JP2010035554A - (1s,2r)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法 - Google Patents
(1s,2r)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法 Download PDFInfo
- Publication number
- JP2010035554A JP2010035554A JP2009161114A JP2009161114A JP2010035554A JP 2010035554 A JP2010035554 A JP 2010035554A JP 2009161114 A JP2009161114 A JP 2009161114A JP 2009161114 A JP2009161114 A JP 2009161114A JP 2010035554 A JP2010035554 A JP 2010035554A
- Authority
- JP
- Japan
- Prior art keywords
- chloro
- fluorocyclopropanecarboxylic acid
- acid
- content
- fluorocyclopropanecarboxylic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/377—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C67/347—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by addition to unsaturated carbon-to-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P41/00—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture
- C12P41/003—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture by ester formation, lactone formation or the inverse reactions
- C12P41/005—Processes using enzymes or microorganisms to separate optical isomers from a racemic mixture by ester formation, lactone formation or the inverse reactions by esterification of carboxylic acid groups in the enantiomers or the inverse reaction
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/40—Preparation of oxygen-containing organic compounds containing a carboxyl group including Peroxycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Genetics & Genomics (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Microbiology (AREA)
- Analytical Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【解決手段】バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼを用いて(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを加水分解する(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法である。
【選択図】なし
Description
項1.バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼを用いて(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを加水分解する(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法。
項2.不斉錯体の存在下、1−クロロ−1−フルオロエチレンとジアゾ酢酸エステルとを反応させて(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを得、次いで、バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼを用いて該(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを加水分解する上記項1に記載の製造方法。
項3.不斉錯体が、銅化合物と光学活性な配位子とを接触させてなる不斉銅錯体である上記項2に記載の製造方法。
項4.光学活性な配位子が、一般式(1):
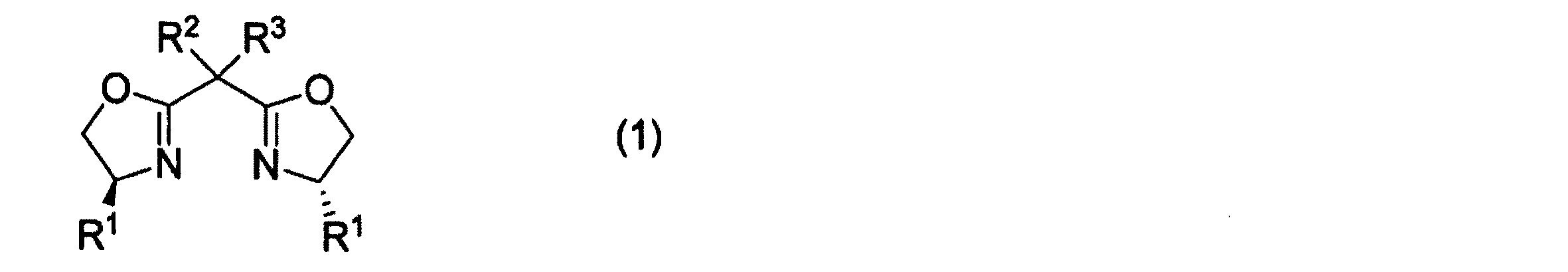
で示される光学活性なビスオキサゾリン化合物である上記項3に記載の製造方法。
項5.光学活性な配位子が、2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパンである上記項3に記載の製造方法。
項6.バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼが、配列番号1または2で示されるアミノ酸配列を有するエステラーゼである上記項1〜5のいずれかに記載の製造方法。
項7.バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼが、配列番号1で示されるアミノ酸配列を有するエステラーゼである上記項1〜5のいずれかに記載の製造方法。
項8.上記項1〜7のいずれかに記載の製造方法により(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を得、次いで、該(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を還元する(1S,2S)−2−フルオロシクロプロパンカルボン酸の製造方法。
項9.還元が、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸存在下、ニッケル−アルミニウム合金に塩基を作用させて行われる上記項8に記載の製造方法。
まず、本発明に用いる(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルの製造方法について説明する。
で示される光学活性なビスオキサゾリン化合物(以下、光学活性ビスオキサゾリン(1)と略記する)や下記一般式(2):
で示されるサルドイミン化合物(以下、光学活性サルドイミン(2)と略記する)等が挙げられ、これらの中でも光学活性ビスオキサゾリン(1)が好ましい。
次に、バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼを用いて(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを加水分解する(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法(本明細書において、「本発明の酵素加水分解」と記載することもある)について説明する。
最後に、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を還元する、(1S,2S)−2−フルオロシクロプロパンカルボン酸の製造方法について説明する。
展開還元は、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸の存在下、ニッケル−アルミニウム合金に塩基を作用させて行われる。
水素添加は、通常、溶媒の存在下、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸とスポンジニッケルとを混合し、得られた混合物を水素雰囲気下で攪拌することにより行われる。また、塩基を適宜使用することもできる。
収率:ガスクロマトグラフ分析
カラム:DB−WAX
0.53mm×30m、膜厚1.0μm
(アジレント・テクノロジー株式会社製)
収率およびanti体/syn体比:ガスクロマトグラフ分析
カラム:DB−WAX
0.25mm×30m、膜厚0.25μm
(アジレント・テクノロジー株式会社製)
光学純度:ガスクロマトグラフ分析
カラム:InertCap(登録商標) CHIRAMIX
0.25mm×30m、膜厚0.25μm
(ジーエルサイエンス株式会社製)
または、
CP−Cyclodextrin−β−2,3,6−M−19
0.25mm×50m、膜厚0.25μm
(ジーエルサイエンス株式会社製)
収率およびanti体/syn体比:高速液体クロマトグラフ分析
カラム:L−column2(登録商標)
4.6mm×250mm、5μm
(財団法人化学物質評価研究機構製)
展開液:KH2PO4水溶液(5mmol/L)にリン酸を加えて
pH2.5に調整した水溶液およびアセトニトリル
光学純度:ガスクロマトグラフ分析
カラム:InertCap(登録商標) CHIRAMIX
0.25mm×30m、膜厚0.25μm
(ジーエルサイエンス株式会社製)
方法:(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を
トリメチルシリルジアゾメタンでメチルエステルに誘導体化してから分析。
収率およびシス体/トランス体比:高速液体クロマトグラフ分析
カラム:L−column2(登録商標)
4.6mm×250mm、5μm
(財団法人化学物質評価研究機構製)
展開液:KH2PO4水溶液(5mmol/L)にリン酸を加えて
pH2.5に調整した水溶液およびアセトニトリル
光学純度:ガスクロマトグラフ分析
カラム:InertCap(登録商標) CHIRAMIX
0.25mm×30m、膜厚0.25μm
(ジーエルサイエンス株式会社製)
方法:(1S,2S)−2−フルオロシクロプロパンカルボン酸を
トリメチルシリルジアゾメタンでメチルエステルに誘導体化してから分析。
化学純度およびシス体/トランス体比:ガスクロマトグラフ分析
カラム:HR−20M
0.53mm×30m、膜厚1.0μm
(信和化工株式会社製)
窒素雰囲気下、水318g、グリシンエチルエステル塩酸塩188g(1.35mol)、n−ヘプタン188gを順次仕込んだ後、得られた混合物を10℃に冷却した。そこに、28重量%水酸化ナトリウム水溶液1.74gを加えてpHを4.7に調整し、内温10±2℃に保ちながら、40重量%亜硝酸ナトリウム水溶液279g(純分112g、1.62mol)とクエン酸・1水和物4.54g(0.0216mol)および水65.8gからなるクエン酸水溶液とを同時並行的に3時間かけて滴下した。得られた混合物を10℃で6時間保温した後、そこに、炭酸ナトリウム6.58g(0.0621mol)および水87.5gからなる炭酸ナトリウム水溶液を滴下した。得られた混合物を内温10±5℃に保ちながら分液し、得られた有機層をモレキュラーシーブス4A6.2gで乾燥させた後、ろ過することにより、ジアゾ酢酸エチルのn−ヘプタン溶液341g(含量:38.0重量%、純分:129g、収率:84.1%)を得た。
窒素雰囲気下、水212g、グリシンエチルエステル塩酸塩126g(0.900mol)、n−ヘキサン126gを順次仕込んだ後、得られた混合物を10℃に冷却した。そこに、28重量%水酸化ナトリウム水溶液1.99gを加えてpHを4.9に調整し、内温10±2℃に保ちながら、40重量%亜硝酸ナトリウム水溶液186g(純分74.5g、1.08mol)とクエン酸・1水和物3.03g(0.0144mol)および水43.9gからなるクエン酸水溶液とを同時並行的に2時間かけて滴下した。得られた混合物を10℃で7時間保温した後、そこに、炭酸ナトリウム4.39g(0.0414mol)および水58.3gからなる炭酸ナトリウム水溶液を滴下した。得られた混合物を内温10±5℃に保ちながら分液し、ジアゾ酢酸エチルのn−ヘキサン溶液213g(含量:41.0重量%、純分:87.2g、収率:84.9%)を得た。
製造例2で得たジアゾ酢酸エチルのn−ヘキサン溶液213g(含量:41.0重量%、純分:87.2g)を温度38〜42℃、減圧度200〜370hPaで濃縮し、得られた残渣を温度52〜60℃、減圧度12.0〜31hPaで蒸留することにより、黄色の油状物として、ジアゾ酢酸エチル70.1g(含量:96.5重量%、純分:67.7g)を得た。
窒素雰囲気下、水637g、グリシンエチルエステル塩酸塩377g(2.70mol)、n−ヘプタン377gを順次仕込んだ後、得られた混合物を10℃に冷却した。そこに、26重量%水酸化ナトリウム水溶液3.14gを加えてpHを4.7に調整し、内温10±2℃に保ちながら、40重量%亜硝酸ナトリウム水溶液559g(純分224g、3.24mol)とクエン酸・1水和物17.0g(0.0810mol)および水247gからなるクエン酸水溶液とを同時並行的に15時間かけて滴下した。得られた混合物を10℃で4時間保温した後、そこに、炭酸ナトリウム24.0g(0.227mol)および水277gからなる炭酸ナトリウム水溶液を滴下した。得られた混合物を内温10±5℃に保ちながら分液し、得られた有機層をモレキュラーシーブス4A12.4gで乾燥させた後、ろ過することにより、ジアゾ酢酸エチルのn−ヘプタン溶液672g(含量:38.0重量%、純分:255g、収率:82.9%)を得た。
窒素雰囲気下、常温で、1300mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン1.79g(6.06mmol)、トリフルオロメタンスルホン酸銅(II)1.98g(5.50mmol)およびn−ヘプタン35.5gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン177g(2.19mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに、製造例1で得たジアゾ酢酸エチルのn−ヘプタン溶液327g(含量:38.0重量%、純分:124g、1.09mol)を5時間かけて滴下した後、得られた混合物を同温度で1時間攪拌した。なお、滴下および保温中、内圧が1MPaを越えた際、パージ操作を行い、圧力を0.9〜1MPaの範囲に保持した。復圧後、昇温し、窒素置換を行った。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水27gで洗浄、分液することにより、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物453g(含量:27.5重量%、純分:125g、収率:68.6%(対 ジアゾ酢酸エチル)、anti体/syn体比=62.5/37.5、anti体光学純度=98.2%ee、syn体光学純度=97.4%ee)を得た。反応に用いたオートクレーブなど使用器具の付着分をアセトニトリルに溶解し含量分析を行ったところ、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルの合計量は、収率換算で1.3%であった。したがって、反応収率は69.9%であった。
窒素雰囲気下、常温で、1300mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン811mg(2.75mmol)、トリフルオロメタンスルホン酸銅(II)903mg(2.50mmol)およびn−ヘプタン16.3gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン203g(2.52mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに、製造例1に準じて得たジアゾ酢酸エチルのn−ヘプタン溶液149g(含量:38.3重量%、純分:57.0g、0.500mol)を5時間かけて滴下した後、得られた混合物を同温度で1時間攪拌した。滴下に従い内圧が上昇し、最終的に1.3MPaに達した。復圧後、昇温し、窒素置換を行った。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水13gで洗浄、分液することにより、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物206g(含量:32.5重量%、純分:66.9g、収率:80.2%(対 ジアゾ酢酸エチル)、anti体/syn体比=62.4/37.6、anti体光学純度=98.3%ee、syn体光学純度=97.5%ee)を得た。反応に用いたオートクレーブなど使用器具の付着分をアセトニトリルに溶解し含量分析を行ったところ、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルの合計量は、収率換算で1.5%であった。したがって、反応収率は81.7%であった。
窒素雰囲気下、常温で、1300mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン1.03g(3.48mmol)、トリフルオロメタンスルホン酸銅(II)1.14g(3.16mmol)およびn−ヘプタン20.5gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン154g(1.91mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに、製造例1に準じて得たジアゾ酢酸エチルのn−ヘプタン溶液189g(含量:38.1重量%、純分:71.9g、0.630mol)を5時間かけて滴下した後、得られた混合物を同温度で1時間攪拌した。滴下に従い内圧が上昇し、最終的に1.5MPaに達した。復圧後、昇温し、窒素置換を行った。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水15.8gで洗浄、分液することにより、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物269g(含量:27.1重量%、純分:73.0g、収率:69.5%(対 ジアゾ酢酸エチル)、anti体/syn体比=62.6/37.4、anti体光学純度=98.2%ee、syn体光学純度=97.5%ee)を得た。反応に用いたオートクレーブなど使用器具の付着分をアセトニトリルに溶解し含量分析を行ったところ、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルの合計量は、収率換算で0.9%であった。したがって、反応収率は70.4%であった。
窒素雰囲気下、常温で、1300mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン0.809g(2.75mmol)、トリフルオロメタンスルホン酸銅(II)0.903g(2.50mmol)およびn−ヘプタン16.3gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン202g(2.51mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに、製造例1に準じて得たジアゾ酢酸エチルのn−ヘプタン溶液146g(含量:39.2重量%、純分:57.0g、0.500mol)を5時間かけて滴下した後、得られた混合物を同温度で1時間攪拌した。滴下に従い内圧が上昇し、最終的に1.2MPaに達した。復圧後、昇温し、窒素置換を行った。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水12.5gで洗浄、分液することにより、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物201g(含量:30.8重量%、純分:62.0g、収率:74.4%(対 ジアゾ酢酸エチル)、anti体/syn体比=62.2/37.8、anti体光学純度=98.2%ee、syn体光学純度=97.5%ee)を得た。反応に用いたオートクレーブなど使用器具の付着分をアセトニトリルに溶解し含量分析を行ったところ、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルの合計量は、収率換算で6.5%であった。したがって、反応収率は80.9%であった。
窒素雰囲気下、常温で、260mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン0.520g(1.77mmol)、トリフルオロメタンスルホン酸銅(II)0.579g(1.61mmol)およびトリフルオロトルエン42.0gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン13.0g(0.162mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに、製造例3で得たジアゾ酢酸エチル9.49g(含量:96.5重量%、純分:9.16g、0.0802mol)およびトリフルオロトルエン42.0gからなる溶液を5時間かけて滴下した後、得られた混合物を同温度で1時間攪拌した。滴下に従い内圧が上昇し、最終的に1.0MPaに達した。復圧後、昇温し、窒素置換を行った。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水8.0gで洗浄、分液することにより、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物107g(含量:3.96重量%、純分:4.22g、収率:31.6%(対 ジアゾ酢酸エチル)、anti体/syn体比=60.0/40.0、anti体光学純度=97.9%ee、syn体光学純度=97.1%ee)を得た。
窒素雰囲気下、常温で、50mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン0.025g(0.085mmol)、トリフルオロメタンスルホン酸銅(II)0.027g(0.075mmol)およびトリフルオロトルエン7.9gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン2.43g(0.0302mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに、製造例3で得たジアゾ酢酸エチル1.77g(含量:96.5重量%、純分:1.71g、0.0150mol)およびトリフルオロトルエン7.9gからなる溶液を5時間かけて滴下した後、得られた混合物を同温度で1時間攪拌した。滴下に従い内圧が上昇し、最終的に1.0MPaに達した。復圧後、昇温し、窒素置換を行った。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水0.38gで洗浄、分液することにより、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物19.7g(含量:5.14重量%、純分:1.01g、収率:40.5%(対 ジアゾ酢酸エチル)、anti体/syn体比=60.4/39.6、anti体光学純度=96.5%ee、syn体光学純度=95.8%ee)を得た。
窒素雰囲気下、常温で、50mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン0.025g(0.085mmol)、トリフルオロメタンスルホン酸銅(II)0.027g(0.075mmol)およびトリフルオロトルエン7.9gを仕込み、得られた混合物を攪拌した。反応容器を密閉し、そこに、製造例3で得たジアゾ酢酸エチル1.77g(含量:96.5重量%、純分:1.71g、0.0150mol)およびトリフルオロトルエン7.9gからなる溶液のうち、10%相当を30分間かけて滴下し、5分間攪拌した後、反応容器を0℃に冷却した。そこに、1−クロロ−1−フルオロエチレン2.46g(0.0306mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに、先に調整したジアゾ酢酸エチル溶液の残りの90%を4.5時間かけて滴下した後、得られた混合物を同温度で1時間攪拌した。滴下に従い内圧が上昇し、最終的に0.9MPaに達した。復圧後、昇温し、窒素置換を行った。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水0.38gで洗浄、分液することにより、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物19.7g(含量:4.47重量%、純分:0.88g、収率:35.4%(対 ジアゾ酢酸エチル)、anti体/syn体比=60.3/39.7、anti体光学純度=94.6%ee、syn体光学純度=93.6%ee)を得た。
窒素雰囲気下、常温で、1300mLオートクレーブに2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパン1.40g(4.75mmol)、トリフルオロメタンスルホン酸銅(II)1.56g(4.32mmol)およびn−ヘプタン35.7gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン173g(2.15mol)を圧入し、内温を7±2℃とした。内温を7±2℃に保ちながら、そこに、製造例4に準じて得たジアゾ酢酸エチルのn−ヘプタン溶液315g(含量:39.1重量%、純分123g、1.08mol)を24時間かけて滴下した後、得られた混合物を同温度で1時間撹拌した。なお、滴下および保温中、内圧が1MPaを越えた際、パージ操作を行い、圧力を0.9〜1MPaの範囲に保持した。復圧後、昇温し、窒素置換を行い、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物396g(含量:26.7重量%、純分:106g、収率:58.8%(対 ジアゾ酢酸エチル)、anti体/syn体比=61.5/38.5、anti体光学純度=97.8%ee、syn体光学純度=96.9%ee)を得た。反応に用いたオートクレーブなど使用器具の付着分をtert−ブチルメチルエーテルに溶解し含量分析を行ったところ、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルの合計量は、収率換算で10.6%であった。したがって、反応収率は69.4%であった。
実施例1とほぼ同様の条件で繰り返し反応を行い、得られた(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物を合一し、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル混合物1278g(含量:25.4重量%、純分:325g、anti体/syn体比=62.6/37.4、anti体光学純度=98.2%ee)を得た。この溶液を温度40〜42℃、減圧度19〜106hPaで濃縮し、得られた残渣を温度70〜88℃、減圧度5.3〜14hPaで蒸留することにより、無色の油状物として、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル294g(含量:98.1重量%、純分:289g、anti体/syn体比=61.1/38.9、anti体光学純度=98.2%ee)を得た。
実施例2で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物206g(含量:32.5重量%、純分:66.9g、anti体/syn体比=62.4/37.6、anti体光学純度=98.3%ee)を温度40〜42℃、減圧度40〜133hPaで濃縮し、得られた残渣を温度67〜88℃、減圧度6.7〜13hPaで蒸留することにより、無色の油状物として、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル59.4g(含量:98.4重量%、純分:58.4g、anti体/syn体比=62.5/37.5、anti体光学純度=98.3%ee)を得た。
実施例3で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物269g(含量:27.1重量%、純分:72.8g、anti体/syn体比=62.6/37.4、anti体光学純度=98.2%ee)を温度40〜42℃、減圧度40〜133hPaで濃縮し、得られた残渣を温度67〜88℃、減圧度6.7〜13hPaで蒸留することにより、無色の油状物として、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル66.6g(含量:97.4重量%、純分:64.9g、anti体/syn体比=62.4/37.6、anti体光学純度=98.2%ee)を得た。
実施例4で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物201g(含量:30.8重量%、純分:62.0g、anti体/syn体比=62.2/37.8、anti体光学純度=98.2%ee)を温度40〜41℃、減圧度27〜101hPaで濃縮し、得られた残渣を温度70〜85℃、減圧度8.0〜27hPaで蒸留することにより、無色の油状物として、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル57.7g(含量:100重量%、純分:57.7g、anti体/syn体比=61.7/38.3、anti体光学純度=98.2%ee)を得た。
実施例5〜7で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物を合一し、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル混合物121g(含量:4.1重量%、純分:5.0g、anti体/syn体比=60.4/39.6、anti体光学純度=96.5%ee)を得た。この溶液を温度35〜40℃、減圧度20〜133hPaで濃縮し、得られた残渣を温度82〜91℃、減圧度8.0〜20hPaで蒸留することにより、無色の油状物として、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル3.8g(含量:86.2重量%、純分:3.3g、anti体/syn体比=58.4/41.6、anti体光学純度=96.3%ee)を得た。
実施例7−1とほぼ同様の条件で繰り返し反応を行い、得られた(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物を合一し、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル混合物2331g(含量:22.7重量%、純分:528g、anti体/syn体比=62.8/37.2、anti体光学純度=98.2%ee)を得た。この混合物に、モレキュラーシーブス4A30.9gを加え、温度39〜42℃、減圧度47〜373hPaで775g(含量:64.2%、純分:498g)まで濃縮した。このうち、385gを温度73〜107℃、減圧度5.3〜13hPaで蒸留することにより、無色の油状物として、(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル247g(含量:97.4重量%、純分:241g、anti体/syn体比=62.0/38.0、anti体光学純度=98.2%ee)を得た。
室温下、3000mL丸底セパラブルフラスコに、リン酸2水素ナトリウム52.3gおよび水1760gからなる0.2mol/Lリン酸緩衝液1812gを仕込み、そこに、10重量%水酸化ナトリウム水を加えてpHを6.5に調整した。そこに、実施例8で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル224g(含量:98.1重量%、純分:220g、1.32mol、anti体/syn体比=61.1/38.9、anti体光学純度=98.2%ee)、さらに、配列番号1で示されるアミノ酸配列からなる加水分解酵素(商品名:リパーゼAH(天野エンザイム株式会社製、Lot.LAHG0150707R))11.0gを順に仕込んだ後、得られた混合物を35℃で50時間攪拌した。攪拌中、10重量%水酸化ナトリウム水を用いて、混合物のpHを6.5に調整した。反応終了後、反応混合物にtert−ブチルメチルエーテル1100gおよび35重量%塩酸を加えて、そのpHを2.0とした後、有機層と水層を分離した。次いで、tert−ブチルメチルエーテル440gを用いて該水層を抽出処理し、得られた有機層を先に得られた有機層と合一した。得られた有機層にラヂオライト(登録商標、昭和化学工業株式会社製)を11g添加し、室温で攪拌した後、ガラスフィルターを用いて固形物をろ別した。ろ液中のわずかに生じた水層を分離した後、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を含む有機層1806g(含量:5.79重量%、純分:104.5g、収率:57.1%、anti体/syn体比=98.6/1.4、anti体光学純度=98.1%ee)を得た。この有機層を5℃に冷却した。温度を5〜15℃に保ちながら、そこに、27重量%水酸化ナトリウム水溶液を117g(0.787mol)滴下することによりpHを13に調整した後、内温を20℃にし、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸をナトリウム塩として水層側に抽出することにより、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウムを含む水層285g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウムとして、含量:41.4重量%、純分:118g、収率:55.7%、anti体/syn体比=98.1/1.9、anti体光学純度=98.1%ee)を得た。
室温下、2000mL丸底セパラブルフラスコに、リン酸2水素ナトリウム14.3gおよび水600gからなる0.2mol/Lリン酸緩衝液614.3gを仕込み、そこに、10重量%水酸化ナトリウム水を加えてpHを6.5に調整した。そこに、実施例10で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル61.2g(含量:97.4重量%、純分60.0g、0.360mol、anti体/syn体比=62.4/37.6、anti体光学純度=98.2%ee)、さらに、配列番号1で示されるアミノ酸配列からなる加水分解酵素(商品名:リパーゼAH(天野エンザイム株式会社製、Lot.LAHG0150707R))3.0gを順に仕込んだ後、得られた混合物を35℃で47時間攪拌した。攪拌中、10重量%水酸化ナトリウム水を用いて、混合物のpHを6.5に調整した。反応終了後、反応混合物にtert−ブチルメチルエーテル300gおよび35重量%塩酸を加えて、そのpHを2.3とした後、有機層と水層を分離した。次いで、tert−ブチルメチルエーテル120gを2回用いて該水層を抽出処理し、得られた有機層を先に得られた有機層と合一した。得られた有機層にラヂオライト(登録商標、昭和化学工業株式会社製)を6g添加し、室温で攪拌した後、ラヂオライト(登録商標、昭和化学工業株式会社製)をろ過剤とし、グラスフィルターを用いて固形物をろ別した。ろ液中にわずかに生じた水層を分離した後、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を含む有機層539g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸として、含量:9.20重量%、純分:29.9g、収率:61.2%、anti体/syn体比=98.6/1.4、anti体光学純度=98.1%ee)を得た。
室温下、500mL丸底セパラブルフラスコに、リン酸2水素ナトリウム9.6gおよび水390.4gからなる0.2mol/Lリン酸緩衝液300gを仕込み、そこに、28重量%水酸化ナトリウム水を加えてpHを6.9に調整した。そこに、実施例11で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル30.0g(含量:100重量%、純分30.0g、0.180mol、anti体/syn体比=61.7/38.3、anti体光学純度=98.2%ee)、配列番号2で示されるアミノ酸配列からなる加水分解酵素(商品名:リパーゼPS(天野エンザイム株式会社製、Lot.LPSAP11522))6.0gを仕込んだ後、得られた混合物を35℃で48時間攪拌した。攪拌中、4重量%水酸化ナトリウム水を用いて、混合物のpHを6.9に調整した。反応終了後、反応混合物にtert−ブチルメチルエーテル150gおよび10重量%塩酸を加えて、そのpHを2.0とした後、有機層と水層を分離した。次いで、tert−ブチルメチルエーテル150gを2回用いて該水層を抽出処理し、得られた有機層を先に得られた有機層と合一した。得られた有機層にラヂオライト(登録商標、昭和化学工業株式会社製)を9g添加し、室温で攪拌した後、ラヂオライト(登録商標、昭和化学工業株式会社製)をろ過剤とし、グラスフィルターを用いて固形物をろ別した。ろ液中にわずかに生じた水層を分離し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を含む有機層480g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸として、含量:3.13重量%、純分:15.0g、収率:60.1%、anti体/syn体比=95.5/4.5、anti体光学純度=98.0%ee)を得た。
室温下、100mL反応容器に、リン酸2水素ナトリウム2.4gおよび水100gからなる0.2mol/Lリン酸緩衝液30gを仕込み、そこに、28重量%水酸化ナトリウム水を加えて、混合物のpHを6.5に調整した。そこに、実施例9で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル1.02g(含量:98.4重量%、純分1.00g、6.00mmol、anti体/syn体比=62.5/37.5、anti体光学純度=98.3%ee)、さらに、配列番号1で示されるアミノ酸配列からなる加水分解酵素(商品名:リパーゼAH(天野エンザイム株式会社製、Lot.LAHG0150707R))50mgを順に仕込んだ後、得られた混合物を30℃で48時間攪拌した。攪拌中、4重量%水酸化ナトリウム水を用いて、混合物のpHを6.5〜7.0に調整した。反応終了後、反応混合物にtert−ブチルメチルエーテル20gおよび5重量%塩酸を加えて、そのpHを2.0とした後、有機層と水層を分離した。次いで、tert−ブチルメチルエーテル20gを用いて該水層を抽出処理し、得られた有機層を先に得られた有機層と合一した。得られた有機層にラヂオライト(登録商標、昭和化学工業株式会社製)を少量添加し、室温で攪拌した後、ラヂオライト(登録商標、昭和化学工業株式会社製)をろ過剤とし、グラスフィルターを用いて固形物をろ別した。ろ液中のわずかに生じた水層を分離し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を含む有機層49.2g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸として、含量:0.87重量%、純分:0.428g、収率:51.5%、anti体/syn体比=99.7/0.3、anti体光学純度=98.0%ee)を得た。
室温下、300mL丸底セパラブルフラスコに、炭酸水素ナトリウム7.60gおよび水120gからなる0.75mol/L炭酸水素ナトリウム緩衝液127.6gを仕込み、そこに、実施例9で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル15.2g(含量:98.4重量%、純分15.0g、90.0mmol、anti体/syn体比=62.5/37.5、anti体光学純度=98.3%ee)、さらに、配列番号1で示されるアミノ酸配列からなる加水分解酵素(商品名:リパーゼAH(天野エンザイム株式会社製、Lot.LAHG0150707R))750mgを順に仕込んだ後、得られた混合物を35℃で48時間攪拌した。反応終了後、反応混合物にtert−ブチルメチルエーテル30gおよび35重量%塩酸を加えて、そのpHを2.0とした後、有機層と水層を分離した。次いで、tert−ブチルメチルエーテル30gを用いて該水層を抽出処理し、得られた有機層を先に得られた有機層と合一した。得られた有機層にラヂオライト(登録商標、昭和化学工業株式会社製)を0.8g添加し、室温で攪拌した後、ガラスフィルターを用いて固形物をろ別した。ろ液中のわずかに生じた水層を分離した後、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を含む有機層59.4g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸として、含量:12.7重量%、純分:7.56g、収率:60.6%、anti体/syn体比=98.2/1.8、anti体光学純度=98.2%ee)を得た。
室温下、1000mL丸底セパラブルフラスコに、炭酸水素ナトリウム42.4gおよび水480gからなる0.97mol/L炭酸水素ナトリウム緩衝液522.4gを仕込み、そこに、実施例12−1に準じて得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル123g(含量:97.3重量%、純分120g、0.720mol、anti体/syn体比=62.2/37.8、anti体光学純度=98.2%ee)、さらに、配列番号1で示されるアミノ酸配列からなる加水分解酵素(商品名:リパーゼAH(天野エンザイム株式会社製、Lot.LAHG0951102R))8.40gを順に仕込んだ後、得られた混合物を35℃で62時間攪拌した。反応終了後、反応混合物にtert−ブチルメチルエーテル240gおよび35重量%塩酸を加えて、そのpHを2.0とした後、有機層と水層を分離した。次いで、tert−ブチルメチルエーテル60gを用いて該水層を抽出処理し、得られた有機層を先に得られた有機層と合一した。得られた有機層にラヂオライト(登録商標、昭和化学工業株式会社製)を6.0g添加し、室温で攪拌した後、ガラスフィルターを用いて固形物をろ別した。ろ液中のわずかに生じた水層を分離した後、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を含む有機層400g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸として、含量:15.1重量%、純分:60.4g、収率:60.4%、anti体/syn体比=98.8/1.2、anti体光学純度=98.1%ee)を得た。この有機層を5℃に冷却した。温度を5〜15℃に保ちながら、そこに、26重量%水酸化ナトリウム水溶液55.4g(0.360mol)、続けて10重量%水酸化ナトリウム水溶液35.4gを滴下してpHを13に調整した後、内温を20℃とし、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸をナトリウム塩として水層側に抽出した。35%塩酸を加えてpHを7に調整し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウムを含む水溶液185g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウムとして、含量:36.0重量%、純分:66.6g、収率:57.4%、anti体/syn体比=98.8/1.2、anti体光学純度=98.1%ee)を得た。
室温下、200mL丸底セパラブルフラスコに、炭酸水素ナトリウム8.83gおよび水100gからなる0.97mol/L炭酸水素ナトリウム緩衝液108.8gを仕込み、そこに、実施例12−1に準じて得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル25.7g(含量:97.3重量%、純分25.0g、0.150mol、anti体/syn体比=62.2/37.8、anti体光学純度=98.2%ee)、さらに、配列番号1で示されるアミノ酸配列からなる加水分解酵素(商品名:リパーゼAH(天野エンザイム株式会社製、Lot.LAHH0250804R))1.75gを順に仕込んだ後、得られた混合物を35℃で40時間攪拌した。反応終了後、反応混合物にtert−ブチルメチルエーテル50.0gおよび35重量%塩酸を加えて、そのpHを2.0とした後、有機層と水層を分離した。次いで、tert−ブチルメチルエーテル12.5gを用いて該水層を抽出処理し、得られた有機層を先に得られた有機層と合一した。得られた有機層にラヂオライト(登録商標、昭和化学工業株式会社製)を1.3g添加し、室温で攪拌した後、ガラスフィルターを用いて固形物をろ別した。ろ液中のわずかに生じた水層を分離した後、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を含む有機層82.6g((1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸として、含量:14.6重量%、純分:12.1g、収率:57.9%、anti体/syn体比=98.9/1.1、anti体光学純度=98.1%ee)を得た。
実施例13で得た(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウム水溶液282g(含量:41.4重量%、純分:117g、0.728mol、anti体/syn体比=98.1/1.9、anti体光学純度=98.1%ee)を常温で撹拌しながら、そこに、ニッケル−アルミニウム合金30.7g(ニッケル含量:49.7重量%、アルミ含量:50.2重量%、アルミニウム純分:15.4g、0.571mol)を加え、混合物の内温を25〜30℃に保ちながら、そこに、エチレンジアミン2.19g(0.036mol)と27重量%水酸化ナトリウム水溶液5.40g(0.036mol)を30分かけて加えた。得られた混合物の内温を29〜42℃に保ちながら、そこに、エチレンジアミン43.8g(0.729mol)と27重量%水酸化ナトリウム水溶液113g(0.765mol)を3.5時間かけて加えた。次いで、得られた混合物の内温を1時間かけて50℃まで昇温し、同温度で3時間攪拌した後、そこに水73gを加え、得られた混合物を同温度で15分間攪拌した。攪拌後、65±5℃で保温されたろ過器を用いて、該混合物からニッケル−アルミニウム合金由来の触媒をろ別した。ろ過器上に残存した触媒を70℃の温水36gを用いて3回洗浄することにより、(1S,2S)−2−フルオロシクロプロパンカルボン酸ナトリウムを含む溶液550g(含量:15.9重量%、純分:87.6g、収率:95.4%、シス体/トランス体比=98.1/1.9、シス体光学純度=98.0%ee)を得た。かかるシス体/トランス体比は高速液体クロマトグラフ分析にて求めた。
実施例17−1で得た(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウム水溶液77.9g(含量:36.0重量%、純分:28.0g、0.174mol、anti体/syn体比=98.8/1.2、anti体光学純度=98.1%ee)を常温で撹拌しながら、そこに、ニッケル−アルミニウム合金7.35g(ニッケル含量:49.7重量%、アルミ含量:50.2重量%、アルミニウム純分:3.67g、0.136mol)を加え、混合物の内温を10〜20℃に保ちながら、そこに、エチレンジアミン1.05g(0.017mol)と26重量%水酸化ナトリウム水溶液2.68g(0.017mol)を2時間かけて加えた。同温度で1時間撹拌した後、得られた混合物の内温を35〜45℃に保ちながら、そこに、エチレンジアミン10.0g(0.166mol)と26重量%水酸化ナトリウム水溶液26.8g(0.174mol)を16時間かけて加えた。次いで、得られた混合物を、内温40℃で1時間撹拌した後、そこに水17gを加え65℃まで昇温し、得られた混合物を同温度で15分間撹拌した。攪拌後、65±5℃で保温されたろ過器を用いて、該混合物からニッケル−アルミニウム合金由来の触媒をろ別した。ろ過器上に残存した触媒を70℃の温水8.7gを用いて3回洗浄することにより、(1S,2S)−2−フルオロシクロプロパンカルボン酸ナトリウムを含む溶液137g(含量:14.8重量%、純分:20.3g、収率:92.2%、シス体/トランス体比=97.2/2.8、シス体光学純度=97.8%ee)を得た。かかるシス体/トランス体比は高速液体クロマトグラフ分析にて求めた。
実施例18−1に準じて得た(1S,2S)−2−フルオロシクロプロパンカルボン酸ナトリウムを含む溶液283g(含量:12.1重量%、純分:34.2g、シス体/トランス体比=97.3/2.7、シス体光学純度=98.0%ee)に、35重量%塩酸97gを加え、そのpHを1以下に調整した。tert−ブチルメチルエーテル102gを3回用いて、得られた混合物を抽出処理し、得られた各有機層を合一した。得られた有機層の全量304gを減圧濃縮し、(1S,2S)−2−フルオロシクロプロパンカルボン酸を含む混合物67.2g(含量:42.1重量%、純分:28.3g、シス体/トランス体比=96.9/3.1)を得た。該混合物にトルエンを44g加えた後、得られた混合物を減圧濃縮した。さらに、トルエン44gを加えて、再度、減圧濃縮し(1S,2S)−2−フルオロシクロプロパンカルボン酸を含む混合物45.4g(含量:58.6重量%、純分:26.6g)を得た。
(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸1.11g(含量:93.7重量%、純分:1.04g、7.5mmol、anti体/syn体比=96.1/3.9、anti体光学純度=97.7%ee)を0〜15℃で撹拌しながら、そこに、10重量%水酸化ナトリウム水溶液2.88g(7.2mmol)を加え、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウム水溶液を調製した。50mLオートクレーブの内部を0〜10℃にし、そこに、展開済スポンジニッケル(川研ファインケミカル株式会社製、NDHT−90)沈殿物1.2mL(ニッケル純分1.0g相当)、エチレンジアミン1.30g(0.0216mol)と水2.20gを攪拌しながら仕込んだ。得られた混合物の内温を10〜30℃に保ちながら、そこに、先に調製した(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸ナトリウム水溶液の全量を滴下した。オートクレーブを密閉し、水素を封入して0.8MPaとした。得られた混合物を35℃まで昇温し、6時間攪拌した。反応終了後、ラヂオライト(登録商標、昭和化学工業株式会社製)を用いて、反応混合物から展開済スポンジニッケル由来の触媒をろ別した。水10g、エタノール10gを順に用い、得られた濾過残渣を洗浄し、得られた洗液と先に得たろ液とを合一した。得られた溶液を減圧濃縮し、その全量を18gとした後、35重量%塩酸を用いて、そのpHを2.0に調整した。tert−ブチルメチルエーテル10gを2回用いて、該溶液を抽出処理し、(1S,2S)−2−フルオロシクロプロパンカルボン酸を含む有機層20.7g(含量:2.29重量%、純分:0.47g、収率:60.5%、シス体/トランス体比=98.3/1.7、シス体光学純度=97.9%ee)を得た。かかるシス体/トランス体比は高速液体クロマトグラフ分析にて求めた。また、水層を分析したところ、水層中に含まれていた(1S,2S)−2−フルオロシクロプロパンカルボン酸は、収率に換算して4.1%であった。よって、反応収率は64.6%であった。
窒素雰囲気下、常温で、260mLオートクレーブに[(R)−N−(5−ニトロサリチリデン)−2−アミノ−1,1−ジ(5−tert−ブチル−2−ブトキシフェニル)−1−プロパノール]銅錯体278mg(0.40mmol)およびジクロロメタン45.6gを仕込み、得られた混合物を攪拌しながら、反応容器を0℃に冷却した。反応容器を密閉し、そこに、1−クロロ−1−フルオロエチレン6.4g(0.080mol)を圧入し、内温を5±2℃とした。内温を5±2℃に保ちながら、そこに製造例3に準じて得たジアゾ酢酸エチル9.60g(含量:95.3重量%、純分:9.13g、0.080mol)をジクロロメタン45.6gに溶解した溶液を5時間かけて
滴下した後、得られた混合物を同温度で1時間攪拌した。復圧後、昇温し、窒素置換をおこなった。得られた反応混合物を0.5モル/Lエチレンジアミン四酢酸水2gで洗浄、分液することにより、(1R)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む混合物を得た。収率は8.5%(対 ジアゾ酢酸エチル)、anti体/syn体比=47/53、anti体光学純度=92.7%ee、syn体光学純度=94.7%eeであった。
リン酸2水素ナトリウム9.6gを水390.4gに溶解した後、28重量%水酸化ナトリウム水を加えてpHを7.0とし、0.2mol/Lリン酸緩衝液400gを調製した。室温下、20mLサンプル管にこのリン酸緩衝液2.5mL、実施例12で得た(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチル25mg(含量:86.2重量%、純分21.6mg、0.129mmol、anti体/syn体比=58.4/41.6、anti体光学純度=96.3%ee)、加水分解酵素(商品名:プロテアーゼA「アマノ」(天野エンザイム株式会社製、Aspergillus oryzae属))5mgを仕込んだ後、得られた混合物を35℃で21時間攪拌した。反応終了後、反応混合物にtert−ブチルメチルエーテル3gおよび5重量%炭酸水素ナトリウム1.0mLを加え、攪拌後、GLクロマトディスク(ジーエルサイエンス株式会社製)を用いて固形物をろ別した。得られた有機層と水層を分離し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸エチルを含む有機層Aを得た。また、該水層をtert−ブチルメチルエーテル2gおよび5重量%塩酸0.8mLを用いて抽出処理した。得られた有機層と水層を分離し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸(収率53.6%、anti体/syn体比=56.5/43.5、anti体光学純度=85.8%ee)を含む有機層Bを得た。
加水分解酵素として、プロテアーゼP「アマノ」3(天野エンザイム株式会社製、Aspergillus melleus属))を用いた以外は比較例1と同様に実施し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸(収率12.8%、anti体/syn体比=50.9/49.1、anti体光学純度=81.0%ee)を含む有機層Bを得た。
加水分解酵素として、プロテアーゼS「アマノ」(天野エンザイム株式会社製、Bacillus stearothermophilus属))を用いた以外は比較例1と同様に実施し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸(収率58.8%、anti体/syn体比=56.3/43.7、anti体光学純度=56.8%ee以上(検出限界))を含む有機層Bを得た。
加水分解酵素として、パパインW−40(天野エンザイム株式会社製、Carica papaya属)を用いた以外は比較例1と同様に実施し、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸(収率77.9%、anti体/syn体比=57.8/42.2、anti体光学純度=58.5%ee以上(検出限界))を含む有機層Bを得た。
anti体/syn体比:ガスクロマトグラフ分析
カラム:DB−WAX
0.53mm×30m、膜厚1.0μm
(アジレント・テクノロジー株式会社製)
方法:(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を
トリメチルシリルジアゾメタンでメチルエステルに誘導体化してから分析。
光学純度:ガスクロマトグラフ分析
カラム:InertCap(登録商標) CHIRAMIX
0.25mm×30m、膜厚0.25μm
(ジーエルサイエンス株式会社製)
方法:(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を
トリメチルシリルジアゾメタンでメチルエステルに誘導体化してから分析。
Claims (9)
- バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼを用いて(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを加水分解する(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法。
- 不斉錯体の存在下、1−クロロ−1−フルオロエチレンとジアゾ酢酸エステルとを反応させて(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを得、次いで、バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼを用いて該(1S)−2−クロロ−2−フルオロシクロプロパンカルボン酸エステルを加水分解する請求項1に記載の製造方法。
- 不斉錯体が、銅化合物と光学活性な配位子とを接触させてなる不斉銅錯体である請求項2に記載の製造方法。
- 光学活性な配位子が、一般式(1):
で示される光学活性なビスオキサゾリン化合物である請求項3に記載の製造方法。 - 光学活性な配位子が、2,2−ビス[2−[(4S)−tert−ブチルオキサゾリン]]プロパンである請求項3に記載の製造方法。
- バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼが、配列番号1または2で示されるアミノ酸配列を有するエステラーゼである請求項1〜5のいずれかに記載の製造方法。
- バークホルデリア・セパシア(Burkholderia cepacia)由来のエステラーゼが、配列番号1で示されるアミノ酸配列を有するエステラーゼである請求項1〜5のいずれかに記載の製造方法。
- 請求項1〜7のいずれかに記載の製造方法により(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を得、次いで、該(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸を還元する(1S,2S)−2−フルオロシクロプロパンカルボン酸の製造方法。
- 還元が、(1S,2R)−2−クロロ−2−フルオロシクロプロパンカルボン酸の存在下、ニッケル−アルミニウム合金に塩基を作用させて行われる請求項8に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009161114A JP5414395B2 (ja) | 2008-07-11 | 2009-07-07 | (1s,2r)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008182066 | 2008-07-11 | ||
| JP2008182066 | 2008-07-11 | ||
| JP2009161114A JP5414395B2 (ja) | 2008-07-11 | 2009-07-07 | (1s,2r)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010035554A true JP2010035554A (ja) | 2010-02-18 |
| JP5414395B2 JP5414395B2 (ja) | 2014-02-12 |
Family
ID=41507116
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009161114A Active JP5414395B2 (ja) | 2008-07-11 | 2009-07-07 | (1s,2r)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8409831B2 (ja) |
| EP (1) | EP2311974B1 (ja) |
| JP (1) | JP5414395B2 (ja) |
| CN (1) | CN102089439B (ja) |
| ES (1) | ES2401415T3 (ja) |
| WO (1) | WO2010005003A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021020458A1 (ja) * | 2019-08-01 | 2021-02-04 | 天野エンザイム株式会社 | 新規リパーゼ及びその用途 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09124556A (ja) * | 1995-08-30 | 1997-05-13 | Dai Ichi Seiyaku Co Ltd | ハロゲン化シクロプロパン誘導体の製造方法 |
| WO1998010091A1 (fr) * | 1996-09-09 | 1998-03-12 | Sumitomo Chemical Company, Limited | Procede de preparation d'isomeres optiquement actifs d'acides 2-halo-2-fluorocyclopropanecarboxyliques |
| JP2001000176A (ja) * | 1999-06-18 | 2001-01-09 | Dai Ichi Seiyaku Co Ltd | バチルス属微生物 |
| JP2004217608A (ja) * | 2002-02-13 | 2004-08-05 | Dai Ichi Seiyaku Co Ltd | (1s,2s)−2−フルオロシクロプロパンカルボン酸誘導体の製造方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3079276B2 (ja) | 1988-02-28 | 2000-08-21 | 天野製薬株式会社 | 組換え体dna、それを含むシュードモナス属菌及びそれを用いたリパーゼの製造法 |
| IL90062A (en) | 1988-04-27 | 1994-10-07 | Daiichi Seiyaku Co | Pyridonecarboxylic acid derivatives, their preparation and pharmaceutical compositions containing them |
| JP3410128B2 (ja) | 1992-11-27 | 2003-05-26 | 天野エンザイム株式会社 | 組換えdnaを有するシュードモナス属菌及びそれを用いるリパーゼの製造法 |
| JP4635343B2 (ja) | 2000-01-25 | 2011-02-23 | 住友化学株式会社 | 光学活性な銅錯体ならびに光学活性なサリチリデンアミノアルコール化合物およびそれを用いる光学活性なシクロプロパンカルボン酸誘導体の製造方法 |
| JP2005015468A (ja) | 2003-06-04 | 2005-01-20 | Sumitomo Chemical Co Ltd | シクロプロパン化合物の有害節足動物防除用途 |
| JP5002916B2 (ja) | 2004-07-01 | 2012-08-15 | 住友化学株式会社 | 不斉銅錯体およびそれを用いた光学活性なシクロプロパンカルボン酸エステル化合物の製造方法 |
-
2009
- 2009-07-07 CN CN2009801271084A patent/CN102089439B/zh active Active
- 2009-07-07 WO PCT/JP2009/062388 patent/WO2010005003A1/ja not_active Ceased
- 2009-07-07 EP EP09794447A patent/EP2311974B1/en active Active
- 2009-07-07 US US13/003,499 patent/US8409831B2/en active Active
- 2009-07-07 JP JP2009161114A patent/JP5414395B2/ja active Active
- 2009-07-07 ES ES09794447T patent/ES2401415T3/es active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09124556A (ja) * | 1995-08-30 | 1997-05-13 | Dai Ichi Seiyaku Co Ltd | ハロゲン化シクロプロパン誘導体の製造方法 |
| WO1998010091A1 (fr) * | 1996-09-09 | 1998-03-12 | Sumitomo Chemical Company, Limited | Procede de preparation d'isomeres optiquement actifs d'acides 2-halo-2-fluorocyclopropanecarboxyliques |
| JP2001000176A (ja) * | 1999-06-18 | 2001-01-09 | Dai Ichi Seiyaku Co Ltd | バチルス属微生物 |
| JP2004217608A (ja) * | 2002-02-13 | 2004-08-05 | Dai Ichi Seiyaku Co Ltd | (1s,2s)−2−フルオロシクロプロパンカルボン酸誘導体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010005003A1 (ja) | 2010-01-14 |
| US20110117616A1 (en) | 2011-05-19 |
| EP2311974A4 (en) | 2012-02-08 |
| CN102089439A (zh) | 2011-06-08 |
| EP2311974A1 (en) | 2011-04-20 |
| CN102089439B (zh) | 2013-11-13 |
| ES2401415T3 (es) | 2013-04-19 |
| US8409831B2 (en) | 2013-04-02 |
| EP2311974B1 (en) | 2012-12-19 |
| JP5414395B2 (ja) | 2014-02-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102099482B (zh) | 5-甲基-3-硝基甲基-己酸酯的立体选择性酶水解方法 | |
| US9061991B2 (en) | Method for producing 1-amino-1-alkoxycarbonyl-2-vinylcyclopropane | |
| US9139513B2 (en) | Process for preparing lacosamide | |
| TW200846307A (en) | Preparation of pregabalin and related compounds | |
| SG187652A1 (en) | Novel hydrolase protein | |
| US7928253B2 (en) | Method of producing 6,6-dimethyl-3-oxabicyclo[3.1.0]hexan-2-one | |
| JP5414395B2 (ja) | (1s,2r)−2−クロロ−2−フルオロシクロプロパンカルボン酸の製造方法 | |
| AU2001230192B2 (en) | Method for the enzymatic resolution of the racemates of aminomethyl-aryl-cyclohexanol derivatives | |
| KR100424832B1 (ko) | 광학활성아미드의분리법 | |
| KR20080086463A (ko) | 라미프릴의 제조를 위한 개선된 방법 | |
| KR102791352B1 (ko) | (2s)-2-[(4r)-2-옥소-4-프로필-피롤리딘-1-일]부틸산 및 이를 브리바라세탐으로 변환하기 위한 효소적 방법 | |
| JP4319847B2 (ja) | (1s,2s)−2−フルオロシクロプロパンカルボン酸誘導体の製造方法 | |
| WO2012176715A1 (ja) | 1-アミノ-2-ビニルシクロプロパンカルボン酸アミドおよびその塩、ならびにその製造方法 | |
| CN1643158A (zh) | 由外消旋N-酰基化β-氨基羧酸制备旋光性β-氨基羧酸的方法 | |
| WO2025041039A1 (en) | Immobilised whole cell nitrilases for biocatalysis of gabapentin intermediate | |
| JP2023172821A (ja) | ブリバラセタムの合成のための重要な中間体である(r)-4-プロピルピロリジン-2-オンの調製のための改良されたプロセス | |
| JPH10248592A (ja) | 光学活性2−ベンジルコハク酸およびその誘導体の製法 | |
| JP2006021999A (ja) | 光学活性β−アミノニトリル化合物およびその対掌体アミド化合物の製造方法 | |
| HK1104579A (en) | Method for producing the enantiomer forms of cis-configured 3-hydroxycyclohexane carboxylic acid derivatives using hydrolases | |
| JP2008295302A (ja) | 光学活性エステル誘導体および/または光学活性カルボン酸の製造方法 | |
| HK1125916A (en) | Preparation of gamma-amino acids having affinity for the alpha-2-delta protein |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120321 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20131022 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131112 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5414395 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |