ES2923184T3 - Sales y profármacos de 1-metil-D-triptófano - Google Patents
Sales y profármacos de 1-metil-D-triptófano Download PDFInfo
- Publication number
- ES2923184T3 ES2923184T3 ES19188078T ES19188078T ES2923184T3 ES 2923184 T3 ES2923184 T3 ES 2923184T3 ES 19188078 T ES19188078 T ES 19188078T ES 19188078 T ES19188078 T ES 19188078T ES 2923184 T3 ES2923184 T3 ES 2923184T3
- Authority
- ES
- Spain
- Prior art keywords
- acid
- indoximod
- methyl
- prodrug
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D209/24—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with an alkyl or cycloalkyl radical attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D209/20—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals substituted additionally by nitrogen atoms, e.g. tryptophane
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/28—Compounds containing heavy metals
- A61K31/285—Arsenic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/03—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C309/04—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton containing only one sulfo group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/20—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic unsaturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6571—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and oxygen atoms as the only ring hetero atoms
- C07F9/6574—Esters of oxyacids of phosphorus
- C07F9/65742—Esters of oxyacids of phosphorus non-condensed with carbocyclic rings or heterocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06026—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atom, i.e. Gly or Ala
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06043—Leu-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/0606—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing heteroatoms not provided for by C07K5/06086 - C07K5/06139, e.g. Ser, Met, Cys, Thr
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06086—Dipeptides with the first amino acid being basic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06104—Dipeptides with the first amino acid being acidic
Abstract
Actualmente se proporcionan profármacos y compuestos de sal de indoximod y composiciones farmacéuticas que comprenden sales y profármacos de indoximod, que producen una mayor concentración plasmática y exposición a indoximod en comparación con la administración directa de indoximod, en pacientes que necesitan tratamiento de inmunosupresión mediada por la indoleamina-2,3 -vía de la dioxigenasa, como pacientes con cáncer o enfermedades infecciosas crónicas. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Sales y profármacos de 1-metil-D-triptófano
Referencia cruzada a solicitudes relacionadas
La presente solicitud reivindica prioridad a la solicitud provisional de EE. UU. N° de serie 62/196.671 presentada el 24 de julio de 2015 y la solicitud provisional de EE. UU. N° de serie 62/305.748 presentada el 9 de marzo de 2016.
ANTECEDENTES DE LA INVENCIÓN
Campo de la invención
La presente divulgación está relacionada con compuestos para la inhibición de la vía de la indolamina-2,3-dioxigenasa, en particular profármacos de indoximod con propiedades farmacocinéticas potenciadas con respecto a indoximod
Sumario de la técnica relacionada
La degradación del triptófano en quinurenina está mediada por la indolamina-2,3-dioxigenasa (IDO1) expresada por células dendríticas plasmacitoides, células placentarias, epiteliales y tumorales y por la triptófano-2,3-dioxigenasa (TDO2) expresada principalmente por el hígado y las células tumorales.
La IDO1 desempeña una función importante en la regulación de las respuestas inmunitarias provocando la anergia en linfocitos T efectores reactivos y modulando la diferenciación y la activación de linfocitos T reguladores (Treg). Desde un punto de vista más general, la enzima IDO participa en la vía que comprende todas las proteínas que contribuyen directa o indirectamente a modular las funciones inmunosupresoras dependientes de la actividad de IDO, que incluyen proteínas que median en la inducción de la expresión de IDO, la activación de la actividad enzimática por reductasas, las modificaciones postraduccionales que regulan la actividad, la degradación de proteínas y la interpretación y la transmisión de las señales causadas por bajas concentraciones de Trp y la presencia de catabolitos de Trp [conocidos conjuntamente como quinureninas (Kyn)] que incluyen sensores de estrés catabólico integrados en la vía no desrreprimida de control general real-2 (GCN2), la vía de receptor de hidrocarburos de arilo (AhR) y las vías de diana de rapamicina en mamíferos (mTOR). Este concepto de vías reguladoras integradas aguas abajo con IDO en el centro ha surgido de estudios en múltiples sistemas modelo por muchos grupos de investigación y esta noción puede ser críticamente importante para el entendimiento de cómo la vía de IDO se induce, cómo la IDO ejerce efectos aguas abajo y el mecanismo de acción de los inhibidores de la vía IDO que se dirigen a IDO directamente o se dirigen a otros componentes de la vía de IDO [1,2].
Por lo tanto, la inhibición farmacológica directa de la actividad enzimática de IDO1 o la inhibición de los factores aguas arriba que activan la enzima IDO1 o la inhibición de los efectos aguas abajo de la actividad enzimática de IDO1 deben estimular una respuesta inmunitaria por múltiples mecanismos que pueden implicar la prevención de anergia de linfocitos T efectores, la reactivación de linfocitos T efectores anérgicos, la prevención de la activación de linfocitos T reguladores, la promoción de la conversión fenotípica de Treg en células TH17 proinflamatorias y la promoción de la reprogramación genotípica de células dendríticas inmunosupresoras en células dendríticas inmunoestimulantes.
Por estos motivos, se han descrito numerosos inhibidores enzimáticos de IDO y están siendo desarrollados para tratar o prevenir enfermedades relacionadas con IDO, tales como cáncer y enfermedades infecciosas. Se han descrito en la bibliografía numerosas moléculas que inhiben la actividad enzimática de IDO, ya sean inhibidores competitivos o no competitivos, por ejemplo en las solicitudes de patente WO2012142237, WO2014159248, WO2011056652, WO2009132238, WO2009073620, WO2008115804, WO 2014150646, WO 2014150677, WO 2015002918, WO 2015006520, WO 2014141110, WO 2014/186035, WO 2014/081689, US 7714139, US 8476454, US 7705022, US 8993605, US 8846726, US 8951536, US7598287.
Uno de los primeros inhibidores de la vía de IDO estudiados en los modelos preclínicos ha sido el 1-metil-DL-triptófano (1 mT), una mezcla racémica de enantiómeros, que se mostró que mediaba en el rechazo dependiente de la inmunidad de fetos alógenos en ratones [3] y en la mejora dependiente de la inmunidad de la actividad antitumoral de quimioterapia y radioterapia [4]. Cada uno de estos enantiómeros muestra diferentes propiedades biológicas. Se ha mostrado que el 1 -metil-L-triptófano (L1mT) inhibe la actividad enzimática de IDO1 (Ki=34 pM, [5]) en ensayos sin células usando la enzima IDO1 recombinante purificada, y en células tumorales tratadas con INFy o en estirpes celulares de tumor transfectadas con vectores de expresión que codifican IDO1 bajo el control de un promotor heterólogo, mientras que el isómero D (indoximod) no inhibe la actividad enzimática en este tipo de ensayos [6]. Sin embargo, ambos isómeros son capaces de restaurar la proliferación de linfocitos T en un ensayo de MLR con células dendríticas IDO+ como células estimuladoras, o en ensayos de proliferación de linfocitos T dependientes de antígeno singénico usando DC IDO+ aisladas de ganglios linfáticos drenantes de tumor [6]. En este tipo de ensayo, donde las DC IDO+ están presentes, los linfocitos T no proliferan. Sin embargo, la inhibición de la vía de IDO por estos inhibidores restaura la capacidad proliferativa de los linfocitos T. Es interesante señalar que ambos isómeros muestran diferente potencia en este ensayo, siendo el indoximod más potente (CE50=30 pM) que L1 mT (CE50= 80-100 pM) o la mezcla racémica (80-100 pM) [6]. Además, a pesar del hecho de que el indoximod no muestra inhibición de la actividad
enzimática en otros tipos de ensayos, muestra inhibición de la actividad enzimática en este ensayo de cocultivo, como se observa por la reducida degradación de Trp y la síntesis de Kyn.
Una cuestión un tanto desconcertante ha sido el hecho de que el indoximod no muestre inhibición de la actividad enzimática de IDO1 in vitro, sino que imita de algún modo las consecuencias biológicas de la inhibición de IDO1 in vivo o en ensayos basados en células. La evidencia experimental de varios laboratorios de investigación apunta a la conclusión de que el indoximod está participando en la inhibición de la vía de IDO1. Varios posibles mecanismos por los que esto podría estar teniendo lugar son: 1) inhibición de las isoformas de IDO1,2) inhibición de IDO2, 3) formación alternativa de los metabolitos derivados de indoximod, 4) racemización de indoximod en L1mT, 5) inhibición del transporte de Trp, 6) inhibición de la vía de GCN2 por formación de complejos de indoximod-ARNt, 7) inhibición de enzimas implicadas en la detección de Trp, tales como WARS1 o WARS2, 8) alteración de la autofagia en condiciones de estrés inducido por la privación de aminoácidos o 9) mecanismos de derivación que inactivan el mTOR en condiciones de deficiencia de aminoácidos [7]. Estos mecanismos no son necesariamente mutuamente excluyentes, y hasta la fecha son compatibles con los actuales datos experimentales. Se necesitan más investigaciones para aclarar que de estos mecanismos bioquímicos es responsable la actividad biológica de indoximod.
La actividad biológica del indoximod para aliviar la inmunosupresión in vivo e in vitro está soportada por estudios realizados en varios laboratorios en modelos preclínicos murinos. El indoximod ha mostrado actividad en los siguientes ensayos biológicos:
1. En combinación con quimioterapia, el indoximod muestra efectos antitumorales en modelos animales de melanoma ectópico, tumores de colon y de pulmón, y en modelos de tumor de mama ortotópico y autóctono. El efecto antitumoral del indoximod se pierde en ratones atímicos e IDO1-KO [6].
2. El indoximod puede prevenir el proceso de activación de Treg maduros in vivo, y facilitar la trans-diferenciación in vitro e in vivo de Treg en linfocitos T de tipo TH17 proinflamatorios [8, 9].
3. En protocolos de vacunación contra tumores, la combinación de dos vacunas antitumorales diferentes con indoximod fue eficaz en convertir una mayor proporción de células Treg en linfocitos T de tipo TH17, con efecto antitumoral simultáneo [9].
4. En modelos de melanoma, la combinación de anti-CTLA4 (ipilimumab) e indoximod produce un efecto antitumoral sinérgico [10].
5. In vivo, el indoximod fue más eficaz que un agente antineoplásico en los ciclos de quimio-inmunoterapia usando ciclofosfamida, paclitaxel o gemcitabina, cuando se probó en modelos de ratón de melanoma trasplantable y cáncer de mama trasplantable (4T1) y autóctono (mmTV-neu) [6].
6. La IDO1 también participa en la diferenciación de linfocitos T CD4 intactos en Tregs, por el efecto combinado de privación de Trp y presencia de catabolitos de Trp, mediante un mecanismo que depende de GCN2 [11, 12]. Esta conversión se interrumpe in vivo en presencia de indoximod.
7. Similarmente, las DC IDO+ también participan en la activación de Treg maduros in vivo, que también requirió una vía de GCN2 intacta en la población de Treg. Este fenómeno se podría prevenir por el exceso de Trp o por indoximod [8].
8. Además de prevenir la activación de linfocitos Treg maduros, el indoximod puede mediar en la conversión de Treg FoxP3+ supresores en células TH17 proinflamatorias in vitro e in vivo. Esta conversión de Treg en células TH17 requirió la presencia de antígeno o interacción de B7 en las pDC, y la presencia de genes IDO1 y GCN2 funcionales en las pDC. El indoximod fue capaz de imitar las consecuencias fenotípicas de la ablación génica de IDO1 o GCN2 [9], soportando, por lo tanto, su función en la inhibición de la vía de IDO.
9. Estudios antitumorales e inmunogénicos usando ratones IDO1-KO o pDC derivadas de ratones IDO1-KO mostraron que los efectos beneficiosos del indoximod se pierden en el contexto de un acervo genético que carece de una IDO1 funcional [6]. En particular, se observó que los ratones IDO1-KO desarrollan tumores, que no son sensibles al tratamiento con indoximod en combinación con quimioterapia. Además, las pDC derivadas de ganglios linfáticos drenantes del tumor de ratones IDO1-KO son capaces de estimular la proliferación de linfocitos T en cultivo, al mismo grado que las APC IDO(-). Estas observaciones fueron interpretadas como una validación genética de IDO1 como la diana farmacológica de indoximod. Sin embargo, esto también se podría interpretar como que el indoximod bloquea algún otro punto de acción dentro de la vía de IDO.
10. Las observaciones antitumorales e inmunológicas hechas por la administración de indoximod también fueron reproducidas por la administración de otros inhibidores de IDO1 bien documentados (es decir, moléculas que inhiben la actividad enzimática de IDO1 in vitro y en ensayos basados en células), tales como 5-Br-brassinina, menadiona, metil-tiohidantoin-triptófano y análogos de fenilimidazol (no publicado), lo que valida así la vía de IDO1 como diana farmacológica [4, 13, 14].
11. En modelos animales preclínicos, los efectos farmacodinámicos in vivo de indoximod se observan principalmente en ganglios linfáticos drenantes del tumor, donde el efecto se observa como una activación y proliferación de células CD8a+, reducción en el número de Tregs FoxP3+, reprogramación de Tregs (CD40L-) en linfocitos T inmunoestimulantes (CD40L+) y reprogramación de células presentadoras de antígenos IDO+ del fenotipo CD11c+/CD80/86- a CD80/86+.
Por estos motivos, el indoximod está siendo investigado en ensayos clínicos humanos para indicaciones para el cáncer. El indoximod está siendo estudiado en varias indicaciones para el cáncer en combinación con diferentes agentes quimioterapéuticos e inmunoterapéuticos biológicos, tales como docetaxel, paclitaxel, gemcitabina, Nabpaclitaxel, temozolomida, ipilimumab, sipuleucel-T, o vacunas.
El indoximod está biodisponible por vía oral con un perfil farmacocinético (FC) favorable (Tmáx: ~ 3 h; semivida: ~10 h) y un excelente perfil de seguridad. En los estudios farmacocinéticos en pacientes se ha demostrado que el indoximod muestra un perfil FC lineal en dosis de hasta 800 mg/dosis, con concentración plasmática máxima (Cmáx) de 15 pM y niveles de exposición al fármaco (ABC(ü-última)) de ~100 pM.h. Sin embargo, dosis crecientes por encima de 800 mg/dosis hasta 2000 mg/dosis no producen un aumento lineal o proporcional ni en Cmáx ni en la exposición al fármaco, lo que posiblemente limita la actividad terapéutica de este fármaco en investigación. Los ejemplos de profármacos de indoximod se proporcionan en los documentos de patente WO0168591, JPH08217791, WO2010132601, WO2004091490, WO9611927 e Y. Bennani et al., SYNLETT 1998, no. 7, 754-756.
El ensayo de proliferación de linfocitos T en una respuesta mixta de linfocitos (MLR) muestra que los linfocitos T que están en un entorno IDO+ restauran ~50 % de su capacidad proliferativa en concentraciones de indoximod superiores a 30 pM. Los experimentos antitumorales murinos muestran que los efectos biológicos del indoximod se observan cuando a los ratones se les administra indoximod en el agua de beber a 3 mg/mL (~500 mg/kg/día), o se les administra por vía oral a 200 mg/kg dos veces al día, que produce una Cmáx superior a 20 pM y exposiciones superiores a 300 pM.h. Por estos motivos, es conveniente aumentar la Cmáx y la exposición a indoximod en ensayos clínicos humanos de manera que se puedan alcanzar los niveles necesarios para la actividad terapéutica. Sin embargo, el perfil farmacocinético no lineal de este fármaco hace que sea poco probable que esto se pueda resolver aumentando la dosis administrada a los pacientes.
Por los motivos anteriormente mencionados, los presentes inventores investigaron si una formulación diferente de indoximod, tal como dispersiones secas en espray o sales o profármacos de indoximod en diferentes formas de sal, aumentaría la solubilidad y la velocidad de absorción o reduciría la depuración en sangre hasta niveles que aumentaran la máxima concentración y exposición a indoximod. Además, los presentes inventores buscaron profármacos y sus sales que pudieran producir aumentos en los parámetros de exposición cuando se administraran por vía oral y en formulación para administración en píldora (cápsula o comprimido).
Los resultados de estas investigaciones mostraron que algunos profármacos seleccionados produjeron aumentos en los parámetros de exposición; y que se podrían lograr aumentos en la solubilidad in vitro y exposición in vivo por algunas sales de indoximod tras la administración por vía oral.
SUMARIO DE LA INVENCIÓN
En un aspecto, la presente divulgación describe compuestos y composiciones farmacéuticas que comprenden los compuestos según la fórmula 1 a y 1 b

en donde A-pn es un anión inorgánico u orgánico y C+pm es un catión inorgánico como se define en el presente documento.
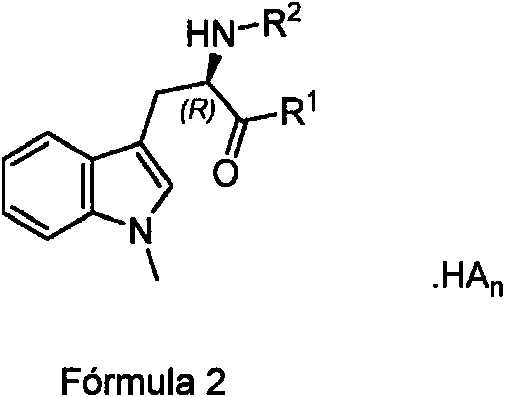
La invención comprende compuestos y composiciones farmacéuticas que comprenden compuestos según la fórmula (2 )

en donde:
(a) R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2, -O-alquil C1-3-R3, -O-alquil C1-6-R6, -O alquil C1-2-C^s^ H(NH2)(COOH) o -O-alquil C1-2-C^H(NH 2)(COOH);
(b) R2 es H, -C(O)C(^ H(NH2)R4, -C(O)(R)H(NH2)R4, -C(O)CH2C^H(NH2)C(O)OCH3 o -C(O)NHR5;
(c) R3 es tetrahidropirano o

(d) R4 es H, -alquilo C1-5, -(CH2)1-2SH, alquil C1-5-S-alquilo C1-5, alquil C1-5-O-alquilo C1-5, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)1-2C(O)NH2, -(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2;
(e) C(S) y C(R) son carbonos con la estereoquímica S o R, respectivamente, cuando R4 no es H;
(f) R5 es H, alquil C1-6-R6 o R6;
(g) R6 es arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo, en donde el arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7;
(h) cada R7 es independientemente halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, -alquilo C1-6, -haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C( O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2, en donde R es H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es H; y
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico, en donde la relación estequiométrica n del ácido es 0, 0,5, 1 o 2 de forma que el profármaco sea de carga neutra.
En otro aspecto, la presente divulgación proporciona
a) una composición farmacéutica que comprende compuestos de la fórmula 2 descrita anteriormente.
b) el profármaco o composición farmacéutica descrito anteriormente para su uso en métodos, para modular la actividad de la vía de la indolamina-2,3-dioxigenasa en un sujeto en necesidad del mismo, que comprende la administración por vía oral de cantidades suficientes de dichas composiciones a dicho sujeto en una forma farmacéutica o vehículo apropiado.
c) el profármaco o composición farmacéutica descrito anteriormente para su uso en métodos de tratamiento de inmunosupresión específica de tumor asociada a cáncer, en un sujeto en necesidad del mismo, que comprende la administración por vía oral de cantidades suficientes de dichas composiciones a dicho sujeto en una forma farmacéutica o vehículo apropiado.
e) el profármaco o composición farmacéutica descrito anteriormente para su uso en métodos de tratamiento de inmunosupresión asociada a enfermedades infecciosas (por ejemplo, infección por el VIH-1, gripe), en un sujeto en necesidad del mismo, que comprende la administración por vía oral de cantidades suficientes de dichas composiciones a dicho sujeto en una forma farmacéutica o vehículo apropiado.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La Figura 1 muestra el espectro de XRPD de indoximod en base libre y en su forma de sal de clorhidrato.
La Figura 2 muestra los análisis termogravimétricos (TGA) y de calorimetría diferencial de barrido (DSC) de la sal de clorhidrato de indoximod.
La Figura 3 muestra el espectro de XRPD de indoximod en base libre y en su forma de sal de fosfato.
La Figura 4 muestra los análisis termogravimétricos (TGA) y de calorimetría diferencial de barrido (DSC) de la sal de fosfato de indoximod.
La Figura 5 muestra el perfil de solubilidad medido frente al pH de indoximod y sus sales en diversas disoluciones de disolvente y líquidos biológicos simulados.
La Figura 6 muestra la concentración plasmática máxima (Cmáx) y la exposición (ABC0-inf) de indoximod frente a la dosis molar de indoximod, clorhidrato de indoximod o fosfato de indoximod administrado a ratas en forma de cápsula oral.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
El indoximod (1 -metil-D-triptófano, D1 mT) es un inhibidor en investigación de la vía de la indolamina-2,3-dioxigenasa (IDO) que está siendo probado en varios ensayos clínicos humanos para múltiples indicaciones para el cáncer, en combinación con agentes quimioterapéuticos e inmunomoduladores habituales y experimentales e inmunoterapias activas.
En presencia de células dendríticas IDO+, los linfocitos T efectores CD8+ se vuelven anérgicos e incapaces de proliferar. Además, los linfocitos T reguladores (CD4+ CD25+ FoxP3+) se activan en presencia de DC IDO+ y son capaces de mediar en la inmunosupresión sistémica a antígenos tumorales o víricos. El indoximod es capaz de revertir estos procesos, lo que permite que los linfocitos T efectores proliferen y dirijan la reprogramación de Treg hacia un fenotipo de tipo TH17 auxiliar. En ensayos in vitro, estos efectos están mediados por indoximod con una CE50 de ~ 30 |uM [6]. En modelos preclínicos de tumor murino, los efectos antitumores, la estimulación de linfocitos T efectores y la reprogramación de Treg en los ganglios linfáticos drenantes requiere dosis diarias de ~ 500 mg/kg, con exposiciones > 300 gM.h.
Los experimentos farmacocinéticos humanos en dosis orales que varían entre 200 mg y 2000 mg/dosis han mostrado que los parámetros farmacocinéticos Cmáx y la exposición (ABCü-inf) aumentan linealmente con la dosis, hasta un intervalo de ~ 800 mg/dosis. En estas dosis, la Cmáx en plasma alcanza un promedio de ~15 |uM y el ABC0-¡nf alcanza ~ 100 |uM.h. Los parámetros Cmáx y ABC no aumentan significativamente por encima de estos valores en dosis más altas de hasta 2000 mg/dosis. Por lo tanto, para lograr niveles de concentración y exposición de indoximod que sean comparables a los que produjeron efectos terapéuticos inmunomoduladores y antitumorales en modelos murinos, sería útil aumentar los niveles de Cmáx y de exposición de indoximod.
La presente divulgación describe compuestos de la fórmula 1a y 1b y la presente invención describe compuestos de la fórmula 2 que producen una mayor exposición y concentración máxima en suero de indoximod tras la administración por vía oral, en comparación con la administración por vía oral de dosis molares equivalentes de indoximod.
Sales de indoximod
Se desvela una sal de indoximod. En un ejemplo, la sal tiene una estructura según la fórmula 1 a:

en donde A-pn es un anión inorgánico u orgánico en un estado de ionización -p. El anión puede estar presente en una relación estequiométrica n que garantiza la neutralidad de carga molecular.
El anión A-pn se puede seleccionar del grupo que consiste en cloruro, fosfato, sulfato, mesilato, besilato, acetato, ascorbato, aspartato, glutamato, glutarato, lactato, maleato, malonato, oxalato, succinato, fumarato, tartrato y citrato. En una realización, el anión se presenta en una relación estequiométrica n de forma que la sal resultante sea de carga neutra. Por consiguiente, el anión puede tener un estado de ionización p de -1, -2 o -3 y se presenta en una relación estequiométrica n de 1, 1/2 o 1/3, respectivamente, de forma que se cumplan las condiciones estequiométricas de neutralidad de carga. En un ejemplo, el fosfato es HPO4-2, y el HPO4-2 está presente en una relación estequiométrica n de 0,5. En un ejemplo, el fosfato es HPO4-, y el HPO4- está presente en una relación estequiométrica n de 1. En un
ejemplo, el sulfato es SO4'2, y SO4'2 está presente en una relación estequiométrica n de 0,5. En un ejemplo, el mesilato es CH3SO3', y el CH3SO3' está presente en una relación estequiométrica n de 0,5.
El anión A-pn 'puede ser Cl- en una relación estequiométrica n de 1.
En otro ejemplo, el anión A-pn es Cl- en una relación estequiométrica n de 1 y la forma cristalina es una isoforma anhidra de la forma 1.
En un ejemplo, la sal tiene una estructura según la fórmula 1b:

en donde C+Pm es un catión en un estado de ionización p. En un ejemplo, el catión está presente en una relación estequiométrica m que garantiza la neutralidad de carga molecular. En un ejemplo, C+Pm se selecciona del grupo que consiste en Li+, Na+, K+, Mg+2 y Ca+2. En un ejemplo, cuando p es 1, m es 1, y cuando p es 2, m es 1A Profármacos de indoximod
En una realización reivindicada, se desvela un profármaco de indoximod. En una realización reivindicada, la estructura del profármaco, en forma de base libre o de sal, se proporciona en la fórmula 2:

En una realización reivindicada, R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2, -O-alquil C1-3-R3, -O-alquil C1-6-R6, -O-alquil C1-2-C^H(NH2)(COOH) o -O-alquil C1-2-CRH(NH2)(COOH).
En una realización reivindicada, R2 es -H, -C(O)C(S)H(NH2)R4, -C(O)C(R)H(NH2)R4,-C(O)CH2C^H(NH2)-C(O)OCH3, o -C(O)NHR5.
En una realización reivindicada, R3 es tetrahidropirano o

En una realización reivindicada, R4 es -H, -alquilo C1-5, -(CH2)1-2SH, -alquil C1-5-S-alquilo C1-5, -alquil C1-5-O-alquilo C1-5, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)1-2C(O)NH2,-(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2.
En una realización reivindicada, cuando R4 no es -H, C(S) y C(R) son carbonos con la estereoquímica S o R, respectivamente.
En una realización reivindicada, R5 es -H, alquil C1-6-R6 o R6. En esta realización, R6 se selecciona del grupo que consiste en -H, arilo, alquilarilo, heteroarilo, cicloalquilo y heterocicloalquilo, en donde el arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7.
En una realización reivindicada, cada R7 es independientemente halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, alquilo C1-6, haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R, -N(R)C(O)OR, o -N(R)C(O)N(R)2, en donde R es H o alquilo C1-4. En una realización reivindicada del profármaco de la fórmula 2, R1 no puede ser -OH cuando R2 es H.
Además, en todas las realizaciones, el profármaco no puede ser Na-terc-butox¡carbonil-1-met¡l-D-tr¡ptófano, Na-bencil-1-metil-D-triptofanato de etilo o Na-('ferc-butoxicarbonil)-1-metil-D-triptofanato de bencilo.
En una realización, HAn es un ácido. En una realización reivindicada, el ácido HAn se selecciona del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico.
En una realización reivindicada, el ácido HAn está presente en una relación estequiométrica n de forma que el profármaco resultante sea de carga neutra. Por consiguiente, en esta realización, la relación estequiométrica n del ácido HAn es 0, 0,5, 1 o 2 de forma que el profármaco sea de carga neutra.
La invención también proporciona profármacos de indoximod, en su forma de base libre o de sal. En una realización, los profármacos de indoximod se representan por compuestos de la fórmula 2

en donde
R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2, -O-alquil C1-3-R3, -NHC(S)HR4(COOH), -NHC(R)HR4(COOH), -O-alquil C1-6-R6, -O-alquilo C1-2, -C^H(NH2)(COOH) o -O-alquil C1-2-CRH(NH2 )(COOH); R2 es -H, -C(O)C(S)H(NH2)R4, -C(O)C(R)H(NH2)R4, -C(O)CH2C^H(NH2)-C(O)OCH3, -C(O)OR5 o -C(O)NHR5, R3 es tetrahidropirano o

en donde R4 es h, -alquilo C1-5, -(CH2K 2SH, alquil C^-S-alquilo C1-5, alquil C1-5-O-alquilo C1-5, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)1-2C(O)NH2, -(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2;
en donde C(S) y C(R) representa un carbono con la estereoquímica S o R, respectivamente, cuando R4 no es -H; en donde R5 es -H, alquil C1-6-R6; o R6
en donde R6 es H, arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo, en donde dicho arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7;
en donde cada R7 se selecciona independientemente de halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, alquilo C1-6, haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2;
en donde R es -H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es -H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico; y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En otra realización, la invención proporciona profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2 ,

en donde R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3, -R2 es H, o -C(O)C(3H(NH2)R4,
R3 es tetrahidropirano o

en donde R4 es H, -alquilo C1-5, -(CH2)1-2SH, -(CH2)1-3SCH3, -(CH2)1-3OCH3, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)1-2C(O)NH2, -(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2;
en donde C(S) representa un carbono con la estereoquímica S, cuando R4 no es H;
en donde R6 es H, arilo, alquilarilo, heteroarilo, cicloalquilo, heterocicloalquilo, en donde dicho arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7;
en donde cada R7 es independientemente halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, -alquilo C1-6, -haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2;
en donde R es H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico; y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En una realización preferida, la invención proporciona profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2 ,

en donde
R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3,
R2 es H, o -C(O)C(S)H(NH2)R4,
R3 es tetrahidropirano o

en donde R4 es H, -alquilo C1-5, -CH2-R6, -(CH2)1-2C(O)NH2, -(CH2)2SCH3, -(CH2)1-3C(O)OH o -(CH2)1-4NH2;
en donde C(S) representa un carbono con la estereoquímica S, cuando R4 no es -H;
en donde R6 es -H, arilo, alquilarilo, o heteroarilo, en donde dicho arilo, alquilarilo o heteroarilo se sustituye opcionalmente con un grupo R7;
en donde R7 se selecciona de halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, alquilo C1-6, haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N( R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2;
en donde R es -H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo de PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico) o C6H5SO3H (ácido bencilsulfónico); y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En otra realización preferida, la invención proporciona profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2

en donde
R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3,
R2 es H, o -C(O)C(S^ H(NH2)R4,
R3 es tetrahidropirano o

en donde R4 es -CH2CH(CH3)2, -C^H(CH)3CH2CH3, -(CH2)2SCH3, -CH2-R6, -(CH2)2C(O)NH2, -(CH2)3C(O)OH o -(CH2)4NH2;
en donde C(S) representa un carbono con la estereoquímica S;
en donde R6 es fenilo;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico) HSO3CH3 (ácido metilsulfónico), y C6H5SO3H (ácido bencilsulfónico), y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En una realización más preferida, la invención proporciona profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2

en donde
R1 es -O-alquilo C2-3 o -OCH2CH(OH)CH2OH,
R2 es H o -C(O)C(S)H(NH2)R4,
en donde R4 es -CH2CH(CH3)2, -(CH2)2SCH3 o -(CH2)2C(O)NH2;
en donde C(S) representa un carbono con la estereoquímica S
con la condición de que R1 no pueda ser -OH cuando R2 es H,
HAn es un ácido seleccionado del grupo de PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico) HSO3CH3 (ácido metilsulfónico) o C6H5SO3H (ácido bencilsulfónico); y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
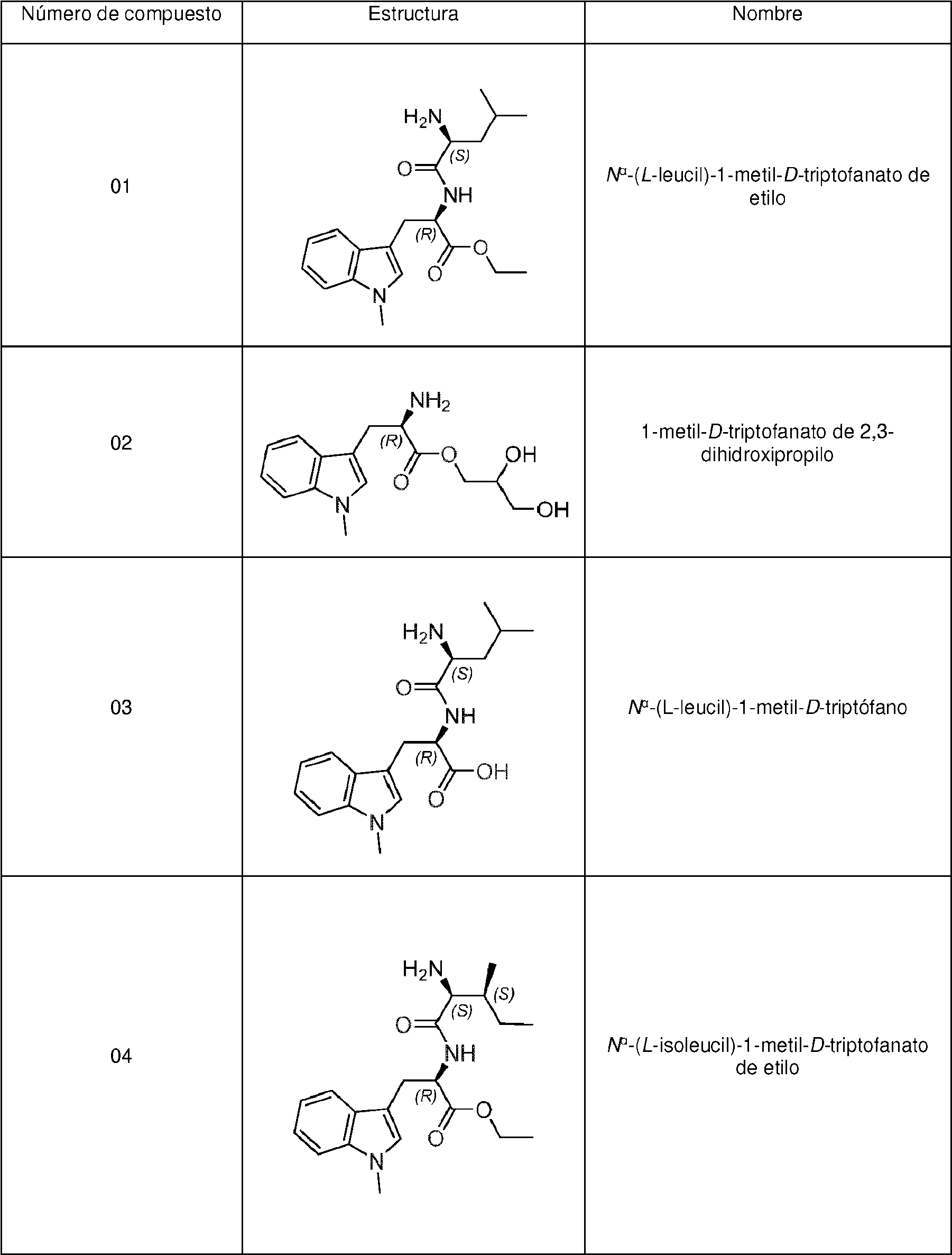
En una realización preferida, la invención proporciona profármacos de indoximod, en su forma de base libre o como una forma de sal farmacéuticamente apropiada, como se representa por los compuestos de la fórmula 2 representados en la Tabla 1.
En una realización, el profármaco incluye sustancialmente al menos uno de los siguientes compuestos: (i) N'-(L-leucil)-1-metil-D-triptofanato de etilo; (ii) 1-metil-D-triptofanato de 2,3-dihidroxipropilo; (iii) W-(L-leucil)-1-metil-D-triptófano; (iv) W-(L-isoleucil)-1-metil-D-triptofanato de etilo; (v) W-(L-glicil)-1-metil-D-triptófano; (vi) ácido (S)-5-amino-6-(((R)-1-carboxi-2-(1 -metil-1 H-indol-3-il)etil)amino)-6-oxohexanoico; (vii) W-(L-lisil)-1 -metil-D-triptófano; (viii) W-(L-fenilalanil)-1-metil-D-triptófano; (ix) W-(L-glutaminil)-1-metil-D-triptofanato de etilo; (x) 1-metil-D-triptofanato de 2-(dimetilamino)etilo; (xi) 1-metil-D-triptofanato de (2-etoxi-2-oxido-1,3,2-dioxafosfolan-4-il)metilo; (xii) 1-metil-D-triptofanato de 2-(tetrahidro-2H-piran-4-il)etilo; (xiii) 1-metil-D-triptofanato de etilo; (xiv) 1-metil-D-triptofanato de isopropilo; (xv) W-(L-metionil)-1-metil-D-triptófano; o (xvi) W-(L-metionil)-1-metil-D-triptofanato de etilo.
Composiciones farmacéuticas de sales y profármacos de indoximod
Se desvela una composición farmacéutica (no englobada por las reivindicaciones) que comprende sales de indoximod, como se representa por los compuestos de la fórmula 1 a y 1 b

en donde A-n es un anión inorgánico u orgánico y C+pm es un catión inorgánico en un estado de ionización y en una relación estequiométrica que garantiza la neutralidad de carga molecular.
A-pn puede ser un anión seleccionado del grupo que consiste en cloruro, fosfato, sulfato, mesilato, besilato, acetato, ascorbato, aspartato, glutamato, glutarato, lactato, maleato, malonato, oxalato, succinato, fumarato, tartrato y citrato, en donde la carga negativa p es -1, -2 o -3 en una relación estequiométrica n de 1, / o 1/3, respectivamente, de manera que cumpla las condiciones estequiométricas de neutralidad de carga.
Se desvela una composición farmacéutica (no englobada por las reivindicaciones) que comprende sales de indoximod, como se representa por los compuestos de la fórmula 1b, en donde C+pm es un catión seleccionado del grupo de Li+, Na+, K+, Mg+2 o Ca+2, en donde la carga positiva p es 1 o 2 en una relación estequiométrica m de 1 o / , respectivamente, de manera que cumpla las condiciones estequiométricas de neutralidad de carga.
Se desvela una composición farmacéutica (no englobada por las reivindicaciones) que comprende sales de indoximod, como se representa por los compuestos de la fórmula 1a, en donde A-pn es un anión seleccionado del grupo que consiste en HPO4-2 (fosfato), SO4-2 (sulfato), H2PO4-(fosfato), Cl- y CH3SO3-(mesilato), en una relación estequiométrica n de 0,5, 0,5, 1 o 1, respectivamente.
En la fórmula 1 a, A-pn puede ser Cl- en una relación estequiométrica n de 1.
En la fórmula 1 a, A-pn puede ser Cl- en una relación estequiométrica n de 1 y la forma cristalina puede ser una isoforma anhidra de la forma 1.
La invención proporciona una composición farmacéutica que comprende profármacos de indoximod, en su forma de base libre o de sal. En una realización, los profármacos de indoximod se representan por los compuestos de la fórmula 2 ,

en donde
R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2, -O-alquil C1-3-R3, -O-alquil C1-6-R6, -O-alquilo C1-2, -C^H(NH2)(COOH) o -O-alquil C1-2-CR^H(NH2 )(COOH);
R2 es -H, -C(O)C(S>H(NH2)R4, -C(O)C(R)H(NH2)R4, -C(O)CH2C^H(NH2)-C(O)OCH3, o -C(O)NHR5,
R3 es tetrahidropirano o

en donde R4 es H, -alquilo C1-5, -(CH2)1-2SH, alquil C1-5-S-alquilo C1-5, alquil C1-5-O-alquilo C1-5, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)1-2C(O)NH2, -(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2;
en donde C(S) y C(R) representa un carbono con la estereoquímica S o R, respectivamente,
cuando R4 no es -H; en donde R5 es -H, alquil C1-6-R6; o R6
en donde R6 es H, arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo, en donde dicho arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7;
en donde cada R7 se selecciona independientemente de halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, alquilo C1-6, haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2;
en donde R es -H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es -H,
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico; y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En otra realización, la invención proporciona una composición farmacéutica que comprende profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2
en donde R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3, -R2 es H o -C(O)C(S)H(NH2)R4,
R3 es tetrahidropirano o

en donde R4 es H, -alquilo C1-5, -(CH2)i -2SH, -(CH2)i -3SCH3, -(CH2)i -3ÜCH3, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)i -2C(0)NH2, -(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2;
en donde C(S) representa un carbono con la estereoquímica S, cuando R4 no es H;
en donde R6 es H, arilo, alquilarilo, heteroarilo, cicloalquilo, heterocicloalquilo, en donde dicho arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7;
en donde cada R7 es independientemente halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, alquilo C1-6, haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2;
en donde R es H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico; y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En una realización preferida, la invención proporciona una composición farmacéutica que comprende profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2,

en donde
R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3,
R2 es H o -C(O)C(S)H(NH2)R4,
R3 es tetrahidropirano o

en donde R4 es H, -alquilo C1-5, -CH2-R6, -(CH2)1-2C(O)NH2, -(CH2)2SCH3, -(CH2)1-3C(O)OH o -(CH2)1-4NH2 en donde C(S) representa un carbono con la estereoquímica S, cuando R4 no es -H;
en donde R6 es -H, arilo, alquilarilo o heteroarilo, en donde dicho arilo, alquilarilo o heteroarilo se sustituye opcionalmente con un grupo R7;
en donde R7 se selecciona de halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, alquilo C1-6, haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N( R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2;
en donde R es -H o alquilo C1-4;
con la condición de que R1 no puede ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo de PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico) o C6H5SO3H (ácido bencilsulfónico); y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En una realización más preferida, la invención proporciona una composición farmacéutica que comprende profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2

en donde
R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3,
R2 es H, o -C(O)C(3H(NH2)R4,
R3 es tetrahidropirano o

en donde R4 es -CH2CH(CH3)2, -C^H(CH)3CH2CH3, -(CH2)2SCH3, -CH2-R6, -(CH2)2C(O)NH2, -(CH2)3C(O)OH o -(CH2)4NH2;
en donde C(S) representa un carbono con la estereoquímica S;
en donde R6 es fenilo;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico) HSO3CH3 (ácido metilsulfónico) y C6H5SO3H (ácido bencilsulfónico), y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En una realización más preferida, la invención proporciona una composición farmacéutica que comprende profármacos de indoximod, en su forma de base libre o de sal, como se representa por los compuestos de la fórmula 2,

en donde
R1 es -O-alquilo C2-3 o -OCH2CH(OH)CH2OH,
R2 es H o -C(O)C(S)H(NH2)R4,
en donde R4 es -CH2CH(CH3)2, -(CH2)2SCH3 o -(CH2)2C(O)NH2;
en donde C(S) representa un carbono con la estereoquímica S
con la condición de que R1 no pueda ser -OH cuando R2 es H,
HA es un ácido seleccionado del grupo de PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico) HSO3CH3 (ácido metilsulfónico) o C6H5SO3H (ácido bencilsulfónico); y n es la relación estequiométrica de 0, 0,5, 1 o 2 que garantiza la neutralidad de carga de la sal resultante.
En una realización preferida, la invención proporciona una composición farmacéutica que comprende profármacos de indoximod, en su forma de base libre o como una forma de sal farmacéuticamente apropiada, como se representa por los compuestos de la fórmula 2 representados en la Tabla 1.
Tabla 1. Pro fármacos de indoximod



En otro aspecto, la divulgación proporciona métodos de uso de las composiciones de las fórmulas 1 y 2, para modular la actividad de la vía de la indolamina-2,3-dioxigenasa en un sujeto en necesidad de los mismos, que comprenden la administración por vía oral de cantidades terapéuticamente eficaces de dichas composiciones a dicho sujeto en una forma farmacéutica apropiada o vehículo.
En otro aspecto, la divulgación proporciona métodos de uso de las composiciones de las fórmulas 1a, 1b y 2, para el tratamiento de cáncer en un sujeto en necesidad de los mismos, que comprenden la administración por vía oral de cantidades terapéuticamente eficaces de dichas composiciones a dicho sujeto en una forma farmacéutica apropiada o vehículo.
En otro aspecto, la divulgación proporciona métodos de uso de las composiciones de las fórmulas 1a, 1b y 2, para el tratamiento de inmunosupresión específica de tumor asociada al cáncer, en un sujeto en necesidad de los mismos, que comprenden la administración por vía oral de cantidades suficientes de dichas composiciones a dicho sujeto en una forma farmacéutica apropiada o vehículo.
En otro aspecto, la divulgación proporciona métodos de uso de las composiciones de las fórmulas 1a, 1b y 2, para tratar inmunosupresión asociada a enfermedades infecciosas (por ejemplo, infección por el VIH-1, gripe), en un sujeto en necesidad de los mismos, que comprenden la administración por vía oral de cantidades suficientes de dichas composiciones a dicho sujeto en una forma farmacéutica apropiada o vehículo.
En una realización, un profármaco de indoximod como se expone anteriormente se incluye en una composición farmacéutica, y la composición se incluye en una cápsula sólida, cápsula de gelatina, comprimido o píldora. En una realización, la sal y/o el profármaco se incluyen en una cápsula disolvible.
En realizaciones específicas, las composiciones de la presente invención pueden contener además otros componentes auxiliares convencionalmente encontrados en las composiciones farmacéuticas, en sus niveles de uso establecidos en la técnica. Así, por ejemplo, las composiciones pueden contener materiales adicionales útiles en la formulación física de diversas formas farmacéuticas de las composiciones de la presente invención, tales como colorantes, aromatizantes, conservantes, antioxidantes, opacificantes, espesantes y estabilizadores. Las formulaciones se pueden esterilizar y, si se desea, mezclar con agentes auxiliares, por ejemplo, lubricantes, conservantes, estabilizadores, humectantes, emulsionantes, sales para influir en la presión osmótica, tampones, colorantes, aromatizantes y/o sustancias aromáticas y similares que no interactúan perjudicialmente con el (los) oligonucleótido(s) de la formulación.
En ciertas realizaciones, las composiciones farmacéuticas de la presente invención comprenden uno o más excipientes. En ciertas de dichas realizaciones, los excipientes se seleccionan de agua, disoluciones salinas, alcohol, polietilenglicoles, gelatina, lactosa, lactosa monohidratada, amilasa, estearato de magnesio, talco, ácido silícico, parafina viscosa, hidroximetilcelulosa, celulosa microcristalina y polivinilpirrolidona.
En ciertas realizaciones, una composición farmacéutica de la presente invención se prepara usando técnicas conocidas, que incluyen, pero no se limitan a, procesos de mezcla, disolución, granulación, formación de comprimidos recubiertos de azúcar, trituración, emulsión, encapsulación, atrapamiento o formación de comprimidos.
Realizaciones adicionales se refieren a las formulaciones farmacéuticas en donde la formulación se selecciona del grupo que consiste en un sólido, polvo, líquido y un gel. En ciertas realizaciones, una composición farmacéutica de la presente invención es un líquido (por ejemplo, una suspensión, elixir y/o disolución). En ciertas de dichas realizaciones, una composición farmacéutica líquida se prepara usando componentes conocidos en la técnica, que incluyen, pero no se limitan a, agua, glicoles, aceites, alcoholes, aromatizantes, conservantes y colorantes.
En ciertas realizaciones, una composición farmacéutica de la presente invención es un sólido (por ejemplo, un polvo, comprimido y/o cápsula). En ciertas de dichas realizaciones, una composición farmacéutica sólida que comprende uno o más componentes conocidos en la técnica, que incluyen, pero no se limitan a, almidones, azúcares, diluyentes, agentes de granulación, lubricantes, aglutinantes y disgregantes.
En ciertas realizaciones, una composición farmacéutica de la presente invención comprende un sistema de administración. Los ejemplos de sistemas de administración incluyen, pero no se limitan a, liposomas y emulsiones. Ciertos sistemas de administración son útiles para preparar ciertas composiciones farmacéuticas que incluyen las que comprenden compuestos hidrófobos. En ciertas realizaciones se usan ciertos disolventes orgánicos, tales como sulfóxido de dimetilo.
En ciertas realizaciones, una composición farmacéutica de la presente invención comprende un sistema de codisolventes. Ciertos de dichos sistemas de codisolventes comprenden, por ejemplo, alcohol bencílico, un tensioactivo no polar, un polímero orgánico miscible con agua y una fase acuosa. En ciertas realizaciones, dichos sistema de codisolventes se usan para compuestos hidrófobos. Un ejemplo no limitante de dicho sistema de codisolventes es el sistema de codisolventes VPD, que es una disolución de etanol absoluto que comprende 3 % p/vol de alcohol bencílico, 8 % p/vol del tensioactivo no polar polisorbato 80 y 65 % p/vol de polietilenglicol 300. Las proporciones de dichos sistemas de codisolventes se pueden variar considerablemente sin alterar significativamente sus características de solubilidad y toxicidad. Además, se puede variar la identidad de los componentes codisolventes: por ejemplo, se pueden usar otros tensioactivos en lugar del polisorbato 80; se puede variar el tamaño de la fracción de polietilenglicol; otros polímeros biocompatibles pueden sustituir al polietilenglicol, por ejemplo, polivinilpirrolidona; y otros azúcares o polisacáridos pueden sustituir a la dextrosa.
En ciertas realizaciones, una composición farmacéutica de la presente invención comprende un sistema de liberación sostenida. Un ejemplo no limitante de dicho sistema de liberación sostenida es una matriz semipermeable de polímeros hidrófobos sólidos. En ciertas realizaciones, los sistemas de liberación sostenida pueden liberar, dependiendo de su naturaleza química, agentes farmacéuticos durante un periodo de horas, días, semanas o meses.
En ciertas realizaciones, una composición farmacéutica de la presente invención se prepara para administración por vía oral. En ciertas de dichas realizaciones, una composición farmacéutica se formula combinando uno o más agentes y vehículos farmacéuticamente aceptables. Ciertos de dichos vehículos permiten la formulación de composiciones farmacéuticas como comprimidos, píldoras, comprimidos recubiertos de azúcar, cápsulas, líquidos, geles, jarabes, lechadas, suspensiones y similares, para ingestión oral por un sujeto. Los excipientes adecuados incluyen, pero no se limitan a, cargas, tales como azúcares, que incluyen lactosa, lactosa monohidratada, sacarosa, manitol o sorbitol; preparaciones de celulosa, tales como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de patata, gelatina, goma tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa sódica, celulosa microcristalina y/o polivinilpirrolidona (PVP). En ciertas realizaciones, dicha mezcla se tritura opcionalmente y se añaden opcionalmente auxiliares. En ciertas realizaciones, las composiciones farmacéuticas se forman para obtener comprimidos o núcleos de comprimidos recubiertos de azúcar. En ciertas realizaciones, se añaden disgregantes (por ejemplo, carboximetilcelulosa reticulada, tal como croscarmelosa sódica, polivinilpirrolidona reticulada, agar o ácido algínico o una sal del mismo, tal como alginato sódico).
En ciertas realizaciones, se proporcionan núcleos de comprimidos recubiertos de azúcar con recubrimientos. En ciertas de tales realizaciones, se pueden usar disoluciones concentradas de azúcar, que pueden contener opcionalmente goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol y/o dióxido de titanio, disoluciones de laca y disolventes orgánicos adecuados o mezclas de disolventes. Se pueden añadir tintas o pigmentos a comprimidos o recubrimientos de comprimidos recubiertos de azúcar.
En ciertas realizaciones, las composiciones farmacéuticas para administración por vía oral son cápsulas duras hechas de gelatina. Ciertas de dichas cápsulas duras comprenden uno o más agentes farmacéuticos de la presente invención en mezcla con una o más cargas, tales como lactosa, aglutinantes, tales como almidones, y/o lubricantes, tales como talco o estearato de magnesio y, opcionalmente, estabilizadores. En ciertas realizaciones, las composiciones farmacéuticas para administración por vía oral son cápsulas selladas blandas hechas de gelatina y un plastificante, tal como glicerol o sorbitol. En ciertas cápsulas blandas, uno o más agentes farmacéuticos de la presente invención se van a disolver o suspender en líquidos adecuados, tales como aceites grasos, parafina líquida o polietilenglicoles líquidos. Además, se pueden añadir estabilizadores.
En ciertas realizaciones, las composiciones farmacéuticas se preparan para administración por vía oral. Ciertas de dichas composiciones farmacéuticas son comprimidos o pastillas para chupar formuladas de manera convencional.
En ciertas realizaciones, una composición farmacéutica se prepara para administración por inyección (por ejemplo, intravenosa, subcutánea, intramuscular, etc.). En ciertas de dichas realizaciones, una composición farmacéutica comprende un vehículo y se formula en disolución acuosa, tal como agua o tampones fisiológicamente compatibles, tales como disolución de Hanks, disolución de Ringer o tampón de solución salina fisiológica. En ciertas realizaciones, se incluyen otros componentes (por ejemplo, componentes que ayudan en la solubilidad o sirven de conservantes). En ciertas realizaciones, se preparan suspensiones inyectables usando vehículos líquidos apropiados, agentes de suspensión y similares. Se presentan ciertas composiciones farmacéuticas para inyección en forma farmacéutica unitaria, por ejemplo, en ampollas o en envases multidosis. Ciertas composiciones farmacéuticas para inyección son suspensiones, disoluciones o emulsiones en vehículos aceitosos o acuosos, y pueden contener agentes de formulación, tales como agentes de suspensión, estabilización y/o dispersantes. Ciertos disolventes adecuados para su uso en composiciones farmacéuticas para inyección incluyen, pero no se limitan a, disolventes lipófilos y aceites grasos, tales como aceite de sésamo, ésteres de ácidos grasos sintéticos, tales como oleato de etilo o triglicéridos, y liposomas. Las suspensiones para inyección acuosa pueden contener sustancias que aumentan la viscosidad de la suspensión, tales como carboximetilcelulosa sódica, sorbitol o dextrano. Opcionalmente, dichas suspensiones también pueden contener estabilizadores adecuados o agentes que aumentan la solubilidad de los agentes farmacéuticos para permitir la preparación de disoluciones altamente concentradas.
En ciertas realizaciones, una composición farmacéutica de la presente invención puede ser un comprimido efervescente o granulado. Los comprimidos efervescentes consisten lo más comúnmente en una fuente soluble de ácido y una fuente de carbonato para producir dióxido de carbono gaseoso, sirviendo el último de disgregante. La acidez necesaria para la reacción efervescente puede derivar de ácidos alimentarios, anhídridos de ácido y sales de ácido. El ácido alimentario puede ser, por ejemplo, ácido cítrico, ácido tartárico, ácido málico, ácido fumárico, ácido adípico o ácido succínico. El anhídrido de ácido puede ser anhídrido succínico o anhídrido cítrico o similares. Las sales de ácido pueden ser, por ejemplo, dihidrogenofosfato de sodio (monofosfato de sodio), dihidrogenopirofosfato de disodio (pirofosfato ácido de sodio), sales de ácido cítrico (dihidrogenocitrato de sodio e hidrogenocitrato de disodio), sulfito ácido de sodio (bisulfito de sodio). Las fuentes de carbonato adecuadas son, por ejemplo, bicarbonato sódico, carbonato sódico, bicarbonato potásico, carbonato potásico, sesquicarbonato sódico (mezcla de cantidades molares iguales de carbonato sódico y bicarbonato sódico), carbonato de glicina, carbonato de L-lisina, carbonato de arginina, carbonato cálcico.
La efervescencia también se puede inducir por la formación de otros gases, tales como oxígeno, por ejemplo liberado de perborato de sodio o de una combinación de, por ejemplo, un compuesto de peroxígeno que produce oxígeno activo tras ser mezclado con agua (por ejemplo, perborato de sodio monohidratado o percarbonato sódico) y un compuesto de cloro que libera el hipoclorito tras el contacto con agua (por ejemplo, dicloroisocianurato de sodio o hipoclorito cálcico).
La composición farmacéutica de la presente invención se puede fabricar según métodos convencionales conocidos en la técnica. Los gránulos y comprimidos efervescentes según la invención se pueden obtener por compactación en seco o granulación en húmedo. Estos gránulos se pueden mezclar posteriormente con, por ejemplo, disgregantes, deslizantes y lubricantes adecuados y comprimirse en comprimidos o envasarse en, por ejemplo, sobres de tamaño adecuado. También se pueden obtener comprimidos efervescentes por compresión directa de una mezcla en polvo adecuada, es decir, sin granulación previa de los excipientes.
También se pueden obtener mezclas en polvo o de gránulos adecuadas según la invención secando por pulverización (por ejemplo, por un secado por pulverización en proceso caliente o por secado por pulverización básico), liofilización, extrusión del fundido, estratificación de pellas, recubrimiento del principio activo farmacéutico o cualquier otro método adecuado. Preferentemente, las condiciones se eligen de tal manera que se prevenga la amortización del principio activo farmacéutico. Los polvos o gránulos así obtenidos se pueden mezclar con uno o más componentes adecuados y las mezclas resultantes se pueden o comprimir para formar comprimidos efervescentes o envasar en sobres.
DEFINICIONES
Los términos usados en el presente documento pueden ir precedidos y/o seguidos por un guión sencillo, "-", o un guión doble, "=", para indicar el orden de enlace del enlace entre el sustituyente mencionado y su resto original; un único guión indica un enlace sencillo y un guión doble indica un doble enlace o un par de enlaces sencillos en el caso de un espiro-sustituyente. En ausencia de un guión sencillo o doble, se entiende que se forma un enlace sencillo entre el sustituyente y su resto parental; además, se pretende que los sustituyentes se lean de "izquierda a derecha", a menos que un guión indique de otro modo. Por ejemplo, alcoxi Ci-6-carboniloxi y -OC(O)-alquilo Ci-6 indican la misma funcionalidad; similarmente arilalquilo, arilalquil- y -alquilarilo indican la misma funcionalidad.
Además, se pueden usar ciertos términos en el presente documento como radicales de enlace tanto monovalentes como divalentes, como sería conocido por los expertos en la técnica, y por su presentación que enlaza otros dos restos. Por ejemplo, un grupo alquilo puede ser tanto un radical monovalente como un radical divalente; en el último caso, sería evidente para un experto en la técnica que un átomo de hidrógeno adicional se retira de un radical monovalente alquilo para proporcionar un resto divalente adecuado.
El término "alquenilo", como se usa en el presente documento, significa un hidrocarburo de cadena lineal o ramificada que contiene desde 2 hasta 10 carbonos, a menos que se especifique de otro modo, y que contiene al menos un doble enlace carbono-carbono. Los ejemplos representativos de alquenilo incluyen, pero no se limitan a, etenilo, 2-propenilo, 2- metil-2-propenilo, 3-butenilo, 4-pentenilo, 5-hexenilo, 2-heptenilo, 2-metil-1-heptenilo, 3-decenilo y 3,7-dimetilocta-2,6-dienilo.
El término "alcoxi", como se usa en el presente documento, significa un grupo alquilo, como se define en el presente documento, unido al resto molecular parental por un átomo de oxígeno. Los ejemplos representativos de alcoxi incluyen, pero no se limitan a, metoxi, etoxi, propoxi, 2-propoxi, butoxi, terc-butoxi, pentiloxi y hexiloxi.
El término "alquilo", como se usa en el presente documento, significa un hidrocarburo de cadena lineal o ramificada que contiene desde 1 hasta 10 átomos de carbono, a menos que se especifique de otro modo. Los ejemplos representativos de alquilo incluyen, pero no se limitan a, metilo, etilo, n-propilo, iso-propilo, n-butilo, sec-butilo, isobutilo, terc-butilo, n-pentilo, isopentilo, neopentilo, n-hexilo, 3-metilhexilo, 2,2-dimetilpentilo, 2,3-dimetilpentilo, nheptilo, n-octilo, n-nonilo y n-decilo. Cuando un grupo "alquilo" es un grupo de enlace entre otros dos restos, entonces también puede ser de cadena lineal o ramificado; los ejemplos incluyen, pero no se limitan a, -CH2-, -CH2CH2-, -CH2CH2CHC(CH3)-, -CH2CH(CH2CH3)CH2-.
El término alquilo C1-5 se refiere a un alquilo lineal o ramificado de 1 a 5 átomos de carbono.
El término alquilo C1-6 se refiere a un alquilo lineal o ramificado de 1 a 6 átomos de carbono.
El término "arilo", como se usa en el presente documento, significa un fenilo (es decir, arilo monocíclico), o un sistema bicíclico de anillos que contiene al menos un anillo de fenilo o un anillo bicíclico aromático que contiene solo átomos de carbono en el sistema bicíclico de anillos aromáticos. El arilo bicíclico puede ser azulenilo, naftilo, o un fenilo condensado con un cicloalquilo monocíclico, un cicloalquenilo monocíclico o un heterociclilo monocíclico. El arilo bicíclico está unido al resto molecular parental por cualquier átomo de carbono contenido dentro de la porción de fenilo del sistema bicíclico, o cualquier átomo de carbono con el anillo naftilo o azulenilo. Las porciones condensadas de cicloalquilo monocíclico o heterociclilo monocíclico del arilo bicíclico se sustituyen opcionalmente con uno o dos grupos oxo y/o tia. Los ejemplos representativos de los arilos bicíclicos incluyen, pero no se limitan a, azulenilo, naftilo, dihidroinden-1 -ilo, dihidroinden-2-ilo, dihidroinden-3-ilo, dihidroinden-4-ilo, 2,3-dihidroindol-4-ilo, 2,3-dihidroindol-5-ilo, 2,3-dihidroindol-6-ilo, 2,3-dihidroindol-7-ilo, inden-1-ilo, inden-2-ilo, inden-3-ilo, inden-4-ilo, dihidronaftalen-2-ilo, dihidronaftalen-3-ilo, dihidronaftalen-4-ilo, dihidronaftalen-1 -ilo, 5,6,7,8-tetrahidronaftalen-1-ilo, 5,6,7,8-tetrahidronaftalen-2-ilo, 2,3-dihidrobenzofuran-4-ilo, 2,3-dihidrobenzofuran-5-ilo, 2,3-dihidrobenzofuran-6-ilo, 2,3-dihidrobenzofuran-7-ilo, benzo[d][1,3]dioxol-4-ilo, benzo[d][1,3]dioxol-5-ilo, 2H-cromen-2-on-5-ilo, 2H-cromen-2-on-6-ilo, 2H-cromen-2-on-7-ilo, 2H-cromen-2-on-8-ilo, isoindolin-1,3-dion-4-ilo, isoindolin-1,3-dion-5-ilo, inden-1-on-4-ilo, inden-1-on-5-ilo, inden-1-on-6-ilo, inden-1-on-7-ilo, 2,3-dihidrobenzo[b][1,4]dioxin-5-ilo, 2,3-dihidrobenzo[b][1,4]dioxin-6- ilo, 2H-benzo[b][1,4]oxazin-3(4H)-on-5-ilo, 2H-benzo[b][1,4]oxazin-3(4H)-on-6-ilo, 2H-benzo[b][1,4]oxazin-3(4H)-on-7- ilo, 2H-benzo[b][1,4]oxazin-3(4H)-on-8-ilo, benzo[d]oxazin-2(3H)-on-5-ilo, benzo[d]oxazin-2(3H)-on-6-ilo, benzo[d]oxazin-2(3H)-on-7-ilo, benzo[d]oxazin-2(3H)-on-8-ilo, quinazolin-4(3H)-on-5-ilo, quinazolin-4(3H)-on-6-ilo, quinazolin-4(3H)-on-7-ilo, quinazolin-4(3H)-on-8-ilo, quinoxalin-2(1H)-on-5-ilo, quinoxalin-2(1H)-on-6-ilo, quinoxalin-2(1H)-on-7-ilo, quinoxalin-2(1H)-on-8-ilo, benzo[d]tiazol-2(3H)-on-4-ilo, benzo[d]tiazol-2(3H)-on-5-ilo, benzo[d]tiazol-2(3H)-on-6-ilo y benzo[d]tiazol-2(3H)-on-7-ilo. En ciertas realizaciones, el arilo bicíclico es (i) naftilo o (ii) un anillo de fenilo condensado con un cicloalquilo monocíclico de 5 o 6 miembros, un cicloalquenilo monocíclico de 5 o 6 miembros o un heterociclilo monocíclico de 5 o 6 miembros, en donde los cicloalquilo, cicloalquenilo y heterociclilo condensados se sustituyen opcionalmente con uno o dos grupos que son independientemente oxo o tia.
El término "arilalquilo", "alquilarilo" y "arilalquil-", como se usa en el presente documento, significa un grupo arilo, como se define en el presente documento, unido al resto molecular parental mediante un grupo alquilo, como se define en el presente documento. Los ejemplos representativos de arilalquilo incluyen, pero no se limitan a, bencilo, 2-feniletilo, 3- fenilpropilo y 2-naft-2-iletilo.
Los términos "ciano" y "nitrilo", como se usan en el presente documento, significan un grupo -CN.
El término "cicloalquilo", como se usa en el presente documento, significa un sistema de anillos de cicloalquilo monocíclico o bicíclico. Los sistemas de anillos monocíclicos son grupos de hidrocarburo cíclico que contienen desde 3 hasta 8 átomos de carbono, donde dichos grupos están saturados o insaturados, pero no son aromáticos. Los grupos cicloalquilo están completamente saturados. Los ejemplos de cicloalquilos monocíclicos incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclopentenilo, ciclohexilo, ciclohexenilo, cicloheptilo y ciclooctilo. Los sistemas de anillos de cicloalquilo bicíclico son anillos monocíclicos puenteados o anillos bicíclicos fusionados. Los anillos monocíclicos puenteados contienen un anillo de cicloalquilo monocíclico donde dos átomos de carbono no adyacentes del anillo monocíclico están unidos por un puente de alquileno de entre uno y tres átomos de carbono adicionales (es decir, un grupo de puente de la forma -(CH2V , donde w es 1, 2 o 3). Los ejemplos representativos de sistemas de anillos bicíclicos incluyen, pero no se limitan a, biciclo[3.1.1]heptano, biciclo[2.2.1]heptano, biciclo[2.2.2]octano, biciclo[3.2.2]nonano, biciclo[3.3.1]nonano y biciclo[4.2.1]nonano. Los sistemas de anillos de cicloalquilo bicíclico condensados contienen un anillo de cicloalquilo monocíclico condensado con un cicloalquilo monocíclico. El cicloalquilo bicíclico puenteado o fusionado está fijado al resto molecular parental mediante cualquier átomo de
carbono contenido dentro del anillo de cicloalquilo monocíclico. Los grupos cicloalquilo se sustituyen opcionalmente con uno o dos grupos que son independientemente oxo o tia. En ciertas realizaciones, el cicloalquilo bicíclico condensado es un anillo de cicloalquilo monocíclico de 5 o 6 miembros condensado con un anillo de fenilo, un cicloalquilo monocíclico de 5 o 6 miembros, un cicloalquenilo monocíclico de 5 o 6 miembros, un heterociclilo monocíclico de 5 o 6 miembros o un heteroarilo monocíclico de 5 o 6 miembros, en donde el cicloalquilo bicíclico condensado se sustituye opcionalmente con uno o dos grupos que son independientemente oxo o tia.
"Cicloalquenilo", como se usa en el presente documento, se refiere a un sistema de anillos de cicloalquenilo monocíclico o bicíclico. Los sistemas de anillos monocíclicos son grupos de hidrocarburo cíclico que contienen desde 3 hasta 8 átomos de carbono, donde dichos grupos son insaturados (es decir, que contienen al menos un doble enlace carbono-carbono anular), pero no aromáticos. Los ejemplos de sistemas de anillos monocíclicos incluyen ciclopentenilo y ciclohexenilo. Los anillos de cicloalquenilo bicíclico son anillos monocíclicos puenteados o anillos bicíclicos condensados. Los anillos monocíclicos puenteados contienen un anillo de cicloalquenilo monocíclico donde dos átomos de carbono no adyacentes del anillo monocíclico están unidos por un puente de alquileno de entre uno y tres átomos de carbono adicionales (es decir, un grupo de puente de la forma -(CH2)w-, donde w es 1, 2 o 3). Los ejemplos representativos de cicloalquenilos bicíclicos incluyen, pero no se limitan a, norbornenilo y biciclo[2.2.2]oct-2-enilo. Los sistemas de anillos de cicloalquenilo bicíclico condensados contienen un anillo de cicloalquenilo monocíclico condensado con un fenilo, un cicloalquilo monocíclico, un cicloalquenilo monocíclico, un heterociclilo monocíclico o un heteroarilo monocíclico. El cicloalquenilo bicíclico puenteado o condensado está unido al resto molecular parental mediante cualquier átomo de carbono contenido dentro del anillo de cicloalquenilo monocíclico. Los grupos cicloalquenilo se sustituyen opcionalmente con uno o dos grupos que son independientemente oxo o tia.
El término "halo" o "halógeno", como se usa en el presente documento, significa Cl, Br, I o F.
El término "haloalquilo", como se usa en el presente documento, significa al menos un halógeno, como se define en el presente documento, unido al resto molecular parental mediante un grupo alquilo, como se define en el presente documento. Los ejemplos representativos de haloalquilo incluyen, pero no se limitan a, clorometilo, 2-fluoroetilo, trifluorometilo, pentafluoroetilo y 2-cloro-3-fluoropentilo.
El término "heteroarilo", como se usa en el presente documento, significa un sistema de anillos de heteroarilo monocíclico o bicíclico que contiene al menos un anillo heteroaromático. El heteroarilo monocíclico puede ser un anillo de 5 o 6 miembros. El anillo de 5 miembros consiste en dos dobles enlaces y uno, dos, tres o cuatro átomos de nitrógeno y opcionalmente un átomo de oxígeno o de azufre. El anillo de 6 miembros consiste en tres dobles enlaces y uno, dos, tres o cuatro átomos de nitrógeno. El heteroarilo de 5 o 6 miembros está conectado al resto molecular parental mediante cualquier átomo de carbono o cualquier átomo de nitrógeno contenido dentro del heteroarilo. Los ejemplos representativos de heteroarilo monocíclico incluyen, pero no se limitan a, furilo, imidazolilo, indolilo, 1-metilindolilo, isoxazolilo, isotiazolilo, oxadiazolilo, oxazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, pirazolilo, pirrolilo, tetrazolilo, tiadiazolilo, tiazolilo, tienilo, triazolilo y triazinilo. El heteroarilo bicíclico consiste en un heteroarilo monocíclico condensado con un fenilo, un cicloalquilo monocíclico, un cicloalquenilo monocíclico, un heterociclilo monocíclico o un heteroarilo monocíclico. La porción condensada de cicloalquilo o heterociclilo del grupo heteroarilo bicíclico se sustituye opcionalmente con uno o dos grupos que son independientemente oxo o tia. Cuando el heteroarilo bicíclico contiene un anillo condensado de cicloalquilo, cicloalquenilo o heterociclilo, entonces el grupo de heteroarilo bicíclico está conectado al resto molecular parental mediante cualquier átomo de carbono o nitrógeno contenido dentro de la porción de heteroarilo monocíclico del sistema de anillos bicíclico. Cuando el heteroarilo bicíclico es un heteroarilo monocíclico condensado con un anillo de fenilo o un heteroarilo monocíclico, entonces el grupo de heteroarilo bicíclico está conectado al resto molecular parental mediante cualquier átomo de carbono o átomo de nitrógeno dentro del sistema de anillos bicíclicos. Los ejemplos representativos de heteroarilo bicíclico incluyen, pero no se limitan a, bencimidazolilo, benzofuranilo, benzotienilo, benzoxadiazolilo, benzoxatiadiazolilo, benzotiazolilo, cinnolinilo, 5,6-dihidroquinolin-2-ilo, 5,6-dihidroisoquinolin-1-ilo, furopiridinilo, indazolilo, indolilo, isoquinolinilo, naftiridinilo, quinolinilo, purinilo, 5,6,7,8-tetrahidroquinolin-2-ilo, 5,6,7,8-tetrahidroquinolin-3-ilo, 5,6,7,8-tetrahidroquinolin-4-ilo, 5,6,7,8-tetrahidroisoquinolin-1-ilo, tienopiridinilo, 4,5,6,7-tetrahidrobenzo[c][1,2,5]oxadiazolilo y 6,7-dihidrobenzo[c][1,2,5]oxadiazol-4(5H)-onilo. En ciertas realizaciones, el heteroarilo bicíclico condensado es un anillo de heteroarilo monocíclico de 5 o 6 miembros condensado con un anillo de fenilo, un cicloalquilo monocíclico de 5 o 6 miembros, un cicloalquenilo monocíclico de 5 o 6 miembros, un heterociclilo monocíclico de 5 o 6 miembros o un heteroarilo monocíclico de 5 o 6 miembros, en donde los grupos cicloalquilo, cicloalquenilo y heterociclilo condensados se sustituyen opcionalmente con uno o dos grupos que son independientemente oxo o tia.
El término "heteroarilalquilo" y "alquilheteroarilo", como se usa en el presente documento, significa un heteroarilo, como se define en el presente documento, unido al resto molecular parental mediante un grupo alquilo, como se define en el presente documento. Los ejemplos representativos de heteroarilalquilo incluyen, pero no se limitan a, fur-3-ilmetilo, 1H-imidazol-2-ilmetilo, 1H-imidazol-4-ilmetilo, 1-(piridin-4-il)etilo, piridin-3-ilmetilo, piridin-4-ilmetilo, pirimidin-5-ilmetilo, 2-(pirimidin-2-il)propilo, tien-2-ilmetilo y tien-3-ilmetilo.
Los términos "heterociclilo" o "heterocicloalquilo", como se usan en el presente documento, significan un heterociclo monocíclico o un heterociclo bicíclico. El heterociclo monocíclico es un anillo de 3, 4, 5, 6 o 7 miembros que contiene al menos un heteroátomo independientemente seleccionado del grupo que consiste en O, N y S, donde el anillo está saturado o insaturado, pero no es aromático. El anillo de 3 o 4 miembros contiene 1 heteroátomo seleccionado del
grupo que consiste en O, N y S. El anillo de 5 miembros puede contener cero o un doble enlace y uno, dos o tres heteroátomos seleccionados del grupo que consiste en O, N y S. El anillo de 6 o 7 miembros contiene cero, uno o dos dobles enlaces y uno, dos o tres heteroátomos seleccionados del grupo que consiste en O, N y S. El heterociclo monocíclico está conectado al resto molecular parental mediante cualquier átomo de carbono o cualquier átomo de nitrógeno contenido dentro del heterociclo monocíclico. Los ejemplos representativos de heterociclo monocíclico incluyen, pero no se limitan a, azetidinilo, azepanilo, aziridinilo, diazepanilo, 1,3-dioxanilo, 1,3-dioxolanilo, 1,3-ditiolanilo, 1,3-ditianilo, imidazolinilo, imidazolidinilo, isotiazolinilo, isotiazolidinilo, isoxazolinilo, isoxazolidinilo, morfolinilo, oxadiazolinilo, oxadiazolidinilo, oxazolinilo, oxazolidinilo, piperazinilo, piperidinilo, piranilo, pirazolinilo, pirazolidinilo, pirrolinilo, pirrolidinilo, tetrahidrofuranilo, tetrahidrotienilo, tiadiazolinilo, tiadiazolidinilo, tiazolinilo, tiazolidinilo, tiomorfolinilo, 1,1 -dioxidotiomorfolinilo (tiomorfolina sulfona), tiopiranilo y tritianilo. El heterociclo bicíclico es un heterociclo monocíclico condensado con un fenilo, un cicloalquilo monocíclico, un cicloalquenilo monocíclico, un heterociclo monocíclico o un heteroarilo monocíclico. El heterociclo bicíclico está conectado al resto molecular parental mediante cualquier átomo de carbono o cualquier átomo de nitrógeno contenido dentro de la porción de heterociclo monocíclico del sistema de anillos bicíclicos. Los ejemplos representativos de heterociclilos bicíclicos incluyen, pero no se limitan a, 2,3-dihidrobenzofuran-2-ilo, 2,3-dihidrobenzofuran-3-ilo, indolin-1-ilo, indolin-2-ilo, indolin-3-ilo, 2,3-dihidrobenzotien-2-ilo, decahidroquinolinilo, decahidroisoquinolinilo, octahidro-1H-indolilo y octahidrobenzofuranilo. Los grupos heterociclilo se sustituyen opcionalmente con uno o dos grupos que son independientemente oxo o tia. En ciertas realizaciones, el heterociclilo bicíclico es un anillo de heterociclilo monocíclico de 5 o 6 miembros condensado con el anillo de fenilo, un cicloalquilo monocíclico de 5 o 6 miembros, un cicloalquenilo monocíclico de 5 o 6 miembros, un heterociclilo monocíclico de 5 o 6 miembros, o un heteroarilo monocíclico de 5 o 6 miembros, en donde el heterociclilo bicíclico se sustituye opcionalmente con uno o dos grupos que son independientemente oxo o tia.
El término "hidroxi", como se usa en el presente documento, significa un grupo -OH.
El término "nitro", como se usa en el presente documento, significa un grupo -NO2.
El término "oxo", como se usa en el presente documento significa un grupo =O.
El término "tia", como se usa en el presente documento significa un grupo -S-.
El término "saturado", como se usa en el presente documento, significa que la estructura química referenciada no contiene múltiples enlaces carbono-carbono. Por ejemplo, un grupo cicloalquilo saturado como se define en el presente documento incluye ciclohexilo, ciclopropilo y similares.
El término "insaturado", como se usa en el presente documento, significa que la estructura química referenciada contiene al menos múltiples enlaces carbono-carbono, pero no es aromático. Por ejemplo, un grupo cicloalquilo insaturado, como se define en el presente documento, incluye ciclohexenilo, ciclopentenilo, ciclohexadienilo y similares.
Como se usa en el presente documento, el término "individuo" o "paciente", usados indistintamente, se refiere a cualquier animal, que incluye mamíferos, preferentemente ratones, ratas, otros roedores, conejos, perros, gatos, cerdos, ganado vacuno, ovejas, caballos o primates, y lo más preferentemente seres humanos.
Como se usa en el presente documento, la expresión "cantidad terapéuticamente eficaz" se refiere a la cantidad de compuesto activo o agente farmacéutico que provoca la respuesta biológica o medicinal que está siendo buscada en un tejido, sistema, animal, individuo o humano por un investigador, veterinario, médico u otro profesional clínico.
En ciertas realizaciones, una cantidad terapéuticamente eficaz puede ser una cantidad adecuada para
(1) prevenir la enfermedad; por ejemplo, prevenir una enfermedad, afección o trastorno en un individuo que puede tener predisposición a la enfermedad, afección o trastorno, pero que todavía no padece o muestra la patología o sintomatología de la enfermedad;
(2) inhibir la enfermedad; por ejemplo, inhibir una enfermedad, afección o trastorno en un individuo que padece o presenta la patología o sintomatología de la enfermedad, afección o trastorno; o
(3) mejorar la enfermedad; por ejemplo, mejorar una enfermedad, afección o trastorno en un individuo que padece o presenta la patología o sintomatología de la enfermedad, afección o trastorno (es decir, revertir la patología y/o sintomatología), tal como disminuir la intensidad de la enfermedad.
Como se usa aquí, los términos "tratamiento" y "tratar" significan (i) mejorar el estado de enfermedad referenciado, por ejemplo, mejorar una enfermedad, afección o trastorno en un individuo que padece o presenta la patología o sintomatología de la enfermedad, afección o trastorno (es decir, revertir o mejorar la patología y/o sintomatología), tal como disminuir la intensidad de la enfermedad; o (ii) provocar el efecto biológico referenciado (por ejemplo, modulación de IDO o inhibición de la degradación de triptófano).
La manifestación de la mejora de una patología con inmunosupresión mediada por IDO subyacente puede requerir la administración concomitante o secuencial de agentes terapéuticos adicionales, tales como agentes antineoplásicos
en el caso de cáncer, o agentes antirretrovíricos en el caso de enfermedades víricas. Por ejemplo, la administración de inhibidores de IDO para el tratamiento del cáncer no siempre produce un efecto antitumoral directo cuando se usa como un único agente. Sin embargo, cuando se combina con fármacos quimioterapéuticos (antineoplásicos), el efecto antitumoral observado es superior a la suma de efectos de cada agente solo.
Como se usa en el presente documento, los términos "bolsillo catalítico", "sitio catalítico", "sitio activo" se refieren conjuntamente e indistintamente a una región de la enzima que contiene restos de aminoácidos responsables de la unión al sustrato (carga, hidrofobia, impedimento estérico) y restos de aminoácidos catalíticos que actúan de donantes o aceptores de protones o son responsables de la unión de un cofactor y participan en la catálisis de una reacción química.
Como se usa en el presente documento, la expresión "sal farmacéuticamente aceptable" se refiere tanto a sales de adición de ácido como de base farmacéuticamente aceptables y a solvatos. Dichas sales farmacéuticamente aceptables incluyen sales de ácidos, tales como clorhídrico, fosfórico, bromhídrico, sulfúrico, sulfínico, fórmico, toluenosulfónico, metanosulfónico, nítrico, benzoico, cítrico, tartárico, maleico, yodhídrico, alcanoico, tal como acético, HOOC-(CH2)n-COOH donde n es 0-4, y similares. Las sales de adición de base farmacéuticas no tóxicas incluyen sales de bases, tales como sodio, potasio, calcio, amonio y similares. Los expertos en la técnica reconocerán una amplia variedad de sales de adición farmacéuticamente aceptables no tóxicas.
Como se usa en el presente documento, el término "indoximod" se refiere a 1-metil-D-triptófano, también denominado D-1MT o D1mT.
Como se usa en el presente documento, el término "profármaco de indoximod" se refiere a cualquier sustancia que después de la administración in vivo sea metabolizada para producir indoximod como uno de los principales metabolitos.
EJEMPLOS
Ejemplo 1: Reactivos y métodos de síntesis
Todos los reactivos y disolventes se compraron de fuentes comerciales. Todos los reactivos y disolventes comerciales se usaron como se recibieron sin más purificación. Las reacciones se monitorizaron usando cromatografía en capa fina (CCF) analítica con placas de gel de sílice de 0,25 mm de EM Science (60F-254). Las placas de CCF desarrolladas se visualizaron por luz UV de onda corta (254 nm) o inmersión en disolución de permanganato de potasio, seguido por calentamiento en una placa caliente. La cromatografía ultrarrápida se realizó con el gel de sílice Selecto Scientific, tamaños de partículas 32-63 pm. Todas las reacciones se realizaron en material de vidrio secado a la llama o en estufa bajo una atmósfera de nitrógeno. Todas las reacciones se agitaron magnéticamente a temperatura ambiente, a menos que se indique lo contrario. Los espectros de RMN 1H se obtuvieron con un Bruker DRX400, Varian VXR400 o VXR300. Los espectros de RMN 1H fueron notificados en partes por millón (5) con respecto a TEM (0,0), DMSO-d6 (2,50) o CD3OD (4,80) como una referencia interna. Todos los espectros de RMN 1H se midieron en CDCh, a menos que se indique lo contrario.
Síntesis de clorhidrato de 1-metil-D-triptofanato de etilo (NLG-1283)

A una suspensión de D-1MT (4,00 g, 18,3 mmoles) en etanol (50 mL) a 0 °C se añadió SOCl2 (1,34 mL, 18,3 mmoles) y la mezcla se agitó a 80 °C durante la noche. Después del enfriamiento hasta ta, el disolvente se separó por destilación y el bruto se diluyó con dietil éter (100 mL), el sólido blanco se separó por filtración y se lavó con éter seco proporcionando el producto deseado (5,1 g, 98 %).
Síntesis de clorhidrato de 1-metil-D-triptofanato de isopropilo (NLG-1284)

A una suspensión de D-1MT (0,500 g, 2,29 mmoles) en isopropanol (15 mL) a 0 °C ta se añadió SOCI2 (0,167 mL, 2,29 mmoles) y la mezcla se agitó a 80 °C durante la noche. Después del enfriamiento hasta ta, el disolvente se separó por destilación y el bruto se basificó con 25 % de NaHCO3 ac. (20 mL), el producto se extrajo con CH2Cl2, el extracto orgánico combinado se secó sobre Na2SO4 y el disolvente se separó por destilación a presión reducida. La base libre se convirtió en su sal de HCl añadiendo HCl seco en dioxano, el disolvente se retiró a presión reducida proporcionando el producto deseado como un sólido blanco (0,252 g, 37%).
Método general para la síntesis de ésteres de carbamato (proporcionado para referencia)

A una disolución con agitación de D-1MT (0,150 g, 0,687 mmoles) en 1:1 de THF/1 M NaHCO3 (2,75 mL, 2,75 mmoles) se añadió el cloroformiato apropiado gota a gota. La mezcla se dejó con agitación durante 30 min y la disolución se diluyó con agua y se extrajo con éter 2x. La fase acuosa se enfrió hasta 0 °C y se añadió disolución conc. de HCl para ajustar el pH a ~1. La fase acuosa fría se extrajo inmediatamente con acetato de etilo y las fases orgánicas combinadas se lavaron con agua, salmuera y se secaron. El disolvente se retiró a presión reducida proporcionando el carbamato en bruto. El bruto se purificó por cromatografía en columna y se trató con carbón vegetal activado, proporcionando el carbamato puro.


Síntesis de Na-(ferc-butoxicarbonil)-1-metil-D-triptófano (proporcionado para referencia)

A una mezcla de D-1MT (3,0 g, 13,75 mmoles) en dioxano (70 mL) a 0 °C se añadió NaOH (550 mg disuelto en 30 mL de agua DI), seguido por la adición de Boc2O. La reacción se agitó a 0 °C durante 4 h y se agitó durante la noche a ta. La disolución se concentró a presión reducida hasta aproximadamente un tercio del volumen original. La reacción se acidificó con HCl 1 N a 0 °C y el producto se extrajo con EtOAc. El extracto orgánico se lavó con salmuera y se secó sobre Na2SO4, el disolvente se evaporó a presión reducida proporcionando el producto que se usó directamente en la siguiente etapa sin más purificación (4,3 g, 98 %).
Síntesis de Na-(íerc-butoxicarbonil)-1 -metil-D-triptofanato de bencilo (proporcionado para referencia)

En 60 mL de DMF se disolvió Na-(terc-butoxicarbonil)-1-metil-D-triptófano (3,00 g, 9,42 mmoles), al que se añadió Cs2CO3 (1,78 g, 5,47 mmoles) y bromuro de bencilo (1,61 mL, 9,42 mmoles). La suspensión resultante se dejó con agitación a temperatura ambiente durante 2 horas. Después del final de la reacción (CCF), la DMF se retiró a presión reducida, seguido por suspensión del residuo en tolueno/acetato de etilo antes de lavar con agua destilada (3 x 50 mL) y salmuera. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró a vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (3,5 g, 91 %).
Síntesis de clorhidrato de 1-metil-D-triptofanato de bencilo (NLG-1338)

Se enfriaron acetato de etilo (26,9 mL) y MeOH (8,9 mL) en un matraz redondo equipado con un tapón y una ventilación por aguja en un baño de hielo con agitación. Se añadió lentamente cloruro de acetilo (14,22 mL). La disolución resultante se agitó a 0 °C durante 20 minutos y se añadió MeOH (0,5 mL). Se puso en un baño de hielo un matraz que contenía Na-(terc-butoxicarbonil)-1-metil-D-triptofanato de bencilo (3,5 g, 8,6 mmoles) y el HCl recién preparado frío (4 M en EtOAc) se vertió lentamente en el matraz que contenía Na-(terc-butoxicarbonil)-1 -metil-D-triptofanato de bencilo. La disolución se agitó vigorosamente a 0 °C durante 15 min, donde se observó la formación de una suspensión blanca y el matraz se sacó del baño de hielo. La suspensión se dejó con agitación vigorosamente durante 2,5 h. La disolución se enfrió en un baño de hielo diluido con éter (50 mL) y la suspensión se filtró y la torta sólida se lavó con éter frío. El sólido se dejó secar a alto vacío y el producto deseado se aisló como un sólido incoloro (6,45 g, 88 %). 1H RMN (d6-dmdso); 3,28 (dd, 2H, J = 5,6, 15,2 Hz), 3,70 (s, 3H), 4,26-4,29 (m, 1H), 5,08 (d, 1H, J = 12,4 Hz), 5,13 (d, 1H, J = 12,4 Hz), 7,04 (t, 1 H, J = 7,6 Hz), 7,06 (s, 1H), 7,10-7,18 (m, 3H), 7,30-7,35 (m, 3H), 7,42 (d, 1H, J = 8 Hz), 7,53 (d, 1H, J = 8 Hz).
Esquema general para la derivatización del grupo -COOH de D-1MT

A una disolución de N-(terc-butoxicarbonil)-1-metil-D-triptófano (3,14 mmoles), alcohol o amina apropiado (3,14 mmoles) y HATU (3,14 mmoles) en acetonitrilo (30 mL) a 0 °C se añadió DIPEA (9,42 mmoles) y la disolución se dejó calentar hasta ta. Después de agitar durante la noche (17 h), la reacción se diluyó con agua (50 mL) y el producto se extrajo con CH2G 2 (3 x 50 mL). El extracto orgánico combinado se lavó con agua (25 mL x 1), salmuera (25 mL x 1) se secó sobre Na2SO4 y se concentró a presión reducida proporcionando el bruto. La purificación cromatográfica dio el producto deseado. Los compuestos con números marcados con * en la tabla que sigue se proporcionan para referencia.



Síntesis de Na-(ferc-butoxicarbonil)-1-metil-D-triptofilglicina (NLG-1579-A-E44) (proporcionado para referencia)

A una disolución de NLG-1578-A-E43 (300 mg, 0,770 mmoles) en THF (10 mL) se añadió agua (2 mL) y monohidrato de litio (49 mg, 1,16 mmoles) y la mezcla se agitó a temperatura ambiente durante 2,0 h. La mezcla se neutralizó con HCl 1 M (a 0 °C) y se vertió en agua helada (20 mL). La fase acuosa se extrajo con EtOAc (3 x 35 mL). Las fases orgánicas combinadas se secaron sobre Na2SO4 y se concentraron. El producto en bruto se purificó por cromatografía ultrarrápida en columna proporcionando el producto deseado como un sólido blanco (260 mg, 90 %). 1H RMN: 1,25 y
1,39 (dos s, 9H). 3,18-3,24 (m, 2H), 3,70 (s, 3H), 3,81 -4,05 (m, 2H), 4,55 (s, 1H), 5,20 - 5,33 (m, 1H), 6,63 (s, 1H), 6,92 (s, 1H), 7,10 (t, 1 H, J = 7,2 Hz), 7,15 - 7,25 (m, 2H), 7,59 (dt, 1H, J = 7,9 Hz)

A una mezcla de amina protegida con t-Boc (1,57 mmoles) en dioxano (15 mL) a ta se añadió HCl (4 mL, disolución 4,0 M en dioxano). Después de agitar durante 2,5 h, el disolvente se separó por destilación a presión reducida. El residuo se agitó con metil terc-butil éter (10 mL), el sólido se filtró y se secó a presión reducida proporcionando el producto deseado.
Los siguientes compuestos se sintetizaron siguiendo los procedimientos descritos en las secciones anteriores. Los números de compuesto marcados con * se proporcionan para referencia.


Síntesis de diclorhidrato de O-(1-metil-D-triptofil)-L-serina (NL-G1551)

A una disolución de NLG-1551-B.1-E15 (0,450 g, 824,66 mmoles) en CH2G 2 (10 mL) se añadió HCl (2 mL, disolución 4 M en dioxano) a 0 °C y la disolución se dejó calentar hasta ta. Después de agitar durante 5 h, el disolvente se evaporó y la reacción se diluyó con ácido trifluoroacético (8 mL) y la disolución se agitó durante 7 h a ta. Después de evaporar el ácido trifluoroacético, la reacción se diluyó con disolución de HCl seco (1 mL, disolución 4 M en dioxano) y la mezcla se agitó durante 10 min. El disolvente se evaporó a presión reducida, el producto se trituró con etanol:éter (10:90, 15 mL) y el producto se filtró y se lavó con éter seco (10 mL). El producto se secó a presión reducida (0,190 g, 61 %).
1H RMN (400 MHz, CD3OD): 3,22-3,28 (m, 1H), 3,43 (dd, 1H, J = 15,4, 4,7 Hz), 3,70 (s, 3H), 4,23 (t, 1H, J = 3,9 Hz), 4,35 (dd, 1 H, J = 8,0, 4,9 Hz), 4,60 (d, 2H, J = 3,8 Hz), 6,99-7,04 (m, 1 H), 7,05 (s, 1H), 7,09-7,16 (m, 1 H), 7,29 (d, 1H, J = 8,3 Hz), 7,50 (d, 1H, J = 7,9 Hz).
Síntesis de clorhidrato de 1-metil-D-triptofil-L-valina (NLG-1556) (proporcionado para referencia)

Se enfrió dioxano (7 mL) y MeOH (1,20 mL, 28,6 mmoles) en un matraz redondo equipado con un tapón y una ventilación por aguja en un baño de hielo con agitación. Se añadió lentamente cloruro de acetilo (2,00 mL, 28,6 mmoles). La disolución resultante se agitó a 0 °C durante 20 minutos y se añadió MeOH (0,1 mL). Se dispuso un matraz que contenía NLG-1556-A-E22 (678 mg, 1,43 mmoles) en un baño de hielo y el HCl recién preparado frío (4 M en dioxano) se vertió lentamente en el matraz que contenía NLG-1556-A-E22. La disolución se dejó calentar hasta ta y se agitó vigorosamente durante 18 h. El disolvente se retiró usando un evaporador rotatorio proporcionando el sólido blanco puro (205 mg, 40 %). (DMSO-d6) 0,71 -0,77 (m, 6 H), 1,91 -2,00 (m, 1H), 3,08 (dd, 1H, J = 14,4, 8,4 Hz), 3,23 (dd, 1H, J = 14,4, 8,4 Hz), 3,73 (s, 3H), 4,12-4,17 (m, 2H), 7,06 (t, 1H, J = 7,4 Hz), 7,17 (t, 1H, J = 7,8 Hz), 7,20 (s, 1 H), 7,40 (d, 1 H, J = 8,4 Hz), 7,74 (d, 1H, J = 8,0 Hz), 8,2 (s a, 3H), 8,74 (d, 1H, J = 8,4 Hz)
Síntesis de clorhidrato de 1-metil-D-triptofanato de 2,3-dihidroxipropilo (NLG-1558)

A una disolución de NLG1558-A-E23 (11,5 g, 26,59 mmoles) en THF (100 mL) a 0 °C se añadió TFA (16,3 mL, 212,7 mmoles) y agua (0,958 g, 53,18 mmoles) y el baño de refrigeración se retiró, la mezcla se agitó a ta durante 2 h. Se añadió HCl (13,3 mL, 53,18 mmoles; disolución 4,0 M en dioxano) y la agitación continuó durante 1 h. La reacción se agitó a 40 °C durante 45 minutos. El sólido blanco precipitado se filtró y se lavó con MTBE proporcionando la sal de clorhidrato (4,5 g, 51 %). 1H RMN (400 MHz, DMSO-afe): 3,32-3,40 (m, 1H), 3,44-3,52 (m, 3H), 3,76-3,86 (m, 4H), 4,16-4,37 (m, 3H), 7,10 (t, 1H, J = 7,4 Hz), 7,14 (s, 1H), 7,19 (t, 1H, J = 7,6 Hz), 7,38 (d, 1H, J = 8,2 Hz), 7,58 (d, 1H, J = 7,9 Hz).
Esquema general para la derivatización del grupo -NH2 y -COOH de D-1MT

Se agitaron éster de clorhidrato de D-triptofanato apropiado (1,0 g, 3,54 mmoles) y ácido apropiado (3,54 mmoles) en acetonitrilo (50 mL) a 0 °C. Se añadieron HATU (1,48 g, 3,89 mmoles) e iPr2NEt (2,46 mL, 14,15 mmoles) y la reacción se agitó durante la noche a temperatura ambiente. El disolvente se retiró a presión reducida y el bruto se diluyó con agua (50 mL) y diclorometano (50 mL). Se separó la fase orgánica y la fase acuosa se extrajo con diclorometano (3 x 50 mL). La fase orgánica combinada se lavó con salmuera (50 mL), se secó sobre Na2SO4 y se concentró a presión reducida. El producto en bruto se purificó por cromatografía ultrarrápida en columna proporcionando el producto deseado. Los números de compuesto marcados con * se proporcionan para referencia.




Síntesis de Na-((S)-5-(terc-butoxi)-2-((terc-butoxicarbonil)amino)-5-oxopentanoil)-1-metil-D-triptófano (NLG-1547-E.2-E17) (proporcionado para referencia)

Se suspendió (S)-5-(((R)-1 -(benciloxi)-3-(1 -metil-1 H-indol-3-il)-1 -oxopropan-2-il)amino)-4-((tercbutoxicarbonil)amino)-5-oxopentanoato de terc-butilo (800 mg, 1,38 mmoles) en MeOH (8 mL) y THF (8 mL). Después del enfriamiento hasta 0 °C, se añadió disolución de NaOH (2,4 mL, 2 M) y la reacción se agitó durante 1 h. La disolución se acidificó con HCl 1 M hasta pH = 4 y los disolventes se concentraron a presión reducida (40 °C). La disolución se repartió entre agua y DCM en un embudo de decantación y se recogió la fase orgánica. La fase acuosa se extrajo con DCM (2 x 15 mL) y la fase orgánica combinada se lavó con agua y salmuera. La purificación cromatográfica proporcionó el producto deseado (0,502 g, 72 %). 1H RMN(Cloroformo-d, 400 MHz): 5 = 1,38 (s, 9H), 1,44 (s, 9H), 1,68 - 1,81 (m, 1H), 1,84 - 1,99 (m, 1 H), 2,12-2,33 (m, 3H), 3,23 - 3,42 (m, 2H), 4,23 (s, 3H), 4,86 (d, 1H, J = 6,9 Hz), 5,41 (d, 1 H, J = 8,6 Hz), 6,83 (d, 1H, J = 7,5 Hz), 6,93 (s, 1H), 7,09 (dt, 1H, J = 8,0, 1,2 Hz), 7,18 (t, 1 H, J = 7,8 Hz), 7,23 (d aparente solapado con CdCI3, 1H,), 7,60 (d, 1H, J = 7,9 Hz).
Síntesis de clorhidrato de ácido (S)-4-amino-5-(((R)-1-carboxi-2-(1-metil-1H-indol-3-il)etil)amino)-5-oxopentanoico (NLG-1547)

A Na-((S)-5-(terc-butoxi)-2-((terc-butoxicarbonil)amino)-5-oxopentanoil)-1-metil-D-triptófano (470 mg, 0,93 mmoles) se añadió HCl (4 M en dioxano) (4,7 mL). La disolución resultante se dejó con agitación a temperatura ambiente durante 5 horas. La disolución se concentró y el sólido se disolvió en MeOH y se trató con carbón vegetal activado y se calentó hasta 60 °C durante 1 h. La disolución se filtró a través de Celite y el filtrado se concentró proporcionando el producto deseado como un sólido beis (0,304, 85 %). 1H RMN (DMSO-afe, 400 MHz): (mezcla de rotámeros) 1,73 - 2,21 (m, 4H), 2,93 - 3,12 (m, 1H), 3,14 - 3,27 (m, 1H), 3,70 (s, 3H), 3,83 (c, 1H, J = 5,8 Hz), 4,53 - 4,72 (m, 1H), 7,01 (tt, 1H, J = 7,3, 3,7 Hz), 7,07 - 7,19 (m, 2H), 7,35 (dt, 1H, J = 7,5, 3,5 Hz), 7,44 - 7,61 (m, 1 H), 8,42 (s a, 3H), 8,83 - 9,10 (m, 1H).
Método general para la hidrólisis de ásteres etílicos de D-1MT sustituidos

A una disolución de amida apropiada (0,991 mmoles) en THF (10 mL) se añadió agua (3 mL) y monohidrato de litio (67 mg, 1,59 mmoles) y la mezcla se agitó a temperatura ambiente durante 2 h. La mezcla se neutralizó con HCl 1 M (a 0 °C) y se vertió en agua helada (20 mL). La fase acuosa se extrajo con EtOAc (3 x 35 mL). Las fases orgánicas combinadas se secaron sobre Na2SO4 y se concentraron. El producto en bruto se purificó por cromatografía ultrarrápida en columna proporcionando el producto deseado. Los números de compuesto marcados con * se proporcionan para referencia.




Método general para la desprotección de tBoc.
A una disolución de amina protegida con ’Boc apropiada (0,707 mmoles) en dioxano (2 mL) se añadió disolución de HCl (1,77 mL, disolución 4,0 M en dioxano) a 0 °C. La disolución se dejó calentar hasta ta y se agitó vigorosamente durante 2,5-18 h. El disolvente se retiró usando un evaporador rotatorio. El sólido se diluyó con éter seco (15 mL) y el producto se filtró proporcionando el producto en bruto. El bruto se secó a alto vacío proporcionando el producto deseado.





Síntesis de clorhidrato de 1-metil-D-triptofanato de (2-etoxi-2-oxido-1,3,2-dioxafosfolan-4-il)metilo (NLG-1559)

Na-(terc-butoxicarbonil)-1-metil-D-triptofanato de 2,3-dihidroxipropilo (NLG-1559-A-E24) (proporcionado para referencia)
A una disolución de base libre de NLG-1558 (0,750 mg, 2,57 mmoles) en acetonitrilo (10 mL) a 0 °C se añadió B0C2O (560 mg, 2,57 mmoles) y la reacción se dejó calentar hasta ta y se agitó durante 4 h. El disolvente se retiró a presión reducida y el bruto se purificó por cromatografía en columna proporcionando el producto deseado (760 mg, 75 %). 1H RMN: 1,34 (s, 9H), 3,13-3,23 (m, 2H, 3,35-3,38 (m, 1H), 3,42-3,45 (m, 1 H), 3,67-3,72 (m, 4H), 4,01 -4,08 (m, 2H), 5,01 -5,04 (m, 1 H), 6,83 (s, 1H), 7,05 (t, 1H, J = 7,4 Hz), 7,16 (t, 1H, J = 7,3 Hz), 7,23 (d, 1H, J = 8,2 Hz), 7,49 (d, 1H, J = 7,9 Hz).
Na-(terc-butoxicarbonil)-1-metil-D-triptofanato de (2-etoxi-2-oxido-1,3,2-dioxafosfolan-4-il)metilo (NLG-1559-B-E24) (proporcionado para referencia)
A una disolución de NLG-1559-A-E24 (650 mg, 1,66 mmoles) en piridina seca (2 mL) a 0 °C se añadió POCI3 y la disolución se dejó calentar hasta ta. Después de agitar durante la noche (18 h), se añadió etanol (1,5 mL) y la reacción continuó durante 4 h. El disolvente se retiró a presión reducida y el bruto se purificó por cromatografía en columna (460 mg, 57 %). 1H RMN: 1,13 (t, 3H, J = 7,0 Hz), 1,30 (s, 9H), 3,10-3,20 (m, 2H), 3,47-3,55 (m, 1H), 3,60 (s, 3H), 41,9-4,44 (m, 3H), 4,55-4,57 (m, 1 H), 5,23-5,27 (m, 1H), 6,79 y 6,83 (dos s, 1H), 7,01 (t, 1H, J = 7,4 Hz), 7,12 (t, 1H, J = 7,2 Hz), 7,18 (d, 1H, J = 9,2 Hz), 7,46 (d, 1 H, J = 7,7 Hz).
Clorhidrato de 1-metil-D-triptofanato de (2-etoxi-2-oxido-1,3,2-dioxafosfolan-4-il)metilo (NLG-1559)
A una disolución de NLG-1559-B-E24 (550 mg, 1,14 mmoles) en CH2G 2 seca (10 mL) a 0 °C se añadió HCl anhidro (1,4 mL, disolución 4 M en dioxano) y la mezcla se dejó calentar hasta ta. Después de agitar durante 2 h, el disolvente se retiró a presión reducida y el bruto se lavó con éter seco (3 x 15 mL). El sólido blanco se filtró y el producto se secó a presión reducida (0,241 g, 61 %). (CD3OD-d4) 1,20 (td, 3H, J = 7,1,4,3 Hz), 3,26-3,42 (m, 2H), 3,44 (dd, 1H, J = 5,1, 3,0 Hz), 3,48-3,56 (m, 1H), 3,71 (s, 3H), 3,95 (h, 2H, J = 7,1 Hz), 4,21 -4,36 (m, 3H), 4,37-4,53 (m, 1H), 7,02 (t, 1H, J = 7,4 Hz), 7,07 (d, 1H, J = 4,0 Hz), 7,10-7,17 (m, 1H), 7,30 (d, 1H, J = 8,2 Hz), 7,49 (d, 1H, J = 7,4 Hz).
Composición (composiciones) de sal farmacéuticamente aceptable (proporcionadas para referencia)
Síntesis de cloruro de (R)-1-carboxi-2-(1-metil-1H-indol-3-il)etan-1-aminio (NLG-1607)

A una disolución acuosa helada de HCl (15,5 mL, 30,9 mmoles; 2 M) se añadió D1MT (4,5 g, 20,6 mmoles). Después de agitar durante 30 minutos, la disolución transparente se evaporó a presión reducida y el bruto se evaporó tres veces con etanol (40 mL). El bruto se agitó en etanol y terc-butil metil éter y se filtró proporcionando el producto deseado (4,25 g, 81 %).
Se desarrolló un método alternativo donde se suspendieron ~ 10 g de D-1 MT en una botella de vidrio de 250 mL con 100 mL de acetonitrilo. Se añadió 10 mL de disolución de HCl previamente disuelta en acetonitrilo (511,2 mg/mL) en la disolución de forma libre de D-1 MT según una relación molar 1:1 entre base libre:ácido, y luego se mantuvo agitando a temperatura ambiente durante la noche para formar la sal. Se secó a vacío el sólido filtrado a 30 °C durante la noche. Se obtuvo un polvo blanco (11,1 g) por el proceso anterior, y se caracterizó por XRPD, DSC y TGA (Figuras 1 -2). La pureza fue del 99,7 % de área basada en el análisis de HPLC, y la estequiometría se analizó por ELSD, la relación molar calculada (API:ácido HCl) fue 1:1,0. El polvo fue cristalino como se evaluó por microscopía óptica polarizada (PLM) y por espectrometría de dispersión de rayos X de polvo (XRPD, Figura 1). La sal fue anhidra como se evaluó por análisis termogravimétrico (TGA) y calorimetría diferencial de barrido (DSC) (Figura 2).
Síntesis de metanosulfonato de (R)-1-carboxi-2-(1-metil-1H-indol-3-il)etan-1-aminio (NLG-1619)

A una disolución con agitación de ácido metanosulfónico (1,50 mL, 22,9 mmoles) en agua DI (50 mL) se añadió D-1MT (1,0 g, 4,48 mmoles) en porciones de 100 mg. La disolución se agitó vigorosamente durante 3 h a 75 °C hasta que la disolución fue homogénea. La disolución se concentró a presión reducida y se recogió el sólido (1,38 g, 96 %).
1H RMN(Metanol-o4, 400 MHz): 5 = 2,69 (s, 3H), 3,32 - 3,39 (m, 1 H), 3,49 (dd, 1H, J = 15,3, 4,9 Hz), 3,80 (s, 3H), 4,25 (dd, 1H, J = 7,8, 4,9 Hz), 7,10 (ddd, 1H, J = 8,0, 7,0, 1,0 Hz), 7,14 (s, 1H), 7,21 (ddd, 1H, J = 8,2, 7,0, 1,1 Hz), 7,38 (dd, 1H, J=8,3, 1,1 Hz), 7,62 (dt, 1H, J = 8,0, 0,9 Hz)
Síntesis de dihidrogenofosfato de (R)-1-carboxi-2-(1-metil-1H-indol-3-il)etan-1-aminio (NLG-1660)

A la disolución de ácido fosfórico (0,673 g, 6,87 mmoles) en agua desionizada (30 mL) a 50 °C se añadió D-1MT (0,5 g, 2,29) en porciones y la mezcla se agitó a 50 °C durante la noche. Entonces se concentró la disolución hasta la mitad de su volumen original y se dejó que reposara a temperatura ambiente durante la noche. El precipitado resultante se filtró, se lavó con etanol frío y se secó dando NLG-1660 como un sólido blanco (0,250, 34 %). 1H RMN (400 MHz, DMSO-ofe) ó 2,95 (dd, 1H, J = 15,1,8,6 Hz), 3,22 - 3,29 (m, 1H), 3,46 (dd, 1H, J = 8,6, 4,2 Hz), 3,71 (s, 3H), 7,00 (ddd, 1H, J = 8,0, 7,1, 1,0 Hz), 7,09-7,15 (m, 2H), 7,37 (d, 1 H, J = 8,4 Hz), 7,55 (d, 1H, J = 7,9 Hz).
Se desarrolló un método alternativo donde ~ 10 g de D-1 MT se suspendieron en una botella de vidrio de 500 mL con 100 mL de THF. Se añadieron 20 mL de disolución de H3PO4 previamente disuelta en THF (792,3 mg/mL) en la disolución de forma libre de D-1 MT según la relación molar 1:3 entre base libre:ácido, y luego se mantuvo la agitación a temperatura ambiente durante la noche para formar la sal. Se secó a vacío el sólido filtrado a 30 °C durante la noche, se comprobó por XRPD, DSC, TGA y ELSD. Se obtuvo un polvo blanco (11,1 g), que mostró que era cristalino por patrón de PLM y XRPD (Figura 3). La sal fue anhidra basándose en datos de d Sc y TGA (Figura 4). La pureza fue del 99,8 % y la estequiometría se analizó por ELSD, la relación molar calculada (base libre:ácido fosfórico) fue 1:0,57.
Síntesis de hidrogenosulfato de (R)-1-carboxi-2-(1-metil-1H-indol-3-il)etan-1-aminio (NLG-1667)

A una suspensión de D-1 MT (1,00 g, 4,58 mmoles) en agua/THF (4:1, 100 mL) a ta se añadió H2SO40,5 M (9,16 mL, 4,58 mmoles) y la mezcla se agitó a ta durante la noche. El sólido blanco se separó por filtración y se lavó con THF frío proporcionando la sal de sulfato de D-1MT (0,429 g, 34 %). (DMSO-d6) 3,17 (dd, 1H, J = 15,1,7,2 Hz), 3,27 (dd, 1H, J = 15,0, 5,3 Hz), 3,74 (s, 3H), 3,96 (t, 1H, J = 6,2 Hz), 7,04 (t, 1H, J = 7,4 Hz), 7,12-7,21 (m, 2H), 7,41 (d, 1H, J = 8,2 Hz), 7,58 (d, 1H, J = 8,0 Hz), 8,52 (s a, 4H).
Método general para la generación de sales de mono- y difosfato de profármacos de indoximod.
A una disolución de base libre (0,747 mmoles) en EtOH (5 mL) a 0 °C se añadió ácido fosfórico (0,747 mmoles; una disolución en 1 mL de EtOH) o (1,494 mmoles en caso de diamina) y la mezcla se dejó calentar hasta ta y se agitó durante 5-18 h. El disolvente se retiró a presión reducida y el residuo se diluyó con metil terc-butil éter (10 mL), después de agitar durante 1-5 h el sólido se filtró y se secó a presión reducida proporcionando el producto deseado. Para NLG-03380-02, la base libre se generó a partir de NLG-03380-01 usando resina de intercambio iónico.



Método general para la generación de sales de mono- y di-metanosulfonato y bencenosulfonato de profármacos de indoximod.
A una disolución de base libre (0,25 g, 0,723 mmoles) en etanol (10 mL) a ta se añadió ácido metanosulfónico o bencenosulfónico (0,723 mmoles o 1,446 mmoles en caso de diaminas) y la mezcla se agitó a ta durante la noche. Se evaporó el etanol y el producto en bruto se agitó en metil terc-butil éter durante 1-5 h. El precipitado se filtró y se secó dando la sal de metanosulfonato o bencenosulfonato correspondiente.


Método general para la generación de sales de mono-, di-sulfato e hidrogenosulfato de indoximod y profármacos de indoximod.
A una disolución de base libre (1,22 mmoles) en THF seco (10 mL) a 0 °C se añadió ácido sulfúrico (0,611 mmoles o 1,22 mmoles) como una disolución en THF (2 mL) y la disolución se dejó calentar hasta ta. Después de agitar durante 2-6 h, el disolvente se separó por destilación y el bruto se agitó con metil terc-butil éter, el sólido se filtró y se secó a vacío dando el producto deseado.
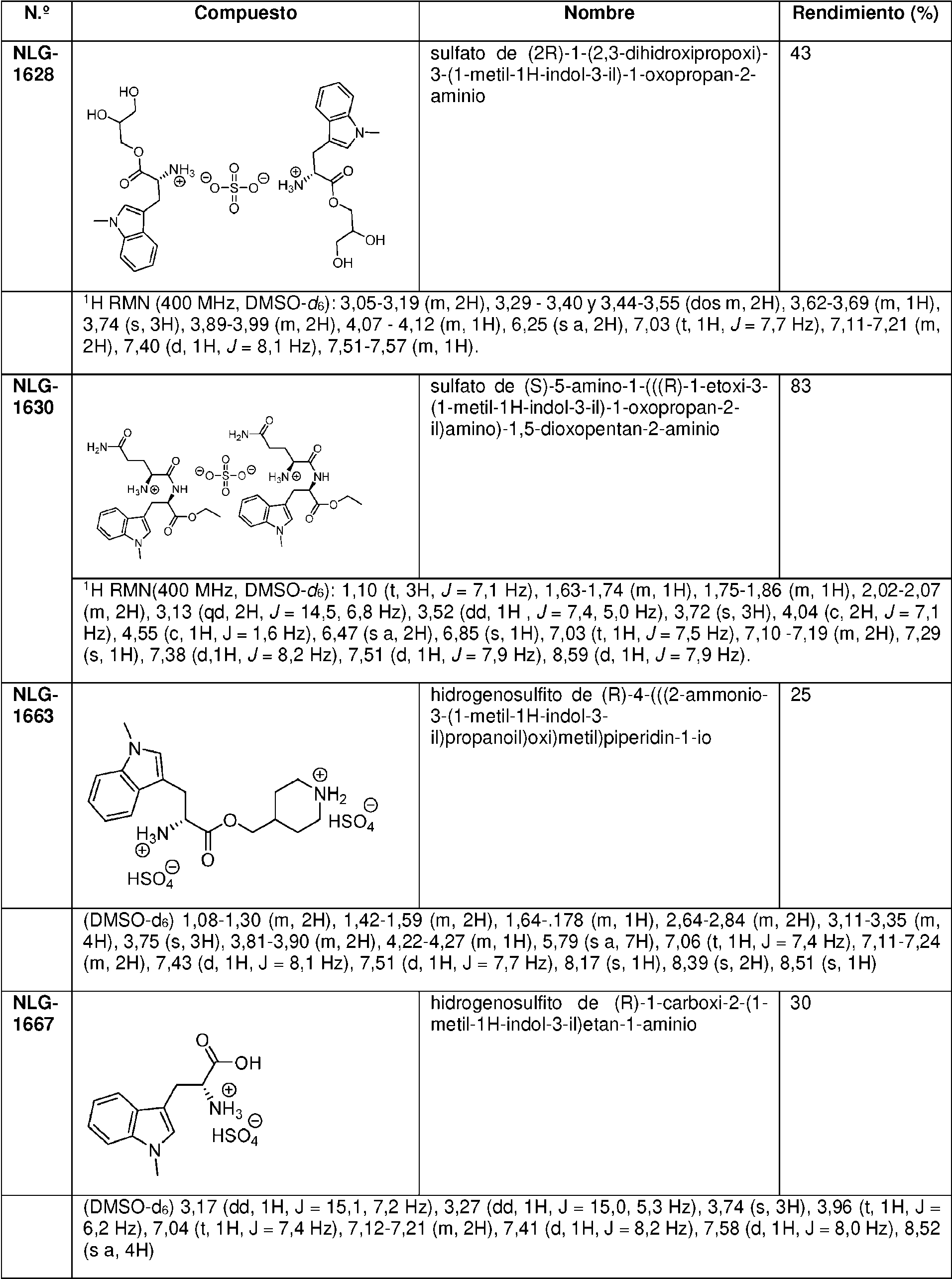
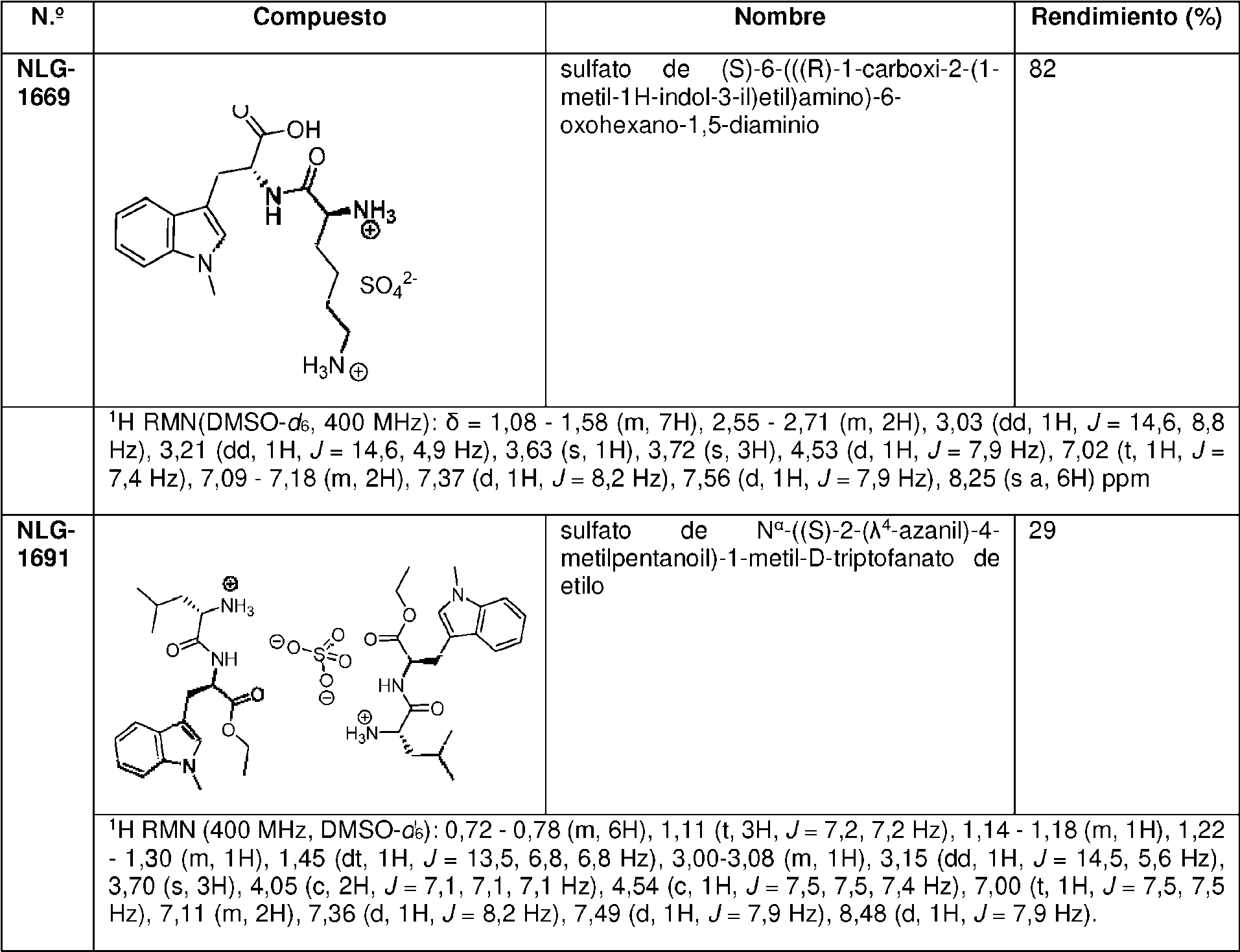

Síntesis de 2-(((2-(1HLimidazol-4-il)fenoxi)carbonil)amino)-3-(1-metil-1H-indol-3-il)propanoato de (fl)-metilo (NLG-1264)
A una disolución de 2-(1H-imidazol-4-il)fenol (1,0 mmol) (preparada según J. Med. Chem., 2008, 51 (16), pp 4968 4977) en DMF (3 mL) se añadió trietilamina (1,1 mmoles). Después de agitar durante 10 min, se añadió gota a gota una disolución de cloruro de 4,4'-dimetoxitritilo (1,0 mmol) en DMF (2 mL). Después de agitar durante la noche bajo una atmósfera de nitrógeno, la mezcla de reacción se vertió en agua con hielo (10 mL). El sólido se separó por filtración, se lavó con agua fría y se disolvió en acetato de etilo. La fase orgánica se secó sobre Na2SO4 y el producto en bruto concentrado se llevó a la siguiente etapa sin más purificación. A una suspensión de 2-amino-3-(1-metil-1H-indol-3-il)propanoato de (R)-metilo (0,5 mmoles) (preparada como se describe por Paul Cox, Donald Craig, Stephanos Ioannidis, Volker S. Rahn, Tetrahedron Letters 2005, 46, 4687) en DCM (3 mL) se añadió trifosgeno (0,5 mmoles) y Et3N (2,0 mmoles) a 0 °C. La disolución se dejó con agitación durante 1 h y se concentró a sequedad. El residuo en bruto se usó inmediatamente en la siguiente etapa sin purificación. El residuo en bruto se disolvió en DCM (5 mL), se añadieron el derivado de fenilimidazol (0,5 mmoles) y DMAP (1,5 mmoles). La disolución resultante se dejó con agitación a ta durante la noche. El disolvente se retiró a presión reducida y el residuo en bruto se filtró a través de un tapón de gel de sílice y se concentró. Al residuo se añadió MeOH (3 mL) y AcOH (2 mL) y la disolución se agitó a ta durante 30 min. La disolución se diluyó con agua y se basificó con K2CO3 sólido (pH ~ 8-9). La acuosa se extrajo con EtOAc y las fases orgánicas combinadas se lavaron con agua, salmuera y se secaron (Na2SO4). El residuo en bruto se purificó por cromatografía en columna sobre gel de sílice dando el compuesto (21 % de rendimiento). 1H RMN:
3,20-3,48 (m, 2H), 3,66 (s,3H), 3,70 (s,3H), 4,61-4,75 (m, 1H), 6,57 (d, 1H, J = 7,2 Hz), 6,90-7,30 (m, 7 H), 7,50-7,58 (m, 1H), 7,10-7,76 (m, 2H).
Ejemplo comparativo 2: Caracterización de la forma sólida de base libre de indoximod
La base libre de D-1MT (pureza por HPLC 99,6 %) es un polvo blanco y muestra birrefringencia, forma de aguja y aspecto cristalino bajo el microscopio óptico polarizado (PLM) y por espectroscopía de dispersión de rayos X de polvo (XRPD) (Figura 1). Solo muestra un único pico endotérmico de fusión con aparición a 293,8 °C por análisis termogravimétrico (TGA) y calorimetría diferencial de barrido (DSC) y ~0,01 % de pérdida de peso desde 30-200 °C, que indica que es una forma de anhidrato. Esta forma cristalina no es higroscópica (0,09 % de aumento de peso desde 0-80 % de HR) y no muestra cambios después del método de sorción dinámica de vapor (DVS). Además, estudios de estabilidad de la forma sólida de polvo indican que D-1MT es químicamente estable en las condiciones probadas (25 °C/60 % de HR, 40°C, 40 °C/75 % de HR, 60 °C y 70 °C) durante 4 semanas. Además, también es estable en disolución en HCl 0,1 N, y tampones fosfato 50 mM a pH 2-8 a 25 °C durante 24 horas, mientras que muestra menor degradación (0,45 %-3,3 %) en tampones de pH 2 y pH 8 con 0,3 % de H2O2 (la mayor impureza era RRT=0,58).
Ejemplo comparativo 3: Caracterización de la solubilidad de la base de indoximod
La solubilidad de indoximod como base libre en disoluciones tamponadas o no tamponadas, así como en líquidos biológicos simulados (SGF, FaSSIF o FeSSIF) se muestra en la Figura 5 (símbolos abiertos). La solubilidad de indoximod en disoluciones acuosas de pH 2-8 es 1,8-2,0 mg/mL, con mayor solubilidad a pH <1,5 o > 10. Es probable esta baja solubilidad en el intervalo de pH neutro debido a la alta energía de empaquetamiento molecular de indoximod en el cristal, que se refleja por el punto de fusión muy alto de 293,8 °C. Esta baja solubilidad de indoximod en el intervalo de pH correspondiente al pH intestinal puede explicar en parte la absorción de dosis limitante a dosis superiores a 800 mg en seres humanos. Por lo tanto, los presentes inventores estudiaron si las sales o las dispersiones secas pulverizadas de indoximod podrían aumentar la solubilidad y la exposición después de la administración por vía oral.
Ejemplo comparativo 4: Caracterización de las sales de indoximod y su solubilidad
Se fabricaron varias sales de indoximod y se evaluaron sus propiedades fisicoquímicas (Tabla 2). Las sales de clorhidrato, sulfato, fosfato, hemi-fosfato, mesilato y hemi-mesilato fueron polvos blancos sólidos que mostraron propiedades cristalinas por PLM y XRPD y fueron anhidras por TGA. Estas sales mostraron menor punto de fusión que la base libre, lo que sugiere una elevada solubilidad en agua en el intervalo de pH entre >1,5 y <10. La mayoría de estas sales mostraron aumentos de solubilidad hasta ~4,7-8,6 mg/mL en agua y 5,5-10,6 mg/mL en SGF, mostrando la sal de clorhidrato una aumento muy significativo de hasta >200 mg/mL en agua o SGF.
Otra sal de indoximod probada fue la sal de ácido maleico, que mostró un bajo punto de fusión de 194 °C y mala cristalinidad por PLM y XRPD. Esta sal tiene el aspecto de un polvo blanco pegajoso de forma de hidrato o solvato (4,5 % de pérdida de peso por TGA).
La sal de tosilato muestra el aspecto de un aceite marrón, que puede ser ventajoso ya que podría aumentar la absorción intestinal del principio activo.
Otras sales tuvieron propiedades fisicoquímicas menos favorables. Por ejemplo, el lactato y la N-metil-glucamina no formaron una sal con indoximod, y el cristal mostró una mezcla de cristales de base libre de indoximod y cristales de N-metilglucamina o lactato.
La sal de sodio no mostró morfología cristalina, fue un hidrato o solvato con fusión muy baja y múltiples picos de descomposición por TGA o DSC y entonces no se caracterizó más.

Ejemplo comparativo 4: Dispersiones secadas por pulverización de indoximod
Se preparó una lista de formulaciones en dispersión secada por pulverización (SDD) de indoximod para evaluar si cualquier formulación de SDD era capaz de aumentar la absorción molecular generando y manteniendo un estado supersaturado de indoximod en líquido gastrointestinal de manera que su absorción se pudiera potenciar. En este estudio, se prepararon formulaciones de SDD por dos métodos: disolución de formulación secada por pulverización en proceso en caliente calentada hasta 110 °C antes del secado por pulverización, y formulación secada por pulverización básica de pH aumentado hasta ~ 11,5 (temperatura ambiente) antes del secado por pulverización. El rendimiento de cada formulación de SDD se investigó por una prueba de disolución in vitro en tampón gástrico simulado (GB) y líquido intestinal simulado (SIF). Como se muestra en la Tabla 3, CmáxGB representó la máxima concentración de indoximod en disolución cuando se disolvió suficiente de la formulación de SDD en GB durante 30 min; Cmáx90 representa la máxima concentración de indoximod cuando SDD se disolvió en SIF durante 90 min; UltraCg0 representa la concentración en SIF después de 90 min de disolución seguido por ultracentrifugación para retirar cualquier partícula y UltraC1200 representa la concentración en SIF después de 1200 min de disolución seguido por ultracentrifugación para retirar cualquier partícula. Se esperaba que las potenciadas concentraciones de indoximod en GB y SIF aumentaran la absorción de indoximod cuando la formulación de SDD se administró en animales, así como seres humanos. Otro criterio para evaluar estas formulaciones de SDD fue la estabilidad física y química de indoximod en estas formulaciones. Se encontró que las formulaciones de SDD hechas por el método de fármacos por pulverización en proceso caliente fueron en general más estables que las hechas por el secado por pulverización del proceso básico. Además, se prefirió mayor carga de fármaco en el polvo puesto que podría disminuir la cantidad de dosis de la formulación final. Basándose en todos estos criterios, se seleccionaron dos formulaciones de SDD para estudios FC in vivo adicionales en monos. El primero fue el 50 % de indoximod/ 50 % de PVPVA-64, que mostró una concentración intestinal predicha 1,8 veces mayor que la de indoximod (UltraC903293 ng/mL frente a 1849 ng/mL); y la segunda fue 50 % de indoximod/ 50 % de Affinisol 126, que mostró una concentración intestinal predicha 2,3 veces mayor que indoximod (UltraC90 4340 ng/mL frente a 1849 ng/mL). Estas SDD se prepararon por secado por pulverización en proceso en caliente, que mostraron mejores propiedades de estabilidad.
Tabla 3: Pruebas de disolución para las formulaciones de dispersión secadas por pulverización de indoximod


Ejemplo comparativo 5: Comparación farmacocinética de base libre de indoximod, sales de indoximod y SDD de indoximod en macacos cangrejeros
Para determinar si las sales o SDD que muestran un aumento en la solubilidad en comparación con la base libre de indoximod producen un aumento en la concentración máxima (Cmáx) y exposición total (ABCü->~) de indoximod, los presentes inventores llevaron a cabo un estudio farmacocinético comparativo de grupos cruzados en macacos cangrejeros, que es una especie común usada para predecir la biodisponibilidad oral humana. Se administró por vía oral a dos grupos de 4 monos cada uno (todos machos) a 275 gmol/kg (grupo 1) o 825 gmol/kg (grupo 2) con: 1) cápsulas de base libre de indoximod; 2) cápsulas de clorhidrato de indoximod; 3) cápsulas de hemifosfato de indoximod; 4) suspensión de SDD1 (indoximod 50 %/50 % de PVPVA-64, (p/p)) y 5) suspensión de SDD2 (indoximod 50 %/Affinisol 12650 % (p/p)). Cada mono se administró con cada una de las 5 dosis de formulaciones una vez cada 7 días, y se tomaron muestras de sangre a las 0, 0,25 h, 0,5 h, 1 h, 2 h, 4 h, 6 h, 8 h, 12 h, 24 h, 36 h y 48 h. Se determinó la concentración de indoximod a partir del plasma por un método analítico validado de CL-EM/EM. Se calculó Cmáx y ABC(0-48h) por análisis no compartimental usando el software WinNonLin (Certara). Para indoximod en la formulación en cápsulas, los animales en el grupo 1 se administraron por vía oral con 3 cápsulas A y los animales en el grupo 2 se administraron con 4 cápsulas B. Las composiciones de cápsulas A y B se muestran en la Tabla 4. Para indoximod en la formulación de SDD, los animales en el grupo 1 se administraron con 4 mL/kg de una suspensión de 15 mg de indoximod/mL y los animales en el grupo 2 se administraron con 4 mL/kg de una suspensión de 45 mg de indoximod/mL. Las formulaciones de SDD en suspensión se prepararon en 0,5 % de metilcelulosa (Methocel).
Tabla 4: Composición de cápsulas que contienen indoximod en sus formas de base libre o de sal para administración oral a macacos cangrejeros

Los valores promedio de los parámetros Cmáx y ABC(0-48h) observados en cada grupo obtenido después de la administración con cada formulación de indoximod se muestran en la Tabla 5. El porcentaje de aumento en estos valores, así como el valor de P obtenido para la comparación de cada formulación frente a la base libre de indoximod, se muestra en la Tabla 5. La administración de cápsulas de HCl de indoximod produce un aumento significativo en la Cmáx (31 -65 %) y la exposición (37-53 %) en ambos niveles de dosis probados en comparación con la administración de las cápsulas de base libre de indoximod. Similarmente, las cápsulas de hemifosfato de indoximod produjeron un aumento significativo en la Cmáx (7-44 %) y la exposición (27-34 %). Por el contrario, el indoximod en la formulación SDD1 o SDD2 produjo un aumento significativo en la Cmáx (15-94 %), pero dejó de aumentar la exposición global con respecto a las cápsulas de base libre de indoximod. Por estos motivos, se prefieren las sales de indoximod en sus sales de clorhidrato, hemifosfato o fosfato con respecto a indoximod en su forma de base libre, ya sea en cápsulas o en dispersiones secadas por pulverización.
Tabla 5: Comparación de Cmáx y exposición total (ABC0->~) entre la base libre de indoximod frente a sus sales o dispersiones secadas por pulverización en monos

Este estudio muestra que las sales de clorhidrato y de fosfato de indoximod pueden producir un aumento en los parámetros farmacocinéticos de Cmáx y ABC con respecto a la base libre, en el intervalo de dosis entre 275 825 pmol/kg.
Ejemplo comparativo 6: Prueba farmacocinética de sales de indoximod en la formulación en cápsulas en ratas
Para determinar si la formación de sal aumentó la concentración máxima (Cmáx) y la exposición total (ABCo->~) de indoximod en ratas, los presentes inventores probaron las sales de clorhidrato, fosfato, sulfato y mesilato de indoximod, y formularon éstas en cápsulas mezclándolas con excipientes apropiados. Se investigaron tres niveles de dosis: 37, 185 o 500 pmol/kg.
Se prepararon cápsulas de gelatina (Torpac, 20 mg de capacidad) que contenían 11,4, 28,6 o 50 pmol/cápsula de indoximod o sus sales, con o sin excipientes, que consistían en celulosa microcristalina, lactosa monohidratada, croscarmelosa sódica y estearato de magnesio, en las proporciones mostradas en la Tabla 6.1-6.3. Las cápsulas se llenaron manualmente y la uniformidad de la composición de una muestra representativa de cápsulas de cada lote se verificó en peso y por CL-EM/EM para determinar el contenido promedio de indoximod.
Tabla 6.1: Composición de cápsulas A que contienen indoximod en su base libre o formas de sal para administración por vía oral de ratas a 37 pmol/kg

Tabla 6.2: Composición de cápsulas B que contienen indoximod en su base libre o formas de sal para administración por vía oral de ratas a 185 pmol/kg
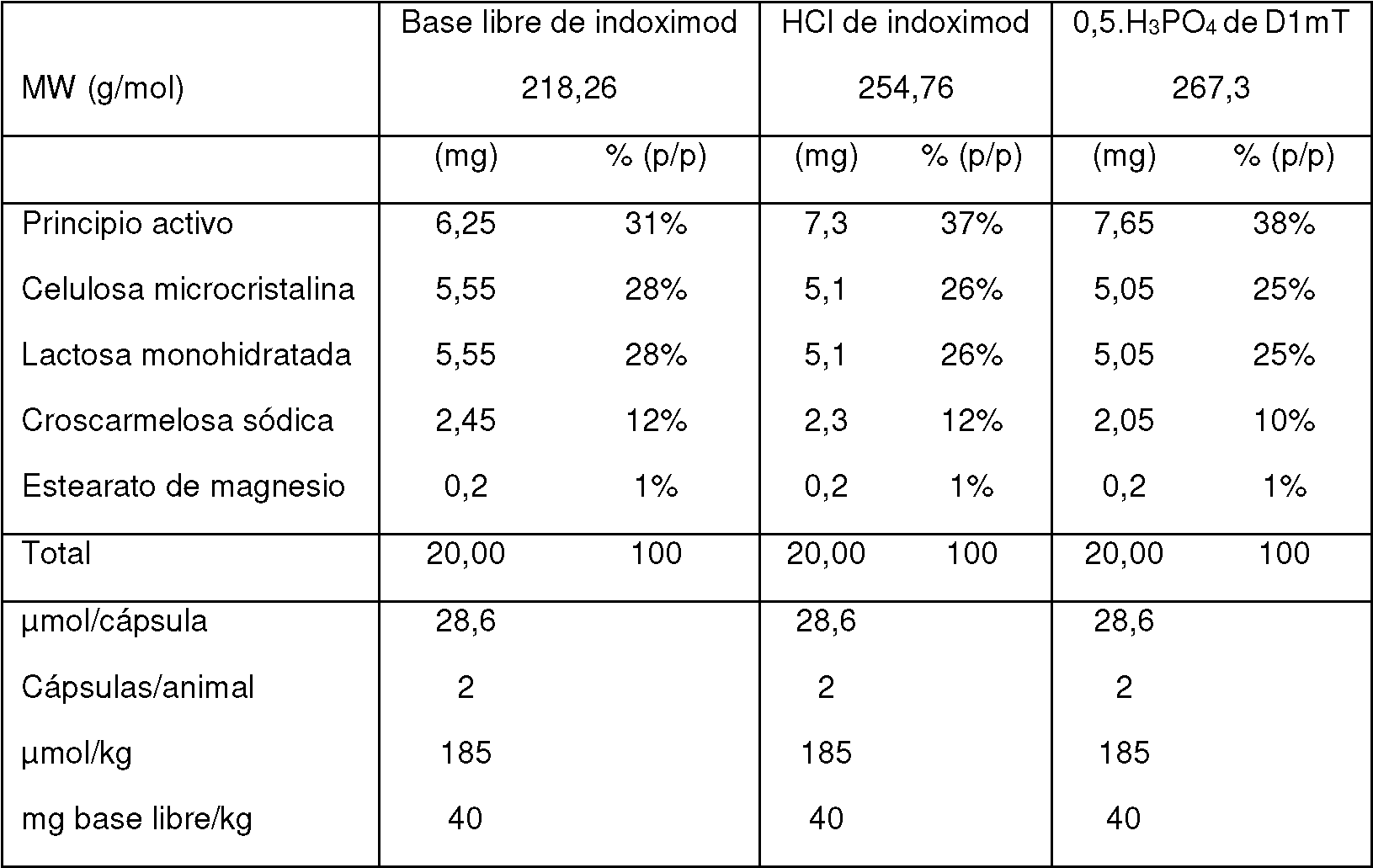
Tabla 6.3: Composición de cápsulas C que contienen indoximod en su base libre o formas de sal para la administración por vía oral de ratas a 500 pmol/kg

Para probar el perfil farmacocinético alcanzado por la administración de indoximod en sus formas de base libre o de sal, las ratas se administraron por administración intraestomacal con 1 cápsula A, 2 cápsulas B o 3 cápsulas C para lograr niveles de dosis de 37, 185 y 500 pmol/kg (equivalentes a 8, 40 y 110 mg/kg de indoximod, respectivamente). Las ratas estuvieron en ayunas 16 h antes de la administración para eliminar cualquier efecto alimenticio de confusión, y se les devolvió la comida 2 h después de la administración. Se tomaron muestras de sangre de cada rata 0, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 10 h, 24 h, 48 h y 72 h después de la administración. La concentración de indoximod en plasma se determinó por CL-EM/EM y se calcularon los parámetros farmacocinéticos usando el software WinNonLin (Certara).
Los parámetros farmacocinéticos más relevantes que se evaluaron fueron la concentración máxima de indoximod (Cmáx) y la exposición total (ABCü->~). La Tablas 7.1-7.3 y la Figura 6 muestran un resumen de los resultados experimentales.
La forma de sal de clorhidrato de indoximod da como resultado una disminución no estadísticamente significativa en Cmáx a bajo nivel de dosis, un aumento estadísticamente significativo a la dosis intermedia y una disminución estadísticamente significativa al nivel alto. La exposición al fármaco (ABC) para la sal de clorhidrato no mostró un cambio significativo en el nivel de dosis baja y alta, pero mostró un aumento significativo en el nivel intermedio. El diferente comportamiento del clorhidrato de indoximod en roedores en comparación con primates es inesperado, basándose en el perfil de solubilidad y de disolución de esta sal, y no sigue una tendencia dependiente de la dosis, que subraya la importancia de realizar pruebas específicas de especie y dependientes de la dosis para la predicción de perfiles farmacocinéticos en seres humanos.
El fosfato y el hemifosfato de indoximod mostraron un aumento significativo en Cmáx y el ABC a los niveles de dosis bajo e intermedio, pero una disminución significativa en Cmáx y una disminución no estadísticamente significativa en la exposición al mayor nivel de dosis.
La correlación dependiente de la dosis para la Cmáx y el ABC para las formas de base libre, HCl y PO4H3 de indoximod se muestra en la Figura 6. Esta figura muestra un aumento en Cmáx para las sales de HCl y PO4H3 con respecto a la base libre a los niveles de dosis bajo e intermedio, pero una saturación en la curva de dosis a Cmáx-respuesta al mayor nivel de dosis, que no se observa para la base libre. La curva de dosis-respuesta para ABC muestra un aumento más lineal de ABC con la dosis, excepto por la sal de PO4H3 que parece aumentar de una forma inferior a proporcional en dosis al mayor nivel de dosis probada.
Similarmente, otras formas de sal de indoximod, tales como el sulfato o el mesilato, aumentan la Cmáx y el ABC ~30-40 % cuando se prueban a 37 pmol/kg.
Estas pruebas indican que las sales de clorhidrato y de fosfato de indoximod tienen una elevada solubilidad con respecto a la forma de base libre y muestran un aumento de los valores de los parámetros de Cmáx y ABC.
Tabla 7.1: Comparación de Cmáx y exposición total(ABC
o
- > ~ )
entre la base libre de indoximod frente a sus formas de sal en ratas administradas a 37 pmol/kg

Tabla 7.2: Comparación de Cmáx y exposición total (ABC
o
->«) entre la base libre de indoximod frente a sus formas de sal en ratas administradas a 185 pmol/kg

Tabla 7.3: Comparación de Cmáx y exposición total (ABC
o
->~) entre la base libre de indoximod frente a sus formas de sal en ratas administradas a 500 pmol/kg

Ejemplo 7: Prueba farmacocinética de profármacos de indoximod en la formulación líquida
Se probó el perfil farmacocinético de indoximod obtenido después de la administración por vía oral de varios profármacos de indoximod de tal forma que reflejara solo diferencias en la permeabilidad intestinal y la conversión de profármaco en indoximod in vivo sin reflejar diferencias en la forma en estado sólido, tal como diferencias en cristales polimórficos o sólidos amorfos que pueden afectar la tasa de solubilidad o solubilización para los diferentes profármacos. Por lo tanto, el indoximod y cada uno de sus profármacos se solubilizó en un vehículo apropiado que fue o solución salina, Cremaphor®:etanol:solución salina (10:10:80) o Cremaphor:EtOH:solución salina:HCl (10:10:80:0,1 N). El indoximod o sus profármacos se disolvieron a una concentración de 1 mg/mL y se administraron a las ratas por sonda nasogástrica oral a 10 mL/kg para lograr una dosis final de 10 mg/kg; o se disolvieron a 25 mg/mL y se administraron a las ratas por sonda nasogástrica oral a 2 mL/kg para lograr una dosis final de 50 mg/kg; o se disolvieron a una concentración de 10 mg/mL y se administraron por vía oral a ratones por sonda nasogástrica oral a 5 mL/kg para lograr una dosis final de 50 mg/kg. Se recogieron muestras de sangre (0,1-0,2 mL) del puerto de la arteria femoral de ratas o por sangrado retrorbitario de ratones y el plasma se recogió inmediatamente por centrifugación y se almacenó en nieve carbónica para evitar la hidrólisis de profármacos después de la recogida del plasma. Se recogieron muestras de sangre 0, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 10 h, 24 h, 48 h y 72 h después de la administración de las ratas o 0, 30 min, 1 h, 2 h, 4 h, 6 h, 16 h y 24 h después de la administración de los ratones. La concentración de indoximod y de cada profármaco en plasma se determinó por CL-EM/EM, y se calcularon los parámetros farmacocinéticos para indoximod y sus profármacos. Los parámetros farmacocinéticos reflejan el promedio de valores de parámetros individuales obtenidos de cada rata individual (n) o un parámetro común de una única curva farmacocinética derivada de muestras de sangre obtenidas de un grupo de ratones (n).
Las Tablas 8.1 y 8.2 muestran la Cmáx y el ABC(o->~) de indoximod obtenidos después de la administración de ya sea indoximod o cada uno de los profármacos de prueba. Puesto que todas las ratas se administraron por vía oral a la misma dosis de 10 mg/kg, pero cada profármaco tiene un peso molecular diferente, para comparar los valores de Cmáx y ABC(o->~) obtenidos después de administrar cada profármaco frente a la administración de indoximod como una base libre, la Cmáx y el ABC(o->~) medidos se normalizaron multiplicándolos por la relación de MWprofármaco/MWindoximod, lo que supone así la farmacocinética lineal en un intervalo de dosis de ~2 veces.
La Tabla 8.1 muestra que algunos profármacos producen un aumento eficaz en cualquiera de Cmáx, ABC o ambos parámetros farmacocinéticos. Puesto que los profármacos se administraron en forma completamente soluble, esto sugiere que los profármacos que muestran Cmáx y/o ABC potenciadas de indoximod en plasma lo hacen por un mecanismo que implica una combinación de factores que incluyen la permeabilidad potenciada del profármaco a través de la pared celular intestinal, reducida depuración del profármaco con respecto a indoximod y buena tasa de conversión del profármaco en indoximod in vivo. No toda forma de profármaco de indoximod produjo una concentración máxima potenciada y exposición de indoximod en comparación con la administración de indoximod. En particular, parece que la exposición (ABC) a indoximod se potencia cuando se administra NLG-1563, NLG-1564, NLG-1566, NLG-1548, NLG-1572, NLG-1557, NLG-1559, NLG-1570, NLG-1565, NLG-1554, NLG-1558, NLG-1551 y NLG-1547, mientras que parece que la Cmáx de indoximod se potencia cuando se administra NLG-1557, NLG-1558, NLG-1554, NLG-1566, NLG-1570, NLG-1283 y NLG-1263.
La Tabla 8.2 muestra profármacos que no produjeron un aumento eficaz en Cmáx de indoximod ni la exposición a indoximod cuando se administró por vía oral a ratas a 10 mg/kg, lo que indica que algunas de estas sustituciones químicas pueden o disminuir la permeabilidad o la tasa de conversión en indoximod o aumentar la tasa de depuración de profármaco por vías que no producen la conversión en indoximod, o una combinación de los efectos.
La Tabla 8.3 muestra profármacos que se probaron por administración oral a las ratas a 50 mg/kg. NLG-1283 provoca un aumento en la Cmáx y el ABC cuando se administró a las ratas a 50 mg/kg. Sin embargo, este profármaco produce una disminución en la Cmáx y el ABC cuando se administra a ratones a 50 mg/kg. En cambio, la molécula altamente similar NLG-1284 no produce un aumento significativo ni en la Cmáx ni el ABC cuando se administra a 50 mg/kg a ratas, pero no produce un aumento significativo en la Cmáx y el ABC en ratones, lo que indica que diferentes especies tienen diferentes tasas de absorción, eliminación y metabolización de estos profármacos y que cambios mínimos en la estructura molecular pueden afectar el resultado en diferentes especies. Se llevó a cabo una FC dependiente de la dosis en ratones, a los que se les administró 10, 50 y 100 mg/kg de indoximod, o en dosis similares para el profármaco NLG-1626 o NLG-1665. Una advertencia de la comparación entre la administración de profármacos frente a indoximod como base libre fue que los profármacos fueron completamente solubles en la formulación de administración, mientras que el indoximod fue insoluble en dosis de 50 y 100 mg/kg. Esto puede producir un efecto de liberación controlada dependiente del tiempo para indoximod que podría producir menor Cmáx pero mayores ABC que cuando se administró en forma completamente soluble. NLG-1626 y NLG-1665 produjeron un aumento significativo en la Cmáx de indoximod en comparación con lo que se observa cuando se administra indoximod en suspensión, en todas las dosis probadas. Sin embargo, NLG-1626 mostró un aumento dependiente de la dosis en el ABC para indoximod, donde el porcentaje de aumento en el ABC disminuye a mayores dosis. La Tabla 8.3 también indica que la formación de carbamatos en el grupo amino de indoximod produce profármacos con una marcada reducción en los parámetros farmacocinéticos para indoximod.
Ejemplo 8: Prueba farmacocinética de sales de profármaco de indoximod en formulación en cápsula sólida en ratas
Para probar que los profármacos tienen el mejor conjunto combinado de propiedades farmacológicas (tasa de solubilización, solubilidad, permeabilidad intestinal, tasa de depuración y tasa de metabolización a indoximod) necesarias para lograr mayores concentraciones plasmáticas de indoximod y una elevada exposición a indoximod después de la administración por vía oral en una formulación en cápsula, los profármacos que mostraron una Cmáx o exposición potenciada a indoximod cuando se administraron en disolución se prepararon en varias formas de sal y se mezclaron con excipientes para formar una mezcla en polvo. Estas mezclas se formularon de manera que cada cápsula contuviera la misma dosis molar de cada profármaco. Se prepararon cápsulas de gelatina (Torpac, 20 mg capacidad) que contenían 11 gmol/cápsula A, 28 gmol/cápsula B o 50 gmol/cápsula C de base libre de indoximod (2,5, 6,3 o 11,4 mg/cápsula, respectivamente) o sus profármacos en diversas formas de sal, en una mezcla de excipientes que consiste en celulosa microcristalina, lactosa monohidratada, croscarmelosa sódica y estearato de magnesio, en las proporciones mostradas en las Tablas 9.1a y 9.1b. La composición y la uniformidad de una muestra representativa de cápsulas de cada lote se verificó en peso y por CL-EM/EM para determinar el contenido promedio de indoximod o de profármaco.
Para probar el perfil farmacocinético logrado por la administración de profármacos de indoximod en diferentes formas de sal, se administró 1 cápsula A (11 gmol/cápsula) o 2 cápsulas B (28 gmol/cápsula) o 3 cápsulas C (50 gmol/cápsula) a las ratas por administración intraestomacal. Los niveles de dosis de prueba fueron equivalentes a 8 mg/kg (37 gmol/kg) de equivalente de indoximod cuando se administra 1 cápsula A de 11 gmol/cápsula, 40 mg/kg (185 gmol/kg) de equivalente de indoximod cuando se administran 2 cápsulas B de 28 gmol/cápsula y 110 mg/kg (500 gmol/kg) de equivalente de indoximod cuando se administran 3 cápsulas C de 50 gmol/cápsula. Las ratas estuvieron en ayunas 16 h antes de la administración para eliminar cualquier efecto alimentario de confusión, y se les devolvió la comida 2 h después de la administración. Se tomaron muestras de sangre de cada rata 0, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 10 h, 24 h, 48 h y 72 h después de la administración. La concentración de indoximod en plasma se determinó por CL-EM/EM y se calcularon los parámetros farmacocinéticos usando el software WinNonLin (Certara).
Los parámetros farmacocinéticos más relevantes evaluados fueron la concentración máxima de indoximod (Cmáx) y la exposición total a indoximod (ABCü->~). Las Tablas 10.1 y 10.2 muestran un resumen de los resultados experimentales.
La comparación estadística de parámetros farmacocinéticos indicó que el N°-(L-leucil)-1-metil-D-triptofanato de etilo en sus formas de sal de clorhidrato (NLG-1564), fosfato (NLG-1665), mesilato (NLG-1666) o besilato (NLG-1671) administrado a 37-185 gmol/kg fue capaz de aumentar significativamente (p<0,05) la exposición de indoximod en el 33-127 %, mientras que su sal de sulfato (NLG-1691) no dio como resultado un aumento significativo en la Cmáx o el ABC a esas dosis. Similarmente, se observaron aumentos significativos en Cmáx para NLG-1564, NLG-1665 y NLG-1666. A dosis de 500 gmol/kg, el clorhidrato NLG-1564, mostró un menor aumento en la Cmáx y el ABC en comparación con indoximod.
La Tabla 10.2 muestra que el 1-metil-D-triptofanato de 2,3-dihidroxipropilo en su forma de fosfato (NLG-1626) produjo un aumento significativo en la Cmáx (37-153 %) y el ABC (46-75 %), mientras que sus sales de clorhidrato (NLG-1558) y sulfato (NLG-1628) produjeron aumentos menos significativos en Cmáx y ABC. Es interesante señalar que la sal de mesilato de 1-metil-D-triptofanato de 2,3-dihidroxipropilo (NLG-1627) produjo una disminución en Cmáx y ABC, aunque esta disminución no fue estadísticamente significativa.
La Tabla 10.2 también muestra que el Na-(L-metionil)-1-metil-D-triptofanato de etilo (sales de HCl y de fosfato, NLG-3272) muestra un aumento estadísticamente significativo en la Cmáx y el ABC en dosis de 37-500 gmol/kg.
Otros profármacos que se estudiaron incluyeron: a) Na-(L-glutaminil)-1-metil-D-triptofanato de etilo (base libre, sales de HCl, fosfato o mesilato), b) Na-glicil-1 -metil-D-triptófano (sal de HCl o de fosfato), c) W4-((R)-1-etoxi-3-(1-metil-1H-indol-3-il)-1-oxopropan-2-il)-L-asparaginato de metilo (forma de HCl) y d) Na-(L-lisil)-1 -metil-D-triptófano (base libre, sales de HCl, de sulfato o de fosfato). Estos profármacos produjeron variaciones menores y estadísticamente no significativas en la Cmáx o el ABC para indoximod en comparación con una dosis molar equivalente de indoximod (Tabla 10.3).
Es interesante señalar que el 1-metil-D-triptofanato de piperidin-4-ilmetilo en sus formas de sal de HCl o de fosfato (NLG-1563 y NLG-1664) produjo una disminución estadísticamente significativa en Cmáx (69-79 %, p<0,004) y ABC (54-64 %, p<0,014) para indoximod. Puesto que este compuesto mostró un aumento en Cmáx (24 %) y ABC (75 %) cuando se administró por disolución oral, la diferencia en la tasa de solubilización o la solubilidad final puede explicar las diferencias observadas cuando se administra en forma de polvo.
Ejemplo 9: Prueba farmacocinética de sales de profármaco de indoximod en formulación en cápsula sólida en macacos cangrejeros
Puesto que la rata muestra un aumento lineal no saturable en la exposición a dosis de indoximod de hasta 100 mg/kg, mientras que los seres humanos muestran una exposición saturable por encima de la dosis de 10 mg/kg, los presentes inventores decidieron evaluar dos de los profármacos en primates, que pueden constituir un mejor modelo para
predecir la farmacocinética humana que las ratas. Se les administró a monos cangrejeros (4,5-5 kg) indoximod, NLG-1564 HCl o NLG-3272 HCl en dosis de 92, 275 o 875 gmol/kg en un diseño del estudio con grupos cruzados donde cada animal recibió la misma dosis molar de indoximod, NLG1564 HCl o NLG-3272 HCl cada 7 días. Las cápsulas se prepararon según la formulación descrita en la Tabla 9.2. Los monos se administraron por dosis oral con 1 o 3 cápsulas A (458 gmol/cápsula) o 4 cápsulas B (1032 gmol/cápsula). Se recogieron muestras de sangre 0, 5 min, 15 min, 30 min, 1, 2, 4, 8, 12, 24, 26 y 48 h después de la administración, y las concentraciones de profármaco e indoximod se analizaron por métodos validados de CL-EM/EM.
Los datos en la Tabla 11.1 muestran que NLG-1564 HCl aumenta la Cmáx de indoximod desde ~ 230-500 % y el ABC desde 195-518 % en un modo estadísticamente significativo. Similarmente, NLG-3272 HCl aumenta la Cmáx de indoximod desde ~ 305-411 % y el ABC desde 136-393 % en un modo estadísticamente significativo. El aumento en los indicadores farmacodinámicos en primates fue inesperadamente superior a partir de los resultados observados en ratas, que indica que en primates, los profármacos de indoximod de la presente divulgación pueden proporcionar una mejora significativa en la concentración máxima y exposición a indoximod y se espera que mejoren la exposición al fármaco y la eficacia terapéutica en pacientes humanos.




Tabla 9.1a: Composiciones de cápsulas - Administración por vía oral a ratas


Tabla 9.1b: Composiciones de cápsulas - Administración por vía oral a ratas
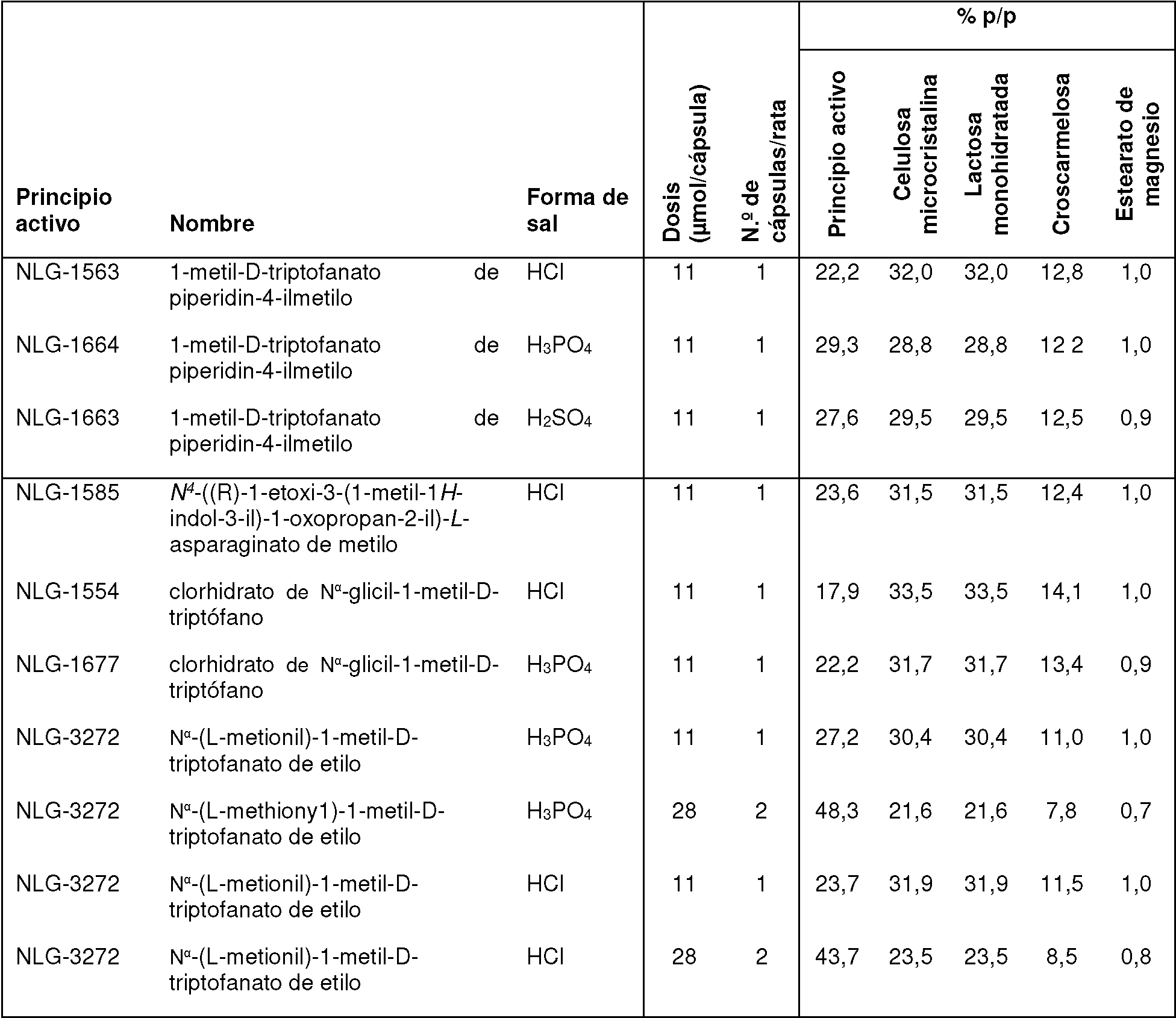

Tabla 9.2: Composiciones de cápsulas - Administración por vía oral a monos





Referencias
1. McGaha, T.L., et al., Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol Rev, 2012.
249(1): p. 135-57.
2. Li, L., et al., Altered tryptophan metabolism as a paradigm for good and bad aspects of immune privilege in chronic inflammatory diseases. Front Immunol, 2012. 3: p. 109.
3. Munn, D.H., et al., Prevention of allogeneic fetal rejection by tryptophan catabolism. science, 1998. 281(5380): p.
1191-3.
4. Muller, A.J., et al., Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med, 2005. 11(3): p. 312-9.
5. Peterson, A.C., et al., Evaluation of functionalized tryptophan derivatives and related compounds as competitive inhibitors of indoleamine 2,3-dioxygenase. Medicinal Chemistry Research, 1994. 3: p. 531-544.
6. Hou, D.Y., et al., Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1 -methyl-tryptophan correlates with antitumor responses. Cancer Res, 2007. 67(2): p. 792-801.
7. Metz, R., et al., IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology, 2012. 1(9): p. 1460-1468.
8. Sharma, M.D., et al., Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest, 2007. 117(9): p. 2570-82.
9. Sharma, M.D., et al., Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumordraining lymph nodes. Blood, 2009.
10. Holmgaard, R.B., et al., Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med, 2013. 210(7): p. 1389-402.
11. Munn, D.H., et al., GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity, 2005. 22(5): p. 633-42.
12. Fallarino, F., et al., The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol, 2006. 176(11): p. 6752-61.
13. Kumar, S., et al., Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase. J Med Chem, 2008. 51(16): p. 4968-77.
14. Banerjee, T., et al., A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene, 2008. 27(20): p. 2851-7.
Claims (12)
1. Un profármaco de indoximod, en forma de base libre o de sal, según la fórmula 2:

en donde:
(a) R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2, -O-alquil C1-3-R3, -O-alquil C1-6-R6, -O alquil C1-2-C<3H(NH2)(COOH) o -O-alquil C1-2-C(R)H(NH2)(COOH);
(b) R2 es H, -C(O)C(S)H(NH2)R4, -C(O)C(R)H(NH2)R4, -C(O)CH2C(SiH(NH2)C(O)OCH3 o -C(O)NHR5;
(c) R3 es tetrahidropirano o

(d) R4 es H, -alquilo C1-5, -(CH2)1-2SH, alquil C1-5-S-alquilo C1-5, alquil C1-5-O-alquilo C1-5, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)1-2C(O)NH2, -(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2;
(e) C(S) y C(R) son carbonos con la estereoquímica S o R, respectivamente, cuando R4 no es H;
(f) R5 es H, alquil C1-6-R6 o R6;
(g) R6 es arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo, en donde el arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7;
(h) cada R7 es independientemente halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, -alquilo C1-6, -haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S(O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C( O)R, -N(R)C(O)OR o -N(R)C(O)N(R)2, en donde R es H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es H; y
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico, en donde la relación estequiométrica n del ácido es 0, 0,5, 1 o 2 de forma que el profármaco sea de carga neutra.
2. El profármaco de la reivindicación 1, en donde:
(a) R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3;
(b) R2 es H o -C(O)C(S)H(NH2)R4,
(c) R3 es tetrahidropirano o

(d) R4 es H, -alquilo C1-5, -(CH2)1-2SH, -(CH2)1-3SCH3, -(CH2)1-3OCH3, -CH2-R6, -CH2OH, -CH(OH)CH3, -(CH2)1-2C(O)NH2, -(CH2)1-3C(O)OH, -(CH2)1-4NH2 o -(CH2)1-3NC(=NH2)NH2;
(e) C(S) es un carbono con la estereoquímica S, cuando R4 no es H;
(f) R6 es arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo, en donde el arilo, alquilarilo, heteroarilo, cicloalquilo o heterocicloalquilo se sustituye opcionalmente con uno, dos o tres grupos R7;
(g) cada R7 es independientemente halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, -alquilo Ci-6, -haloalquilo Ci-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S( O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R , -N(R)C(O)OR o -N(R)C(O)N(R)2, en donde R es H o alquilo C1-4;
con la condición de que Ri no pueda ser -OH cuando R2 es H; y
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico), C6H5SO3H (ácido bencilsulfónico), ácido acético, ácido ascórbico, ácido aspártico, ácido glutámico, ácido glutárico, ácido láctico, ácido maleico, ácido malónico, ácido oxálico, ácido succínico, ácido fumárico, ácido tartárico y ácido cítrico, en donde la relación estequiométrica n es 0, 0,5, 1 o 2 de forma que el profármaco sea de carga neutra.
3. El profármaco de la reivindicación 1, en donde:
(a) R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3;
(b) R2 es H o -C(O)C(S)H(NH2)R4;
(c) R3 es tetrahidropirano o

(d) R4 es H, -alquilo C1-5, -(CH2)2SCH3, -CH2-R6, -(CH2)1-2C(O)NH2, -(CH2)1-3C(O)OH o -(CH2)1-4NH2;
(e) C(S) es un carbono con la estereoquímica S, cuando R4 no es H;
(f) R6 es arilo, alquilarilo o heteroarilo, en donde el arilo, alquilarilo o heteroarilo se sustituye opcionalmente con un grupo R7;
(g) cada R7 es independientemente halógeno, ciano, nitro, -OR, -N(R)2, -SR, -C(O)OR, -alquilo C1-6, -haloalquilo C1-6, -C(O)N(R)2, -C(O)R, -S(O)R, -S(O)OR, -S(O)N(R)2, -S( O)2R, -S(O)2OR, -S(O)2N(R)2, -OC(O)R, -OC(O)OR, -OC(O)N(R)2, -N(R)C(O)R , -N(R)C(O)OR o -N(R)C(O)N(R)2, en donde R es H o alquilo C1-4;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico) y C6H5SO3H (ácido bencilsulfónico), en donde la relación estequiométrica n del ácido es 0, 0,5, 1 o 2 de forma que el profármaco sea de carga neutral.
4. El profármaco de la reivindicación 1, en donde
(a) R1 es -OH, -O-alquilo C2-3, -OCH2CH(OH)CH2OH, -O(CH2)2N(CH3)2 o -O-alquil C1-3-R3;
(b) R2 es H o -C(O)C(S)H(NH2)R4;
(c) R3 es tetrahidropirano o

(d) R4 es -CH2CH(CH3)2, -(CH2)2SCH3, -C(S)H(CH3)CH2CH3, -CH2-R6, -(CH2)2C(O)NH2, -(CH2)3C(O)OH o -(CH2)4NH2;
(e) C(S) es un carbono con la estereoquímica S;
(f) R6 es fenilo;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico) y C6H5SO3H (ácido bencilsulfónico), en donde la relación estequiométrica n del ácido es 0, 0,5, 1 o 2 de forma que el profármaco sea de carga neutral.
5. El profármaco de la reivindicación 1, en donde:
(a) R1 es -O-alquil C2-3 o -OCH2CH(OH)CH2OH;
(b) R2 es H o -C(O)C^H(NH2)R4;
(c) R4 es -CH2CH(CH3)2, -(CH2)2SCH3 o -(CH2)2C(O)NH2;
(d) C(S) es un carbono con la estereoquímica S;
con la condición de que R1 no pueda ser -OH cuando R2 es H;
HAn es un ácido seleccionado del grupo que consiste en PO4H3 (ácido fosfórico), SO4H2 (ácido sulfúrico), HCl (ácido clorhídrico), HSO3CH3 (ácido metilsulfónico) y C6H5SO3H (ácido bencilsulfónico), en donde la relación estequiométrica n del ácido es de 0, 0,5, 1 o 2 de forma que el profármaco sea de carga neutral.
6. El profármaco de la reivindicación 1, en donde el profármaco comprende uno de los siguientes compuestos:
A/3-(L-leucil)-1-metil-D-triptofanato de etilo;
/V3-(L-metionil)-1-metil-D-triptofanato de etilo;
1-metil-D-triptofanato de 2,3-dihidroxipropilo;
Af'-(L-leucil)-1-metil-D-triptófano;
A/3-(L-met¡on¡l)-1-met¡l-D-tr¡ptófano;
/V3-(L-isoleucil)-1-metil-D-triptofanato de etilo;
Af'-(L-glic¡l)-1-met¡l-D-tr¡ptófano;
ácido (S)-5-amino-6-(((R)-1 -carbox¡-2-(1 -metil-1 H-indol-3-il)etil)amino)-6-oxohexanoico;
Wa-(L-lisil)-1-metil-D-triptófano;
Wa-(L-fenilalanil)-1-metil-D-triptófano;
Wa-(L-glutaminil)-1-metil-D-triptofanato de etilo;
1-metil-D-triptofanato de 2-(dimetilamino)etilo;
1 -metil-D-triptofanato de (2-etoxi-2-oxido-1,3,2-dioxafosfolan-4-il)metilo;
1-metil-D-triptofanato de 2-(tetrahidro-2H-piran-4-il)etilo;
1-metil-D-triptofanato de etilo; o
1-metil-D-triptofanato de isopropilo; o una sal farmacéuticamente aceptable de los mismos.
7. El profármaco de la reivindicación 1, en donde el profármaco es Wa-(L-leucil)-1 -metil-D-triptofanato de etilo o una sal farmacéuticamente aceptable del mismo, o la sal de HCl de Wa-(L-leucil)-1-metil-D-triptofanato de etilo.
8. Una composición farmacéutica que comprende el profármaco de una cualquiera de las reivindicaciones 1-7.
9. La composición farmacéutica de la reivindicación 8, en donde la composición es una cápsula sólida, comprimido o píldora.
10. La composición farmacéutica de la reivindicación 8, en donde la composición es una cápsula disolvible.
11. El profármaco de una cualquiera de las reivindicaciones 1-7, o la composición farmacéutica de una cualquiera de las reivindicaciones 8-10, para su uso en un método de tratamiento de cáncer en un sujeto en necesidad del mismo, comprendiendo el método administrar una cantidad terapéuticamente eficaz del profármaco o composición.
12. El profármaco de una cualquiera de las reivindicaciones 1-7 o la composición farmacéutica de una cualquiera de las reivindicaciones 8-10,
(a) para su uso en un método para modular la actividad de la vía de la indolamina-2,3-dioxigenasa en un sujeto en necesidad del mismo, que comprende administrar por vía oral una cantidad terapéuticamente eficaz del profármaco o composición al sujeto en una forma farmacéutica apropiada o vehículo;
(b) para su uso en un método de tratamiento de inmunosupresión específica de tumor asociada al cáncer en un sujeto en necesidad del mismo, que comprende administrar por vía oral una cantidad suficiente del profármaco o composición al sujeto en una forma farmacéutica apropiada o vehículo; o
(c) para su uso en un método de tratamiento de inmunosupresión asociada a enfermedades infecciosas, en un sujeto en necesidad del mismo, que comprende administrar por vía oral una cantidad suficiente del profármaco o composición al sujeto en una forma farmacéutica apropiada o vehículo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562196671P | 2015-07-24 | 2015-07-24 | |
| US201662305748P | 2016-03-09 | 2016-03-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2923184T3 true ES2923184T3 (es) | 2022-09-26 |
Family
ID=57836622
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16830974T Active ES2894334T3 (es) | 2015-07-24 | 2016-06-02 | Sales y profármacos de 1-metil-D-triptófano |
| ES19188078T Active ES2923184T3 (es) | 2015-07-24 | 2016-06-02 | Sales y profármacos de 1-metil-D-triptófano |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16830974T Active ES2894334T3 (es) | 2015-07-24 | 2016-06-02 | Sales y profármacos de 1-metil-D-triptófano |
Country Status (24)
| Country | Link |
|---|---|
| US (4) | US9732035B2 (es) |
| EP (3) | EP3613420B1 (es) |
| JP (4) | JP6842429B2 (es) |
| KR (2) | KR20180030825A (es) |
| CN (2) | CN107847486A (es) |
| AU (1) | AU2016298471C1 (es) |
| BR (1) | BR112018000225A2 (es) |
| CA (3) | CA2992016C (es) |
| CL (1) | CL2018000082A1 (es) |
| CO (1) | CO2017013724A2 (es) |
| CR (1) | CR20180023A (es) |
| DO (1) | DOP2018000011A (es) |
| EA (1) | EA201792256A1 (es) |
| EC (1) | ECSP18002561A (es) |
| ES (2) | ES2894334T3 (es) |
| HK (2) | HK1247837A1 (es) |
| IL (2) | IL255625B (es) |
| MA (1) | MA43294A1 (es) |
| MX (1) | MX2018001014A (es) |
| NZ (1) | NZ736978A (es) |
| PE (1) | PE20180928A1 (es) |
| PH (1) | PH12017502046A1 (es) |
| WO (1) | WO2017019175A1 (es) |
| ZA (1) | ZA201800208B (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112018000225A2 (pt) | 2015-07-24 | 2018-09-04 | Newlink Genetics Corporation | ?sais e pró-fármacos de 1-metil-d-triptofano? |
| CN109400517A (zh) * | 2017-08-17 | 2019-03-01 | 上海时莱生物技术有限公司 | 1-甲基-色氨酸类化合物及其制备方法和用途 |
| US20200261414A1 (en) * | 2017-09-14 | 2020-08-20 | Lankenau Institute For Medical Research | Methods and compositions for the treatment of cancer |
| CN109646683A (zh) * | 2019-02-27 | 2019-04-19 | 武汉理工大学 | 一种1-mt-羧甲基壳聚糖药物的制备方法 |
| AU2021340521A1 (en) | 2020-09-10 | 2023-05-25 | Nammi Therapeutics, Inc. | Formulated and/or co-formulated liposome compositions containing pd-1 antagonist prodrugs useful in the treatment of cancer and methods thereof |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1330573A (en) | 1970-09-12 | 1973-09-19 | Ajinomoto Kk | Optical resolution of dl-tryptophan derivatives |
| US4072691A (en) | 1974-01-12 | 1978-02-07 | Tanabe Seiyaku Co., Ltd. | Process for the resolution of DL-6-chlorotryptophan |
| US5185157A (en) | 1990-05-02 | 1993-02-09 | Caston John C | Treatment of refractory Eosinophilia-Myalgia Syndrome with L-tryptophan composition |
| WO1996011927A1 (en) | 1994-10-12 | 1996-04-25 | Abbott Laboratories | Endothelin antagonists |
| AU1628699A (en) | 1997-12-05 | 1999-06-28 | Medical College Of Georgia Research Institute, Inc. | High-affinity tryptophan transporter |
| KR100523119B1 (ko) * | 2000-03-16 | 2005-10-20 | 에프. 호프만-라 로슈 아게 | Ip 길항제로서의 카복실산 유도체 |
| EP1392688B1 (en) * | 2001-06-05 | 2008-06-04 | Lilly Icos LLC | Pyrazino[1',2':1,6]-pyrido[3,4-b] indole-1,4-dione derivatives |
| US8008281B2 (en) | 2003-03-27 | 2011-08-30 | Lankenau Institute For Medical Research | Methods for the treatment of cancer |
| US7598287B2 (en) | 2003-04-01 | 2009-10-06 | Medical College Of Georgia Research Institute, Inc. | Use of inhibitors of indoleamine-2,3-dioxygenase in combination with other therapeutic modalities |
| JO2625B1 (en) | 2003-06-24 | 2011-11-01 | ميرك شارب اند دوم كوربوريشن | Phosphoric acid salts of dipeptidyl betidase inhibitor 4 |
| EP1707560A4 (en) | 2003-12-25 | 2007-10-17 | Kureha Corp | HYDROXAMIC ACID DERIVATIVE AND THE DERIVATIVE INHIBITOR OF THE GENERATION OF AGE |
| AU2006244068B9 (en) | 2005-05-10 | 2012-10-25 | Incyte Holdings Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| US7705022B2 (en) | 2005-10-27 | 2010-04-27 | Lankenau Institute For Medical Research | IDO inhibitors and methods of use thereof |
| CA2634198C (en) | 2005-12-20 | 2014-06-03 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| EP1981534A4 (en) * | 2006-01-07 | 2012-04-04 | Med College Georgia Res Inst | INDOLEAMINE 2,3-DIOXYGENASE PATHWAYS IN T REGULATORY LYMPHOCYTE PRODUCTION |
| CA2569204A1 (en) | 2006-11-28 | 2008-05-28 | Apotex Technologies Inc. | Crystalline d-isoglutamyl-d-tryptophan and the mono ammonium salt of d-isoglutamyl-d-tryptophan |
| WO2008100562A2 (en) * | 2007-02-14 | 2008-08-21 | Medical College Of Georgia Research Institute, Inc. | Indoleamine 2,3-dioxygenase, pd-1/pd-l pathways, and ctla4 pathways in the activation of regulatory t cells |
| US8389568B2 (en) | 2007-03-16 | 2013-03-05 | Lankenau Institute For Medical Research | IDO inhibitors and methods of use thereof |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| EP2247564B1 (en) * | 2008-02-01 | 2011-06-29 | Firmenich S.A. | Substituted cyclohexenones |
| CA2722159C (en) | 2008-04-24 | 2016-04-05 | Newlink Genetics Corporation | Substituted phenylimidazole compounds and their use as ido inhibitors |
| PL2315756T3 (pl) | 2008-07-08 | 2015-02-27 | Incyte Holdings Corp | 1,2,5-oksadiazole jako inhibitory 2,3-dioksygenazy indoloaminy |
| MX360800B (es) | 2009-05-13 | 2018-11-16 | Gilead Pharmasset Llc Star | Compuestos antivirales. |
| US8722720B2 (en) * | 2009-10-28 | 2014-05-13 | Newlink Genetics Corporation | Imidazole derivatives as IDO inhibitors |
| EP2533779A4 (en) * | 2010-02-09 | 2013-08-21 | Georgia Health Sciences University Res Inst Inc | ALPHA-METHYL-TRYPTOPHANE AS INHIBITOR OF INDOLE AMINE DIOXYGENASE |
| NO2694640T3 (es) | 2011-04-15 | 2018-03-17 | ||
| JO3353B1 (ar) | 2012-04-20 | 2019-03-13 | Ono Pharmaceutical Co | شكل صلب معزول من أحادي هيدروكلوريد أناموريلين بنسبة مولارية منخفضة من الكلوريد: أناموريلين ومحتوى منخفض من مذيب عضوي متبقي |
| CA2891412A1 (en) | 2012-11-20 | 2014-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase |
| RU2667509C2 (ru) | 2013-03-14 | 2018-09-21 | Ньюлинк Джинетикс Корпорейшин | Трициклические соединения в качестве ингибиторов иммуносупрессии, опосредуемой метаболизированием триптофана |
| KR102189529B1 (ko) | 2013-03-14 | 2020-12-11 | 큐라데브 파마 프라이버트 리미티드 | 키누레닌 경로의 억제제 |
| WO2014141110A2 (en) | 2013-03-14 | 2014-09-18 | Curadev Pharma Pvt. Ltd. | Aminonitriles as kynurenine pathway inhibitors |
| JP6313416B2 (ja) | 2013-03-15 | 2018-04-18 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Ido阻害剤 |
| US9624188B2 (en) | 2013-03-15 | 2017-04-18 | Bristol-Myers Squibb Company | IDO inhibitors |
| US9765018B2 (en) | 2013-07-01 | 2017-09-19 | Bristol-Myers Squibb Company | IDO inhibitors |
| ES2707961T3 (es) | 2013-07-11 | 2019-04-08 | Bristol Myers Squibb Co | Inhibidores de IDO |
| BR112018000225A2 (pt) | 2015-07-24 | 2018-09-04 | Newlink Genetics Corporation | ?sais e pró-fármacos de 1-metil-d-triptofano? |
| JP7126947B2 (ja) | 2016-03-09 | 2022-08-29 | エモリー ユニバーシティー | 抗ウイルス剤によるb型肝炎ウイルスの排除 |
| WO2018006035A1 (en) | 2016-06-30 | 2018-01-04 | Timpson Electrical & Aerial Services, LLC | High voltage training device and system and method thereof |
-
2016
- 2016-06-02 BR BR112018000225-9A patent/BR112018000225A2/pt not_active Application Discontinuation
- 2016-06-02 MA MA43294A patent/MA43294A1/fr unknown
- 2016-06-02 EP EP19188078.0A patent/EP3613420B1/en active Active
- 2016-06-02 ES ES16830974T patent/ES2894334T3/es active Active
- 2016-06-02 KR KR1020187000919A patent/KR20180030825A/ko active IP Right Grant
- 2016-06-02 CN CN201680036956.4A patent/CN107847486A/zh active Pending
- 2016-06-02 CA CA2992016A patent/CA2992016C/en active Active
- 2016-06-02 EA EA201792256A patent/EA201792256A1/ru unknown
- 2016-06-02 ES ES19188078T patent/ES2923184T3/es active Active
- 2016-06-02 AU AU2016298471A patent/AU2016298471C1/en active Active
- 2016-06-02 PE PE2018000117A patent/PE20180928A1/es unknown
- 2016-06-02 CN CN201911070622.0A patent/CN111004167B/zh active Active
- 2016-06-02 JP JP2017561946A patent/JP6842429B2/ja active Active
- 2016-06-02 KR KR1020187021125A patent/KR20180086300A/ko active IP Right Grant
- 2016-06-02 EP EP16830974.8A patent/EP3324958B1/en active Active
- 2016-06-02 CA CA3051388A patent/CA3051388C/en active Active
- 2016-06-02 NZ NZ736978A patent/NZ736978A/en not_active IP Right Cessation
- 2016-06-02 WO PCT/US2016/035391 patent/WO2017019175A1/en active Application Filing
- 2016-06-02 CR CR20180023A patent/CR20180023A/es unknown
- 2016-06-02 US US15/171,031 patent/US9732035B2/en active Active
- 2016-06-02 CA CA3132620A patent/CA3132620C/en active Active
- 2016-06-02 EP EP21187070.4A patent/EP3954369A1/en active Pending
- 2016-06-02 MX MX2018001014A patent/MX2018001014A/es unknown
-
2017
- 2017-06-27 US US15/634,610 patent/US10207990B2/en active Active
- 2017-11-09 PH PH12017502046A patent/PH12017502046A1/en unknown
- 2017-11-13 IL IL255625A patent/IL255625B/en active IP Right Grant
- 2017-12-28 CO CONC2017/0013724A patent/CO2017013724A2/es unknown
-
2018
- 2018-01-10 CL CL2018000082A patent/CL2018000082A1/es unknown
- 2018-01-11 DO DO2018000011A patent/DOP2018000011A/es unknown
- 2018-01-11 ZA ZA2018/00208A patent/ZA201800208B/en unknown
- 2018-01-15 EC ECIEPI20182561A patent/ECSP18002561A/es unknown
- 2018-06-06 HK HK18107364.3A patent/HK1247837A1/zh unknown
- 2018-06-07 HK HK18107413.4A patent/HK1247844A1/zh unknown
- 2018-10-30 JP JP2018204025A patent/JP7286299B2/ja active Active
-
2019
- 2019-02-15 US US16/277,248 patent/US20190248739A1/en not_active Abandoned
- 2019-07-14 IL IL268048A patent/IL268048A/en unknown
-
2020
- 2020-05-22 US US16/881,709 patent/US11485705B2/en active Active
-
2021
- 2021-05-26 JP JP2021088493A patent/JP2021121633A/ja active Pending
-
2023
- 2023-03-10 JP JP2023037970A patent/JP2023060349A/ja not_active Withdrawn
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2923184T3 (es) | Sales y profármacos de 1-metil-D-triptófano | |
| ES2891734T3 (es) | Derivados de bencimidazol como inhibidores de tirosina quinasa ERBB para el tratamiento del cáncer | |
| US20230233562A1 (en) | Heteroaryl compounds, pharmaceutical compositions thereof, and their therapeutic use | |
| WO2018143403A1 (ja) | 複素環化合物 | |
| WO2022266249A1 (en) | Kras protein degraders, pharmaceutical compositions thereof, and their therapeutic applications | |
| EA040751B1 (ru) | Соли и пролекарства 1-метил-d-триптофана | |
| WO2021047672A1 (en) | Triterpenoid compounds, pharmaceutical compositions thereof, and their use for treating nuclear receptor subfamily 4 group member 1-mediated disease | |
| WO2024012557A1 (en) | Anti-apoptotic bcl-2 family protein degraders, pharmaceutical compositions, and therapeutic applications | |
| AU2021233433A1 (en) | Crystal of imidazopyridinone compound or salt thereof |