JP2007016190A - 多孔性微粒子分散体 - Google Patents
多孔性微粒子分散体 Download PDFInfo
- Publication number
- JP2007016190A JP2007016190A JP2005201885A JP2005201885A JP2007016190A JP 2007016190 A JP2007016190 A JP 2007016190A JP 2005201885 A JP2005201885 A JP 2005201885A JP 2005201885 A JP2005201885 A JP 2005201885A JP 2007016190 A JP2007016190 A JP 2007016190A
- Authority
- JP
- Japan
- Prior art keywords
- silica
- dispersion
- fine particles
- porous fine
- porous
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/10—Process efficiency
- Y02P20/129—Energy recovery, e.g. by cogeneration, H2recovery or pressure recovery turbines
Landscapes
- Pigments, Carbon Blacks, Or Wood Stains (AREA)
- Silicon Compounds (AREA)
Abstract
【課題】多孔性微粒子を分散媒に分散させた分散体であって、多孔性微粒子の問題がなく、長期保存安定性に優れた新規な多孔性微粒子分散体を提供する。
【解決手段】多孔性微粒子が分散媒に分散してなる分散体において、多孔性微粒子の細孔容積をTPV(ml/g)、分散体中の多孔性微粒子濃度をC(重量%)としたとき、50/(1.8+TPV)≦C≦100/(1.8+TPV)の関係を満たす多孔性微粒子分散体。分散体の固形分濃度を多孔性微粒子の細孔容積に応じた濃度とすることにより、多孔性微粒子が沈殿を形成せず、保存安定性に優れた多孔性微粒子分散体を得ることができる。
【選択図】なし
【解決手段】多孔性微粒子が分散媒に分散してなる分散体において、多孔性微粒子の細孔容積をTPV(ml/g)、分散体中の多孔性微粒子濃度をC(重量%)としたとき、50/(1.8+TPV)≦C≦100/(1.8+TPV)の関係を満たす多孔性微粒子分散体。分散体の固形分濃度を多孔性微粒子の細孔容積に応じた濃度とすることにより、多孔性微粒子が沈殿を形成せず、保存安定性に優れた多孔性微粒子分散体を得ることができる。
【選択図】なし
Description
本発明は、多孔性微粒子が分散媒中に分散してなる多孔性微粒子分散体に関する。詳しくは、多孔性微粒子の沈降の問題がなく、保存安定性に優れた新規な多孔性微粒子分散体に関する。
一般に、物質を微粒子化することにより、基体への表面被覆性、他の物質との混合性等が向上することから、従来より、様々な母物質について微粒子化が施され、各種用途に適用されてきた。
微粒子化される母物質としては、例えば、酸化チタン、酸化亜鉛、シリカをはじめとする白色顔料、シリカゲル、アルミノケイ酸塩をはじめとする乾燥剤、銀をはじめとする抗菌剤、アクリル系高分子をはじめとする塗料バインダーが挙げられる。
微粒子化した母物質は凝集を起こすおそれがあるため、安定な保存形態として、これを分散媒に分散させた分散体が用いられる。ここで、微粒子を分散させる分散媒としては、水、アルコールをはじめとする極性溶媒、もしくはトルエン、ヘキサン等の非極性溶媒がしばしば用いられる。
多孔性微粒子を分散媒に分散させた分散体は、製造直後は極めて分散性のよい状態を維持する。しかし、分散微粒子表面の安定化エネルギーが重力による位置エネルギーの減少による安定化に勝ると分散微粒子の沈降が起こる。
沈降により生じた沈降物は互いに強固に結合しており、振揺、攪拌では容易に再分散するものではない。これを解砕するためには超音波の照射、粉砕等の工程が必要となり、作業性が悪いという課題がある。
例えば、水を分散媒として用いてシリカ微粒子を分散させたシリカ微粒子分散体の場合を例として、この分散体中のシリカ微粒子の状態を説明すると、以下の通りである。
シリカ微粒子の表面にはシラノール基に由来するOH基が存在し、その一部が解離し、負に帯電したO−の状態となっている。その結果、微粒子は負に帯電し、微粒子間は静電的反発力にて反発している。この反発力は微粒子の電荷の積に比例し、微粒子間距離の2乗に反比例する。そのため微粒子間は一定の距離を保とうとする。このため、安定に分散した分散体を得ることができる。
シリカ微粒子の表面にはシラノール基に由来するOH基が存在し、その一部が解離し、負に帯電したO−の状態となっている。その結果、微粒子は負に帯電し、微粒子間は静電的反発力にて反発している。この反発力は微粒子の電荷の積に比例し、微粒子間距離の2乗に反比例する。そのため微粒子間は一定の距離を保とうとする。このため、安定に分散した分散体を得ることができる。
しかし、地球上のような重力場の中においては質量mを持った粒子は重力加速度による力mgを受ける。これは重力場の中においては重力方向に物は引っ張られ、重力方向により低い場所にあるものが安定であることを意味する。
微粒子間の静電的反発力よりも、この重力加速度による力により引っ張られることによる安定化が勝る場合、例えば、微粒子の質量mの大きい場合や、微粒子の電荷が小さい場合もしくは微粒子間の電荷の符号が異なる場合には、微粒子は沈降する。
そして、沈降粒子間には、シリカ微粒子表面のOH基同士の水素結合もしくは水による水素結合を仲立ちとした結合が起こる。この結合反応は時間とともに進行し、沈降物は強固に結合する。このため、沈降物は超音波、衝撃波等の物理的外力を与えなければ分散し得ないものとなる。
一方で、例えば、シリカ微粒子分散体に樹脂、有機溶媒等の添加剤を加えて塗料として使用する場合、分散体に含有されているシリカ微粒子と樹脂、有機溶媒とが充分に混合される必要があることから、シリカ微粒子が充分に均一に分散している分散体が必要となる。ここで、シリカの分散性が悪いシリカ微粒子分散体を用いると、凝集したシリカ微粒子が塗布時に残り、平滑な塗布面が得られない。このため、分散性に優れたシリカ微粒子分散体が求められる。
本発明は、上述の課題に鑑みてなされたものである。
即ち、本発明の目的は、多孔性微粒子を分散媒に分散させた分散体であって、多孔性微粒子の沈降の問題がなく、長期保存安定性に優れた新規な多孔性微粒子分散体を提供することにある。
即ち、本発明の目的は、多孔性微粒子を分散媒に分散させた分散体であって、多孔性微粒子の沈降の問題がなく、長期保存安定性に優れた新規な多孔性微粒子分散体を提供することにある。
本発明者らは上記の課題を解決すべく鋭意検討した結果、分散体の固形分濃度を多孔性微粒子の細孔容積に応じた濃度とすることにより、多孔性微粒子が沈殿を形成せず、保存安定性に優れた多孔性微粒子分散体が得られるという知見を得、本発明を完成するに至った。
即ち、本発明は以下を要旨とする。
[1] 多孔性微粒子が分散媒に分散してなる分散体において、該多孔性微粒子の細孔容積をTPV(ml/g)、分散体中の多孔性微粒子濃度をC(重量%)としたとき、
50/(1.8+TPV)≦C≦100/(1.8+TPV)
の関係を満たすことを特徴とする多孔性微粒子分散体。
[2] 多孔性微粒子の平均粒径X50が0.01μm以上であることを特徴とする[1]に記載の多孔性微粒子分散体。
[3] 多孔性微粒子の細孔容積TPVが0.01ml/g以上であることを特徴とする[1]又は[2]に記載の多孔性微粒子分散体。
[4] 多孔性微粒子がシリカであることを特徴とする[1]〜[3]に記載の多孔性微粒子分散体。
[1] 多孔性微粒子が分散媒に分散してなる分散体において、該多孔性微粒子の細孔容積をTPV(ml/g)、分散体中の多孔性微粒子濃度をC(重量%)としたとき、
50/(1.8+TPV)≦C≦100/(1.8+TPV)
の関係を満たすことを特徴とする多孔性微粒子分散体。
[2] 多孔性微粒子の平均粒径X50が0.01μm以上であることを特徴とする[1]に記載の多孔性微粒子分散体。
[3] 多孔性微粒子の細孔容積TPVが0.01ml/g以上であることを特徴とする[1]又は[2]に記載の多孔性微粒子分散体。
[4] 多孔性微粒子がシリカであることを特徴とする[1]〜[3]に記載の多孔性微粒子分散体。
本発明者によれば、分散体の固形分濃度を多孔性微粒子の細孔容積に応じて調整することにより、多孔性微粒子が沈殿を形成することなく、安定な分散状態を長期間維持する保存安定性に優れた多孔性微粒子分散体が提供される。
本発明による多孔性微粒子の沈殿防止効果の作用機構の詳細については明らかではないが、多孔性微粒子の細孔容積に応じて分散体の固形分濃度を適度な濃度とすることにより、水被覆微粒子として後述するように、多孔性微粒子の表面が分散媒分子の極薄層で覆われ、この分散媒分子で被覆された多孔性微粒子同士が分散体内で静電気力に反発する一方で、最近傍の多孔性微粒子同士がわずかな点で接触し、この接触力と静電気反発力との均衡により、多孔性微粒子同士が互いに支え合い、沈降することなく、安定な分散状態を維持することによると推察される。
このような本発明の効果は、平均粒径X50が0.01μm以上の多孔性微粒子を分散させる場合に特に有効である(請求項2)。
また、本発明は、細孔容積TPVが0.01ml/g以上であるような多孔性微粒子を分散させる場合において、分散安定性の向上に特に有効である(請求項3)。
本発明の多孔性微粒子分散体としては、特に多孔性シリカ微粒子分散体が工業的に有用である(請求項4)。
また、本発明は、細孔容積TPVが0.01ml/g以上であるような多孔性微粒子を分散させる場合において、分散安定性の向上に特に有効である(請求項3)。
本発明の多孔性微粒子分散体としては、特に多孔性シリカ微粒子分散体が工業的に有用である(請求項4)。
以下に本発明の多孔性微粒子分散体の実施の形態を詳細に説明する。
なお、以下においては、産業上最もよく使われ、乾燥剤等として効果の高い多孔性シリカ微粒子の水分散体を例に、本発明を詳細に説明するが、本発明は何ら多孔性シリカ微粒子の水分散体に限らず、例えば、シリカ、酸化チタン、酸化亜鉛、アルミノケイ酸塩、酸化ニッケル等の酸化物、炭酸カルシウム、硫酸カルシウム、硫酸鉛、硫化鉛等の各種多孔性金属塩の分散体であっても良く、これらの1種又は2種以上の多孔性微粒子を、水、アルコール、アセトン、メチルエチルケトンのようなケトン類、ジメチルフォルムアミド、テトラヒドロキシフラン、N−メチルピロリドン等の極性溶媒や、トルエン、ヘキサン、ベンゼン、酢酸エチル、酢酸ブチルのようなエステル類、ジエチルエーテル等のエーテル類等の非極性溶媒の1種又は2種以上に分散させてなる各種の多孔性微粒子分散体に適用することができる。
本発明に係る多孔性微粒子として例示する「シリカ」とは、無水ケイ酸と含水ケイ酸の両方を示す。例えば無水ケイ酸としては、石英、トリディマイト、クリストバル石、コーサイト、スティショフ石、石英ガラスなどが挙げられる。そして含水ケイ酸としては、シリカヒドロゾルをゲル化し乾燥させて得られる、いわゆる非晶質の「シリカゲル」以外に、コロイダルシリカ、シリケートオリゴマー、そして有機物等を鋳型として形成された、例えばモービル社製:MCM−41のようなタイプのシリカ(いわゆる、ミセルテンプレート型シリカ)等が挙げられる。また「シリカゲル」の原料としては、水ガラスやアルコキシシラン類が挙げられる。
本発明に係る多孔性微粒子として例示する「シリカ」とは、無水ケイ酸と含水ケイ酸の両方を示す。例えば無水ケイ酸としては、石英、トリディマイト、クリストバル石、コーサイト、スティショフ石、石英ガラスなどが挙げられる。そして含水ケイ酸としては、シリカヒドロゾルをゲル化し乾燥させて得られる、いわゆる非晶質の「シリカゲル」以外に、コロイダルシリカ、シリケートオリゴマー、そして有機物等を鋳型として形成された、例えばモービル社製:MCM−41のようなタイプのシリカ(いわゆる、ミセルテンプレート型シリカ)等が挙げられる。また「シリカゲル」の原料としては、水ガラスやアルコキシシラン類が挙げられる。
[1]多孔性シリカ微粒子分散体
本発明の多孔性シリカ微粒子分散体(以下、単に「本発明のシリカ分散体」と略記する場合がある。)は、多孔性シリカ微粒子(以下、単に「シリカ」と略記する場合がある。)を、その細孔容積TPV(ml/g)に対して、分散媒中の分散濃度(以下「固形分濃度」と称す場合がある。)C(重量%)が
50/(1.8+TPV)≦C≦100/(1.8+TPV)
となる固形分濃度で水中に分散したものである。
本発明の多孔性シリカ微粒子分散体(以下、単に「本発明のシリカ分散体」と略記する場合がある。)は、多孔性シリカ微粒子(以下、単に「シリカ」と略記する場合がある。)を、その細孔容積TPV(ml/g)に対して、分散媒中の分散濃度(以下「固形分濃度」と称す場合がある。)C(重量%)が
50/(1.8+TPV)≦C≦100/(1.8+TPV)
となる固形分濃度で水中に分散したものである。
なお、本発明の多孔性微粒子分散体が細孔容積の異なる2種以上の多孔性微粒子を含む場合、各々の多孔性微粒子の細孔容積と分散濃度が上記式を満たすものであれば良い。
〔分散媒〕
本発明のシリカ分散体に用いられる分散媒の種類は特に制限されない。分散媒の具体例としては、水、アセトン等のケトン類、ジメチルホルムアミド等のアミド類、メタノール、エタノール等のアルコール類等が挙げられるが、中でも水、ジメチルホルムアミド、メタノール、エタノールが好ましく、水、メタノール、エタノールが特に好ましい。これらの分散媒は1種を単独で用いてもよく、2種以上を任意の組み合わせ、任意の比率で混合して用いてもよい。
本発明のシリカ分散体に用いられる分散媒の種類は特に制限されない。分散媒の具体例としては、水、アセトン等のケトン類、ジメチルホルムアミド等のアミド類、メタノール、エタノール等のアルコール類等が挙げられるが、中でも水、ジメチルホルムアミド、メタノール、エタノールが好ましく、水、メタノール、エタノールが特に好ましい。これらの分散媒は1種を単独で用いてもよく、2種以上を任意の組み合わせ、任意の比率で混合して用いてもよい。
〔シリカ〕
本発明のシリカ分散体に含有されるシリカは、ケイ酸の水和物であり、SiO2・nH2Oの組成式で表される。
本発明に係るシリカの細孔容積TPV(ml/g)は、通常0.01ml/g以上、中でも0.015ml/g以上、特に0.02ml/g以上であることが好ましい。細孔容積TPVが0.01ml/g以上であると、多孔質として細孔内に分散媒を含有することができ、好ましい。なお、ここで、シリカの細孔容積TPVとしては、本発明のシリカ分散体を乾燥して得られたサンプルにつきBET窒素吸着等温線を求め、P/P0=0.98の時の値を用いる。シリカの細孔容積TPVの上限については特に制限はないが、通常10ml/g以下、好ましくは8ml/g以下、より好ましくは5ml/g以下である。シリカの細孔容積TPVがこの上限を超えると、構造が軟弱となり安定な分散体の形成が不可能である。
本発明のシリカ分散体に含有されるシリカは、ケイ酸の水和物であり、SiO2・nH2Oの組成式で表される。
本発明に係るシリカの細孔容積TPV(ml/g)は、通常0.01ml/g以上、中でも0.015ml/g以上、特に0.02ml/g以上であることが好ましい。細孔容積TPVが0.01ml/g以上であると、多孔質として細孔内に分散媒を含有することができ、好ましい。なお、ここで、シリカの細孔容積TPVとしては、本発明のシリカ分散体を乾燥して得られたサンプルにつきBET窒素吸着等温線を求め、P/P0=0.98の時の値を用いる。シリカの細孔容積TPVの上限については特に制限はないが、通常10ml/g以下、好ましくは8ml/g以下、より好ましくは5ml/g以下である。シリカの細孔容積TPVがこの上限を超えると、構造が軟弱となり安定な分散体の形成が不可能である。
本発明の分散体に含有されるシリカの平均粒径X50は、通常0.01μm以上、中でも0.03μm以上、特に0.05μm以上が好ましい。平均粒径X50がこの下限より小さいと、先に述べた重力により引っ張られる力より、静電気力による静電気反発による安定化が増大するが、この下限以上であると、沈降性が高くなるため、本発明による効果が有効に発揮される。なお、ここで、シリカの平均粒径X50としては、粒度分布計を用いて測定したシリカの粒度分布における体積基準積算粒度分布50%の粒径の値を用いる。シリカの平均粒径X50の上限については特に制限はないが、通常100μm以下、好ましくは80μm以下、より好ましくは50μm以下である。シリカの平均粒径X50がこの上限を超えると、微粒子としての性質が大幅に低下し、先に述べた表面被覆性、他物質との混合性が低下する。
また、本発明のシリカ分散体に含有されるシリカは、その三次元構造を見るに、非晶質であること、即ち、結晶性構造が認められないことが好ましい。このことは、本発明のシリカ分散体に含有されるシリカをX線回折で分析した場合に、結晶性ピークが実質的に認められないことを意味する。なお、本明細書において非晶質のシリカとは、X線回折パターンで6オングストローム(Å Units d-spacing)を超えた位置に、結晶構造に起因するピークを一つも示さないものを指す。この様なシリカの材料としては、有機テンプレートを用いて細孔を形成するミセルテンプレートシリカが挙げられる。非晶質のシリカは、結晶性のシリカに較べて、極めて生産性に優れている。
また、本発明のシリカ分散体に含有されるシリカの最頻細孔径(Dmax)は、通常30nm以下、中でも29nm以下、更には28nm以下が好ましい。最頻細孔径(Dmax)は、気体や液体の吸着や吸収に関する特性であり、最頻細孔径(Dmax)が小さいほど吸着や吸収性能が高い。従って、種々の特性の中で最頻細孔径(Dmax)は、特に触媒担体や吸着剤として使用するシリカゲル微粒子凝集体に重要な物性である。最頻細孔径(Dmax)の下限は特に制限されないが、通常は2nm以上である。最頻細孔径(Dmax)は、窒素ガス吸脱着によるBET法で測定した等温脱着曲線から、E.P.Barrett,L.G.Joyner,P.H.Haklenda,J.Amer.Chem.Soc.,vol.73,373(1951)に記載のBJH法により算出される細孔分布曲線をプロットして求められる。ここで、細孔分布曲線とは、微分細孔容積、即ち、細孔径d(nm)に対する微分窒素ガス吸着量(ΔV/Δ(logd))を言う。なお、上記のVは窒素ガス吸着容積を表す。
また、本発明のシリカ分散体に含有されるシリカは、上記の最頻細孔径(Dmax)の値を中心として±20%の範囲内の径を有する細孔の容積の、全細孔容積に対する割合(以下、この容積割合を「Dmax±20%容積比」と称す場合がある。)が通常20%以上、中でも20.5%以上、更には21%以上であることが好ましい。このことは、本発明のシリカ分散体に含有されるシリカが有する細孔の径が、最頻細孔径(Dmax)付近で揃っていることを意味する。なお、Dmax±20%容積比の上限は特に制限されないが、通常は90%以下である。
かかる特徴に関連して、本発明のシリカ分散体に含有されるシリカは、上記のBJH法により算出された最頻細孔径(Dmax)における微分細孔容積ΔV/Δ(logd)(d:細孔直径(nm)、V:窒素ガス吸着容積)(以下、この値を「Dmax微分細孔容積ΔV/Δ(logd)」と称す場合がある。)が、通常2ml/g以上、また、通常20ml/g以下、中でも12ml/g以下の範囲であることが好ましい。Dmax微分細孔容積ΔV/Δ(logd)が前記範囲に含まれるシリカは、最頻細孔径(Dmax)付近に径が揃っている細孔の絶対量が極めて多いものと言える。
〔固形分濃度範囲〕
本発明のシリカ分散体の固形分濃度(重量%)範囲は、シリカの細孔容積TPV(ml/g)に対して次の式で表される。
50/(1.8+TPV)≦C≦100/(1.8+TPV)
固形分濃度Cがこの範囲である場合、分散体中のシリカ微粒子は極薄層の水分子で覆われた状態となる(以下、この水分子で覆われたシリカ微粒子を「水被覆微粒子」と称す場合がある。)。この水被覆微粒子同士は静電気力により反発する。また、水被覆微粒子は最近傍の水被覆微粒子とわずかな点で接触する。この接触力及び静電気反発力により、水被覆微粒子は互いに支え合い、沈降現象を起こすことのない、安定な分散微粒子となる。
この領域よりも希薄になると、即ち固形分濃度Cが50/(1.8+TPV)よりも小さいと静電気力による反発及び、点接触による支え合いの力が働かず沈降現象が生じる。また、この領域よりも濃厚になると、即ち固形分濃度Cが100/(1.8+TPV)よりも大きいと表面の水の層がうまく形成できないため、分散性が著しく劣り、分散の不均一化ひいては不均一に結合した大粒子を形成し、使用に耐えないものとなる。
本発明のシリカ分散体の固形分濃度(重量%)範囲は、好ましくは
50/(1.8+TPV)≦C≦90/(1.8+TPV)
である。
本発明は、特にシリカ濃度が上記条件を満たすと共に、1重量%以上、例えば10〜50重量%であるような比較的高濃度のシリカ分散体において、その優れた沈降防止効果が有効に発揮される。
本発明のシリカ分散体の固形分濃度(重量%)範囲は、シリカの細孔容積TPV(ml/g)に対して次の式で表される。
50/(1.8+TPV)≦C≦100/(1.8+TPV)
固形分濃度Cがこの範囲である場合、分散体中のシリカ微粒子は極薄層の水分子で覆われた状態となる(以下、この水分子で覆われたシリカ微粒子を「水被覆微粒子」と称す場合がある。)。この水被覆微粒子同士は静電気力により反発する。また、水被覆微粒子は最近傍の水被覆微粒子とわずかな点で接触する。この接触力及び静電気反発力により、水被覆微粒子は互いに支え合い、沈降現象を起こすことのない、安定な分散微粒子となる。
この領域よりも希薄になると、即ち固形分濃度Cが50/(1.8+TPV)よりも小さいと静電気力による反発及び、点接触による支え合いの力が働かず沈降現象が生じる。また、この領域よりも濃厚になると、即ち固形分濃度Cが100/(1.8+TPV)よりも大きいと表面の水の層がうまく形成できないため、分散性が著しく劣り、分散の不均一化ひいては不均一に結合した大粒子を形成し、使用に耐えないものとなる。
本発明のシリカ分散体の固形分濃度(重量%)範囲は、好ましくは
50/(1.8+TPV)≦C≦90/(1.8+TPV)
である。
本発明は、特にシリカ濃度が上記条件を満たすと共に、1重量%以上、例えば10〜50重量%であるような比較的高濃度のシリカ分散体において、その優れた沈降防止効果が有効に発揮される。
〔その他〕
本発明のシリカ分散体は、シリカ、分散媒の他に、必要に応じて各種の成分を含有していてもよい。この様な成分の例としては、界面活性剤、分散剤、酸、アルカリ等が挙げられる。
本発明のシリカ分散体は、シリカ、分散媒の他に、必要に応じて各種の成分を含有していてもよい。この様な成分の例としては、界面活性剤、分散剤、酸、アルカリ等が挙げられる。
なお、シリカ微粒子について上述したシリカの物性、即ち細孔容積TPV、平均粒径X50、非晶性、最頻細孔径(Dmax)、Dmax±20%容積比、Dmax微分細孔容積ΔV/Δ(logd)は、本発明の多孔性微粒子分散体のシリカ以外の多孔性微粒子にも好適に適用される。また、上記のその他の成分についても、同様に、本発明の多孔性微粒子分散体のシリカ以外の多孔性微粒子にも好適に適用される。
[2]多孔性シリカ微粒子分散体の製造方法
本発明の多孔性微粒子分散体の製造方法は、多孔性微粒子の細孔容積TPV(ml/g)に対応して前述の固形分濃度を満足するような分散体が得られる方法であれば良く、その製造方法には特に制限はない。
本発明の多孔性微粒子分散体の製造方法は、多孔性微粒子の細孔容積TPV(ml/g)に対応して前述の固形分濃度を満足するような分散体が得られる方法であれば良く、その製造方法には特に制限はない。
本発明の多孔性微粒子分散体の原料となる多孔性微粒子(多孔性物質原料)の物性は特に制限されないが、上述の各種物性を満たしていることが好ましく、従って、非晶質であること、細孔容積TPVが0.01ml/g以上であること、平均粒径X50が0.01μm以上であること、最頻細孔径(Dmax)が30nm未満であること、Dmax±20%容積比が20%以上であること、Dmax微分細孔容積ΔV/Δ(logd)が2ml/g以上であることが好ましい。
以下に、産業上有用なシリカを例に、分散体原料の多孔性シリカの製造方法、及びシリカ分散体の製造方法を説明するが、本発明は何ら以下の記載に限定されるものではない。
シリカゲルの製造方法としては、従来、最も一般的には、ケイ酸ソーダ等のケイ酸アルカリ塩を鉱酸で加水分解し、得られるシリカヒドロゾルをゲル化して乾燥する方法が用いられており、シリカゲルの性能を改良するために、この製造方法の詳細につき多くの提案がなされている。
例えば、特開昭62−113713号公報では、ケイ酸アルカリ水溶液と鉱酸との反応により生成したシリカヒドロゾルをゲル化し、これをpH2.5以下の酸溶液で処理し、水洗後、緩衝作用を有する水溶液中でpH4〜9に調整して水熱処理することにより、細孔分布の狭いシリカゲルを製造する方法が提案されている。また、特開平9−30809号公報では、シリカヒドロゲルの乾燥を回分式流動乾燥、次いで水熱処理する方法が提案されている。
これらの製造方法によれば、得られるシリカゲルの性能には確かに変化が認められ、よりシャープな細孔分布を有するシリカゲルが製造できる。しかし、得られるシリカゲルの細孔容積、比表面積及び平均細孔径を十分に変化させるまでには至らず、耐熱性や耐水性も充分ではないため、所望の物性範囲のシリカゲルを得る方法としては不十分である。
〔原料多孔性シリカ微粒子の製造方法〕
本発明の分散体の原料多孔性シリカ微粒子は、従来のゾル−ゲル法とは異なり、好ましくは、シリコンアルコキシドを加水分解する加水分解工程と共に得られたシリカヒドロゾルを縮合する縮合工程を経てシリカヒドロゲルを形成する加水分解・縮合工程と、当該加水分解・縮合工程に引き続き、シリカヒドロゲルを熟成することなく水熱処理することにより、所望の物性範囲の原料多孔性シリカを得る物性調節工程と、得られた原料シリカを微粉砕して一次粒子とし、これを凝集体の形状に造粒する微粉砕・造粒工程とを、ともに包含する方法で製造することができる。
本発明の分散体の原料多孔性シリカ微粒子は、従来のゾル−ゲル法とは異なり、好ましくは、シリコンアルコキシドを加水分解する加水分解工程と共に得られたシリカヒドロゾルを縮合する縮合工程を経てシリカヒドロゲルを形成する加水分解・縮合工程と、当該加水分解・縮合工程に引き続き、シリカヒドロゲルを熟成することなく水熱処理することにより、所望の物性範囲の原料多孔性シリカを得る物性調節工程と、得られた原料シリカを微粉砕して一次粒子とし、これを凝集体の形状に造粒する微粉砕・造粒工程とを、ともに包含する方法で製造することができる。
〈加水分解・縮合工程〉
原料シリカの材料として使用されるシリコンアルコキシドとしては、トリメトキシシラン、テトラメトキシシラン、トリエトキシシラン、テトラエトキシシラン、テトラプロポキシシラン、テトラブトキシシラン等の炭素数1〜4の低級アルキル基を有するトリ又はテトラアルコキシシラン或いはそれらのオリゴマーが挙げられるが、好ましくはテトラメトキシシラン、テトラエトキシシラン及びそれらのオリゴマーである。これらは1種を単独で用いても良く、2種以上を混合して用いても良い。
以上のシリコンアルコキシドは蒸留により容易に精製し得るので、高純度シリカの原料として好適である。
シリコンアルコキシド中の金属不純物の総含有量は、通常100ppm以下、中でも50ppm以下、更には30ppm以下、特に10ppm以下が好ましい。これらの金属不純物の含有率は、一般的なシリカ中の不純物含有率の測定法と同じ方法で測定できる。
原料シリカの材料として使用されるシリコンアルコキシドとしては、トリメトキシシラン、テトラメトキシシラン、トリエトキシシラン、テトラエトキシシラン、テトラプロポキシシラン、テトラブトキシシラン等の炭素数1〜4の低級アルキル基を有するトリ又はテトラアルコキシシラン或いはそれらのオリゴマーが挙げられるが、好ましくはテトラメトキシシラン、テトラエトキシシラン及びそれらのオリゴマーである。これらは1種を単独で用いても良く、2種以上を混合して用いても良い。
以上のシリコンアルコキシドは蒸留により容易に精製し得るので、高純度シリカの原料として好適である。
シリコンアルコキシド中の金属不純物の総含有量は、通常100ppm以下、中でも50ppm以下、更には30ppm以下、特に10ppm以下が好ましい。これらの金属不純物の含有率は、一般的なシリカ中の不純物含有率の測定法と同じ方法で測定できる。
シリコンアルコキシドの加水分解は、シリコンアルコキシド1モルに対して、通常2〜20モル、好ましくは3〜10モル、特に好ましくは4〜8モルの水を用いて行なう。シリコンアルコキシドの加水分解により、目的異元素をドープしたシリカのヒドロゲルとアルコールとが生成する。
この加水分解時の温度は、通常30℃以上、好ましくは40℃以上、更に好ましくは50℃以上、また、通常100℃以下、好ましくは90℃以下、中でも好ましくは80℃以下、更に好ましくは70℃以下である。この加水分解反応は、加圧下で液相を維持することで、より高い温度で行なうことも可能である。
また、加水分解時には必要に応じて、水と相溶性のあるアルコール類等の溶媒を添加してもよい。具体的には、炭素数1〜3の低級アルコール類、ジメチルホルムアミド、ジメチルスルホキシド、アセトン、テトラヒドロフラン、メチルセロルブ、エチルセロルブ、メチルエチルケトン、その他の水と任意に混合できる有機溶媒の1種又は2種以上を任意に用いることができるが、中でも強い酸性や塩基性を示さないものが、均一なシリカヒドロゲルを生成できる理由から好ましい。
これらの溶媒を使用しない場合、原料シリカの製造のためには、特に加水分解の際の攪拌速度が重要である。即ち、シリコンアルコキシドと加水分解用の水は初期には分液しているため、攪拌によりエマルジョン化し、反応を促進させる。
この際、攪拌を充分に行なうことが重要となる。例えば、回転軸に攪拌翼を備えた攪拌装置を用いた場合、その攪拌速度(回転軸の回転数)としては、攪拌翼の形状・枚数・液との接触面積等にもよるが、通常は30rpm以上、好ましくは40rpm以上である。また、この攪拌速度は、一般的に速過ぎると、槽内で生じた飛沫が各種のガスラインを閉塞させたり、また反応器内壁に付着して熱伝導を悪化させ、物性制御に重要な温度管理に影響を及ぼしたりする場合がある。更に、この内壁の付着物が剥離し、製品に混入して品質を悪化させる場合もある。この様な理由から、攪拌速度は1000rpm以下、中でも500rpm以下が好ましい。
この際、攪拌を充分に行なうことが重要となる。例えば、回転軸に攪拌翼を備えた攪拌装置を用いた場合、その攪拌速度(回転軸の回転数)としては、攪拌翼の形状・枚数・液との接触面積等にもよるが、通常は30rpm以上、好ましくは40rpm以上である。また、この攪拌速度は、一般的に速過ぎると、槽内で生じた飛沫が各種のガスラインを閉塞させたり、また反応器内壁に付着して熱伝導を悪化させ、物性制御に重要な温度管理に影響を及ぼしたりする場合がある。更に、この内壁の付着物が剥離し、製品に混入して品質を悪化させる場合もある。この様な理由から、攪拌速度は1000rpm以下、中でも500rpm以下が好ましい。
本発明において、分液している二液相(水相、及びシリコンアルコキシド相)の攪拌方法は、反応を促進させる方法であれば任意の攪拌方法を用いることができる。中でも、この二液相をより混合させるような装置としては、例えば以下の(i)、(ii)が挙げられる。
(i)回転軸が液面に対し垂直又は僅かに角度を持って挿入され、上下に液の流動が生じる攪拌翼を有する装置。
(ii)回転軸方向を二液相の界面と略平行に設け、二液相間に攪拌を生じさせる攪拌翼を有する攪拌装置。
上述した(i)、(ii)の様な装置を用いた際の攪拌翼の回転速度は、攪拌翼の周速度(攪拌翼先端速度)で、0.05〜10m/s、中でも0.1〜5m/s、さらには0.1〜3m/sであることが好ましい。
(i)回転軸が液面に対し垂直又は僅かに角度を持って挿入され、上下に液の流動が生じる攪拌翼を有する装置。
(ii)回転軸方向を二液相の界面と略平行に設け、二液相間に攪拌を生じさせる攪拌翼を有する攪拌装置。
上述した(i)、(ii)の様な装置を用いた際の攪拌翼の回転速度は、攪拌翼の周速度(攪拌翼先端速度)で、0.05〜10m/s、中でも0.1〜5m/s、さらには0.1〜3m/sであることが好ましい。
攪拌翼の形状や長さ等は任意であり、攪拌翼としては例えばプロペラ型、平羽根型、角度付平羽根型、ピッチ付平羽根型、平羽根ディスクタービン型、湾曲羽根型、ファウドラー型、ブルマージン型等が挙げられる。
翼の幅、枚数、傾斜角等は反応器の形状、大きさ、目的とする攪拌動力に応じて適宜選定すればよい。例えば反応器の槽内径(回転軸方向に対して垂直面を形成する液相面の最長径)に対する翼幅(回転軸方向の翼の長さ)の比率(b/D)は0.05〜0.2、傾斜角(θ)90゜±10゜、翼枚数3〜10枚の攪拌装置が好適な例として挙げられる。
中でも、上述の回転軸を反応容器内の液面よりも上に設け、この回転軸から伸ばした軸の先端部分に攪拌翼を設ける構造が、攪拌効率及び設備メンテナンスの観点から好適に使用される。
翼の幅、枚数、傾斜角等は反応器の形状、大きさ、目的とする攪拌動力に応じて適宜選定すればよい。例えば反応器の槽内径(回転軸方向に対して垂直面を形成する液相面の最長径)に対する翼幅(回転軸方向の翼の長さ)の比率(b/D)は0.05〜0.2、傾斜角(θ)90゜±10゜、翼枚数3〜10枚の攪拌装置が好適な例として挙げられる。
中でも、上述の回転軸を反応容器内の液面よりも上に設け、この回転軸から伸ばした軸の先端部分に攪拌翼を設ける構造が、攪拌効率及び設備メンテナンスの観点から好適に使用される。
かかる攪拌条件を満足しない場合には、原料シリカを得るのが困難になる。なお、加水分解によりアルコールが生成して液が均一液となり、発熱が収まった後には、均一なヒドロゲルを形成させるために攪拌を停止することが好ましい。
結晶性を有するシリカは、水中熱安定性に乏しくなる傾向にあり、ゲル中に細孔を形成するのに用いられる界面活性剤等のテンプレートの存在下でシリコンアルコキシドを加水分解すると、ゲルは容易に結晶性を示すものとなる。従って、本発明においては、界面活性剤等のテンプレートの非存在下で、即ち、これらがテンプレートとしての機能を発揮するほどの量は存在しない条件下で加水分解するのが好ましい。
反応時間は、反応液組成(シリコンアルコキシドの種類や、水とのモル比)並びに反応温度に依存し、ゲル化するまでの時間が異なるので、一概には規定されない。なお、反応系に触媒として、酸、アルカリ、塩類などを添加することで加水分解を促進させることができる。しかしながら、かかる添加物の使用は、後述するように、生成したヒドロゲルの熟成を引き起こすことになるので、本発明のシリカの製造においてはあまり好ましくない。
〈物性調節工程〉
上記のシリコンアルコキシドの加水分解反応では、シリコンアルコキシドが加水分解してシリケートが生成するが、引き続いて該シリケートの縮合反応が起こり、反応液の粘度が上昇し、最終的にゲル化してシリカヒドロゲルとなる。原料シリカを製造するためには、上記の加水分解により生成したシリカのヒドロゲルの硬さが上昇しないように、実質的に熟成することなく、直ちに水熱処理を行なうことが重要である。シリコンアルコキシドを加水分解すると、軟弱なシリカヒドロゲルが生成するが、このヒドロゲルを安定した熟成、あるいは乾燥させ、更にこれに水熱処理を施す方法では、最終的に細孔特性の制御された、本発明で規定する物性範囲のシリカを製造することは困難である。
上記のシリコンアルコキシドの加水分解反応では、シリコンアルコキシドが加水分解してシリケートが生成するが、引き続いて該シリケートの縮合反応が起こり、反応液の粘度が上昇し、最終的にゲル化してシリカヒドロゲルとなる。原料シリカを製造するためには、上記の加水分解により生成したシリカのヒドロゲルの硬さが上昇しないように、実質的に熟成することなく、直ちに水熱処理を行なうことが重要である。シリコンアルコキシドを加水分解すると、軟弱なシリカヒドロゲルが生成するが、このヒドロゲルを安定した熟成、あるいは乾燥させ、更にこれに水熱処理を施す方法では、最終的に細孔特性の制御された、本発明で規定する物性範囲のシリカを製造することは困難である。
上記にある、加水分解により生成したシリカヒドロゲルを、実質的に熟成することなく、直ちに水熱処理を行なうということは、シリカヒドロゲルが生成した直後の軟弱な状態が維持されたままで、次工程の水熱処理に供するようにするということを意味する。
具体的には、シリカヒドロゲルが生成した時点から、一般的には10時間以内に水熱処理することが好ましく、中でも8時間以内、更には6時間以内、特に4時間以内にシリカヒドロゲルを水熱処理することが好ましい。
また、工業用プラント等においては、大量に生成したシリカヒドロゲルを一旦サイロ等に貯蔵し、その後水熱処理を行なう場合が考えられる。この様な場合、シリカヒドロゲルは、シリカヒドロゲルが生成してから水熱処理に供されるまでの時間、いわゆる放置時間が、上述の範囲を超える場合が考えられる。この様な場合には、熟成が実質的に生じないように、サイロ内での静置中に、例えばシリカヒドロゲル中の液体成分が乾燥しないようにすればよい。
具体的には、サイロ内を密閉したり、湿度を調節したりすればよい。また、水やその他の溶媒にシリカヒドロゲルを浸した状態で、シリカヒドロゲルを静置してもよい。
静置の際の温度は、できるだけ低くすることが好ましく、例えば50℃以下、中でも35℃以下、特に30℃以下で静置することが好ましい。また、熟成が実質的に生じないようにする別の方法としては、シリカヒドロゲル中のシリカ濃度が低くなるように、予め原料組成を制御してシリカヒドロゲルを調製する方法が挙げられる。
具体的には、サイロ内を密閉したり、湿度を調節したりすればよい。また、水やその他の溶媒にシリカヒドロゲルを浸した状態で、シリカヒドロゲルを静置してもよい。
静置の際の温度は、できるだけ低くすることが好ましく、例えば50℃以下、中でも35℃以下、特に30℃以下で静置することが好ましい。また、熟成が実質的に生じないようにする別の方法としては、シリカヒドロゲル中のシリカ濃度が低くなるように、予め原料組成を制御してシリカヒドロゲルを調製する方法が挙げられる。
シリカヒドロゲルを実質的に熟成せずに水熱処理することにより奏される効果と、この効果が得られる理由を考察すると、以下のことが考えられる。
まず、シリカヒドロゲルを熟成させると、−Si−O−Si−結合によるマクロ的網目構造が、シリカヒドロゲル全体に形成されると考えられる。この網目構造がシリカヒドロゲル全体に有ることで、水熱処理の際、この網目構造が障害となり、メソポーラスの形成が困難となることが考えられる。またシリカヒドロゲル中のシリカ濃度が低くなるように、予め原料組成を制御して得られたシリカヒドロゲルは、静置中に生ずるシリカヒドロゲルにおける架橋の進行を抑制できる。その為、シリカヒドロゲルが熟成しないと考える。
よって、本発明では、シリカヒドロゲルを熟成することなく、水熱処理を行なうことが重要である。
まず、シリカヒドロゲルを熟成させると、−Si−O−Si−結合によるマクロ的網目構造が、シリカヒドロゲル全体に形成されると考えられる。この網目構造がシリカヒドロゲル全体に有ることで、水熱処理の際、この網目構造が障害となり、メソポーラスの形成が困難となることが考えられる。またシリカヒドロゲル中のシリカ濃度が低くなるように、予め原料組成を制御して得られたシリカヒドロゲルは、静置中に生ずるシリカヒドロゲルにおける架橋の進行を抑制できる。その為、シリカヒドロゲルが熟成しないと考える。
よって、本発明では、シリカヒドロゲルを熟成することなく、水熱処理を行なうことが重要である。
シリコンアルコキシドの加水分解反応系に酸、アルカリ、塩類等を添加すること、又は該加水分解反応の温度を厳しくし過ぎることなどは、ヒドロゲルの熟成を進行させるため好ましくない。また、加水分解後の後処理における水洗、乾燥、放置などにおいて、必要以上に温度や時間をかけるべきではない。
ヒドロゲルの熟成状態を具体的に確認する手段としては、後述の実施例に示すような方法で測定したヒドロゲルの硬度(圧壊強度)を参考にすることができる。即ち、破壊応力が、通常6MPa以下、好ましくは3MPa以下、更に好ましくは2MPa以下の柔らかい状態のヒドロゲルを水熱処理することで、本発明で規定する物性範囲のシリカを得ることができる。
この水熱処理の条件としては、水の状態が液体、気体のいずれでもよく、溶媒や他の気体によって希釈されていてもよいが、好ましくは液体の水が使われる。シリカのヒドロゲルに対して、通常0.1〜10重量倍、好ましくは0.5〜5重量倍、特に好ましくは1〜3重量倍の水を加えて分散体状とし、通常40〜250℃、好ましくは50〜200℃の温度で、通常0.1〜100時間、好ましくは1〜10時間実施される。
水熱処理に使用される水には低級アルコール類、メタノール、エタノール、プロパノールや、ジメチルホルムアミド(DMF)やジメチルスルホキシド(DMSO)、その他の有機溶媒などが含まれてもよい。
なお、加水分解反応の反応器を用い、続けて温度条件変更により水熱処理を行なうことも可能であるが、加水分解反応とその後の水熱処理とでは通常は最適条件が異なっているため、この方法で本発明の原料シリカを得ることは一般的には難しい。
以上の水熱処理条件において温度を高くすると、得られるシリカの細孔径、細孔容積が大きくなる傾向がある。水熱処理温度としては、100〜200℃の範囲であることが好ましい。また、処理時間とともに、得られるシリカの比表面積は、一度極大に達した後、緩やかに減少する傾向がある。以上の傾向を踏まえて、所望の物性値に応じて条件を適宜選択する必要があるが、水熱処理は、シリカの物性を変化させる目的なので、通常、前記の加水分解の反応条件より高温条件とすることが好ましい。
水熱処理の温度、時間を上記範囲外に設定すると、本発明に好適な原料シリカを得ることが困難となる。例えば、水熱処理の温度が高すぎると、シリカゲルの細孔径、細孔容積が大きくなりすぎ、また、細孔分布も広がる。逆に、水熱処理の温度が低過ぎると、生成するシリカは、架橋度が低く、熱安定性に乏しくなり、細孔分布にピークが発現しなくなったり、前述した固体Si−NMRにおけるQ4/Q3値が極端に小さくなったりする。
特に、水熱処理の際に、反応系内の温度が5時間以内に目的温度に達する様に、速い昇温速度条件とすることが好ましく、具体的には、槽に充填して処理される場合、昇温開始から目標温度到達までの平均昇温速度を0.1〜100℃/min、中でも0.1〜30℃/min、特に0.2〜10℃/minとするのが好ましい。熱交換器などを利用した昇温方法、予め作っておいた熱水を仕込む方法なども、昇温速度を短縮することができるので好ましい。また、昇温速度が上記範囲ならば、段階的に昇温を行なってもよい。反応系内の温度が目的温度に達するまでに長期間を要した場合は、昇温中にシリカヒドロゲルの熟成が進んで、ミクロ構造的な均質性が低下する虞がある。上記の目的温度に達するまでの昇温時間は、好ましくは4時間以内、更に好ましくは3時間以内である。昇温時間の短縮のため、水熱処理に使用する水を予熱することもできる。
なお、水熱処理をアンモニア水中で行なうと、純水中で行なう場合よりも低温で同様の効果が得られる。また、アンモニア水中で水熱処理すると、純水中で処理する場合と比較して、最終的に得られるシリカは一般に疎水性となるが、通常30〜250℃、好ましくは40〜200℃という比較的高温で水熱処理すると、特に疎水性が高くなる。ここでのアンモニア水のアンモニア濃度としては、好ましくは0.001〜10重量%、特に好ましくは0.005〜5重量%である。
水熱処理されたシリカヒドロゲルは、通常40〜200℃、好ましくは60〜120℃で乾燥する。乾燥方法は特に限定されるものではなく、バッチ式でも連続式でもよく、且つ、常圧でも減圧下でも乾燥することができる。ただし、次工程の粉砕工程が湿式法である場合には、乾燥を省略する場合もある。必要に応じ、原料のシリコンアルコキシドに由来する炭素分が含まれている場合には、通常400〜600℃での焼成により除去することができる。また、表面状態をコントロールするため、最高900℃の温度で焼成することもある。
〈微粉砕・造粒工程〉
分散体の原料多孔性シリカ微粒子の平均粒径X50が0.01μm〜0.1μmと極めて微細である場合には、上述した様に、本発明の効果が顕著であるため好ましい。このような微細なシリカを得るためには、上述の手順で得られたシリカを微粉砕する必要がある。
分散体の原料多孔性シリカ微粒子の平均粒径X50が0.01μm〜0.1μmと極めて微細である場合には、上述した様に、本発明の効果が顕著であるため好ましい。このような微細なシリカを得るためには、上述の手順で得られたシリカを微粉砕する必要がある。
シリカを微粉砕する方法としては、公知のいかなる装置・器具を用いてもよいが、平均粒径10μm以下の微粉(シリカ微粒子)を得るためには、ボールミル(転動ミル、振動ボールミル、遊星ミル等)、攪拌ミル(塔式粉砕器、攪拌槽型ミル、流通管型ミル、アニュラー(環状)ミル等)、高速回転微粉砕機(スクリーンミル、ターボ型ミル、遠心分級型ミル)、ジェット粉砕機(循環ジェットミル、衝突タイプミル、流動層ジェットミル)、せん断ミル(擂解機、オングミル)、コロイドミル、乳鉢などの装置・器具を用いることができる。これらの中で、平均粒径2μm以下の超微粒子を得る際には、ボールミル、攪拌ミルがより好ましい。また、粉砕時の状態としては、湿式法及び乾式法があり、何れも選択可能であるが、超微粒子を得るためには湿式法がより好ましい。湿式法の場合、使用する分散媒としては、水及びアルコール等の有機溶媒の何れを用いても、また2種以上の混合溶媒としてもよく、目的に応じて使い分ける。また、湿式法の場合には、次工程の造粒工程に入る前に、必要に応じて乾燥を行なうことがある。微粉砕時に不必要に強い圧力や剪断力を長時間かけ続けることは、原料シリカの細孔特性を損なうことがあり好ましくない。
〔多孔性シリカ微粒子分散体の調製〕
以上の手順で得られたシリカを分散媒に分散させる手段は特に制限されず、公知の各種の分散手段を用いることが可能である。具体例としては、アトライタ、ビーズミル、ボールミル等を用いることができる。
以上の手順で得られたシリカを分散媒に分散させる手段は特に制限されず、公知の各種の分散手段を用いることが可能である。具体例としては、アトライタ、ビーズミル、ボールミル等を用いることができる。
また、本発明のシリカ分散体に水を含有させるための手法は特に制限されず、単に水と混合する方法の他、例えば、次のようにして親水性有機溶媒を併用する方法であっても良い。
(I)親水性有機溶媒と水を予め混合し、これを分散媒としてシリカを分散させる手法
(II)親水性有機溶媒を分散媒として、これに水を含有しているシリカを加えて分散させる手法
(III)親水性有機溶媒を分散媒として、これにシリカを加えて分散させ、更に水を加えて混合する手法
また、(I)〜(III)のうち複数の手法を組み合わせて用いてもよい。
何れの場合も、最終的に得られる分散体の固形物濃度が、本発明の規定の範囲内となればよい。
(I)親水性有機溶媒と水を予め混合し、これを分散媒としてシリカを分散させる手法
(II)親水性有機溶媒を分散媒として、これに水を含有しているシリカを加えて分散させる手法
(III)親水性有機溶媒を分散媒として、これにシリカを加えて分散させ、更に水を加えて混合する手法
また、(I)〜(III)のうち複数の手法を組み合わせて用いてもよい。
何れの場合も、最終的に得られる分散体の固形物濃度が、本発明の規定の範囲内となればよい。
なお、シリカ分散体の調製後に、前述の手法でシリカの微粉砕を行なうことにより、微細粒径のシリカを含有するシリカ分散体を得てもよい。
[3]多孔性シリカ微粒子分散体の用途
以上説明した本発明の多孔性シリカ微粒子分散体は、保存安定性に優れているという効果を有する。特に、平均粒径X50が0.01μm以上のシリカ微粒子を分散させる場合に、その効果は顕著である。
こうした効果を有する本発明のシリカ分散体は、従来のシリカ分散体の用途を始めとする、各種の用途に利用することができる。このうち従来の用途としては、以下のものが挙げられる。
以上説明した本発明の多孔性シリカ微粒子分散体は、保存安定性に優れているという効果を有する。特に、平均粒径X50が0.01μm以上のシリカ微粒子を分散させる場合に、その効果は顕著である。
こうした効果を有する本発明のシリカ分散体は、従来のシリカ分散体の用途を始めとする、各種の用途に利用することができる。このうち従来の用途としては、以下のものが挙げられる。
例えば、産業用設備で製品の製造及び処理に用いられる用途分野においては、各種触媒及び触媒担体(酸塩基触媒、光触媒、貴金属触媒等)、廃水・廃油処理剤、臭気処理剤、ガス分離剤、工業用乾燥剤、バイオリアクター、バイオセパレーター、メンブランリアクター等の用途が挙げられる。
建材用途では、調湿剤、防音・吸音材、耐火物、断熱材等の用途が挙げられる。
空調分野の用途では、デシカント空調機用調湿剤、ヒートポンプ用蓄熱剤等が挙げられる。
塗料・インク用途分野においては、艶消し剤、粘度調整剤、色度調整剤、沈降防止剤、消泡剤、インク裏抜け防止剤、スタンピングホイル用、壁紙用等の用途が挙げられる。
樹脂用添加剤用途分野においては、フィルム用アンチブロッキング剤(ポリオレフィンフィルム等)、プレートアウト防止剤、シリコーン樹脂用補強剤、ゴム用補強剤(タイヤ用・一般ゴム用等)、流動性改良材、パウダー状樹脂の固結防止剤、印刷適性改良剤、合成皮革やコーティングフィルム用の艶消し剤、接着剤・粘着テープ用充填剤、透光性調整剤、防眩性調整剤、多孔性ポリマーシート用フィラー等の用途が挙げられる。
製紙用途分野においては、感熱紙用フィラー(カス付着防止剤等)、インクジェット紙画像向上用フィラー(インク吸収剤等)、ジアゾ感光紙用フィラー(感光濃度向上剤等)、トレーシングペーパー用筆記性改良剤、コート紙用フィラー(筆記性、インク吸収性、アンチブロッキング性改良剤等)、静電記録用フィラー等の用途が挙げられる。
食品用途分野においては、ビール用濾過助剤、醤油・清酒・ワイン等発酵製品のおり下げ剤、各種発酵飲料の安定化剤(混濁因子タンパクや酵母の除去等)、食品添加剤、粉末食品の固結防止剤等の用途が挙げられる。
医農薬分野においては、薬品等の打錠助剤、粉砕助剤、分散・医薬用担体(分散・徐放・デリバリー性改善等)、農薬用担体(油状農薬キャリア・水和分散性改善、徐放・デリバリー性改善等)、医薬用添加剤(固結防止剤・粉粒性改良剤等)・農薬用添加剤(固結防止剤・沈降防止剤等)等が挙げられる。
分離材料分野では、クロマトグラフィー用充填剤、分離剤、フラーレン分離剤、吸着剤(タンパク質・色素・臭等)、脱湿剤等の用途が挙げられる。
農業用分野では、飼料用添加剤、肥料用添加剤が挙げられる。
さらにその他の用途として、生活関連分野では、調湿剤、乾燥剤、化粧品添加剤、抗菌剤、消臭・脱臭・芳香剤、洗剤用添加剤(界面活性剤粉末化等)、研磨剤(歯磨き用等)、粉末消火剤(粉粒性改良剤・固結防止剤等)、消泡剤、バッテリーセパレーター等が挙げられる。
更に、これら各種の分野に広く用いられる用途として、調湿繊維としての用途が挙げられる。
建材用途では、調湿剤、防音・吸音材、耐火物、断熱材等の用途が挙げられる。
空調分野の用途では、デシカント空調機用調湿剤、ヒートポンプ用蓄熱剤等が挙げられる。
塗料・インク用途分野においては、艶消し剤、粘度調整剤、色度調整剤、沈降防止剤、消泡剤、インク裏抜け防止剤、スタンピングホイル用、壁紙用等の用途が挙げられる。
樹脂用添加剤用途分野においては、フィルム用アンチブロッキング剤(ポリオレフィンフィルム等)、プレートアウト防止剤、シリコーン樹脂用補強剤、ゴム用補強剤(タイヤ用・一般ゴム用等)、流動性改良材、パウダー状樹脂の固結防止剤、印刷適性改良剤、合成皮革やコーティングフィルム用の艶消し剤、接着剤・粘着テープ用充填剤、透光性調整剤、防眩性調整剤、多孔性ポリマーシート用フィラー等の用途が挙げられる。
製紙用途分野においては、感熱紙用フィラー(カス付着防止剤等)、インクジェット紙画像向上用フィラー(インク吸収剤等)、ジアゾ感光紙用フィラー(感光濃度向上剤等)、トレーシングペーパー用筆記性改良剤、コート紙用フィラー(筆記性、インク吸収性、アンチブロッキング性改良剤等)、静電記録用フィラー等の用途が挙げられる。
食品用途分野においては、ビール用濾過助剤、醤油・清酒・ワイン等発酵製品のおり下げ剤、各種発酵飲料の安定化剤(混濁因子タンパクや酵母の除去等)、食品添加剤、粉末食品の固結防止剤等の用途が挙げられる。
医農薬分野においては、薬品等の打錠助剤、粉砕助剤、分散・医薬用担体(分散・徐放・デリバリー性改善等)、農薬用担体(油状農薬キャリア・水和分散性改善、徐放・デリバリー性改善等)、医薬用添加剤(固結防止剤・粉粒性改良剤等)・農薬用添加剤(固結防止剤・沈降防止剤等)等が挙げられる。
分離材料分野では、クロマトグラフィー用充填剤、分離剤、フラーレン分離剤、吸着剤(タンパク質・色素・臭等)、脱湿剤等の用途が挙げられる。
農業用分野では、飼料用添加剤、肥料用添加剤が挙げられる。
さらにその他の用途として、生活関連分野では、調湿剤、乾燥剤、化粧品添加剤、抗菌剤、消臭・脱臭・芳香剤、洗剤用添加剤(界面活性剤粉末化等)、研磨剤(歯磨き用等)、粉末消火剤(粉粒性改良剤・固結防止剤等)、消泡剤、バッテリーセパレーター等が挙げられる。
更に、これら各種の分野に広く用いられる用途として、調湿繊維としての用途が挙げられる。
本発明のシリカ分散体は、これらの用途の中でも特に安定性の求められる用途、更には、制御された細孔特性が要求されるとともに、長期にわたって物性変化の少ないことが要求される用途において、好適に用いることができる。
具体的には、シリカ分散体に樹脂、有機溶媒等の添加剤を加え塗料として使用する場合、分散体に含有されているシリカと樹脂、有機溶媒とが充分に混合される必要があることから、シリカが充分に分散している分散体が必要となる。ここで、分散性が悪いシリカ分散体を用いると、前述の如く、凝集したシリカ粒子が塗布時に残り平滑な塗布面が得られない。このため、分散性に優れた本発明のシリカ分散体が有効に適用される。特に、平均粒径X50が2μm以下の微細なシリカを用いる用途や、シリカ含有率が1重量%以上という高濃度のシリカ分散体が要求される用途において、好適に利用することが可能である。
以下、本発明を実施例により詳細に説明するが、本発明はその要旨を逸脱しない範囲において、以下の実施例に制限されること無く、任意に変形して実施することができる。
(1)シリカ分散体の分析方法
1−1)細孔容積
対象となる分散体を90℃で通風乾燥し、得られたサンプルを用いて次に示す方法で測定した。
カンタクローム社製AS−6にてBET窒素吸着等温線を測定し、細孔容積を求めた。具体的には、P/P0=0.98の時の値を採用した。
また、BJH法で細孔分布曲線及び最頻細孔径(Dmax)における微分細孔容積を求めた。測定する相対圧の各点の間隔は0.025とした。
1−2)比表面積
対象となる分散体を90°で乾燥し、得られたサンプルについて、カンタクローム社製AS−6にてBET窒素吸着等温線を測定し、比表面積を求めた。比表面積は、P/P0=0.1,0.2,0.3の3点の窒素吸着量よりBET多点法を用いて算出した。
1−3)粒度分布
レーザー回折式粒度分布計(堀場製作所製LA−920)を用いて測定を行なった。得られた粒度分布から体積基準積算粒度分布50%の粒径X50を算出し、これを平均粒径とした。
1−4)圧壊強度
島津製作所製オートグラフAG−1を用いて圧壊強度測定を行った。
1−1)細孔容積
対象となる分散体を90℃で通風乾燥し、得られたサンプルを用いて次に示す方法で測定した。
カンタクローム社製AS−6にてBET窒素吸着等温線を測定し、細孔容積を求めた。具体的には、P/P0=0.98の時の値を採用した。
また、BJH法で細孔分布曲線及び最頻細孔径(Dmax)における微分細孔容積を求めた。測定する相対圧の各点の間隔は0.025とした。
1−2)比表面積
対象となる分散体を90°で乾燥し、得られたサンプルについて、カンタクローム社製AS−6にてBET窒素吸着等温線を測定し、比表面積を求めた。比表面積は、P/P0=0.1,0.2,0.3の3点の窒素吸着量よりBET多点法を用いて算出した。
1−3)粒度分布
レーザー回折式粒度分布計(堀場製作所製LA−920)を用いて測定を行なった。得られた粒度分布から体積基準積算粒度分布50%の粒径X50を算出し、これを平均粒径とした。
1−4)圧壊強度
島津製作所製オートグラフAG−1を用いて圧壊強度測定を行った。
(2)シリカ分散体の製造、評価
[実施例1]
・原料シリカの製造
ガラス製で、上部に大気開放の水冷コンデンサが取り付けてある5Lセパラブルフラスコ(ジャケット付き)に、純水1000gを仕込んだ。80rpmで撹拌しながら、これにテトラメトキシシラン(金属不純物の総含有量0.1ppm)1400gを3分間かけて仕込んだ。水/テトラメトキシシランのモル比は約6である。セパラブルフラスコのジャケットには50℃の温水を通水した。引き続き撹拌を継続し、内容物が沸点に到達した時点で、撹拌を停止した。引き続き約0.5時間、ジャケットに50℃の温水を通水して生成したゾルをゲル化させた。その後、速やかにゲルを取り出し、目開き600ミクロンのナイロン製網を通してゲルを粉砕し、粉体状のウェットゲル(シリカヒドロゲル)を得た。このヒドロゲルの圧壊強度は3MPaであった。
このヒドロゲル300gと純水450gを1Lのガラス製オートクレーブに仕込み、b/D=0.2、傾斜角90°、翼枚数3の平羽根型攪拌翼を用い、2m/sの周速度で攪拌しながら、昇温速度10℃/minで昇温して、160℃、3時間の条件で水熱処理を実施した。水熱処理後、No.5A濾紙で濾過し、得られたシリカゲルを水洗することなく100℃で恒量となるまで減圧乾燥して、原料となるシリカを得た。原料シリカの諸物性を下記の表1に示す。
[実施例1]
・原料シリカの製造
ガラス製で、上部に大気開放の水冷コンデンサが取り付けてある5Lセパラブルフラスコ(ジャケット付き)に、純水1000gを仕込んだ。80rpmで撹拌しながら、これにテトラメトキシシラン(金属不純物の総含有量0.1ppm)1400gを3分間かけて仕込んだ。水/テトラメトキシシランのモル比は約6である。セパラブルフラスコのジャケットには50℃の温水を通水した。引き続き撹拌を継続し、内容物が沸点に到達した時点で、撹拌を停止した。引き続き約0.5時間、ジャケットに50℃の温水を通水して生成したゾルをゲル化させた。その後、速やかにゲルを取り出し、目開き600ミクロンのナイロン製網を通してゲルを粉砕し、粉体状のウェットゲル(シリカヒドロゲル)を得た。このヒドロゲルの圧壊強度は3MPaであった。
このヒドロゲル300gと純水450gを1Lのガラス製オートクレーブに仕込み、b/D=0.2、傾斜角90°、翼枚数3の平羽根型攪拌翼を用い、2m/sの周速度で攪拌しながら、昇温速度10℃/minで昇温して、160℃、3時間の条件で水熱処理を実施した。水熱処理後、No.5A濾紙で濾過し、得られたシリカゲルを水洗することなく100℃で恒量となるまで減圧乾燥して、原料となるシリカを得た。原料シリカの諸物性を下記の表1に示す。
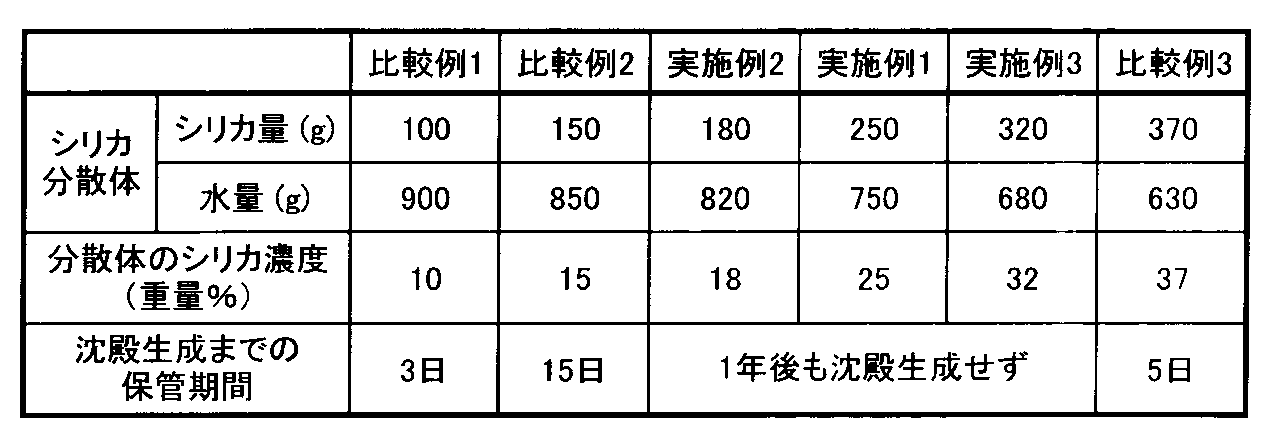
上述の原料シリカを、スクリューフィーダーを用いて供給速度2.5kg/hrでホソカワミクロン製AFG200型ジェットミルに供給し、粉砕圧力0.6MPa、分級ローター回転数11,500rpmにて予備粉砕した。予備粉砕後のシリカの諸物性を下記の表2に示す。
得られたシリカを濾布にて回収し、このうち250gを、分散媒として750gの水に投入し、攪拌して分散させることにより、シリカ分散体を得た。分散体濃度Cは(25重量%)。この分散体を、三井鉱山製SC−50型ビーズミルにて、回転数2400rpm、ジルコニアビーズ径0.2mmにて1時間粉砕を行ない、平均粒径X501.1μmのシリカを含有する分散体(実施例1のシリカ分散体)を得た。
得られた実施例1のシリカ分散体を目視で観察したところ、沈殿が観察されず、且つ、流動性があることが分かった。また、粒度分布測定を行なったところ、シャープな粒度分布が得られた。これらの点から、シリカがよく分散されていることが確認された。この調整直後の実施例1のシリカ分散体のシリカの最頻細孔径Dmax(nm)、Dmax微分細孔容積ΔV/Δ(logd)、比表面積(m2/g)、細孔容積TPV(ml/g)、Dmax±20%容積比(%)と、目視観察による沈殿の有無を表3に示す。
・評価
実施例1のシリカ分散体を500mlポリエチレン製容器に入れて蓋をし、室温にて保管を行なったところ、3日目でやわらかいゲル状になっていることが確認された。これが前述の水被覆微粒子が互いに支えあった状態であるが、このものは、軽く振揺することで容易にもとの液状に戻ることが確認された。
シリカ分散体の調製から3時間、1日、3日、1週間、2週間、1ヶ月、2ヶ月、3ヶ月、6ヶ月、9ヶ月、1年の各時間経過後における外観検査、粒度分布、細孔分布等の測定、沈殿の生成の有無の検査を行なったが、何れの時点でも変化が見られなかった。このうち、調製から3時間後、3日後、6ヶ月後のシリカの最頻細孔径Dmax(nm)、Dmax微分細孔容積ΔV/Δ(logd)、比表面積(m2/g)、細孔容積TPV(ml/g)、Dmax±20%容積比(%)と、目視観察による沈殿の有無を表3に示す。
実施例1のシリカ分散体を500mlポリエチレン製容器に入れて蓋をし、室温にて保管を行なったところ、3日目でやわらかいゲル状になっていることが確認された。これが前述の水被覆微粒子が互いに支えあった状態であるが、このものは、軽く振揺することで容易にもとの液状に戻ることが確認された。
シリカ分散体の調製から3時間、1日、3日、1週間、2週間、1ヶ月、2ヶ月、3ヶ月、6ヶ月、9ヶ月、1年の各時間経過後における外観検査、粒度分布、細孔分布等の測定、沈殿の生成の有無の検査を行なったが、何れの時点でも変化が見られなかった。このうち、調製から3時間後、3日後、6ヶ月後のシリカの最頻細孔径Dmax(nm)、Dmax微分細孔容積ΔV/Δ(logd)、比表面積(m2/g)、細孔容積TPV(ml/g)、Dmax±20%容積比(%)と、目視観察による沈殿の有無を表3に示す。
なお、シリカの細孔容積TPVを、前述の本発明に係る固形分濃度の式に代入して求めた固形分濃度濃度C範囲は、17.1〜34.1重量%であり、シリカ濃度25重量%の実施例1のシリカ分散体は本発明の固形分濃度条件を満たすものである。
[比較例1]
シリカを100g、分散媒の水を900gとした他は、実施例1と同様の作業を行なうことにより、シリカ分散体(シリカ濃度10重量%)を作製した(比較例1のシリカ分散体)。得られた比較例1のシリカ分散体を実施例1と同様の手順で評価したところ、保管3日経過時点で下部に沈殿が生成していることが認められた。
シリカを100g、分散媒の水を900gとした他は、実施例1と同様の作業を行なうことにより、シリカ分散体(シリカ濃度10重量%)を作製した(比較例1のシリカ分散体)。得られた比較例1のシリカ分散体を実施例1と同様の手順で評価したところ、保管3日経過時点で下部に沈殿が生成していることが認められた。
[実施例2,3、比較例2,3]
シリカと分散媒の水の量を表4に示す通りとした他は、実施例1と同様の作業を行なうことにより、各々シリカ分散体を作製し、得られたシリカ分散体について実施例1と同様の手順で評価を行い、沈殿が生成するまでの保管期間を調べ、結果を、実施例1と比較例1の結果と共に表4に示した。
シリカと分散媒の水の量を表4に示す通りとした他は、実施例1と同様の作業を行なうことにより、各々シリカ分散体を作製し、得られたシリカ分散体について実施例1と同様の手順で評価を行い、沈殿が生成するまでの保管期間を調べ、結果を、実施例1と比較例1の結果と共に表4に示した。
以上説明したように、本発明の多孔性微粒子分散体は、保存安定性に優れていることから、従来の多孔性微粒子分散体の用途を始めとする各種の用途に利用することが可能であり、その産業上の利用可能性は極めて高い。
Claims (4)
- 多孔性微粒子が分散媒に分散してなる分散体において、
該多孔性微粒子の細孔容積をTPV(ml/g)、分散体中の多孔性微粒子濃度をC(重量%)としたとき、
50/(1.8+TPV)≦C≦100/(1.8+TPV)
の関係を満たすことを特徴とする多孔性微粒子分散体。 - 多孔性微粒子の平均粒径X50が0.01μm以上であることを特徴とする請求項1に記載の多孔性微粒子分散体。
- 多孔性微粒子の細孔容積TPVが0.01ml/g以上であることを特徴とする請求項1又は2に記載の多孔性微粒子分散体。
- 多孔性微粒子がシリカであることを特徴とする請求項1ないし3の何れか1項に記載の多孔性微粒子分散体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005201885A JP2007016190A (ja) | 2005-07-11 | 2005-07-11 | 多孔性微粒子分散体 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005201885A JP2007016190A (ja) | 2005-07-11 | 2005-07-11 | 多孔性微粒子分散体 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2007016190A true JP2007016190A (ja) | 2007-01-25 |
Family
ID=37753630
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005201885A Pending JP2007016190A (ja) | 2005-07-11 | 2005-07-11 | 多孔性微粒子分散体 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2007016190A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020530868A (ja) * | 2017-08-03 | 2020-10-29 | ダブリュー・アール・グレース・アンド・カンパニー−コーンW R Grace & Co−Conn | シリカ系艶消し剤及びそれを作製及び使用する方法 |
-
2005
- 2005-07-11 JP JP2005201885A patent/JP2007016190A/ja active Pending
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020530868A (ja) * | 2017-08-03 | 2020-10-29 | ダブリュー・アール・グレース・アンド・カンパニー−コーンW R Grace & Co−Conn | シリカ系艶消し剤及びそれを作製及び使用する方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5218465B2 (ja) | シリカ及びその製造方法 | |
| US20050047985A1 (en) | Silica | |
| CN102295290A (zh) | 二氧化硅颗粒的制造方法 | |
| CN101796144A (zh) | 经环状物处理的金属氧化物 | |
| WO2006123433A1 (ja) | 微粒子状シリカ | |
| Lazareva et al. | Synthesis of high-purity silica nanoparticles by sol-gel method | |
| WO2010082442A1 (ja) | 非晶質シリカ及びその製造法 | |
| Yang et al. | Disperse ultrafine amorphous SiO2 nanoparticles synthesized via precipitation and calcination | |
| CN1318300C (zh) | 无机氧化物 | |
| JP2003226516A (ja) | シリカ及びその製造方法 | |
| JP4314077B2 (ja) | シリカ及びその製造方法 | |
| JP4160350B2 (ja) | シリカ、及びシリカの製造方法 | |
| JP2007016190A (ja) | 多孔性微粒子分散体 | |
| JP4160348B2 (ja) | シリカ、及びシリカの製造方法 | |
| JP2008273834A (ja) | シリカ | |
| JP4314076B2 (ja) | シリカ及びその製造方法 | |
| JP2008222552A (ja) | シリカ | |
| JP4160349B2 (ja) | シリカヒドロゲル及びシリカ、並びにシリカヒドロゲルの製造方法 | |
| JP2003160326A (ja) | シリカゲル | |
| JP5701087B2 (ja) | 球状シリカ−チタニア複合酸化物粒子及びその製造方法 | |
| JP2003226515A (ja) | シリカ及びその製造方法 | |
| JP2003171112A (ja) | シリカ | |
| JP2003238142A (ja) | シリカ微粒子凝集体 | |
| JP4163919B2 (ja) | シリカ、及びシリカの製造方法 | |
| JPH0764548B2 (ja) | 単分散されたシリカ粒子の製造方法 |