JP2009535344A - 神経炎症性疾患の処置のためのピリダジン化合物を含む処方物 - Google Patents
神経炎症性疾患の処置のためのピリダジン化合物を含む処方物 Download PDFInfo
- Publication number
- JP2009535344A JP2009535344A JP2009507823A JP2009507823A JP2009535344A JP 2009535344 A JP2009535344 A JP 2009535344A JP 2009507823 A JP2009507823 A JP 2009507823A JP 2009507823 A JP2009507823 A JP 2009507823A JP 2009535344 A JP2009535344 A JP 2009535344A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- alkyl
- aryl
- formula
- iii
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 Cc(c(/C1=*\C*/*=C(\CC(C(*I)C(*)C(C*)C2N)C2O)/C(/*)=C1\*)c1*)c(*)c(C)c1I Chemical compound Cc(c(/C1=*\C*/*=C(\CC(C(*I)C(*)C(C*)C2N)C2O)/C(/*)=C1\*)c1*)c(*)c(C)c1I 0.000 description 9
- SGNDKEUAZAKOHQ-IRDSKBQXSA-N CC(C[C@H](CC(CC1)=CCC(C2)C2N1C1C=CCC1)N1CCCCC1)C1=CCCC=C1 Chemical compound CC(C[C@H](CC(CC1)=CCC(C2)C2N1C1C=CCC1)N1CCCCC1)C1=CCCC=C1 SGNDKEUAZAKOHQ-IRDSKBQXSA-N 0.000 description 1
- VLHTWZCWNAMULL-UHFFFAOYSA-N CC1C2[N](C)(C)C12 Chemical compound CC1C2[N](C)(C)C12 VLHTWZCWNAMULL-UHFFFAOYSA-N 0.000 description 1
Images

































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/20—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Pain & Pain Management (AREA)
- Immunology (AREA)
- Endocrinology (AREA)
- Dermatology (AREA)
- Ophthalmology & Optometry (AREA)
- Psychology (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Otolaryngology (AREA)
- Pulmonology (AREA)
- Hospice & Palliative Care (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Emergency Medicine (AREA)
Abstract
Description
本発明は、化合物、組成物ならびにその作製方法および使用方法に関する。特に、本発明は、選択されたピリダジン化合物、該化合物を含む組成物、ならびに細胞経路のモジュレーションのため、炎症性疾患の処置もしくは予防のため、神経系の病状(neurological conditions)の処置もしくは予防のため、研究、薬物スクリーニングおよび治療適用のための該化合物および組成物の使用方法を提供する。
神経系の病状および障害の処置は医学において非常に重要であり、このような病状および障害の進行を抑制および逆転させるための新規な薬物および処置の必要性が存在する。神経炎症は、多くの神経系の病状および疾患の病態において顕著な特徴として認識される。神経炎症は、主に、グリア(小グリア細胞および星状細胞)の異常に高いまたは慢性的な活性化に起因する過程である。このグリア過剰活性状態により炎症性および酸化的ストレス分子のレベル上昇がもたらされ、それにより、ニューロンの損傷または死滅がもたらされ得る。また、ニューロンの損傷または死滅によりグリアの活性化が誘導され得、限局性の有害な神経炎症サイクルの伝播が促進され得る(非特許文献1)。該炎症サイクルは、炎症性疾患を処置するための新規なアプローチの開発における潜在的な治療標的として提案されている。しかしながら、これまでに開発されたほとんどの抗炎症治療剤は、待機的であり、もたらされる症状の軽減は最小限で短期間であり、炎症性疾患の進行に対する効果は限定的である。したがって、疾患の進行または予防に影響を与える抗炎症治療剤の必要性が存在する。
Griffin、WSTら、Brain Pathol 8:65−72、1998
本発明は、ある種のピリダジン化合物、該化合物を含む組成物、ならびに細胞経路(例えば、シグナル伝達経路)のモジュレーションのため、炎症性疾患の処置もしくは予防のため、神経系の疾患および病状の処置または予防のため、研究、薬物スクリーニングおよび治療適用のための該化合物および組成物の使用方法を提供する。特に、本発明は、一般的に、特に神経炎症性疾患において、副作用のリスクの低下をもたらすため、および/または有益な薬物動態プロフィールをもたらすための投薬形態、製剤および方法を提供する。

の化合物、その異性体、薬学的に許容され得る塩または誘導体を含む、処置後の副作用のリスクの低下および/または有益な薬物動態プロフィールをもたらすのに有効な組成物、特に製剤または投薬形態が想定される。
の化合物またはその異性体、薬学的に許容され得る塩もしくは誘導体を含む、処置後の副作用のリスクの低下および/または有益な薬物動態プロフィールをもたらすのに有効な組成物、製剤または投薬形態が提供される。
である。
の化合物を含む、処置後の副作用のリスクの低下および/または有益な薬物動態プロフィールをもたらすのに有効な組成物、特に製剤または投薬形態が想定される。
便宜上、本明細書、実施例および添付の特許請求の範囲で用いる一部の用語をここに集める。
n)神経炎症性疾患(例えば、アルツハイマー病)の症状を有する被験体の生存の増加。
アルブミン、抗トリプシン、マクログロブリン、ハプトグロビン、セルロプラスミン(caeruloplasm)、トランスフェリン(transferring)、α−もしくはβ−リポタンパク質、β−もしくはγ−グロブリンまたはフィブリノゲンなどであってもよい。
本明細書で用いる場合、用語「シアノ」は、窒素原子によって共有される4つの共有結合のうち3つを有する炭素残基、特に、−C≡Nをいう。シアノ基は、本明細書に記載の置換基で置換されたものであってもよい。
で表される基が挙げられる。
で表され得る基が挙げられる。
カルボニル基の2つの非共有結合の一方に結合されたアミノ、モノアルキルアミノ、ジアルキルアミノ、モノシクロアルキルアミノ、アルキルシクロアルキルアミノおよびジシクロアルキルアミノ残基をいう。
本発明では、式中、R1、R4、R5、R6、R7、R8、R9、R12、R13およびR14が、独立して、水素、ヒドロキシル、アルキル、アルケニル、アルキニル、アルキレン、アルケニレン、アルコキシ、アルケニルオキシ、シクロアルキル、シクロアルケニル、シクロアルキニル、シクロアルコキシ、アリール、アリールオキシ、アリールアルコキシ、アロイル、ヘテロアリール、複素環式基、アシル、アシルオキシ、アミノ、イミノ、アジド、チオール、チオアルキル、チオアルコキシ、チオアリール、ニトロ、シアノ、ハロ、硫酸基、スルフェニル、スルフィニル、スルホニル、スルホン酸基、スルホキシド、シリル、シリルオキシ、シリルアルキル、シリルチオ、=O、=S、ホスホン酸基、ウレイド、カルボキシル、カルボニル、カルバモイル、またはカルボキサミドであり、Xが、任意に置換されたピリミジニルまたはピリダジニルである、単離された純粋な、特に実質的に純粋な式Iの化合物、その異性体、薬学的に許容され得る塩または誘導体の使用が想定される。
である。
本発明のある態様において、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、R16およびR17は、独立して、水素、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C1〜C6アルコキシ、C2〜C6アルケニルオキシ、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C3〜C10シクロアルコキシ、C6〜C10アリール、C6〜C10アリールオキシ、C6〜C10アリール−C1〜C3アルコキシ、C6〜C10アロイル、C6〜C10ヘテロアリール、C3〜C10複素環式基、C1〜C6アシル、C1〜C6アシルオキシ、−NH2、−NHR28、−NR28R29、=NR28、−S(O)2R28、−SH、−SO3H、ニトロ、シアノ、ハロ、ハロアルキル、ハロアルコキシ、ヒドロキシアルキル、−CO2H、−CO2R28、−NHC(O)R28、−C(O)NH2、−C(O)NHR28、−C(O)NR28R29、−NHS(O)2R28から選択され、ここで、R28およびR29は、独立して、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C6〜C10アリール、C6〜C10アリールC1〜C3アルキル、C6〜C10ヘテロアリールおよびC3〜C10複素環式基から選択される。
一態様において、本発明は、R11が水素であり、R10が、1〜4個の窒素原子を含有する5〜6員の不飽和ヘテロモノシクリル基、特に、ピロリル、ピロリニル、イミダゾリル、ピラゾリル、2−ピリジル、3−ピリジル、4−ピリジル、ピリジニル、ピリミジニル、ピラジニル、ピリダジニル、トリアゾリル、またはテトラゾリル、より特別にはピリジニルである式IIIの化合物の調製方法であって、R10がハロ、特にクロロであり、R11が水素である式IIIの化合物を、1〜4個の窒素原子を含有する5〜6員の不飽和ヘテロモノシクリル基、特に、ピロリル、ピロリニル、イミダゾリル、ピラゾリル、2−ピリジル、3−ピリジル、4−ピリジル、ピリジニル、ピリミジニル、ピラジニル、ピリダジニル、トリアゾリル、またはテトラゾリル(より特別には、ピリジニル)で置換されたボロン酸と、R11が水素であり、R10が、1〜4個の窒素原子を含有する5〜6員の不飽和ヘテロモノシクリル基、特に、ピロリル、ピロリニル、イミダゾリル、ピラゾリル、2−ピリジル、3−ピリジル、4−ピリジル、ピリジニル、ピリミジニル、ピラジニル、ピリダジニル、トリアゾリル、またはテトラゾリル、より特別にはピリジニルである式IIIの化合物が調製されるのに適当な条件下で反応させることを含む方法を提供する。
本発明は、利点をもたらす投薬形態、製剤および方法、特に、副作用のリスクの低下(例えば、QT関連副作用のリスクの低下)および/または有益な薬物動態プロフィール、より詳しくは、徐放性薬物動態プロフィールをもたらす投薬形態、製剤および方法を提供する。式I、IIまたはIIIの化合物は、純粋または実質的に純粋な形態、その薬学的に許容され得る塩の形態、また、無水形態または水和物形態などの他の形態の投薬形態で利用され得る。
ピリダジン化合物の合成
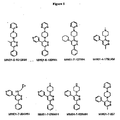
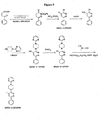
MW01−2−151SRM、MW01−6−189WH、MW01−7−107WH、MW01−4−179LKM、WM01−7−084WH、MH01−7−085WH、MW01−7−133WH、およびMW01−7−057の構造を図1に示し、該化合物作製のための合成スキームを以下に記載する。
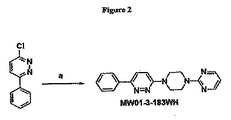
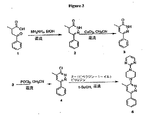
4−メチル−6−フェニル−3−(4−ピリミジン−2−イルピペラジン−1−イル)ピリダジン(MW01−2−151SRM)を、図3(スキーム1)、図4(スキーム2)、図5(スキーム3)および図6(スキーム4)に示すいくつかの合成スキームによって調製し、本明細書に詳細に記載しているようにして行なった。種々の反応スキーム(スキーム1、2および3)が本発明の化合物に一般的に適用可能であり、使用はMW01−2−151SRMの調製のみに限定されない。
4,5−ジヒドロ−4−メチル−6−フェニルピリダジン−3(2H)−オン(2)
温度プローブおよび冷却器を取り付けた250mL容の三ッ口丸底フラスコに、7.7g(40mmol)の2−メチル−4−オキソ−4−フェニルブタン酸1および20mlのエタノール(95%)を仕込む。懸濁液を10℃未満まで冷却し、2.2ml(42mmol、1.05当量)のヒドラジン一水和物を含有する10mLのエタノールを、溶液が20℃未満の温度に維持される速度で滴下する。添加すると、懸濁液は、薄黄色溶液に変化する。添加後、反応混合物を還流加熱し、2時間攪拌し、20分間の加熱後、固形物が混合物中に見られる。反応が修了したら、フラスコを油浴から取り出し、周囲温度まで冷却させる。冷却すると、フラスコ内で白色結晶が形成され、これを、濾過によって回収する。固形物を、最初に30mLの2N NaHCO3で洗浄した後、60mLのMilli−Q水で3回洗浄し、中(medium)フリット焼結ガラス漏斗上で真空乾燥させると、所望の生成物2が96.1%収率で得られた[Hansen,KBら Organic process research & development,2005,9,634−639;Nelson,DA.US 20050137397A1.Coudert,Pら Journal of Heterocyclic Chemistry,1988,25(3)、799−802を参照のこと]。
7.0g(35mmol)の2を250ml容一ッ口丸底フラスコに入れた後、30mLのアセトニトリルを入れる。混合物を攪拌して2を溶解させる。11.3g(84mmol、2.4当量)の無水塩化銅(II)をこの溶液に添加すると、黄緑色の懸濁液が得られる。還流冷却器をフラスコに接続し、無水CaCl2を充填した乾燥チューブを冷却器の上部に取り付ける。反応過程中に形成されるHClガスを制御するため、NaOH溶液を用いて、乾燥チューブから放出されるHClを吸収させる。反応混合物を還流加熱し、加熱すると、反応懸濁液の色が暗緑色に変化する。反応が終了したとき(2時間還流後)、フラスコを油浴から取り出し、周囲温度まで冷却させる。反応物は氷水浴中で冷却させ、150mLの氷水を添加して反応をクエンチする。混合物を10分間激しく攪拌すると、灰色沈殿物と、塩化銅(I)を含有する青色の液体とが得られる。沈殿物を濾過によって回収し(濾液のpHは0〜1である)、100mLの1N HCl溶液で洗浄し、次いで、100mLの水で5回洗浄する。固形物内に取り込まれた残留銅副生成物を除去するため、濾過ケークを150mLの1N HCl溶液中で0.5時間攪拌し、濾過する。続いて、濾過ケークを、濾液がpH7になるまでMilli−Q水で洗浄する(だいたい7回洗浄)。固形物を中フリット焼結ガラス漏斗上で真空乾燥させると、3が淡灰色粉末として93.8%収率で得られる[Eddy,Sら Synthetic Communications,2000,30(1)、l−7.Csende,Fら Synthesis,1995,1240−1242を参照のこと]。
6.0g(32mmol)の3を250mL容一ッ口丸底フラスコに入れ、30mLのアセトニトリルを添加して、薄黄色スラリーを生成させる。6.0ml(64mmol、2当量)のオキシ塩化リンを添加すると、スラリーがより暗色に変化する。フラスコに還流冷却器を取り付け、無水CaCl2を充填した乾燥チューブを冷却器の上部に取り付ける。反応混合物を還流加熱すると、暗赤色液体となる。反応終了後(2.5時間)、混合物を周囲温度まで冷却させ、氷水浴中に入れる。氷水(150mL)を、攪拌下の反応混合物中にゆっくりと注入してオキシ塩化リンをHClとH3PO4に分解させると、ピンク色の固形物の形成がもたらされる。固形物を濾過によって回収し、50mLのMilli−Q水で3回洗浄する。固形物を250mL容ビーカーに移した後、100mLの水を添加して懸濁液を形成する。続いて、この水性懸濁液がpH=8になるまで1N NaOH添加し、混合物を5分間攪拌してあらゆる微量の出発材料混入物質を除去する。固形物を濾過し、100mLの水で3回洗浄して過剰の塩基を洗い流す。固形物を中フリット焼結ガラス漏斗上で真空乾燥させると、4が淡いピンク色粉末として96%収率で得られる[Contreras,JMら Journal of Medicinal Chemistry,2001,44(17)、2707−2718;Nelson,DA.US20050137397A1を参照のこと]。
7.5g(36.6mmol)の4を250mL容一ッ口丸底フラスコに入れ、125mLの水中に懸濁させる。60.17g(366.0mmol、10当量)の2−(ピペラジン−1−イル)ピリミジンを添加し、フラスコに冷却器を取り付ける。反応混合物を高速攪拌下で60時間還流加熱し、連続的にアミンを添加すると、反応速度を促進することが可能である。終了したら、反応混合物を周囲温度まで冷却させ、橙色水層およびフラスコ底面に沈降した褐色油状物からなる2層がフラスコ内に観察される。水をデカンテーションすると、油状物が残り、これは生成物5である。次いで、油状物を最少容量のイソプロパノールに溶解し、還流加熱する。10分間の還流後、溶液を周囲温度まで冷却し、0℃まで冷却して結晶化を誘導する。薄黄色結晶をイソプロパノールから濾過し、最少の冷エーテルでリンス処理し、5を得る。結晶の回収は50%であるが、化合物の反復結晶化により増加させ得る[Contreras,JMら Journal of Medicinal Chemistry,1999,42(4)、730−741.Chayer,Sら Tetrahedron Letters,1998,39,841−844]。
3−クロロ−6−フェニルピリダジン−4−オールを、Coudert,P.ら(前掲)に記載の手順に従って合成した。
この化合物を、3−クロロ−4−ヒドロキシ−6−フェニルピリダジン(14g、68mmol)から、以下に記載のようにして調製し、白色固形物(22.1g、66mmol、97.3%)を得た。ESI−MS:m/z 335.2(M+H+)。
6−フェニル−3−(4−ピリミジン−2−イルピペラジン−1−イル)ピリダジン−4−オール(22.0g、66mmol)を75mlのオキシ塩化リン中に懸濁し、攪拌しながら100℃で3時間加熱した。室温まで冷却後、混合物を粉砕氷上に注入した。次いで、混合物をNaOH溶液で中和し、白色懸濁液を得た。沈殿を濾別し、水で洗浄し、濾過漏斗上で乾燥させると、白色固形物が得られた(21.3g、60.3mmol、91.4%)。ESI−MS:m/z 353.4(M+H+)。
反応チューブ内に、MW01−6−127WH(1.4g、4.0mmol)、K2CO3粉末(1.7g、12.4mmol)、Pd(dppf)Cl2(326mg、0.4mmol)、酸化銀(2.3g、10mmol)、メチルボロン酸(324mg、5.4mmol)および20mlのTHFを添加した。次いで、チューブ内をアルゴンで3分間フラッシュ洗浄した。次いで、チューブをしっかりと密封し、攪拌しながら80度で12時間加熱した。冷却後、混合物を10%NaOH溶液でクエンチし、酢酸エチルで抽出した。有機相を真空濃縮し、残渣をカラムクロマトグラフィーによって精製した(1:4, 酢酸エチル:石油エーテルで溶出)。白色粉末固形物が得られた(0.60g、1.8mmol、収率45.2%)。ESI−MS:m/z 333.4(M+H+)。
反応チューブ内に、MW01−6−127WH(1.4g、4.0mmol)、K2CO3粉末(1.7g、12.4mmol)、Pd(PPh3)4(240mg、0.2mmol)、酸化銀(2.3g、10mmol)、メチルボロン酸(324mg、5.4mmol)および20mlのDMEを添加した。次いで、チューブ内をアルゴンで3分間フラッシュ洗浄した。次いで、チューブをしっかりと密封し、攪拌しながら120℃で24時間加熱した。冷却後、混合物をアセライト土(acelite earth)に通して濾過(was filter)し、次いで、濾液を濃縮し、残渣をカラムクロマトグラフィーによって精製した(1:4, 酢酸エチル:石油エーテルで溶出)。白色粉末固形物が得られた(0.64g、1.93mmol、収率48.1%)。ESI−MS:m/z 333.4(M+H+)。
4.5−ジヒドロ−4−メチル−6−フェニルピリダジン−3(2H)−オン(MW01−8−004WH)
7.7g(40mmol)の2−メチル−4−オキソ−4−フェニルブタン酸を、100ml容一ッ口丸底フラスコに添加した後、3.0ml(60mmol)のヒドラジン一水和物、次いで、20mlの試薬等級エタノール(100%、95%のエタノールでもよい)を添加した。フラスコに還流冷却器を取り付け、反応混合物を、110℃(油浴の温度)の油浴中で還流加熱し、2時間攪拌した。次いで、フラスコを油浴から取り出し、反応混合物周囲温度まで冷却させた。攪拌バーを取りだし、溶媒を45℃の水浴中で真空蒸発させた。次いで、残渣を50mlのMilli−Q水で処理し、10分間攪拌して懸濁液を得た。沈殿物を濾過によって回収し、100mlの2N NaHCO3で洗浄し、次いで、60mlのMilli−Q水で3回洗浄し、中フリット焼結ガラス漏斗上で真空乾燥させると、7.15gの白色結晶が得られた(Syn.ID,WH−8−004)。収率95%(ESI−MSによって確認)、ESI−MS:m/z 189.2(M+H+)。
7.0g(35mmol)のMW01−8−004WHを、100ml容の一ッ口丸底フラスコ内に入れた後、9.4g(70mmol)の無水塩化銅(II)、次いで、30mlのアセトニトリルを入れ、黄褐色懸濁液を得た。還流冷却器をフラスコに接続し、CaCl2を充填した乾燥チューブを冷却器の上部に取り付けた。反応混合物を油浴(110℃)中で3時間還流加熱した。還流を開始すると、反応懸濁液の色が暗黄色に変化した。反応終了後(HPLCによってモニター)、フラスコを油浴から取り出し、周囲温度まで冷却させた。混合物を、300gの粉砕氷上に注入し、10分間激しく攪拌すると、灰色沈殿物および青色の液体が得られた。次いで、沈殿物を濾過によって回収し(濾液のpHは1.5〜2.0であった)、100mlの1N HCl溶液で洗浄し、残留(あれば)銅副生成物である固形物を除去した。この後、100mlのMilli−Q水で洗浄して固形物中の酸を除去し、濾液のpH値を確認することによりモニターする。固形物を、濾液がpH7を示すまで洗浄した(だいたい5回洗浄後)。固形物を、中フリット焼結ガラス漏斗上で真空乾燥させると、6.3gの青灰色固形物が得られた。収率は96.7%であった(ESI−MSによって確認)。ESI−MS:m/z 187.3(M+H+)。
6.0g(32mmol)のMW01−8−008WHおよび30ml(320mmol)のオキシ塩化リンを100ml容の一ッ口丸底フラスコに入れた。フラスコを還流冷却器に接続し、無水CaCl2を充填した乾燥チューブを冷却器の上部に取り付けた(反応中にHClガスが形成されるため、大規模合成ではHClを吸収させるために、NaOHなどの塩基性溶液が必要とされ得る)。反応混合物を油浴(90℃)中で2時間攪拌し、次いで、周囲温度まで冷却し、粉砕氷上に注入した(オキシ塩化リンは水によって分解され、HClおよびH3PO4が生成され得る)。次いで、混合物を10分間激しく攪拌し、白色懸濁液を得た。懸濁液を2N NaOH溶液で、懸濁液のpHがpH=7になるまで中和した。沈殿物を濾過し、100mlのMilli−Q水で3回洗浄し、中フリット焼結ガラス漏斗上で真空乾燥させると、5.9gの淡いピンク色粉末が得られた(Syn.ID,WH−8−012)。収率は89.4%であった(ESI−MSによって確認)。ESI−MS:m/z 205.4(M+H+)。
0.82g(4.0mmol)のWH−8−012を30ml容の加圧容器内に入れた後、2.6g(16.0mmol)の1−(2−ピリミジル)ピペラジンを添加し、次いで、15mlの1−BuOHを添加した。容器をしっかりと密封し、油浴中に入れ、130℃(油浴の温度)で2.5日間攪拌した。次いで、反応混合物を周囲温度まで冷却し、一ッ口フラスコに移し、減圧下でエバポレーションした。溶媒の除去により赤褐色の残渣が得られ、これを30mlの水で処理すると、褐色の粘性の油状物が得られた。混合物を一晩周囲温度に維持し、その間、油状物は徐々に固化した。次いで、形成された固形物をスチール製のスパチュラで小片に粉砕した。固形物を濾過によって回収し、50mlのMilli−Q水で3回洗浄し、濾過漏斗上で真空乾燥させると、1.25gの淡黄色固形物が得られた(Syn.ID,WH−8−020)。収率は94%であった(択一的な分離は、固化プロセスの代わりに沈殿手順を使用することである。固化は、簡単で廉価な操作であるが、時間がかかる。沈殿は、時間効率はよいが、前者よりも高価である。そのため、どの手順を製造に採用するかの判断は、プロセス化学者次第である。沈殿プロセスは以下のとおりである。油状生成物を10mlの試薬等級エタノールまたはアセトンに完全に溶解させ、溶液を形成した。次いで、この溶液を、激しく攪拌しながら150mlの氷水に滴下した。すると、淡黄色懸濁液が徐々に形成された。固形物を濾過によって回収し、Milli−Q水で洗浄し、濾過漏斗上で真空乾燥させると、所望の生成物が得られた)。最終化合物は、ESI−MSおよびNMRによって確認した。ESI−MS:m/z 333.8(M+H+)。
3−クロロ−6−フェニルピリダジン−4−オールを、Coudert,P.ら(前掲)に記載の手順に従って合成した。
該化合物を、3−クロロ−4−ヒドロキシ−6−フェニルピリダジン(14g、68mmol)から調製した。3−クロロ−4,6−ジフェニルピリダジン(267mg、1.0mmol)、1−(2−ピリミジル)ピペラジン(656mg、4.0mmol)の混合物を含む3mlの1−BuOHを、攪拌しながら130℃で3日間加熱した。溶媒をエバポレーションによって真空除去し、残渣を水で処理して懸濁液を得た。次いで、固形物を濾別し、水で洗浄し、濾過漏斗上で真空乾燥させると、淡いピンク色の固形物が得られた。白色固形物を得た(22.1g、66mmol、97.3%)。ESI−MS:m/z 335.2(M+H+)。
6−フェニル−3−(4−ピリミジン−2−イルピペラジン−1−イル)ピリダジン−4−オール(22.0g、66mmol)を75mlのオキシ塩化リンに懸濁し、攪拌しながら100℃で3時間加熱した。室温まで冷却後、混合物を粉砕氷上に注入した。次いで、混合物をNaOH溶液で中和し、白色懸濁液を得た。沈殿を濾別し、水で洗浄し、濾過漏斗上で乾燥させると、白色固形物が得られた(21.3g、60.3mmol、91.4%)。ESI−MS:m/z 353.4(M+H+)。
3−クロロ−4,6−ジフェニルピリダジン(267mg、1.0mmol)、1−(2−ピリミジル)ピペラジン(656mg、4.0mmol)の混合物を含む3mlの1−BuOHを、攪拌しながら130℃で3日間加熱した。溶媒をエバポレーションによって真空除去し、残渣を水で処理して懸濁液を得た。次いで、固形物を濾別し、水で洗浄し、濾過漏斗上で真空乾燥させると、淡いピンク色の固形物が得られた(320mg、O.81mmol、収率81.1%)。ESI−MS:m/z 395.5(M+H+)。HRMS計算値395.1979、実測値395.1973;
4,5−ジヒドロ−6−フェニル−4−フェニルピリダジン−3(2H)−オン
135ml(135mmol)の臭化フェニルマグネシウム(1M)のTHF溶液を、6−フェニルピリダジノン化合物7.8g(45mmol)を含む乾燥トルエン(50ml)の高温懸濁液に添加した。混合物を8時間還流し、周囲温度で一晩放置し、次いで、塩化アンモニウムの飽和溶液で分解した。有機層を分離し、水層を100mlの酢酸エチルで抽出した。溶媒を除去し、残渣をエタノールから結晶化させた。結晶を濾過によって回収し、中フリット焼結ガラス漏斗上で真空乾燥させると、5.6gの白色結晶が得られた。収率は50%であった(ESI−MSによって確認)。ESI−MS:m/z 250.1(M+H+)。
上記で得られた4.4g(17.5mmol)の6−ピリダジノンを、50ml容の一ッ口丸底フラスコ内に入れた後、4.7g(35mmol)の無水塩化銅(II)を入れ、次いで20mlのアセトニトリルを入れ、黄褐色懸濁液を得た。還流冷却器をフラスコに接続し、CaCl2を充填した乾燥チューブを冷却器の上部に取り付けた。反応混合物を油浴(110℃)中で3時間還流加熱した。還流を開始すると、反応懸濁液の色が暗黄色に変化した。反応終了後(HPLCによってモニター)、フラスコを油浴から取り出し、周囲温度まで冷却させた。混合物を200gの粉砕氷上に注入し、10分間激しく攪拌すると、灰色沈殿物および青色の液体が得られた。次いで、沈殿物を濾過によって回収し(濾液のpHは1.5〜2.0であった)、50mlの1N HCl溶液で洗浄して残留(あれば)銅副生成物である固形物を除去した。この後、100mlのMilli−Q水で洗浄して固形物中の酸を除去し、濾液のpH値を確認することによりモニターする。固形物を、濾液がpH7を示すまで洗浄した(だいたい5回洗浄後)。固形物を、中フリット焼結ガラス漏斗上で真空乾燥させると、3.9gの青灰色固形物が得られた。収率は90%であった(ESI−MSによって確認)。ESI−MS:m/z 248.1(M+H+)。
上記で得られた2.0g(8mmol)の6−フェニルピリダジノンおよび10ml(54mmol)のオキシ塩化リン(試薬等級、Aldrich)を50ml容の一ッ口丸底フラスコに入れた。フラスコを還流冷却器に接続し、CaCl2を充填した乾燥チューブを冷却器の上部に取り付けた(反応中にHClガスが形成されるため、大規模合成ではHClを吸収させるために、NaOHなどの塩基性溶液が必要とされ得る)。反応混合物を油浴(90℃)中で2時間攪拌し、次いで、周囲温度まで冷却し、粉砕氷上に注入した(オキシ塩化リンは水によって分解され、HClおよびH3PO4が生成され得る)。次いで、混合物を10分間激しく攪拌し、白色懸濁液を得た。懸濁液を2N NaOH溶液で、懸濁液のpHがpH=7になるまで中和した。沈殿物を濾過し、100mlの水で3回洗浄し、中フリット焼結ガラス漏斗上で真空乾燥させると、1.8gの淡いピンク色粉末が得られた。収率は85%であった(ESI−MSによって確認)。ESI−MS:m/z 266.4(M+H+)。
上記で得られた1.1g(4.0mmol)の3−クロロピリダジンを、30ml容の加圧容器内に入れた後、2.6g(16.0mmol)の1−(2−ピリミジル)ピペラジンを添加し、次いで、15mlの1−BuOH(試薬等級)を添加した。容器をしっかりと密封し、油浴中に入れ、130℃(油浴の温度)で3日間攪拌した。次いで、反応混合物を周囲温度まで冷却し、一ッ口フラスコに移し、減圧下でエバポレーションした。溶媒の除去により赤褐色の残渣が得られ、これを30mlの水で処理し、褐色の懸濁液を得た。固形物を濾過によって回収し、50mLの水で3回洗浄し、濾過漏斗上で真空乾燥させると、0.96gの淡黄色固形物が得られた。収率は90%であった。ESI−MS:m/z 395.5(M+H+)。HRMS計算値395.1979、実測値395.1973;
3−クロロ−6−フェニルピリダジン−4−オールを、Coudert, P.ら、(前掲)に記載の手順に従って合成した。
3−クロロ−4,6−ジフェニルピリダジン(267mg、1.0mmol)、1−(2−ピリミジル)ピペラジン(656mg、4.0mmol)の混合物を含む3mlの1−BuOHを、攪拌しながら130℃で3日間加熱した。溶媒をエバポレーションによって真空除去し、残渣を水で処理して懸濁液を得た。次いで、固形物を濾別し、水で洗浄し、濾過漏斗上で真空乾燥させると、淡いピンク色の固形物が得られた(320mg、0.81mmol、収率81.1%)。ESI−MS:m/z 395.5(M+H+)。HRMS計算値395.1979、実測値395.1973;
4−ピリジル−6−フェニル3−(4−ピリミジン−2−イルピペラジン−1−イル)ピリダジン(MW01−6−189WH)を、図10Aおよび10Bに示す2つの合成スキームによって調製し、本明細書に詳細に記載しているようにして行なった。種々の反応スキーム(スキーム1および2)が本発明の化合物に一般的に適用可能であり、使用はMW01−2−189WHの調製のみに限定されない。
3−クロロ−6−フェニルピリダジン−4−オールを、Coudert,P.ら(前掲)に記載の手順に従って合成した。
この化合物を、3−クロロ−4−ヒドロキシ−6−フェニルピリダジン(14g、68mmol)から調製した。3−クロロ−4,6−ジフェニルピリダジン(267mg、1.0mmol)、1−(2−ピリミジル)ピペラジン(656mg、4.0mmol)の混合物を含む3mlの1−BuOHを、攪拌しながら130℃で3日間加熱した。溶媒をエバポレーションによって真空除去し、残渣を水で処理して懸濁液を得た。次いで、固形物を濾別し、水で洗浄し、濾過漏斗上で真空乾燥させると、淡いピンク色の固形物が得られた。白色固形物を得た(22.1g、66mmol、97.3%)。ESI−MS:m/z 335.2(M+H+)。
6−フェニル−3−(4−ピリミジン−2−イルピペラジン−1−イル)ピリダジン−4−オール1h(22.0g、66mmol)を、75mlのオキシ塩化リンに懸濁させ、攪拌しながら100℃で3時間加熱した。室温まで冷却後、混合物を粉砕氷上に注入した。次いで、混合物をNaOH溶液で中和し、白色懸濁液を得た。沈殿を濾別し、水で洗浄し、濾過漏斗上で乾燥させると、白色固形物が得られた(21.3g、60.3mmol、91.4%)。ESI−MS:m/z 353.4(M+H+)。
反応チューブ内に、WH−6−127(1.4g、4.0mmol)、K2CO3粉末(1.7g、12.4mmol)、Pd(PPh3)4(240mg、0.2mmol)、4−ピリジンボロン酸(664mg、5.4mmol)および20mlのDMEを添加した。次いで、チューブ内をアルゴンで3分間フラッシュ洗浄した。次いで、チューブをしっかりと密封し、攪拌しながら120度で24時間加熱した。冷却後、混合物をセライト土に通して濾過し、次いで、濾液を濃縮し、残渣をカラムクロマトグラフィーによって精製した(1:4,酢酸エチル:石油エーテルで溶出)。淡黄色針状結晶が得られた(0.65g、1.65mmol、収率41.2%)。ESI−MSおよびNMRによって確認。ESI−MS:m/z 396.2(M+H+)。
4,5−ジヒドロ−6−フェニル−4−(ピリジン−4−イル)ピリダジン−3(2H)−オン
磁気攪拌バー、150ml容の均圧滴下漏斗、還流冷却器およびガラス栓を備えた200ml容の三ッ口丸底フラスコに、21g(135mmol)の4−ブロモピリジンおよび70の無水THFを添加した。使用前に、系をオーブン乾燥させ、アルゴンでフラッシュ洗浄した。臭化フェニルマグネシウム(1M)の135ml(135mmol)のTHF溶液を均圧滴下漏斗に入れた。次いで、グリニャール溶液を10分間にわたって滴下した。滴下後、15分間攪拌して反応を終了させた。これにより、グリニャール試薬の溶液が得られた。上記で得られた臭化4−ピリジルマグネシウムの溶液を、6−フェニルピリダジノン化合物7.8g(45mmol)の乾燥トルエン(50ml)高温懸濁液に添加した。混合物を8時間還流し、周囲温度で一晩放置し、次いで、塩化アンモニウムの飽和溶液で分解した。有機層を分離し、水層を100mlの酢酸エチルで抽出した。溶媒を除去し、残渣をエタノールから結晶化させた。結晶を濾過によって回収し、中フリット焼結ガラス漏斗上で真空乾燥させると、5.6gの白色結晶が得られた。収率は50%であった(ESI−MSによって確認)。ESI−MS:m/z 252.1(M+H+)。
上記で得られた4.4g(17.5mmol)の6−ピリダジノンを、50ml容の一ッ口丸底フラスコ内に入れた後、4.7g(35mmol)の無水塩化銅(II)を入れ、次いで、20mlのアセトニトリルを入れ、黄褐色懸濁液を得た。還流冷却器をフラスコに接続し、CaCl2を充填した乾燥チューブを冷却器の上部に取り付けた。反応混合物を油浴(110℃)中で3時間還流加熱した。還流を開始すると、反応懸濁液の色が暗黄色に変化した。反応終了後(HPLCによってモニター)、フラスコを油浴から取り出し、周囲温度まで冷却させた。混合物を200gの粉砕氷上に注入し、10分間激しく攪拌すると、灰色沈殿物および青色の液体が得られた。次いで、沈殿物を濾過によって回収し(濾液のpHは1.5〜2.0であった)、50mlの1N HCl溶液で洗浄して残留(あれば)銅副生成物である固形物を除去した。この後、100mlのMilli−Q水で洗浄して固形物中の酸を除去し、濾液のpH値を確認することによりモニターする。固形物を、濾液がpH7を示すまで洗浄した(だいたい5回洗浄後)。固形物を、中フリット焼結ガラス漏斗上で真空乾燥させると、3.9gの青灰色固形物が得られた。収率は90%であった(ESI−MSによって確認)。ESI−MS:m/z 250.1(M+H+)。
上記で得られた2.0g(8mmol)の6−フェニルピリダジノンおよび10ml(54mmol)のオキシ塩化リン(試薬等級、Aldrich)を50ml容の一ッ口丸底フラスコに入れた。フラスコを還流冷却器に接続し、CaCl2を充填した乾燥チューブを冷却器の上部に取り付けた(反応中にHClガスが形成されるため、大規模合成ではHClを吸収させるために、NaOHなどの塩基性溶液が必要とされ得る)。反応混合物を油浴(90℃)中で2時間攪拌し、次いで、周囲温度まで冷却し、粉砕氷上に注入した(オキシ塩化リンは水によって分解され、HClおよびH3PO4が生成され得る)。次いで、混合物を10分間激しく攪拌し、白色懸濁液を得た。懸濁液を2N NaOH溶液で、懸濁液のpHがpH=7になるまで中和した。沈殿物を濾過し、100mlの水で3回洗浄し、中フリット焼結ガラス漏斗上で真空乾燥させると、1.8gの淡いピンク色粉末が得られた。収率は85%であった(ESI−MSによって確認)。ESI−MS:m/z 268.4(M+H+)。
上記で得られた1.1g(4.0mmol)の3−クロロピリダジンを、30ml容の加圧容器内に入れた後、2.6g(16.0mmol)の1−(2−ピリミジル)ピペラジンを添加し、次いで、15mlの1−BuOH(試薬等級)を添加した。容器をしっかりと密封し、油浴中に入れ、130℃(油浴の温度)で3日間攪拌した。次いで、反応混合物を周囲温度まで冷却し、一ッ口フラスコに移し、減圧下でエバポレーションした。溶媒の除去により赤褐色の残渣が得られ、これを30mlの水で処理すると、褐色の懸濁液が得られた。固形物を濾過によって回収しし、50mLの水で3回洗浄し、濾過漏斗上で真空乾燥させると、0.96gの淡黄色固形物が得られた。収率は90%であった(ESI−MSおよびNMRによって確認)ESI−MS:m/z 396.2(M+H+)。
4−クロロ−6−フェニルピリダジン−3(2H)−オンを、Coudert,P.[18]に記載の手順に従って合成した。
クロロピリダジノン1(25.5g、0.12mol)、4−N,N−ジメチルアミノピリジン(0.20g)およびi−Pr2NEt(26.7g、0.21mol)の混合物を含む無水CH2Cl2(30OmL)を、0℃(氷浴)で30分間攪拌した。塩化メトキシメチル(25g、0.31mol)を添加し、混合物を0℃で1時間攪拌し、次いで、室温まで昇温させた。反応が終了するまで室温で攪拌した。次いで、溶媒を真空除去し、残渣を水で処理し、希Na2CO3溶液で洗浄し、EtOAcで抽出した。有機層を無水Na2SO4上で乾燥させ、濾過し、エバポレーションした。次いで、残渣を95%エタノールからの再結晶によって精製すると、20.1の淡黄色固形物が得られた。収率66.9%。
保護ピリダジノンMW01−7−053WH(1.0当量)を、アリールボロン酸(1.37当量)、Pd(PPh3)4(0.05当量)およびK2CO3(3.1当量)ならびに200mLのDMEと、350mlの加圧容器内で混合し、アルゴンで3分間フラッシュ洗浄し、次いで、出発材料が消失するまで混合物を攪拌および還流した(油浴、120℃)。冷却後、溶液を減圧下で濃縮乾固し、残渣を水で処理し、濾別した。濾過ケークを濾過漏斗において水で洗浄し、次いで、次の工程に直接使用した。上記で得られた残渣を200mlのEtOHに溶解し、6N HCl(200mL)を添加し、反応混合物を6時間還流し(油浴、120℃)、次いで室温まで放冷し、減圧下で濃縮乾固した。残渣を希NaOH溶液で中和した。次いで、懸濁液を濾別し、水で洗浄し、濾過漏斗上で乾燥させた。90%エタノールからの再結晶により、黄褐色固形物が得られた。収率80.4%. ESI−MS:m/z 294.3(M+H+)
3−クロロ−6−フェニル−4−(ピリジン−4−イル)ピリダジン(MW01−7−076WH)
3−クロロ−6−フェニル−4−(ピリジン−4−イル)ピリダジン(MW01−7−076 WH)(66mmol)を、75mlのオキシ塩化リンに懸濁させ、攪拌しながら100℃で3時間加熱した。室温まで冷却後、混合物を粉砕氷上に注入した。次いで、混合物をNaOH溶液で中和し、白色懸濁液を得た。沈殿を濾別し、水で洗浄し、濾過漏斗上で乾燥させると、淡黄色固形物が得られた。ESI−MS:m/z 268.4(M+H+)。
N−(シクロプロピルメチル)−6−フェニル−4−(ピリジン−4−イル)ピリダジン−3−アミン(MW01−7−084WH)( 0.5mmol)、C−シクロプロピル−メチルアミン(2.0mmol)の混合物を含む3mlの1−BuOHを、攪拌しながら130℃で7日間加熱した。溶媒をエバポレーションによって真空除去し、残渣を水で処理して懸濁液を得た。次いで、固形物を濾別し、水で洗浄し、次いで1:3の酢酸エチル:石油エーテルで洗浄し、濾過漏斗上で真空乾燥させると、灰色固形物が得られた。ESI−MS:m/z 330.4(M+H+)。
4,6−ジフェニル3−ピペラジニルピリダジン(MW01−7−133WH)の調製のための合成反応を図13に示す。合成は、本明細書に記載のようにして行なった。該化合物を3−クロロ−4,6−ジフェニルピリダジン(533mg、20mmole)から、MW01−7−057WHについて記載したのと同様にして調製し、淡黄色固形物を得た(550mg、17.4mmol、収率86.9%)。ESI−MS:m/z 317.3(M+H+)。
2−(4−(6−フェニル−4−(ピペリジン−1−イル)ピリダジン−3−イル)ピペラジン−1−イル)ピリミジン(MW01−7−107WH)の調製のための合成反応を図14に示す。合成は、本明細書に記載のようにして行なった。該化合物をMW01−6−127WH(200mg、0.57mmol)から、MW01−7−057WHについて記載したのと同様にして調製し、淡黄色固形物を得た(220mg、0.55mmol、収率96.3%)。ESI−MS:m/z 402.5(M+H+)。
6−メチル−4−フェニル−3−(4−ピリミジン−2−イルピペラジン−1−イル)ピリダジン(MW01−7−057)の調製のための合成反応を図15に示す。合成は、本明細書に記載のようにして行なった。3−クロロ−6−メチル−4−フェニルピリダジン(100mg、0.5mmol)、1−(2−ピリミジル)ピペラジン(400mg、2.0mmol)の混合物を含む3mlの1−BuOHを、攪拌しながら130℃で7日間加熱した。溶媒をエバポレーションによって真空除去し、残渣を水で処理して懸濁液を得た。次いで、固形物を濾別し、水で洗浄し、次いで1:3の酢酸エチル:石油エーテルで洗浄し、濾過漏斗上で真空乾燥させると、淡黄色固形物が得られた(68mg、0.20mmol、収率41.7%)。純度>95%;ESI−MS:m/z 333.1(M+H+)。
ピリダジン化合物の活性を確認するためのアッセイ
以下のアッセイは、ピリダジン化合物の活性を確認するために使用され得る。
Tg6799 5X FADマウスモデルにおける有効性
MW01−2−151SRMを5、10および25mg/kgで、Tg6799マウスにおいて試験する。上記のように、神経炎症およびシナプス機能障害の生化学的評価項目およびY字型迷路行動評価項目を調べる。緊張の特徴に基づいてすでに病態の徴候を示す動物への投与の開始に基づいて、高容量が提案される。注入モデルと比べた有意性には、採血によるコロニーの拡大培養が必要とされるため、より多くの動物およびより長期間が必要とされる。
薬物リード化合物の選択
以下の8種類の化合物:MW01−4−179LKM;MW01−2−151SRM;MW01−7−107WH;MW01−6−189WH;MW01−7−084WH;MW01−7−085WH7)MW01−7−133WH;およびMW01−7−057WHを合成した(図1〜15および実施例1を参照)。
以下のもの:MW01−2−151SRM;MW01−4−179LKM;MW01−6−189WH;MW01−7−084WH;MW01−7−085WH;MW01−7−133WH;およびMW01−7−057WHは、選択的な化合物であった。
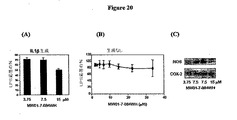
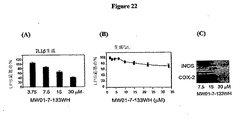
1つの化合物MW01−7−107WHは、同じ濃度範囲において、NO、iNOSおよびCOX−2の生成も阻害したため、非選択的であった(BV2小グリア細胞におけるMW01−2−151SRM;MW01−6−189WH;MW01−4−107WH;MW01−4−179LKM;MW01−7−084WH;MW01−7−085WH;MW01−7−133WH;およびMW01−7−057WHの細胞の活性の結果を示す図16〜23を参照)。
hERGチャネル阻害アッセイおよび心臓QT間隔アッセイ
所望でない(off−target)毒性のために後の試験で心臓QT間隔の延長を誘導する可能性の高い化合物(あれば)をプロセスの初期において排除するため、化合物をhERG(ヒトエーテル−a−go−go)カリウムイオンチャネルの結合および阻害についてスクリーニングした。hERGチャネルは、心臓再分極に決定的に寄与する整流体であるカリウムの流れの遅滞の活性化を速やかに行なう。hERGチャネル遺伝子における変異および該流れの薬物誘導型ブロックは、QT間隔の延長をもたらす活動電位の再分極の遅滞と関連している(Finlaysonら、2004;Recanatiniら、2005;Roden,2004)。QT延長は、新規な薬物の心臓安定性の重要なリスクファクターとみなされている。したがって、hERGチャネル阻害について試験することによる開発プロセスの早期での心臓安定性の考慮により、潜在的な化合物心臓安定性に対する障害を評価するための効率的で予測的な手段が提供される。また、FDA(USA)は、これを、将来的な承認基準とみなしており、特に推奨している。これまでに行なわれたアッセイは、商業施設(MDS PharmaService)によるものである。
競合結合アッセイ:1〜2mg
パッチクランプアッセイ:1〜2mg
QT間隔アッセイ:5mg/動物/用量=25mg/アッセイ、15mg/kg用量
エキソビボ活性アッセイは、偽陽性および偽陰性を示し易いため、FDA方針書のガイドラインに従ってインビボQT間隔アッセイ試験を終了するほうがよいと考えられる。
競合阻害アッセイ:
MW01−5−188WH、MW01−2−151SRM,およびMW01−6−127WHは、10μM 濃度で試験した。
MW01−2−151SRMおよびMW01−6−189WHを、3つの濃度(0.1、1、10μM)で試験した。これらの化合物は最小の阻害を示し、IC50値は、MW01−6−189WHで4.81μMおよびMW01−2−151SRMで9.21μMであった。
結果ならびに材料および方法の概要を、以下に示す。
試験物質(例えば、MW01−2−151SRM)は、麻酔モルモットで測定したリードIIの心電図のQT間隔に対してもたらされ得る効果について評価した。QT間隔(QTc)は、バゼット式およびフリデリシアの式の両方を用いて心拍数の変化で補正した。ビヒクル処理対照群において、2つの連続する観察時間の対応する時間点で変化の95%信頼上限を超えるベースライン値より上のQTc値の増加(あれば)は、個々に処置した動物における有意なQTc延長を示す。15mg/kg POの試験物質では、投与後60分の間、5匹の処置動物すべてにおいて、QTc間隔有意な延長はなんら引き起こされなかった(図29および31)。他方、0.3mg/kgでのソタロールの静脈内投与では、すべての(5.5)動物において、QTc間隔の有意な延長が引き起こされた(図30および32)。QT補正にバゼット式またはフリデリシアの式のいずれかを使用することにより、結果は同様の結論にに達した。
陽性対照化合物であるソタロールは、心臓QTc間隔の有意な増加を誘導する。
試験物質を2%Tween 80に溶解し、経口投与によって投与した。該物質は15mg/kgで処置し、投薬容量は10ml/kgとし、投薬容量は10ml/kgとした。MDS Pharma Services−Taiwan Ltdから得たDuncan Hartley系モルモットを使用した。ソタロールはSigma,USAから入手した。
急性および慢性毒性アッセイ
肝臓毒性は、肝臓が初期薬物代謝の主要な部位であり、動物の代謝全体およびホメオスタシスに不可欠であるため、経口投与される化合物に関する特に重要な初期考慮事項である。また、肝臓障害は、ある種の長期投与薬物で見られる特発性組織障害の要素である。したがって、マウスへの化合物の経口投与後に肝臓毒性の初期評価を行なうことは重要である。
標準的なアプローチは、化合物を2つの初期インビボ毒性アッセイ:短期間漸増用量パラダイムおよび長期間治療用量レジメンで試験することである。漸増用量短期間毒性アッセイでは、マウス(5匹/実験群)に、化合物またはビヒクルのいずれかを0.5%カルボキシメチルセルロースにて(あるいはまた、ヒマシ油もしくはゴマ油が使用され得る)、経口強制投与によって1日1回3日間投与する。標準的な化合物の用量は、3.1、12.5および50mg/kgであり、最大用量は、治療用量の20倍である。第4日に、マウスを致死させ、肝臓を採取し、組織学検査のために固定する。パラフィン包埋し、ヘマトキシリン&エオシン(H&E)染色した肝臓組織切片を、損傷について、処置群について知らされていない2名の個人が顕微鏡により解析する。0(最良)〜9(最低)の半定量的組織学的スコア化システムを適用し、これは、構造的特徴(正常から広範な線維化)、細胞の特徴(正常から広範な浮腫および広範な壊死)、ならびに炎症性浸潤の程度(正常から広範な浸潤)を考慮したものである。各短期間の毒性アッセイに対し、15mgの化合物が必要である。
毒性試験の結果を図34に示す。
インビトロ安定性、経口バイオアベイラビリティおよび脳内取込み。ラット肝臓ミクロソーム(BD Biosciences,Bedford,MA)との標準的なインキュベーションおよびNADPH再生系におけるMW01−5−188WH(1μM)の安定を、37℃で30分間および120分間行なった。反応をアセトニトリルによって停止させ、反応混合物を16,000μgで10分間遠心分離した。10マイクロリットルの上清みを較正HPLCによって解析し、インキュベーション後に残留するMW01−5−188WH初期量に対するパーセントを定量した。HPLC系(Dionex,Sunnyvale,CA)は、Dionex P680ポンプ、ガードカラムを有するPhenomenex(Torrance,CA)Luna C18カラム(250×2.0mm;5μm)およびDionex UVD340U紫外線検出器を含む。移動相は、試薬Aとしての0.1%ギ酸および試薬Bとしての0.08%ギ酸/水含有80%アセトニトリルからなるものとし、0.2ml/分の流速とした。勾配は、以下の試薬Bにおける線形および定組成勾配溶出変化からなるものとした。すなわち、0〜5分で60%の定組成、5〜39分で60〜90%、44分まで90%の定組成である。ピーク定量は、MW01−5−188WHの連続希釈を用いて得られた標準曲線に関して260nmで測定した吸光度に基づいて行なった。経口バイオアベイラビリティ(経口投与後の時間の関数としての血中の化合物の濃度)を評価するため、および潜在的な脳内取込みにおける見識を得るため、MW01−5−188WH(2.5mg/kg)を0.5%(w/v)カルボキシメチルセルロース懸濁液にて、マウスに経口強制投与によって投与した。化合物投与の5、15、60および120分後、動物をペントバルビタール(50mg/kg)で麻酔した。心臓内穿刺によって採血し、ヘパリン加チューブ内に回収し、遠心分離によって血漿を得た。マウスにPBSを灌流した。脳ホモジネートを12,000μgで10分間遠心分離し、上清みを0.1%ギ酸(Fluka, Sigma− Aldrich,St. Louis,MO)で1:3に希釈することにより酸性化した。固相抽出の後HPLC解析を使用し、脳上清み中の化合物の量を定量した。簡単には、カートリッジ(Sep−Pak C18;Waters Associates,Milford, MA)を、1mlのアセトニトリル(HPLC grade;EMD Biosciences,San Diego,CA)で条件設定し、1mlの水で平衡化した。MW01−5−188WHの構造類縁化合物を内部標準として使用した。酸性化した脳上清みを、カートリッジに添加した後、30%アセトニトリルを含む1mlの洗浄液を添加した。MW01−5−188WHをカートリッジから、80%アセトニトリルを用いて溶出した。溶出液を蒸発乾固し、0.08%ギ酸/水含有80%アセトニトリル中で再構成し、HPLCによって解析した(試薬Bにおいて以下の勾配、すなわち2〜5分で0〜60%、7分まで65%の定組成、7〜12分で65〜80%、15分まで80%の定組成、15〜18分で89〜100%、および23分まで100%の定組成を使用)。血漿試料を0.1Mの過塩素酸中でタンパク除去し、12,000μgで10分間遠心分離した。上清みを1M NaOHで中和し、次いで、ジクロロメタンで抽出し、3000μgで5分間層分離させた。3回の連続抽出による有機相をプールし、次いで、減圧下で蒸発乾固した。乾燥残渣を50μlの試薬B中で再構成し、10μlのこの再構成物質をHPLCによって解析した(脳上清みについて上記の勾配を使用)。
MW01−5−188WHの経口バイオアベイラビリティおよび脳内取込み
神経科学のための統合(integrative)生物化学的ツールおよびCNS標的化薬物は、適切なバイオアベイラビリティおよび脳内取込みまたは血液脳関門の通過を示すものであるのがよい。毎日の経口投与は、動物モデルを用いた長期間および時限的インビボ試験のための投与の好ましい方法であり、さまざまな理由(例えば、より良好な患者コンプライアンス)で薬物開発の好ましい様式である。これに関連して、インヒビターのバイオアベイラビリティおよび適切な初期脳内取込み割合を実証し、インビボ試験の結果を充分に解釈することは重要である。したがって、経口投与後の血中のMW01−5−188WH濃度の変化の割合(経口バイオアベイラビリティ)およびその脳内での変化の割合を調べた。生物学的試料から抽出したMW01−5−188WHの定量的解析で上記のプロトコルを使用し、マウスへの低用量経口投与(2.5mg/kg)後の血中および脳内の出現割合を調べた。血中のMW01−5−188WHの出現(図35A)は、可能な最も早い時間点(5分)以内に容易に検出され、15分以内にピーク濃度に達し、大量クリアランスは経口投与後120分以内に起こる。これは、MW01−5−188WHが、良好な経口バイオアベイラビリティ特性を有することを示す。同様の時間依存性濃度変化パターンが脳でも見られ(図35B)、MW01−5−188WHの初期脳内取込みが血中のものを反映することを示す。しかしながら、MW01−5−188WHのピーク脳内/血中濃度比は、>3.3であり、臨床使用におけるCNS薬物のものと同等である。例えば、ミナプリン(6−フェニルアミノピリダジンCNS薬物)の脳内/血中比は、約2である(Cacciaら、1985 Xenobiotica,15(12):1111−9)。この結果は、MW01−5−188WHが、細胞培養物において活性な多くの化合物はインビボ研究での使用から典型的に排除されるという基準を満たすことを示し、経口投与後、ヒトAβ ICV注入モデルによって課される実験の制約内でインビボで作用する可能性を示す。
選択されたグリア活性化経路の抑制への新たな着目および経口投与されたMW01−5−188WHの優れた脳内取込み特性により、該化合物が末梢組織による炎症誘発性サイトカイン生成抑制に対するCNS炎症誘発性サイトカイン抑制に関して選択性を示すかもしれないという可能性が生じた。この可能性を調べるため、MW01−5−188WHを標準的な治療用量(2.5mg/kg)で、経口強制投与によって2週間毎日投与し、次いで、マウスに細菌LPSの腹腔内注射で抗原刺激した。LPS刺激の6時間後、IL−1βおよびTNF−αの血清および脳レベルを測定した。予測どおり、LPS刺激により、生理食塩水を注射した対照マウスと比べて、血清(図35C、D)および脳(図35E、F)において、IL−1βおよびTNF−αのレベルの増加が誘導された。興味深い所見は、MW01−5−188WHでの2週間の処置により、脳内ではIL−1βおよびTNF−α生成のLPS誘導型上方調節が抑制されるが(図35E、F)、血清応答は抑制されないことであった(図35C、D)。MW01−5−188WHによる脳サイトカイン応答の抑制は、活性化グリアにより炎症誘発性サイトカイン生成を抑制するその能力、ならびに上述したその経口バイオアベイラビリティおよび脳内取込み特性と整合する。
薬物動態試験
イヌおよび/またはラットにおける血漿薬物動態および絶対バイオアベイラビリティ
2つの群(1群あたり3匹の動物;雄動物)に、PO投与およびIV投与する。用量レベルは1つとし(2.5mg/kg)、クロスオーバー計画を使用し、投薬期間毎に1週間の洗い流しを行なう。血漿薬物濃度は、投与後24時間を超えない8回以上の時点(例えば、単回用量の投与の15、30、60、90、120、240および480分後ならびに24時間後)で測定する。誘導されるPKパラメータとしては、Cmax、Tmax、t1/2、AUC、CI/F、VdおよびMRTが挙げられる。投与製剤:経口強制投与/CMC溶液。
試験は、14C標識ミノザック(minozac)(MW01−2−151SRM)を用いて行ない、排泄(尿、便)および血漿分布が解析され得る。
試験のフェーズAは、単回用量MTDである(各用量レベルについて3M/3F、n=測定されるMTDまたはMFDまで)。利用可能なデータ(あれば)に基づいて設計される用量レベル;以下に示す用量は、例示の目的のためだけに使用され得る。投与は、CMC溶液での経口強制投与によるものである。
用量レベル1:10mg/kg;用量レベル2:100mg/kg;用量レベル3:500mg/kg;用量レベル4:1000mg/kg;用量レベル5:3000mg/kg;
結果:推定単回用量MTD/MFD(sdMTD)
試験のフェーズBが行なわれ得る。このフェーズは、7日間の用量範囲確認試験(各群において3M/3F、n=24)を含む。対照+1つの用量レベル(sdMTDの一部)を存在させ、さらなる用量レベル(1つまたは複数)を、初期7日間の用量範囲確認試験の結果で必要に応じて組み込む。
イヌにおける用量範囲確認試験
この試験では、単回用量MTD(5Mクロスオーバー試験、n=測定されるMTDまたはMFDまで)を用いる。用量レベルは、利用可能なデータに基づいて設計する。用量の例を以下に示す。投与は、CMC溶液での経口強制投与によるものであるのが好ましい。あるいはまた、充填ゼラチンカプセル剤を使用する。
IV用量1:100mg/kg;IV用量2:300mg/kg;経口用量レベル4:1000mg/kg;経口用量レベル5:3000mg/kg
その後の各投与は、適切な洗い流し期間後(IV曝露の2日後または5日後)に行なう。薬物動態および絶対バイオアベイラビリティは、用量レベル1および2に対して測定する。血漿薬物濃度は、投与後24時間を超えない8回の時点(例えば、単回経口用量の投与の15、30、60、120、240および480分後)で測定する。誘導されるPKパラメータとしては、Cmax、Tmax、t1/2、AUC、CI/F、VdおよびMRTが挙げられる。
主な試験は、各処置群n=80において10M/10Fを伴う。投与は、CMC溶液での経口強制投与によるものである。対照、低用量、中用量および高用量を存在させる。結果:PK(血漿およびCSF薬物レベル)を第1日および第28日に測定する。処置終了後に剖検を行なう。死亡率、臨床観察、体重、食物消費量、臨床病態、検眼鏡検査、肉眼病理検査および臓器重量を調べる。組織病理学検査を、対照および高用量群において行なう。
各処置群n=24で3M/3Fを用いる主な試験を行なう。投与は、CMC溶液での経口強制投与によるものが好ましい。あるいはまた、必要に応じて充填ゼラチンカプセル剤を使用する。対照、低用量、中用量および高用量を存在させる。結果:PK(血漿およびCSF薬物レベル)を第1日および第28日に測定する。処置終了後に剖検を行なう。死亡率、臨床観察、体重、食物消費量、臨床病態、肉眼病理検査および臓器重量を調べる。組織病理学検査を、すべての用量群において行なう。
一般的な方法:
科学薬品は、おおむね、Aldrich (Milwaukee, WI)またはVWR Internationalから購入し、受領したまま使用した。すべての溶媒は、本文中で特に記載のない限り、受領したまま使用した。すべての有機溶液は、最終のエバポレーション前に硫酸マグネシウムで乾燥させた。マイクロ波照射は、CEM−Discoverマイクロ波合成システム(Matthews, NC)を用いて行なった。
2−ベンジル−6−フェニル−4,5−ジヒドロピリダジン−3(2H)−オン(18)
3−ベンゾイルプロピオン酸17(17.8g、0.1mol)、ベンジルヒドラジンジヒドロクロリド(19.5g、0.1mol)および酢酸ナトリウム(74.9g、0.55mol)を500mLのエタノール(95%)に懸濁した。白色懸濁液を、29時間還流加熱した。エタノールを減圧除去し、残渣を水(300mL)で処理した。水層のpHを炭酸ナトリウムの濃縮溶液でpH=8に調整し、酢酸エチルで抽出した(1×200mL)。有機層をブラインで洗浄し、減圧下で濃縮乾固した。生成物18が黄色油状物として78%収率で得られ、さらなる精製をせずに次の工程で使用した。HPLC(tr/純度):23.4分、80%.
3,4−ジクロロ−6−フェニルピリダジン(19)。
19(158g、0.7mol)および酢酸(700mL)の混合物を5時間還流加熱した。反応混合物を室温まで冷却し、沈殿物を濾過し、明黄色濾過ケークを水(5×500mL)で洗浄した。濾過ケークを酢酸エチル(200mL)から再結晶化させ、濾過し、中フリット焼結ガラス漏斗上で真空乾燥させると、所望の生成物20が32%収率で得られた。HPLC(tr/純度):15.37分、>95%. ESI m/z(MeOH):207.3(MH+)。
化合物20(14g、0.068mol)を、反応チューブ内に、1−ブタノール(30mL)および4当量の1−(2−ピリミジル)ピペラジン(45g、0.27mol、4当量)とともに入れた。フラスコにキャップをし、130℃で41時間加熱した。反応混合物を周囲温度まで冷却し、1−ブタノールを減圧除去すると、暗色油状残渣が得られた。この油状物を水で処理して懸濁液を得、次いで、これを濾過し、水でで洗浄する。濾過ケークを、中フリット焼結ガラス漏斗上で真空乾燥させると、所望の生成物21が97%収率で得られた。HPLC(tr/純度):17.30分、>99%. ESI m/z(MeOH):334.38(MH+)。
化合物21(22g、0.066mol)を、オキシ塩化リン(80mL)に懸濁させた。反応混合物を100℃で3時間加熱し、室温まで冷却し、粉砕氷(2kg)上で精製した。水性混合物をNaOH溶液で中和し、白色懸濁液を得た。沈殿物を濾過し、中フリット焼結ガラス漏斗上で真空乾燥させると、所望の生成物6が91%収率(21g)で得られた。
Zouら(Jet Lett.2001 42:7213−7215)の手順に従い、化合物6(100mg、0.28mmol)を、1.37当量のベンジルボロン酸(42mg、0.31mmol)、0.2当量のPd(dppf)Cl2CH2Cl2(23mg、0.02mmol)、2.5当量の酸化銀(164mg、0.71mmol)および3当量の炭酸カリウム(117mg、0.85mmol)を含むTHFに懸濁させた。混合物にアルゴンをパージし、密封チューブ内で120℃で16時間加熱した。次いで、反応混合物を周囲温度まで冷却し、33%過酸化水素または10%水酸化ナトリウムのいずれかでクエンチした。水層をエーテルで抽出し(3×30mL)、エーテル層を合わせ、減圧下でエバポレーションした。粗製混合物をシリカゲルカラムに供し、ヘキサン:酢酸エチル(1:1 v/v)で溶出する。生成物2が、薄ピンク色の固形物として45%収率で得られる。
化合物6(700mg、2.0mmol)を反応容器内に、3.1当量の炭酸カリウム(851mg、6.2mmol)、1.37当量(330mg、2.7mmol)の4−ピリジニルボロン酸および0.05当量のPd(PPh3)4(120mg、0.1mmol)とともに入れた。DME (10mL)を添加し、混合物にアルゴンをパージした。反応混合物を密封し、110℃で20時間加熱した。溶液を周囲温度まで冷却し、セライトに通して濾過した。濾液を減圧濃縮し、酢酸エチル(30mL)に溶解し、2N HCl(50mL)で洗浄した。有機層を減圧濃縮し、酢酸エチル/石油エーテル混合物で再結晶させると、生成物3が淡黄色針状物として41%収率で得られた。
Zouら(前掲)の手順に従い、化合物6(200mg、0.56mmol)を、1.37当量の(2−メチルプロピル)ボロン酸(79mg、0.77mmol)、0.2当量のPd(dppf)Cl2CH2Cl2(92.5mg、0.11mmol)、2.5当量の酸化銀(328mg、1.41mmol)および3当量の炭酸カリウム(234mg、1.7mmol)を含むTHFに懸濁させた。混合物にアルゴンをパージし、密封チューブ内で120℃で42時間加熱した。反応物を周囲温度まで冷却し、反応を水酸化ナトリウム水溶液(10%)でクエンチし、エーテルで抽出した(3×50ml)。エーテル層を合わせ、硫酸マグネシウムで乾燥させ、減圧下でエバポレーションすると、粘性固形物が残留した。粗製混合物をカラムクロマトグラフィーで精製し、40%酢酸エチル含有ヘキサンを用いて溶出すると、4が白色粉末として52.5%収率で得られた。
Zouら(前掲)の手順に従い、化合物6(250mg、0.71mmol)を、1.37当量のメチルボロン酸(59mg、0.97mmol)、0.25当量のPd(dppf)Cl2CH2Cl2(144mg、0.18mmol)、2.5当量の酸化銀(410mg、1.78mmol)および3当量の炭酸(arbonate)カリウム(294mg、2.1mmol)を含むTHFに懸濁させた。混合物にアルゴンをパージし、密封チューブ内で120℃で18.5時間加熱した。周囲温度まで冷却後、反応を水性水酸化ナトリウム(10%)でクエンチし、エーテルで抽出した(3×75ml)。化合物をカラムクロマトグラフィーによって精製し、酢酸エチルヘキサン(1:3 v/v)の混合物を用いて溶出した。化合物5は、白色の結晶化固形物であり、45.8%収率で得られた。
4−メチル−6−フェニル−3−(4−ピラジン−2−イル)ピペラジン−1−イル)ピリダジン(7)
化合物15(500mg、2.4mmol)をキャップ付きフラスコに入れ、20mLの水に懸濁した。2.5当量(1g、6mmol)の1−(2−ピラジニル)ピペラジンおよび5当量(1.69mL、12mmol)のトリエチルアミンを添加し、フラスコにキャップをし、130℃まで160時間加熱した。反応物を周囲温度まで冷却すると、暗褐色油状物がフラスコの底に得られた。水をデカンテーションして油状物を分離し、油状物を最少のイソプロパノールに溶解し、70℃まで加熱した。冷却すると、褐色固形物が形成され、焼結ガラス漏斗において濾過し、ヘキサンでリンス処理すると、生成物7が褐色粉末として28.8%収率で得られた。
化合物15(190mg、0.93mmol)を反応チューブ内に、1−ブタノールおよび4当量の1−(ピリジン−2−イル)ピペラジン(605mg、3.7mmol)とともに入れ、キャップをし、140℃で48時間加熱した。
反応混合物を周囲温度まで冷却し、1−ブタノールを減圧除去すると、暗油状残渣が得られた。油状物を水で処理して懸濁液を得、これを、濾過し、まず水で、次いで、酢酸エチル:ヘキサン(1:6 v/v)の混合物で洗浄すると、生成物8が黄褐色粉末として54.5%収率で得られる。
化合物15(190mg、0.93mmol)を反応チューブ内に、1−ブタノールおよび4当量の4−ピペラジンo−ピリダジン(605mg、3.7mmol)とともに入れた。フラスコにキャップをし、140℃で72時間加熱した。反応混合物を周囲温度まで冷却し、1−ブタノールを減圧除去すると、暗赤色油状残渣が得られた。油状物を20mLの水で処理し、次いで10mLの酢酸エチルで抽出した。褐色の懸濁液が有機層に形成された。沈殿物を濾過によって回収し、10mLの水で次いで10mLの酢酸エチルで洗浄すると、生成物9が黄褐色粉末として34.1%収率で得られた。
化合物15(200mg、0.96mmol)を、10mL容マイクロ波用ガラス容器内で4当量のシクロヘキシルピペラジン(651.5mg、3.87mmol)を含む5mLの水に懸濁させ、栓でキャップをした。75Wのマイクロ波を使用し、温度は、室温から175℃まで傾斜をつけた。175℃に達したら、反応混合物をこの温度に3時間保持した。反応混合物を室温まで放冷し、暗褐色溶液を水上に注入して懸濁液を得、これを濾過するとベージュ色の固形物が得られた。この固形物を20mLの飽和重炭酸ナトリウムで洗浄すると、10が95%収率で得られた。
化合物15(500mg、2.4mmol)を、キャップ付きフラスコ内で、4当量の1−メチル−ピペラジン(961mg、9.6mmol)を含む20mLの水に懸濁させた。容器にキャップをし、終了まで120℃で120時間加熱した。混合物を周囲温度まで冷却すると、白色固形物沈殿物を有する薄黄色溶液が得られた。反応物を濾過し、水性濾液をエーテルで洗浄して微量の出発材料を除去し、次いで、酢酸エチルで抽出した(5×10mL)。有機洗浄席を合わせ、硫酸マグネシウムで乾燥させ、酢酸エチルを減圧除去する。残留した油状物をエーテルで処理し、冷却すると、生成物11が黄色針状物として38.7%収率で得られた。
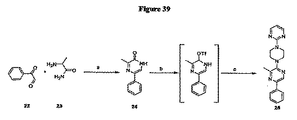
3−メチル−5−フェニルピラジン−2(1H)−オン(24)
この化合物は、Jones(J.Amer.Chem.Soc.1949,71,78−81)の手順に従って調製した。簡単には、市販のフェニルグリオキサル22(1.02g、7.62mmol)をメタノールに溶解し、−41℃まで冷却した。市販のアラニンアミド23(672mg、7.62mmol)を25mlのメタノールに溶解し、反応混合物に添加した。12.5N NaOH(0.760mL、9.53mmol)溶液を攪拌下に滴下し、その間、反応温度は−10℃未満に維持した。滴下終了時、反応物を−5℃で2時間置いた。次いで、反応物を室温まで昇温させ、12N HCl溶液(0.76mL)でクエンチした後、重炭酸ナトリウムで溶液を中和した。メタノールを減圧除去し、残渣をクロロホルムで抽出し、酢酸エチルで沈殿させた。化合物を白色粉末として単離し、24が18%収率で得られた。HPLC(tr/純度):15.91分、>97%. ESI m/z(MeOH):187.35(MH+)。
この化合物は、ピラジントリフレートを経由し、1−(2−ピリミジル)ピペラジンをアミンとして使用し、Adamsら(Synlett 2004,11,2031−2033)の手順に従って調製した。ピリジンは、確実な密封瓶(Aldrich)内でアルゴン下に維持した無水試薬として使用した。化合物24(100mg、0.52mmol)およびDMAP(65.7、0.52mmol)をピリジンと塩化メチレン(0.5:4ml v/v)に溶解し、0℃まで冷却した。トリフルオロメタンスルホン酸(0.8mmol、135.5μL)を滴下し、0℃で15分間、次いで室温で3時間攪拌した。トリフレートは、ESI(363.7(MH+))およびHPLC(tR=25.33分)によって確認した。反応混合物をジクロロメタンで希釈し、20mlの水、重炭酸ナトリウムおよびブラインで1回ずつ洗浄した。ジクロロメタンを減圧除去し、残留残渣をDMSOに直接溶解させた。1−(2−ピリミジル)ピペラジン(5.3mmol、750μL)を添加し、反応物を60℃まで加熱し、2時間攪拌した。終了したら、反応物を酢酸エチルで希釈し、1N HClで洗浄し、ブラインおよび水で洗浄した後、残留ピリジンは除去された。次いで、有機物を乾燥させ、真空蒸発させると、25が黄色固形物として得られた(63%収率)。HPLC(tr/純度):24.74分、>98%. ESI m/z(CH2Cl2)333.29(MH+)。
4,6−ジフェニル3−(4−ピリミジン−2−イル)ピペラジン−1−イル)ピリダジンジクロロ一水和物塩(26)。
4,5−ジヒドロ−4−メチル−6−フェニルピリダジン−3(2H)−オン(13)(Hansen, KBら、Org.Process Res.Dev.,2005,9,634−639,Nelson,DA.US 20050137397A1)。温度プローブおよび冷却器を取り付けた250ml容の三ッ口丸底フラスコに、7.7g(40mmol)の2−メチル−4−オキソ−4−フェニルブタン酸12および20mlのエタノール(95%)を仕込んだ。懸濁液を10℃未満に冷却し、2.2ml(42mmol、1.05当量)のヒドラジン一水和物を含有する10mlのエタノールを滴下した。滴下後、反応混合物を還流加熱し、2時間攪拌した。反応混合物を周囲温度まで冷却し、形成された白色結晶を濾過によって回収した。次いで、固形物を2N NaHCO3(1×30mL)、Milli−Q水(3×60mL)で洗浄し、中フリット焼結ガラス漏斗上で真空乾燥させると、所望の生成物13が96.1%収率で得られた。
7.0g(35mmol)の13を、250ml容一ッ口丸底フラスコ内で30mlのアセトニトリルに溶解した。11.3g(84mmol、2.4当量)の無水塩化銅(II)をこの溶液に添加し、反応混合物を2時間還流加熱した。反応過程中に形成されるHClガスを制御するため、NaOH溶液を用い、乾燥チューブから放出されるHClを吸収させた。反応混合物を周囲温度まで冷却し、氷水浴中に入れた。150mLの氷水を添加して反応をクエンチした。混合物を10分間激しく攪拌すると、灰色沈殿物および青色の塩化銅(I)含有液が得られた。次いで、沈殿物を濾過によって回収し(濾液のpHは0〜1である)、まず1N HCl(100mL)で、次いでMilli−Q水(5×100mL)で洗浄した。残留銅副生成物を除去するため、濾過ケークを1N HCl(150mL)中で0.5時間攪拌し、次いで濾過した。濾過ケークを、濾液がpH7になるまで(だいたい7回洗浄)Milli−Q水で洗浄した。固形物を、中フリット焼結ガラス漏斗上で真空乾燥させると、14が淡灰色粉末として93.8%収率で得られた。
7.5g(36.6mmol)の15を、125mLのMilli−Q水に懸濁させた。60.17g(366.0mmol、10当量)の1−(2−ピリミジル)ピペラジンを添加し、反応混合物を高速攪拌下で60時間、還流加熱した。終了したら、反応混合物を周囲温度まで冷却すると、フラスコ内に、橙色水層とフラスコ底面にに沈降した褐色油状物からなる2層が観察された。
水をデカンテーションし、油状物を最少容量のイソプロパノールに溶解し、還流加熱した。10分間の還流後、溶液をゆっくりと0℃まで冷却して結晶化を誘導した。薄黄色結晶をイソプロパノールから濾過し、最少の冷エーテルでリンス処理すると、5が54%収率で得られた。
物理化学的特性
材料/方法:
HPLC系(Dionex Corp.,Sunnyvale,CA)は、以下の要素:Dionex P680 Pump、Dionex ASI−100自動試料採取装置、ガードカラムを有するPhenomenex(Torrance,CA)Luna C18カラム(250×2.0mm;5μM)、およびDionex UVD 170U検出器からなるものとした。移動相は、溶媒Aとして0.1%ギ酸(Fluka)含有Milli−Q水および溶媒Bとして80%アセトニトリル(Burdick & Jackson)とともに0.08%ギ酸含有Milli−Q水からなるものとした。ピーク定量は、化合物の連続希釈によって得られた標準曲線に関して254nmにおける吸光度に基づいて行なった。
乾燥させた清浄なホウケイ酸塩製毛細管を、化学天秤を用いて計量した。17〜30mgの16を秤量し、チューブに添加した。蒸留精製Milli−Q水をチューブに添加し、1〜2g/mlの範囲の濃度を有する溶液を作製した。試験チューブを手動で混合し、充分な濡れを確実にし、37℃に設定したインキュベータ内に一晩入れた。試料を各チューブから回収し、10,000rpmで10分間遠心分離し、逆相HPLCにインジェクトした。
乾燥させた清浄なガラス製三角フラスコを、化学天秤を用いて計量した。30mgまでの26をフラスコに添加した。蒸留精製水をフラスコに添加し、飽和溶液を作製した。フラスコを回転式撹拌器/インキュベータ(37℃、175rpm)内に72時間入れた。試料を24時間間隔で取り出し、10,000rpmで10分間遠心分離して粒状物を除去し、逆相HPLCシステムにインジェクトした。
16および26の分配係数を、1−オクタノール(Sigma)および水を用いて決定した。0.5〜1mg/mlの各化合物をMilli−Q水に溶解し、事前に飽和させた(presaturated)オクタノールに分配させた。試料を回転式撹拌器/インキュベータ(37℃)内に1時間、水平に置いた。1時間後、試料を1500rpmで5分間遠心分離し、水相を分離した。水相とオクタノール相両方の化合物の濃度を測定した。
細胞培養アッセイ。化合物の濃度依存性活性のグリア細胞系アッセイを、既報のようにして行なった(Hu W、Ralay Ranaivoら、Current Alzheimer’s Research 2005,2:197−205;Mirzoeva Sら、J Med Chem 2002,45:563−566;Ralay Ranaivo Hら、J Neurosci 2006,26:662−670)。BV−2マウス小グリア細胞を、マルチウェルプレート内で1日培養し、次いで、無血清培地中で16時間、希釈剤または種々の濃度の化合物の存在下、対照バッファーまたは標準的なグリア活性化刺激剤であるリポ多糖(LPS、ネズミチフス菌由来;100ng/ml)のいずれかで処理した。既報(Hu W、Ralay Ranaivoら、Current Alzheimer’s Research 2005,2:197−205;Mirzoeva Sら、J Med Chem 2002,45:563−566;Mirzoeva Sら、Brain Res 1999,844:126−134)のグリースアッセイでは、一酸化窒素(NO)の安定な代謝産物である亜硝酸塩の蓄積がBV−2馴化培地において測定された。細胞ライセート中のIL−1β、TNFα、MCP−1およびIL−10のレベルを、Mesoscale Discoveryシステムにより、製造業者の使用説明書のとおりに測定した。細胞ライセートを、ウエスタンブロットによって既報のとおりに解析し(Mirzoeva Sら、J Med Chem 2002,45:563−566;Ralay Ranaivo Hら,J Neurosci 2006,26:662−670)、誘導一酸化窒素シンターゼ(iNOS)およびシクロオキシゲナーゼ−2(COX−2)のレベルを測定した。本発明の化合物の結果を表1に示す。
経口バイオアベイラビリティ(経口投与後の時間の関数としての血中の化合物の濃度)を推定するため、および潜在的な脳内取込みにおける見識を得るため、化合物5(2.5mg/kg)を0.5%(w/v)カルボキシメチルセルロース懸濁液にて、マウスに経口強制投与によって投与した(Ralay Ranaivo Hら、J Neurosci 2006,26:662−670)。経口投与の5、15、30、60および120分後、マウスを致死させ、灌流し、その血液および脳を回収した。脳をアセトニトリル中でホモジナイズし、次いで、12000×gで10分間遠心分離した。次に、血漿および脳上清みを、0.1%ギ酸(Fluka)で、それぞれ、1:1および1:3に希釈することによって酸性化した。固相抽出の後HPLC解析を使用し、血漿脳上清み中の化合物の量を定量した。簡単には、カートリッジ(Sep−Pak(登録商標)C18、Waters)を1mlのアセトニトリル(HPLC等級、EMD Biosciences)で条件設定し、1mlの水で平衡化した。構造類縁化合物である6−メチル−4−フェニル−3−(4(ピリミジン−2−イル)ピペラジン−1−イル)ピリダジン(MW01−7−057WH)を、回収内部標準として使用した。酸性化した試料をカートリッジに負荷した後、10%アセトニトリルを含む1mlの洗浄液を負荷した。化合物5をカートリッジから、80%アセトニトリルを用いて溶出した。溶出液を蒸発乾固し、0.08%ギ酸/水含有80%アセトニトリル中で再構成し、HPLCによって解析した(試薬Aとして0.1%ギ酸含有水および試薬Bとして0.1%ギ酸含有アセトニトリルを使用し、試薬Bにおける以下の勾配を用いた。すなわち、3分まで0%〜50%、6分まで50%の定組成、6〜10分で50%〜70%、13分まで70%の定組成、13〜18分で70%〜80%、21分まで80%の定組成、21〜23分で80%〜70%を使用し、最後に23〜28分で70%〜0%に戻した)。
logSは化合物の中性型の固有溶解度
PSA:1,2,4−7=58.0≒3=70.93;8,9=45.15;10,11=32.26
50%の阻害に必要な濃度(μM)
IL−Iβ=インターロイキン−Iβ;NO=一酸化窒素。
Claims (26)
- 治療有効量の式I:
の化合物、その異性体、薬学的に許容され得る塩または誘導体を含む、神経炎症性疾患に苦しむ被験体の処置後に副作用のリスクの低下および/または有益な薬物動態プロフィールをもたらすのに有効な組成物。 - R1、R4、R5、R6、R7、R8、R9、R12、R13およびR14が、独立して、水素、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C1〜C6アルコキシ、C2〜C6アルケニルオキシ、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C3〜C10シクロアルコキシ、C6〜C10アリール、C6〜C10アリールオキシ、C6〜C10アリール−C1〜C3アルコキシ、C6〜C10アロイル、C6〜C10ヘテロアリール、C3〜C10複素環式基、C1〜C6アシル、C1〜C6アシルオキシ、−NH2、−NHR28、−NR28R29、=NR28、−S(O)2R28、−SH、−SO3H、ニトロ、シアノ、ハロ、ハロアルキル、ハロアルコキシ、ヒドロキシアルキル、−CO2H、−CO2R28、−NHC(O)R28、−C(O)NH2、−C(O)NHR28、−C(O)NR28R29、−NHS(O)2R28(式中、R28およびR29は、独立して、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C6〜C10アリール、C6〜C10アリールC1〜C3アルキル、C6〜C10ヘテロアリールおよびC3〜C10複素環式基から選択される)である、請求項1に記載の組成物。
- 式II:
の化合物またはその異性体、薬学的に許容され得る塩もしくは誘導体の治療有効量を含む、請求項1に記載の組成物。 - R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、R16およびR17が、独立して、水素、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C1〜C6アルコキシ、C2〜C6アルケニルオキシ、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C3〜C10シクロアルコキシ、C6〜C10アリール、C6〜C10アリールオキシ、C6〜C10アリール−C1〜C3アルコキシ、C6〜C10アロイル、C6〜C10ヘテロアリール、C3〜C10複素環式基、C1〜C6アシル、C1〜C6アシルオキシ、−NH2、−NHR28、−NR28R29、=NR28、−S(O)2R28、−SH、−SO3H、ニトロ、シアノ、ハロ、ハロアルキル、ハロアルコキシ、ヒドロキシアルキル、−CO2H、−CO2R28、−NHC(O)R28、−C(O)NH2、−C(O)NHR28、−C(O)NR28R29、−NHS(O)2R28(式中、R28およびR29は、独立して、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C6〜C10アリール、C6〜C10アリールC1〜C3アルキル、C6〜C10ヘテロアリールおよびC3〜C10複素環式基から選択される)から選択される、請求項3に記載の組成物。
- R1が、アルキル、シクロアルキルまたはヘテロアリールである、請求項1または2に記載の組成物。
- R1が、
である、請求項1または2に記載の組成物。 - R15、R16およびR17が、独立して、水素、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C1〜C6アルコキシ、C2〜C6アルケニルオキシ、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C3〜C10シクロアルコキシ、C6〜C10アリール、C6〜C10アリールオキシ、C6〜C10アリール−C1〜C3アルコキシ、C6〜C10アロイル、C6〜C10ヘテロアリール、C3〜C10複素環式基、C1〜C6アシル、C1〜C6アシルオキシ、−NH2、−NHR28、−NR28R29、=NR28、−S(O)2R28、−SH、−SO3H、ニトロ、シアノ、ハロ、ハロアルキル、ハロアルコキシ、ヒドロキシアルキル、−CO2H、−CO2R28、−NHC(O)R28、−C(O)NH2、−C(O)NHR28、−C(O)NR28R29、−NHS(O)2R28(式中、R28およびR29は、独立して、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C6〜C10アリール、C6〜C10アリールC1〜C3アルキル、C6〜C10ヘテロアリールおよびC3〜C10複素環式基から選択される)から選択される、請求項6に記載の組成物。
- 式III:
の化合物またはその異性体、薬学的に許容され得る塩もしくは誘導体、またはその異性体、薬学的に許容され得る塩もしくは誘導体の治療有効量を含む、神経炎症性疾患に苦しむ被験体において処置後に副作用のリスクの低下および/または有益な薬物動態プロフィールをもたらすのに有効な組成物。 - R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、R16およびR17は、独立して、水素、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C1〜C6アルコキシ、C2〜C6アルケニルオキシ、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C3〜C10シクロアルコキシ、C6〜C10アリール、C6〜C10アリールオキシ、C6〜C10アリール−C1〜C3アルコキシ、C6〜C10アロイル、C6〜C10ヘテロアリール、C3〜C10複素環式基、C1〜C6アシル、C1〜C6アシルオキシ、−NH2、−NHR28、−NR28R29、=NR28、−S(O)2R28、−SH、−SO3H、ニトロ、シアノ、ハロ、ハロアルキル、ハロアルコキシ、ヒドロキシアルキル、−CO2H、−CO2R28、−NHC(O)R28、−C(O)NH2、−C(O)NHR28、−C(O)NR28R29、−NHS(O)2R28(式中、R28およびR29は、独立して、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、C3〜C10シクロアルキル、C4〜C10シクロアルケニル、C6〜C10アリール、C6〜C10アリールC1〜C3アルキル、C6〜C10ヘテロアリールおよびC3〜C10複素環式基から選択される)である、請求項8に記載の組成物。
- 式IIIの化合物または異性体もしくはその薬学的に許容され得る塩において、R4、R5、R6、R7、R8、R9、R12、R13、R14、R15、R16およびR17が、水素、ヒドロキシル、アルキルであり、R10およびR11の一方または両方が、独立して、置換もしくは非置換の、水素、ヒドロキシル、アルキル、アルケニル、アルキニル、アルキレン、アルケニレン、アルコキシ、アルケニルオキシ、シクロアルキル、シクロアルケニル、アリール、アリールオキシ、アリールアルコキシ、アロイル、ヘテロアリール、複素環式基、アシル、アシルオキシ、スルホニル、スルフィニル、スルフェニル、アミノ、イミノ、アジド、チオール、チオアルキル、チオアルコキシ、チオアリール、ニトロ、ウレイド、シアノ、ハロ、シリル、シリルアルキル、シリルオキシ、シリルチオ、=O、=S、カルボキシル、カルボニル、またはカルバモイルである、請求項8に記載の組成物。
- 式IIIの化合物において、R10およびR11の一方が、アルキル、特にC1〜C6アルキルであり、R10およびR11の他方が水素である、請求項8に記載の組成物。
- 式IIIの化合物において、R10およびR11の一方がアリールであり、R10およびR11の他方が水素である、請求項8に記載の組成物。
- 式IIIの化合物において、R10およびR11の一方が、ヘテロアリール、特に、1〜4個の窒素原子を含有する5〜6員の不飽和ヘテロモノシクリル基であり、R10およびR11の他方が水素である、請求項8に記載の組成物。
- 式IIIの化合物において、R4、R5、R6、R7、R8、R9、R10、R12、R13、R14、R15、R16およびR17が水素であり、そしてR11が、アルキル、アルケニル、アルキニル、アルキレン、アルコキシ、アリール、または1〜4個の窒素原子を含有する5〜6員の不飽和ヘテロモノシクリル基である、請求項8に記載の組成物。
- 式IIIの化合物において、R4、R5、R6、R7、R8、R9、R10、R12、R13、R14、R15、R16およびR17が水素であり、R11がアルキルまたはピリジニルである、請求項8に記載の組成物。
- 式IIIの化合物が4−メチル−6−フェニル−3−(4−ピリミジン−2−イルピペラジン−1−イル)ピリダジンである、請求項8に記載の組成物。
- IL−1βおよびS100Bの上方調節を選択的に低減またはブロックする、ならびに/またはPSD−95および/もしくはシナプトフィシンの減損を低減または抑制する化合物の治療有効量を含む、請求項1〜16のいずれか1項に記載の組成物。
- hERGカリウムチャネルにおける阻害活性を低下させるとともに神経炎症性疾患を処置するための式I、IIまたはIIIの化合物の治療有効量を含む、請求項1〜17のいずれか1項に記載の組成物。
- hERG阻害を低減させるとともに神経炎症性疾患を処置するための式I、IIまたはIIIの化合物の治療有効量を含む、請求項1〜18のいずれか1項に記載の組成物。
- 前記治療有効量が、投与期間中、IL−1βおよびS100Bの上方調節を選択的に低減またはブロックするのに有効であり、PSD−95および/またはシナプトフィシンの減損を低減または抑制するのに有効なものである、請求項1〜19のいずれか1項に記載の組成物。
- 式I、IIまたはIIIの化合物の、使用環境における有効濃度、または本明細書に開示した疾患の症状の予防、処置もしくは制御において治療効果をもたらす有効用量を提供するための被験体への投与に適した該化合物の治療有効量を含む、請求項1〜20のいずれか1項に記載の組成物。
- 前記疾患が神経炎症性疾患である、請求項21に記載の組成物。
- 約0.1〜100mg/kg、0.1〜50mg/kg、0.1〜25mg/kg、0.1〜20mg/kg、0.1〜15mg/kg、0.1〜10mg/kg、0.1〜5mg/kg、0.1〜4mg/kg、0.1〜3mg/kg、0.1〜2mg/kg、または0.1〜1mg/kgの用量の式I、IIまたはIIIの化合物を含有する、請求項1〜22のいずれか1項に記載の組成物。
- 請求項1〜23のいずれか1項に記載の組成物を被験体に投与することを含む、被験体において神経炎症性疾患を処置する方法。
- 神経炎症性疾患の処置において副作用のリスクの低下および/または有益な薬物動態プロフィールをもたらすための医薬を調製するための、請求項1〜23のいずれか1項に記載の式I、IIまたはIIIの化合物の少なくとも1種類の使用。
- 請求項1〜23のいずれか1項に記載の組成物の1種類以上、容器、および使用説明書を備えるキット。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US79632806P | 2006-04-28 | 2006-04-28 | |
| PCT/US2007/010248 WO2007127375A2 (en) | 2006-04-28 | 2007-04-27 | Formulations containing pyridazine compounds for treating neuroinflammatory diseases |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009535344A true JP2009535344A (ja) | 2009-10-01 |
| JP2009535344A5 JP2009535344A5 (ja) | 2011-06-16 |
Family
ID=38626233
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009507823A Pending JP2009535344A (ja) | 2006-04-28 | 2007-04-27 | 神経炎症性疾患の処置のためのピリダジン化合物を含む処方物 |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US20090325973A1 (ja) |
| EP (1) | EP2063894B1 (ja) |
| JP (1) | JP2009535344A (ja) |
| CN (1) | CN101754762A (ja) |
| AU (1) | AU2007243280A1 (ja) |
| BR (1) | BRPI0710938A2 (ja) |
| CA (1) | CA2650625A1 (ja) |
| IL (1) | IL194968A0 (ja) |
| MX (1) | MX2008013843A (ja) |
| WO (1) | WO2007127375A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014530902A (ja) * | 2011-10-28 | 2014-11-20 | インヒビタクシン リミテッド | 治療に有用なピリダジン誘導体 |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2264014A1 (en) | 2001-08-31 | 2010-12-22 | Université Louis Pasteur | Substituted pyridazines as anti-inflammatory agents and protein kinase inhibitors |
| ES2543813T3 (es) | 2004-11-02 | 2015-08-24 | Northwestern University | Compuestos de piridazina para el tratamiento de enfermedades inflamatorias |
| EP1812007B1 (en) | 2004-11-02 | 2011-09-07 | Northwestern University | Pyridazine compounds and methods |
| AU2007243280A1 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Formulations containing pyridazine compounds for treating neuroinflammatory diseases |
| CA2650711A1 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Compositions and treatments using pyridazine compounds and cholinesterase inhibitors |
| US8536168B2 (en) | 2007-03-15 | 2013-09-17 | Novartis Ag | Benzyl and pyridinyl derivatives as modulators of the hedgehog signaling pathway |
| US20100041663A1 (en) | 2008-07-18 | 2010-02-18 | Novartis Ag | Organic Compounds as Smo Inhibitors |
| US20100227793A1 (en) * | 2009-03-04 | 2010-09-09 | Scott Thomas Brady | Compositions and Methods for Treating Amyotrophic Lateral Sclerosis |
| CA2788355C (en) | 2010-02-18 | 2018-03-06 | Devi Reddy Gohimukkula | Phenyl-heteroaryl derivatives and methods of use thereof |
| WO2011156901A2 (en) * | 2010-06-17 | 2011-12-22 | Waratah Pharmaceuticals Inc. | Compounds, compositions and methods for treatment of multiple sclerosis |
| WO2012031383A1 (zh) | 2010-09-06 | 2012-03-15 | 中国科学院广州生物医药与健康研究院 | 酰胺类化合物 |
| CA3030167A1 (en) | 2016-07-12 | 2018-01-18 | Revolution Medicines, Inc. | 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric shp2 inhibitors |
| EP4230623A3 (en) | 2017-01-23 | 2023-10-11 | Revolution Medicines, Inc. | Pyridine compounds as allosteric shp2 inhibitors |
| IL296456A (en) | 2017-01-23 | 2022-11-01 | Revolution Medicines Inc | Bicyclics as allosteric shp2 inhibitors |
| WO2019051084A1 (en) | 2017-09-07 | 2019-03-14 | Revolution Medicines, Inc. | SHP2 INHIBITOR COMPOSITIONS AND METHODS OF TREATING CANCER |
| EP3694848A1 (en) * | 2017-10-12 | 2020-08-19 | Revolution Medicines, Inc. | Pyridine, pyrazine, and triazine compounds as allosteric shp2 inhibitors |
| EP3724189B1 (en) | 2017-12-15 | 2023-10-04 | Revolution Medicines, Inc. | Polycyclic compounds as allosteric shp2 inhibitors |
| WO2023163951A1 (en) * | 2022-02-23 | 2023-08-31 | Immunochem Therapeutics, Llc | Treatment of intracranial hemorrhage |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2857384A (en) * | 1955-08-19 | 1958-10-21 | Ciba Pharm Prod Inc | New pyridazone compounds |
| WO2005009976A1 (en) * | 2003-07-29 | 2005-02-03 | Novo Nordisk A/S | Pyridazinyl- piperazines and their use as histamine h3 receptor ligands |
| JP2005533747A (ja) * | 2002-02-05 | 2005-11-10 | ノボ ノルディスク アクティーゼルスカブ | 新規なアリールおよびへテロアリールピペラジン |
| JP2008518958A (ja) * | 2004-11-02 | 2008-06-05 | ノースウェスタン ユニバーシティ | ピリダジン化合物、組成物および方法 |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1345880A (en) * | 1971-06-18 | 1974-02-06 | Cepbepe | Pyridazine derivatives |
| FR2510998B1 (fr) * | 1981-08-07 | 1986-01-10 | Sanofi Sa | Nouveaux derives amines de la pyridazine, leur procede de preparation et les medicaments, a action desinhibitrice, qui en comportent |
| FR2511366A1 (fr) * | 1981-08-11 | 1983-02-18 | Sanofi Sa | Nouveaux derives de la pyridazine, leur procede de preparation et les medicaments, actifs sur le systeme nerveux central, qui en contiennent |
| DE3217325A1 (de) | 1982-05-08 | 1983-11-10 | Hoechst Ag, 6230 Frankfurt | 3-amino-6-aryl-1,2,4-triazolo(4,3-b)-pyridazine, ihre herstellung und ihre verwendung |
| FR2540113A1 (fr) * | 1983-01-27 | 1984-08-03 | Sanofi Sa | Acides derives de la pyridazine actifs sur le systeme nerveux central |
| ES8802151A1 (es) | 1985-07-31 | 1988-04-01 | Janssen Pharmaceutica Nv | Un procedimiento para la preparacion de nuevos piridazinaminas. |
| US4654343A (en) * | 1985-10-31 | 1987-03-31 | American Cyanamid Company | N-substituted-N[3-(1,2,4-triazolo[4,3-b]pyridazin-6-yl)phenyl]alkanamides, carbamates and ureas |
| FR2601011B1 (fr) * | 1986-07-03 | 1988-10-28 | Sanofi Sa | Nouveaux derives tricycliques agonistes des recepteurs cholinergiques et medicaments en contenant |
| DE3882982T2 (de) * | 1987-11-02 | 1993-11-25 | Yoshitomi Pharmaceutical | Kondensierte pyridazin-verbindungen und deren verwendung als arzneimittel. |
| PT93060B (pt) | 1989-02-07 | 1995-12-29 | Sanofi Sa | Processo para a obtencao de derivados de piridazina e de composicoes farmaceuticas que os contem |
| DK0628550T3 (da) | 1993-06-08 | 1998-09-28 | Vertex Pharma | Pyridazin som interleukin-1 beta-omdannede enzyminhibitorer |
| US5484940A (en) * | 1994-11-28 | 1996-01-16 | Grant; Francine S. | Substituted 3-indolyl-5-pyrazolone compounds |
| GB9707693D0 (en) | 1997-04-16 | 1997-06-04 | Smithkline Beecham Plc | Novel method of treatment |
| TWI241295B (en) | 1998-03-02 | 2005-10-11 | Kowa Co | Pyridazine derivative and medicine containing the same as effect component |
| US6602872B1 (en) | 1999-12-13 | 2003-08-05 | Merck & Co., Inc. | Substituted pyridazines having cytokine inhibitory activity |
| EP2264014A1 (en) * | 2001-08-31 | 2010-12-22 | Université Louis Pasteur | Substituted pyridazines as anti-inflammatory agents and protein kinase inhibitors |
| GB0129260D0 (en) | 2001-12-06 | 2002-01-23 | Eisai London Res Lab Ltd | Pharmaceutical compositions and their uses |
| FR2847253B1 (fr) | 2002-11-19 | 2007-05-18 | Aventis Pharma Sa | Nouveaux derives de pyridazinones a titre de medicaments et compositions pharmaceutiques les renfermant |
| US20040167226A1 (en) * | 2002-12-16 | 2004-08-26 | Serafini Tito A. | Methods for the treatment of pain and traumatic injury using benzamides and compositions containing the same |
| TW200528455A (en) | 2003-12-19 | 2005-09-01 | Bristol Myers Squibb Co | Azabicyclic heterocycles as cannabinoid receptor modulators |
| DE602004006165T2 (de) | 2003-12-19 | 2008-01-17 | Bristol-Myers Squibb Co. | Azabicyclische heterocyclen als modulatoren des cannabinoidrezeptors |
| US7220858B2 (en) * | 2003-12-23 | 2007-05-22 | Barbeau Pharma, Inc. | Synthesis of hydrazine and chlorinated derivatives of bicyclic pyridazines |
| MY145822A (en) * | 2004-08-13 | 2012-04-30 | Neurogen Corp | Substituted biaryl piperazinyl-pyridine analogues |
| EP1812007B1 (en) * | 2004-11-02 | 2011-09-07 | Northwestern University | Pyridazine compounds and methods |
| AU2007243280A1 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Formulations containing pyridazine compounds for treating neuroinflammatory diseases |
| WO2007130383A2 (en) | 2006-04-28 | 2007-11-15 | Northwestern University | Compositions and treatments using pyridazine compounds and secretases |
| WO2007127475A2 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Pyridazines for demyelinating diseases and neuropathic pain |
| CA2650704A1 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Salts of pyridazine compounds |
| CA2650711A1 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Compositions and treatments using pyridazine compounds and cholinesterase inhibitors |
| EP2131839A2 (en) | 2007-03-02 | 2009-12-16 | Northwestern University | Compositions comprising derivatives of 3-phenylpyridazine for treating seizure-related disorders |
-
2007
- 2007-04-27 AU AU2007243280A patent/AU2007243280A1/en not_active Abandoned
- 2007-04-27 JP JP2009507823A patent/JP2009535344A/ja active Pending
- 2007-04-27 BR BRPI0710938-5A patent/BRPI0710938A2/pt not_active IP Right Cessation
- 2007-04-27 US US12/298,652 patent/US20090325973A1/en not_active Abandoned
- 2007-04-27 CA CA002650625A patent/CA2650625A1/en not_active Abandoned
- 2007-04-27 CN CN200780023749A patent/CN101754762A/zh active Pending
- 2007-04-27 WO PCT/US2007/010248 patent/WO2007127375A2/en active Application Filing
- 2007-04-27 EP EP07776351.4A patent/EP2063894B1/en active Active
- 2007-04-27 MX MX2008013843A patent/MX2008013843A/es not_active Application Discontinuation
-
2008
- 2008-10-28 IL IL194968A patent/IL194968A0/en unknown
-
2012
- 2012-10-25 US US13/660,671 patent/US9408845B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2857384A (en) * | 1955-08-19 | 1958-10-21 | Ciba Pharm Prod Inc | New pyridazone compounds |
| JP2005533747A (ja) * | 2002-02-05 | 2005-11-10 | ノボ ノルディスク アクティーゼルスカブ | 新規なアリールおよびへテロアリールピペラジン |
| WO2005009976A1 (en) * | 2003-07-29 | 2005-02-03 | Novo Nordisk A/S | Pyridazinyl- piperazines and their use as histamine h3 receptor ligands |
| JP2008518958A (ja) * | 2004-11-02 | 2008-06-05 | ノースウェスタン ユニバーシティ | ピリダジン化合物、組成物および方法 |
Non-Patent Citations (3)
| Title |
|---|
| JPN5009005466; HU WENHUI: CURRENT ALZHEIMER RESEARCH V2 N2, 2005, P197-205, BENTHAM SCIENCE PUBLISHERS * |
| JPN5009005467; RANAIVO H R: THE JOURNAL OF NEUROSCIENCE V26 N2, 20060111, P662-670 * |
| JPN5009005468; NELSON: LANCET NEUROLOGY V5 N3, 200603, P210, LANCET PUBLISHING GROUP * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014530902A (ja) * | 2011-10-28 | 2014-11-20 | インヒビタクシン リミテッド | 治療に有用なピリダジン誘導体 |
Also Published As
| Publication number | Publication date |
|---|---|
| IL194968A0 (en) | 2009-08-03 |
| CN101754762A (zh) | 2010-06-23 |
| MX2008013843A (es) | 2008-11-10 |
| EP2063894A2 (en) | 2009-06-03 |
| CA2650625A1 (en) | 2007-11-08 |
| AU2007243280A1 (en) | 2007-11-08 |
| US20090325973A1 (en) | 2009-12-31 |
| BRPI0710938A2 (pt) | 2012-06-26 |
| US20130072496A1 (en) | 2013-03-21 |
| WO2007127375A3 (en) | 2008-02-07 |
| EP2063894B1 (en) | 2019-08-28 |
| WO2007127375A2 (en) | 2007-11-08 |
| US9408845B2 (en) | 2016-08-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2009535344A (ja) | 神経炎症性疾患の処置のためのピリダジン化合物を含む処方物 | |
| US8158627B2 (en) | Compositions and treatments using pyridazine compounds and cholinesterase inhibitors | |
| US9663493B2 (en) | Pyridazine compounds, compositions and methods | |
| WO2007130383A2 (en) | Compositions and treatments using pyridazine compounds and secretases | |
| US20100168120A1 (en) | Salts of pyridazine compounds | |
| US8063047B2 (en) | Pyridazine compounds and methods | |
| AU2012216322A1 (en) | Pyridazine compounds, compositions and methods |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100423 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100423 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110418 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120827 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20121030 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20121106 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20130430 |