JP2010001352A - 新規なポリイミド前駆体組成物及びその利用 - Google Patents
新規なポリイミド前駆体組成物及びその利用 Download PDFInfo
- Publication number
- JP2010001352A JP2010001352A JP2008160323A JP2008160323A JP2010001352A JP 2010001352 A JP2010001352 A JP 2010001352A JP 2008160323 A JP2008160323 A JP 2008160323A JP 2008160323 A JP2008160323 A JP 2008160323A JP 2010001352 A JP2010001352 A JP 2010001352A
- Authority
- JP
- Japan
- Prior art keywords
- solution
- bis
- polyimide precursor
- precursor composition
- polyimide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Materials For Photolithography (AREA)
- Paints Or Removers (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
Abstract
【解決手段】 少なくとも、分子内に少なくとも2つのイミド結合を含有し、重量平均分子量が1000以上15000以下であり、酸価が50〜150mgKOH/gである酸末端化合物及び鎖延長剤とを含むポリイミド前駆体組成物により、上記課題を解決する。
【選択図】 なし
Description
(A)分子内に少なくとも2つのイミド結合を含有し、重量平均分子量が1000以上15000以下であり、酸価が50〜150mgKOH/gである酸末端化合物とは、例えば、下記一般式(1)
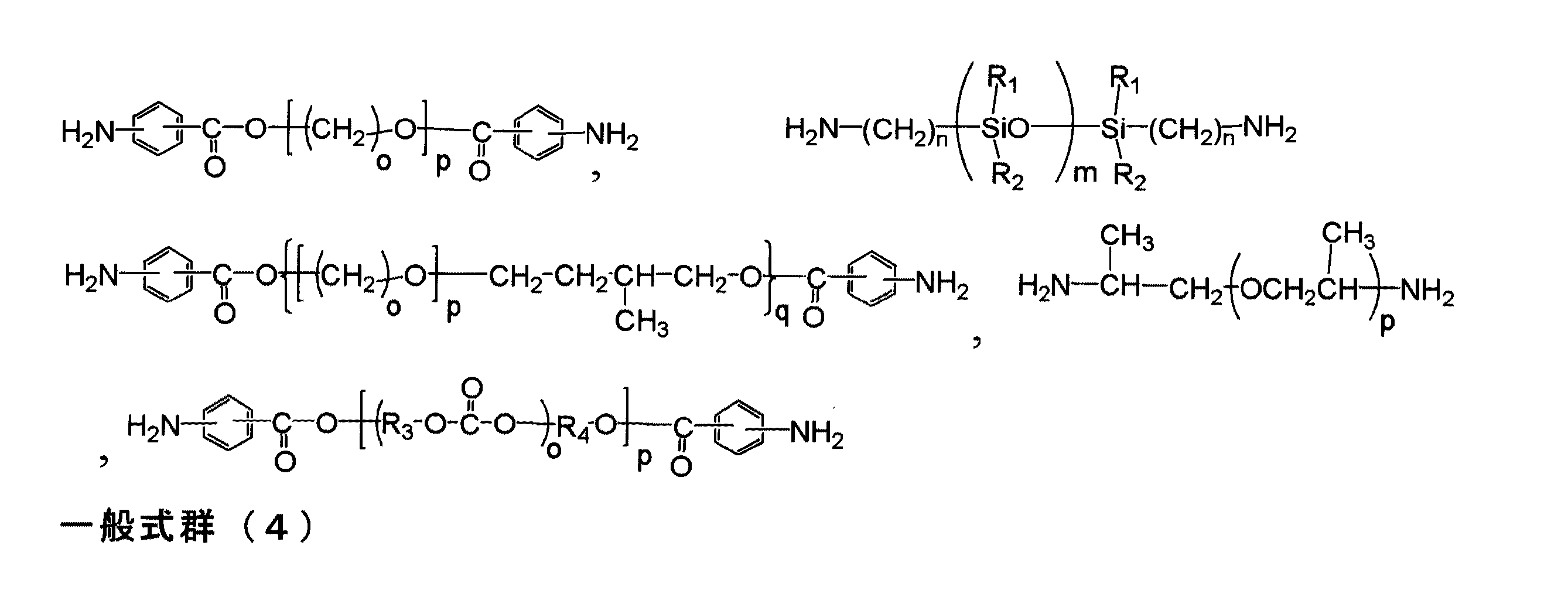
で示されるように、構造式中に少なくとも2つのイミド結合を有しており、末端がテトラカルボン酸になっている構造を持つイミド化したテトラカルボンが挙げられるが、これに限定されるものではない。
上記一部イミド化したテトラカルボン酸の製造方法としては、種々の方法が挙げられる。
本願発明で用いられる鎖延長剤とは、(A)分子内に少なくとも2つのイミド結合を含有し、重量平均分子量が1000以上15000以下であり、酸価が50〜150mgKOH/gである酸末端化合物と反応し、イミド結合を生成しながら(A)成分を高分子量化させることができる化合物である。例えば、ジアミノ化合物やイソシアネート系化合物を用いることができるが、これらに限定されるものではない。
本願発明で用いられるジアミノ化合物とは、下記一般式(6)で示される、分子内にアミノ基を2つ有する化合物である。
本願発明で用いられるイソシアネート系化合物とは、イソシアネート基を2つ以上有する化合物である。
本発明のポリイミド前駆体組成物溶液の調製方法について記載する。(A)分子内に少なくとも2つのイミド結合を含有し、重量平均分子量が1000以上15000以下であり、酸価が50〜150mgKOH/gである酸末端化合物を合成した溶液中でポリイミド前駆体組成物溶液を調整する場合には、そのままその溶液を用いて、その溶液に(B)鎖延長剤を投入してポリイミド前駆体組成物溶液を得ることが好ましく、一度、固形として分離したポリイミド前駆体組成物については、溶剤で希釈して用いることが好ましい。
本願発明のポリイミド前駆体組成物の利用の1例として、感光性樹脂組成物が挙げられる。以下において、感光性樹脂組成物について詳述する。なお、本願発明のポリイミド前駆体組成物の利用の例としては、これに限られることは言うまでもない。感光性樹脂組成物の構成は、次のとおりである。すなわち、上記ポリイミド前駆体組成物と、少なくとも感光性樹脂、及び、光重合開始剤を含有することを特徴とする感光性樹脂組成物である。なお、感光性樹脂組成物に使用するポリイミド前駆体組成物については、上記ポリイミド前駆体組成物であれば、特に限定する事無く使用可能である。
本願発明における感光性樹脂とは、光もしくは熱によって発生したラジカル、酸、塩基、プロトン、アミン等によって、重合するモノマー、オリゴマーもしくは高分子樹脂である。より好ましくは、少なくとも不飽和二重結合を1つ有する樹脂である。さらには、前記不飽和二重結合は、アクリル基(CH2=CH−基)、メタアクリロイル基(CH2=C(CH3)−基)もしくはビニル基(−CH=CH−基)であることが好ましい。下記に本願発明で好適に用いられる感光性樹脂を例示するが、上記不飽和二重結合を少なくとも1つ有する樹脂であればどのような樹脂を用いても良い。
光重合開始剤としては、光の照射により、ラジカル、酸、塩基、プロトン、アミン等を発生するものであればどのような構造のものも使用することができる。例えば、ミヒラ−ズケトン、4,4'−ビス(ジエチルアミノ)ベンゾフェノン、4,4',4''−トリス(ジメチルアミノ)トリフェニルメタン、2,2'−ビス(2−クロロフェニル)−4,4',5,5'−テトラフェニル−1,2'−ジイミダゾール、アセトフェノン、ベンゾイン、2−メチルベンゾイン、ベンゾインメチルエ−テル、ベンゾインエチルエ−テル、ベンゾインイソプロピルエ−テル、ベンゾインイソブチルエ−テル、2−t−ブチルアントラキノン、1,2−ベンゾ−9,10−アントラキノン、メチルアントラキノン、チオキサントン、2,4−ジエチルチオキサントン、2−イソプロピルチオキサントン、1−ヒドロキシシクロヘキシルフェニルケトン、ジアセチルベンジル、ベンジルジメチルケタ−ル、ベンジルジエチルケタ−ル、2(2'−フリルエチリデン)−4,6−ビス(トリクロロメチル)−S−トリアジン、2[2'(5''−メチルフリル)エチリデン]−4,6−ビス(トリクロロメチル)−S−トリアジン、2(p−メトキシフェニル)−4,6−ビス(トリクロロメチル)−S−トリアジン、2,6−ジ(p−アジドベンザル)−4−メチルシクロヘキサノン、4,4’−ジアジドカルコン、ジ(テトラアルキルアンモニウム)−4,4'−ジアジドスチルベン−2,2'−ジスルフォネ−ト、2,2−ジメトキシ−1,2−ジフェニルエタン−1−オン、1−ヒドロキシ−シクロヘキシル−フェニル−ケトン、2−ヒドロキシ−2−メチル−1−フェニル−プロパン−1−オン、1−[4−(2−ヒドロキシエトキシ)−フェニル]−2−ヒドロキシ−2−メチル−1−プロパン−1−オン、2−メチル−1−[4−(メチルチオ)フェニル]−2−モルフォリノプロパン−1−オン、2−ベンジル−2−ジメチルアミノ−1−(4−モルフォリノフェニル)−ブタン−1、ビス(2,4,6−トリメチルベンゾイル)−フェニルフォスフィンオキサイド、ビス(2,6−ジメトキシベンゾイル)−2,4,4−トリメチル−ペンチルフォスフィンオキサイド、2,4,6−トリメチルベンゾイル−ジフェニル−フォスフィンオキサイド、2−ヒドロキシ−2−メチル−1−フェニル−プロパン−1−ケトン、ビス(n5−2,4−シクロペンタジエン−1−イル)−ビス(2,6−ジフルオロ−3−(1H−ピロール−1−イル)−フェニル)チタニウム、1,2−オクタノンジオン,1−[4−(フェニルチオ)−,2−(O−ベンゾイルオキシム)]、ヨード二ウム,(4−メチルフェニル)[4−(2−メチルプロピル)フェニル]−ヘキサフルオロフォスフェート(1−)、エチル−4−ジメチルアミノベンゾエート、2−エチルヘキシル−4−ジメチルアミノベンゾエート、エタノン,1−[9−エチル−6−(2−メチルベンゾイル)−9H−カルバゾール−3−イル]−,1−(O−アセチルオキシオム)などが挙げられる。上記光重合開始剤は適宜選択することが好ましく、1種以上を混合させて用いることが好ましい。
本願発明の感光性樹脂組成物は、硬化後の耐熱性(半田耐熱性等)、耐薬品性(アルカリ溶液耐性、耐酸性、耐溶剤性等)、耐湿環境安定性、耐熱環境安定性に優れる樹脂組成物にするために、熱硬化性樹脂を配合することが好ましい。
この発明の感光性樹脂組成物には、さらに必要に応じて難燃剤、消泡剤、カップリング剤、充填剤、接着助剤、レベリング剤、重合禁止剤等の各種添加剤を加えることができる。充填剤としては、シリカ、マイカ、タルク、硫酸バリウム、ワラストナイト、炭酸カルシウムなどの微細な無機充填剤、微細な有機ポリマ−充填剤を含有させてもよい。含有量は適宜選定することが好ましい。
本願発明の感光性樹脂組成物は、取扱いの観点から有機溶剤に溶解した感光性樹脂組成物溶液として用いることが好ましい。本願発明の感光性樹脂組成物は、種々の有機溶剤に溶解性が高く、例えば、ジメチルスルホキシド、ジエチルスルホキシドなどのスルホキシド系溶媒、N,N−ジメチルホルムアミド、N,N−ジエチルホルムアミドなどのホルムアミド系溶媒、N,N−ジメチルアセトアミド、N,N−ジエチルアセトアミドなどのアセトアミド系溶媒、N−メチル−2−ピロリドン、N−ビニル−2−ピロリドンなどのピロリドン系溶媒、フェノール、o−、m−またはp−クレゾール、キシレノール、ハロゲン化フェノール、カテコールなどのフェノール系溶媒、あるいはヘキサメチルホスホルアミド、γ−ブチロラクトン、メチルモノグライム(1,2-ジメトキシエタン)、メチルジグライム(ビス(2-メトキシエテル)エーテル)、メチルトリグライム(1,2-ビス(2-メトキシエトキシ)エタン)、メチルテトラグライム(ビス[2-(2-メトキシエトキシエチル)]エーテル)、エチルモノグライム(1,2-ジエトキシエタン)、エチルジグライム(ビス(2-エトキシエチル)エーテル)、ブチルジグライム(ビス(2-ブトキシエチル)エーテル)等の対称グリコールジエーテル類、γ―ブチロラクトンやN−メチル−2−ピロリドン、メチルアセテート、エチルアセテート、イソプロピルアセテート、n―プロピルアセテート、ブチルアセテート、プロピレングリコールモノメチルエーテルアセテート、エチレングリコールモノブチルエーテルアセテート、ジエチレングリコールモノエチルエーテルアセテート(別名、カルビトールアセテート、酢酸2-(2-ブトキシエトキシ)エチル))、ジエチレングリコールモノブチルエーテルアセテート、3−メトキシブチルアセテート、エチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテート、ジプロピレングリコールメチルエーテルアセテート、プロピレングリコールジアセテート、1,3―ブチレングリコールジアセテート等のアセテート類や、ジプロピレングリコールメチルエーテル、トリプロピレングリコールメチルエーテル、プロピレングリコールn−プロピルエーテル、ジプロピレングリコールn−プロピルエーテル、プロピレングリコールn−ブチルエーテル、ジプロピレングリコールn−ブチルエーテル、トリピレングリコールn−プロピルエーテル、プロピレングリコールフェニルエーテル、ジプロピレングリコールジメチルエーテル、1,3―ジオキソラン、エチレングリコールモノブチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノブチルエーテル、エチレングリコールものエチルエーテル等のエーテル類の溶剤を用いることもできる。尚、必要に応じて低沸点のヘキサン、アセトン、トルエン、キシレン等を用いて感光性樹脂組成物溶液とすることができる。
この発明の感光性樹脂組成物は、上記感光性樹脂組成物に配合される各種原料を均一に混合して得られる。均一に混合する方法としては、例えば3本ロール、ビーズミル装置等の一般的な混練装置を用いて混合すればよい。また、溶液の粘度が低い場合には、一般的な攪拌装置を用いて混合してもよい。
この発明の感光性樹脂組成物を直接に、もしくは、上記感光性樹脂組成物溶液を調整した後に、以下のようにしてパタ−ンを形成することができる。先ず上記の感光性樹脂組成物を基板に塗布し、乾燥して有機溶媒を除去する。基板への塗布はスクリ−ン印刷、カ−テンロ−ル、リバ−スロ−ル、スプレーコーティング、スピンナーを利用した回転塗布等により行うことができる。塗布膜(好ましくは厚み:5〜100μm、特に10〜100μm)の乾燥は120℃以下、好ましくは40〜100℃で行う。乾燥後、乾燥塗布膜にネガ型のフォトマスクを置き、紫外線、可視光線、電子線などの活性光線を照射する。次いで、未露光部分をシャワー、パドル、浸漬または超音波等の各種方式を用い、現像液で洗い出すことによりレリ−フパタ−ンを得ることができる。なお、現像装置の噴霧圧力や流速、エッチング液の温度によりパターンが露出するまでの時間が異なる為、適宜最適な装置条件を見出すことが好ましい。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン183gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、下記一般式(7)のシリコンジアミン、
上記ポリイミド前駆体組成物溶液を用いて、ベーカー式アプリケーターを用いて、75μmのポリイミドフィルム(株式会社カネカ製:商品名75NPI)に最終乾燥厚みが25μmになるように流延・塗布し、120℃で1時間乾燥した後、窒素雰囲気下160℃で30分加熱してイミド化を行った。
このポリイミドフィルムの接着強度をJIS K5400に従って碁盤目テープ法で評価した。
碁盤目テープ法で剥がれの無いものを○、
升目の半分以上が残存している場合を△、
升目の残存量が半分未満のものを×とした。
ポリイミドフィルムのイミド化が充分でないと、環境試験装置内での安定性が低下する。そのため、環境試験装置内での安定性を測定した。
環境試験装置は、エスペック株式会社製恒温高湿器 型式:PR−1Kを用いて85℃/85%RH 1000時間試験後のポリイミドフィルム上の塗膜の状態で判断した。
ポリイミド樹脂が変化無いものを〇、
ポリイミド樹脂が一部溶解しているものを△、
ポリイミド樹脂が完全に溶解しているもの×とした。
ポリイミドフィルム表面の耐薬品性の評価を行った。評価方法は下記評価項目1〜3の評価条件でフィルムを浸漬した後にフィルム表面の状態を観察して評価を行った。
評価項目1:25℃のイソプロパノール中に10分浸漬した後、風乾した。
評価項目2:25℃の2Nの塩酸溶液中に10分間浸漬した後、純水で洗浄して風乾燥した。
評価項目3:25℃の2Nの水酸化ナトリウム溶液中に浸漬した後、純水で洗浄して風乾した。
ポリイミド樹脂が変化無いものを〇、
ポリイミド樹脂が一部溶解しているものを△、
ポリイミド樹脂が完全に溶解しているもの×とした。
25μm厚みのポリイミドフィルム(株式会社カネカ製アピカル25NPI)表面にポリイミド樹脂溶液を最終フィルム厚みが25μmになるように塗布して、120℃で90分、160℃で30分乾燥してポリイミドフィルム積層体を得た。本ポリイミドフィルム積層体を30mm×10mmの短冊に切り出して、15mmのところで180°に10回折り曲げて塗膜を目視で確認してクラックの確認を行った。
○:硬化膜にクラックが無いもの
△:硬化膜に若干クラックがあるもの
×:硬化膜にクラックがあるもの
上記の評価結果を表1に記載する。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン169gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、下記一般式(7)のシリコンジアミン、
更に、ポリイミド前駆体組成物から得られる硬化被膜の特性を実施例1と同様の方法で行った。その評価結果を表1に記載する。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン193gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名X−22−9409S、分子量1492、下記一般式(7)のシリコンジアミン、
更に、ポリイミド前駆体組成物から得られる硬化被膜の特性を実施例1と同様の方法で行った。その評価結果を表1に記載する。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン207gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名X−22−9409S、分子量1492、下記一般式(7)のシリコンジアミン、
更に、ポリイミド前駆体組成物から得られる硬化被膜の特性を実施例1と同様の方法で行った。その評価結果を表1に記載する。
4,4'―オキシジフタル酸二無水物20.0g(0.0645mol)を1,2-ビス(2-メトキシエトキシ)エタン52.7gに分散し、20℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、下記一般式(7)のシリコンジアミン、
更に、ポリイミド前駆体組成物から得られる硬化被膜の特性を実施例1と同様の方法で行った。その評価結果を表1に記載する。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物300g(0.576mol)を1,2-ビス(2-メトキシエトキシ)エタン510gに分散し、80℃に保った。これにポリカーボネートジオールビス(4−アミノベンゾエート)(下記一般式(8)で表されるジアミンであり、
更に、ポリイミド前駆体組成物から得られる硬化被膜の特性を実施例1と同様の方法で行った。その評価結果を表1に記載する。
ヘキサメチレンジアミン2.73g(23.5mmol)をジメチルアセトアミド24.0gに溶解し、これに3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物3.78g(11.75mmol)を30分間にわたり徐々に加え、ポリアミド結合を持ったオリゴマーを得た。1時間均一攪拌した後、3,3’,4,4’−ベンゾフェノンテトラカルボン酸3.02g(9.40mmol)を加え1時間撹拌を続けたところ、粘調な溶液が得られた(溶質濃度28重量%)。この溶液の粘度を測定したところ、3100ポイズであった。得られた溶液の重量平均分子量は、GPC法により測定した結果、ポリスチレン換算で103000、酸価は固形分換算で84mgKOH/gであった。
実施例1と同様の方法で評価を行った。評価結果を表2に記載する。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン183gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、下記一般式(7)のシリコンジアミン、
実施例1と同様の評価方法を行った結果を表2に記載する。耐環境試験安定性が悪く、耐溶剤性、耐アルカリ性が悪いことが明らかになった。
4,4'−ジアミノジフェニルエーテル8.22g(41.1mmol)をN,N−ジメチルアセトアミド55.0gに溶解し、室温下で攪拌した。これにピロメリット酸二無水物11.9g(54.8mmol)を添加して、室温下で2時間攪拌した。メタノールを1.32g(41.1mmol)及びジメチルアミノエタノール0.066gを加えて、70℃湯浴上で2時間加熱攪拌してイミド基を分子内に有さないカルボン酸化合物を得た。得られたイミド基を分子内に有さないカルボン酸化合物の重量平均分子量は、GPC法により測定した結果、ポリスチレン換算で2300、酸価は固形分換算で90mgKOH/gであった。室温まで冷却した後、鎖延長剤として、4,4'−ジアミノジフェニルエーテル2.74g(13.7mmol)を加え、更に1時間攪拌を続けたところ、均一な溶液が得られた。この溶液の粘度は23℃で18ポイズであった。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン183gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、下記一般式(7)のシリコンジアミン、
ピロメリット酸二無水物7.00g(32.1mmol)を1,2-ビス(2-メトキシエトキシ)エタン31.3gに分散して、水2.31gを添加して、80℃で10時間攪拌して、ピロメリット酸溶液を得た。この溶液に、4,4−ジアミノジフェニルエーテル6.43g(32.1mmol)を添加して溶液を調製した。
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン183gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、下記一般式(7)のシリコンジアミン、
2,2−ビス[4−(3,4−ジカルボキシフェノキシ)フェニル]プロパン二無水物(以下BPADAと略す)200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン140gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、一般式(7)中のR1、R2がメチル基、n=3、m=6〜11である。)を128g(0.154mol)投入し、30分間均一攪拌を行った。次いで、140℃に加熱して1時間均一攪拌を行い、次いで180℃に昇温させて3時間加熱還流を行いイミド化反応を行った。次いで、80℃まで冷却し水を27.7g(1.54mol)投入し、5時間加熱還流を行った。このようにしてイミド化したテトラカルボン酸(末端テトラカルボン酸シロキサンイミドオリゴマー)を溶解した溶液を得た。得られたイミド化したテトラカルボン酸の重量平均分子量は、GPC法により測定した結果、ポリスチレン換算で2500、酸価は固形分換算で95mgKOH/gであった。この溶液の固形分濃度は66重量%、溶液の粘度は23℃で140ポイズであった。この末端テトラカルボン酸溶液は、1ヶ月間室温で放置しておいても、粘度の変化は殆ど無く安定的な溶液であった。この合成した化合物を化合物Aと略す。
BPADA200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン159gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名X−22−9409S、分子量1492、一般式(7)中のR1、R2がメチル基もしくはフェニル基、n=3、m=9〜12である。)を172g(0.115mol)投入し、30分間均一攪拌を行った。次いで、140℃に加熱して1時間均一攪拌を行い、次いで、180℃に昇温させて3時間加熱還流を行いイミド化反応を行った。次いで、80℃まで冷却し水を27.7g(1.54mol)投入し、5時間加熱還流を行った。このようにしてイミド化したテトラカルボン酸(末端テトラカルボン酸シロキサンイミドオリゴマー)を溶解した溶液を得た。得られたイミド化したテトラカルボン酸の重量平均分子量は、GPC法により測定した結果、ポリスチレン換算で4500、酸価は固形分換算で90mgKOH/gであった。この溶液の固形分濃度は67重量%、溶液の粘度は23℃で120ポイズであった。この末端テトラカルボン酸溶液は、1ヶ月間室温で放置しておいても、粘度の変化は殆ど無く安定的な溶液であった。この合成した化合物を化合物Bと略す。
BPADA200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン154gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名KF8010、分子量830、一般式(7)中のR1、R2がメチル基、n=3、m=6〜11である。)を159g(0.192mol)投入し、30分間均一攪拌を行った。次いで、140℃に加熱して1時間均一攪拌を行い、180℃に昇温させて3時間加熱還流を行いイミド化反応を行った。80℃まで冷却し水を27.7g(1.54mol)投入し、5時間加熱還流を行った。このようにしてイミド化したテトラカルボン酸(末端テトラカルボン酸シロキサンイミドオリゴマー)を溶解した溶液を得た。得られたイミド化したテトラカルボン酸の重量平均分子量は、GPC法により測定した結果、ポリスチレン換算で3600、酸価は固形分換算で84mgKOH/gであった。この溶液の固形分濃度は66重量%、溶液の粘度は23℃で100ポイズであった。この末端テトラカルボン酸溶液は、1ヶ月間室温で放置しておいても、粘度の変化は殆ど無く安定的な溶液であった。この合成した化合物を化合物Cと略す。
BPADA200g(0.384mol)を1,2-ビス(2-メトキシエトキシ)エタン184gに分散し、80℃に保った。これにシリコンジアミン(シロキサンジアミン)(信越化学社製:商品名X−22−9409S、分子量1492、一般式(7)中のR1、R2がメチル基もしくはフェニル基、n=3、m=9〜12である。)を229g(0.154mol)投入し、30分間均一攪拌を行った。次いで、140℃に加熱して1時間均一攪拌を行い、180℃に昇温させて3時間加熱還流を行いイミド化反応を行った。80℃まで冷却し水を27.7g(1.54mol)投入した。均一に30分間攪拌した後、80℃に加熱して5時間加熱還流を行った。このようにしてイミド化したテトラカルボン酸(末端テトラカルボン酸シロキサンイミドオリゴマー)を溶解した溶液を得た。得られたイミド化したテトラカルボン酸の重量平均分子量は、GPC法により測定した結果、ポリスチレン換算で5600、酸価は固形分換算で80mgKOH/gであった。この溶液の固形分濃度は67重量%、溶液の粘度は23℃で90ポイズであった。この末端テトラカルボン酸溶液は、1ヶ月間室温で放置しておいても、粘度の変化は殆ど無く安定的な溶液であった。この合成した化合物を化合物Dと略す。
合成例1で反応後に投入する水をメタノールに変更し、ハーフエステル化した。この合成した化合物を化合物Eと略す。得られたイミド化したテトラカルボン酸の重量平均分子量は、GPC法により測定した結果、ポリスチレン換算で2500、酸価は固形分換算で40mgKOH/gであった。
合成例1で反応後に水を投入することなく、上記反応を行い末端が無水物基のイミド化した酸無水物を得た。この合成した化合物を化合物Fと略す。
合成例1〜4で得られた末端テトラカルボン酸(表3においては(A)成分と表す。)にジアミノ化合物(表3においては(B)成分と表す。)、感光性樹脂(表3においては(C)成分と表す。)、光重合開始剤(表3においては(D)成分と表す。)、有機溶剤を添加して感光性樹脂組成物溶液を作製した。それぞれの構成原料の樹脂固形分での配合量及び原料の種類を表3に記載する。なお、表中の溶媒である1,2-ビス(2-メトキシエトキシ)エタンは上記感光性樹脂組成物溶液等に含まれる溶剤等も含めた全溶剤量である。
上記感光性樹脂組成物溶液を、ベーカー式アプリケーターを用いて、75μmのポリイミドフィルム(株式会社カネカ製:商品名75NPI)に最終乾燥厚みが25μmになるように100mm×100mmの面積に流延・塗布し、80℃で20分乾燥した。この乾燥フィルムは10枚用意した。9枚は50mm×50mmの面積が完全に透明な、ネガ型のフォトマスクをおいて、1枚には、ライン幅/スペース幅=100μm/100μmのネガ型フォトマスク(30mm長さ×100μm幅のラインが10本残るフォトマスク)を置いて窒素雰囲気下で紫外線を300mJ/cm2露光して感光させた。この感光フィルムに対し、1.0重量%の炭酸ナトリウム水溶液を30℃に加熱した溶液を用いて、1.0kgf/mm2の吐出圧で30秒間、スプレー現像を行った。現像後、純水で十分洗浄した後、170℃のオーブン中で60分加熱乾燥させて感光性樹脂組成物の硬化膜を作製した。
感光性樹脂組成物の感光性の評価は、上記(ポリイミドフィルム上への塗膜の作製)の項目で得られた硬化膜の表面観察を行い判定した。
ポリイミドフィルム表面に
〇:くっきりとしたライン幅/スペース幅=100/100μmの感光パターンが描けており、ライン部の剥離に伴うラインの揺れが発生しておらず、スペース部にも溶解残りが無いもの。
△:くっきりとしたライン幅/スペース幅=100/100μmの感光パターンが描けており、ライン部に剥離に伴うラインの揺れが発生しているが、スペース部には溶解残りが無いもの。
×:くっきりとしたライン幅/スペース幅=100/100μmの感光パターンが描けておらず、ライン部が剥離しており、しかも、スペース部には溶解残りが発生しているもの。
上記(ポリイミドフィルム上への塗膜の作製)の項目で得られた感光性樹脂組成物の硬化膜の接着強度をJIS K5400に従って碁盤目テープ法で評価した。
碁盤目テープ法で剥がれの無いものを○、
升目の95%以上が残存している場合を△、
升目の残存量が80%未満のものを×とした。
上記(ポリイミドフィルム上への塗膜の作製)の項目で得られた感光性樹脂組成物の硬化膜の耐溶剤性の評価を行った。評価方法は25℃のイソプロパノール中に15分間浸漬した後風乾し、フィルム表面の状態を観察した。
○:塗膜に異常がない。
×:塗膜に異常が発生する。
上記(ポリイミドフィルム上への塗膜の作製)の項目で得られた感光性樹脂組成物の硬化膜の耐酸性の評価を行った。評価方法は25℃の2N塩酸溶液中に15分間浸漬した後風乾し、フィルム表面の状態を観察した。
○:塗膜に異常(白化もしくは剥離)がないもの。
×:塗膜に異常(白化もしくは剥離)が発生する。
上記(ポリイミドフィルム上への塗膜の作製)の項目で得られた感光性樹脂組成物の硬化膜の耐アルカリ性の評価を行った。評価方法は25℃の2N水酸化ナトリウム溶液中に15分間浸漬した後風乾し、フィルム表面の状態を観察した。
○:塗膜に異常(白化もしくは剥離)がない。
×:塗膜に異常(白化もしくは剥離)が発生する。
上記(ポリイミドフィルム上への塗膜の作製)の項目と同様の方法で、25μm厚みのポリイミドフィルム(株式会社カネカ製アピカル25NPI)表面に感光性樹脂組成物の硬化膜積層フィルムを作製した。硬化膜積層フィルムを30mm×10mmの短冊に切り出して、15mmのところで180°に10回折り曲げて塗膜を目視で確認してクラックの確認を行った。
○:硬化膜にクラックが無いもの。
△:硬化膜に若干クラックがあるもの。
×:硬化膜にクラックがあるもの。
フレキシブル銅貼り積層版(銅箔の厚み12μm、ポリイミドフィルムは株式会社カネカ製アピカル25NPI、ポリイミド系接着剤で銅箔を接着している)上にライン幅/スペース幅=100μm/100μmの櫛形パターンを作製し、10容量%の硫酸水溶液中に1分間浸漬した後、純水で洗浄し銅箔の表面処理を行った。その後、ポリイミドフィルム上への硬化膜の作製方法と同様の方法で櫛形パターン上に感光性樹脂組成物の硬化膜を作製し試験片の調整を行った。85℃、85%RHの環境試験機中で試験片の両端子部分に100Vの直流電流を印加し、絶縁抵抗値の変化やマイグレーションの発生などを観察した。
○:試験開始後、500時間で10の6乗以上の抵抗値を示し、マイグレーション、デンドライトなどの発生が無いもの。
×:試験開始後、500時間でマイグレーション、デンドライトなどの発生があるもの。
上記実施例7の感光性樹脂組成物の固形分100重量部に対して、エポキシ樹脂(クレゾールノボラック型の多官能エポキシ樹脂であるエピクロンN―665)を5重量部投入した以外は、実施例7と同様の方法で評価を行った。更に、半田耐熱性の試験として、下記評価方法で評価を行った。評価結果を表4に示す。
感光性樹脂組成物溶液を、ベーカー式アプリケーターを用いて、75μmのポリイミドフィルム(株式会社カネカ製:商品名75NPI)に最終乾燥厚みが25μmになるように100mm×100mmの面積に流延・塗布し、80℃で20分乾燥した後、50mm×50mmの面積が完全に透明なネガ型フォトマスクを置いて窒素雰囲気下で紫外線を300mJ/cm2露光して感光させた。この感光フィルムに対し、1.0重量%の炭酸ナトリウム水溶液を30℃に加熱した溶液を用いて、1.0kgf/mm2の吐出圧で30秒間、スプレー現像を行った。現像後、純粋で十分洗浄した後、170℃のオーブン中で60分加熱乾燥させて感光性樹脂組成物の硬化膜を作製した。
碁盤目テープ法で剥がれの無いものを○、
升目の95%以上が残存している場合を△、
升目の残存量が80%未満のものを×とした。
上記実施例8の感光性樹脂組成物の固形分100重量部に対して、エポキシ樹脂(クレゾールノボラック型の多官能エポキシ樹脂であるエピクロンN―665)を5重量部投入した以外は、実施例7と同様の方法で評価を行った。更に、半田耐熱性の試験として、実施例11と同様の評価方法で評価を行った。評価結果を表4に示す。
合成例5で得られたハーフエステル化した化合物を用いた以外は実施例7と同様の方法で感光性樹脂組成物溶液を作製し、実施例7と同様の方法で評価を行った。評価結果を表5に示す。イミド化が充分には進んでおらず、耐湿絶縁性の非常に悪いものになった。
合成例6で合成した化合物は、末端が無水物基のイミドシロキサンオリゴマーが、ジアミノ化合物と反応し、高粘度の溶液となった。評価の際には、使用可能な粘度範囲にまで1,2-ビス(2-メトキシエトキシ)エタンを添加して溶液の粘度を低下させた。評価結果を表5に示す。感光性が悪いものになった。
※2 新中村化学社製 ビスフェノールA EO変性ジアクリレート 分子量が1684
※3 チバ・スペシャルティーケミカルズ社製 光重合開始剤
Claims (6)
- 少なくとも、(A)分子内に少なくとも2つのイミド結合を含有し、重量平均分子量が1000以上15000以下であり、酸価が50〜150mgKOH/gである酸末端化合物及び(B)鎖延長剤を含むことを特徴とする、ポリイミド前駆体組成物。
- 前記(A)分子内に少なくとも2つのイミド結合を含有し、重量平均分子量が1000以上15000以下であり、酸価が50〜150mgKOH/gである酸末端化合物が、イミド化したテトラカルボン酸であり、(B)鎖延長剤が、ジアミノ化合物及び/又はイソシアネート系化合物であることを特徴とする請求項1記載のポリイミド前駆体組成物。
- 請求項1または2に記載のポリイミド前駆体組成物を40〜90重量%の溶質濃度に溶解して得られるポリイミド前駆体組成物溶液。
- 請求項1または2に記載のポリイミド前駆体組成物または請求項3に記載のポリイミド前駆体組成物溶液から得られるポリイミド塗膜。
- 請求項1または2に記載のポリイミド前駆体組成物または請求項3に記載のポリイミド前駆体組成物溶液をプリント配線板に塗工し、加熱してイミド化して得られるポリイミド塗膜付きプリント配線板。
- 請求項1または2に記載のポリイミド前駆体組成物と、少なくとも感光性樹脂、及び、光重合開始剤を含有することを特徴とする感光性樹脂組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008160323A JP5097025B2 (ja) | 2008-06-19 | 2008-06-19 | 新規なポリイミド前駆体組成物及びその利用 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008160323A JP5097025B2 (ja) | 2008-06-19 | 2008-06-19 | 新規なポリイミド前駆体組成物及びその利用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010001352A true JP2010001352A (ja) | 2010-01-07 |
| JP5097025B2 JP5097025B2 (ja) | 2012-12-12 |
Family
ID=41583335
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008160323A Expired - Fee Related JP5097025B2 (ja) | 2008-06-19 | 2008-06-19 | 新規なポリイミド前駆体組成物及びその利用 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5097025B2 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015132832A (ja) * | 2008-11-18 | 2015-07-23 | 住友化学株式会社 | 感光性樹脂組成物及び表示装置 |
| WO2018139558A1 (ja) * | 2017-01-27 | 2018-08-02 | 積水化学工業株式会社 | 硬化性樹脂組成物、接着剤、イミドオリゴマー、イミドオリゴマー組成物、及び、硬化剤 |
| US11421107B2 (en) | 2017-01-27 | 2022-08-23 | Sekisui Chemical Co., Ltd. | Curable resin composition, cured product, adhesive, bonding film, coverlay film, flexible copper-clad laminate and circuit board |
| JP2023552228A (ja) * | 2020-12-11 | 2023-12-14 | アスペン エアロゲルズ,インコーポレイティド | ポリイミドプロセスのための水共触媒 |
| US12139576B2 (en) | 2018-03-28 | 2024-11-12 | Sekisui Chemical Co., Ltd. | Curable resin composition, adhesive agent, adhesive film, circuit substrate, interlayer insulating material, and printed wiring board |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS56143240A (en) * | 1980-04-09 | 1981-11-07 | Nitto Electric Ind Co Ltd | Water-soluble composition |
| JPS61162525A (ja) * | 1985-01-04 | 1986-07-23 | ゼネラル エレクトリツク カンパニイ | 新しいコポリアミドイミドとその製造法及びそのプレポリマ−とその製造法 |
| JPH04114035A (ja) * | 1990-09-04 | 1992-04-15 | Nitto Denko Corp | 熱硬化性樹脂組成物 |
| WO1999006470A1 (en) * | 1997-07-30 | 1999-02-11 | Commonwealth Scientific And Industrial Research Organisation | Aqueous polyimide process |
| JP2001163974A (ja) * | 1999-12-09 | 2001-06-19 | Unitika Ltd | ポリイミド前駆体溶液及びその製造方法、それから得られるポリイミド塗膜並びにポリイミドシームレス管状フィルム及びその製造方法 |
| JP2005113014A (ja) * | 2003-10-08 | 2005-04-28 | Ube Ind Ltd | ポリシロキサン絶縁膜用組成物、絶縁膜、及び、絶縁膜の形成方法 |
| WO2008078620A1 (ja) * | 2006-12-26 | 2008-07-03 | Kaneka Corporation | 新規なポリイミド前駆体組成物、その利用及びそれらの製造方法 |
| WO2008132960A1 (ja) * | 2007-04-19 | 2008-11-06 | Kaneka Corporation | 新規なポリイミド前駆体組成物及びその利用 |
-
2008
- 2008-06-19 JP JP2008160323A patent/JP5097025B2/ja not_active Expired - Fee Related
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS56143240A (en) * | 1980-04-09 | 1981-11-07 | Nitto Electric Ind Co Ltd | Water-soluble composition |
| JPS61162525A (ja) * | 1985-01-04 | 1986-07-23 | ゼネラル エレクトリツク カンパニイ | 新しいコポリアミドイミドとその製造法及びそのプレポリマ−とその製造法 |
| JPH04114035A (ja) * | 1990-09-04 | 1992-04-15 | Nitto Denko Corp | 熱硬化性樹脂組成物 |
| WO1999006470A1 (en) * | 1997-07-30 | 1999-02-11 | Commonwealth Scientific And Industrial Research Organisation | Aqueous polyimide process |
| JP2001163974A (ja) * | 1999-12-09 | 2001-06-19 | Unitika Ltd | ポリイミド前駆体溶液及びその製造方法、それから得られるポリイミド塗膜並びにポリイミドシームレス管状フィルム及びその製造方法 |
| JP2005113014A (ja) * | 2003-10-08 | 2005-04-28 | Ube Ind Ltd | ポリシロキサン絶縁膜用組成物、絶縁膜、及び、絶縁膜の形成方法 |
| WO2008078620A1 (ja) * | 2006-12-26 | 2008-07-03 | Kaneka Corporation | 新規なポリイミド前駆体組成物、その利用及びそれらの製造方法 |
| WO2008132960A1 (ja) * | 2007-04-19 | 2008-11-06 | Kaneka Corporation | 新規なポリイミド前駆体組成物及びその利用 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015132832A (ja) * | 2008-11-18 | 2015-07-23 | 住友化学株式会社 | 感光性樹脂組成物及び表示装置 |
| WO2018139558A1 (ja) * | 2017-01-27 | 2018-08-02 | 積水化学工業株式会社 | 硬化性樹脂組成物、接着剤、イミドオリゴマー、イミドオリゴマー組成物、及び、硬化剤 |
| CN109415509A (zh) * | 2017-01-27 | 2019-03-01 | 积水化学工业株式会社 | 固化性树脂组合物、粘接剂、酰亚胺低聚物、酰亚胺低聚物组合物以及固化剂 |
| US11421107B2 (en) | 2017-01-27 | 2022-08-23 | Sekisui Chemical Co., Ltd. | Curable resin composition, cured product, adhesive, bonding film, coverlay film, flexible copper-clad laminate and circuit board |
| CN115197421A (zh) * | 2017-01-27 | 2022-10-18 | 积水化学工业株式会社 | 固化性树脂组合物、粘接剂、酰亚胺低聚物、酰亚胺低聚物组合物以及固化剂 |
| US11802177B2 (en) | 2017-01-27 | 2023-10-31 | Sekisui Chemical Co., Ltd. | Curable resin composition, adhesive, imide oligomer, imide oligomer composition, and curing agent |
| US12139576B2 (en) | 2018-03-28 | 2024-11-12 | Sekisui Chemical Co., Ltd. | Curable resin composition, adhesive agent, adhesive film, circuit substrate, interlayer insulating material, and printed wiring board |
| JP2023552228A (ja) * | 2020-12-11 | 2023-12-14 | アスペン エアロゲルズ,インコーポレイティド | ポリイミドプロセスのための水共触媒 |
| JP7522321B2 (ja) | 2020-12-11 | 2024-07-24 | アスペン エアロゲルズ,インコーポレイティド | ポリイミドプロセスのための水共触媒 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5097025B2 (ja) | 2012-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5469062B2 (ja) | 新規なポリイミド前駆体組成物、その利用及びそれらの製造方法 | |
| JP5506017B2 (ja) | 新規なポリイミド前駆体組成物、その利用及びそれらの製造方法 | |
| JP5895024B2 (ja) | 新規なポリイミド前駆体組成物及びその利用 | |
| JP5735275B2 (ja) | 新規な樹脂組成物及びその利用 | |
| JP5642961B2 (ja) | 新規なポリイミド前駆体組成物及びその利用 | |
| JP5049175B2 (ja) | 新規な感光性樹脂組成物、それから得られる感光性樹脂組成物溶液、感光性フィルム、絶縁膜及び絶縁膜付きプリント配線板 | |
| JP2009271445A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP2008261921A (ja) | 新規な感光性樹脂組成物、それから得られる硬化膜、絶縁膜及び絶縁膜付きプリント配線板 | |
| JP2009300873A (ja) | 新規な回路基板の製造方法 | |
| JP2009300872A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP5097025B2 (ja) | 新規なポリイミド前駆体組成物及びその利用 | |
| JP2009282172A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP2011084653A (ja) | 新規なポリイミド前駆体組成物及びその利用 | |
| JP2011126922A (ja) | 新規な樹脂組成物及びその利用 | |
| JP2011059340A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP2009288517A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP2009280661A (ja) | 新規なポリイミド前駆体組成物、その利用及びそれらの製造方法 | |
| JP2010002717A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP2010006868A (ja) | 新規な硬化膜及びその利用 | |
| JP2008197545A (ja) | 絶縁被膜を有するフレキシブルプリント配線板 | |
| JP2008261920A (ja) | 新規な感光性樹脂組成物、それから得られる硬化膜、絶縁膜及び絶縁膜付きプリント配線板 | |
| JP2009288518A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP2010001351A (ja) | 新規なポリイミド前駆体組成物、その利用及びそれらの製造方法 | |
| JP2009294252A (ja) | 新規な感光性樹脂組成物及びその利用 | |
| JP2011116848A (ja) | 新規なポリイミド前駆体組成物及びその利用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110420 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120528 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120605 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120803 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20120803 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120904 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120921 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5097025 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150928 Year of fee payment: 3 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150928 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |