JP2006052182A - フッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法 - Google Patents
フッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法 Download PDFInfo
- Publication number
- JP2006052182A JP2006052182A JP2004236246A JP2004236246A JP2006052182A JP 2006052182 A JP2006052182 A JP 2006052182A JP 2004236246 A JP2004236246 A JP 2004236246A JP 2004236246 A JP2004236246 A JP 2004236246A JP 2006052182 A JP2006052182 A JP 2006052182A
- Authority
- JP
- Japan
- Prior art keywords
- group
- general formula
- purine nucleoside
- represented
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 51
- 150000003834 purine nucleoside derivatives Chemical class 0.000 title claims abstract description 47
- 238000000034 method Methods 0.000 claims abstract description 37
- 238000010511 deprotection reaction Methods 0.000 claims abstract description 20
- -1 9-phenylfluorenyl group Chemical group 0.000 claims description 58
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 49
- 125000006239 protecting group Chemical group 0.000 claims description 41
- 125000003277 amino group Chemical group 0.000 claims description 32
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 32
- 125000005843 halogen group Chemical group 0.000 claims description 25
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 24
- 125000001424 substituent group Chemical group 0.000 claims description 20
- 239000003153 chemical reaction reagent Substances 0.000 claims description 16
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 12
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 claims description 11
- 238000006477 desulfuration reaction Methods 0.000 claims description 9
- 230000023556 desulfurization Effects 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 125000004434 sulfur atom Chemical group 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- 239000001257 hydrogen Substances 0.000 claims description 2
- 230000004048 modification Effects 0.000 abstract description 6
- 238000012986 modification Methods 0.000 abstract description 6
- 230000008569 process Effects 0.000 abstract description 5
- 238000009776 industrial production Methods 0.000 abstract description 4
- 150000007523 nucleic acids Chemical class 0.000 abstract description 4
- 102000039446 nucleic acids Human genes 0.000 abstract description 4
- 108020004707 nucleic acids Proteins 0.000 abstract description 4
- 230000003009 desulfurizing effect Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 57
- 150000001875 compounds Chemical class 0.000 description 49
- 238000006243 chemical reaction Methods 0.000 description 37
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 33
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- 238000003786 synthesis reaction Methods 0.000 description 16
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 125000002252 acyl group Chemical group 0.000 description 12
- 125000003710 aryl alkyl group Chemical group 0.000 description 12
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 12
- 239000000203 mixture Substances 0.000 description 11
- 150000003833 nucleoside derivatives Chemical class 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- 238000004587 chromatography analysis Methods 0.000 description 10
- 238000005160 1H NMR spectroscopy Methods 0.000 description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 238000003682 fluorination reaction Methods 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 8
- 239000003480 eluent Substances 0.000 description 8
- 0 *c(nc(nc1[n]23)I)c1nc2SC1C3OC(CO*)C1F Chemical compound *c(nc(nc1[n]23)I)c1nc2SC1C3OC(CO*)C1F 0.000 description 7
- 230000002411 adverse Effects 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 6
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 6
- 239000007868 Raney catalyst Substances 0.000 description 5
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 5
- 229910000564 Raney nickel Inorganic materials 0.000 description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 239000012025 fluorinating agent Substances 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 230000002194 synthesizing effect Effects 0.000 description 5
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 5
- ZQXCQTAELHSNAT-UHFFFAOYSA-N 1-chloro-3-nitro-5-(trifluoromethyl)benzene Chemical compound [O-][N+](=O)C1=CC(Cl)=CC(C(F)(F)F)=C1 ZQXCQTAELHSNAT-UHFFFAOYSA-N 0.000 description 4
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 4
- 125000001118 alkylidene group Chemical group 0.000 description 4
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 4
- 244000309464 bull Species 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 4
- 125000001188 haloalkyl group Chemical group 0.000 description 4
- 125000005462 imide group Chemical group 0.000 description 4
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 4
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 4
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 3
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 3
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 3
- 150000001720 carbohydrates Chemical class 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 238000007086 side reaction Methods 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 2
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 125000000649 benzylidene group Chemical group [H]C(=[*])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- 238000010908 decantation Methods 0.000 description 2
- 125000004663 dialkyl amino group Chemical group 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- MRWXACSTFXYYMV-FDDDBJFASA-N nebularine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC=C2N=C1 MRWXACSTFXYYMV-FDDDBJFASA-N 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- LUYQYZLEHLTPBH-UHFFFAOYSA-N perfluorobutanesulfonyl fluoride Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)S(F)(=O)=O LUYQYZLEHLTPBH-UHFFFAOYSA-N 0.000 description 2
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 239000002212 purine nucleoside Substances 0.000 description 2
- 239000002718 pyrimidine nucleoside Substances 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229960002317 succinimide Drugs 0.000 description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 2
- UFXIRMVZNARBDL-UHFFFAOYSA-N trifluoro(morpholin-4-yl)-$l^{4}-sulfane Chemical compound FS(F)(F)N1CCOCC1 UFXIRMVZNARBDL-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- NBFKXQZHVNHRSB-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,9-nonadecafluorononane-1-sulfonyl fluoride Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)S(F)(=O)=O NBFKXQZHVNHRSB-UHFFFAOYSA-N 0.000 description 1
- JBWYRBLDOOOJEU-UHFFFAOYSA-N 1-[chloro-(4-methoxyphenyl)-phenylmethyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC(OC)=CC=1)C1=CC=CC=C1 JBWYRBLDOOOJEU-UHFFFAOYSA-N 0.000 description 1
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 1
- KZELNMSPWPFAEB-AJFJRRQVSA-N 2-amino-9-[(2r,3s,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-8-sulfanylidene-3,7-dihydropurin-6-one Chemical compound SC1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O KZELNMSPWPFAEB-AJFJRRQVSA-N 0.000 description 1
- RTJUXLYUUDBAJN-KVQBGUIXSA-N 2-amino-9-[(2r,4s,5r)-4-fluoro-5-(hydroxymethyl)oxolan-2-yl]-3h-purin-6-one Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@H]1C[C@H](F)[C@@H](CO)O1 RTJUXLYUUDBAJN-KVQBGUIXSA-N 0.000 description 1
- ASJSAQIRZKANQN-CRCLSJGQSA-N 2-deoxy-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)CC=O ASJSAQIRZKANQN-CRCLSJGQSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HYQXNCDBSALQLB-UHFFFAOYSA-N 9-bromo-9-phenylfluorene Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1(Br)C1=CC=CC=C1 HYQXNCDBSALQLB-UHFFFAOYSA-N 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- KLZUFWVZNOTSEM-UHFFFAOYSA-K Aluminium flouride Chemical compound F[Al](F)F KLZUFWVZNOTSEM-UHFFFAOYSA-K 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- IEDWIIRIBOAJAE-UHFFFAOYSA-N FC1C(N(C(N1C)=O)C)F Chemical compound FC1C(N(C(N1C)=O)C)F IEDWIIRIBOAJAE-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- CTDUJDNZPPZTAV-RRKCRQDMSA-N [(2r,3s,5r)-5-(6-aminopurin-9-yl)-3-fluorooxolan-2-yl]methanol Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](F)[C@@H](CO)O1 CTDUJDNZPPZTAV-RRKCRQDMSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000006242 amine protecting group Chemical group 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- JEPAHPFDUXQBAO-FJFJXFQQSA-N arabinofuranosylguanine Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(NC(=N)N=C2O)=C2N[CH]1 JEPAHPFDUXQBAO-FJFJXFQQSA-N 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229910001512 metal fluoride Inorganic materials 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- HNCYSTNKVUHPRI-UHFFFAOYSA-N n-ethylethanamine;1,1,2,3,3,4-hexafluorohept-1-ene Chemical compound CCNCC.CCCC(F)C(F)(F)C(F)=C(F)F HNCYSTNKVUHPRI-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- 238000001668 nucleic acid synthesis Methods 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000005740 oxycarbonyl group Chemical group [*:1]OC([*:2])=O 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- SUBJHSREKVAVAR-UHFFFAOYSA-N sodium;methanol;methanolate Chemical compound [Na+].OC.[O-]C SUBJHSREKVAVAR-UHFFFAOYSA-N 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- QHMQWEPBXSHHLH-UHFFFAOYSA-N sulfur tetrafluoride Chemical compound FS(F)(F)F QHMQWEPBXSHHLH-UHFFFAOYSA-N 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 238000011282 treatment Methods 0.000 description 1
- 238000005866 tritylation reaction Methods 0.000 description 1
Landscapes
- Saccharide Compounds (AREA)
Abstract
【課題】 工業的生産に適した、経済的かつ効率的な3’位(好ましくはα位)がフッ素化されたフッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法を提供する。
【解決手段】 5’位が保護された新規な、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体をフッ素化することによって、3’位がフッ素化された新規な8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルプリン誘導体(2)が高収率で得られ、さらに、化合物(2)に対し、脱硫工程、R1の脱保護、又は必要に応じて核酸塩基の保護、脱保護、修飾を行うことにより、3’位がフッ素化された所望のプリンヌクレオシド誘導体を容易に合成することができる。
【化1】
【選択図】 なし
【解決手段】 5’位が保護された新規な、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体をフッ素化することによって、3’位がフッ素化された新規な8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルプリン誘導体(2)が高収率で得られ、さらに、化合物(2)に対し、脱硫工程、R1の脱保護、又は必要に応じて核酸塩基の保護、脱保護、修飾を行うことにより、3’位がフッ素化された所望のプリンヌクレオシド誘導体を容易に合成することができる。
【化1】
【選択図】 なし
Description
本発明は、3’位(好ましくは、α位)がフッ素化されたプリンヌクレオシド誘導体の製造方法に関し、更にはこれら誘導体の製造に有用な中間体化合物およびその製造方法に関する。
3’位(好ましくは、α位)がフッ素化されたプリンヌクレオシド誘導体は、抗腫瘍性や抗ウイルス性を有することから注目されている。例えば、2’,3’−ジデオキシ−3’−フルオロアデノシンについては特許文献1、2’,3’−ジデオキシ−3’−フルオログアノシンについては特許文献2、2’,3’−ジデオキシ−3’−フルオロイノシンについては非特許文献1がそれぞれ参照される。
3’−α位がフッ素化されたヌクレオシド誘導体の合成方法としては、例えば、3’−β位にヒドロキシル基を有する誘導体に対して、SN2型置換反応によって立体反転的に3’−α位がフッ素化された誘導体を得る方法(非特許文献2を参照)が知られている。
しかしながら、この方法では、フッ素化工程において、最も効率的なフッ素化剤として知られているジエチルアミノサルファートリフルオリド(DAST)を使用しても脱離反応などの副反応が優先するため35%と低収率であり、満足できるものではなかった。
また、対応する3−α位がフッ素化された糖を合成した後に、核酸塩基とカップリングさせることにより、3’−α位がフッ素化されたヌクレオシド誘導体を得る方法も知られている。この方法に関しては、3−α位がフッ素化された糖の合成方法については、非特許文献3及び非特許文献4が参照され、また、得られた3−α位がフッ素化された糖と核酸塩基とのカップリング方法については、非特許文献5および非特許文献6が参照される。
非特許文献3では、2−デオキシリボースを出発原料として、アノマー水酸基のメチル化、5位への保護基導入、3位ヒドロキシル基の酸化、還元を経て、3−β位がヒドロキシル基である糖に誘導し、この糖に対して、フッ素化剤(DAST)を作用させることにより3−α位がフッ素化された糖が合成されている。しかしながら、この方法では、フッ素化された糖を合成するために多段階の工程が必要であり、フッ素化工程において、脱離反応が副反応として生じることに起因して低収率であり、さらに、核酸塩基とのカップリング反応を要することを考慮すると、本方法は、工業化に適していない。
非特許文献4では、キシロースを出発原料として、3−α位がフッ素化された糖が合成されている。しかしながら、この方法においても、3−α位がフッ素化された糖を得るために、多段階の工程数を経る必要があり、さらに2位のヒドロキシル基をデオキシ化することも困難であるので、本方法も工業化に適していない。
非特許文献5では、3−α位がフッ素化された糖と核酸塩基とを化学的にカップリグさせる方法が記載されている。しかしながら、本カップリング反応では、αアノマーとβアノマーとの生成比が1:1であり、理論上50%以上の収率が見込めない上、一般的に生成比が1:1のα/βアノマー混合物の分離が困難であるので、本方法は工業化に適していない。
非特許文献6では、3−α位がフッ素化された糖の1位にホスファナート基を導入した後、これと核酸塩基とを酵素的にカップリングさせることにより選択的にβ−アノマーを得る方法が記載されている。しかしながら、この方法では、基質である1位ホスファナート体を得るためにメトキシ体からさらに3工程を要するので、工業化に適していない。
したがって、上記の非特許文献3〜6に記載された方法では、3’−α位にフッ素が導入された糖を得る工程および糖と核酸塩基とをカップリングさせる工程の双方に問題を有している。
また、チミジンを原料として3’−α位がフッ素化されたピリミジンヌクレオシド誘導体を合成した後(特許文献3参照)、3’−α位がフッ素化されたピリミジンヌクレオシド誘導体を、酵素的トランスグリコシル化により3’−α位がフッ素化されたプリンヌクレオシド誘導体へと誘導する方法が知られている(特許文献4参照)。
しかしながら、この方法では、フッ素化工程において取扱いの困難なフッ素化水素と三フッ化アルミニウムを併せて用いる必要があり、さらに2段階の酵素反応を要するなどの問題により、工業化に適していない。
さらに、2’−α位にヒドロキシル基またはヒドロキシル基をシリル化したシリルオキシ基を持ち、3’−β位にフッ素原子以外のハロゲン原子を持つ3−デオキシ−3−ハロゲノ−β−D−キシロフラノシル型ヌクレオシド誘導体をジアルキルアミノサルファートリフルオリドと反応させることにより、2’−β位にハロゲン原子が転位し3’−α位がフッ素化された2,3−ジデオキシ−3−フルオロ−2−ハロゲノ−β−D−アラビノフラノシル型ヌクレオシド誘導体に変換し、次いで2’位ハロゲン原子を脱ハロゲン化することにより2,3−ジデオキシ−3−フルオロ−β−D−リボフラノシル型ヌクレオシド誘導体を合成する方法が知られている(非特許文献7参照)。
しかしながら、この方法では、フッ素化工程において、2’位と3’位のフッ素化の位置選択性が十分でないという問題がある。
東独国特許第158903号明細書
東独国特許第209197号明細書
欧州特許出願第495224号明細書
米国特許第5637574号明細書
ジャーナル・オブ・メディシナル・ケミストリー 32巻、1743頁、1989年(J.Med.Chem.1989,32,1743.)
ジャーナル・オブ・メディシナル・ケミストリー 31巻、2040頁、1988年(J.Med.Chem.1988,31,2040)
リービッヒズ・アナーレン・デア・ケミー 1137頁、1990年(Liebigs Ann.Chem.1990,1137)
バイオオーガニック・メディシナル・ケミストリー・レターズ 13巻、817頁、2003年(Bioorg.Med.Chem.Lett.2003,13,817.)
カーボハイドレート・リサーチ 328巻、49頁、200年(Carbohydr.Res.2000,328,49)
テトラヘドロン・レターズ 44巻、2899頁、2003年(Tetrahedron Lett.2003,44,2899.)
ヌクレオシド・ヌクレオチド・アンド・ヌクリックアシッヅ 22巻、711頁、2003年(Nucleosides Nucleotides&Nucleic Acids,2003,22,711)
本発明は、工業的生産に適した、経済的かつ効率的な3’位(好ましくはα位)がフッ素化されたフッ素化ヌクレオシド誘導体の製造方法およびその中間体およびその製造方法を提供することを目的とする。
本発明者らは前記の課題を解決すべく鋭意検討した結果、5’位が保護された新規な、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体をフッ素化試薬と反応させることによって、立体保持でフッ素化され、3’位(好ましくはα位)がフッ素化された新規な8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体が高収率で得られることを見い出した。
更にこの8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体の8位及び2’位に結合している硫黄原子を除去し、5’位ヒドロキシル基の脱保護、又は必要に応じて、核酸塩基の置換基変換等を行うことにより、3’位(好ましくは、α位)がフッ素化された所望のプリンヌクレオシド誘導体を容易に合成できることを見いだした。
本発明者らは以上の知見に基づき本発明を完成させた。
すなわち本発明は以下の内容を含むものである。
(1)一般式(1)
[式中、R1はヒドロキシル基の保護基を示し、X1は水素原子、ハロゲン原子、置換されてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y1は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表されるプリンヌクレオシド誘導体に、フッ素化試薬を作用させることを特徴とする一般式(2)
で表されるプリンヌクレオシド誘導体に、フッ素化試薬を作用させることを特徴とする一般式(2)
[式中、R1、X1及びY1は前記と同義である。]
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。
(2)前記フッ素化試薬がフッ化パーフルオロアルカンスルホニルであり、塩基存在下に作用させる、(1)に記載の製造方法。
(3)(1)または2のいずれか1つに記載の方法に従って、一般式(2)で表されるフッ素化プリンヌクレオシド誘導体を製造した後、
(A)硫黄原子を除去する脱硫工程、
(B)R1を除去する脱保護工程、
(C)X1とX2とが異なる基を示す場合、および/またはY1とY2とが異なる基を示す場合、X1をX2に変換し、および/またはY1をY2に変換する工程
の各工程を順不同に行うことを特徴とする、一般式(3)
(A)硫黄原子を除去する脱硫工程、
(B)R1を除去する脱保護工程、
(C)X1とX2とが異なる基を示す場合、および/またはY1とY2とが異なる基を示す場合、X1をX2に変換し、および/またはY1をY2に変換する工程
の各工程を順不同に行うことを特徴とする、一般式(3)
[式中、X2は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y2は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。
(4)一般式(1)で表されるプリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有し、
一般式(2)で表されるフッ素化プリンヌクレオシド誘導体は、
で表される相対配置を有し、
一般式(2)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有し、
一般式(3)で表されるフッ素化プリンヌクレオシド誘導体は、
で表される相対配置を有し、
一般式(3)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、X2及びY2は、上記と同義である。]
で表される相対配置を有する、(1)〜(3)のいずれか1つに記載の製造方法。
で表される相対配置を有する、(1)〜(3)のいずれか1つに記載の製造方法。
(5)一般式(4)
[式中、R1はヒドロキシル基の保護基を示し、R2及びR2’は、それぞれ独立して水素原子、又はアミンの保護基を示す。]
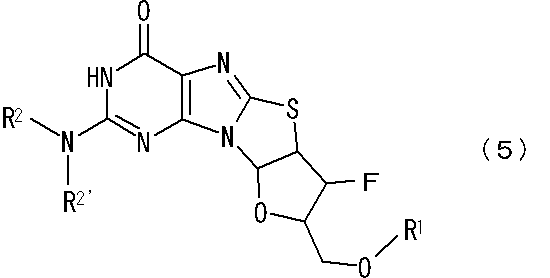
で表されるプリンヌクレオシド誘導体に、フッ素化試薬を作用させることを特徴とする一般式(5)
で表されるプリンヌクレオシド誘導体に、フッ素化試薬を作用させることを特徴とする一般式(5)
[式中、R1、R2及びR2’は前記と同義である。]
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。
(6)前記フッ素化試薬は、フッ化パーフルオロアルカンスルホニルであり、塩基の存在下に作用させる、(5)に記載の製造方法。
(7)(5)または(6)のいずれか1つに記載の方法に従って、一般式(5)で表されるフッ素化プリンヌクレオシド誘導体を製造した後、
(A)硫黄原子を除去する脱硫工程、
(B)R1を除去する脱保護工程
(C’)R2及び/またはR2’が保護基である場合には、脱保護工程
の各工程を順不同に行うことを特徴とする、一般式(6)
(A)硫黄原子を除去する脱硫工程、
(B)R1を除去する脱保護工程
(C’)R2及び/またはR2’が保護基である場合には、脱保護工程
の各工程を順不同に行うことを特徴とする、一般式(6)
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。
(8)一般式(4)で表されるプリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有し、
一般式(5)で表されるフッ素化プリンヌクレオシド誘導体は、
で表される相対配置を有し、
一般式(5)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有し、
一般式(6)で表されるフッ素化プリンヌクレオシド誘導体は、
で表される相対配置を有し、
一般式(6)で表されるフッ素化プリンヌクレオシド誘導体は、
で表される相対配置を有する、(5)〜(7)のいずれか1つに記載の製造方法。
(9)R1及びR2がそれぞれ置換基を有していてもよい、トリチル基、又は9−フェニルフルオレニル基であり、R2’が水素である、(5)〜(8)のいずれか1つに記載の製造方法。
(10)一般式(1)
[式中、R1はヒドロキシル基の保護基を示し、X1は水素原子、ハロゲン原子、置換されてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y1は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表される、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
で表される、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
(11)一般式(1)で表されるプリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有する、(10)に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
で表される相対配置を有する、(10)に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
(12)一般式(4)
[式中、R1はヒドロキシル基の保護基を示し、R2及びR2’は、それぞれ独立して、水素原子、又はアミノ基の保護基を示す。]
で表される、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
で表される、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
(13)一般式(4)で表されるプリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有する、(12)に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
で表される相対配置を有する、(12)に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
(14)R1及びR2が、それぞれ、置換基を有していてもよい、トリチル基、9−フェニルフルオレニル基、又はアセチル基であり、R2’が水素原子である、(12)または(13)に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
(15)一般式(2)
[式中、R1はヒドロキシル基の保護基を示し、X1は水素原子、ハロゲン原子、置換されてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y1は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表される、8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
(16)一般式(2)で表されるフッ素化プリンヌクレオシド誘導体は、
で表される、8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
(16)一般式(2)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有している、(15)に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
で表される相対配置を有している、(15)に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。
(17)一般式(5)
[式中、R1はヒドロキシル基の保護基を示し、R2及びR2’はそれぞれ独立して水素原子、又はアミノ基の保護基を示す。]
で表される、8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
で表される、8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
(18)一般式(5)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有している、(19)に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
で表される相対配置を有している、(19)に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
(19)R1及びR2は、それぞれ、置換基を有していてもよい、トリチル基、9−フェニルフルオレニル基、又はアセチル基であり、R2’は、水素原子である、(17)または(18)に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
(20)R1が置換基を有していてもよい、トリチル基であり、R2’、R2’が一緒になってジメチルアミノメチリデン基である、(12)または(13)に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
(21)R1が置換基を有していてもよい、トリチル基であり、R2’、R2’が一緒になってジメチルアミノメチリデン基である、(17)または(18)に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
本発明は、新規な化合物(1)を立体保持的にフッ素化することによって、3’位(好ましくはα位)がフッ素化された新規な化合物(2)を合成し、この化合物(2)に対して、脱硫、脱保護反応等を行うことにより、3’位(好ましくはα位)がフッ素化された所望のプリンヌクレオシド誘導体を収率よく合成でき、工業的生産に適した、経済的かつ効率的な3’位(好ましくはα位)がフッ素化されたフッ素化ヌクレオシド誘導体の製造方法およびその中間体およびその製造方法を提供することができる。
以下に本発明について詳細に説明する。
まず、本明細書に使用している記号を定義する。
本発明における式中、R1はヒドロキシル基の保護基を示す。ヒドロキシル基の保護基としては、本発明の反応に悪影響を与えず、かつ、反応後容易に脱保護できる基であれば特に限定はなく、適切な保護基としては、例えばアシル基、アルキル基、アラルキル基、シリル基等が挙げられる。アシル基としては、例えばホルミル基、アセチル基、ピバロイル基、アルカノイル基、ベンゾイル基等の炭素数1〜7のものが挙げられる。アルキル基としては、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、tert−ブチル基等の炭素数1〜7のものが挙げられる。アラルキル基としては、例えばベンジル基、トリチル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基、9−フェニルフルオレニル基等の炭素数7〜22のものが挙げられる。シリル基としては、例えばトリメチルシリル基、トリエチルシリル基、tert−ブチルジメチルシリル基等の3置換のものが挙げられる。上記のアシル基等の他、メトキシメチル基、メチルチオメチル基、ベンジルオキシメチル基、メトキシエトキシメチル基、テトラヒドロピラニル基、メトキシカルボニル基、9−フルオレニルメトキシカルボニル基、2,2,2−トリクロロエトキシカルボニル基、ベンジルオキシカルボニル基、tert−ブトキシカルボニル基等のヒドロキシル基の保護基として一般に知られた基を使用してもよい。
上記に挙げた各保護基のうち、好ましいものとしては、アセチル基、トリチル基、9−フェニルフルオレニル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基が挙げられ、特に好ましいものとしては、トリチル基、9−フェニルフルオレニル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基が挙げられる。
本発明における式中、R2、R2’は、水素原子、又はアミノ基の保護基を示す。
アミノ基の保護基としては、本発明の反応に悪影響を与えない基であれば特に限定はなく、例えば、アラルキル基、アシル基、アルコキシカルボニル基等が挙げられる。アラルキル基としては、例えば、ベンジル基、トリチル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基、9−フェニルフルオレニル基等の炭素数7〜22のものが挙げられる。アシル基としては、例えば、ホルミル基、アセチル基、ピバロイル基、ベンゾイル基、フェニルアセチル基等の炭素数1〜10のものが挙げられる。アルコキシカルボニル基としては、例えば、ベンジルオキシカルボニル基、tert−ブトキシカルボニル基、メトキシカルボニル基等が挙げられる。R2とR2’は、一緒になってアミノ基の保護基を形成する態様であってもよい。当該態様の保護基としては、アルキリデン基、アミノ基の窒素原子と一緒になってイミド基を形成する態様等が挙げられる。アルキリデン基としては、例えば、ベンジリデン基、ジフェニルメチリデン基、ビス(メチルチオ)メチリデン基、ジメチルアミノメチリデン基等が挙げられる。イミド基としては、フタルイミド、スクシンイミド、マレイミド等が挙げられる。
上記に挙げた各保護基のうち、好ましいものとしては、アセチル基、トリチル基、9−フェニルフルオレニル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基、ジメチルアミノメチリデン基が挙げられ、特に好ましいものとしては、トリチル基、9−フェニルフルオレニル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基が挙げられる。
本発明における式中、X1、Y1は、各々独立して、水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。ここで、置換されていてもよい上記各基における置換基には、保護基と修飾基とが含まれる。
本明細書においては、保護基は、化合物の他の部分の所望の変換を行った後、除去することが意図されている基を表し、修飾基は、化合物の他の部分の所望の変換を行った後においても、除去が意図されておらず、化学的性質または生理活性の変更を目的として導入される基を表す。
ハロゲン原子としては、塩素原子、フッ素原子、臭素原子、ヨウ素原子が挙げられる。
「置換されていてもよいアミノ基」における置換基に包含される保護基としては、本発明の反応に悪影響を与えない基であれば特に限定はなく、例えば、アラルキル基、アシル基、アルコキシカルボニル基、アルキリデン基(アミノ基の窒素原子と一緒になったイミド基を形成する態様)等が挙げられる。アラルキル基としては、例えば、ベンジル基、トリチル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基、9−フェニルフルオレニル基等の炭素数7〜22のアラルキル基が挙げられる。アシル基としては、例えば、ホルミル基、アセチル基、ピバロイル基等のアルカノイル基、ベンゾイル基、フェニルアセチル基等の炭素数1〜10のものが挙げられる。アルコキシカルボニル基としては、例えば、ベンジルオキシカルボニル基、tert−ブトキシカルボニル基、メトキシカルボニル基が挙げられる。アルキリデン基としては、例えば、ベンジリデン基、ジフェニルメチリデン基、ビス(メチルチオ)メチリデン基、ジメチルアミノメチリデン基等が挙げられる。イミド基の形態としては、フタルイミド、スクシンイミド、マレイミド等が挙げられる。
上記に挙げた各保護基のうち、好ましいものとしては、アセチル基、トリチル基、9−フェニルフルオレニル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基等の炭素数7〜22のアラルキル基、ジメチルアミノメチレン基が挙げられ、特に好ましいものとしては、トリチル、9−フェニルフルオレニル、4−メトキシトリチル、4,4’−ジメトキシトリチルが挙げられる。
「置換されていてもよいアミノ基」における置換基に包含される修飾基としては、本発明の反応に悪影響を与えない基であれば特に限定はなく、例えば、アルキル基、ハロアルキル基が挙げられる。アルキル基としては、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、tert−ブチル基、シクロプロピル基等の炭素数1〜10のアルキル基が挙げられる。ハロアルキル基としては、例えばトリフルオロメチル基等が挙げられる。
「置換されてもよいヒドロキシル基」における置換基に包含される保護基としては、本発明の反応に悪影響を与えない基であれば特に限定はなく、例えば、アシル基、アラルキル基、シリル基等が挙げられる。アシル基としては、例えば、ホルミル基、アセチル基、ピバロイル基、ベンゾイル基等の炭素数1〜7のものが挙げられる。アラルキル基としては、例えば、ベンジル基、トリチル基、4−モノメトキシトリチル基、4,4’−ジメトキシトリチル基、9−フェニルフルオレニル基等の炭素数7〜22のものが挙げられる。シリル基としては、例えば、トリメチルシリル基、トリエチルシリル基、tert−ブチルジメチルシリル基等の3置換シリル基が挙げられる。その他、メトキシメチル基、メチルチオメチル基、ベンジルオキシメチル基、メトキシエトキシメチル基、テトラヒドロピラニル基、メトキシカルボニル基、9−フルオレニルメトキシカルボニル基、2,2,2−トリクロロエトキシカルボニル基、ベンジルオキシカルボニル基、tert−ブトキシカルボニル基等のヒドロキシル基の保護基として一般的なものを使用することができる。
「置換されてもよいヒドロキシル基」における置換基に包含される修飾基としては、本発明の反応に悪影響を与えない基であれば特に限定はなく、例えば、アルキル基、ハロアルキル基が挙げられる。アルキル基としては、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、tert−ブチル基等の炭素数1〜7のものが挙げられる。ハロアルキル基としては、例えば、トリフルオロメチル基等が挙げられる。
本発明における式中、X2、Y2は、各々独立して、水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、上記X1及びY1と同様に定義される。
プリンヌクレオシドのプリン塩基部位は、上記に挙げた具体的なX1、Y1(又はX2、Y2)を組み合わせることによって、種々のものが構成され、X1、Y1(又はX2、Y2)の各組み合わせに対して、それぞれ、具体的な名称が知られている。以下の表にはX1、Y1(又はX2、Y2)の組み合わせ、およびその組み合わせに対するプリン塩基の名称を示している。
X1、Y1の組み合わせのうち好ましいものとしては、(X1=H、Y1=NH2)、(X1=NH2、Y1=OH)、(X1=H、Y1=OH)、(X1=AcNH、Y1=OH)、(X1=Ph3CNH、Y1=OH)、(X1=p−MeOC6H4Ph2CNH、Y1=OH)、(X1=(p−MeOC6H4)2PhCNH、Y1=OH)、(X1=Me2NC=N、Y1=OH)等が挙げられ、特に、(X1=Ph3CNH、Y1=OH)、(X1=p−MeOC6H4Ph2CNH、Y1=OH)、(X1=(p−MeOC6H4)2PhCNH、Y1=OH)が好ましい。
X2、Y2の組み合わせのうち、好ましいものとしては、アデニン(2)、グアニン(3)、ヒポキサンチン(4)が挙げられ、特に好ましいものとしては、グアニンが挙げられる。
X2、Y2の組み合わせのうち、好ましいものとしては、アデニン(2)、グアニン(3)、ヒポキサンチン(4)が挙げられ、特に好ましいものとしては、グアニンが挙げられる。
以下、本発明のプリンヌクレオシドの製造方法について説明する。
(化合物(1)の製造方法)
新規な化合物(1)は、化合物(A)
新規な化合物(1)は、化合物(A)
[式中、X3は水素原子、ハロゲン原子、置換基で置換されていてもよいアミノ基、又は置換基で置換されていてもよいヒドロキシル基を示し、Y3は水素原子、ハロゲン原子、置換基で置換されていてもよいアミノ基、又は置換基で置換されていてもよいヒドロキシル基を示す(該置換基は、保護基または修飾基である)。]
で表されるヌクレオシド誘導体に対して、下記の(A’)、(B’)の各工程を順不同に行うことによって製造することが出来る。
(A’)5’位のヒドロキシル基を保護してR1を導入する工程、
(B’)必要に応じて、X3および/またはY3を保護、脱保護および修飾の少なくとも一つを行う工程。
で表されるヌクレオシド誘導体に対して、下記の(A’)、(B’)の各工程を順不同に行うことによって製造することが出来る。
(A’)5’位のヒドロキシル基を保護してR1を導入する工程、
(B’)必要に応じて、X3および/またはY3を保護、脱保護および修飾の少なくとも一つを行う工程。
式(A)のX3およびY3は、それぞれ上記X1およびY1と同様のものが例示される。
原料の化合物(A)は公知化合物であり、例えば、ケミカル・アンド・ファーマシューティカル・ブレチン 15巻、94頁、1967年(Chem.Pharm.Bull.1967,15,94)(X3=H、Y3=NH2)、ケミカル・アンド・ファーマシューティカル・ブレチン 20巻、635頁、1972年(Chem.Pharm.Bull.1972,20,635)(X3=H、Y3=H)(X3=OH、Y3=OH)、ケミカル・アンド・ファーマシューティカル・ブレチン 29巻、3449頁、1981年(Chem.Pharm.Bull.1981,29,3449)(X3= NH2、Y3= NH2)、カナディアン・ジャーナル・オブ・ケミストリー(Can.J.Chem.1972,50,1100) (X3=NH2、Y3=OH)等に記載された方法に準じて製造することができる。
工程(A’)は、化合物(A)の5’位のヒドロキシル基を保護して、R1で定義されるヒドロキシル基の保護基を導入する工程であり、保護基の種類により従来公知の方法(例えば、プロテクティング・グループス・イン・オーガニック・シンセシス 第3版、ジョン・ウィリー・アンド・ソンズ社、1999年(Protecting Groups in Organic Synthesis 3rd edition, John Wiley&Sons, Inc. 1999)参照)により行うことができる。
以下、好ましい態様である保護基がアラルキル基(トリチル基、9−フェニルフルオレニル基等)である場合について説明するが、工程(A’)は当該態様に限定されるものではない。
保護基のアラルキル基を導入する工程(A’)は、化合物(A)を塩基の存在下、アラルキルハライド(例えば、トリチルクロリド,4,4’-ジメトキシトリチルクロリド、9−ブロモ-9−フェニルフルオレン等)と反応させることにより行なうことができる。
塩基としては、トリエチルアミン、ピリジン、4−ジメチルアミノピリジン、トリブチルアミン、N,N−ジイソプロピルエチルアミン、1,8−ジアザビシクロ[5,4,0]ウンデカ−7−エン等が挙げられ、その使用量としては、化合物(A)に対して通常1〜100当量好ましくは1〜3当量である。
アラルキルハライドの使用量は、化合物(A)に対して1〜10当量、好ましくは1〜3当量である。
アラルキルハライドの使用量は、化合物(A)に対して1〜10当量、好ましくは1〜3当量である。
当該保護反応は、通常溶媒中で行なわれ、溶媒としては反応を阻害しないものであれば特に限定はなく、例えば、N,N−ジメチルホルムアミド、アセトニトリル、ピリジン、N,N−ジメチルアセトアミド、N−メチルピロリドン等の単独または混合溶媒が挙げられ、好ましくはN,N−ジメチルホルムアミド、アセトニトリルである。溶媒の使用量は、通常化合物(A)に対して10〜100重量倍である。
反応温度は、通常10〜60℃の範囲であり、反応時間は、通常1〜24時間である。
工程(B’)の「必要に応じて」とは、例えば、X3がX1と異なる場合、および/またはY3がY1と異なる場合を指し、「保護、脱保護および修飾の少なくとも一つを行なう」とは、X3および/またはY3をそれぞれX1およびY1に変換すること意味する。
工程(B’)の「保護」は、X3および/またはY3がアミノ基および/またはヒドロキシル基である場合に、例えば上記で例示された保護基を導入する工程であり、「脱保護」は、保護基を除去する工程である。当該「保護」および「脱保護」は、従来公知の方法(例えば、プロテクティング・グループス・イン・オーガニック・シンセシス 第3版、ジョン・ウィリー・アンド・ソンズ社、1999年(Protecting Groups in Organic Synthesis 3rd edition John Wiley&Sons, Inc. 1999)参照)で行うことができる。
工程(B’)の「修飾」とは上記の「保護」および「脱保護」とは異なる置換基の変換を示し、例えば、X3および/またはY3がアミノ基および/またはヒドロキシル基である場合に、例えば上記で例示された修飾基を導入する工程またはハロゲン原子、水素原子などに変換する工程等を意味し、核酸合成における従来公知の方法(例えば、「Nucleic acid Chemistry PartII」Leroy B. Townsend, R. Stuart Tipson著、A Wiley-Interscience Publication出版参照)で行うことができる。
ここで、化合物(A)のX3および/またはY3が、保護基で置換されたアミノ基、又は保護基で置換されたヒドロキシル基である場合、これらアミノ基またはヒドロキシル基は、上記工程(A’)を実施する前に予め、工程(B’)を実施することにより保護基が付与されていてもよいが、上記工程(A’)において、5’位のヒドロキシル基の保護化と同時に、工程(B’)に相当する保護化を行ってもよい。この場合、保護試薬等の使用量は、保護される基の数に乗じて使用すればよい。例えば、X3=NH2の場合、トリエチルアミン等の通常用いられる塩基条件下で塩化トリチルと反応させることにより、5’位とX3であるアミノ基との両方を同時にトリチル基で保護することができる。
工程(A’)および(B’)によって、化合物(A)に存在する保護化可能な基(例えば、3’位ヒドロキシル基)に同時に保護基が付与された場合は、選択的に3’位を脱保護することにより化合物(1)を製造することも可能である。例えば、化合物(A)において、5’位ヒドロキシル基、X3であるアミノ基、および3’位ヒドロキシル基の全てを保護(例えば、アセチル基等のアシル基)した後、2級である3’位ヒドロキシル基のみを脱保護することも可能である。
なお、式(1)で表される化合物を得るための反応は、保護基を付与する反応であり、このような保護化反応は、保護基の種類、保護化の条件等一般に知られている方法により行うことができるので、保護化の詳細については、例えば、プロテクティング・グループス・イン・オーガニック・シンセシス 第3版、ジョン・ウィリー・アンド・ソンズ社、1999年(Protecting Groups in Organic Synthesis 3rd edition John Wiley&Sons, Inc. 1999)を参照すればよいとして、ここでは詳細な説明は省略する。例えばトリチル基により保護する場合には、N,N−ジメチルホルムアミドを溶媒として用い、トリエチルアミンを塩基として添加して、塩化トリチルと化合物(A)で表される化合物を所定時間反応させることによりトリチル化体を得ることができる。
(化合物(2)の製造方法)
新規な化合物(2)は、新規な化合物(1)にフッ素化試薬を作用させることにより得ることができる。
新規な化合物(2)は、新規な化合物(1)にフッ素化試薬を作用させることにより得ることができる。
フッ素化に用いられるフッ素化試薬としては、求核的フッ素化試薬を使用することができ、例えば、金属フルオリド(フッ化カリウム、フッ化セシウム等)、アンモニウムフルオリド(フッ化テトラブチルアンモニウム等)、四フッ化硫黄系フッ素化試薬(ジエチルアミノサルファートリフルオリド、モルホリノサルファートリフルオリド等のジアルキルアミノサルファートリフルオリドなど)、フルオロアルキルアミン系フッ素化剤(ジエチルアミン-ヘキサフルオロヘプテン付加物、2,2−ジフルオロ−1,3−ジメチルイミダゾリジノン等)、フッ化パーフルオロアルカンスルホニル(フッ化パーフルオロブタンスルホニル、フッ化パーフルオロノナンスルホニル等)等が挙げられる。なかでもフッ化パーフルオロアルカンスルホニル、ジアルキルアミノサルファートリフルオリドが好ましく、スケールアップへの適用可能性の観点から、特にフッ化パーフルオロアルカンスルホニルが好ましい。ここで、フッ化パーフルオロアルカンスルホニルが含有する「アルカン」部分については、ブタン、ノナン等が挙げられる。また、ジアルキルアミノサルファートリフルオリドが含有する「アルキル」部分については、エチル、2−メトキシエチル等が挙げられる。また「ジアルキル」は一体化して環を形成してもよく、このような例としては、ジアルキルアミノがモルホリノとなったモルホリノサルファートリフルオリド等が挙げられる。
フッ素化剤の使用量は、化合物(1)に対して、通常、1〜30当量、好ましくは、1〜15当量である。また、上記各種のフッ素化試薬は、塩基条件等の他の条件が必要な場合があり、例えば、フッ化パーフルオロアルカンスルホニルの場合、塩基条件にてフッ素化を行う必要がある。この場合の塩基条件にするために使用される塩基としては、例えば、N,N−ジイソプロピルエチルアミン、トリエチルアミン、トリブチルアミン等が挙げられ、好ましくは、N,N−ジイソプロピルアミンである。当該アミンの使用量は、化合物(1)に対して、通常、1〜30当量、好ましくは、1〜15当量である。
フッ素化剤の使用量は、化合物(1)に対して、通常、1〜30当量、好ましくは、1〜15当量である。また、上記各種のフッ素化試薬は、塩基条件等の他の条件が必要な場合があり、例えば、フッ化パーフルオロアルカンスルホニルの場合、塩基条件にてフッ素化を行う必要がある。この場合の塩基条件にするために使用される塩基としては、例えば、N,N−ジイソプロピルエチルアミン、トリエチルアミン、トリブチルアミン等が挙げられ、好ましくは、N,N−ジイソプロピルアミンである。当該アミンの使用量は、化合物(1)に対して、通常、1〜30当量、好ましくは、1〜15当量である。
本反応は、反応溶媒中で行われ、好適な反応溶媒としては、反応に悪影響を与えない限り特に限定はないが、例えば、酢酸エチル、酢酸イソプロピル、テトラヒドロフラン、アセトニトリル、ジクロロメタン等が挙げられ、このうち、好ましくは、酢酸エチル、テトラヒドロフランを挙げることができる。溶媒の使用量は、化合物(1)に対して、通常、5〜50重量倍であり、10〜30重量倍が好ましい。反応時間は、用いるフッ素化試薬によって異なるが、例えば、フッ化パーフルオロアルカンスルホニルを用いた場合には、2〜24時間である。また、反応温度についても、用いるフッ素化剤によって異なるが、例えば、フッ化パーフルオロアルカンスルホニルを用いた場合には、40〜80℃である。
(化合物(3)の製造方法)
化合物(3)は、上記により得られた化合物(2)に対して、下記の各工程を順不同にて行うことにより得ることができる。
(A)硫黄原子を除去する脱硫工程
(B)R1を除去する脱保護工程
(C)X1とX2が異なる基を示す場合、および/またはY1とY2が異なる基を示す場合、X1をX2に変換し、および/またはY1をY2に変換する工程。
化合物(3)は、上記により得られた化合物(2)に対して、下記の各工程を順不同にて行うことにより得ることができる。
(A)硫黄原子を除去する脱硫工程
(B)R1を除去する脱保護工程
(C)X1とX2が異なる基を示す場合、および/またはY1とY2が異なる基を示す場合、X1をX2に変換し、および/またはY1をY2に変換する工程。
(A)の工程に採用される条件としては、目的とする脱硫反応以外の部分に副反応を生ずるおそれがない限り、特に限定されないが、代表的な脱硫方法としては、ラネーニッケルを用いる方法が挙げられる。
ラネーニッケルを用いる方法により脱硫反応を行う場合、好適な溶媒としては、1−プロパノール、1−ブタノール、トルエンが挙げられる。ラネーニッケルの使用量は、その活性グレードにもよるが、通常、化合物(2)に対して、通常、3〜50重量倍、好ましくは、5〜20重量倍である。また、本反応における好適な反応時間は、1〜24時間であり、好適な反応温度は、70〜150℃である。
(B)の工程は、ヒドロキシル基の脱保護工程であり、前述のヒドロキシル基の保護基として挙げた各基に対してそれぞれ脱保護方法が周知である(例えば、プロテクティング・グループス・イン・オーガニック・シンセシス 第3版、ジョン・ウィリー・アンド・ソンズ社、1999年(Protecting Groups in Organic Synthesis 3rd edition John Wiley&Sons, Inc. 1999)参照)。例えば、R1がトリチル基である場合には、酢酸、ギ酸、トリフルオロ酢酸等の適当な酸を使用する条件下に、適当な温度(例えば、10〜100℃)で攪拌することにより、脱保護される。
(C)の工程は、X1とX2が異なる基を示す場合、および/またはY1とY2が異なる基を示す場合、必要に応じて適宜なされる工程であり、具体的には、保護、脱保護、修飾する処理を施して、X2及び/またはY2を形成する工程である。ここで、保護は、上記工程(B’)におけるヒドロキシル基またはアミノ基に対する保護と同様の操作を行うことができ、該保護には種々の基および各基毎に保護化反応が知られおり、また、脱保護反応についても、上記保護基に応じて種々のものが知られているので、例えば、プロテクティング・グループス・イン・オーガニック・シンセシス 第3版、ジョン・ウィリー・アンド・ソンズ社、1999年(Protecting Groups in Organic Synthesis 3rd edition John Wiley&Sons, Inc. 1999)を参照するとして、詳しい説明は省略する。また、修飾も、上記工程(B’)と同様に行うことができる。
工程(C)における保護、脱保護、修飾は、工程(A)および工程(B)の反応条件で、同時に進行してもよい。例えばX1および/またはY1が保護基で置換されたアミノ基および/または保護基で置換されたヒドロキシル基である場合に、、工程(B)の脱保護の反応条件において、当該保護基が除去されて、すなわち、工程(C)の脱保護が同時に進行してもよい。
(化合物(4)の製造方法)
新規な化合物(4)は、前述の化合物(1)において、X1=NR2R2’(R2およびR2’は、前記と同義を示す。)、Y1=OHである場合に該当し、化合物(1)と同様の反応を行うことにより製造することができるので、本化合物(4)の製造方法については、前述の化合物(1)の製造方法を参照すればよいものとして、詳細な説明は省略する。
新規な化合物(4)は、前述の化合物(1)において、X1=NR2R2’(R2およびR2’は、前記と同義を示す。)、Y1=OHである場合に該当し、化合物(1)と同様の反応を行うことにより製造することができるので、本化合物(4)の製造方法については、前述の化合物(1)の製造方法を参照すればよいものとして、詳細な説明は省略する。
(化合物(5)の製造方法)
新規な化合物(5)は、前述の化合物(2)において、X1=NR2R2’(R2およびR2’は、前記と同義を示す。)、Y1=OHである場合に該当し、化合物(2)と同様の反応を行うことにより製造することができるので、本化合物(5)の製造方法については、前述の化合物(2)の製造方法を参照すればよいものとして、詳細な説明は省略する。
新規な化合物(5)は、前述の化合物(2)において、X1=NR2R2’(R2およびR2’は、前記と同義を示す。)、Y1=OHである場合に該当し、化合物(2)と同様の反応を行うことにより製造することができるので、本化合物(5)の製造方法については、前述の化合物(2)の製造方法を参照すればよいものとして、詳細な説明は省略する。
(化合物(6)の製造方法)
化合物(6)は、前述の化合物(3)において、X1=NR2R2’(R2およびR2’は、前記と同義を示す。)、Y1=OHである場合に該当し、化合物(3)と同様の反応を行うことにより製造することができるので、本化合物(6)の製造方法については、前述の化合物(3)の製造方法を参照すればよいものとして、詳細な説明は省略する。なお、工程(C’)は、工程(C)のうち脱保護に相当する工程である。
化合物(6)は、前述の化合物(3)において、X1=NR2R2’(R2およびR2’は、前記と同義を示す。)、Y1=OHである場合に該当し、化合物(3)と同様の反応を行うことにより製造することができるので、本化合物(6)の製造方法については、前述の化合物(3)の製造方法を参照すればよいものとして、詳細な説明は省略する。なお、工程(C’)は、工程(C)のうち脱保護に相当する工程である。
なお、上記各工程は、化学反応において一般に知られている単離・生成方法によって精製することができ、具体的には、各種クロマトグラフィー、抽出、再結晶等を、適宜、単独またはこれらを組み合わせて用いることができる。
以下、フッ素化プリンヌクレオシド誘導体の製造方法に関する実施例について説明する。
(実施例1)
N2,5’-O−ジトリチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
カナディアン・ジャーナル・オブ・ケミストリー(Can.J.Chem.1972,50,1100)に記載の方法に準じて合成した8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(5g,16.8mmol)のN,N−ジメチルホルムアミド(125mL)溶液に、トリエチルアミン(7.03mL,50.4mmol)と塩化トリチル(14.6g,52.4mmol)とを加え、40℃で20時間加熱攪拌した。得られた溶液にメタノール(50mL)を加え反応を停止させ、減圧下濃縮した。濃縮残渣にジクロロメタン(500mL)と水(300mL)とを加えて分層した後、水層にジクロロメタン(300mL)を加えて再抽出し、得られた二つの有機層を混合し、無水硫酸マグネシウムで乾燥させ、減圧下濃縮した。濃縮残渣にトルエン(73mL)を加えて室温で12時間スラリー洗浄し、得られた固体をクロマトグラフィー(抽出液 ジクロロメタン→ジクロロメタン:メタノール=16:1)により精製し、目的物(7.88g,60%収率)を白色固体として得た。
N2,5’-O−ジトリチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
カナディアン・ジャーナル・オブ・ケミストリー(Can.J.Chem.1972,50,1100)に記載の方法に準じて合成した8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(5g,16.8mmol)のN,N−ジメチルホルムアミド(125mL)溶液に、トリエチルアミン(7.03mL,50.4mmol)と塩化トリチル(14.6g,52.4mmol)とを加え、40℃で20時間加熱攪拌した。得られた溶液にメタノール(50mL)を加え反応を停止させ、減圧下濃縮した。濃縮残渣にジクロロメタン(500mL)と水(300mL)とを加えて分層した後、水層にジクロロメタン(300mL)を加えて再抽出し、得られた二つの有機層を混合し、無水硫酸マグネシウムで乾燥させ、減圧下濃縮した。濃縮残渣にトルエン(73mL)を加えて室温で12時間スラリー洗浄し、得られた固体をクロマトグラフィー(抽出液 ジクロロメタン→ジクロロメタン:メタノール=16:1)により精製し、目的物(7.88g,60%収率)を白色固体として得た。
1H−NMR(CDCl3)δ3.12(dd,J=10.2,5.6Hz,1H),3.28(dd,J=10.2,5.2Hz,1H),3.94(br,1H),4.30−4.33(m,1H),4.60−4.63(m,2H),6.33(d,J=5.9Hz,1H),6.41(br,1H),7.04−7.30(m,30H).
ESI MASS m/z 781.9(M+H)+
ESI MASS m/z 781.9(M+H)+
(実施例2)
N2,5’-O−ジトリチル−8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
N2,5’-O−ジトリチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(78.1mg,0.1mmol)の酢酸エチル(2mL)溶液に、フッ化パーフルオロブタンスルホニル(0.224mL,1.25mmol)とN,N−ジイソプロピルエチルアミン(0.218mL,1.25mmol)を加え65℃で20時間加熱攪拌した。室温に冷却した後、酢酸エチル(5mL)と水(2mL)とを加えて分層し、無水硫酸マグネシウムで乾燥させ、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール=32:1)により精製し、目的物(71.8mg,91%収率)を白色固体として得た。
N2,5’-O−ジトリチル−8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
N2,5’-O−ジトリチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(78.1mg,0.1mmol)の酢酸エチル(2mL)溶液に、フッ化パーフルオロブタンスルホニル(0.224mL,1.25mmol)とN,N−ジイソプロピルエチルアミン(0.218mL,1.25mmol)を加え65℃で20時間加熱攪拌した。室温に冷却した後、酢酸エチル(5mL)と水(2mL)とを加えて分層し、無水硫酸マグネシウムで乾燥させ、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール=32:1)により精製し、目的物(71.8mg,91%収率)を白色固体として得た。
1H−NMR(CDCl3)δ3.03−3.09(m,1H),3.18−3.23(m,1H),4.60(dt,J=22.9,6.9Hz,1H),4.83(dd,J=20.4,6.5Hz,1H),5.29(d,J=52.1Hz,1H),6.34(d,J=6.5Hz,1H),6.43(br,1H),7.05−7.40(m,30H).
ESI MASS m/z 784.4(M+H)+
ESI MASS m/z 784.4(M+H)+
(実施例3)
N2,5’-O−ジトリチル−3’α−フルオロ−2’,3’−ジデオキシグアノシンの合成
ラネーニッケル(550mg,川研ファイン製NDHT−90)に1−プロパノール(5mL)を加え、室温で5分間攪拌した後静置し、溶剤をデカンテーションで除いた。この操作をもう一度繰り返した後、トルエン(5mL)を加え、室温で5分間攪拌した後静置し、溶液をデカンテーションで除き、トルエン(1mL)、N2,5’-O−ジトリチル−8,2’−アンヒドロ−3’−デオキシ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニン(39mg,0.05mmol)を加え、アルゴン雰囲気下90℃で12時間加熱撹拌した。冷却後得られた反応液にジクロロメタン(15mL)を加え、ラネーニッケルをろ別し、ろ液を減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン→ジクロロメタン:メタノール=16:1)により精製し、目的物(22.9mg,61%収率)を白色固体として得た。
N2,5’-O−ジトリチル−3’α−フルオロ−2’,3’−ジデオキシグアノシンの合成
ラネーニッケル(550mg,川研ファイン製NDHT−90)に1−プロパノール(5mL)を加え、室温で5分間攪拌した後静置し、溶剤をデカンテーションで除いた。この操作をもう一度繰り返した後、トルエン(5mL)を加え、室温で5分間攪拌した後静置し、溶液をデカンテーションで除き、トルエン(1mL)、N2,5’-O−ジトリチル−8,2’−アンヒドロ−3’−デオキシ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニン(39mg,0.05mmol)を加え、アルゴン雰囲気下90℃で12時間加熱撹拌した。冷却後得られた反応液にジクロロメタン(15mL)を加え、ラネーニッケルをろ別し、ろ液を減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン→ジクロロメタン:メタノール=16:1)により精製し、目的物(22.9mg,61%収率)を白色固体として得た。
1H−NMR(CDCl3)δ2.00−2.30(m,2H),3.20−3.30(m,2H),4.25(dm,J=26.6Hz,1H),5.03(dm,J=54.7Hz,1H),5.57−5.62(m,1H),7.15−7.45(m,31H).ESI MASS m/z 754.3(M+H)+
(実施例4)
3’α−フルオロ−2,3’−ジデオキシグアノシンの合成
N2,5’-O−ジトリチル−3’α−フルオロ−2’,3’−ジデオキシグアノシン(22.6mg,0.03mmol)を80%酢酸水溶液(0.3mL)に加え、80℃で2時間加熱攪拌した。得られた溶液を減圧下濃縮し、得られた固体をクロマトグラフィー(溶出液 ジクロロメタン:メタノール=4:1)により精製し、目的物(5.6mg,69%収率)を白色固体として得た。
3’α−フルオロ−2,3’−ジデオキシグアノシンの合成
N2,5’-O−ジトリチル−3’α−フルオロ−2’,3’−ジデオキシグアノシン(22.6mg,0.03mmol)を80%酢酸水溶液(0.3mL)に加え、80℃で2時間加熱攪拌した。得られた溶液を減圧下濃縮し、得られた固体をクロマトグラフィー(溶出液 ジクロロメタン:メタノール=4:1)により精製し、目的物(5.6mg,69%収率)を白色固体として得た。
1H−NMR(DMSO-d6)δ2.50−2.67(m,1H),2.70−2.90(m,1H),3.50−3.60(m,2H),4.16(dt,J=27.0,4.9Hz,1H),5.15(t,J=5.4Hz,1H),5.38(dd,J=53.6,4.3Hz,1H),6.15(dd,J=9.4,5.6Hz,1H),6.49(br,2H),7.94(s,1H),10.54(br,1H).
ESI MASS m/z 270.2(M+H)+
ESI MASS m/z 270.2(M+H)+
(実施例5)
N2,5’-O−ジアセチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
ケミカル・アンド・ファーマシューティカル・ブレチン 29巻、3449頁、1981年(Chem.Pharm.Bull.1981,29,3449)に記載の方法に準じて合成したN2,3’,5’-O−トリアセチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(1.52g,3.6mmol)のメタノール(30mL)溶液に、28%ナトリウムメトキシドメタノール溶液(トータル0.84mL,4.1mmol)を20分おきに0.3mL、0.3mL、0.12mL、0.06mL、0.06mLずつ、80分かけて加えた。その後、反応液に6mol/L塩酸水溶液(0.7mL)を加えて反応を停止させ、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 16:1→4:1)により精製し、目的物(423mg,31%収率)を白色固体として得た。
N2,5’-O−ジアセチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
ケミカル・アンド・ファーマシューティカル・ブレチン 29巻、3449頁、1981年(Chem.Pharm.Bull.1981,29,3449)に記載の方法に準じて合成したN2,3’,5’-O−トリアセチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(1.52g,3.6mmol)のメタノール(30mL)溶液に、28%ナトリウムメトキシドメタノール溶液(トータル0.84mL,4.1mmol)を20分おきに0.3mL、0.3mL、0.12mL、0.06mL、0.06mLずつ、80分かけて加えた。その後、反応液に6mol/L塩酸水溶液(0.7mL)を加えて反応を停止させ、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 16:1→4:1)により精製し、目的物(423mg,31%収率)を白色固体として得た。
1H−NMR(DMSO-d6)δ1.91(s,3H),2.16(s,3H),3.95−4.00(m,1H),4.10−4.18(m,2H),4.33−4.38(m,1H),4.84(dd,J=6.7,3.0Hz,1H),6.09(br,1H),6.47(d,J=6.7Hz,1H).
ESI MASS m/z 382.0(M+H)+
ESI MASS m/z 382.0(M+H)+
(実施例6)
N2,5’-O−ジアセチル−8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
N2,5’-O−ジアセチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(76.3mg,0.2mmol)のジクロロメタン(4mL)懸濁液にジエチルアミノサルファートリフルオリド(0.99mL,1.0mmol)を室温にて加え、40℃で1時間加熱攪拌した。室温に冷却した後、炭酸水素ナトリウム水溶液(100mg)を加えて反応を停止させ、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 32:1→16:1)により精製し、目的物(35.8mg,47%)を白色固体として得た。
N2,5’-O−ジアセチル−8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
N2,5’-O−ジアセチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(76.3mg,0.2mmol)のジクロロメタン(4mL)懸濁液にジエチルアミノサルファートリフルオリド(0.99mL,1.0mmol)を室温にて加え、40℃で1時間加熱攪拌した。室温に冷却した後、炭酸水素ナトリウム水溶液(100mg)を加えて反応を停止させ、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 32:1→16:1)により精製し、目的物(35.8mg,47%)を白色固体として得た。
1H−NMR(DMSO-d6)δ1.83(s,3H),2.17(s,3H),4.00−4.03(m,2H),4.69(dt,J=23.8,5.8Hz,1H),5.31(dd,J=21.7,6.7Hz,1H),5.59(d,J=50.7Hz,1H),6.66(d,J=6.7Hz,1H).
ESI MASS m/z 383.8(M+H)+
ESI MASS m/z 383.8(M+H)+
(実施例7)
N2−ジメチルアミノメチレン−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
カナディアン・ジャーナル・オブ・ケミストリー(Can.J.Chem.1972,50,1100)に記載の方法に準じて合成した8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(594.6mg,2mmol)のN,N−ジメチルホルムアミド懸濁液(10mL)に、N,N−ジメチルホルムアミドジメチルアセタール(1.0mL,7.5mmol)を加え、室温で13時間攪拌し、得られた反応液を減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 8:1)により精製し、目的物(728.0mg,92%)を白色固体として得た。
N2−ジメチルアミノメチレン−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
カナディアン・ジャーナル・オブ・ケミストリー(Can.J.Chem.1972,50,1100)に記載の方法に準じて合成した8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(594.6mg,2mmol)のN,N−ジメチルホルムアミド懸濁液(10mL)に、N,N−ジメチルホルムアミドジメチルアセタール(1.0mL,7.5mmol)を加え、室温で13時間攪拌し、得られた反応液を減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 8:1)により精製し、目的物(728.0mg,92%)を白色固体として得た。
1H−NMR(DMSO-d6)δ3.03(s,3H),3.16(s,3H),3.35−3.41(m,1H),3.46−3.51(m,1H),3.91−3.95(m,1H),4.33−4.37(m,1H),4.81(dd,J=6.6,2.3Hz,1H),4.92(t,J=5.5Hz,1H),5.87(d,J=4.6Hz,1H),6.34(d,J=6.6Hz,1H),8.51(s,1H).
ESI MASS m/z 352.6(M+H)+
ESI MASS m/z 352.6(M+H)+
(実施例8)
N2−ジメチルアミノメチレン−5’−O−トリチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
N2−ジメチルアミノメチレン−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(710mg,1.8mmol)のN,N−ジメチルホルムアミド(2mL)とピリジン(2mL)混合溶液に、塩化トリチル(669mg,2.4mmol)を三度に分け、10分おきに加え、室温で25時間攪拌し、得られた反応液を減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール8:1)により精製し、目的物(558.8mg,52%)を白色固体として得た。
N2−ジメチルアミノメチレン−5’−O−トリチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニンの合成
N2−ジメチルアミノメチレン−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(710mg,1.8mmol)のN,N−ジメチルホルムアミド(2mL)とピリジン(2mL)混合溶液に、塩化トリチル(669mg,2.4mmol)を三度に分け、10分おきに加え、室温で25時間攪拌し、得られた反応液を減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール8:1)により精製し、目的物(558.8mg,52%)を白色固体として得た。
1H−NMR(DMSO-d6)δ2.98−3.02(m,1H),3.02(s,3H),3.08−3.14(m,1H),3.17(s,3H),4.12−4.17(m,1H),4.37−4.41(m,1H),4.76(dd,J=6.4,3.3Hz,1H),5.91(d,J=4.7Hz,1H),6.44(d,J=6.4Hz,1H),7.10−7.30(m,15H),8.56(s,1H).
ESI MASS m/z 595.0(M+H)+
ESI MASS m/z 595.0(M+H)+
(実施例9)
N2−ジメチルアミノメチレン−5’−O−トリチル−8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニン
N2−ジメチルアミノメチレン−5’−O−トリチルメチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(59.5mg,0.1mmol)のジクロロメタン(1.0mL)懸濁液に、ジエチルアミノサルファートリフルオリド(0.05mL,0.1mmol)を室温にて加え、40℃で1時間加熱攪拌した。得られた反応液をジクロロメタン(4mL)で希釈し、飽和炭酸水素ナトリウム水溶液(1.5mL)と水(1.5mL)との混合溶液に加えて反応を停止し、分層した。水層にジクロロメタン(5mL)を加えて再抽出し、得られた二つの有機層を混合し、無水硫酸マグネシウムで乾燥し、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 32:1→16:1)により精製し、目的物(26.0mg,44%)を白色固体として得た。
N2−ジメチルアミノメチレン−5’−O−トリチル−8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルグアニン
N2−ジメチルアミノメチレン−5’−O−トリチルメチル−8,2’−アンヒドロ−8−メルカプト−9−β−アラビノフラノシルグアニン(59.5mg,0.1mmol)のジクロロメタン(1.0mL)懸濁液に、ジエチルアミノサルファートリフルオリド(0.05mL,0.1mmol)を室温にて加え、40℃で1時間加熱攪拌した。得られた反応液をジクロロメタン(4mL)で希釈し、飽和炭酸水素ナトリウム水溶液(1.5mL)と水(1.5mL)との混合溶液に加えて反応を停止し、分層した。水層にジクロロメタン(5mL)を加えて再抽出し、得られた二つの有機層を混合し、無水硫酸マグネシウムで乾燥し、減圧下濃縮した。クロマトグラフィー(溶出液 ジクロロメタン:メタノール 32:1→16:1)により精製し、目的物(26.0mg,44%)を白色固体として得た。
1H−NMR(DMSO-d6)δ2.96−3.20(m,2H),3.03(s,3H),3.17(s,3H),4.55(dt,J=24.9,6.5Hz,1H),5.24(dd,J=20.2,6.4Hz,1H),5.52(d,J=50.9Hz,1H),6.53(d,J=6.4Hz,1H),7.15−7.30(m,15H),8.55(s,1H),11.40(br,1H).ESI MASS m/z 597.0(M+H)+
本発明は、3’位(好ましくはα位)がフッ素化されたプリンヌクレオシド誘導体の製造方法に関し、更にはこれら誘導体の製造に有用な中間体化合物およびその製造方法に関しており、工業的生産に適した、経済的かつ効率的な3’−α位がフッ素化されたフッ素化ヌクレオシド誘導体の製造方法およびその中間体およびその製造方法を提供することができる。
Claims (21)
- 一般式(1)
[式中、R1はヒドロキシル基の保護基を示し、X1は水素原子、ハロゲン原子、置換されてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y1は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表されるプリンヌクレオシド誘導体に、フッ素化試薬を作用させることを特徴とする一般式(2)
[式中、R1、X1及びY1は前記と同義である。]
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。 - 前記フッ素化試薬がフッ化パーフルオロアルカンスルホニルであり、塩基存在下に作用させる、請求項1に記載の製造方法。
- 請求項1または2のいずれか1つに記載の方法に従って、一般式(2)で表されるフッ素化プリンヌクレオシド誘導体を製造した後、
(A)硫黄原子を除去する脱硫工程、
(B)R1を除去する脱保護工程、
(C)X1とX2とが異なる基を示す場合、および/またはY1とY2とが異なる基を示す場合、X1をX2に変換し、および/またはY1をY2に変換する工程
の各工程を順不同に行うことを特徴とする、一般式(3)
[式中、X2は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y2は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。 - 一般式(1)で表されるプリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有し、
一般式(2)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有し、
一般式(3)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、X2及びY2は、上記と同義である。]
で表される相対配置を有する、請求項1〜3のいずれか1つに記載の製造方法。 - 一般式(4)
[式中、R1はヒドロキシル基の保護基を示し、R2及びR2’は、それぞれ独立して水素原子、又はアミノ基の保護基を示す。]
で表されるプリンヌクレオシド誘導体に、フッ素化試薬を作用させることを特徴とする一般式(5)
[式中、R1、R2及びR2’は前記と同義である。]
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。 - 前記フッ素化試薬は、フッ化パーフルオロアルカンスルホニルであり、塩基の存在下に作用させる、請求項5に記載の製造方法。
- 請求項5または6のいずれか1つに記載の方法に従って、一般式(5)で表されるフッ素化プリンヌクレオシド誘導体を製造した後、
(A)硫黄原子を除去する脱硫工程、
(B)R1を除去する脱保護工程
(C’)R2及び/またはR2’が保護基である場合には、脱保護工程
の各工程を順不同に行うことを特徴とする、一般式(6)
で表されるフッ素化プリンヌクレオシド誘導体の製造方法。 - 一般式(4)で表されるプリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有し、
一般式(5)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有し、
一般式(6)で表されるフッ素化プリンヌクレオシド誘導体は、
で表される相対配置を有する、請求項5〜7のいずれか1つに記載の製造方法。 - R1及びR2がそれぞれ置換基を有していてもよい、トリチル基、又は9−フェニルフルオレニル基であり、R2’が水素である、請求項5〜8のいずれか1つに記載の製造方法。
- 一般式(1)
[式中、R1はヒドロキシル基の保護基を示し、X1は水素原子、ハロゲン原子、置換されてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y1は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表される、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。 - 一般式(1)で表されるプリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有する、請求項10に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。 - 一般式(4)
[式中、R1はヒドロキシル基の保護基を示し、R2及びR2’は、それぞれ独立して、水素原子、又はアミノ基の保護基を示す。]
で表される、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。 - 一般式(4)で表されるプリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有する、請求項12に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。 - R1及びR2が、それぞれ、置換基を有していてもよい、トリチル基、9−フェニルフルオレニル基、又はアセチル基であり、R2’が水素原子である、請求項12または13に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
- 一般式(2)
[式中、R1はヒドロキシル基の保護基を示し、X1は水素原子、ハロゲン原子、置換されてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示し、Y1は水素原子、ハロゲン原子、置換されていてもよいアミノ基、又は置換されていてもよいヒドロキシル基を示す。]
で表される、8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。 - 一般式(2)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、X1及びY1は、上記と同義である。]
で表される相対配置を有している、請求項15に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルプリン誘導体。 - 一般式(5)
[式中、R1はヒドロキシル基の保護基を示し、R2及びR2’はそれぞれ独立して水素原子、又はアミノ基の保護基を示す。]
で表される、8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。 - 一般式(5)で表されるフッ素化プリンヌクレオシド誘導体は、
[式中、R1、R2及びR2’は、上記と同義である。]
で表される相対配置を有している、請求項17に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。 - R1及びR2は、それぞれ、置換基を有していてもよい、トリチル基、9−フェニルフルオレニル基、又はアセチル基であり、R2’は、水素原子である、請求項17または18に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
- R1が置換基を有していてもよい、トリチル基であり、R2’、R2’が一緒になってジメチルアミノメチリデン基である、請求項12または13に記載の8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
- R1が置換基を有していてもよい、トリチル基であり、R2’、R2’が一緒になってジメチルアミノメチリデン基である、請求項17または18に記載の8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−アラビノフラノシルグアニン誘導体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004236246A JP2006052182A (ja) | 2004-08-13 | 2004-08-13 | フッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004236246A JP2006052182A (ja) | 2004-08-13 | 2004-08-13 | フッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2006052182A true JP2006052182A (ja) | 2006-02-23 |
Family
ID=36029882
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004236246A Abandoned JP2006052182A (ja) | 2004-08-13 | 2004-08-13 | フッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2006052182A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1816135A1 (en) * | 2006-01-30 | 2007-08-08 | Ajinomoto Co., Inc. | Production method of fluorinated purine nucleoside derivative, intermediate therefor and production method thereof |
-
2004
- 2004-08-13 JP JP2004236246A patent/JP2006052182A/ja not_active Abandoned
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1816135A1 (en) * | 2006-01-30 | 2007-08-08 | Ajinomoto Co., Inc. | Production method of fluorinated purine nucleoside derivative, intermediate therefor and production method thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3830999B2 (ja) | 2,2−ジフルオロ−3−カルバモイルリボーススルホネート化合物およびβ−ヌクレオシドの製造方法 | |
| CN1086519A (zh) | 立体有择阴离子糖基化方法 | |
| US8648188B2 (en) | Preparation of 2-chloro-9-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)-adenine | |
| JP4593917B2 (ja) | プリンヌクレオシドを調製する方法 | |
| EP2993177A1 (en) | A chemo-enzymatic preparation method for purine nucleosides and their deaza- and aza- analogues | |
| ES2267725T3 (es) | Procedimientos para la sintesis de 2-cloro-9 (-2-desoxi-2-fluoro-beta-d-arabinofuranosil)-9h-purin-6-amina. | |
| JP2006052182A (ja) | フッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法 | |
| EP0653437B1 (en) | Process for preparing AZT and derivatives thereof | |
| EP1550665A1 (en) | A process for the production of purine nucleoside compounds | |
| JP2006500375A (ja) | 9−β−アノマー性ヌクレオシド類似体の調製方法 | |
| JP2007522151A (ja) | ジフルオロヌクレオシド及びその調製方法 | |
| Komatsu et al. | An efficient amination method for manufacturing cytidines | |
| Cooperwood et al. | Synthesis of L-3′-hydroxymethylribonucleosides | |
| EP0945460A1 (en) | Methods for producing nucleoside derivatives and intermediates therefore | |
| EP1816135A1 (en) | Production method of fluorinated purine nucleoside derivative, intermediate therefor and production method thereof | |
| Robles et al. | Highly β-Stereoselective Nucleosidation from α-D-Xylo-and α-D-Ribo-furanose 1, 2-Thiocarbonates | |
| Jabgunde et al. | Synthesis of 3′-fluoro-4′-amino-hexitol nucleosides with a pyrimidine nucleobase as building blocks for oligonucleotides | |
| US20070179290A1 (en) | Production method of fluorinated purine nucleoside derivative, intermediate therefor and production method thereof | |
| JP2002293792A (ja) | ヌクレオシド又は糖のフッ素化誘導体の製造方法 | |
| US20150376219A1 (en) | Selective Preparations of Purine Nucleosides and Nucleotides: Reagents and Methods | |
| JPH0296590A (ja) | 新規核酸類 | |
| JP2005206585A (ja) | プリンヌクレオシド化合物の製造方法 | |
| JPH11322780A (ja) | ヌクレオシド誘導体とその製法 | |
| Haraguchi et al. | 4-Thiofuranoid glycals: versatile synthons for stereoselective synthesis of 4′-thionucleosides | |
| JPH01104092A (ja) | ヌクレオシド誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070626 |
|
| A762 | Written abandonment of application |
Free format text: JAPANESE INTERMEDIATE CODE: A762 Effective date: 20080715 |