ES2942837T3 - Derivados de azaespiro piperazina - Google Patents
Derivados de azaespiro piperazina Download PDFInfo
- Publication number
- ES2942837T3 ES2942837T3 ES19716666T ES19716666T ES2942837T3 ES 2942837 T3 ES2942837 T3 ES 2942837T3 ES 19716666 T ES19716666 T ES 19716666T ES 19716666 T ES19716666 T ES 19716666T ES 2942837 T3 ES2942837 T3 ES 2942837T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- alkoxy
- membered
- heteroaryl
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
- A61P25/10—Antiepileptics; Anticonvulsants for petit-mal
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/08—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
Abstract
La presente invención proporciona, en parte, compuestos de Fórmula I: o un N-óxido de los mismos, o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que: X1, X2, R1, R2, m y n son como se describen Aquí en; procesos para la elaboración de; intermedios utilizados en la preparación de; y composiciones que contienen dichos compuestos, N-óxidos o sales, y sus usos para tratar trastornos mediados por M4 (o asociados a M4) que incluyen, por ejemplo, enfermedad de Alzheimer, enfermedad de Parkinson, esquizofrenia (por ejemplo, sus síntomas cognitivos y negativos), dolor , adicción y un trastorno del sueño. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados de azaespiro piperazina
Campo de la invención
La presente invención se refiere en general a nuevos derivados de azaespiro piperazina, a sales de los mismos, a composiciones farmacéuticas de los mismos, que son agonistas del receptor muscarínico M4, y son útiles en el tratamiento de enfermedades y trastornos mediados por M4 tales como esquizofrenia, enfermedad de Alzheimer, demencia con cuerpos de Lewy, enfermedad de Parkinson y disfunción ejecutiva y de la memoria relacionadas, inquietud y psicosis asociadas con las mismas.
Antecedentes de la invención
Los pacientes con esquizofrenia, enfermedad de Alzheimer, enfermedad de Parkinson, enfermedad de Huntington, depresión y diversas otras enfermedades neurológicas/neurodegenerativas, con frecuencia padecen deterioro conductual y cognitivo lo que da como resultado una alteración debilitante de su vida diaria. A lo largo de los años, se han descubierto muchos tratamientos farmacológicos que proporcionan alguna mejora en el comportamiento y la función cognitiva. Sin embargo, la mejora es, en el mejor de los casos, moderada y, como suele ser el caso, los efectos adversos subyacentes limitantes de la dosis asociados con estos tratamientos, incluyendo efectos secundarios extrapiramidales y metabólicos, conducen a una respuesta parcial y al incumplimiento terapéutico.
En un esfuerzo por descubrir tratamientos farmacológicos nuevos y mejorados, los investigadores comenzaron a considerar el receptor muscarínico de acetilcolina (mAChR) como un mecanismo viable. Hay cinco subtipos de mAChR (M1-M5) identificados y son parte de la superfamilia de receptores acoplados a la proteína G (GPCR). Estos subtipos se distribuyen extensamente por la periferia y el sistema nervioso central (SNC), siendo los subtipos M1 y M4 los que se expresan predominantemente en el SNC.
Desde entonces, los investigadores se han centrado en identificar activadores selectivos del receptor muscarínico del subtipo M4 de acetilcolina. Por ejemplo, se han estudiado moduladores alostéricos positivos (PAM) del receptor muscarínico M4 de acetilcolina. Además de los PAM M4, las investigaciones también se han centrado en identificar agonistas del receptor M4. De hecho, el agonista M4 HTL0016878 que se está desarrollando para el tratamiento de los principales síntomas de la enfermedad de Alzheimer entró en un estudio clínico de fase I. Sin embargo, los nuevos activadores o los mejorados, incluyendo los agonistas de los receptores muscarínicos M4, son necesarios para proporcionar nuevas terapias y terapias mejoradas para tratar enfermedades y trastornos mediados por M4, tales como enfermedad de Parkinson, esquizofrenia, enfermedad de Alzheimer y otras descritas en el presente documento. Sumario de la invención
La presente invención proporciona, en parte, un compuesto de fórmula I:

o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:
cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH,
R' se selecciona de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -N(R6)(R7), alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alquiltio (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde
dicho alquilo (C-i-Ca), alquenilo (C2-C6), alquinilo (C2-C6), alquiltio (Ci-Ca), alcoxi (C-i-Ca), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, -N(R6)(R7), alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);
R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, ciano, hidroxi, -SF5, nitro, -N(R6)(R7), alquilo (C1-C6) y alcoxi (C1-C6), en donde dichos alquilo (C1-C6) y alcoxi (C1-C6) están opcionalmente sustituidos con de 1 a 3 halógeno;
cada uno de R6 y R7 se selecciona independientemente entre hidrógeno, alquilo (C1-C6) o C(O)CH3;
m es 1 o 2; y
n es 1 o 2.
Los compuestos de fórmulas I, Ia, Ib, Ic y I' son útiles para tratar una enfermedad o trastorno mediado por M4 (o asociado a M4 en un paciente, en donde el método comprende administrar a un paciente una cantidad terapéuticamente eficaz de un compuesto de fórmulas I, Ia, Ib, Ic y I' o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido.
La presente invención también se refiere a los compuestos descritos en el presente documento o un N-óxido de los mismos o una sal farmacéuticamente aceptable del compuesto o del N-óxido, para su uso en el tratamiento de una enfermedad o trastorno mediado por M4 (o asociado a M4) en donde la enfermedad o trastorno es enfermedad de Alzheimer, esquizofrenia o psicosis, dolor, adicción, un trastorno del sueño, un trastorno cognitivo (por ejemplo, deterioro cognitivo leve), enfermedad de Parkinson, discinesia inducida por levodopa en la enfermedad de Parkinson, enfermedad de Huntington, discinesia, boca seca, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica (EPOC), asma, incontinencia urinaria, glaucoma, Trisomía 21 (síndrome de Down), angiopatía amiloidea cerebral, demencia, hemorragia cerebral hereditaria con amiloidosis de tipo holandés (HCHWA-D), enfermedad de Creutzfeld-Jakob, trastornos priónicos, esclerosis lateral amiotrófica, parálisis supranuclear progresiva, traumatismo craneal, accidente cerebrovascular, pancreatitis, miositis por cuerpos de inclusión, otras amiloidosis periféricas, diabetes, autismo y ateroesclerosis.
La presente invención también se refiere a formulaciones farmacéuticas que contienen una cantidad terapéuticamente eficaz de un compuesto de fórmulas I, Ia, Ib, Ic y I', o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, y un excipiente farmacéuticamente aceptable.
Debe apreciarse que tanto la descripción general anterior como la descripción detallada a continuación son únicamente a modo de ejemplo y explicación y no son restrictivas de la invención, tal como se reivindica.
Descripción detallada de la invención
Los encabezados en el presente documento se utilizan únicamente para agilizar su revisión por el lector. No deben interpretarse como limitantes de la invención o de las reivindicaciones en modo alguno.
Definiciones y ejemplos
La presente invención se puede entender más rápidamente con referencia a la siguiente descripción detallada de las realizaciones a modo de ejemplo de la invención y los ejemplos incluidos en el presente documento.
Debe apreciarse que esta invención no se limita a los métodos de síntesis específicos de fabricación que, por supuesto, pueden variar. También debe entenderse que la terminología usada en el presente documento es únicamente con el fin de describir realizaciones particulares y no se pretende que sea limitante. En la presente memoria descriptiva y en las reivindicaciones que se presentan a continuación, se hará referencia a diversos términos que se definirán con los significados siguientes:
Como se usa en el presente documento, en la memoria descriptiva, "un" o "una" pueden significar uno/una o más. Como se usa en la presente memoria en las reivindicaciones, cuando se usa junto con la expresión "que comprende", las palabras "un" o "una" pueden significar uno o más de uno. Como se usa en el presente documento, "otro" puede significar al menos un segundo o más.
El término "aproximadamente" se refiere a un término relativo que denota una aproximación de más o menos el 10 % del valor nominal al que se refiere, en una realización, a más o menos el 5 %, en otra realización, a más o menos el 2 %. Para el campo de esta divulgación, este nivel de aproximación es apropiado a menos que se indique de forma específica que el valor refiere un intervalo más estrecho.
Como se usa en el presente documento, la expresión "agonista del receptor muscarínico M4" significa los compuestos de la presente invención inducen un efecto sobre el receptor M4 ausente en ausencia de un ligando natural (por ejemplo, acetilcolina)
Como se usa en el presente documento, la expresión "n miembros", donde n es un número entero, normalmente describe el número de átomos que forman el anillo en una fracción donde el número de átomos que forman el anillo es n. Por ejemplo, la piridina es un ejemplo de un anillo heteroarilo de 6 miembros y el tiofeno es un ejemplo de un anillo heteroarilo de 5 miembros.
En distintos lugares en la presente memoria descriptiva, se divulgan sustituyentes de compuestos de la invención en grupos o en intervalos. Se pretende específicamente que la invención incluya todas y cada una de las subcombinaciones individuales de los miembros de dichos grupos e intervalos. Por ejemplo, la expresión "alquilo C1-6" pretende específicamente incluir alquilo C1 (metilo), alquilo C2 (etilo), alquilo C3, alquilo C4, alquilo C5 y alquilo C6. Para otro ejemplo, la expresión "un grupo heteroarilo de 5 a 10 miembros" pretende específicamente incluir cualquier grupo heteroarilo de 5, 6, 7, 8, 9 o 10 miembros.
La expresión "alquilo (C1-C6)'', como se usa en el presente documento, se refiere a un grupo alquilo saturado, de cadena lineal o ramificada, que contiene de 1 a 6 átomos de carbono, tal como, pero no limitado a, metilo, etilo, npropilo, isopropilo, n-butilo, sec-butilo, isobutilo, ferc-butilo, n-pentilo, isopentilo, neopentilo y n-hexilo. El alquilo (C1-C6) puede estar opcionalmente sustituido en cuyo caso uno o más átomos de hidrógeno se sustituyen con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6). Por ejemplo, un resto alquilo (C1-C6) puede estar sustituido con uno o más átomos de halógeno para formar un "haloalquilo (C1-C6)''. Los ejemplos representativos de un haloalquilo (C1-C6) incluyen, pero no se limitan a, fluorometilo, 2-fluoroetilo, difluorometilo, trifluorometilo y pentafluoroetilo. Otros ejemplos representativos de un alquilo (C1-C6) sustituido incluyen, pero no están limitados a, cianobutilo y etoxietilo.
La expresión "alquenilo (C2-C6)'' se refiere a un hidrocarburo alifático que tiene de 2 a 6 átomos de carbono y que tiene al menos un doble enlace carbono-carbono, incluyendo grupos de cadena lineal o ramificada que tienen al menos un doble enlace carbono-carbono. Los ejemplos representativos incluyen, pero no se limitan a, etenilo, 1-propenilo, 2-propenilo (alilo), isopropenilo, 2-metil-1-propenilo, 1 -butenilo, 2-butenilo y similares. Cuando los compuestos de la invención contienen un grupo alquenilo (C2-C6), el compuesto puede existir como la forma pura E (entgegen), la forma pura Z (zusammen) o cualquier mezcla de las mismas. El alquenilo (C2-C6) puede estar opcionalmente sustituido en el que uno o más átomos de hidrógeno se reemplazan con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6).
La expresión "alquinilo (C2-C6)" se refiere a un hidrocarburo alifático que tiene de dos a seis átomos de carbono y al menos un triple enlace carbono-carbono, incluyendo cadenas lineales y cadenas ramificadas que tienen al menos un triple enlace carbono-carbono. Los ejemplos representativos incluyen, pero no se limitan a, etinilo, propinilo, butinilo, pentinilo y hexinilo. El alquinilo (C2-C6) puede estar opcionalmente sustituido en el que uno o más átomos de hidrógeno se reemplazan con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6).
La expresión "alcoxi (C1-C6)'', como se usa en el presente documento, se refiere a un grupo alquilo (C1-C6), como se ha definido anteriormente, unido al resto molecular precursor a través de un átomo de oxígeno. Los ejemplos representativos de un alcoxi (C1-C6) incluyen, pero no se limitan a, metoxi, etoxi, propoxi, 2-propoxi, butoxi, ferc-butoxi, pentiloxi y hexiloxi. El alcoxi (C1-C6) puede estar opcionalmente sustituido en el que uno o más átomos de hidrógeno se reemplazan con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6). Por ejemplo, un alcoxi (C1-C6) puede estar sustituido con uno o más átomos de halógeno para formar un "haloalcoxi (C1-C6)". Los ejemplos representativos de un haloalcoxi (C1-C6) incluyen, pero no se limitan a, fluorometoxi, difluorometoxi, 2-fluoroetoxi, trifluorometoxi y pentafluoroetoxi.
La expresión "alquitio (C1-C6)", como se usa en el presente documento, se refiere a un grupo alquilo (C1-C6), como se ha definido anteriormente, unido al resto molecular precursor a través de un átomo de azufre. Los ejemplos representativos de un alquiltio (C1-C6) incluyen, pero no se limitan a, metiltio, etiltio, propiltio y similares. El alquiltio (C1-C6) puede estar opcionalmente sustituido en el que uno o más átomos de hidrógeno se reemplazan con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6).
Como se usa en el presente documento, la expresión "cicloalquilo (C3-C6)" se refiere a un sustituyente carbocíclico obtenido eliminando un hidrógeno de una molécula carbocíclica saturada que tiene de 3 a 6 átomos de carbono. Un "cicloalquilo" puede ser un anillo monocíclico, cuyos ejemplos incluyen ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo. El cicloalquilo (C3-C6) puede estar opcionalmente sustituido en el que uno o más átomos de hidrógeno se reemplazan con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6).
Un "heterocicloalquilo", como se usa en el presente documento, se refiere a un cicloalquilo como se ha definido anteriormente, en el que al menos uno de los átomos de carbono en el anillo está reemplazado con un heteroátomo seleccionado de entre nitrógeno, oxígeno o azufre. La expresión "heterocicloalquilo (de 4 a 6 miembros)" significa el sustituyente heterocicloalquilo que contiene un total de 4 a 6 átomos en el anillo, al menos uno de los cuales es un heteroátomo. La expresión "heterocicloalquilo (de 4 a 8 miembros)" significa el sustituyente heterocicloalquilo que contiene un total de 4 a 8 átomos en el anillo, al menos uno de los cuales es un heteroátomo. Un "heterocicloalquilo (de 6 miembros)" significa el sustituyente heterocicloalquilo que contiene un total de 6 átomos en el anillo, al menos uno de los cuales es un heteroátomo. Un "heterocicloalquilo (de 5 miembros)" significa el sustituyente heterocicloalquilo que contiene un total de 5 átomos en el anillo, al menos uno de los cuales es un heteroátomo. El sustituyente heterocicloalquilo puede estar unido a través de un átomo de nitrógeno que tiene la valencia apropiada o a través de cualquier átomo de carbono del anillo. El resto heterocicloalquilo puede estar opcionalmente sustituido con uno o más sustituyentes en un átomo de nitrógeno que tiene la valencia apropiada o en cualquier átomo de carbono disponible.
Los ejemplos de anillos heterocicloalquilo incluyen, pero no se limitan a, azetidinilo, dihidrofuranilo, dihidrotiofenilo, tetrahidrotiofenilo, tetrahidrofuranilo, tetrahidrotriazinilo, tetrahidropirazolilo, tetrahidrooxazinilo, tetrahidropirimidinilo, imidazolidinilo, pirrolidinilo, piperidinilo, piperazinilo, oxazolidinilo, tiazolidinilo, pirazolidinilo, tiomorfolinilo, tetrahidropiranilo, tetrahidrotiazinilo, tetrahidrotiadiazinilo, tetrahidrooxazolilo, morfolinilo, oxetanilo, tetrahidrodiazinilo, oxazinilo, oxatiazinilo. Otros ejemplos de anillos heterocicloalquilo incluyen tetrahidrofuran-2-ilo, tetrahidrofuran-3-ilo, imidazolidin-1-ilo, imidazolidin-2-ilo, imidazolidin-4-ilo, pirrolidin-1-ilo, pirrolidin-2-ilo, pirrolidin-3-ilo, piperidin-1-ilo, piperidin-2-ilo, piperidin-3-ilo, piperidin-4-ilo, piperazin-1-ilo, piperazin-2-ilo, 1,3-oxazolidin-3-ilo, isotiazolidinilo, 1,3-tiazolidin-3-ilo, 1,2-pirazolidin-2-ilo, 1,2-tetrahidrotiazin-2-ilo, 1,3-tiazinan-3-ilo, 1,2-tetrahidrodiazin-2-ilo, 1,3-tetrahidrodiazin-1-ilo, 1,4-oxazin-4-ilo, 2-oxo-piperidinilo (por ejemplo, 2-oxo-piperidin-1-ilo) y similares. El heterocicloalquilo puede estar opcionalmente sustituido en el que uno o más átomos de hidrógeno están reemplazados con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6).
Un "arilo (C6-C10)'' se refiere a un grupo aromático monocíclico completamente de carbono o policíclico de anillo condensado, que tiene un sistema de electrones pi conjugado que contiene de 6 a 10 átomos de carbono, tal como fenilo o naftilo.
Como se usa en el presente documento, el término "heteroarilo" se refiere a un sistema carbocíclico aromático que contiene uno, dos, tres o cuatro heteroátomos seleccionados independientemente entre oxígeno, nitrógeno y azufre y que tiene uno, dos o tres anillos en donde dichos anillos pueden estar condensados, en donde condensado se ha definido anteriormente. Un anillo "heteroarilo (de 5 a 10 miembros)" se refiere a un anillo heteroarilo que tiene de 5 a 10 átomos en el anillo en el que al menos uno de los átomos en el anillo es nitrógeno, estando los átomos restantes del anillo independientemente seleccionados de entre grupo que consiste en carbono, oxígeno, azufre y nitrógeno. Un anillo "heteroarilo (de 5 a 6 miembros)" se refiere a un anillo heteroarilo que tiene de 5 a 6 átomos en el anillo en el que al menos uno de los átomos en el anillo es nitrógeno, estando los átomos restantes del anillo independientemente seleccionados de entre grupo que consiste en carbono, oxígeno, azufre y nitrógeno. Los ejemplos de heteroarilos incluyen, pero no se limitan a, pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo.
Debe entenderse que el heteroarilo puede estar opcionalmente condensado con un grupo cicloalquilo o con un grupo heterocicloalquilo, como se define en el presente documento.
El sustituyente heteroarilo puede estar unido mediante un átomo de nitrógeno que tiene la valencia apropiada o mediante cualquier átomo de carbono. El resto heteroarilo puede estar opcionalmente sustituido con uno o más sustituyentes en un átomo de nitrógeno que tiene la valencia apropiada o en cualquier átomo de carbono disponible. El heteroarilo (de 5 a 10 miembros) puede estar opcionalmente sustituido en el cual uno o más átomos de hidrógeno se reemplazan con un sustituyente seleccionado de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -alcoxi (C1-C6) y -N(R6)(R7), en el que cada uno de R6 y R7 se selecciona independientemente entre hidrógeno y alquilo (C1-C6). El sustituyente puede estar unido al resto heteroarilo en cualquier átomo de carbono disponible o a un heteroátomo cuando el heteroátomo es nitrógeno que tiene la valencia adecuada.
"halo" o "halógeno", como se usa en el presente documento, se refiere a un átomo de cloro, flúor, bromo o yodo.
"Hidroxi" o "hidroxilo", como se usa en el presente documento, significa un grupo -OH.
"ciano", como se usa en el presente documento, significa un grupo -CN, que también puede representarse:
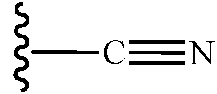
"nitro", como se usa en el presente documento, significa un grupo -NO2.
"Opcionalmente sustituido", como se usa en el presente documento, significa que la sustitución es opcional y, por lo tanto, incluye átomos y restos sin sustituir y sustituidos. Un átomo o resto "sustituido" indica que cualquier hidrógeno en el átomo o resto designado puede reemplazarse con una selección entre el grupo sustituyente indicado (hasta e incluyendo que todos los átomos de hidrógenos en el átomo o resto designado estén reemplazados con una selección de entre el grupo sustituyente indicado), con la condición de que la valencia normal del átomo o resto designado no se supere y que la sustitución dé como resultado un compuesto estable. Por ejemplo, si un grupo metilo (es decir, -CH3) está opcionalmente sustituido, entonces hasta 3 átomos de hidrógeno en el átomo de carbono pueden estar reemplazados con grupos sustituyentes.
"Paciente" se refiere a animales de sangre caliente tales como, por ejemplo, cerdos, vacas, pollos, caballos, cobayas, ratones, ratas, jerbos, gatos, conejos, perros, monos, chimpancés y seres humanos.
"Farmacéuticamente aceptable" indica que la sustancia o composición debe ser compatible, química y/o toxicológicamente, con los otros ingredientes que comprende una formulación y/o con el mamífero que está siendo tratado con la misma.
La expresión "cantidad terapéuticamente eficaz" como se usa en el presente documento se refiere a la cantidad del compuesto (incluyendo un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del W-óxido) que se administra y que aliviará en cierta medida uno o más de los síntomas del trastorno que se está tratando. En referencia al tratamiento de un trastorno mediado por M4 (por ejemplo, enfermedad de Alzheimer o esquizofrenia), una cantidad terapéuticamente eficaz se refiere a la cantidad que tiene el efecto de aliviar en cierta medida (o, por ejemplo, eliminar) uno o más síntomas asociados con el trastorno mediado por M4 (por ejemplo, síntoma positivo, negativo o cognitivo de la esquizofrenia; o síntoma psicótico de la enfermedad de Alzheimer).
El término "tratar", como se usa en el presente documento, a menos que se indique de otro modo, significa revertir, aliviar, inhibir el progreso de, o prevenir, el trastorno o afección al cual se aplica dicho término, o uno o más síntomas de dicho trastorno o afección. El término "tratamiento", como se usa en el presente documento, a menos que se indique de otro modo, se refiere al acto de tratar como se define en el presente documento "tratar". El término "tratar" también incluye el tratamiento adyuvante y neoadyuvante de un sujeto.
"Isómero" significa "estereoisómero" e ''isómero geométrico" como se define más adelante.
"Estereoisómero" se refiere a compuestos que poseen uno o más centros quirales, que pueden existir en la configuración R o S. Los estereoisómeros incluyen todas las formas diastereoméricas, enantioméricas y epiméricas, así como los racematos y mezclas de los mismos.
"Isómero geométrico" se refiere a compuestos que pueden existir en las formas cis, trans, anti, entgegen (E) y zusammen (Z) así como las mezclas de las mismas.
Como se usa en el presente documento, a menos que se especifique, el punto de unión de un sustituyente puede ser desde cualquier posición adecuada del sustituyente. Por ejemplo, piridinilo (o piridilo) puede ser 2-piridinilo (o piridin-2-ilo), 3-piridinilo (o piridin-3-ilo) o 4-piridinilo (o piridin-4-ilo).
Cuando se describe un resto sustituido u opcionalmente sustituido sin indicar el átomo mediante el cual dicho resto se une a un sustituyente, entonces el sustituyente puede estar unido mediante cualquier átomo apropiado en dicho resto. Por ejemplo en un heteroarilo (de 5 a 10 miembros) opcionalmente sustituido, un sustituyente en el heteroarilo puede estar unido a cualquier átomo de carbono en la parte heteroarilo o en el heteroátomo del heteroarilo, si lo permite la valencia. Las combinaciones de sustituyentes y/o variables sólo se permiten si dichas combinaciones dan como resultado compuestos estables.
Esta memoria descriptiva utiliza los términos "sustituyente", "radical" y "grupo" de forma intercambiable.
Si los sustituyentes se describen como "independientemente seleccionados" entre un grupo, cada caso de un sustituyente está seleccionado independientemente de cualquier otro. Por lo tanto, cada sustituyente puede ser idéntico a, o diferente del, otro u otros sustituyentes.
Como se usa en la presente memoria los términos "Fórmula I", "Fórmula I'", "Fórmula Ia", "Fórmula IB" y "Fórmula IC", se pueden denominar en lo sucesivo en el presente documento "compuesto o compuestos de la invención". Dichos términos también se definen para incluir todas las formas de los compuestos de la invención que incluyen, pero no se limitan a, hidratos, solvatos, isómeros (incluyendo por ejemplo, estereoisómeros rotacionales), formas cristalinas y no cristalinas, isomorfos, polimorfos, metabolitos, profármacos de los mismos. Por ejemplo, los compuestos de la invención o las sales farmacéuticamente aceptables de los mismos, pueden existir en formas no solvatadas y solvatadas con disolventes farmacéuticamente aceptables tales como agua, etanol y similares. Cuando el disolvente o el agua están enlazados firmemente, el complejo tendrá una estequiometría bien definida independiente de la humedad. Cuando, sin embargo, el disolvente o el agua estén enlazados débilmente, como en los solvatos de canal
y los compuestos higroscópicos, el contenido de agua/disolvente será dependiente de las condiciones de humedad y secado. En dichos casos, la no estequiometría será la norma. En general, las formas solvatadas se consideran equivalentes a las formas sin solvatar para los propósitos de la presente invención.
Los compuestos de la invención pueden existir en forma de clatratos u otros complejos (por ejemplo, cocristales). Incluidos dentro del alcance de la invención se encuentran complejos tales como los clatratos, complejos de inclusión fármaco-hospedador en donde el fármaco y el hospedador estén presentes en cantidades estequiométricas o no estequiométricas. También se incluyen los complejos de los compuestos de la invención que contienen dos o más componentes orgánicos y/o inorgánicos, los cuales pueden estar en cantidades estequiométricas y no estequiométricas. Los complejos resultantes pueden estar ionizados, parcialmente ionizados o no ionizados. Para una revisión de dichos complejos, véase J. Pharm. Sci., 64 (8), 1269-1288 de Haleblian (agosto de 1975). Normalmente los cocristales se definen como complejos cristalinos de constituyentes moleculares neutros que se unen mediante interacciones no covalentes, pero también pueden ser un complejo de una molécula neutra con una sal. Los cocristales se pueden preparar mediante cristalización en estado fundido, mediante recristalización en disolventes o moliendo físicamente los componentes juntos; véase O. Almarsson y M. J. Zaworotko, Chem. Commun. 2004, 17, 1889-1896. Para una revisión general de los complejos multicomponente, véase J. K. Haleblian, J. Pharm. Sci. 1975, 64, 1269 1288.
Los compuestos de la invención pueden existir en forma de isómeros geométricos, en donde los compuestos tienen átomos de carbono asimétricos y, por tanto, pueden existir como dos o más formas estereoisoméricas. La presente invención incluye todos los estereoisómeros individuales e isómeros geométricos de los compuestos de la invención y las mezclas de los mismos. Los enantiómeros individuales pueden obtenerse mediante separación quiral o utilizando el enantiómero pertinente en la síntesis. Los enlaces carbono-carbono de los compuestos de la invención se pueden representar en el presente documento usando una línea continua (-------- ), una cuña sólida o una línea discontinua ..........111). El uso de una línea continua para representar enlaces a átomos de carbono asimétricos pretende indicar que todos los posibles estereoisómeros (por ejemplo, enantiómeros específicos, mezclas racémicas, etc.) en un átomo de carbono, están incluidos. El uso tanto de una cuña sólida como de una cuña discontinua para representar enlaces a átomos de carbono asimétricos pretende indicar que el estereoisómero mostrado está presente. Cuando está presente en compuestos racémicos, se utilizan cuñas sólidas y discontinuas para definir la estereoquímica relativa, en lugar de la estereoquímica absoluta. Los compuestos racémicos que poseen dicha estereoquímica relativa indicada se pueden marcar con (+/-). Por ejemplo, a menos que se indique de otro modo, se pretende que los compuestos de la invención puedan existir en forma de estereoisómeros, que incluyen isómeros cis y trans, isómeros ópticos tales como enantiómeros R y S, diastereómeros, isómeros geométricos, isómeros rotacionales, isómeros conformacionales, atropisómeros y mezclas de los mismos (tales como racematos y pares diastereoméricos). Los compuestos de la invención pueden mostrar más de un tipo de isomerismo. También están incluidas las sales de adición de ácidos o sales de adición de bases en donde el contraión es ópticamente activo, por ejemplo, D-lactato o L-lisina, o racémico, por ejemplo, DL-tartrato o DL-arginina.
En algunas realizaciones, los compuestos de la presente invención pueden existir y/o aislarse en forma de atropisómeros (por ejemplo, uno o más atropenantiómeros). Los expertos en la materia reconocerán que la atropisomería puede existir en un compuesto que tiene dos o más anillos aromáticos (por ejemplo, dos anillos aromáticos unidos mediante un enlace sencillo). Véase, por ejemplo, Freedman, T. B. et al., Absolute Configuration Determination of Chiral Molecules in the Solution State Using Vibrational Circular Dichroism. Chirality 2003, 15, 743 758; y Bringmann, G. et al., Atroposelective Synthesis of Axially Chiral Biaryl Compounds. Angew. Chem., Int. Ed.
2005, 44, 5384-5427.
Cuando cualquier racemato se cristaliza, son posibles cristales de dos tipos diferentes. El primer tipo es el compuesto racémico (racemato real) mencionado anteriormente, en donde se produce una forma homogénea de cristal que contiene ambos enantiómeros en cantidades equimolares. El segundo tipo es la mezcla racémica o conglomerado en donde se producen dos formas de cristal en cantidades equimolares, comprendiendo cada una un enantiómero individual.
Los compuestos de la presente invención también pueden existir en forma de un N-óxido de los mismos o una sal farmacéuticamente aceptable del compuesto o del N-óxido.
Como sabe el experto en la materia, los compuestos de amina (es decir, aquellos que comprenden uno o más átomos de nitrógeno), por ejemplo aminas terciarias, pueden formar N-óxidos (también conocidos como óxidos de o N-óxidos de amina). Un N-óxido tiene la fórmula de (R100R200R300)N+-O‘ en donde la amina precursora (R100R200R300)N puede ser, por ejemplo, una amina terciaria (por ejemplo, cada uno de R100, R200, R300 es independientemente alquilo, arilalquilo, arilo, heteroarilo o similar), una amina heterocíclica o heteroaromática [por ejemplo, (R100R200R300)N juntos forman 1-alquilpiperidina, 1 -alquilpirrolidina, 1 -bencilpirrolidina o piridina]. Por ejemplo, un nitrógeno de ¡mina, en
especial un nitrógeno de imina heterocíclico o heteroaromático o un átomo de nitrógeno tipo piridina ("^_N ^ ) [tal como un átomo de nitrógeno en piridina, piridazina o pirazina], se puede N-oxidar para formar el N-óxido que comprende el grupo

Por lo tanto, un compuesto de acuerdo con la presente invención que comprende uno o más átomos de nitrógeno (por ejemplo, un átomo de nitrógeno de imina) puede ser capaz de formar un N-óxido del mismo (por ejemplo, mono-N-óxidos, bis-N-óxidos o multi-N-óxidos, o mezclas de los mismos, dependiendo del número de átomos de nitrógeno adecuados para formar N-óxidos estables).
Como se usa en el presente documento, la expresión "N-óxido" se refiere a toda las formas de N-óxido posibles y, en particular, todas las estables, de los compuestos amina (por ejemplo, compuestos que comprenden uno o más átomos de nitrógeno de imina) descritos en el presente documento, tales como mono-W-óxidos (que incluyen los distintos isómeros cuando más de un átomo de nitrógeno de un compuesto amina puede formar un mono-N-óxido) o multi-N-óxidos (por ejemplo, bis-N-óxidos) o sus mezclas en cualquier proporción.
Como se ha indicado anteriormente, los compuestos de la invención (o sus N-óxidos) pueden existir en forma de sales farmacéuticamente aceptables derivadas de ácidos orgánicos o inorgánicos. Dependiendo del compuesto particular, una sal del compuesto puede ser ventajosa debido a una o más de las propiedades físicas de la sal, tales como estabilidad farmacéutica mejorada a diferentes temperaturas y humedades o una solubilidad deseable en agua o en aceite. En algunos casos, también se puede usar una sal de un compuesto como un auxiliar en el aislamiento, purificación y/o resolución del compuesto.
Cuando se pretende administrar una sal a un paciente (en oposición a, por ejemplo, usarse en un contexto in vitro), la sal preferentemente es farmacéuticamente aceptable. La expresión "sal farmacéuticamente aceptable" se refiere a una sal preparada combinando un compuesto de la presente invención con un ácido cuyo anión, o una base cuyo catión, se considera normalmente adecuado para el consumo humano. Las sales farmacéuticamente aceptables son particularmente útiles como productos de los métodos de la presente invención debido a su mayor solubilidad en agua con respecto al compuesto precursor.
Las sales de adición de ácido farmacéuticamente aceptables adecuadas de los compuestos de la presente invención, cuando es posible, incluyen las derivadas de ácidos inorgánicos, tales como, pero no limitados a, los ácidos clorhídrico, bromhídrico, fluorhídrico, bórico, fluorobórico, fosfórico, metafosfórico, nítrico, carbónico, sulfónico y sulfúrico, y de ácidos orgánicos, tales como los ácidos acético, bencenosulfónico, benzoico, cítrico, etanosulfónico, fumárico, glucónico, glicólico, isotiónico, láctico, lactobiónico, maleico, málico, metanosulfónico, trifluorometanosulfónico, succínico, toluenosulfónico, tartárico y trifluoroacético. Los ácidos orgánicos adecuados generalmente incluyen pero no se limitan a las clases alifática, cicloalifática, aromática, aralifática, heterocíclica, carboxílica y sulfónica de ácidos los orgánicos.
Los ejemplos específicos de ácidos orgánicos adecuados incluyen, pero no se limitan a, acetato, trifluoroacetato, formiato, propionato, succinato, glicolato, gluconato, digluconato, lactato, malato, tartrato, citrato, ascorbato, glucuronato, maleato, fumarato, piruvato, aspartato, glutamato, benzoato, antranilato, estearato, salicilato, phidroxibenzoato, fenilacetato, mandelato, embonato (pamoato), metanosulfonato, etanosulfonato, bencenosulfonato, pantotenato, toluenosulfonato, 2-hidroxietanosulfonato, sufanilato, ciclohexilaminosulfonato, ácido algénico, ácido phidroxibutírico, galactarato, galacturonato, adipato, alginato, butirato, alcanforato, canforsulfonato, ciclopentanopropionato, dodecilsulfato, glicoheptanoato, glicerofosfato, heptanoato, hexanoato, nicotinato, 2-naftalenosulfonato, oxalato, palmoato, pectinato, 3-fenilpropionato, picrato, pivalato, tiocianato y undecanoato.
Además, cuando los compuestos de la invención portan un resto ácido, las sales farmacéuticamente aceptables adecuadas de los mismos pueden incluir sales de metales alcalinos, por ejemplo, sales de sodio o potasio; sales de metales alcalinotérreos, por ejemplo, sales de calcio o magnesio; y sales formadas con ligandos orgánicos adecuados, por ejemplo, sales de amonio cuaternario. En otra realización, las sales de bases se forman a partir de bases que forman sales no tóxicas, que incluyen sales de aluminio, arginina, benzatina, colina, dietilamina, diolamina, glicina, lisina, meglumina, olamina, trometamina y cinc.
Las sales orgánicas pueden fabricarse a partir de sales de amina secundaria, terciaria o cuaternaria, tales como trometamina, dietilamina, W,W’-dibenciletilendiamina, cloroprocaína, colina, dietanolamina, etilendiamina, meglumina (N-metilglucamina) y procaína. Los grupos básicos que contienen nitrógeno pueden cuaternizarse con agentes, tales como haluros de alquilo inferior (Ci-Ca) (por ejemplo, cloruros, bromuros y yoduros de metilo, etilo, propilo y butilo), sulfatos de dialquilo (por ejemplo, sulfatos de dimetilo, dietilo, dibutilo y diamilo), haluros de cadena larga (por ejemplo, cloruros, bromuros y yoduros de decilo, laurilo, miristilo y estearilo), haluros de arilalquilo (por ejemplo, bromuros de bencilo y fenetilo) y otros.
En una realización, también pueden formarse hemisales de ácidos y bases, por ejemplo, sales de hemisulfato y hemicalcio.
Para una revisión de las sales adecuadas, véase "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" de Stahl y Wermuth (Wiley-VCH, 2002). El experto en la materia conoce los métodos para preparar sales farmacéuticamente aceptables de los compuestos de la invención.
Los compuestos de la invención pueden existir en un continuo de estados sólidos que van desde completamente amorfos hasta completamente cristalinos. El término "amorfo" se refiere a un estado en el que el material carece de un orden de largo alcance a nivel molecular y, dependiendo de la temperatura, puede presentar las propiedades físicas de un sólido o un líquido. Normalmente, dichos materiales no proporcionan patrones de difracción de rayos X distintivos y, aunque presentan las propiedades de un sólido, se describen más formalmente como un líquido. Tras el calentamiento, tiene lugar un cambio de aparente sólido a un material con propiedades líquidas, que se caracteriza por un cambio de estado, habitualmente de segundo orden ("transición vítrea"). El término 'cristalino' se refiere a una fase sólida en la que el material tiene una estructura interna ordenada regular a nivel molecular y proporciona un patrón de rayos X distintivo con picos definidos. Dichos materiales, cuando se calientan suficientemente, también exhibirán las propiedades de un líquido, pero el cambio de sólido a líquido se caracteriza por un cambio de fase, habitualmente de primer orden ("punto de fusión").
Los compuestos de la invención también pueden existir en un estado mesomórfico (mesofase o cristal líquido) cuando se someten a las condiciones adecuadas. El estado mesomórfico es intermedio entre el verdadero estado cristalino y el verdadero estado líquido (fundido o en solución). El mesoformismo que aparece como resultado de un cambio de temperatura se describe como "termotrópico" y el que resulta de la adición de un segundo componente, tal como agua u otro disolvente, se describe como "liotrópico". Los compuestos que tienen potencial para formar mesofases liotrópicas se describen como "amfifílicos" y consisten en moléculas que poseen un grupo de cabeza polar iónico (tal como -COO'Na+, -COO'K+ o -SO3-Na+) o no iónico (tal como -N'N+(c H3)3). Para más información, véase Crystals and the Polarizing Microscope de N. H. Hartshorne y A. Stuart, 4a edición (Edward Arnold, 1970).
La presente invención también incluye todos los compuestos marcados isotópicamente farmacéuticamente aceptables, que son idénticos a los enumerados en el presente documento, en donde uno o más átomos se reemplazan por un átomo que tiene el mismo número atómico, pero una masa atómica o número másico diferente de la masa atómica o número másico que predomina en la naturaleza. Los ejemplos de isótopos adecuados para su inclusión en los compuestos de la presente invención incluyen, pero no se limitan a, isótopos de hidrógeno, tales como 2H, 3H; carbono, tales como 11C, 13C y 14C; cloro, tales como 36Cl; flúor, tales como 18F; yodo, tales como 123I y 125I; nitrógeno, tales como 13N y 15N; oxígeno, tales como 15O, 17O y 18O; fósforo, tales como 32P; y azufre, tales como 35S. Ciertos compuestos marcados isotópicamente de la presente invención, por ejemplo, los que incorporan un isótopo radiactivo, son útiles en estudios de distribución de fármacos y/o sustratos en tejidos (por ejemplo, ensayos). Los isótopos radiactivos tritio, es decir, 3H y carbono 14, es decir, 14C, son particularmente útiles para este propósito en vista de su facilidad de incorporación y medios de detección sencillos. La sustitución con isótopos más pesados tales como deuterio, es decir, 2H, puede proporcionar ciertas ventajas terapéuticas resultantes de una mayor estabilidad metabólica, por ejemplo, semivida in vivo aumentada o requerimientos de dosificación reducidos y, por tanto, puede preferirse en algunas circunstancias. La sustitución con isótopos emisores de positrones, tales como 11C, 15F, 18F, 15O y 13N, puede ser útil en estudios de tomografía de emisión de positrones (PET) para examinar la ocupación del receptor en el sustrato. Los compuestos marcados isotópicamente de la presente invención pueden prepararse generalmente por técnicas convencionales conocidas por los expertos en la materia o por procesos análogos a los descritos en los esquemas y/o en los ejemplos y preparaciones adjuntas usando un reactivo isotópicamente marcado adecuado en lugar del reactivo no marcado empleado previamente. Los solvatos farmacéuticamente aceptables de acuerdo con la invención incluyen aquellos en donde el disolvente de cristalización puede estar isotópicamente sustituido, por ejemplo, D2O, acetona-cfe, o DMSO-cfe. Los compuestos de la invención, que incluyen los compuestos ejemplificados en los ejemplos 1-51 descritos a continuación, incluyen versiones marcadas isotópicamente de estos compuestos, tales como, pero no limitados a, los isótopos deuterados y tritiados y todos los otros isótopos analizados anteriormente.
En determinadas realizaciones, la presente divulgación se refiere a nuevos agonistas de M4 radiomarcados, selectivos, que son útiles para la obtención de imágenes y cuantificar la distribución de los compuestos M4 en los tejidos (por ejemplo, cerebro), usando tomografía de emisión de positrones (PET).
Compuestos
Los compuestos de fórmula I, como se ha descrito anteriormente, contienen un núcleo de carboxilato de piperazin-1-il-2 -azaespiro en donde la piperazina está unida al heteroarilo de 6 miembros (piridina o pirazina) que está sustituido con R' y con R2; y el resto azaespiro se selecciona de entre un 2-azaespiro[3.4]octano, un 6-azaespiro[3.4]octano o un 2-azaespiro[3.3]heptano.
En una realización, en la fórmula I como se ha descrito anteriormente, X1 es nitrógeno y X2 es CH.
En otra realización, X1 es nitrógeno y X2 es nitrógeno.
En otra realización más, X1 es CH y X2 es nitrógeno.
Debe apreciarse que cualquiera de los subgéneros (realizaciones) de X1 y X2 mencionados anteriormente se pueden combinar junto con cualquiera de los subgéneros para R1, R2, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula I como se ha descrito anteriormente, R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-Ca), alcoxi (CrCa) y cicloalquilo (C3-C6), en donde dicho alquilo (Ci-Ce), alcoxi (C1-C6) y cicloalquilo (C3-C6) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) está cada uno opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).
En ciertas realizaciones, R' es un alcoxi (C1-C6). Cuando R' es un alcoxi (C1-C6), el alcoxi incluye, pero no se limita a, metoxi, trifluoroetoxi, difluorometoxi y trifluorometoxi.
En ciertas realizaciones, R' es un cicloalquilo (C3-C6). Cuando R' es un cicloalquilo (C3-C6), el cicloalquilo incluye, pero no se limita a ciclopropilo.
En otra realización, en la fórmula I como se ha descrito anteriormente, R1 es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -(CH2)2-O-CH2CH3 y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).
En otra realización, en la fórmula I como se ha descrito anteriormente, R' es un heteroarilo (de 5 a 6 miembros).
Cuando R' es un heteroarilo (de 5 a 10 miembros) sustituido o un heteroarilo (de 5 a 6 miembros) sustituido, el sustituyente es un alquilo (C1-C6) o un alcoxi (C1-C6), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo y el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi.
En otra realización, en la fórmula I como se ha descrito anteriormente, R' es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).
Cuando R' es un heterocicloalquilo (de 4 a 8 miembros) sustituido, el sustituyente es un alquilo (C1-C6), un alcoxi (C1-C6) o un heteroarilo (de 5 a 6 miembros), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo, el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi y el sustituyente heteroarilo (de 5 a 6 miembros) es un pirazolilo, que está opcionalmente sustituido con un sustituyente metilo.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R' se puede combinar con cualquiera de los subgéneros para X1, X2, R2, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula I como se ha descrito anteriormente, R2 es hidrógeno.
En otra realización, R2 es flúor.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R2 se puede combinar junto con cualquiera de los subgéneros para X1, X2, R1, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula I como se ha descrito anteriormente, m es 2 y n es 1.
En otra realización, m es 1 y n es 2.
En otra realización, m es 1 y n es 1.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de m y n se puede combinar junto con cualquiera de los subgéneros para X1, X2, R1 y R2 como se ha descrito anteriormente y a continuación en el presente documento.
En otras ciertas realizaciones, la presente invención es un compuesto de fórmula Ia:

o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:
cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH,
R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6); y
R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (C1-C6) y alcoxi (C1-C6), en donde dicho alquilo (C1-C6) y alcoxi (C1-C6) están opcionalmente sustituidos con de 1 a 3 halógenos.
En una realización, en la fórmula Ia como se ha descrito anteriormente, X1 es nitrógeno y X2 es CH.
En otra realización, X1 es nitrógeno y X2 es nitrógeno.
En otra realización más, X1 es CH y X2 es nitrógeno.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de X1 y X2 se puede combinar con cualquiera de los subgéneros para R', R2, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula Ia como se ha descrito anteriormente, R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) está cada uno opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).
En determinadas realizaciones, R' es un alcoxi (Ci-Ca). Cuando R' es un alcoxi (Ci-Ca), el alcoxi incluye, pero no se limita a, metoxi, trifluoroetoxi, difluorometoxi y trifluorometoxi.
En determinadas realizaciones, R' es un cicloalquilo (C3-C6). Cuando R' es un cicloalquilo (C3-Ca), el cicloalquilo incluye, pero no se limita a ciclopropilo.
En otra realización, en la fórmula Ia como se ha descrito anteriormente, R' es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -(CH2)2-O-CH2CH3 y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).
En otra realización, en la fórmula Ia como se ha descrito anteriormente, R' es un heteroarilo (de 5 a a miembros) opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci Ca).
Cuando R' es un heteroarilo (de 5 a 10 miembros) sustituido o un heteroarilo (de 5 a a miembros) sustituido, el sustituyente es un alquilo (Ci-Ca) o un alcoxi (Ci-Ca), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo y el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi.
En otra realización, en la fórmula Ia como se ha descrito anteriormente, R' es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-a-azaespiro[3.3]hept-a-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci Ca).
Cuando R' es un heterocicloalquilo (de 4 a 8 miembros) sustituido, el sustituyente es un alquilo (Ci-Ca), un alcoxi (Ci Ca) o un heteroarilo (de 5 a a miembros), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo, el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi y el sustituyente heteroarilo (de 5 a a miembros) es un pirazolilo, que está opcionalmente sustituido con un sustituyente metilo.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R' se puede combinar con cualquiera de los subgéneros para X1, X2 y R2 como se ha descrito anteriormente y a continuación en el presente documento.
En otra realización, en la fórmula Ia como se ha descrito anteriormente, R2 es hidrógeno.
En otra realización, R2 es flúor.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R2 se puede combinar junto con cualquiera de los subgéneros para X1, X2 y R' como se ha descrito anteriormente y a continuación en el presente documento.
En otras ciertas realizaciones, la presente invención es un compuesto de fórmula IB.

o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:
cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH,
R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ce), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (Ca-C1ü), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dicho alquilo (CrCa), alcoxi (C1-Ca), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-C™), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (C1-Ca), alcoxi (C1-Ca) y heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (CrCa) y alcoxi (C1-Ca);
R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (C1-Ca) y alcoxi (C1-Ca), en donde dicho alquilo (C1-Ca) y alcoxi (CrCa) están opcionalmente sustituidos con de 1 a 3 halógenos.
En una realización, en la fórmula IB como se ha descrito anteriormente, X1 es nitrógeno y X2 es CH.
En otra realización, X1 es nitrógeno y X2 es nitrógeno.
En otra realización más, X1 es CH y X2 es nitrógeno.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de X1 y X2 se puede combinar con cualquiera de los subgéneros para R', R2, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula IB como se ha descrito anteriormente, R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (CrCa), alcoxi (CrCa) y cicloalquilo (C3-Ca), en donde dicho alquilo (CrCa), alcoxi (CrCa) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (CrCa), alcoxi (CrCa), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (CrCa), alcoxi (CrCa) y heteroarilo (de 5 a a miembros) está cada uno opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca) y alcoxi (C1-Ca).
En determinadas realizaciones, R' es un alcoxi (CrCa). Cuando R' es un alcoxi (CrCa), el alcoxi incluye, pero no se limita a, metoxi, trifluoroetoxi, difluorometoxi y trifluorometoxi.
En determinadas realizaciones, R' es un cicloalquilo (C3-Ca). Cuando R' es un cicloalquilo (C3-Ca), el cicloalquilo incluye, pero no se limita a ciclopropilo.
En otra realización, en la fórmula IB como se ha descrito anteriormente, R' es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (CrCa), alcoxi (CrCa), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (CrCa), alcoxi (CrCa) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (CrCa) y alcoxi (C1-Ca).
En otra realización, en la fórmula IB como se ha descrito anteriormente, R' es un heteroarilo (de 5 a 6 miembros) opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C-i-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C-i-Ca) y alcoxi (C1-Ca).
Cuando R' es un heteroarilo (de 5 a 10 miembros) sustituido o un heteroarilo (de 5 a a miembros) sustituido, el sustituyente es un alquilo (C1-Ca) o un alcoxi (C1-Ca), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo y el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi.
En otra realización, en la fórmula IB como se ha descrito anteriormente, R' es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-a-azaespiro[3.3]hept-a-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (C1-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca) y alcoxi (C1-Ca).
Cuando R' es un heterocicloalquilo (de 4 a 8 miembros) sustituido, el sustituyente es un alquilo (C1-Ca), un alcoxi (C1-Ca) o un heteroarilo (de 5 a a miembros), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo, el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi y el sustituyente heteroarilo (de 5 a a miembros) es un pirazolilo, que está opcionalmente sustituido con un sustituyente metilo.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R' se puede combinar con cualquiera de los subgéneros para X1, X2 y R2 como se ha descrito anteriormente y a continuación en el presente documento.
En otra realización, en la fórmula IB como se ha descrito anteriormente, R2 es hidrógeno.
En otra realización, R2 es flúor.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R2 se puede combinar junto con cualquiera de los subgéneros para X1, X2 y R' como se ha descrito anteriormente y a continuación en el presente documento.
En otras ciertas realizaciones, la presente invención es un compuesto de fórmula IC:

o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:
cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH,
R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3
Cq), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (Ce-Cío), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dicho alquilo (Cí-Ce), alcoxi (Cí-Ce), cicloalquilo (C3-Cq), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (Ce-Cío), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, -alquilo (Cí-Ce), alcoxi (Cí-Ce), cicloalquilo (C3-Cq) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Cí-Ce), alcoxi (Cí-Ce) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce) y alcoxi (Cí-Ce); y
R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (Cí-Ce) y alcoxi (Cí-Ce), en donde dicho alquilo (Cí-Ce) y alcoxi (Cí-Ce) están opcionalmente sustituidos con de í a 3 halógenos.
En una realización, en la fórmula IC como se ha descrito anteriormente, Xí es nitrógeno y X2 es CH.
En otra realización, Xí es nitrógeno y X2 es nitrógeno.
En otra realización más, Xí es CH y X2 es nitrógeno.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de Xí y X2 e puede combinar con cualquiera de los subgéneros para Rí y R2 como se ha descrito anteriormente y a continuación en el presente documento.
En otra realización, en la fórmula IC como se ha descrito anteriormente, R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Cí-Ce), alcoxi (Cí-Ce) y cicloalquilo (C3-Cq), en donde dicho alquilo (Cí-Ce), alcoxi (Cí-Ce) y cicloalquilo (C3-Cq) están opcionalmente sustituidos con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce), alcoxi (Cí-Ce), cicloalquilo (C3-Cq) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Cí-Ce), alcoxi (Cí-Ce) y heteroarilo (de 5 a 6 miembros) está cada uno opcionalmente sustituido con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce) y alcoxi (Cí-Ce).
En determinadas realizaciones, R' es un alcoxi (Cí-Ce). Cuando R' es un alcoxi (Cí-Ce), el alcoxi incluye, pero no se limita a, metoxi, trifluoroetoxi, difluorometoxi y trifluorometoxi.
En determinadas realizaciones, R' es un cicloalquilo (C3-Cq). Cuando R' es un cicloalquilo (C3-Cq), el cicloalquilo incluye, pero no se limita a ciclopropilo.
En otra realización, en la fórmula IC como se ha descrito anteriormente, R' es un heteroarilo (de 5 a í0 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a í0 miembros) está opcionalmente sustituido con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce), alcoxi (Cí-Ce), cicloalquilo (C3-Cq), -(CH2)2-O-CH2CH3 y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Cí-Ce), alcoxi (Cí-Ce) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce) y alcoxi (Cí-Ce).
En otra realización, en la fórmula IC como se ha descrito anteriormente, R' es un heteroarilo (de 5 a 6 miembros) opcionalmente sustituido con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce), alcoxi (Cí-Ce), cicloalquilo (C3-Cq) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Cí-Ce), alcoxi (Cí-Ce) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce) y alcoxi (Cí-Ce).
Cuando R' es un heteroarilo (de 5 a í0 miembros) sustituido o un heteroarilo (de 5 a 6 miembros) sustituido, el sustituyente es un alquilo (Cí-Ce) o un alcoxi (Cí-Ce), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo y el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi.
En otra realización, en la fórmula IC como se ha descrito anteriormente, R' es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce), alcoxi (Cí-Ce), cicloalquilo (C3-Cq) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Cí-Ce), alcoxi (Cí-Ce) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de í a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Cí-Ce) y alcoxi (Cí-Ce).
Cuando R' es un heterocicloalquilo (de 4 a 8 miembros) sustituido, el sustituyente es un alquilo (Cí-Ce), un alcoxi (Cí-
í5
Ce) o un heteroarilo (de 5 a 6 miembros), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo, el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi y el sustituyente heteroarilo (de 5 a 6 miembros) es un pirazolilo, que está opcionalmente sustituido con un sustituyente metilo.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R' se puede combinar con cualquiera de los subgéneros para X1, X2 y R2 como se ha descrito anteriormente y a continuación en el presente documento.
En otra realización, en la fórmula IC como se ha descrito anteriormente, R2 es hidrógeno.
En otra realización, R2 es flúor.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R2 se puede combinar junto con cualquiera de los subgéneros para X1, X2 y R' como se ha descrito anteriormente y a continuación en el presente documento.
En otras ciertas realizaciones, la presente invención es un compuesto de fórmula I':

o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:
cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH,
R' se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);
R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (C1-C6) y alcoxi (C1-C6), en donde dichos alquilo (C1-C6) y alcoxi (C1-C6) están opcionalmente sustituidos con de 1 a 3 halógeno;
m es 1 o 2; y
n es 1 o 2.
En una realización, en la fórmula I' como se ha descrito anteriormente, X1 es nitrógeno y X2 es CH.
En otra realización, X1 es nitrógeno y X2 es nitrógeno.
En otra realización más, X1 es CH y X2 es nitrógeno.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de X1 y X2 se puede combinar con cualquiera de los subgéneros para R', R2, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula I' como se ha descrito anteriormente, R' se selecciona de entre el grupo que consiste
en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ce) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y heteroarilo (de 5 a a miembros) está cada uno opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).
En determinadas realizaciones, R' es un alcoxi (Ci-Ca). Cuando R' es un alcoxi (Ci-Ca), el alcoxi incluye, pero no se limita a, metoxi, trifluoroetoxi, difluorometoxi y trifluorometoxi.
En determinadas realizaciones, R' es un cicloalquilo (C3-Ca). Cuando R' es un cicloalquilo (C3-Ca), el cicloalquilo incluye, pero no se limita a ciclopropilo.
En otra realización, en la fórmula I' como se ha descrito anteriormente, R' es un heteroarilo (de 5 a i0 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a i0 miembros) está opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -(CH2)2-O-CH2CH3 y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).
En otra realización, en la fórmula I' como se ha descrito anteriormente, R' es un heteroarilo (de 5 a a miembros) opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci Ca).
Cuando R' es un heteroarilo (de 5 a i0 miembros) sustituido o un heteroarilo (de 5 a a miembros) sustituido, el sustituyente es un alquilo (Ci-Ca) o un alcoxi (Ci-Ca), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo, y el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi.
En otra realización, en la fórmula I' como se ha descrito anteriormente, R' es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-a-azaespiro[3.3]hept-a-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).
Cuando R' es un heterocicloalquilo (de 4 a 8 miembros) sustituido, el sustituyente es un alquilo (Ci-Ca), un alcoxi (Ci Ca) o un heteroarilo (de 5 a a miembros), en donde el sustituyente alquilo incluye, pero no se limita a metilo, etilo, cianobutilo y etoxietilo, el sustituyente alcoxi incluye, pero no se limita a metoxi, etoxi, trifluoroetoxi, difluoroetoxi y fluorometoxi y el sustituyente heteroarilo (de 5 a a miembros) es un pirazolilo, que está opcionalmente sustituido con un sustituyente metilo.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R' se puede combinar con cualquiera de los subgéneros para X1, X2, R2, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula I' como se ha descrito anteriormente, R2 es hidrógeno.
En otra realización, R2 es flúor.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de R2 se puede combinar junto con cualquiera de los subgéneros para X1, X2, R1, m y n como se ha descrito anteriormente y en lo sucesivo en el presente documento.
En otra realización, en la fórmula I' como se ha descrito anteriormente, m es 2 y n es i.
En otra realización, m es i y n es 2.
En otra realización, m es i y n es i.
Debe apreciarse que cualquiera de los subgéneros mencionados anteriormente (realizaciones) de m y n se puede combinar junto con cualquiera de los subgéneros para X1, X2, R1 y R2 como se ha descrito anteriormente y a continuación en el presente documento.
En otras ciertas realizaciones, la presente invención se refiere al uso de los compuestos, o el N-óxido o la sal farmacéuticamente aceptable de uno cualquiera de los compuestos de la presente invención, en el tratamiento de una enfermedad o trastorno mediado por M4-(o asociado a M4).
En otras ciertas realizaciones, la presente invención se refiere a un uso de los compuestos de la invención para tratar una enfermedad o un trastorno mediado por M4 (o asociado a M4) en un paciente, comprendiendo dicho método administrar al paciente una cantidad terapéuticamente eficaz de un compuesto, o un N-óxido o una sal farmacéuticamente aceptable de uno cualquiera de los compuestos de la presente invención.
En determinadas realizaciones, los compuestos de la presente invención son agonistas del receptor M4, en donde el compuesto tiene una afinidad de unión por el receptor M4 e induce un efecto sobre él sin la presencia de un ligando nativo (por ejemplo, acetilcolina).
En otras ciertas realizaciones, la presente invención se refiere al uso mencionado anteriormente en donde la enfermedad o trastorno mediado por M4 (o asociado a M4) es una enfermedad o trastorno seleccionado de entre el grupo que consiste en enfermedad de Alzheimer, esquizofrenia, dolor, adicción, un trastorno del sueño, un trastorno cognitivo (por ejemplo, deterioro cognitivo leve, deterioro cognitivo leve senil y deterioro cognitivo leve amnésico), enfermedad de Parkinson, discinesia inducida por levodopa en la enfermedad de Parkinson (PD-LID), enfermedad de Huntington, discinesia, boca seca, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica (EPOC), asma, incontinencia urinaria, glaucoma, trisomía 21 (síndrome de Down), angiopatía amiloidea cerebral, demencia, hemorragia cerebral hereditaria con amiloidosis de tipo holandés (HCHWA-D), enfermedad de Creutzfeld-Jakob, trastornos priónicos, esclerosis lateral amiotrófica, parálisis supranuclear progresiva, traumatismo craneal, accidente cerebrovascular, pancreatitis, miositis por cuerpos de inclusión, otras amiloidosis periféricas, diabetes, autismo y ateroesclerosis.
En determinadas realizaciones, la enfermedad o trastorno mediado por M4 (o asociado a M4) es una enfermedad o trastorno seleccionado de entre el grupo que consiste en enfermedad de Alzheimer, enfermedad de Parkinson, enfermedad de Huntington, esquizofrenia, dolor, adicción y un trastorno del sueño.
La presente invención también proporciona composiciones (por ejemplo, composiciones farmacéuticas) que comprenden un nuevo compuesto de la presente invención. Por consiguiente, en una realización, la invención proporciona una composición farmacéutica que comprende (una cantidad terapéuticamente eficaz de) un nuevo compuesto de la presente invención y que opcionalmente comprende un transportador farmacéuticamente aceptable. En una realización más, la invención proporciona una composición farmacéutica que comprende (una cantidad terapéuticamente eficaz de) un compuesto de la invención, que opcionalmente comprende un transportador farmacéuticamente aceptable y, opcionalmente, al menos otro medicamento o agente farmacéutico (tal como un agente antipsicótico o un agente contra la esquizofrenia descrito a continuación). En una realización, el otro medicamento o agente farmacéutico es un agente contra la esquizofrenia como se describe a continuación.
Farmacología
El receptor muscarínico M4 de acetilcolina (también conocido como muscarínico 4 o CHRM4) es una proteína humana codificada por el gen CHRM4. Los receptores M4 se expresan predominantemente en el cerebro. Las regiones clave del cerebro donde tiene lugar la expresión del receptor M4 son el cuerpo estriado, la corteza y el hipocampo, teniendo lugar la expresión más alta en el cuerpo estriado (aproximadamente el 46 %), donde M4 es el subtipo muscarínico principal. M4 se expresa esporádicamente en la periferia (p. ej., en los testículos, en la piel y en el colon).
Los receptores M4 están acoplados a las proteínas Gq/i y actúan como autorreceptores inhibidores en el cuerpo estriado y el mesencéfalo (Zhang et al. 2002; Tzavara et al. 2004), y como receptores moduladores postsinápticos en el cuerpo estriado, la neocorteza y el hipocampo (Levy et al. 1991; Zhang et al. 1997). Los receptores M4 también se encuentran presinápticamente en las sinapsis glutamatérgicas de la corteza al cuerpo estriado (Pancani, T., et al., "Allosteric activation of M4 improve behavioral and physiological alterations in early symptomatic YAC128 mice", Proceedings of the National Academy of the Sciences of the United States of America, 10 de nov. de 2015; 112(45): 14078-83) y en neuronas glutamatérgicas del hipocampo (donde el M4 presináptico modula la liberación de glutamato. La mayor expresión de los receptores M4 se encuentra en el cuerpo estriado, Los receptores M4 también poseen un efecto regulador sobre la neurotransmisión dopaminérgica y se coexpresan con los receptores de dopamina D1 en un subconjunto de neuronas espinosas medianas del cuerpo estriado que contienen GABA como neurotransmisor principal (Bernard et al. 1992; Di Chiara et al. 1994; Ince et al. 1997).
Se ha planteado la hipótesis de que la administración de un agonista selectivo de M4 proporcionaría actividad antipsicótica para el tratamiento de la esquizofrenia (Felder et al. "Elucidating the Role of Muscarinic Receptors in Psychosis", Life Sci. 68:2605-2613, 2001). Esta creencia fue respaldada además por estudios que demostraron que
los receptores M4 modulan la dinámica de la neurotransmisión dopaminérgica y colinérgica y que se produce un estado de hiperfunción dopaminérgica con la pérdida de la función M4 (Tzavara et al., "M4 Muscarinic Receptors Regulate the Dynamics of Cholinergic and Dopaminergic Neurotransmission: relevance to the pathophysiology and treatment of related CNS pathologies" FASEB J. 18:1410-1412, 2004).
Los compuestos de la presente invención también pueden ser útiles para el tratamiento/alivio de los síntomas neuropsiquiátricos (es decir, síntomas conductuales) asociados a la enfermedad de Alzheimer y a la esquizofrenia (Foster, Daniel J. et. al., "Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer's disease and schizophrenia", Neuropsychiatric Disease and Treatment, Volumen 2014:10, págs. 183-191). Estos síntomas conductuales incluyen, pero sin limitación, inquietud, ansiedad, irritabilidad, agresividad, desorientación, alucinación, delirio, apatía, depresión, desinhibición, comportamientos motores aberrantes y obsesivo-compulsivos, así como trastornos del sueño (Dillon, Carol, et. al. "Behavioral symptoms related to cognitive impairment", Neuropsychiatric Disease and Treatment 2013:9 1443-1455). Al tratar/aliviar los síntomas conductuales mencionados anteriormente, se cree que los compuestos de la presente invención también potenciarán las funciones intelectuales.
En vista de lo anterior, los compuestos de la presente invención pueden ser útiles para el tratamiento de la esquizofrenia y la enfermedad de Alzheimer. Los compuestos de la presente invención también pueden ser útiles para el tratamiento de la enfermedad de Parkinson, enfermedad de Huntington, adicción, depresión y epilepsia.
Se cree que los activadores selectivos de M4 de la presente invención también pueden tener una amplia gama de otras aplicaciones terapéuticas para el tratamiento de afecciones o enfermedades del sistema nervioso central, que incluyen trastornos neurológicos, neurodegenerativos y/o psiquiátricos. Los trastornos neurológicos, neurodegenerativos y/o psiquiátricos incluyen, pero sin limitación, (1) trastornos del estado de ánimo [afectivos]; (2) trastornos neuróticos, relacionados con el estrés y somatomorfos, incluidos los trastornos de ansiedad; (3) trastornos que comprenden el síntoma de deficiencia cognitiva en un mamífero, incluyendo un ser humano; (4) trastornos que comprenden déficits de atención, déficits de la función ejecutiva (déficit de la memoria de trabajo), disfunción del control de impulsos, síntomas extrapiramidales, trastornos que están basados en una disfunción de los núcleos basales, del hipocampo y la corteza prefrontal; (5) trastornos conductuales y emocionales que suelen aparecer en la infancia y la adolescencia; (6) trastornos del desarrollo psicológico; (7) atrofias sistémicas que afectan principalmente al sistema nervioso central; (8) trastornos extrapiramidales y discinesias; (9) síndromes conductuales asociados a alteraciones fisiológicas y a factores físicos; (10) trastornos de la personalidad y del comportamiento del adulto; (11) esquizofrenia y otros trastornos psicóticos; (12) trastornos mentales y conductuales debido al uso de sustancias psicoactivas; (13) disfunción sexual que comprende una libido excesiva; (14) retraso mental; (15) trastornos facticios, p. ej., manía alucinatoria aguda; (16) trastornos episódicos y paroxísticos, epilepsia; (17) narcolepsia; (18) demencia y (19) esclerosis lateral amiotrófica.
Los ejemplos de trastornos del estado de ánimo [afectivos] que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, trastorno bipolar I, hipomanía (forma maníaca y mixta), trastorno bipolar II; trastornos depresivos tales como un episodio depresivo único o trastorno depresivo mayor recurrente, depresión crónica, depresión psicótica, trastorno depresivo menor, trastorno depresivo de inicio posparto, trastornos depresivos con síntomas psicóticos; trastornos persistentes del estado de ánimo [afectivos] tales como la ciclotimia, distimia, eutimia; síndrome premenstrual (SPM) y trastorno disfórico premenstrual.
Los ejemplos de trastornos neuróticos relacionados con el estrés y somatomorfos que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, trastornos de ansiedad, trastorno de ansiedad social, trastorno de ansiedad generalizada, trastorno de pánico con o sin agorafobia, fobia específica, fobia social, trastornos de ansiedad crónica; trastorno obsesivo compulsivo; reacción al estrés intenso y trastornos de adaptación, tal como el trastorno por estrés postraumático (TEPT), trastorno por estrés agudo; otros trastornos neuróticos tales como el síndrome de despersonalización-desrealización.
La expresión "deficiencia cognitiva" como se usa en el presente documento y "trastornos que comprenden el síntoma de deficiencia cognitiva" se refiere a un funcionamiento por debajo de lo normal o subóptimo en uno o más aspectos cognitivos tales como la memoria, la inteligencia, la capacidad de aprendizaje y lógica, o la atención y la función ejecutiva (memoria de trabajo) en un individuo en particular comparado con otros individuos dentro de la misma población de edad general.
Los ejemplos de "trastornos que comprenden el síntoma de deficiencia cognitiva" que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, déficits cognitivos principalmente pero no exclusivamente relacionados con la amnesia, psicosis (esquizofrenia), enfermedad de Parkinson, enfermedad de Alzheimer, demencia por infarto múltiple, demencia senil, demencia de cuerpos de Lewy, ictus, demencia frontotemporal, parálisis supranuclear progresiva, enfermedad de Huntington, enfermedad por VIH (demencia asociada al VIH), traumatismo cerebral y drogadicción; TDAH de trastorno cognitivo leve, síndrome de Asperger y deterioro senil de la memoria; deterioro cognitivo o delirio postoperatorio o asociado a terapia de cuidados intensivos.
Los ejemplos de trastornos diagnosticados habitualmente por primera vez en la primera infancia, la infancia tardía y la adolescencia que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, trastornos
hipercinéticos, incluyendo alteraciones de la actividad y la atención, trastorno por déficit de atención con hiperactividad (TDAH), trastorno de conducta hipercinética; trastorno por déficit de atención (TDA); trastornos disociales, incluyendo, pero sin limitación, trastorno disocial depresivo; trastornos de tic incluyendo el trastorno de tic transitorio, trastorno de tic motor o vocal crónico, trastorno combinado de tic vocal y motor múltiple (síndrome de Gilles de la Tourette), trastornos de tic inducido por sustancias; trastornos autistas; enfermedad de Batten, masturbación excesiva, onicofagia, hurgarse la nariz y chuparse el dedo.
Los ejemplos de trastornos del desarrollo psicológico que pueden tratarse de acuerdo con la presente invención incluyen, aunque sin limitación, trastornos generalizados del desarrollo, incluyendo, pero sin limitación, síndrome de Asperger y síndrome de Rett, trastornos autistas, autismo infantil y trastorno hiperactivo asociado a retraso mental y movimientos estereotipados, trastorno específico del desarrollo de la función motora, trastornos específicos del desarrollo de competencias académicas.
Los ejemplos de atrofias sistémicas que afectan principalmente al sistema nervioso central que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, atrofias sistémicas por esclerosis múltiple que afectan principalmente a los núcleos basales, incluyendo enfermedad de Huntington y esclerosis lateral amiotrófica.
Los ejemplos de trastornos extrapiramidales y discinesias con disfunción y/o degeneración de los núcleos basales que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, enfermedad de Huntington; enfermedad de Parkinson; parkinsonismo secundario tal como parkinsonismo posencefalítico; parkinsonismo comprendido en otros trastornos; enfermedad de Niemann-Pick, enfermedad por cuerpos de Lewy; enfermedades degenerativas de los núcleos basales; otros trastornos extrapiramidales y discinesias incluyendo temblor, temblor esencial y temblor farmacógeno, mioclonía, corea y corea farmacógena, tics farmacógenos y tics de origen orgánico, distonía aguda farmacógena, discinesia tardía farmacógena, espasmos musculares y trastornos asociados a espasticidad o debilidad muscular incluyendo temblores; deficiencia mental (incluyendo espasticidad, síndrome de Down y síndrome del cromosoma X frágil), discinesia inducida por L-dopa; síndrome de las piernas inquietas y síndrome del hombre rígido.
Otros ejemplos de discinesias con disfunción y/o degeneración de los núcleos basales que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, distonía incluyendo, pero sin limitación, distonía focal, distonía focal múltiple o segmentaria, distonía de torsión, distonía hemisférica, generalizada y tardía (inducida por psicofármacos). La distonía focal incluye distonía cervical (tortícolis), blefaroespasmo (espasmo del párpado), distonía apendicular (calambre en las extremidades, como el calambre del escritor), o distonía mandibular y disfonía espasmódica (calambre de la cuerda vocal); discinesias inducidas por neurolépticos, incluyendo, pero sin limitación, síndrome maligno por neurolépticos (SMN), parkinsonismo inducido por neurolépticos, discinesia aguda o de inicio temprano inducida por neurolépticos, distonía aguda inducida por neurolépticos, acatisia aguda inducida por neurolépticos, discinesia tardía inducida por neurolépticos y temblor inducido por neurolépticos.
Los ejemplos de síndromes conductuales asociados a alteraciones fisiológicas y a factores físicos de acuerdo con la presente invención incluyen, pero sin limitación, trastornos del sueño no orgánicos, incluyendo, pero sin limitación, hipersomnia no orgánica, trastorno no orgánico del horario de sueño-vigilia (trastorno del sueño del ritmo circadiano), insomnio, parasomnia y falta de sueño; trastornos mentales y conductuales asociados al puerperio, incluyendo depresión posnatal y posparto; trastornos de la conducta alimentaria, incluyendo, pero sin limitación, anorexia nerviosa, bulimia nerviosa, trastorno alimentario compulsivo, hiperfagia, obesidad, trastornos compulsivos de la conducta alimentaria y pagofagia.
Los ejemplos de trastornos de la personalidad y el comportamiento de los adultos que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, trastornos de la personalidad, incluyendo, pero sin limitación, trastorno de la personalidad emocionalmente inestable, fronterizo, obsesivo-compulsivo, anancástico, dependiente y pasivo-agresivo; trastornos de hábitos e impulsos (trastorno de control de impulsos), incluyendo trastorno explosivo intermitente, ludopatía, conducta repetitiva de provocar incendios (piromanía), conducta repetitiva de robar (cleptomanía), tricotilomanía; síndrome de Munchausen.
Los ejemplos de esquizofrenia y otros trastornos psicóticos que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, esquizofrenia continuada o episódica de diferentes tipos (por ejemplo, paranoide, hebefrénica, catatónica, indiferenciada, residual y trastornos esquizofreniformes); trastornos esquizotípicos (como fronterizo, latente, prepsicótico, prodrómico, esquizofrenia pseudopsicopática pseudoneurótica y trastorno esquizotípico de la personalidad); trastornos delirantes persistentes; trastornos psicóticos agudos, transitorios y persistentes; trastornos delirantes inducidos; trastornos esquizoafectivos de distinto tipo (por ejemplo, maníaco depresivo o de tipo mixto); psicosis puerperal y otras psicosis no orgánicas inespecíficas, tales como el aislamiento social en la esquizofrenia.
Los ejemplos de trastornos mentales y conductuales debidos al uso de sustancias psicoactivas que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, trastornos mentales y conductuales debido al uso de alcohol, opioides, canabinoides, sedantes o hipnóticos, cocaína; trastornos mentales y conductuales debido al uso de otros estimulantes incluyendo la cafeína, trastornos mentales y conductuales debido a la drogodependencia y la
drogadicción (p. ej., dependencia de estupefacientes, alcoholismo, dependencia de anfetamina y metanfetamina, dependencia de opioides, adicción a la cocaína, dependencia de la nicotina y síndrome de abstinencia y prevención de recurrencia), uso de alucinógenos, tabaquismo (nicotina), disolventes volátiles y trastornos mentales y conductuales debidos al uso de múltiples fármacos y al uso de otras sustancias psicoactivas, incluyendo los siguientes síntomas subtipo: uso nocivo, síndrome de dependencia, abstinencia y abstinencia con delirio confusional.
Los ejemplos de demencia que pueden tratarse de acuerdo con la presente invención incluyen, pero sin limitación, demencia vascular, demencia debida a la enfermedad de Creutzfeld-Jacob, VIH, traumatismo craneal, enfermedad de Parkinson, de Huntington, de Pick, demencia de tipo Alzheimer.
En determinadas realizaciones, la presente invención está dirigida al uso de los compuestos de la presente invención para el tratamiento de la esquizofrenia mediante la administración de una cantidad terapéuticamente eficaz de un compuesto de la presente invención a un paciente que lo necesite.
En otras realizaciones determinadas, la invención se dirige además al uso de los compuestos de la presente invención para el tratamiento del deterioro cognitivo asociado a la esquizofrenia mediante la administración de una cantidad terapéuticamente eficaz de un compuesto de la presente invención a un paciente que lo necesite.
La esquizofrenia o psicosis para la que pueden ser útiles los compuestos de la invención, un N-óxido de los mismos y sales farmacéuticamente aceptables de los anteriores, incluyen una o más de las siguientes afecciones: esquizofrenia (paranoide, desorganizada, catatónica o indiferenciada), trastorno esquizofreniforme, trastorno esquizoafectivo, trastorno delirante, trastorno psicótico breve, trastorno psicótico compartido, trastorno psicótico debido a una afección médica general y trastorno psicótico inducido por sustancias o fármacos (fenciclidina, ketamina y otros anestésicos disociativos, anfetamina y otros psicoestimulantes y cocaína), psicosis asociada a trastornos afectivos, psicosis reactiva breve, psicosis esquizoafectiva, trastornos del "espectro de esquizofrenia" tales como trastornos esquizoides o esquizotípicos de la personalidad o enfermedades asociadas a psicosis (tales como depresión mayor, trastorno maníaco depresivo (bipolar), enfermedad de Alzheimer y síndrome de estrés postraumático), incluyendo síntomas tanto positivos como negativos de la esquizofrenia y otras psicosis; trastornos cognitivos incluyendo demencia (asociada a enfermedad de Alzheimer, isquemia, demencia multiinfarto, traumatismo, problemas vasculares o ictus, enfermedad del VIH, enfermedad de Parkinson, enfermedad de Huntington, enfermedad de Pick, enfermedad de Creutzfeldt-Jacob, hipoxia perinatal, otras afecciones médicas generales o drogadicción); delirios, trastornos amnésicos o deterioro cognitivo senil.
Además de los trastornos del sistema nervioso central mencionados anteriormente, los compuestos de la presente invención pueden usarse para tratar otros trastornos mediados por M4 (o asociados a M4) tales como, pero sin limitación, adicción (p. ej,, adicción a sustancias tal como la adicción a los opioides, cocaína o alcohol), dolor (p. ej., dolor agudo, dolor inflamatorio y dolor neuropático) y un trastorno del sueño (tal como los relacionados con la regulación del sueño REM, por ejemplo, los relacionados con el inicio del sueño REM). Otros trastornos o afecciones mediados por M4 (o asociados a M4) que pueden tratarse con los compuestos de la invención incluyen, xerostomía, un trastorno cognitivo (p. ej., deterioro cognitivo leve), discinesia, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica (EPOC), asma, incontinencia urinaria, glaucoma, trisomía 21 (síndrome de Down), angiopatía amiloidea cerebral, demencia (p. ej., demencia degenerativa), hemorragia cerebral hereditaria con amiloidosis de tipo holandés (HCHWA-D), enfermedad de Creutzfeld-Jakob, trastornos priónicos, esclerosis lateral amiotrófica, parálisis supranuclear progresiva, traumatismo craneal, ictus, pancreatitis, miositis por cuerpos de inclusión, otras amiloidosis periféricas, diabetes, autismo y ateroesclerosis. Véase, por ejemplo, el documento US8.664.234.
Los posibles trastornos del sueño para los que pueden ser útiles los compuestos de la invención, un N-óxido de los mismos y sales farmacéuticamente aceptables de los anteriores incluyen: potenciar la calidad del sueño; mejorar la calidad del sueño; aumentar el mantenimiento del sueño; aumentar el valor que se calcula a partir del tiempo que un sujeto duerme dividido entre el tiempo que un sujeto intenta dormir; disminuir la latencia o inicio del sueño (el tiempo que lleva dormirse); disminuir las dificultades para quedarse dormido; aumentar de la continuidad del sueño; disminuir el número de despertares durante el sueño; disminuir las activaciones cerebrales nocturnas; disminuir el tiempo que se pasa despierto tras el inicio del sueño; aumentar la cantidad total de sueño; reducir la fragmentación del sueño; alterar el tiempo, la frecuencia o la duración de los episodios de sueño REM; alterar el tiempo, la frecuencia o la duración de los episodios de sueño de onda lenta (es decir, fases 3 o 4); aumentar la cantidad y el porcentaje de sueño en la fase 2; promover el sueño de onda lenta; potenciar la actividad EEG-delta durante el sueño; aumentar el estado de alerta diurno; reducir la somnolencia diurna; tratar o reducir la somnolencia diurna excesiva; insomnio; hipersomnia; narcolepsia; sueño interrumpido; apnea del sueño; estado de vigilia; mioclonía nocturna; interrupciones del sueño REM; desfase horario; alteraciones del sueño de los trabajadores por turnos; disomnias; terrores nocturnos; insomnio asociado a la depresión, trastornos emocionales/anímicos, así como el sonambulismo y la enuresis, y los trastornos del sueño que acompañan al envejecimiento; empeoramiento vespertino en el Alzheimer; afecciones asociadas a la ritmicidad circadiana, así como trastornos mentales y físicos asociados a los viajes a través de husos horarios y con horarios rotativos de trabajo por turnos; afecciones debidas a fármacos que provocan reducciones en el sueño REM como un efecto secundario; síndromes que se manifiestan por sueño no reparador y mialgia o apnea del sueño que se asocia a perturbaciones respiratorias durante el sueño; y afecciones resultantes de una disminución de la calidad del sueño.
Los trastornos del dolor para los que pueden ser útiles los compuestos de la invención, un A/-óxido de los mismos y sales farmacéuticamente aceptables de los anteriores incluyen, dolor neuropático (tal como neuralgia posherpética, lesión nerviosa, las "dinias", p. ej., vulvodinia, dolor del miembro fantasma, avulsiones radiculares, neuropatía diabética dolorosa, mononeuropatía traumática dolorosa, polineuropatía dolorosa); síndromes de dolor central (posiblemente causados por prácticamente cualquier lesión en cualquier nivel del sistema nervioso); síndromes de dolor posquirúrgico (p. ej., síndrome de dolor posmastectomía, síndrome de dolor postoracotomía, dolor del muñón); osteodinia y artralgia (artrosis), dolor por movimientos repetitivos, odontalgia, dolor oncológico, dolor miofascial (lesión muscular, fibromialgia); dolor perioperatorio (cirugía general, ginecológica), dolor crónico, dismenorrea, así como dolor asociado a angina de pecho y dolor inflamatorio de diferentes orígenes (por ejemplo, artrosis, artritis reumatoide, enfermedad reumática, tenosinovitis y gota), cefalea, jaqueca y cefalea en brotes, cefalea, hiperalgesia primaria, hiperalgesia secundaria, alodinia primaria, alodinia secundaria u otro dolor provocado por la sensibilización central.
Los compuestos de la invención, N-óxidos de los mismos y sales farmacéuticamente aceptables de los anteriores, pueden utilizarse para disminuir la tolerancia y/o dependencia al tratamiento del dolor con opioides, y para el tratamiento del síndrome de abstinencia, p. ej., de alcohol, opioides y cocaína.
Formulaciones
Los compuestos de la invención pueden administrarse por vía oral. La administración oral puede implicar la deglución, de modo que el compuesto entre en el tracto gastrointestinal o, puede emplearse administración bucal o sublingual, mediante las que el compuesto entra al torrente sanguíneo directamente desde la boca.
En otra realización, los compuestos de la invención también pueden administrarse directamente en el torrente sanguíneo, en el músculo o en un órgano interno. Los medios adecuados para la administración parenteral incluyen intravenosa, intraarterial, intraperitoneal, intratecal, intraventricular, intrauretral, intraesternal, intracraneal, intramuscular y subcutánea. Los dispositivos adecuados para administración parenteral incluyen inyectores de aguja (que incluyen microagujas), inyectores sin aguja y técnicas de infusión.
En otra realización, los compuestos de la invención también pueden formularse de modo que la administración tópica a la piel o mucosa (es decir, por vía dérmica o transdérmica) conduzca a la absorción sistémica del compuesto. En otra realización, los compuestos de la invención también pueden formularse de modo que la administración intranasal 0 por inhalación conduzca a la absorción sistémica del compuesto. En otra realización, los compuestos de la invención pueden formularse de modo que la administración por vía rectal o vaginal conduzca a la absorción sistémica del compuesto.
El régimen de dosificación para los compuestos y/o las composiciones que contienen los compuestos se basa en diversos factores, que incluyen el tipo, la edad, el peso, el sexo y la patología del paciente; la gravedad de la afección; la vía de administración; y la actividad del compuesto particular empleado. Por tanto, el régimen de dosificación puede variar ampliamente. Niveles de dosificación en el orden de aproximadamente 0,01 mg a aproximadamente 100 mg por kilogramo de peso corporal al día son útiles en el tratamiento de las afecciones indicadas anteriormente. En una realización, normalmente la dosis diaria total de un compuesto de la invención (administrada en dosis únicas o divididas) es de aproximadamente 0,01 a aproximadamente 100 mg/kg. En otra realización, la dosis diaria total del compuesto de la invención es de aproximadamente 0,1 a aproximadamente 50 mg/kg y, en otra realización, de aproximadamente 0,5 a aproximadamente 30 mg/kg (es decir, mg de compuesto de la invención por kg de peso corporal). En una realización, la dosificación es de 0,01 a 10 mg/kg/día. En otra realización, la dosificación es de 0,1 a 1,0 mg/kg/día. Las composiciones en unidades de dosificación pueden contener dichas cantidades o submúltiplos de las mismas para componer la dosis diaria. En muchos casos, la administración del compuesto se repetirá una pluralidad de veces al día (habitualmente no más de 4 veces). Normalmente se pueden usar múltiples dosis diarias para aumentar la dosis diaria total, si se desea.
Para la administración oral, las composiciones pueden proporcionarse en forma de comprimidos que contengan 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 75,0, 100, 125, 150, 175, 200, 250 y 500 miligramos del principio activo para el ajuste sintomático de la dosificación del paciente. Habitualmente un medicamento contiene de aproximadamente 0,01 mg a aproximadamente 500 mg del principio activo o, en otra realización, de aproximadamente 1 mg a aproximadamente 100 mg de principio activo. Por vía intravenosa, las dosis pueden variar de aproximadamente 0,1 a aproximadamente 10 mg/kg/minuto durante una infusión a velocidad constante.
Los sujetos adecuados de acuerdo con la presente invención incluyen sujetos mamíferos. Los mamíferos de acuerdo con la presente invención incluyen, pero no se limitan a, caninos, felinos, bovinos, caprinos, equinos, ovinos, porcinos, roedores, lagomorfos, primates y similares e incluyen mamíferos en el útero. En una realización, los seres humanos son sujetos adecuados. Los sujetos humanos pueden ser de cualquier género y pueden estar en cualquier fase del desarrollo.
En otra realización, la invención comprende el uso de uno o más compuestos de la invención para la preparación de un medicamento para el tratamiento de las afecciones enumeradas en el presente documento.
Para el tratamiento de las afecciones mencionadas anteriormente, los compuestos de la invención se pueden administrar en forma de un compuesto per se. Como alternativa, las sales farmacéuticamente aceptables son adecuadas para aplicaciones médicas debido a su mayor solubilidad acuosa con respecto al compuesto precursor.
En otra realización, la presente invención comprende las composiciones farmacéuticas. Dichas composiciones farmacéuticas comprenden un compuesto de la invención presentado con un vehículo farmacéuticamente aceptable. El vehículo puede ser un sólido, un líquido o ambos, y puede estar formulado con el compuesto en forma de una composición de dosis unitaria, por ejemplo, un comprimido, que puede contener del 0,05 % al 95 % en peso de los compuestos activos. Un compuesto de la invención puede acoplarse con polímeros adecuados en forma de vehículos farmacológicos direccionables. Además, pueden estar presentes otras sustancias farmacológicamente activas.
Los compuestos de la invención pueden administrarse mediante cualquier vía adecuada, preferentemente en forma de una composición farmacéutica adaptada a dicha vía y en una dosis eficaz para el tratamiento previsto. Los compuestos y composiciones activas, por ejemplo, pueden administrarse por vía oral, rectal, parenteral o tópica (por ejemplo, intranasal u oftálmica).
La administración oral de una dosis sólida puede, por ejemplo, presentarse en unidades discretas, tales como cápsulas duras o blandas, píldoras, obleas, pastillas para chupar o comprimidos, conteniendo cada uno una cantidad predeterminada de al menos un compuesto de la presente invención. En otra realización, la administración oral puede estar en forma de polvo o gránulos. En otra realización, la forma de dosis oral es sublingual, tal como, por ejemplo, una pastilla para chupar. En dichas formas farmacéuticas sólidas, los compuestos de la presente invención se combinan de forma ordinaria con uno o más adyuvantes. Dichas cápsulas o comprimidos pueden contener una formulación de liberación controlada. En el caso de cápsulas, comprimidos y píldoras, las formas farmacéuticas también pueden comprender agentes tamponantes o pueden prepararse con recubrimientos entéricos.
En otra realización, la administración oral puede estar en forma de una dosis líquida. Las formas farmacéuticas líquidas para administración oral incluyen, por ejemplo, emulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables que contienen diluyentes inertes usados comúnmente en la materia (por ejemplo, agua). Dichas composiciones también pueden comprender adyuvantes, tales como agentes humectantes, emulsionantes, de suspensión, saborizantes (por ejemplo, edulcorantes) y/o aromatizantes.
En otra realización, la presente invención comprende una forma de dosis parenteral. La "administración parenteral" incluye, por ejemplo, inyecciones subcutáneas, inyecciones intravenosas, inyecciones intraperitoneales, inyecciones intramusculares, inyecciones intraesternales e infusiones. Las preparaciones inyectables (es decir, suspensiones estériles inyectables acuosas u oleaginosas) pueden formularse de acuerdo con la técnica conocida usando agentes de dispersión, humectantes y/o de suspensión adecuados, e incluyen formulaciones de depósito.
En otra realización, la presente invención comprende una forma de dosis tópica. La "administración tópica" incluye, por ejemplo, la administración transdérmica, tal como a través de parches transdérmicos o dispositivos de iontoforesis, la administración intraocular o la administración intranasal o por inhalación. Las composiciones para la administración tópica también incluyen, por ejemplo, geles tópicos, pulverizaciones, pomadas y cremas. Una formulación tópica puede incluir un compuesto que potencie la absorción o penetración del principio activo a través de la piel u otras zonas afectadas. Cuando se administran los compuestos de la presente invención mediante un dispositivo transdérmico, la administración se llevará a cabo utilizando un parche tanto de tipo depósito como de tipo membrana porosa, o de diversas matrices sólidas. Las formulaciones habituales para este propósito incluyen geles, hidrogeles, lociones, soluciones, cremas, pomadas, polvos finos, apósitos, espumas, películas, parches cutáneos, sellos, implantes, esponjas, fibras, vendajes y microemulsiones. También pueden usarse liposomas. Los vehículos habituales incluyen alcohol, agua, aceite mineral, vaselina líquida, vaselina blanca, glicerina, polietilenglicol y propilenglicol. Pueden incorporarse potenciadores de la penetración - véase, por ejemplo, Finnin y Morgan, J. Pharm. Sci., 88 (10), 955-958 (1999).
Las formulaciones adecuadas para administración tópica en el ojo incluyen, por ejemplo, gotas oculares en donde el compuesto de esta invención se disuelve o suspende en un vehículo adecuado. Una formulación habitual adecuada para administración ocular o aural puede estar en la forma de gotas de una suspensión o solución micronizada en solución salina estéril isotónica, de pH ajustado. Otras formulaciones adecuadas para administración ocular y aural incluyen pomadas, implantes biodegradables (por ejemplo, esponjas de gel absorbible, colágeno) y no biodegradables (por ejemplo, de silicona), sellos, lentes y sistemas particulados o vesiculares, tales como niosomas o liposomas. Un polímero tal como ácido poliacrílico reticulado, alcohol polivinílico, ácido hialurónico, un polímero celulósico, por ejemplo, hidroxipropilmetilcelulosa, hidroxietilcelulosa o metilcelulosa, o un polímero de heteropolisacárido, por ejemplo, goma gelano, puede incorporarse junto con un conservante, tal como cloruro de benzalconio. Dichas formulaciones también pueden administrarse mediante iontoforesis.
Para la administración intranasal o la administración por inhalación, los compuestos activos de la invención se administran de forma conveniente en forma de una solución o suspensión a partir de un recipiente para pulverización de tipo bomba que el paciente presiona o bombea o en forma de una presentación en aerosol a partir de un recipiente
presurizado o un nebulizador, con el uso de un propulsor adecuado. Las formulaciones adecuadas para la administración intranasal se administran normalmente en forma de un polvo seco (tanto solo; como en una mezcla, por ejemplo, en una combinación seca con lactosa; o como una partícula de componentes mixtos, por ejemplo, mezclado con fosfolípidos, tales como fosfatidilcolina) a partir de un inhalador de polvo seco o en forma de una pulverización en aerosol a partir de un recipiente presurizado, bomba, pulverización, atomizador (preferentemente un atomizador que usa la electrohidrodinámica para producir una niebla fina) o nebulizador, con o sin el uso de un propulsor adecuado, tal como 1,1,1,2-tetrafluoroetano o 1,1,1,2,3,3,3-heptafluoropropano. Para uso intranasal, el polvo puede comprender un agente bioadhesivo, por ejemplo, quitosano o ciclodextrina.
En otra realización, la presente invención comprende una forma de dosis rectal. Dicha forma de dosis rectal puede estar en forma de, por ejemplo, un supositorio. La manteca de cacao es una base de supositorio tradicional, pero pueden usarse varias alternativas según sea apropiado.
Además, pueden utilizarse otros materiales transportadores y modos de administración conocidos en la técnica farmacéutica. Las composiciones farmacéuticas de la invención se pueden preparar mediante cualquier técnica farmacéutica bien conocida, tales como una formulación y procedimientos de administración eficaces. Las consideraciones anteriores con respecto a las formulaciones y procedimientos de administración eficaces se conocen bien en la técnica y se describen en libros de texto convencionales. Las formulaciones de fármacos se analizan en, por ejemplo, Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pensilvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Nueva York, N. Y., 1980; y Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3a ed.), American Pharmaceutical Association, Washington, 1999.
Los compuestos de la presente invención se pueden usar, solos en combinación con otros agentes terapéuticos, en el tratamiento de diversas afecciones o patologías. El compuesto o compuestos de la presente invención y otro agente o agentes terapéuticos pueden administrarse de forma simultánea (ya sea en la misma forma farmacéutica o en formas farmacéuticas distintas) o secuencial. Un agente terapéutico a modo de ejemplo puede ser, por ejemplo, un agonista del receptor metabotrópico de glutamato.
La administración de dos o más compuestos "en combinación" significa que los dos compuestos se administran de manera suficientemente cercana en el tiempo como para que la presencia de uno modifique los efectos biológicos del otro. Los dos o más compuestos pueden administrarse de forma simultánea, coincidente o secuencial. Además, la administración simultánea puede llevarse a cabo mezclando los compuestos antes de la administración o administrando los compuestos en el mismo punto en el tiempo, pero en distintos sitios anatómicos o utilizando distintas vías de administración.
Las expresiones "administración coincidente", "coadministración", "administración simultánea" y "administrado simultáneamente" significan que los compuestos se administran en combinación.
La presente invención incluye el uso de una combinación de un compuesto activador de M4 de la presente invención y uno o más de otros principios farmacéuticamente activos. Si se administra una combinación de principios activos, estos se pueden administrar de forma secuencial o simultánea, en formas farmacéuticas separadas o combinados en una única forma farmacéutica. Por consiguiente, la presente invención también incluye composiciones farmacéuticas que comprenden una cantidad de: (a) un primer agente que comprende un compuesto de la presente invención o una sal farmacéuticamente aceptable del compuesto; (b) un segundo principio farmacéuticamente activo; y (c) un portador, vehículo o diluyente farmacéuticamente aceptable.
Pueden seleccionarse diversos agentes farmacéuticamente activos para su uso junto con los compuestos de la presente invención, dependiendo de la enfermedad, el trastorno o la afección a tratar. Los principios farmacéuticamente activos que pueden usarse en combinación con las composiciones de la presente invención incluyen, sin limitación:
(i) inhibidores de la acetilcolinesterasa, tales como clorhidrato de donepecilo (ARICEPT, MEMAC), salicilato de fisostigmina (ANTILIRIO), sulfato de fisostigmina (ESERINE), metrifonato, neostigmina, ganstigmina, piridostigmina (MESTINON), ambenonio (MYTELASE), demarcario, Debio 9902 (también conocido como ZT-1; Debiopharm), rivastigmina (EXELON), ladostigil, NP-0361, bromhidrato de galantamina (RAZADYNE, RIMINYL, NIVALIN), tacrina (COGNEX), tolserina, maleato de velnacrino, memoquin, hupercina A (HUP-A; NeuroHitech), fenerina, edrofonio (ENLON, TENSILON) e INM-176;
(ii) amiloide p (o fragmentos del mismo), tal como Ap i-15 conjugado con un panepítopo de unión a HLA DR (PADRE, forma siglada de pan HLA DR-binding epitope), ACC-001 (Elan/Wyeth), ACI-01, a C i-24, AN-1792, Affitope AD-01, CAD106 y V-950;
(iii) anticuerpos contra amiloide p (o fragmentos del mismo), tales como ponezumab, solanezumab, bapineuzumab (también conocido como a AB-001), AAB-002 (Wyeth/Elan), ACI-01-Ab7, BAN-2401, Ig intravenosa (GAMMAGARD), LY2062430 (m266 humanizado; Lilly), R1450 (Roche), ACU-5A5, huC091 y los desvelados en las publicaciones de patente internacional N.° WO04/032868, WO05/025616, WO06/036291, WO06/069081, WO06/118959, en las Publicaciones de patente de Estados Unidos N.° US2003/0073655, US2004/0192898, US2005/0048049, US2005/0019328, en las publicaciones de patente europea N.° EP0994728 y 1257584 y en la
patente de Estados Unidos N.° 5.750.349;
(iv) agentes reductores o inhibidores de amiloide (incluyendo los que reducen la producción, acumulación y fibrilización de amiloide) tales como dimebón, davunetida, eprodisato, leuprolida, SK-PC-B70M, celecoxib, lovastatina, anapsos, oxiracetam, pramiracetam, vareniclina, nicergolina, colostrinina, bisnorcimserina (también conocida como BNC), NIC5-15 (Humanetics), E-2012 (Eisai), pioglitazona, clioquinol (también conocido como PBT1), PBT2 (Prana Biotechnology), flurbiprofeno (ANSAID, FROBEN) y sus enantiómeros R tarenflurbil (FLURIZAN), nitroflurbiprofeno, fenoprofeno (FENOPRON, NALFON), ibuprofeno (ADVIL, MOTRIN, NUROFEN), lisinato de ibuprofeno, ácido meclofenámico, meclofenamato de sodio (MECLOMEN), indometacina (Indocin), diclofenaco de sodio (VOLTAREN), diclofenaco de potasio, sulindaco (Clinoril), sulfuro de sulindaco, diflunisal (DOLOBID), naproxeno (NAPROSYN), naproxeno de sodio (ANAPROX, ALEVE), ARC031 (Archer Pharmaceuticals), CAD-106 (Cytos), LY450139 (Lilly), enzima que degrada la insulina (también conocida como insulisina), el extracto de Gingko biloba EGb-761 (ROKAN, TEBONIN), tramiprosato (CEREBRIL, ALZHEMED), eprodisato (FIBRILLEX, KIACTA), compuesto W [ácido 3,5-bis(4-nitrofenoxi)benzoico], NGX-96992, neprilisina (también conocida como endopeptidasa neutra (NEP)), esciloinositol (también conocido como escilitol), atorvastatina (Lipitor), simvastatina (Zocor), KLVFF-(EEX)3, SKF-74652, mesilato de ibutamoreno, inhibidores de BACE tales como ASP-1702, SCH-745966, JNJ-715754, AMG-0683, AZ-12304146, BMS-782450, GSK-188909, NB-533, E2609 y TTP-854; moduladores de gamma secretasa tales como ELND-007; e inhibidores de RAGE (receptor para productos finales de glucación avanzada), tales como TTP488 (Transtech) y TTP4000 (Transtech), y los desvelados en la patente de Estados Unidos N.° 7.285.293, incluyendo PTI-777;
(v) agonistas de receptores adrenérgicos alfa, tales como guanfacina (INTUNIV, TENEX), clonidina (CATAPRES), metaraminol (ARAMINE), metildopa (ALDOMET), DOPAMET, NOVOMEDOPA), tizanidina (ZANAFLEX), fenilefrina (también conocida como neosinefrina), metoxamina, cirazolina, guanfacina (INTUNIV), lofexidina, xilazina, modafinilo (PROVIGIL), adrafinilo y armodafinilo (NUVIGIL);
(vi) agentes bloqueantes de receptores adrenérgicos beta (bloqueantes beta), tales como carteolol, esmolol (BREVIBLOC), labetalol (NORMODYNE, TRANDATE), oxprenolol (LARACOR, TRASACOR), pindolol (VISKEN), propanolol (INDERAL), sotalol (BETAPACE, SOTALEX, SOTACOR), timolol (BLOCADREN, TIMOPTIC), acebutolol (SECTRAL, PRENT), nadolol (CORGARD), tartrato de metoprolol (LOPRESSOR), succinato de metoprolol (TOPROL-XL), atenolol (TENORMIN), butoxamina y SR 59230A (Sanofi);
(vii) anticolinérgicos, tales como amitriptilina (ELAVIL, ENDEP), butriptilina, mesilato de benzotropina (c Og ENTIN), trihexifenidilo (ARTANE), difenhidramina (BENADRYL), orfenadrina (NORFLEX), hiosciamina, atropina (ATROPEN), escopolamina (TRANSDERM-s Co P), metilbromuro de escopolamina (PARMINE), dicicloverina (BENTYL, BYCLOMINE, DIBENT, DILOMINE), tolterodina (DETROL), oxibutinina (DITROPAN, LYRINEL XL, OXYTROL), bromuro de pentienato, propantelina (PRO-BANTHINE), ciclicina, clorhidrato de imipramina (TOFRANIL), maleato de imipramina (SURMONTIL), lofepramina, desipramina (NORPRAMIN), doxepina (SINEQUAN, ZONALON), trimipramina (SURMONTIL) y glicopirrolato (ROBINUL);
(viii) anticonvulsivos, tales como carbamacepina (TEGRETOL, CARBATROL), oxcarbacepina (TRILEPTAL), fenitoína de sodio (PHENYTEK), fosfenitoína (CEREBYX, PRODILANTIN), divalproex de sodio (DEPAKOTE), gabapentina (NEu Ro NTIN), pregabalina (LYRICA), topirimato (TOPAMAX), ácido valproico (DEPAKENE), valproato de sodio (DEPACON), 1-bencil-5-bromouracilo, progabida, beclamida, zonisamida (TRERIEF, EXCEGRAN), CP-465022, retigabina, talampanel y primidona (My SOLINE);
(ix) antipsicóticos, tales como lurasidona (LATUDA, también conocida como SM-13496; Dainippon Sumitomo), aripiprazol (ABILIFY), clorpromacina (THo Ra ZINE), haloperidol (HALDOL), iloperidona (FANAPt A), decanoato de flupentixol (DEPIx Ol , FLUANXOL), reserpina (SEr PlAN), pimocida (ORAP), decanoato de flufenazina, clorhidrato de flufenazina, proclorperacina (COMPRO), asenapina (SAPHRIS), loxapina (LOXITANE), molindona (MOBAN), perfenazina, tioridazina, tiotixina, trifluoperacina (STELAZINE), ramelteón, clozapina (CLOZARIL), norclozapina (ACP-104), risperidona (RISPERDAL), paliperidona (INVEGA), melperona, olanzapina (ZYPREXA), quetiapina (SEROQUEL), talnetant, amisulprida, ciprasidona (GEODON), blonanserina (LONASEN) y ACP-103 (Acadia Pharmaceuticals);
(x) bloqueadores de los canales de calcio tales como lomerizina, ziconotida, nilvadipina (ESCOR, NIVADIL), diperdipina, amlodipina (NORVASC, ISTIN, AMLODIN), felodipina (PLENDIL), nicardipina (CARDENO), nifedipina (ADALAT, PROCARDiA), MEM 1003 y su compuesto precursor nimodipina (NIMOTOp ), nisoldipina (SUlA r ), nitrendipina, lacidipina (LACIPIL, MOTENS), lercanidipina (ZANIDIP), lifaricina, diltiazem (CARDIZEM), verapamilo (CALAN, VERELAN), AR-R 18565 (AstraZeneca) y enecadina;
(xi) inhibidores de la catecol O-metiltransferasa (COMT), tales como nitecapona, tolcapona (TASMAR), entacapona (COMTAN), y tropolona;
(xii) estimulantes del sistema nervioso central, tales como atomoxetina, reboxetina, yohimbina, cafeína, fenmetrazina, fendimetrazina, pemolina, fencamfamina (GLUCOENERGAN, REACTIVAN), fenetilina (CAPTAGON), pipradol (MERETRAN), deanol (también conocido como dimetilaminoetanol), metilfenidato (DAYTRANA), clorhidrato de metilfenidato (RITALIN), dexmetilfenidato (FOCALIN), anfetamina (sola o junto con otros estimulantes del SNC, p. ej., ADDERALL (aspartato de anfetamina, sulfato de anfetamina, sacarato de dextroanfetamina y sulfato de dextroanfetamina)), sulfato de dextroanfetamina (DEXEDRINE, DEXTROSTAT), metanfetamina (d Es OXYN), lisdexamfetamina (v Yv ANSE) y benzfetamina (DIDREX);
(xiii) corticoesteroides, tales como prednisona (STERAPRED, DELTASONE), prednisolona (PRELONE), acetato de prednisolona (OMNIPRED, Pr Ed MILD, PRED FORTE), fosfato de prednisolona sódica (ORAPr Ed ODT), metilprednisolona (MEDROL); acetato de metilprednisolona (DEPO-MEDROL) y succinato de metilprednisolona de sodio (A-METHAPRED, SOLU-MEDROL);
(xiv) agonistas del receptor de dopamina, tales como apomorfina (APOKYN), bromocriptina (PARLODEL), cabergolina (DOSTINEX), dihidrexidina, dihidroergocriptina, fenoldopam (CORLOPAM), lisurida (d OPERGIn ), espergolida de tergurida (PERMAX), piribedil (TRIVASTAL, TRASTAL), pramipexol (MIrA p EX), quinpirol, ropinirol (REQUIP), rotigotina (NEUPRO), Sk F-82958 (GlaxoSmithKline), cariprazina, pardoprunox y sarizotan;
(xv) antagonistas del receptor de dopamina, tales como clorpromacina, flufenazina, haloperidol, loxapina, risperidona, tioridazina, tiotixeno, trifluoperazina, tetrabenacina (Nitoman, Xenazina, 7-hidroxamoxapina, droperidol (IN-APSINE, DRIDOL, DROPLETAN), domperidona (MOTILIO), L-741742, L-745870, racloprida, SB-277011A, SCH-23390, ecopipam, SKF-83566 y metoclopramida (REGLAN);
(xvi) inhibidores de la recaptación de dopamina tales como bupropión, safinamida, maleato de nomifensina (MERITAL), vanoxerina (también conocida como GBR-12909) y su éster de decanoato DBL-583, y amineptina; (xvii) agonistas del receptor del ácido gamma aminobutírico (GABA), tales como baclofeno (LIORESAL, KEMSTRO), siclofeno, pentobarbital (NEMBUTAL), progabida (Ga Br ENE) y clometiazol;
(xviii) antagonistas de histamina 3 (H3) tales como ciproxifano, tiprolisant, S-38093, irdabisant, pitolisant, GSK-239512, GSK-207040, JNJ-5207852, JNJ-17216498, HPP-404, SAR-110894, trans-N-etil-3-fluoro-3-[3-fluoro-4-(pirrolidin-1-ilmetil)fenil]-ciclobutanocarboxamida (PF-3654746 y los desvelados en las publicaciones de patente de Estados Unidos N.° US2005-0043354, US2005-0267095, US2005-0256135, US2008-0096955, US2007-1079175 y US2008-0176925; en las publicaciones de patente internacional N.° WO2006/136924, WO2007/063385, WO2007/069053, WO2007/088450, WO2007/099423, WO2007/105053, WO2007/138431 y WO2007/088462; y en la patente de Estados Unidos N.° 7.115.600);
(xix) inmunomoduladores tales como acetato de glatiramer (también conocido como copolímero-1; COPAXONE), MBP-8298 (péptido sintético de la proteína básica de mielina), fumarato de dimetilo, fingolimod (también conocido como FTY720), roquinimex (LINo M iDE), laquinimod (también conocido como ABR-215062 y sA iK-MS), ABT-874 (anticuerpo humano anti-IL-12; Abbott), rituximab (RITUXAN), alemtuzumab (CAMPATH), daclizumab (Ze NAPAX) y natalizumab (TYSABRI);
(xx) inmunosupresores tales como metotrexato (TREXALL, RHEUMATREX), mitoxantrona (NOVANTRONE), micofenolato de mofetilo (CELLCEPT), micofenolato de sodio (MYFORTIC), azatioprina (Az As AN, IMURAn ), mercaptopurina (PURI-NETHOL), ciclofosfamida (NEOSAR, CYTOXAN), clorambucilo (LEUKERAN), cladribina (LEUSTATIN, MYLINAX), alfa-fetoproteína, etanercept (ENBREL) y 4-(benciloxi)-5-[(5-undecil-2H-pirrol-2-ilideno)metil]-1H,1'H-2,2'-bipirrol (también conocido como PNU-156804);
(xxi) interferones, incluyendo interferón beta-1a (AVONEX, REBIF) e interferón beta-1b (BETASERON, BETAFER-ON);
(xxii) levodopa (o su éster de metilo o de etilo), sola o junto con un inhibidor de la DOPA descarboxilasa (p. ej., carbidopa (SINEMET, CARBILEV, PARCOPA), benseracida (MADOPAR), a-metildopa, monoflurometildopa, difluorometildopa, brocresina o m-hidroxibencilhidrazina);
(xxiii) antagonistas del receptor de N-metil-D-aspartato (NMDA), tales como memantina (NAMENDA, AXURA, EBIXA), amantadina (SYMMETREL), acamprosato (CAm PrAL), besonprodilo, ketamina (KETALAR), delucemina, dexanabinol, dexefaroxano, dextrometorfano, dextrorfano, traxoprodilo, CP-283097, himantano, idantadol, ipenoxazona, L-701252 (Merck), lancicemina, levorfanol (DROMOr An ), LY-233536 y LY-235959 (ambos de Lilly), metadona, (DOLOPHINE), neramexano, percinfotel, fenciclidina, tianeptina (STABLON), dizocilpina (también conocida como MK-801), EAB-318 (Wyeth), ibogaína, voacangina, tiletamina, riluzol (RILUTEk ), aptiganel (CERES0TAT), gavestinel y remacimida;
(xxiv) inhibidores de la monoaminooxidasa (MAO), tales como selegilina (EMSAM), clorhidrato de selegilina (I-deprenilo, EL-DEPRYL, ZELAPAR), dimetilselegileno, brofaromina, fenelcina (NARDIL), tranilcipromina (pAr NATE), moclobemida (AURoRlX, MANERIX), befloxatona, safinamida, isocarboxacida (Ma Rp LAN), nialamida (NIAMID), rasagilina (AZILECT), iproniacida (MARSILID, IPROZID, IPRONID), CHF-3381 (Chiesi Farmaceutici), iproclocida, toloxatona (HUMORYL, PERENUM), bifemelano, desoxipeganina, harmina (también conocida como telepatina o banasterina), harmalina, linezolida (ZYVOX, ZYVOXlD) y pargilina (EUDATIN, SUPIRDYL)];
(xxv) agonistas del receptor muscarínico (particularmente del subtipo M1), tales como cevimelina, levetiracetam, cloruro de betanecol (d Uv OID, URECHOLINE), itamelina, pilocarpina (SALAGEN), NGX267, arecolina, L-687306 (Merck), L-689660 (Merck), yoduro de furtretonio (FURa Mo N, Fu RANOL), bencenosulfonato de furtretonio, ptoluenosulfonato de furtretonio, McN-A-343, oxotremorina, sabcomelina, AC-90222 (Acadia Pharmaceuticals) y carbacol (CARBASTAT, MIOSTAT, CARBOPTIC);
(xxvi) fármacos neuroprotectores tales como bosutinib, condoliasa, airmoclomol, lamotrigina, perampanel, aniracetam, minaprime, riluzol, N-hidroxi-1,2,4,9-tetrahidro-3H-carbazol-3-imina, desmoteplasa, anatibant, astaxantina, neuropéptido NAP (por ejemplo, AL-108 y AL-208; ambos de Allon Therapeutics), neurostrol, perampenel, isproniclina, bis(4-p-D-glucopiranosiloxibencil)-2-p-D-glucopiranosil-2-isobutiltartrato (también conocido como dacticlorina B o DHB), formobactina, xaliproden (XAPRILA), lactacistina, clorhidrato de dimebolina (DIMEBON), disufenton (CEROVIVE), ácido arúndico (ONO-2506, PROGLlA, CEREACT), citicolina (también conocida como citidina 5'-difosfocolina), edaravona (RADlCUT), AEOL-10113 y AEOL-10150 (ambos de Aeolus Pharmaceuticals), AGY-94806 (también conocido como SA-450 y Msc-1), factor estimulante de colonias de granulocitos (también conocido como AX-200), BAY-38-7271 (también conocido como KN-387271); Bayer AG), ancrod (VIPRINEX, ARWIN), DP-b99 (D-Pharm Ltd), HF-0220 (17-p-hidroxiepiandrosterona; Newron Pharmaceuticals), HF-0420 (también conocido como oligotropina), piridoxal 5'-fosfato (también conocido como MC-1), microplasmina, S-18986, piclozotan, NP031112, tacrolimus, L-seril-L-metionil-L-alanil-L-lisil-L-glutamil-glicil-L-valina, a C-184897 (Acadia Pharmaceuticals), ADNF-14 (National Institutes of Health), estilbazulenil nitrona, SUN
N8075 (Daiichi Suntory Biomedical Research) y zonampanel;
(xxvii) agonistas del receptor nicotínico, tales como epibatidina, bupropión, CP-601927, vareniclina, ABT-089 (Abbott), ABT-594, AZD-0328 (AstraZeneca), EVP-6124, R3487 (también conocido como MEM3454); Roche/Memory Pharmaceuticals), R4996 (también conocido como MEM63908); Roche/Memory Pharmaceuticals), TC-4959 y TC-5619 (ambos Targacept) y RJR-2403;
(xxviii) inhibidores de la recaptación de norepinefrina (noradrenalina), tales como atomoxetina (STRATTERA), doxepina (APONAL, ADAPIN, SINEQUAN), nortriptilina (AVENTYL, PAMELOR, NORTRILEN), amoxapina (ASENDIN, DEMOLOX, MOXIDIL), reboxetina (EDRONAX, VESTRA), viloxazina (VIVALAN), maprotilina (DEPRILEPT, LUDIOMIL, PSYMION), bupropión (WELLBUTRIN) y radaxafina;
(xxix) inhibidores de la fosfodiesterasa (PDE), incluyendo pero sin limitación, (a) inhibidores de PDE1 (p. ej., vinpocetina (CAVINTON, CERACTIN, INTe Le CTOL) y los desvelados en la patente de Estados Unidos N.° 6.235.742, (b) inhibidores de PDE2 (p. ej., eritro-9-(2-hidroxi-3-nonil)adenina (EHNA), BAY 60-7550 y los descritos en la patente de Estados Unidos N.° 6.174.884), (c) inhibidores de PDE3 (p. ej., anagrelida, cilostazol, milrinona, olprinona, parogrelil y pimobendan), (d) inhibidores de PDE4 (p. ej., apremilast, ibudilastroflumilast, rolipram, Ro 20-1724, ibudilast (KETAS), piclamilast (también conocido como RP73401), CDP840, cilomilast (ARIFLO), roflumilast, tofimilast, oglemilast (también conocido como GRC 3886), tetomilast (también conocido como OpC-6535), lirimifast, teofilina (UNIPHYL, THEOLAIR), arofilina (también conocida como LAS-31025), doxofilina, RPR-122818, o mesembrina) y (e) inhibidores de p De 5 (por ejemplo, sildenafilo (VIAGRA, REVATIo ), tadalafilo (CIALIS), vardenafilo (LEVITRA, VIVANZA), udenafilo, avanafilo, dipiridamol (PERSANTINE), E-4010, E-4021, E-8010, zaprinast, yodenafilo, mirodenafilo, DA-8159 y los desvelados en las solicitudes de patente internacional WO2002/020521, WO2005/049616, WO2006/120552, WO2006/126081, WO2006/126082, WO2006/126083 y WO2007/122466), (f) inhibidores de PDE7; (g) inhibidores de PDE8; (h) inhibidores de PDE9 (p. ej., BAY 73-6691 (Bayer AG) y los desvelados en las publicaciones de patente de Estados Unidos N.° US2003/0195205, US2004/0220186, US2006/0111372, US2006/0106035 y USSN 12/118.062 (presentada el 9 de mayo de 2008)), (i) inhibidores de PDE10 tales como 2-({4-[1-metil-4-(piridin-4-il)-1H-pirazol-3-il]fenoxi}metil)quinolin-3(4H)-ona y SCH-1518291; y (j) inhibidores de PDE11;
(xxx) quinolinas, tales como quinina (incluido su clorhidrato, diclorhidrato, sulfato, sales de bisulfato y gluconato), cloroquina, sontoquina, hidroxicloroquina (PLAQUENIL), mefloquina (LARIAM) y amodiaquina (CAMOQUIN, FLAVOQUINE);
(xxxi) inhibidores de p-secretasa, tales como ASP-1702, SCH-745966, JNJ-715754, AMG-0683, AZ-12304146, BMS-782450, GSK-188909, NB-533, LY-2886721, E-2609, HPP-854, tartrato de (+)-fenserina (POSIPHEN), LSN-2434074 (también conocido como LY-2434074), KMI-574, SCH-745966, Ac-rER (N2-acetil-D-arginil-L-arginina), loxistatina (también conocida como E64d) y CA074Me;
(xxxii) inhibidores y moduladores de Y-secretasa, tales como BMS-708163 (Avagacest), W020060430064 (Merck), DSP8658 (Dainippon), ITI-009, L-685458 (Merck), ELAN-G, ELAN-Z, 4-cloro-A/-[(2S)-3-et¡l-1-hidrox¡pentan-2-il]bencenosulfonamida;
(xxxiii) antagonistas del receptor de serotonina (5-hidroxitriptamina) 1A (5-HT1a), tales como espiperona, levopindolol, BMY 7378, NAD-299, S-(-)-UH-301, NAN 190, lecozotan;
(xxxiv) agonistas del receptor de serotonina (5-hidroxitriptamina) 2C (5-HT2c), tales como vabicaserina y zicronapina;
(xxxv) agonistas del receptor de serotonina (5-hidroxitriptamina) 4 (5-HT4), tales como PRX-03140 (Epix);
(xxxvi) antagonistas del receptor de serotonina (5-hidroxitriptamina) 6 (5-HT6), tales como A-964324, AVI-101, Av N-211, mianserina (TOr Vo L, BOLVIDON, NORVAL), metiotepina (también conocida como metitepina), ritanserina, ALX-1161, ALX-1175, MS-245, LY-483518 (también conocido como SGS518); Lilly), MS-245, Ro 04 6790, Ro 43-68544, Ro 63-0563, Ro 65-7199, Ro 65-7674, SB-399885, SB-214111, SB-258510, SB-271046, SB-357134, SB-699929, SB-271046, SB-742457 (GlaxoSmithKline), Lu AE58054 (Lundbeck A/S) y PRX-07034 (Epix); (xxxvii) inhibidores de la recaptación de serotonina (5-HT) tales como alaproclato, citalopram (CELEXA, CIPRAMIL), escitalopram (LEXAPRO, CIPRALEX), clomipramina (ANAFRANIL), duloxetina (CYMBALTA), femoxetina (MALEXlL), fenfluramina (PONDIMIN), norfenfluramina, fluoxetina (PROZAC), fluvoxamina (LUVOX), indalpina, milnacipran (IXEL), paroxetina (PAXIL, SEROXAT), sertralina (ZOLOFT, LUSTRAL), trazodona (DESYREL, MOLlPAXIN), venlafaxina (EFFEXOR), zimelidina (NORMUD, ZELMID), bicifadina, desvenlafaxina (PRISTIQ), brasofensina, vilazodona, cariprazina, neuralstem y tesofensina;
(xxxviii) factores tróficos, tales como factor de crecimiento nervioso (NGF), factor básico de crecimiento de fibroblastos (bFGF; ERSOFERMIN), neurotrofina-3 (NT-3), cardiotrofina-1, factor neurotrófico derivado de cerebro (BDNF), neublastina, meteorina y factor neurotrófico derivado de neurogliocitos (GDNF) y agentes que estimulan la producción de factores tróficos, tales como propentofilina, idebenona, PYM50028 (COGANE; Phytopharm) y AlT-082 (NEOTROFIN);
(xxxix) inhibidores del transportador de glicina-1 tales como paliflutina, ORG-25935, JNJ-17305600 y ORG-26041; (xl) moduladores del receptor de glutamato de tipo AMPA tales como perampanel, mibampator, selurampanel, g Sk -729327, N-{(3S, 4S)-4-[4-(5-cianotiofen-2-il)fenoxi]tetrahidrofuran-3-il}propan-2-sulfonamida, y similares. (xli) inhibidores de la cinasa Jano (JAK) tales como, pero sin limitación, tofacitinib, ruxolitinib, baricitinib, CYT387, GLPG0634, lestaurtinib, pacritinib y TG101348.
(xlii) inhibidores de cinasa 4 asociada al receptor de interleucina 1 (IRAK4) tales como, pero sin limitación, PF-06650833.
La presente invención comprende además kits que son adecuados para su uso en la realización de los métodos de tratamiento descritos anteriormente. En una realización, el kit contiene una primera forma de dosificación que
comprende uno o más de los compuestos de la presente invención y un recipiente para la dosificación, en cantidades suficientes para llevar a cabo los métodos de la presente invención.
En otra realización, el kit de la presente invención comprende uno o más compuestos de la invención.
Un ejemplo de dicho kit es el denominado envase de tipo blíster. Los envases de tipo blíster son bien conocidos en la industria del envasado y se están usando extensamente para el envasado de formas farmacéuticas unitarias (comprimidos, cápsulas y similares). Los envases de tipo blíster consisten en una lámina de material relativamente rígido cubierta con una hoja de un material plástico transparente. Durante el procedimiento de envasado, se forman rebajes en la lámina de plástico. Los rebajes tienen el tamaño y la forma de los comprimidos o cápsulas que se van a envasar. A continuación, los comprimidos o cápsulas se colocan en los rebajes y la lámina de material relativamente rígido se sella contra la hoja de plástico en la cara de la hoja que es opuesta a la dirección en la que se formaron los rebajes. Como resultado, los comprimidos o las cápsulas se sellan en los rebajes entre la hoja de plástico y la lámina. En algunas realizaciones, la resistencia de la lámina es tal que los comprimidos o las cápsulas pueden extraerse del envase de tipo blíster aplicando manualmente presión sobre los rebajes, mediante la cual se forma una abertura en la lámina en el lugar de los rebajes. El comprimido o la cápsula pueden entonces extraerse a través de dicha abertura.
Puede ser conveniente proporcionar un recordatorio en el kit, por ejemplo, en forma de números próximos a los comprimidos o cápsulas, donde los números corresponden a los días de la pauta en que deben ingerirse los comprimidos o cápsulas así especificados. Otro ejemplo de este tipo de recordatorio es un calendario impreso en el cartón, por ejemplo, por ejemplo, como sigue: "Primera semana, lunes, martes, etc... Segunda semana, lunes, martes,..." etc. Otras variaciones de los recordatorios serán fácilmente evidentes. Una "dosis diaria" puede ser un solo comprimido o cápsula o varias pastillas o cápsulas a tomar en un día dado. Además, una dosis diaria del compuesto de fórmula I puede consistir en un comprimido o cápsula mientras que una dosis diaria del segundo compuesto puede consistir en varios comprimidos o cápsulas, y viceversa. El recordatorio debería reflejar esto.
En otra realización específica de la invención, se proporciona un dosificador diseñado para dispensar las dosis diarias de una en una en el orden de su uso previsto. Por ejemplo, el dosificador está equipado con un recordatorio, para facilitar aún más el cumplimiento del régimen. Un ejemplo de tal recordatorio es un contador mecánico que indica el número de dosis diarias que se han dispensado. Otro ejemplo de dicho recordatorio es una memoria de microchip alimentada por batería acoplada a una lectura en cristal líquido o una señal audible de recordatorio que, por ejemplo, lea la fecha en que se tomó la última dosis diaria y/o recuerde cuándo se debe tomar la siguiente dosis.
Como se ha indicado anteriormente, los compuestos de la presente invención se pueden usar junto con uno o más de otros agentes contra la esquizofrenia que se describen en el presente documento. Cuando se usa una terapia de combinación, el uno o más de otros agentes contra la esquizofrenia se pueden administrar de forma secuencial o simultánea con el compuesto de la invención. En una realización, el otro agente contra la esquizofrenia se administra a un mamífero (por ejemplo, un ser humano) antes de la administración del compuesto de la invención. En otra realización, el otro agente contra la esquizofrenia se administra al mamífero después de la administración del compuesto de la invención. En otra realización, el otro agente contra la esquizofrenia se administra al mamífero (por ejemplo, un ser humano) de forma simultánea a la administración del compuesto de la invención (o un N-óxido del mismo o una sal farmacéuticamente aceptable de los anteriores).
La invención también proporciona una composición farmacéutica para el tratamiento de la esquizofrenia en un mamífero, incluyendo un ser humano, que comprende una cantidad de un compuesto de la presente invención (incluido un N-óxido del mismo o una sal del compuesto o del N-óxido), como se define anteriormente (incluyendo hidratos, solvatos y polimorfos de dicho compuesto o sales farmacéuticamente aceptables de los mismos), en combinación con uno o más (por ejemplo, de uno a tres) agentes contra la esquizofrenia tales como ciprasidona, risperidona, olanzapina, quetiapina, aripiprazol, asenapina, blonanserina o iloperidona, en donde las cantidades del principio activo y la combinación, cuando se toman en conjunto, son terapéuticamente eficaces para el tratamiento de la esquizofrenia.
La invención también proporciona una composición farmacéutica para tratar una enfermedad o trastorno mediado por M4 (o asociado a M4) en un mamífero, incluyendo un ser humano, que comprende una cantidad de un compuesto de la presente invención (incluido un N-óxido del mismo o una sal del compuesto o del N-óxido), como se define anteriormente (incluyendo hidratos, solvatos y polimorfos de dicho compuesto, N-oxido o sales farmacéuticamente aceptables de los anteriores), en combinación con uno o más (por ejemplo, de uno a tres) de otros agentes para el tratar la enfermedad o trastorno mediado por M4 (o asociado a M4), en donde la cantidad de principios activos y la combinación, cuando se toman en conjunto, son terapéuticamente eficaces para el tratamiento de la enfermedad o trastorno mediado por M4 (o asociado a M4).
Se entenderá que los compuestos de la presente invención representados anteriormente (Fórmula I, fórmula Ia y fórmula Ib) no se limitan a un estereoisómero particular (por ejemplo, enantiómero o atropisómero) mostrado, sino que también incluyen todos los estereoisómeros y mezclas de los mismos.
ESQUEMAS GENERALES
Los compuestos de fórmula I, Ia, Ib, Ic y I' se pueden preparar mediante los métodos descritos a continuación, junto con los métodos de síntesis conocidos en la materia de la química orgánica o las modificaciones y transformaciones que son familiares para los expertos habituales en la materia. Los materiales de partida usados en el presente documento están disponibles en el mercado o se pueden fabricar por métodos conocidos en la materia [tales como los métodos divulgados en los libros de referencia convencionales tales como el Compendium of Organic Synthetic Methods, vol. I-XIII (publicado por Wiley-Interscience)]. Los métodos preferidos incluyen, pero no se limitan a, los descritos a continuación.
Durante cualquiera de las secuencias de síntesis siguientes, puede ser necesario o deseable proteger grupos sensibles o reactivos de cualquiera de las moléculas implicadas. Esto se puede lograr por medio de grupos protectores convencionales, tales como los descritos en T. W. Greene, Protective Groups in Organic Chemistry, John Wiley & Sons, 1981; T. W. Greene y P. G. M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1991; y T. W. Greene y P. G. M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1999.
Los compuestos de fórmula I, Ia, Ib, Ic y I' o sus sales farmacéuticamente aceptables, se pueden preparar de acuerdo con los esquemas de reacción analizados más adelante en el presente documento. A menos que se indique de otro modo, los sustituyentes en los esquemas se definen como anteriormente. Por ejemplo, en la fórmula Ia m es 2 y n es 1. En la fórmula IB m es 1 y n es 2. En la fórmula IC m y n son ambos 1. El aislamiento y la purificación de los productos se realiza mediante procedimientos convencionales, que son conocidos para un químico con experiencia ordinaria.
Un experto en la materia entenderá que los diferentes símbolos, superíndices y subíndices usados en los esquemas, métodos y ejemplos, se usan por conveniencia de la representación y/o para reflejar el orden en el que se introducen en los esquemas, y no pretenden necesariamente corresponder a los símbolos, superíndices o subíndices de las reivindicaciones adjuntas. Además, un experto en la materia reconocerá que en muchos casos, estos compuestos serán mezclas y enantiómeros que se pueden separar en diferentes etapas de los esquemas de síntesis usando técnicas convencionales, tal como, pero no limitadas a, cristalización, cromatografía de fase normal, cromatografía de fase inversa y cromatografía quiral, para proporcionar los enantiómeros individuales. Los esquemas son representativos de los métodos útiles para sintetizar los compuestos de la presente invención. No tienen por qué limitar el alcance de la invención en modo alguno.
Esquema 1

El esquema 1 se refiere a una secuencia de síntesis para la preparación de compuestos de fórmula I. Con referencia al esquema 1, el compuesto II se puede acoplar a un bromuro de heteroarilo, en donde los sustituyentes X1, X2 y R2 de la fórmula II deben estar representados por los mismos restos que se desean en el producto fina o una variación protegida de los mismos, y R1 se refiere a un arilo o heteroarilo de 5 o 6 miembros, para producir el compuesto III mediante una reacción de acoplamiento de Suzuki catalizada con paladio usando una selección convencional de fuente de paladio, ligando y base, en un disolvente convencional, por ejemplo, pero no limitado a, acetonitrilo, tolueno o etanol. Los ejemplos de la combinación Pd/ligando/base incluyen, pero no se limitan a, tetraquis(trifenilfosfina)paladio (0) mas carbonato sódico y tris(dibencilidenoacetona)dipaladio (0) mas diciclohexilfosfino-2',4',6'-triisopropilbifenilo mas carbonato potásico. La eliminación de grupo protector P1 da como resultado el compuesto IV. En este caso, el grupo protector P1 se refiere a grupos bien conocidos por los expertos en la materia de la protección de amina. Por
ejemplo, P1 puede ser un ferc-butoxicarbonilo (BOC), que se puede escindir en condiciones ácidas en un disolvente apropiado, que incluyen, pero no se limitan a, tratamiento con una solución de HCl en 1,4-dioxano. Como alternativa P1 puede ser uno de muchos otros grupos protectores adecuados para las aminas, que incluyen grupos carboxibencilo (Cbz) o benzoílo (Bz) y se puede escindir en condiciones convencionales conocidas por un experto en la materia. Los compuestos IV y V, en donde m y n se representan independientemente por un número entero seleccionado de entre 1 o 2, se pueden acoplar para producir compuestos racémicos de fórmula I usando un procedimiento convencional de aminación reductora usando, por ejemplo, pero sin limitarse a, una combinación de cianoborohidruro sódico y etóxido de titanio (IV) en un disolvente adecuado. La separación quiral mediante, por ejemplo, un método cromatográfico quiral tal como HPLC o cromatografía de fluidos supercríticos, puede producir los compuestos de fórmula I’.
Esquema 2

El esquema 2 se refiere rutas de síntesis alternativas para la preparación de compuestos de fórmula I y I'. Con respecto al esquema 2a, el compuesto IV, en donde los sustituyentes X1, X2, R1 y R2 de la fórmula IV deben estar representados por los mismos restos que se desean en el producto final o una variación protegida de los mismos, puede desplazar el sulfonato del compuesto VI enantioméricamente puro, donde R3 es un sustituyente arilo o alquilo, por ejemplo metilo o 4-metilfenilo y m y n se representan independientemente por un número entero seleccionado de entre 1 o 2, en presencia de una base tal como carbonato potásico en un disolvente apropiado, que incluye, pero no se limita a acetonitrilo. Con respecto al esquema 2b, el compuesto IV, en donde X1, X2, R1 y R2 deben estar representados por los mismos restos que se desean en el producto final o una variación protegida de los mismos, puede desplazar de manera similar el alquil sulfonato del compuesto quiral VII, donde R3 es un sustituyente arilo o alquilo y m y n se representan independientemente por un número entero seleccionado de entre 1 o 2, para producir el compuesto VIII. La eliminación del grupo BOC, que se puede escindir en condiciones ácidas en un disolvente apropiado, que incluye, pero no se limita a ácido trifluoroacético en diclorometano, seguida de tratamiento con cloroformiato de etilo en diclorometano u otros disolventes apropiados, produce los compuestos de fórmula I'. Como alternativa, como se muestra en el esquema 2c, el compuesto IV, en donde los sustituyentes X1, X2, R1 y R2 de la fórmula IV deben estar representados por los mismos restos que se desean en el producto final o una variación protegida de los mismos, se puede acoplar al compuesto IX, en donde m y n se representan independientemente por un número entero
seleccionado de entre 1 o 2, para producir compuestos de fórmula general X usando un procedimiento convencional de aminación reductora usando, por ejemplo, pero sin limitarse a, una combinación de cianoborohidruro sódico y etóxido de titanio (IV) en un disolvente adecuado. La eliminación del grupo BOC, que se puede escindir en condiciones ácidas en un disolvente apropiado, que incluye, pero no se limita a ácido trifluoroacético en diclorometano, seguido de tratamiento con cloroformiato de etilo en diclorometano u otro disolvente apropiado, produce compuestos racémicos de fórmula general I. Los compuestos de fórmula I' se pueden aislar después de separación quiral, por ejemplo por cromatografía quiral de fluidos supercríticos o HPLC.
Esquema 3
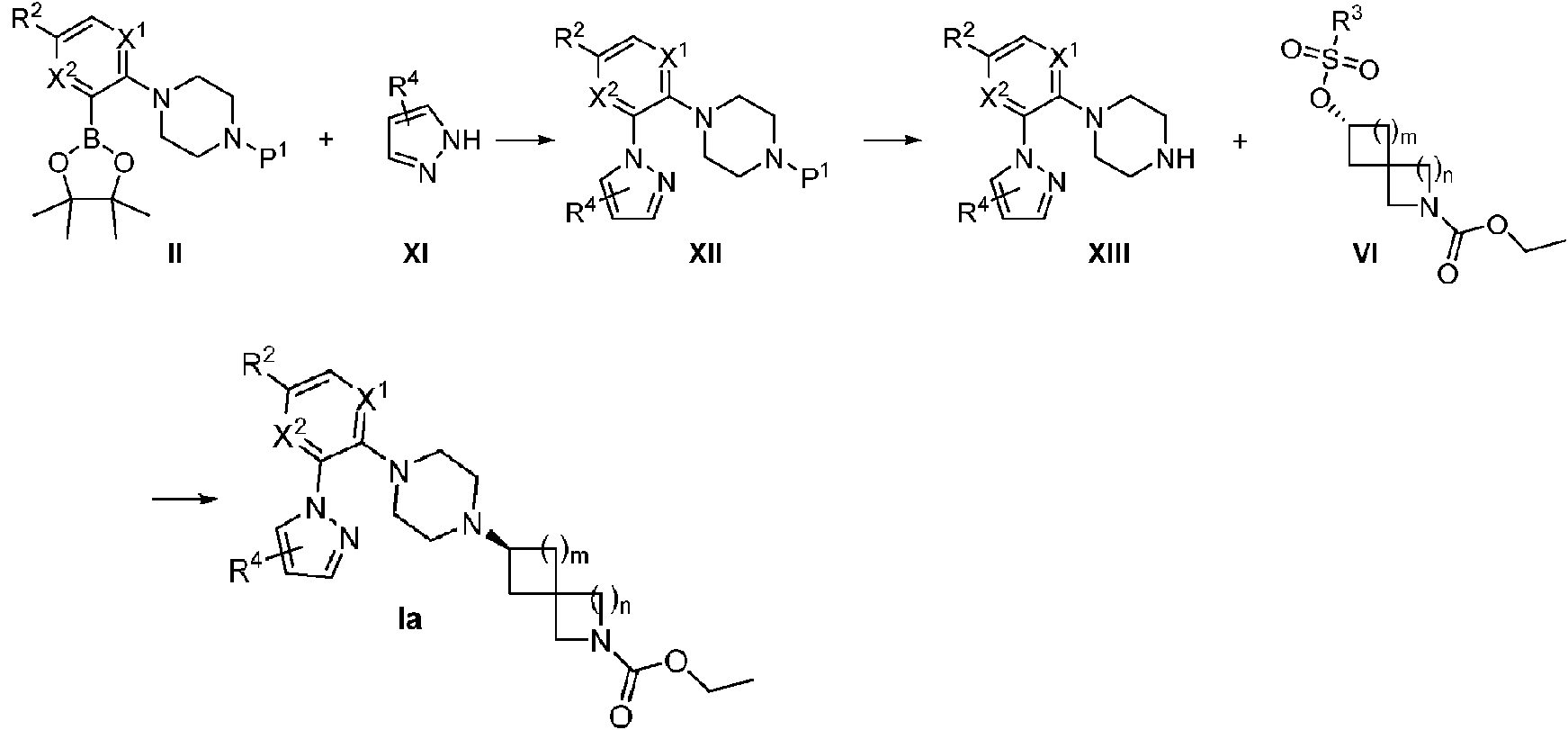
El esquema 3 se refiere a la preparación de compuestos de Fórmula la. El compuesto II, en donde los sustituyentes X1, X2 y R2 de la fórmula II deben estar representados por los mismos restos que se desean en el producto final o una variación protegida de los mismos y XI, donde R4 es un alquilo o alquiloxi pequeño, se puede acoplar para formar el compuesto XII mediante un acoplamiento de Ullman usando, por ejemplo, un catalizador de cobre tal como yoduro de cobre (I), un ligando tal como (1R,2R)-N,N-dimetilciclohexan-1,2-diamina y una base tal como fosfato potásico en un disolvente adecuado tal como NMP. La eliminación de grupo protector P1 da como resultado el compuesto XIII. P1 en este caso se refiere a grupos bien conocidos por los expertos en la materia para la protección de aminas. Por ejemplo, P1 puede ser un ferc-butoxicarbonilo (BOC), que se puede escindir en condiciones ácidas en un disolvente apropiado, que incluyen, pero no se limitan a, tratamiento con una solución de HCl en 1,4-dioxano. La síntesis de los compuestos de fórmula Ia puede realizarse mediante la reacción del sulfonato quiral compuesto VI, donde R3 es un sustituyente arilo o alquilo y m y n están representados independientemente mediante un número entero seleccionado de entre 1 o 2, con el compuesto XIII en presencia de una base tal como carbonato potásico en un disolvente apropiado, que incluye, pero no se limita a acetonitrilo.
Esquema 4

El esquema 4 se refiere a la preparación de compuestos de fórmula Ib. Una reacción de acoplamiento cruzado entre el compuesto XIV, en donde el sustituyente R2 debe estar representado por los mismos restos que se desean en el producto final o una variación protegida de los mismos y piperazin-1-carboxilato de terc-butilo para producir el compuesto XV puede realizarse usando una fuente de paladio, tal como tris(dibencilidenoacetona)dipaladio (0), un ligando tal como RuPhos ([2',6'-bis(propan-2-iloxi)bifenil-2-il](diciclohexil)fosfano) y una base adecuada tal como terc-butóxido sódico en 1,4-dioxano u otro disolvente adecuado. La eliminación del grupo BOC usando condiciones ácidas, tales como, pero no limitadas a HCl en 1,4-dioxano, da como resultado la formación del compuesto XVI. Los compuestos XVI y V, en donde m y n se representan independientemente por un número entero seleccionado de entre 1 o 2, se pueden acoplar para producir el compuesto XVII usando un procedimiento de aminación reductora convencional usando, por ejemplo, pero sin limitarse a, una combinación de cianoborohidruro sódico y etóxido de titanio (IV) en un disolvente adecuado. El desplazamiento nucleófilo del flúor del compuesto XVII con un alcohol, donde R5 es como se ha descrito anteriormente, usando una base adecuada tal como hidruro sódico en un disolvente adecuado tal como DMF, proporciona los compuestos de fórmula Ib.
Esquema 5

El esquema 5 se refiere a la síntesis de compuestos de fórmula I'. El compuesto XVIII, en donde los sustituyentes X1, X2 y R2 deben estar representados por los mismos restos que se desean en el producto final o una variación protegida de los mismos, se pueden acoplar con ácidos aril o heteroarilborónicos, para producir los compuestos de fórmula XIX mediante reacción de acoplamiento de Suzuki usando una selección convencional de fuente de paladio, ligando y base, en un disolvente convencional, por ejemplo, pero no limitado a, acetonitrilo, tolueno o etanol. Los ejemplos de la combinación de Pd/ligando/base incluyen, pero no se limitan a, [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) y carbonato sódico. La eliminación de grupo protector P1 da como resultado el compuesto XX. P1 puede ser un terc-butoxicarbonilo (BOC), que se puede escindir en condiciones ácidas en un disolvente apropiado, que incluyen, pero no se limitan a, tratamiento con una solución de HCl en 1,4-dioxano. La síntesis de los compuestos de fórmula I' se puede llevar a cabo mediante el desplazamiento del sulfonato del compuesto VI, donde R3 es un sustituyente arilo o alquilo y m y n están representados independientemente mediante un número entero seleccionado de entre 1 o 2, con el compuesto XX en presencia de una base adecuada tal como carbonato potásico en un disolvente apropiado, que incluye, pero no se limita a acetonitrilo.
Esquema 6
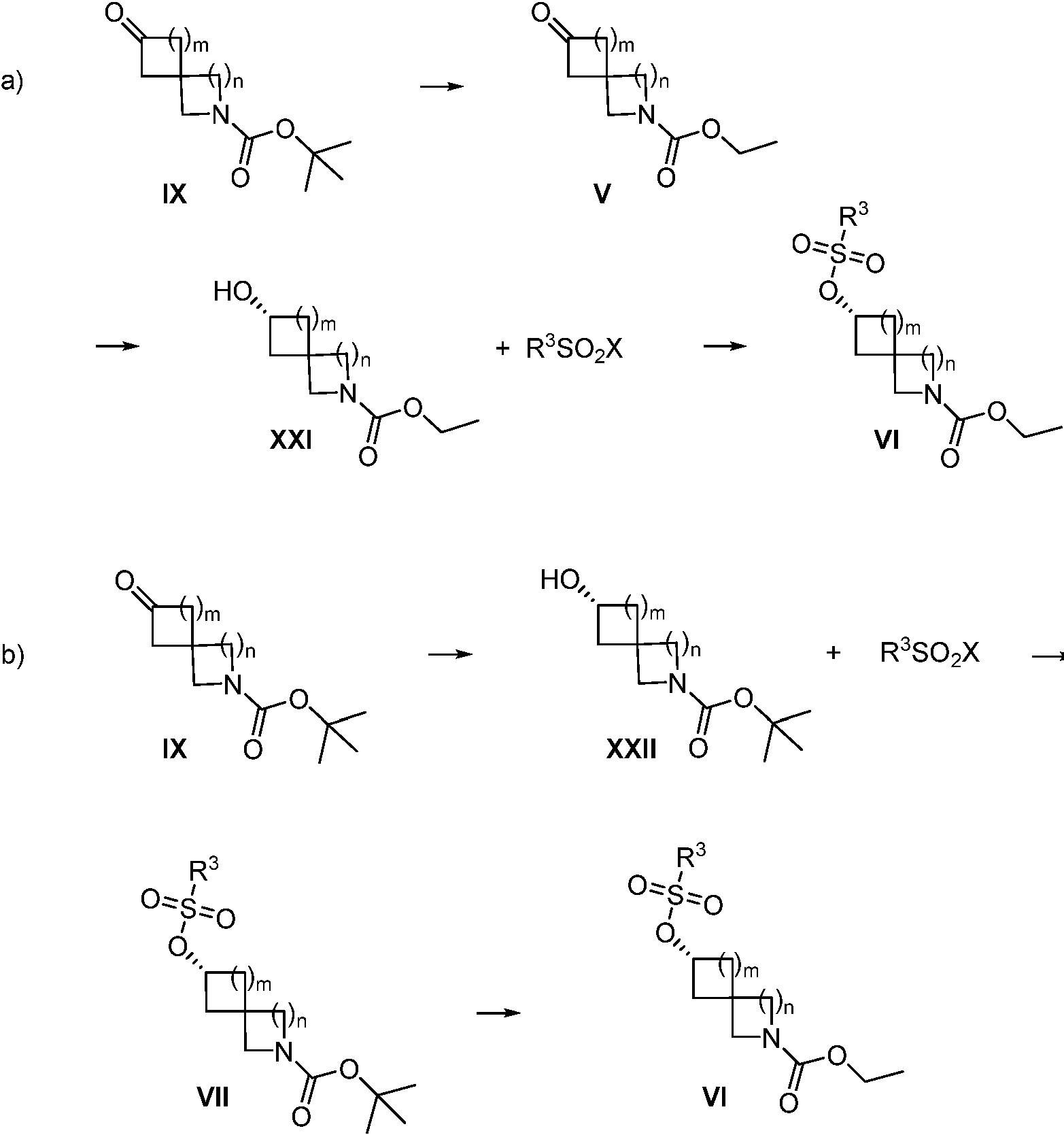
El esquema 6 se refiere a la preparación de las fórmulas generales VI y VII. Con respecto al esquema 6a, el grupo protector BOC en el compuesto IX, en donde m y n se representan independientemente por un número entero seleccionado de entre 1 o 2, se puede eliminar usando medios ácidos, por ejemplo, pero sin limitarse a, HCl en metanol. El material en bruto se puede combinar con cloroformiato de etilo y una base, tal como trietilamina, en un disolvente tal como diclorometano para formar los carbamatos de etilo de fórmula V. La reducción de la cetona al alcohol XXI enantioméricamente puro se puede conseguir usando un reactivo enzimático, tal como la cetorreductasa KRED-P3-G09 Codex® y NADP+ (nicotinamida adenina dinucleótido fosfato) en un tampón apropiado. Como alternativa, también se puede llevar a cabo una reducción racémica con un reductor tal como borohidruro sódico, por ejemplo, y se puede realizar separación quiral en una etapa posterior. La combinación de XXI con un cloruro o anhídrido de alquil o arilsulfonilo activado en un disolvente apropiado, en presencia de una base, tal como trietilamina, proporciona los compuestos de fórmula VI. Con respecto al esquema 6b, la reducción de cetona del compuesto IX al alcohol XXII enantioméricamente puro se puede conseguir usando un reactivo enzimático, tal como la cetorreductasa KRED-P3-G09 Codex® y NADP+ (nicotinamida adenina dinucleótido fosfato) en un tampón apropiado. La adición de un cloruro o anhídrido de alquil o arilsulfonilo con una base apropiada, tal como trietilamina y 4-(dimetilamino)piridina (DMAP) en un disolvente adecuado tal como diclorometano da como resultado la formación de compuestos de fórmula VII. La eliminación del grupo BOC se puede realizar en condiciones ácidas apropiadas, por ejemplo, pero sin limitarse a, ácido trifluoroacético en diclorometano, después el tratamiento del material en bruto resultante con cloroformiato de etilo en condiciones básicas, por ejemplo trietilamina en diclorometano, da como resultado la formación de los compuestos de fórmula VI.
Como se usa en el presente documento, el término "reaccionar" (o "reacción" o "reaccionado") se refiere a la unión de los reactivos químicos designados de manera que tenga lugar una transformación química generando un compuesto diferente de cualquiera introducido inicialmente en el sistema. Las reacciones pueden tener lugar en presencia o ausencia de disolvente.
Los compuestos de fórmula I pueden existir en forma de estereoisómeros, tales como atropisómeros, racematos,
enantiómeros o diastereómeros. Las técnicas convencionales para la preparación/aislamiento de enantiómeros individuales incluyen la síntesis quiral a partir de un precursor ópticamente puro adecuado o la resolución del racemato usando, por ejemplo, cromatografía líquida de alta resolución (HPLC) quiral. Como alternativa, el racemato (o un precursor racémico) puede hacerse reaccionar con un compuesto adecuado ópticamente activo, por ejemplo, un alcohol o, en el caso en que el compuesto contenga un resto ácido o básico, un ácido o una base tal como ácido tartárico o 1 -feniletilamina. La mezcla diastereomérica resultante puede separarse mediante cromatografía y/o cristalización fraccionada y uno o los dos diastereoisómeros convertirse en el o los enantiómeros puros correspondientes por medios bien conocidos por un experto en la materia. Pueden obtenerse compuestos quirales de fórmula I (y precursores quirales de los mismos) en forma enantioméricamente enriquecida usando cromatografía, normalmente HPLC, con sistemas de disolventes mixtos, tales como, pero limitados a acuoso más acetonitrilo, cualquiera o ambos de los cuales pueden contener aditivos tales como ácido trifluoroacético, ácido fórmico, hidróxido de amonio concentrado, o con cromatografía de fluidos supercríticos, llevada a cabo usando una combinación de dióxido de carbono y un disolvente orgánico tal como metanol o acetonitrilo, que contiene opcionalmente un aditivo tal como dietilamina o hidróxido de amonio, en una resina asimétrica con una fase móvil compuesta por un hidrocarburo, normalmente heptano o hexano, que contiene del 0 % al 50 % de 2-propanol, normalmente del 2 % al 20 % y del 0 % al 5% de una alquilamina, normalmente el 0,1 % de dietilamina. La concentración del eluato produce la mezcla enriquecida. Los conglomerados estereoisoméricos pueden separarse mediante técnicas convencionales conocidas por los expertos en la materia. Véase, por ejemplo, Stereochemistry of Organic Compounds de E. L. Eliel y S. H. Wilen (Wiley, Nueva York, 1994), cuya divulgación se incorpora en la presente memoria como referencia en su totalidad. Las técnicas estereoselectivas adecuadas son bien conocidas por los expertos habituales en la materia.
La invención se describirá con más detalle por medio de ejemplos específicos. Los siguientes ejemplos se ofrecen con fines ilustrativos y no pretenden limitar la invención en modo alguno. Los expertos en la materia reconocerán fácilmente diversos parámetros no críticos que pueden cambiarse o modificarse para producir esencialmente los mismos resultados. Pueden prepararse otros compuestos dentro del ámbito de esta invención usando los métodos ilustrados en estos ejemplos, tanto solos como en combinación con técnicas generalmente conocidas en la materia. En los siguientes ejemplos y preparaciones, "DMSO" significa dimetilsulfóxido, "N" donde se refiere a concentración significa Normal, "M" significa molar, "ml" significa mililitro, "mmol" significa milimoles, "pmol" significa micromoles, "eq." significa equivalente, "°C" significa grados Celsius, "MHz" significa megahercio, "HPLC" significa cromatografía líquida de alto rendimiento.
PROCEDIMIENTOS EXPERIMENTALES
A continuación se ilustra la síntesis de diversos compuestos de la presente invención. Pueden prepararse otros compuestos dentro del ámbito de esta invención usando los métodos ilustrados en estos ejemplos, tanto solos como en combinación con técnicas generalmente conocidas en la materia.
Los experimentos se llevaron a cabo normalmente en atmósfera inerte (nitrógeno o argón), en particular en casos donde se emplearon productos intermedios o reactivos sensibles al oxígeno o la humedad. Los disolventes y reactivos comerciales se usaron generalmente sin más purificación. Cuando fue adecuado, se emplearon disolventes anhidros, generalmente productos AcroSeal® de Acros Organics, Aldrich® Sure/Seal™ de Sigma-Aldrich o productos DriSolv® de EMD Chemicals. En otros casos, se pasaron disolventes comerciales a través de columnas empaquetadas con tamices moleculares 4 A, hasta que se alcanzaron los siguientes estándares de CC para el agua: a) <100 ppm para diclorometano, tolueno, W,W-dimetilformamida y tetrahidrofurano; b) <180 ppm para metanol, etanol, 1,4-dioxano y diisopropilamina. Para reacciones muy sensibles, los disolventes se trataron además con sodio metálico, hidruro cálcico o tamices moleculares y se destilaron justo antes de su uso. Por lo general, los productos se secaron al vacío antes de llevarse a otras reacciones o someterse a ensayos biológicos. Los datos de espectrometría de masas se obtienen mediante cromatografía líquida-espectrometría de masas (LCMS), instrumentación de ionización química a presión atmosférica (APCI) o cromatografía de gases y espectrometría de masas (CGMS). Los datos de los desplazamientos químicos para la resonancia magnética nuclear (RMN) se expresan en partes por millón (ppm, 8) en referencia a los picos residuales referenciados para los picos residuales de los disolventes deuterados empleados. En algunos ejemplos, se llevaron a cabo separaciones quirales para separar enantiómeros de determinados compuestos de la invención (en algunos ejemplos, los enantiómeros separados se denominan ENT-1 y ENT-2, de acuerdo con su orden de elución). En algunos ejemplos, la rotación óptica de un enantiómero se midió usando un polarímetro. De acuerdo con sus datos de rotación observados (o sus datos de rotación específicos), un enantiómero con rotación horaria se denominó enantiómero (+) y un enantiómero con rotación antihoraria se denominó enantiómero (-). Los compuestos racémicos se indican mediante la presencia de (+/-) adyacente a la estructura; en estos casos, la estereoquímica indicada representa la configuración relativa (en lugar de absoluta) de los sustituyentes del compuesto.
Las reacciones que tienen lugar mediante productos intermedios detectables generalmente se controlaron por LCMS y se dejaron proceder hasta la conversión completa antes de la adición de los reactivos posteriores. Para las síntesis que hace referencia a procedimientos en otros ejemplos o métodos, las condiciones de reacción (tiempo y temperatura de reacción) pueden variar. En general, las reacciones fueron seguidas por cromatografía de capa fina o espectrometría de masas y se sometieron a procesamiento cuando fue adecuado. Las purificaciones pueden variar entre experimentos: en general, los disolventes y las proporciones de disolventes usados para los eluyentes/gradientes se seleccionaron para proporcionar los Fr o tiempos de retención apropiados. Todos los materiales de partida en estas
preparaciones y ejemplos están disponibles en el mercado o se pueden preparar por métodos conocidos en la materia o como se describen en el presente documento.
Los compuestos y productos intermedios descritos a continuación se nombraron usando la convención de denominación proporcionada con ACD/ChemSketch 2012, File Version C10H41, Build 69045 (Advanced Chemistry Development, Inc., Toronto, Ontario, Canadá). Los expertos en la materia conocen bien la convención de denominación proporcionada con ACD/ChemSketch 2012 y se cree que la convención de denominación proporcionada con ACD/ChemSketch 2012 en general cumple con las recomendaciones de la IUPAC (International Union for Pure and Applied Chemistry) sobre la nomenclatura de la química orgánica y las reglas del Índice CAS.
Preparación P1
6-oxo-2-azaespiro[3.4]octan-2-carboxilato de etilo ( P1 )

Se añadió cloruro de acetilo (88 ml, 1,24 mol) a metanol (500 ml) a 0 °C y la solución resultante se agitó en un recipiente cerrado herméticamente durante 1 hora. A esta solución de cloruro de hidrógeno en metanol se le añadió 6-oxo-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo (20,0 g, 88,8 mmol) y sulfato de magnesio (20 g). La mezcla de reacción se agitó a 55 °C durante 2 horas, tras lo cual se enfrió a temperatura ambiente y se concentró al vacío. El residuo (18,4 g) se mezcló con diclorometano (700 ml) y se enfrió a 0 °C con agitación vigorosa. Después de la adición gota a gota de cloroformiato de etilo (30 ml, 310 mmol), la mezcla de reacción se trató gota a gota con trietilamina (60 ml, 430 mmol) y se agitó a 0 °C durante 1,5 horas. Se dejó calentar a temperatura ambiente y se agitó durante una noche. Se añadió ácido clorhídrico (1 M; 200 ml, 200 mmol) y la agitación continuó a temperatura ambiente durante 10 minutos. La capa orgánica se lavó con una solución acuosa saturada de cloruro sódico (200 ml), se secó sobre sulfato sódico, se filtró y se concentró a presión reducida. La cromatografía sobre gel de sílice (Gradiente: del 0 % al 100 % de acetato de etilo en heptano) proporcionó el producto en forma de un aceite de color ámbar. Rendimiento: 13,2 g, 66,9 mmol, 75 %. RMN 1H (400 MHz, CDCla) 84,13 (c, J = 7,2 Hz, 2H), 3,92 (cuadruplete AB, Jab = 8,6 Hz, Avab = 9,1 Hz, 4H), 2,46 (s, 2H), 2,33-2,27 (m, 2H), 2,23-2,17 (m, 2H), 1,25 (t, J = 7,2 Hz, 3H).
Preparación P2
(6S)-6-[(metilsulfonil)oxi]-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo ( P2 )

Este experimento se realizó en 2 lotes. Una mezcla de 6-oxo-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo (40,0 g, 178 mmol) en 2-propanol (64 ml, 840 mmol) se calentó a 50 °C hasta que se formó una solución. Se cargó un reactor Mettler EasyMax con tampón [fosfato potásico acuoso, pH 7,5 (0,1 M, que contenía cloruro de magnesio 2 mM)]
(280 ml) a 30 °C y agitación a 600 rpm. Se añadieron cetorreductasa KRED-P3-G09 Codex® (800 mg) y NADP+ (nicotinamida adenina dinucleótido fosfato) (80 mg) y la mezcla resultante se agitó durante 10 minutos. Se añadió lentamente la solución caliente de sustrato, mientras la temperatura de reacción se mantuvo por debajo de 33 °C. Se usó más 2-propanol (10 ml y 6 ml) para aclarar el matraz del sustrato. Se aumentó la velocidad de agitación de la reacción a 600 rpm y se aplicó una aguja de rociado de nitrógeno a un caudal de 100 cc/minuto. Se recogieron alícuotas de forma periódica: aproximadamente 80 pl de la mezcla de reacción se mezclaron con deuterocloroformo (920 pl) y la muestra se agitó en vórtice, se centrifugó y la capa orgánica se analizó por RMN 1H. Después de 23 horas, se añadieron más cetorreductasa KRED-P3-G09 Codex® (200 mg) y NADP+ (20 mg) en forma de una solución en el tampón de pH 7,5 (4 ml), seguido de la adición de 2-propanol (20 ml). Después de otras 22 horas, la mezcla de reacción se diluyó con acetato de etilo (400 ml) y se agitó durante 50 minutos, tras lo cual se añadió tierra de diatomeas (25 g) y la mezcla se agitó durante 10 minutos más. Después se filtró a través de tierra de diatomeas (25 g) y el lecho del filtro se aclaró con acetato de etilo (200 ml). Este filtrado de 200 ml se usó para extraer la capa acuosa de la filtración inicial y las capas orgánicas combinadas se lavaron con una solución acuosa saturada de cloruro sódico, se secaron sobre sulfato sódico, se filtraron y se concentraron al vacío para proporcionar un aceite de color pardo (39,6 g). Los dos lotes se combinaron después en diclorometano (600 ml), se trataron con gel de sílice (150 g) y se concentraron al vacío para la cromatografía. La cromatografía sobre gel de sílice (Gradiente: del 0 % al 100 % de acetato de etilo en heptano; el producto comenzó a eluir a aproximadamente el 50 % de acetato de etilo) proporcionó el producto en forma de un sólido. Rendimiento combinado: 63,4 g, 279 mmol, 78 %. RMN 1H (400 MHz, CDCh) 84,39-4,33 (m, 1H), 3,87 (cuadruplete AB, Jab = 8,4 Hz, Avab = 41,1 Hz, 2H), 3,80-3,74 (m, 2H), 2,12-2,01 (m, 2H), 1,97-1,77 (m, 3H), 1,69 1,59 (m, 1H), 1,44 (s, 9H). El análisis proporción un ee (exceso enantiomérico) de >99 % [Cromatografía de fluidos supercríticos. Columna: Chiral Technologies Chiralpak AD-3, 100 x 3,0 mm, 3 pm; Fase móvil A: dióxido de carbono; Fase móvil B: [metanol que contenía 0,2 % (amoniaco 7 M en metanol)]; Gradiente: al 5 % de B durante 1,0 minuto, después del 5 % al 15 % de B durante 7,0 minutos; Caudal: 2,0 ml/minuto; Contrapresión: 6,89 kPa (1800 psi)].
Véase a continuación [Correlación estereoquímica de (6S)-6-hidroxi-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo (C1) con (6S)-6-hidroxi-2-azaespiro[3.4]octan-2-carboxilato de etilo (C2)] para la asignación de la estereoquímica absoluta indicada de C1.
Etapa 2. Síntesis de (6S)-6-[(metilsulfonil)oxi]-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo (P2).
Se añadieron trietilamina (13,5 ml, 96,9 mmol) y 4-(dimetilamino)piridina (295 mg, 2,41 mmol) a una solución de C1 (11,0 g, 48,4 mmol) en diclorometano (400 ml). Después se añadió cloruro de metanosulfonilo (8,40 ml, 108 mmol) {Precaución: exotérmico} y la mezcla de reacción se dejó en agitación durante una noche. Después de eliminar el disolvente al vacío, el residuo se mezcló con diclorometano y se filtró; el filtrado se concentró a presión reducida y se purificó por cromatografía sobre gel de sílice (Gradiente: del 0% al 100% de acetato de etilo en heptano) para proporcionar el producto en forma de un aceite. Rendimiento: 14,6 g, 47,8 mmol, 99 %. LCMS m/z 328,2 [M+Na+] RMN 1H (400 MHz, CDCh) 85,19-5,14 (m, 1H), 3,93 (d, medio cuadruplete AB, J = 8,2 Hz, 1H), 3,85-3,78 (m, 3H), 3,00 (s, 3H), 2,26 (d a, medio cuadruplete AB, J = 14,4 Hz, 1H), 2,16 (dd, componente del patrón ABX, J = 14,6, 6,0 Hz, 1H), 2,13-1,99 (m, 3H), 1,92-1,83 (m, 1H), 1,44 (s, 9H).
Preparación P3
(6S)-6-[(metilsulfonil)oxi]-2-azaespiro[3.4]octan-2-carboxilato de etilo (P3)

Una mezcla de P2 (14,6 g, 47,8 mmol) en diclorometano (250 ml) y ácido trifluoroacético (55 ml, 710 mmol) se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se concentró al vacío y el residuo se diluyó con diclorometano (250 ml) y se trató secuencialmente con cloroformiato de etilo (9,10 ml, 95,2 mmol) y trietilamina (26,7 ml, 192 mmol). Después de esto la mezcla de reacción se agitó a temperatura ambiente durante 2 horas, se concentró a presión reducida y se purificó usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 100 % de acetato de etilo en heptano). El producto, que según análisis por RMN 1H no era totalmente puro, se obtuvo en forma de un aceite de color amarillo. Rendimiento: 13,1 g, 47,2 mmol, 99 %. LCMS m/z 278,2 [M+H]+. RMN 1H (400 MHz, CDCla), únicamente picos del producto: 85,20-5,15 (m, 1H), 4,12 (c, J = 7,1 Hz, 2H), 4,00 (d, medio cuadruplete AB, J = 8,6 Hz, 1H), 3,91-3,85 (m, 3H), 3,00 (s, 3H), 2,29 (d a, medio cuadruplete AB, J = 14,8 Hz, 1H), 2,20-2,02 (m, 4H), 1,93-1,85 (m, 1H), 1,25 (t, J = 7,0 Hz, 3H).
Preparación P4
(6S)-6-{[(4-metilfenil)sulfonil]oxi}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( P4 )

Se cargó un reactor Mettler EasyMax con tampón [fosfato potásico acuoso, pH 7,0 (0,1 M, que contenía cloruro de magnesio 2 mM)] (8,0 ml) que contenía cetorreductasa KRED-P3-G09 Codex® (60 mg) y NADP+ (nicotinamida adenina dinucleótido fosfato) (6 mg). Se usó más tampón (2,5 ml) para enjuagar el recipiente de vidrio y se añadió a la mezcla de reacción. Después se añadió una solución de P1 (1,5 g, 7,6 mmol) en 2-propanol (1,5 ml), junto con un enjuagado con 2-propanol 1,5 ml). La mezcla de reacción se agitó a 300 rpm y 30 °C con una corriente de nitrógeno de 10SCCM (centímetros cúbicos estándar por minuto). Se recogieron alícuotas de forma periódica: aproximadamente 50 |jl de la mezcla de reacción se mezclaron con deuterocloroformo (0,75 ml) y la muestra se agitó en vórtice, se centrifugó y la capa orgánica se analizó por RMN 1H. Cuando la reacción alcanzó aproximadamente el 80 % de conversión, el caudal del nitrógeno se aumentó a 25 SCCM y se dejó que la reacción prosiguiera durante la noche. Se añadió acetato de etilo (15 ml) y la mezcla resultante se agitó vigorosamente durante 10 minutos, tras lo cual se trató con tierra de diatomeas (1,5 g) y se filtró a través de un lecho humedecido de tierra de diatomeas (1,5 g). Después de lavar el lecho de filtro con acetato de etilo (5 ml), la capa acuosa de los filtrados combinados se mezcló con acetato de etilo (15 ml), se agitó vigorosamente durante 5 minutos y se vertió a través del lecho de filtro. Se usó de nuevo acetato de etilo (5 ml) para lavar el lecho de filtro y la capa acuosa de estos filtrados se extrajo del mismo modo. Las capas orgánicas combinadas procedentes de estas manipulaciones se secaron sobre sulfato sódico, se filtraron y se concentraron al vacío. La cromatografía sobre gel de sílice (Gradiente: del 20 % al 80 % de acetato de etilo en heptano) proporcionó el producto en forma de un aceite de color amarillo claro. El análisis proporcionó un ee (exceso enantiomérico) de >99 % {Cromatografía de fluidos supercríticos. Columna: Chiral Technologies ChiralpakAD, 250 x 4,6 mm, 5 jm ; Fase móvil: 85:15 dióxido de carbono/[metanol que contenía 0,2 % (amoniaco 7 M en metanol)]; Caudal de 3,0 ml/minuto; Contrapresión: 12 MPa (120 bar)}. Rendimiento: 1,20 g, 6,02 mmol, 79%. RMN 1H (400 MHz, CDCla) 54,41-4,33 (m, 1H), 4,10 (c, J = 7,0 Hz, 2H), 3,93 (cuadruplete AB, Jab = 8,4 Hz, Avab = 42,3 Hz, 2h ), 3,86-3,80 (m, 2H), 2,13-2,02 (m, 2h ), 1,98-1,78 (m, 3H), 1,70-1,6 (m, 1H, asumido; parcialmente oscurecido por el pico de agua), 1,42 (d, J = 3,1 Hz, 1H), 1,24 (t, J = 7,0 Hz, 3H). La estereoquímica absoluta indicada se asignó basándose en la conversión de C2 a P4 y después a 6 (véase Síntesis alternativa del ejemplo 6, a continuación).
Etapa 2. Síntesis de (6S)-6-{[(4-metilfenil)sulfonil]oxi}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( P4 ).
Se añadió anhídrido de 4-metilbencenosulfónico (6,29 g, 19,3 mmol) a una mezcla a 0 °C de C2 (sintetizado mediante la biorreducción de P1, véase la etapa anterior; 3,20 g, 16,1 mmol) en piridina (80 ml). Después de la adición de 4-(dimetilamino)piridina (196 mg, 1,60 mmol), la mezcla de reacción se dejó en agitación durante una noche, mientras el baño de hielo se fundía. El análisis por Lc MS en este momento indicó la presencia del producto: LCMS m/z 354,3 [M+H]+. Después de que la mezcla de reacción se hubiese concentrado al vacío, esta se diluyó con solución acuosa de hidrógeno sulfato sódico (10 %; 100 ml); la capa acuosa se extrajo después secuencialmente con éter dietílico (150 ml) y con diclorometano (150 ml). Las capas orgánicas combinadas se concentraron a presión reducida y se purificaron por cromatografía sobre gel de sílice (Gradiente: del 0% al 100% de acetato de etilo en heptano), proporcionando el producto en forma de un aceite. Rendimiento: 4,54 g, 12,8 mmol, 80 %. RMN 1H (400 MHz, CDCh) 5 7,78 (d, J = 8,2 Hz, 2H), 7,35 (d, J = 7,8 Hz, 2H), 5,01-4,92 (m a, 1H), 4,09 (c, J = 7,2 Hz, 2H), 3,87 (cuadruplete AB, Jab = 8,6 Hz, A^ab=32,4 Hz, 2H), 3,82-3,76 (m, 2H), 2,46 (s, 3H), 2,12 (d a, medio cuadruplete AB, J = 14,8 Hz, 1H), 2,09-1,96 (m, 2H), 1,94-1,86 (m, 2H), 1,85-1,76 (m, 1H), 1,24 (t, J = 7,2 Hz, 3H). La configuración absoluta indicada de este material se estableció mediante su uso a continuación en la Síntesis alternativa del ejemplo 6. Se mostró que
esa muestra de 6 era idéntica al material usado en la determinación de la estructura cristalina de rayos X descrita a continuación.
Correlación estereoquímica de (6S)-6-hidroxi-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo ( C1 ) con (6S)-6-hidroxi-2-azaespiro[3.4]octan-2-carboxilato de etilo ( C2 )

Se añadió borohidruro sódico (95 mg, 2,5 mmol) en una porción a una solución de P1 (280 mg, 1,42 mmol) en metanol (10 ml) {Precaución: exotérmico}. Después de agitar la mezcla de reacción durante 2 horas, esta se diluyó con ácido clorhídrico (1 M; 5 ml) y se agitó a temperatura ambiente durante 5 minutos. Después se añadió agua (5 ml) y la capa acuosa se extrajo con diclorometano (2 x 30 ml). Las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron y se concentraron al vacío para proporcionar el producto en forma de un aceite. Rendimiento: 233 mg, 1,17 mmol, 82 %. RMN 1H (400 MHz, CDCh) 84,41-4,34 (m, 1H), 4,10 (c, J = 7,2 Hz, 2H), 3,93 (cuadruplete AB, Jab = 8,4 Hz, Avab = 41,9 Hz, 2H), 3,86-3,80 (m, 2H), 2,14-2,02 (m, 2H), 1,98-1,78 (m, 3H), 1,70-1,6 (m, 1H, asumido; parcialmente oscurecido por el pico de agua), 1,45-1,32 (s a, 1H), 1,24 (t, J = 7,2 Hz, 3H).
Etapa 1. Síntesis de (6S)-2-azaespiro[3.4]octan-6-ol, sal clorhidrato ( C4 ).
Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 8 ml, 32 mmol) a una mezcla de C1 (512 mg, 2,25 mmol) en acetato de etilo (12 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 3 horas. La eliminación de los disolventes al vacío proporcionó el producto, que se usó en la reacción siguiente sin purificación.
Etapa 2. Síntesis de (6S)-6-hidroxi-2-azaespiro[3.4]octan-2-carboxilato de etilo ( C2 a p a rtir de C1 ).
Se añadió cloroformiato de etilo (0,258 ml, 2,70 mmol) gota a gota a una mezcla de C4 (de la etapa anterior, <2,25 mmol) y trietilamina (0,943 ml, 6,76 mmol) en diclorometano (10 ml). Después de que la mezcla de reacción se hubiese agitado a temperatura ambiente durante 1 hora, esta se diluyó con ácido clorhídrico (1 M; 10 ml) y se agitó a temperatura ambiente durante 5 minutos. La capa acuosa se extrajo con diclorometano (15 ml) y las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron y se concentraron al vacío. La cromatografía sobre gel de sílice (Gradiente: del 0 % al 100 % de acetato de etilo en heptano) proporcionó el producto en forma de un aceite. Según análisis por RMN 1H, este material no era completamente puro. Rendimiento: 100 mg, 0,502 mmol, 22 % en 2 etapas. RMN 1H (400 MHz, CDCla), únicamente picos del producto: 84,34-4,26 (m a, 1H), 4,06 (c, J = 7,0 Hz, 2H), 3,89 (cuadruplete AB, Jab = 8,6 Hz, Avab = 44,8 Hz, 2H), 3,81-3,75 (m, 2H), 2,09-1,96 (m, 2H), 1,93-1,82 (m, 2H), 1,82 1,73 (m, 1H), 1,66-1,56 (m, 1H), 1,20 (t, J = 7,2 Hz, 3H).
Se demostró que las estereoquímicas absolutas de C1 y C2 a partir de reducción con KRED-P3-G09 (Preparación P4) eran iguales del siguiente modo. El compuesto C1 se convirtió en la muestra C2 a partir de C1 (Etapas 1 y 2 anteriores). El racemato de C2 (C3) se examinó mediante cromatografía de fluidos supercríticos [Columna: Chiral Technologies Chiralpak AD-H, 250 x 4,6 mm, 5 pm; Fase móvil A: dióxido de carbono; Fase móvil B: metanol que contenía 0,2 % (amoniaco 7 M en metanol); Gradiente: al 5 % de B durante 1,0 minuto, después del 5 % al 40 % de B durante 8,0 minutos; Caudal: 3,0 ml/minuto; Contrapresión: 6,89 kPa (1800 psi)]. Los dos enantiómeros eluyeron en los tiempos de retención de 4,57 y 4,94 minutos. Una muestra de C2 procedente de la reducción de P1 con KRED-P3-G09 (Preparación P4) proporcionó un tiempo de retención de 4,9 minutos en las mismas condiciones cromatográficas, al igual que la muestra de C2 a partir de C1.
Ejem plos 1, 2 y 3
6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( 1 ), 6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 ( 2 ) y 6-{4-[3-(5- metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2 (3)
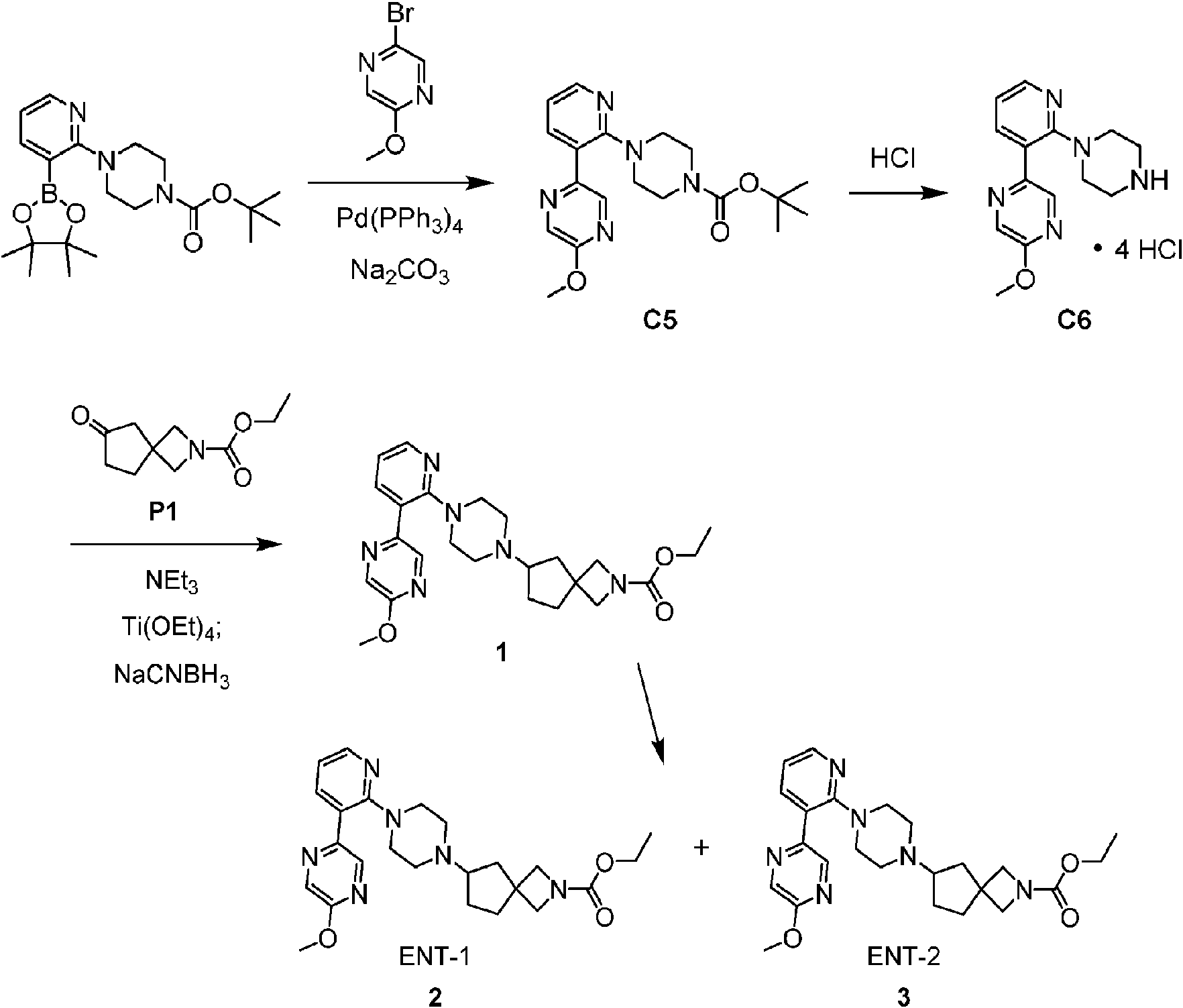
Etapa 1. Síntesis de 4-(3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (C5).
Se añadió una solución de tetraquis(trifenilfosfina)paladio (0) (89 mg, 77 ^mol) en tolueno (5 ml) y etanol (2 ml) a una mezcla de 4-[3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (300 mg, 0,771 mmol), 2-bromo-5-metoxipirazina (146 mg, 0,772 mmol) y solución acuosa de carbonato sódico (2 M, 10 ml). La mezcla de reacción se agitó a 100 °C con irradiación con microondas durante 3 horas, tras lo cual se concentró al vacío. [Los experimentos a los que hace referencia este método a menudo usaron calentamiento convencional, a 60 °C o más.] La purificación del residuo usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 40 % de acetato de etilo en éter de petróleo) proporcionó el producto en forma de un sólido de color amarillo. Rendimiento: 260 mg, 0,700 mmol, 91 %. LCMS m/z 372,2 [M+H]+. RMN 1H (400 MHz, CDCls) 58,80-8,72 (s a, 1H), 8,33 (d, J = 1,5 Hz, 1H), 8,33-8,28 (m, 1H), 7,86 (d a, J = 6,8 Hz, 1H), 7,08-7,01 (m, 1H), 4,03 (s, 3H), 3,50-3,41 (m a, 4H), 3,21-3,05 (m a, 4H), 1,46 (s, 9H).
Etapa 2. Síntesis de 2-metoxi-5-[2-(piperazin-1-il)piridin-3-il]pirazina, sal tetraclorhidrato (C6). Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 3 ml, 12 mmol) a una solución de C5 (260 mg, 0,700 mmol) en acetonitrilo (6 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La eliminación de los disolventes al vacío proporcionó el producto en forma de un aceite de color amarillo, que se usó directamente en la etapa siguiente. Según análisis por r Mn 1H, este material no era completamente puro. Rendimiento: 290 mg, 0,695 mmol, 99 %. LCMS m/z 272,2 [M+H]+. RMN 1H (400 MHz, DMSO-de), solo picos del producto: 59,57-9,42 (m a, 2H), 8,77 (d, J = 1,2 Hz, 1H), 8,45 (d, J = 1,5 Hz, 1H), 8,30 (dd, J = 5,1, 1,7 Hz, 1H), 7,98 (dd, J = 7,3, 1,5 Hz, 1H), 7,22 (dd, J = 7,5, 5,3 Hz, 1H), 3,98 (s, 3H), 3,36-3,28 (m a, 4H), 3,12-3,04 (m a, 4H).
Etapa 3. Síntesis de 6-{4-(3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( 1 ).
Una mezcla de C6 (220 mg, 0,527 mmol) y trietilamina (1,0 ml, 7,2 mmol) en diclorometano (40 ml) se agitó durante 30 minutos a temperatura ambiente, tras lo cual se añadió P1 (156 mg, 0,791 mmol), seguido de etóxido de titanio (IV) (1,5 ml, 7,2 mmol). Después de agitar la mezcla de reacción durante 16 horas a temperatura ambiente, se añadió cianoborohidruro sódico (490 mg, 7,80 mmol), seguido de metanol (6 ml) y la agitación continuó durante otras 4 horas a temperatura ambiente. Después se añadió agua (4 ml) y la mezcla resultante se concentró al vacío. Se llevó a cabo la purificación usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 10 % de metanol en diclorometano), seguido de cromatografía de fase inversa (Columna: Agela Technologies C18; Fase móvil A: agua que contenía hidróxido de amonio al 0,05 %; Fase móvil B: acetonitrilo; Gradiente: del 45 % al 60 % de B). El producto se aisló en forma de un sólido de color amarillo claro. Rendimiento: 170 mg, 0,376 mmol, 71 %. LCMS m/z 453,3 [M+H]+. RMN 1H (400 MHz, CDCls) 58,76 (s a, 1H), 8,32 (d, J = 1,2 Hz, 1H), 8,28 (dd, J = 4,9, 1,7 Hz, 1H), 7,81 (dd, J = 7,5, 1,8 Hz, 1H), 6,99 (dd, J = 7,5, 5,0 Hz, 1H), 4,10 (c, J = 7,1 Hz, 2H), 4,04 (s, 3H), 3,86 (cuadruplete AB, el doblete de campo bajo se ensancha, Jab = 8,3 Hz, Avab = 22,5 Hz, 2H), 3,81-3,74 (m, 2H), 3,29-3,10 (m a, 4H), 2,67-2,40 (m a, 5H), 2,11 (dd, J = 12, 7 Hz, 1H), 2,02-1,45 (m, 5H, asumido; parcialmente oscurecido por el pico de agua), 1,24 (t, J = 7,1 Hz, 3H).
Etapa 4. Aislamiento de 6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 ( 2 ) y 6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2 ( 3 ).
La separación de 1 (150 mg, 0,331 mmol) en sus enantiómeros componentes se llevó a cabo mediante HPLC de fase inversa [Columna: Chiral Technologies ChiralCel OD, 10 ^m; Fase móvil: 4:1 de hexano/etanol]. Los enantiómeros aislados se sometieron de manera individual a cromatografía de fase inversa (Columna: Agela Technologies C18; Fase móvil A: agua que contenía hidróxido de amonio al 0,1 %; Fase móvil B: acetonitrilo; Gradiente: del 0 % al 60 % de B) para proporcionar los productos en forma de sólidos de color amarillo. El primer enantiómero en eluir se denominó 2 y el segundo enantiómero en eluir se denominó 3.
Rendimiento de 2: 40 mg, 88 |jmol, 27 %. LCMS m/z 453,3 [M+H]+. RMN 1H (400 MHz, CDCls) 58,76 (s a, 1H), 8,32 (d, J = 1,2 Hz, 1H), 8,28 (dd, J = 4,8, 1,8 Hz, 1H), 7,81 (dd, J = 7,5, 1,8 Hz, 1H), 6,99 (dd, J = 7,3, 4,9 Hz, 1H), 4,10 (c, J = 7,1 Hz, 2 H), 4,03 (s, 3H), 3,86 (cuadruplete A b , el doblete de campo bajo se ensancha, JAB = 8,3 Hz, AvAB = 22,5 Hz, 2H), 3,80-3,74 (m, 2H), 3,30-3,10 (m a, 4H), 2,69-2,41 (m a, 5H), 2,11 (dd, J = 12,5, 6,8 Hz, 1H), 2,00-1,48 (m, 5H, asumido; parcialmente oscurecido por el pico de agua), 1,24 (t, J = 7,1 Hz, 3H). Tiempo de retención: 3,52 minutos (Condiciones analíticas. Columna: Chiral Technologies ChiralCel OD-H, 150 x 4,6 mm, 5 ^m; Fase móvil: 80:20:0,1 hexano/etanol/dietilamina; Caudal: 1,0 ml/minuto).
Rendimiento de 3: 43 mg, 95 |jmol, 29 %. LCMS m/z 453,3 [M+H]+. RMN 1H (400 MHz, CDCh) 58,75 (s a, 1H), 8,32 (d, J = 1,2 Hz, 1H), 8,28 (dd, J = 4,9, 2,0 Hz, 1H), 7,81 (dd, J = 7,5, 1,8 Hz, 1H), 6,99 (dd, J = 7,3, 4,9 Hz, 1H), 4,10 (c, J = 7,1 Hz, 2H), 4,04 (s, 3H), 3,93-3,86 (m a, 1H), 3,84 (d, medio cuadruplete AB, J = 8,3 Hz, 1H), 3,80-3,75 (m, 2H), 3,33-3,06 (m a, 4H), 2,71-2,35 (m a, 5H), 2,18-2,06 (m, 1 H), 2,04-1,45 (m, 5H, asumido; parcialmente oscurecido por el pico de agua), 1,24 (t, J = 7,1 Hz, 3H). Tiempo de retención: 4,53 minutos (Condiciones analíticas idénticas a las usadas para 2).
E jem plo 4
(6R)-6-{4-[3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( 4 )


Etapa 1. Síntesis de 4-[3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (C7).
Se añadieron etanol (30 ml) y una solución de carbonato sódico (8,85 g, 83,5 mmol) en agua (33 ml) a una mezcla de 4-[3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (13,0 g, 33,4 mmol) y 4-bromo-1,3-tiazol (6,57 g, 40,1 mmol) en tolueno (180 ml). Después se añadió tetrakis(trifenilfosfina)paladio (0) (2,69 g, 2,33 mmol) y la mezcla de reacción se agitó durante 12 horas a 90 °C. Después de eliminar los disolventes al vacío, el residuo se purificó usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 60 % de acetato de etilo en éter de petróleo) para proporcionar el producto en forma de un sólido de color amarillo claro. Rendimiento: 7,00 g, 20,2 mmol, 60 %. LCMS m/z 347,1 [M+H]+.
Etapa 2. Síntesis de 1-[3-(1,3-tiazol-4-il)piridin-2-il]piperazina (C8). Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 50 ml, 200 mmol) a una solución a 0°C de C7 (11,0 g, 31,8 mmol) en acetonitrilo (100 ml). La mezcla de reacción se agitó durante 16 horas a temperatura ambiente, tras lo cual se filtró. Los sólidos recogidos se lavaron con acetato de etilo y después se suspendieron en una mezcla de diclorometano (150 ml) y metanol (25 ml). Se añadió carbonato potásico (20 g, 145 mmol) y la mezcla se agitó durante 16 horas a temperatura ambiente, después se filtró. La torta de filtro se lavó con una mezcla de diclorometano y metanol (10:1,60 ml) y los filtrados combinados se secaron sobre sulfato sódico, se filtraron y se concentraron al vacío para proporcionar el producto en forma de un aceite de color amarillo. Según análisis por RMN 1H, este material no era completamente puro. Rendimiento: 7,5 g, 30 mmol, 94 %. LCMS m/z 247,1 [M+H]+. RMN 1H (400 MHz, DMSO-d6), solo picos del producto: 59,19 (d, J = 2,0 Hz, 1H), 8,24 (d, J = 2,0 Hz, 1H), 8,22 (dd, J = 4,8, 1,8 Hz, 1H), 8,10 (dd, J = 7,5, 1,8 Hz, 1H), 7,04 (dd, J = 7,5, 4,8 Hz, 1H), 2,97 2,91 (m, 4H), 2,80-2,73 (m, 4H).
Etapa 3. Síntesis de (6R)-6-{4-[3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de tercbutilo (C9).
Este experimento se llevó a cabo en dos lotes idénticos. Una mezcla de C8 (1,10 g, 4,47 mmol), P2 (1,91 g, 6,25 mmol) y carbonato potásico (1,54 g, 11,1 mmol) en acetonitrilo (20 ml) se puso en un recipiente cerrado herméticamente y se calentó a 95 °C durante 16 horas, tras lo cual se concentró al vacío. La cromatografía sobre gel de sílice (Gradiente: del 0 % al 10 % de metanol en diclorometano) proporcionó el producto en forma de un aceite de color amarillo claro. Rendimiento combinado: 1,70 g, 3,73 mmol, 42 %. LCMS m/z 456,2 [M+H]+. RMN 1H (400 MHz, DMSO-d6) 59,20 (d, J = 1,5 Hz, 1H), 8,23 (dd, J = 4,6, 1,5 Hz, 1H), 8,20 (s a, 1H), 8,10 (d a, J = 7,6 Hz, 1H), 7,06 (dd, J = 7,3, 4,6 Hz, 1H), 3,76-3,58 (m a, 4H), 3,07-2,96 (m a, 4H), 2,64-2,39 (m, 5H, asumido; en gran parte oscurecido por el pico del disolvente), 2,07-1,96 (m, 1H), 1,87-1,69 (m, 3H), 1,69-1,57 (m, 1H), 1,50-1,4 (m, 1H), 1,36 (s, 9H).
Etapa 4. Síntesis de (6R)-6-{4-[3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-i}-2-azaespiro[3.4]octano (C10).
Se añadió ácido trifluoroacético (15 ml) a una solución de C9 (6,00 g, 13,2 mmol) en diclorometano (120 ml) y la mezcla de reacción se agitó durante 16 horas a temperatura ambiente. Después se concentró al vacío y el residuo se disolvió en una mezcla de diclorometano y metanol (9:1, 150 ml); se añadió carbonato sódico (15 g) y la mezcla resultante se agitó durante 3 horas a temperatura ambiente. Después se filtró la mezcla y el filtrado se concentró a presión reducida, proporcionando el producto en forma de un aceite de color amarillo. Rendimiento: 4,50 g, 12,7 mmol, 96 %. LCMS m/z 356,2 [M+H]+. RMN 1H (400 MHz, CDsOD) 59,15-9,13 (m, 1H), 8,31-8,27 (m, 1H), 8,16-8,13 (m, 1H), 8,07 (d a, J = 7,6 Hz, 1H), 7,18-7,12 (m, 1H), 3,69 (d a, medio cuadruplete AB, J = 12 Hz, 1H), 3,65-3,49 (m, 5H), [3,40-3,24 (m) y 3,16-3,07 (m), 7H total, asumido; parcialmente oscurecido por el pico del disolvente], 2,41-2,28 (m, 2H), 2,02-1,81 (m, 3H), 1,72-1,63 (m a, 1H).
Etapa 5. Síntesis de (6R)-6-{4-[3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo (4). Se añadió cloroformiato de etilo (3,66 g, 33,7 mmol) a una mezcla a 0 °C de C10 (4,00 g, 11,2 mmol) y N,N-diisopropiletilamina (8,73 g, 67,5 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 3 horas. Después de verter la mezcla de reacción en agua, esta se extrajo con diclorometano (2 x 50 ml). Las capas orgánicas combinadas se concentraron y se purificaron por cromatografía sobre gel de sílice (Gradiente: del 0 % al 10 % de metanol en diclorometano) para proporcionar una goma de color amarillo claro (2,1 g); este material se volvió a purificar usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 20 % de metanol en diclorometano) para proporcionar el producto en forma de una espuma de color castaño. Rendimiento: 1,77 g, 4,14 mmol, 37 %. LCMS m/z 428,4 [M+H]+. RMN 1H (400 MHz, CDCla) 58,88 (d, J = 2,0 Hz, 1H), 8,26 (dd, J = 4,7, 2,0 Hz, 1H), 8,11 (dd, J = 7,4, 2,0 Hz, 1H), 8,02 (d, J = 2,0 Hz, 1H), 6,98 (dd, J = 7,6, 4,9 Hz, 1H), 4,10 (c, J = 7,0 Hz, 2H), 3,86 (cuadruplete AB, Jab = 8,4 Hz, Avab = 20,8 Hz, 2H), 3,79 (cuadruplete AB, Jab = 8,4 Hz, Avab = 6,0 Hz, 2H), 3,21-3,15 (m, 4H), 2,62-2,48 (m a, 5H), 2,12 (dd, J = 12,7, 6,8 Hz, 1H), 1,98-1,78 (m, 3H), 1,72 (dd, J = 12,7, 9,6 Hz, 1H), 1,61-1,50 (m, 1H), 1,24 (t, J = 7,2 Hz, 3H). Ejemplo 5
(6R)-6-{4-[3-(1,3,4-tiadiazol-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo (5)

Etapa 1. Síntesis de 4-[3-(1,3,4-tiadiazol-2-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (C11). Este experimento se llevó a cabo en 8 lotes idénticos.
A una mezcla de 4-[3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (400 mg, 1,03 mmol) en acetonitrilo (20 ml) se le añadió 2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo (XPhos; 147 mg, 0,308 mmol), seguido de 2-bromo-1,3,4-tiadiazol (203 mg, 1,23 mmol), carbonato sódico (163 mg, 1,54 mmol), agua (4 ml) y tris(dibencilidenoacetona)dipaladio (0) (94,0 mg, 0,103 mmol). La mezcla de reacción se agitó durante 7 horas a 100 °C en un recipiente cerrado herméticamente, tras lo cual se concentró al vacío y se purificó por cromatografía sobre gel de sílice (Gradiente: del 0 % al 80 % de acetato de etilo en éter de petróleo) para proporcionar un aceite de color amarillo claro (550 mg). Los productos de las 8 reacciones se combinaron y se sometieron a cromatografía sobre gel de sílice (Gradiente: del 0 % al 70 % de acetato de etilo en éter de petróleo), proporcionando el producto en forma de un sólido de color amarillo claro. Rendimiento combinado: 1,20 g, 3,45 mmol, 42 %. LCMS m/z 348,1 [M+H]+. RMN 1H (400 MHz, CDCh) 59,20 (s, 1H), 8,50 (dd, J = 7,7, 1,8 Hz, 1H), 8,47 (dd, J = 4,8, 1,8 Hz, 1H), 7,19 (dd, J = 7,7, 4,8 Hz, 1H), 3,61 (dd, J = 5, 5 Hz, 4H), 3,10 (dd, J = 5, 5 Hz, 4H), 1,48 (s, 9H). Etapa 2. Síntesis de 1-[3-(1,3,4-tiadiazol-2-il)piridin-2-il]piperazina (C12). Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 15 ml, 60 mmol) a una mezcla de C11 (1,10 g, 3,17 mmol) en acetonitrilo (30 ml) y la mezcla de reacción se agitó durante 3 horas a temperatura ambiente. Después de eliminar los disolventes al vacío, el
residuo se trituró con acetato de etilo para proporcionar un sólido de color blanco (1,0 g). Este material se recogió en una mezcla de diclorometano y metanol (10:1, 150 ml) y se trató con carbonato potásico (5,0 g, 36,2 mmol); después de agitar esta mezcla durante 16 horas a temperatura ambiente, se filtró. El filtrado se secó sobre sulfato sódico y se concentró a presión reducida para proporcionar el producto en forma de un sólido de color amarillo claro. Rendimiento: 700 mg, 2,83 mmol, 89 %. LCMS m/z 248,1 [M+H]+. RMN 1H (400 MHz, CDCls) 59,20 (s, 1H), 8,52-8,45 (m, 2H), 7,17 (dd, J = 7,6, 4,9 Hz, 1H), 3,14-3,04 (m, 8H).
Etapa 3. Síntesis de (6R)-6-{4-[3-(1,3,4-tiadiazol-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( 5 ).
Una mezcla de C12 (700 mg, 2,83 mmol), P3 (1,23 g, 4,43 mmol) y carbonato potásico (511 mg, 3,70 mmol) en acetonitrilo (20 ml) se puso en un recipiente cerrado herméticamente y se agitó a 100 °C durante 16 horas. Después, la mezcla de reacción se concentró al vacío y se purificó usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 10 % de metanol en diclorometano) para proporcionar el producto en forma de un sólido de color blanco. Rendimiento: 400 mg, 0,933 mmol, 33 %. LCMS m/z 429,2 [M+H]+. RMN 1H (400 MHz, CDCls) 59,20 (s, 1H), 8,47 8,41 (m, 2H), 7,14 (dd, J = 7,6, 4,9 Hz, 1H), 4,10 (c, J = 7,1 Hz, 2H), 3,88 (cuadruplete AB, Jab = 8,3 Hz, Avab = 21,7 Hz, 2H), 3,83-3,76 (m, 2H), 3,25-3,12 (m a, 4H), 2,73-2,55 (m a, 5H), 2,13 (dd, J = 12,6, 7,0 Hz, 1H), 2,02-1,51 (m, 5H, asumido; parcialmente oscurecido por el pico de agua), 1,24 (t, J = 7,1 Hz, 3H).
Ejem plos 6 y 7
(6R)-6-[4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo (6) y (6S)-6-(4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo ( 7 )

Etapa 1. Síntesis de 6-[4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo ( C13 ). Una suspensión de 6-oxo-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo (2,00 g, 8,88 mmol), 1-(3-metoxipiridin-2-il)piperazina, sal triclorhidrato (2,71 g, 8,96 mmol), trietilamina (7,38 ml, 52,9 mmol), cianoborohidruro sódico (3,35 g, 53,3 mmol) y sulfato de magnesio (3,21 g, 26,7 mmol) en etanol (50 ml) se agitó a 45 °C durante 16 horas. Después, la mezcla de reacción se concentró a sequedad al vacío, la cromatografía sobre gel de sílice (Eluyente: 1:10 metanol/diclorometano) proporcionó el producto en forma de un aceite de color amarillo claro, que se usó en la etapa siguiente sin purificación. LCMS m/z 403,1 [M+H]+.
Etapa 2. Síntesis de 6-[4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octano, sal trifluoroacetato ( C14 ).
Se añadió ácido trifluoroacético (20 ml) gota a gota a una solución de C13 (de la etapa anterior; <8,88 mmol) en diclorometano (80 ml). La mezcla de reacción se agitó a 10 °C durante 2 horas, tras lo cual se concentró a sequedad
a presión reducida, proporcionando el producto en forma de un aceite de color amarillo claro, que se usó directamente en la etapa siguiente. LCMS m/z 302,9 [M+H]+.
Etapa 3. Síntesis de 6-[4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo (C15). A una solución de C14 (de la etapa anterior, <8,88 mmol) y trietilamina (12,3 ml, 88,2 mmol) en diclorometano (100 ml) se le añadió cloroformiato de etilo (2,89 g, 26,6 mmol). La mezcla de reacción se agitó a 10 °C durante 16 horas, tras lo cual se concentró a sequedad al vacío. Se purificó mediante cromatografía sobre gel de sílice (Gradiente: del 0 % al 9 % de metanol en diclorometano) seguido de HPLC de fase inversa (Columna: Phenomenex Gemini C18, 10 pm; Fase móvil A: hidróxido de amonio al 0,05 % en agua; Fase móvil B: acetonitrilo; Gradiente: del 25 % al 44 % de B). El material resultante se sometió una vez más a cromatografía sobre gel de sílice (eluyente acetato de etilo, seguido de un gradiente del 0 % al 9 % de metanol en diclorometano) para proporcionar el producto en forma de un sólido de color blanco. Rendimiento: 1,68 g, 4,49 mmol, 51 % en 3 etapas. LCMS m/z 375,2 [M+H]+. RMN 1H (400 MHz, CD3OD) 8 7,80 (dd, J = 5,0, 1,5 Hz, 1H), 7,29 (dd, J = 8,0, 1,0 Hz, 1H), 6,98 (dd, J = 8,0, 5,0 Hz, 1H), 4,08 (c, J = 7,0 Hz, 2H), 3,99-3,78 (m, 4H), 3,87 (s, 3H), 3,68-3,44 (m a, 4H), 3,41-3,3 (m, 1H, asumido; parcialmente oscurecido por el pico de disolvente), 3,28-3,11 (m a, 4H), 2,40 (dd, J = 13,0, 8,0 Hz, 1H), 2,22-2,11 (m, 1H), 2,10-2,00 (m, 1H), 2,00-1,90 (m, 2H), 1,83-1,70 (m, 1H), 1,23 (t, J = 7,0 Hz, 3H).
Etapa 4. Aislamiento de (6R)-6-[4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo (6) y (6S)-6-[4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo (7).
La separación de C15 (1,67 g, 4,46 mmol) en sus enantiómeros componentes se realizó mediante cromatografía de fluidos supercríticos {Columna: Phenomenex Lux Amylose-1, 5 pm; Fase móvil: 4:1 de dióxido de carbono/[etanol que contenía 0,2% (amoniaco 7M en etanol)]; Contrapresión: 12 MPa (120 bar)}. El primer enantiómero en eluir se denominó 6 y el segundo enantiómero en eluir se denominó 7. Las configuraciones absolutas indicadas se asignaron basándose en la determinación estructural de rayos X realizada sobre la sal clorhidrato de 6 (véase a continuación).
Rendimiento de 6: 394 mg, 1,05 mmol, 24 %. Tiempo de retención: 5,80 minutos {Condiciones analíticas. Columna: Phenomenex Lux Amylose-1, 250 x 4,6 mm, 5 pm; Fase móvil A: dióxido de carbono; Fase móvil B: [etanol que contenía 0,2 % (amoniaco 7 M en etanol)]; Gradiente: 5% durante 1 minuto, después del 5 % al 60 % de B durante 8,0 minutos; Caudal: 3,0 ml/minuto; Contrapresión: 12 MPa (120 bar)}.
Rendimiento de 7: 453 mg, 1,21 mmol, 27 %. LCMS m/z 375,3 [M+H]+. RMN 1H (400 MHz, CDCh) 87,87 (d a, J = 4,7 Hz, 1H), 7,03 (d a, J = 7,8 Hz, 1H), 6,83 (dd, J = 8,0, 4,9 Hz, 1H), 4,10 (c, J = 7,2 Hz, 2H), 3,90 (d, medio cuadruplete AB, J = 8,6 Hz, 1H), 3,86-3,82 (m, 1H), 3,84 (s, 3H), 3,79 (cuadruplete AB, Jab = 8,4 Hz, Avab = 8,4 Hz, 2H), 3,47-3,39 (m a, 4H), 2,71-2,57 (m a, 5H), 2,14 (dd, J = 12,9, 7,0 Hz, 1H), 1,99-1,81 (m, 3H), 1,77 (dd, J = 12,5, 9,8 Hz, 1H), 1,67 1,55 (m, 1H), 1,24 (t, J = 7,0 Hz, 3H). Tiempo de retención: 6,68 minutos (Condiciones analíticas idénticas a las usadas para 6).
Conversión de 6 en su sal clorhidrato (6 • HCl) para la determinación estructural de rayos X de monocristal
Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 6,3 pl, 25 pmol) a una solución de 6 (9,5 mg, 25 pmol) en etanol (254 pl) en un vial de 1 dram. Después de agitar el vial a mano durante 30 segundos, se dejó reposar, sin cubrir, durante 18 horas. En este momento, el etanol se había evaporado, dejando pequeños cristales con forma de aguja de 6 • HCl, uno de los cuales se analizó mediante determinación estructural de rayos X de monocristal, como se describe a continuación.
Determinación estructural de rayos X de monocristal de 6 • HCl
Análisis de rayos X de monocristal
La recogida de datos se realizó en un difractómetro Bruker APEX a -150 °C. La recogida de datos consistió en exploraciones omega y phi.
La estructura se resolvió por métodos directos usando el paquete de programas informáticos SHELX en el grupo espacial de clase ortorrómbica P2 -|2 -|21. La estructura se refinó posteriormente mediante el método de mínimos cuadrados de matriz completa. Se encontraron todos los átomos distintos de hidrógeno y se refinaron usando parámetros de desplazamiento anisotrópico.
Los átomos de hidrógeno ubicados en el nitrógeno como aceptor de protones se encontraron a partir del mapa de diferencias de Fourier se refinaron con distancias restringidas. Los átomos de hidrógeno se pusieron en posiciones calculadas y se les permitió montar en sus átomos portadores. El refinamiento final incluyó parámetros de desplazamiento isotrópicos para todos los átomos de hidrógeno.
Se aplicó el algoritmo Squeeze mediante el programa Platon para eliminar la densidad de electrones observada del disolvente de acetato de etilo probablemente desordenado que se encuentra en el centro de simetría. El factor de concordancia mejoró en un 1,7 %.
El análisis de la estructura absoluta usando métodos de probabilidad (Hooft, 2008) se realizó usando el programa PLATON (Spek). Los resultados indican que la estructura absoluta se ha asignado correctamente. El método calcula que la probabilidad de que la estructura sea correcta es de 1,000. El parámetro Hooft se indica como 0,035 con una esd de 0,011.
La unidad asimétrica comprendió dos moléculas de 6 protonado (2+), dos iones cloruro (2-) y una molécula de agua (semiocupada). El índice R final fue 5,3 %. Una diferencia final de Fourier reveló que hay ninguna densidad electrónica faltante o extraviada.
El cristal pertinente, los datos recogidos y la información del refinado se resumen en la tabla A. Las coordenadas atómicas, longitudes de enlace, ángulos de enlace y parámetros de desplazamiento se enumeran en las tablas B-D.
Software y Referencias
SHELXTL, versión 5.1, Bruker AXS, 1997.
PLATON, A. L. Spek, J. Appl. Cryst. 2003, 36, 7-13.
MERCURY, C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler y J. van de Streek, J. Appl. Cryst. 2006, 39, 453-457.
OLEX2, O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard y H. Puschmann, J. Appl. Cryst. 2009, 42, 339 341.
R. W. W. Hooft, L. H. Straver y A. L. Spek, J. Appl. Cryst. 2008, 41, 96-103.
H. D. Flack, Acta Cryst. 1983, A39, 867-881.
Tabla A. Datos del cristal y el refinado de la estructura para 6 • HCl.
Fórmula empírica C20H32N4O3,5Cl
Peso de la fórmula 419,95
Temperatura 150,15(2) °C (123(2) °K)
Longitud de onda 1,54178 A
Sistema cristalino Ortorrómbico
Grupo espacial P212-|21
Dimensiones de la celda unitaria a = 7,0692(2) A a 90°
b = 18,3143(4) A P 90°
c = 35,5680(8) A Y 90°
Volumen 4604,90(19) A3
Z 8
Densidad (calculada) 1,209 Mg/m3
Coeficiente de absorción 1,705 mm-1
F(000) 1792
Tamaño del cristal 0,160 x 0,140 x 0,040 mm3
Intervalo de theta para la recopilación de datos de 2,484 a 68,745°
Intervalos de índice -8<=h<=8, -21<=k<=22 -42<=/<=42
Reflexiones recogidas 52301
Reflexiones independientes 8415 [Rm = 0,0833]
Completitud de theta = 67,679° 100,0 %
Método de refinamiento Mínimos cuadrados de matriz completa en F2 Datos/restricciones/parámetros 8415 / 2 / 524
Bondad de ajuste en F2 0,997
Índices R finales [l>2a(l)] R1 = 0,0531, wR2 = 0,1262
Índices R (todos los datos) R1 = 0,0695, wR2 = 0,1342
Parámetro de estructura absoluta 0,035(11)
Coeficiente de extinción n/a
Mayor diferencia de pico y orificio 0,454 y -0,281 e.A'3
Tabla B. Coordenadas atómicas (* 104) y parámetros de desplazamiento isotrópico equivalentes (A2 * 103) para 6 • HCl. U(eq.) se define como un tercio de la traza del tensor Uij ortogonalizado.
x y z U(eq.) Cl(1) 10393(2) 8029(1) 7707(1) 38(1) Cl(2) 4410(2) 5729(1) 7217(1) 38(1)
N(1) -89(6) 6045(2) 8702(1) 40(1)
N(2) -240(5) 5431(2) 8129(1) 29(1)
N(3) 188(5) 5589(2) 7322(1) 23(1)
N(4) 1284(7) 6561(3) 5832(1) 52(1)
N(5) 4825(5) 2854(2) 8717(1) 35(1)
N(6) 4757(5) 3402(2) 8132(1) 28(1)
N(7) 5288(5) 3142(2) 7333(1) 26(1)
N(8) 4441(8) 4052(3) 5849(1) 58(1)
O(1) -562(5) 4113(2) 8526(1) 40(1)
O(2) 664(8) 6759(3) 5219(1) 86(2)
O(3) 3569(7) 6414(3) 5416(1) 68(1)
O(4) 4476(5) 4777(2) 8501(1) 36(1)
O(5) 5152(8) 4376(3) 5246(1) 81(2)
O(6) 2155(8) 4430(3) 5476(1) 73(1)
O(1W) 9873(7) 2630(2) 6413(1) 78(1)
C(1) -350(7) 4742(3) 8727(1) 36(1)
C(2) -408(8) 3438(3) 8723(2) 48(1)
C(3) -342(7) 4772(3) 9115(1) 44(1)
C(4) -231(8) 5440(3) 9295(1) 53(1)
C(5) -139(8) 6056(3) 9079(1) 45(1)
C(6) -180(6) 5408(3) 8522(1) 33(1)
C(7) 845(6) 4904(2) 7905(1) 29(1)
C(8) 33(6) 4869(2) 7515(1) 29(1)
C(9) -767(6) 6154(2) 7556(1) 29(1)
C(10) -58(7) 6160(2) 7960(1) 30(1)
C(11) 137(6) 4964(2) 6689(1) 31(1)
C(12) -437(8) 5233(2) 6301(1) 37(1)
C(13) 148(7) 6039(2) 6306(1) 33(1)
C(14) -263(7) 6279(2) 6713(1) 35(1)
C(15) -649(7) 5572(2) 6933(1) 28(1)
C(16) 2171(7) 6173(3) 6154(1) 41(1)
C(17) -623(8) 6504(3) 5979(1) 44(1)
C(18) 1720(10) 6591(4) 5468(2) 61(2)
C(19) 4199(12) 6547(6) 5033(2) 103(3)
C(20) 6119(11) 6413(5) 5019(2) 92(3)
C(21) 4643(6) 4160(2) 8713(1) 32(1)
C(22) 4652(7) 5465(2) 8686(1) 41(1)
C(23) 4631(7) 4162(3) 9100(1) 40(1)
C(24) 4699(8) 3508(3) 9300(1) 45(1)
C(25) 4763(7) 2877(3) 9097(1) 44(1)
C(26) 4782(6) 3473(2) 8526(1) 29(1)
C(27) 5791(6) 3919(2) 7892(1) 31(1)
C(28) 5009(6) 3885(2) 7494(1) 28(1)
C(29) 4384(7) 2595(2) 7587(1) 30(1)
C(30) 5060(6) 2666(2) 7988(1) 32(1)
(continuación)
x y z U(eq.) C(31) 4556(6) 3090(2) 6938(1) 29(1) C(32) 5513(7) 3619(2) 6667(1) 34(1) C(33) 5014(8) 3304(2) 6283(1) 41(1) C(34) 5028(8) 2479(2) 6339(1) 46(1) C(35) 4957(7) 2340(2) 6764(1) 35(1) C(36) 3199(8) 3641(3) 6112(1) 49(1) C(38) 4012(11) 4292(3) 5496(2) 63(2) C(37) 6098(9) 3636(3) 5945(1) 50(1) C(39) 1589(12) 4712(5) 5107(2) 83(2) C(40) -417(15) 4838(6) 5139(2) 110(3)
Tabla C. Longitudes [Al y ángulos [°] de enlace para 6 • HCl.
N(1)-C(6) 1,334(6)
N(1)-C(5) 1,342(6)
N(2)-C(6) 1,397(5)
N(2)-C(7) 1,469(5)
N(2)-C(10) 1,469(5)
N(3)-C(8) 1,489(5)
N(3)-C(9) 1,490(5)
N(3)-C(15) 1,508(5)
N(4)-C(18) 1,334(7)
N(4)-C(17) 1,449(7)
N(4)-C(16) 1,485(6)
N(5)-C(26) 1,323(5)
N(5)-C(25) 1,351(6)
N(6)-C(26) 1,407(5)
N(6)-C(30) 1,458(5)
N(6)-C(27) 1,470(5)
N(7)-C(28) 1,489(5)
N(7)-C(29) 1,493(5)
N(7)-C(31) 1,501(5)
N(8)-C(38) 1,364(7)
N(8)-C(37) 1,438(7)
N(8)-C(36) 1,488(7)
O(1)-C(1) 1,364(6)
O(1)-C(2) 1,425(5)
O(2)-C(18) 1,200(8)
O(3)-C(18) 1,359(8)
O(3)-C(19) 1,453(7)
O(4)-C(21) 1,364(5)
O(4)-C(22) 1,426(5)
O(5)-C(38) 1,211(7)
O(6)-C(38) 1,339(8)
O(6)-C(39) 1,465(7)
C(1)-C(3) 1,383(6)
C(1)-C(6) 1,425(6)
C(3)-C(4) 1,384(8)
C(4)-C(5) 1,367(7)
(continuación)
C(7)-C(8) 1,502(6)
C(9)-C(10) 1,521(5)
C(11)-C(15) 1,516(6)
C(11)-C(12) 1,522(6)
C(12)-C(13) 1,534(6)
C(13)-C(17) 1,542(6)
C(13)-C(14) 1,539(6)
C(13)-C(16) 1,549(7)
C(14)-C(15) 1,536(6)
C(19)-C(20) 1,380(12)
C(21)-C(23) 1,375(6)
C(21)-C(26) 1,427(6)
C(23)-C(24) 1,394(7)
C(24)-C(25) 1,363(7)
C(27)-C(28) 1,519(6)
C(29)-C(30) 1,509(6)
C(31)-C(32) 1,525(6)
C(31)-C(35) 1,532(6)
C(32)-C(33) 1,522(6)
C(33)-C(34) 1,523(7)
C(33)-C(36) 1,549(7)
C(33)-C(37) 1,551(7)
C(34)-C(35) 1,533(6)
C(39)-C(40) 1,440(12)
C(6)-N(1)-C(5) 119,5(4)
C(6)-N(2)-C(7) 120,5(3)
C(6)-N(2)-C(10) 115,7(3)
C(7)-N(2)-C(10) 109,2(3)
C(8)-N(3)-C(9) 108,9(3)
C(8)-N(3)-C(15) 112,1(3)
C(9)-N(3)-C(15) 110,5(3)
C(18)-N(4)-C(17) 124,5(5)
C(18)-N(4)-C(16) 132,1(6)
C(17)-N(4)-C(16) 94,7(4)
C(26)-N(5)-C(25) 119,1(4)
C(26)-N(6)-C(30) 115,7(3)
C(26)-N(6)-C(27) 120,9(3)
C(30)-N(6)-C(27) 108,5(3)
C(28)-N(7)-C(29) 108,8(3)
C(28)-N(7)-C(31) 112,0(3)
C(29)-N(7)-C(31) 112,1(3)
C(38)-N(8)-C(37) 124,7(5)
C(38)-N(8)-C(36) 127,6(6)
C(37)-N(8)-C(36) 93,6(4)
C(1)-O(1)-C(2) 117,8(3)
C(18)-O(3)-C(19) 112,5(6)
C(21)-O(4)-C(22) 117,9(3)
C(38)-O(6)-C(39) 112,6(5)
O(1)-C(1)-C(3) 123,8(4)
O(1)-C(1)-C(6) 117,6(4)
C(3)-C(1)-C(6) 118,5(4)
(continuación)
C(1)-C(3)-C(4) 119,8(5)
C(5)-C(4)-C(3) 118,2(4)
N(1)-C(5)-C(4) 123,4(5)
N(1)-C(6)-N(2) 117,1(4)
N(1)-C(6)-C(1) 120,5(4)
N(2)-C(6)-C(1) 122,3(4)
N(2)-C(7)-C(8) 109,3(3)
N(3)-C(8)-C(7) 111,1(3)
N(3)-C(9)-C(10) 112,5(3)
N(2)-C(10)-C(9) 110,5(3)
C(15)-C(11)-C(12) 100,6(3)
C(11)-C(12)-C(13) 103,2(3)
C(12)-C(13)-C(17) 115,3(4)
C(12)-C(13)-C(14) 103,6(3)
C(17)-C(13)-C(14) 119,1(4)
C(12)-C(13)-C(16) 113,3(4)
C(17)-C(13)-C(16) 88,6(4)
C(14)-C(13)-C(16) 117,2(4)
C(15)-C(14)-C(13) 105,8(3)
N(3)-C(15)-C(11) 113,4(3)
N(3)-C(15)-C(14) 112,4(3)
C(11)-C(15)-C(14) 105,2(3)
N(4)-C(16)-C(13) 87,4(4)
N(4)-C(17)-C(13) 89,0(4)
O(2)-C(18)-N(4) 125,8(7)
O(2)-C(18)-O(3) 124,0(6)
N(4)-C(18)-O(3) 110,2(5)
C(20)-C(19)-O(3) 107,8(7)
O(4)-C(21)-C(23) 123,4(4)
O(4)-C(21)-C(26) 118,5(4)
C(23)-C(21)-C(26) 118,1(4)
C(21)-C(23)-C(24) 120,4(5)
C(25)-C(24)-C(23) 117,4(4)
N(5)-C(25)-C(24) 123,8(4)
N(5)-C(26)-N(6) 115,7(4)
N(5)-C(26)-C(21) 121,1(4)
N(6)-C(26)-C(21) 123,1(4)
N(6)-C(27)-C(28) 109,5(3)
N(7)-C(28)-C(27) 110,4(3)
N(7)-C(29)-C(30) 112,3(3)
N(6)-C(30)-C(29) 111,4(3)
N(7)-C(31)-C(32) 113,5(3)
N(7)-C(31)-C(35) 111,8(3)
C(32)-C(31)-C(35) 103,4(3)
C(33)-C(32)-C(31) 102,8(4)
C(32)-C(33)-C(34) 105,0(4)
C(32)-C(33)-C(36) 113,1(4)
C(34)-C(33)-C(36) 116,9(5)
C(32)-C(33)-C(37) 115,6(4)
C(34)-C(33)-C(37) 119,1(4)
C(36)-C(33)-C(37) 87,0(4)
(continuación)
C(33)-C(34)-C(35) 107,0(4)
C(31)-C(35)-C(34) 104,8(3)
N(8)-C(36)-C(33) 87,7(4)
O(5)-C(38)-O(6) 126,0(6)
O(5)-C(38)-N(8) 124,8(7)
O(6)-C(38)-N(8) 109,2(5)
N(8)-C(37)-C(33) 89,4(4)
C(40)-C(39)-O(6) 104,8(7)
Transformaciones de simetría usadas para generar átomos equivalentes.
Tabla D. Parámetros de desplazamiento anisotrópico (A2 x 103) para 6 • HCl. El exponente del factor de desplazamiento anisotrópico toma la forma: -2n2[h2 a*2U11 ... 2 h k a* b* U12 ].
U11 U22 U33 U23 U13 U12 Cl(1) 25(1) 30(1) 61(1) 2(1) 0(1) -2(1) Cl(2) 23(1) 30(1) 63(1) 2(1) 3(1) -2(1) N(1) 44(2) 46(2) 29(2) -6(2) 3(2) 7(2) N(2) 33(2) 31(2) 23(2) 2(1) 1(2) -1(2) N(3) 22(2) 24(2) 25(2) -2(1) 1(1) -1(1) N(4) 71(3) 62(3) 24(2) 4(2) 1(2) -10(2) N(5) 34(2) 38(2) 32(2) 11(2) 4(2) 0(2) N(6) 33(2) 25(2) 25(2) 5(1) -2(2) 0(2) N(7) 24(2) 23(2) 31(2) 2(1) -2(2) -1(2) N(8) 83(3) 58(3) 33(2) 10(2) 5(2) 7(3) O(1) 45(2) 38(2) 37(2) 10(1) 2(2) -1(2) O(2) 101(4) 128(4) 29(2) 14(2) -10(2) -33(3) O(3) 84(3) 84(3) 35(2) -12(2) 9(2) -35(3) O(4) 49(2) 31(2) 28(2) -1(1) 1(2) 4(2) O(5) 123(4) 85(3) 34(2) 12(2) 19(3) 2(3) O(6) 106(4) 78(3) 35(2) 5(2) -6(2) 20(3) O(1W)82(3) 71(3) 82(3) 19(2) 13(3) 2(3) C(1) 29(2) 47(3) 31(2) 5(2) 4(2) 4(2) C(2) 47(3) 44(3) 52(3) 21(2) -2(3) -5(3) C(3) 42(3) 61(3) 31(2) 14(2) 4(2) 5(3) C(4) 55(3) 80(4) 23(2) -1(2) 2(2) 8(3) C(5) 50(3) 51(3) 34(2) -7(2) 1(2) 7(3) C(6) 29(2) 42(2) 28(2) 2(2) 0(2) 4(2) C(7) 32(2) 25(2) 29(2) 1(2) 3(2) 4(2) C(8) 36(2) 22(2) 27(2) 1(2) 4(2) -1(2) C(9) 33(2) 26(2) 28(2) -2(2) 3(2) 5(2) C(10)38(3) 26(2) 27(2) -2(2) 0(2) 0(2) C(11)34(2) 30(2) 28(2) -1(2) 1(2) -1(2) C(12)44(3) 36(2) 31(2) -4(2) -1(2) -1(2) C(13)41(3) 33(2) 25(2) 1(2) -2(2) 4(2) C(14)44(3) 30(2) 30(2) 1(2) 1(2) 8(2) C(15)29(2) 32(2) 22(2) -1(2) -4(2) -2(2) C(16)47(3) 46(3) 29(2) 3(2) 0(2) -1(2) C(17)63(3) 38(2) 30(2) 1(2) -7(2) -1(3) C(18)86(5) 71(4) 27(3) 4(3) -4(3) -30(4)
(continuación)
U11 U22 U33 U23 U13 U12
C(19)87(6) 183(9) 38(4) -28(5) 15(3) -50(6) C(20)82(5) 155(8) 37(3) -22(4) 10(3) -30(5) C(21)28(2) 37(2) 33(2) 5(2) 2(2) -1(2) C(22)41(3) 37(2) 45(3) -4(2) 2(2) 5(2) C(23)39(3) 53(3) 29(2) 1(2) 4(2) 1(3) C(24)46(3) 65(3) 24(2) 10(2) 2(2) 2(3) C(25)42(3) 50(3) 39(3) 20(2) 2(2) -3(3) C(26)25(2) 31(2) 31(2) 5(2) 2(2) -4(2) C(27)35(2) 26(2) 32(2) 1(2) 1(2) -4(2) C(28)34(2) 23(2) 29(2) 2(2) 2(2) 1(2) C(29)27(2) 24(2) 39(2) 5(2) 1(2) -2(2) C(30)33(2) 28(2) 34(2) 7(2) 0(2) -2(2) C(31)26(2) 33(2) 28(2) -5(2) -4(2) 2(2) C(32)42(3) 33(2) 28(2) 2(2) -2(2) 1(2) C(33)56(3) 37(2) 29(2) -4(2) -1(2) 3(2) C(34)63(4) 35(2) 41(3) -10(2) 0(3) 5(2) C(35)35(3) 33(2) 37(2) -4(2) -2(2) 1(2) C(36)60(3) 53(3) 33(3) -1(3) 0(2) 8(3) C(38)91(5) 48(3) 49(4) 0(3) 5(3) 8(4) C(37)71(4) 46(3) 34(3) 0(2) 7(2) 4(3) C(39)104(6) 95(6) 50(4) 18(4) -15(4) 18(5) C(40)113(7) 153(8) 66(5) 0(5) -11(5) 20(7)
Síntesis alternativa de l ejem plo 6
(6R)-6-[4-(3-metoxipiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo (6)

1-(3-metoxipiridin-2-il)piperazina, sal clorhidrato (130 mg, 0,566 mmol), P4 (240 mg, 0,679 mmol), carbonato potásico (313 mg, 2,26 mmol) y acetonitrilo (2,3 ml) se pusieron en un recipiente cerrado herméticamente y se calentaron a 90 °C durante una noche. Después de que la mezcla de reacción se hubiese enfriado a temperatura ambiente, esta se adsorbió sobre gel de sílice y se purificó por cromatografía sobre gel de sílice (Gradiente: del 0 % al 20 % de metanol en diclorometano), proporcionando el producto en forma de un aceite de color pardo claro. Rendimiento: 80 mg, 0,21 mmol, 37 %. LCMS m/z 375,1 [M+H]+. RMN 1H (400 MHz, CDCla) 87,88 (dd, J = 4,9, 1,4 Hz, 1H), 7,03 (dd, J = 7,8, 1,2 Hz, 1H), 6,84 (dd, J = 8,0, 4,9 Hz, 1H), 4,10 (c, J = 7,2 Hz, 2H), 3,91 (d, medio cuadruplete AB, J = 8,2 Hz, 1H), 3,87-3,83 (m, 1H), 3,85 (s, 3H), 3,80 (cuadruplete AB, Jab = 8,2 Hz, Avab = 8,4 Hz, 2H), 3,48-3,39 (m a, 4H), 2,69 2,56 (m a, 5H), 2,15 (dd, J = 12,9, 7,0 Hz, 1H), 2,00-1,80 (m, 3H), 1,76 (dd, J = 12,7, 9,6 Hz, 1H), 1,65-1,53 (m, 1H, asumido; parcialmente oscurecido por el pico de agua), 1,24 (t, J = 7,0 Hz, 3H).
La configuración absoluta indicada de este material sintetizado (6 - Síntesis alternativa) se estableció comparándolo con el material (6 - Preparación de rayos X) usado para la preparación de la muestra de estructura cristalina de rayos
X descrita anteriormente, como sigue. El racemato C15 se examinó usando cromatografía de fluidos supercríticos {Columna: Phenomenex Lux Amylose-1,250 x 4,6 mm, 5 ^m; Fase móvil A: dióxido de carbono; Fase móvil B: [metanol que contenía 0,2 % (amoniaco 7 M en metanol); Gradiente: al 5 % de B durante 1,0 minuto, después del 5 % al 60 % de B durante 8,0 minutos; Caudal: 3,0 ml/minuto; Contrapresión: 12 MPa (120 bar)}. Se observaron dos picos: uno a los 5,82 minutos y uno a los 6,54 minutos, para los dos enantiómeros. En condiciones idénticas, 6 - Síntesis alternativa dio un tiempo de retención de 5,83 minutos y 6 - Preparación de rayos X dio un tiempo de retención de 5,83 minutos, estableciéndose que las dos muestras poseen la misma estereoquímica absoluta.
Ejemplo 8
(6R)-6-{4-[3-(pirazin-2-il)piridin-2-il]piperazin- 1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo (8)

Etapa 1. Síntesis de 4-[3-(pirazin-2-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (C16). Una mezcla de 4-[3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-il]piperazin-1-carboxilato de terc-butilo (6,00 g, 15,4 mmol), 2-bromopirazina (2,7 g, 17 mmol), tetraquis(trifenilfosfina)paladio (0) (1,78 g, 1,54 mmol) y carbonato potásico (6,39 g, 46,2 mmol) en una mezcla de tolueno (80 ml), etanol (30 ml) y agua (3 ml) se agitó a 100 °C durante 16 horas. Después, la mezcla de reacción se concentró al vacío y el residuo se purificó por cromatografía sobre gel de sílice (Gradiente: del 0 % al 50 % de acetato de etilo en éter de petróleo) para proporcionar el producto en forma de una goma de color amarillo. Rendimiento: 5,00 g, 14,6 mmol, 95 %. Lc MS m/z 342,2 [M+H]+. Rm N 1H (400 MHz, CDCb) 5 9,22 (s a, 1H), 8,68 (dd, J = 2,4, 1,5 Hz, 1H), 8,51 (d, J = 2,4 Hz, 1H), 8,35 (dd, J = 4,9, 1,7 Hz, 1H), 7,90 (dd, J = 7,6, 1,7 Hz, 1H), 7,06 (dd, J = 7,6, 4,9 Hz, 1H), 3,46-3,39 (m, 4H), 3,16-3,07 (m a, 4H), 1,45 (s, 9H).
Etapa 2. Síntesis de 2-[2-(piperazim-1-il)piridim-3-il]pirazima (C17). Una mezcla de C16 (5,30 g, 15,5 mmol) y una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 15,5 ml, 62 mmol) en diclorometano (60 ml) y metanol (20 ml) se agitó a temperatura ambiente durante 2 horas y posteriormente se calentó a 40 °C durante 1 hora. Después de eliminar el disolvente al vacío, el residuo se disolvió en metanol (100 ml), se trató con carbonato potásico (12,0 g, 86,8 mmol) y se agitó a temperatura ambiente durante 1 hora. La mezcla se concentró a presión reducida y el residuo se purificó usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 10 % de metanol en diclorometano) para proporcionar el producto. Rendimiento: 2,90 g, 12,0 mmol, 77 %. LCMS m/z 242,2 [M+H]+. RMN 1H (400 MHz, DMSO-de) 59,18 (d, J = 1,7 Hz, 1H), 8,74 (dd, J = 2,6, 1,6 Hz, 1H), 8,57 (d, J = 2,7 Hz, 1H), 8,30 (dd, J = 4,9, 2,0 Hz, 1H), 7,85 (dd, J = 7,6, 2,0 Hz, 1H), 7,07 (dd, J = 7,5, 4,8 Hz, 1H), 2,95-2,90 (m, 4H), 2,67-2,62 (m, 4H).
Etapa 3. Síntesis de (6R)-6-{4-[3-(pirazim-2-il)piridim-2-il]piperazim-1-il}-2-azaespiro[3.4]octam-2-carboxilato de etilo (8).
Una mezcla de C17 (2,50 g, 10,4 mmol), P4 (5,13 g, 14,5 mmol) y carbonato potásico (4,3 g, 31,1 mmol) en acetonitrilo (25 ml) se puso en un recipiente cerrado herméticamente y se agitó a 100 °C durante 48 horas. Después de eliminar el disolvente al vacío, el residuo se diluyó con agua (150 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron, se concentraron al vacío y se purificaron por cromatografía sobre gel de sílice (Gradiente: del 0% al 10% de metanol en diclorometano) para proporcionar el
producto en forma de una goma de color amarillo. Rendimiento: 2,90 g, 6,86 mmol, 66 %. LCMS m/z 423,2 [M+H]+. RMN 1H (400 MHz, CDCls) 59,23 (s a, 1H), 8,66 (dd, J = 2,4, 1,5 Hz, 1H), 8,48 (d, J = 2,7 Hz, 1H), 8,33 (dd, J = 4,8, 1,8 Hz, 1H), 7,88 (dd, J = 7,5, 1,8 Hz, 1H), 7,03 (dd, J = 7,3, 4,9 Hz, 1H), 4,10 (c, J = 7,1 Hz, 2H), 3,85 (cuadruplete AB, el doblete de campo bajo se ensancha, Jab = 8,3 Hz, Avab = 22,7 Hz, 2H), 3,79-3,74 (m, 2H), 3,32-3,09 (m a, 4H), 2,69-2,38 (m a, 5H), 2,10 (dd, J = 12,5, 6,8 Hz, 1H), 2,01-1,46 (m, 5H, asumido; parcialmente oscurecido por el pico de agua), 1,24 (t, J = 7,1 Hz, 3H).
E jem plo 9
(6R)-6-{4-[3-(4-metil-1H-pirazol-1-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( 9 )
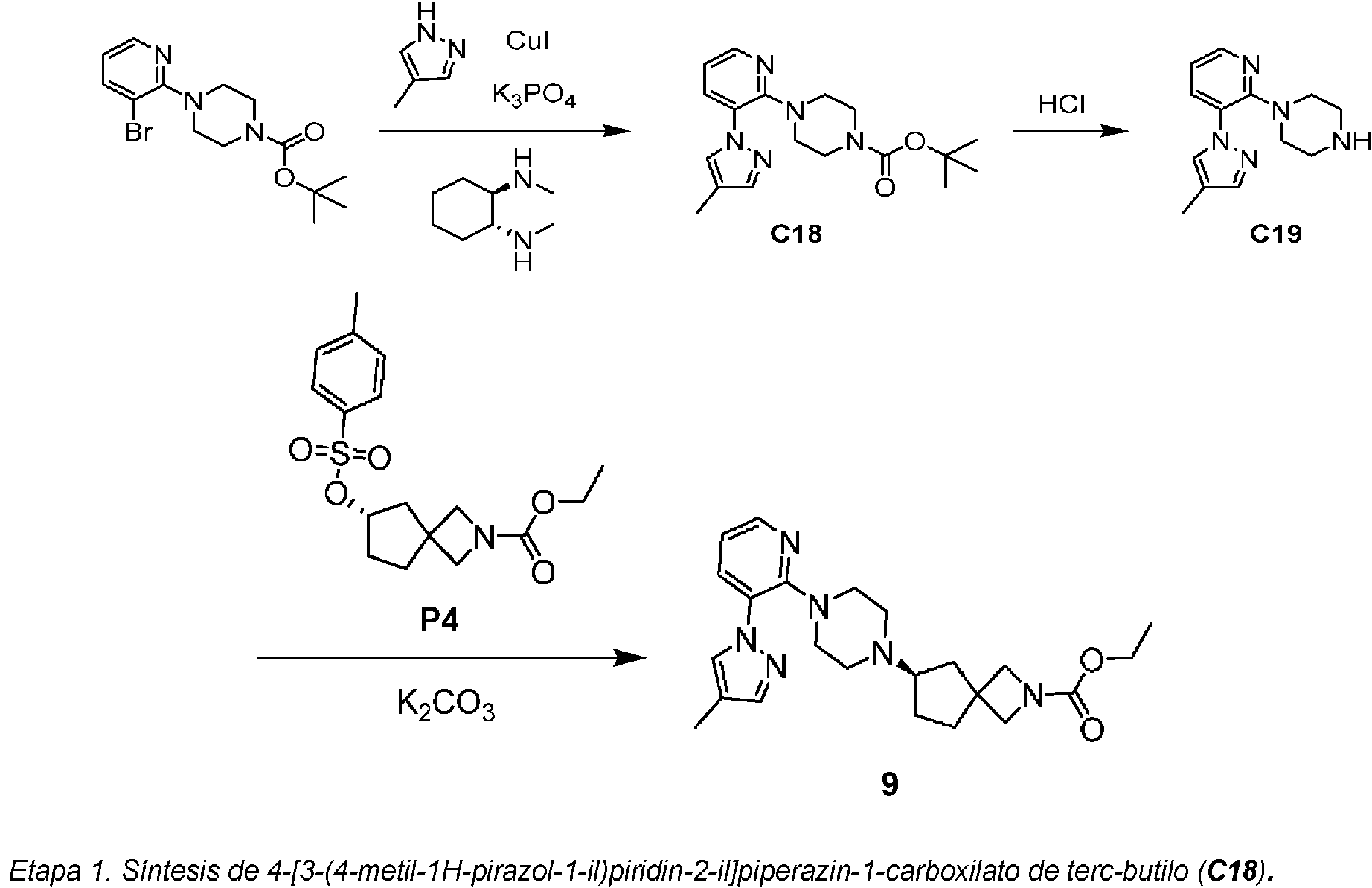
Una mezcla de 4-(3-bromopiridin-2-il)piperazin-1-carboxilato de terc-butilo (400 mg, 1,17 mmol), 4-metil-1H-pirazol (144 mg, 1,75 mmol), yoduro de cobre (I) (22 mg, 0,12 mmol), (1R,2R)-N,N'-dimetilciclohexan-1,2-diamina (34 mg, 0,24 mmol) y fosfato potásico (746 mg, 3,51 mmol) en 1 -metilpirrolidin-2-ona (4 ml) se puso en un recipiente cerrado herméticamente y se agitó a 140 °C durante 16 horas. La mezcla de reacción se concentró al vacío y el residuo se sometió a cromatografía sobre gel de sílice (Gradiente: del 0 % al 100 % de acetato de etilo en éter de petróleo), proporcionando el producto en forma de un sólido de color amarillo claro (160 mg), que se usó directamente en la etapa siguiente. Según análisis por RMN 1H, este material no era completamente puro. LCMS m/z 344,2 [M+H]+. RMN 1H (400 MHz, CDCls), únicamente picos del producto: 58,24 (dd, J = 4,9, 1,7 Hz, 1H), 7,78-7,76 (m, 1H), 7,71 (dd, J = 7,8, 1,7 Hz, 1H), 7,54 (s a, 1H), 6,97 (dd, J = 7,7, 4,8 Hz, 1H), 3,46-3,38 (m, 4H), 2,97-2,89 (m, 4H), 2,16 (s, 3H), 1,46 (s, 9H).
Etapa 2. Síntesis de 1-[3-(4-metil-1H-pirazol-1-il)piridin-2-il]piperazina ( C19 ).
Una mezcla de C18 (de la etapa anterior; 160 mg, <0,466 mmol) y una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 0,5 ml, 2 mmol) en diclorometano (10 ml) se agitó a temperatura ambiente durante 3 horas, tras lo cual se concentró al vacío. El residuo se disolvió en metanol (20 ml), se trató con carbonato potásico (200 mg, 1,45 mmol), se agitó a temperatura ambiente durante 20 minutos y se concentró a presión reducida. La purificación por cromatografía sobre gel de sílice (Gradiente: del 0 % al 70 % de metanol en diclorometano) proporcionó el producto en forma de un sólido de color amarillo claro. Rendimiento: 60 mg, 0,25 mmol, 21 % en dos etapas. LCMS m/z 244,2 [M+H]+. RMN 1H (400 MHz, CDCls) 58,24 (dd, J = 4,8, 1,6 Hz, 1H), 7,70 (dd, J = 7,6, 1,7 Hz, 1H), 7,66 (s a, 1H), 7,54 (s a, 1H), 7,01 (dd, J = 7,6, 4,9 Hz, 1H), 3,27-3,20 (m, 4H), 3,16-3,08 (m, 4H), 2,17 (s, 3H).
Etapa 3. Síntesis de (6R)-6-{4-[3-(4-metil-1H-pirazol-1-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo ( 9 ).
Una mezcla de C19 (60 mg, 0,25 mmol), P4 (131 mg, 0,371 mmol) y carbonato potásico (102 mg, 0,738 mmol) en
acetonitrilo (3 ml) se puso en un recipiente cerrado herméticamente y se agitó a 100 °C durante 48 horas. Después de eliminar el disolvente al vacío, el residuo se purificó por cromatografía sobre gel de sílice (Gradiente: del 0 % al 20 % de metanol en diclorometano), seguido de HPLC de fase inversa (Columna: Waters XBridge C18, 5 ^m; Fase móvil A: agua que contenía hidróxido de amonio al 0,05 %; Fase móvil B: acetonitrilo; Gradiente: del 40 % al 50 % de B). El producto se aisló en forma de un sólido de color amarillo claro. Rendimiento: 25 mg, 59 ^mol, 24 %. LCMS m/z 425,3 [M+H]+. RMN 1H (400 MHz, CDCls) 58,23 (dd, J = 4,8, 1,8 Hz, 1H), 7,73 (s a, 1H), 7,68 (dd, J = 7,8, 1,7 Hz, 1H), 7,52 (s a, 1H), 6,93 (dd, J = 7,7, 4,8 Hz, 1H), 4,10 (c, J = 7,1 Hz, 2H), 3,86 (cuadruplete AB, el doblete de campo bajo se ensancha, Jab = 8,3 Hz, Avab = 21,4 Hz, 2H), 3,78 (cuadruplete A b , Jab = 8,3 Hz, Avab = 6,0 Hz, 2H), 3,09-2,94 (m a, 4H), 2,62-2,39 (m a, 5H), 2,17 (s, 3H), 2,11 (dd, J = 12,7, 6,8 Hz, 1H), 1,99-1,48 (m, 5H, asumido; parcialmente oscurecido por el pico de agua), 1,24 (t, J = 7,1 Hz, 3H).
Ejemplo 10
6-{4-[2-(2,2,2-trifluoroetoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo (10)
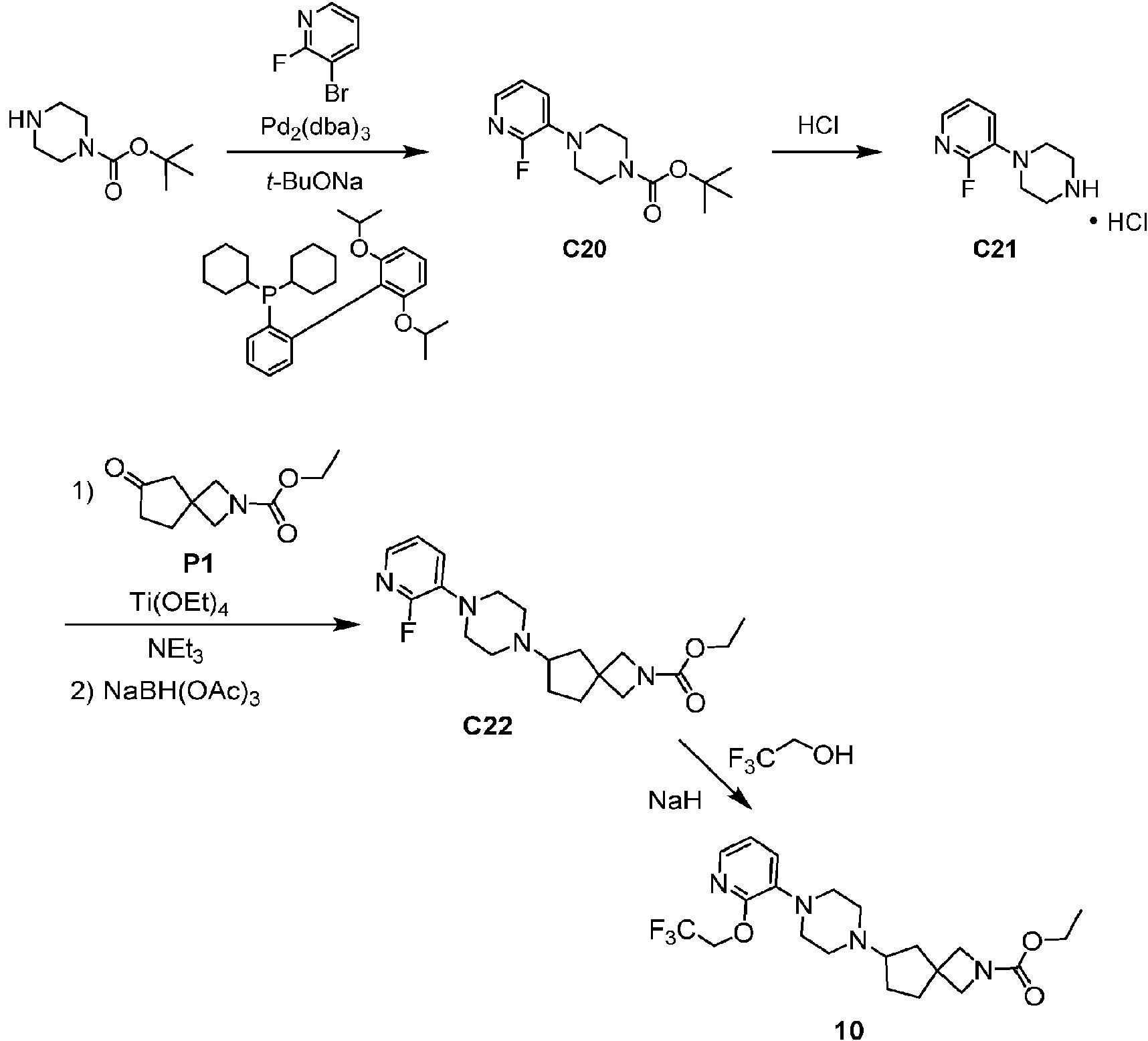
Etapa 1. Síntesis de 4-(2-fluoropiridin-3-il)piperazin-1-carboxilato de terc-butilo (C20). Una mezcla de 3-bromo-2-fluoropiridina (10,0 g, 56,8 mmol), piperazin-1-carboxilato de terc-butilo (12,7 g, 68,2 mmol), tris(dibencilidenoacetona)dipaladio (0) (2,60 g, 2,84 mmol), [2',6'-bis(propan-2-iloxi)bifenil-2-il](diciclohexil)fosfano (RuPhos; 2,67 g, 5,72 mmol) y terc-butóxido sódico (11,0 g, 114 mmol) en 1,4-dioxano (150 ml) se agitó a 110 °C durante 16 horas. Después de que la mezcla de reacción se hubiese concentrado al vacío, el residuo se diluyó con acetato de etilo (300 ml), se lavó secuencialmente con agua (2 x 150 ml) y una solución acuosa saturada de cloruro sódico (150 ml), se secó sobre sulfato sódico, se filtró y se concentró a presión reducida. La cromatografía sobre gel de sílice (Gradiente: del 50 % al 100 % de acetato de etilo en éter de petróleo) proporcionó el producto en forma de una goma de color pardo. Rendimiento: 5,20 g, 18,5 mmol, 33 %. LCMS m/z 282,2 [M+H]+. RMN 1H (400 MHz, CDCls) 5 7,79 (ddd, J = 4,9, 1,7, 1,5 Hz, 1H), 7,30-7,23 (m, 1H), 7,12 (ddd, J = 7,8, 4,8, 1,4 Hz, 1H), 3,61 (dd a, J = 5,1,5,1 Hz, 4H), 3,06 (dd a, J = 5,1,4,9 Hz, 4H), 1,50 (s, 9H).
Etapa 2. Síntesis de 1-(2-fluoropiridin-3-il)piperazina, sal clorhidrato (C21).
A una solución de C20 (5,20 g, 18,5 mmol) en diclorometano (20 ml) se le añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4,0 M; 18,5 ml, 74,0 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 4 horas, tras lo cual se concentró al vacío para proporcionar el producto en bruto en forma de un sólido de color pardo claro, que se usó en la etapa siguiente sin purificación. Según análisis por RMN 1H, este material no era completamente puro. LCMS m/z 182,2 [M+H]+. RMN 1H (400 MHz, DMSO-da), solo picos del producto: 89,7-9,4 (m a, 2H), 7,80 (d a, J = 4,9 Hz, 1H), 7,58 (ddd, J = 10,9, 7,9, 1,5 Hz, 1H), 7,29 (ddd, J = 7,7, 4,9, 1,2 Hz, 1H), 3,33-3,15 (m, 8H).
Etapa 3. Síntesis de 6-[4-(2-fluoropiridin-3-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo (C22). Una mezcla de C21 (de la etapa anterior; <18,5 mmol), P1 (4,00 g, 18,4 mmol), etóxido de titanio (IV) (16,8 g, 73,6 mmol) y trietilamina (9,3 g, 92 mmol) en diclorometano (80 ml) y metanol (80 ml) se agitó a temperatura ambiente durante una noche. Después se añadió triacetoxiborohidruro sódico (19,5 g, 92,0 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante otras 3 horas. La reacción se interrumpió con agua (10 ml), lo que produjo un precipitado de color blanco; después la mezcla se secó sobre sulfato sódico y se filtró. El lecho de filtro se lavó con acetato de etilo (100 ml) y los filtrados combinados se concentraron al vacío. La cromatografía sobre gel de sílice (Gradiente: del 50 % al 100 % de acetato de etilo en éter de petróleo) proporcionó el producto en forma de una goma de color blanco. Rendimiento: 2,4 g, 6,6 mmol, 36 % en 2 etapas. LCMS m/z 363,2 [M+H]+. RMN 1H (400 MHz, CDCh) 87,81 (ddd, J = 4,9, 1,7, 1,5 Hz, 1H), 7,29 (ddd, J = 10,3, 7,8, 1,7 Hz, 1H), 7,13 (ddd, J = 7,8, 4,8, 1,3 Hz, 1H), 4,10 (c, J = 7,1 Hz, 2H), 3,90 (cuadruplete AB, Jab = 8,6 Hz, Avab = 30,4 Hz, 2H), 3,80 (cuadruplete AB, Jab = 8,3 Hz, Avab = 7,6 Hz, 2H), 3,31 (dd a, J = 4,9, 4,6 Hz, 4H), 3,11-2,98 (m a, 5H), 2,24 (dd, mitad del patrón de ABX, J = 13,1, 7,7 Hz, 1H), 2,18 2,09 (m, 1H), 2,08-1,82 (m, 4H), 1,24 (t, J = 7,1 Hz, 3H).
Etapa 4. Síntesis de 6-{4-[2-(2,2,2-trifluoroetoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo (10). Se añadió hidruro sódico (dispersión al 60 % en aceite mineral; 32 mg, 0,80 mmol) en tetrahidrofurano (1 ml) a una solución a 0 °C de 2,2,2-trifluoroetanol (75 mg, 0,75 mmol) en W,W-dimetilformamida (1 ml) y la mezcla resultante se agitó a 0 °C durante 30 minutos. Se añadió una solución de C22 (90 mg, 0,25 mmol) en tetrahidrofurano (1 ml) y la mezcla de reacción se agitó a 50 °C durante 16 horas, tras lo cual se diluyó con acetato de etilo (30 ml), se lavó secuencialmente con agua (2 x 10 ml) y una solución acuosa saturada de cloruro sódico (10 ml), se secó sobre sulfato sódico, se filtró y se concentró al vacío. La HPLC de fase inversa (Columna: Phenomenex Gemini C18, 5 pm; Fase móvil A: hidróxido de amonio al 0,05% en agua; Fase móvil B: acetonitrilo; Gradiente: del 0% al 100% de B) proporcionó el producto en forma de una goma de color amarillo claro. Rendimiento: 29,5 mg, 66,7 pmol, 27 %. LCMS m/z 443,2 [M+H]+. RMN 1H (400 MHz, CDCla) 87,76 (dd, J = 4,9, 1,5 Hz, 1H), 7,14 (dd, J = 7,7, 1,6 Hz, 1H), 6,94 (dd, J = 7,6, 4,9 Hz, 1H), 4,81 (c, Jhf = 8,6 Hz, 2H), 4,11 (c, J = 7,1 Hz, 2H), 3,89 (cuadruplete AB, el doblete de campo bajo se ensancha, Jab = 8,4 Hz, Avab = 23,3 Hz, 2H), 3,81 (cuadruplete AB, Jab = 8,2 Hz, Avab = 9,6 Hz, 2H), 3,25-3,07 (m a, 4H), 2,79-2,57 (m a, 5H), 2,18 (dd a, J = 12, 7 Hz, 1 H), 2,04-1,52 (m, 5H, asumido; parcialmente oscurecido por el pico de agua), 1,25 (t, J = 7,1 Hz, 3H).
Ejemplo 11
(6R)-6-{4-[3-(1,3-tiazol-5-il)pirazin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo (11)

Etapa 1. Síntesis de 4-[3-(1,3-tiazol-5-il)pirazin-2-il]piperazin-1-carboxilato de terc-butilo (C23). A una mezcla de 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,3-tiazol (848 mg, 4,02 mmol) y 4-(3-cloropirazin-2-il)piperazin-1-carboxilato de terc-butilo (1,00 g, 3,35 mmol) en tolueno (35 ml) se le añadió agua (5 ml) y carbonato sódico (1,06 g, 10,0 mmol), seguido de [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) (245 mg, 0,335 mmol). Después el recipiente de reacción se cerró herméticamente y se calentó a 100 °C durante 16 horas. Después de eliminar los disolventes al vacío, el residuo se purificó por cromatografía sobre gel de sílice (Gradiente: del 0 % al 90 % de acetato de etilo en éter de petróleo) para proporcionar el producto en forma de un aceite de color amarillo. Rendimiento: 500 mg, 1,44 mmol, 43 %. LCMS m/z 348,1 [M+H]+. RMN 1H (400 MHz, CDCh) 89,0-8,8 (s a, 1H), 8,8-8,6 (s a, 1H), 8,23 (d, J = 2,4 Hz, 1H), 8,17 (d, J = 2,4 Hz, 1H), 3,68-3,57 (m a, 4H), 3,24-3,14 (m a, 4H), 1,48 (s, 9H).
Etapa 2. Síntesis de 2-(piperazin-1-il)-3-(1,3-tiazol-5-il)pirazina, sal clorhidrato (C24).
Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M; 3 ml, 12 mmol), a una solución de C23 (500 mg, 1,44 mmol) en acetonitrilo (9 ml) y la mezcla de reacción se agitó durante 16 horas a temperatura ambiente. Después se concentró al vacío y el residuo se trituró con acetato de etilo para proporcionar el producto en forma de un sólido de color amarillo. Rendimiento: 330 mg, 1,16 mmol, 81 %. LCMS m/z 248,1 [M+H]+. RMN 1H (400 MHz, DMSO-d6), picos característicos: 88,36 (d, J = 2,4 Hz, 1H), 8,32 (d, J = 2,4 Hz, 1H), 3,40-3,33 (m a, 4H), 3,32-3,23 (m a, 4H).
Etapa 3. Síntesis de (6R)-6-{4-[3-(1,3-tiazol-5-il)pirazin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo (11). Una mezcla de C24 (base libre; 120 mg, 0,485 mmol), P3 (242 mg 0,873 mmol) y carbonato potásico (67 mg, 0,48 mmol) en acetonitrilo (4 ml) se agitó en un recipiente cerrado herméticamente durante 16 horas a 95 °C. Después de concentrarlo al vacío, el residuo se purificó usando cromatografía sobre gel de sílice (Gradiente: del 0 % al 10 % de metanol en diclorometano), seguido de HPLC de fase inversa (Columna: Phenomenex Gemini C18, 5 pm; Fase móvil A: ácido fórmico al 0,1 % en agua; Fase móvil B: acetonitrilo; Gradiente: del 12 % al 20 % de B). El producto se aisló en forma de una goma de color amarillo claro. Rendimiento: 30 mg, 70 pmol, 14 %. LCMS m/z 429,2 [M+H]+. RMN 1H (400 MHz, CDCla) 88,88 (s, 1H), 8,68 (s, 1H), 8,22 (d, J = 2,4 Hz, 1H), 8,16 (d, J = 2,4 Hz, 1H), 4,11 (c, J = 7,2 Hz, 2H), 3,89 (cuadruplete AB, Jab = 8,4 Hz, Avab = 27,7 Hz, 2H), 3,83-3,77 (m, 2H), 3,44-3,29 (m a, 4H), 2,90-2,77 (m a, 5H), 2,18 (dd, J = 13,0, 7,3 Hz, 1H), 2,05-1,70 (m, 5H), 1,24 (t, J = 7,1 Hz, 3H).
Usando la metodología descrita anteriormente para los ejemplos 1-11 y materiales de partida análogos, como se indica en la tabla, se sintetizaron los ejemplos 12-53. Véase la tabla 1 para los métodos específicos utilizados, así como para los datos de caracterización de estos ejemplos.
T l 1. M r r i n r r fi i ími l m l 12- .

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

1. El 2-oxo-6-azaespiro[3.4]octan-6-carboxilato de terc-butilo se sometió a cloruro de hidrógeno en 1,4-dioxano para eliminar el grupo protector; la reacción posterior con cloroformiato de etilo proporcionó el 2-oxo-6-azaespiro[3.4]octan-6-carboxilato de etilo requerido. RMN 1H (400 MHz, CDCh) 84,15 (c, J = 7,2 Hz, 2H), 3,63 3,43 (m a, 4H), 3,05 (cuartete AB a, Jab = 17 Hz, Avab = 38 Hz, 4H), 2,08 (dd, J = 6,8, 6,8 Hz, 2H), 1,27 (t, J = 7,1 Hz, 3H).
2. En este caso, se llevó a cabo acoplamiento de Suzuki usando [1,1'-bis(di-tercbutilfosfino)ferroceno]dicloropaladio (II) (Pd-118) y carbonato de cesio o carbonato potásico.
3. En este caso, la aminación reductora se llevó a cabo con cianoborohidruro sódico, W,A/-diisopropiletilamina y sulfato de magnesio.
4. Condiciones para HPLC analítica. Columna: Waters XBridge C18, 2,1 x 50 mm, 5 pm; Fase móvil A: ácido trifluoroacético al 0,0375 % en agua; Fase móvil B: ácido trifluoroacético al 0,01875 % en acetonitrilo; Gradiente: del 1 % al 5 % de B durante 0,6 minutos; del 5 % al 100 % de B durante 3,4 minutos; Caudal: 0,8 ml/minuto. 5. En este caso, se usó 6-oxo-2-azaespiro[3.4]octan-2-carboxilato de ferc-butilo en lugar de P1. El 6-{4-[3-(pirimidin-5-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de terc-butilo se desprotegió con ácido trifluoroacético y después se hizo reaccionar con carbonato de 1 -cloroetil etilo para proporcionar el ejemplo 14.
6. En este caso, se llevó a cabo acoplamiento de Suzuki usando [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) y carbonato potásico.
7. Se sintetizó la 1-[2-(difluorometoxi)piridin-3-il]piperazina requerida a partir de 3-bromo-2-(difluorometoxi)piridina, usando el método descrito en el ejemplo 10 para la conversión de 3-bromo-2-fluoropiridina en C21.
8. El producto racémico se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralpak IG; Fase móvil: 7:3 de hexano/etanol) y el primer enantiómero en eluir se denominó Ejemplo 19. Ambos enantiómeros se sometieron después de manera individual a cromatografía de fase inversa (Columna: Agela Technologies C18; Fase móvil A: hidróxido de amonio al 0,05% en agua; Fase móvil B: acetonitrilo; Gradiente: del 0 % al 100 % de B). En HPLC analítica (Columna: Chiral Technologies Chiralpak IG, 4.6 x 150 mm, 5 pm; Fase móvil: 7:3 de hexano/etanol; Caudal: 1,0 ml/minuto), El ejemplo 19 mostró un tiempo de retención de 5,25 minutos. El enantiómero del ejemplo 19, 6-{4-[2-(difluorometoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2, tuvo un tiempo de retención de 6,08 minutos en las mismas condiciones. El enantiómero del ejemplo 19, LCMS m/z 411,2 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, 201 nM (3 determinaciones); Emáx de M474 % (3 determinaciones).
9. Se sintetizó la 1-[2-(trifluorometoxi)piridin-3-il]piperazina requerida a partir de 3-bromo-2-(trifluorometoxi)piridina, usando el método descrito en el ejemplo 10 para la conversión de 3-bromo-2-fluoropiridina en C21.
10. El producto racémico se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralcel OD, 10 pm; Fase móvil: 90:10 de hexano/etanol). El primer enantiómero en eluir se denominó Ejemplo 20. Ambos enantiómeros se sometieron después de manera individual a cromatografía de fase inversa (Columna: Agela Technologies C18; Fase móvil A: hidróxido de amonio al 0,05 % en agua; Fase móvil B: acetonitrilo; Gradiente: del 0 % al 100 % de B). En HPLC analítica (Columna: Chiral Technologies Chiralcel OD-H, 4.6 x 150 mm, 5 pm; Fase móvil: 90:10 de hexano/etanol; Caudal: 1,0 ml/minuto), El ejemplo 20 mostró un tiempo de retención de 4,31 minutos. El enantiómero del ejemplo 20, 6-{4-[2-(trifluorometoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2, tuvo un tiempo de retención de 4,93 minutos en las mismas condiciones. El enantiómero del ejemplo 20, LCMS m/z 429,2 [M+H]+, mostró los datos biológicos siguientes: EC50 de M4, 3600 nM (2 determinaciones); Emáx de M495,3 % (2 determinaciones).
11. En este caso, el material de partida 1-(3-metilpiridin-2-il)piperazina estaba disponible en el mercado.
12. El producto racémico se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralcel OZ-H, 5 pm; Fase móvil: 80:20:0,1 hexano/etanol/dietilamina). El primer enantiómero en eluir se denominó Ejemplo 21. Ambos enantiómeros se sometieron después de manera individual a HPLC de fase inversa (Columna: C18; Fase móvil A: hidróxido de amonio al 0,05 % en agua; Fase móvil B: acetonitrilo; Gradiente:
del 70 % al 75 % de B). En HPLC analítica (Columna: Chiral Technologies Chiralcel OZ-H, 4,6 x 150 mm, 5 |jm; Fase móvil: 80:20:0,1 hexano/etanol/dietilamina; Caudal: 1,0 ml/minuto), El ejemplo 21 mostró un tiempo de retención de 5,29 minutos. El enantiómero del ejemplo 21, 6-[4-(3-metilpiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2, tuvo un tiempo de retención de 6,04 minutos en las mismas condiciones. El enantiómero del ejemplo 21, LCMS m/z 359,3 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, >241 nM (4 determinaciones); Emáx de M476,2 % (3 determinaciones).
13. La reacción de Suzuki de 4-(3-bromopiridin-2-il)piperazin-1-carboxilato de ferc-butilo y ácido ciclopropilborónico, en presencia de diclorobis(triciclohexil fosfina)paladio (II) y fosfato tripotásico, proporcionó el 4-(3-ciclopropilpiridin-2-il)piperazin-1 -carboxilato de ferc-butilo requerido.
14. El producto racémico se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralpak AD-H, 5 jm ; Fase móvil: 100:0,1 de etanol/dietilamina). El primer enantiómero en eluir se denominó Ejemplo 22. Ambos enantiómeros se sometieron después de manera individual a cromatografía de fase inversa (Columna: Agela Technologies C18; Fase móvil A: hidróxido de amonio al 0,05 % en agua; Fase móvil B: metanol; Gradiente: del 5 % al 80 % de B). En HPLC analítica (Columna: Chiral Technologies Chiralpak AD-H, 4,6 x 250 mm, 5 jm ; Fase móvil: 100:0,1 de etanol/dietilamina; Caudal: 1,0 ml/minuto), El ejemplo 22 mostró un tiempo de retención de 12,00 minutos. El enantiómero del ejemplo 22, 6-[4-(3-ciclopropilpiridin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2, tuvo un tiempo de retención de 14,94 minutos en las mismas condiciones. El enantiómero del ejemplo 22, LCMS m/z 385,2 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, 11,6 nM (6 determinaciones); Emáx de M4103 % (6 determinaciones).
15. En este caso, la aminación reductora se realizó con triacetoxiborohidruro sódico.
16. El producto racémico se separó en sus enantiómeros mediante cromatografía de fluidos supercríticos [Columna: Chiral Technologies Chiralpak IG, 5 jm ; Fase móvil 7:3 de dióxido de carbono/(2-propanol que contenía 1-aminopropan-2-ol al 0,2%)]. El segundo enantiómero en eluir se denominó Ejemplo 23. En HPLC analítica [Columna: Phenomenex Lux Cellulose-4, 4,6 x 250 mm, 5 jm ; Fase móvil A: dióxido de carbono; Fase móvil B: etanol que contenía 0,2 % (amoniaco 7 M en metanol); Gradiente: al 5 % de B durante 1,00 minuto, después del 5 % al 60 % de B durante 8,00 minutos; Caudal: 3,0 ml/minuto; Contrapresión: 12 MPa (120 bar)], El ejemplo 23 mostró un tiempo de retención de 7,31 minutos. El enantiómero del ejemplo 23, 2-[4-(5-ciano-2,3'-bipiridin-2'-il)piperazin-1-il]-6-azaespiro[3.4]octan-6-carboxilato de etilo, ENT-1, tuvo un tiempo de retención de 7,01 minutos en las mismas condiciones. El enantiómero del ejemplo 23, LCMS m/z 447,3 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, >10.000 nM (1 determinación); Emáx de M4, no determinado.
17. En este caso, se usó 6-oxo-2-azaespiro[3.3]heptan-2-carboxilato de ferc-butilo en lugar de P1. El 6-[4-(5-ciano-2,3'-bipiridin-2'-il)piperazin-1-il]-2-azaespiro[3.3]heptan-2-carboxilato de ferc-bufilo resultante se desprotegió con ácido trifluoroacético y después se hizo reaccionar con cloroformiato de etilo para proporcionar el ejemplo 24. 18. La reacción de piperazin-1-carboxilato de ferc-butilo con 2,3-dibromo-5-fluoropiridina y carbonato potásico proporcionó 4-(3-bromo-5-fluoropiridin-2-il)piperazin-1-carboxilato de ferc-bufilo. Este material se sometió a acoplamiento de Stille con 4-(tributilestananil)-1,3-tiazol en presencia de tetraquis(trifenilfosfina)paladio (0) y fluoruro de cesio para proporcionar el 4-[5-fluoro-3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-carboxilato de ferc-butilo requerido.
19. El producto racémico se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralpak AD-H, 5 jm ; Fase móvil: 1:1 de hexano/etanol). El primer enantiómero en eluir se denominó Ejemplo 26. En HPLC analítica (Columna: Chiral Technologies Chiralpak AD-H, 4,6 x 250 mm, 5 jm ; Fase móvil: 1:1 de hexano/etanol; Caudal: 1,0 ml/minuto), El ejemplo 26 mostró un tiempo de retención de 7,81 minutos. El enantiómero del ejemplo 26, 6-{4-[5-fluoro-3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2, tuvo un tiempo de retención de 16,42 minutos en las mismas condiciones. El enantiómero del ejemplo 26, LCMS m/z 446,2 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, 152 nM (4 determinaciones); Emáx de M467,1 % (4 determinaciones).
20. El piperazin-1-carboxilato de ferc-butilo 4-sustituido requerido se sintetizó mediante reacción de piperazin-1-carboxilato de ferc-bufilo con el reactivo heteroaromático sustituido con cloro apropiado.
21. El producto racémico se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralcel OJ, 10 jm ; Fase móvil: 90:10:0,1 hexano/etanol/dietilamina). El segundo enantiómero en eluir se denominó Ejemplo 27. Ambos enantiómeros se sometieron después de manera individual a cromatografía de fase inversa (Columna: Agela Technologies C18; Fase móvil A: hidróxido de amonio al 0,05 % en agua; Fase móvil B: metanol; Gradiente: del 0 % al 100 % de B). En HPLC analítica (Columna: Chiral Technologies Chiralcel OJ-H, 4,6 x 150 mm, 5 jm ; Fase móvil: 90:10:0,1 hexano/etanol/dietilamina; Caudal: 1,0 ml/minuto), El ejemplo 27 mostró un tiempo de retención de 5,19 minutos. El enantiómero del ejemplo 27, 6-[4-(3-metoxipirazin-2-il)piperazin-1-il]-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1, tuvo un tiempo de retención de 4,42 minutos en las mismas condiciones. El enantiómero del ejemplo 27, LCMS m/z 376,2 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, >10.000 nM (1 determinación); Emáx de M4, no determinado.
22. La reacción de 4-(3-bromopiridin-2-il)piperazin-1-carboxilato de ferc-butilo con la amina apropiada, ferc-butóxido sódico, tris(dibencilidenoacetona)dipaladio (0) y 4,5-bis(difenilfosfino)-9,9-dimetilxanteno (Xantphos) proporcionó el producto acoplado, que se desprotegió usando ácido trifluoroacético. La amina secundaria resultante se sometió a aminación reductora con P1, triacetoxiborohidruro sódico y N,N-diisopropiletilamina para proporcionar el ejemplo.
23. Condiciones para HPLC analítica. Columna: Waters Atlantis dC18, 4,6 x 50 mm, 5 jm ; Fase móvil A: ácido trifluoroacético al 0,05 % en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,05 % en acetonitrilo (v/v); Gradiente: del 5,0 % al 95 % de B, lineal durante 4,0 minutos; Caudal: 2 ml/minuto.
24. La reacción de 6-oxo-2-azaespiro[3.3]heptan-2-carboxilato de ferc-butilo con cloruro de acetilo en metanol proporcionó 6,6-dimetoxi-2-azaespiro[3.3]heptano. El tratamiento con cloroformiato de etilo y trietilamina, seguido de desprotección de cetal con ácido clorhídrico, proporcionó el 6-oxo-2-azaespiro[3.3]heptan-2-carboxilato de etilo requerido. RMN 1H (400 MHz, CDCla) 84,19 (s, 4H), 4,14 (c, J = 7,0 Hz, 2H), 3,31 (s, 4H), 1,26 (t, J = 7,0 Hz, 3H).
25. El ejemplo racémico 33 se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralpak IG, 5 pm; Fase móvil: 50/50/0,1 de hexano/etanol/dietilamina). El primer enantiómero en eluir se denominó Ejemplo 34. Ambos enantiómeros se sometieron de forma individual a cromatografía sobre gel de sílice (Gradiente: del 0% al 10% de metanol en diclorometano). En HPLC analítica (Columna: Chiral Technologies Chiralpak IG, 4,6 x 150 mm, 5 pm; Fase móvil: 1:1 de hexano/etanol; Caudal: 1,0 ml/minuto), El ejemplo 34 mostró un tiempo de retención de 9,66 minutos. El enantiómero del ejemplo 34, 6-{4-[3-(1,2,5-tiadiazol-3-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2, tuvo un tiempo de retención de 15,53 minutos en las mismas condiciones. El enantiómero del ejemplo 34, LCMS m/z 429,2 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, 347 nM (3 determinaciones); Emáx de M472,3 % (3 determinaciones).
26. Condiciones para HPLC analítica. Columna: Waters XBridge C18, 2,1 x 50 mm, 5 pm; Fase móvil A: ácido trifluoroacético al 0,0375 % en agua; Fase móvil B: ácido trifluoroacético al 0,01875 % en acetonitrilo; Gradiente: del 10 % al 100 % de B durante 4,0 minutos; Caudal: 0,8 ml/minuto.
27. El producto racémico se separó en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralcel OD, 10 pm; Fase móvil: 3:2 de hexano/etanol). El segundo enantiómero en eluir se denominó Ejemplo 41. Ambos enantiómeros se sometieron después de manera individual a cromatografía de fase inversa (Columna: Agela Technologies C18; Fase móvil A: hidróxido de amonio al 0,1 % en agua; Fase móvil B: acetonitrilo; Gradiente: del 10 % al 60 % de B). En HPLC analítica (Columna: Chiral Technologies Chiralcel OD-H, 4,6 x 150 mm, 5 pm; Fase móvil: 7:3 de hexano/etanol; Caudal: 1,0 ml/minuto), El ejemplo 41 mostró un tiempo de retención de 6,02 minutos. El enantiómero del ejemplo 41, 6-{4-[3-(5-cianopirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1, tuvo un tiempo de retención de 4,96 minutos en las mismas condiciones. El enantiómero del ejemplo 41, LCMS m/z 448,3 [M+H]+, mostró los datos biológicos siguientes: CE50 de M4, 268 nM (3 determinaciones); Emáx de M452,7 % (3 determinaciones).
28. La reacción de 4-(3-cloropirazin-2-il)piperazin-1-carboxilato de ferc-butilo con morfolina y carbonato potásico proporcionó el 4-[3-(morfolin-4-il)pirazin-2-il]piperazin-1-carboxilato de ferc-butilo requerido.
29. La reacción de 4-(3-cloropirazin-2-il)piperazin-1-carboxilato de ferc-bufilo con 4-metoxipiperidina y carbonato potásico proporcionó el 4-[3-(4-metoxipiperidin-1-il)pirazin-2-il]piperazin-1-carboxilato de ferc-butilo requerido. 30. El acoplamiento de 4-(3-bromopiridin-2-il)piperazin-1-carboxilato de ferc-bufilo y 2-oxa-6-azaespiro[3.3]heptano se realizó usando rac-BINAP-Pd-G3 (Aldrich, número de catálogo 804967), 1,1'-binaftalen-2,2'-diilbis(difenilfosfano) y ferc-butóxido sódico. El producto se desprotegió con ácido trifluoroacético para proporcionar el 6-[2-(piperazin-1-il)piridin-3-il]-2-oxa-6-azaespiro[3.3]heptano requerido.
31. En este caso, el acoplamiento de Suzuki se realizó usando [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) y bicarbonato sódico.
32. En este caso, se introdujo la hidroxipirazina se introdujo como derivado de (4-metoxibencil)oxi. Se hizo reaccionar 5-bromopirazin-2-ol con 1-(clorometil)-4-metoxibenceno y carbonato de plata para proporcionar 2-bromo-5-[(4-metoxiben-zil)oxi]pirazina, que se usó en la reacción de acoplamiento. Se empleó una desprotección mediada por ácido trifluoroacético para eliminar el grupo ferc-butoxicarbonilo; esto también eliminó el resto 4-metoxibencilo.
33. La reacción de 4-yodo-1H-pirazol con el reactivo haloalquilo apropiado, en presencia de carbonato de cesio y yoduro potásico, proporcionó el 4-yodo-1H-pirazol 1 -sustituido requerido.
34. Se llevó a cabo reacción de Suzuki usando [1,1'-bis(di-ferc-butilfosfino)ferroceno]dicloropaladio (II) (Pd-118) y fosfato tripotásico.
35. Condiciones para HPLC analítica. Columna: Waters Atlantis dC18, 4,6 x 50 mm, 5 pm; Fase móvil A: ácido trifluoroacético al 0,05 % en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,05 % en acetonitrilo (v/v); Gradiente: del 5,0% al 80% de B, lineal durante 3,75 minutos, después del 80% al 95% de B durante 0,25 minutos, después al 95 % de B durante 1,0 minuto; Caudal: 2 ml/minuto.
36. La reacción de 4-[3-(etoxicarbonil)piridin-2-il]piperazin-1-carboxilato de ferc-bufilo con hidrazina, seguido de acetilación con cloruro de acetilo y W,A/-diisopropiletilamina, proporcionó 4-{3-[(2-acetilhidrazinil)carbonil]piridin-2-il}piperazin-1-carboxilato de ferc-bufilo. Someter este material a cloruro de p-toluenosulfonilo y trietilamina proporcionó el 4-[3-(5-metil-1,3,4-oxadiazol-2-il)piridin-2-il]piperazin-1-carboxilato de ferc-bufilo requerido.
37. Se sintetiza la 1-[2-(metoxi)piridin-3-il]piperazina requerida a partir de 3-bromo-2-(metoxi)piridina, usando el método descrito en el ejemplo 10 para la conversión de 3-bromo-2-fluoropiridina en C21.
38. El producto racémico se separa en sus enantiómeros mediante HPLC de fase inversa (Columna: Chiral Technologies Chiralcel OD, 10 pm) usando una mezcla disolvente de hidrocarburo/etanol como fase móvil. Si se necesita más purificación, los enantiómeros se someten de forma individual a cromatografía de fase inversa.
La afinidad de unión al agonista de M4 para los compuestos de la presente invención se determinó utilizando los ensayos biológicos siguientes:
Métodos de ensayo con GloSensor APMc del agonista muscarínico M4h
Los compuestos se prepararon antes del ensayo. Los compuestos de prueba se solubilizaron en sulfóxido de dimetilo al 100 % (DMSO, Sigma D8418) a una concentración de 30 mM. Se creó una serie de dilución intermedia de 10 puntos utilizando diluciones semilogarítmicas en DMSO al 100 % con una concentración máxima de 4 mM. Los compuestos diluidos en serie se aplicaron como 200 nl/pocillo en una placa de 384 pocillos (número de catálogo Matrix 4325). El intervalo de concentración final del compuesto en el ensayo fue de 10 pM a 0,3 nM con una concentración final de DMSO de 0,25 %.
Se generaron líneas celulares estables que expresaban el M4 mAChR humano (subtipo M4 del receptor muscarínico de acetilcolina) utilizando una célula HEK precursora que expresaba la construcción GloSensor (Clon celular Promega Sor-L9 HEK293 humano/M4/GloSensor número 40). Las células se cultivaron en medio Eagle modificado de Dulbecco al 90 % (DMEM, Gibco 11960), suero bovino fetal al 10 % (FBS, Hyclone CH.30160-03), Penicilina/Estreptomicina al 1 % (Gibco 15070-063), Geneticina 500 pg/ml (Gibco 10131-027), Higromicina B 200 pg/ml (Invitrogen 10687-010) y Glutamax al 1 % (Thermo Fisher 35050061).
Un día antes del ensayo, las células se extrajeron utilizando tampón de disociación (Gibco 13151-014) y se centrifugaron en una centrífuga a 250 veces la gravedad durante 5 minutos a temperatura ambiente. Se eliminó el sobrenadante y se resuspendió el sedimento celular en medio de crecimiento a una concentración de 6,25 x 106 células/ml. A continuación, las células se añadieron a placas blancas recubiertas con poli-d-lisina (Becton Dickinson 356661) a razón de 40 pl por pocillo (25000 células) y se incubaron durante la noche (20-24 horas) en una incubadora humidificada a 37 °C con dióxido de carbono (CO2) al 5 %.
Al día siguiente, el medio de cultivo se retiró de las placas con células y se reemplazó con 40 pl de medio de equilibrio que contenía medio independiente de CO2 al 88 % (Invitrogen 18045088), FBS al 10 % y reactivo GloSensor AMPc al 2 % (Promega E1291) que se había calentado a 37 °C. A continuación, las placas se cubrieron y se incubaron durante 2 horas a temperatura ambiente mientras se protegían de la luz.
A las placas anteriormente preparadas con los compuestos diluidos en serie, se añadieron 200 nl de ACh 4 mM (Sigma A2661, 10 pM final) o 200 nL de DMSO al 100% (0,25% final) a los pocillos de control positivo y negativo, respectivamente. A continuación, las placas con los compuestos se diluyeron añadiendo 16 pl de medio independiente de CO2 que contenía FBS al 10 % y una concentración CEs0 de isoproterenol (Sigma 16504). Antes de realizar las pruebas con el compuesto, se procesaron curvas de respuesta a la concentración con respecto al isoproterenol para determinar la concentración c Es0. Al final de un tiempo de equilibrio de 2 horas, se transfirieron 10 pl de las placas con el compuesto a las placas con células. Las placas con células se incubaron durante 7 minutos más a temperatura ambiente y después, utilizando un lector de placas EnVision de etiquetas múltiples (Perkin Elmer), se realizó la lectura para determinar la luminiscencia.
Utilizando Activity Base (IDBS), se analizaron los datos sin procesar expresados en unidades relativas de luz. El efecto porcentual a cada concentración de compuesto se calculó basándose en, y en relación con, la cantidad de AMPc producida en los pocillos de control positivo y negativo contenidos en cada placa de ensayo. Los pocillos de control positivo contenían una concentración CE100 de ACh y los pocillos de control negativo contenían solo DMSO. Los valores de concentración y el efecto % (porcentual) se ajustaron utilizando una ecuación logística de respuesta a la dosis de cuatro parámetros, y para definir la eficacia, se determinó la concentración necesaria para obtener un efecto del 50 % (CE50) así como la asíntota máxima de la curva de respuesta a la concentración.
T l 2. A ivi i l i n m r I PA r l m l 1- .

(continuación)

(continuación)

(continuación)

Otras realizaciones de la invención serán evidentes para los expertos en la materia a partir de la consideración de la memoria descriptiva y la práctica de la invención divulgada en el presente documento. Se pretende que la memoria descriptiva y los ejemplos se consideren únicamente a modo de ejemplo.
Claims (1)
- REIVINDICACIONES1. Un compuesto de fórmula I:
 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la quecada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -N(R6)(R7), alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alquiltio (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alquiltio (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, -N(R6)(R7), alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, ciano, hidroxi, -SF5, nitro, -N(R6)(R7), alquilo (C1-C6) y alcoxi (C1-C6), en donde dichos alquilo (C1-C6) y alcoxi (C1-C6) están opcionalmente sustituidos con de 1 a 3 halógenos;cada uno de R6 y R7 se selecciona independientemente de entre hidrógeno, alquilo (C1-C6) o C(O)-CH3;m es 1 o 2; yn es 1 o 2.2. El compuesto de acuerdo con la reivindicación 1 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) X1 es nitrógeno y X2 es CH;ii) X1 es nitrógeno y X2 es nitrógeno; oiii) X1 es CH y X2 es nitrógeno.3. El compuesto de acuerdo con las reivindicaciones 1 o 2 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están cada uno opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);ii) R1 es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -(CH2)2-O-CH2CH3 y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).4. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-3 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde R2 es hidrógeno.5. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-4 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) m es 2 y n es 1;ii) m es 1 y n es 2; oiii) m es 1 y n es 1.a. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula Ia:
o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la quecada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, hidroxi, -SF5, nitro, -N(R6)(R7), alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alquiltio (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alquiltio (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C10), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, -N(R6)(R7), alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, ciano, hidroxi, -SF5, nitro, -N(R6)(R7), alquilo (C1-C6) y alcoxi (C1-C6), en donde dichos alquilo (C1-C6) y alcoxi (C1-C6) están opcionalmente sustituidos con de 1 a 3 halógenos;cada uno de R6 y R7 se selecciona independientemente de entre hidrógeno, alquilo (C1-C6) o C(O)-CH3;m es 1 o 2; yn es 1 o 2.2. El compuesto de acuerdo con la reivindicación 1 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) X1 es nitrógeno y X2 es CH;ii) X1 es nitrógeno y X2 es nitrógeno; oiii) X1 es CH y X2 es nitrógeno.3. El compuesto de acuerdo con las reivindicaciones 1 o 2 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están cada uno opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);ii) R1 es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -(CH2)2-O-CH2CH3 y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).4. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-3 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde R2 es hidrógeno.5. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-4 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) m es 2 y n es 1;ii) m es 1 y n es 2; oiii) m es 1 y n es 1.a. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula Ia: o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-C1o), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-C1o), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (C1-Ca), alcoxi (C1-Ca) y heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca) y alcoxi (C1-Ca); yR2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (C1-Ca) y alcoxi (C1-Ca), en donde dichos alquilo (C1-Ca) y alcoxi (CrCa) están opcionalmente sustituidos con de 1 a 3 halógenos.7. El compuesto de acuerdo con la reivindicación a o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) X1 es nitrógeno y X2 es CH;ii) X1 es nitrógeno y X2 es nitrógeno; oiii) X1 es CH y X2 es nitrógeno.7a8. El compuesto de acuerdo con una cualquiera de las reivindicaciones 6-7 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-C6), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (C i Ca) y heteroarilo (de 5 a a miembros) están cada uno opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca);ii) R1 es un heteroarilo (de 5 a i0 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a i0 miembros) está opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-a-azaespiro[3.3]hept-a-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).9. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula IB:
o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-C1o), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-C1o), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca), alcoxi (C1-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (C1-Ca), alcoxi (C1-Ca) y heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-Ca) y alcoxi (C1-Ca); yR2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (C1-Ca) y alcoxi (C1-Ca), en donde dichos alquilo (C1-Ca) y alcoxi (CrCa) están opcionalmente sustituidos con de 1 a 3 halógenos.7. El compuesto de acuerdo con la reivindicación a o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) X1 es nitrógeno y X2 es CH;ii) X1 es nitrógeno y X2 es nitrógeno; oiii) X1 es CH y X2 es nitrógeno.7a8. El compuesto de acuerdo con una cualquiera de las reivindicaciones 6-7 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-C6), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (C i Ca) y heteroarilo (de 5 a a miembros) están cada uno opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca);ii) R1 es un heteroarilo (de 5 a i0 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a i0 miembros) está opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-a-azaespiro[3.3]hept-a-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).9. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula IB: o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci Ca); yR2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (Ci-Ca) y alcoxi (Ci-Ca), en donde dichos alquilo (Ci-Ca) y alcoxi (Ci-Ca) están opcionalmente sustituidos con de 1 a 3 halógenos.10. El compuesto de acuerdo con la reivindicación 9 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde X1 es nitrógeno y X2 es CH.11. El compuesto de acuerdo con una cualquiera de las reivindicaciones 9-10 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-C6), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (C i Ca) y heteroarilo (de 5 a a miembros) están cada uno opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca);ii) R1 es un heteroarilo (de 5 a i0 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a i0 miembros) está opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-a-azaespiro[3.3]hept-a-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -(CH2)2-O-CH2CHa y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).12. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula IC:
o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci Ca); yR2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (Ci-Ca) y alcoxi (Ci-Ca), en donde dichos alquilo (Ci-Ca) y alcoxi (Ci-Ca) están opcionalmente sustituidos con de 1 a 3 halógenos.10. El compuesto de acuerdo con la reivindicación 9 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde X1 es nitrógeno y X2 es CH.11. El compuesto de acuerdo con una cualquiera de las reivindicaciones 9-10 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-C6), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (C i Ca) y heteroarilo (de 5 a a miembros) están cada uno opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca);ii) R1 es un heteroarilo (de 5 a i0 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a i0 miembros) está opcionalmente sustituido con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de i a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-a-azaespiro[3.3]hept-a-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -(CH2)2-O-CH2CHa y heteroarilo (de 5 a a miembros), en donde dicho alquilo (Ci-Ca), alcoxi (Ci-Ca) y dicho heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca).12. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula IC: o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, -alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca); yR2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (Ci-Ca) y alcoxi (Ci-Ca), en donde dichos alquilo (Ci-Ca) y alcoxi (Ci-Ca) están opcionalmente sustituidos con de 1 a 3 halógenos.13. El compuesto de acuerdo con la reivindicación 12 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde X1 es nitrógeno y X2 es CH.14. El compuesto de acuerdo con una cualquiera de las reivindicaciones 12-13 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (Ci-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (Ci-C6), alcoxi (C-i-C6) y heteroarilo (de 5 a 6 miembros) están cada uno opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6) y alcoxi (Ci-C6);ii) R1 es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6) y alcoxi (Ci-C6); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6), alcoxi (Ci-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).15. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula I':
o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca), -O-heterocicloalquilo (de 4 a a miembros), arilo (Ca-Cio), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, -alquilo (Ci-Ca), alcoxi (Ci-Ca), cicloalquilo (C3-Ca) y heteroarilo (de 5 a a miembros), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y heteroarilo (de 5 a a miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-Ca) y alcoxi (Ci-Ca); yR2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (Ci-Ca) y alcoxi (Ci-Ca), en donde dichos alquilo (Ci-Ca) y alcoxi (Ci-Ca) están opcionalmente sustituidos con de 1 a 3 halógenos.13. El compuesto de acuerdo con la reivindicación 12 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde X1 es nitrógeno y X2 es CH.14. El compuesto de acuerdo con una cualquiera de las reivindicaciones 12-13 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca), en donde dichos alquilo (Ci-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-Ca) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (Ci-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (Ci-C6), alcoxi (C-i-C6) y heteroarilo (de 5 a 6 miembros) están cada uno opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6) y alcoxi (Ci-C6);ii) R1 es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6) y alcoxi (Ci-C6); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6), alcoxi (Ci-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).15. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto es de fórmula I': o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C i0), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C i0), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6), alcoxi (Ci-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (C1-C6) y alcoxi (C1-C6), en donde dichos alquilo (Ci-C6) y alcoxi (Ci-C6) están opcionalmente sustituidos con de 1 a 3 halógeno;m es 1 o 2; yn es 1 o 2.16. El compuesto de acuerdo con la reivindicación 15 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) X1 es nitrógeno y X2 es CH;ii) X1 es nitrógeno y X2 es nitrógeno; oiii) X1 es CH y X2 es nitrógeno.17. El compuesto de acuerdo con una cualquiera de las reivindicaciones 15-16 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6), en donde dichos alquilo (C-i-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-C6) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están cada uno opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);ii) R1 es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).18. El compuesto de acuerdo con la reivindicación 1 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde dicho compuesto se selecciona de entre:6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (2); 6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2 (3); (6R)-6-{4-[3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;(6R)-6-{4-[3-(1,3,4-tiadiazol-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;(aR)-a-[4-(3-metox¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(as)-a-[4-(3-metox¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(aR)-a-{4-[3-(p¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(6R)-6-{4-[3-(4-metiMH-pirazoM-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[2-(2,2,2-trifluoroetoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;(6R)-6-{4-[3-(1,3-tiazol-5-il)pirazin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;2-[4-(3-metox¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo;2-{4-[3-(1-met¡MH-p¡razol-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo;a-{4-[3-(p¡r¡m¡d¡n-5-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(3-met¡lp¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;2-{4-[3-(1,3-t¡azol-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo;a-{4-[3-(p¡r¡daz¡n-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(1,3-oxazol-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[2-(difluorometoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (Ejemplo 19); a-{4-[2-(trifluorometoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (Ejemplo 20); a-[4-(3-met¡lp¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-1 (Ejemplo 21);a-[4-(3-c¡cloprop¡lp¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-1 (Ejemplo 22); 2-[4-(5-c¡ano-2,3'-b¡p¡r¡d¡n-2'-¡l)p¡peraz¡n-1"-¡l]-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo, ENT-2 (Ejemplo 23); a-[4-(5-c¡ano-2,3'-b¡p¡r¡d¡n-2'-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.3]heptan-2-carbox¡lato de etilo;a-{4-[2-(oxetan-3-¡lox¡)p¡r¡d¡n-3-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[5-fluoro-3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (Ejemplo 26);a-[4-(3-metox¡p¡raz¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-2 (Ejemplo 27);6-(4-{3-[4-(1 -metil-1 W-pirazol-5-il)piperidin-1 -il]piridin-2-il}piperazin-1-il)-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[3-(3,3-d¡fluorop¡rrol¡d¡n-1-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(3-c¡anoazet¡d¡n-1-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(4-metil-1,2-tiazol-5-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.3]heptan-2-carboxilato de etilo;a-{4-[3-(3-metil-1,2-tiazol-5-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.3]heptan-2-carboxilato de etilo;a-{4-[3-(1,2,5-t¡ad¡azol-3-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(1,2,5-t¡ad¡azol-3-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-1 (Ejemplo 34);a-{4-[3-(p¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.3]heptan-2-carbox¡lato de etilo;a-{4-[3-(2,4-dimetil-1,3-tiazol-5-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[5'-(d¡fluorometox¡)-3,3'-b¡p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-[4-(a'-metox¡-3,3'-b¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(imidazo[1,2-a]piridin-a-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[3-([1,2,4]tr¡azolo[4,3-a]p¡r¡d¡n-a-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo; 6-{4-[3-(5-cianopirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2 (Ejemplo 41);(6R)-6-{4-[3-(morfol¡n-4-¡l)p¡raz¡n-2-¡l]p¡perazin-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(6R)-6-{4-[3-(4-metox¡p¡per¡d¡n-1-¡l)p¡raz¡n-2-¡l]p¡peraz¡n-1-il}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-{4-[3-(2-oxa-6-azaesp¡ro[3.3]hept-6-¡l)p¡r¡d¡n-2-¡l]piperaz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; 6-{4-[3-(p¡r¡daz¡n-3-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-{4-[3-(p¡r¡m¡d¡n-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-{4-[3-(2-met¡lp¡r¡m¡d¡n-5-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;6-{4-[3-(5-h¡drox¡p¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-(4-{3-[1-(4-c¡anobut¡l)-1H-p¡razol-4-¡l]p¡r¡d¡n-2-¡l}piperaz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; (6R)-6-(4-{3-[1-(2-etox¡et¡l)-1H-p¡razol-4-¡l]p¡r¡d¡n-2-¡l}piperaz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; (6R)-6-{4-[3-(5-met¡l-1,3,4-oxad¡azol-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; 6-(4-(3-(4-acetam¡dofen¡l)p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;6-(4-(3-(4-c¡anofen¡l)p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;6-(4-(2-metox¡p¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-(4-(2-metox¡p¡r¡d¡n-3-¡l)piperaz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo y(6S)-6-(4-(2-metox¡p¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo.19. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-{4-[3-(1,3-t¡azol-4-¡l)pir¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un A/-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.20. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-{4-[3-(1,3,4-t¡ad¡azol-2-¡l)pir¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.21. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-{4-[3-(p¡raz¡n-2-¡l)pir¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.22. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es 6-(4-(2-metox¡p¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del m¡smo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.23. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-(4-(2-metoxip¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.24. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6S)-6-(4-(2-metoxip¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.25. Una formulación farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de fórmulas I, Ia, Ib, Ic o I' de acuerdo con una cualquiera de las reivindicaciones 1-24 o un N-óxido del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do y un exc¡p¡ente farmacéut¡camente aceptable.26. El compuesto o el N-óx¡do o la sal farmacéut¡camente aceptable de acuerdo con una cualqu¡era de las reivindicaciones 1-24, o la formulación farmacéutica de acuerdo con la reivindicación 25, para su uso en el tratamiento de una enfermedad o de un trastorno mediados por M4 (o asociados a M4) en un paciente, comprendiendo dicho método administrar al paciente una cantidad terapéuticamente eficaz del compuesto, del N-óxido, de la sal farmacéuticamente aceptable o de la formulación farmacéutica, en donde: i)i) la enfermedad o el trastorno mediados por M4 (o asociados a M4) son una enfermedad o un trastorno seleccionados de entre el grupo que consiste en enfermedad de Alzheimer, esquizofrenia o psicosis, dolor, adicción, un trastorno del sueño, un trastorno cognitivo (por ejemplo, deterioro cognitivo leve), enfermedad de Parkinson, discinesia inducida por levodopa en la enfermedad de Parkinson, enfermedad de Huntington, discinesia, boca seca, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica (EPOC), asma, incontinencia urinaria, glaucoma, Trisomía 21 (síndrome de Down), angiopatía amiloidea cerebral, demencia, hemorragia cerebral hereditaria con amiloidosis de tipo holandés (HCHWA-D), enfermedad de Creutzfeld-Jakob, trastornos priónicos, esclerosis lateral amiotrófica, parálisis supranuclear progresiva, traumatismo craneal, accidente cerebrovascular, pancreatitis, miositis por cuerpos de inclusión, otras amiloidosis periféricas, diabetes, autismo y ateroesclerosis; o ¡¡) la enfermedad o el trastorno med¡ados por M4 (o asoc¡ados a M4) son una enfermedad o un trastorno selecc¡onados de entre el grupo que cons¡ste en enfermedad de Alzhe¡mer, esqu¡zofren¡a, dolor, ad¡cc¡ón, enfermedad de Park¡nson, d¡sc¡nes¡a ¡nduc¡da por levodopa en la enfermedad de Park¡nson y un trastorno del sueño.
o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en la que:cada uno de X1 y X2 es independientemente CH o nitrógeno, con la condición de que X1 y X2 no pueden ser ambos CH;R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C i0), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6), -O-heterocicloalquilo (de 4 a 6 miembros), arilo (C6-C i0), heteroarilo (de 5 a 10 miembros) y heterocicloalquilo (de 4 a 8 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (Ci-C6), alcoxi (Ci-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);R2 se selecciona de entre el grupo que consiste en hidrógeno, halógeno, alquilo (C1-C6) y alcoxi (C1-C6), en donde dichos alquilo (Ci-C6) y alcoxi (Ci-C6) están opcionalmente sustituidos con de 1 a 3 halógeno;m es 1 o 2; yn es 1 o 2.16. El compuesto de acuerdo con la reivindicación 15 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) X1 es nitrógeno y X2 es CH;ii) X1 es nitrógeno y X2 es nitrógeno; oiii) X1 es CH y X2 es nitrógeno.17. El compuesto de acuerdo con una cualquiera de las reivindicaciones 15-16 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde:i) R1 se selecciona de entre el grupo que consiste en halógeno, ciano, alquilo (C1-C6), alcoxi (C1-C6) y cicloalquilo (C3-C6), en donde dichos alquilo (C-i-Ca), alcoxi (Ci-Ca) y cicloalquilo (C3-C6) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dichos alquilo (C1-C6), alcoxi (C1-C6) y heteroarilo (de 5 a 6 miembros) están cada uno opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6);ii) R1 es un heteroarilo (de 5 a 10 miembros) seleccionado de entre el grupo que consiste en pirazolilo, pirimidinilo, piridazinilo, tiazolilo, pirazinilo, oxazolilo, tiadiazolilo, piridinilo, imidazopiridinilo, triazolopiridinilo y oxadiazolilo, en donde dicho heteroarilo (de 5 a 10 miembros) está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6); oiii) R1 es un heterocicloalquilo (de 4 a 8 miembros) seleccionado de entre el grupo que consiste en oxetanilo, morfolino, 2-oxa-6-azaespiro[3.3]hept-6-ilo, tetrahidrofuranilo, tetrahidropiranilo, azetidinilo, pirrolidinilo y piperidinilo, en donde dicho heterocicloalquilo está opcionalmente sustituido con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6), alcoxi (C1-C6), cicloalquilo (C3-C6) y heteroarilo (de 5 a 6 miembros), en donde dicho alquilo (C1-C6), alcoxi (C1-C6) y dicho heteroarilo (de 5 a 6 miembros) están opcionalmente sustituidos con de 1 a 3 sustituyentes seleccionados de entre el grupo que consiste en halógeno, ciano, hidroxi, alquilo (C1-C6) y alcoxi (C1-C6).18. El compuesto de acuerdo con la reivindicación 1 o un N-óxido del mismo o una sal farmacéuticamente aceptable del compuesto o del N-óxido, en donde dicho compuesto se selecciona de entre:6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (2); 6-{4-[3-(5-metoxipirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2 (3); (6R)-6-{4-[3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;(6R)-6-{4-[3-(1,3,4-tiadiazol-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;(aR)-a-[4-(3-metox¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(as)-a-[4-(3-metox¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(aR)-a-{4-[3-(p¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(6R)-6-{4-[3-(4-metiMH-pirazoM-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[2-(2,2,2-trifluoroetoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;(6R)-6-{4-[3-(1,3-tiazol-5-il)pirazin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;2-[4-(3-metox¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo;2-{4-[3-(1-met¡MH-p¡razol-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo;a-{4-[3-(p¡r¡m¡d¡n-5-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(3-met¡lp¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;2-{4-[3-(1,3-t¡azol-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo;a-{4-[3-(p¡r¡daz¡n-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(1,3-oxazol-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[2-(difluorometoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (Ejemplo 19); a-{4-[2-(trifluorometoxi)piridin-3-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (Ejemplo 20); a-[4-(3-met¡lp¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-1 (Ejemplo 21);a-[4-(3-c¡cloprop¡lp¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-1 (Ejemplo 22); 2-[4-(5-c¡ano-2,3'-b¡p¡r¡d¡n-2'-¡l)p¡peraz¡n-1"-¡l]-a-azaesp¡ro[3.4]octan-a-carbox¡lato de etilo, ENT-2 (Ejemplo 23); a-[4-(5-c¡ano-2,3'-b¡p¡r¡d¡n-2'-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.3]heptan-2-carbox¡lato de etilo;a-{4-[2-(oxetan-3-¡lox¡)p¡r¡d¡n-3-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[5-fluoro-3-(1,3-tiazol-4-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-1 (Ejemplo 26);a-[4-(3-metox¡p¡raz¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-2 (Ejemplo 27);6-(4-{3-[4-(1 -metil-1 W-pirazol-5-il)piperidin-1 -il]piridin-2-il}piperazin-1-il)-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[3-(3,3-d¡fluorop¡rrol¡d¡n-1-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(3-c¡anoazet¡d¡n-1-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(4-metil-1,2-tiazol-5-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.3]heptan-2-carboxilato de etilo;a-{4-[3-(3-metil-1,2-tiazol-5-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.3]heptan-2-carboxilato de etilo;a-{4-[3-(1,2,5-t¡ad¡azol-3-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(1,2,5-t¡ad¡azol-3-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo, ENT-1 (Ejemplo 34);a-{4-[3-(p¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.3]heptan-2-carbox¡lato de etilo;a-{4-[3-(2,4-dimetil-1,3-tiazol-5-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[5'-(d¡fluorometox¡)-3,3'-b¡p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-[4-(a'-metox¡-3,3'-b¡p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l]-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;a-{4-[3-(imidazo[1,2-a]piridin-a-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo;a-{4-[3-([1,2,4]tr¡azolo[4,3-a]p¡r¡d¡n-a-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo; 6-{4-[3-(5-cianopirazin-2-il)piridin-2-il]piperazin-1-il}-2-azaespiro[3.4]octan-2-carboxilato de etilo, ENT-2 (Ejemplo 41);(6R)-6-{4-[3-(morfol¡n-4-¡l)p¡raz¡n-2-¡l]p¡perazin-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de etilo;(6R)-6-{4-[3-(4-metox¡p¡per¡d¡n-1-¡l)p¡raz¡n-2-¡l]p¡peraz¡n-1-il}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-{4-[3-(2-oxa-6-azaesp¡ro[3.3]hept-6-¡l)p¡r¡d¡n-2-¡l]piperaz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; 6-{4-[3-(p¡r¡daz¡n-3-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-{4-[3-(p¡r¡m¡d¡n-4-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-{4-[3-(2-met¡lp¡r¡m¡d¡n-5-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;6-{4-[3-(5-h¡drox¡p¡raz¡n-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-(4-{3-[1-(4-c¡anobut¡l)-1H-p¡razol-4-¡l]p¡r¡d¡n-2-¡l}piperaz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; (6R)-6-(4-{3-[1-(2-etox¡et¡l)-1H-p¡razol-4-¡l]p¡r¡d¡n-2-¡l}piperaz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; (6R)-6-{4-[3-(5-met¡l-1,3,4-oxad¡azol-2-¡l)p¡r¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo; 6-(4-(3-(4-acetam¡dofen¡l)p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;6-(4-(3-(4-c¡anofen¡l)p¡r¡d¡n-2-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;6-(4-(2-metox¡p¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo;(6R)-6-(4-(2-metox¡p¡r¡d¡n-3-¡l)piperaz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo y(6S)-6-(4-(2-metox¡p¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo.19. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-{4-[3-(1,3-t¡azol-4-¡l)pir¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un A/-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.20. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-{4-[3-(1,3,4-t¡ad¡azol-2-¡l)pir¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.21. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-{4-[3-(p¡raz¡n-2-¡l)pir¡d¡n-2-¡l]p¡peraz¡n-1-¡l}-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.22. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es 6-(4-(2-metox¡p¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del m¡smo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.23. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6R)-6-(4-(2-metoxip¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.24. El compuesto de acuerdo con la re¡v¡nd¡cac¡ón 1, en donde el compuesto es (6S)-6-(4-(2-metoxip¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)-2-azaesp¡ro[3.4]octan-2-carbox¡lato de et¡lo o un N-óx¡do del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do.25. Una formulación farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de fórmulas I, Ia, Ib, Ic o I' de acuerdo con una cualquiera de las reivindicaciones 1-24 o un N-óxido del mismo o una sal farmacéut¡camente aceptable del compuesto o del N-óx¡do y un exc¡p¡ente farmacéut¡camente aceptable.26. El compuesto o el N-óx¡do o la sal farmacéut¡camente aceptable de acuerdo con una cualqu¡era de las reivindicaciones 1-24, o la formulación farmacéutica de acuerdo con la reivindicación 25, para su uso en el tratamiento de una enfermedad o de un trastorno mediados por M4 (o asociados a M4) en un paciente, comprendiendo dicho método administrar al paciente una cantidad terapéuticamente eficaz del compuesto, del N-óxido, de la sal farmacéuticamente aceptable o de la formulación farmacéutica, en donde: i)i) la enfermedad o el trastorno mediados por M4 (o asociados a M4) son una enfermedad o un trastorno seleccionados de entre el grupo que consiste en enfermedad de Alzheimer, esquizofrenia o psicosis, dolor, adicción, un trastorno del sueño, un trastorno cognitivo (por ejemplo, deterioro cognitivo leve), enfermedad de Parkinson, discinesia inducida por levodopa en la enfermedad de Parkinson, enfermedad de Huntington, discinesia, boca seca, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica (EPOC), asma, incontinencia urinaria, glaucoma, Trisomía 21 (síndrome de Down), angiopatía amiloidea cerebral, demencia, hemorragia cerebral hereditaria con amiloidosis de tipo holandés (HCHWA-D), enfermedad de Creutzfeld-Jakob, trastornos priónicos, esclerosis lateral amiotrófica, parálisis supranuclear progresiva, traumatismo craneal, accidente cerebrovascular, pancreatitis, miositis por cuerpos de inclusión, otras amiloidosis periféricas, diabetes, autismo y ateroesclerosis; o ¡¡) la enfermedad o el trastorno med¡ados por M4 (o asoc¡ados a M4) son una enfermedad o un trastorno selecc¡onados de entre el grupo que cons¡ste en enfermedad de Alzhe¡mer, esqu¡zofren¡a, dolor, ad¡cc¡ón, enfermedad de Park¡nson, d¡sc¡nes¡a ¡nduc¡da por levodopa en la enfermedad de Park¡nson y un trastorno del sueño.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862647106P | 2018-03-23 | 2018-03-23 | |
| PCT/US2019/023916 WO2019183636A1 (en) | 2018-03-23 | 2019-03-25 | Piperazine azaspiro derivaves |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2942837T3 true ES2942837T3 (es) | 2023-06-07 |
Family
ID=66102243
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19716666T Active ES2942837T3 (es) | 2018-03-23 | 2019-03-25 | Derivados de azaespiro piperazina |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US11524954B2 (es) |
| EP (2) | EP3768669B1 (es) |
| JP (2) | JP2021519269A (es) |
| CN (1) | CN112154145B (es) |
| AR (1) | AR115015A1 (es) |
| CA (1) | CA3094366A1 (es) |
| DK (1) | DK3768669T3 (es) |
| ES (1) | ES2942837T3 (es) |
| FI (1) | FI3768669T3 (es) |
| HU (1) | HUE061533T2 (es) |
| PL (1) | PL3768669T3 (es) |
| PT (1) | PT3768669T (es) |
| TW (1) | TWI801540B (es) |
| WO (1) | WO2019183636A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4041737A1 (en) | 2019-10-09 | 2022-08-17 | Novartis AG | 5-oxa-2-azaspiro[3.4]octane derivatives as m4 agonists |
| CA3156320A1 (en) | 2019-10-09 | 2021-04-15 | Novartis Ag | 2-AZASPIRO[3,4] OCTANE DERIVATIVES USED AS M4 AGONISTS |
| KR20210067005A (ko) * | 2019-11-28 | 2021-06-08 | 제이투에이치바이오텍 (주) | 에다라본 전구체 약물 화합물 및 이들의 신경퇴행성 또는 운동신경 질환의 치료 또는 개선을 위한 의약 용도 |
| TW202140462A (zh) * | 2020-02-05 | 2021-11-01 | 美商紐羅克里生物科學有限公司 | 蕈毒受體4拮抗劑及使用方法 |
| KR20240042472A (ko) * | 2021-07-30 | 2024-04-02 | 뉴로크린 바이오사이언시즈 인코퍼레이티드 | 무스카린성 수용체 4 길항제 및 사용 방법 |
| WO2024059249A1 (en) * | 2022-09-16 | 2024-03-21 | Cerevel Therapeutics, Llc | M4 activators/modulators and uses thereof |
Family Cites Families (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5750349A (en) | 1993-01-25 | 1998-05-12 | Takeda Chemical Industries Ltd. | Antibodies to β-amyloids or their derivatives and use thereof |
| DE19709877A1 (de) | 1997-03-11 | 1998-09-17 | Bayer Ag | 1,5-Dihydro-pyrazolo[3,4-d]-pyrimidinon-derivate |
| AU743827B2 (en) | 1997-04-09 | 2002-02-07 | Intellect Neurosciences, Inc. | Recombinant antibodies specific for beta-amyloid ends, DNA encoding and methods of use thereof |
| US8173127B2 (en) | 1997-04-09 | 2012-05-08 | Intellect Neurosciences, Inc. | Specific antibodies to amyloid beta peptide, pharmaceutical compositions and methods of use thereof |
| GB9722520D0 (en) | 1997-10-24 | 1997-12-24 | Pfizer Ltd | Compounds |
| TWI239847B (en) | 1997-12-02 | 2005-09-21 | Elan Pharm Inc | N-terminal fragment of Abeta peptide and an adjuvant for preventing and treating amyloidogenic disease |
| US6743427B1 (en) | 1997-12-02 | 2004-06-01 | Neuralab Limited | Prevention and treatment of amyloidogenic disease |
| DE60106394T3 (de) | 2000-02-24 | 2013-07-25 | Washington University St. Louis | Humanisierte antikörper, die amyloid beta peptid sequestrieren |
| DE10045112A1 (de) | 2000-09-11 | 2002-03-21 | Merck Patent Gmbh | Verwendung von Indolderivaten zur Behandlung von Erkrankungen des zentralen Nervensystems |
| EP1335738A4 (en) | 2000-11-03 | 2004-09-08 | Proteotech Inc | METHODS OF ISOLATING AMYLOID INHIBITOR COMPOUNDS AND USE OF COMPOUNDS USED FROM UNCARIA TOMENTOSA AND PARENT PLANTS |
| EP1432444A4 (en) | 2001-08-17 | 2005-11-02 | Lilly Co Eli | ANTI-BETA ANTIBODIES |
| US20030195205A1 (en) | 2001-11-02 | 2003-10-16 | Pfizer Inc. | PDE9 inhibitors for treating cardiovascular disorders |
| DE10238724A1 (de) | 2002-08-23 | 2004-03-04 | Bayer Ag | Alkyl-substituierte Pyrazolpyrimidine |
| DE10238723A1 (de) | 2002-08-23 | 2004-03-11 | Bayer Ag | Phenyl-substituierte Pyrazolyprimidine |
| KR20050071564A (ko) | 2002-10-09 | 2005-07-07 | 리나트 뉴로사이언스 코퍼레이션 | 아밀로이드 베타 펩티드에 대한 항체를 사용하여알츠하이머병을 치료하는 방법 및 그의 조성물 |
| US20040220186A1 (en) | 2003-04-30 | 2004-11-04 | Pfizer Inc. | PDE9 inhibitors for treating type 2 diabetes,metabolic syndrome, and cardiovascular disease |
| BRPI0410630A (pt) | 2003-06-19 | 2006-06-13 | Pfizer Prod Inc | antagonista de nk1 |
| EP1666061A1 (en) | 2003-09-09 | 2006-06-07 | Takeda Pharmaceutical Company Limited | Use of antibody |
| UY28538A1 (es) * | 2003-09-26 | 2005-04-29 | Vertex Pharma | Derivados de fenil-piperazina como moduladores de receptores muscarínicos |
| GB0327319D0 (en) | 2003-11-24 | 2003-12-24 | Pfizer Ltd | Novel pharmaceuticals |
| BRPI0507374A (pt) | 2004-02-02 | 2007-07-10 | Pfizer Prod Inc | moduladores do receptor de histamina-3 |
| US7456164B2 (en) | 2004-05-07 | 2008-11-25 | Pfizer, Inc | 3- or 4-monosubtituted phenol and thiophenol derivatives useful as H3 ligands |
| EP1595881A1 (en) | 2004-05-12 | 2005-11-16 | Pfizer Limited | Tetrahydronaphthyridine derivates useful as histamine H3 receptor ligands |
| EP1753429A1 (en) * | 2004-05-28 | 2007-02-21 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| NZ552480A (en) | 2004-07-30 | 2010-01-29 | Rinat Neuroscience Corp | Antibodies directed against amyloid-beta peptide and methods of using same |
| GB0423356D0 (en) | 2004-10-21 | 2004-11-24 | Merck Sharp & Dohme | Therapeutic agents |
| US20090074775A1 (en) | 2004-12-22 | 2009-03-19 | David Michael Holtzman | Use Of Anti-AB Antibody To Treat Traumatic Brain Injury |
| PE20061323A1 (es) | 2005-04-29 | 2007-02-09 | Rinat Neuroscience Corp | Anticuerpos dirigidos contra el peptido amiloide beta y metodos que utilizan los mismos |
| PL1881985T3 (pl) | 2005-05-12 | 2011-05-31 | Pfizer | Bezwodne krystaliczne postacie N-[1-(2-etoksyetylo)-5-(N-etylo-N-metyloamino)-7-(4-metylopirydyn-2-yloamino)-1H-pirazolo[4,3-d]pirymidyno-3-karbonylo]metano-sulfonoamidu |
| WO2006126083A1 (en) | 2005-05-24 | 2006-11-30 | Pharmacia & Upjohn Company Llc | Pyridine [3 , 4-b] pyrazinone compounds as pde-5 inhibitors |
| CA2603830A1 (en) | 2005-05-24 | 2006-11-30 | Pharmacia & Upjohn Co Llc | PYRIDINE [2,3-B] PYRAZINONES |
| WO2006126082A2 (en) | 2005-05-24 | 2006-11-30 | Pharmacia & Upjohn Company Llc | Pyridine [3,4-b] pyrazinones as pde-5 inhibitors |
| US7812040B2 (en) | 2005-06-22 | 2010-10-12 | Pfizer Inc. | Histamine-3 receptor antagonists |
| TW200713011A (en) | 2005-09-30 | 2007-04-01 | Hon Hai Prec Ind Co Ltd | System, method and electrical apparatus for data transferring |
| US8158673B2 (en) | 2005-10-27 | 2012-04-17 | Pfizer Inc. | Histamine-3 receptor antagonists |
| US20070107917A1 (en) | 2005-11-14 | 2007-05-17 | Doherty Brian J | Multifunctional robot tool |
| WO2007063385A2 (en) | 2005-12-01 | 2007-06-07 | Pfizer Products Inc. | Spirocyclic amine histamine-3 receptor antagonists |
| WO2007069053A1 (en) | 2005-12-14 | 2007-06-21 | Pfizer Products Inc. | Benzimidazole antagonists of the h-3 receptor |
| WO2007088462A1 (en) | 2006-02-01 | 2007-08-09 | Pfizer Products Inc. | Spirochromane antagonists of the h-3 receptor |
| WO2007088450A2 (en) | 2006-02-01 | 2007-08-09 | Pfizer Products Inc. | Chromane antagonist of the h-3 receptor |
| WO2007099423A1 (en) | 2006-03-02 | 2007-09-07 | Pfizer Products Inc. | 1-pyrrolidine indane derivatives as histamine-3 receptor antagonists |
| CA2643055A1 (en) | 2006-03-13 | 2007-09-20 | Pfizer Products Inc. | Tetralines antagonists of the h-3 receptor |
| SI2013208T1 (sl) | 2006-04-21 | 2011-09-30 | Pfizer Prod Inc | Piridin(3,4-b)pirazinoni |
| WO2007138431A2 (en) | 2006-05-30 | 2007-12-06 | Pfizer Products Inc. | Azabicyclic ether histamine-3 antagonists |
| WO2008090429A1 (en) | 2007-01-22 | 2008-07-31 | Pfizer Products Inc. | Tosylate salt of a therapeutic compound and pharmaceutical compositions thereof |
| EP2624697B1 (en) | 2010-10-04 | 2015-11-25 | Merck Sharp & Dohme Corp. | Dihydrobenzoquinazolinone m1 receptor positive allosteric modulators |
| US11497586B2 (en) | 2014-03-21 | 2022-11-15 | Align Technology, Inc. | Segmented orthodontic appliance with elastics |
| EP3406609A1 (en) * | 2014-02-06 | 2018-11-28 | Heptares Therapeutics Limited | Bicyclic aza compounds as muscarinic receptor agonists |
| GB201504675D0 (en) * | 2015-03-19 | 2015-05-06 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
| GB201513742D0 (en) | 2015-08-03 | 2015-09-16 | Heptares Therapeutics Ltd | Muscarinic agonists |
| GB201513743D0 (en) * | 2015-08-03 | 2015-09-16 | Heptares Therapeutics Ltd | Muscarinic agonists |
| GB201513740D0 (en) * | 2015-08-03 | 2015-09-16 | Heptares Therapeutics Ltd | Muscarinic agonist |
| GB201709652D0 (en) * | 2017-06-16 | 2017-08-02 | Heptares Therapeutics Ltd | Pharmaceutical compounds |
-
2019
- 2019-03-25 TW TW108110350A patent/TWI801540B/zh active
- 2019-03-25 DK DK19716666.3T patent/DK3768669T3/da active
- 2019-03-25 HU HUE19716666A patent/HUE061533T2/hu unknown
- 2019-03-25 FI FIEP19716666.3T patent/FI3768669T3/fi active
- 2019-03-25 CN CN201980033911.5A patent/CN112154145B/zh active Active
- 2019-03-25 AR ARP190100746A patent/AR115015A1/es unknown
- 2019-03-25 US US17/040,479 patent/US11524954B2/en active Active
- 2019-03-25 WO PCT/US2019/023916 patent/WO2019183636A1/en active Application Filing
- 2019-03-25 PL PL19716666.3T patent/PL3768669T3/pl unknown
- 2019-03-25 CA CA3094366A patent/CA3094366A1/en active Pending
- 2019-03-25 JP JP2020551327A patent/JP2021519269A/ja active Pending
- 2019-03-25 EP EP19716666.3A patent/EP3768669B1/en active Active
- 2019-03-25 EP EP23151669.1A patent/EP4219464A1/en active Pending
- 2019-03-25 ES ES19716666T patent/ES2942837T3/es active Active
- 2019-03-25 PT PT197166663T patent/PT3768669T/pt unknown
-
2022
- 2022-10-19 US US17/969,479 patent/US20230192654A1/en active Pending
-
2023
- 2023-12-20 JP JP2023214561A patent/JP2024037979A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| US20210024497A1 (en) | 2021-01-28 |
| JP2021519269A (ja) | 2021-08-10 |
| CN112154145B (zh) | 2023-10-17 |
| US11524954B2 (en) | 2022-12-13 |
| CA3094366A1 (en) | 2019-09-26 |
| TW202003491A (zh) | 2020-01-16 |
| US20230192654A1 (en) | 2023-06-22 |
| EP3768669B1 (en) | 2023-01-25 |
| WO2019183636A1 (en) | 2019-09-26 |
| EP3768669A1 (en) | 2021-01-27 |
| TWI801540B (zh) | 2023-05-11 |
| JP2024037979A (ja) | 2024-03-19 |
| HUE061533T2 (hu) | 2023-07-28 |
| AR115015A1 (es) | 2020-11-18 |
| PL3768669T3 (pl) | 2023-07-03 |
| DK3768669T3 (da) | 2023-04-24 |
| FI3768669T3 (fi) | 2023-04-26 |
| PT3768669T (pt) | 2023-04-24 |
| CN112154145A (zh) | 2020-12-29 |
| EP4219464A1 (en) | 2023-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2942837T3 (es) | Derivados de azaespiro piperazina | |
| US11905284B2 (en) | 5,7-dihydro-pyrrolo-pyridine derivatives | |
| ES2742078T3 (es) | Piridonas bicíclicas novedosas como moduladores de gamma-secretasa | |
| ES2818806T3 (es) | Novedosas ciclopropabenzofuranil piridopirazindionas | |
| ES2937236T3 (es) | Derivados de dihidro-pirrolo-piridina | |
| EA044379B1 (ru) | Производные 5,7-дигидропирролопиридина |