下であると、重合反応時に触媒が加水分解を受けにくく、触媒活性の持続性の観点から好ましい。
これらのチタン含有触媒(t7)は、例えば、市販されているチタンテトラアルコキシドと芳香族カルボン酸を、酢酸エチル中で70〜90℃にて反応させることで得ることができる。
これらの(t)の中で好ましいのは、チタンジケトンエノレート(t2)、カルボン酸チタン(t3)、カルボン酸チタニル塩(t5)、一般式(I)又は(II)で表されるチタン含有化合物(t6)、及び一般式(III)で表されるチタン含有化合物(t7)であり、更に好ましいのはカルボン酸チタン(t3)、一般式(I)又は(II)で表されるチタン含有化合物(t6)、及び一般式(III)で表されるチタン含有化合物(t7)である。
ポリオールとポリカルボン酸の反応比率は、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、好ましくは2/1〜1/2、更に好ましくは1.5/1〜1/1.3、特に好ましくは1.3/1〜1/1.2である。
ポリエステル樹脂(p1)を構成単位として有する樹脂(p2)としては、(p1)と後述するポリイソシアネート(15)から得られるポリウレタン樹脂、(p1)と後述するポリエポキシド(18)から得られるエポキシ樹脂、(p1)と後述するポリアミン(16)から得られるポリアミド樹脂等があげられる。
(p1)と(p2)は併用してもよい。
これら(p2)のうち好ましいものは、ポリウレタン樹脂及びエポキシ樹脂であり、更に好ましいものは、ポリウレタン樹脂である。
(p1)と(p2)は併用してもよい。
これら(p2)のうち好ましいものは、ポリウレタン樹脂及びエポキシ樹脂であり、更に好ましいものは、ポリウレタン樹脂である。
樹脂(b)がポリエステル樹脂(p1)及び/又はポリエステル樹脂(p1)を構成単位として有する樹脂(p2)を含有する場合、ポリエステル樹脂(p1)又はポリエステル樹脂(p1)を構成単位として有する樹脂(p2)として好ましいものは、各種被着体との密着性が良好である点から、ポリエステル樹脂(p1)及び/又はポリエステル樹脂(p1)を構成単位として有するポリウレタン樹脂である。
樹脂(b)は、ポリエステル樹脂(p1)又はポリエステル樹脂(p1)を構成単位として有する樹脂(p2)以外に、必要によりポリウレタン樹脂、エポキシ樹脂、ビニル樹脂及びポリエステル樹脂(p1)以外のポリエステル樹脂からなる群から選ばれる1種以上の樹脂を含有してもよい。また、樹脂(b)には、上記の樹脂以外にもポリアミド樹脂、ポリイミド樹脂、ケイ素樹脂、フェノール樹脂、メラミン樹脂、ユリア樹脂、アニリン樹脂、アイオノマー樹脂、ポリカーボネート樹脂等を含有していてもよい。
ビニル樹脂、ポリウレタン樹脂、エポキシ樹脂及びポリエステル樹脂(p1)以外のポリエステル樹脂について説明するが、他の樹脂についてもこれらの樹脂と同様にして使用できる。
ビニル樹脂は、ビニルモノマーを単独重合又は共重合したポリマーである。重合には、公知の重合触媒等が使用できる。
ビニルモノマーとしては、下記(5)〜(14)等が挙げられる。
ビニル樹脂は、ビニルモノマーを単独重合又は共重合したポリマーである。重合には、公知の重合触媒等が使用できる。
ビニルモノマーとしては、下記(5)〜(14)等が挙げられる。
(5)ビニル炭化水素:
(5−1)脂肪族ビニル炭化水素:炭素数2〜12のアルケン(例えばエチレン、プロピレン、ブテン、イソブチレン、ペンテン、ヘプテン、ジイソブチレン、オクテン、ドデセン、オクタデセン及び炭素数3〜24のα−オレフィン);炭素数4〜12のアルカジエン(例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン及び1,7−オクタジエン)。
(5−2)脂環式ビニル炭化水素:炭素数6〜15のモノ−又はジ−シクロアルケン(例えばシクロヘキセン、ビニルシクロヘキセン及びエチリデンビシクロヘプテン等)、炭素数5〜12のモノ−又はジ−シクロアルカジエン(例えば、(ジ)シクロペンタジエン等);及びテルペン(例えばピネン、リモネン及びインデン等)等。
(5−3)芳香族ビニル炭化水素:スチレン;スチレンの炭化水素(炭素数1〜24の、アルキル、シクロアルキル、アラルキル及び/又はアルケニル)置換体(例えばα−メチルスチレン、ビニルトルエン、2,4−ジメチルスチレン、エチルスチレン、イソプロピルスチレン、ブチルスチレン、フェニルスチレン、シクロヘキシルスチレン、ベンジルスチレン、クロチルベンゼン、ジビニルベンゼン、ジビニルトルエン、ジビニルキシレン及びトリビニルベンゼン);及びビニルナフタレン等。
(5−1)脂肪族ビニル炭化水素:炭素数2〜12のアルケン(例えばエチレン、プロピレン、ブテン、イソブチレン、ペンテン、ヘプテン、ジイソブチレン、オクテン、ドデセン、オクタデセン及び炭素数3〜24のα−オレフィン);炭素数4〜12のアルカジエン(例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン及び1,7−オクタジエン)。
(5−2)脂環式ビニル炭化水素:炭素数6〜15のモノ−又はジ−シクロアルケン(例えばシクロヘキセン、ビニルシクロヘキセン及びエチリデンビシクロヘプテン等)、炭素数5〜12のモノ−又はジ−シクロアルカジエン(例えば、(ジ)シクロペンタジエン等);及びテルペン(例えばピネン、リモネン及びインデン等)等。
(5−3)芳香族ビニル炭化水素:スチレン;スチレンの炭化水素(炭素数1〜24の、アルキル、シクロアルキル、アラルキル及び/又はアルケニル)置換体(例えばα−メチルスチレン、ビニルトルエン、2,4−ジメチルスチレン、エチルスチレン、イソプロピルスチレン、ブチルスチレン、フェニルスチレン、シクロヘキシルスチレン、ベンジルスチレン、クロチルベンゼン、ジビニルベンゼン、ジビニルトルエン、ジビニルキシレン及びトリビニルベンゼン);及びビニルナフタレン等。
(6)カルボキシル基含有ビニルモノマー及びそれらの塩:
炭素数3〜30の不飽和モノカルボン酸(例えば(メタ)アクリル酸(アクリル酸及び/又はメタクリル酸を表す。以下同様である。)、クロトン酸イソクロトン酸及び桂皮酸);炭素数3〜30の不飽和ジカルボン酸(無水物)(例えば、(無水)マレイン酸、フマル酸、イタコン酸、(無水)シトラコン酸及びメサコン酸);及び炭素数3〜30の不飽和ジカルボン酸のモノアルキル(炭素数1〜24)エステル(例えば、マレイン酸モノメチルエステル、マレイン酸モノオクタデシルエステル、フマル酸モノエチルエステル、イタコン酸モノブチルエステル、イタコン酸グリコールモノエーテル及びシトラコン酸モノエイコシルエステル)等。
カルボキシル基含有ビニルモノマーの塩としては、例えばアルカリ金属塩(ナトリウム塩、カリウム塩等)、アルカリ土類金属塩(カルシウム塩、マグネシウム塩等)、アンモニウム塩、アミン塩若しくは4級アンモニウム塩が挙げられる。アミン塩としては、アミン化合物であれば特に限定されないが、例えば、1級アミン塩(エチルアミン塩、ブチルアミン塩、オクチルアミン塩等)、2級アミン(ジエチルアミン塩、ジブチルアミン塩等)、3級アミン(トリエチルアミン塩、トリブチルアミン塩等)が挙げられる。4級アンモニウム塩としては、テトラエチルアンモニウム塩、トリエチルラウリルアンモニウム塩、テトラブチルアンモニウム塩、トリブチルラウリルアンモニウム塩等)が挙げられる。
カルボキシル基含有ビニルモノマーの塩の具体例としては、アクリル酸ナトリウム、メタクリル酸ナトリウム、マレイン酸モノナトリウム、マレイン酸ジナトリウム、アクリル酸カリウム、メタクリル酸カリウム、マレイン酸モノカリウム、アクリル酸リチウム、アクリル酸セシウム、アクリル酸アンモニウム、アクリル酸カルシウム及びアクリル酸アルミニウム等が挙げられる。
炭素数3〜30の不飽和モノカルボン酸(例えば(メタ)アクリル酸(アクリル酸及び/又はメタクリル酸を表す。以下同様である。)、クロトン酸イソクロトン酸及び桂皮酸);炭素数3〜30の不飽和ジカルボン酸(無水物)(例えば、(無水)マレイン酸、フマル酸、イタコン酸、(無水)シトラコン酸及びメサコン酸);及び炭素数3〜30の不飽和ジカルボン酸のモノアルキル(炭素数1〜24)エステル(例えば、マレイン酸モノメチルエステル、マレイン酸モノオクタデシルエステル、フマル酸モノエチルエステル、イタコン酸モノブチルエステル、イタコン酸グリコールモノエーテル及びシトラコン酸モノエイコシルエステル)等。
カルボキシル基含有ビニルモノマーの塩としては、例えばアルカリ金属塩(ナトリウム塩、カリウム塩等)、アルカリ土類金属塩(カルシウム塩、マグネシウム塩等)、アンモニウム塩、アミン塩若しくは4級アンモニウム塩が挙げられる。アミン塩としては、アミン化合物であれば特に限定されないが、例えば、1級アミン塩(エチルアミン塩、ブチルアミン塩、オクチルアミン塩等)、2級アミン(ジエチルアミン塩、ジブチルアミン塩等)、3級アミン(トリエチルアミン塩、トリブチルアミン塩等)が挙げられる。4級アンモニウム塩としては、テトラエチルアンモニウム塩、トリエチルラウリルアンモニウム塩、テトラブチルアンモニウム塩、トリブチルラウリルアンモニウム塩等)が挙げられる。
カルボキシル基含有ビニルモノマーの塩の具体例としては、アクリル酸ナトリウム、メタクリル酸ナトリウム、マレイン酸モノナトリウム、マレイン酸ジナトリウム、アクリル酸カリウム、メタクリル酸カリウム、マレイン酸モノカリウム、アクリル酸リチウム、アクリル酸セシウム、アクリル酸アンモニウム、アクリル酸カルシウム及びアクリル酸アルミニウム等が挙げられる。
(7)スルホ基含有ビニルモノマー及びそれらの塩:
炭素数2〜14のアルケンスルホン酸(例えばビニルスルホン酸、(メタ)アリルスルホン酸及びメチルビニルスルホン酸);スチレンスルホン酸及びこのアルキル(炭素数2〜24)誘導体(例えばα−メチルスチレンスルホン酸);炭素数5〜18のスルホ(ヒドロキシ)アルキル−(メタ)アクリレート(例えば、スルホプロピル(メタ)アクリレート、2−ヒドロキシ−3−(メタ)アクリロキシプロピルスルホン酸、2−(メタ)アクリロイルオキシエタンスルホン酸及び3−(メタ)アクリロイルオキシ−2−ヒドロキシプロパンスルホン酸);炭素数5〜18のスルホ(ヒドロキシ)アルキル(メタ)アクリルアミド(例えば、2−(メタ)アクリロイルアミノ−2,2−ジメチルエタンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸及び3−(メタ)アクリルアミド−2−ヒドロキシプロパンスルホン酸);アルキル(炭素数3〜18)アリルスルホコハク酸(例えば、プロピルアリルスルホコハク酸、ブチルアリルスルホコハク酸、2−エチルヘキシル−アリルスルホコハク酸);ポリ〔n(重合度、以下同様)=2〜30〕オキシアルキレン(オキシエチレン、オキシプロピレン、オキシブチレン:単独、ランダム、ブロックでもよい)モノ(メタ)アクリレートの硫酸エステル[例えば、ポリ(n=5〜15)オキシエチレンモノメタクリレート硫酸エステル、ポリ(n=5〜15)オキシプロピレンモノメタクリレート硫酸エステル];下記一般式(7−1)〜(7−3)で表される化合物;及びこれらの塩等が挙げられる。
尚、塩としては、(6)カルボキシル基含有ビニルモノマーの塩と同様の塩等が用いられる。
炭素数2〜14のアルケンスルホン酸(例えばビニルスルホン酸、(メタ)アリルスルホン酸及びメチルビニルスルホン酸);スチレンスルホン酸及びこのアルキル(炭素数2〜24)誘導体(例えばα−メチルスチレンスルホン酸);炭素数5〜18のスルホ(ヒドロキシ)アルキル−(メタ)アクリレート(例えば、スルホプロピル(メタ)アクリレート、2−ヒドロキシ−3−(メタ)アクリロキシプロピルスルホン酸、2−(メタ)アクリロイルオキシエタンスルホン酸及び3−(メタ)アクリロイルオキシ−2−ヒドロキシプロパンスルホン酸);炭素数5〜18のスルホ(ヒドロキシ)アルキル(メタ)アクリルアミド(例えば、2−(メタ)アクリロイルアミノ−2,2−ジメチルエタンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸及び3−(メタ)アクリルアミド−2−ヒドロキシプロパンスルホン酸);アルキル(炭素数3〜18)アリルスルホコハク酸(例えば、プロピルアリルスルホコハク酸、ブチルアリルスルホコハク酸、2−エチルヘキシル−アリルスルホコハク酸);ポリ〔n(重合度、以下同様)=2〜30〕オキシアルキレン(オキシエチレン、オキシプロピレン、オキシブチレン:単独、ランダム、ブロックでもよい)モノ(メタ)アクリレートの硫酸エステル[例えば、ポリ(n=5〜15)オキシエチレンモノメタクリレート硫酸エステル、ポリ(n=5〜15)オキシプロピレンモノメタクリレート硫酸エステル];下記一般式(7−1)〜(7−3)で表される化合物;及びこれらの塩等が挙げられる。
尚、塩としては、(6)カルボキシル基含有ビニルモノマーの塩と同様の塩等が用いられる。
(8)ホスホノ基含有ビニルモノマー及びその塩:
(メタ)アクリロイルオキシアルキルリン酸モノエステル(アルキル基の炭素数1〜24)(例えば、2−ヒドロキシエチル(メタ)アクリロイルホスフェート及びフェニル−2−アクリロイロキシエチルホスフェート)、(メタ)アクリロイルオキシアルキルホスホン酸(アルキル基の炭素数1〜24)(例えば2−アクリロイルオキシエチルホスホン酸)。
尚、塩としては、(6)カルボキシル基含有ビニルモノマーの塩と同様の塩等が用いられる。
(メタ)アクリロイルオキシアルキルリン酸モノエステル(アルキル基の炭素数1〜24)(例えば、2−ヒドロキシエチル(メタ)アクリロイルホスフェート及びフェニル−2−アクリロイロキシエチルホスフェート)、(メタ)アクリロイルオキシアルキルホスホン酸(アルキル基の炭素数1〜24)(例えば2−アクリロイルオキシエチルホスホン酸)。
尚、塩としては、(6)カルボキシル基含有ビニルモノマーの塩と同様の塩等が用いられる。
(9)ヒドロキシル基含有ビニルモノマー:
ヒドロキシスチレン、N−メチロール(メタ)アクリルアミド、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、(メタ)アリルアルコール、クロチルアルコール、イソクロチルアルコール、1−ブテン−3−オール、2−ブテン−1−オール、2−ブテン−1,4−ジオール、プロパルギルアルコール、2−ヒドロキシエチルプロペニルエーテル、庶糖アリルエーテル等。
ヒドロキシスチレン、N−メチロール(メタ)アクリルアミド、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、(メタ)アリルアルコール、クロチルアルコール、イソクロチルアルコール、1−ブテン−3−オール、2−ブテン−1−オール、2−ブテン−1,4−ジオール、プロパルギルアルコール、2−ヒドロキシエチルプロペニルエーテル、庶糖アリルエーテル等。
(10)含窒素ビニルモノマー:
(10−1)アミノ基含有ビニルモノマー:
アミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、t−ブチルアミノエチルメタクリレート、N−アミノエチル(メタ)アクリルアミド、(メタ)アリルアミン、モルホリノエチル(メタ)アクリレート、4−ビニルピリジン、2−ビニルピリジン、クロチルアミン、N,N−ジメチルアミノスチレン、メチルα−アセトアミノアクリレート、ビニルイミダゾール、N−ビニルピロール、N−ビニルチオピロリドン、N−アリールフェニレンジアミン、アミノカルバゾール、アミノチアゾール、アミノインドール、アミノピロール、アミノイミダゾール、アミノメルカプトチアゾール、これらの塩等。
(10−1)アミノ基含有ビニルモノマー:
アミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、t−ブチルアミノエチルメタクリレート、N−アミノエチル(メタ)アクリルアミド、(メタ)アリルアミン、モルホリノエチル(メタ)アクリレート、4−ビニルピリジン、2−ビニルピリジン、クロチルアミン、N,N−ジメチルアミノスチレン、メチルα−アセトアミノアクリレート、ビニルイミダゾール、N−ビニルピロール、N−ビニルチオピロリドン、N−アリールフェニレンジアミン、アミノカルバゾール、アミノチアゾール、アミノインドール、アミノピロール、アミノイミダゾール、アミノメルカプトチアゾール、これらの塩等。
(10−2)アミド基(カルバモイル基)含有ビニルモノマー:
(メタ)アクリルアミド、N−メチル(メタ)アクリルアミド、N−ブチルアクリルアミド、ジアセトンアクリルアミド、N−メチロール(メタ)アクリルアミド、N,N’−メチレン−ビス(メタ)アクリルアミド、桂皮酸アミド、N,N−ジメチルアクリルアミド、N,N−ジベンジルアクリルアミド、メタクリルホルムアミド、N−メチルN−ビニルアセトアミド、N−ビニルピロリドン等。
(10−3)炭素数3〜10のニトリル基(シアノ基)含有ビニルモノマー:
(メタ)アクリロニトリル、シアノスチレン、シアノアクリレート等。
(10−4)4級アンモニウムカチオンを有する基(第4級アンモニオ基)を含有するビニルモノマー:
トリメチルアンモニオエチル(メタ)アクリレートクロライド、メチルジエチルアンモニオエチル(メタ)アクリレートブロマイド、トリメチルアンモニオエチル(メタ)アクリルアミドメトサルフェート、ベンジルジエチルアンモニオエチル(メタ)アクリルアミドカーボネート、ジメチルジアリルアンモニウムクロライド、トリメチルアリルアンモニウムクロライド等。
(10−5)炭素数8〜12のニトロ基含有ビニルモノマー:
ニトロスチレン等。
(メタ)アクリルアミド、N−メチル(メタ)アクリルアミド、N−ブチルアクリルアミド、ジアセトンアクリルアミド、N−メチロール(メタ)アクリルアミド、N,N’−メチレン−ビス(メタ)アクリルアミド、桂皮酸アミド、N,N−ジメチルアクリルアミド、N,N−ジベンジルアクリルアミド、メタクリルホルムアミド、N−メチルN−ビニルアセトアミド、N−ビニルピロリドン等。
(10−3)炭素数3〜10のニトリル基(シアノ基)含有ビニルモノマー:
(メタ)アクリロニトリル、シアノスチレン、シアノアクリレート等。
(10−4)4級アンモニウムカチオンを有する基(第4級アンモニオ基)を含有するビニルモノマー:
トリメチルアンモニオエチル(メタ)アクリレートクロライド、メチルジエチルアンモニオエチル(メタ)アクリレートブロマイド、トリメチルアンモニオエチル(メタ)アクリルアミドメトサルフェート、ベンジルジエチルアンモニオエチル(メタ)アクリルアミドカーボネート、ジメチルジアリルアンモニウムクロライド、トリメチルアリルアンモニウムクロライド等。
(10−5)炭素数8〜12のニトロ基含有ビニルモノマー:
ニトロスチレン等。
(11)炭素数6〜18のエポキシ基含有ビニルモノマー:
グルシジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート、p−ビニルフェニルフェニルオキサイド等。
グルシジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート、p−ビニルフェニルフェニルオキサイド等。
(12)炭素数2〜16のハロゲン原子含有ビニルモノマー:
塩化ビニル、臭化ビニル、塩化ビニリデン、アリルクロライド、クロルスチレン、ブロムスチレン、ジクロルスチレン、クロロメチルスチレン、テトラフルオロスチレン、クロロプレン等。
塩化ビニル、臭化ビニル、塩化ビニリデン、アリルクロライド、クロルスチレン、ブロムスチレン、ジクロルスチレン、クロロメチルスチレン、テトラフルオロスチレン、クロロプレン等。
(13)ビニルエステル、ビニル(チオ)エーテル、ビニルケトン、ビニルスルホン:
(13−1)炭素数4〜16のビニルエステル;
酢酸ビニル、ビニルブチレート、プロピオン酸ビニル、酪酸ビニル、ジアリルフタレート、ジアリルアジペート、イソプロペニルアセテート、ビニルメタクリレート、メチル4−ビニルベンゾエート、シクロヘキシルメタクリレート、ベンジルメタクリレート、フェニル(メタ)アクリレート、ビニルメトキシアセテート、ビニルベンゾエート、エチルα−エトキシアクリレート、炭素数1〜50のアルキル基を有するアルキル(メタ)アクリレート[メチル(メタ)アクリレート、エチル(メタ)アクリレート、プロピル(メタ)アクリレート、ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ドデシル(メタ)アクリレート、ヘキサデシル(メタ)アクリレート、ヘプタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート等]、ジアルキルフマレート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖若しくは脂環式の基である)、ジアルキルマレエート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖若しくは脂環式の基である)、ポリ(メタ)アリロキシアルカン[ジアリロキシエタン、トリアリロキシエタン、テトラアリロキシエタン、テトラアリロキシプロパン、テトラアリロキシブタン、テトラメタアリロキシエタン等]、ポリアルキレングリコール鎖を有するビニルモノマー[ポリエチレングリコール(数平均分子量300)モノ(メタ)アクリレート、ポリプロピレングリコール(数平均分子量500)モノアクリレート、メチルアルコールEO10モル付加物(メタ)アクリレート、ラウリルアルコールEO30モル付加物(メタ)アクリレート等]、ポリ(メタ)アクリレート[多価アルコールのポリ(メタ)アクリレート:エチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等]等。
(13−1)炭素数4〜16のビニルエステル;
酢酸ビニル、ビニルブチレート、プロピオン酸ビニル、酪酸ビニル、ジアリルフタレート、ジアリルアジペート、イソプロペニルアセテート、ビニルメタクリレート、メチル4−ビニルベンゾエート、シクロヘキシルメタクリレート、ベンジルメタクリレート、フェニル(メタ)アクリレート、ビニルメトキシアセテート、ビニルベンゾエート、エチルα−エトキシアクリレート、炭素数1〜50のアルキル基を有するアルキル(メタ)アクリレート[メチル(メタ)アクリレート、エチル(メタ)アクリレート、プロピル(メタ)アクリレート、ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ドデシル(メタ)アクリレート、ヘキサデシル(メタ)アクリレート、ヘプタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート等]、ジアルキルフマレート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖若しくは脂環式の基である)、ジアルキルマレエート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖若しくは脂環式の基である)、ポリ(メタ)アリロキシアルカン[ジアリロキシエタン、トリアリロキシエタン、テトラアリロキシエタン、テトラアリロキシプロパン、テトラアリロキシブタン、テトラメタアリロキシエタン等]、ポリアルキレングリコール鎖を有するビニルモノマー[ポリエチレングリコール(数平均分子量300)モノ(メタ)アクリレート、ポリプロピレングリコール(数平均分子量500)モノアクリレート、メチルアルコールEO10モル付加物(メタ)アクリレート、ラウリルアルコールEO30モル付加物(メタ)アクリレート等]、ポリ(メタ)アクリレート[多価アルコールのポリ(メタ)アクリレート:エチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等]等。
(13−2)炭素数3〜16のビニル(チオ)エーテル;
ビニルメチルエーテル、ビニルエチルエーテル、ビニルプロピルエーテル、ビニルブチルエーテル、ビニル2−エチルヘキシルエーテル、ビニルフェニルエーテル、ビニル2−メトキシエチルエーテル、メトキシブタジエン、ビニル2−ブトキシエチルエーテル、3,4−ジヒドロ1,2−ピラン、2−ブトキシ−2’−ビニロキシジエチルエーテル、ビニル2−エチルメルカプトエチルエーテル、アセトキシスチレン、フェノキシスチレン。
(13−3)炭素数4〜12のビニルケトン;
ビニルメチルケトン、ビニルエチルケトン、ビニルフェニルケトン。
(13−4)炭素数2〜16のビニルスルホン;
ジビニルサルファイド、p−ビニルジフェニルサルファイド、ビニルエチルサルファイド、ビニルエチルスルホン、ジビニルスルホン及びジビニルスルホキサイド等。
ビニルメチルエーテル、ビニルエチルエーテル、ビニルプロピルエーテル、ビニルブチルエーテル、ビニル2−エチルヘキシルエーテル、ビニルフェニルエーテル、ビニル2−メトキシエチルエーテル、メトキシブタジエン、ビニル2−ブトキシエチルエーテル、3,4−ジヒドロ1,2−ピラン、2−ブトキシ−2’−ビニロキシジエチルエーテル、ビニル2−エチルメルカプトエチルエーテル、アセトキシスチレン、フェノキシスチレン。
(13−3)炭素数4〜12のビニルケトン;
ビニルメチルケトン、ビニルエチルケトン、ビニルフェニルケトン。
(13−4)炭素数2〜16のビニルスルホン;
ジビニルサルファイド、p−ビニルジフェニルサルファイド、ビニルエチルサルファイド、ビニルエチルスルホン、ジビニルスルホン及びジビニルスルホキサイド等。
(14)その他のビニルモノマー:
イソシアナトエチル(メタ)アクリレート、m−イソプロペニル−α,α−ジメチルベンジルイソシアネート等。
イソシアナトエチル(メタ)アクリレート、m−イソプロペニル−α,α−ジメチルベンジルイソシアネート等。
ビニル樹脂のうち、ビニルモノマーを共重合したポリマー(ビニルモノマーの共重合体)としては、上記(5)〜(14)の任意のモノマー同士を、2元又はそれ以上の個数で、任意の割合で共重合したポリマーが用いらる。このようなポリマーとしては例えばスチレン−(メタ)アクリル酸エステル共重合体、スチレン−ブタジエン共重合体、(メタ)アクリル酸−(メタ)アクリル酸エステル共重合体、スチレン−アクリロニトリル共重合体、スチレン−(無水)マレイン酸共重合体、スチレン−(メタ)アクリル酸共重合体、スチレン−(メタ)アクリル酸−ジビニルベンゼン共重合体及びスチレン−スチレンスルホン酸−(メタ)アクリル酸エステル共重合体等が挙げられる。
ポリウレタン樹脂としては、ポリイソシアネート(15)と活性水素化合物(D){水、ポリオール[前記ジオール(1)及び3〜8価のポリオール(2)]、前記ジカルボン酸(3)、前記3〜6価のポリカルボン酸(4)、ポリアミン(16)、ポリチオール(17)等}との重付加物等が挙げられる。
重付加には、公知の重合触媒等が使用できる。
重付加には、公知の重合触媒等が使用できる。
ポリイソシアネート(15)としては、炭素数(NCO基中の炭素を除く、以下同様)6〜20の芳香族ポリイソシアネート、炭素数2〜18の脂肪族ポリイソシアネート、炭素数4〜15の脂環式ポリイソシアネート、炭素数8〜15の芳香脂肪族ポリイソシアネート及びこれらのポリイソシアネートの変性物及びこれらの2種以上の混合物が挙げられる。
上記芳香族ポリイソシアネートの具体例としては、1,3−又は1,4−フェニレンジイソシアネート、2,4−又は2,6−トリレンジイソシアネート(TDI)、粗製TDI、2,4’−又は4,4’−ジフェニルメタンジイソシアネート(MDI)、粗製MDI[粗製ジアミノフェニルメタン〔ホルムアルデヒドと芳香族アミン(アニリン)又はその混合物との縮合生成物;ジアミノジフェニルメタンと少量(たとえば5〜20%)の3官能以上のポリアミンとの混合物〕のホスゲン化物:ポリアリルポリイソシアネート(PAPI)]、1,5−ナフチレンジイソシアネート、4,4’,4”−トリフェニルメタントリイソシアネート、m−又はp−イソシアナトフェニルスルホニルイソシアネート等が挙げられる。
上記脂肪族ポリイソシアネートの具体例としては、エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)、ドデカメチレンジイソシアネート、1,6,11−ウンデカントリイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、リジンジイソシアネート、2,6−ジイソシアナトメチルカプロエート、ビス(2−イソシアナトエチル)フマレート、ビス(2−イソシアナトエチル)カーボネート、2−イソシアナトエチル−2,6−ジイソシアナトヘキサノエート等が挙げられる。
上記脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)、ビス(2−イソシアナトエチル)−4−シクロヘキセン−1,2−ジカルボキシレート、2,5−又は2,6−ノルボルナンジイソシアネート等が挙げられる。
上記芳香脂肪族ポリイソシアネートの具体例としては、m−又はp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)等が挙げられる。
上記脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)、ビス(2−イソシアナトエチル)−4−シクロヘキセン−1,2−ジカルボキシレート、2,5−又は2,6−ノルボルナンジイソシアネート等が挙げられる。
上記芳香脂肪族ポリイソシアネートの具体例としては、m−又はp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)等が挙げられる。
また、上記ポリイソシアネートの変性物には、ウレタン変性、カルボジイミド変性、アロファネート変性、ウレア変性、ビューレット変性、ウレトジオン変性、ウレトイミン変性、イソシアヌレート変性、又はオキサゾリドン変性による変性物等が挙げられる。変性物の具体例としては、変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDI等)、ウレタン変性TDI等及びこれらの2種以上の混合物[たとえば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]が含まれる。
これらのうちで好ましいものは6〜15の芳香族ポリイソシアネート、炭素数4〜12の脂肪族ポリイソシアネート、及び炭素数4〜15の脂環式ポリイソシアネートであり、特に好ましいものはTDI、MDI、HDI、水添MDI、及びIPDIである。
これらのうちで好ましいものは6〜15の芳香族ポリイソシアネート、炭素数4〜12の脂肪族ポリイソシアネート、及び炭素数4〜15の脂環式ポリイソシアネートであり、特に好ましいものはTDI、MDI、HDI、水添MDI、及びIPDIである。
ポリアミン(16)の例としては、炭素数2〜18の脂肪族ポリアミン、炭素数4〜15の脂環式ポリアミン、炭素数4〜15の複素環式ポリアミン、炭素数6〜20の芳香族ポリアミン、ポリアミドポリアミン及びポリエーテルポリアミン等が挙げられる。
炭素数2〜18の脂肪族ポリアミンとしては、以下の〔1〕〜〔4〕のポリアミンが挙げられる。
〔1〕炭素数2〜6の脂肪族ポリアミン
アルキレンジアミン(エチレンジアミン、プロピレンジアミン、トリメチレンジアミン、テトラメチレンジアミン、ヘキサメチレンジアミン等)、ポリアルキレン(アルキレンの炭素数2〜6)ポリアミン〔ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン,トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン等〕。
〔2〕脂肪族ポリアミンのアルキル(アルキルの炭素数1〜4)又はヒドロキシアルキル(アルキルの炭素数2〜4)置換体
ジアルキル(アルキルの炭素数1〜3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン、メチルイミノビスプロピルアミン等。
〔3〕脂環又は複素環含有脂肪族ポリアミン
3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカン等。
〔4〕炭素数8〜15の芳香環含有脂肪族アミン
キシリレンジアミン、テトラクロル−p−キシリレンジアミン等。
〔1〕炭素数2〜6の脂肪族ポリアミン
アルキレンジアミン(エチレンジアミン、プロピレンジアミン、トリメチレンジアミン、テトラメチレンジアミン、ヘキサメチレンジアミン等)、ポリアルキレン(アルキレンの炭素数2〜6)ポリアミン〔ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン,トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン等〕。
〔2〕脂肪族ポリアミンのアルキル(アルキルの炭素数1〜4)又はヒドロキシアルキル(アルキルの炭素数2〜4)置換体
ジアルキル(アルキルの炭素数1〜3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン、メチルイミノビスプロピルアミン等。
〔3〕脂環又は複素環含有脂肪族ポリアミン
3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカン等。
〔4〕炭素数8〜15の芳香環含有脂肪族アミン
キシリレンジアミン、テトラクロル−p−キシリレンジアミン等。
炭素数4〜15の脂環式ポリアミンとしては、1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4’−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)等が挙げられる。
炭素数4〜15の複素環式ポリアミンとしては、ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジン等が挙げられる。
炭素数4〜15の複素環式ポリアミンとしては、ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジン等が挙げられる。
炭素数6〜20の芳香族ポリアミンとしては、以下の〔1〕〜〔4〕のポリアミンが挙げられる。
〔1〕非置換芳香族ポリアミン
1,2−、1,3−又は1,4−フェニレンジアミン、2,4’−又は4,4’−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、ビス(3,4−ジアミノフェニル)スルホン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4’,4”−トリアミン、ナフチレンジアミン等。
〔1〕非置換芳香族ポリアミン
1,2−、1,3−又は1,4−フェニレンジアミン、2,4’−又は4,4’−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、ビス(3,4−ジアミノフェニル)スルホン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4’,4”−トリアミン、ナフチレンジアミン等。
〔2〕核置換アルキル基〔炭素数1〜4のアルキル(メチル,エチル,n−及びi−プロピル、ブチル等)を有する芳香族ポリアミン〕
2,4−又は2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4’−ジアミノ−3,3’−ジメチルジフェニルメタン、4,4’−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、1,3−ジエチル−2,4−ジアミノベンゼン、1,3−ジメチル−2,6−ジアミノベンゼン、1,4−ジエチル−2,5−ジアミノベンゼン、1,4−ジイソプロピル−2,5−ジアミノベンゼン、1,4−ジブチル−2,5−ジアミノベンゼン、2,4−ジアミノメシチレン、1,3,5−トリエチル−2,4−ジアミノベンゼン、1,3,5−トリイソプロピル−2,4−ジアミノベンゼン、1−メチル−3,5−ジエチル−2,4−ジアミノベンゼン、1−メチル−3,5−ジエチル−2,6−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン、2,6−ジメチル−1,5−ジアミノナフタレン、2,6−ジイソプロピル−1,5−ジアミノナフタレン、2,6−ジブチル−1,5−ジアミノナフタレン、3,3’,5,5’−テトラメチルベンジジン、3,3’,5,5’−テトライソプロピルベンジジン、3,3’,5,5’−テトラメチル−4,4’−ジアミノジフェニルメタン、3,3’,5,5’−テトラエチル−4,4’−ジアミノジフェニルメタン、3,3’,5,5’−テトライソプロピル−4,4’−ジアミノジフェニルメタン、3,3’,5,5’−テトラブチル−4,4’−ジアミノジフェニルメタン、3,5−ジエチル−3’−メチル−2’,4−ジアミノジフェニルメタン,3,5−ジイソプロピル−3’−メチル−2’,4−ジアミノジフェニルメタン、3,3’−ジエチル−2,2’−ジアミノジフェニルメタン、4,4’−ジアミノ−3,3’−ジメチルジフェニルメタン、3,3’,5,5’−テトラエチル−4,4’−ジアミノベンゾフェノン、3,3’,5,5’−テトライソプロピル−4,4’−ジアミノベンゾフェノン、3,3’,5,5’−テトラエチル−4,4’−ジアミノジフェニルエーテル、3,3’,5,5’−テトライソプロピル−4,4’−ジアミノジフェニルスルホン等〕、及びこれらの異性体の種々の割合の混合物。
2,4−又は2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4’−ジアミノ−3,3’−ジメチルジフェニルメタン、4,4’−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、1,3−ジエチル−2,4−ジアミノベンゼン、1,3−ジメチル−2,6−ジアミノベンゼン、1,4−ジエチル−2,5−ジアミノベンゼン、1,4−ジイソプロピル−2,5−ジアミノベンゼン、1,4−ジブチル−2,5−ジアミノベンゼン、2,4−ジアミノメシチレン、1,3,5−トリエチル−2,4−ジアミノベンゼン、1,3,5−トリイソプロピル−2,4−ジアミノベンゼン、1−メチル−3,5−ジエチル−2,4−ジアミノベンゼン、1−メチル−3,5−ジエチル−2,6−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン、2,6−ジメチル−1,5−ジアミノナフタレン、2,6−ジイソプロピル−1,5−ジアミノナフタレン、2,6−ジブチル−1,5−ジアミノナフタレン、3,3’,5,5’−テトラメチルベンジジン、3,3’,5,5’−テトライソプロピルベンジジン、3,3’,5,5’−テトラメチル−4,4’−ジアミノジフェニルメタン、3,3’,5,5’−テトラエチル−4,4’−ジアミノジフェニルメタン、3,3’,5,5’−テトライソプロピル−4,4’−ジアミノジフェニルメタン、3,3’,5,5’−テトラブチル−4,4’−ジアミノジフェニルメタン、3,5−ジエチル−3’−メチル−2’,4−ジアミノジフェニルメタン,3,5−ジイソプロピル−3’−メチル−2’,4−ジアミノジフェニルメタン、3,3’−ジエチル−2,2’−ジアミノジフェニルメタン、4,4’−ジアミノ−3,3’−ジメチルジフェニルメタン、3,3’,5,5’−テトラエチル−4,4’−ジアミノベンゾフェノン、3,3’,5,5’−テトライソプロピル−4,4’−ジアミノベンゾフェノン、3,3’,5,5’−テトラエチル−4,4’−ジアミノジフェニルエーテル、3,3’,5,5’−テトライソプロピル−4,4’−ジアミノジフェニルスルホン等〕、及びこれらの異性体の種々の割合の混合物。
〔3〕核置換電子吸引基{ハロゲン(フッ素、塩素、臭素、ヨウ素等)原子、アルコキシ(メトキシ、エトキシ等)基を有する芳香族ポリアミン}
メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロル−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロル−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン、3−ジメトキシ−4−アミノアニリン、4,4’−ジアミノ−3,3’−ジメチル−5,5’−ジブロモ−ジフェニルメタン、3,3’−ジクロロベンジジン、3,3’−ジメトキシベンジジン、ビス(4−アミノ−3−クロロフェニル)オキサイド、ビス(4−アミノ−2−クロロフェニル)プロパン、ビス(4−アミノ−2−クロロフェニル)スルホン、ビス(4−アミノ−3−メトキシフェニル)デカン、ビス(4−アミノフェニル)スルフイド、ビス(4−アミノフェニル)テルリド、ビス(4−アミノフェニル)セレニド、ビス(4−アミノ−3−メトキシフェニル)ジスルフイド、4,4’−メチレンビス(2−ヨードアニリン)、4,4’−メチレンビス(2−ブロモアニリン)、4,4’−メチレンビス(2−フルオロアニリン)、4−アミノフェニル−2−クロロアニリン等。
メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロル−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロル−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン、3−ジメトキシ−4−アミノアニリン、4,4’−ジアミノ−3,3’−ジメチル−5,5’−ジブロモ−ジフェニルメタン、3,3’−ジクロロベンジジン、3,3’−ジメトキシベンジジン、ビス(4−アミノ−3−クロロフェニル)オキサイド、ビス(4−アミノ−2−クロロフェニル)プロパン、ビス(4−アミノ−2−クロロフェニル)スルホン、ビス(4−アミノ−3−メトキシフェニル)デカン、ビス(4−アミノフェニル)スルフイド、ビス(4−アミノフェニル)テルリド、ビス(4−アミノフェニル)セレニド、ビス(4−アミノ−3−メトキシフェニル)ジスルフイド、4,4’−メチレンビス(2−ヨードアニリン)、4,4’−メチレンビス(2−ブロモアニリン)、4,4’−メチレンビス(2−フルオロアニリン)、4−アミノフェニル−2−クロロアニリン等。
〔4〕2級アミノ基を有する芳香族ポリアミン〔上記〔1〕〜〔3〕の芳香族ポリアミンの−NH2の一部又は全部が−NH−R’(R’はアルキル基たとえばメチル,エチル等の低級アルキル基)で置き換ったもの〕
4,4’−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼン等〕 ポリアミドポリアミン:
ジカルボン酸(ダイマー酸等)と過剰の(酸1モル当り2モル以上の)ポリアミン(上記アルキレンジアミン,ポリアルキレンポリアミン等)との縮合により得られる低分子量ポリアミドポリアミン等。
4,4’−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼン等〕 ポリアミドポリアミン:
ジカルボン酸(ダイマー酸等)と過剰の(酸1モル当り2モル以上の)ポリアミン(上記アルキレンジアミン,ポリアルキレンポリアミン等)との縮合により得られる低分子量ポリアミドポリアミン等。
ポリエーテルポリアミンとしては、ポリエーテルポリオール(ポリアルキレングリコール等)のシアノエチル化物の水素化物等が挙げられる。
ポリチオール(17)としては、エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオール等が挙げられる。
エポキシ樹脂としては、ポリエポキシド(18)の開環重合物、ポリエポキシド(18)と前記活性水素化合物(D)との重付加物、又はポリエポキシド(18)と前記ジカルボン酸(3)又は3〜6価のポリカルボン酸(4)の酸無水物との硬化物等が挙げられる。
ポリエポキシド(18)は、分子中に2個以上のエポキシ基を有していれば、特に限定されない。ポリエポキシド(18)として好ましいものは、硬化物の機械的性質の観点から、分子中にエポキシ基を2〜6個有するものである。ポリエポキシド(18)のエポキシ当量(エポキシ基1個当たりの分子量)は、65〜1000が好ましく、更に好ましいのは90〜500である。この範囲であると、硬化物の耐水性、耐薬品性、機械的強度が更に良好となる。尚、エポキシ当量が65未満のポリエポキシドを合成するのは困難である。
ポリエポキシド(18)の例としては、芳香族ポリエポキシド、複素環ポリエポキシド、脂環族ポリエポキシド及び脂肪族ポリエポキシドが挙げられる。
芳香族ポリエポキシドとしては、多価フェノールのグリシジルエーテル、多価フェノールグリシジルエステル、グリシジル芳香族ポリアミン、及びアミノフェノールのグリシジル化物等が挙げられる。
芳香族ポリエポキシドとしては、多価フェノールのグリシジルエーテル、多価フェノールグリシジルエステル、グリシジル芳香族ポリアミン、及びアミノフェノールのグリシジル化物等が挙げられる。
多価フェノールのグリシジルエーテルとしては、ビスフェノールFジグリシジルエーテル、ビスフェノールAジグリシジルエーテル、ビスフェノールBジグリシジルエーテル、ビスフェノールADジグリシジルエーテル、ビスフェノールSジグリシジルエーテル、ハロゲン化ビスフェノールAジグリシジルエーテル、テトラクロロビスフェノールAジグリシジルエーテル、カテキンジグリシジルエーテル、レゾルシノールジグリシジルエーテル、ハイドロキノンジグリシジルエーテル、ピロガロールトリグリシジルエーテル、1,5−ジヒドロキシナフタリンジグリシジルエーテル、ジヒドロキシビフェニルジグリシジルエーテル、オクタクロロ−4,4’−ジヒドロキシビフェニルジグリシジルエーテル、テトラメチルビフェニルジグリシジルエーテル、ジヒドロキシナフチルクレゾールトリグリシジルエーテル、トリス(ヒドロキシフェニル)メタントリグリシジルエーテル、ジナフチルトリオールトリグリシジルエーテル、テトラキス(4−ヒドロキシフェニル)エタンテトラグリシジルエーテル、p−グリシジルフェニルジメチルトリールビスフェノールAグリシジルエーテル、トリスメチル−tret−ブチル−ブチルヒドロキシメタントリグリシジルエーテル、9,9’−ビス(4−ヒドキシフェニル)フロオレンジグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)テトラクレゾールグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)フェニルグリシジルエーテル、ビス(ジヒドロキシナフタレン)テトラグリシジルエーテル、フェノール又はクレゾールノボラック樹脂のグリシジルエーテル、リモネンフェノールノボラック樹脂のグリシジルエーテル、ビスフェノールA2モルとエピクロロヒドリン3モルの反応から得られるジグリシジルエーテル、フェノールとグリオキザール、グルタールアルデヒド、又はホルムアルデヒドの縮合反応によって得られるポリフェノールのポリグリシジルエーテル、及びレゾルシンとアセトンの縮合反応によって得られるポリフェノールのポリグリシジルエーテル等が挙げられる。
多価フェノールのグリシジルエステルとしては、フタル酸ジグリシジルエステル、イソフタル酸ジグリシジルエステル、テレフタル酸ジグリシジルエステル等が挙げられる。
グリシジル芳香族ポリアミンとしては、N,N−ジグリシジルアニリン、N,N,N’,N’−テトラグリシジルキシリレンジアミン、N,N,N’,N’−テトラグリシジルジフェニルメタンジアミン等が挙げられる。
アミノフェノールのグリシジル化物としては、P−アミノフェノールのトリグリシジルエーテル等が挙げられる。
グリシジル芳香族ポリアミンとしては、N,N−ジグリシジルアニリン、N,N,N’,N’−テトラグリシジルキシリレンジアミン、N,N,N’,N’−テトラグリシジルジフェニルメタンジアミン等が挙げられる。
アミノフェノールのグリシジル化物としては、P−アミノフェノールのトリグリシジルエーテル等が挙げられる。
芳香族ポリエポキシ化合物には、トリレンジイソシアネート又はジフェニルメタンジイソシアネートとグリシドールとの付加反応によって得られるジグリシジルウレタン化合物、トリレンジイソシアネート又はジフェニルメタンジイソシアネートとグリシドールとポリオールとを反応させて得られるグリシジル基含有ポリウレタン(プレ)ポリマー及びビスフェノールAのアルキレンオキサイド付加物のジグリシジルエーテルも含まれる。
複素環ポリエポキシドとしては、トリスグリシジルメラミン等が挙げられる。
脂環族ポリエポキシドとしては、ビニルシクロヘキセンジオキサイド、リモネンジオキサイド、ジシクロペンタジエンジオキサイド、ビス(2,3−エポキシシクロペンチル)エーテル、エチレングリコールビスエポキシジシクロペンチルエーテル、3,4−エポキシ−6−メチルシクロヘキシルメチル−3’,4’−エポキシ−6’−メチルシクロヘキサンカルボキシレート、ビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)アジペート、及びビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)ブチルアミン、ダイマー酸ジグリシジルエステル及び芳香族ポリエポキシドの核水添化物等が挙げられる。
脂肪族ポリエポキシドとしては、多価脂肪族アルコールのポリグリシジルエーテル、多価脂肪酸のポリグリシジルエステル、及びグリシジル脂肪族アミンが挙げられる。
脂環族ポリエポキシドとしては、ビニルシクロヘキセンジオキサイド、リモネンジオキサイド、ジシクロペンタジエンジオキサイド、ビス(2,3−エポキシシクロペンチル)エーテル、エチレングリコールビスエポキシジシクロペンチルエーテル、3,4−エポキシ−6−メチルシクロヘキシルメチル−3’,4’−エポキシ−6’−メチルシクロヘキサンカルボキシレート、ビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)アジペート、及びビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)ブチルアミン、ダイマー酸ジグリシジルエステル及び芳香族ポリエポキシドの核水添化物等が挙げられる。
脂肪族ポリエポキシドとしては、多価脂肪族アルコールのポリグリシジルエーテル、多価脂肪酸のポリグリシジルエステル、及びグリシジル脂肪族アミンが挙げられる。
多価脂肪族アルコールのポリグリシジルエーテルとしては、エチレングリコールジグリシジルエーテル、プロピレングリコールジグリシジルエーテル、テトラメチレングリコールジグリシジルエーテル、1,6−ヘキサンジオールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、ポリテトラメチレングリコールジグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、トリメチロールプロパンポリグリシジルエーテル、グリセロールポリグリシジルエーテル、ペンタエリスリトールポリグリシジルエーテル、ソルビトールポリグリシジルエーテル及びポリグリセロールンポリグリシジルエーテル等が挙げられる。
多価脂肪酸のポリグリシジルエステルとしては、ジグリシジルオキサレート、ジグリシジルマレート、ジグリシジルスクシネート、ジグリシジルグルタレート、ジグリシジルアジペート、ジグリシジルピメレート等が挙げられる。
グリシジル脂肪族アミンとしては、N,N,N’,N’−テトラグリシジルヘキサメチレンジアミン等が挙げられる。
脂肪族ポリエポキシドには、ジグリシジルエーテル、グリシジル(メタ)アクリレートの(共)重合体も含まれる。
これらのうち、好ましいのは、脂肪族ポリエポキシド及び芳香族ポリエポキシドである。ポリエポキシドは、2種以上併用しても差し支えない。
グリシジル脂肪族アミンとしては、N,N,N’,N’−テトラグリシジルヘキサメチレンジアミン等が挙げられる。
脂肪族ポリエポキシドには、ジグリシジルエーテル、グリシジル(メタ)アクリレートの(共)重合体も含まれる。
これらのうち、好ましいのは、脂肪族ポリエポキシド及び芳香族ポリエポキシドである。ポリエポキシドは、2種以上併用しても差し支えない。
ポリエステル樹脂(p1)以外のポリエステル樹脂の具体例としては、チタン触媒(t)を用いない点以外は、前述のポリエステル樹脂(p1)として例示したものと同様の原料から得られるものが挙げられ、好ましいものも同様である。
本発明の製造方法において、樹脂(a)としては、水性分散液(W)を形成しうる樹脂であればいかなる樹脂であっても使用でき、熱可塑性樹脂であっても熱硬化性樹脂であってもよいが、例えばビニル樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂、ポリアミド樹脂、ポリイミド樹脂、ケイ素系樹脂、フェノール樹脂、メラミン樹脂、ユリア樹脂、アニリン樹脂、アイオノマー樹脂及びポリカーボネート樹脂等が挙げられる。樹脂(a)としては、上記樹脂の2種以上を併用しても差し支えない。このうち好ましいのは、微細球状樹脂粒子の水性分散体が得られやすいという観点からビニル樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂及びこれらの併用であり、更に好ましいのはビニル樹脂である。
ビニル樹脂、ポリウレタン樹脂、エポキシ樹脂及びポリエステル樹脂の具体例としては、上述の樹脂(b)に使用されるビニル樹脂、ポリウレタン樹脂、エポキシ樹脂及びポリエステル樹脂と同様のものが挙げられる。
樹脂(a)は、水性分散液(W)中で樹脂粒子(A)として存在している必要があることから、少なくとも水性分散体(X1)を形成する条件下(5〜90℃が好ましい。)で、樹脂(a)は水に完全に溶解していないことが必要である。そのため、ビニル樹脂が共重合体である場合、ビニル樹脂を構成する疎水性モノマーと親水性モノマーの比率は、選ばれるモノマーの種類にもよるが、一般に疎水性モノマーが10%以上であることが好ましく、30%以上であることがより好ましい。疎水性モノマーの比率が、10%未満になるとビニル樹脂が水溶性になりやすく、樹脂粒子(C)の粒径均一性が損なわれることがある。
ここで、親水性モノマーとは、25℃の水100gに100g以上溶解するモノマーをいい、疎水性モノマーとは、それ以外のモノマー(25℃の水100gに100g以上溶解しないモノマー)をいう。
ここで、親水性モノマーとは、25℃の水100gに100g以上溶解するモノマーをいい、疎水性モノマーとは、それ以外のモノマー(25℃の水100gに100g以上溶解しないモノマー)をいう。
本発明の製造方法において、樹脂(a)を含有する樹脂粒子(A)の水性分散液(W)と、樹脂(b)及び/又はその前駆体(b0)、並びに必要により有機溶剤(u)を含有する油性液(OL)とを混合し、水性分散液(W)中に油性液(OL)を分散させて、前駆体(b0)を使用する場合は更に(W)中で(b0)を反応させて、樹脂(b)を含有する樹脂粒子(B)が形成される際に、樹脂粒子(A)が樹脂粒子(B)の表面に吸着されるため、樹脂粒子(B)同士あるいは樹脂粒子(C)同士が合一しににくなる。また、このため、高剪断条件下においても、樹脂粒子(C)は分裂されにくくなる。このような現象により、樹脂粒子(C)の粒径は一定の値に収斂するようになり、結果として粒径の均一な樹脂粒子が得られる。そのため、樹脂粒子(A)は、油性液(OL)を分散する際の温度において、剪断により破壊されない程度の強度を有すること、水に溶解したり、膨潤したりしにくいこと、油性液(OL)に溶解したり、膨潤したりしにくいことが好ましい。
樹脂(a)のガラス転移温度(Tg)は、樹脂粒子(C)の粒径の均一性、粉体流動性、保存時の耐熱性、耐ストレス性の観点から、0〜300℃が好ましく、更に好ましくは20〜250℃、より好ましくは50〜200℃である。尚、水性分散体(X1)を作製する温度よりTgが低いと、合一を防止したり、分裂を防止したりする効果が小さくなり、粒径の均一性を高める効果が小さくなる。
樹脂粒子(A)が水性溶剤{水と必要により有機溶剤(u)を含有する溶剤}に対して、溶解したり、膨潤したりするのを低減する観点から、樹脂(a)の分子量、SP値、結晶性、架橋点間分子量等を適宜調整するのが好ましい。
樹脂(a)のMnは、200〜500万が好ましく、更に好ましくは2,000〜500,000である。また、樹脂(a)のSP値は、好ましくは7〜18、更に好ましくは8〜14である。樹脂(a)の融点(DSCにて測定)は、好ましくは50℃以上、更に好ましくは80℃以上である。また、樹脂粒子(C)の耐熱性、耐水性、耐薬品性及び粒径の均一性等を向上させたい場合、樹脂(a)に、たとえば、3官能以上のモノマーを原料として用いて架橋構造を導入させてもよい。かかる架橋構造は、共有結合性、配位結合性、イオン結合性、水素結合性等、いずれの架橋形態であってもよい。樹脂(a)に架橋構造を導入する場合の架橋点間分子量は、30以上が好ましく、更に好ましくは50以上である。
樹脂(a)を樹脂粒子(A)の水性分散液(W)にする方法は、特に限定されないが、以下の〔1〕〜〔8〕が挙げられる。
〔1〕ビニル樹脂の場合、モノマーを出発原料として、懸濁重合法、乳化重合法、シード重合法又は分散重合法等の重合反応により、直接、樹脂粒子(A)の水性分散液を製造する方法。
〔2〕ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂等の重付加あるいは縮合樹脂の場合、前駆体(モノマー、オリゴマー等:液体であることが好ましく、加熱により液状化してもよい)又はその有機溶剤(u)溶液を適当な分散剤存在下で水性溶剤[水と必要により有機溶剤(u)を含有する溶剤]中に分散させ、その後に加熱したり、硬化剤を加えたりして、前駆体を硬化させて樹脂粒子(A)の水性分散液を製造する方法。
〔1〕ビニル樹脂の場合、モノマーを出発原料として、懸濁重合法、乳化重合法、シード重合法又は分散重合法等の重合反応により、直接、樹脂粒子(A)の水性分散液を製造する方法。
〔2〕ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂等の重付加あるいは縮合樹脂の場合、前駆体(モノマー、オリゴマー等:液体であることが好ましく、加熱により液状化してもよい)又はその有機溶剤(u)溶液を適当な分散剤存在下で水性溶剤[水と必要により有機溶剤(u)を含有する溶剤]中に分散させ、その後に加熱したり、硬化剤を加えたりして、前駆体を硬化させて樹脂粒子(A)の水性分散液を製造する方法。
〔3〕ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂等の重付加あるいは縮合樹脂の場合、前駆体(モノマー、オリゴマー等:液体であることが好ましく、加熱により液状化してもよい)又はその有機溶剤(u)溶液中に適当な乳化剤を溶解させた後、水を加えて転相乳化した後、加熱したり、硬化剤を加えたりして前駆体を硬化させて、樹脂粒子(A)の水性分散液を製造する方法。
〔4〕あらかじめ重合反応(付加重合、開環重合、重付加、付加縮合、縮合重合等いずれの重合反応様式であってもよい)により作製した樹脂を機械回転式又はジェット式等の微粉砕機を用いて粉砕し、次いで、分級することによって樹脂粒子を得た後、適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(A)の樹脂分散液を製造する方法。
〔4〕あらかじめ重合反応(付加重合、開環重合、重付加、付加縮合、縮合重合等いずれの重合反応様式であってもよい)により作製した樹脂を機械回転式又はジェット式等の微粉砕機を用いて粉砕し、次いで、分級することによって樹脂粒子を得た後、適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(A)の樹脂分散液を製造する方法。
〔5〕あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液を霧状に噴霧することにより樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(A)の樹脂分散液を製造する方法。
〔6〕あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液に貧溶剤を添加するか、又はあらかじめ有機溶剤(u)に加熱溶解した樹脂溶液を冷却することにより樹脂粒子を析出させ、次いで、有機溶剤(u)を除去して樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(A)の樹脂分散液を製造する方法。
〔6〕あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液に貧溶剤を添加するか、又はあらかじめ有機溶剤(u)に加熱溶解した樹脂溶液を冷却することにより樹脂粒子を析出させ、次いで、有機溶剤(u)を除去して樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水性溶剤中に分散させて、樹脂粒子(A)の樹脂分散液を製造する方法。
〔7〕あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液を、適当な分散剤存在下で水性溶剤中に分散させ、これを加熱又は減圧等によって有機溶剤(u)を除去して、樹脂粒子(A)の樹脂分散液を製造する方法。
〔8〕あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液中に適当な乳化剤を溶解させた後、水性溶剤を加えて転相乳化して樹脂粒子(A)の樹脂分散液を製造する方法。
〔8〕あらかじめ重合反応により作製した樹脂を有機溶剤(u)に溶解した樹脂溶液中に適当な乳化剤を溶解させた後、水性溶剤を加えて転相乳化して樹脂粒子(A)の樹脂分散液を製造する方法。
上記〔1〕〜〔8〕の方法において、併用する乳化剤又は分散剤としては、公知の界面活性剤(s)、水溶性ポリマー(q)等を用いることができる。また、乳化又は分散の助剤として有機溶剤(u)、可塑剤(v)等を併用することができる。
界面活性剤(s)としては、アニオン界面活性剤(s−1)、カチオン界面活性剤(s−2)、両性界面活性剤(s−3)、非イオン界面活性剤(s−4)等が挙げられる。界面活性剤(s)は2種以上の界面活性剤を併用したものであってもよい。
アニオン界面活性剤(s−1)としては、カルボン酸又はその塩、硫酸エステル塩、カルボキシメチル化物の塩、スルホン酸塩及びリン酸エステル塩が挙げられる。
カルボン酸又はその塩としては、炭素数8〜22の飽和又は不飽和脂肪酸又はその塩が挙げられ、具体的にはカプリン酸、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、アラキジン酸、ベヘン酸、オレイン酸、リノール酸、リシノール酸及びヤシ油、パーム核油、米ぬか油、牛脂等をケン化して得られる高級脂肪酸の混合物があげられる。塩としてはそれらのナトリウム、カリウム、アンモニウム、アルカノールアミン等の塩があげられる。
硫酸エステル塩としては、高級アルコール硫酸エステル塩(炭素数8〜18の脂肪族アルコールの硫酸エステル塩)、高級アルキルエーテル硫酸エステル塩(炭素数8〜18の脂肪族アルコールのEO1〜10モル付加物の硫酸エステル塩)、硫酸化油(天然の不飽和油脂又は不飽和のロウをそのまま硫酸化して中和したもの)、硫酸化脂肪酸エステル(不飽和脂肪酸の低級アルコールエステルを硫酸化して中和したもの)及び硫酸化オレフィン(炭素数12〜18のオレフィンを硫酸化して中和したもの)が挙げられる。塩としては、ナトリウム塩、カリウム塩、アンモニウム塩、アルカノールアミン塩が挙げられる。
高級アルコール硫酸エステル塩の具体例としては、オクチルアルコール硫酸エステル塩、デシルアルコール硫酸エステル塩、ラウリルアルコール硫酸エステル塩、ステアリルアルコール硫酸エステル塩、チーグラー触媒を用いて合成されたアルコール(例えば、ALFOL 1214:CONDEA社製)の硫酸エステル塩、オキソ法で合成されたアルコール(たとえばトリデカノール:協和発酵工業株式会社製、オキソコール1213、1215、1415:日産化学工業株式会社製、ドバノール23、25、45、ダイヤドール115−L、115H、135:三菱化学株式会社製)の硫酸エステル塩が挙げられる。
高級アルキルエーテル硫酸エステル塩の具体例としては、ラウリルアルコールEO2モル付加物硫酸エステル塩、オクチルアルコールEO3モル付加物硫酸エステル塩等が挙げられる。
硫酸化油の具体例としては、ヒマシ油、落花生油、オリーブ油、ナタネ油、牛脂、羊脂等の硫酸化物の塩(ナトリウム塩、カリウム塩、アンモニウム塩、アルカノールアミン塩)等が挙げられる。
硫酸化脂肪酸エステルの具体例としては、オレイン酸ブチル、リシノレイン酸ブチル等の硫酸化物の塩(ナトリウム塩、カリウム塩、アンモニウム塩、アルカノールアミン塩)等が挙げられる。
硫酸化オレフィンの具体例としては、ティーポール(シェル社製)等が挙げられる。
硫酸化油の具体例としては、ヒマシ油、落花生油、オリーブ油、ナタネ油、牛脂、羊脂等の硫酸化物の塩(ナトリウム塩、カリウム塩、アンモニウム塩、アルカノールアミン塩)等が挙げられる。
硫酸化脂肪酸エステルの具体例としては、オレイン酸ブチル、リシノレイン酸ブチル等の硫酸化物の塩(ナトリウム塩、カリウム塩、アンモニウム塩、アルカノールアミン塩)等が挙げられる。
硫酸化オレフィンの具体例としては、ティーポール(シェル社製)等が挙げられる。
カルボキシメチル化物の塩としては、炭素数8〜16の脂肪族アルコールのカルボキシメチル化物の塩及び炭素数8〜16の脂肪族アルコールのEO1〜10モル付加物のカルボキシメチル化物の塩が挙げられる。
脂肪族アルコールのカルボキシメチル化物の塩の具体例としては、オクチルアルコールカルボキシメチル化ナトリウム塩、デシルアルコールカルボキシメチル化ナトリウム塩、ラウリルアルコールカルボキシメチル化ナトリウム塩、sec−トリデカノール23カルボキシメチル化ナトリウム塩、トリデカノールカルボキシメチル化ナトリウム塩等が挙げられる。
脂肪族アルコールのEO1〜10モル付加物のカルボキシメチル化物の塩の具体例としては、オクチルアルコールEO3モル付加物カルボキシメチル化ナトリウム塩、ラウリルアルコールEO4モル付加物カルボキシメチル化ナトリウム塩、sec−トリデカノール23EO3モル付加物カルボキシメチル化ナトリウム塩、トリデカノールEO5モル付加物カルボキシメチル化ナトリウム塩等が挙げられる。
脂肪族アルコールのカルボキシメチル化物の塩の具体例としては、オクチルアルコールカルボキシメチル化ナトリウム塩、デシルアルコールカルボキシメチル化ナトリウム塩、ラウリルアルコールカルボキシメチル化ナトリウム塩、sec−トリデカノール23カルボキシメチル化ナトリウム塩、トリデカノールカルボキシメチル化ナトリウム塩等が挙げられる。
脂肪族アルコールのEO1〜10モル付加物のカルボキシメチル化物の塩の具体例としては、オクチルアルコールEO3モル付加物カルボキシメチル化ナトリウム塩、ラウリルアルコールEO4モル付加物カルボキシメチル化ナトリウム塩、sec−トリデカノール23EO3モル付加物カルボキシメチル化ナトリウム塩、トリデカノールEO5モル付加物カルボキシメチル化ナトリウム塩等が挙げられる。
スルホン酸塩としては、アルキルベンゼンスルホン酸塩、アルキルナフタレンスルホン酸塩、スルホコハク酸ジエステル型、α−オレフィンスルホン酸塩(テーポール)、イゲポンT型、その他芳香環含有化合物のスルホン酸塩が挙げられる。
アルキルベンゼンスルホン酸塩の具体例としては、ドデシルベンゼンスルホン酸ナトリウム塩等が挙げられる。
アルキルナフタレンスルホン酸塩の具体例としては、ドデシルナフタレンスルホン酸ナトリウム塩等が挙げられる。
スルホコハク酸ジエステル型の具体例としては、スルホコハク酸ジ−2−エチルヘキシルエステルナトリウム塩等が挙げられる。
その他芳香環含有化合物のスルホン酸塩としては、アルキル化ジフェニルエーテルのモノ又はジスルホン酸塩、スチレン化フェノールスルホン酸塩等が挙げられる。
アルキルベンゼンスルホン酸塩の具体例としては、ドデシルベンゼンスルホン酸ナトリウム塩等が挙げられる。
アルキルナフタレンスルホン酸塩の具体例としては、ドデシルナフタレンスルホン酸ナトリウム塩等が挙げられる。
スルホコハク酸ジエステル型の具体例としては、スルホコハク酸ジ−2−エチルヘキシルエステルナトリウム塩等が挙げられる。
その他芳香環含有化合物のスルホン酸塩としては、アルキル化ジフェニルエーテルのモノ又はジスルホン酸塩、スチレン化フェノールスルホン酸塩等が挙げられる。
リン酸エステル塩としては、高級アルコールリン酸エステル塩及び高級アルコールEO付加物リン酸エステル塩が挙げられる。
高級アルコールリン酸エステル塩の具体例としては、ラウリルアルコールリン酸モノエステルジナトリウム塩、ラウリルアルコールリン酸ジエステルナトリウム塩等が挙げれる。
高級アルコールEO付加物リン酸エステル塩の具体例としては、オレイルアルコールEO5モル付加物リン酸モノエステルジナトリウム塩が挙げられる。
高級アルコールEO付加物リン酸エステル塩の具体例としては、オレイルアルコールEO5モル付加物リン酸モノエステルジナトリウム塩が挙げられる。
カチオン界面活性剤(s−2)としては、第4級アンモニウム塩型、アミン塩型等が挙げられる。
第4級アンモニウム塩型としては、ラウリルトリメチルアンモニウムクロライド、ジデシルジメチルアンモニウムクロライド、ジオクチルジメチルアンモニウムブロマイド、ステアリルトリメチルアンモニウムブロマイド、ラウリルジメチルベンジルアンモニウムクロライド(塩化ベンザルコニウム)、セチルピリジニウムクロライド、ポリオキシエチレントリメチルアンモニウムクロライド、ステアラミドエチルジエチルメチルアンモニウムメトサルフェート等が挙げられる。
第4級アンモニウム塩型としては、ラウリルトリメチルアンモニウムクロライド、ジデシルジメチルアンモニウムクロライド、ジオクチルジメチルアンモニウムブロマイド、ステアリルトリメチルアンモニウムブロマイド、ラウリルジメチルベンジルアンモニウムクロライド(塩化ベンザルコニウム)、セチルピリジニウムクロライド、ポリオキシエチレントリメチルアンモニウムクロライド、ステアラミドエチルジエチルメチルアンモニウムメトサルフェート等が挙げられる。
アミン塩型としては、1〜3級アミンを無機酸(塩酸、硝酸、硫酸、ヨウ化水素酸等)又は有機酸(酢酸、ギ酸、蓚酸、乳酸、グルコン酸、アジピン酸、アルキル燐酸等)で中和することにより得られる。第1級アミン塩型としては、脂肪族高級アミン(ラウリルアミン、ステアリルアミン、セチルアミン、硬化牛脂アミン、ロジンアミン等の高級アミン)の無機酸塩又は有機酸塩;低級アミンの高級脂肪酸(ステアリン酸、オレイン酸等)塩等が挙げられる。第2級アミン塩型としては、脂肪族アミンのEO付加物等の無機酸塩又は有機酸塩が挙げられる。また、第3級アミン塩型としては、脂肪族アミン(トリエチルアミン、エチルジメチルアミン、N,N,N’,N’−テトラメチルエチレンジアミン等)、脂肪族アミンのEO(2モル以上)付加物、脂環式アミン(N−メチルピロリジン、N−メチルピペリジン、N−メチルヘキサメチレンイミン、N−メチルモルホリン、1,8−ジアザビシクロ(5,4,0)−7−ウンデセン等)、含窒素ヘテロ環芳香族アミン(4−ジメチルアミノピリジン、N−メチルイミダゾール、4,4’−ジピリジル等)の無機酸塩又は有機酸塩等が挙げれる。
両性界面活性剤(s−3)としては、アミノ酸型両性界面活性剤、ベタイン型両性界面活性剤、イミダゾリン型両性界面活性剤等のカルボン酸塩型両性界面活性剤等が挙げられる。
アミノ酸型両性界面活性剤は、分子内にアミノ基とカルボキシル基を持っている両性界面活性剤で、例えば、下記一般式で示される化合物が挙げられる。
[R−NH−(CH2)n−COO]mM
[式中、Rは1価の炭化水素基;nは1又は2;mは1又は2;Mは水素原子、アルカリ金属原子、アルカリ土類金属原子、アンモニウムカチオン、アミンカチオン、アルカノールアミンカチオン等である。]
アミノ酸型両性界面活性剤の具体例としては、例えば、アルキルアミノプロピオン酸型両性界面活性剤(ステアリルアミノプロピオン酸ナトリウム、ラウリルアミノプロピオン酸ナトリウム等);アルキルアミノ酢酸型両性界面活性剤(ラウリルアミノ酢酸ナトリウム等);グリシン型両性界面活性剤(ナトリウムラウロイルグリシン、ナトリウムラウリルジアミノエチルグリシン、ラウリルジアミノエチルグリシン塩酸塩、ジオクチルジアミノエチルグリシン塩酸塩等)等が挙げられる。
[R−NH−(CH2)n−COO]mM
[式中、Rは1価の炭化水素基;nは1又は2;mは1又は2;Mは水素原子、アルカリ金属原子、アルカリ土類金属原子、アンモニウムカチオン、アミンカチオン、アルカノールアミンカチオン等である。]
アミノ酸型両性界面活性剤の具体例としては、例えば、アルキルアミノプロピオン酸型両性界面活性剤(ステアリルアミノプロピオン酸ナトリウム、ラウリルアミノプロピオン酸ナトリウム等);アルキルアミノ酢酸型両性界面活性剤(ラウリルアミノ酢酸ナトリウム等);グリシン型両性界面活性剤(ナトリウムラウロイルグリシン、ナトリウムラウリルジアミノエチルグリシン、ラウリルジアミノエチルグリシン塩酸塩、ジオクチルジアミノエチルグリシン塩酸塩等)等が挙げられる。
ベタイン型両性界面活性剤は、分子内に第4級アンモニウム塩型のカチオン部分とカルボン酸型のアニオン部分を持っている両性界面活性剤で、例えば、アルキルジメチルベタイン(ステアリルジメチルアミノ酢酸ベタイン、ラウリルジメチルアミノ酢酸ベタイン等)、アミドベタイン(ヤシ油脂肪酸アミドプロピルベタイン等)、アルキルジヒドロキシアルキルベタイン(ラウリルジヒドロキシエチルベタイン等)等が挙げられる。
イミダゾリン型両性界面活性剤としては、例えば、2−ウンデシル−N−カルボキシメチル−N−ヒドロキシエチルイミダゾリニウムベタイン等が挙げられる。
非イオン界面活性剤(s−4)としては、アルキレンオキサイド付加型非イオン界面活性剤及び多価アルコ−ル型非イオン界面活性剤等が挙げられる。
アルキレンオキサイド付加型非イオン界面活性剤は、高級アルコ−ル、高級脂肪酸又はアルキルアミン等に直接アルキレンオキサイドを付加させるか、グリコ−ルにアルキレンオキサイドを付加させて得られるポリアルキレングリコ−ルに高級脂肪酸等を反応させるか、あるいは多価アルコ−ルと高級脂肪酸とを反応して得られたエステル化物にアルキレンオキサイドを付加させるか、高級脂肪酸アミドにアルキレンオキサイドを付加させることにより得られる。
アルキレンオキサイドのうち好ましいものは、エチレンオキサイド(以下、EOと略記)及びEOとプロピレンオキサイド(以下、POと略記)のランダム又はブロック付加物である。
アルキレンオキサイドの付加モル数としては10〜50モルが好ましく、該アルキレンオキサイドのうち50〜100%がEOであるものが好ましい。
アルキレンオキサイドの付加モル数としては10〜50モルが好ましく、該アルキレンオキサイドのうち50〜100%がEOであるものが好ましい。
アルキレンオキサイド付加型非イオン界面活性剤の具体例としては、オキシアルキレンアルキルエ−テル(例えば、オクチルアルコールEO付加物、ラウリルアルコールEO付加物、ステアリルアルコールEO付加物、オレイルアルコールEO付加物、ラウリルアルコールEO・POブロック付加物等);ポリオキシアルキレン高級脂肪酸エステル(例えば、ステアリル酸EO付加物、ラウリル酸EO付加物等);ポリオキシアルキレン多価アルコ−ル高級脂肪酸エステル(例えば、ポリエチレングリコールのラウリン酸ジエステル、ポリエチレングリコールのオレイン酸ジエステル、ポリエチレングリコールのステアリン酸ジエステル等);ポリオキシアルキレンアルキルフェニルエ−テル(例えば、ノニルフェノールEO付加物、ノニルフェノールEO・POブロック付加物、オクチルフェノールEO付加物、ビスフェノールA・EO付加物、ジノニルフェノールEO付加物、スチレン化フェノールEO付加物等);ポリオキシアルキレンアルキルアミノエ−テル及び(例えば、ラウリルアミンEO付加物,ステアリルアミンEO付加物等);ポリオキシアルキレンアルキルアルカノ−ルアミド(例えば、ヒドロキシエチルラウリン酸アミドのEO付加物、ヒドロキシプロピルオレイン酸アミドのEO付加物、ジヒドロキシエチルラウリン酸アミドのEO付加物等)が挙げられる。
多価アルコ−ル型非イオン界面活性剤としては、多価アルコール脂肪酸エステル、多価アルコール脂肪酸エステルアルキレンオキサイド付加物、多価アルコールアルキルエーテル、多価アルコールアルキルエーテルアルキレンオキサイド付加物が挙げられる。
多価アルコール脂肪酸エステルの具体例としては、ペンタエリスリトールモノラウレート、ペンタエリスリトールモノオレート、ソルビタンモノラウレート、ソルビタンモノステアレート、ソルビタンモノラウレート、ソルビタンジラウレート、ソルビタンジオレート、ショ糖モノステアレート等が挙げられる。
多価アルコール脂肪酸エステルアルキレンオキサイド付加物の具体例としては、エチレングリコールモノオレートEO付加物、エチレングリコールモノステアレートEO付加物、トリメチロールプロパンモノステアレートEO・POランダム付加物、ソルビタンモノラウレートEO付加物、ソルビタンモノステアレートEO付加物、ソルビタンジステアレートEO付加物、ソルビタンジラウレートEO・POランダム付加物等が挙げられる。
多価アルコール脂肪酸エステルアルキレンオキサイド付加物の具体例としては、エチレングリコールモノオレートEO付加物、エチレングリコールモノステアレートEO付加物、トリメチロールプロパンモノステアレートEO・POランダム付加物、ソルビタンモノラウレートEO付加物、ソルビタンモノステアレートEO付加物、ソルビタンジステアレートEO付加物、ソルビタンジラウレートEO・POランダム付加物等が挙げられる。
多価アルコールアルキルエーテルの具体例としては、ペンタエリスリトールモノブチルエーテル、ペンタエリスリトールモノラウリルエーテル、ソルビタンモノメチルエーテル、ソルビタンモノステアリルエーテル、メチルグリコシド、ラウリルグリコシド等が挙げられる。
多価アルコールアルキルエーテルアルキレンオキサイド付加物の具体例としては、ソルビタンモノステアリルエーテルEO付加物、メチルグリコシドEO・POランダム付加物、ラウリルグリコシドEO付加物、ステアリルグリコシドEO・POランダム付加物等が挙げられる。
多価アルコールアルキルエーテルアルキレンオキサイド付加物の具体例としては、ソルビタンモノステアリルエーテルEO付加物、メチルグリコシドEO・POランダム付加物、ラウリルグリコシドEO付加物、ステアリルグリコシドEO・POランダム付加物等が挙げられる。
水溶性ポリマー(q)としては、セルロース化合物(例えば、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、エチルヒドロキシエチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルセルロース及びそれらのケン化物等)、ゼラチン、デンプン、デキストリン、アラビアゴム、キチン、キトサン、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレングリコール、ポリエチレンイミン、ポリアクリルアミド、アクリル酸(塩)含有ポリマー(ポリアクリル酸ナトリウム、ポリアクリル酸カリウム、ポリアクリル酸アンモニウム、ポリアクリル酸の水酸化ナトリウム部分中和物、アクリル酸ナトリウム−アクリル酸エステル共重合体)、スチレン−無水マレイン酸共重合体の水酸化ナトリウム(部分)中和物、水溶性ポリウレタン(ポリエチレングリコール、ポリカプロラクトンジオール等とポリイソシアネートの反応生成物等)等が挙げられる。
有機溶剤(u)は、樹脂(a)の乳化分散の際に必要に応じて水性溶剤中に加えてもよいし、被乳化分散体中[前躯体(b0)を含む油性液(OL)中]に加えてもよい。
本発明における有機溶剤(u)の具体例としては、芳香族炭化水素溶剤{トルエン、キシレン、エチルベンゼン、テトラリン等};脂肪族又は脂環式炭化水素溶剤{n−ヘキサン、n−ヘプタン、ミネラルスピリット、シクロヘキサン等};ハロゲン溶剤{塩化メチル、臭化メチル、ヨウ化メチル、メチレンジクロライド、四塩化炭素、トリクロロエチレン、パークロロエチレン等};エステル又はエステルエーテル溶剤{酢酸エチル、酢酸ブチル、メトキシブチルアセテート、メチルセロソルブアセテート、エチルセロソルブアセテート等};エーテル溶剤{ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチルセロソルブ、ブチルセロソルブ、プロピレングリコールモノメチルエーテル等};ケトン溶剤{アセトン、メチルエチルケトン、メチルイソブチルケトン、ジ−n−ブチルケトン、シクロヘキサノン等};アルコール溶剤{メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、t−ブタノール、2−エチルヘキシルアルコール、ベンジルアルコール等};アミド溶剤{ジメチルホルムアミド、ジメチルアセトアミド等};スルホキシド溶剤{ジメチルスルホキシド等};複素環式化合物溶剤{N−メチルピロリドン等}並びにこれらの2種以上の混合溶剤が挙げられる。
尚、前記水性溶剤中に用いる有機溶剤(u)としては、これらのうち、25℃で水と任意の割合で混和する溶剤{アセトン及びメタノール等}が好ましい。
尚、前記水性溶剤中に用いる有機溶剤(u)としては、これらのうち、25℃で水と任意の割合で混和する溶剤{アセトン及びメタノール等}が好ましい。
可塑剤(v)は、樹脂(a)の乳化分散の際に必要に応じて水性溶剤中に加えてもよいし、被乳化分散体中[前駆体(b0)を含む油性液(OL)中]に加えてもよい。
可塑剤(v)としては、何ら限定されず、(v1)フタル酸エステル[フタル酸ジブチル、フタル酸ジオクチル、フタル酸ブチルベンジル、フタル酸ジイソデシル等];(v2)脂肪族2塩基酸エステル[アジピン酸ジ−2−エチルヘキシル、セバシン酸−2−エチルヘキシル等];(v3)トリメリット酸エステル[トリメリット酸トリ−2−エチルヘキシル、トリメリット酸トリオクチル等];(v4)燐酸エステル[リン酸トリエチル、リン酸トリ−2−エチルヘキシル、リン酸トリクレジール等];(v5)脂肪酸エステル[オレイン酸ブチル等];(v6)及びこれらの2種以上の混合物が挙げられる。
可塑剤(v)としては、何ら限定されず、(v1)フタル酸エステル[フタル酸ジブチル、フタル酸ジオクチル、フタル酸ブチルベンジル、フタル酸ジイソデシル等];(v2)脂肪族2塩基酸エステル[アジピン酸ジ−2−エチルヘキシル、セバシン酸−2−エチルヘキシル等];(v3)トリメリット酸エステル[トリメリット酸トリ−2−エチルヘキシル、トリメリット酸トリオクチル等];(v4)燐酸エステル[リン酸トリエチル、リン酸トリ−2−エチルヘキシル、リン酸トリクレジール等];(v5)脂肪酸エステル[オレイン酸ブチル等];(v6)及びこれらの2種以上の混合物が挙げられる。
樹脂粒子(A)の粒径は、通常、樹脂粒子(B)の粒径よりも小さいことが必要であり、得られる樹脂粒子(C)又は(B)の粒径の均一性の観点から、粒径比[樹脂粒子(A)の体積平均粒径/樹脂粒子(C)の体積平均粒径]の値が0.001〜0.3の範囲であるのが好ましい。粒径比の下限は更に好ましくは0.003、特に好ましくは0.005であり、上限は更に好ましくは0.25、特に好ましくは0.1である。かかる粒径比が、0.3より大きいと樹脂粒子(A)が樹脂粒子(B)の表面に効率よく吸着しないため、得られる樹脂粒子(C)の粒度分布が広くなる傾向がある。
樹脂粒子(A)の体積平均粒径は、所望の粒径の樹脂粒子(C)を得るのに適した粒径になるように、上記粒径比の範囲で適宜調整することができる。
樹脂粒子(A)の体積平均粒径は、0.0005〜30μmが好ましい。上限は、更に好ましくは20μm、特に好ましくは10μm、最も好ましくは2μmであり、下限は、更に好ましくは0.01μm、特に好ましくは0.02μm、最も好ましくは0.04μmである。ただし、例えば、体積平均粒径1μmの樹脂粒子(C)を得たい場合、好ましくは0.0005〜0.3μm、特に好ましくは0.001〜0.2μmの範囲、10μmの樹脂粒子(C)を得たい場合、好ましくは0.005〜3μm、特に好ましくは0.04〜2μm、最も好ましくは0.05〜1μm、100μmの樹脂粒子(C)を得たい場合、好ましくは0.05〜30μm、特に好ましくは0.1〜20μmである。尚、体積平均粒径は、レーザー式粒度分布測定装置{たとえば、LA−920(株式会社堀場製作所製)}やコールターカウンター〔例えば、商品名:マルチサイザーIII(コールター社製)〕で測定できる。
尚、上記粒径比が得やすいことから、後述する樹脂粒子(B)の体積平均粒径は、0.1〜300μmが好ましい。この上限は、更に好ましくは250μm、特に好ましくは200μm、最も好ましくは20μmであり、下限は、更に好ましくは0.5μm、特に好ましくは1μm、最も好ましくは4μmである。
樹脂粒子(A)の体積平均粒径は、0.0005〜30μmが好ましい。上限は、更に好ましくは20μm、特に好ましくは10μm、最も好ましくは2μmであり、下限は、更に好ましくは0.01μm、特に好ましくは0.02μm、最も好ましくは0.04μmである。ただし、例えば、体積平均粒径1μmの樹脂粒子(C)を得たい場合、好ましくは0.0005〜0.3μm、特に好ましくは0.001〜0.2μmの範囲、10μmの樹脂粒子(C)を得たい場合、好ましくは0.005〜3μm、特に好ましくは0.04〜2μm、最も好ましくは0.05〜1μm、100μmの樹脂粒子(C)を得たい場合、好ましくは0.05〜30μm、特に好ましくは0.1〜20μmである。尚、体積平均粒径は、レーザー式粒度分布測定装置{たとえば、LA−920(株式会社堀場製作所製)}やコールターカウンター〔例えば、商品名:マルチサイザーIII(コールター社製)〕で測定できる。
尚、上記粒径比が得やすいことから、後述する樹脂粒子(B)の体積平均粒径は、0.1〜300μmが好ましい。この上限は、更に好ましくは250μm、特に好ましくは200μm、最も好ましくは20μmであり、下限は、更に好ましくは0.5μm、特に好ましくは1μm、最も好ましくは4μmである。
樹脂(b)のMn、融点、Tg、SP値は、樹脂粒子(C)又は樹脂粒子(B)の用途によって好ましい範囲に適宜調整すればよい。例えば、樹脂粒子(C)、樹脂粒子(B)をスラッシュ成形用樹脂、粉体塗料として用いる場合、樹脂(b)のMnは、2,000〜50万が好ましく、更に好ましくは4,000〜20万である。樹脂(b)の融点は、0〜200℃が好ましく、更に好ましくは、35〜150℃である。樹脂(b)のTgは、−60〜100℃が好ましく、更に好ましくは、−30〜60℃である。樹脂(b)のSP値は、7〜18が好ましく、更に好ましくは8〜14である。
樹脂粒子(C)又は樹脂粒子(B)を電子部品(液晶ディスプレイ等)製造用スペーサー、電子測定機の標準粒子として用いる場合、樹脂(b)のMnは、2万〜1,000万が好ましく、更に好ましくは4万〜200万である。樹脂(b)の融点は、40〜300℃が好ましく、更に好ましくは、70〜250℃である。樹脂(b)のTgは、−0〜250℃が好ましく、更に好ましくは、50〜200℃である。樹脂(b)のSP値は、8〜18が好ましく、更に好ましくは9〜14である。
樹脂粒子(C)又は樹脂粒子(B)をトナーの母体粒子{電子写真、静電記録、静電印刷等に使用されるトナーの母体粒子}として用いる場合、樹脂(b)のMnは、1,000〜500万が好ましく、更に好ましくは2,000〜50万である。樹脂(b)の融点は、20〜300℃が好ましく、更に好ましくは、80〜250℃である。樹脂(b)のTgは、20〜200℃が好ましく、更に好ましくは、40〜100℃である。樹脂(b)のSP値は、8〜16が好ましく、更に好ましくは9〜14である。
樹脂粒子(C)又は樹脂粒子(B)を電子部品(液晶ディスプレイ等)製造用スペーサー、電子測定機の標準粒子として用いる場合、樹脂(b)のMnは、2万〜1,000万が好ましく、更に好ましくは4万〜200万である。樹脂(b)の融点は、40〜300℃が好ましく、更に好ましくは、70〜250℃である。樹脂(b)のTgは、−0〜250℃が好ましく、更に好ましくは、50〜200℃である。樹脂(b)のSP値は、8〜18が好ましく、更に好ましくは9〜14である。
樹脂粒子(C)又は樹脂粒子(B)をトナーの母体粒子{電子写真、静電記録、静電印刷等に使用されるトナーの母体粒子}として用いる場合、樹脂(b)のMnは、1,000〜500万が好ましく、更に好ましくは2,000〜50万である。樹脂(b)の融点は、20〜300℃が好ましく、更に好ましくは、80〜250℃である。樹脂(b)のTgは、20〜200℃が好ましく、更に好ましくは、40〜100℃である。樹脂(b)のSP値は、8〜16が好ましく、更に好ましくは9〜14である。
本発明の製造方法においては、樹脂(a)を含有する樹脂粒子(A)の水性分散液(W)と、樹脂(b)及び/又はその前駆体(b0)、及び必要により有機溶剤(u)を含有する油性液(OL)とを混合し、水性分散液(W)中に油性液(OL)を分散させて、前駆体(b0)を使用する場合は更に(W)中で(b0)を反応させて、樹脂粒子(A)の水性分散液中で、樹脂(b)を含有する樹脂粒子(B)を形成させることにより、樹脂粒子(B)の表面に樹脂粒子(A)が付着してなる構造の樹脂粒子(C)の水性分散体(X1)を得る。
樹脂(b)及び/又はその前駆体(b0)、及び必要により有機溶剤(u)を含有する油性液(OL)を水性分散液(W)に分散させる際、分散装置を用いることができる。
分散装置としては、一般に乳化機や、分散機として市販されているものであれば特に限定されず、例えば、バッチ式乳化機{ホモジナイザー(IKA社製)、ポリトロン(キネマティカ社製)、TKオートホモミキサー(特殊機化工業株式会社製)等}、連続式乳化機{エバラマイルダー(株式会社荏原製作所製)、TKフィルミックス、TKパイプラインホモミキサー(特殊機化工業株式会社製)、コロイドミル(神鋼パンテック株式会社製)、スラッシャー、トリゴナル湿式微粉砕機(サンテック株式会社製)、キャピトロン(ユーロテック社製)、ファインフローミル(太平洋機工株式会社製)等}、高圧乳化機{マイクロフルイダイザー(みずほ工業株式会社製)、ナノマイザー(エス・ジーエンジニアリング株式会社製)、APVガウリン(ガウリン社製)等}、膜乳化機{膜乳化機(冷化工業株式会社製)等}、振動式乳化機{バイブロミキサー(冷化工業株式会社製)等}、超音波乳化機{超音波ホモジナイザー(ブランソン社製)等}等が挙げられる。これらのうち、粒径の均一化の観点で好ましいものは、バッチ式乳化機、連続式乳化機、高圧乳化機、更に好ましくはAPVガウリン、ホモジナイザー、TKオートホモミキサー、エバラマイルダー、TKフィルミックス、TKパイプラインホモミキサーである。
分散装置としては、一般に乳化機や、分散機として市販されているものであれば特に限定されず、例えば、バッチ式乳化機{ホモジナイザー(IKA社製)、ポリトロン(キネマティカ社製)、TKオートホモミキサー(特殊機化工業株式会社製)等}、連続式乳化機{エバラマイルダー(株式会社荏原製作所製)、TKフィルミックス、TKパイプラインホモミキサー(特殊機化工業株式会社製)、コロイドミル(神鋼パンテック株式会社製)、スラッシャー、トリゴナル湿式微粉砕機(サンテック株式会社製)、キャピトロン(ユーロテック社製)、ファインフローミル(太平洋機工株式会社製)等}、高圧乳化機{マイクロフルイダイザー(みずほ工業株式会社製)、ナノマイザー(エス・ジーエンジニアリング株式会社製)、APVガウリン(ガウリン社製)等}、膜乳化機{膜乳化機(冷化工業株式会社製)等}、振動式乳化機{バイブロミキサー(冷化工業株式会社製)等}、超音波乳化機{超音波ホモジナイザー(ブランソン社製)等}等が挙げられる。これらのうち、粒径の均一化の観点で好ましいものは、バッチ式乳化機、連続式乳化機、高圧乳化機、更に好ましくはAPVガウリン、ホモジナイザー、TKオートホモミキサー、エバラマイルダー、TKフィルミックス、TKパイプラインホモミキサーである。
樹脂(b)及び/又はその前駆体(b0)を樹脂粒子(A)の水性分散液(W)に分散させる際、前駆体(b0)は液体であることが好ましい。樹脂(b)及び/又はその前駆体(b0)が常温で固体である場合、それらの融点以上の温度で分散させてもよい。
樹脂(b)及び/又はその前駆体(b0)、及び必要により有機溶剤(u)を含有する油性液(OL)の粘度は、粒径均一性の観点から、好ましくは10〜5万mPa・s(B型粘度計で測定、分散時の温度)、更に好ましくは100〜1万mPa・sである。
分散時の温度としては、0〜150℃(加圧下)が好ましく、更に好ましくは5〜98℃である。上記の粘度が高い場合、温度を上げて粘度を上記好ましい範囲まで低下させて、乳化分散を行うことが好ましい。
樹脂(b)及び/又はその前駆体(b0)、及び必要により有機溶剤(u)を含有する油性液(OL)の粘度は、粒径均一性の観点から、好ましくは10〜5万mPa・s(B型粘度計で測定、分散時の温度)、更に好ましくは100〜1万mPa・sである。
分散時の温度としては、0〜150℃(加圧下)が好ましく、更に好ましくは5〜98℃である。上記の粘度が高い場合、温度を上げて粘度を上記好ましい範囲まで低下させて、乳化分散を行うことが好ましい。
油性液(OL)に用いる有機溶剤(u)として好ましいものは、樹脂(b)及びその前駆体(b0)の種類によって異なるが、樹脂(b)又は前駆体(b0)とのSP値差が3以下であることが好適である。また、樹脂粒子(C)の粒径均一性の観点からは、樹脂(b)を溶解させるが、樹脂(a)を含有する樹脂粒子(A)を溶解・膨潤させにくい溶剤が好ましい。
樹脂(b)の前駆体(b0)としては、化学反応により樹脂(b)になりうるものであれば特に限定されず、例えば、樹脂(b)がポリエステル樹脂(p1)をビニル樹脂である場合、前駆体(b0)は、先述のビニルモノマー(単独で用いても、混合して用いてもよい)が挙げられる。また、樹脂(b)が縮合樹脂(例えば、ポリウレタン樹脂、エポキシ樹脂、及びポリエステル樹脂)である場合、前駆体(b0)は、反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせが例示される。
ポリエステル樹脂(p1)をビニルモノマーを前駆体(b0)として用いた場合、前駆体(b0)を反応させて樹脂(b)にする方法としては、例えば、油溶性開始剤、ビニルモノマー及び必要により有機溶剤(u)を水溶性ポリマー(q)存在下、樹脂粒子(A)の水性分散液(W)中に分散懸濁させ、加熱によりラジカル重合反応を行わせる方法(いわゆる懸濁重合法);ビニルモノマー及び必要により有機溶剤(u)を乳化剤[界面活性剤(s)と同様のものが例示される]、水溶性開始剤を含む樹脂粒子(A)の水性分散液(W)中に乳化させ、加熱によりラジカル重合反応を行わせる方法(いわゆる乳化重合法)等が挙げられる。
尚、ポリエステル樹脂(p1)の有する官能基(水酸基、カルボキシル基等)と反応可能な官能基を有するビニルモノマーを用い、予めポリエステル樹脂(p1)と反応させることにより、ポリエステル樹脂(p1)を構成単位として有するビニル樹脂を得ることができる。
尚、ポリエステル樹脂(p1)の有する官能基(水酸基、カルボキシル基等)と反応可能な官能基を有するビニルモノマーを用い、予めポリエステル樹脂(p1)と反応させることにより、ポリエステル樹脂(p1)を構成単位として有するビニル樹脂を得ることができる。
上記油溶性又は水溶性開始剤としては、パーオキサイド系重合開始剤(I)、アゾ系重合開始剤(II)等が挙げられる。また、パーオキサイド系重合開始剤(I)と還元剤とを併用してレドックス系重合開始剤(III)を形成してもよい。更には、(I)〜(III)のうちから2種以上を併用してもよい。
(I)パーオキサイド系重合開始剤
(I−1)油溶性パーオキサイド系重合開始剤:アセチルシクロヘキシルスルホニルパーオキサイド、イソブチリルパーオキサイド、ジイソプロピルパーオキシジカーボネート、ジ−2−エチルヘキシルパーオキシジカーボネート、2,4−ジクロロベンゾイルパーオキサイド、t−ブチルパーオキシビバレート、3,5,5−トリメチルヘキサノニルパーオキサイド、オクタノイルパーオキサイド、デカノイルパーオキサイド、ラウロイルパーオキサイド、ステアロイルパーオキサイド、プロピオニトリルパーオキサイド、サクシニックアシッドパーオキサイド、アセチルパーオキサイド、t−ブチルパーオキシ−2−エチルヘキサノエート、ベンゾイルパーオキサイド、パラクロロベンゾイルパーオキサイド、t−ブチルパーオキシイソブチレート、t−ブチルパーオキシマレイックアシッド、t−ブチルパーオキシラウレート、シクロヘキサノンパーオキサイド、t−ブチルパーオキシイソプロピルカーボネート、2,5−ジメチル−2,5−ジベンゾイルパーオキシヘキサン、t−ブチルパーオキシアセテート、t−ブチルパーオキシベンゾエート、ジイソブチルジパーオキシフタレート、メチルエチルケトンパーオキサイド、ジクミルパーオキサイド、2,5−ジメチル−2,5−ジt−ブチルパーオキシヘキサン、t−ブチルクミルパーオキサイド、t−ブチルヒドロパーオキサイド、ジt−ブチルパーオキサイド、ジイソプロピルベンゼンヒドロパーオキサイド、パラメンタンヒドロパーオキサイド、ピナンヒドロパーオキサイド、2,5−ジメチルヘキサン−2,5−ジヒドロパーオキサイド、クメンパーオキサイド等。
(I−2)水溶性パーオキサイド系重合開始剤:過酸化水素、過酢酸、過硫酸アンモニウム、過硫酸ナトリウム等。
(I−1)油溶性パーオキサイド系重合開始剤:アセチルシクロヘキシルスルホニルパーオキサイド、イソブチリルパーオキサイド、ジイソプロピルパーオキシジカーボネート、ジ−2−エチルヘキシルパーオキシジカーボネート、2,4−ジクロロベンゾイルパーオキサイド、t−ブチルパーオキシビバレート、3,5,5−トリメチルヘキサノニルパーオキサイド、オクタノイルパーオキサイド、デカノイルパーオキサイド、ラウロイルパーオキサイド、ステアロイルパーオキサイド、プロピオニトリルパーオキサイド、サクシニックアシッドパーオキサイド、アセチルパーオキサイド、t−ブチルパーオキシ−2−エチルヘキサノエート、ベンゾイルパーオキサイド、パラクロロベンゾイルパーオキサイド、t−ブチルパーオキシイソブチレート、t−ブチルパーオキシマレイックアシッド、t−ブチルパーオキシラウレート、シクロヘキサノンパーオキサイド、t−ブチルパーオキシイソプロピルカーボネート、2,5−ジメチル−2,5−ジベンゾイルパーオキシヘキサン、t−ブチルパーオキシアセテート、t−ブチルパーオキシベンゾエート、ジイソブチルジパーオキシフタレート、メチルエチルケトンパーオキサイド、ジクミルパーオキサイド、2,5−ジメチル−2,5−ジt−ブチルパーオキシヘキサン、t−ブチルクミルパーオキサイド、t−ブチルヒドロパーオキサイド、ジt−ブチルパーオキサイド、ジイソプロピルベンゼンヒドロパーオキサイド、パラメンタンヒドロパーオキサイド、ピナンヒドロパーオキサイド、2,5−ジメチルヘキサン−2,5−ジヒドロパーオキサイド、クメンパーオキサイド等。
(I−2)水溶性パーオキサイド系重合開始剤:過酸化水素、過酢酸、過硫酸アンモニウム、過硫酸ナトリウム等。
(II)アゾ系重合開始剤:
(II−1)油溶性アゾ系重合開始剤:2,2’−アゾビスイソブチロニトリル、1,1’−アゾビスシクロヘキサン1−カーボニトリル、2,2’−アゾビス−4−メトキシ−2,4−ジメチルバレロニトリル、2,2’−アゾビス−2,4−ジメチルバレロニトリル、ジメチル−2,2’−アゾビス(2−メチルプロピオネート)、1,1’−アゾビス(1−アセトキシ−1−フェニルエタン)、2,2’−アゾビス(4−メトキシ−2,4−ジメチルバレロニトリル)等。
(II−2)水溶性アゾ系重合開始剤:アゾビスアミジノプロパン塩、アゾビスシアノバレリックアシッド(塩)、2,2’−アゾビス[2−メチル−N−(2−ヒドロキシエチル)プロピオンアミド]等。
(II−1)油溶性アゾ系重合開始剤:2,2’−アゾビスイソブチロニトリル、1,1’−アゾビスシクロヘキサン1−カーボニトリル、2,2’−アゾビス−4−メトキシ−2,4−ジメチルバレロニトリル、2,2’−アゾビス−2,4−ジメチルバレロニトリル、ジメチル−2,2’−アゾビス(2−メチルプロピオネート)、1,1’−アゾビス(1−アセトキシ−1−フェニルエタン)、2,2’−アゾビス(4−メトキシ−2,4−ジメチルバレロニトリル)等。
(II−2)水溶性アゾ系重合開始剤:アゾビスアミジノプロパン塩、アゾビスシアノバレリックアシッド(塩)、2,2’−アゾビス[2−メチル−N−(2−ヒドロキシエチル)プロピオンアミド]等。
(III)レドックス系重合開始剤
(III−1)非水系レドックス系重合開始剤:油溶性過酸化物{ヒドロペルオキサイド、過酸化ジアルキル、過酸化ジアシル等}と、油溶性還元剤{第三アミン、ナフテン酸塩、メルカプタン、有機金属化合物(トリエチルアルミニウム、トリエチルホウ素、ジエチル亜鉛等)等}との組合せ等。
(III−2)水系レドックス系重合開始剤:水溶性過酸化物{過硫酸塩、過酸化水素、ヒドロペルオキサイド等}と、水溶性の無機若しくは有機還元剤(2価鉄塩、亜硫酸水素ナトリウム、アルコール、ポリアミン等)との組合せ等。
(III−1)非水系レドックス系重合開始剤:油溶性過酸化物{ヒドロペルオキサイド、過酸化ジアルキル、過酸化ジアシル等}と、油溶性還元剤{第三アミン、ナフテン酸塩、メルカプタン、有機金属化合物(トリエチルアルミニウム、トリエチルホウ素、ジエチル亜鉛等)等}との組合せ等。
(III−2)水系レドックス系重合開始剤:水溶性過酸化物{過硫酸塩、過酸化水素、ヒドロペルオキサイド等}と、水溶性の無機若しくは有機還元剤(2価鉄塩、亜硫酸水素ナトリウム、アルコール、ポリアミン等)との組合せ等。
前駆体(b0)としては、反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせを用いることもできる。ここで「反応性基」とは硬化剤(β)と反応可能な基のことをいう。樹脂(b)がポリエステル樹脂(p1)を構成単位として有する場合、前駆体(b0)のうち、プレポリマー(α)がポリエステル樹脂(p1)を構成単位として有することが好ましい。
この場合、前駆体(b0)を反応させて樹脂(b)を形成する方法としては、反応性基含有プレポリマー(α)、硬化剤(β)及び必要により有機溶剤(u)を、樹脂粒子(A)の水性分散液(W)中に分散させ、加熱により反応性基含有プレポリマー(α)と硬化剤(β)とを反応させて樹脂(b)を含有する樹脂粒子(B)を形成させる方法;反応性基含有プレポリマー(α)又はその溶剤溶液を樹脂粒子(A)の水性分散液(W)中に分散させ、ここに水溶性の硬化剤(β)を加え反応させて、樹脂(b)を含有する樹脂粒子(B)を形成させる方法;反応性基含有プレポリマー(α)が水と反応して硬化するものである場合は、反応性基含有プレポリマー(α)又はその溶剤溶液を樹脂粒子(A)の水性分散液(W)に分散させることで水と反応させて、樹脂(b)を含有する樹脂粒子(B)を形成させる方法等が例示できる。
この場合、前駆体(b0)を反応させて樹脂(b)を形成する方法としては、反応性基含有プレポリマー(α)、硬化剤(β)及び必要により有機溶剤(u)を、樹脂粒子(A)の水性分散液(W)中に分散させ、加熱により反応性基含有プレポリマー(α)と硬化剤(β)とを反応させて樹脂(b)を含有する樹脂粒子(B)を形成させる方法;反応性基含有プレポリマー(α)又はその溶剤溶液を樹脂粒子(A)の水性分散液(W)中に分散させ、ここに水溶性の硬化剤(β)を加え反応させて、樹脂(b)を含有する樹脂粒子(B)を形成させる方法;反応性基含有プレポリマー(α)が水と反応して硬化するものである場合は、反応性基含有プレポリマー(α)又はその溶剤溶液を樹脂粒子(A)の水性分散液(W)に分散させることで水と反応させて、樹脂(b)を含有する樹脂粒子(B)を形成させる方法等が例示できる。
反応性基含有プレポリマー(α)が有する反応性基と、硬化剤(β)の組み合わせとしては、下記〔1〕、〔2〕等が挙げられる。
〔1〕:反応性基含有プレポリマー(α)が有する反応性基が、活性水素化合物と反応可能な官能基(α1)であり、硬化剤(β)が活性水素化合物(β1)であるという組み合わせ。
〔2〕:反応性基含有プレポリマー(α)が有する反応性基が活性水素含有基(α2)であり、硬化剤(β)が活性水素含有基と反応可能な化合物(β2)であるという組み合わせ。
これらのうち、水性溶剤中での反応率の観点から、〔1〕の組合せがより好ましい。
〔1〕:反応性基含有プレポリマー(α)が有する反応性基が、活性水素化合物と反応可能な官能基(α1)であり、硬化剤(β)が活性水素化合物(β1)であるという組み合わせ。
〔2〕:反応性基含有プレポリマー(α)が有する反応性基が活性水素含有基(α2)であり、硬化剤(β)が活性水素含有基と反応可能な化合物(β2)であるという組み合わせ。
これらのうち、水性溶剤中での反応率の観点から、〔1〕の組合せがより好ましい。
上記組合せ〔1〕において、活性水素化合物と反応可能な官能基(α1)としては、イソシアネート基(α1a)、ブロック化イソシアネート基(α1b)、エポキシ基(α1c)、酸無水物基(1,3−オキソ−2−オキサプロピレン基)(α1d)及び酸ハライド基(ハロカルボニル基)(α1e)等が挙げられる。これらのうち好ましいものは、(α1a)、(α1b)及び(α1c)であり、特に好ましいものは、(α1a)及び(α1b)である。
ブロック化イソシアネート基(α1b)は、ブロック化剤によりブロックされたイソシアネート基のことをいう。上記ブロック化剤としては、オキシム[アセトオキシム、メチルイソブチルケトオキシム、ジエチルケトオキシム、シクロペンタノンオキシム、シクロヘキサノンオキシム、メチルエチルケトオキシム等];ラクタム[γ−ブチロラクタム、ε−カプロラクタム、γ−バレロラクタム等];炭素数1〜20の脂肪族アルコール[エタノール、メタノール、オクタノール等];フェノール[フェノール、m−クレゾール、キシレノール、ノニルフェノール等];活性メチレン化合物[アセチルアセトン、マロン酸エチル、アセト酢酸エチル等];塩基性窒素含有化合物[N,N−ジエチルヒドロキシルアミン、2−ヒドロキシピリジン、ピリジンN−オキサイド、2−メルカプトピリジン等];及びこれらの2種以上の混合物が挙げられる。これらのうち好ましいのはオキシムであり、特に好ましいものはメチルエチルケトオキシムである。
反応性基含有プレポリマー(α)の構成単位としては、ポリエーテル(αw)、ポリエステル(αx)、エポキシ樹脂(αy)及びポリウレタン(αz)等が挙げられる。これらのうち好ましいものは、(αx)、(αy)及び(αz)であり、特に好ましいものは(αx)及び(αz)である。ポリエーテル(αw)としては、ポリエチレンオキサイド、ポリプロピレンオキサイド、ポリブチレンオキサイド、ポリテトラメチレンオキサイド等が挙げられる。
ポリエステル(αx)としては、前述のポリエステル樹脂(p1)のプレポリマー及び/又はポリエステル樹脂(p1)以外のポリエステル樹脂のプレポリマー、ポリラクトン(ε−カプロラクトンの開環重合物等)等が挙げられる。
エポキシ樹脂(αy)としては、ビスフェノール(ビスフェノールA、ビスフェノールF、ビスフェノールS等)とエピクロルヒドリンとの付加縮合物等が挙げられる。
ポリウレタン(αz)としては、ジオール(1)及び/又は3〜8価のポリオール(2)とポリイソシアネート(15)との重付加物、ポリエステル(αx)とポリイソシアネート(15)の重付加物等が挙げられる。
エポキシ樹脂(αy)としては、ビスフェノール(ビスフェノールA、ビスフェノールF、ビスフェノールS等)とエピクロルヒドリンとの付加縮合物等が挙げられる。
ポリウレタン(αz)としては、ジオール(1)及び/又は3〜8価のポリオール(2)とポリイソシアネート(15)との重付加物、ポリエステル(αx)とポリイソシアネート(15)の重付加物等が挙げられる。
ポリエステル(αx)、エポキシ樹脂(αy)、ポリウレタン(αz)等に反応性基を含有させる方法としては、次の2つの方法等が挙げられる。
〔1〕:二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させる方法。
〔2〕:二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させ、更に残存した該官能基と反応可能な官能基及び反応性基を含有する化合物とを反応させる方法。
上記方法〔1〕では、水酸基含有ポリエステルプレポリマー、カルボキシル基含有ポリエステルプレポリマー、酸ハライド基(ハロカルボニル基)含有ポリエステルプレポリマー、水酸基含有エポキシ樹脂プレポリマー、エポキシ基含有エポキシ樹脂プレポリマー、水酸基含有ポリウレタンプレポリマー、イソシアネート基含有ポリウレタンプレポリマー等が得られる。
〔1〕:二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させる方法。
〔2〕:二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させ、更に残存した該官能基と反応可能な官能基及び反応性基を含有する化合物とを反応させる方法。
上記方法〔1〕では、水酸基含有ポリエステルプレポリマー、カルボキシル基含有ポリエステルプレポリマー、酸ハライド基(ハロカルボニル基)含有ポリエステルプレポリマー、水酸基含有エポキシ樹脂プレポリマー、エポキシ基含有エポキシ樹脂プレポリマー、水酸基含有ポリウレタンプレポリマー、イソシアネート基含有ポリウレタンプレポリマー等が得られる。
二以上の構成成分の比率は、例えば、水酸基含有ポリエステルプレポリマーの場合、ポリオール成分とポリカルボン酸成分の比率が、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、2/1〜1.01/1が好ましく、更に好ましくは1.5/1〜1.01/1、特に好ましくは1.3/1〜1.02/1である。他の骨格、末端基のプレポリマーの場合も、構成成分が変わるだけで比率は同様である。
上記方法〔2〕では、上記方法〔1〕で得られたプレプリマーに、ポリイソシアネートを反応させることでイソシアネート基含有プレポリマーが得られ、ブロック化ポリイソシアネートを反応させることでブロック化イソシアネート基含有プレポリマーが得られ、ポリエポキサイドを反応させることでエポキシ基含有プレポリマーが得られ、カルボニル基を4以上有する酸無水物を反応させることで酸無水物基(1,3−オキソ−2−オキサプロピレン基)含有プレポリマーが得られる。
官能基及び反応性基を含有する化合物の使用量は、例えば、水酸基含有ポリエステルにポリイソシアネートを反応させてイソシアネート基含有ポリエステルプレポリマーを得る場合、ポリイソシアネートの比率が、イソシアネート基[NCO]と、水酸基含有ポリエステルの水酸基[OH]の当量比[NCO]/[OH]として、5/1〜1.01/1が好ましく、更に好ましくは4/1〜1.2/1、特に好ましくは2.5/1〜1.5/1である。他の骨格、末端基を有するプレポリマーの場合も、構成成分が変わるだけで比率は同様である。
反応性基含有プレポリマー(α)中の1分子当たりに含有する反応性基は、通常1個以上、好ましくは、平均1.5〜3個、更に好ましくは、平均1.8〜2.5個である。上記範囲にすることで、硬化剤(β)と反応させて得られる硬化物の分子量が高くなる。
反応性基含有プレポリマー(α)のMnは、500〜30,000が好ましく、更に好ましくは1,000〜20,000、特に好ましくは2,000〜10,000である。
反応性基含有プレポリマー(α)の重量平均分子量(以下、Mwと記載)は、1,000〜50,000が好ましく、更に好ましくは2,000〜40,000、特に好ましくは4,000〜20,000である。
反応性基含有プレポリマー(α)の粘度は、100℃において、2,000ポイズ以下が好ましく、更に好ましくは1,000ポイズ以下である。2,000ポイズ以下にすることで粒度分布のシャープな樹脂粒子(C)が得られる点で好ましい。
反応性基含有プレポリマー(α)のMnは、500〜30,000が好ましく、更に好ましくは1,000〜20,000、特に好ましくは2,000〜10,000である。
反応性基含有プレポリマー(α)の重量平均分子量(以下、Mwと記載)は、1,000〜50,000が好ましく、更に好ましくは2,000〜40,000、特に好ましくは4,000〜20,000である。
反応性基含有プレポリマー(α)の粘度は、100℃において、2,000ポイズ以下が好ましく、更に好ましくは1,000ポイズ以下である。2,000ポイズ以下にすることで粒度分布のシャープな樹脂粒子(C)が得られる点で好ましい。
活性水素化合物(β1)としては、脱離可能な化合物でブロック化されていてもよいポリアミン(β1a)、ポリオール(β1b)、ポリメルカプタン(β1c)及び水(β1d)等が挙げられる。これらのうち好ましいものは、(β1a)、(β1b)及び(β1d)であり、更に好ましいもは、(β1a)及び(β1d)であり、特に好ましいもは、ブロック化されたポリアミン及び(β1d)である。
ポリアミン(β1a)としては、ポリアミン(16)と同様のものが例示される。これらのうち、好ましいものは、4,4’−ジアミノジフェニルメタン、キシリレンジアミン、イソホロンジアミン、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン及びそれらの混合物である。
ポリアミン(β1a)としては、ポリアミン(16)と同様のものが例示される。これらのうち、好ましいものは、4,4’−ジアミノジフェニルメタン、キシリレンジアミン、イソホロンジアミン、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン及びそれらの混合物である。
ポリアミン(β1a)が脱離可能な化合物でブロック化されたポリアミンである場合、この例としては、前記ポリアミンと炭素数3〜8のケトン(アセトン、メチルエチルケトン、メチルイソブチルケトン等)から得られるケチミン、炭素数2〜8のアルデヒド(ホルムアルデヒド、アセトアルデヒド)から得られるアルジミン、エナミン、及びオキサゾリジン等が挙げられる。
ポリオール(β1b)としては、前記のジオール(1)及び3〜8価のポリオール(2)と同様のものが例示される。ジオール(1)単独、又はジオール(1)と少量のポリオール(2)の混合物が好ましい。
ポリメルカプタン(β1c)としては、エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオール等が挙げられる。
ポリメルカプタン(β1c)としては、エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオール等が挙げられる。
必要により、活性水素化合物(β1)と共に反応停止剤(βs)を用いることができる。反応停止剤を活性水素化合物(β1)と一定の比率で併用することにより、樹脂(b)を所定の分子量に調整することが可能である。
反応停止剤(βs)としては、モノアミン(ジエチルアミン、ジブチルアミン、ブチルアミン、ラウリルアミン、モノエタノールアミン、ジエタノールアミン等);モノアミンをブロックしたもの(ケチミン化合物等);モノオール(メタノール、エタノール、イソプロパノール、ブタノール、フェノール;モノメルカプタン(ブチルメルカプタン、ラウリルメルカプタン等);モノイソシアネート(ラウリルイソシアネート、フェニルイソシアネート等);モノエポキサイド(ブチルグリシジルエーテル等)等が挙げられる。
反応停止剤(βs)としては、モノアミン(ジエチルアミン、ジブチルアミン、ブチルアミン、ラウリルアミン、モノエタノールアミン、ジエタノールアミン等);モノアミンをブロックしたもの(ケチミン化合物等);モノオール(メタノール、エタノール、イソプロパノール、ブタノール、フェノール;モノメルカプタン(ブチルメルカプタン、ラウリルメルカプタン等);モノイソシアネート(ラウリルイソシアネート、フェニルイソシアネート等);モノエポキサイド(ブチルグリシジルエーテル等)等が挙げられる。
上記組合せ〔2〕における反応性基含有プレポリマー(α)が有する活性水素含有基(α2)としては、アミノ基(α2a)、水酸基(アルコール性水酸基及びフェノール性水酸基)(α2b)、メルカプト基(α2c)、カルボキシル基(α2d)及びそれらが脱離可能な化合物でブロック化された有機基(α2e)等が挙げられる。これらのうち好ましいものは、(α2a)、(α2b)及びアミノ基が脱離可能な化合物でブロック化された有機基(α2e)であり、特に好ましいものは、(α2b)である。尚、アミノ基が脱離可能な化合物でブロック化された有機基としては、前記(β1a)の場合と同様のものが例示できる。
活性水素含有基と反応可能な化合物(β2)としては、ポリイソシアネート(β2a)、ポリエポキシド(β2b)、ポリカルボン酸(β2c)、ポリ酸無水物(β2d)及びポリ酸ハライド(β2e)等が挙げられる。これらのうち好ましいものは、(β2a)及び(β2b)であり、更に好ましいものは、(β2a)である。
ポリイソシアネート(β2a)としては、ポリイソシアネート(15)と同様のものが例示され、好ましいものも同様である。
ポリエポキシド(β2b)としては、ポリエポキシド(18)と同様のものが例示され、好ましいものも同様である。
ポリエポキシド(β2b)としては、ポリエポキシド(18)と同様のものが例示され、好ましいものも同様である。
ポリカルボン酸(β2c)としては、ジカルボン酸(β2c−1)及び3価以上のポリカルボン酸(β2c−2)が挙げられ、(β2c−1)単独、及び(β2c−1)と少量の(β2c−2)の混合物が好ましい。
ジカルボン酸(β2c−1)としては、前記ジカルボン酸(3)と、ポリカルボン酸としては、前記3〜6価のポリカルボン酸(4)と同様のものが例示され、好ましいものも同様である。
ジカルボン酸(β2c−1)としては、前記ジカルボン酸(3)と、ポリカルボン酸としては、前記3〜6価のポリカルボン酸(4)と同様のものが例示され、好ましいものも同様である。
ポリカルボン酸無水物(β2d)としては、ピロメリット酸無水物等が挙げられる。
ポリ酸ハライド(β2e)としては、前記(β2c)の酸ハライド(酸クロライド、酸ブロマイド、酸アイオダイド)等が挙げられる。
更に、必要により(β2)と共に反応停止剤(βs)を用いることができる。
ポリ酸ハライド(β2e)としては、前記(β2c)の酸ハライド(酸クロライド、酸ブロマイド、酸アイオダイド)等が挙げられる。
更に、必要により(β2)と共に反応停止剤(βs)を用いることができる。
硬化剤(β)の比率は、反応性基含有プレポリマー(α)中の反応性基の当量[α]と、硬化剤(β)中の活性水素含有基[β]の当量の比[α]/[β]として、1/2〜2/1が好ましく、更に好ましくは1.5/1〜1/1.5、特に好ましくは1.2/1〜1/1.2である。尚、硬化剤(β)が水(β1d)である場合は水は2価の活性水素化合物として取り扱う。
反応性基含有プレポリマー(α)と硬化剤(β)で構成される前駆体(b0)を水性溶剤中で反応させて得られた樹脂(b)が樹脂粒子(B)及び樹脂粒子(C)の構成成分となる。反応性基含有プレポリマー(α)と硬化剤(β)とを反応させた樹脂(b)のMwは、3,000以上が好ましく、更に好ましくは3,000〜1000万、特に好ましくは,5000〜100万である。
また、水性分散液(W)中において、反応性基含有プレポリマー(α)と硬化剤(β)との反応時に、反応性基含有プレポリマー(α)及び硬化剤(β)と反応しないポリマー[いわゆるデッドポリマー]を系内に含有させることもできる。この場合樹脂(b)は、反応性基含有プレポリマー(α)と硬化剤(β)とを反応させて得られた樹脂と、デットポリマーとの混合物となる。
デッドポリマーとしては、ビニル樹脂及びポリエステル樹脂が好ましく、ポリエステル樹脂が更に好ましく、ポリエステル樹脂(p1)が特に好ましい。
樹脂(b)中のデッドポリマー〔前躯体(b0)が反応して得られた樹脂以外のポリマー〕の含有量は、好ましくは0〜80%、更に好ましくは5〜70%ある。
デッドポリマーとしては、ビニル樹脂及びポリエステル樹脂が好ましく、ポリエステル樹脂が更に好ましく、ポリエステル樹脂(p1)が特に好ましい。
樹脂(b)中のデッドポリマー〔前躯体(b0)が反応して得られた樹脂以外のポリマー〕の含有量は、好ましくは0〜80%、更に好ましくは5〜70%ある。
樹脂粒子(A)及び/又は樹脂粒子(B)中に他の添加物(顔料、充填剤、帯電防止剤、着色剤、離型剤、荷電制御剤、紫外線吸収剤、酸化防止剤、ブロッキング防止剤、耐熱安定剤、難燃剤等)を混合しても差し支えない。樹脂粒子(A)又は樹脂粒子(B)中に他の添加物する方法としては、水性分散液(W)中で水性分散体(X1)を形成させる際に混合してもよいが、あらかじめ樹脂(a)又は樹脂(b)の前躯体(b0)を含有する油性液(OL)と添加物とを混合した後、水性分散液(W)中にその混合物を加えて分散させることがより好ましい。
また、本発明においては、添加剤は、必ずしも、水性分散液(W)中で粒子を形成させる時に混合しておく必要はなく、粒子を形成せしめた後、添加してもよい。たとえば、着色剤を含まない樹脂粒子を形成させた後、公知の染着の方法で着色剤を添加したり、有機溶剤(u)及び/又は可塑剤(v)とともに上記添加物を樹脂粒子に含浸させることもできる。
また、本発明においては、添加剤は、必ずしも、水性分散液(W)中で粒子を形成させる時に混合しておく必要はなく、粒子を形成せしめた後、添加してもよい。たとえば、着色剤を含まない樹脂粒子を形成させた後、公知の染着の方法で着色剤を添加したり、有機溶剤(u)及び/又は可塑剤(v)とともに上記添加物を樹脂粒子に含浸させることもできる。
樹脂(b)100重量部に対する水性分散液(W)の使用量は、50〜2,000重量部が好ましく、更に好ましくは100〜1,000重量部である。50重量部以上では樹脂(b)の分散状態が良好である。2,000重量部以下であると経済的である。
伸長及び/又は架橋反応時間は、プレポリマー(α)の有する反応性基の構造と硬化剤(β)の組み合わせによる反応性により選択されるが、好ましくは10分〜40時間、更に好ましくは30分〜24時間である。反応温度は、0〜150℃が好ましく、更に好ましくは50〜120℃である。また、必要に応じて公知の触媒を使用することができる。具体的には、例えばイソシアネートと活性水素化合物の反応の場合には、ジブチルチンラウレート、ジオクチルチンラウレート等が挙げられる。
樹脂粒子(C)は、水性分散体(X1)から水性溶剤を除去することにより得られる。水性溶剤を除去する方法としては、以下の方法等が例示される。
〔1〕水性分散体(X1)を減圧下又は大気圧下で乾燥する方法。
〔2〕遠心分離器、スパクラフィルター、フィルタープレス等により、水性分散体(X1)を固液分離し、得られた粉末を乾燥する方法。
〔3〕水性分散体(X1)を凍結させて乾燥させる方法(いわゆる凍結乾燥)。
上記〔1〕又は〔2〕の方法において、得られた粉末を乾燥する際、流動層式乾燥機、減圧乾燥機、循風乾燥機等公知の設備を用いて行うことができる。また、必要に応じ、風力分級器等を用いて分級し、所定の粒度分布とすることもできる。
〔1〕水性分散体(X1)を減圧下又は大気圧下で乾燥する方法。
〔2〕遠心分離器、スパクラフィルター、フィルタープレス等により、水性分散体(X1)を固液分離し、得られた粉末を乾燥する方法。
〔3〕水性分散体(X1)を凍結させて乾燥させる方法(いわゆる凍結乾燥)。
上記〔1〕又は〔2〕の方法において、得られた粉末を乾燥する際、流動層式乾燥機、減圧乾燥機、循風乾燥機等公知の設備を用いて行うことができる。また、必要に応じ、風力分級器等を用いて分級し、所定の粒度分布とすることもできる。
樹脂粒子(A)と樹脂粒子(B)との付着力を強めたいとき、水性分散液(W)中に油性液(OL)を分散した際に、樹脂粒子(A)と樹脂粒子(B)が正負逆の電荷を持つようにしたり、樹脂粒子(A)と樹脂粒子(B)が同一の電荷持つ場合、樹脂粒子(A)及び樹脂粒子(B)と逆電荷をもつ界面活性剤(s)又は水溶性ポリマー(q)を使用したり、樹脂(a)と樹脂(b)とのSP値差を2以下にしたりすることが有効である。
樹脂粒子(C)の粒径均一性、保存安定性等の観点から、樹脂粒子(C)は、0.1〜50(好ましくは0.2〜40)%の樹脂粒子(A)と50〜99.9(好ましくは60〜99.8)%の樹脂粒子(B)とから構成されることが好ましい。
樹脂粒子(C)の粒径均一性、粉体流動性、保存安定性等の観点から、樹脂粒子(B)の表面の5(好ましくは30、更に好ましくは80)%以上が樹脂粒子(A)で覆われているのが好ましい。尚、表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めることができる。
表面被覆率(%)=(SA)×100/(SA)+(SB)
(SA):樹脂粒子(A)に覆われている部分の面積
(SB):樹脂粒子(B)が露出している部分の面積
表面被覆率(%)=(SA)×100/(SA)+(SB)
(SA):樹脂粒子(A)に覆われている部分の面積
(SB):樹脂粒子(B)が露出している部分の面積
粒径均一性から、樹脂粒子(C)の[体積平均粒径/個数平均粒径]は、1.0〜1.5が好ましく、更に好ましくは1.0〜1.45、特に好ましくは1.05〜1.2である。
樹脂粒子(C)の体積平均粒径は、用途により異なるが、0.1〜300μmが好ましい。上限は、更に好ましくは250μm、特に好ましくは200μm、最も好ましくは20μmであり、下限は、更に好ましくは0.5μm、特に好ましくは1μm、最も好ましくは4μmである。
尚、体積平均粒径及び個数平均粒径は、コールターカウンターで同時に測定することができる。
樹脂粒子(C)の体積平均粒径は、用途により異なるが、0.1〜300μmが好ましい。上限は、更に好ましくは250μm、特に好ましくは200μm、最も好ましくは20μmであり、下限は、更に好ましくは0.5μm、特に好ましくは1μm、最も好ましくは4μmである。
尚、体積平均粒径及び個数平均粒径は、コールターカウンターで同時に測定することができる。
粉体流動性を向上させたい場合、樹脂粒子(C)のBET値比表面積が0.5〜5.0m2/gであるのが好ましい。BET比表面積は、比表面積計、例えばQUANTASORB(ユアサアイオニクス製)を用いて測定(測定ガス:He/Kr=99.9/0.1vol%、検量ガス:窒素)することができる。
同様に粉体流動性の観点から、樹脂粒子(C)の表面平均中心線粗さRaが0.01〜0.8μmであるのが好ましい。Raは、粗さ曲線とその中心線との偏差の絶対値を算術平均した値のことであり、例えば、走査型プローブ顕微鏡システム(東陽テクニカ製)で測定することができる。
同様に粉体流動性の観点から、樹脂粒子(C)の表面平均中心線粗さRaが0.01〜0.8μmであるのが好ましい。Raは、粗さ曲線とその中心線との偏差の絶対値を算術平均した値のことであり、例えば、走査型プローブ顕微鏡システム(東陽テクニカ製)で測定することができる。
樹脂粒子(C)の形状は、粉体流動性、溶融レベリング性等の観点から球状であるのが好ましい。その場合、樹脂粒子(A)及び樹脂粒子(B)も球状であるのが好ましい。樹脂粒子(C)はWadellの実用球形度が0.85〜1.00であるのが好ましく、更に好ましくは0.90〜1.00である。尚、Wadellの実用球形度は、粒子の投影面積に等しい面積を持つ円の直径と粒子の投影像に外接する最小面積の円との直径の比から求められる。粒子の投影像は、例えば走査電子顕微鏡(SEM)によって撮影することができる。
樹脂粒子(A)の樹脂粒子(B)に対する粒径比、及び、水性分散体(X1)中における樹脂粒子(A)による樹脂粒子(B)表面の被覆率、水性分散体(X1)中における樹脂粒子(B)/水性溶剤界面上で樹脂粒子(A)が樹脂粒子(B)側に埋め込まれている深さ、を変えることにより、樹脂粒子(B)の表面を平滑にしたり、表面に凹凸を付与したりすることができる。
樹脂粒子(A)による樹脂粒子(B)表面の被覆率や、樹脂粒子(A)が樹脂粒子(B)側に埋め込まれている深さは、以下のような方法で制御することができる。
樹脂粒子(A)による樹脂粒子(B)表面の被覆率や、樹脂粒子(A)が樹脂粒子(B)側に埋め込まれている深さは、以下のような方法で制御することができる。
〔1〕水性分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)とが正負逆の電荷を持つようにすると被覆率、深さが大きくなる。この場合、樹脂粒子(A)、樹脂粒子(B)各々の電荷を大きくするほど、被覆率、深さが大きくなる。
〔2〕水性分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)とが同極性(どちらも正、又はどちらも負)の電荷を持つようにすると、被覆率は下がり、深さが小さくなる傾向にある。この場合、一般に界面活性剤(s)及び/又は水溶性ポリマー(t)[特に樹脂粒子(A)及び樹脂粒子(B)と逆電荷を有するもの]を使用すると被覆率が上がる。また、水溶性ポリマー(t)を使用する場合には、水溶性ポリマー(t)の分子量が大きいほど深さが小さくなる。
〔2〕水性分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)とが同極性(どちらも正、又はどちらも負)の電荷を持つようにすると、被覆率は下がり、深さが小さくなる傾向にある。この場合、一般に界面活性剤(s)及び/又は水溶性ポリマー(t)[特に樹脂粒子(A)及び樹脂粒子(B)と逆電荷を有するもの]を使用すると被覆率が上がる。また、水溶性ポリマー(t)を使用する場合には、水溶性ポリマー(t)の分子量が大きいほど深さが小さくなる。
〔3〕水性分散体(X1)を製造する際に、樹脂(a)がカルボキシル基、ホスホノ基、スルホ基等の酸性官能基を有する樹脂(一般に酸性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性溶剤のpHが低いほど被覆率、深さが大きくなる。逆に、pHを高くするほど被覆率、深さが小さくなる。
〔4〕水性分散体(X1)を製造する際に、樹脂(a)が1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニオ基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性溶剤のpHが高いほど被覆率、深さが大きくなる。逆に、pHを低くするほど被覆率、深さが小さくなる。
〔5〕樹脂(a)と樹脂(b)とのSP値差を小さくするほど被覆率、深さが大きくなる。
〔4〕水性分散体(X1)を製造する際に、樹脂(a)が1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニオ基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性溶剤のpHが高いほど被覆率、深さが大きくなる。逆に、pHを低くするほど被覆率、深さが小さくなる。
〔5〕樹脂(a)と樹脂(b)とのSP値差を小さくするほど被覆率、深さが大きくなる。
樹脂粒子(B)の体積平均粒径は、用いられる用途により異なるが、0.1〜300μmが好ましい。上限は、更に好ましくは250μm、特に好ましくは200μm、最も好ましくは20μmであり、下限は、更に好ましくは0.5μm、特に好ましくは1μm、最も好ましくは4μmである。また、粒径均一性から、樹脂粒子(B)の[体積平均粒径/個数平均粒径]は、1.0〜1.5であるのが好ましく、更に好ましくは1.0〜1.45、特に好ましくは1.05〜1.15である。
粉体流動性を向上させたい場合には、樹脂粒子(B)のBET値比表面積を0.5〜5.0m2/gとすることが好ましく、表面平均中心線粗さRaを0.01〜0.8μmと
することが好ましい。 樹脂粒子(B)の形状は、粉体流動性、溶融レベリング性等の観点から球状であるのが好ましく、Wadellの実用球形度が0.85〜1.00であるのが好ましく、より好ましくは0.90〜1.00である。
することが好ましい。 樹脂粒子(B)の形状は、粉体流動性、溶融レベリング性等の観点から球状であるのが好ましく、Wadellの実用球形度が0.85〜1.00であるのが好ましく、より好ましくは0.90〜1.00である。
以下実施例により本発明を更に説明するが、本発明はこれに限定されるものではない。以下の記載において、「部」は重量部を意味する。
実施例における樹脂物性の測定条件は、以下のとおりである。
1.ガラス転移温度(Tg)
ASTM D3418−82に規定の方法(DSC法)。
装置:セイコー電子工業(株)製 DSC20,SSC/5803.
2.酸価及び水酸基価
JIS K0070−1992に規定の方法。
1.ガラス転移温度(Tg)
ASTM D3418−82に規定の方法(DSC法)。
装置:セイコー電子工業(株)製 DSC20,SSC/5803.
2.酸価及び水酸基価
JIS K0070−1992に規定の方法。
3.Mn及びMw
ポリウレタン樹脂以外の樹脂{ポリエステル樹脂を含む}のMn及びMwは、テトラヒドロフラン(THF)可溶分について、ゲルパーミエーションクロマトグラフィー(GPC)を用いて以下の条件で測定した。
装置 : 東ソー製 HLC−8120
カラム : TSKgelGMHXL(2本)
TSKgelMultiporeHXL−M(1本)
測定温度 : 40℃
試料溶液 : 0.25%のTHF溶液
溶液注入量 : 100μl
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点 (分子量 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000 4480000)
また、ポリウレタン樹脂のMn及びMwは、GPCを用いて以下の条件で測定した。
装置 : 東ソー製 HLC−8220GPC
カラム : Guardcolumn α
TSKgel α−M
流量 : 1ml/分
試料溶液 : 0.125%のジメチルホルムアミド溶液
溶液注入量 : 100μl
温度 : 40℃
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000)
ポリウレタン樹脂以外の樹脂{ポリエステル樹脂を含む}のMn及びMwは、テトラヒドロフラン(THF)可溶分について、ゲルパーミエーションクロマトグラフィー(GPC)を用いて以下の条件で測定した。
装置 : 東ソー製 HLC−8120
カラム : TSKgelGMHXL(2本)
TSKgelMultiporeHXL−M(1本)
測定温度 : 40℃
試料溶液 : 0.25%のTHF溶液
溶液注入量 : 100μl
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点 (分子量 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000 4480000)
また、ポリウレタン樹脂のMn及びMwは、GPCを用いて以下の条件で測定した。
装置 : 東ソー製 HLC−8220GPC
カラム : Guardcolumn α
TSKgel α−M
流量 : 1ml/分
試料溶液 : 0.125%のジメチルホルムアミド溶液
溶液注入量 : 100μl
温度 : 40℃
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000)
<製造例1>
[チタン触媒(t−1)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、チタニウムジイソプロポキシビス(トリエタノールアミネート)1617部とイオン交換水126部を入れ、窒素にて液中バブリング下、90℃まで徐々に昇温し、90℃で4時間反応(加水分解)させることで、チタニウムジヒドロキシビス(トリエタノールアミネート)を得た。更に、100℃にて2時間、400Paの減圧下で反応(脱水縮合)させることで、分子内重縮合物であるチタン触媒(t−1)を得た。
[チタン触媒(t−1)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、チタニウムジイソプロポキシビス(トリエタノールアミネート)1617部とイオン交換水126部を入れ、窒素にて液中バブリング下、90℃まで徐々に昇温し、90℃で4時間反応(加水分解)させることで、チタニウムジヒドロキシビス(トリエタノールアミネート)を得た。更に、100℃にて2時間、400Paの減圧下で反応(脱水縮合)させることで、分子内重縮合物であるチタン触媒(t−1)を得た。
<製造例2>
[水性分散体[樹脂粒子(A−1)分散体]の合成]
撹拌棒及び温度計をセットした反応容器に、水682部、メタクリル酸EO付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業株式会社製)11部、スチレン138部、メタクリル酸138部、過硫酸アンモニウム1部を仕込み、25℃、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。この乳濁液を加熱して75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成してビニル樹脂(スチレン−メタクリル酸−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の水性分散体[樹脂粒子(A−1)分散体]を得た。[樹脂粒子(A−1)分散体]の体積平均粒径は、0.10μmであった。また、[樹脂粒子(A−1)分散体]の一部を乾燥して樹脂粒子(A−1)を単離した。樹脂粒子(A)のTgは148℃であった。
[水性分散体[樹脂粒子(A−1)分散体]の合成]
撹拌棒及び温度計をセットした反応容器に、水682部、メタクリル酸EO付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業株式会社製)11部、スチレン138部、メタクリル酸138部、過硫酸アンモニウム1部を仕込み、25℃、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。この乳濁液を加熱して75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成してビニル樹脂(スチレン−メタクリル酸−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の水性分散体[樹脂粒子(A−1)分散体]を得た。[樹脂粒子(A−1)分散体]の体積平均粒径は、0.10μmであった。また、[樹脂粒子(A−1)分散体]の一部を乾燥して樹脂粒子(A−1)を単離した。樹脂粒子(A)のTgは148℃であった。
<製造例3>
[ポリエステル樹脂(p−1)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール(以下、単にプロピレングリコールと記載する)675部(99.5モル部)、ネオペンチルグリコール2.3部(0.25モル部)、テレフタル酸519部(35モル部)、イソフタル酸208部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは300部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−1)とした。
ポリエステル樹脂(p−1)のMnは1900、Mwは4800、酸価は25であった。
[ポリエステル樹脂(p−1)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール(以下、単にプロピレングリコールと記載する)675部(99.5モル部)、ネオペンチルグリコール2.3部(0.25モル部)、テレフタル酸519部(35モル部)、イソフタル酸208部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは300部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−1)とした。
ポリエステル樹脂(p−1)のMnは1900、Mwは4800、酸価は25であった。
<製造例4>
[ポリエステル樹脂(p−2)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール671部(99モル部)、ネオペンチルグリコール6.5部(0.7モル部)、テレフタル酸519部(35モル部)、イソフタル酸207部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは298部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−2)とした。ポリエステル樹脂(p−2)のMnは2000、Mwは4900、酸価は25であった。
[ポリエステル樹脂(p−2)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール671部(99モル部)、ネオペンチルグリコール6.5部(0.7モル部)、テレフタル酸519部(35モル部)、イソフタル酸207部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは298部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−2)とした。ポリエステル樹脂(p−2)のMnは2000、Mwは4900、酸価は25であった。
<製造例5>
[ポリエステル樹脂(p−3)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール609部(96モル部)、ネオペンチルグリコール13部(1.5モル部)、フェノールノボラック樹脂(平均重合度5.6)のEO5.6モル付加体74部(1.1モル部)、テレフタル酸485部(35モル部)、イソフタル酸194部(14モル部)、アジピン酸12部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは279部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.5モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−3)とした。
ポリエステル樹脂(p−3)のMnは1900、Mwは5000、酸価は25であった。
[ポリエステル樹脂(p−3)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール609部(96モル部)、ネオペンチルグリコール13部(1.5モル部)、フェノールノボラック樹脂(平均重合度5.6)のEO5.6モル付加体74部(1.1モル部)、テレフタル酸485部(35モル部)、イソフタル酸194部(14モル部)、アジピン酸12部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは279部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.5モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−3)とした。
ポリエステル樹脂(p−3)のMnは1900、Mwは5000、酸価は25であった。
<製造例6>
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール644部(95モル部)、ネオペンチルグリコール46部(5モル部)、テレフタル酸652部(44モル部)、アジピン酸78部(6モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは300部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−4)とした。
ポリエステル樹脂(p−4)のMnは1700、Mwは4500、酸価は25であった。
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール644部(95モル部)、ネオペンチルグリコール46部(5モル部)、テレフタル酸652部(44モル部)、アジピン酸78部(6モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは300部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p−4)とした。
ポリエステル樹脂(p−4)のMnは1700、Mwは4500、酸価は25であった。
<比較製造例1>
[ポリエステル樹脂(p’−5)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール678部(100モル部)、テレフタル酸518部(35モル部)、イソフタル酸207部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは298部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p’−5)とした。
ポリエステル樹脂(p’−5)のMnは1800、Mwは4800、酸価は25であった。
[ポリエステル樹脂(p’−5)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール678部(100モル部)、テレフタル酸518部(35モル部)、イソフタル酸207部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは298部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p’−5)とした。
ポリエステル樹脂(p’−5)のMnは1800、Mwは4800、酸価は25であった。
<比較製造例2>
[ポリエステル樹脂(p’−6)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール555部(93.5モル部)、フェノールノボラック樹脂(平均重合度5.6)のEO5.6モル付加体158部(2.5モル部)、テレフタル酸454部(35モル部)、イソフタル酸182部(14モル部)、アジピン酸11部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは261部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p’−6)とした。
ポリエステル樹脂(p’−6)のMnは2000、Mwは5200、酸価は25であった。
[ポリエステル樹脂(p’−6)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール555部(93.5モル部)、フェノールノボラック樹脂(平均重合度5.6)のEO5.6モル付加体158部(2.5モル部)、テレフタル酸454部(35モル部)、イソフタル酸182部(14モル部)、アジピン酸11部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは261部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p’−6)とした。
ポリエステル樹脂(p’−6)のMnは2000、Mwは5200、酸価は25であった。
<比較製造例3>
[ポリエステル樹脂(p’−7)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール606部(91モル部)、ネオペンチルグリコール77部(8.5モル部)、テレフタル酸509部(35モル部)、イソフタル酸204部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは293部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p’−7)とした。
ポリエステル樹脂(p’−7)のMnは2000、Mwは4700、酸価は25であった。
[ポリエステル樹脂(p’−7)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、プロピレングリコール606部(91モル部)、ネオペンチルグリコール77部(8.5モル部)、テレフタル酸509部(35モル部)、イソフタル酸204部(14モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは293部(44モル部)であった。次いで180℃まで冷却し、無水トリメリット酸39部(2.3モル部)を加え、密閉下2時間反応後、220℃、常圧で反応させ、取り出した。これをポリエステル樹脂(p’−7)とした。
ポリエステル樹脂(p’−7)のMnは2000、Mwは4700、酸価は25であった。
<製造例7>
[[ポリエステル樹脂(p−8)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール629部(98モル部)、ネオペンチルグリコール9部(1モル部)、フェノールノボラック樹脂(平均重合度5.6)のEO5.6モル付加体21部(0.3モル部)、テレフタル酸464部(33モル部)、イソフタル酸267部(19モル部)、アジピン酸12部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、これをポリエステル樹脂(p−8)とした。回収されたプロピレングリコールは283部(44モル部)であった。
ポリエステル樹脂(p−8)のMnは4800、Mwは18000、酸価は0.5であった。
[[ポリエステル樹脂(p−8)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール629部(98モル部)、ネオペンチルグリコール9部(1モル部)、フェノールノボラック樹脂(平均重合度5.6)のEO5.6モル付加体21部(0.3モル部)、テレフタル酸464部(33モル部)、イソフタル酸267部(19モル部)、アジピン酸12部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、これをポリエステル樹脂(p−8)とした。回収されたプロピレングリコールは283部(44モル部)であった。
ポリエステル樹脂(p−8)のMnは4800、Mwは18000、酸価は0.5であった。
<製造例8>
[[ポリエステル樹脂(p−9)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール661部(100モル部)、ネオペンチルグリコール2.7部(0.31モル部)、テレフタル酸477部(33モル部)、イソフタル酸275部(19モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、これをポリエステル樹脂(p−9)とした。回収されたプロピレングリコールは315部(46モル部)であった。
ポリエステル樹脂(p−9)のMnは5000、Mwは18000、酸価は0.8であった。
[[ポリエステル樹脂(p−9)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール661部(100モル部)、ネオペンチルグリコール2.7部(0.31モル部)、テレフタル酸477部(33モル部)、イソフタル酸275部(19モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、これをポリエステル樹脂(p−9)とした。回収されたプロピレングリコールは315部(46モル部)であった。
ポリエステル樹脂(p−9)のMnは5000、Mwは18000、酸価は0.8であった。
<比較製造例4>
[[ポリエステル樹脂(p’−10)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール685部(100モル部)、テレフタル酸494部(33モル部)、イソフタル酸284部(19モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、これをポリエステル樹脂(p’−10)とした。回収されたプロピレングリコールは305部(44.5モル部)であった。ポリエステル樹脂(p’−10)のMnは4900、Mwは18500、酸価は0.7であった。
[[ポリエステル樹脂(p’−10)の合成]
冷却管、攪拌機及び窒素導入管の付いた反応槽中に、1,2−プロピレングリコール685部(100モル部)、テレフタル酸494部(33モル部)、イソフタル酸284部(19モル部)、アジピン酸13部(1モル部)、重合触媒としてチタン触媒(t−1)0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら12時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、更に5〜20mmHgの減圧下に反応させ、これをポリエステル樹脂(p’−10)とした。回収されたプロピレングリコールは305部(44.5モル部)であった。ポリエステル樹脂(p’−10)のMnは4900、Mwは18500、酸価は0.7であった。
<製造例9>
[ポリエステル樹脂(p−8)を構成単位として有する[ウレタンプレポリマー1]の合成]
オートクレーブに、上記ポリエステル樹脂(p−8)422部、IPDI61部、酢酸エチル517部を投入し、密閉状態で100℃、8時間反応を行い、ポリエステル樹脂(p−8)を構成単位として有し、かつ分子末端にイソシアネート基を有する[ウレタンプレポリマー1]の溶液を得た。[ウレタンプレポリマー1]の溶液のNCO含量は0.8%であった。
[ポリエステル樹脂(p−8)を構成単位として有する[ウレタンプレポリマー1]の合成]
オートクレーブに、上記ポリエステル樹脂(p−8)422部、IPDI61部、酢酸エチル517部を投入し、密閉状態で100℃、8時間反応を行い、ポリエステル樹脂(p−8)を構成単位として有し、かつ分子末端にイソシアネート基を有する[ウレタンプレポリマー1]の溶液を得た。[ウレタンプレポリマー1]の溶液のNCO含量は0.8%であった。
<製造例10>
[ポリエステル樹脂(p−9)を構成単位として有する[ウレタンプレポリマー2]の合成]
ポリエステル樹脂(p−8)をポリエステル樹脂(p−9)に代える以外は製造例9の[ウレタンプレポリマー1]と同様に反応させ、ポリエステル樹脂(p−9)を構成単位として有し、かつ分子末端にイソシアネート基を有する[ウレタンプレポリマー2]の溶液を得た。[ウレタンプレポリマー2]の溶液のNCO含量は0.7%であった。
[ポリエステル樹脂(p−9)を構成単位として有する[ウレタンプレポリマー2]の合成]
ポリエステル樹脂(p−8)をポリエステル樹脂(p−9)に代える以外は製造例9の[ウレタンプレポリマー1]と同様に反応させ、ポリエステル樹脂(p−9)を構成単位として有し、かつ分子末端にイソシアネート基を有する[ウレタンプレポリマー2]の溶液を得た。[ウレタンプレポリマー2]の溶液のNCO含量は0.7%であった。
<比較製造例5>
[ポリエステル樹脂(p’−10)を構成単位として有する[ウレタンプレポリマー3’]の合成]
ポリエステル樹脂(p−8)をポリエステル樹脂(p’−10)に代える以外は製造例9の[ウレタンプレポリマー1]と同様に反応させ、ポリエステル樹脂(p’−10)を構成単位として有し、かつ分子末端にイソシアネート基を有する[ウレタンプレポリマー3’]の溶液を得た。[ウレタンプレポリマー3’]の溶液のNCO含量は0.7%であった。
[ポリエステル樹脂(p’−10)を構成単位として有する[ウレタンプレポリマー3’]の合成]
ポリエステル樹脂(p−8)をポリエステル樹脂(p’−10)に代える以外は製造例9の[ウレタンプレポリマー1]と同様に反応させ、ポリエステル樹脂(p’−10)を構成単位として有し、かつ分子末端にイソシアネート基を有する[ウレタンプレポリマー3’]の溶液を得た。[ウレタンプレポリマー3’]の溶液のNCO含量は0.7%であった。
<製造例11>
[[硬化剤1]の合成]
撹拌機、脱溶剤装置、及び温度計をセットした反応容器に、イソホロンジアミン50部とメチルエチルケトン300部を投入し、50℃で5時間反応を行った後、脱溶剤してケチミン[硬化剤1]を得た。[硬化剤1]の全アミン価は415であった。
[[硬化剤1]の合成]
撹拌機、脱溶剤装置、及び温度計をセットした反応容器に、イソホロンジアミン50部とメチルエチルケトン300部を投入し、50℃で5時間反応を行った後、脱溶剤してケチミン[硬化剤1]を得た。[硬化剤1]の全アミン価は415であった。
<製造例12>
[樹脂粒子(A−1)分散体]を含む[水性分散液W−1]の作製]
水634部、[樹脂粒子(A−1)分散体]286部、カルボキシメチルセルロース(「CMCダイセル1170」、ダイセル化学工業株式会社製)2部、及びドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(「エレミノールMON−7」、三洋化成工業株式会社製)154部を混合攪拌し、乳白色の液体[水性分散液W−1]を得た。
[樹脂粒子(A−1)分散体]を含む[水性分散液W−1]の作製]
水634部、[樹脂粒子(A−1)分散体]286部、カルボキシメチルセルロース(「CMCダイセル1170」、ダイセル化学工業株式会社製)2部、及びドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(「エレミノールMON−7」、三洋化成工業株式会社製)154部を混合攪拌し、乳白色の液体[水性分散液W−1]を得た。
<実施例1>
ビーカー内に[ポリエステル樹脂(p−1)]177部、酢酸エチル181部、[ウレタンプレポリマー1]の溶液39.2部、[硬化剤1]0.9部を投入して溶解・混合均一化し、[樹脂溶液1]を得た。この[樹脂溶液1]中に[水性分散液W−1]600部を添加し、TKホモミキサー(特殊機化工業株式会社製)を使用し、回転数12000rpmで25℃で1分間分散操作を行い、更にフィルムエバポレータで減圧度−0.05MPa(ゲージ圧)、温度40℃、回転数100rpmの条件で240分間脱溶剤し、水性分散体(X−1)を得た。水性分散体(X−1)100部を遠心分離し、更に水60部を加えて遠心分離して固液分離する工程を2回繰り返した後、35℃で1時間乾燥して樹脂粒子(C1)を得た。
ビーカー内に[ポリエステル樹脂(p−1)]177部、酢酸エチル181部、[ウレタンプレポリマー1]の溶液39.2部、[硬化剤1]0.9部を投入して溶解・混合均一化し、[樹脂溶液1]を得た。この[樹脂溶液1]中に[水性分散液W−1]600部を添加し、TKホモミキサー(特殊機化工業株式会社製)を使用し、回転数12000rpmで25℃で1分間分散操作を行い、更にフィルムエバポレータで減圧度−0.05MPa(ゲージ圧)、温度40℃、回転数100rpmの条件で240分間脱溶剤し、水性分散体(X−1)を得た。水性分散体(X−1)100部を遠心分離し、更に水60部を加えて遠心分離して固液分離する工程を2回繰り返した後、35℃で1時間乾燥して樹脂粒子(C1)を得た。
<実施例2>
上記<実施例1>において、[ポリエステル樹脂(p−1)]の替わりに[ポリエステル樹脂(p−2)]を、[ウレタンプレポリマー1]の替わりに[ウレタンプレポリマー2]を使用する以外は同様の方法により、水性分散体(X−2)及び樹脂粒子(C2)を得た。
上記<実施例1>において、[ポリエステル樹脂(p−1)]の替わりに[ポリエステル樹脂(p−2)]を、[ウレタンプレポリマー1]の替わりに[ウレタンプレポリマー2]を使用する以外は同様の方法により、水性分散体(X−2)及び樹脂粒子(C2)を得た。
<実施例3>
上記<実施例1>において、[ポリエステル樹脂(p−1)]177部の替わりに[ポリエステル樹脂(p−3)]196部、酢酸エチル181部を201部とし、[硬化剤1]及び[ウレタンプレポリマー1]を使用しない以外は同様の方法により、水性分散体(X−3)及び樹脂粒子(C3)を得た。
上記<実施例1>において、[ポリエステル樹脂(p−1)]177部の替わりに[ポリエステル樹脂(p−3)]196部、酢酸エチル181部を201部とし、[硬化剤1]及び[ウレタンプレポリマー1]を使用しない以外は同様の方法により、水性分散体(X−3)及び樹脂粒子(C3)を得た。
<実施例4>
上記<実施例1>において、[ポリエステル樹脂(p−1)]の替わりに[ポリエステル樹脂(p−4)]を使用する以外は同様の方法により、水性分散体(X−4)、樹脂粒子(C4)を得た。
上記<実施例1>において、[ポリエステル樹脂(p−1)]の替わりに[ポリエステル樹脂(p−4)]を使用する以外は同様の方法により、水性分散体(X−4)、樹脂粒子(C4)を得た。
<比較例1>
ビーカー内に[ポリエステル樹脂(p’−5)]196部、酢酸エチル201部、を投入して溶解・混合均一化し、[樹脂溶液5]を得た。この[樹脂溶液5]中に[水性分散液W−1]600部を添加し、TKホモミキサー(特殊機化工業株式会社製)を使用し、回転数12000rpmで25℃で1分間分散操作を行い、更にフィルムエバポレータで減圧度−0.05MPa(ゲージ圧)、温度40℃、回転数100rpmの条件で240分間脱溶剤し、比較の水性分散体(X’−5)を得た。水性分散体(X’−5)100部を遠心分離し、更に水60部を加えて遠心分離して固液分離する工程を2回繰り返した後、35℃で1時間乾燥して比較の樹脂粒子(CP1)を得た。
ビーカー内に[ポリエステル樹脂(p’−5)]196部、酢酸エチル201部、を投入して溶解・混合均一化し、[樹脂溶液5]を得た。この[樹脂溶液5]中に[水性分散液W−1]600部を添加し、TKホモミキサー(特殊機化工業株式会社製)を使用し、回転数12000rpmで25℃で1分間分散操作を行い、更にフィルムエバポレータで減圧度−0.05MPa(ゲージ圧)、温度40℃、回転数100rpmの条件で240分間脱溶剤し、比較の水性分散体(X’−5)を得た。水性分散体(X’−5)100部を遠心分離し、更に水60部を加えて遠心分離して固液分離する工程を2回繰り返した後、35℃で1時間乾燥して比較の樹脂粒子(CP1)を得た。
<比較例2>
上記<比較例1>において、[ポリエステル樹脂(p’−5)]196部の代わりに[ポリエステル樹脂(p’−6)]177部、酢酸エチル201部を181部とし、[硬化剤1]0.9部、[ウレタンプレポリマー3’]39.2部を使用する以外は同様の方法により、比較の水性分散体(X’−6)及び樹脂粒子(CP2)を得た。
上記<比較例1>において、[ポリエステル樹脂(p’−5)]196部の代わりに[ポリエステル樹脂(p’−6)]177部、酢酸エチル201部を181部とし、[硬化剤1]0.9部、[ウレタンプレポリマー3’]39.2部を使用する以外は同様の方法により、比較の水性分散体(X’−6)及び樹脂粒子(CP2)を得た。
<比較例3>
上記<比較例1>において、[ポリエステル樹脂(p’−5)]196部の代わりに[ポリエステル樹脂(p’−7)]177部、酢酸エチル201部を181部とし、[硬化剤1]0.9部、[ウレタンプレポリマー3’]39.2部を使用する以外は同様の方法により、比較の水性分散体(X’−7)及び樹脂粒子(CP3)を得た。
上記<比較例1>において、[ポリエステル樹脂(p’−5)]196部の代わりに[ポリエステル樹脂(p’−7)]177部、酢酸エチル201部を181部とし、[硬化剤1]0.9部、[ウレタンプレポリマー3’]39.2部を使用する以外は同様の方法により、比較の水性分散体(X’−7)及び樹脂粒子(CP3)を得た。
<物性測定例>
製造例2で得た樹脂粒子(A−1)の体積平均粒径、並びに実施例1〜4及び比較例1〜3で得た樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の体積平均粒径、個数平均粒径、表面被覆率、低温溶融性及び耐ブロッキング性を以下の測定方法により測定した結果を表1に示す。
<体積平均粒径及び個数平均粒径>
製造例2で得た樹脂粒子(A−1)の体積平均粒径、並びに実施例1〜4及び比較例1〜3で得た樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の体積平均粒径、個数平均粒径、表面被覆率、低温溶融性及び耐ブロッキング性を以下の測定方法により測定した結果を表1に示す。
<体積平均粒径及び個数平均粒径>
樹脂粒子(A−1)の体積平均粒径は、レーザー式粒度分布測定装置LA−920(堀場製作所製)で測定した{1%のイオン交換水の分散液、25℃}。
樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の体積平均粒径及び粒度分布{体積平均粒径/個数平均粒径}は、コールカウンター{マルチサイザーIII、コールター社製}で測定した{0.5%のイオン交換水の分散液、25℃}。
<表面被覆率>
樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めた。
表面被覆率(%)=(SA)×100/(SA)+(SB)
(SA):樹脂粒子(A)に覆われている部分の面積
(SB):樹脂粒子(B)が露出している部分の面積
<低温溶融性>
樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の各々0.1gを、縦5cm×横5cmのガラス片にのせ、90℃から5℃刻みで160℃まで温調されたホットプレート上で加熱しながら、もう一枚のガラスを上から乗せた後に10kg/cm2の圧力をかけて樹脂膜を作製した。得られた樹脂膜のヘイズを測定し、ヘイズが20以下となるホットプレートの最低温度をこの樹脂の低温溶融性とした。この温度以下では樹脂粒子は十分に溶融せず、ヘイズは20以上を示す。
<耐ブロッキング性>
50℃に温調された乾燥機に樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)それぞれ50gを15時間静置し、ブロッキングの程度により下記の基準で評価した。
○ : ブロッキングが発生しない。
△ : ブロッキングが発生するが、力を加えると容易に分散する。
× : ブロッキングが発生し、力を加えても分散しない。
樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の体積平均粒径及び粒度分布{体積平均粒径/個数平均粒径}は、コールカウンター{マルチサイザーIII、コールター社製}で測定した{0.5%のイオン交換水の分散液、25℃}。
<表面被覆率>
樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めた。
表面被覆率(%)=(SA)×100/(SA)+(SB)
(SA):樹脂粒子(A)に覆われている部分の面積
(SB):樹脂粒子(B)が露出している部分の面積
<低温溶融性>
樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)の各々0.1gを、縦5cm×横5cmのガラス片にのせ、90℃から5℃刻みで160℃まで温調されたホットプレート上で加熱しながら、もう一枚のガラスを上から乗せた後に10kg/cm2の圧力をかけて樹脂膜を作製した。得られた樹脂膜のヘイズを測定し、ヘイズが20以下となるホットプレートの最低温度をこの樹脂の低温溶融性とした。この温度以下では樹脂粒子は十分に溶融せず、ヘイズは20以上を示す。
<耐ブロッキング性>
50℃に温調された乾燥機に樹脂粒子(C1)〜(C4)及び(CP1)〜(CP3)それぞれ50gを15時間静置し、ブロッキングの程度により下記の基準で評価した。
○ : ブロッキングが発生しない。
△ : ブロッキングが発生するが、力を加えると容易に分散する。
× : ブロッキングが発生し、力を加えても分散しない。
本発明の樹脂粒子は、粒径が均一で、帯電特性、耐ブロッキング性等に優れるため、スラッシュ成形用樹脂、粉体塗料、液晶等の電子部品製造用スペーサー、電子測定機器の標準粒子、電子写真、静電記録、静電印刷等に用いられるトナー母体粒子、各種ホットメルト接着剤、その他成形材料等に用いる樹脂粒子として極めて有用である。
Claims (11)
- 樹脂(a)を含有する樹脂粒子(A)の水性分散液(W)と、樹脂(b)及び/又はその前駆体(b0)並びに必要により有機溶剤(u)を含有する油性液(OL)とを混合し、(W)中に(OL)を分散させ、前駆体(b0)を使用する場合は更に(W)中で(b0)を反応させて、(b)を含有する樹脂粒子(B)を形成させることにより、(B)の表面に(A)が付着された構造の樹脂粒子(C)の水性分散体(X1)を得る工程を含み、樹脂(b)が、ポリオール成分(x)とポリカルボン酸成分(y)とが重縮合されてなるポリエステル樹脂(p1)であって、(y)中にその70モル%以上の、テレフタル酸、イソフタル酸及び/又はそれらの低級アルキルエステル(アルキル基の炭素数:1〜4)を含有し、かつ、(x)から重縮合反応中に系外に留去されるポリオールを除いた(p1)を構成するポリオールの80〜99.99モル%が1,2−プロピレングリコールであり、0.01〜11モル%がネオペンチルグリコールであるポリエステル樹脂(p1)、及び/又は、(p1)を構成単位として有する樹脂(p2)を含有することを特徴とする水性分散体(X1)の製造方法。
- ポリエステル樹脂(p1)において、ポリカルボン酸成分(y)中に、更に炭素数4〜18のアルカンジカルボン酸及び/又は3〜6価の炭素数9〜20の芳香族ポリカルボン酸若しくはその酸無水物を含有する請求項1記載の製造方法。
- ポリエステル樹脂(p1)が、チタン含有触媒(t)の存在下、ポリオール成分(x)とポリカルボン酸成分(y)とが重縮合されて得られたものである請求項1又は2記載の製造方法。
- 樹脂(b)が、ポリエステル樹脂(p1)及び(p1)を構成単位として有する樹脂(p2)以外に、ポリウレタン樹脂、エポキシ樹脂、ビニル樹脂、及び(p1)以外のポリエステル樹脂からなる群から選ばれる1種以上の樹脂を含有する請求項1〜3のいずれか記載の製造方法。
- 前躯体(b0)が、ポリエステル樹脂(p1)の構成単位及び反応性基を有するプレポリマー(α)と硬化剤(β)とで構成される請求項1〜4のいずれか記載の製造方法。
- 反応性基含有プレポリマー(α)が、イソシアネート基、ブロック化イソシアネート基及びエポキシ基からなる群から選ばれる少なくとも1種の反応性基を有し、かつ硬化剤(β)が活性水素基を有する請求項5記載の製造方法。
- 樹脂(a)が、ビニル樹脂、ポリウレタン樹脂、エポキシ樹脂及びポリエステル樹脂からなる群から選ばれる少なくとも1種である請求項1〜6のいずれか記載の製造方法。
- 請求項1〜7のいずれか記載の製造方法により得られた水性分散体(X1)から水性溶剤を除去して樹脂粒子(C)を得る工程を含む樹脂粒子の製造方法。
- 請求項8記載の製造方法により得られた樹脂粒子。
- 以下の〔1〕〜〔4〕を満たす請求項9記載の樹脂粒子;
〔1〕[樹脂粒子(A)の体積平均粒径/樹脂粒子(C)の体積平均粒径]が0.001〜0.3であり、
〔2〕樹脂粒子(A)の体積平均粒径が0.0005〜30μm、且つ樹脂粒子(C)の体積平均粒径が0.1〜300μmであり、
〔3〕樹脂粒子(B)の表面の5%以上が樹脂粒子(A)で覆われており、
〔4〕樹脂粒子(C)の[体積平均粒径/個数平均粒径]が1.0〜1.5である。 - スラッシュ成形用樹脂、粉体塗料、電子部品製造用スペーサー、電子測定機器の標準粒子、電子写真トナー用母体粒子、静電記録トナー用母体粒子、静電印刷トナー用母体粒子又はホットメルト接着剤に使用するための請求項9又は10記載の樹脂粒子。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010010887A JP2010189633A (ja) | 2009-01-21 | 2010-01-21 | 樹脂分散体の製造方法及び樹脂粒子 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009011435 | 2009-01-21 | ||
| JP2010010887A JP2010189633A (ja) | 2009-01-21 | 2010-01-21 | 樹脂分散体の製造方法及び樹脂粒子 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2010189633A true JP2010189633A (ja) | 2010-09-02 |
Family
ID=42815994
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010010887A Pending JP2010189633A (ja) | 2009-01-21 | 2010-01-21 | 樹脂分散体の製造方法及び樹脂粒子 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2010189633A (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011070751A1 (ja) | 2009-12-10 | 2011-06-16 | 三洋化成工業株式会社 | 熱可塑性ウレタン樹脂 |
| JP2012097253A (ja) * | 2010-10-06 | 2012-05-24 | Sanyo Chem Ind Ltd | 非水性樹脂分散液及びその製造方法 |
| JP2013203815A (ja) * | 2012-03-27 | 2013-10-07 | Fujifilm Corp | 樹脂及びその製造方法、並びにこれ用いたトナー |
| KR20130132839A (ko) | 2010-11-12 | 2013-12-05 | 산요가세이고교 가부시키가이샤 | 우레탄 수지 입자 |
| JP2014016551A (ja) * | 2012-07-10 | 2014-01-30 | Kao Corp | トナー用結着樹脂 |
| CN105566610A (zh) * | 2016-01-15 | 2016-05-11 | 苏州珍展科技材料有限公司 | 一种冲击韧性改进剂及其改性环氧树脂组合物的制备方法 |
-
2010
- 2010-01-21 JP JP2010010887A patent/JP2010189633A/ja active Pending
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011070751A1 (ja) | 2009-12-10 | 2011-06-16 | 三洋化成工業株式会社 | 熱可塑性ウレタン樹脂 |
| JP2012097253A (ja) * | 2010-10-06 | 2012-05-24 | Sanyo Chem Ind Ltd | 非水性樹脂分散液及びその製造方法 |
| KR20130132839A (ko) | 2010-11-12 | 2013-12-05 | 산요가세이고교 가부시키가이샤 | 우레탄 수지 입자 |
| US9023941B2 (en) | 2010-11-12 | 2015-05-05 | Sanyo Chemical Industries, Ltd. | Urethane resin particles |
| KR101971742B1 (ko) | 2010-11-12 | 2019-04-23 | 산요가세이고교 가부시키가이샤 | 우레탄 수지 입자 |
| JP2013203815A (ja) * | 2012-03-27 | 2013-10-07 | Fujifilm Corp | 樹脂及びその製造方法、並びにこれ用いたトナー |
| JP2014016551A (ja) * | 2012-07-10 | 2014-01-30 | Kao Corp | トナー用結着樹脂 |
| CN105566610A (zh) * | 2016-01-15 | 2016-05-11 | 苏州珍展科技材料有限公司 | 一种冲击韧性改进剂及其改性环氧树脂组合物的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4843565B2 (ja) | 樹脂分散体の製造方法及び樹脂粒子 | |
| JP4130639B2 (ja) | 樹脂分散体の製造方法及び樹脂粒子 | |
| JP5183519B2 (ja) | 樹脂粒子 | |
| JP4718392B2 (ja) | 樹脂粒子及び樹脂分散体 | |
| JP5020841B2 (ja) | 樹脂粒子 | |
| JP4598807B2 (ja) | 樹脂粒子及び樹脂分散体 | |
| JP5497516B2 (ja) | 樹脂粒子及びその製造方法 | |
| JP2010254896A (ja) | 樹脂粒子およびその水性分散体、並びにその製造方法 | |
| JP4976237B2 (ja) | 樹脂粒子および樹脂粒子の製造方法 | |
| JP2006206851A (ja) | 樹脂分散体および樹脂粒子 | |
| JP4170349B2 (ja) | 樹脂粒子および樹脂分散体 | |
| JP2010189633A (ja) | 樹脂分散体の製造方法及び樹脂粒子 | |
| JP4874907B2 (ja) | 樹脂分散体の製造方法及び樹脂粒子 | |
| JP4431122B2 (ja) | 樹脂分散体及び樹脂粒子 | |
| JP4025844B2 (ja) | 粒径が均一である樹脂分散体およびその製造方法 | |
| JP2008208346A (ja) | 樹脂粒子 | |
| JP4616218B2 (ja) | 樹脂粒子の製造方法および樹脂粒子 | |
| JP4625275B2 (ja) | 樹脂分散体の製造方法及び樹脂粒子 | |
| JP5101208B2 (ja) | 樹脂粒子および樹脂粒子の製造方法 | |
| JP2007246676A (ja) | 樹脂粒子の製造方法及び樹脂粒子 | |
| JP2008208354A (ja) | 樹脂粒子 | |
| JP2007009074A (ja) | 樹脂粒子の製造法 | |
| JP4964834B2 (ja) | 樹脂粒子 | |
| JP2009035610A (ja) | 樹脂粒子および樹脂粒子の製造方法 | |
| JP5020529B2 (ja) | 着色樹脂粒子 |