JP2006160902A - 高分子電解質膜及びその製造方法 - Google Patents
高分子電解質膜及びその製造方法 Download PDFInfo
- Publication number
- JP2006160902A JP2006160902A JP2004355264A JP2004355264A JP2006160902A JP 2006160902 A JP2006160902 A JP 2006160902A JP 2004355264 A JP2004355264 A JP 2004355264A JP 2004355264 A JP2004355264 A JP 2004355264A JP 2006160902 A JP2006160902 A JP 2006160902A
- Authority
- JP
- Japan
- Prior art keywords
- polymer
- electrolyte membrane
- group
- fluorine
- film
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
Landscapes
- Manufacture Of Macromolecular Shaped Articles (AREA)
- Conductive Materials (AREA)
- Fuel Cell (AREA)
Abstract
【課題】メタノールのクロスオーバーが少なく、直接メタノール燃料電池の電解質膜として有用な電解質膜を提供する。
【解決手段】−SO2X基を有するポリマー(Xはフッ素原子又は塩素原子)と、該ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーとを混合する工程、前記含フッ素ポリマーが溶融する温度で混練する工程、押し出し成形する工程、圧延する工程及び−SO2X基をスルホン酸基、スルホンイミド基等のプロトン導電性の基に変換する工程を経て高分子電解質膜を得る。
【選択図】図2
【解決手段】−SO2X基を有するポリマー(Xはフッ素原子又は塩素原子)と、該ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーとを混合する工程、前記含フッ素ポリマーが溶融する温度で混練する工程、押し出し成形する工程、圧延する工程及び−SO2X基をスルホン酸基、スルホンイミド基等のプロトン導電性の基に変換する工程を経て高分子電解質膜を得る。
【選択図】図2
Description
本発明は高分子電解質膜、特に直接メタノール燃料電池に有用な高分子電解質膜に関する。
直接メタノール燃料電池(以下、DMFCという)は、アノードに燃料としてメタノール又はメタノール水溶液を供給し、カソードには酸素又は空気を供給することにより、電力を得る電気化学的エネルギー変換システムである。DMFCでは改質器を必要としないため、水素を燃料とする固体高分子型燃料電池に比べて、システム全体を小型化することができる。また、燃料を液体として供給することができるため、高いエネルギー密度が期待できる。このため、携帯機器用から車載用等移動用電源としての幅広い応用を目指し、活発に研究が進められている。
DMFCの理論的な出力電圧としては、水素−酸素燃料電池とほぼ同等の約1.2Vを期待できるが、分極抵抗が大きいため、現状では約0.8V程度しか得られていない。このため、メタノールの陽極酸化反応について多くの研究がなされているものの、いまだに十分な特性は得られていない。この理由としては、主に、十分な活性を有するメタノール酸化触媒が見出されていないこと、通常電解質として多く用いられるイオン交換膜ではメタノール透過性が高いため、燃料であるメタノールの利用効率が低く、また対極のカソードに到達したメタノールがカソード表面で反応することにより出力電圧が低下すること、が挙げられる。後者はメタノールのクロスオーバー現象と言われ、DMFCの実用上、最も大きな課題として取り上げられている。
DMFCの電解質膜としては通常、電導性、化学的安定性、機械的特性の観点から、スルホン酸基を有するパーフルオロ化したポリマーからなる膜(例えば旭硝子社製フレミオン(商品名)、デュポン社製ナフィオン(商品名)等)等のプロトン伝導性固体高分子膜が多く用いられている。しかし、これらの膜ではメタノール透過性が高いという問題がある。これらの膜においてメタノール透過性が高い理由は明らかではないが、これらの膜は膨潤しやすく、メタノールと水の相互作用でさらに膨潤しやすくなることが原因の1つとして考えられる。また、プロトンがアノードからカソードへ移動する際にはプロトン1原子あたり数分子の水を伴うが、この同伴水の移動に伴ってメタノールも移動すること等も原因として考えられる。
これに対し、電池出力の低下の要因であるメタノール透過を抑制するために、以下のような膜が提案されている。従来のスルホン酸基を有するパーフルオロカーボン重合体からなる膜の表面に電子線を照射した膜(特許文献1参照)、メタノール不透過性有機材料にSiO2やP2O5等の無機系プロトン伝導材料を含有させた膜、イオン伝導性ポリマーにグアニジン、トリアゾール等の窒素含有化合物を添加又はコートした膜(特許文献2、3参照)、ホスホネート又はスルホネート官能基を有するアルキル基若しくはアリール基で置換されたポリベンズイミダゾール樹脂に水や酸媒体を含浸させた膜(特許文献4参照)等。しかし、これらの検討結果は多くの場合、膜のメタノール透過を抑制すると膜自体の比抵抗が増大し、現在までに十分なメタノール遮蔽性と電池の電解質として実用可能な導電性を兼ね備えた膜は見出されていない。
また、上述のスルホン酸基を有するパーフルオロカーボン重合体からなる電解質膜は含水時に膜の長さ方向に寸法が増大しやすく、様々な弊害を生じやすい。例えば固体高分子型燃料電池では反応により生成した水や燃料ガスとともに供給される水蒸気により、またさらにDMFCではメタノールを希釈する水により、又はメタノール自体により、膜が膨潤し、膜の寸法が増大する。通常、膜と電極は接合しているので電極も膜の寸法変化に追従する。膜電極接合体は通常ガスの流路として溝が形成されたセパレータ等で拘束されているため、膜の寸法増大分は「しわ」となる。そして、そのしわがセパレータの溝を埋めてガスの流れを阻害することがある。
上記の問題を解決する方法として、ポリテトラフルオロエチレン(以下、PTFEという。)多孔膜にスルホン酸基を有するフッ素系イオン交換ポリマーを含浸する方法が提案されている(特許文献5参照)。しかしPTFEの多孔質体はその材質に由来し比較的軟質であるために補強効果が十分でなく、上記課題を解決するに至っていない。
上述のとおり、従来のDMFCでは、電解質膜として用いられるプロトン伝導性固体高分子電解質膜が、導電性等の電解質膜として必要な性能を保持しながらメタノールのクロスオーバーを十分に抑制することはできていなかったため、充分な出力をとり出すことができなかった。また、強度の点でも水やメタノールに長時間曝されても寸法変化が少なく安定した形状が維持できる膜が必要とされている。
そこで本発明は、上記問題点を解決し、プロトン伝導性を維持しつつ、メタノールの透過を抑制でき、かつ好ましくは寸法安定性に優れるイオン伝導性の高分子電解質膜を提供することを目的とする。
本発明は、−SO2X基を有するポリマー(Xはフッ素原子又は塩素原子)と、該ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーとを混合する工程、前記含フッ素ポリマーが溶融する温度で混練する工程、フィルム化する工程、フィルムを積層する工程及び−SO2X基を−SO3H基に変換する工程を経て製造することを特徴とする高分子電解質膜の製造方法を提供する。
上記製造方法により得られる高分子電解質膜は、従来の電解質膜に比べメタノールの透過を充分に抑制できるため、例えばDMFCの電解質膜として使用すると、有用である。
また、本発明は、上述の製造方法において、前記フィルム化する工程では押し出し成形を行ってフィルムを2枚作製し、得られた2枚のフィルムを互いの押し出し方向が直行するように重ねて積層する工程をさらに含む高分子電解質膜の製造方法を提供する。
押し出し成形によりフィルム化した場合、押し出し方向(以下、MD方向という)の引張弾性率、引裂き強度が高まって強度が充分に高まり、水やメタノールに含浸させても膜の寸法変化が少ないが、MD方向と垂直な方向(以下、TD方向という)については強度が高まらず、寸法変化も少なくならず、膜強度に異方性が生じやすい。ところが2枚のフィルムのMD方向が直行するように重ねて積層する工程を行うことにより、膜強度の異方性は解消され、いずれの方向についても膜強度が高まる。
また、本発明は、プロトン伝導性のイオン導電性ポリマーからなる連続相と、該イオン導電性ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーを主体とする分散相とからなる海島構造を有することを特徴とする高分子電解質膜を提供する。
本発明による高分子電解質膜は、従来の高分子電解質膜に比べ、メタノールのクロスオーバーが抑制されるという効果を有する。
本発明の高分子電解質膜は、イオン伝導性ポリマーと該イオン伝導性ポリマーに非相溶でありかつ溶融成形可能な含フッ素ポリマーとからなる。そして、溶融成形可能な含フッ素ポリマーは電解質膜全面においては連続しておらず、電解質膜は相分離構造を有するプロトン導電性のイオン交換膜である。すなわち、電解質膜の面内では、イオン伝導性ポリマーが連続相となっていて、溶融成形可能な含フッ素ポリマーは膜の全面では連続しておらず、溶融成形可能な含フッ素ポリマーを主体とする分散相からなる海島構造を有している。分散相にはイオン伝導性ポリマーが含まれていてもよい。
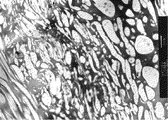
本発明の高分子電解質膜について透過型電子顕微鏡(TEM)による写真を図2に示す。図2は後述する実施例の例1で得られた膜の断面のTEM写真である。図2中、丸い形状や短いひも状になった部分が上述の分散相であり、分散相間の連続している部分が上述の連続相である。
本発明では、イオン伝導性ポリマーに非相溶かつ溶融成形可能なポリマーとして、化学的安定性、機械的強度、溶融成型性等の観点から、含フッ素ポリマーを使用する。具体的には、テトラフルオロエチレン−ヘキサフルオロプロピレン共重合体(FEP)、テトラフルオロエチレン−エチレン共重合体(ETFE)、ポリビニリデンフルオライド(PVdF)、テトラフルオロエチレン−パーフルオロ(アルキルビニルエーテル)重合体、ポリクロロトリフルオロエチレン、エチレン−クロロトリフルオロエチレン共重合体、及びポリビニルフルオライドからなる群から選ばれる1種以上が好ましい。なかでも、溶融成形性の観点からFEPが特に好ましく用いられる。
これらのポリマーは、分子量が20000〜90000であることが好ましく、さらには25000〜65000であることが好ましい。分子量が低すぎると強度が低下する傾向にあり、分子量が高すぎるとフィルム成形性が悪くなる傾向にある。
また、イオン伝導性ポリマーとしては、従来より公知の重合体が広く採用される。なかでもスルホン酸基又はスルホンイミド基を有する含フッ素重合体が好ましく、特に耐久性の観点からスルホン酸基を有するパーフルオロカーボン重合体(エーテル結合性の酸素原子等を含んでもよい)が好ましい。スルホン酸基又はスルホンイミド基を有するイオン伝導性ポリマーは、−SO2F基又は−SO2Cl基を有する含フッ素重合体を置換して得られる。スルホン酸基を有するパーフルオロカーボン重合体の場合は、−SO2F基を有するパーフルオロカーボン重合体を加水分解及び酸型化して得ることが好ましい。
−SO2F基を有するパーフルオロカーボン重合体である、上記前駆体としては、CF2=CF−(OCF2CFX)m−Op−(CF2)n−SO2Fで表されるフルオロビニル化合物(式中、Xはフッ素原子又はトリフルオロメチル基であり、mは0〜3の整数、nは0〜12の整数、pは0又は1であり、n=0のときはp=0かつmは1〜3である。)に基づく重合単位と、テトラフルオロエチレン、ヘキサフルオロプロピレンのようなパーフルオロオレフィン、クロロトリフルオロエチレン、又はパーフルオロ(アルキルビニルエーテル)に基づく重合単位とを含む共重合体が好ましい。特に上記フルオロビニル化合物に基づく重合単位とテトラフルオロエチレンに基づく重合単位とを含む共重合体が好ましい。
上記フルオロビニル化合物の好ましい例としては、以下のいずれかの式で表される化合物が挙げられる。ただし、下記式中、qは1〜8の整数、rは1〜8の整数、sは1〜8の整数、tは1〜5の整数を示す。

本発明におけるイオン伝導性ポリマーのイオン交換容量としては、0.4〜1.5ミリ当量/g乾燥樹脂、特に0.9〜1.3ミリ当量/g乾燥樹脂であることが好ましい。イオン交換容量が低すぎると膜の抵抗が大きくなる傾向にあり、高すぎると膜の機械的強度が弱くなる傾向にある。
また、高分子電解質膜において、イオン伝導性ポリマーと溶融成形可能なポリマーとの含有比は、電導性とメタノール透過性の観点から質量比で30/70〜70/30、特には40/60〜60/40が好ましい。溶融成形可能なポリマーが多すぎると抵抗が高くなり、少なすぎるとメタノールの透過が充分には抑制されないおそれがある。
また、本発明の高分子電解質膜の厚さは、強度、取り扱い性及び膜抵抗の観点から、5〜200μm、特には20〜100μmであるのが好ましい。膜厚が薄すぎると機械的な強度が弱くなる傾向にある。膜厚が厚すぎると膜の抵抗が高くなる傾向にある。
また、本発明の高分子電解質膜の厚さは、強度、取り扱い性及び膜抵抗の観点から、5〜200μm、特には20〜100μmであるのが好ましい。膜厚が薄すぎると機械的な強度が弱くなる傾向にある。膜厚が厚すぎると膜の抵抗が高くなる傾向にある。
次に、本発明の高分子電解質膜の好ましい製造方法について詳細に説明する。本発明における電解質膜は、−SO2X基を有するポリマー(Xはフッ素原子又は塩素原子)と、該ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーとを混合する工程、前記含フッ素ポリマーが溶融する温度で混練する工程、フィルム化する工程及び−SO2X基をスルホン酸基又はスルホンイミド基に変換する工程を経て得られる。
高分子電解質膜に含まれるイオン伝導性ポリマーとしてスルホン酸基を有するパーフルオロカーボン重合体を用いる場合を例にとって、具体的な好ましい手順を説明すると、以下のとおりである。
(1)溶融成形可能な含フッ素ポリマーと−SO2F基を有するパーフルオロカーボン重合体との混合。
(2)(1)で得られた混合物の、上記含フッ素ポリマーが溶融する温度での混練、ペレット化。
(3)(2)で得られたペレットを用いて押出し成形によるフィルム化。
(4)(3)で得られたフィルム2枚を、互いのMD方向が直行するように重ねて積層。
(5)加水分解、酸型化処理、洗浄、乾燥。
(1)溶融成形可能な含フッ素ポリマーと−SO2F基を有するパーフルオロカーボン重合体との混合。
(2)(1)で得られた混合物の、上記含フッ素ポリマーが溶融する温度での混練、ペレット化。
(3)(2)で得られたペレットを用いて押出し成形によるフィルム化。
(4)(3)で得られたフィルム2枚を、互いのMD方向が直行するように重ねて積層。
(5)加水分解、酸型化処理、洗浄、乾燥。
上記(1)〜(5)の工程をさらに具体的に説明する。
まず、溶融成形可能な含フッ素ポリマーと−SO2F基を有するパーフルオロカーボン重合体とを充分に混合する((1)の工程)。この混合には例えばVブレンダー、リボンミキサー、ヘンシェルミキサー等の混合機等が使用される。次に、得られた混合物を、溶融成形可能な含フッ素ポリマーが溶融する温度で混練する((2)の工程)。この際、例えば2軸押出し機、コ・ニーダ、非スクリュー型押出し機(インスタ・メルト)等により混練、ペレット化することができる。
まず、溶融成形可能な含フッ素ポリマーと−SO2F基を有するパーフルオロカーボン重合体とを充分に混合する((1)の工程)。この混合には例えばVブレンダー、リボンミキサー、ヘンシェルミキサー等の混合機等が使用される。次に、得られた混合物を、溶融成形可能な含フッ素ポリマーが溶融する温度で混練する((2)の工程)。この際、例えば2軸押出し機、コ・ニーダ、非スクリュー型押出し機(インスタ・メルト)等により混練、ペレット化することができる。
次いで得られたペレットは、(3)の工程で、好ましくは加熱下で単軸押出し成形されフィルム化される。また、(2)のペレット化する工程を経ずに、上記混合物を直接押出し成形し、この押出し成形の工程でフィルム化してもよい。加熱下で単軸押出し成形する場合は、成形(ダイス)温度が260〜310℃程度となるように成形することが好ましい。成形温度が260℃未満の場合は、フィルムの表面が荒れて厚さが不均一になり、また溶融伸度も低下し薄膜成形性が悪くなるおそれがあり好ましくない。また、成形(ダイス)温度が320℃を超えるとイオン伝導性ポリマーの熱分解が起こるおそれがあり好ましくない。このようにして(3)の工程を終えて得られるフィルムの厚さは5〜200μm程度となる。
膜の面内強度を均一化させたい場合は、次いで(4)の工程を行う。すなわち、2枚のフィルムを、互いのMD方向が直行するように重ねて積層する。積層は、金属又はゴムロールが対になった熱ロールプレス、平板熱プレス等により行うことができる。
次に、パーフルオロカーボン重合体の−SO2F基をスルホン酸基(−SO3H基)に変換するため、加水分解、酸型化処理、洗浄、乾燥を行う((5)の工程)。
次に、パーフルオロカーボン重合体の−SO2F基をスルホン酸基(−SO3H基)に変換するため、加水分解、酸型化処理、洗浄、乾燥を行う((5)の工程)。
なお、スルホンイミド基を有するイオン導電性ポリマーにより電解質膜を構成する場合は、(5)の工程のかわりに−SO2F基をスルホンイミド基に変換する工程を行う。この工程には公知の方法を用いることができる。例えば、トリフルオロメタンスルホンアミド、ヘプタフルオロエタンスルホンアミド、ノナフルオロブタンスルホンアミド等のパーフルオロスルホンアミドをアルカリ金属フッ化物や有機アミン等の塩基性化合物存在下に接触させる方法や、該スルホンアミドのアルカリ金属、アルカリ土類金属塩、該金属塩をさらにシリル化した化合物と接触させる方法により、ポリマー中の−SO2F基を反応させ、塩基由来の塩型のスルホンイミド基に変換した後、さらに塩酸や硫酸等の水溶液で酸型化することでスルホンイミド基に変換できる。ここで上記化合物と膜中の−SO2F基を接触させる方法としては、上記化合物が可溶な溶媒に溶解した溶液を膜と必要により加熱しながら接触させることが好ましい。
上記溶媒としては、メタノール、エタノール、ペンタフルオロプロパノール、テトラフルオロプロパノール等のアルコール類、アセトン、メチルエチルケトン等のケトン系溶剤、テトラヒドロフラン、グライム、ジオキサン等のエーテル系溶剤、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリジノン、ジメチルスルホキシド等が挙げられるが、これらに限られない。また、これらの溶媒と含フッ素溶媒との混合溶媒も、膜と溶液との接触時の濡れ性、膜の膨潤等の効果により反応をより効率的に進行させることができる場合があり、好ましい。
上記のように作製することで最終的にスルホン酸基又はスルホンイミド基を有するイオン伝導性ポリマー部と溶融成形可能なポリマー部が緻密な相分離構造をとる電解質膜とすることができる。さらに、イオン伝導性ポリマーの溶融粘度ηm、含有比φm、溶融成形可能なポリマーの溶融粘度ηd、含有比φdとした時、(ηm/ηd)×(φd/φm)の値が1に近くなるようにすることが好ましい。この値を1に近づけることで、より緻密な、アスペクト比の大きな分散相を形成する相分離構造とすることができる。
この電解質膜において、メタノール透過性が抑制される要因は明らかではないが、緻密な相分離構造をとることによって、分散相が、スルホン酸基によるクラスターネットワーク部が水又はメタノールによって膨潤することを抑制する機能を発揮することが考えられる。また、分散相はメタノール不透過性なので、膜内を通過するメタノールの実質的な拡散距離が長くなること等も考えられる。
本発明の電解質膜は、メタノール透過性が抑制されているので、DMFC用の電解質膜として使用すると、出力の高いDMFCが得られる。また、本発明の好ましい態様として(4)の工程等を行うことにより、水やメタノールに浸漬しても寸法変化が少ない強度の高い電解質膜が得られるため、この電解質膜を用いるとDMFCを長期間運転しても安定して高い出力を得ることが可能となる。また、このような電解質膜は水に対する寸法変化率も低いので、水素/酸素型の固体高分子電解質膜としても有用である。
以下、本発明について、実施例及び比較例により具体的に説明するが、本発明はこれらに限定されない。
[例1(実施例)]
テトラフルオロエチレンに基づく重合単位とCF2=CF−OCF2CF(CF3)O(CF2)2SO2Fに基づく重合単位とからなる共重合体粉末(酸型化したときのイオン交換容量1.1ミリ当量/g乾燥樹脂、以下、共重合体Aという。)とFEP(372℃におけるメルトインデックス7)とを質量比で50:50(容量比で52:48)になるようにして粉体混合機により十分混合した。この混合物を二軸押出し機(池貝社製、PCM30)を用い、300℃で混練した後、直径2.5mmのストランドを押出し、これを長さ2.5mmに切断してペレットを得た。このペレットを口径30mmの単軸押出し機(田辺プラスチックス社製、VS30)に供給し、350mmの口金幅を有するフラットダイを用い、ダイス温度300℃、押出し量1.3kg/hにて押出した。
テトラフルオロエチレンに基づく重合単位とCF2=CF−OCF2CF(CF3)O(CF2)2SO2Fに基づく重合単位とからなる共重合体粉末(酸型化したときのイオン交換容量1.1ミリ当量/g乾燥樹脂、以下、共重合体Aという。)とFEP(372℃におけるメルトインデックス7)とを質量比で50:50(容量比で52:48)になるようにして粉体混合機により十分混合した。この混合物を二軸押出し機(池貝社製、PCM30)を用い、300℃で混練した後、直径2.5mmのストランドを押出し、これを長さ2.5mmに切断してペレットを得た。このペレットを口径30mmの単軸押出し機(田辺プラスチックス社製、VS30)に供給し、350mmの口金幅を有するフラットダイを用い、ダイス温度300℃、押出し量1.3kg/hにて押出した。
得られた押出し物を表面温度が30〜50℃になるように調整したロールにより1.71m/分の速度で引き取ることにより、共重合体AとFEPとからなる厚さ20μmのブレンドフィルムを得た。次に、金属ロールと厚さ10mmのゴムを被覆したロールとの一対からなるロールを、金属ロール温度200℃、ゴム被覆ロール温度135℃に調整した。上記ブレンドフィルム2枚を、それらの押し出し方向(以下、MD方向という)どうしが直行するように重ね、フィルム幅で換算した線圧力が40kg/cmとなるように加圧して上記ロール間を押し出し速度0.4m/分で通すことにより、40μmの積層ブレンドフィルムを得た。
次いで、この積層ブレンドフィルムをジメチルスルホキシド(以下、DMSOという)、水酸化カリウム(KOH)を質量比で30:15の割合で含む水溶液を用い、温度95℃、浸漬時間30分で加水分解した後、イオン交換水にて十分に洗浄した。次に、1モル/Lの硫酸にて、温度25℃、浸漬時間60分で酸型化処理して−SO3H基に変換した後、脱イオン水で十分洗浄した。次いで、表面の水分をろ紙で除去した後、温度25℃、相対湿度50%の条件で予備乾燥を行い、さらに65℃のオーブン中で60分間加熱乾燥し、電解質膜を得た。
[例2(実施例)]
共重合体AとFEPの組成比を質量比で60:40(容量比で62:38)になるようにした以外は、例1と同様にしてフィルム化し、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの電解質膜を得た。
共重合体AとFEPの組成比を質量比で60:40(容量比で62:38)になるようにした以外は、例1と同様にしてフィルム化し、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの電解質膜を得た。
[例3(実施例)]
共重合体AとETFE(旭硝子社製、製品名:フルオンLM−730AP。メルトインデックス35)を質量比で55:45(容量比で52:48)になるように混合して原料として使用した以外は、例1と同様にしてフィルム化し、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの電解質膜を得た。
共重合体AとETFE(旭硝子社製、製品名:フルオンLM−730AP。メルトインデックス35)を質量比で55:45(容量比で52:48)になるように混合して原料として使用した以外は、例1と同様にしてフィルム化し、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの電解質膜を得た。
[例4(実施例)]
共重合体AとFEPの組成比を質量比で50:50(容量比で52:48)になるようにし、例1と同様にしてフィルム化し、厚さ60μmの膜を得た。次いで、フィルムのMD方向のみに温度95℃、速度2m/分で、延伸倍率3倍の延伸を行い、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの電解質膜を得た。
共重合体AとFEPの組成比を質量比で50:50(容量比で52:48)になるようにし、例1と同様にしてフィルム化し、厚さ60μmの膜を得た。次いで、フィルムのMD方向のみに温度95℃、速度2m/分で、延伸倍率3倍の延伸を行い、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの電解質膜を得た。
[例5(比較例)]
共重合体Aのみを用い、例1と同様にしてフィルム化し、厚さ20μmのフィルムを得た。このフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの重合体Aのみからなる電解質膜を得た。
共重合体Aのみを用い、例1と同様にしてフィルム化し、厚さ20μmのフィルムを得た。このフィルム2枚を例1と同様に直交積層、加水分解、酸型化、乾燥して厚さ40μmの重合体Aのみからなる電解質膜を得た。
[例6(実施例)]
共重合体AとFEPの組成比を質量比で50:50(容量比で52:48)になるようにし、例1と同様にしてフィルム化し、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を膜のMD方向が一致するように積層した以外は、例1と同様に加水分解、酸型化、乾燥して厚さ40μmのポリマーブレンド電解質膜を得た。
共重合体AとFEPの組成比を質量比で50:50(容量比で52:48)になるようにし、例1と同様にしてフィルム化し、厚さ20μmのブレンドフィルムを得た。このブレンドフィルム2枚を膜のMD方向が一致するように積層した以外は、例1と同様に加水分解、酸型化、乾燥して厚さ40μmのポリマーブレンド電解質膜を得た。
[例7(実施例)]
共重合体AをDMSO、水酸化カリウムを質量比で30:15の割合で含む水溶液で加水分解し、1モル/Lの塩酸に室温にて16時間浸漬して酸型化処理を行い、水洗、乾燥した。これを、エタノールに分散させ、固形分濃度が溶液全体の質量の10%の、スルホン酸基を含有するイオン交換体ポリマー分散液を得た。次いで、FEP(粉体)を1000メッシュのふるいで分級し、20μm以下の微粉末を得た。得られたイオン交換体ポリマー分散液と微粉末FEPを質量比が50:50(容量比が52:48)になるよう混合し、FEP微粉末含有イオン交換体ポリマー分散液を得た。これをPETフィルム上にバーコーターで塗工し、90℃のオーブン中で30分間加熱乾燥し、40μmの電解質膜を得た。
共重合体AをDMSO、水酸化カリウムを質量比で30:15の割合で含む水溶液で加水分解し、1モル/Lの塩酸に室温にて16時間浸漬して酸型化処理を行い、水洗、乾燥した。これを、エタノールに分散させ、固形分濃度が溶液全体の質量の10%の、スルホン酸基を含有するイオン交換体ポリマー分散液を得た。次いで、FEP(粉体)を1000メッシュのふるいで分級し、20μm以下の微粉末を得た。得られたイオン交換体ポリマー分散液と微粉末FEPを質量比が50:50(容量比が52:48)になるよう混合し、FEP微粉末含有イオン交換体ポリマー分散液を得た。これをPETフィルム上にバーコーターで塗工し、90℃のオーブン中で30分間加熱乾燥し、40μmの電解質膜を得た。
例1〜7の各例で得られた膜について、下記の諸特性を測定した。その結果を表1に示す。
[引張弾性率]
引張弾性率はJIS K7127に準じて測定した。まず、各例で得られたそれぞれの電解質膜について、幅1cm、長さ15cmの短冊状サンプルを切り出した。このサンプルは、長さ方向が電解質膜のMD方向と一致するサンプルと、長さ方向がMD方向と直交するサンプルの2種類についてそれぞれ作製した。なお、MD方向が直行するように2枚のフィルムを積層した積層膜の場合は、積層膜を構成するフィルムの一方のフィルムのMD方向を長さ方向としたサンプルについての測定値をX1、もう一方のフィルムのMD方向を長さ方向としたサンプルについての測定値をY1とした。測定は、標点間距離50mm、チャック間距離100mm、試験速度50mm/分で引張って行い、得られた変位(歪)と応力より、初期の4%歪における引張弾性率を求めた。測定は、25℃、相対湿度50%の条件において、5サンプルについて行い、その平均値を求めた。
引張弾性率はJIS K7127に準じて測定した。まず、各例で得られたそれぞれの電解質膜について、幅1cm、長さ15cmの短冊状サンプルを切り出した。このサンプルは、長さ方向が電解質膜のMD方向と一致するサンプルと、長さ方向がMD方向と直交するサンプルの2種類についてそれぞれ作製した。なお、MD方向が直行するように2枚のフィルムを積層した積層膜の場合は、積層膜を構成するフィルムの一方のフィルムのMD方向を長さ方向としたサンプルについての測定値をX1、もう一方のフィルムのMD方向を長さ方向としたサンプルについての測定値をY1とした。測定は、標点間距離50mm、チャック間距離100mm、試験速度50mm/分で引張って行い、得られた変位(歪)と応力より、初期の4%歪における引張弾性率を求めた。測定は、25℃、相対湿度50%の条件において、5サンプルについて行い、その平均値を求めた。
[引裂き強さ]
引裂き強さはJIS K7128に準じて測定した。まず、各例で得られたそれぞれの電解質膜について、幅5cm、長さ15cmの短冊状サンプルを切り出した。このサンプルは、引張弾性率の測定サンプルと同様に、長さ方向がフィルムのMD方向と一致するサンプルと、MD方向と直交する方向と一致するサンプルの2種類をそれぞれ作製し、積層膜の場合も引張弾性率の測定サンプルと同様にした。すなわち前者の測定値をX2、後者の測定値をY2とした。各サンプルは、長さ方向に沿って2等分するように、短辺の中央から長さ15cmの半分の7.5cmまで切れ目を入れる。次いで、切れ目部分から引裂かれるように、切れ端の一方を引張り試験機の上部チャックに、もう一方を下部チャックに取り付け、25℃、相対湿度50%の条件で200mm/分の速度でチャック間を広げ、引裂き荷重を測定する。引裂き強さは、引裂き荷重をサンプルの厚さで除した値(N/mm)として算出する。各方向について5サンプルずつ測定し、その平均値を引裂き強さとする。
なお、例6ではY2の引裂き強さの測定中に、引裂き方向がX方向に移ってしまい、Y方向についての引裂き強さの評価ができなかった。
引裂き強さはJIS K7128に準じて測定した。まず、各例で得られたそれぞれの電解質膜について、幅5cm、長さ15cmの短冊状サンプルを切り出した。このサンプルは、引張弾性率の測定サンプルと同様に、長さ方向がフィルムのMD方向と一致するサンプルと、MD方向と直交する方向と一致するサンプルの2種類をそれぞれ作製し、積層膜の場合も引張弾性率の測定サンプルと同様にした。すなわち前者の測定値をX2、後者の測定値をY2とした。各サンプルは、長さ方向に沿って2等分するように、短辺の中央から長さ15cmの半分の7.5cmまで切れ目を入れる。次いで、切れ目部分から引裂かれるように、切れ端の一方を引張り試験機の上部チャックに、もう一方を下部チャックに取り付け、25℃、相対湿度50%の条件で200mm/分の速度でチャック間を広げ、引裂き荷重を測定する。引裂き強さは、引裂き荷重をサンプルの厚さで除した値(N/mm)として算出する。各方向について5サンプルずつ測定し、その平均値を引裂き強さとする。
なお、例6ではY2の引裂き強さの測定中に、引裂き方向がX方向に移ってしまい、Y方向についての引裂き強さの評価ができなかった。
[寸法変化率]
寸法変化率は、以下のようにして測定した。まず、各例で得られたそれぞれの電解質膜について、一辺がMD方向と平行になるように、5cm角の正方形サンプルを切り出した。このサンプルを25℃、相対湿度50%の環境で4時間保持した後、その環境中でMD方向と平行な一辺の長さx1(cm)と、その辺と直交する一辺の長さy1(cm)とを測定した。次いで、サンプルを90℃の脱イオン水に16時間浸漬した後、水中下で同様にMD方向と平行な一辺の長さx2(cm)と、その辺と直交する一辺の長さy2(cm)とを測定した。寸法変化率は、以下のように算出した。
MD方向の寸法変化率X3(%):(x2−x1)/x1×100
MD方向と垂直な方向の寸法変化率Y3(%):(y2−y1)/y1×100
寸法変化率は、以下のようにして測定した。まず、各例で得られたそれぞれの電解質膜について、一辺がMD方向と平行になるように、5cm角の正方形サンプルを切り出した。このサンプルを25℃、相対湿度50%の環境で4時間保持した後、その環境中でMD方向と平行な一辺の長さx1(cm)と、その辺と直交する一辺の長さy1(cm)とを測定した。次いで、サンプルを90℃の脱イオン水に16時間浸漬した後、水中下で同様にMD方向と平行な一辺の長さx2(cm)と、その辺と直交する一辺の長さy2(cm)とを測定した。寸法変化率は、以下のように算出した。
MD方向の寸法変化率X3(%):(x2−x1)/x1×100
MD方向と垂直な方向の寸法変化率Y3(%):(y2−y1)/y1×100
[ポリマーブレンド電解質膜断面のTEM観察]
膜の断面についてTEMを用いて相分離構造を観察した。拡大倍率10000倍にて観察し、相分離の有無を確認した。例1についてのTEM写真を図2に示す。
膜の断面についてTEMを用いて相分離構造を観察した。拡大倍率10000倍にて観察し、相分離の有無を確認した。例1についてのTEM写真を図2に示す。
[例8(実施例)]
例1で得られた厚さ20μmのブレンドフィルムに対し、積層せずに例1で得られた積層ブレンドフィルムと同様に加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
例1で得られた厚さ20μmのブレンドフィルムに対し、積層せずに例1で得られた積層ブレンドフィルムと同様に加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
[例9(実施例)]
重合体AとFEPの組成比を質量比で40:60(容量比で42:58)になるようにした以外は例1と同様にしてブレンドフィルムを得て、例8と同様に積層せずに加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
重合体AとFEPの組成比を質量比で40:60(容量比で42:58)になるようにした以外は例1と同様にしてブレンドフィルムを得て、例8と同様に積層せずに加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
[例10(実施例)]
FEPのかわりにETFEを用い、共重合体AとETFE(旭硝子社製、LM−730AP)を質量比で55:45(容量比で52:48)になるようにして、例1と同様にしてブレンドフィルムを得て、例8と同様に積層せずに加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
FEPのかわりにETFEを用い、共重合体AとETFE(旭硝子社製、LM−730AP)を質量比で55:45(容量比で52:48)になるようにして、例1と同様にしてブレンドフィルムを得て、例8と同様に積層せずに加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
[例11(比較例)]
重合体Aのみを用い、例7と同様にしてフィルム化し、厚さ20μmのフィルムを得て、例8と同様に積層せずに加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
重合体Aのみを用い、例7と同様にしてフィルム化し、厚さ20μmのフィルムを得て、例8と同様に積層せずに加水分解、酸型化、洗浄、乾燥の処理を行い、厚さ20μmの電解質膜を得た。
例8〜11で得られた膜について、以下のように測定を行い、メタノール透過係数と比抵抗を求めた。その結果を表2に示す。
[メタノール透過係数]
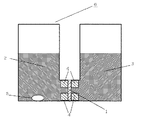
メタノール透過係数は、以下のようにして測定、算出した。まず、測定する膜を2cm角に切り出し、膜の厚さL1(cm)を測定した。次いで、この膜1を、図1に示すメタノール透過測定用セル6に挟みこみ、膜の片面の容器(水用)2には水、もう一方の面の容器(メタノール用)3には30%メタノール水溶液を同時に入れ、時間経過とともにメタノールが膜1を透過した量をガスクロマトグラフィーを用いて測定した。具体的には、ガスクロマトグラフィーにより、t(sec)経過した後の容器2内のメタノール濃度a(質量割合)を測定する。
メタノール透過係数は、以下のようにして測定、算出した。まず、測定する膜を2cm角に切り出し、膜の厚さL1(cm)を測定した。次いで、この膜1を、図1に示すメタノール透過測定用セル6に挟みこみ、膜の片面の容器(水用)2には水、もう一方の面の容器(メタノール用)3には30%メタノール水溶液を同時に入れ、時間経過とともにメタノールが膜1を透過した量をガスクロマトグラフィーを用いて測定した。具体的には、ガスクロマトグラフィーにより、t(sec)経過した後の容器2内のメタノール濃度a(質量割合)を測定する。
なお、膜1はガスケット4により固定し、容器2内には、膜を透過するメタノールを均一に分散させるために撹拌子5を入れて液を撹拌した。t(sec)経過したときのメタノール透過量V(cm3)を算出することにより、以下のとおりメタノール透過係数を算出した。ただし、ここでdはメタノールの密度(g/cm3)であり、S1(cm2)はメタノールが透過可能な膜の面積である。ここで、各例において、dは0.285g/cm3としてメタノール透過係数を求めた。
V=a×容器2内の液体の質量(g)/d
メタノール透過係数(cm2/sec)=V/t×L1/S1。
V=a×容器2内の液体の質量(g)/d
メタノール透過係数(cm2/sec)=V/t×L1/S1。
[比抵抗(膜厚方向)]
比抵抗は、以下のようにして測定した。まず、膜を3cm×4cmの大きさに切り出し、温度80℃、0.1モル/Lの硫酸水溶液に1時間浸漬させる。次いで、硫酸水溶液を含浸させた膜の厚みL2(cm)を測定する。膜を抵抗測定用セルにセットした後、セル内を温度80℃の0.1モル/Lの硫酸水溶液で満たす。この状態(膜を挟んだ状態)で白金電極間の抵抗R1(Ω)を測定し、続いて膜をセットしていない状態での抵抗R0(Ω)を測定する。白金電極間の距離と膜の幅の積により通電面積S2(cm2)を求めると、下式により比抵抗R(Ω・cm)が求められる。なお、抵抗の測定は、交流インピーダンス法(10kHz)にて行った。
実行抵抗(Ω・cm2)=(R1−R0)×S2、
比抵抗R(Ω・cm)=実行抵抗/膜厚L2。
比抵抗は、以下のようにして測定した。まず、膜を3cm×4cmの大きさに切り出し、温度80℃、0.1モル/Lの硫酸水溶液に1時間浸漬させる。次いで、硫酸水溶液を含浸させた膜の厚みL2(cm)を測定する。膜を抵抗測定用セルにセットした後、セル内を温度80℃の0.1モル/Lの硫酸水溶液で満たす。この状態(膜を挟んだ状態)で白金電極間の抵抗R1(Ω)を測定し、続いて膜をセットしていない状態での抵抗R0(Ω)を測定する。白金電極間の距離と膜の幅の積により通電面積S2(cm2)を求めると、下式により比抵抗R(Ω・cm)が求められる。なお、抵抗の測定は、交流インピーダンス法(10kHz)にて行った。
実行抵抗(Ω・cm2)=(R1−R0)×S2、
比抵抗R(Ω・cm)=実行抵抗/膜厚L2。
本発明によれば、従来技術の電解質膜に比べ、メタノールの透過が大幅に低減された電解質膜が得られる。したがって、本発明の電解質膜を電解質として備えるDMFCは、高出力が得られる。
1:膜
2:容器(水用)
3:容器(メタノール用)
4:ガスケット
5:撹拌子
6:メタノール透過測定用セル
2:容器(水用)
3:容器(メタノール用)
4:ガスケット
5:撹拌子
6:メタノール透過測定用セル
Claims (10)
- −SO2X基を有するポリマー(Xはフッ素原子又は塩素原子)と、該ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーとを混合する工程、前記含フッ素ポリマーが溶融する温度で混練する工程、フィルム化する工程及び−SO2X基をスルホン酸基又はスルホンイミド基に変換する工程を経て製造することを特徴とする高分子電解質膜の製造方法。
- 前記フィルム化する工程では押し出し成形を行ってフィルムを2枚作製し、得られた2枚のフィルムを互いの押し出し方向が直行するように重ねて積層する工程をさらに含む請求項1に記載の高分子電解質膜の製造方法。
- 前記−SO2X基を有するポリマーと前記含フッ素ポリマーは、質量比で30/70〜70/30の割合で混合する請求項1又は2に記載の高分子電解質膜の製造方法。
- 前記−SO2X基を有するポリマーは、−SO2F基を有するパーフルオロカーボン重合体(エーテル結合性の酸素原子を含んでいてもよい)であり、前記含フッ素ポリマーが、エチレン−テトラフルオロエチレン共重合体、テトラフルオロエチレン−パーフルオロ(アルキルビニルエーテル)共重合体、テトラフルオロエチレン−ヘキサフルオロプロピレン共重合体、ポリクロロトリフルオロエチレン、エチレン−クロロトリフルオロエチレン共重合体、ポリビニリデンフルオライド、及びポリビニルフルオライドからなる群から選ばれる1種以上からなる請求項1〜3のいずれかに記載の高分子電解質膜の製造方法。
- 前記高分子電解質膜は、プロトン伝導性のイオン導電性ポリマーからなる連続相と、該イオン導電性ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーを主体とする分散相とからなる海島構造を有する請求項1〜4のいずれかに記載の高分子電解質膜の製造方法。
- プロトン伝導性のイオン導電性ポリマーからなる連続相と、該イオン導電性ポリマーと非相溶でありかつ溶融成形可能な含フッ素ポリマーを主体とする分散相とからなる海島構造を有することを特徴とする高分子電解質膜。
- 前記イオン導電性ポリマーと前記含フッ素ポリマーは、質量比で30/70〜70/30の割合で含まれる請求項6に記載の高分子電解質膜。
- 前記分散相における粒子は、透過型電子顕微鏡で観察される平均アスペクト比が2以上である請求項6又は7に記載の高分子電解質膜。
- 前記イオン導電性ポリマーは、スルホン酸基又はスルホンイミド基を有するパーフルオロカーボン重合体(エーテル結合性の酸素原子を含んでいてもよい)であり、前記含フッ素ポリマーが、エチレン−テトラフルオロエチレン共重合体、テトラフルオロエチレン−パーフルオロ(アルキルビニルエーテル)共重合体、テトラフルオロエチレン−ヘキサフルオロプロピレン共重合体、ポリクロロトリフルオロエチレン、エチレン−クロロトリフルオロエチレン共重合体、ポリビニリデンフルオライド、及びポリビニルフルオライドからなる群から選ばれる1種以上からなる請求項6〜8のいずれかに記載の高分子電解質膜。
- 直接メタノール燃料電池に用いられる請求項6〜9のいずれかに記載の高分子電解質膜。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004355264A JP2006160902A (ja) | 2004-12-08 | 2004-12-08 | 高分子電解質膜及びその製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004355264A JP2006160902A (ja) | 2004-12-08 | 2004-12-08 | 高分子電解質膜及びその製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2006160902A true JP2006160902A (ja) | 2006-06-22 |
Family
ID=36663286
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004355264A Withdrawn JP2006160902A (ja) | 2004-12-08 | 2004-12-08 | 高分子電解質膜及びその製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2006160902A (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008001923A1 (fr) * | 2006-06-26 | 2008-01-03 | Toyota Jidosha Kabushiki Kaisha | Film poreux pour film d'électrolyte dans une pile à combustible et procédé de production de celui-ci |
| JP2011003358A (ja) * | 2009-06-17 | 2011-01-06 | Toyota Motor Corp | 燃料電池電解質膜用の多孔質膜の製造方法 |
| JP2013527572A (ja) * | 2010-05-10 | 2013-06-27 | ユーティーシー パワー コーポレイション | 交差積層された電気化学電池膜 |
| JP2014067605A (ja) * | 2012-09-26 | 2014-04-17 | Nitto Denko Corp | 高分子電解質膜およびそれを用いた燃料電池 |
| US9413019B2 (en) | 2011-08-18 | 2016-08-09 | Audi Ag | Fuel cell and membrane therefore |
| EP3021395A4 (en) * | 2013-07-09 | 2017-02-08 | JSR Corporation | Electrolyte membrane, membrane-electrode assembly, and solid polymer fuel cell |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004030132A1 (ja) * | 2002-09-30 | 2004-04-08 | Asahi Glass Company, Limited | 電解質膜、その製造方法及び固体高分子型燃料電池 |
| JP2004319442A (ja) * | 2003-04-01 | 2004-11-11 | Toray Ind Inc | 高分子固体電解質膜およびその製造方法 |
-
2004
- 2004-12-08 JP JP2004355264A patent/JP2006160902A/ja not_active Withdrawn
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004030132A1 (ja) * | 2002-09-30 | 2004-04-08 | Asahi Glass Company, Limited | 電解質膜、その製造方法及び固体高分子型燃料電池 |
| JP2004319442A (ja) * | 2003-04-01 | 2004-11-11 | Toray Ind Inc | 高分子固体電解質膜およびその製造方法 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008001923A1 (fr) * | 2006-06-26 | 2008-01-03 | Toyota Jidosha Kabushiki Kaisha | Film poreux pour film d'électrolyte dans une pile à combustible et procédé de production de celui-ci |
| JP2008004500A (ja) * | 2006-06-26 | 2008-01-10 | Toyota Motor Corp | 燃料電池電解質膜用の多孔質膜とその製造方法 |
| JP2011003358A (ja) * | 2009-06-17 | 2011-01-06 | Toyota Motor Corp | 燃料電池電解質膜用の多孔質膜の製造方法 |
| JP2013527572A (ja) * | 2010-05-10 | 2013-06-27 | ユーティーシー パワー コーポレイション | 交差積層された電気化学電池膜 |
| US9413019B2 (en) | 2011-08-18 | 2016-08-09 | Audi Ag | Fuel cell and membrane therefore |
| JP2014067605A (ja) * | 2012-09-26 | 2014-04-17 | Nitto Denko Corp | 高分子電解質膜およびそれを用いた燃料電池 |
| EP3021395A4 (en) * | 2013-07-09 | 2017-02-08 | JSR Corporation | Electrolyte membrane, membrane-electrode assembly, and solid polymer fuel cell |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102112648B1 (ko) | 고분자 전해질막 | |
| KR102112645B1 (ko) | 고분자 전해질막 | |
| US6692858B2 (en) | Electrolyte membrane for polymer electrolyte fuel cell and producing method thereof | |
| CN100560612C (zh) | 通过直接氟化交联的聚合物电解质膜 | |
| JP6034200B2 (ja) | レドックスフロー二次電池 | |
| US8003732B2 (en) | Gradient reinforced proton exchange membrane | |
| WO2012046777A1 (ja) | フッ素系高分子電解質膜 | |
| JP5331122B2 (ja) | 燃料電池用補強型電解質膜、燃料電池用膜−電極接合体、及びそれを備えた固体高分子形燃料電池 | |
| JP2002260705A (ja) | 固体高分子電解質材料、液状組成物、固体高分子型燃料電池、含フッ素ポリマー及び含フッ素ポリマーからなる固体高分子電解質膜 | |
| JP6328355B1 (ja) | 電解質膜およびその製造方法 | |
| WO2018061838A1 (ja) | ポリマー、固体高分子電解質膜および膜電極接合体 | |
| JP5489945B2 (ja) | フッ素系高分子電解質膜 | |
| CN101542795A (zh) | 固体高分子型燃料电池用固体高分子电解质膜及膜电极接合体 | |
| JP2007095433A (ja) | 固体高分子形燃料電池用電解質膜及びその製造方法 | |
| JP5189394B2 (ja) | 高分子電解質膜 | |
| JP2006160902A (ja) | 高分子電解質膜及びその製造方法 | |
| Kim et al. | 10.36-polymers in membrane electrode assemblies | |
| JP2006164777A (ja) | 直接メタノール燃料電池用膜電極接合体及びその製造方法 | |
| JP2014110232A (ja) | フッ素系高分子電解質膜 | |
| JP2002343380A (ja) | 固体高分子型燃料電池用電解質膜及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20071025 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20100611 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100622 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20100630 |