ES2929876T3 - Replegamiento y purificación de moxetumomab pasudotox - Google Patents
Replegamiento y purificación de moxetumomab pasudotox Download PDFInfo
- Publication number
- ES2929876T3 ES2929876T3 ES18200555T ES18200555T ES2929876T3 ES 2929876 T3 ES2929876 T3 ES 2929876T3 ES 18200555 T ES18200555 T ES 18200555T ES 18200555 T ES18200555 T ES 18200555T ES 2929876 T3 ES2929876 T3 ES 2929876T3
- Authority
- ES
- Spain
- Prior art keywords
- refolding
- buffer
- immunoconjugate
- column
- purification
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39591—Stabilisation, fragmentation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2851—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the lectin superfamily, e.g. CD23, CD72
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/107—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides
- C07K1/113—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides without change of the primary structure
- C07K1/1136—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides without change of the primary structure by reversible modification of the secondary, tertiary or quarternary structure, e.g. using denaturating or stabilising agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/36—Extraction; Separation; Purification by a combination of two or more processes of different types
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/21—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Pseudomonadaceae (F)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/10—Immunoglobulins specific features characterized by their source of isolation or production
- C07K2317/14—Specific host cells or culture conditions, e.g. components, pH or temperature
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/55—Fusion polypeptide containing a fusion with a toxin, e.g. diphteria toxin
Abstract
La presente invención proporciona métodos para preparar inmunoconjugados activos, incluidos los inmunoconjugados anti-CD22. Adecuadamente, los métodos incluyen un proceso de replegamiento por lotes alimentados y/o un proceso de extracción en columna que dan como resultado un aumento en el rendimiento del inmunoconjugado sobre otros procesos que no utilizan los métodos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Replegamiento y purificación de moxetumomab pasudotox
Antecedentes de la invención
Campo de la invención
La presente invención proporciona métodos de preparación de inmunoconjugados activos, que incluyen inmunoconjugados anti-CD22. De manera adecuada, los métodos incluyen un proceso semicontinuo y/o un proceso de elución en columna que da como resultado un aumento en el rendimiento del inmunoconjugado sobre otros procesos que no utilizan los métodos.
Antecedentes de la técnica
La purificación económica a gran escala de proteínas es un factor crítico en la producción en la industria biofarmacéutica. Las proteínas terapéuticas se producen normalmente utilizando líneas celulares procariotas o eucariotas que están diseñadas para expresar la proteína de interés de un plásmido recombinante que contiene el gen que codifica la proteína. La separación de la proteína deseada de la mezcla de componentes alimentados a las células y subproductos celulares a una pureza adecuada, por ejemplo, suficiente para su uso como agente terapéutico humano, plantea un desafío formidable para los fabricantes de productos biológicos por varias razones.
Los fabricantes de productos farmacéuticos basados en proteínas deben cumplir con estrictos estándares regulatorios, incluidos los requisitos de pureza extremadamente estrictos. Para garantizar la seguridad, las agencias reguladoras, como la Administración de Alimentos y fármacos (FDA, de sus siglas en inglés), requieren que los productos farmacéuticos basados en proteínas estén sustancialmente libres de impurezas, incluidos los contaminantes relacionados con el producto, tales como los agregados, fragmentos y variantes de la proteína recombinante y los contaminantes relacionados con el proceso, tales como proteínas, componentes de medios, virus, ADN y endotoxinas de la célula hospedadora. Si bien varios esquemas de purificación de proteínas están disponibles y se utilizan ampliamente en la industria biofarmacéutica, normalmente incluyen una etapa de purificación por afinidad, tal como la purificación de proteína A en el caso de los anticuerpos, para alcanzar un grado de pureza farmacéuticamente aceptable.
El documento WO 2012/015912 describe métodos para aislar un inmunoconjugado o polipéptido activo mediante la purificación de una solución que contiene tanto el inmunoconjugado como el polipéptido activo y una variante ácida del mismo, tal como una variante desamidada, utilizando cromatografía de intercambio aniónico.
El desarrollo de un esquema de purificación aplicable tanto a una biomolécula particular como a varias biomoléculas que sea escalable, controlable y proporcione un alto rendimiento de una biomolécula purificada, permitirá su integración en el desarrollo del producto en una etapa muy temprana en el desarrollo general del fármaco. Por lo tanto, es deseable y ventajoso proporcionar un proceso simple y eficaz que pueda producir un fármaco de alta calidad y seguridad.
Breve sumario de la invención
La presente invención está definida por las reivindicaciones adjuntas. La presente invención se refiere a un método para preparar un inmunoconjugado activo, en el que dicho inmunoconjugado está compuesto por dos cadenas polipeptídicas unidas por un enlace disulfuro, y en el que el inmunoconjugado es la inmunotoxina moxetumomab pasudotox que comprende la subunidad VH-PE38 de SEQ ID NO: 1 y la subunidad VL de SEQ ID NO:2, comprendiendo el método replegamiento de dicho inmunoconjugado en un proceso semicontinuo de replegamiento y purificación del inmunoconjugado replegado en una o más columnas de cromatografía.
En realizaciones de los métodos, el replegamiento del inmunoconjugado comprende un tampón de replegamiento que tiene un pH de 9,5 o menos.
De manera adecuada, el inmunoconjugado comprende un anticuerpo o fragmento de unión a antígeno del mismo, por ejemplo, un anticuerpo o fragmento de unión a antígeno comprende un Fab, un Fab', un F(ab')2, un Fd, un Fv monocatenario o scFv, un Fv ligado a disulfuro, un dominio V-NAR, una IgNar, un intracuerpo, una IgGACH2, un minicuerpo, un F(ab')3, un tetracuerpo, un triacuerpo, un diacuerpo, un anticuerpo de dominio único, DVD-IG, Fcab, AcM2, un (scFv)2, o un scFv-Fc.
El anticuerpo o fragmento de unión a antígeno se une a un receptor de la superficie celular, adecuadamente, CD22.
Adecuadamente, el inmunoconjugado comprende una toxina, por ejemplo, toxinas que incluyen, pero sin limitación, exotoxina de Pseudomonas, ricina, abrina, toxina de la difteria y subunidades de la misma, así como toxinas botulínicas A a F y variantes, y derivados de las mismas.
La toxina es una exotoxina de Pseudomonas, o una variante de la misma, que tiene, adecuadamente, una secuencia de aminoácidos seleccionada del grupo que consiste en las SEQ ID NO: 16-22.
El anticuerpo o fragmento de unión a antígeno del mismo comprende una secuencia VH y una secuencia VL, adecuadamente, la secuencia VH se selecciona del grupo que consiste en las SEQ ID NO: 6-11, y la secuencia VL se selecciona del grupo que consiste en las SEQ ID NO: 2, y 12-15.
El inmunoconjugado de acuerdo con la invención es la inmunotoxina Moxetumomab pasudotox que comprende la subunidad VH-PE38 de SEQ ID NO: 1 y la subunidad VL de SEQ ID NO: 2.
El proceso semicontinuo utiliza una velocidad de adición de aproximadamente 52 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora a aproximadamente 13 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora, más, adecuadamente, una velocidad de adición de aproximadamente 35 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora a aproximadamente 17 ml cuerpos de inclusión solubilizados por l de tampón replegado por hora, o una velocidad de adición de aproximadamente 30 ml de cuerpos de inclusión solubilizados por l de tampón replegado por hora a aproximadamente 18 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora, o una velocidad de adición de aproximadamente 26 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora.
De manera adecuada, el tampón de separación para su uso en los diversos métodos comprende etanolamina de aproximadamente 30-60 mM, arginina de aproximadamente 0,25 a aproximadamente 0,75 M, EDTA de aproximadamente 1-3 mM, urea de aproximadamente 7-9 M y DTT de aproximadamente 9-11 mM.
La presente invención se refiere a una composición que comprende un inmunoconjugado que tiene menos de entre aproximadamente el 25 % y aproximadamente el 1 % de especies desamidadas, en la que el inmunoconjugado es la inmunotoxina Moxetumomab pasudotox que comprende la subunidad VH-PE38 de SEQ ID NO: 1 y la subunidad VL de SEQ ID NO: 2, y en la que dicho inmunoconjugado se prepara mediante el método de cualquiera de las reivindicaciones 1-11.
De manera adecuada, una cantidad del inmunoconjugado recuperado del método de preparación es al menos trescientos % (300 %) mayor que una cantidad del inmunoconjugado recuperado utilizando un método que no comprende un proceso de replegamiento semicontinuo.
Otras realizaciones, características y ventajas de las realizaciones, así como la estructura y el funcionamiento de las diversas realizaciones, se describen en detalle a continuación con referencia a los dibujos adjuntos.
Breves descripciones de los dibujos
La Figura 1 muestra un flujo adecuado del proceso de renaturalización y purificación para Moxetumomab pasudotox como se describe en el presente documento.
La Figura 2 muestra los resultados de una etapa de captura con Fractogel TMAE (M) (Ciclo 1).
La Figura 3 muestra los resultados de una cromatografía con hidroxiapatita.
La Figura 4 muestra los resultados de una cromatografía con Phenyl 650 M.
La Figura 5 muestra los resultados de una cromatografía con Q Sepharose HP. La carga máxima de la columna fue de 10,4 g/l.
La Figura 6 muestra los resultados de un análisis de IEC de Moxetumomab pasudotox parcialmente purificado. La Figura 7 muestra los resultados de una etapa de captura con Fractogel TMAE (M) (Ciclo 1).
La Figura 8 muestra los resultados de una cromatografía con hidroxiapatita.
La Figura 9 muestra los resultados de una cromatografía con Phenyl 650 M.
La Figura 10 muestra los resultados de una cromatografía con Q Sepharose HP.
La Figura 11 muestra los resultados de un cromatograma de la etapa de captura con Fractogel TMAE (M).
La Figura 12 muestra los resultados de un cromatograma de transferencia con Fractogel TMAE (M).
La Figura 13 muestra los resultados de un cromatograma con Fractogel TMAE (M) que utiliza el tampón de solubilización de cuerpos de inclusión (CI) de Moxetumomab pasudotox para la limpieza de la columna.
La Figura 14 muestra los resultados de un cromatograma de transferencia con Fractogel TMAE (M) con tampón de solubilización de CI.
La Figura 15 muestra los resultados de un cromatograma de tampón en blanco con Fractogel TMAE (M) con tampón de solubilización de CI.
La Figura 16 muestra los resultados de un cromatograma de transferencia con Fractogel TMAE (M) después de 9 ciclos de purificación.
La Figura 17 muestra los resultados de un cromatograma representativo con Capto Blue Sepharose.
La Figura 18 muestra los resultados de un análisis de SDS-PAGE no reducido de las fracciones de purificación con Capto Blue Sepharose mostradas en la Figura 17.
La Figura 19 muestra los resultados de un cromatograma representativo de la etapa de captura con Capto Blue Sepharose.
Descripción detallada de la invención
Tal como se utiliza en la presente memoria descriptiva, las formas singulares "un", "una" y "el", "la" específicamente también abarcan las formas plurales de los términos a los que se refieren, a menos que el contenido indique claramente lo contrario. El término "aproximadamente" como se utiliza en el presente documento significa aproximadamente, en la región de, prácticamente, o alrededor. Cuando el término "aproximadamente" se utiliza junto con un intervalo numérico, modifica ese intervalo al extender los límites por encima y por debajo de los valores numéricos establecidos. En general, el término "aproximadamente" se utiliza en el presente documento para modificar un valor numérico por encima y por debajo del valor establecido por una variación del 20 %.
Los términos técnicos y científicos utilizados en el presente documento tienen el significado comúnmente entendido por un experto en la materia a la que se refiere la presente solicitud, a menos que se defina lo contrario. En el presente documento se hace referencia a diversas metodologías y materiales conocidos por los expertos en la materia. Los trabajos de referencia estándar que establecen los principios generales de la tecnología de ADN recombinante incluyen Sambrook et al., "Molecular Cloning: A Laboratory Manual," 2a Ed., Cold Spring Harbor Laboratory Press, Nueva York (1989); Kaufman et al., Eds., "Handbook of Molecular and Cellular Methods in Biology in Medicine," CRC Press, Boca Raton (1995); y McPherson, Ed., "Directed Mutagenesis: A Practical Approach," IRL Press, Oxford (1991).
Los términos "polipéptido", "péptido", "proteína", y "fragmento de proteína" se utilizan indistintamente en el presente documento para referirse a un polímero de restos de aminoácidos. Los términos se aplican a los polímeros de aminoácidos en donde uno o más restos de aminoácidos es un mimético químico artificial de un aminoácido correspondiente de origen natural, así como a polímeros de aminoácidos de origen natural y a polímeros de aminoácidos de origen no natural.
El término "aminoácido" se refiere a aminoácidos de origen natural y sintético, así como a análogos de aminoácidos y miméticos de aminoácidos que funcionan de una manera similar a los aminoácidos de origen natural. Los aminoácidos de origen natural son los codificados mediante el código genético, así como aquellos aminoácidos que se modifican posteriormente, por ejemplo, hidroxiprolina, gamma-carboxiglutamato, y O-fosfoserina. Análogos de aminoácidos se refieren a los compuestos que tienen la misma estructura química básica que la de un aminoácido de origen natural, por ejemplo, un carbono alfa que está unido a un hidrógeno, un grupo carboxilo, un grupo amino, y un grupo R, por ejemplo, homoserina, norleucina, metionina sulfóxido, metionina metil sulfonio. Dichos análogos pueden tener grupos R modificados (por ejemplo, norleucina) o cadenas peptídicas modificadas, pero retienen la misma estructura química básica que un aminoácido que se produce naturalmente. Miméticos de aminoácidos se refiere a compuestos químicos que tienen una estructura que es diferente de la estructura química general de un aminoácido, pero que funcionan de manera similar a un aminoácido de origen natural. Los aminoácidos cargados negativamente incluyen ácido aspártico (o aspartato) y ácido glutámico (o glutamato). Los aminoácidos cargados positivamente incluyen arginina, histidina y lisina.
La "composición" a purificar en el presente documento comprende el polipéptido de interés y una o más impurezas. La composición puede estar "parcialmente purificada" (es decir, haber sido sometida a una o más etapas de purificación, o puede obtenerse directamente de una célula u organismo hospedador que produce el polipéptido (por ejemplo, la composición puede comprender líquido de cultivo celular recogido).
Una "variante ácida" es una variante de un polipéptido o inmunoconjugado que es más ácido (por ejemplo, según lo determinado mediante cromatografía de intercambio catiónico) que el polipéptido de interés. Un ejemplo de una variante ácida es una variante desamidada. Las proteínas desamidadas son aquellas que han tenido algunos o todos los grupos funcionales amida libres hidrolizados en ácidos carboxílicos, como la conversión de glutaminas en ácido glutámico. La velocidad de esta reacción depende de la secuencia primaria, estructura tridimensional, pH, temperatura, tipo de tampón, fuerza iónica y otras propiedades de la solución. De manera importante, la reacción de desamidación introduce una carga negativa en la molécula. Como se describe adicionalmente a continuación, la desamidación de proteínas puede tener un impacto negativo en la actividad de las proteínas.
Como se utiliza en el presente documento, los términos "anticuerpo" e "inmunoglobulina" se utilizan indistintamente en el sentido más amplio e incluyen anticuerpos monoclonales (por ejemplo, anticuerpos monoclonales de longitud completa o intactos), anticuerpos policlonales, anticuerpos multivalentes, anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos siempre que exhiban la actividad biológica deseada) y fragmentos de anticuerpos como se describe en el presente documento. La expresión "anticuerpo biespecífico" pretende incluir cualquier anticuerpo que
tenga dos especificidades de unión diferentes, es decir, el anticuerpo se une a dos epítopos diferentes, que pueden ubicarse en el mismo antígeno diana o, más habitualmente, en diferentes antígenos diana.
Los anticuerpos e inmunoglobulinas nativas son generalmente glucoproteínas heterotetramé ricas de aproximadamente 150.000 Dalton, compuestas de dos cadenas ligeras idénticas (L) y dos cadenas pesadas idénticas (H). Cada cadena ligera está unida a una cadena pesada mediante un enlace disulfuro covalente, mientras que el número de enlaces disulfuro varía entre las cadenas pesadas de diferentes isotipos de inmunoglobulina. Cada cadena pesada y ligera tiene también puentes disulfuro intracatenarios a espacios regulares. Cada cadena pesada tiene en un extremo un dominio variable (VH) seguido de varios dominios constantes. Cada cadena ligera tiene un dominio variable en un extremo (VH) y un dominio constante en su otro extremo. El dominio constante de la cadena ligera está alineado con el primer dominio constante de la cadena pesada y el dominio variable de cadena ligera está alineado con el dominio variable de la cadena pesada. Se cree que restos de aminoácidos particulares forman una interfaz entre los dominios variables de cadena ligera y cadena pesada (Clothia et al., J. Mol. Biol. 186, 651-66, 1985); Novotny y Haber, Proc. Natl. Acad. Sci. utiliza 82, 4592-4596 (1985)). Cinco clases de inmunoglobulina humana se definen en base a su composición de cadena pesada, y se denominan IgG, IgM, IgA, IgG e IgD. Los anticuerpos clase IgG y clase IgA se dividen además en subclases, concretamente, IgG1, IgG2, IgG3, e IgG4, e IgAl e IgA2. Las cadenas pesadas en los anticuerpos IgG, IgA e IgD tienen tres dominios de región constante, que se designan como CHI, CH2 y CH3, y las cadenas pesadas en los anticuerpos IgM e IgE tienen cuatro dominios de región constante, CHI, CH2, CH3 y CH4. Por tanto, las cadenas pesadas tienen una región variable y tres o cuatro regiones constantes. Se revisan la estructura y la función de la inmunoglobulina, por ejemplo, en Harlow et al., Eds., Antibodies: A Laboratory Manual, Capítulo 14, Cold Spring Harbor Laboratory, Cold Spring Harbor (1988).
La expresión "fragmento de anticuerpo" se refiere a una porción de un anticuerpo intacto y se refiere a las regiones variables determinantes antigénicas de un anticuerpo intacto. Los ejemplos de fragmentos de anticuerpo incluyen, pero, sin limitación, a Fab, Fab', F(ab')2, Fv y fragmentos de Fv de cadena sencilla, anticuerpos lineales, anticuerpos monocatenarios y anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpo.
La expresión "anticuerpo monoclonal" tal como se utiliza en el presente documento, se refiere a un anticuerpo obtenido a partir de una población de anticuerpos sustancialmente homogéneos, es decir, los anticuerpos individuales que comprenden la población son idénticos salvo por las posibles mutaciones que tienen lugar de manera natural que pueden estar presentes en pequeñas cantidades. Los anticuerpos monoclonales son altamente específicos y se unen a un solo antígeno. Además, a diferencia de las preparaciones de anticuerpos policlonales que normalmente incluyen anticuerpos diferentes dirigidos contra determinantes diferentes (epítopos), cada anticuerpo monoclonal se dirige contra un único determinante del antígeno. Que un anticuerpo "se une selectivamente" o "se une específicamente" significa que el anticuerpo reacciona o se asocia con mayor frecuencia, más rápidamente, con mayor duración, con mayor afinidad, o con alguna combinación de lo anterior con un epítopo que con sustancias alternativas, incluso proteínas no relacionadas. "Se une selectivamente" o "se une específicamente" significa, por ejemplo, que un anticuerpo se une a una proteína con una Kd de al menos aproximadamente 0,1 mM, pero más generalmente al menos aproximadamente 1 |jM. "Se une selectivamente" o "se une específicamente" significa en ocasiones que un anticuerpo se une a una proteína a veces con una Kd de al menos aproximadamente 0,1 j M o mejor, y otras veces al menos aproximadamente 0,01 j M o mejor. Debido a la identidad de secuencia entre proteínas homólogas en diferentes especies, la unión específica puede incluir un anticuerpo que reconoce una proteína marcadora de células tumorales en más de una especie.
Los anticuerpos del presente documento incluyen específicamente anticuerpos “quiméricos" en los que una porción de la cadena pesada y/o ligera es idéntica u homóloga a las secuencias correspondientes en los anticuerpos procedentes de una especie concreta o pertenecientes a una clase o subclase de anticuerpo concreto, mientras que el grupo de la(s) cadena(s) es idéntico u homólogo a las secuencias correspondientes en los anticuerpos procedentes de otra especie o pertenecientes a otra clase o subclase de anticuerpos, así como fragmentos de dichos anticuerpos, siempre que muestren la actividad biológica deseada (patente de e E.UU. n.° 4.816.567; y Morrison et al., Proc. Natl. Acad. Sci. utiliza 57:6851-6855 (1984)).
Las formas "humanizadas" de anticuerpos no humanos (por ejemplo, murinos) son anticuerpos quiméricos que contienen una secuencia mínima procedente de inmunoglobulina no humana. En su mayoría, los anticuerpos humanizados son inmunoglobulinas humanas (anticuerpo receptor) en la que los restos de una región hipervariable del receptor son sustituidos por restos de una región hipervariable de una especie no humana (anticuerpo donante), tal como ratón, rata, conejo o primate no humano que tiene la especificidad, afinidad y capacidad deseadas. En algunos casos, los restos de la región marco (FR) de la inmunoglobulina humana están sustituidos por los correspondientes restos no humanos. Además, los anticuerpos humanizados pueden comprender restos que no se encuentran en el anticuerpo receptor o en el anticuerpo donante. Estas modificaciones se efectúan para refinar adicionalmente el funcionamiento del anticuerpo. En general, el anticuerpo humanizado comprenderá sustancialmente la totalidad de al menos uno, y normalmente dos, dominios variables, en los que todos o sustancialmente todos los bucles hipervariables corresponden a los de una inmunoglobulina no humana y todas, o sustancialmente todas, las FR son las de una secuencia de inmunoglobulina humana. El anticuerpo humanizado también comprenderá al menos una porción de una región constante de inmunoglobulina (Fc), normalmente de una inmunoglobulina humana. Para más detalles, véase Jones et al., Nature 327:522-525 (1986); Riechmann et al., Nature 332:323-329 (1988); y Presta, Curr. Op. Struct. Biol.
2:593-596 (1992). Véanse también los siguientes artículos de revisión y referencias citadas en los mismos: Vaswani y
Hamilton, Ann. Allergy, Asthma & Immunol. 7: 105-1 15 (1998); Harris, Biochem. Soc. Transactions 23: 1035-1038 (1995); Hurle y Gross, Curr. Op. Biotech. 5:428-433 (1994).
Un "anticuerpo humano" es uno que posee una secuencia de aminoácidos que corresponde a la de un anticuerpo producido por un ser humano y/o que ha sido fabricado utilizando cualquiera de las técnicas para producir anticuerpos humanos como se desvela en el presente documento. Esta definición de un anticuerpo humano excluye específicamente un anticuerpo humanizado que comprenda restos de unión a antígeno no humanos.
El término "inmunoconjugado" o "conjugado" o "inmunotoxina" como se utiliza en el presente documento se refiere a un compuesto o un derivado del mismo que está unido a un agente de unión celular (por ejemplo, un anticuerpo anti-CD22 o fragmento del mismo) y se define mediante una fórmula genérica: C-L-A, en donde C = citotoxina, L = conector y A = agente de unión celular (por ejemplo, anticuerpos anti-CD22 o fragmento de anticuerpo). Los inmunoconjugados también pueden definirse mediante la fórmula genérica en orden inverso: A-L-C.
El término "citotoxina" o "agente citotóxico", tal como se utiliza en el presente documento, se refiere a una sustancia que inhibe o evita la función de las células y/o produce la destrucción de las células. El término pretende incluir isótopos radiactivos (por ejemplo, At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32 e isótopos radioactivos de Lu), agentes quimioterapéuticos, por ejemplo, metotrexato, adriamicina, alcaloides de la vinca (vincristina, vinblastina, etopósido), doxorrubicina, melfalán, mitomicina C, clorambucilo, daunorrubicina u otros agentes intercalantes, enzimas y fragmentos de las mismas, tales como enzimas nucleolíticas, antibióticos y toxinas tales como toxinas de moléculas pequeñas o toxinas enzimáticamente activas de origen bacteriano, fúngico, vegetal o animal, incluyendo fragmentos y/o variantes de los mismos, y los diversos agentes antitumorales o anticancerígenos desvelados a continuación. Los ejemplos de agentes citotóxicos incluyen, pero sin limitación, abrina, ricina, exotoxina de Pseudomonas (PE), toxina diftérica (DT), toxina botulínica, o toxinas modificadas de la misma. Por ejemplo, PE y DT son compuestos altamente tóxicos que normalmente causan la muerte por toxicidad hepática. PE y Dt , sin embargo, pueden modificarse en una forma para su uso como inmunotoxina mediante la eliminación del componente de direccionamiento nativo de la toxina (por ejemplo, el dominio la de PE o la cadena B de DT) y reemplazarlo con una fracción de direccionamiento diferente, tal como un anticuerpo.
La toxina es exotoxina de Pseudomonas. La exotoxina A de Pseudomonas (PE) es una proteína monomérica extremadamente activa (peso molecular 66 kD), secretada por Pseudomonas aeruginosa, que inhibe la síntesis de proteínas en células eucariotas mediante la inactivación del factor de alargamiento 2 (EF-2) mediante la catalización de su ADP-ribosilación (catalización de la transferencia de la fracción ribosilo ADP de NAD oxidado en EF-2).
Un "inmunoconjugado de PE" o "inmunotoxina de PE" es un inmunoconjugado o inmunotoxina que comprende un anticuerpo o fragmento de unión a antígeno del mismo y una toxina de PE o una variante de la misma.
"Purificar" un polipéptido o inmunoconjugado de una composición que comprende el polipéptido y una o más impurezas, significa aumentar el grado de pureza del polipéptido en la composición mediante la eliminación (total o parcialmente) de al menos una impureza de la composición. De acuerdo con la presente invención, la purificación se realiza sin el uso de una etapa de cromatografía de afinidad.
El término "cromatografía" se refiere al proceso por el cual un soluto de interés en una mezcla se separa de otros solutos en una mezcla como resultado de diferencias en las tasas a las cuales los solutos individuales de la mezcla migran a través de un medio estacionario bajo la influencia de una fase en movimiento, o en procesos de unión y elución.
El término "intercambio iónico" y "cromatografía de intercambio iónico" se refiere al proceso cromatográfico en el que un soluto de interés (como una proteína) en una mezcla interactúa con un compuesto cargado unido (por unión covalente) a un material de intercambio iónico de fase sólida tal que el soluto de interés interactúa no específicamente con el compuesto cargado más o menos que las impurezas de soluto o contaminantes en la mezcla. Los solutos contaminantes en la mezcla se eluyen de una columna del material de intercambio iónico más rápido o más lento que el soluto de interés o se unen o excluyen de la resina con respecto al soluto de interés. La "cromatografía de intercambio iónico" incluye específicamente intercambio de cationes, intercambio de aniones y la cromatografía de modo mixto.
La frase "material de intercambio iónico" se refiere a una fase sólida que está cargada negativamente (es decir, una resina de intercambio catiónico) o cargada positivamente (es decir, una resina de intercambio aniónico). La carga puede proporcionarse mediante la unión de uno o más ligandos cargados a la fase sólida, por ejemplo, mediante enlace covalente. Como alternativa, además, la carga puede ser una propiedad inherente de la fase sólida (por ejemplo, como es el caso de la sílice, que tiene una carga negativa global).
Una "resina de intercambio aniónico" se refiere a una fase sólida que está cargada positivamente, teniendo así uno o más ligandos cargados positivamente unidos a la misma. Se puede utilizar cualquier ligando cargado positivamente unido a la fase sólida adecuado para formar la resina de intercambio aniónico, tal como grupos amino cuaternarios. Las resinas de intercambio aniónico disponibles comercialmente incluyen celulosa DEAE, Poros PI 20, PI 50, HQ 10, HQ 20, HQ 50, D 50 de Applied Biosystems, Sartobind Q de Sartorius, MonoQ, MiniQ, Source 15Q y 30Q, Q, DEAE y ANX Sepharose Fast Flow, Q Sepharose de alto rendimiento, QAE SEPHADEX™ y FAST Q SEPHAROSE™ (GE
Healthcare), WP PEI, WP DEAM, WP QUAT de J. T. Baker, Hydrocell DEAE y Hydrocell QA de Biochrom Labs Inc., U Osphere Q, Macro-Prep DEAE y Macro-Prep High Q de Biorad, Ceramic HyperD Q, ceramic HyperD DEAE, T risacryl M y LS DEAE, Spherodex LS DeAe , QMA Spherosil LS, QMA Spherosil M y Mustang Q de Pall Technologies, resinas aniónicas DOw Ex de malla fina de base fuerte de tipo I y tipo II y DOWEX MONOSPHER E 77, anión de base débil de las separaciones de líquidos Dow, membrana Intercept Q, Matrex Cellufine A200, A500, Q500 y Q800, de Millipore, Fractogel EMD TMAE, Fractogel EMD DEAE y Fractogel EMD DMAE de EMD, intercambiadores de aniones Amberlite fuertes y débiles tipo I y II, intercambiadores de aniones DOWEX fuertes y débiles tipo I y II, intercambiadores de aniones Diaion fuertes y débiles tipo I y II, Duolite de Sigma-Aldrich, TSK gel Q y DEAE 5PW y 5PW-HR, Toyopearl SuperQ-650S, 650M y 650C, QAE-550C y 650S, DEAE-650M y 650C de Tosoh, QA52, DE23, DE32, DE51, DE52, DE53, Express-Ion D y Express-Ion Q de Whatman.
Por "fase sólida" se entiende una matriz no acuosa a la que pueden adherirse uno o más ligandos argados. La fase sólida puede ser una columna de purificación, una fase discontinua de partículas discretas, una membrana o un filtro, etc. Los ejemplos de materiales para formar la fase sólida incluyen polisacáridos (tal como agarosa y celulosa); y otras matrices mecánicamente estables tales como sílice (por ejemplo, vidrio de poro controlado), poli(estirenodivinil)benceno, poliacrilamida, partículas cerámicas y derivados de cualquiera de los anteriores.
El término "unión específica" como se utiliza en el presente documento, tal como para describir las interacciones entre una molécula de interés y un ligando unido a una matriz de fase sólida, se refiere a la unión generalmente reversible de una proteína de interés a un ligando a través de los efectos combinados de complementariedad espacial de las estructuras de proteínas y ligandos en un sitio de unión junto con fuerzas electrostáticas, enlaces de hidrógeno, fuerzas hidrófobas y/o fuerzas de van der Waals en el sitio de unión. Cuanto mayor sea la complementariedad espacial y cuanto más fuertes sean las otras fuerzas en el sitio de unión, mayor será la especificidad de unión de una proteína para su ligando respectivo. Los ejemplos no limitantes de unión específica incluyen unión de anticuerpo-antígeno, unión de enzima-sustrato, unión de enzima-cofactor, quelación de ión metálico, unión de ADN-proteína de unión a ADN, interacciones proteína-proteína reguladoras, y similares.
La expresión "unión no específica" como se utiliza en el presente documento, tal como para describir las interacciones entre una molécula de interés y un ligando u otro compuesto unido a una matriz de fase sólida, se refiere a la unión de una proteína de interés al ligando o compuesto en una matriz de fase sólida a través de fuerzas electrostáticas, enlaces de hidrógeno, fuerzas hidrófobas y/o fuerzas de van der Waals en el sitio de unión, pero que carece de complementariedad estructural que aumenta los efectos de las fuerzas no estructurales. Los ejemplos de interacciones no específicas incluyen, pero sin limitación, fuerzas electrostáticas, hidrófobas y de van der Waals, así como también enlaces de hidrógeno.
Un "tampón" utilizado en la presente invención es una solución que resiste cambios en el pH mediante la adición de ácido o base por la acción de sus componentes conjugados ácido-base. Pueden emplearse diversos tampones en un método de la presente invención dependiendo del pH deseado del tampón y la etapa particular en el proceso de purificación [véase Buffers. A Guide for the Preparation and Use of Buffers in Biological Systems, Gueffroy, D., ed. Calbiochem Corporation (1975)]. Los ejemplos no limitantes de componentes de tampón que pueden utilizarse para controlar el intervalo de pH deseable para un método de la invención incluyen acetato, citrato, histidina, fosfato, tampones de amonio tales como acetato de amonio, succinato, MES, CHAPS, MOPS, MOPSO, HEPES, Tris, y similares, así como combinaciones de estos TRIS-ácido málico-NaOH, maleato, cloroacetato, formiato, benzoato, propionato, piridina, piperazina, ADA, PIPES, ACES, BES, TES, tricina, bicina, TAPS, etanolamina, CHES, CAPS, metilamina, piperidina, ácido O-bórico, ácido carbónico, ácido láctico, ácido butanoandioico, ácido dietilmalónico, glicilglicina, HEPPS, HEPPSO, imidazol, fenol, POPSO, succinato, TAPS, a base de amina, bencilamina, trimetil o dimetil o etil o fenil amina, etilendiamina o mofolina. Los componentes adicionales (aditivos) pueden estar presentes en un tampón según sea necesario, por ejemplo, se pueden utilizar sales para ajustar la fuerza iónica del tampón, tal como cloruro de sodio, sulfato de sodio y cloruro de potasio; y otros aditivos tal como aminoácidos (tal como glicina e histidina), caótropos (tal como urea), alcoholes (tales como metanol, manitol, glicerol y alcohol bencílico), detergentes (véase supra.) y azúcares (tal como sacarosa, manitol, maltosa, trehalosa, glucosa y fructosa). Los componentes y aditivos del tampón, y las concentraciones utilizadas, pueden variar de acuerdo con el tipo de cromatografía practicada en la invención.
El "tampón de carga" es el que se utiliza para cargar la composición que comprende la molécula polipeptídica de interés y una o más impurezas en la resina de intercambio iónico. El tampón de carga tiene una conductividad y/o pH de modo que la molécula polipeptídica de interés (y generalmente una o más impurezas) se une a la resina de intercambio iónico o dicha proteína de interés fluye a través de la columna mientras que las impurezas se unen a la resina.
La expresión "tampón de lavado" cuando se utiliza aquí se refiere a un tampón utilizado para lavar o re-equilibrar la resina de intercambio iónico, antes de eluir la molécula polipeptídica de interés. De manera conveniente, el tampón de lavado y el tampón de carga pueden ser los mismos, pero esto no es necesario.
El "tampón de elución" se utiliza para eluir el polipéptido de interés de la fase sólida. La conductividad y/o el pH del tampón de elución son tales que el polipéptido de interés se eluye de la resina de intercambio iónico.
El "pi" o "punto isoeléctrico" de un polipéptido se refiere al pH en el cual la carga positiva del polipéptido equilibra su carga negativa, pudiéndose calcular pi a partir de la carga neta de los restos de aminoácidos o restos de ácido siálico de los carbohidratos unidos del polipéptido o se puede determinar mediante enfoque isoeléctrico.
Por "unión" de una molécula a un material de intercambio iónico, se entiende la exposición de la molécula al material de intercambio iónico en condiciones apropiadas (pH/conductividad) de modo que la molécula se inmoviliza de manera reversible en o sobre el material de intercambio iónico en virtud de interacciones iónicas entre la molécula y un grupo cargado o grupos cargados del material de intercambio iónico.
Por "lavado" del material de intercambio iónico se entiende el paso de un tampón apropiado a través o sobre el material de intercambio iónico.
Para "eluir" una molécula (por ejemplo, polipéptido o impureza) de un material de intercambio iónico se entiende eliminar la molécula de la misma mediante la alteración de la fuerza iónica del tampón que rodea el material de intercambio iónico de modo que el tampón compita con la molécula por los sitios cargados en el material de intercambio iónico.
Como se utiliza en la presente divulgación y en las reivindicaciones, las formas singulares "un", "uno", "una", y "el" o "la", incluyen formas en plural a menos que el contexto dicte claramente otra cosa.
Se entiende que siempre que se describan realizaciones en el presente documento con el lenguaje "que comprende", se proporcionan también realizaciones análogas descritas en términos de "que consisten en y/o" que consisten esencialmente en ".
La expresión "y/o" como se utiliza en una frase tal como "A y/o B" en el presente documento pretende incluir tanto "A como B", "A o B", "A," y "B." Del mismo modo, la expresión "y/o" tal como se utiliza en una frase tal como "A, B y/o C" pretende abarcar cada una de las siguientes realizaciones: A, B, y C; A, B, o C; A o C; A o B; B o C; A y C; A y B; B y C; A (solo); B (solo); y C (solo).
Exotoxinas de Pseudomonas y otras toxinas
Las toxinas pueden emplearse con anticuerpos de la presente invención para producir inmunotoxinas. Las toxinas ejemplares incluyen ricina, abrina, toxina de la difteria y subunidades de la misma, así como las toxinas botulínicas de la A a la F. Estas toxinas están fácilmente disponibles en fuentes comerciales (por ejemplo, Sigma Chemical Company, St. Louis, Mo.). La toxina diftérica se aísla de Corynebaclerium diphtheriae. La ricina es la lectina RCA60 de Ricinus communis (semilla de ricino). El término también hace referencia a variantes tóxicas de la misma. Por ejemplo, véase, las Patentes de EE.UU. n.° 5.079.163 y 4.689.401. La aglutinina de Ricinus communis (RCA, de sus siglas en inglés) se presenta en dos formas designadas RCA 60 y RCA 120 de acuerdo con sus pesos moleculares de aproximadamente 65 y 120 kD, respectivamente (Nicholson y Blaustein, J. Biochem. Biophys. Acta 266:543 (1972)). La cadena A es responsable de inactivar la síntesis de proteínas y matar las células. La cadena B se une a la ricina con los restos de galactosa de la superficie celular y facilita el transporte de la cadena A en el citosol (Olsnes, et al., Nature 249: 621 631 (1974) y en la Patente de EE.UU. N° 3.060.165).
La abrina incluye lectinas tóxicas de Abrus precalorius. Los principios tóxicos, abrina a, b, c, y d, tienen un peso molecular de aproximadamente 63 y 67 kD y están compuestos por dos cadenas polipeptídicas A y B unidas por disulfuro. La cadena A inhibe la síntesis de proteínas; la cadena B (abrina-b) se une a los restos de D-galactosa (véase, Funatsu, et al., Agr. Biol. Chem. 52: 1095 (1988); y Olsnes, Methods Enzymol. 50:330-335 (1978)).
La toxina es la exotoxina de Pseudomonas (PE). La exotoxina de Pseudomonas (o exotoxina A) es una exotoxina producida por Pseudomonas aeruginosa. La expresión "exotoxina de Pseudomonas", como se utiliza en el presente documento, se refiere a una PE nativa de longitud completa (de origen natural) o una PE que ha sido modificada. Dichas modificaciones pueden incluir, pero sin limitación, eliminación del dominio la, varias deleciones de aminoácidos en los dominios Ib, II y III, sustituciones de aminoácidos individuales y la adición de una o más secuencias en el extremo carboxilo, tal como KDEL (SEQ ID NO: 3) y REDL (SEQ ID NO: 4). Véase Siegall, et al., J. Biol. Chem. 264: 14256-14261 (1989). El fragmento citotóxico de PE retiene al menos el 50 %, preferentemente el 75 %, más preferentemente al menos el 90 %, y lo más preferentemente el 95 % de la citotoxicidad de la PE nativa. El fragmento citotóxico es más tóxico que la PE nativa.
La exotoxina A de Pseudomonas (PE) nativa es una proteína monomérica extremadamente activa (peso molecular 66 kD), secretada por Pseudomonas aeruginosa, que inhibe la síntesis de proteínas en células eucariotas. La secuencia de PE nativa se proporciona en la Patente de Ee .UU. n.° 5.602.095 cedida de manera común. El método de acción es la inactivación de la ADP-ribosilación del factor de elongación 2 (EF-2). La exotoxina contiene tres dominios estructurales que actúan en concierto para causar citotoxicidad. El dominio Ia (aminoácidos 1-252) media la unión celular. El dominio II (aminoácidos 253-364) es responsable de la translocación al citosol y el dominio III (aminoácidos 400-613) media la ribosilación de ADP del factor de elongación 2. La función del dominio b (aminoácidos 365-399) permanece indefinida, aunque una gran parte de ella, aminoácidos 365-380, pueden eliminarse sin pérdida de citotoxicidad. Véase Siegall, et al., (1989), supra.
La PE empleada en la presente invención incluye la secuencia nativa, los fragmentos citotóxicos de la secuencia nativa y las variantes modificadas conservativamente de la PE nativa y sus fragmentos citotóxicos. Las variantes de PE útiles en la invención se describen en los documentos US 7.355.012 y WO 2007/016150 y WO 2009/032954. Los fragmentos citotóxicos de PE incluyen aquellos que son citotóxicos con o sin posterior procesamiento proteolítico u otro proceso en la célula diana (por ejemplo, como una proteína o pre-proteína). Los fragmentos citotóxicos de PE incluyen PE40, PE38 y PE35.
La PE se ha modificado para reducir o eliminar la unión celular no específica, frecuentemente mediante la eliminación del dominio la como se enseña en la Patente de EE.UU. n.° 4.892.827, aunque esto también se puede lograr, por ejemplo, mediante la mutación de ciertos restos del dominio la. La Patente de EE.UU n.° 5.512.658, por ejemplo, desvela que una PE mutada en la que está presente el dominio la, pero en la que los restos básicos del dominio la en las posiciones 57, 246, 247 y 249 se reemplazan con restos ácidos (ácido glutámico o "E"). )) exhibe una citotoxicidad no específica muy disminuida. Esta forma mutante de PE a veces se denomina PE4E.
PE40 es un derivado truncado de PE como se describió anteriormente en la materia, con una eliminación del dominio la de la molécula de PE nativa. Véase, Pai, et al., Proc. Nat'l Acad. Sci utiliza 55:3358-62 (1991); y Kondo, et al., J. Biol. Chem. 263:9470-9475 (1988). PE35 es un fragmento carboxiterminal de 35 kD de PE en la que se han eliminado los restos de aminoácidos 1-279 y la molécula comienza con una met en la posición 280 seguida por los aminoácidos 281-364 y 381-613 de PE nativa. PE35 y PE40 se desvelan, por ejemplo, en las patentes de los Estados Unidos N.° 5.602.095 y 4.892.827. PE4E es una forma de PE donde están presentes todos los dominios de PE nativa, pero donde los restos básicos del dominio la en las posiciones 57, 246, 247 y 249 se reemplazan con restos ácidos (ácido glutamina, o "E").
Se emplea el fragmento citotóxico PE38. PE38 es una pro-proteína de PE truncada compuesta por los aminoácidos 253-364 y 381-613 que se activa a su forma citotóxica al procesarse dentro de una célula (véase, por ejemplo, las patentes de EE. UU. Números 5.608.039, 7.355.012, y Pastan et al., Biochim. Biophys. Acta 1333:C1-C6 (1997)).
Como se ha señalado anteriormente, algunos o todos los dominios lb pueden eliminarse, y las porciones restantes se unen mediante un conector o directamente mediante un enlace peptídico. Parte de la porción amino del dominio II puede ser eliminada. Y, el extremo C-terminal puede contener la secuencia nativa de los restos 609-613 (REDLK) (SEQ ID NO: 5), o puede contener una variación encontrada para mantener la capacidad de la construcción para translocarse en el citosol, como REDL (SEQ ID NO: 4) o KDEL (SEQ ID NO: 3), y repeticiones de estas secuencias. Véase, por ejemplo, las patentes de EE.UU. n.° 5.854.044; 5.821.238; y 5.602.095 y el documento WO 99/51643. El PE es PE4E, PE40 o PE38, o cualquier forma de PE en la que se haya eliminado o reducido la citotoxicidad no específica a niveles en los que no se produzca una toxicidad significativa para las células no diana, puede utilizarse en las inmunotoxinas de la presente invención, siempre que siga siendo capaz de la translocación y ribosilación de EF-2 en una célula diana.
Variantes de PE modificadas conservativamente
Las variantes modificadas conservativamente de PE o fragmentos citotóxicos de las mismas tienen al menos un 80 % de similitud de secuencia, preferentemente al menos un 85 % de similitud de secuencia, más preferentemente al menos un 90 % de similitud de secuencia, y lo más preferentemente al menos un 95 % de similitud de secuencia a nivel de aminoácidos, con la PE de interés, tal como PE38.
La expresión "variantes modificadas de forma conservativa" se aplica a secuencias tanto de aminoácidos como de ácido nucleico. Con respecto a secuencias de ácidos nucleico concretas, las variantes modificadas de manera conservativa se refieren a aquellas secuencias de ácido nucleico que codifican secuencias de aminoácidos idénticas o esencialmente idénticas, o si el ácido nucleico no codifica una secuencia de aminoácidos, a secuencias de ácido nucleico esencialmente idénticas. A causa de la degeneración del código genético, un gran número de ácidos nucleicos funcionalmente idénticos codifican cualquier polipéptido dado. Por ejemplo, los codones GCA, GCC, GCG y GCU codifican, todos ellos, el aminoácido alanina. Por tanto, en cada posición donde una alanina se especifica mediante un codón, el codón se puede alterar a cualquiera de los correspondientes codones descritos sin alterar el polipéptido codificado. Dichas variaciones de ácido nucleico se denominan "variaciones silenciosas", que son un tipo de variaciones modificadas de forma conservativa. Cada secuencia de ácido nucleico del presente documento que codifica un polipéptido también describe todas y cada una de las posibles variaciones silenciosas del ácido nucleico. Un experto reconocerá que cada codón en un ácido nucleico (excepto AUG, que normalmente es el único codón para la metionina) puede modificarse para producir una molécula funcionalmente idéntica. En consecuencia, todas las variaciones silenciosas de un ácido nucleico que codifica un polipéptido están implícitas en cada secuencia descrita.
Como secuencias de aminoácidos, un experto en la materia reconocerá que las sustituciones, deleciones o adiciones individuales a un ácido nucleico, péptido, polipéptido o secuencia de proteínas que altera, añada o elimine un único aminoácido o un pequeño porcentaje de aminoácidos de la secuencia codificada es una "variante modificada de manera conservativa" donde la alteración da como resultado la sustitución de un aminoácido por un aminoácido químicamente similar.
Las exotoxinas de Pseudomonas empleadas en la invención pueden ensayarse para el nivel deseado de citotoxicidad
mediante ensayos bien conocidos por los expertos en la materia. Por tanto, los fragmentos citotóxicos de PE y las variantes modificadas de manera conservativa de dichos fragmentos pueden analizarse fácilmente para determinar la citotoxicidad. Un gran número de moléculas de PE candidatas pueden analizarse simultáneamente para determinar la citotoxicidad mediante métodos bien conocidos en la materia. Por ejemplo, los subgrupos de las moléculas candidatas pueden analizarse para determinar la citotoxicidad. Los subgrupos que reaccionan positivamente de las moléculas candidatas pueden subdividirse y reexaminarse continuamente hasta que se identifiquen el (los) fragmento(s) citotóxico(s) deseado(s). Dichos métodos permiten la detección rápida de grandes cantidades de fragmentos citotóxicos o variantes conservadoras de PE.
Inmunoconjugados anti-CD22/PE
El polipéptido de interés comprende un anticuerpo que se une específicamente a CD22. "CD22" se refiere a un antígeno de linfocitos B con restricción de linaje que pertenece a la superfamilia de Ig. Se expresa en el 60-70 % de los linfomas y leucemias de linfocitos B y no está presente en la superficie celular en las etapas iniciales del desarrollo de los linfocitos B o en las células madre. Véase, por ejemplo, Vaickus et al., Crit. Rev. Oncol/Hematol. 77:267-297 (1991). El polipéptido de interés puede ser un fragmento de anticuerpo que se une a CD22 (por ejemplo, Fab, o scFv).
Como se utiliza en el presente documento, el término "anti-CD22" en referencia a un anticuerpo, se refiere a un anticuerpo que se une específicamente a CD22 e incluye una referencia a un anticuerpo que se genera contra CD22. En algunas realizaciones, el CD22 es un CD22 de primate tal como CD22 humano. En una realización, el anticuerpo se genera contra CD22 humano sintetizado mediante un mamífero no primate después de la introducción en el animal de ADNc que codifica CD22 humano. El polipéptido de interés es un inmunoconjugado de anticuerpo CD22 que comprende la exotoxina PE38.
Un ejemplo de un inmunoconjugado CD22/PE38 es Moxetumomab pasudotox descrito en las Publicaciones de Solicitud de Patente Internacional N° WO 2012/015912, WO 98/41641 y WO2003/27135, las patentes de EE.UU. n.° 7.541.034, 7.355.012 y la publicación de EE.UU. n.° 2007/0189962. Moxetumomab pasudotox (CAT-8015) es una proteína de inmunotoxina recombinante compuesta por un fragmento Fv de anticuerpo basado en el anticuerpo anti-CD22 RFB4 fusionado a una forma truncada de la proteína de exotoxina de Pseudomonas, PE38. El fragmento Fv anti-CD22 consiste en dos dominios, un VL y un VH, donde este último se modificó para mejorar la unión a la diana CD22 humano. La proteína de Moxetumomab pasudotox está compuesta por dos polipéptidos independientes, la cadena VL (SEQ ID NO: 2) y la cadena VH, fusionadas en el extremo C al dominio PE38 (VH-PE38) (SEQ ID NO: 1). Otras secuencias VL y VH-PE38 útiles en esta invención se describen en los documentos US 7.541.034, 7.355.012, 2007/0189962 y WO 2012/015912. Ambos dominios se diseñaron para que cada uno contenga restos de cisteína diseñados para permitir la formación de un enlace disulfuro intermolecular. Esta característica aumenta la estabilidad de la proteína de fusión.
La secuencia de aminoácidos de la subunidad VH-P38 (SEQ ID NO: 1) de Moxetumomab pasudotox es la siguiente:
MEVQLVESGGGLVKPGGSLKLSCAASGFAFSIYDMSWVRQTPEKCLEWVAYISS
GGGTTYYPDTVKGRFTISRDNAKNTLYLQMSSLKSEDTAMYYCARHSGYGTHW
GVLFAYWGOGTLVSAKASGGPEGGSLAALTAHOACHLPLETFTRHROPRGW
EQLEQCGYPVQRLVALYIAARLSWNQVDQVIRALASPGSGGDLGEAIREQPE
QARLALTLAAAESERFVRQGTGNDEAGAANGPADSGDALLERNYPTGAEFL
GDGGDVSFSTRGTQNWTVERLLQAHRQLEERGYVFVGYHGTFLEAAQSIVF
GGVRARSQDLDAIWRGFYIAGDPALAYGYAQDQEPDARGRIRNGALLRVYV
PRSSLPGFYRTSLTLAAPEAAGEVERLIGHPLPLRLDAITGPEEEGGRLETILG
WPLAERTWIPSAIPTDPRNVGGDLDPSSIPDKEQAISALPDYASQPGKPPRED
LK (SEQ ID NO : 1)
La secuencia PE38 se muestra en negrita, y los cinco conectores de aminoácidos entre el dominio VH y el dominio PE38 se muestran subrayados.
La secuencia de aminoácidos de la subunidad VL (SEQ ID NO: 2) de Moxetumomab pasudotox es la siguiente:
MDIQMTQTTSSLSASLGDRVTISCRASQDISNYLNWYQQKPDGTVKLLIYYTSILH
SGVPSRFSGSGSGTDYSLTISNLEQEDFATYFCQQGNTLPWT FGCGTKLEIK (SEQ
ID NO : 2 )
La toxina PE del inmunoconjugado es una PE o una variante de la misma seleccionada entre las siguientes: PE nativa
AEEAFDLWNECAKACVLDLKDGVRSSRMSVDPAIADTNGQGVLHYSMVLEGGN
DALKLAIDNALSITSDGLTIRLEGGVEPNKPVRYSYTRQARGSWSLNWLVPIGHE
KPSNIKVF1HELNAGNQLSHMSPIYTIEMGDELLAKLARDATFFVRAHESNEMQP
TLAISHAGVSWMAQTQPRREKRWSEWASGKVLCLLDPLDGVYNYLAQQRCNL
DDTWEGKIYRVLAGNPAKHDLDIKPTVISHRLHFPEGGSLAALTAHQACHLPLET
ETRHRQPRGWEQLEQCGYPVQRLVALYLAARLSWNQVDQVIRNALASPGSGGD
LGEAIREQPEQARLALTLAAAESERFVRQGTGNDEAGAANGPADSGDALLERNY
PTGAEFLGDGGDVSFSTRGTQNWTVERLLQAHRQLEERGYVFVGYHGTFLEAA
QSIVFGGVRARSQDLDAIWRGFYIAGDPALAYGYAQDQEPDARGRIRNGALLRV
YVPRSSLPGFYRTSLTLAAPEAAGEVERLIGHPLPLRLDAITGPEEEGGRLETILGW
PLAERTWIPS AIPTDPRNV GGDLDPS SIPDKEQAIS ALPD Y ASQPGKPPREDLK
(SEQ ID NO: 16)
PE40
GGSLAALTAHQACHLPLETFTRHRQPRGWEQLEQCGYPVQRLVALYLAARLSW
NQVDQVIRNALASPGSGGDLGEAIREQPEQARLALTLAAAESERFVRQGTGNDE
AGAANADVVSLTCPVAAGECAGPADSGDALLERNYPTGAEFLGDGGDVSFSTR
GTQNWTVERLLQAHRQLEERGYVFVGYHGTFLEAAQSIVFGGVRARSQDLDAI
WRGFYIAGD PALAY G Y AQDQEPD ARGRIRN G ALLR Y Y VPRS S LPGFYRTS LTLA
APEAAGEVERLIGHPLPLRLDAITGPEEEGGRLETILGWPLAERTVVIPSAIPTDPR
NVGGDLDPSSIPDKEQAISALPDYAS QPGKPPREDLK (SEQ ID NO: 17)
PE38
GGSLAALTAHQACHLPLETFTRHRQPRGWEQLEQCGYPVQRLVALYLAARLSW
NQVDQVIRNALASPGSGGDLGEAIREQPEQARLALTLAAAESERFVRQGTGNDE
AGAANGPADSGDALLERNYPTGAEFLGDGGDVSFSTRGTQNWTVERLLQAHRQ
LEERGYVFVGYHGTFLEAAQSIVFGGVRARSQDLDAIWRGFYIAGDPALAYGYA
QDQEPD ARGRIRN GALLRV Y VPRS S LPGFYRTS LTLA APE A AGE VERLIGHPLPLR
LD AITGPEEEGGRLETILGWPLAERTWIPS AIPTDPRNV GGDLDPS SIPDKEQAIS A
LPDYASQPGKPPREDLK (SEQ ID NO: 18)
PE35
MWEQLEQCGYPVQRLVALYLAARLSWNQVDQVIRNALASPGSGGDLGEAIREQ
PEQARLALTLAAAESERFVRQGTGNDEAGAANGPADSGDALLERNYPTGAEFLG
DGGDVSFSTRGTQNWTVERLLQAHRQLEERGYVFVGYHGTFLEAAQSIVFGGVR
ARSQDLDAIWRGFYIAGDPALAYGYAQDQEPDARGRIRNGALLRVYVPRSSLPG
FYRTSLTLAAPEAAGEVERLIGHPLPLRLDAITGPEEEGGRLETILGWPLAERTVVI
PSAIPTDPRNVGGDLDPSSIPDKEQAISALPDYASQPGKPPREDLK (SEQ ID NO:
19)
PE-LR
RHRQPRGWEQLPTGAEFLGDGGDVSFSTRGTQNWTVERLLQAHRQLEERGYVF
VGYHGTFLEAAQSIVFGGVRARSQDLDAIWRGFYIAGDPALAYGYAQDQEPDAR
GRIRNGALLRVYVPRSSLPGFYRTSLTLAAPEAAGEVERLIGHPLPLRLDAITGPEE
EGGRLETILGWPLAERTVVIPS AIPTDPRNV GGDLDPS SIPDKEQAIS ALPDYASQP
GKPPREDLK (SEQ ID NO:20)
PE-LR-6X
RHRQPRGWEQLPTGAEFLGDGGDVSFSTRGTQNWTVERLLQAHRQLEEGGYVF
VGYHGTFLEAAQSIVFGGVRARSQDLDAIWAGFYIAGDPALAYGYAQDQEPDAA
GRIRNGALLRVYVPRSSLPGFYATSLTLAAPEAAGEVERLIGHPLPLRLDAITGPEE
AGGRLETILGWPLAERTVVIPSAIPTDPRNVGGDLDPSSIPDSEQAISALPDYASQP
GKPPREDLK (SEQ ID NO:21)
PE-38 (Moxetumomab pasudotox)
PEGGSLAALTAHQACHLPLETFTRHRQPRGWEQLEQCGYPVQRLVALYLAARLS
WNQVDQVIRNALASPGSGGDLGEAIREQPEQARLALTLAAAESERFVRQGTGND
EAGAANGPADSGDALLERNYPTGAEFLGDGGDVSFSTRGTQNWTVERLLQAHR
QLEERGYVFVGYHGTFLEAAQSIVFGGVRARSQDLDAIWRGFYIAGDPALAYGY
AQDQEPDARGRIRNGALLRVYVPRSSLPGFYRTSLTLAAPEAAGEVERLIGHPLPL
RLDAITGPEEEGGRLETILGWPLAERTVVIPSAIPTDPRNVGGDLDPSSIPDKEQAIS
ALPDYASQPGKPPREDLK (SEQ ID NO:22)
La toxina PE del inmunoconjugado se fusiona o conjuga con el dominio VH o VL directamente o mediante un conector en el extremo N o en el extremo C del dominio VH o VL. Un ejemplo de un conector se describe anteriormente para Moxetumomab pasudotox y corresponde a la secuencia de aminoácidos KASGG (SEQ ID NO: 23). Se pueden generar fácilmente conectores adicionales mediante técnicas conocidas en la materia.
Expresión de un inmunoconjugado de PE
El inmunoconjugado de PE de la presente invención se expresa, adecuadamente, en células, tales como células Bacterianas, y luego se aísla de cuerpos de inclusión. El inmunoconjugado de PE aislado de cuerpos de inclusión luego se purifica/aísla adicionalmente utilizando pasos posteriores como se describe en este documento.
Se puede utilizar una variedad de sistemas de vectores de expresión en el hospedador para expresar el inmunoconjugado de PE de la presente invención. Dichos sistemas de expresión en el hospedador representan vehículos mediante los cuales las secuencias codificantes de interés pueden producirse y posteriormente purificarse, pero también representan células que, cuando se transforman o transfectan con las secuencias que codifican los nucleótidos adecuados, expresan una molécula de anticuerpo de la invención in situ. Estos incluyen pero no se limitan a microorganismos tales como bacterias (por ejemplo,, E. coli, B. subtilis) transformadas con ADN de bacteriófago recombinante, ADN plásmido o vectores de expresión de ADN cósmido que contienen secuencias codificantes de anticuerpos; levadura (por ejemplo, Saccharomyces, Pichia) transformadas con vectores de expresión en levadura recombinantes que contienen secuencias codificantes de anticuerpo; sistemas de células de insecto infectadas con vectores de expresión de virus recombinante (por ejemplo, baculovirus) que contienen secuencias codificantes de anticuerpo; sistemas de células vegetales infectadas con vectores de expresión de virus recombinante (por ejemplo, virus del mosaico de la coliflor, CaMV; virus del mosaico del tabaco, TMV) o transformadas con vectores de expresión de plásmido recombinante que contienen secuencias codificantes de anticuerpo; o sistemas de células de mamíferos (por ejemplo, células COS, CHO, BLK, 293, 3T3) que albergan construcciones de expresión recombinantes que contienen promotores procedentes del genoma de células de mamífero (por ejemplo, promotor de metalotioneína) o de virus de mamífero (por ejemplo, el promotor tardío de adenovirus; el promotor 7.5K del virus vaccinia).
El ADN que codifica cada uno de los polipéptidos de la toxina VL y VH-PE (por ejemplo, VH-PE38) puede introducirse en un vector de expresión mediante técnicas bien conocidas en la materia.
Un "vector" se refiere a cualquier vehículo para la clonación y/o transferencia de un ácido nucleico a una célula hospedadora. Un vector puede ser un replicón al que se puede unir otro segmento de ADN para lograr la replicación del segmento adjunto. Un "replicón" se refiere a cualquier elemento genético (por ejemplo, plásmido, fago, cósmido, cromosoma, virus) que funciona como una unidad autónoma de replicación de ADN in vivo, es decir, capaz de replicarse bajo su propio control. El término "vector" incluye vehículos para introducir el ácido nucleico en una célula in vitro, ex vivo o in vivo. Se puede utilizar un gran número de vectores conocidos en la materia para manipular ácidos nucleicos, incorporar elementos de respuesta y promotores, tales como promotores inducibles, en genes, etc. Los vectores posibles incluyen, por ejemplo, plásmidos tales como pBR322 o derivados plásmidos pUC, o el vector Bluescript. Por ejemplo, la inserción de los fragmentos de ADN correspondientes a elementos de respuesta y promotores en un vector adecuado se puede lograr mediante la unión de los fragmentos de ADN apropiados en un vector elegido que tiene extremos cohesivos complementarios. Como alternativa, los extremos de las moléculas de ADN pueden modificarse enzimáticamente o cualquier sitio puede producirse mediante la unión de secuencias de nucleótidos (conectores) en los extremos del ADN. Dichos vectores pueden diseñarse para contener genes marcadores seleccionables que proporcionan la selección de células. Dichos marcadores permiten la identificación y/o selección de células hospedadoras que expresan las proteínas codificadas por el marcador.
La expresión "vector de expresión" se refiere a un vector, plásmido o vehículo diseñado para permitir la expresión de
una secuencia de ácido nucleico insertada después de la transformación en el hospedador. El gen clonado, es decir, la secuencia de ácido nucleico insertada, por ejemplo, un gen que codifica un VH anti-CD22, VL anti-CD22 o VH o VL anti-CD22 fusionado a una toxina PE, generalmente se coloca bajo el control de los elementos de control tales como un promotor, un promotor mínimo, un potenciador o similar. Las regiones de control de iniciación o promotores, que son útiles para dirigir la expresión de un ácido nucleico en la célula hospedadora deseada, son numerosos y familiares para los expertos en la materia. Prácticamente cualquier promotor capaz de dirigir la expresión de estos genes puede utilizarse en un vector de expresión, incluyendo, pero sin limitación, promotores virales, promotores bacterianos, promotores animales, promotores de mamíferos, promotores sintéticos, promotores constitutivos, promotores específicos de tejido, promotores relacionados con patogénesis o enfermedades, promotores específicos del desarrollo, promotores inducibles, promotores regulados por luz; incluyendo, pero sin limitación, la región promotora temprana de SV40 (SV40), el promotor contenido en la repetición terminal larga (LTR, de sus siglas en inglés) de 3' del virus del sarcoma de Rous (RSV, de sus siglas en inglés), el EIA o promotor tardío principal (MLP, de sus siglas en inglés) de adenovirus (Ad), el promotor temprano inmediato de citomegalovirus humano (HCMV, de sus siglas en inglés), el promotor timidina quinasa (TK) del virus herpes simplex (HSV, de sus siglas en inglés), el promotor IE1 baculovirus, el promotor del factor de elongación 1 alfa (EF1), promotor de gliceraldehído-3-fosfato deshidrogenasa (GAPDH), el promotor de la fosfoglicerato quinasa (PGK), el promotor de la ubiquitina C (Ube), el promotor de albúmina, las secuencias reguladoras del promotor metalotioneína-L de ratón y las regiones de control de la transcripción, los promotores ubicuos (HPRt , vimentina, p-actina, tubulina y similares), los promotores de los filamentos intermedios (desmina, neurofilamentos, queratina, GFAP, y similares), los promotores de genes terapéuticos (del tipo MDR, CFTR o factor VIII, y similares), promotores relacionados con patogénesis o enfermedades. Además, estas secuencias de expresión pueden modificarse mediante la adición de secuencias potenciadoras o reguladoras y similares.
El término "expresión" se refiere a la producción biológica de un producto codificado por una secuencia codificante. En la mayoría de los casos, una secuencia de ADN, incluida la secuencia codificante, se transcribe para formar un ARN mensajero (ARNm). El ARN mensajero luego se traduce para formar un producto polipeptídico que tiene una actividad biológica relevante. Además, el proceso de expresión puede implicar etapas de procesamiento adicionales para el producto de transcripción del ARN, como el empalme para eliminar intrones, y/o el procesamiento posterior a la traducción de un producto polipeptídico.
Los polipéptidos VL y VH-PE38 se expresan, adecuadamente, en células, por ejemplo, células Bacterianas, tal como E. coli. Los polipéptidos se expresan, por ejemplo, en células de E. coli y se aíslan de cuerpos de inclusión. En determinadas realizaciones, las subunidades VL y VH-PE38 se expresan en diferentes células. Por ejemplo, el VL se expresa en una célula en un primer vector y el VH-PE38 se expresa en una célula diferente en un segundo vector. En otras realizaciones, las subunidades VL y VH-PE38 se expresan en la misma célula en diferentes vectores, por ejemplo, el VL se expresa en una célula en un primer vector y el VH-PE38 se expresa en la misma célula en un vector diferente. En otras realizaciones determinadas, las subunidades VL y VH-PE38 se expresan en el mismo vector en la misma célula. Los cuerpos de inclusión de las células se recuperan, se solubilizan y las subunidades VL y VH-PE38 se combinan para formar un inmunoconjugado, como se describe en el presente documento.
Métodos de preparación de inmunoconjugados
En el presente documento se proporcionan métodos para preparar inmunoconjugados activos, es decir, inmunoconjugados capaces de unirse a una diana deseada y suministrar el compuesto (por ejemplo, inmunotoxina) que está unida al agente de direccionamiento celular (por ejemplo, un anticuerpo, fragmento de anticuerpo u otra proteína).
Los métodos descritos en el presente documento se utilizan, adecuadamente, para preparar inmunoconjugados que se desamidan en uno o más restos. Tal como se describe en el presente documento, tal desamidación a menudo da como resultado una inhibición de la potencia de un inmunoconjugado, y por lo tanto, los métodos proporcionados son beneficiosos para preparar inmunoconjugados activos adecuados para entornos clínicos.
Tal como se describe en el presente documento, los inmunoconjugados se preparan, adecuadamente, utilizando sistemas de expresión de bacterias, incluyendo E. coli. Los cuerpos de inclusión de las células se recuperan, se solubilizan y las proteínas se recuperan. En realizaciones, las subunidades de toxina VL y VH diseñadas para formar un inmunoconjugado, como se describe en el presente documento, se preparan en las células Bacterianas.
Tal como se describe en el presente documento, los cuerpos de inclusión que comprenden las subunidades de inmunoconjugado deseados se solubilizan, se concentran y se clarifican. Los métodos adecuados de aclaramiento se describen en el presente documento, así como en la divulgación y los ejemplos del documento WO2012/015912. Tras el aclaramiento, se lleva a cabo el replegamiento del inmunoconjugado.
En realizaciones, los métodos para preparar inmunoconjugados comprenden, adecuadamente, replegar un inmunoconjugado utilizando un proceso semicontinuo. Después del replegamiento, el inmunoconjugado replegado se purifica con una o más columnas de cromatografía como se describe en el presente documento, así como en el documento WO2012/015912.
Como se utiliza en el presente documento, "repliegue" se refiere al proceso bajo el cual una proteína, aislada de cuerpos de inclusión, se pliega en su estructura tridimensional característica y funcional a partir de una orientación aleatoria previa.
Un proceso "semicontinuo" se refiere a un proceso de replegamiento en el que se agrega una mezcla de cuerpo de inclusión solubilizada (que contiene el inmunoconjugado deseado) (es decir, se inyecta o mezcla) a un tampón de replegamiento adecuado a una velocidad controlada durante un período de tiempo. De manera adecuada, la adición se produce a una velocidad constante durante todo el curso del tiempo, aunque la velocidad también puede variar durante el proceso, si se desea. Este curso de tiempo de adición se denomina aquí como la "tasa de adición", y se expresa, adecuadamente, en l/h.
Se ha descubierto inesperadamente que el uso de un proceso semicontinuo en el que las subunidades de un inmunoconjugado están inicialmente presentes en una concentración baja, y la concentración aumenta luego durante un período de tiempo relativamente prolongado (adecuadamente, a una velocidad de adición fija durante el curso de 2, 3, 4, 5, 6 horas, etc.), da como resultado un aumento del rendimiento del inmunoconjugado en comparación con los procesos no semicontinuos que utilizan una adición masiva de subunidades, en las que la concentración es inicialmente mayor y no cambia con el tiempo. Este descubrimiento es particularmente sorprendente cuando se utilizan las subunidades VL y VH-PE38 descritas en el presente documento, que deben replegarse juntas (es decir, unirse en una solución diluida) para formar el inmunoconjugado final, en contraste con una proteína de un solo componente.
El rendimiento de inmunoconjugado recuperado es, adecuadamente, al menos aproximadamente 20 % mayor, al menos aproximadamente 30 % mayor, al menos aproximadamente 40 % mayor, al menos aproximadamente 50 % mayor, al menos aproximadamente 60 % mayor, al menos aproximadamente 70 % mayor, al menos aproximadamente 80 % mayor, al menos aproximadamente 90 % mayor, al menos aproximadamente 100 % mayor, al menos aproximadamente 125 % mayor, al menos aproximadamente 150 % mayor, al menos aproximadamente 175 % mayor, al menos aproximadamente 200 % mayor, al menos aproximadamente 300 % mayor, al menos aproximadamente 400 % mayor, al menos aproximadamente 500 % mayor, al menos aproximadamente 600 % mayor, al menos aproximadamente 700 % mayor, al menos aproximadamente 800 % mayor, al menos aproximadamente 900 % mayor, etc., en comparación con los procesos no semicontinuos que utilizan una adición masiva de subunidades y una concentración constante de subunidades.
En determinadas realizaciones adicionales, el rendimiento de inmunoconjugado recuperado es, adecuadamente, al menos aproximadamente 20 % mayor, al menos aproximadamente 30 % mayor, al menos aproximadamente 40 % mayor, al menos aproximadamente 50 % mayor, al menos aproximadamente 60 % mayor, al menos aproximadamente 70 % mayor, al menos aproximadamente 80 % mayor, al menos aproximadamente 90 % mayor, al menos aproximadamente 100 % mayor, al menos aproximadamente 125 % mayor, al menos aproximadamente 150 % mayor, al menos aproximadamente 175 % mayor, al menos aproximadamente 200 % mayor, al menos aproximadamente 300 % mayor, al menos aproximadamente 400 % mayor, al menos aproximadamente 500 % mayor, al menos aproximadamente 600 % mayor, al menos aproximadamente 700 % mayor, al menos aproximadamente 800 % mayor, al menos aproximadamente 900 % mayor, etc., en donde el inmunoconjugado se repliega en un proceso semicontinuo en un tampón de replegamiento que tiene un pH de 9,5 o menos. El inmunoconjugado replegado se purifica utilizando una columna de intercambio aniónico, en donde la columna se separa entre ciclos de columna con un tampón de separación que contiene, arginina, urea y ditiotreitol (DTT). El proceso de recuperación de dicho inmunoconjugado corresponde al de un proceso no semicontinuo, excepto que el replegamiento se realiza en un proceso semicontinuo en un tampón de replegamiento que tiene un pH de 9,5 o menos y/o la purificación de dicho inmunoconjugado utiliza una columna de intercambio iónico, en donde la columna se separa entre ciclos de columna con un tampón de separación que contiene arginina, urea y ditiotreitol (DTT).
De manera adecuada, la velocidad de adición (l/h) de los cuerpos de inclusión solubilizados se ajusta de manera que se agrega una mezcla de cuerpos de inclusión solubilizados a un tampón de replegamiento (adecuadamente pre enfriado) en el transcurso de aproximadamente 2-8 horas, adecuadamente, 3-6 horas, 3-5 horas, o más adecuadamente, en el transcurso de aproximadamente 4 horas.
La adición se realiza en el transcurso de 1 a 10 horas, utilizando una velocidad de adición de aproximadamente 100 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora a una velocidad de adición de aproximadamente 5 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora (aproximadamente 100 ml/l/h a aproximadamente 5 ml/l/h). La adición se realiza en el transcurso de 2 a 8 horas, utilizando una velocidad de adición de aproximadamente 52 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora a una velocidad de adición de aproximadamente 13 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora (aproximadamente 52 ml/l/h a aproximadamente 13 ml/l/h). La adición se realiza en el transcurso de 3 a 6 horas, utilizando una velocidad de adición de aproximadamente 35 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora a una velocidad de adición de aproximadamente 17 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora (aproximadamente 35 ml/l/h a aproximadamente 17 ml/l/h). La adición se realiza en el transcurso de 3,5 a 5 horas, utilizando una velocidad de adición de aproximadamente 30 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora a una velocidad de adición de aproximadamente 18 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora (aproximadamente 30 ml/l/h a aproximadamente 18 ml/l/h). La adición se realiza en el
transcurso de aproximadamente 4 horas, utilizando una velocidad de adición de aproximadamente 26 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora (aproximadamente 26 ml/l/h).
Adecuadamente, los métodos comprenden replegar el inmunoconjugado utilizando cualquiera de los métodos descritos en el presente documento o como se desvela en el documento WO 2012/0152912.
Tal como se describe en el presente documento, para la limpieza y reutilización de la columna, de manera adecuada, la columna se separa entre ciclos operativos (es decir, carga de la columna, lavado, elución) del inmunoconjugado replegado utilizando un tampón de separación. En realizaciones ilustrativas, el tampón de separación que se utiliza en los métodos descritos en el presente documento comprende arginina, urea y ditiotreitol (DTT) tamponados. Los métodos descritos en el presente documento también pueden utilizar ciclos de 3, 4-, 5-, 6-, 7-, 8-, 9-, 10-, etc., columnas, según se desee, con separación con el tampón de separación descrito entre cada ciclo consecutivo.
Se ha encontrado sorprendentemente que el uso de un tampón de separación entre ciclos consecutivos que tienen la composición descrita en el presente documento, que comprende, adecuadamente, urea y arginina, da como resultado un aumento del rendimiento del inmunoconjugado final, en comparación con los métodos de elución y las columnas que no utilizan un tampón de separación como se describe en el presente documento entre eluciones consecutivas.
El rendimiento de inmunoconjugado recuperado es, adecuadamente, al menos aproximadamente 20 % mayor, al menos aproximadamente 30 % mayor, al menos aproximadamente 40 % mayor, al menos aproximadamente 50 % mayor, al menos aproximadamente 60 % mayor, al menos aproximadamente 70 % mayor, al menos aproximadamente 80 % mayor, al menos aproximadamente 90 % mayor, al menos aproximadamente 100 % mayor, al menos aproximadamente 125 % mayor, al menos aproximadamente 150 % mayor, al menos aproximadamente 175 % mayor, al menos aproximadamente 200 % mayor, al menos aproximadamente 300 % mayor, al menos aproximadamente 400 % mayor, al menos aproximadamente 500 % mayor, al menos aproximadamente 600 % mayor, al menos aproximadamente 700 % mayor, al menos aproximadamente 800 % mayor, al menos aproximadamente 900 % mayor, etc., en comparación con los métodos de elución y las columnas que no utilizan un tampón de separación como se describe en el presente documento entre eluciones consecutivas
El tampón de separación útil en los métodos descritos en el presente documento comprende arginina de aproximadamente 0,10 a aproximadamente 0,9 M, urea de aproximadamente 5-10 M y DTT de aproximadamente 7 15 mM. Más adecuadamente, el tampón de separación comprende, arginina de aproximadamente 0,25 a aproximadamente 0,75 M, urea de aproximadamente 7-9 M y DTT de aproximadamente 9-11 mM. Más adecuadamente, aproximadamente arginina de 0,45 a aproximadamente 0,55 M, urea de aproximadamente 7,5-8,5 M y DTT de aproximadamente 9,5-10,5 mM. De forma más adecuada, el tampón de separación comprende aproximadamente arginina 0,50 M, aproximadamente urea 8,0 M y aproximadamente DTT 10,0 mM.
El tampón de replegamiento que se utiliza en las etapas de replegamiento descritas en el presente documento tiene un pH menor de o aproximadamente 10,0, adecuadamente, menor de o aproximadamente 9,5, y más adecuadamente, menor de o aproximadamente 9,4 (por ejemplo, un pH de aproximadamente 10,0, aproximadamente 9,9, aproximadamente 9,8, aproximadamente 9,7, aproximadamente 9,6, aproximadamente 9,5, aproximadamente 9,3, aproximadamente 9,2, aproximadamente 9,1 o aproximadamente 9,0). Se ha encontrado sorprendentemente que el uso de un tampón de replegamiento que tiene un pH menor de o aproximadamente 10,0, menor de o aproximadamente 9,5 y lo más adecuadamente, menor de aproximadamente 9,4, da como resultado un aumento del rendimiento del inmunoconjugado final, en comparación con los procesos que utilizan tampones de replegamiento que tienen un pH mayor que estos valores citados.
El rendimiento de inmunoconjugado recuperado es, adecuadamente, al menos aproximadamente 20 % mayor, al menos aproximadamente 30 % mayor, al menos aproximadamente 40 % mayor, al menos aproximadamente 50 % mayor, al menos aproximadamente 60 % mayor, al menos aproximadamente 70 % mayor, al menos aproximadamente 80 % mayor, al menos aproximadamente 90 % mayor, al menos aproximadamente 100 % mayor, al menos aproximadamente 125 % mayor, al menos aproximadamente 150 % mayor, al menos aproximadamente 175 % mayor, al menos aproximadamente 200 % mayor, al menos aproximadamente 300 % mayor, al menos aproximadamente 400 % mayor, al menos aproximadamente 500 % mayor, al menos aproximadamente 600 % mayor, al menos aproximadamente 700 % mayor, al menos aproximadamente 800 % mayor, al menos aproximadamente 900 % mayor, etc., en comparación con los procesos que utilizan tampones de replegamiento que tienen un pH mayor que estos valores citados.
En los diversos métodos de preparación descritos en el presente documento, adecuadamente, el inmunoconjugado comprende un anticuerpo o fragmento de unión a antígeno del mismo. Como se describe en todo el documento, adecuadamente, el anticuerpo o fragmento de unión a antígeno comprende un Fab, un Fab', un F(ab')2, un Fd, un Fv monocatenario o scFv, un Fv ligado a disulfuro, un dominio V-NAR, una IgNar, un intracuerpo, una IgGACH2, un minicuerpo, un F(ab')3, un tetracuerpo, un triacuerpo, un diacuerpo, un anticuerpo de dominio único, DVD-IG, Fcab, AcM 2 , un (scFv) 2 , o un scFv-Fc.
Tal como se describe en el presente documento, de manera adecuada, el anticuerpo o fragmento de unión a antígeno del inmunoconjugado se une a un receptor de la superficie celular. Un receptor de superficie celular ejemplar incluye
CD22.
El inmunoconjugado que se prepara de acuerdo con los métodos descritos en el presente documento comprende una toxina. Toxinas ejemplares y métodos de preparación de dichas toxinas se describen en todo el documento. De manera adecuada, la toxina se selecciona entre el grupo que consiste en: exotoxina de Pseudomonas, ricina, abrina, toxina de la difteria y subunidades de la misma, así como toxinas botulínicas A a F o variantes, o derivados de las mismas. En realizaciones, la toxina es una exotoxina de Pseudomonas, o una variante de la misma. Los métodos ejemplares de preparación de exotoxina de Pseudomonas (PE) se describen en el presente documento en detalle así como en el documento WO2012/015912.
La exotoxina de Pseudomonas para su uso en los inmunoconjugados descritos en el presente documento tiene una secuencia de aminoácidos seleccionada del grupo que consiste en las SEQ ID NO: 16-22. De manera adecuada, la exotoxina de Pseudomonas, o variante de la misma, tiene la secuencia de aminoácidos de la SEQ ID NO: 22.
De manera adecuada, el anticuerpo o fragmento de unión a antígeno del mismo que es un componente de la inmunotoxina comprende una secuencia VH y una secuencia VL. De manera adecuada, la secuencia VH se selecciona del grupo que consiste en las SEQ ID NO: 6-11, y la secuencia VL se selecciona del grupo que consiste en las SEQ ID NO: 2, y 12-15.
Como se describe en todo el documento, los métodos de preparación de inmunoconjugados se utilizan, adecuadamente, para preparar inmunoconjugados que comprenden un anticuerpo anti-CD22 o fragmento de unión a antígeno del mismo y una PE o variante de la misma. En realizaciones adecuadas, el inmunoconjugado que se prepara mediante los diversos métodos descritos en el presente documento es la inmunotoxina de Moxetumomab pasudotox que comprende la subunidad VH-PE38 de la s Eq ID NO: 1 y la subunidad VL de la SEQ ID NO: 2.
Tal como se describe en el presente documento, dichos métodos comprenden, adecuadamente, replegar un inmunoconjugado con un proceso semicontinuo en un tampón de replegamiento que tiene un pH inferior a 9,5 y purificar el inmunoconjugado replegado utilizando una columna de intercambio iónico, en la que la columna se separa entre ciclos de columna con un tampón de separación que comprende arginina, urea y ditiotreitol (DTT).
La cantidad de inmunoconjugado recuperado es de manera adecuada al menos aproximadamente al menos aproximadamente 300 % mayor, al menos aproximadamente 400 % mayor, al menos aproximadamente 500 % mayor, al menos aproximadamente 600 % mayor, al menos aproximadamente 700 % mayor, al menos aproximadamente 800 % mayor, al menos aproximadamente 900 % mayor, etc., en comparación con los procesos que no comprenden un proceso de replegamiento semicontinuo y/o columna de intercambio aniónico que se ha separado utilizando el tampón de separación descrito y/o no utiliza un tampón de replegamiento con un pH inferior a 9,4.
Tal como se describe en el presente documento, adecuadamente, el inmunoconjugado comprende un anticuerpo o fragmento de unión a antígeno del mismo, que incluye un anticuerpo o fragmento de unión a antígeno del inmunoconjugado se une a un receptor de la superficie celular tal como CD22.
El inmunoconjugado comprende una toxina, de manera adecuada, exotoxina de Pseudomonas (PE). El anticuerpo o fragmento de unión a antígeno del mismo que es un componente de la inmunotoxina comprende una secuencia Vh y una secuencia VL. De manera adecuada, la secuencia VH se selecciona del grupo que consiste en las SEQ ID NO: 6-11, y la secuencia VL se selecciona del grupo que consiste en las SEQ ID NO: 2, y 12-15. Como se describe en todo el documento, los métodos de preparación de inmunoconjugados se utilizan, adecuadamente, para preparar inmunoconjugados que comprenden un anticuerpo anti-CD22 o fragmento de unión a antígeno del mismo y una PE o variante de la misma. El inmunoconjugado que se prepara mediante los diversos métodos descritos en el presente documento es la inmunotoxina de Moxetumomab pasudotox que comprende la subunidad VH-PE38 de la SEQ ID NO: 1 y la subunidad VL de la SEQ ID NO: 2.
Se proporcionan composiciones que comprenden un inmunoconjugado preparado mediante los diversos métodos descritos en el presente documento. De manera adecuada, los inmunoconjugados preparados mediante dichos métodos tienen menos de entre aproximadamente el 25 % y aproximadamente el 1 % de especies desamidadas. Más adecuadamente, menos de aproximadamente el 25 % de las especies desamidadas está presente, o menos de aproximadamente el 20 % de las especies desamidadas está presente, o menos de aproximadamente el 10 % de las especies desamidadas está presente, o menos de aproximadamente el 5 % de las especies desamidadas está presente, o menos de aproximadamente el 3 % de las especies desamidadas está presente, o menos de aproximadamente el 2 % de las especies desamidadas está presente, o menos de aproximadamente el 1 % de las especies desamidadas está presente.
Los siguientes ejemplos se incluyen aquí con fines ilustrativos únicamente y no pretenden ser limitativos.
Ejemplos
Ejemplo 1: Renaturalización y purificación de Moxetumomab pasudotox
Introducción
CAT-8015 (Moxetumomab pasudotox) es una inmunotoxina recombinante producida en cuerpos de inclusión de E. coli. La generación de Moxetumomab pasudotox activo utiliza, adecuadamente, el replegamiento de los precursores inactivos y la purificación del producto replegado mediante un proceso de 4 columnas. La Figura 1 proporciona una visión general de los procesos de renaturalización y purificación.
El fin del proceso de solubilización es extraer y transferir VH y VL, adecuadamente, de cuerpos de inclusión insolubles a la fase líquida y desnaturalizar ambas subunidades antes del replegamiento. La filtración en profundidad elimina los restos celulares insolubles y los componentes de cuerpos de inclusión de VH y VL solubilizados. El filtrado se concentra posteriormente mediante filtración de flujo tangencial a un peso fijo de retenido, que se determina mediante el factor de dilución del retenido y el peso de replegamiento final. La función de la etapa de concentración es asegurar condiciones de inicio de replegamiento consistentes en términos de concentraciones de VH y VL y ditiotreitol (DTT) a la proporción de glutatión oxidado.
El objetivo del proceso de purificación de 4 columnas es separar el de Moxetumomab pasudotox activo correctamente plegado de los contaminantes relacionados con el producto, tales como las variantes del producto mal plegadas, los agregados, los fragmentos y las isoformas de carga del producto inactivo biológicamente, así como los contaminantes relacionados con el proceso, incluido el ADN de la célula hospedadora, las endotoxinas y las proteínas de la célula hospedadora.
Para lograr un proceso comercialmente viable, los rendimientos de replegamiento y purificación se maximizan al tiempo que se mantiene la calidad, la actividad y la seguridad del producto. Los métodos y procedimientos descritos en el presente documento se han desarrollado para la fabricación de inmunoconjugados, incluida el fármaco Moxetumomab pasudotox.
A. Materiales y métodos
1. Solubilización de cuerpos de inclusión
Los cuerpos de inclusión de VH-PE38 (VH) y VL producidos a partir de células adecuadas se descongelan durante 12-24 horas a temperatura ambiente. La concentración inicial de la solubilización de VH y VL CI es de 0,3 g de VH por litro de replegamiento y 0,07 g de VL por litro de replegamiento. Los cuerpos de inclusión se combinan en una relación molar 1:1 de VH a VL y se ajustan a una concentración final de VH de 10 g/l mediante la adición de tampón Tris/EDTA (Tris 50 mM, EDTA 20 mM, pH 7,4). Los cuerpos de inclusión se solubilizan mediante la adición de 6 kg de tampón de solubilización de cuerpos de inclusión (etanolamina 50 mM, urea 8 M, arginina 0,5 M, EDTA 2 mM, DTT 10 mM, pH 9,3 ± 0,1) a cada kg de solución de cuerpos inclusión ajustada a la concentración. La solubilización se lleva a cabo durante 90 ± 15 minutos a temperatura ambiente con agitación constante.
2. Aclaramiento y ultrafiltración 1 de cuerpos de inclusión
Los cuerpos de inclusión solubilizados se clarifican mediante filtración a través de una serie de filtros de profundidad (véase documento WO 2012/059212). El filtrado clarificado se concentra mediante filtración de flujo tangencial a 1/10 del peso de replegamiento final utilizando una membrana de ultrafiltración de corte de peso molecular de 5 kDa (MWCO).
3. Replegamiento y Ultrafiltración/Diafiltración 2
Moxetumomab pasudotox se repliega mediante una dilución de 10 veces (p/p) del filtrado de cuerpos de inclusión clarificado y concentrado en un tampón de replegamiento preenfriado (2-8 °C) (etanolamina 50 mM, arginina 1 M, EDTA 2 mM, glutatión oxidado 1,0 mM, pH 9,4). La adición (l/h) se ajusta de manera que el filtrado de cuerpos de inclusión clarificado y concentrado se agregue al tampón de replegamiento preenfriado en el transcurso de 4 horas (adecuadamente 26 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora). La reacción de replegamiento se deja proceder durante 48-72 horas a 2-8 °C con mezclado continuo y se calienta a temperatura ambiente antes de la concentración y la diafiltración.
La solución de replegamiento se concentra mediante filtración de flujo tangencial con una membrana de MWCO de 10 kDa y luego se diafiltra con 10 volúmenes de tampón de equilibrio TMAE (fosfato 20 mM, pH 7,4).
4. Cromatografía en columna
a. Cromatografía con Fractogel TMAE (M)
La solución de replegamiento concentrada y diafiltrada se filtra de forma estéril a través de un filtro de 0,2 |jm y se carga en una columna Fractogel TMAE (EMD Biosciences o equivalente) equilibrada con 10 volúmenes de columna (VC) del tampón de equilibrio TMAE (tampón fosfato 20 mM, pH 7,4). Las etapas de cromatografía se realizan a un caudal lineal de 200 cm/h, a menos que se indique lo contrario. Después de cargar, se lava primero la columna con 4 VC de tampón de equilibrio TMAE (fosfato 20 mM, pH 7,4), seguido de un lavado de 6 VC con tampón de lavado 1 (fosfato 20 mM, Triton X-100 al 0,1 %) y un lavado de 8 v C con tampón de lavado 2 (fosfato 20 mM, NaCl 100 mM, pH 7,4). El producto se eluye de la columna con 3 VC de tampón de elución (fosfato 20 mM, cloruro sódico 200 mM,
pH 7,4). Después de la elución, la columna se separa con 3 VC de tampón de separación (etanolamina 50 mM, arginina 0,5 M, EDTA 2 mM, urea 8 M, DTT 10 mM, pH 9,3). El caudal puede reducirse durante el paso de separación. A continuación, la columna se lava con 3 VC de agua para inyección (WFI) y se regenera con 3 VC de solución de regeneración (NaCl 2 M). La columna se sanitiza con al menos 3 VC de solución de sanitización (hidróxido de sodio 1 N) y se almacena con 3 VC de solución de almacenamiento a corto plazo (hidróxido de sodio 0,1 N) o 3 VC de solución de almacenamiento a largo plazo (fosfato 20 mM, etanol al 20 % (p/v), pH 7,4).
b. Cromatografía con hidroxiapatita
La etapa de cromatografía con hidroxiapatita se opera como una etapa de cromatografía de flujo continuo. Las etapas de cromatografía se realizan a un caudal lineal de 250 cm/h, a menos que se indique lo contrario. El producto de la etapa de captura se carga en una columna de hidroxiapatita cerámica (Bio-Rad Laboratories o equivalente) equilibrada con 5 VC de tampón de preequilibrio (fosfato 400 mM, cloruro de sodio 200 mM, pH 7,4) y 5 VC de tampón de equilibrio (fosfato de potasio 20 mM, cloruro de sodio 200 mM, pH 7,4). El producto se recoge en la fracción a través del flujo. Después de cargar, se lava la columna con 3 VC de tampón de equilibrio. La columna se regenera con 3 VC de tampón de pre-equilibrio, se sanitiza con 3 VC de un tampón de sanitización (hidróxido de sodio 1 N) y se almacena en 3 VC de un tampón de almacenamiento (fosfato 10 mM, hidróxido de sodio 0,1 N) a temperatura ambiente.
c. Cromatografía con Phenyl 650 M y Ultrafiltración/Diafiltración 3
El producto de hidroxiapatita se diluye en una proporción 1:1 (p/p) con tampón de preparación de carga (fosfato 20 mM, sulfato de sodio 1,2 M, pH 7,4) y se carga en una columna de Phenyl 650 M (Tosoh o equivalente) equilibrada con 5 VC de tampón de equilibrio (fosfato 20 mM, sulfato de sodio 0,6 M, pH 7,4). Después de cargar, se lava la columna con 1 VC de tampón de equilibrio. El producto se eluye con 20 VC de un tampón de elución con gradiente lineal de 0 a 100 % (fosfato de sodio 20 mM, pH 7,4). La columna se separa con 2 VC de agua para inyección y se regenera con 2 VC de urea 8 M. La columna se sanitiza con 3 VC de tampón de sanitización (hidróxido de sodio 0,5 N) y se almacena con 3 VC de tampón de almacenamiento (fosfato 20 mM, etanol al 20 % (p/p), pH 7,4) a temperatura ambiente.
El conjunto de productos Phenyl 650 M se diafiltró con 10 volúmenes de Tris 10 mM, pH 8,0, utilizando filtración de flujo tangencial con una membrana de MWCO de 10 kDa.
d. Cromatografía con Q Sepharose HP y Ultrafiltración/Diafiltración 4
El producto Phenyl 650 M diafiltrado se carga en una columna de Q Sepharose HP (GE Healthcare o equivalente), se equilibra previamente con 5 VC de tampón de pre-equilibrio (Tris 10 mM, cloruro de sodio 1 M, pH 8,0 y se equilibra con 5 VC de tampón de equilibrio (Tris 10 mM, pH 8,0). La columna se lava con 1 VC de tampón de equilibrio y luego se lava con 3 VC de tampón de equilibrio al 65 % (v/v), tampón de elución al 35 % (v/v) (Tris 10 mM, cloruro de sodio 0,5 M, pH 8,0). El producto se eluye con un gradiente lineal de 10 VC desde tampón de equilibrio al 65 % (v/v), tampón de elución al 35 % (v/v) hasta tampón de equilibrio al 45 % (v/v), tampón de elución al 55 % (v/v). La columna se separa con 2 VC de tampón de pre-equilibrio y se sanitiza con 3 VC de tampón de sanitización (hidróxido de sodio 1 N). La columna se almacena en 3 VC de una solución de almacenamiento a corto plazo (hidróxido de sodio 0,1 N) o en 3 VC de una solución de almacenamiento a largo plazo (fosfato 20 mM, etanol al 20 % (p/v), pH 7,4).
El producto de Q Sepharose HP se concentra mediante filtración de flujo tangencial utilizando una membrana de MWCO de 10 kDa a una concentración de proteína diana de 1,3-2 mg/ml. El producto concentrado de Q Sepharose se diafiltra con al menos seis volúmenes de tampón de formulación (fosfato de sodio 25 mM, sacarosa al 4 % (p/v), glicina al 8 % (p/v), pH, 7,40). El producto de Q Sepharose HP diafiltrado se diluye con tampón de formulación hasta una concentración de proteína final de 0,95-1,05 mg/ml.
5. Formación del fármaco (SF) Moxetumomab pasudotox
El producto de Q Sepharose HP diafiltrado se diluye con tampón de formulación hasta una concentración final de proteína de 0,95-1,05 mg/ml y luego se ajusta con al 0,02 % (p/v) de polisorbato-80 con solución de picos de formulación (polisorbato-80 al 10 % (p/v)) para hacer el fármaco. el fármaco es filtra en botellas de HDPE estériles de 0. 2.|jm y almacenadas a < -70 °C.
Ejemplo 2: Replegamiento de 250 litros de Moxetumomab pasudotox, Ensayo 1
A. Materiales y métodos
1. Filtros y membranas
Los filtros Millistak+ HC POD C0HC y X0HC (de 0,55 m2 cada uno) fueron de Millipore. Las membranas de filtración de flujo tangencial Pellicon 2 BioMax-5 (pantalla V, 2 m2) y Pellicon 2 BioMax-10 (pantalla A, 0,5 y 2,5 m2) fueron de Millipore. Los filtros Durapore Millipak 20, SHC Opticap XL150 y SHC Opticap XL300 fueron de Millipore.
2. Medios e Instrumentación de cromatografía
Fractogel TMAE (M) fue de EMD. La hidroxiapatita, tipo 1,40 |jm, fue de BioRad. Phenyl 650 M era de Tosoh. Q Sepharose HP era de GE Healthcare. La purificación con Fractogel TMAE se realizó en una columna BPG 140x500 (GE Healthcare). La purificación con hidroxiapatita y Phenyl 650 M se realizó en una columna BPG 100x500. La purificación con Q Sepharose HP se realizó en una columna Millipore QuikScale 70x550 (Millipore). Todas las purificaciones se realizaron en un sistema de cromatografía AKTA Pilot.
3. Renaturalización en la escalera de replegamiento de 250 litros
Se diluyeron 3,39 kg de suspensión de cuerpos de inclusión (CI) del VH y 0,33 kg de suspensión de CI del VL con 3,77 kg de tampón TE hasta una concentración final de VH de 10 g/l. Los CI se solubilizaron con 44,9 kg de tampón de solubilización de CI durante 90 minutos a temperatura ambiente. La solución de CI solubilizada se aclaró con un filtro de profundidad C0HC (0,55 m2) conectado en serie con un filtro de profundidad X0HC (0,55 m2). La solución de CI solubilizada y aclarada se concentró hasta un peso de retenido de ultrafiltrado final (UF) 1 de 25,5 kg mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-5 (pantalla V, 2 m2). Veinticinco kg de retenido de UF 1 se ajustaron a la temperatura a 2-8 °C y se agregaron en el transcurso de 4 horas a 225 kg de tampón de replegamiento previamente enfriado (pH 9,4) con mezcla constante (adecuadamente 26 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora). El replegamiento se terminó después de 66 horas mediante el aumento de la temperatura de la solución de replegamiento a temperatura ambiente. La solución de replegamiento se concentró a 24,9 kg mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-10 (pantalla A, 2,5 m2) y posteriormente se diafiltró con 10 volúmenes de tampón de equilibrio TMAE.
4. Purificación en la escalera de replegamiento de 200 litros
La purificación de Moxetumomab pasudotox se realizó como se describe anteriormente. 9,9 l y 9,3 l de solución de replegamiento concentrada y diafiltrada se cargaron en una columna Fractogel TMAE (M) empaquetada (altura del lecho: 18cm, volumen: 2,77 l). Se realizaron dos ciclos de purificación (con tampón de separación utilizado entre eluciones consecutivas). Las fracciones de eluato de Fractogel TMAE (M) se combinaron en un único conjunto de productos de TMAE (7,1 kg) y se cargaron en una columna de hidroxiapatita (altura del lecho: 21,8 cm; volumen: 1,71 l). El flujo de hidroxiapatita a través del conjunto se diluyó 1:1 con 7,91 kg de tampón de preparación de carga y se cargó en una columna Phenyl 650M (altura del lecho: 17,5 cm; volumen: 1,37 l). El conjunto de productos Phenyl 650M (11,6 kg) se concentró hasta 8,1 kg mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-10 (pantalla A, 0,5 m2) y posteriormente se diafiltró con 10 volúmenes de tampón de equilibrio Q Sepharose HP. El producto Phenyl 650 M concentrado y diafiltrado se cargó en una columna de Q Sepharose HP (altura del lecho: 18,2 cm; volumen 0,70 l). El conjunto de productos Q Sepharose HP (2,8 kg) se concentró hasta 2 g/l mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-10 (pantalla A, 0,5 m2) y posteriormente se diafiltró con 7 volúmenes de tampón de formulación. El producto de Q Sepharose HP concentrado y diafiltrado se diluyó con tampón de formulación hasta una concentración final de proteína de 1,02 g/l (volumen: 4,7 kg) y posteriormente se ajustó a un 0,02 % (p/v) de polisorbato-80 con solución de picos de formulación. El Moxetumomab pasudotox formulado se filtró de forma estéril con filtros Durapore Millipak 20 en botellas PETG y se almacenó a < -70 °C.
B. Resultados y discusión
1. Renaturalización
La solubilización, el aclaramiento y los rendimientos en UF 1 se evaluaron mediante RP-HPLC y se muestran en las Tablas I y II. La eficacia del aclaramiento se evaluó mediante mediciones de turbidez antes y después de la filtración en profundidad (Tabla III).
Tabla I Recuperación de Vh (%) para operaciones de unidades de pre-repleglamiento

Tabla II Recuperación de Vl (%) para operaciones de unidades de pre-repleglamiento

Tabla III Aclaramiento de la eficacia de la operación de unidades de filtración en profundidad

Los datos en las Tablas IV y II demuestran la eficacia de la composición del tampón de solubilización de CI en la extracción y transferencia de VH y VL desde cuerpos de inclusión a la fase líquida. Ambas subunidades se recuperaron cuantitativamente en la solución de CI solubilizada. La serie de filtros de profundidad COHC-X0HC logró una reducción de 8 veces en la turbidez de la solución de CI solubilizada y produjo una solución ópticamente transparente adecuada para su posterior procesamiento mediante filtración de flujo tangencial. Los rendimientos de la etapa de aclaramiento fueron comparables para VH y VL (89,4 y 78,8 % respectivamente) a pesar de la diferencia de 4 veces en el peso molecular de las dos subunidades. El rendimiento óptimo de las operaciones de las unidades de replegamiento previo se demuestra por el hecho de que el rendimiento final de VH de UF 1 cumplió con la cantidad requerida para lograr una concentración de replegamiento diana de 0,3 g de VH por l de replegamiento.
El replegamiento de Moxetumomab pasudotox se inició mediante una dilución de 10 veces de UF 1 retenido en tampón de replegamiento. El retenido de Uf 1 se añadió en el transcurso de 4 horas al tampón de replegamiento para maximizar los rendimientos de replegamiento. El título de replegamiento de Moxetumomab pasudotox se determinó mediante LC-MS, la concentración de Moxetumomab pasudotox en el conjunto de UFDF 2 mediante RP-HPLC. El rendimiento de replegamiento se calculó basándose en la concentración de Vh inicial en la reacción de replegamiento, en función de la relación de dilución y la concentración de VH en RP-HPLC en el grupo de retenido de Uf 1. El título de replegamiento, la concentración de Moxetumomab pasudotox en UFDF2 y los rendimientos por etapas se muestran en la Tabla IV.
Tabla IV Replegamientos y Rendimientos por etapas en UFDF

El replegamiento de proteínas heterodiméricas representa un desafío importante debido al hecho de que las subunidades separadas pueden avanzar a lo largo de múltiples vías de plegamiento improductivas y formar agregados insolubles o variantes de productos inactivos mal plegados. Por lo tanto, el replegamiento de proteínas heterodiméricas se caracteriza a menudo por títulos de replegamiento y rendimientos por etapas bajos. Las condiciones de replegamiento descritas en el presente documento se han optimizado cuidadosamente para lograr una utilización eficaz de los materiales de partida de los cuerpos de inclusión y maximizar los títulos de replegamiento (Tabla IV). Como resultado, los títulos de replegamiento y los rendimientos por etapas fueron aproximadamente 2-3 veces más altos en comparación con los procesos de replegamiento anteriores.
La función de la operación de las unidades de filtración de flujo tangencial posterior al replegamiento es terminar el replegamiento mediante la eliminación de los componentes del tampón de replegamiento y la preparación del material replegado para la purificación de la etapa de captura. Un rendimiento por etapas de casi el 87 % de Moxetumomab pasudotox replegado estuvo dentro del intervalo esperado para este tipo de operación unitaria y material de partida.
Los datos en las Tablas III-IV demuestran el rendimiento de los métodos desvelados y la capacidad de este proceso para generar Moxetumomab pasudotox biológicamente activo a partir de precursores inactivos adecuados para la fabricación del fármaco Mabetumomab pasoxotoum.
2. Purificación
Moxetumomab pasudotox se purificó a partir de solución de replegamiento concentrada y diafiltrada a la escala de replegamiento de 200 l mediante cromatografía con Fractogel t Ma E (M), hidroxiapatita, Phenyl 650 M y Q Sepharose HP. Los cromatogramas para cada etapa de purificación se muestran en las Figuras 2-5.
El cromatograma con Fractogel TMAE (M) muestra que no solo el Moxetumomab pasudotox replegado, sino también la mayoría de las impurezas relacionadas con el producto y el proceso, se unen a la columna con poca o ninguna proteína detectada en el flujo de la columna a través de la fracción. Algunas impurezas unidas de manera hidrófoba se eliminaron de la columna con el detergente no iónico Triton X-100 (lavado 1), mientras que las impurezas unidas de manera iónica débiles se eliminaron con un lavado de sal de baja concentración (lavado 2). La elución de Moxetumomab pasudotox plegado se logró con cloruro de sodio 200 mM tamponado. La mayor parte de las impurezas unidas se separaron de la columna Fractogel TMAE con tampón de solubilización de CI (separación 1). La eficacia de este tampón para la limpieza de la columna se demuestra mediante la observación de que muy poca proteína se eluyó de la columna durante una segunda separación con cloruro de sodio 2 M (separación 2).
El cromatograma en la figura 3 muestra que Moxetumomab pasudotox no se unió a hidroxiapatita en presencia de fosfato 20 mM y se recuperó en el flujo a través de la fracción. Los contaminantes relacionados con el proceso, tales como las proteínas, el ADN y la endotoxina de la célula hospedadora se unieron estrechamente a la hidroxiapatita en estas condiciones y posteriormente se eliminaron de la resina con un tampón de separación fosfato 400 mM, cloruro de sodio 200 mM.
El cromatograma muestra que Moxetumomab pasudotox y las impurezas relacionadas con el producto y el proceso se unen a la resina Phenyl 650 M con poca o ninguna proteína detectada en el flujo de la columna a través de la fracción. Moxetumomab pasudotox se eluyó con un gradiente de sal decreciente y se recuperó de la columna en el intervalo de conductividad 60-30 mS/cm. Las impurezas relacionadas con el producto y el proceso se separaron de la columna con un lavado con agua (Separación) y solución de urea 8 M (Regeneración). La eficacia del protocolo de limpieza posterior a la elución se demuestra mediante la observación de que no se observa un aumento de la absorbancia a 280 nm durante la etapa de sanitización con hidróxido de sodio 0,5 N.
El cromatograma con Q Sepharose HP en la Figura 5 muestra que bajo las condiciones de unión actuales Moxetumomab pasudotox y las impurezas relacionadas con el producto y el proceso se unen a la columna. No se detectó proteína en el flujo de la columna a través de la fracción (Carga, Persecución) o fracción de lavado (Lavado). El Moxetumomab pasudotox se recuperó de la columna con un gradiente salino tamponado creciente desde cloruro de sodio 175 mM a cloruro de sodio 275 mM a pH 8,0. Moxetumomab pasudotox se eluyó fuera de la columna en un intervalo de conductividad muy estrecho entre 21 y 24 mS/cm. Las impurezas restantes se eluyeron de la columna con una separación con cloruro sódico 1 M y una solución de hidróxido sódico 1 N.
Se determinaron los rendimientos de la proteína total mediante medidas de absorbancia a 280 nm. En la tabla V se muestran los rendimientos por etapas de proteínas totales para el lote de purificación 250L3.
Tabla V Tabla de rendimiento por etapas de proteína total

La solución de replegamiento concentrada y diafiltrada (producto UFDF2, muestra de carga de TMAE) contiene Moxetumomab pasudotox replegado, pero también otras proteínas, incluidas las variantes de productos agregados y mal plegados y las proteínas de la célula hospedadora. Por lo tanto, las mediciones de la concentración de proteínas basadas en la absorbancia a 280 nm no son específicas para Moxetumomab pasudotox correctamente plegado y, como consecuencia, no reflejan el rendimiento específico de Moxetumomab pasudotox en la etapa de captura con Fractogel TMAE (M). Por el contrario, los rendimientos por etapas de la proteína total y de Moxetumomab pasudotox para la etapa de purificación con hidroxiapatita están mucho más alineados entre sí debido al aumento de la pureza del conjunto de productos Fractogel TMAE (M) (carga de hidroxiapatita) en el que la mayoría de las impurezas relacionadas con el producto y el proceso se han eliminado. También se obtuvieron altas recuperaciones para las etapas posteriores de purificación e intercambio de tampón. El rendimiento de purificación final fue de 4,6 g de fármaco a la escala de replegamiento y purificación de 200 l.
La Tabla VI muestra la depuración de contaminantes relacionados con el producto, incluidos los agregados y fragmentos de Moxetumomab pasudotox desamidado.
Tabla VI Depuración de contaminantes relacionados con el producto en el proceso de purificación

La actividad biológica de Moxetumomab pasudotox depende de la amplitud de la desamidación de la asparagina 358 en la subunidad VH. La desamidación de Moxetumomab pasudotox se analizó mediante cromatografía de intercambio iónico de alto rendimiento (IEC, de sus siglas en inglés) y se correlaciona con el % medido del área antes del pico (véase Figura 6).
Los datos en la Tabla VI demuestran que el área antes del pico en la IEC de Moxetumomab pasudotox en el producto TMAE fue menor al 5 % y que la cromatografía con Q Sepharose HP redujo el área antes del pico de la IEC a menos del 2 %, proporcionando un producto biológicamente activo.
La cromatografía con Fractogel TMAE (M) produjo un aumento significativo en la pureza del monómero y eliminó la mayoría de las proteínas y fragmentos de bajo peso molecular de la corriente del proceso, según lo medido mediante cromatografía de exclusión de tamaño de alto rendimiento (HPSEQ, de sus siglas en inglés). La cromatografía con Phenyl 650 M proporcionó una depuración adicional de fragmentos y agregados y generó un conjunto de productos de Moxetumomab pasudotox que tenía una pureza del 99 % del monómero mediante análisis HPSEQ.
La Tabla VII muestra la depuración de contaminantes relacionados con el proceso, incluidas las proteínas, el ADN y las endotoxinas de la célula hospedadora.
Tabla VII Depuración de contaminantes relacionados con el proceso en el proceso de purificación

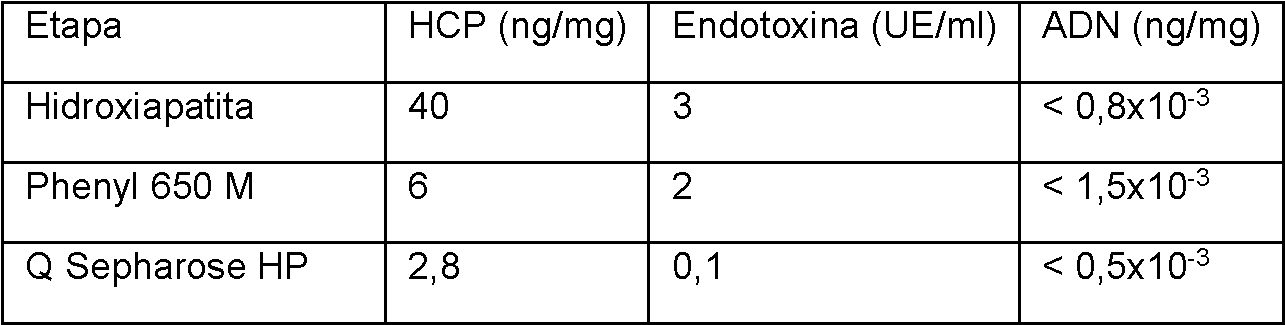
La Tabla VII ilustra la eficacia de la etapa de captura de Fractogel TMAE (M) y la cromatografía con hidroxiapatita para eliminar los contaminantes relacionados con el proceso de la corriente del proceso Moxetumomab pasudotox. La cromatografía con Fractogel TMAE (M) redujo las concentraciones de proteína de la célula hospedadora en 5 veces y las concentraciones de endotoxinas en 600 veces. La cromatografía con hidroxiapatita redujo además las concentraciones de proteínas de la célula hospedadora aproximadamente 8 veces, las concentraciones de endotoxinas en más de 36 veces y las concentraciones de ADN residual en más de 1000 veces por debajo del límite de cuantificación. La cromatografía con Phenyl 650 M logró una reducción adicional de 6,5 veces en la concentración de proteínas de la célula hospedadora mientras que la cromatografía con Q Sepharose HP proporcionó una reducción adicional de 20 veces en la concentración de endotoxinas.
Los datos en las Tablas VI y VII demuestran el rendimiento de los métodos de purificación descritos en el presente documento y la capacidad de estos procesos para generar un fármaco de alta calidad adecuada para ensayos clínicos.
Ejemplo 3: Replegamiento de 250 litros de Moxetumomab pasudotox, Ensayo 2
A. Materiales y métodos
1. Filtros y membranas
Los filtros Millistak+ HC POD C0HC y X0HC (de 0,55 m2 cada uno) fueron de Millipore. Las membranas de filtración de flujo tangencial Pellicon 2 BioMax-5 (pantalla V, 2 m2) y Pellicon 2 BioMax-10 (pantalla A, 0,5 y 2,5 m2) fueron de Millipore. Los filtros Durapore Millipak 20, SHC Opticap XL150 y SHC Opticap XL300 fueron de Millipore.
2. Medios e Instrumentación de cromatografía
Fractogel TMAE (M) fue de EMD. La hidroxiapatita, tipo 1,40 |jm, fue de BioRad. Phenyl 650 M era de Tosoh. Q Sepharose HP era de GE Healthcare. La purificación con Fractogel TMAE se realizó en una columna BPG 140x500 (GE Healthcare). Las purificaciones con hidroxiapatita, Phenyl 650 M y Q Sepharose HP se realizaron en una columna BPG 100x500. Todas las purificaciones se realizaron en un sistema de cromatografía AKTA Pilot.
3. Renaturalización en la escalera de replegamiento de 250 litros
Se diluyeron 3,40 kg de suspensión de CI VH y 0,33 kg de suspensión de CI VL con 3,77 kg de tampón TE hasta una concentración final de VH de 10 g/l. Los CI se solubilizaron con 45,0 kg de tampón de solubilización de CI durante 90 minutos a temperatura ambiente. La solución de CI solubilizada se aclaró con un filtro de profundidad C0HC (0,55 m2) conectado en serie con un filtro de profundidad X0HC (0,55 m2). La solución de CI solubilizada y aclarada se concentró hasta un peso de retenido de UF 1 de 25,5 kg mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-5 V, 2 m2). Veinticinco kg de retenido de UF 1 se ajustaron a la temperatura a 2-8 °C y se agregaron en el transcurso de 4 horas a 225 kg de tampón de replegamiento previamente enfriado (pH 9,4) con mezcla constante (adecuadamente 25 ml de cuerpos de inclusión solubilizados por l de tampón de replegamiento por hora). El replegamiento se terminó después de 66 horas mediante el aumento de la temperatura de la solución de replegamiento a temperatura ambiente. La solución de replegamiento se concentró a 22,6 kg mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-10 (pantalla A, 2,5 m2) y posteriormente se diafiltró con 10 volúmenes de tampón de equilibrio TMAE.
4. Purificación en la escalera de replegamiento de 200 litros
La purificación de Moxetumomab pasudotox se realizó como se describe anteriormente. Nueve y 8,7 l de solución de replegamiento concentrada y diafiltrada se cargaron en una columna Fractogel TMAE (M) empaquetada (altura del lecho: 18cm, volumen: 2,77 l). Se realizaron dos ciclos de purificación, separando con el tampón de separación desvelado entre eluciones consecutivas. Las fracciones de eluato de Fractogel TMAE (M) se combinaron en un único conjunto de productos de TMAE (6,2 kg) y se cargaron en una columna de hidroxiapatita (altura del lecho: 21,8 cm; volumen: 1,71 l). El flujo de hidroxiapatita a través del conjunto se diluyó 1:1 con 6,7 kg de tampón de preparación de carga y se cargó en una columna Phenyl 650M (altura del lecho: 17,5 cm; volumen: 1,37 l). El conjunto de productos Phenyl 650M (10,1 kg) se concentró hasta 8,0 kg mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-10 (pantalla A, 0,5 m2) y posteriormente se diafiltró con 10 volúmenes de tampón de equilibrio Q Sepharose HP. El producto Phenyl 650 M concentrado y diafiltrado se cargó en una columna de Q Sepharose HP (altura del lecho: 18,2cm; volumen 1,4 l). El conjunto de productos Q Sepharose HP (5,1 kg) se concentró hasta 2 g/l mediante filtración de flujo tangencial utilizando una membrana Pellicon 2 BioMax-10 (pantalla A, 0,5 m2) y
posteriormente se diafiltró con 7 volúmenes de tampón de formulación. El producto de Q Sepharose HP concentrado y diafiltrado se diluyó con tampón de formulación hasta una concentración final de proteína de 1,05 g/l (volumen: 5,4 kg) y posteriormente se ajustó a un 0,02 % (p/v) de polisorbato-80 con solución de picos de formulación. El Moxetumomab pasudotox formulado se filtró de forma estéril con filtros Durapore Millipak 20 en botellas PETG y se almacenó a < -70 °C.
B. Resultados y discusión
1. Renaturalización
La solubilización, el aclaramiento y los rendimientos en etapas de UF 1 se evaluaron mediante RP-HPLC y se muestran en las Tablas VIII y IX. La eficacia del aclaramiento se evaluó mediante mediciones de turbidez antes y después de la filtración (Tabla 3.2.2.1-3).
Tabla VIII Recuperación de Vh (%) para operaciones de unidades de pre-repleglamiento
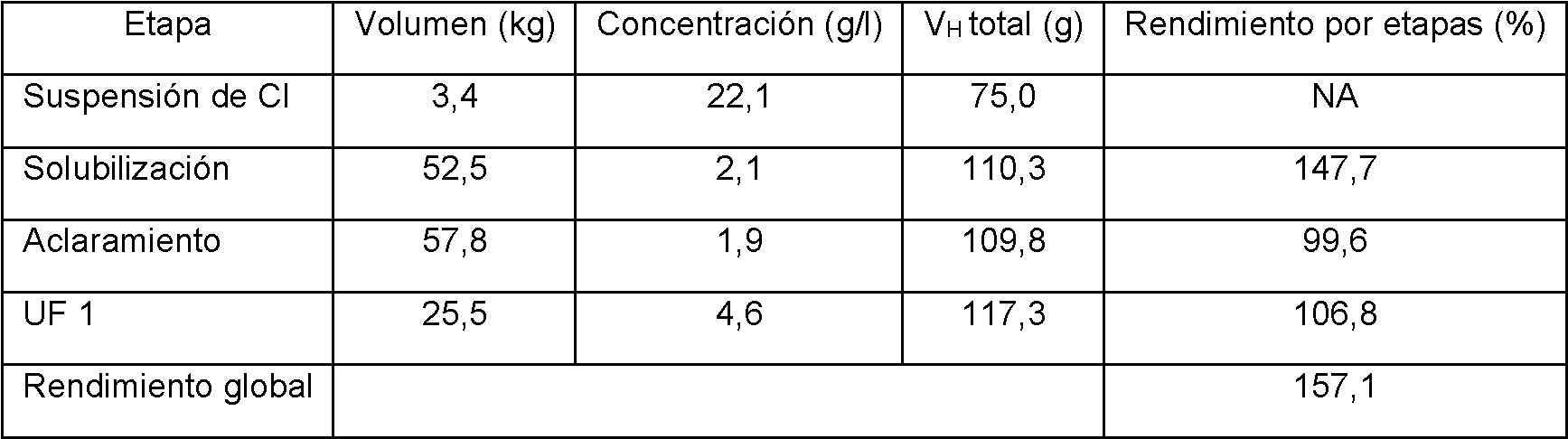
Tabla IX Recuperación de Vl (%) para operaciones de unidades de pre-repleglamiento

Tabla X Eficacia de aclaramiento de la operación de unidades de filtración en profundidad

Los datos en las tablas VIII y IX muestran que VH y VL se recuperaron cuantitativamente de la suspensión inicial de CI en el conjunto de retenido UF 1 según se midió mediante análisis de RP-HPLC. La filtración en profundidad redujo la turbidez de la solución de CI solubilizada aproximadamente 6 veces y produjo una solución transparente óptica que se concentró adicionalmente mediante filtración de flujo tangencial antes del replegamiento.
El replegamiento de Moxetumomab pasudotox se inició mediante una dilución de 10 veces de retenido de UF 1 en un tampón de replegamiento previamente enfriado en el transcurso de 4 horas. El retenido de UF 1 se mantuvo a 2-8 °C durante la adición al tampón de replegamiento para minimizar la generación de variantes de productos desamidados.
El título de replegamiento de Moxetumomab pasudotox se determinó mediante LC-MS. La concentración de Moxetumomab pasudotox en el conjunto de UFDF 2 se determinó mediante RP-HPLC. El rendimiento por etapas de replegamiento se calculó basándose en la concentración de VH inicial en la reacción de replegamiento, determinada mediante la concentración de UF 1 VH y el factor de dilución. Los rendimientos por etapas de replegamiento y UFDF2 se muestran en la Tabla XI.
Tabla XI Rendimientos en etapas de UFDF y replegamientos

El título de replegamiento fue aproximadamente de 2 a 3 veces más alto en comparación con los procesos de replegamiento anteriores de Moxetumomab pasudotox. El rendimiento por etapas del 10 % fue comparable a los procesos de replegamiento anteriores de Moxetumomab pasudotox.
La función de la operación de las unidades de filtración de flujo tangencial posterior al replegamiento es terminar el replegamiento mediante la eliminación de los componentes del tampón de replegamiento y la preparación del material replegado para la purificación de la etapa de captura. Los datos de rendimiento por etapas en la tabla XI muestran que el Moxetumomab pasudotox replegado se recuperó cuantitativamente de la operación de la unidad UFDF2.
Los datos de las tablas VIII a IX demuestran el rendimiento de los métodos desvelados y la capacidad de estos procesos para generar inmunoconjugados biológicamente activos (adecuadamente, Moxetumomab pasudotox) a partir de los precursores inactivos adecuados para la fabricación de fármacos.
2. Purificación
Moxetumomab pasudotox se purificó a partir de solución de replegamiento concentrada y diafiltrada a la escala de replegamiento de 200 l mediante cromatografía con Fractogel t Ma E (M), hidroxiapatita, Phenyl 650 M y Q Sepharose h P. Los cromatogramas para cada etapa de purificación se muestran en las Figuras 7-10.
El cromatograma con Fractogel TMAE (M) muestra que Moxetumomab pasudotox replegado y la mayoría de las impurezas relacionadas con el producto y el proceso se unen a la columna. La progresión de la proteína se observó al final de la etapa de carga; a diferencia de la purificación en el Ejemplo 2, en el que no se detectó la progresión de proteínas al final de la etapa de carga de la columna.
El cromatograma en la figura 8 muestra que Moxetumomab pasudotox no se unió a hidroxiapatita en presencia de fosfato 20 mM y se recuperó en el flujo a través de la fracción.
El cromatograma en la Figura 9 muestra que Moxetumomab pasudotox y las impurezas relacionadas con el producto y el proceso se unen a la resina Phenyl 650 M con poca o ninguna proteína detectada en el flujo de la columna a través de la fracción. Moxetumomab pasudotox se eluyó con un gradiente de sal decreciente y se recuperó de la columna en el intervalo de conductividad 60-30 mS/cm.
El cromatograma con Q Sepharose HP en la Figura 10 muestra que Moxetumomab pasudotox y las impurezas relacionadas con el producto y el proceso se unen a la columna. No se detectó proteína en el flujo de la columna a través de la fracción (Carga, Persecución) o fracción de lavado (Lavado). El Moxetumomab pasudotox se recuperó de la columna con un gradiente salino tamponado creciente desde cloruro de sodio 175 mM a cloruro de sodio 275 mM a pH 8,0. El Moxetumomab pasudotox activo se eluyó fuera de la columna en un intervalo de conductividad muy estrecho entre 21 y 24 mS/cm. Las impurezas restantes se eluyeron de la columna con una separación con cloruro sódico 1 M y una solución de hidróxido sódico 1 N.
Se determinaron los rendimientos de la proteína total mediante medidas de absorbancia a 280 nm. Los rendimientos por etapas de proteínas totales se muestran en la Tabla XII.
Tabla XII Tabla de rendimiento por etapas de proteína y Moxetumomab pasudotox

Al igual que en el caso anterior, la solución de replegamiento concentrada y diafiltrada (producto UFDF2, muestra de carga de TMAE) contiene Moxetumomab pasudotox replegado, pero también otras proteínas, incluidas las variantes de productos agregados y mal plegados y las proteínas de la célula hospedadora. Por lo tanto, las mediciones de la concentración de proteínas basadas en la absorbancia a 280 nm no son específicas para Moxetumomab pasudotox correctamente plegado y, como consecuencia, no reflejan el rendimiento específico de Moxetumomab pasudotox de la etapa de captura con Fractogel TMAE (M). Por el contrario, los rendimientos por etapas de la proteína total y de Moxetumomab pasudotox para la etapa de purificación con hidroxiapatita están mucho más alineados entre sí debido al aumento de la pureza del conjunto de productos Fractogel TMAE (M) (carga de hidroxiapatita) en el que la mayoría de las impurezas relacionadas con el producto y el proceso se han eliminado. Los rendimientos por etapas de Phenyl 650 M y UFDF3 para el Ejemplo 3 fueron comparables a los rendimientos por etapas logrados en el Ejemplo 2 para las mismas operaciones de unidades. Aquí, el volumen de la columna Q Sepharose HP se incrementó de 0,7 l a 1,4 l para disminuir la carga máxima de columna para esta etapa de purificación. Como resultado, la carga máxima de columna disminuyó de 10,4 g/l de resina en el Ejemplo 2 a 4,5 g/l de resina en el Ejemplo 3. El rendimiento por etapas de Q Sepharose HP mejoró en casi un 20 % desde el 69 % en el Ejemplo 2 al 88 % en el Ejemplo 3. Los rendimientos por etapas de Q Sepharose HP fueron 2-3 veces más altos en comparación con los rendimientos anteriores del proceso de Moxetumomab pasudotox con Q Sepharose HP. El rendimiento de purificación final fue de 5,6 g de fármaco a la escala de replegamiento y purificación de 200 l.
La Tabla XIII muestra la depuración de los contaminantes relacionados con el producto, incluidos los agregados y fragmentos de Moxetumomab pasudotox desamidado.
Tabla XIII Depuración de contaminantes relacionados con el producto en el proceso de purificación

La correlación de % del área de pre-pico en la IEC, desamidación y actividad biológica de Moxetumomab pasudotox ha sido discutida anteriormente. Los datos en la tabla XIII muestran que el área antes del pico en IEC de Moxetumomab pasudotox en el conjunto de productos de hidroxiapatita del Ejemplo 3 fue 2 veces mayor que en el Ejemplo 2. La cromatografía con Phenyl 650M y Q Sepharose HP redujo el área de pre-pico en IEC en aproximadamente un 5 % a
menos de un 3,5 %, proporcionando un producto biológicamente activo.
La Tabla XIII demuestra que la cromatografía con Fractogel TMAE (M) aumenta significativamente la pureza del monómero y elimina la mayoría de las proteínas y fragmentos de bajo peso molecular de la corriente del proceso, medida mediante cromatografía de exclusión de tamaño de alto rendimiento (HPSEQ). La cromatografía con Phenyl 650 M proporcionó una depuración adicional de fragmentos y agregados y generó un conjunto de productos de Moxetumomab pasudotox que tenía una pureza del 99,5 % del monómero mediante análisis HPSEQ.
La Tabla XIV muestra la depuración de contaminantes relacionados con el proceso, incluidas las proteínas, el ADN y las endotoxinas de la célula hospedadora.
Tabla XIV Depuración de contaminantes relacionados con el proceso en el proceso de purificación

Los datos en la Tabla XIV demuestran la eficacia de la etapa de captura con Fractogel TMAE (M) y la cromatografía con hidroxiapatita en la eliminación de contaminantes relacionados con el proceso de la corriente del proceso Moxetumomab pasudotox. La cromatografía con Fractogel TMAE (M) redujo las concentraciones de proteína de la célula hospedadora en aproximadamente 20 veces y las concentraciones de endotoxinas en más de 800 veces. La cromatografía con Fractogel TMAE (M) logró la depuración del ADN de la célula hospedadora por debajo del límite de cuantificación. La cromatografía con hidroxiapatita redujo además la concentración de proteína de la célula hospedadora aproximadamente 2 veces y las concentraciones de endotoxinas en más de 10 veces. La cromatografía con Phenyl 650 M logró una reducción de casi 9 veces en la concentración de proteínas de la célula hospedadora mientras que la cromatografía con Q Sepharose HP proporcionó una reducción adicional de 30 veces en la concentración de endotoxinas.
3. Resumen
Se logró un rendimiento de fármaco comparable para las dos purificaciones descritas en los Ejemplos 2 y 3. Los datos demuestran la reproducibilidad de los métodos de renaturalización y purificación descritos en el presente documento y la capacidad de estos métodos para generar un fármaco de alta calidad adecuada para ensayos clínicos.
Los métodos descritos en el presente documento también contribuyen a una mejora significativa en los rendimientos generales del proceso en comparación con los métodos de renaturalización y purificación de Moxetumomab pasudotox previos. Estas mejoras en el proceso desempeñarán un papel importante en la fabricación de Moxetumomab pasudotox en un proceso económicamente viable.
Ejemplo 4: Método de limpieza con Fractogel TMAE (M)
La operación de un proceso de purificación económicamente viable dicta que las resinas de cromatografía se utilizan para múltiples ciclos de producción. Esto requiere eficaces
Tabla XIX Resumen del rendimiento de la columna Capto Blue


El avance de las impurezas no unidas se observó durante la carga, seguido de un pico de elución durante el gradiente de elución del cloruro de sodio. La Tabla XIX muestra que el conjunto de UF/DF 2 contenía 583,7 miligramos de proteína total (medida mediante absorbancia a 280 nm) pero solo 58,4 miligramos de producto de Moxetumomab pasudotox (medido mediante RP-HPLC). El conjunto de Capto Blue contenía 58,4 miligramos de proteína total, de los cuales 53,5 miligramos fueron Moxetumomab pasudotox, lo que dio como resultado un rendimiento total de proteínas de solo el 9,4 %, pero un rendimiento de Moxetumomab pasudotox del 91,7 %. Se logró una reducción de 4,6 veces en HCP mediante la etapa de cromatografía con Capto Blue. También se añadió propilenglicol a los tampones de elución en combinación con cloruro de sodio para modular la unión de Moxetumomab pasudotox. La elución con cloruro de sodio solo dio como resultado un pico de elución amplio; sin embargo, la presencia de propilenglicol en la elución del gradiente de cloruro de sodio agudizó el pico de elución y aumentó el rendimiento del producto.
Las Figuras 17-19 demuestran la reproducibilidad de los métodos de purificación con Capto Blue Sepharose descritos en el presente documento y su capacidad para capturar selectivamente inmunoconjugados replegados, incluyendo Moxetumomab pasudotox, de una solución compleja que consiste en un producto intacto, así como en variantes de productos agregadas y mal plegadas e impurezas relacionadas con el proceso.
Aunque la presente invención se ha descrito completamente junto con varias realizaciones de la misma con referencia a los dibujos adjuntos, debe
Tabla XV Secuencia de purificación con Fractogel TMAE (M) para la Figura 11

Se observaron varios picos durante las etapas de limpieza de la columna. Posteriormente, se realizó una ejecución en blanco sin carga de proteína ni tampón de lavado 1 en la misma columna. La Tabla XVI describe la secuencia de
purificación con Fractogel TMAE para la ejecución en blanco.
Tabla XVI Secuencia de purificación con Fractogel TMAE (M) para la Figura 12

La Figura 12 muestra que se observaron picos de transferencia significativos cuando la columna limpiada previamente se separó nuevamente con NaCl 2 M (Separación 1) y urea 8 M (Separación 2). Se observó el mismo perfil durante al menos dos ensayos adicionales ejecutados de acuerdo con la tabla XVI, lo que demuestra que el método de limpieza y sanitización descrito anteriormente no eliminó de manera eficaz los contaminantes relacionados con el producto y el proceso de la resina.
Basándose en la observación de que un tampón que contenía urea 8 M, arginina 0,5 M y DTT 10 mM era capaz de disolver cuerpos de inclusión de E. coli, el tampón de solubilización de CI de Moxetumomab pasudotox (etanolamina 50 mM, urea 8 M, arginina 0,5 M, EDTA 2 mM, DTT 10 mM, pH 9,5) se probó como solución de limpieza de la columna en la etapa de captura. La secuencia de purificación se describe en la tabla XVII.
Tabla XVII Secuencia de purificación con Fractogel TMAE (M) para la Figura 13


La Figura 13 muestra un cromatograma de captura con Fractogel TMAE M con una separación de 5 VC que utiliza el tampón de solubilización de CI de Moxetumomab pasudotox. Se observó un pico muy fuerte cuando la columna se limpió con tampón de solubilización de CI (Separación 1); con poca o ninguna proteína adicional que se eluya de la columna en el subsiguiente lavado con alto contenido de sal (Separación 2) o paso de sanitización.
La Figura 14 muestra el cromatograma con Fractogel TMAE (M) posterior sin carga de proteína. La secuencia de purificación se describe en la tabla XVIII.
Tabla XVIII Secuencia de purificación con Fractogel TMAE (M) para las Figuras 14 a 16

No se observó transferencia de contaminantes relacionados con el producto y el proceso. Los picos de absorbancia observados durante las etapas de separación y sanitización se debieron a la absorbancia de fondo de los componentes del tampón de solubilización de CI (véase Figura 15 para el cromatograma de tampón en blanco con resina nueva). El cromatograma transferido con Fractogel TMAE (M) después de 9 ciclos de purificación observado en la Figura 16 era idéntico al cromatograma de tampón en blanco con nueva resina observada en la Figura 15, lo que demuestra que el tampón de solubilización de CI limpia y regenera eficazmente la resina a su estado original.
Ejemplo 4: Captura con Capto Blue
La cromatografía con colorante azul de Cibacron se usó para capturar Moxetumomab pasudotox del conjunto de UF/DF 2 post-replegamiento en condiciones ligeramente ácidas como alternativa a la captura de intercambio aniónico. La interacción de Moxetumomab pasudotox con el ligando de colorante azul de Cibacron es de naturaleza multimodal y bastante fuerte. La unión selectiva de Moxetumomab pasudotox en resinas azul de Cibacron se puede modular con componentes iónicos, así como con componentes hidrófobos como el propilenglicol. Captura de Moxetumomab pasudotox mediante colorante azul de Cibacron como se describe en el presente documento.
A. Materiales y métodos
1. Cromatografía e instrumentación
Capto Blue Sepharose era de GE Healthcare. Las purificaciones con Capto Blue Sepharose se realizaron en columnas XK16/20 (GE Healthcare). Todas las purificaciones se realizaron en el sistema de cromatografía AKTA Explorer.
Claims (12)
1. Un método para preparar un inmunoconjugado activo, en el que dicho inmunoconjugado está compuesto por dos cadenas polipeptídicas unidas por un enlace disulfuro, y en el que el inmunoconjugado es la inmunotoxina Moxetumomab pasudotox que comprende la subunidad VH-PE38 de SEQ ID NO: 1 y la subunidad VL de SEQ ID NO:2, comprendiendo el método replegamiento de dicho inmunoconjugado en un proceso semicontinuo de replegamiento y purificación del inmunoconjugado replegado en una o más columnas de cromatografía.
2. El método de la reivindicación 1, comprendiendo el método purificar el inmunoconjugado replegado usando una columna de intercambio aniónico, en el que la columna se separa entre ciclos de columna con un tampón de separación que comprende arginina, urea y ditiotreitol (DTT).
3. El método de la reivindicación 1 o 2, en el que el replegamiento de dicho inmunoconjugado comprende un tampón de replegamiento que tiene un pH de 9,5 o menos.
4. El método de la reivindicación 1, comprendiendo el método replegamiento de dicho inmunoconjugado en un proceso semicontinuo de replegamiento en un tampón de replegamiento que tiene un pH de 9,5 o menos, y purificación del inmunoconjugado replegado en una columna de intercambio iónico, en el que la columna se separa entre ciclos de columna con un tampón de separación que comprende arginina, urea y ditiotreitol (DTT).
5. El método de la reivindicación 4, en el que la cantidad del inmunoconjugado recuperado del método de preparación es al menos un trescientos % (300 %) mayor que la cantidad del inmunoconjugado recuperado utilizando un método que no comprende un proceso semicontinuo de replegamiento y/o purificación en una columna de intercambio iónico que ha sido separada utilizando el tampón de separación.
6 . El método de una cualquiera de las reivindicaciones 1 a 5, en el que el proceso semicontinuo utiliza una tasa de adición de aproximadamente 52 ml de cuerpos de inclusión solubilizados por L de tampón de replegado por hora a aproximadamente 13 ml de cuerpos de inclusión solubilizados por L de tampón de replegado por hora.
7. El método de la reivindicación 6 , en el que el proceso semicontinuo utiliza una tasa de adición de aproximadamente 35 ml de cuerpos de inclusión solubilizados por L de tampón de replegado por hora a aproximadamente 17 ml de cuerpos de inclusión solubilizados por L de tampón de replegado por hora.
8 . El método de la reivindicación 7, en el que el proceso semicontinuo utiliza una tasa de adición de aproximadamente 30 ml de cuerpos de inclusión solubilizados por L de tampón de replegado por hora a aproximadamente 18 ml de cuerpos de inclusión solubilizados por L de tampón de replegado por hora.
9. El método de la reivindicación 8 , en el que el proceso semicontinuo utiliza una tasa de adición de aproximadamente 26 ml de cuerpos de inclusión solubilizados por L de tampón de replegamiento por hora.
10. El método de la reivindicación 2 o 4, en el que el tampón de separación comprende aproximadamente 0,25 a aproximadamente 0,75 M de arginina, aproximadamente 7-9 M de urea y aproximadamente 9-11 mM de DTT.
11. El método de una cualquiera de las reivindicaciones 1-10, en el que el proceso semicontinuo se produce durante un período de aproximadamente 2 a aproximadamente 8 horas.
12. Una composición que comprende un inmunoconjugado que tiene menos de entre aproximadamente el 25 % y aproximadamente el 1 % de especies desamidadas, en la que el inmunoconjugado es la inmunotoxina Moxetumomab pasudotox que comprende la subunidad VH-PE38 de SEQ ID NO: 1 y la subunidad VL de SEQ ID NO: 2, y en la que dicho inmunoconjugado se prepara mediante el método de cualquiera de las reivindicaciones 1 - 11 .
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261740111P | 2012-12-20 | 2012-12-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2929876T3 true ES2929876T3 (es) | 2022-12-02 |
Family
ID=50979399
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13863999T Active ES2710314T3 (es) | 2012-12-20 | 2013-12-19 | Métodos de producción de inmunoconjugados |
| ES18200555T Active ES2929876T3 (es) | 2012-12-20 | 2013-12-19 | Replegamiento y purificación de moxetumomab pasudotox |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13863999T Active ES2710314T3 (es) | 2012-12-20 | 2013-12-19 | Métodos de producción de inmunoconjugados |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US9920124B2 (es) |
| EP (2) | EP3494992B1 (es) |
| JP (1) | JP6553515B2 (es) |
| CN (2) | CN105007937B (es) |
| AU (2) | AU2013361235B2 (es) |
| CA (1) | CA2894908C (es) |
| DK (1) | DK2934597T3 (es) |
| ES (2) | ES2710314T3 (es) |
| HK (2) | HK1215547A1 (es) |
| WO (1) | WO2014100443A2 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG10202101105XA (en) * | 2015-08-13 | 2021-03-30 | Amgen Inc | Charged depth filtration of antigen-binding proteins |
| JP2023544457A (ja) * | 2020-09-29 | 2023-10-23 | クンミン シノウェイ ナチュラル ファーマシューティカルズ カンパニー リミテッド | ヒト化抗cd22組換え免疫毒素およびその用途 |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3060165A (en) | 1962-10-23 | Preparation of toxic ricin | ||
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US5079163A (en) | 1985-03-29 | 1992-01-07 | Cetus Corporation | Recombinant ricin toxin fragments |
| US4689401A (en) | 1986-03-06 | 1987-08-25 | Cetus Corporation | Method of recovering microbially produced recombinant ricin toxin a chain |
| US4892827A (en) | 1986-09-24 | 1990-01-09 | The United States Of America As Represented By The Department Of Health And Human Services | Recombinant pseudomonas exotoxins: construction of an active immunotoxin with low side effects |
| DE69131449T2 (de) | 1990-05-11 | 1999-11-25 | Us Health | Verbesserte exotoxine aus pseudomonas mit geringer toxität bei tieren und hoher zelltötender aktivität |
| US5608039A (en) | 1990-10-12 | 1997-03-04 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Single chain B3 antibody fusion proteins and their uses |
| CA2136724A1 (en) | 1992-06-18 | 1993-12-23 | Ira H. Pastan | Recombinant pseudomonas exotoxin with increased activity |
| ATE383430T1 (de) | 1997-03-20 | 2008-01-15 | Us Gov Health & Human Serv | Rekombinante antikörper und immunkonjugate gezielt auf cd22-tragende zellen und tumoren |
| US6296843B1 (en) | 1998-04-03 | 2001-10-02 | The Penn State Research Foundation | Mutagenized IL 13-based chimeric molecules |
| US7355012B2 (en) | 2001-09-26 | 2008-04-08 | United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Mutated anti-CD22 antibodies with increased affinity to CD22-expressing leukemia cells |
| SI1648507T1 (sl) * | 2003-07-24 | 2017-07-31 | Innate Pharma S.A. | Metode in sestavki za povečanje učinkovitosti terapevtskih protiteles z uporabo potenciranja spojin nk celice |
| AU2004262023B2 (en) * | 2003-08-01 | 2011-03-10 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services, National Institutes Of Health | Methods for expression and purification of immunotoxins |
| ES2343965T3 (es) * | 2003-11-25 | 2010-08-13 | The Government Of The United States, As Represented By The Secretary Of Health And Human Services | Anticuerpos anti-cd22 e inmunocongujados mutados. |
| CN1676531A (zh) * | 2004-04-02 | 2005-10-05 | 北京安波特基因工程技术有限公司 | 一种含(Arg) 9肽段的基于假单胞菌外毒素的重组免疫毒素 |
| ES2660026T3 (es) | 2005-07-29 | 2018-03-20 | The Government Of The United States Of America, As Represented By The Secretary Of Health And Human Services | Exotoxinas de pseudomonas mutadas con antigenicidad reducida |
| EP1845103B1 (en) | 2006-04-10 | 2015-02-25 | Boehringer Ingelheim RCV GmbH & Co KG | Method for refolding a protein |
| UA95958C2 (en) * | 2006-05-30 | 2011-09-26 | Дженентек, Инк. | Antibody that binds to cd22, immunoconjugates and uses therefor |
| AU2007272412B2 (en) * | 2006-07-14 | 2013-11-07 | Genentech, Inc. | Refolding of recombinant proteins |
| ES2642516T3 (es) | 2007-09-04 | 2017-11-16 | The Government Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Supresiones en el dominio II de la exotoxina A de Pseudomonas que reducen la toxicidad no específica |
| EP2475398B1 (en) * | 2009-09-11 | 2015-05-20 | The Government of the United States of America as represented by The Secretary of the Department of Health and Human Services | Improved pseudomonas exotoxin a with reduced immunogenicity |
| CA2806314C (en) | 2010-07-30 | 2019-09-24 | Medimmune, Llc | Method for purifying active polypeptides or immunoconjugates |
| EP2451238B1 (en) | 2010-11-05 | 2015-01-07 | Alcatel Lucent | Control message encoding |
| WO2012152912A1 (en) | 2011-05-12 | 2012-11-15 | Newvectys | Genetically modified pig as a cancer prone model |
-
2013
- 2013-12-19 AU AU2013361235A patent/AU2013361235B2/en active Active
- 2013-12-19 CN CN201380067573.XA patent/CN105007937B/zh active Active
- 2013-12-19 JP JP2015549722A patent/JP6553515B2/ja not_active Expired - Fee Related
- 2013-12-19 WO PCT/US2013/076625 patent/WO2014100443A2/en active Application Filing
- 2013-12-19 EP EP18200555.3A patent/EP3494992B1/en active Active
- 2013-12-19 DK DK13863999.2T patent/DK2934597T3/en active
- 2013-12-19 ES ES13863999T patent/ES2710314T3/es active Active
- 2013-12-19 US US14/654,384 patent/US9920124B2/en active Active
- 2013-12-19 EP EP13863999.2A patent/EP2934597B1/en active Active
- 2013-12-19 CA CA2894908A patent/CA2894908C/en active Active
- 2013-12-19 CN CN201911117704.6A patent/CN110950955A/zh not_active Withdrawn
- 2013-12-19 ES ES18200555T patent/ES2929876T3/es active Active
-
2016
- 2016-03-29 HK HK16103571.3A patent/HK1215547A1/zh unknown
- 2016-04-08 HK HK16104008.4A patent/HK1216142A1/zh unknown
-
2018
- 2018-09-20 AU AU2018232979A patent/AU2018232979A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| ES2710314T3 (es) | 2019-04-24 |
| AU2013361235B2 (en) | 2018-07-26 |
| JP2016503062A (ja) | 2016-02-01 |
| CA2894908A1 (en) | 2014-06-26 |
| DK2934597T3 (en) | 2019-02-11 |
| AU2013361235A1 (en) | 2015-07-02 |
| CN110950955A (zh) | 2020-04-03 |
| EP3494992A1 (en) | 2019-06-12 |
| US20150337040A1 (en) | 2015-11-26 |
| EP3494992B1 (en) | 2022-07-27 |
| EP2934597A4 (en) | 2016-07-20 |
| US9920124B2 (en) | 2018-03-20 |
| EP2934597B1 (en) | 2018-10-17 |
| WO2014100443A3 (en) | 2015-07-16 |
| CN105007937B (zh) | 2019-11-19 |
| AU2018232979A1 (en) | 2018-10-11 |
| JP6553515B2 (ja) | 2019-07-31 |
| WO2014100443A2 (en) | 2014-06-26 |
| HK1216142A1 (zh) | 2016-10-21 |
| EP2934597A2 (en) | 2015-10-28 |
| HK1215547A1 (zh) | 2016-09-02 |
| CN105007937A (zh) | 2015-10-28 |
| CA2894908C (en) | 2022-07-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11136396B2 (en) | Method for purifying active polypeptides or immunoconjugates | |
| KR20080111487A (ko) | 단백질 정제 방법 | |
| ES2929876T3 (es) | Replegamiento y purificación de moxetumomab pasudotox | |
| AU2011282774B9 (en) | Method for purifying active polypeptides or immunoconjugates |