ES2946551T3 - Síntesis de compuestos de 4-aminopirimidina - Google Patents
Síntesis de compuestos de 4-aminopirimidina Download PDFInfo
- Publication number
- ES2946551T3 ES2946551T3 ES18785637T ES18785637T ES2946551T3 ES 2946551 T3 ES2946551 T3 ES 2946551T3 ES 18785637 T ES18785637 T ES 18785637T ES 18785637 T ES18785637 T ES 18785637T ES 2946551 T3 ES2946551 T3 ES 2946551T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- process according
- iii
- amine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
La presente invención se refiere a un proceso para fabricar 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina de Fórmula (I) que comprende partir de 6-cloro-2-isobutilpirimidin-4- amina y terc-butil azetidin-3-il(metil)carbamato, u otra N-metilazetidin-3-amina N-protegida, y realizando las siguientes etapas: (a) reacción de acoplamiento de ambos compuestos en sulfóxido de dimetilo en presencia de carbonato de potasio para proporcionar un compuesto intermedio protegido; y (b) desprotección del compuesto protegido para producir 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina. La invención también se refiere a un proceso para fabricar el compuesto intermedio protegido, en el que se omite el paso de desprotección (b). La invención también se refiere a los compuestos obtenidos a partir de los procedimientos según la invención. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Síntesis de compuestos de 4-aminopirimidina
Campo de la invención
La presente invención pertenece al campo de la fabricación de compuestos orgánicos. La presente invención se refiere especialmente a la síntesis de 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina.
Antecedentes de la invención
Los derivados de 4-aminopirimidina son una clase de compuestos de fórmula general (A):

que son útiles como ingredientes terapéuticamente activos, especialmente como antagonistas del receptor de histamina H4.
Entre los derivados de 4-aminopirimidina, 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina de fórmula (I) (en lo sucesivo "Compuesto (I)", abreviado "(I)") es de particular interés:

La solicitud de patente WO 2009/080721 describe el compuesto de fórmula (I) y varios métodos sintéticos diferentes del mismo, especialmente el acoplamiento asistido por base de un compuesto aromático clorado con una diamina secundaria heterocíclica protegida con ferf-butiloxicarbonilo (Boc) como se representa en el esquema 1 a continuación:

Esta reacción se lleva a cabo en presencia de N,N-diisopropiletilamina (DIPEA) como base en etanol (EtOH) a reflujo. El compuesto intermedio protegido con Boc se somete luego a una etapa de desprotección para proporcionar el compuesto (I). La etapa de desprotección se realiza mediante ácido clorhídrico (HCl) en metanol (MeOH) a temperatura ambiente, directamente sobre el compuesto intermedio no aislado previamente.
El procedimiento del documento WO 2009/080721 tiene muchos inconvenientes que limitan significativamente la síntesis industrial del compuesto (I). Especialmente, durante la etapa de acoplamiento:
- la conversión es lenta, por lo que se requiere un tiempo de reacción prolongado (al menos 2 días) para alcanzar una conversión suficiente de compuesto aromático clorado en compuesto intermedio;
- la conversión es incompleta, de modo que el compuesto intermedio sintetizado se mezcla con el compuesto aromático clorado sin reaccionar y/o la diamina secundaria heterocíclica sin reaccionar; y
- la diamina secundaria heterocíclica se degrada en el medio de reacción, de modo que sus productos de degradación están presentes con el compuesto intermedio sintetizado, y de modo que se tiene que agregar un alto exceso de diamina secundaria heterocíclica en porciones para compensar la degradación de este compuesto.
Todos estos factores coinciden con el alto coste de operación de los procesos de fabricación de la técnica anterior, aunque simplemente logran un rendimiento bajo y una pureza deficiente del compuesto (I). La pureza insuficiente requiere una etapa de purificación extenso por cromatografía. Por lo tanto, los procesos existentes no satisfacen las necesidades de la industria moderna en términos de costes e impacto ambiental.
El solicitante realizó una investigación en profundidad para mejorar el método de fabricación del compuesto (I) y, sorprendentemente, descubrió que la selección de condiciones experimentales muy específicas supera las limitaciones del procedimiento de la técnica anterior, especialmente las limitaciones de la etapa de acoplamiento.
Sumario
Esta invención se refiere a un procedimiento de fabricación de 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina de fórmula (I), como se describe a continuación, el procedimiento comprende las siguientes etapas:
(a) hacer reaccionar 6-cloro-2-isobutilpirimidin-4-amina de fórmula (III), como se describe a continuación, con una N-metilazetidin-3-amina N-protegida de fórmula (IV), como se describe a continuación,
en un disolvente orgánico que es dimetilsulfóxido (DMSO);
en presencia de una base que es carbonato de potasio (K2CO3);
a una temperatura que oscila entre 85 y 125 °C;
oscilando la proporción molar inicial entre el compuesto de fórmula (IV) y el compuesto de fórmula (III) entre 0.85 y 1.25;
para obtener un compuesto intermedio protegido de fórmula (II), como se describe a continuación;
y
(b) realizar una desprotección de N del compuesto de fórmula (II); para obtener el compuesto de fórmula (I).
Según la invención, el grupo protector se selecciona de carbobenciloxi (Cbz), p-metoxibencil carbonilo (Moz), tertbutiloxicarbonilo (Boc), 9-fluorenilmetiloxicarbonilo (Fmoc) y 2,2,2-tricloroetoxicarbonilo (Troc). Según una realización, el grupo protector preferiblemente es ferf-butiloxicarbonilo (Boc).
Según una realización, la proporción molar inicial en la etapa (a) de acoplamiento entre el compuesto de fórmula (IV) y el compuesto de fórmula (III) oscila entre 0.90 y 1.20, preferiblemente desde 0.95 a 1.10, más preferiblemente desde 0.99 a 1.09. En una realización, la proporción molar inicial en la etapa (a) de acoplamiento entre el compuesto de fórmula (IV) y el compuesto de fórmula (III) es 1.04.
Según una realización, la temperatura durante la etapa (a) de acoplamiento oscila entre 90 a 120 °C, preferiblemente desde 100 a 120 °C, más preferiblemente desde 105 a 115 °C. En una realización, la temperatura durante la etapa (a) de acoplamiento es de 110 °C.
Según una realización, el tiempo de reacción de la etapa (a) de acoplamiento oscila entre 16 h y 32 h, preferiblemente desde 20 a 24 h.
Según una realización, la etapa (a) de acoplamiento va seguida por una etapa (a') de tratamiento y aislamiento que comprende las etapas: (a'-1) dilución de la mezcla de reacción con agua y con un primer disolvente orgánico aprótico polar; (a'-2) separación de fases, y opcionalmente lavado con agua; (a'-3) cambio de disolvente del primer disolvente a un segundo disolvente orgánico aprótico polar; (a'-4) recristalización por enfriamiento; y (a'-5) filtración; para proporcionar el compuesto de fórmula (II). En una realización, el disolvente orgánico aprótico polar en la etapa (a'-1) de dilución es acetato de etilo. En una realización, el disolvente orgánico aprótico polar en la etapa (a'-3) de cambio de disolvente es acetato de isopropilo.
Según una realización, la etapa (a) de acoplamiento o etapa (a') de tratamiento y aislamiento va seguida por una etapa (a”) de purificación que comprende una etapa de recristalización del compuesto de fórmula (II) en un disolvente orgánico aprótico polar, preferiblemente acetato de isopropilo. Según una realización, la etapa (b) de desprotección se
realiza poniendo en contacto el compuesto de fórmula (II) con una solución acuosa diluida de ácido clorhídrico, preferiblemente una solución acuosa de ácido clorhídrico al 30 % p/p.
Según una realización, la proporción molar inicial entre el compuesto de fórmula (II) y el ácido clorhídrico oscila entre 6 y 10, preferiblemente desde 7 a 9, más preferiblemente es 7.9.
La invención también se refiere a un procedimiento de fabricación de un compuesto de fórmula (II), como se describe a continuación, el procedimiento que comprende:
hacer reaccionar 6-cloro-2-isobutilpirimidin-4-amina de fórmula (III), como se describe a continuación, con una N-metilazetidin-3-amina N-protegida de fórmula (IV), como se describe a continuación, en el que el grupo protector se selecciona de carbobenciloxi (Cbz), p-metoxibencil carbonilo (Moz), tert-butiloxicarbonilo (Boc), 9-fluorenilmetiloxicarbonilo (Fmoc) y 2,2,2-tricloroetoxicarbonilo (Troc);
en un disolvente orgánico que es dimetilsulfóxido;
en presencia de una base que es carbonato de potasio;
a una temperatura que oscila entre 85 y 125 °C;
oscilando la proporción molar inicial entre el compuesto de fórmula (IV) y el compuesto de fórmula (III) entre 0.85 y 1.25;
para obtener el compuesto de fórmula (II).
Definiciones
En la presente invención, los siguientes términos tienen los siguientes significados:
- "Aproximadamente" precediendo a una cifra significa más o menos el 10 % del valor de dicha cifra.
- "Proporción molar" se refiere a las proporciones relativas de los reactivos que se usan en una reacción química. La proporción molar entre un reactivo A y otro reactivo B se calcula dividiendo la cantidad molar del reactivo A por la cantidad molar del reactivo B. A menos que se especifique lo contrario, la proporción molar se refiere a la proporción molar inicial, es decir, con la proporción molar entre las cantidades molares introducidas de reactivos, antes de cualquier reacción de los mismos.
- "grupo protector" se refiere a una unidad estructural orgánica apropiada utilizado para proteger un determinado grupo funcional en una síntesis química. En la presente invención, el grupo funcional protegido es un grupo amino secundario y el grupo protector se refiere a un grupo protector de amina secundaria, preferiblemente ferf-butiloxicarbonilo (Boc).
- "solvato" se usa en este documento para describir un complejo molecular que comprende el compuesto de fórmula I divulgado en este documento y una o más moléculas de disolvente farmacéuticamente aceptables, por ejemplo, etanol. El término "hidrato" se emplea cuando dicho disolvente es agua.
- Por "farmacéuticamente aceptable" se entiende que los ingredientes de una composición farmacéutica son compatibles entre sí y no son perjudiciales para el sujeto al que se administra.
- "Excipiente farmacéuticamente aceptable" se refiere a un excipiente que no produce reacciones adversas, alérgicas u otras reacciones no deseadas cuando se administra a un animal, preferiblemente a un ser humano. Incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardadores de la absorción y similares. Para la administración humana, las preparaciones deben cumplir con los estándares de esterilidad, pirogenicidad, seguridad general, calidad y pureza requeridos por las oficinas reguladoras, tales como, por ejemplo, la oficina de FDA o EMA.
- "Sujeto" se refiere a un mamífero, preferiblemente un ser humano. En una realización, el sujeto es una mascota, que incluye, sin limitación, un perro, un gato, un conejillo de indias, un hámster, una rata, un ratón, un hurón, un conejo, un pájaro o un anfibio. En una realización, un sujeto puede ser un "paciente", es decir, una mujer o un hombre, un adulto o un niño, que/quien está a la espera de recibir, o está recibiendo atención médica o fue/es/será objeto de un procedimiento médico, o es monitoreado para el desarrollo de una enfermedad mediada por el receptor de histamina H4 o de una enfermedad, trastorno o afección del αdo.
- "Cantidad terapéuticamente eficaz" se refiere al nivel o cantidad del agente a la que está dirigido, sin causar efectos secundarios negativos o adversos significativos a la diana, (1) retrasar o prevenir la aparición de la afección o trastorno patológico diana; (2) ralentizar o detener la progresión, el agravamiento o el deterioro de uno o más síntomas de la afección o trastorno patológico diana; (3) provocar mejoras de los síntomas de la afección o trastorno patológico diana; (4) reducir la gravedad o incidencia de la afección o trastorno patológico diana; o (5) curar la afección o trastorno patológico diana. Puede administrarse una cantidad terapéuticamente eficaz antes de la aparición de la afección o trastorno patológico diana, para una acción profiláctica o preventiva. Como alternativa o adicionalmente, la cantidad
terapéuticamente eficaz se puede administrar después de la aparición de la afección o trastorno patológico diana, para una acción terapéutica. En una realización, una cantidad terapéuticamente eficaz de la composición es una cantidad que es eficaz para reducir al menos un síntoma de la afección o trastorno patológico diana.
- "Tratar" o "tratamiento" o "alivio" se refiere tanto al tratamiento terapéutico como a las medidas profilácticas o preventivas; en los que el objetivo es prevenir o ralentizar (disminuir) la afección o trastorno patológico diana. Aquellos que necesitan tratamiento incluyen aquellos que ya padecen el trastorno, así como aquellos propensos a tener el trastorno o aquellos en quienes se debe prevenir el trastorno. Un sujeto o mamífero es "tratado" con éxito para el trastorno patológico diana sí, después de recibir una cantidad terapéutica del compuesto o composición divulgada en este documento, el sujeto o mamífero muestra efectos observables en uno o más de los siguientes: (i) alivio hasta cierto punto de uno o más de los síntomas asociados con el trastorno o afección específica (tales como, por ejemplo, vértigo, mareos y tinnitus en el caso de una enfermedad del αdo); (ii) reducción de la morbilidad y mortalidad, y (iii) mejora en los aspectos de calidad de vida. En una realización, la afección o trastorno patológico diana es un trastorno del αdo, y el término "tratado" puede referirse a la promoción del funcionamiento del αdo; o disminución de la pérdida auditiva. Los parámetros anteriores para evaluar el éxito del tratamiento y la mejora del trastorno se pueden medir fácilmente mediante procedimientos de rutina familiares para un médico.
- "Trastornos vestibulares lesionales" se refiere a trastornos vestibulares en los que están presentes o aparecerán lesiones de las células del αdo interno y/o del nervio vestibular durante el transcurso del trastorno. En este caso, la funcionalidad del vestíbulo se ve afectada. Sin embargo, las alteraciones morfofuncionales de los órganos terminales vestibulares no pueden evaluarse directamente (a excepción de las lesiones grandes que pueden detectarse mediante MRI). Por el contrario, actualmente se usan métodos de evaluación indirecta para evaluar la pérdida de funcionalidad del vestíbulo. Estos métodos de prueba generalmente se llevan a cabo en clínicas/hospitales ENT Ejemplos de tales métodos incluyen, pero no se limitan a, la videonistagmografía (VNG) y la evaluación del reflejo vestíbulo-ocular (VOR) usando pruebas calóricas o rotacionales, prueba de impulso de cabeza de video (vHIT) y potenciales miogénicos evocados vestibulares (VEMP). Los trastornos vestibulares lesionales incluyen, pero no se limitan a:
° trastornos vestibulares en los que una inflamación del αdo interno y/o del nervio vestibular induce daños reversibles y/o irreversibles. Ejemplos de afecciones de este grupo incluyen, pero no se limitan a, neuritis vestibular, vestibulopatía unilateral aguda y neuronitis vestibular;
° trastornos vestibulares en los que se ven afectados los fluidos del αdo interno (anomalías en la cantidad, composición y/o presión de la endolinfa), estos trastornos suelen desarrollar lesiones durante el transcurso de la enfermedad. Ejemplos de afecciones de este grupo son la enfermedad de Meniére y la hidropesía endolinfática secundaria. Están asociados con tinnitus y pérdida de audición;
° trastornos vestibulares inducidos por agresiones o lesiones de los órganos terminales vestibulares. Ejemplos de dichas afecciones son vértigo causado por isquemia local, excitotoxicidad, traumatismo que afecta a los huesos temporales o agresión ototóxica a las células ciliadas vestibulares por fármacos tales como gentamicina y cisplatino;
° trastornos vestibulares iterativos de origen desconocido que conducen a déficits vestibulares permanentes, pero sin tinnitus ni pérdida auditiva. Un ejemplo de una afección de este grupo es la migraña vestibular (o vértigo migrañoso).
- "Trastornos vestibulares no lesionales" se refiere a los trastornos vestibulares apoyados por una crisis de vértigo transitoria y, a menudo, iterativa en los que no se puede observar ninguna lesión en las células del αdo interno y/o el nervio vestibular. En este caso, la funcionalidad del vestíbulo evaluada entre las crisis de vértigo mediante pruebas funcionales (VOR, VNG) no difiere del vestíbulo sano. Los trastornos vestibulares no lesionales incluyen, pero no se limitan a:
o trastornos vestibulares en los que se han acumulado residuos en una parte del αdo interno. Estos desechos, llamados otoconias, están formados por pequeños cristales de carbonato de calcio y, cuando se desplazan, envían señales falsas al cerebro. Los ejemplos de dichas afecciones incluyen, pero no se limitan a, vértigos posicionales y, en particular, vértigo posicional paroxístico benigno (BPPV);
o trastornos vestibulares iterativos de origen desconocido sin tinnitus, pérdida de audición o déficits vestibulares permanentes.
En la presente invención, las siguientes abreviaturas tienen los siguientes significados:
- 2-MeTHF: 2-metiltetrahidrofurano;
-ACN: acetonitrilo;
- AcOH: ácido acético;
- AL: capa acuosa;
- Boc: tert-butiloxicarbonilo;
- DIPEA: N,N'-diisopropiletilamina;
- DMSO: dimetilsulfóxido;
- EtOAc: acetato de etilo;
- EtOH: etanol;
- GC: cromatografía de gases;
- HCl: ácido clorhídrico;
- HLPC: cromatografía líquida de alta resolución;
- H2O: agua;
- iPAc: acetato de isopropilo;
- MeOH: metanol;
- MTBE: metil tert-butil éter;
- NaOEt: etóxido de sodio;
- NH3: amoníaco;
- OL: capa de aceite;
- POCl3: cloruro de fosforilo;
- TA: temperatura ambiente.
Esta invención se refiere a un procedimiento de fabricación de 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina ("Compuesto (I)", abreviado "(I)") de fórmula (I):

el procedimiento que comprende las siguientes etapas:
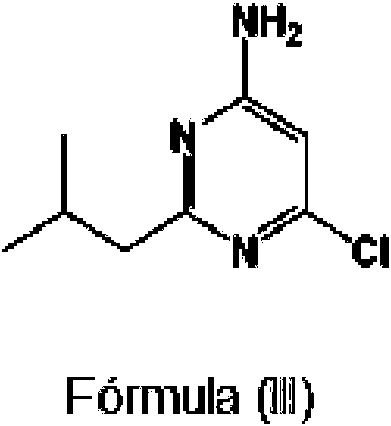
(a) hacer reaccionar 6-cloro-2-isobutilpirimidin-4-amina ("Compuesto 1 )", abreviado )") de fórmula (III):

con una N-metilazetidin-3-amina N-protegida (en adelante "Compuesto (IV)", abreviado "(IV)") de fórmula (IV):

en la que RP es un grupo protector seleccionado de carbobenciloxi (Cbz), p-metoxibencil carbonilo (Moz), tertbutiloxicarbonilo (Boc), 9-fluorenilmetiloxicarbonilo (Fmoc) y 2,2,2-tricloroetoxicarbonilo (Troc);
en dimetilsulfóxido (DMSO) como disolvente orgánico;
en presencia de carbonato de potasio (K2CO3) como base;
a una temperatura que oscila entre 85 y 125 °C;
oscilando la proporción molar inicial entre el compuesto (IV) y el compuesto (III) entre 0.85 y 1.25;
para obtener un compuesto intermedio protegido ("Compuesto (II)", abreviado "(II)") de fórmula (II):

en la que RP es un grupo protector seleccionado de carbobenciloxi (Cbz), p-metoxibencil carbonilo (Moz), tertbutiloxicarbonilo (Boc), 9-fluorenilmetiloxicarbonilo (Fmoc) y 2,2,2-tricloroetoxicarbonilo (Troc); y
(b) realizar una desprotección del compuesto (II);
para obtener el compuesto (I).
Este procedimiento según la invención se representa en el esquema 2, a contionuación:

Según la invención, el grupo protector RP es una unidad estructural orgánica seleccionada de carbobenciloxi (CBz), pmetoxibencil carbonilo (Moz), ferf-butiloxicarbonilo (Boc), 9-fluorenilmetiloxicarbonilo (FMOC) y 2,2,2-tricloroetoxicarbonilo (Troc).
En una realización preferida, el grupo protector RP es ferf-butiloxicarbonilo (Boc). En esta realización preferida, el compuesto (IV) es ferf-butil azetidin-3-il(metil)carbamato de fórmula (IV'):

y el compuesto (II) es tert-butil(1-(6-amino-2-isobutilpirimidin-4-il)azetidin-3-il)(metil)carbamato de fórmula (II'):

Según una realización, la proporción molar inicial en la etapa (a) de acoplamiento entre el compuesto (IV) y el compuesto (III) oscila entre 0.90 y 1.20, preferiblemente desde 0.95 a 1.10, más preferiblemente desde 0.99 a 1.09, más preferiblemente es 1.04.
Según una realización, la proporción molar inicial en la etapa (a) de acoplamiento entre K2CO3 y el compuesto (III) oscila entre 1.05 y 1.5, preferiblemente desde 1.1 a 1.3, más preferiblemente desde 1.15 a 1.25, más preferiblemente es 1.2.
Según una realización, la temperatura durante la etapa (a) de acoplamiento oscila entre 90 a 120 °C, preferiblemente desde 100 a 120 °C, más preferiblemente desde 105 a 115 °C, más preferiblemente es 110 °C.
Según una realización, el tiempo de reacción de la etapa (a) de acoplamiento es de al menos 12 h, preferiblemente de al menos 16 h, más preferiblemente de al menos 20 h. En una realización, el tiempo de reacción de la etapa (a) de acoplamiento no es superior a 48 h, preferiblemente no superior a 32 h, más preferiblemente no superior a 24 h. En una realización preferida, el tiempo de reacción oscila entre 20 y 24 h.
Ventajosamente, la conversión de los compuestos (III) y (IV) en el compuesto (II) después de la etapa (a) de acoplamiento es de al menos el 90 %, preferiblemente al menos el 95 %, más preferiblemente al menos el 98 %. Según una realización, la etapa (a) de acoplamiento va seguida por una etapa (a') de tratamiento y aislamiento que comprende las siguientes subetapas:
(a'-1) dilución de la mezcla de reacción con agua y con un primer disolvente orgánico aprótico polar, preferiblemente acetato de etilo (EtOAc);
(a'-2) separación de fases, y opcionalmente lavado con agua;
(a'-3) cambio de disolvente del primer disolvente a un segundo disolvente orgánico aprótico polar, preferiblemente acetato de isopropilo (iPAc);
(a'-4) recristalización por enfriamiento; y
(a'-5) filtración;
para obtener el compuesto (II) aislado.
Después de la etapa (a') de tratamiento y aislamiento, el compuesto (II) se aísla como un sólido. Ventajosamente, el compuesto (II) se obtiene después de la etapa (a') de tratamiento y aislamiento con un rendimiento de al menos el 80 %, preferiblemente al menos el 85 % de rendimiento, más preferiblemente al menos el 90 % de rendimiento.
Según una realización, la etapa (a) de acoplamiento o la etapa (a') de tratamiento y aislamiento va seguida de una etapa (a”) de purificación que comprende una etapa de recristalización del compuesto (II) en un disolvente orgánico aprótico polar, preferiblemente acetato de isopropilo (iPAc), para obtener el compuesto (II) purificado. Ventajosamente, el compuesto (II) se obtiene después de la etapa (a”) de purificación con al menos un 75 % de rendimiento, preferiblemente al menos un 80 % de rendimiento, más preferiblemente al menos un 85 % de rendimiento. Ventajosamente, el compuesto (II) se obtiene después de la etapa (a”) de purificación con al menos un 95 % de pureza, preferiblemente al menos un 99 % de pureza, más preferiblemente al menos un 99.5 % de pureza. Especialmente, el
compuesto (II) se obtiene ventajosamente después de la etapa (a”) de purificación con menos de 100 ppm de compuesto (IV).
Según una realización, la etapa (b) de desprotección se realiza mediante acidificación, preferiblemente mediante la puesta en contacto del compuesto (II) con ácido clorhídrico (HCl) diluido, más preferiblemente con solución acuosa de HCl; para obtener el compuesto (I). En una realización, la concentración de HCl en la solución de HCl oscila entre 20 y 40 % p/p, preferiblemente desde 25 a 35 % p/p, más preferiblemente la concentración de HCl es de 30 % p/p. En una realización, la proporción molar inicial entre el compuesto (II) y el HCl oscila entre 6 y 10, preferiblemente desde 7 a 9, más preferiblemente la proporción molar inicial entre el compuesto (II) y el HCl es 7.9. En una realización, el tiempo de reacción de la etapa (b) de desprotección es de al menos 15 min, preferiblemente de al menos 30 min. En una realización, el tiempo de reacción de la etapa (b) de desprotección no es superior a 4 h, preferiblemente no superior a 2 h, más preferiblemente no superior a 1 h. En una realización preferida, el tiempo de reacción oscila entre 30 y 60 min. En una realización, la etapa (b) de desprotección se realiza en un disolvente acuoso, preferiblemente agua. Ventajosamente, la conversión del compuesto (II) en el compuesto (I) después de la etapa (b) de desprotección es de al menos el 95 %, preferiblemente al menos 98 %, más preferiblemente al menos 99 %.
Según una realización, la etapa (b) de desprotección va seguida por una etapa (b') de tratamiento y aislamiento que comprende las siguientes subetapas:
(b'-1) extracción mediante un primer disolvente orgánico aprótico polar, preferiblemente 2-metiltetrahidrofurano (2-MeTHF);
(b'-2) separación de fases a una temperatura que oscila entre 35 a 50 °C, preferiblemente desde 40 a 45 °C, y opcionalmente lavado con solución de NaCl;
(b'-3) concentración y secado de la fase orgánica;
(b'-4) cambio de disolvente del primer disolvente a un segundo disolvente orgánico aprótico polar, preferiblemente acetato de isopropilo (iPAc);
(b'-5) recristalización por enfriamiento; y
(b'-6) filtración y, opcionalmente, lavado con metil tert-butil éter (MTBE);
para obtener el compuesto (I) aislado.
Ventajosamente, el compuesto (I) se obtiene después de la etapa (b') de tratamiento y aislamiento con un rendimiento de al menos 85 %, preferiblemente al menos 90 % de rendimiento, más preferiblemente al menos 95 % de rendimiento. Ventajosamente, el compuesto (I) se obtiene después de la etapa (b') de tratamiento y aislamiento con una pureza de al menos 98 %, preferiblemente una pureza de al menos 99 %, más preferiblemente una pureza de al menos 99.9 %. Ventajosamente, no se forma sal de adición de ácido del compuesto (IV) con ácido clorhídrico (HCl) durante la etapa (a) de acoplamiento. Sin estar ligado a ninguna teoría, el solicitante cree que la protonación del compuesto (IV) impacta negativamente en la conversión de la etapa (a) de acoplamiento.
La invención también se refiere a un procedimiento de fabricación de un compuesto de fórmula (II) como se describió previamente, preferiblemente ferf-butil (1-(6-amino-2-isobutilpirimidin-4-il)azetidin-3-il)(metil)carbamato de fórmula (II') como se ha descrito previamente. El compuesto de fórmula (II) o (II') se fabrica mediante un procedimiento según la invención como se describió previamente, en el que se omite la etapa (b) de desprotección, para obtener el compuesto (II) o (II').
Un compuesto de fórmula (III) como se ha descrito anteriormente puede fabricarse mediante cualquier método apropiado de la técnica, conocido por un experto en la técnica.
Sin embargo, el compuesto (III) se fabrica preferiblemente mediante un procedimiento que comprende las siguientes etapas:
(i) síntesis de 6-amino-2-isobutilpirimidin-4-ol (Compuesto (V), abreviado "(V)") por cicloadición de acetato de 3-metilbutanimidamida (o acetato de isobutilamidina) (Compuesto (VI), abreviado "(VI)") con 2-cianoacetato de etilo (Compuesto (VII), abreviado "(VII)"); y
(ii) síntesis del compuesto (III) por cloración del grupo hidroxilo del compuesto (V).
La síntesis del compuesto (III) por el procedimiento divulgado en este documento se representa en el esquema 3 a continuación:

Según una realización, la etapa (i) de cicloadición se realiza en presencia de una base, preferiblemente una base que es etóxido de sodio (NaOEt), en un disolvente, preferiblemente en etanol (EtOH). En una realización, la temperatura durante la etapa (i) de cicloadición oscila entre 65 y 80 °C, preferiblemente desde 70 a 75 °C. En una realización, el compuesto (VII) se agrega a una solución que comprende el compuesto (VI) y NaOEt. En una realización, la proporción molar inicial en la etapa (i) de cicloadición entre el compuesto (VII) y el compuesto (VI) oscila entre 1.0 y 1.25, preferiblemente desde 1.05 a 1.20, más preferiblemente es 1.15. En una realización, la proporción molar inicial de 16 en la etapa (i) de cicloadición entre NaOEt y el compuesto (VI) oscila entre 1.25 y 1.75, preferiblemente desde 1.4 a 1.6, más preferiblemente es 1.5.
En una realización, la etapa (i) de cicloadición va seguida por una etapa (i') de tratamiento que comprende una etapa de neutralización por ácido acético (AcOH). En una realización, la etapa (i) de cicloadición o la etapa (i') de tratamiento va seguida por una etapa (i”) de aislamiento que comprende una etapa de filtración del compuesto (V) fuera del agua, y que comprende opcionalmente una etapa de lavado del compuesto (V) con acetonitrilo (ACN).
Según una realización, la etapa (ii) de cloración se realiza mediante cloruro de fosforilo (POCh). En una realización, la etapa (ii) de cloración se realiza en un disolvente, preferiblemente en acetonitrilo (ACN). En una realización, la temperatura durante la etapa (ii) de cloración oscila entre 60 y 90 °C, preferiblemente desde 65 a 85 °C, más preferiblemente desde 70 a 80 °C. En una realización, la proporción molar inicial en la etapa (ii) de cloración entre POCla y el compuesto (V) oscila entre 4 y 6, preferiblemente desde 4.5 a 5.5, más preferiblemente es 5.
En una realización, la etapa (ii) de cloración va seguida de una etapa de extinción (ii') que comprende las siguientes subetapas:
(ii'-1) concentración a volumen mínimo;
(ii'-2) extracción sucesiva con un disolvente, preferiblemente tolueno;
(ii'-3) inactivar el cloruro de fosforilo restante mediante la adición de agua;
(ii'-4) agitación de la mezcla durante al menos 1 hora a una temperatura que oscila entre 55 y 65 °C, preferiblemente 60 °C; y
(ii'-5) aislamiento de la capa acuosa que comprende el compuesto (III).
En una realización, la etapa (ii) de cloración o la etapa (ii') de inactivación va seguida por una etapa (ii”) de aislamiento que comprende las siguientes etapas:
(ii"-1) adición de un disolvente, preferiblemente 2-metiltetrahidrofurano (2-MeTHF), preferiblemente en 10 volúmenes; (ii"-2) basificación, preferiblemente a pH 9;
(ii"-3) separación de fases y cambio de disolvente a un disolvente no polar no prótico, preferiblemente n-heptano; (ii"-4) filtración del compuesto (III) y lavado opcional con un disolvente no polar no prótico, preferiblemente n-heptano.
En una realización preferida, la etapa (ii”-2) de basificación se realiza mediante amoníaco (NH3), preferiblemente solución de NH3/H2O con una cantidad de amoníaco que oscila entre el 25 y el 30 % p/p.
Según una realización, el procedimiento no comprende ninguna etapa de fabricación de una sal de Pinner a partir del compuesto (VII) antes de la reacción con el compuesto (VI). Una "sal de Pinner" es un producto de una reacción de Pinner en un nitrilo, es decir, una sal de ácido mineral de un éster imino. En una realización, el procedimiento no comprende ninguna etapa de fabricación de un compuesto intermedio a partir del compuesto (VII) antes de la reacción con el compuesto (VI).
El compuesto (IV), y preferiblemente el compuesto (IV'), se pueden fabricar mediante cualquier método apropiado de la técnica, conocido por un experto en la técnica. Por ejemplo, el compuesto (IV') puede fabricarse mediante el procedimiento representado en el esquema 4, a continuación.

Este procedimiento comprende las siguientes etapas: [1] mesilación de 1 -benzhidrilazetidin-3-ol; [2] aminación del compuesto resultante; [3] Protección Boc del compuesto resultante; y [4] hidrogenación del compuesto resultante para obtener el compuesto (IV').
Un ejemplo de síntesis del compuesto (IV') se divulga en la Solicitud de Patente WO 2009/080721 (Ejemplo de referencia 2, página 72-73).
La conversión se puede controlar mediante cualquier método apropiado de la técnica, conocido por un experto en la técnica. Preferiblemente, la conversión se mide mediante cromatografía de gases (GC) o cromatografía líquida de alta resolución (HPLC). La pureza de los compuestos sintetizados se puede determinar mediante cualquier método apropiado de la técnica, conocido por un experto en la técnica. Preferiblemente, la pureza se determina por GC o por HPLC.
En este documento también se divulga una composición que comprende, consiste en o consiste esencialmente en al menos un compuesto de fórmula (I) una sal o solvato farmacéuticamente aceptable del mismo.
En este documento también se divulga una composición farmacéutica que comprende, consiste o consiste esencialmente en al menos un compuesto de fórmula (I) una sal o solvato farmacéuticamente aceptable del mismo y uno o más excipientes farmacéuticamente aceptables.
En este documento también se divulga un medicamento que comprende, consiste o consiste esencialmente en al menos un compuesto de fórmula (I) una sal o solvato farmacéuticamente aceptable del mismo.
Como se usa en este documento, "que consiste esencialmente en", con referencia a una composición, composición farmacéutica o medicamento, significa que al menos un compuesto de fórmula (I) una sal o solvato farmacéuticamente aceptable del mismo es el único agente terapéutico o agente con una actividad biológica dentro de dicha composición, composición farmacéutica o medicamento.
Los compuestos de fórmula (I) divulgados en este documento pueden estar en forma de sales farmacéuticamente aceptables. Los ejemplos de sales farmacéuticamente aceptables de los compuestos de fórmula I incluyen, pero no se limitan a, las sales de adición de ácido de los mismos. Las sales de adición de ácido apropiadas se forman a partir de ácidos que forman sales no tóxicas. Los ejemplos de sales de adición de ácido incluyen sales de besilato,
clorhidrato/cloruro, malato, benzoato, etano-1,2-disulfonato, fumarato, tartrato, acetato, adipato, ascorbato, aspartato, bicarbonato/carbonato, bisulfato/sulfato, borato, camsilato, citrato, ciclamato, edisilato, esilato, etanosulfonato, formiato, gluceptato, gluconato, glucuronato, glutamato, hexafluorofosfato, hibenzato, bromhidrato/bromuro, yodhidrato/yoduro, isetionato, lactato, maleato, malonato, mesilato, metilsulfato, naftilato, 2-napsilato, nicotinato, nitrato, orotato, oxalato, palmitato, pamoato, fosfato/hidrógeno fosfato/dihidrógeno fosfato, piroglutamato, sacarato, estearato, succinato, tanato, p-toluenosulfonato, tosilato, trifluoroacetato y xinofoato.
Los ejemplos de excipientes farmacéuticamente aceptables incluyen, pero no se limitan a, agua, solución salina, solución de Ringer, solución de dextrosa y soluciones de etanol, almidón, glucosa, sacarosa, dextrano, manosa, manitol, sorbitol, polietilenglicol (PEG), fosfato, acetato, gelatina, colágeno, Carbopol®, aceites vegetales y similares. Además, se pueden incluir conservantes, estabilizantes, antioxidantes, antimicrobianos y agentes reguladores apropiados, como, por ejemplo, hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), ácido cítrico, ácido ascórbico, tetraciclina y similares.
Carbopol® o "carbómero" se refiere a polímeros de alto peso molecular de ácido poliacrílico reticulados con alil sacarosa o pentaeritritol alil éter, comprendiendo dicho polímero homopolímeros y copolímeros. Ejemplos de Carbopol® incluyen, pero no se limitan a, Carbopol® 910, carbopol® 934, carbopol® 940, carbopol® 941, carbopol® 974, Carbopol® 981, Carbopol® Ultrez y policarbofilo.
Otros ejemplos de excipientes farmacéuticamente aceptables que se pueden usar en la composición divulgada en este documento incluyen, pero no se limitan a, intercambiadores de iones, alúmina, estearato de magnesio, estearato de aluminio, lecitina, proteínas séricas, tales como albúmina sérica humana, sustancias reguladoras tales como fosfatos, glicina, ácido sórbico, sorbato de potasio, mezclas de glicéridos parciales de ácidos grasos vegetales saturados, agua, sales o electrolitos, tales como sulfato de protamina, disodiohidrógeno fosfato, hidrógenofosfato de potasio, cloruro de sodio, sales de zinc, sílice coloidal, trisilicato de magnesio, polivinilo pirrolidona, sustancias a base de celulosa, polietilenglicol, carboximetilcelulosa de sodio, poliacrilatos, ceras, polímeros en bloque de polietilenopolioxipropileno, polietilenglicol y grasa de lana.
Además, los excipientes farmacéuticamente aceptables también pueden comprender, sin limitación, surfactantes (por ejemplo, hidroxipropilcelulosa); portadores apropiados, tales como, por ejemplo, disolventes y medios de dispersión que contengan, por ejemplo, agua, etanol, poliol (por ejemplo, glicerol, propilenglicol y polietilenglicol líquido, y similares), mezclas apropiadas de los mismos, y aceites vegetales, tales como, por ejemplo, aceite de cacahuete y aceite de sésamo; agentes isotónicos, tales como por ejemplo, azúcares o cloruro de sodio; agentes de recubrimiento tales como, por ejemplo, hidroxipropilmetilcelulosa (HPMC), polietilenglicol (PEG), polisorbato 80, dióxido de titanio y lecitina; agentes retardadores de la absorción, tales como, por ejemplo, monoestearato de aluminio y gelatina; conservantes, tales como, por ejemplo, cloruro de benzalconio, cloruro de bencetonio, clorobutanol, timerosal y similares; soluciones reguladoras, tales como, por ejemplo, ácido bórico, bicarbonato de sodio y potasio, boratos de sodio y potasio, carbonato de sodio y potasio, acetato de sodio, bifosfato de sodio y similares; agentes de tonicidad, tales como, por ejemplo, dextrosa, cloruro de potasio, propilenglicol, cloruro de sodio; antioxidantes y estabilizantes, tales como, por ejemplo, bisulfito de sodio, metabisulfito de sodio, tiosulfito de sodio, tiourea y similares; agentes humectantes o clarificantes no iónicos, tales como, por ejemplo, polisorbato 80, polisorbato 20, poloxámero 282 y tiloxapol; agentes modificadores de la viscosidad, tales como, por ejemplo, dextrano 40, dextrano 70, gelatina, glicerina, hidroxietilcelulosa, hidroximetilpropilcelulosa, lanolina, metilcelulosa, vaselina, polietilenglicol, alcohol polivinílico, polivinilpirrolidona, carboximetilcelulosa; diluyentes, adyuvantes y similares.
En este documento también se divulga un compuesto de fórmula (I) para tratar (o para usar en el tratamiento de) una enfermedad mediada por el receptor de histamina H4.
En este documento también se divulga un método para tratar una enfermedad mediada por el receptor de histamina H4 en un sujeto que lo necesite, preferiblemente en un sujeto humano, que comprende administrar a dicho sujeto al menos un compuesto de fórmula (I) o una sal o solvato farmacéuticamente aceptable del mismo.
En una realización, el método divulgado en este documento es para prevenir una enfermedad mediada por el receptor de histamina H4. En otra realización, el método divulgado en este documento es para aliviar al menos un síntoma de una enfermedad mediada por el receptor de histamina H4 o para curar una enfermedad mediada por el receptor de histamina H4.
En una realización, se administra o se va a administrar al sujeto una cantidad terapéuticamente eficaz del al menos un compuesto de fórmula (I).
En este documento también se divulga el uso de un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables para la fabricación de un medicamento para el tratamiento de una enfermedad mediada por el receptor de histamina H4.
En una realización, dicha enfermedad mediada por el receptor de histamina H4 es una enfermedad o dolor alérgico, inmunológico o inflamatorio.
Los ejemplos de enfermedades alérgicas, inmunológicas o inflamatorias incluyen, pero no se limitan a, enfermedades respiratorias, enfermedades oculares, enfermedades de la piel, enfermedades inflamatorias del intestino, diabetes, artritis reumatoide, esclerosis múltiple, lupus cutáneo, lupus eritematoso sistémico y rechazo de trasplantes.
En una realización, la enfermedad alérgica, inmunológica o inflamatoria se selecciona del grupo que comprende asma, rinitis alérgica, enfermedad pulmonar obstructiva crónica (COPD), rinoconjuntivitis alérgica, ojo seco, cataratas, dermatitis (por ejemplo, dermatitis atópica), psoriasis, urticaria, prurito, colitis ulcerosa, enfermedad de Crohn, nefropatía diabética, artritis reumatoide, esclerosis múltiple, lupus cutáneo, lupus eritematoso sistémico y rechazo de trasplantes.
Los ejemplos de dolores incluyen, pero no se limitan a, dolor inflamatorio, hiperalgesia inflamatoria, hiperalgesia, dolor posquirúrgico, migraña, dolor por cáncer, dolor visceral, dolor por osteoartritis y dolor neuropático.
En una realización, la enfermedad mediada por el receptor de histamina H4 es un trastorno del αdo.
En una realización, la enfermedad mediada por el receptor de histamina H4 es un trastorno vestibular. En una realización, dicho trastorno vestibular es un trastorno vestibular lesional. En otra realización, dicho trastorno vestibular es un trastorno vestibular no lesional.
Los ejemplos de trastornos vestibulares incluyen, sin limitación, migraña vestibular, neuritis vestibular (que incluye, sin limitación, neuronitis vestibular, neuronitis viral, laberintitis, laberintitis endolinfática viral, laberintitis serosa, laberintitis supurativa), vestibulopatía unilateral aguda, vértigo y mareos (que incluyen, sin limitación,, vértigo asociado con migraña, vértigo episódico espontáneo, vértigo paroxístico posicional benigno, vértigo episódico familiar, mareos y desequilibrio relacionados con la edad, cinetosis, mal de débarquement), ototoxicidad vestibular (que incluye, sin limitación, ototoxicidad inducida por compuestos e inducida por fármacos, es decir., que induce el deterioro de la función del vestíbulo que conduce a déficits vestibulares e inducida por los compuestos enumerados sin limitación anteriormente), deterioros del vestíbulo tóxicos, hidropesía (incluyendo, sin limitación, hidropesía endolinfática, hidropesía endolinfática secundaria), enfermedad de Méniére (incluyendo, sin limitación, episodio de la enfermedad de Méniére, enfermedad de Méniére crónica), fístula (incluyendo, sin limitación, fístula perilinfática, fístula laberíntica), traumatismo (incluyendo, sin limitación, traumatismo craneoencefálico con hemorragia laberíntica, barotrauma), infecciones (incluyendo, sin limitación, crónica o infección laberíntica aguda), enfermedad autoinmune del αdo interno, tumores benignos o malignos (incluyendo, sin limitación, schwannomas vestibulares, neuroma acústico), síndromes presbivestibulares, vestibulares después de tratamientos quirúrgicos del αdo medio, canalopatías, dehiscencia del canal semicircular superior, saco endolinfático o ángulo pontocerebeloso, ataxia (incluyendo, sin limitación, ataxia episódica), acueducto vestibular dilatado, hipofunción vestibular bilateral, vestibulopatía neurotóxica, trastorno vestibular pediátrico, síndrome de Cogan, hiperacusia vestibular, insuficiencia vertebrobasilar.
Los ejemplos de trastornos vestibulares lesionales incluyen, sin limitación, migraña vestibular, neuritis vestibular (que incluye, sin limitación, neuronitis vestibular, neuronitis viral, laberintitis, laberintitis endolinfática viral, laberintitis serosa, laberintitis supurativa), vestibulopatía unilateral aguda, vértigo y mareos (que incluyen, sin limitación, vértigo asociado con migraña, vértigo episódico espontáneo, vértigo episódico familiar, mareos y desequilibrio relacionados con la edad), ototoxicidad vestibular (incluyendo, sin limitación, ototoxicidad inducida por compuestos e inducida por fármacos, es decir, induciendo el deterioro de la función del vestíbulo que conduce a déficits vestibulares, e inducido por los compuestos enumerados sin limitación anteriormente), deterioros del vestíbulo-tóxicos, hidropesía (incluyendo, sin limitación, hidropesía endolinfática, hidropesía endolinfática secundaria), enfermedad de Méniére (incluyendo, sin limitación, episodio de la enfermedad de Méniére, enfermedad de Méniére crónica), fístula (que incluye, sin limitación, fístula perilinfática, fístula laberíntica), traumatismo (que incluye, sin limitación, traumatismo craneoencefálico con hemorragia laberíntica, barotrauma), infecciones (que incluyen, sin limitación, infección laberíntica crónica o aguda), enfermedad autoinmune del αdo interno, tumores benignos o malignos (incluyendo, sin limitación, schwannomas vestibulares, neuroma acústico), síndromes presbivestibulares, vestibulares después de tratamientos quirúrgicos del αdo medio, canalopatías, dehiscencia del canal semicircular superior, saco endolinfático o ángulo pontocerebeloso, ataxia (incluyendo, sin limitación, ataxia episódica), acueducto vestibular dilatado, hipofunción vestibular bilateral, vestibulopatía neurotóxica, trastorno vestibular pediátrico, síndrome de Cogan, hiperacusia vestibular, insuficiencia vertebrobasilar.
Ejemplos de trastornos vestibulares no lesionales incluyen, sin limitación, vértigo posicional paroxístico benigno, cinetosis y mal de débarquement.
En este documento también se divulga un método para restaurar la funcionalidad vestibular en un sujeto que lo necesite, preferiblemente en un sujeto afectado con una enfermedad vestibular, más preferiblemente con una enfermedad vestibular lesional, que comprende o consiste en administrar al sujeto una cantidad terapéuticamente efectiva de al menos un compuesto de fórmula (I) una sal o solvato farmacéuticamente aceptable del mismo.
En una realización, el método divulgado en este documento es para prevenir un trastorno vestibular, preferiblemente una enfermedad vestibular lesional. En otra realización, el método divulgado en este documento es para aliviar un
síntoma de un trastorno vestibular, preferiblemente una enfermedad vestibular lesional o para curar un trastorno vestibular, preferiblemente una enfermedad vestibular lesional.
En una realización, la enfermedad mediada por el receptor de histamina H4 es tinnitus.
En una realización, la enfermedad mediada por el receptor de histamina H4 es la pérdida de audición.
En una realización, la enfermedad mediada por el receptor de histamina H4 es vértigo o mareo. En una realización, la enfermedad mediada por el receptor de histamina H4 se selecciona del grupo que comprende vértigo asociado con migraña, vértigo episódico espontáneo, vértigo episódico familiar, mareos y desequilibrio relacionados con la edad.
En una realización, el compuesto, composición, composición farmacéutica o medicamento divulgado en este documento debe administrarse sistémica o localmente.
En una realización, el compuesto, composición, composición farmacéutica o medicamento divulgado en este documento debe administrarse por inyección, por vía oral, tópica, nasal, por inhalación, por vía bucal, rectal, intratraqueal, transmucosa, transtimpánica, por administración percutánea, intramuscular o por administración parenteral. .
En una realización, el compuesto, la composición, la composición farmacéutica o el medicamento divulgado en este documento debe administrarse mediante inyección, preferiblemente se inyectará sistémicamente. Los ejemplos de formulaciones adaptadas a inyecciones sistémicas incluyen, pero no se limitan a, soluciones o suspensiones líquidas, formas sólidas apropiadas para solución o suspensión en líquido antes de la inyección. Los ejemplos de inyecciones sistémicas incluyen, pero no se limitan a, inyección o perfusión intravenosa, subcutánea, intramuscular, intradérmica, intravítrea e intraperitoneal. En otra realización, cuando se inyecta, el compuesto, composición, composición farmacéutica o medicamento divulgado en este documento es estéril. Los métodos para obtener una composición farmacéutica estéril incluyen, sin limitación, filtración estéril, esterilización terminal (calor seco, radiación, calor húmedo, gases, radiación gamma) o esterilización mediante procesamiento aséptico.
En una realización, el compuesto, composición, composición farmacéutica o medicamento divulgado en este documento debe administrarse sistémicamente, preferiblemente debe administrarse por vía oral. Los ejemplos de formulaciones adaptadas a la administración oral incluyen, pero no se limitan a: formas sólidas, formas líquidas y geles. Ejemplos de formas sólidas adaptadas a la administración oral incluyen, pero no se limitan a, píldora, comprimido, cápsula, cápsula de gelatina blanda, cápsula de gelatina dura, comprimido oblongo, comprimido preparado por compresión, sello, oblea, píldora recubierta de azúcar, comprimido recubierto de azúcar o comprimido que se dispersa y/o se desintegra, polvo, formas sólidas apropiadas para solución o suspensión en líquido antes de la administración oral y comprimido efervescente. Los ejemplos de formas líquidas adaptadas a la administración oral incluyen, pero no se limitan a, soluciones, suspensiones, soluciones bebibles, elixires, ampollas selladas, pociones, empapados, jarabes y licores.
En otra realización, el compuesto, composición, composición farmacéutica o medicamento divulgado en este documento debe administrarse por vía tópica. Los ejemplos de formulaciones adaptadas a la administración tópica incluyen, pero no se limitan a, barras, ceras, cremas, lociones, ungüentos, bálsamos, geles, mascarillas, lavados sin aclarado y/o similares.
En una realización, el compuesto, composición, composición farmacéutica o medicamento divulgado en este documento debe administrarse directamente en el αdo, en particular, en el αdo interno, en el αdo medio, en el αdo externo, en la cóclea o en el vestíbulo mediante administración transtimpánica o intratimpánica. Esta vía de administración puede preferirse para introducir un efecto directo y a largo plazo en el αdo. Dicha administración se puede realizar por vía tópica o por inyección. Las técnicas de administración para dicha administración pueden incluir el uso de dispositivos o vehículos de fármacos para transportar y/o administrar el principio activo al αdo, donde se difunde en el αdo, se infunde activamente o se inyecta. Ejemplos de formulaciones adaptadas a tal administración incluyen, pero no se limitan a, otowicks, catéteres de ventana redonda, diversos tipos de geles, espumas, fibrinas, emulsiones, soluciones, parches u otros portadores de fármacos, que se colocan en el αdo y se cargan con la composición divulgada en este documento para liberación sostenida. También puede incluir dispositivos que se insertan en el conducto coclear o en cualquier otra parte de la cóclea.
La difusión de la composición a través de las estructuras tisulares de la interfase entre el αdo medio e interno, en particular la membrana de la ventana redonda, depende de una variedad de factores, tales como el peso molecular, la concentración, la liposolubilidad, la carga eléctrica y el espesor de la membrana.
En una realización, el compuesto de fórmula (I) debe administrarse en una forma de liberación inmediata.
En una realización, el compuesto de fórmula (I) debe administrarse en una forma de liberación sostenida. En una realización, la composición, la composición farmacéutica o el medicamento divulgado en este documento comprende
un sistema de administración que controla la liberación de al menos un compuesto de fórmula I o de una sal o solvato del mismo.
En una realización, el compuesto de fórmula (I) se formula como una formulación de depósito, es decir, como formulación parenteral de acción prolongada (por ejemplo., inyectable) diseñada para proporcionar una acción lenta, sostenida y prolongada. Las formulaciones de depósito se pueden colocar por vía subcutánea o intramuscular, por ejemplo. La liberación del compuesto de fórmula (I) puede ser pulsátil o continua dependiendo de la estructura del dispositivo y las características del polímero. Las formulaciones de depósito pueden estar en forma de micropartículas, implantes (por ejemplo, en forma de barra) o bolos sólidos que forman in situ. La mayoría de las formulaciones de depósito contienen excipientes poliméricos biodegradables. El excipiente de polímero controla la tasa de liberación del fármaco y se reabsorbe durante y/o después de la liberación del fármaco. Los ejemplos de polímeros biodegradables incluyen, pero no se limitan a, polímeros de lactida/glicolida. Estos polímeros reabsorbibles son biocompatibles y tienen un largo historial de seguridad en humanos. Se reabsorben únicamente por hidrólisis, inicialmente a ácido láctico y ácido glicólico, y finalmente a dióxido de carbono y agua.
Se entenderá que el uso diario total del compuesto de fórmula (I) una sal o solvato farmacéuticamente aceptable del mismo; o de la composición, composición farmacéutica o medicamento que la comprende será decidido por el médico tratante dentro del alcance del sano juicio médico. El nivel de dosis terapéuticamente eficaz específico para cualquier paciente en particular dependerá de una variedad de factores que incluyen el trastorno que se está tratando y la gravedad del trastorno; actividad del compuesto específico empleado; la composición específica empleada, la edad, el peso corporal, el estado de salud general, el sexo y la dieta del paciente; el tiempo de administración, vía de administración y tasa de excreción del compuesto específico empleado; la duración del tratamiento; fármacos usados en combinación o coincidentes con la composición específica empleada; y como factores bien conocidos en las artes médicas. Por ejemplo, está bien dentro de la experiencia en la técnica comenzar las dosis del compuesto a niveles más bajos que los requeridos para lograr el efecto terapéutico deseado y aumentar gradualmente la dosificación hasta que se logre el efecto deseado. Sin embargo, la dosificación diaria del compuesto de fórmula (I) una sal o solvato farmacéuticamente aceptable del mismo puede variar en un amplio intervalo desde aproximadamente 0.1 a aproximadamente 10000 mg por adulto por día, preferiblemente de 0.1 a aproximadamente 2000, más preferiblemente de aproximadamente 0.1 a aproximadamente 500 mg por adulto por día, más preferiblemente de aproximadamente 1 a aproximadamente 200 o 300 mg por adulto por día.
Ejemplos
La presente invención se ilustra además mediante los siguientes ejemplos.
Materiales y métodos
Material
Se preparó el tert-butil azetidin-3-il(metil)carbamato de fórmula (IV') [Compuesto (IV')] según los métodos de la literatura. Acetato de 3-metilbutanimidamida [Compuesto (VI)], 2-cianoacetato de etilo [Compuesto (VII)] y todos los demás reactivos y disolventes se adquirieron de fuentes comerciales y se usaron sin purificación adicional.
Métodos
La conversión se midió por GC o por HPLC. La pureza de los compuestos sintetizados se determinó mediante GC.
Ejemplo 1: Síntesis del compuesto (V) [6-amino-2-isobutilpirimidin-4-ol]
Reacción: Se suspendió acetato de 3-metilbutanimidamida [Compuesto (VI)] (20.0 g; 125 mmol; 1.0 eq) en EtOH (40 ml; 2 volúmenes relativos). Se cargó NaOEt al 21 % en peso (60.7 g; 187 mmol; 1.5 eq.) y se calentó a 70 °C. Una solución de 2-cianoacetato de etilo [Compuesto (VII)] (16.2 g; 144 mmol; 1.15 eq.) en EtOH (20 ml; 2 volúmenes) se dosifica lentamente (aproximadamente 4 horas). La conversión se comprobó por GC después de la adición del 40 % [Compuesto (V) 45 %] y la adición del 100 % [Compuesto (V) 69 %]. La conversión se comprobó mediante GC después de la reacción durante 21 horas a 70 °C [85 % del compuesto (V)]. La mezcla se agitó durante la noche a 70 °C.
Tratamiento y aislamiento: La mezcla se enfrió a 50 °C y luego se inactivó con AcOH (0.55 eq.; 4.13 g). Se cargó agua (8 volúmenes). La mezcla se concentra a 50-55 °C hasta aproximadamente 7 volúmenes. La suspensión se extrajo con ACN (3 volúmenes) hasta un volumen final de aproximadamente 6 volúmenes, luego se enfrío a 0-5 °C. La mezcla se diluyó adicionalmente con agua (2 volúmenes) y se agitó durante 2 horas. Los sólidos se filtraron, se lavaron con agua/ACN 1/1 (3 volúmenes), ACN (2 volúmenes) y se secaron bajo flujo de nitrógeno durante 1 hora.
Resultados: Se aisló 6-amino-2-isobutilpirimidin-4-ol [Compuesto (V)] con buen rendimiento (17.9 g, 86 %) y alta pureza (100 %).
Ejemplo 2: Síntesis del compuesto (III) r6-doro-2-isobutilpirimidin-4-amina 1
Reacción: Se suspendió 6-amino-2-isobutilpirimidin-4-ol [Compuesto (V)] (30.0 g; 179 mmol; 1.0 eq) del ejemplo 1 en ACN (150 ml; 5 volúmenes) a temperatura ambiente (TA). Se cargó POCl3 (138 g; 897 mmol; 5.0 eq.). La suspensión se calentó por etapas hasta 60 °C, 65 °C, luego 70 °C y se agitó a 70 °C, durante 15 minutos. La suspensión se calentó adicionalmente a 80 °C y se agitó durante la noche. La conversión se comprobó con HPLC después de 19 horas a 80 °C ([Compuesto (III)] 98.9 %).
Tratamiento: La mezcla se concentró a 60-70 °C hasta aproximadamente 3 volúmenes relativos. La mezcla se extrajo 3 veces con tolueno (3 volúmenes) a 60-70 °C, se diluyó con tolueno (2 volúmenes) y luego la temperatura se ajustó a ± 50 °C. La mezcla se inactivó con agua (5 volúmenes). La temperatura se ajustó a 60 °C y la mezcla se agitó durante al menos 30 minutos a 55-65 °C. Separación de fases: AL se aisló y se transfirió de nuevo al reactor y se diluyó con agua (1 vol.). La temperatura se ajustó a 20 - 25 °C.
Aislamiento: Se cargó 2-MeTHF (10 volúmenes). La dosis controlada ajustó el pH a 9-10 con NH3/h2O 25 - 30 % (alrededor de 2,5 volúmenes). Separación de fases: AL fue eliminado y descartado; la capa de aceite (OL) se mantuvo en el reactor. OL se lavó con agua (2 volúmenes) y se concentró a 40 - 50 °C a 4 volúmenes rel. OL se destiló 2 veces con n-heptano (3 volúmenes) a 40-50 °C. La suspensión se enfrió a 0 - 5 °C y se agitó durante 1 hora. Los sólidos se filtraron, se lavaron dos veces con n-heptano (2 volúmenes) y se secaron bajo flujo de nitrógeno.
Resultados: Se aisló 6-cloro-2-isobutilpirimidin-4-amina [Compuesto (III)] con buen rendimiento (29.2 g, 88 %) y alta pureza (99.8 %).
Ejemplo 3: Síntesis del compuesto (II') [ferf-butil (1-(6-amino-2-isobutilpirimidin-4-il)azetidin-3-il)(metil)carbamato]
Reacción: 6-cloro-2-isobutilpirimidin-4-amina [Compuesto (III)] (25.0 g; 135 mmol; 1.0 eq) del ejemplo 2 y K2CO3 (22.3 g; 162 mmol; 1.2 eq.) se cargaron al reactor. Se disolvió el ferf-butil azetidin-3-il(metil)carbamato [Compuesto (IV')] (26.1 g, 140 mmol, 1.04 eq.) en DMSO (msolución = 148 g, 17.6 % en peso) y la mezcla se calentó a 110 °C. La conversión se midió después de 24 horas (Compuesto (II') 93 %).
Tratamiento y aislamiento: La mezcla se enfrió a 50-60 °C, luego se diluyó con agua (6 volúmenes) y EtOAc (10 volúmenes) y se agitó durante 5 min. Separación de fases: se aisló el OL y luego se lavó con agua (2 volúmenes). El OL se concentró a aproximadamente 6 volúmenes relativos a 65-75 °C. La solución se extrajo 3 veces con iPAc (4 volúmenes). La solución se enfrió lentamente a TA. La suspensión se enfrió más a 0-5 °C. Después de 30 min de agitación, los sólidos se filtraron, se lavaron dos veces con MTBE (2 volúmenes) y se secaron con purga de nitrógeno.
Purificación: El producto aislado se suspendió en iPAc (100 ml, 4 volúmenes) y se calentó hasta disolución. La mezcla se enfrió a TA. La suspensión se enfrió adicionalmente a 0-5 °C. Después de 1 hora de agitación, los sólidos se filtraron, se lavaron dos veces con MTBE (2 volúmenes) y se secaron con purga de nitrógeno.
Resultados: Se obtuvo el ferf-butil (1-(6-amino-2-isobutilpirimidin-4-il)azetidina-3-il)(metil)carbamato [Compuesto (II')] con alto rendimiento (40.6 g, 89.9 %) y en alta pureza (99.8 %) después de la etapa de "tratamiento y aislamiento" como se describe anteriormente. El compuesto (II') se aisló con alto rendimiento (38.7 g, 85.6 %) y con alta pureza (99.9 %) después de una etapa de "purificación" adicional como se describe anteriormente.
Ejemplo 4: Síntesis del compuesto (I) [2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina]
Reacción: Se suspendió ferf-butil(1-(6-amino-2-isobutilpirimidin-4-il)azetidina-3-il)(metil)carbamato [Compuesto (II')] (20.0 g; 59.6 mmol; 1.0 eq) del ejemplo 3 se suspendió en agua (50 ml; 2.5 volúmenes) a TA. Se dosificó HCl al 30 % (50 ml; 470 mmol; 7.9 eq.) durante 15 min por debajo de 30 °C. La mezcla se agitó a 25 - 30 °C. La conversión se midió con HPLC después de 30 min a 25-30 °C (Compuesto (I) 99.1 %).
Tratamiento y aislamiento: La mezcla de reacción se enfrió a 20 °C. Se cargó 2-MeTHF (15 volúmenes). El pH se ajustó a 11-12 con una solución acuosa de NaOH (33 %, aproximadamente 63 g). Se dejó que la temperatura subiera hasta 40 °C. Separación de fases a 40-45 °C: AL fue eliminado y descartado; OL se mantuvo en el reactor. OL se lavó con una solución de NaCl (20 %, 1 vol) a 40 °C. OL se concentró a aproximadamente 10 volúmenes y se secó con un aparato Dean-Stark. Se realizó una filtración de cribado y se limpió el reactor. OL se concentró a 60-65 °C hasta aproximadamente 5 volúmenes. La solución se extrajo atmosféricamente 3 veces con iPAc (3 volúmenes), Tfin = 88 -89 °C. La suspensión se enfrió lentamente hasta la cristalización. Los sólidos se filtraron, se lavaron dos veces con MTBE (2 volúmenes) y se secaron.
Resultados: Se aisló el 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina [Compuesto (I)] con alto rendimiento (13.2 g, 94 %) y alta pureza (99.9 %).
Claims (15)
1. Un procedimiento de fabricación de 2-isobutil-6-(3-(metilamino)azetidin-1-il)pirimidin-4-amina de fórmula (I):

comprendiendo dicho procedimiento las siguientes etapas:
(a) hacer reaccionar 6-cloro-2-isobutilpirimidin-4-amina de fórmula (III):
con una N-metilazetidin-3-amina N-protegida de fórmula (IV):

en la que RP es un grupo protector seleccionado de carbobenciloxi (Cbz), p-metoxibencil carbonilo (Moz), tertbutiloxicarbonilo (Boc), 9-fluorenilmetiloxicarbonilo (Fmoc) y 2,2,2-tricloroetoxicarbonilo (Troc);
en un disolvente orgánico que es dimetilsulfóxido;
en presencia de una base que es carbonato de potasio;
a una temperatura que oscila entre 85 y 125 °C;
oscilando la proporción molar inicial entre dicho compuesto de fórmula (IV) y dicho compuesto de fórmula (III) entre 0.85 y 1.25;
para obtener un compuesto intermedio protegido de fórmula (II):

y
(b) realizar una desprotección en N de dicho compuesto de fórmula (II);
para obtener dicho compuesto de fórmula (I).
2. El procedimiento según la reivindicación 1, en el que el grupo protector RP es terf-butiloxicarbonilo.
3. El procedimiento según la reivindicación 1 o la reivindicación 2, en el que la proporción molar inicial en la etapa (a) de acoplamiento entre dicho compuesto de fórmula (IV) y dicho compuesto de fórmula (III) oscila entre 0.90 y 1.20.
4. El procedimiento según la reivindicación 3, en el que la proporción molar inicial en la etapa (a) de acoplamiento entre dicho compuesto de fórmula (IV) y dicho compuesto de fórmula (III) oscila entre 0.95 y 1.10.
5. El procedimiento según la reivindicación 4, en el que la proporción molar inicial en la etapa (a) de acoplamiento entre dicho compuesto de fórmula (IV) y dicho compuesto de fórmula (III) es 1.04.
6. El procedimiento según una cualquiera de las reivindicaciones 1 a 5, en el que la temperatura durante la etapa (a) de acoplamiento oscila entre 90 y 120 °C.
7. El procedimiento según la reivindicación 6, en el que la temperatura durante la etapa (a) de acoplamiento oscila entre 100 y 120 °C.
8. El procedimiento según la reivindicación 7, en el que la temperatura durante la etapa (a) de acoplamiento es de 110 °C.
9. El procedimiento según una cualquiera de las reivindicaciones 1 a 8, en el que el tiempo de reacción de la etapa (a) de acoplamiento oscila entre 16 horas y 32 horas.
10. El procedimiento según una cualquiera de las reivindicaciones 1 a 9, en el que a la etapa (a) de acoplamiento le sigue una etapa (a') de tratamiento y aislamiento que comprende las siguientes etapas:
(a'-1) dilución de la mezcla de reacción con agua y con un primer disolvente orgánico aprótico polar;
(a'-2) separación de fases, y opcionalmente lavado con agua;
(a'-3) cambio de disolvente del primer disolvente a un segundo disolvente orgánico aprótico polar;
(a'-4) recristalización por enfriamiento; y
(a'-5) filtración;
para proporcionar dicho compuesto de fórmula (II).
11. El procedimiento según la reivindicación 10, en el que dicho disolvente orgánico aprótico polar en la etapa (a'-1) de dilución es acetato de etilo y/o en el que dicho disolvente orgánico aprótico polar en la etapa (a'-3) de cambio de disolvente es acetato de isopropilo.
12. El procedimiento según una cualquiera de las reivindicaciones 1 a 11, en el que la etapa (a) de acoplamiento o la etapa (a') de tratamiento y aislamiento va seguida de una etapa (a”) de purificación que comprende una etapa de recristalización de dicho compuesto de fórmula (II) en un disolvente orgánico aprótico polar.
13. El procedimiento según una cualquiera de las reivindicaciones 1 a 12, en el que la etapa (b) de desprotección se realiza mediante la puesta en contacto de dicho compuesto de fórmula (II) con una solución acuosa diluida de ácido clorhídrico.
14. El procedimiento según la reivindicación 13, en el que la proporción molar inicial entre dicho compuesto de fórmula (II) y el ácido clorhídrico oscila entre 6 y 10.
15. Un procedimiento de fabricación de un compuesto de fórmula (II):

en la que RP es un grupo protector seleccionado de carbobenciloxi (Cbz), p-metoxibencil carbonilo (Moz), tert-butiloxicarbonilo (Boc), 9-fluorenilmetiloxicarbonilo (Fmoc) y 2,2,2-tricloroetoxicarbonilo (Troc);
comprendiendo dicho procedimiento hacer reaccionar 6-cloro-2-isobutilpirimidin-4-amina de fórmula (III):

con una N-metilazetidin-3-amina N-protegida de fórmula (IV):

en un disolvente orgánico que es dimetilsulfóxido;
en presencia de una base que es carbonato de potasio;
a una temperatura que oscila entre 85 y 125 °C;
oscilando la proporción molar inicial entre dicho compuesto de fórmula (IV) y dicho compuesto de fórmula (III) entre 0.85 a 1.25;
para obtener dicho compuesto de fórmula (II).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17306404 | 2017-10-17 | ||
| PCT/EP2018/078385 WO2019076974A1 (en) | 2017-10-17 | 2018-10-17 | SYNTHESIS OF 4-AMINOPYRIMIDINE COMPOUNDS |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2946551T3 true ES2946551T3 (es) | 2023-07-20 |
Family
ID=60191299
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18785637T Active ES2946551T3 (es) | 2017-10-17 | 2018-10-17 | Síntesis de compuestos de 4-aminopirimidina |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US11180480B2 (es) |
| EP (1) | EP3697776B1 (es) |
| JP (1) | JP7274473B2 (es) |
| CN (1) | CN111465602B (es) |
| ES (1) | ES2946551T3 (es) |
| TW (1) | TWI797176B (es) |
| WO (1) | WO2019076974A1 (es) |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000169475A (ja) * | 1998-07-24 | 2000-06-20 | Dai Ichi Seiyaku Co Ltd | ピラゾ―ル誘導体およびその塩 |
| DE102008020113A1 (de) * | 2008-04-23 | 2009-10-29 | Bayer Schering Pharma Aktiengesellschaft | Substituierte Dihydropyrazolone und ihre Verwendung |
| CL2008000467A1 (es) * | 2007-02-14 | 2008-08-22 | Janssen Pharmaceutica Nv | Compuestos derivados de 2-aminopirimidina, moduladores del receptor histamina h4; su procedimiento de preparacion; composicion farmaceutica que comprende a dichos compuestos; y su uso para tratar un trastorno inflamatorio seleccionado de alegia, asma |
| DE102007026341A1 (de) | 2007-06-06 | 2008-12-11 | Merck Patent Gmbh | Benzoxazolonderivate |
| DE102007032507A1 (de) | 2007-07-12 | 2009-04-02 | Merck Patent Gmbh | Pyridazinonderivate |
| DE102007061963A1 (de) | 2007-12-21 | 2009-06-25 | Merck Patent Gmbh | Pyridazinonderivate |
| AR069814A1 (es) * | 2007-12-21 | 2010-02-17 | Palau Pharma Sa | Derivados de 4 amino-pirimidina, proceso de preparacion y composiciones farmaceuticas que los contienen |
| DE102008019907A1 (de) | 2008-04-21 | 2009-10-22 | Merck Patent Gmbh | Pyridazinonderivate |
| DE102008028905A1 (de) | 2008-06-18 | 2009-12-24 | Merck Patent Gmbh | 3-(3-Pyrimidin-2-yl-benzyl)-[1,2,4]triazolo[4,3-b]pyridazinderivate |
| PE20120016A1 (es) | 2008-12-22 | 2012-01-24 | Merck Patent Gmbh | Nuevas formas polimorficas de dihidrogeno-fosfato de 6-(1-metil-1h-pirazol-4-il)-2-{3-[5-(2-morfolin-4-il-etoxi)-pirimidin-2-il]-bencil}-2h-piridazin-3-ona y procesos para su preparacion |
| EP2201982A1 (en) | 2008-12-24 | 2010-06-30 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Histamine H4 receptor antagonists for the treatment of vestibular disorders |
| ES2629006T3 (es) | 2009-10-29 | 2017-08-07 | Vectura Limited | Derivados que contienen N-heteroarilo como inhibidores de la quinasa jak3 |
| CA2785353C (en) | 2009-12-23 | 2017-10-31 | Palau Pharma, S.A. | Aminoalkylpyrimidine derivatives as histamine h4 receptor antagonists |
| PL2858647T3 (pl) | 2012-06-08 | 2018-10-31 | Sensorion | Inhibitory receptora h4 do leczenia szumów usznych |
| MY182082A (en) * | 2013-05-01 | 2021-01-18 | Hoffmann La Roche | Biheteroaryl compounds and uses thereof |
-
2018
- 2018-10-17 TW TW107136532A patent/TWI797176B/zh active
- 2018-10-17 CN CN201880080973.7A patent/CN111465602B/zh active Active
- 2018-10-17 EP EP18785637.2A patent/EP3697776B1/en active Active
- 2018-10-17 WO PCT/EP2018/078385 patent/WO2019076974A1/en unknown
- 2018-10-17 ES ES18785637T patent/ES2946551T3/es active Active
- 2018-10-17 JP JP2020522003A patent/JP7274473B2/ja active Active
- 2018-10-17 US US16/757,008 patent/US11180480B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| TW201932457A (zh) | 2019-08-16 |
| WO2019076974A1 (en) | 2019-04-25 |
| US11180480B2 (en) | 2021-11-23 |
| JP7274473B2 (ja) | 2023-05-16 |
| TWI797176B (zh) | 2023-04-01 |
| EP3697776B1 (en) | 2023-04-19 |
| JP2020537673A (ja) | 2020-12-24 |
| CN111465602B (zh) | 2023-05-23 |
| US20200270228A1 (en) | 2020-08-27 |
| CN111465602A (zh) | 2020-07-28 |
| EP3697776A1 (en) | 2020-08-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10577337B2 (en) | Compositions and methods of treatment with prodrugs of tizoxanide, an analogue or salt thereof | |
| ES2552712T3 (es) | Inhibidores de VHC | |
| AU2021201251B2 (en) | Antimicrobial compounds, compositions, and uses thereof | |
| CN108164474A (zh) | 作为免疫调节剂的1,2,4-*二唑衍生物 | |
| CN105849092A (zh) | 作为免疫调节剂的1,3,4-*二唑和1,3,4-噻二唑衍生物 | |
| RU2013108857A (ru) | Соединения для лечения/профилактики воспалительных глазных заболевний | |
| ES2932194T3 (es) | Compuestos terapéuticos y composiciones para el tratamiento de trastornos sociales y trastornos por uso de sustancias | |
| US10183925B2 (en) | Substituted naphtho[2,3-b]furans as water-soluble prodrugs for preventing and/or treating cancer | |
| US11135234B2 (en) | Effective aminoglycoside antibiotic for multidrug-resistant bacteria | |
| US20200109166A1 (en) | Polyamide compound and use thereof | |
| BRPI0912604A2 (pt) | quinolinilmetilimidazóis como agentes terapêuticos | |
| WO2008087461B1 (en) | Use of kynurenic acid and derivatives thereof in the treatment of conditions of the gastrointestinal tract accompanied by hypermotility and inflammation or gout or multiple sclerosis | |
| EP2508522A1 (en) | Azilsartan organic amine salts, preparation method and use thereof | |
| ES2946551T3 (es) | Síntesis de compuestos de 4-aminopirimidina | |
| US11325943B2 (en) | Crystalline salt forms of SBT-20 | |
| RU2515557C9 (ru) | Фармацевтическая соль 8-метил-7-[5-метил-6-(метиламино)-3-пиридинил]-1-циклопропил-4-оксо-1,4-дигидро-3-хинолинкарбоновой кислоты, ее содержащая фармацевтическая композиция, лекарственное средство и способ лечения или профилактики бактериальных инфекций с помощью вышеуказанной соли | |
| ES2296618T3 (es) | Derivados opticamente activos de acidos quinolin-carboxilicos que tienen sustituyentes de 7-pirrolidina que provocan actividad optica y un procedimiento para la preparacion de estos. | |
| US20110003751A1 (en) | Novel Immunoregulatory Peptides, Compositions and Uses Thereof | |
| EP1686990B1 (en) | 2-guanidinylimidazolidinedione compounds and methods of making and using thereof | |
| CN111315758A (zh) | 短肽季铵盐化合物及其用途 | |
| US10822310B2 (en) | Prevention and treatment of human metabolic syndrome including type 2 diabetes, steatohepititis and related conditions using non-absorbable, orally administered compounds | |
| US20240246920A1 (en) | Triazole derivatives with antifungal activity | |
| WO2020159588A1 (en) | Methods of treating diabetic neuropathy with a thiazoline anti-hyperalgesic agent |