ES2909486T3 - Sistemas de administración para liberación controlada de fármaco - Google Patents
Sistemas de administración para liberación controlada de fármaco Download PDFInfo
- Publication number
- ES2909486T3 ES2909486T3 ES16812576T ES16812576T ES2909486T3 ES 2909486 T3 ES2909486 T3 ES 2909486T3 ES 16812576 T ES16812576 T ES 16812576T ES 16812576 T ES16812576 T ES 16812576T ES 2909486 T3 ES2909486 T3 ES 2909486T3
- Authority
- ES
- Spain
- Prior art keywords
- group
- optionally substituted
- alkyl
- optionally
- agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000013267 controlled drug release Methods 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 184
- 150000003839 salts Chemical class 0.000 claims abstract description 99
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 96
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 79
- 239000000126 substance Substances 0.000 claims abstract description 65
- -1 vinylcarbonyl group Chemical group 0.000 claims abstract description 62
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 56
- 125000006850 spacer group Chemical group 0.000 claims abstract description 54
- 239000012634 fragment Substances 0.000 claims abstract description 52
- 230000027455 binding Effects 0.000 claims abstract description 51
- 239000012453 solvate Substances 0.000 claims abstract description 50
- 125000003107 substituted aryl group Chemical group 0.000 claims abstract description 44
- 125000006575 electron-withdrawing group Chemical group 0.000 claims abstract description 42
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 42
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 38
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims abstract description 34
- 150000001413 amino acids Chemical class 0.000 claims abstract description 24
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims abstract description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims abstract description 23
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 claims abstract description 21
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 21
- 239000002260 anti-inflammatory agent Substances 0.000 claims abstract description 20
- 125000002228 disulfide group Chemical group 0.000 claims abstract description 17
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 17
- 125000005030 pyridylthio group Chemical group N1=C(C=CC=C1)S* 0.000 claims abstract description 17
- 125000004069 aziridinyl group Chemical group 0.000 claims abstract description 16
- ZBKFYXZXZJPWNQ-UHFFFAOYSA-N isothiocyanate group Chemical group [N-]=C=S ZBKFYXZXZJPWNQ-UHFFFAOYSA-N 0.000 claims abstract description 16
- 239000003112 inhibitor Substances 0.000 claims abstract description 15
- NMHMNPHRMNGLLB-UHFFFAOYSA-N phloretic acid Chemical group OC(=O)CCC1=CC=C(O)C=C1 NMHMNPHRMNGLLB-UHFFFAOYSA-N 0.000 claims abstract description 15
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 claims abstract description 14
- 239000000137 peptide hydrolase inhibitor Substances 0.000 claims abstract description 14
- 239000005556 hormone Substances 0.000 claims abstract description 13
- 229940088597 hormone Drugs 0.000 claims abstract description 13
- 239000000824 cytostatic agent Substances 0.000 claims abstract description 12
- 239000003242 anti bacterial agent Substances 0.000 claims abstract description 11
- 230000001741 anti-phlogistic effect Effects 0.000 claims abstract description 11
- 239000003429 antifungal agent Substances 0.000 claims abstract description 11
- 239000003443 antiviral agent Substances 0.000 claims abstract description 11
- 239000003018 immunosuppressive agent Substances 0.000 claims abstract description 11
- 229940125721 immunosuppressive agent Drugs 0.000 claims abstract description 11
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 claims abstract description 10
- 102000004127 Cytokines Human genes 0.000 claims abstract description 10
- 108090000695 Cytokines Proteins 0.000 claims abstract description 10
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 claims abstract description 10
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 claims abstract description 10
- 229940079156 Proteasome inhibitor Drugs 0.000 claims abstract description 10
- 230000000202 analgesic effect Effects 0.000 claims abstract description 10
- 239000004037 angiogenesis inhibitor Substances 0.000 claims abstract description 10
- 229940121369 angiogenesis inhibitor Drugs 0.000 claims abstract description 10
- 229940121363 anti-inflammatory agent Drugs 0.000 claims abstract description 10
- 230000003356 anti-rheumatic effect Effects 0.000 claims abstract description 10
- 239000003435 antirheumatic agent Substances 0.000 claims abstract description 10
- 229940076005 apoptosis modulator Drugs 0.000 claims abstract description 10
- 230000003115 biocidal effect Effects 0.000 claims abstract description 10
- 229940076006 cell cycle modulator Drugs 0.000 claims abstract description 10
- 229940127089 cytotoxic agent Drugs 0.000 claims abstract description 10
- 239000002254 cytotoxic agent Substances 0.000 claims abstract description 10
- 231100000599 cytotoxic agent Toxicity 0.000 claims abstract description 10
- 239000002532 enzyme inhibitor Substances 0.000 claims abstract description 10
- 229940125532 enzyme inhibitor Drugs 0.000 claims abstract description 10
- 239000003207 proteasome inhibitor Substances 0.000 claims abstract description 10
- 239000000941 radioactive substance Substances 0.000 claims abstract description 10
- 229940121375 antifungal agent Drugs 0.000 claims abstract description 9
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims abstract description 9
- 230000019491 signal transduction Effects 0.000 claims abstract description 9
- 239000003688 hormone derivative Substances 0.000 claims abstract description 7
- 229940122954 Transcription factor inhibitor Drugs 0.000 claims abstract description 5
- 125000000217 alkyl group Chemical group 0.000 claims description 187
- 206010028980 Neoplasm Diseases 0.000 claims description 98
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 claims description 74
- 229960005277 gemcitabine Drugs 0.000 claims description 72
- 229910052736 halogen Inorganic materials 0.000 claims description 64
- 150000002367 halogens Chemical class 0.000 claims description 64
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 44
- 235000018102 proteins Nutrition 0.000 claims description 36
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 33
- 239000002253 acid Substances 0.000 claims description 29
- 201000010099 disease Diseases 0.000 claims description 29
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 29
- 201000011510 cancer Diseases 0.000 claims description 27
- 102000009027 Albumins Human genes 0.000 claims description 25
- 108010088751 Albumins Proteins 0.000 claims description 25
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 25
- 125000001188 haloalkyl group Chemical group 0.000 claims description 25
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 25
- 235000001014 amino acid Nutrition 0.000 claims description 23
- 125000001424 substituent group Chemical group 0.000 claims description 23
- 229950010159 nemorubicin Drugs 0.000 claims description 20
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 20
- 229910001413 alkali metal ion Inorganic materials 0.000 claims description 19
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 claims description 19
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 claims description 18
- CTMCWCONSULRHO-UHQPFXKFSA-N nemorubicin Chemical compound C1CO[C@H](OC)CN1[C@@H]1[C@H](O)[C@H](C)O[C@@H](O[C@@H]2C3=C(O)C=4C(=O)C5=C(OC)C=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)C1 CTMCWCONSULRHO-UHQPFXKFSA-N 0.000 claims description 17
- 125000002252 acyl group Chemical group 0.000 claims description 16
- RVFGKBWWUQOIOU-NDEPHWFRSA-N lurtotecan Chemical compound O=C([C@]1(O)CC)OCC(C(N2CC3=4)=O)=C1C=C2C3=NC1=CC=2OCCOC=2C=C1C=4CN1CCN(C)CC1 RVFGKBWWUQOIOU-NDEPHWFRSA-N 0.000 claims description 16
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 claims description 15
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims description 14
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 claims description 13
- 229960004679 doxorubicin Drugs 0.000 claims description 13
- 229960003048 vinblastine Drugs 0.000 claims description 13
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 claims description 13
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 claims description 12
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 11
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 claims description 10
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 claims description 10
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 claims description 10
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 claims description 10
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 claims description 10
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 claims description 10
- 108010044540 auristatin Proteins 0.000 claims description 10
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 claims description 10
- 229940127093 camptothecin Drugs 0.000 claims description 10
- 235000018417 cysteine Nutrition 0.000 claims description 10
- 229960000975 daunorubicin Drugs 0.000 claims description 10
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 10
- 229960001904 epirubicin Drugs 0.000 claims description 10
- 229960000908 idarubicin Drugs 0.000 claims description 10
- 229960001156 mitoxantrone Drugs 0.000 claims description 10
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 10
- IEJSCSAMMLUINT-NRFANRHFSA-N (2s)-2-[[4-[(2,7-dimethyl-4-oxo-1h-quinazolin-6-yl)methyl-prop-2-ynylamino]-2-fluorobenzoyl]amino]-4-(2h-tetrazol-5-yl)butanoic acid Chemical compound C([C@H](NC(=O)C1=CC=C(C=C1F)N(CC#C)CC=1C=C2C(=O)N=C(NC2=CC=1C)C)C(O)=O)CC=1N=NNN=1 IEJSCSAMMLUINT-NRFANRHFSA-N 0.000 claims description 9
- SLURUCSFDHKXFR-WWMWMSKMSA-N (7s,9s)-7-[[(1s,3r,4as,9s,9ar,10as)-9-methoxy-1-methyl-3,4,4a,6,7,9,9a,10a-octahydro-1h-pyrano[1,2][1,3]oxazolo[3,4-b][1,4]oxazin-3-yl]oxy]-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound O=C1C2=CC=CC(OC)=C2C(=O)C(C(O)=C23)=C1C(O)=C3C[C@@](O)(C(=O)CO)C[C@@H]2O[C@H]1C[C@@H]2N3CCO[C@H](OC)[C@H]3O[C@@H]2[C@H](C)O1 SLURUCSFDHKXFR-WWMWMSKMSA-N 0.000 claims description 9
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 claims description 9
- HAWSQZCWOQZXHI-FQEVSTJZSA-N 10-Hydroxycamptothecin Chemical compound C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-FQEVSTJZSA-N 0.000 claims description 9
- ZZVDXRCAGGQFAK-UHFFFAOYSA-N 2h-oxazaphosphinine Chemical class N1OC=CC=P1 ZZVDXRCAGGQFAK-UHFFFAOYSA-N 0.000 claims description 9
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 claims description 9
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 claims description 9
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 claims description 9
- FUXVKZWTXQUGMW-FQEVSTJZSA-N 9-Aminocamptothecin Chemical compound C1=CC(N)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 FUXVKZWTXQUGMW-FQEVSTJZSA-N 0.000 claims description 9
- 108010006654 Bleomycin Proteins 0.000 claims description 9
- HAWSQZCWOQZXHI-UHFFFAOYSA-N CPT-OH Natural products C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-UHFFFAOYSA-N 0.000 claims description 9
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 claims description 9
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 9
- 108010092160 Dactinomycin Proteins 0.000 claims description 9
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 claims description 9
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 claims description 9
- 229930012538 Paclitaxel Natural products 0.000 claims description 9
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 claims description 9
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 claims description 9
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 claims description 9
- 229960002707 bendamustine Drugs 0.000 claims description 9
- 229960001561 bleomycin Drugs 0.000 claims description 9
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 claims description 9
- 229930195731 calicheamicin Natural products 0.000 claims description 9
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 claims description 9
- 229960004630 chlorambucil Drugs 0.000 claims description 9
- 229960002436 cladribine Drugs 0.000 claims description 9
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 9
- 229960000684 cytarabine Drugs 0.000 claims description 9
- 229960000640 dactinomycin Drugs 0.000 claims description 9
- 229960003668 docetaxel Drugs 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 9
- 229930013356 epothilone Natural products 0.000 claims description 9
- 229960000390 fludarabine Drugs 0.000 claims description 9
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 claims description 9
- 229960002949 fluorouracil Drugs 0.000 claims description 9
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 claims description 9
- 229960004768 irinotecan Drugs 0.000 claims description 9
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 claims description 9
- 229960001924 melphalan Drugs 0.000 claims description 9
- 229960000485 methotrexate Drugs 0.000 claims description 9
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 claims description 9
- 229960001592 paclitaxel Drugs 0.000 claims description 9
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 claims description 9
- 229960005079 pemetrexed Drugs 0.000 claims description 9
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 claims description 9
- 229960002340 pentostatin Drugs 0.000 claims description 9
- HRGDZIGMBDGFTC-UHFFFAOYSA-N platinum(2+) Chemical class [Pt+2] HRGDZIGMBDGFTC-UHFFFAOYSA-N 0.000 claims description 9
- 229950001461 plevitrexed Drugs 0.000 claims description 9
- 229960003171 plicamycin Drugs 0.000 claims description 9
- 229960004432 raltitrexed Drugs 0.000 claims description 9
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 9
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 claims description 9
- 229960003087 tioguanine Drugs 0.000 claims description 9
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 claims description 9
- 229960000303 topotecan Drugs 0.000 claims description 9
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 claims description 9
- 229960004528 vincristine Drugs 0.000 claims description 9
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 claims description 9
- 229960004355 vindesine Drugs 0.000 claims description 9
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 claims description 9
- 229960002066 vinorelbine Drugs 0.000 claims description 9
- FFGSXKJJVBXWCY-UHFFFAOYSA-N 1,4-bis[2-(2-hydroxyethylamino)ethylamino]anthracene-9,10-dione Chemical compound O=C1C2=CC=CC=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO FFGSXKJJVBXWCY-UHFFFAOYSA-N 0.000 claims description 8
- WRZBVYYRSDNCLD-APTPUMFKSA-N NC1=NC(N(C=C1F)C[C@@H]1O[C@@H]([C@H]([C@H]1O)O)C)=O Chemical compound NC1=NC(N(C=C1F)C[C@@H]1O[C@@H]([C@H]([C@H]1O)O)C)=O WRZBVYYRSDNCLD-APTPUMFKSA-N 0.000 claims description 8
- 229950011363 ametantrone Drugs 0.000 claims description 8
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 claims description 8
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 claims description 8
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 claims description 8
- 229950002654 lurtotecan Drugs 0.000 claims description 8
- 108010059074 monomethylauristatin F Proteins 0.000 claims description 8
- XKIDBQHJMYSFPX-NRFANRHFSA-N 80758-83-4 Chemical compound C1=CC=C2C(C=O)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 XKIDBQHJMYSFPX-NRFANRHFSA-N 0.000 claims description 7
- 208000023275 Autoimmune disease Diseases 0.000 claims description 7
- 241000233866 Fungi Species 0.000 claims description 7
- 230000001154 acute effect Effects 0.000 claims description 7
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 claims description 7
- 244000005700 microbiome Species 0.000 claims description 7
- 230000003612 virological effect Effects 0.000 claims description 7
- 241000894006 Bacteria Species 0.000 claims description 6
- 125000003368 amide group Chemical group 0.000 claims description 6
- 208000030090 Acute Disease Diseases 0.000 claims description 5
- 108091023040 Transcription factor Proteins 0.000 claims description 5
- 102000040945 Transcription factor Human genes 0.000 claims description 5
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 claims description 5
- 208000037976 chronic inflammation Diseases 0.000 claims description 5
- 208000037893 chronic inflammatory disorder Diseases 0.000 claims description 5
- 125000004185 ester group Chemical group 0.000 claims description 5
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 5
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 5
- QWPXBEHQFHACTK-KZVYIGENSA-N (10e,12e)-86-chloro-12,14,4-trihydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-15,16-dihydro-14h-7-aza-1(6,4)-oxazina-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-6-one Chemical compound CN1C(=O)CC(O)C2(C)OC2C(C)C(OC(=O)N2)CC2(O)C(OC)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 QWPXBEHQFHACTK-KZVYIGENSA-N 0.000 claims description 4
- LGNCNVVZCUVPOT-FUVGGWJZSA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-methoxy-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 LGNCNVVZCUVPOT-FUVGGWJZSA-N 0.000 claims description 4
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 claims description 4
- WOWDZACBATWTAU-FEFUEGSOSA-N (2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-n-[(3r,4s,5s)-1-[(2s)-2-[(1r,2r)-3-[[(1s,2r)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-n,3-dimethylbutanamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)C1=CC=CC=C1 WOWDZACBATWTAU-FEFUEGSOSA-N 0.000 claims description 4
- BLUGYPPOFIHFJS-UUFHNPECSA-N (2s)-n-[(2s)-1-[[(3r,4s,5s)-3-methoxy-1-[(2s)-2-[(1r,2r)-1-methoxy-2-methyl-3-oxo-3-[[(1s)-2-phenyl-1-(1,3-thiazol-2-yl)ethyl]amino]propyl]pyrrolidin-1-yl]-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]-3-methyl-2-(methylamino)butanamid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C=1SC=CN=1)CC1=CC=CC=C1 BLUGYPPOFIHFJS-UUFHNPECSA-N 0.000 claims description 4
- LQDGLTOVYOUCRG-QMMMGPOBSA-N (6as)-3-hydroxy-2-methoxy-6a,7,8,9-tetrahydropyrrolo[2,1-c][1,4]benzodiazepin-11-one Chemical compound N1=C[C@@H]2CCCN2C(=O)C2=C1C=C(O)C(OC)=C2 LQDGLTOVYOUCRG-QMMMGPOBSA-N 0.000 claims description 4
- UQVNRKBFAXNOGA-OHLDGCSVSA-N (Z)-tomaymycin Chemical compound CO[C@H]1NC2=CC(O)=C(OC)C=C2C(=O)N2C\C(=C/C)C[C@@H]12 UQVNRKBFAXNOGA-OHLDGCSVSA-N 0.000 claims description 4
- FONKWHRXTPJODV-DNQXCXABSA-N 1,3-bis[2-[(8s)-8-(chloromethyl)-4-hydroxy-1-methyl-7,8-dihydro-3h-pyrrolo[3,2-e]indole-6-carbonyl]-1h-indol-5-yl]urea Chemical compound C1([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)NC=3C=C4C=C(NC4=CC=3)C(=O)N3C4=CC(O)=C5NC=C(C5=C4[C@H](CCl)C3)C)=C2C=C(O)C2=C1C(C)=CN2 FONKWHRXTPJODV-DNQXCXABSA-N 0.000 claims description 4
- BOHCOUQZNDPURZ-ICNZIKDASA-N 2-[(1R,4S,8R,10S,13S,16S,27R,34S)-34-[(2S)-butan-2-yl]-13-[(2R,3R)-3,4-dihydroxybutan-2-yl]-8-hydroxy-2,5,11,14,27,30,33,36,39-nonaoxo-27lambda4-thia-3,6,12,15,25,29,32,35,38-nonazapentacyclo[14.12.11.06,10.018,26.019,24]nonatriaconta-18(26),19,21,23-tetraen-4-yl]acetamide Chemical compound CC[C@H](C)[C@@H]1NC(=O)CNC(=O)[C@@H]2Cc3c([nH]c4ccccc34)[S@](=O)C[C@H](NC(=O)CNC1=O)C(=O)N[C@@H](CC(N)=O)C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H]([C@@H](C)[C@@H](O)CO)C(=O)N2 BOHCOUQZNDPURZ-ICNZIKDASA-N 0.000 claims description 4
- YSNABXSEHNLERR-ZIYNGMLESA-N 5'-Deoxy-5-fluorocytidine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)N=C(N)C(F)=C1 YSNABXSEHNLERR-ZIYNGMLESA-N 0.000 claims description 4
- OHSHVNCARFVERY-QFIPXVFZSA-N 7-acetyl camptothecin Chemical compound C1=CC=C2C(C(C)=O)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 OHSHVNCARFVERY-QFIPXVFZSA-N 0.000 claims description 4
- QCZXQEYEVLCQHL-UHFFFAOYSA-N Amanin Natural products O=C1NC(CC(O)=O)C(=O)N2CC(O)CC2C(=O)NC(C(C)C(O)CO)C(=O)NC(C2)C(=O)NCC(=O)NC(C(C)CC)C(=O)NCC(=O)NC1CS(=O)C1=C2C2=CC=CC=C2N1 QCZXQEYEVLCQHL-UHFFFAOYSA-N 0.000 claims description 4
- 231100000729 Amatoxin Toxicity 0.000 claims description 4
- 229960005532 CC-1065 Drugs 0.000 claims description 4
- OFDNQWIFNXBECV-UHFFFAOYSA-N Dolastatin 10 Natural products CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)CC)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-UHFFFAOYSA-N 0.000 claims description 4
- AZVARJHZBXHUSO-UHFFFAOYSA-N Duocarmycin A Natural products COC1=C(OC)C(OC)=C2NC(C(=O)N3CC4CC44C5=C(C(C=C43)=O)NC(C5=O)(C)C(=O)OC)=CC2=C1 AZVARJHZBXHUSO-UHFFFAOYSA-N 0.000 claims description 4
- VQNATVDKACXKTF-UHFFFAOYSA-N Duocarmycin SA Natural products COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C(C64CC6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-UHFFFAOYSA-N 0.000 claims description 4
- XXPXYPLPSDPERN-UHFFFAOYSA-N Ecteinascidin 743 Natural products COc1cc2C(NCCc2cc1O)C(=O)OCC3N4C(O)C5Cc6cc(C)c(OC)c(O)c6C(C4C(S)c7c(OC(=O)C)c(C)c8OCOc8c37)N5C XXPXYPLPSDPERN-UHFFFAOYSA-N 0.000 claims description 4
- 229930126263 Maytansine Natural products 0.000 claims description 4
- QWPXBEHQFHACTK-UHFFFAOYSA-N Maytansinol Natural products CN1C(=O)CC(O)C2(C)OC2C(C)C(OC(=O)N2)CC2(O)C(OC)C=CC=C(C)CC2=CC(OC)=C(Cl)C1=C2 QWPXBEHQFHACTK-UHFFFAOYSA-N 0.000 claims description 4
- VGQOVCHZGQWAOI-UHFFFAOYSA-N UNPD55612 Natural products N1C(O)C2CC(C=CC(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-UHFFFAOYSA-N 0.000 claims description 4
- BYRVKDUQDLJUBX-JJCDCTGGSA-N adozelesin Chemical compound C1=CC=C2OC(C(=O)NC=3C=C4C=C(NC4=CC=3)C(=O)N3C[C@H]4C[C@]44C5=C(C(C=C43)=O)NC=C5C)=CC2=C1 BYRVKDUQDLJUBX-JJCDCTGGSA-N 0.000 claims description 4
- 229950004955 adozelesin Drugs 0.000 claims description 4
- 229910052783 alkali metal Inorganic materials 0.000 claims description 4
- QCZXQEYEVLCQHL-MIBTZWEZSA-N amanin Chemical compound O=C1N[C@@H](CC(O)=O)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@@H]([C@@H](C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@H]1CS(=O)C1=C2C2=CC=CC=C2N1 QCZXQEYEVLCQHL-MIBTZWEZSA-N 0.000 claims description 4
- BOHCOUQZNDPURZ-UHFFFAOYSA-N amaninamide Natural products O=C1NC(CC(N)=O)C(=O)N2CC(O)CC2C(=O)NC(C(C)C(O)CO)C(=O)NC(C2)C(=O)NCC(=O)NC(C(C)CC)C(=O)NCC(=O)NC1CS(=O)C1=C2C2=CC=CC=C2N1 BOHCOUQZNDPURZ-UHFFFAOYSA-N 0.000 claims description 4
- 108010004258 amaninamide Proteins 0.000 claims description 4
- VGQOVCHZGQWAOI-HYUHUPJXSA-N anthramycin Chemical compound N1[C@@H](O)[C@@H]2CC(\C=C\C(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-HYUHUPJXSA-N 0.000 claims description 4
- 108010008739 auristatin PHE Proteins 0.000 claims description 4
- 108010009065 auristatin PYE Proteins 0.000 claims description 4
- 229950006844 bizelesin Drugs 0.000 claims description 4
- BBZDXMBRAFTCAA-AREMUKBSSA-N carzelesin Chemical compound C1=2NC=C(C)C=2C([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)C3=CC4=CC=C(C=C4O3)N(CC)CC)=C2C=C1OC(=O)NC1=CC=CC=C1 BBZDXMBRAFTCAA-AREMUKBSSA-N 0.000 claims description 4
- 229950007509 carzelesin Drugs 0.000 claims description 4
- OFDNQWIFNXBECV-VFSYNPLYSA-N dolastatin 10 Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-VFSYNPLYSA-N 0.000 claims description 4
- 108010045524 dolastatin 10 Proteins 0.000 claims description 4
- 229960005501 duocarmycin Drugs 0.000 claims description 4
- 229930184221 duocarmycin Natural products 0.000 claims description 4
- 229960005519 duocarmycin A Drugs 0.000 claims description 4
- 229960005513 duocarmycin B1 Drugs 0.000 claims description 4
- NIADGRRCOZRRQF-UHFFFAOYSA-N duocarmycin B1 Natural products COC(=O)C1(C)NC2=C(C3CC(Br)CN(C(=O)c4cc5cc(OC)c(OC)c(OC)c5[nH]4)C3=CC2=O)C1=O NIADGRRCOZRRQF-UHFFFAOYSA-N 0.000 claims description 4
- 229960005514 duocarmycin B2 Drugs 0.000 claims description 4
- UQPQXFUURNIVNJ-UHFFFAOYSA-N duocarmycin B2 Natural products COC1=C(OC)C(OC)=C2NC(C(=O)N3CC(CBr)C=4C5=C(C(=CC=43)O)NC(C5=O)(C)C(=O)OC)=CC2=C1 UQPQXFUURNIVNJ-UHFFFAOYSA-N 0.000 claims description 4
- 229960005510 duocarmycin SA Drugs 0.000 claims description 4
- UFNVPOGXISZXJD-XJPMSQCNSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-XJPMSQCNSA-N 0.000 claims description 4
- 229960003649 eribulin Drugs 0.000 claims description 4
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 claims description 4
- AZVARJHZBXHUSO-DZQVEHCYSA-N methyl (1R,4R,12S)-4-methyl-3,7-dioxo-10-(5,6,7-trimethoxy-1H-indole-2-carbonyl)-5,10-diazatetracyclo[7.4.0.01,12.02,6]trideca-2(6),8-diene-4-carboxylate Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C[C@H]4C[C@]44C5=C(C(C=C43)=O)N[C@@](C5=O)(C)C(=O)OC)=CC2=C1 AZVARJHZBXHUSO-DZQVEHCYSA-N 0.000 claims description 4
- UQPQXFUURNIVNJ-MZHQLVBMSA-N methyl (2r,8s)-8-(bromomethyl)-4-hydroxy-2-methyl-1-oxo-6-(5,6,7-trimethoxy-1h-indole-2-carbonyl)-7,8-dihydro-3h-pyrrolo[3,2-e]indole-2-carboxylate Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C[C@@H](CBr)C=4C5=C(C(=CC=43)O)N[C@@](C5=O)(C)C(=O)OC)=CC2=C1 UQPQXFUURNIVNJ-MZHQLVBMSA-N 0.000 claims description 4
- SUWUAMDOMCWKCL-GWQKEKGPSA-N methyl (2r,8s)-8-bromo-4-hydroxy-2-methyl-1-oxo-6-(5,6,7-trimethoxy-1h-indole-2-carbonyl)-3,7,8,9-tetrahydropyrrolo[3,2-f]quinoline-2-carboxylate Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C[C@@H](Br)CC=4C5=C(C(=CC=43)O)N[C@@](C5=O)(C)C(=O)OC)=CC2=C1 SUWUAMDOMCWKCL-GWQKEKGPSA-N 0.000 claims description 4
- 108010093470 monomethyl auristatin E Proteins 0.000 claims description 4
- 229930014626 natural product Natural products 0.000 claims description 4
- YUOCYTRGANSSRY-UHFFFAOYSA-N pyrrolo[2,3-i][1,2]benzodiazepine Chemical compound C1=CN=NC2=C3C=CN=C3C=CC2=C1 YUOCYTRGANSSRY-UHFFFAOYSA-N 0.000 claims description 4
- RAGFPHFDFVNLCG-INYQBOQCSA-N sibiromycin Chemical compound O[C@@H]1[C@@](O)(C)[C@@H](NC)[C@H](C)O[C@H]1OC(C(=C1O)C)=CC(C2=O)=C1N[C@H](O)[C@H]1N2C=C(\C=C\C)C1 RAGFPHFDFVNLCG-INYQBOQCSA-N 0.000 claims description 4
- RAGFPHFDFVNLCG-UHFFFAOYSA-N sibiromycin Natural products OC1C(O)(C)C(NC)C(C)OC1OC(C(=C1O)C)=CC(C2=O)=C1NC(O)C1N2C=C(C=CC)C1 RAGFPHFDFVNLCG-UHFFFAOYSA-N 0.000 claims description 4
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 claims description 4
- 229960000977 trabectedin Drugs 0.000 claims description 4
- 125000001033 ether group Chemical group 0.000 claims description 3
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 3
- 150000003863 ammonium salts Chemical class 0.000 claims description 2
- 125000001302 tertiary amino group Chemical group 0.000 claims description 2
- 125000002947 alkylene group Chemical group 0.000 claims 3
- 125000005677 ethinylene group Chemical group [*:2]C#C[*:1] 0.000 claims 3
- HHJUWIANJFBDHT-KOTLKJBCSA-N vindesine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(N)=O)N4C)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 HHJUWIANJFBDHT-KOTLKJBCSA-N 0.000 claims 2
- PXYUMIVMMHXGBH-OALUTQOASA-N (6as)-3-[3-[[(6as)-2-methoxy-11-oxo-6a,7,8,9-tetrahydropyrrolo[2,1-c][1,4]benzodiazepin-3-yl]oxy]propoxy]-2-methoxy-6a,7,8,9-tetrahydropyrrolo[2,1-c][1,4]benzodiazepin-11-one Chemical compound N1=C[C@@H]2CCCN2C(=O)C(C=C2OC)=C1C=C2OCCCOC1=CC(N=C[C@H]2N(CCC2)C2=O)=C2C=C1OC PXYUMIVMMHXGBH-OALUTQOASA-N 0.000 claims 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 abstract description 13
- 125000006702 (C1-C18) alkyl group Chemical group 0.000 abstract 21
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 abstract 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract 2
- 239000000203 mixture Substances 0.000 description 134
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 114
- 235000002639 sodium chloride Nutrition 0.000 description 77
- 239000000243 solution Substances 0.000 description 74
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 54
- 239000007787 solid Substances 0.000 description 50
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 48
- 229940125758 compound 15 Drugs 0.000 description 48
- 239000003814 drug Substances 0.000 description 47
- 238000000034 method Methods 0.000 description 37
- 239000002904 solvent Substances 0.000 description 36
- 238000006243 chemical reaction Methods 0.000 description 33
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 32
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 31
- 241001465754 Metazoa Species 0.000 description 31
- 238000004128 high performance liquid chromatography Methods 0.000 description 31
- 239000000427 antigen Substances 0.000 description 30
- 108091007433 antigens Proteins 0.000 description 30
- 102000036639 antigens Human genes 0.000 description 30
- 229940079593 drug Drugs 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 30
- 125000003118 aryl group Chemical group 0.000 description 28
- 230000015572 biosynthetic process Effects 0.000 description 28
- 238000003786 synthesis reaction Methods 0.000 description 26
- 125000005647 linker group Chemical group 0.000 description 24
- 238000003756 stirring Methods 0.000 description 23
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 22
- 239000000546 pharmaceutical excipient Substances 0.000 description 22
- 238000011282 treatment Methods 0.000 description 22
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 21
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 20
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 20
- 238000012377 drug delivery Methods 0.000 description 20
- 229910052799 carbon Inorganic materials 0.000 description 19
- 241000699670 Mus sp. Species 0.000 description 18
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 18
- 239000002244 precipitate Substances 0.000 description 18
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 17
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 17
- 102000008100 Human Serum Albumin Human genes 0.000 description 17
- 108091006905 Human Serum Albumin Proteins 0.000 description 17
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 17
- 239000000562 conjugate Substances 0.000 description 17
- 239000000047 product Substances 0.000 description 17
- 229940024606 amino acid Drugs 0.000 description 16
- 239000012064 sodium phosphate buffer Substances 0.000 description 16
- 208000031648 Body Weight Changes Diseases 0.000 description 15
- 206010033128 Ovarian cancer Diseases 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 230000004579 body weight change Effects 0.000 description 15
- 210000004027 cell Anatomy 0.000 description 15
- 230000004614 tumor growth Effects 0.000 description 15
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- 239000000872 buffer Substances 0.000 description 14
- 230000000694 effects Effects 0.000 description 14
- 230000037396 body weight Effects 0.000 description 13
- 239000011575 calcium Substances 0.000 description 13
- 239000012535 impurity Substances 0.000 description 13
- 239000007788 liquid Substances 0.000 description 13
- 239000011734 sodium Substances 0.000 description 13
- 229940124597 therapeutic agent Drugs 0.000 description 13
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 12
- 108060003951 Immunoglobulin Proteins 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- 238000009472 formulation Methods 0.000 description 12
- 125000000623 heterocyclic group Chemical group 0.000 description 12
- 125000005597 hydrazone group Chemical group 0.000 description 12
- 150000007857 hydrazones Chemical group 0.000 description 12
- 102000018358 immunoglobulin Human genes 0.000 description 12
- 238000002360 preparation method Methods 0.000 description 12
- 239000003381 stabilizer Substances 0.000 description 12
- 239000004094 surface-active agent Substances 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- 239000004067 bulking agent Substances 0.000 description 11
- 238000013270 controlled release Methods 0.000 description 11
- 125000004122 cyclic group Chemical group 0.000 description 11
- 239000003085 diluting agent Substances 0.000 description 11
- 238000001990 intravenous administration Methods 0.000 description 11
- 102000004196 processed proteins & peptides Human genes 0.000 description 11
- 238000002560 therapeutic procedure Methods 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 229940049595 antibody-drug conjugate Drugs 0.000 description 10
- 230000009286 beneficial effect Effects 0.000 description 10
- 239000008393 encapsulating agent Substances 0.000 description 10
- 125000005842 heteroatom Chemical group 0.000 description 10
- 239000012074 organic phase Substances 0.000 description 10
- 208000008443 pancreatic carcinoma Diseases 0.000 description 10
- 239000006228 supernatant Substances 0.000 description 10
- 101150065749 Churc1 gene Proteins 0.000 description 9
- 206010009944 Colon cancer Diseases 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 229940045799 anthracyclines and related substance Drugs 0.000 description 9
- 125000004429 atom Chemical group 0.000 description 9
- 239000012043 crude product Substances 0.000 description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 9
- 239000012071 phase Substances 0.000 description 9
- 229920001184 polypeptide Polymers 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- 239000004034 viscosity adjusting agent Substances 0.000 description 9
- 201000009030 Carcinoma Diseases 0.000 description 8
- 229910019142 PO4 Inorganic materials 0.000 description 8
- 239000000611 antibody drug conjugate Substances 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 239000000969 carrier Substances 0.000 description 8
- 239000002577 cryoprotective agent Substances 0.000 description 8
- 125000000753 cycloalkyl group Chemical group 0.000 description 8
- 239000008103 glucose Substances 0.000 description 8
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 8
- 239000010452 phosphate Substances 0.000 description 8
- 238000005406 washing Methods 0.000 description 8
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 7
- 229940126657 Compound 17 Drugs 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- 206010061535 Ovarian neoplasm Diseases 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 150000001721 carbon Chemical group 0.000 description 7
- 235000019439 ethyl acetate Nutrition 0.000 description 7
- 230000006870 function Effects 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 239000003002 pH adjusting agent Substances 0.000 description 7
- 239000008363 phosphate buffer Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 235000000346 sugar Nutrition 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- 231100000419 toxicity Toxicity 0.000 description 7
- 230000001988 toxicity Effects 0.000 description 7
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 7
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 6
- 150000007513 acids Chemical class 0.000 description 6
- 125000004414 alkyl thio group Chemical group 0.000 description 6
- 230000000890 antigenic effect Effects 0.000 description 6
- 239000003963 antioxidant agent Substances 0.000 description 6
- 235000006708 antioxidants Nutrition 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 229940126142 compound 16 Drugs 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 125000001183 hydrocarbyl group Chemical group 0.000 description 6
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 201000002528 pancreatic cancer Diseases 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 230000003442 weekly effect Effects 0.000 description 6
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 5
- 108090000531 Amidohydrolases Proteins 0.000 description 5
- 102000004092 Amidohydrolases Human genes 0.000 description 5
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 5
- 108090000371 Esterases Proteins 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 230000009471 action Effects 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 150000001720 carbohydrates Chemical class 0.000 description 5
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 5
- 239000006260 foam Substances 0.000 description 5
- 210000004408 hybridoma Anatomy 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 150000007523 nucleic acids Chemical class 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 208000000649 small cell carcinoma Diseases 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 150000005846 sugar alcohols Chemical class 0.000 description 5
- 150000008163 sugars Chemical class 0.000 description 5
- 230000032258 transport Effects 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 206010006417 Bronchial carcinoma Diseases 0.000 description 4
- 229920000858 Cyclodextrin Polymers 0.000 description 4
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 230000000259 anti-tumor effect Effects 0.000 description 4
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 4
- 235000014633 carbohydrates Nutrition 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 208000029742 colonic neoplasm Diseases 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 235000013305 food Nutrition 0.000 description 4
- 201000010175 gallbladder cancer Diseases 0.000 description 4
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical compound C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 208000032839 leukemia Diseases 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 201000001441 melanoma Diseases 0.000 description 4
- 102000039446 nucleic acids Human genes 0.000 description 4
- 108020004707 nucleic acids Proteins 0.000 description 4
- 150000002894 organic compounds Chemical class 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 4
- 125000003367 polycyclic group Polymers 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- 239000011550 stock solution Substances 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 3
- NQLRKKYEAIYVMB-HPZKVCJDSA-N (3ar)-7-[3-[[(6as)-11-oxo-6a,7,8,9-tetrahydropyrrolo[2,1-c][1,4]benzodiazepin-3-yl]oxy]propoxy]-8-methoxy-2,3,3a,10a-tetrahydro-1h-pyrrolo[3,2-c][1]benzazepin-10-one Chemical compound C1=C2N=C[C@@H]3CCCN3C(=O)C2=CC=C1OCCCOC1=CC(N=C[C@H]2C(NCC2)C2=O)=C2C=C1OC NQLRKKYEAIYVMB-HPZKVCJDSA-N 0.000 description 3
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 description 3
- QBHDSQZASIBAAI-UHFFFAOYSA-N 4-acetylbenzoic acid Chemical compound CC(=O)C1=CC=C(C(O)=O)C=C1 QBHDSQZASIBAAI-UHFFFAOYSA-N 0.000 description 3
- GGHVZKXIPULPLP-UHFFFAOYSA-N 6-(2,5-dioxopyrrol-1-yl)hexanoyl chloride Chemical compound ClC(=O)CCCCCN1C(=O)C=CC1=O GGHVZKXIPULPLP-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- 206010002091 Anaesthesia Diseases 0.000 description 3
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Natural products OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 3
- 101710201075 Carboxylesterase 2 Proteins 0.000 description 3
- 102100021864 Cocaine esterase Human genes 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 3
- 208000017604 Hodgkin disease Diseases 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 3
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 206010025323 Lymphomas Diseases 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical class ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 3
- 238000011786 NMRI nude mouse Methods 0.000 description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 3
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 3
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- 229940122803 Vinca alkaloid Drugs 0.000 description 3
- 229960004308 acetylcysteine Drugs 0.000 description 3
- 125000004423 acyloxy group Chemical group 0.000 description 3
- 230000002776 aggregation Effects 0.000 description 3
- 238000004220 aggregation Methods 0.000 description 3
- 150000001340 alkali metals Chemical class 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 3
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 3
- 150000001409 amidines Chemical class 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 3
- 230000037005 anaesthesia Effects 0.000 description 3
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 235000005911 diet Nutrition 0.000 description 3
- 230000037213 diet Effects 0.000 description 3
- 235000019441 ethanol Nutrition 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 235000020680 filtered tap water Nutrition 0.000 description 3
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 150000002466 imines Chemical class 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 229960002725 isoflurane Drugs 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- AWIJRPNMLHPLNC-UHFFFAOYSA-N methanethioic s-acid Chemical compound SC=O AWIJRPNMLHPLNC-UHFFFAOYSA-N 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 3
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 3
- 230000002035 prolonged effect Effects 0.000 description 3
- 239000008213 purified water Substances 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 125000006413 ring segment Chemical group 0.000 description 3
- 239000007974 sodium acetate buffer Substances 0.000 description 3
- 239000001488 sodium phosphate Substances 0.000 description 3
- 229910000162 sodium phosphate Inorganic materials 0.000 description 3
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- DKACXUFSLUYRFU-UHFFFAOYSA-N tert-butyl n-aminocarbamate Chemical compound CC(C)(C)OC(=O)NN DKACXUFSLUYRFU-UHFFFAOYSA-N 0.000 description 3
- 150000007970 thio esters Chemical class 0.000 description 3
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 3
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 3
- 229960000575 trastuzumab Drugs 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 3
- 235000019786 weight gain Nutrition 0.000 description 3
- ZPGDWQNBZYOZTI-SFHVURJKSA-N (2s)-1-(9h-fluoren-9-ylmethoxycarbonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 ZPGDWQNBZYOZTI-SFHVURJKSA-N 0.000 description 2
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 2
- ADFXKUOMJKEIND-UHFFFAOYSA-N 1,3-dicyclohexylurea Chemical compound C1CCCCC1NC(=O)NC1CCCCC1 ADFXKUOMJKEIND-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 2
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- SAJYSJVBNGUWJK-UHFFFAOYSA-N 4-amino-2-nitrobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C([N+]([O-])=O)=C1 SAJYSJVBNGUWJK-UHFFFAOYSA-N 0.000 description 2
- WOJKKJKETHYEAC-UHFFFAOYSA-N 6-Maleimidocaproic acid Chemical compound OC(=O)CCCCCN1C(=O)C=CC1=O WOJKKJKETHYEAC-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 206010000830 Acute leukaemia Diseases 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- 206010061424 Anal cancer Diseases 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 206010003571 Astrocytoma Diseases 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 102000004506 Blood Proteins Human genes 0.000 description 2
- 108010017384 Blood Proteins Proteins 0.000 description 2
- 206010005949 Bone cancer Diseases 0.000 description 2
- 208000018084 Bone neoplasm Diseases 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- 208000011691 Burkitt lymphomas Diseases 0.000 description 2
- 208000017897 Carcinoma of esophagus Diseases 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- 208000009738 Connective Tissue Neoplasms Diseases 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 206010014733 Endometrial cancer Diseases 0.000 description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 description 2
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 102100021888 Helix-loop-helix protein 1 Human genes 0.000 description 2
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 2
- 101000897691 Homo sapiens Helix-loop-helix protein 1 Proteins 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- 206010023825 Laryngeal cancer Diseases 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 2
- 208000000172 Medulloblastoma Diseases 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- DIKAGCUVAYMPMG-UHFFFAOYSA-N NC1=CC(=C(C(=O)NNC(=O)OC(C)(C)C)C=C1)F Chemical compound NC1=CC(=C(C(=O)NNC(=O)OC(C)(C)C)C=C1)F DIKAGCUVAYMPMG-UHFFFAOYSA-N 0.000 description 2
- UIPDRQRERPNNOR-UHFFFAOYSA-N NC1=CC(=C(C(=O)NNC(=O)OC(C)(C)C)C=C1)[N+](=O)[O-] Chemical compound NC1=CC(=C(C(=O)NNC(=O)OC(C)(C)C)C=C1)[N+](=O)[O-] UIPDRQRERPNNOR-UHFFFAOYSA-N 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 2
- 201000010133 Oligodendroglioma Diseases 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 102000057297 Pepsin A Human genes 0.000 description 2
- 108090000284 Pepsin A Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- 208000006265 Renal cell carcinoma Diseases 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- 102000007562 Serum Albumin Human genes 0.000 description 2
- 108010071390 Serum Albumin Proteins 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 description 2
- 206010057644 Testis cancer Diseases 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 206010062129 Tongue neoplasm Diseases 0.000 description 2
- 206010046431 Urethral cancer Diseases 0.000 description 2
- 206010046458 Urethral neoplasms Diseases 0.000 description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 description 2
- 201000005969 Uveal melanoma Diseases 0.000 description 2
- 208000014070 Vestibular schwannoma Diseases 0.000 description 2
- 208000004064 acoustic neuroma Diseases 0.000 description 2
- 208000009956 adenocarcinoma Diseases 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 201000007434 ampulla of Vater carcinoma Diseases 0.000 description 2
- 201000007538 anal carcinoma Diseases 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 210000000628 antibody-producing cell Anatomy 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 208000011825 carcinoma of the ampulla of vater Diseases 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 201000010989 colorectal carcinoma Diseases 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 150000002016 disaccharides Chemical class 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 201000003914 endometrial carcinoma Diseases 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 201000005619 esophageal carcinoma Diseases 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 238000007710 freezing Methods 0.000 description 2
- 230000008014 freezing Effects 0.000 description 2
- 201000007487 gallbladder carcinoma Diseases 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 210000004907 gland Anatomy 0.000 description 2
- 239000000174 gluconic acid Substances 0.000 description 2
- 235000012208 gluconic acid Nutrition 0.000 description 2
- 201000009277 hairy cell leukemia Diseases 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 201000005787 hematologic cancer Diseases 0.000 description 2
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 2
- 239000004312 hexamethylene tetramine Substances 0.000 description 2
- 235000010299 hexamethylene tetramine Nutrition 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 150000002454 idoses Chemical class 0.000 description 2
- 229940072221 immunoglobulins Drugs 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- 150000002596 lactones Chemical class 0.000 description 2
- 206010023841 laryngeal neoplasm Diseases 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 210000002751 lymph Anatomy 0.000 description 2
- 235000018977 lysine Nutrition 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 206010027191 meningioma Diseases 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 239000013580 millipore water Substances 0.000 description 2
- FVLVBVSILSHUAF-UHFFFAOYSA-N n-benzyl-3,5-dimethyl-n-propan-2-ylbenzamide Chemical compound C=1C(C)=CC(C)=CC=1C(=O)N(C(C)C)CC1=CC=CC=C1 FVLVBVSILSHUAF-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 208000025440 neoplasm of neck Diseases 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 230000000010 osteolytic effect Effects 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 229940111202 pepsin Drugs 0.000 description 2
- 229920001983 poloxamer Polymers 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 150000004804 polysaccharides Chemical class 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 238000002390 rotary evaporation Methods 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 201000002314 small intestine cancer Diseases 0.000 description 2
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 2
- 235000010378 sodium ascorbate Nutrition 0.000 description 2
- 229960005055 sodium ascorbate Drugs 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- BBMHARZCALWXSL-UHFFFAOYSA-M sodium dihydrogenphosphate monohydrate Chemical compound O.[Na+].OP(O)([O-])=O BBMHARZCALWXSL-UHFFFAOYSA-M 0.000 description 2
- 235000011008 sodium phosphates Nutrition 0.000 description 2
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 201000003120 testicular cancer Diseases 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- KCIXCKZANDLARF-UHFFFAOYSA-N toluene;2,2,2-trifluoroacetic acid Chemical compound CC1=CC=CC=C1.OC(=O)C(F)(F)F KCIXCKZANDLARF-UHFFFAOYSA-N 0.000 description 2
- 201000006134 tongue cancer Diseases 0.000 description 2
- WVLBCYQITXONBZ-UHFFFAOYSA-N trimethyl phosphate Chemical compound COP(=O)(OC)OC WVLBCYQITXONBZ-UHFFFAOYSA-N 0.000 description 2
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 206010046766 uterine cancer Diseases 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- QXVUNVYPQOAFMQ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-acetylbenzoate Chemical compound C1=CC(C(=O)C)=CC=C1C(=O)ON1C(=O)CCC1=O QXVUNVYPQOAFMQ-UHFFFAOYSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- OEANUJAFZLQYOD-CXAZCLJRSA-N (2r,3s,4r,5r,6r)-6-[(2r,3r,4r,5r,6r)-5-acetamido-3-hydroxy-2-(hydroxymethyl)-6-methoxyoxan-4-yl]oxy-4,5-dihydroxy-3-methoxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](OC)O[C@H](CO)[C@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](OC)[C@H](C(O)=O)O1 OEANUJAFZLQYOD-CXAZCLJRSA-N 0.000 description 1
- ZFTFOHBYVDOAMH-XNOIKFDKSA-N (2r,3s,4s,5r)-5-[[(2r,3s,4s,5r)-5-[[(2r,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxymethyl]-3,4-dihydroxy-2-(hydroxymethyl)oxolan-2-yl]oxymethyl]-2-(hydroxymethyl)oxolane-2,3,4-triol Chemical class O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@@H]1[C@@H](O)[C@H](O)[C@](CO)(OC[C@@H]2[C@H]([C@H](O)[C@@](O)(CO)O2)O)O1 ZFTFOHBYVDOAMH-XNOIKFDKSA-N 0.000 description 1
- QZNNVYOVQUKYSC-JEDNCBNOSA-N (2s)-2-amino-3-(1h-imidazol-5-yl)propanoic acid;hydron;chloride Chemical compound Cl.OC(=O)[C@@H](N)CC1=CN=CN1 QZNNVYOVQUKYSC-JEDNCBNOSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- YWNKNCQONKYNJB-UHFFFAOYSA-N 1-phenylethanone;hydrochloride Chemical compound Cl.CC(=O)C1=CC=CC=C1 YWNKNCQONKYNJB-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- AENZGWONVTXLRC-UHFFFAOYSA-N 2-(2,5-dioxopyrrol-1-yl)benzoic acid Chemical class OC(=O)C1=CC=CC=C1N1C(=O)C=CC1=O AENZGWONVTXLRC-UHFFFAOYSA-N 0.000 description 1
- SXGZJKUKBWWHRA-UHFFFAOYSA-N 2-(N-morpholiniumyl)ethanesulfonate Chemical compound [O-]S(=O)(=O)CC[NH+]1CCOCC1 SXGZJKUKBWWHRA-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-GASJEMHNSA-N 2-amino-2-deoxy-D-galactopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@H](O)[C@@H]1O MSWZFWKMSRAUBD-GASJEMHNSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- UZPDKLGPANSZTM-UHFFFAOYSA-N 4-acetylbenzoyl chloride Chemical compound CC(=O)C1=CC=C(C(Cl)=O)C=C1 UZPDKLGPANSZTM-UHFFFAOYSA-N 0.000 description 1
- QHERSCUZBKDVOC-UHFFFAOYSA-N 4-amino-2-fluorobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C(F)=C1 QHERSCUZBKDVOC-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- RZVHIXYEVGDQDX-UHFFFAOYSA-N 9,10-anthraquinone Chemical group C1=CC=C2C(=O)C3=CC=CC=C3C(=O)C2=C1 RZVHIXYEVGDQDX-UHFFFAOYSA-N 0.000 description 1
- NUCXYBQPKQTHRU-UHFFFAOYSA-N 9h-fluoren-1-ylmethyl 2,5-dioxopyrrolidine-1-carboxylate Chemical compound C=1C=CC(C2=CC=CC=C2C2)=C2C=1COC(=O)N1C(=O)CCC1=O NUCXYBQPKQTHRU-UHFFFAOYSA-N 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 101800002638 Alpha-amanitin Proteins 0.000 description 1
- 229920000945 Amylopectin Polymers 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 206010051779 Bone marrow toxicity Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- FSYLQGBSXWUKFI-XNRPHZJLSA-N C(C)(=O)C1=CC=C(C(=O)NC2=NC(N(C=C2)[C@@H]2O[C@@H]([C@H](C2(F)F)O)CO)=O)C=C1 Chemical compound C(C)(=O)C1=CC=C(C(=O)NC2=NC(N(C=C2)[C@@H]2O[C@@H]([C@H](C2(F)F)O)CO)=O)C=C1 FSYLQGBSXWUKFI-XNRPHZJLSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 101710201076 Carboxylesterase 1 Proteins 0.000 description 1
- 108090000863 Carboxylic Ester Hydrolases Proteins 0.000 description 1
- 102000004308 Carboxylic Ester Hydrolases Human genes 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229920002567 Chondroitin Polymers 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 208000028698 Cognitive impairment Diseases 0.000 description 1
- 208000009798 Craniopharyngioma Diseases 0.000 description 1
- 102100026846 Cytidine deaminase Human genes 0.000 description 1
- 108010031325 Cytidine deaminase Proteins 0.000 description 1
- YTBSYETUWUMLBZ-UHFFFAOYSA-N D-Erythrose Natural products OCC(O)C(O)C=O YTBSYETUWUMLBZ-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-CBPJZXOFSA-N D-Gulose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@H](O)[C@H]1O WQZGKKKJIJFFOK-CBPJZXOFSA-N 0.000 description 1
- WQZGKKKJIJFFOK-IVMDWMLBSA-N D-allopyranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@H](O)[C@@H]1O WQZGKKKJIJFFOK-IVMDWMLBSA-N 0.000 description 1
- YTBSYETUWUMLBZ-IUYQGCFVSA-N D-erythrose Chemical compound OC[C@@H](O)[C@@H](O)C=O YTBSYETUWUMLBZ-IUYQGCFVSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-YMDCURPLSA-N D-galactopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-YMDCURPLSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- MNQZXJOMYWMBOU-VKHMYHEASA-N D-glyceraldehyde Chemical compound OC[C@@H](O)C=O MNQZXJOMYWMBOU-VKHMYHEASA-N 0.000 description 1
- CIWBSHSKHKDKBQ-DUZGATOHSA-N D-isoascorbic acid Chemical compound OC[C@@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-DUZGATOHSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-VANFPWTGSA-N D-mannopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@@H]1O AEMOLEFTQBMNLQ-VANFPWTGSA-N 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- ZAQJHHRNXZUBTE-NQXXGFSBSA-N D-ribulose Chemical compound OC[C@@H](O)[C@@H](O)C(=O)CO ZAQJHHRNXZUBTE-NQXXGFSBSA-N 0.000 description 1
- ZAQJHHRNXZUBTE-UHFFFAOYSA-N D-threo-2-Pentulose Natural products OCC(O)C(O)C(=O)CO ZAQJHHRNXZUBTE-UHFFFAOYSA-N 0.000 description 1
- YTBSYETUWUMLBZ-QWWZWVQMSA-N D-threose Chemical compound OC[C@@H](O)[C@H](O)C=O YTBSYETUWUMLBZ-QWWZWVQMSA-N 0.000 description 1
- ZAQJHHRNXZUBTE-WUJLRWPWSA-N D-xylulose Chemical compound OC[C@@H](O)[C@H](O)C(=O)CO ZAQJHHRNXZUBTE-WUJLRWPWSA-N 0.000 description 1
- 229920000045 Dermatan sulfate Polymers 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 206010056474 Erythrosis Diseases 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical class C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 229920002670 Fructan Polymers 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- 229920000855 Fucoidan Polymers 0.000 description 1
- 101000766307 Gallus gallus Ovotransferrin Proteins 0.000 description 1
- 229920001503 Glucan Polymers 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229920002527 Glycogen Polymers 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000822020 Homo sapiens Equilibrative nucleoside transporter 1 Proteins 0.000 description 1
- 206010021033 Hypomenorrhoea Diseases 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 102000012745 Immunoglobulin Subunits Human genes 0.000 description 1
- 108010079585 Immunoglobulin Subunits Proteins 0.000 description 1
- 229920001202 Inulin Polymers 0.000 description 1
- 102000011782 Keratins Human genes 0.000 description 1
- 108010076876 Keratins Proteins 0.000 description 1
- 208000007976 Ketosis Diseases 0.000 description 1
- LKDRXBCSQODPBY-AMVSKUEXSA-N L-(-)-Sorbose Chemical compound OCC1(O)OC[C@H](O)[C@@H](O)[C@@H]1O LKDRXBCSQODPBY-AMVSKUEXSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VSOAQEOCSA-N L-altropyranose Chemical compound OC[C@@H]1OC(O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-VSOAQEOCSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 1
- XVOYSCVBGLVSOL-UHFFFAOYSA-N L-cysteine sulfonic acid Natural products OC(=O)C(N)CS(O)(=O)=O XVOYSCVBGLVSOL-UHFFFAOYSA-N 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 102100030817 Liver carboxylesterase 1 Human genes 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 229920004011 Macrolon® Polymers 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 208000032271 Malignant tumor of penis Diseases 0.000 description 1
- 229920000057 Mannan Polymers 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100060131 Mus musculus Cdk5rap2 gene Proteins 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- DZTHIGRZJZPRDV-UHFFFAOYSA-N N-acetyltryptophan Chemical compound C1=CC=C2C(CC(NC(=O)C)C(O)=O)=CNC2=C1 DZTHIGRZJZPRDV-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 206010034299 Penile cancer Diseases 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002560 Polyethylene Glycol 3000 Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 208000028017 Psychotic disease Diseases 0.000 description 1
- 229920001218 Pullulan Polymers 0.000 description 1
- 239000004373 Pullulan Substances 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- RXGJTYFDKOHJHK-UHFFFAOYSA-N S-deoxo-amaninamide Natural products CCC(C)C1NC(=O)CNC(=O)C2Cc3c(SCC(NC(=O)CNC1=O)C(=O)NC(CC(=O)N)C(=O)N4CC(O)CC4C(=O)NC(C(C)C(O)CO)C(=O)N2)[nH]c5ccccc35 RXGJTYFDKOHJHK-UHFFFAOYSA-N 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical class O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001323 aldoses Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 239000004007 alpha amanitin Substances 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- SRBFZHDQGSBBOR-STGXQOJASA-N alpha-D-lyxopyranose Chemical compound O[C@@H]1CO[C@H](O)[C@@H](O)[C@H]1O SRBFZHDQGSBBOR-STGXQOJASA-N 0.000 description 1
- CIORWBWIBBPXCG-SXZCQOKQSA-N alpha-amanitin Chemical group O=C1N[C@@H](CC(N)=O)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@@H]([C@@H](C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@H]1C[S@@](=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-SXZCQOKQSA-N 0.000 description 1
- CIORWBWIBBPXCG-UHFFFAOYSA-N alpha-amanitin Natural products O=C1NC(CC(N)=O)C(=O)N2CC(O)CC2C(=O)NC(C(C)C(O)CO)C(=O)NC(C2)C(=O)NCC(=O)NC(C(C)CC)C(=O)NCC(=O)NC1CS(=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- RGHILYZRVFRRNK-UHFFFAOYSA-N anthracene-1,2-dione Chemical compound C1=CC=C2C=C(C(C(=O)C=C3)=O)C3=CC2=C1 RGHILYZRVFRRNK-UHFFFAOYSA-N 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000009831 antigen interaction Effects 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003972 antineoplastic antibiotic Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 229960003589 arginine hydrochloride Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- XSCHRSMBECNVNS-UHFFFAOYSA-N benzopyrazine Natural products N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 231100000366 bone marrow toxicity Toxicity 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- BPKIGYQJPYCAOW-FFJTTWKXSA-I calcium;potassium;disodium;(2s)-2-hydroxypropanoate;dichloride;dihydroxide;hydrate Chemical compound O.[OH-].[OH-].[Na+].[Na+].[Cl-].[Cl-].[K+].[Ca+2].C[C@H](O)C([O-])=O BPKIGYQJPYCAOW-FFJTTWKXSA-I 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000012829 chemotherapy agent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000004775 chlorodifluoromethyl group Chemical group FC(F)(Cl)* 0.000 description 1
- DLGJWSVWTWEWBJ-HGGSSLSASA-N chondroitin Chemical compound CC(O)=N[C@@H]1[C@H](O)O[C@H](CO)[C@H](O)[C@@H]1OC1[C@H](O)[C@H](O)C=C(C(O)=O)O1 DLGJWSVWTWEWBJ-HGGSSLSASA-N 0.000 description 1
- WCZVZNOTHYJIEI-UHFFFAOYSA-N cinnoline Chemical compound N1=NC=CC2=CC=CC=C21 WCZVZNOTHYJIEI-UHFFFAOYSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000009295 crossflow filtration Methods 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 150000001945 cysteines Chemical class 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 231100000050 cytotoxic potential Toxicity 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 235000019621 digestibility Nutrition 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- OGGXGZAMXPVRFZ-UHFFFAOYSA-M dimethylarsinate Chemical compound C[As](C)([O-])=O OGGXGZAMXPVRFZ-UHFFFAOYSA-M 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- WBZKQQHYRPRKNJ-UHFFFAOYSA-L disulfite Chemical compound [O-]S(=O)S([O-])(=O)=O WBZKQQHYRPRKNJ-UHFFFAOYSA-L 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 150000003883 epothilone derivatives Chemical class 0.000 description 1
- 235000010350 erythorbic acid Nutrition 0.000 description 1
- UQPHVQVXLPRNCX-UHFFFAOYSA-N erythrulose Chemical compound OCC(O)C(=O)CO UQPHVQVXLPRNCX-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 230000037433 frameshift Effects 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- FBPFZTCFMRRESA-GUCUJZIJSA-N galactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-GUCUJZIJSA-N 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229960005144 gemcitabine hydrochloride Drugs 0.000 description 1
- 229940020967 gemzar Drugs 0.000 description 1
- 229950006191 gluconic acid Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 108010016102 glutamine transport proteins Proteins 0.000 description 1
- 229960005150 glycerol Drugs 0.000 description 1
- 229940096919 glycogen Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 150000002337 glycosamines Chemical class 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 150000002386 heptoses Chemical class 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 150000002402 hexoses Chemical class 0.000 description 1
- 102000045714 human SLC29A1 Human genes 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical class C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 239000005414 inactive ingredient Substances 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 150000004001 inositols Chemical class 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- JYJIGFIDKWBXDU-MNNPPOADSA-N inulin Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@]1(OC[C@]2(OC[C@]3(OC[C@]4(OC[C@]5(OC[C@]6(OC[C@]7(OC[C@]8(OC[C@]9(OC[C@]%10(OC[C@]%11(OC[C@]%12(OC[C@]%13(OC[C@]%14(OC[C@]%15(OC[C@]%16(OC[C@]%17(OC[C@]%18(OC[C@]%19(OC[C@]%20(OC[C@]%21(OC[C@]%22(OC[C@]%23(OC[C@]%24(OC[C@]%25(OC[C@]%26(OC[C@]%27(OC[C@]%28(OC[C@]%29(OC[C@]%30(OC[C@]%31(OC[C@]%32(OC[C@]%33(OC[C@]%34(OC[C@]%35(OC[C@]%36(O[C@@H]%37[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O%37)O)[C@H]([C@H](O)[C@@H](CO)O%36)O)[C@H]([C@H](O)[C@@H](CO)O%35)O)[C@H]([C@H](O)[C@@H](CO)O%34)O)[C@H]([C@H](O)[C@@H](CO)O%33)O)[C@H]([C@H](O)[C@@H](CO)O%32)O)[C@H]([C@H](O)[C@@H](CO)O%31)O)[C@H]([C@H](O)[C@@H](CO)O%30)O)[C@H]([C@H](O)[C@@H](CO)O%29)O)[C@H]([C@H](O)[C@@H](CO)O%28)O)[C@H]([C@H](O)[C@@H](CO)O%27)O)[C@H]([C@H](O)[C@@H](CO)O%26)O)[C@H]([C@H](O)[C@@H](CO)O%25)O)[C@H]([C@H](O)[C@@H](CO)O%24)O)[C@H]([C@H](O)[C@@H](CO)O%23)O)[C@H]([C@H](O)[C@@H](CO)O%22)O)[C@H]([C@H](O)[C@@H](CO)O%21)O)[C@H]([C@H](O)[C@@H](CO)O%20)O)[C@H]([C@H](O)[C@@H](CO)O%19)O)[C@H]([C@H](O)[C@@H](CO)O%18)O)[C@H]([C@H](O)[C@@H](CO)O%17)O)[C@H]([C@H](O)[C@@H](CO)O%16)O)[C@H]([C@H](O)[C@@H](CO)O%15)O)[C@H]([C@H](O)[C@@H](CO)O%14)O)[C@H]([C@H](O)[C@@H](CO)O%13)O)[C@H]([C@H](O)[C@@H](CO)O%12)O)[C@H]([C@H](O)[C@@H](CO)O%11)O)[C@H]([C@H](O)[C@@H](CO)O%10)O)[C@H]([C@H](O)[C@@H](CO)O9)O)[C@H]([C@H](O)[C@@H](CO)O8)O)[C@H]([C@H](O)[C@@H](CO)O7)O)[C@H]([C@H](O)[C@@H](CO)O6)O)[C@H]([C@H](O)[C@@H](CO)O5)O)[C@H]([C@H](O)[C@@H](CO)O4)O)[C@H]([C@H](O)[C@@H](CO)O3)O)[C@H]([C@H](O)[C@@H](CO)O2)O)[C@@H](O)[C@H](O)[C@@H](CO)O1 JYJIGFIDKWBXDU-MNNPPOADSA-N 0.000 description 1
- 229940029339 inulin Drugs 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 229940026239 isoascorbic acid Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- BJHIKXHVCXFQLS-PQLUHFTBSA-N keto-D-tagatose Chemical compound OC[C@@H](O)[C@H](O)[C@H](O)C(=O)CO BJHIKXHVCXFQLS-PQLUHFTBSA-N 0.000 description 1
- BQINXKOTJQCISL-GRCPKETISA-N keto-neuraminic acid Chemical compound OC(=O)C(=O)C[C@H](O)[C@@H](N)[C@@H](O)[C@H](O)[C@H](O)CO BQINXKOTJQCISL-GRCPKETISA-N 0.000 description 1
- 150000002584 ketoses Chemical class 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 150000002605 large molecules Chemical class 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- AIHDCSAXVMAMJH-GFBKWZILSA-N levan Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@@H]1[C@@H](O)[C@H](O)[C@](CO)(CO[C@@H]2[C@H]([C@H](O)[C@@](O)(CO)O2)O)O1 AIHDCSAXVMAMJH-GFBKWZILSA-N 0.000 description 1
- 229920006008 lipopolysaccharide Polymers 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 239000012931 lyophilized formulation Substances 0.000 description 1
- 150000002669 lysines Chemical class 0.000 description 1
- 230000005291 magnetic effect Effects 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- LUEWUZLMQUOBSB-GFVSVBBRSA-N mannan Chemical class O[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@H]3[C@H](O[C@@H](O)[C@@H](O)[C@H]3O)CO)[C@@H](O)[C@H]2O)CO)[C@H](O)[C@H]1O LUEWUZLMQUOBSB-GFVSVBBRSA-N 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- HOVAGTYPODGVJG-ZFYZTMLRSA-N methyl alpha-D-glucopyranoside Chemical compound CO[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O HOVAGTYPODGVJG-ZFYZTMLRSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 108091005573 modified proteins Proteins 0.000 description 1
- 102000035118 modified proteins Human genes 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- CERZMXAJYMMUDR-UHFFFAOYSA-N neuraminic acid Natural products NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO CERZMXAJYMMUDR-UHFFFAOYSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical group NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 238000013421 nuclear magnetic resonance imaging Methods 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 230000001151 other effect Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- QUBQYFYWUJJAAK-UHFFFAOYSA-N oxymethurea Chemical compound OCNC(=O)NCO QUBQYFYWUJJAAK-UHFFFAOYSA-N 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 230000005298 paramagnetic effect Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 210000003899 penis Anatomy 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 150000002972 pentoses Chemical class 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- CCTIOCVIZPCTGO-BYPYZUCNSA-N phosphoarginine Chemical compound OC(=O)[C@@H](N)CCCNC(=N)NP(O)(O)=O CCTIOCVIZPCTGO-BYPYZUCNSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical compound C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940068977 polysorbate 20 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- OTYBMLCTZGSZBG-UHFFFAOYSA-L potassium sulfate Chemical compound [K+].[K+].[O-]S([O-])(=O)=O OTYBMLCTZGSZBG-UHFFFAOYSA-L 0.000 description 1
- 229910052939 potassium sulfate Inorganic materials 0.000 description 1
- 235000011151 potassium sulphates Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 235000019423 pullulan Nutrition 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 235000017709 saponins Nutrition 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000009919 sequestration Effects 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- GJPYYNMJTJNYTO-UHFFFAOYSA-J sodium aluminium sulfate Chemical compound [Na+].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O GJPYYNMJTJNYTO-UHFFFAOYSA-J 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 238000000967 suction filtration Methods 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical group 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- 125000006168 tricyclic group Chemical group 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
- 229920001221 xylan Polymers 0.000 description 1
- 150000004823 xylans Chemical class 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
- 229960005502 α-amanitin Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/643—Albumins, e.g. HSA, BSA, ovalbumin or a Keyhole Limpet Hemocyanin [KHL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/475—Quinolines; Isoquinolines having an indole ring, e.g. yohimbine, reserpine, strychnine, vinblastine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/07—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/545—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/26—Acyclic or carbocyclic radicals, substituted by hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06052—Val-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06104—Dipeptides with the first amino acid being acidic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06191—Dipeptides containing heteroatoms different from O, S, or N
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Communicable Diseases (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Virology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Hematology (AREA)
- Transplantation (AREA)
- Cell Biology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Saccharide Compounds (AREA)
Abstract
Un compuesto que tiene la estructura de la fórmula (I): **(Ver fórmula)** Fórmula (I) o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo; en donde: a) el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores; **(Ver fórmula)** el espaciador está ausente, o se selecciona del grupo que consiste en **(Ver fórmula)** y **(Ver fórmula)** n es 0 o 1; X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con - OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido; R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido; Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)- , -C(O)-NH-, -C(O)-O-, y -O-C(O)-; R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con - OCH2CH2-; y alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-, o R1 es un aminoácido natural o no natural, o R1 tiene la siguiente fórmula: **(Ver fórmula)** en donde: está ausente o se selecciona del grupo que consiste en: **(Ver fórmula)** y**(Ver fórmula)** R es: OPO3M1 en donde M1 = Mg2+, 2 Na+, 2K+, 2H+, y/o 2NH4+ o SO3M2 en donde M2 = Na+, K+, H+, NH4+ R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido; Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, un grupo aceptor de electrones, y un grupo soluble en agua; PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N- hidroxisuccinimida opcionalmente sustituido, un anticuerpo o un fragmento del mismo, y un anticuerpo derivatizado o fragmento derivatizado del mismo; en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; y en donde al menos uno de Z1, Z2, Z3 y Z4 es un grupo aceptor de electrones; o b) el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores; **(Ver fórmula)** el espaciador está ausente, n es 0 o 1; X se selecciona del grupo que consiste en: alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con - OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido; R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido; Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)- , -C(O)-NH-, -C(O)-O-, y -O-C(O)-; R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con - OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-, R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido; Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H y un grupo aceptor de electrones; PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N- hidroxisuccinimida opcionalmente sustituido, y un anticuerpo o fragmento del mismo; en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; y en donde al menos uno de Z1, Z2, Z3 y Z4 es un grupo aceptor de electrones.
Description
DESCRIPCIÓN
Sistemas de administración para liberación controlada de fármaco
Solicitud relacionada
Esta solicitud reivindica el beneficio de y la prioridad de las solicitudes de patente provisionales en EE UU 62/182.219, presentada el 19 de junio, 2015, 62/261.213, presentada el 30 de noviembre, 2015, y 62/261.563, presentada el 1 de diciembre, 2015.
Antecedentes de la invención
Muchos fármacos, en particular agentes terapéuticos contra el cáncer, tienen una ventana terapéutica estrecha, en donde sus efectos secundarios limitan sus efectos beneficiosos. La administración sistémica de tales fármacos con frecuencia produce un efecto terapéutico limitado porque la dosis requerida para provocar un efecto más robusto produce efectos secundarios inaceptables al paciente. Esto es particularmente crítico en el caso de esos fármacos que poseen un alto potencial citotóxico, tal como agentes citostáticos, agentes virostáticos o agentes inmunosupresores. Numerosos esfuerzos de investigación han estudiado administrar un fármaco particular en un sitio particular de acción. Con frecuencia, este enfoque produce una mayor concentración del fármaco en el sitio de acción de lo que se alcanzaría por administración sistémica, mientras limita los efectos secundarios.
La administración de fármacos en oncología es de interés particular debido a la estrecha ventana terapéutica de agentes usados en tal indicación. Se han concentrado numerosos esfuerzos de investigación en conjugar fármacos anticáncer con un amplio espectro de portadores de bajo y alto peso molecular incluyendo azúcares, factores de crecimiento, vitaminas, péptidos, anticuerpos, polisacáridos, lectinas, proteínas séricas, y polímeros sintéticos. En la mayoría de tales sistemas de administración de fármacos, el fármaco está unido al portador a través de un espaciador que incorpora un punto de ruptura predeterminado que permite que el fármaco unido se libere en el sitio diana celular (Kratz et al., ChemMedChem, 3:20-53 (2008)). Se conocen conjugados en los que agentes citostáticos están unidos a proteínas séricas, predominantemente a moléculas portadoras específicas tal como seroalbúmina humana y transferrina sérica humana, y después se administran. En algunos casos, los conjugados comprenden una sustancia terapéuticamente eficaz y, tras la administración, produce el transporte de la sustancia terapéuticamente eficaz al sitio diana donde se libera (documento US 7.387.771). En otros casos, conjugados que comprenden una sustancia terapéuticamente eficaz, una molécula espaciadora y una molécula que se une a proteínas, se unen covalentemente a seroalbúmina circulante tras la administración, lo que produce el transporte de la sustancia terapéuticamente eficaz al sitio diana donde se libera (documento US 7.387.771). En aún otros casos, conjugados de fármaco y anticuerpo (ADC) pueden transportar el fármaco al sitio diana para liberación local (Kratz et al., ChemMedChem, 3:20-53 (2008); Panowski et al., mAbs, 6, 34-45 (2014); Chari et al., Angewandte Chem. Int. Ed., 53, 3796-3827 (2014)).
Sin embargo, cuando se diseñan sistemas de administración de fármacos, se debe lograr el equilibrio apropiado entre conservar las propiedades de direccionamiento del portador del fármaco mientras se permite una liberación controlada del fármaco. La construcción de administración del fármaco debe tener suficiente estabilidad en el torrente sanguíneo, y aún permitir la liberación eficaz del fármaco en el sitio tumoral por corte enzimático, reducción o, de una manera dependiente del pH (Kratz et al., ChemMedChem, 3:20-53 (2008)). Por ejemplo, gemcitabina (Gemzar®) es un agente quimioterapéutico nucleosídico anticáncer que se usa mucho para tratar tumores sólidos. Desafortunadamente, a su dosis recomendada de ~1000 mg/m2, aproximadamente el 90% se desactiva por citidina desaminasa al metabolito uridina inactivo y se excreta en la orina. Una desventaja adicional que produce quimiorresistencia es el bajo nivel de expresión del transportador de nucleósido equilibrador 1 humano (hENTI) en la superficie celular de células cancerosas, que previene así la absorción sustancial de gemcitabina.
Por tanto, todavía hay una necesidad para sistemas de administración y liberación de fármacos eficaces con el fin de alcanzar administración y liberación controlada más eficaz del fármaco.
Compendio de la invención
La presente invención proporciona sistemas de administración para la administración y liberación eficaz y controlada de agentes terapéuticos.
La presente invención se refiere a un sistema de administración de fármacos que comprende una fracción hidrazona que se corta en un medio ácido de manera controlada, para proporcionar la liberación controlada de agentes terapéuticos. La presente invención se refiere a un sistema de administración de fármacos que comprende un enlace amida, un enlace carbamato, y/o un enlace éster que se corta enzimáticamente, por ejemplo, por esterasas o amidasas y/o hidrolíticamente, para proporcionar liberación controlada de agentes terapéuticos. En algunas formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende un enlace amida que se corta enzimáticamente, por ejemplo, por esterasas o amidasas y/o hidrolíticamente, para proporcionar liberación controlada de agentes terapéuticos. En otras formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende un enlace carbamato que se corta enzimáticamente y/o
hidrolíticamente. En aún otras formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende un enlace éster que se corta enzimáticamente y/o hidrolíticamente.
La presente invención se refiere a un sistema de administración de fármacos que comprende: (i) una fracción hidrazona que se corta en un medio ácido de manera controlada, y opcionalmente (ii) un enlace amida, un enlace carbamato, y/o un enlace éster que se corta enzimáticamente, por ejemplo, por esterasas o amidasas y/o hidrolíticamente, para proporcionar liberación controlada de agentes terapéuticos. En algunas formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende: (i) una fracción hidrazona que se corta en un medio ácido de manera controlada, y opcionalmente (ii) un enlace amida que se corta enzimáticamente, por ejemplo, por esterasas o amidasas y/o hidrolíticamente, para proporcionar liberación controlada de agentes terapéuticos. En algunas formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende: (i) una fracción hidrazona que se corta en un medio ácido de manera controlada, y opcionalmente (ii) un enlace carbamato que se corta enzimáticamente y/o hidrolíticamente, para proporcionar liberación controlada de agentes terapéuticos. En algunas formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende: (i) una fracción hidrazona que se corta en un medio ácido de manera controlada, y opcionalmente (ii) un enlace éster que se corta enzimáticamente y/o hidrolíticamente, para proporcionar liberación controlada de agentes terapéuticos. En algunas formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende: (i) una fracción hidrazona que se corta en un medio ácido de manera controlada, y opcionalmente (ii) un enlace amida que se corta selectivamente por carboxilesterasa 1 y/o 2, para proporcionar liberación controlada de agentes terapéuticos. En algunas formas de realización, la presente invención se refiere a un sistema de administración de fármacos que comprende: (i) una fracción hidrazona que se corta en un medio ácido de manera controlada, y (ii) un enlace amida que se corta selectivamente por carboxilesterasa 2, para proporcionar liberación controlada de agentes terapéuticos.
La presente invención proporciona un compuesto que tiene la estructura representada por la fórmula (I):

en donde:
el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un proteasoma o inhibidor de proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de proteasa, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;
el espaciador está ausente 
n es 0 o 1;
X se selecciona del grupo que consiste en: alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;
R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;
Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;
R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; y alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-,
o R1 es un aminoácido natural o no natural,
o R1 tiene la siguiente fórmula:

en donde:


R es: -O P O 3M1 en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, 2NH4+ o
~ S 03M2 en donde M2 = Na+, K+, H+, NH4+
R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;
Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, un grupo aceptor de electrones, y un grupo soluble en agua;
PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido, un anticuerpo o un fragmento del mismo, y un anticuerpo derivatizado o fragmento derivatizado del mismo;
en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; y
en donde al menos uno de Z1, Z2, Z3 y Z4 es un grupo aceptor de electrones.
En algunas formas de realización, PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, y un grupo éster de N-hidroxisuccinimida opcionalmente sustituido.
En algunas formas de realización, PBG está asociado con un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está covalentemente unido a un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está asociado con albúmina. En otras formas de realización, PBG está covalentemente unido a
albúmina endógena o exógena. En otras formas de realización, PBG está covalentemente unido a la cisteína 34 de albúmina endógena o exógena.
La presente invención proporciona un compuesto que tiene la estructura representada por la fórmula (I):

o una sal, hidrato, solvato, o isómero farmacéuticamente aceptable del mismo; en donde
el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un proteasoma o inhibidor de proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de proteasa, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;
el espaciador está ausente 
n es 0 o 1;
X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;
R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;
Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;
R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; y alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-,
R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;
Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H o un grupo aceptor de electrones;
PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido, y un anticuerpo o un fragmento del mismo;
en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; y
en donde al menos uno de Z1, Z2, Z3 y Z4 es un grupo aceptor de electrones.
En algunas formas de realización, PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, y un grupo éster de N-hidroxisuccinimida opcionalmente sustituido.
En algunas formas de realización, PBG está asociado con un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está covalentemente unido a un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está asociado con albúmina. En otras formas de realización, PBG está covalentemente unido a albúmina endógena o exógena. En otras formas de realización, PBG está covalentemente unido a la cisteína 34 de albúmina endógena o exógena.
En ciertas formas de realización, la invención proporciona un compuesto representado por la fórmula (II):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde agente, PBG, Y, R1, Z1, Z2 , Z3 y Z4 son como se ha definido para un compuesto de fórmula (I).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (III):

en donde agente, espaciador, PBG, Y, R1, Z2, Z3 y Z4 son como se ha definido para un compuesto de fórmula (I); y
en donde Zi es un grupo aceptor de electrones.
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (IV):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde agente, PBG, Y, R1, Z2, Z3 y Z4 son como se ha definido para un compuesto de fórmula (I); y
en donde Zi es un grupo aceptor de electrones.
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (V):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde agente, PBG, n, Y, Ri, R2, Zi, Z2, Z3 y Z4 son como se ha definido para un compuesto de fórmula (I). En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (VIa):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde:
M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno.
X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3.
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3.
R' es -R3 o -CH2R3 ;
en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y
en donde X, n, Zi, Z2, Z3 , Z4, Y, Ri, R2, y PBG, son como se ha definido para un compuesto de fórmula (V).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (VIb):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo; en donde
M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario.
Xi y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3.
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3.
R3 se selecciona de -OH, -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y
en donde X, n, Zi, Z2, Z3 , Z4, Y, R1, R2, y PBG, son como se ha definido para un compuesto de fórmula (V).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (VlIa):
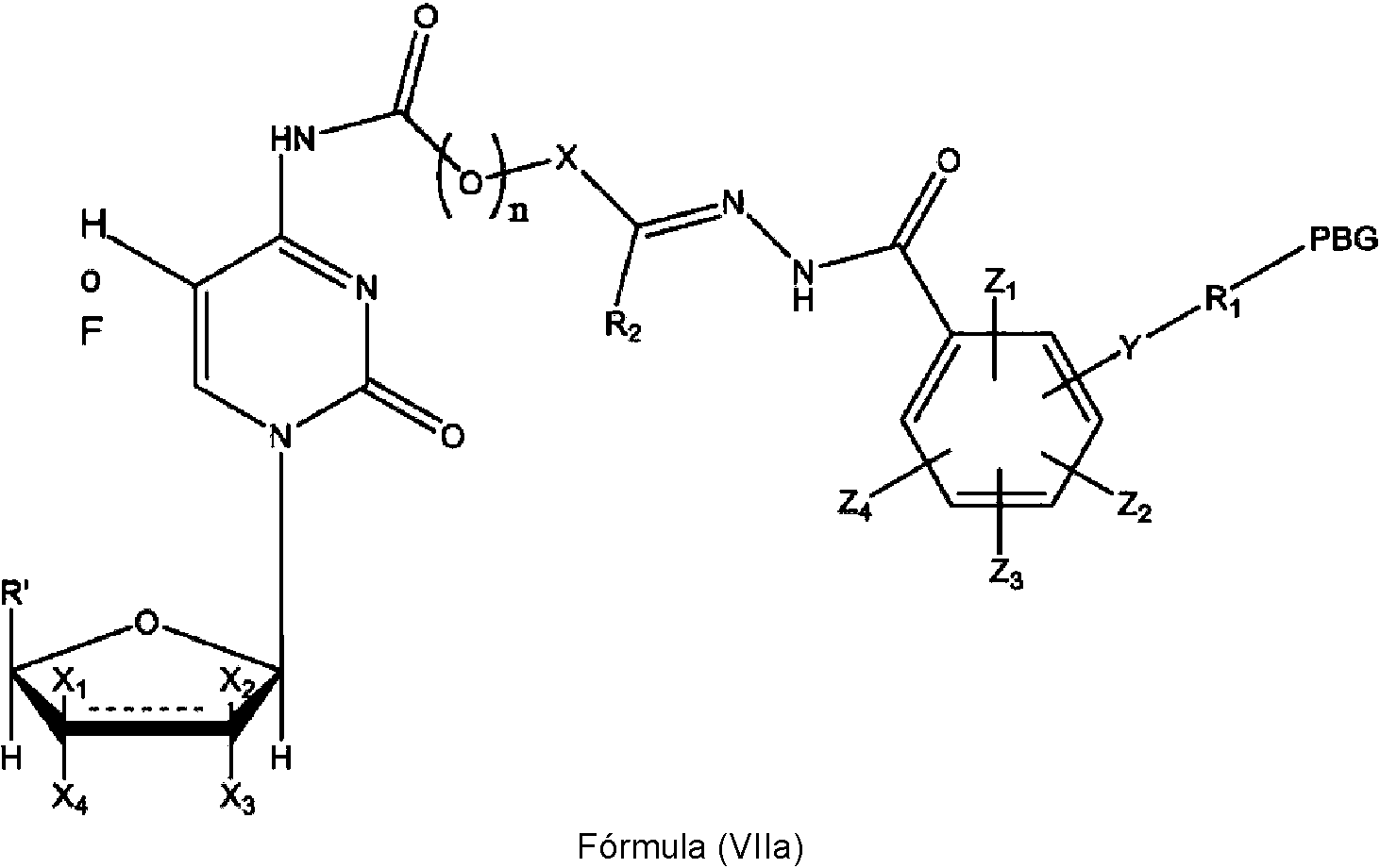
o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde R' es -R3 o -CH2R3 ; y X, Xi, X2, X3 ,  y PBG, son c un compuesto de fórmula (VI).
y PBG, son c un compuesto de fórmula (VI).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (Vllb):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde X, Xi, X2 , X3 , X4 , n, Y, Ri, R2 , R3, Zi, Z2 , Z3 , Z4 y PBG, son como se ha definido para un compuesto de fórmula (Vl).
En ciertas formas de realización, la invención proporciona un compuesto que tiene una estructura representada por la fórmula (VIII):

Z1 es un grupo aceptor de electrones; y
en donde agente, X, n, R2, PBG, Y, R1, Z2 , Z3 y Z4 son como se ha definido para un compuesto de fórmula (V). En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (IXa):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde:
M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno;
Xi y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
R' es -R3 o -CH2R3 ;
en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y
en donde X, n, Y, Zi, Z2, Z3 , Z4, Ri, R2, y PBG, son como se ha definido para un compuesto de fórmula (VIII).
En ciertas formas de realización, la invención proporciona un compuesto que tiene una estructura representada por la fórmula (IXb):

o una sal, hidrato, solvato o isómero del mismo farmacéuticamente aceptable; en donde
M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario;
X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
R3 se selecciona de -OH, -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y en donde X, n, Y, Zi, Z2, Z3 , Z4, Ri, R2, y PBG, son como se ha definido para un compuesto de fórmula (VIII).
En algunas formas de realización, Xi, X2 , X3 , y X4 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, -CH3, -F, -Cl, -Br, -I, y -N3.
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (Xa):

i i
Fórmula (Xa)
o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde R' es -R3 o -CH2R3;  3 , Z4, R1 y compuesto de fórmula (IX).
3 , Z4, R1 y compuesto de fórmula (IX).
En ciertas formas de realización, la invención proporciona un compuesto que tiene una estructura representada por la fórmula (Xb):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde X1, X2, X3 , X4 , R3, X, n, Y, Z1, Z2, Z3 , Z4 , R1 y R2 son como se ha definido para un compuesto de fórmula (IX).
En algunas formas de realización, el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 5'-desoxi-5-fluorocitidina, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 7-acetilcamptotecina, 9-formilcamptotecina, 9-acetilcamptotecina, 9-formil-10-hidroxicamptotecina, 10-formilcamptotecina, 10-acetilcamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansina, maitansinol; auristatina (incluyendo, pero no limitado a auristatina D, auristatina E, auristatina F, monometil auristatina D, monometil auristatina E, monometil auristatina F, éster metílico de monometil auristatina F, auristatina PYE auristatina PHE, el producto natural relacionado dolastatina 10, y derivados de las mismas); amatoxinas (incluyendo, pero no limitado a a-amanitina, p-amanitina, Y-amanitina, £-amanitin, amanina, amaninamida, arnanulina, y ácido amanulínico y derivados de los mismos); duocarmicina A, duocarmicina B1, duocarmicina B2, duocarmicina C, duocarmicina SA, CC1065, adozelesina, bizelesina, carzelesina; eribulina; trabectedina; pirrolobenzodiazepina, antramicina, tomaimicina, sibiromicina, DC-81, DSB-120; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.
En algunas formas de realización, el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 9-formilcamptotecina, 9-formil-10-hidroxicamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansinoides; auristatinas; epotilonas; bleomicina;
dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.
En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -Cn . En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y -CN. En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3. En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo. En algunas formas de realización, al menos uno de Z1, Z2, Z3 y Z4 no es -H.
En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -Cn . En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y -CN. En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3. En algunas formas de realización, al menos uno de Z1, Z2, Z3 y Z4 no es -H.
En algunas formas de realización, Z1 se selecciona del grupo que consiste en halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -CN; y Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6 , y -CN. En algunas formas de realización, Z1 se selecciona del grupo que consiste en -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN; y Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3, y -CN. En algunas formas de realización, Z1 se selecciona del grupo que consiste en -Cl, -F, y -NO2 , y Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2 , y -CF3.
En algunas formas de realización, la fracción compuesto de la invención se selecciona del grupo que consiste en:
compuesto de la invención se selecciona del grupo que consiste en:

En algunas formas de realización, la fracción compuesto de la invención se selecciona del grupo que consiste en:
compuesto de la invención se selecciona del grupo que consiste en:

En algunas formas de realización, Y es -C(O)-NH-. En algunas formas de realización, Y es -C(O)-O-. En algunas formas de realización, Y está ausente.
En algunas formas de realización R1 se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-.
En algunas formas de realización R1 se selecciona del grupo que consiste en
En algunas formas de realización Ri está ausente.
En algunas formas de realización, PBG es un grupo maleimida opcionalmente sustituido. En algunas formas de
realización, 
En algunas formas de realización, la fracción compuesto de la invención se selecciona del grupo que consiste en:
compuesto de la invención se selecciona del grupo que consiste en:

M2 = Na+, K+, H+, NH4+,

Mi = Mg2+, 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+

En algunas formas de realización, el espaciador se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido; y R2 es como se ha definido para la fórmula I.
se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido; y R2 es como se ha definido para la fórmula I.
En algunas formas de realización, el espaciador 
que consiste en: -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido y heteroarilo opcionalmente sustituido; W 1, W 2 , W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6 , y -CN. En algunas formas de realización, W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -Br, -I, -F, C(O)OH, -NO2 , -CF3 , y -CN. En algunas formas de realización, W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -F, -NO2 , y -CF3. En algunas formas de realización, W 1, W 2, W3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: un grupo fenoxi, un grupo amino primario, secundario o terciario, un grupo éter, un grupo fenol, un grupo amida, un grupo éster, un grupo alquilo, un grupo alquilo sustituido, un grupo fenilo, y un grupo vinilo. En algunas formas de realización, W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo. En algunas formas de realización, al menos uno de W 1, W 2, W 3 y W 4 no es -H.
En algunas formas de realización, W 1 se selecciona del grupo que consiste en: halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -CN; y W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -Cn . En algunas formas de realización, W 1 se selecciona del grupo que consiste en: -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y
-CN; y W 2 , W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3, y -CN. En algunas formas de realización, W 1 se selecciona del grupo que consiste en: -Cl, -F, y -NO2, y Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -F, -NO2, y -CF3.
En algunas formas de realización, el espaciador es en donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido y heteroarilo opcionalmente sustituido.
en donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido y heteroarilo opcionalmente sustituido.
En algunas formas de realización, el espaciador es
En algunas formas de realización, el espaciador es  y m es 1, 2, 3, 4, 5, o 6.
y m es 1, 2, 3, 4, 5, o 6.
En algunas formas de realización, el espaciador es 
En algunas formas de realización, el compuesto de la invención se selecciona del grupo que consiste en:

o una sal, hidrato, solvato, o isómero farmacéuticamente aceptable del mismo,
en donde:


R es ~>0P03Mi en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, 2NH4+, Na+, K+, NH4+, y/o H+, o
~ S 03M2 en donde M2 = Na+, K+, H+, NH4+
En algunas formas de realización, el compuesto de la invención se selecciona del grupo que consiste en:






o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo.
En algunas formas de realización, el agente es a-amanitina, y el compuesto de la presente invención se selecciona del grupo que consiste en:

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo.
En algunas formas de realización, el agente es una auristatina o derivado de la misma, y el compuesto de la presente invención se selecciona del grupo que consiste en:

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo.
En ciertas formas de realización, la invención proporciona una composición farmacéutica que comprende un compuesto divulgado en el presente documento, y un soporte farmacéuticamente aceptable.
En ciertas formas de realización, la invención proporciona un método para tratar una enfermedad o afección seleccionada del grupo que consiste en un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, enfermedad inflamatoria aguda o crónica, y una enfermedad producida por bacterias, hongos, u otros microorganismos, que comprende administrar a un paciente en necesidad de ello una cantidad terapéuticamente eficaz de un compuesto descrito en el presente documento o una composición farmacéutica descrita en el presente documento.
En algunas formas de realización, la invención proporciona compuestos y composiciones para uso como un medicamento. En ciertas formas de realización, la invención proporciona compuestos y composiciones para uso en tratar una enfermedad o afección seleccionada del grupo que consiste en un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, enfermedad inflamatoria aguda o crónica, y una enfermedad producida por bacterias, hongos, u otros microorganismos.
En algunas formas de realización, el compuesto divulgado en el presente documento se puede usar en la fabricación o preparación de un medicamento para el tratamiento de una enfermedad o afección seleccionada del grupo que consiste en un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, enfermedad inflamatoria aguda o crónica, y una enfermedad producida por bacterias, hongos, u otros microorganismos.
Breve descripción de los dibujos
La figura 1 muestra el efecto del compuesto 15 y gemcitabina en crecimiento tumoral en un modelo de xenoinjerto de NSCL LXFE 397.
La figura 2 muestra el efecto del compuesto 15 y gemcitabina en crecimiento tumoral en un modelo de xenoinjerto de NSCL LXFE 397.
La figura 3 muestra el efecto del compuesto 15 y gemcitabina en el cambio de peso corporal en un modelo de xenoinjerto de carcinoma de células no microcíticas humano LXFE 397.
La figura 4 muestra el efecto del compuesto 15 y gemcitabina en crecimiento tumoral en un modelo de xenoinjerto OVXF 899 de cáncer ovárico.
La figura 5 muestra el efecto del compuesto 15 y gemcitabina en el cambio de peso corporal en un modelo de xenoinjerto OVXF 899 de cáncer ovárico.
Las figuras 6(A) y 6(B) muestran gráficos de dispersión para los volúmenes de tumores individuales después de tratamiento con el compuesto 15 o gemcitabina en un modelo de xenoinjerto OVXF 899 de cáncer ovárico. La figura 6(A) muestra el volumen tumoral absoluto el día 0; la figura 6(B) muestra el volumen tumoral absoluto el día 67.
La figura 7 muestra el efecto del compuesto 15 y gemcitabina en crecimiento tumoral en un modelo de xenoinjerto Panc11159 de cáncer pancreático.
La figura 8 muestra el efecto del compuesto 15 y gemcitabina en el cambio de peso corporal en un modelo de xenoinjerto Panc11159 de cáncer pancreático.
Descripción detallada de la invención
A menos que se definan de otra manera en el presente documento, los términos técnicos y científicos usados en esta solicitud tendrán los significados que entienden comúnmente los expertos en la materia. En general, la nomenclatura usada en relación con, y técnicas de, química, biología molecular, biología celular y de cáncer, inmunología, microbiología, farmacología, y química de proteínas y ácidos nucleicos, descritas en el presente documento, son esas bien conocidas y comúnmente usadas en la técnica.
A lo largo de esta especificación, la palabra “comprender” o variaciones tales como “comprende” o “que comprende” se entenderá que implica la inclusión de un número entero (o componentes) o grupo de números enteros (o componentes) expuestos, pero no la exclusión de ningún otro número entero (o componentes) o grupo de números enteros (o componentes).
A lo largo de esta solicitud, donde se describe un compuesto o composición como que tiene, incluye o comprende, componentes específicos, se contempla que tal compuesto o composición también pueda consistir esencialmente en, o consistir en, los componentes enumerados. De forma similar, donde se describen métodos o procesos como que tienen, incluyen, o comprenden etapas de proceso específicas, los procesos también pueden consistir esencialmente en, o consistir en, las etapas de procesamiento enumeradas. Además, se debe entender que el orden de las etapas o el orden de realizar ciertas acciones es irrelevante siempre que los compuestos, composiciones y métodos descritos en el presente documento permanezcan operables. Además, dos o más etapas o acciones se pueden realizar simultáneamente.
Las formas singulares “un”, “una”, “el” y “la” incluyen los plurales a menos que el contexto claramente dicte otra cosa.
El término “ incluir” se usa para significar “incluir, pero no limitado a”. “ Incluir” e “ incluir, pero no limitado a” se usan de forma intercambiable.
El término “o” como se usa en el presente documento se debe entender que significa “y/o”, a menos que el contexto claramente indique otra cosa.
Los términos “fármaco”, “agente”, “agente terapéutico” o “sustancia terapéuticamente eficaz” se usan para significar cualquier compuesto que produce un efecto farmacológico ya sea por si mismo o después de su conversión en el organismo en cuestión, y por tanto también incluye los derivados de estas conversiones. El efecto farmacológico de los fármacos de la composición según la presente invención puede ser un efecto único solo, por ejemplo, un efecto citostático, o un amplio espectro farmacológico de acciones, tal como un efecto inmunosupresor o antiflogístico al mismo tiempo.
El término “antraciclina” se refiere a una clase de antibióticos antineoplásicos que tienen una unidad estructural antracenodiona (también llamada antraquinona o dioxoantraceno). Por ejemplo, se pretende específicamente que el término “antraciclina” incluya individualmente doxorrubicina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, valrubicina, pirarrubicina, zorurrubicina, aclarrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetoxialquilantraciclina, PNU-159682, caminomicina, mitoxantrona, y ametantrona.
Los términos “paciente”, “sujeto”, o “ individuo” se usan de forma intercambiable y se refieren a un humano o un animal no humano. Estos términos incluyen mamíferos tal como seres humanos, primates, animales de ganadería (por ejemplo, bovinos, porcinos), animales de compañía (por ejemplo, caninos, felinos) y roedores (por ejemplo, ratones y ratas). En ciertas formas de realización, el paciente o sujeto es un paciente o sujeto humano, tal como un paciente humano que tiene una afección en necesidad de tratamiento.
El término “composición farmacéutica” se refiere a una composición adecuada para uso farmacéutico en un sujeto animal, incluyendo seres humanos y mamíferos, por ejemplo, combinada con uno o más soportes, excipientes o solventes farmacéuticamente aceptables. Tal composición también puede contener diluyentes, rellenos, sales, tampones, estabilizantes, solubilizantes, y otros materiales bien conocidos en la técnica. En ciertas formas de realización, una composición farmacéutica abarca una composición que comprende el principio(s) activo(s), y el ingrediente(s) inerte(s) que hace(n) el excipiente, soporte o diluyente, así como cualquier producto que resulte, directa o indirectamente, de la combinación, complejación o agregación de cualesquiera dos o más de los ingredientes, o de la disociación de uno o más de los ingredientes. Según esto, las composiciones farmacéuticas de la presente divulgación abarcan cualquier composición hecha mezclando un compuesto de la divulgación y uno o más excipiente(s), soporte(s) y/o diluyente(s) farmacéuticamente aceptable(s).
El término “soporte farmacéuticamente aceptable” se refiere a un soporte no tóxico que se puede administrar a un paciente, junto con una sustancia terapéuticamente eficaz de esta invención, y que no destruye la actividad farmacológica del agente. El término “excipiente” se refiere a un aditivo en una formulación o composición que no es
un ingrediente farmacéuticamente activo. En ciertas formas de realización, una sustancia “farmacéuticamente aceptable” es adecuada para uso en contacto con células, tejidos u órganos de animales o seres humanos sin excesiva toxicidad, irritación, respuesta alérgica, inmunogenicidad u otras reacciones adversas, en la cantidad usada en la forma farmacéutica según el plan de dosificación, y proporcional con un cociente riesgo/beneficio razonable. En ciertas formas de realización, una sustancia “farmacéuticamente aceptable” que es un componente de una composición farmacéutica es, además, compatible con el/los otro(s) ingrediente(s) de la composición. En ciertas formas de realización, los términos “excipiente farmacéuticamente aceptable”, “soporte farmacéuticamente aceptable” y “diluyente farmacéuticamente aceptable” abarcan, sin limitación, ingredientes inactivos, materiales, composiciones y vehículos farmacéuticamente aceptables, tales como rellenos líquidos, rellenos sólidos, diluyentes, excipientes, soportes, solventes y materiales de encapsulación. Los soportes, diluyentes y excipientes también incluyen todos los medios de dispersión, recubrimientos, tampones, agentes isotónicos, estabilizantes, agentes de retraso de absorción, agentes antimicrobianos, agentes antibacterianos, agentes antifúngicos, adyuvantes, y demás farmacéuticamente aceptables. Excepto en cuanto a que cualquier excipiente, soporte o diluyente convencional sea incompatible con el principio activo, la presente divulgación abarca el uso de excipientes, soportes y diluyentes convencionales en composiciones farmacéuticas. Véase, por ejemplo, Remington: The Science and Practice of Pharmacy, 21a Ed., Lippincott Williams & Wilkins (Filadelfia, Pensilvania, 2005); Handbook of Pharmaceutical Excipients, 5a Ed., Rowe et al., Eds., The Pharmaceutical Press and the American Pharmaceutical Association (2005); Handbook of Pharmaceutical Additives, 3a Ed., Ash and Ash, Eds., Gower Publishing Co. (2007); y Pharmaceutical Preformulation and Formulation, Gibson, Ed., CRC Press LLC (Boca Raton, Florida, 2004).
Los términos “cantidad farmacéuticamente eficaz”, “cantidad terapéuticamente eficaz”, o “dosis terapéuticamente eficaz” se refieren a una cantidad eficaz para tratar una enfermedad en un paciente, por ejemplo, efectuando una alteración beneficiosa y/o deseable en la salud general de un paciente que padece una enfermedad (por ejemplo, cáncer), tratamiento, curación, inhibición o mejora de una respuesta fisiológica o afección, etc. El efecto terapéutico completo no se produce necesariamente por la administración de una dosis, y se puede producir solo después de la administración de una serie de dosis. Por tanto, una cantidad terapéuticamente eficaz se puede administrar en una o más administraciones. La cantidad eficaz precisa necesaria para un sujeto dependerá de, por ejemplo, el tamaño la salud y edad, del sujeto, la naturaleza y grado de enfermedad, el agente terapéutico o combinación de agentes terapéuticos seleccionados para administración, y el modo de administración. El experto en la materia puede determinar fácilmente la cantidad eficaz para una situación determinada por experimentación rutinaria. El experto en la materia reconocerá que tratar cáncer incluye, pero no está limitado a, destruir células cancerosas, prevenir el crecimiento de nuevas células cancerosas, producir regresión del tumor (una disminución en el tamaño tumoral), producir una disminución en metástasis, mejorar las funciones vitales de un paciente, mejorar el bienestar del paciente, disminuir el dolor, mejorar el apetito, mejorar el peso del paciente, y cualquier combinación de las mismas. Los términos “cantidad farmacéuticamente eficaz”, “cantidad terapéuticamente eficaz”, o “dosis terapéuticamente eficaz” también se refieren a la cantidad requerida para mejorar los síntomas clínicos de un paciente. Los métodos terapéuticos o métodos de tratar cáncer descritos en el presente documento no se deben interpretar o limitar de otra manera a “curar” cáncer.
Como se usa en el presente documento, el término “tratar” o “tratamiento” incluye revertir, reducir, o parar los síntomas, signos clínicos, y patología subyacente de una afección de forma que mejore o estabilice el estado de un paciente. Como se usa en el presente documento, y como se entiende bien en la técnica, “tratamiento” es un enfoque para obtener resultados beneficiosos o deseados, incluyendo resultados clínicos. Los resultados clínicos beneficiosos o deseados pueden incluir, pero no están limitados a, alivio, mejora, o ralentizar la evolución, de uno o más síntomas o estados asociados con una afección, por ejemplo, cáncer, disminución del grado de la enfermedad, estado estabilizado (es decir, sin empeoramiento) de la enfermedad, retraso o ralentización de la evolución de la enfermedad, mejora o alivio del estado de enfermedad, y remisión (sea parcial o total), sea detectable o indetectable. “Tratamiento” también puede significar prolongar la supervivencia en comparación con la supervivencia esperada si no se recibe tratamiento. Los resultados clínicos beneficiosos ejemplares se describen en el presente documento.
“Administrar” o “administración de” una sustancia, un compuesto o un agente a un sujeto se puede llevar a cabo usando uno de una variedad de métodos conocidos por los expertos en la materia. Por ejemplo, un compuesto o un agente se puede administrar por vía intravenosa, arterial, intradérmica, intramuscular, intraperitoneal, subcutánea, ocular, sublingual, oral (por ingestión), intranasal (por inhalación), intrarraquídea, intracerebral, y transdérmica (por absorción, por ejemplo, a través de un conducto de la piel). Un compuesto o agente también se puede introducir apropiadamente por dispositivos poliméricos recargables o biodegradables u otros dispositivos, por ejemplo, parches y bombas, o formulaciones, que proporcionan la liberación extendida, lenta o controlada del compuesto o agente. Administrar también se puede realizar, por ejemplo, una vez, una pluralidad de veces, y/o a lo largo de uno o más periodos extendidos. En algunos aspectos, la administración incluye tanto administración directa, que incluye autoadministración, como administración indirecta, que incluye el acto de recetar un fármaco. Por ejemplo, como se usa en el presente documento, un médico que instruye a un paciente a autoadministrase un fármaco, o a que otro le administre un fármaco y/o que proporciona una receta a un paciente para un fármaco administra el fármaco al paciente. Cuando un método es parte de una pauta terapéutica que implica más de un agente o modalidad de tratamiento, la divulgación contempla que los agentes se puedan administrar al mismo tiempo o diferentes y a través de la misma o diferentes rutas de administración. Los métodos apropiados de administrar una sustancia, un compuesto o un agente a un sujeto también dependerán, por ejemplo, de la edad del sujeto, si el sujeto es activo o inactivo en el momento de
la administración, si el sujeto tiene deterior cognitivo en el momento de la administración, el grado de deterioro, y las propiedades químicas y biológicas del compuesto o agente (por ejemplo, solubilidad, digestibilidad, biodisponibilidad, estabilidad y toxicidad).
El término “sustituido” se refiere a fracciones que tienen sustituyentes que reemplazan un hidrógeno en uno o más carbonos del esqueleto de un compuesto químico. Se entenderá que “sustitución” o “sustituido con” incluye la condición implícita de que tal sustitución es según la valencia permitida del átomo sustituido y el sustituyente, y que la sustitución produce un compuesto estable, por ejemplo, que no experimenta espontáneamente transformación tal como por reorganización, ciclación, eliminación, etc. Como se usa en el presente documento, se contempla que el término “sustituido” incluya todos los sustituyentes permisibles de compuestos orgánicos. En un aspecto amplio, los sustituyentes permisibles incluyen sustituyentes acíclicos y cíclicos, ramificados y sin ramificar, carbocíclicos y heterocíclicos, aromáticos y no aromáticos de compuestos orgánicos. Los sustituyentes permisibles pueden ser uno o más e iguales o diferentes para compuestos orgánicos apropiados. Para los fines de la invención, los heteroátomos tal como nitrógeno pueden tener sustituyentes hidrógeno y/o cualquier sustituyente permisible de compuestos orgánicos descritos en el presente documento que satisfagan las valencias de los heteroátomos. Los sustituyentes pueden incluir cualquier sustituyente descrito en el presente documento, por ejemplo, un halógeno, un hidroxilo, un carbonilo (tal como un carboxilo, un alcoxicarbonilo, un formilo, o un acilo), un tiocarbonilo (tal como un tioéster, un tioacetato, o un tioformiato), un alcoxilo, un alquiltio, un aciloxi, un fosforilo, un fosfato, un fosfonato, un amino, un amido, una amidina, una imina, un ciano, un nitro, un azido, un sulfhidrilo, un alquiltio, un sulfato, un sulfonato, un sulfamoilo, un sulfonamido, un sulfonilo, un heterociclilo, un aralquilo, o una fracción aromática o heteroaromática.
“Opcional” u “opcionalmente” significa que la circunstancia posteriormente descrita puede o no suceder, de modo que la solicitud incluye casos donde la circunstancia sucede y casos donde no. Por ejemplo, la frase “opcionalmente sustituido” significa que un sustituyente no hidrógeno puede o no estar presente en un átomo determinado, y, por tanto, la solicitud incluye estructuras donde un sustituyente no hidrógeno está presente y estructuras conde un sustituyente no hidrógeno no está presente.
A menos que específicamente se manifieste como “sin sustituir”, se entiende que referencias a fracciones químicas en el presente documento incluyen variantes sustituidas. Por ejemplo, la referencia a un grupo o fracción “alquilo” implícitamente incluye variantes tanto sustituidas como sin sustituir. Los ejemplos de sustituyentes en fracciones químicas incluyen, pero no están limitados a, halógeno, hidroxilo, carbonilo (tal como carboxilo, alcoxicarbonilo, formilo, o acilo), tiocarbonilo (tal como tioéster, tioacetato, o tioformiato), alcoxilo, alquiltio, aciloxi, fosforilo, fosfato, fosfonato, amino, amido, amidina, imina, ciano, nitro, azido, sulfhidrilo, alquiltio, sulfato, sulfonato, sulfamoilo, sulfonamido, sulfonilo, heterociclilo, aralquilo, o fracción arilo o heteroarilo.
El término “acilo” está reconocido en la técnica y se refiere a un grupo representado por la fórmula general hidrocarbil-C(O)-, preferiblemente alquilo-C(O)-.
El término “alquilo” se refiere al radical de grupos alifáticos saturados, incluyendo grupos alquilo de cadena lineal, y grupos alquilo de cadena ramificada. En formas de realización preferidas, un alquilo de cadena lineal o cadena ramificada tiene 30 o menos átomos de carbono en su esqueleto (por ejemplo, C1-C30 para cadenas lineales, C3-C30 para cadenas ramificadas), y más preferiblemente 20 o menos. En ciertas formas de realización, los grupos alquilo son grupos alquilo inferiores, por ejemplo, metilo, etilo, n-propilo, i-propilo, n-butilo, y n-pentilo. Además, el término “alquilo” como se usa a lo largo de la especificación, ejemplos y reivindicaciones se pretende que incluya tanto “alquilos sin sustituir” como “alquilos sustituidos”, el último de los cuales se refiere a fracciones alquilo que tienen sustituyentes reemplazando un hidrógeno en uno o más carbonos del esqueleto hidrocarburo. En ciertas formas de realización, un alquilo de cadena lineal o cadena ramificada tiene 30 o menos átomos de carbono en su esqueleto (por ejemplo, C1-C30 para cadenas lineales, C3-C30 para cadenas ramificadas). En formas de realización preferidas, la cadena tiene diez o menos (C1-C10) átomos en su esqueleto. En otras formas de realización, la cadena tiene seis o menos (C1-C6) átomos en su esqueleto.
Tales sustituyentes pueden incluir, por ejemplo, un halógeno, un hidroxilo, un carbonilo (tal como un carboxilo, un alcoxicarbonilo, un formilo, o un acilo), un tiocarbonilo (tal como un tioéster, un tioacetato, o un tioformiato), un alcoxilo, un alquiltio, un aciloxi, un fosforilo, un fosfato, un fosfonato, un amino, un amido, una amidina, una imina, un ciano, un nitro, un azido, un sulfhidrilo, un alquiltio, un sulfato, un sulfonato, un sulfamoilo, un sulfonamido, un sulfonilo, un heterociclilo, un aralquilo, o una fracción arilo o heteroarilo.
El término “amida”, como se usa en el presente documento, se refiere a un grupo representado por

en donde RXy RY representan cada uno independientemente un hidrógeno o un grupo hidrocarbilo, o RXy RY tomados juntos con el átomo de N al que están unidos completan un heterociclo que tiene de 4 a 8 átomos en la estructura de anillo.
En algunas formas de realización, la amida es -NH-C(O)- o -C(O)-NH-.
Los términos “amina” y “amino” están reconocidos en la técnica y se refieren a aminas tanto sin sustituir como sustituidas y sales de las mismas, por ejemplo, una fracción que puede estar representada por
en donde RX, RY y RZ representan cada uno independientemente un hidrógeno o un grupo hidrocarbilo, o RX y RY tomados juntos con el átomo de N al que están unidos completan un heterociclo que tiene de 4 a 8 átomos en la estructura de anillo.
El término “arilo”, como se usa en el presente documento, incluye grupos aromáticos de anillo único sustituidos o sin sustituir en los que cada átomo del anillo es carbono. Preferiblemente el anillo es un anillo de 5 a 7 miembros, más preferiblemente un anillo de 6 miembros. Los grupos arilo incluyen fenilo, fenol, anilina, y similares. El término “arilo” también incluye “policiclilo”, “policiclo”, y sistemas de anillos “policíclicos” que tienen dos o más anillos en los que dos o más átomos son comunes a dos anillos contiguos, por ejemplo, los anillos son “anillos fusionados”, en donde al menos uno de los anillos es aromático, por ejemplo, los otros anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos. En algunas formas de realización preferidas, los policiclos tienen 2-3 anillos. En ciertas formas de realización preferidas, los sistemas de anillos policíclicos tienen dos anillos cíclicos en los que ambos anillos son aromáticos. Cada uno de los anillos del policiclo puede estar sustituido o sin sustituir. En ciertas formas de realización, cada anillo del policiclo contiene de 3 a 10 átomos en el anillo, preferiblemente de 5 a 7. Por ejemplo, los grupos arilo incluyen, pero no están limitados a, fenilo, tolilo, antracenilo, fluorenilo, indenilo, azulenilo, y naftilo, así como fracciones carbocíclicas benzofusionadas tal como 5,6,7,8-tetrahidronaftilo y similares.
En algunas formas de realización, el arilo es un grupo aromático de anillo único. En algunas formas de realización, el arilo es un grupo aromático de dos anillos. En algunas formas de realización, el arilo es un grupo aromático de tres anillos.
El término “cicloalquilo”, como se usa en el presente documento, se refiere al radical de un anillo alifático saturado. En formas de realización preferidas, los cicloalquilos tienen de 3-10 átomos de carbono en su estructura de anillo, y más preferiblemente de 5-7 átomos de carbono en su estructura de anillo. En algunas formas de realización, los dos anillos cíclicos pueden tener dos o más átomos en común, por ejemplo, los anillos son “anillos fusionados”. Los cicloalquilos adecuados incluyen cicloheptilo, ciclohexilo, ciclopentilo, ciclobutilo y ciclopropilo.
En algunas formas de realización, el cicloalquilo es un grupo monocíclico. En algunas formas de realización, el cicloalquilo es un grupo bicíclico. En algunas formas de realización, el cicloalquilo es un grupo tricíclico.
El término “haloalquilo”, como se usa en el presente documento, significa un grupo alquilo sustituido con uno o más halógenos. Cuando más de un halógeno está presente, los halógenos pueden ser iguales o diferentes. Por ejemplo, los grupos haloalquilo incluyen, pero no están limitados a, fluorometilo, difluorometilo, trifluorometilo, clorodifluorometilo, 2,2,2-trifluoroetilo, pentafluoroetilo, y similares.
Los términos “halo” y “halógeno”, como se usan en el presente documento, significan halógeno e incluye cloro, fluoro, bromo, y yodo.
El término “heteroarilo” incluye estructuras de anillo único aromáticas sustituidas o sin sustituir, preferiblemente anillos de 5 a 7 miembros, más preferiblemente anillos de 5 a 6 miembros, cuyas estructuras de anillos incluyen al menos un heteroátomo (por ejemplo, O, N, o S), preferiblemente de uno o cuatro o de uno a 3 heteroátomos, más preferiblemente uno o dos heteroátomos. Cuando dos o más heteroátomos están presentes en un anillo heteroarilo, pueden ser iguales o diferentes. El término “heteroarilo” también incluye “policiclilo”, “policiclo”, y sistemas de anillos “policíclicos” que tienen dos o más anillos en los que dos o más carbonos son comunes a dos anillos contiguos, por ejemplo, los anillos son “anillos fusionados”, en donde al menos uno de los anillos es heteroaromático, por ejemplo, los otros anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos, heteroarilos y/o heterociclilos. En algunas formas de realización preferidas, los policiclos tienen 2-3 anillos. En ciertas formas de realización, los sistemas de anillos policíclicos preferidos tienen dos anillos cíclicos en los que ambos anillos son aromáticos. En ciertas formas de realización, cada anillo del policiclo contiene de 3 a 10 átomos en el anillo, preferiblemente de 5 a 7. Por ejemplo, los grupos heteroarilo incluyen, pero no están limitados a, pirrol, furano, tiofeno, imidazol, oxazol, tiazol, pirazol, piridina,
pirazina, piridazina, quinolina, pirimidina, indolizina, indol, indazol, benzimidazol, benzotiazol, benzofurano, benzotiofeno, cinolina, ftalazina, quinazolina, carbazol, fenoxazina, quinolina, purina y similares.
En algunas formas de realización, el heteroarilo es un grupo aromático de anillo único. En algunas formas de realización, el heteroarilo es un grupo aromático de dos anillos. En algunas formas de realización, el heteroarilo es un grupo aromático de tres anillos.
Los términos “heterociclilo”, “heterociclo” y “heterocíclico” se refieren a estructuras de anillos no aromáticos sustituidos o sin sustituir, preferiblemente anillos de 3 a 10 miembros, más preferiblemente anillos de 3 a 7 miembros, cuyas estructuras de anillo incluyen al menos un heteroátomo, preferiblemente de uno o cuatro heteroátomos, más preferiblemente uno o dos heteroátomos. En ciertas formas de realización, la estructura de anillo puede tener dos anillos cíclicos. En algunas formas de realización, los dos anillos cíclicos pueden tener dos o más átomos en común, por ejemplo, los anillos son “anillos fusionados”. Los grupos heterociclilo incluyen, por ejemplo, piperidina, piperazina, pirrolidina, morfolina, lactonas, lactamas, y similares.
El término “hidrocarbilo”, como se usa en el presente documento, se refiere a un grupo que está unido a través de un átomo de carbono que no tiene un sustituyente =O o =S, y típicamente tiene al menos un enlace carbono-hidrógeno y un esqueleto principalmente de carbono, pero puede opcionalmente incluir heteroátomos. Por tanto, grupos como metilo, etoxietilo, 2-piridilo, y trifluorometilo se considera que son hidrocarbilo para los fines de esta solicitud, pero sustituyentes tal como acetilo (que tiene un sustituyente =O en el carbono de enlace) y etoxi (que está unido a través de oxígeno, no carbono) no son. Los grupos hidrocarbilo incluyen, pero no están limitados a arilo, heteroarilo, carbociclo, heterociclo, alquilo, alquenilo, alquinilo, y combinaciones de los mismos.
Los términos “fracción hidrazona” o “hidrazona” se refieren  . La estereoquímica de la fracción hidrazona puede ser E o Z. El término hidrazona como se usa en el presente documento incluye los isómeros tanto E como Z.
. La estereoquímica de la fracción hidrazona puede ser E o Z. El término hidrazona como se usa en el presente documento incluye los isómeros tanto E como Z.
En varios sitios en la presente especificación los sustituyentes de los compuestos de la divulgación se divulgan en grupos o en intervalos. Se pretende específicamente que la divulgación incluya cada una y todas las subcombinaciones individuales de los miembros de tales grupos e intervalos. Por ejemplo, se pretende específicamente que el término “alquilo de Ci-Ca” divulgue individualmente metilo, etilo, propilo, isopropilo, n-butilo, sec-butilo, isobutilo, etc.
Una “sal farmacéuticamente aceptable” es una sal de un compuesto que es adecuada para uso farmacéutico, incluyendo, pero no limitada a sales metálicas (por ejemplo, sodio, potasio, magnesio, calcio, etc.), sales de adición ácida (por ejemplo, ácidos minerales, ácidos carboxílicos, etc.), y sales de adición de base (por ejemplo, amoniaco, aminas orgánicas, etc.). La forma de sal de adición ácida de un compuesto que se produce en su forma libre como una base se puede obtener tratando dicha forma de base libre con un ácido apropiado tal como un ácido inorgánico, por ejemplo, halhídrico, tal como clorhídrico o bromhídrico, sulfúrico, nítrico, fosfórico y similares; o un ácido orgánico, tal como, por ejemplo, acético, hidroxiacético, propanoico, láctico, pirúvico, malónico, succínico, maleico, fumárico, málico, tartárico, cítrico, metanosulfónico, etanosulfónico, bencenosulfónico, p-toluenosulfónico, cíclico, salicílico, paminosalicílico, pamoico y similares. Véase, por ejemplo, el documento WO 01/062726. Algunas sales farmacéuticamente aceptables enumeradas por Berge et al., Journal of Pharmaceutical Sciences, 66: 1-19 (1977).
Los compuestos que contienen protones ácidos se pueden convertir a su forma de sal de adición de base no tóxica terapéuticamente activa, por ejemplo, sales metálicas o de aminas, por tratamiento con bases orgánicas e inorgánicas apropiadas. Las formas de sal de base apropiada incluyen, por ejemplo, sales de amonio, sales o iones de metales alcalinos y alcalinotérreos, por ejemplo, sales de litio, sodio, potasio, magnesio, calcio, y similares, sales con bases orgánicas, por ejemplo, N-metil-D-glucamina, sales de hidrabamina, y sales con aminoácidos tal como, por ejemplo, arginina, lisina y similares. Al contrario, dichas formas de sal se pueden convertir en las formas libres por tratamiento con una base o ácido apropiados. Los compuestos y sus sales pueden estar en forma de un solvato, que se incluye en el ámbito de la presente divulgación. Tales solvatos incluyen, por ejemplo, hidratos, alcoholatos y similares. Véase, por ejemplo, el documento WO 01/062726.
La divulgación proporciona además composiciones farmacéuticas que comprenden uno o más compuestos de la divulgación junto con un soporte o excipiente farmacéuticamente aceptable. Los compuestos o composiciones farmacéuticas de la divulgación se pueden usar in vitro o in vivo.
El término “isómero”, como se usa en el presente documento, incluye, pero no está limitado a, tautómeros, isómeros cis y trans (E (entgegen), Z (zusammen)), enantiómeros R y S (dicha notación R y S se usa en correspondencia con las reglas descritas en Pure Appl. Chem. (1976), 45, 11-30), diastereómeros, isómeros (D), isómeros (L),
estereoisómeros, las mezclas racémicas de los mismos, y otras mezclas de los mismos. Los tautómeros, mientras no se indican explícitamente en las fórmulas descritas en el presente documento, se pretende que estén incluidos en el ámbito de la presente invención.
Compuestos de la invención
La presente invención proporciona un compuesto que tiene la estructura representada por la fórmula (I):

en donde:
el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de proteasa, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;
el espaciador está ausente; o se selecciona del grupo que consiste  y
y

n es 0 o 1;
X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;
R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;
Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;
R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-,
o R1 es un aminoácido natural o no natural,
o R1 tiene la siguiente fórmula:

en donde:
está ausente o se selecciona del grupo que consiste en:

R es: ~ 0 P 03Mi en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, 2NH4+ o
- S O 3M2 en donde M2 = Na+, K+, H+, NH4+
R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;
Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente de -H, un grupo aceptor de electrones, y/o un grupo soluble en agua;
PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido, un anticuerpo o un fragmento del mismo, y un anticuerpo derivatizado o fragmento derivatizado del mismo;
en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; y
en donde al menos uno de Z1, Z2, Z3 y Z4 es un grupo aceptor de electrones.
En algunas formas de realización de los compuestos descritos en el presente documento, PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, y un grupo éster de N-hidroxisuccinimida opcionalmente sustituido.
En algunas formas de realización, PBG está asociado con un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está covalentemente unido a un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está asociado con albúmina. En otras formas de realización, PBG está covalentemente unido a albúmina endógena o exógena. En otras formas de realización, PBG está covalentemente unido a la cisteína 34 de albúmina endógena o exógena.
En ciertas formas de realización, la presente invención proporciona un compuesto que tiene la estructura representada por la fórmula (I):
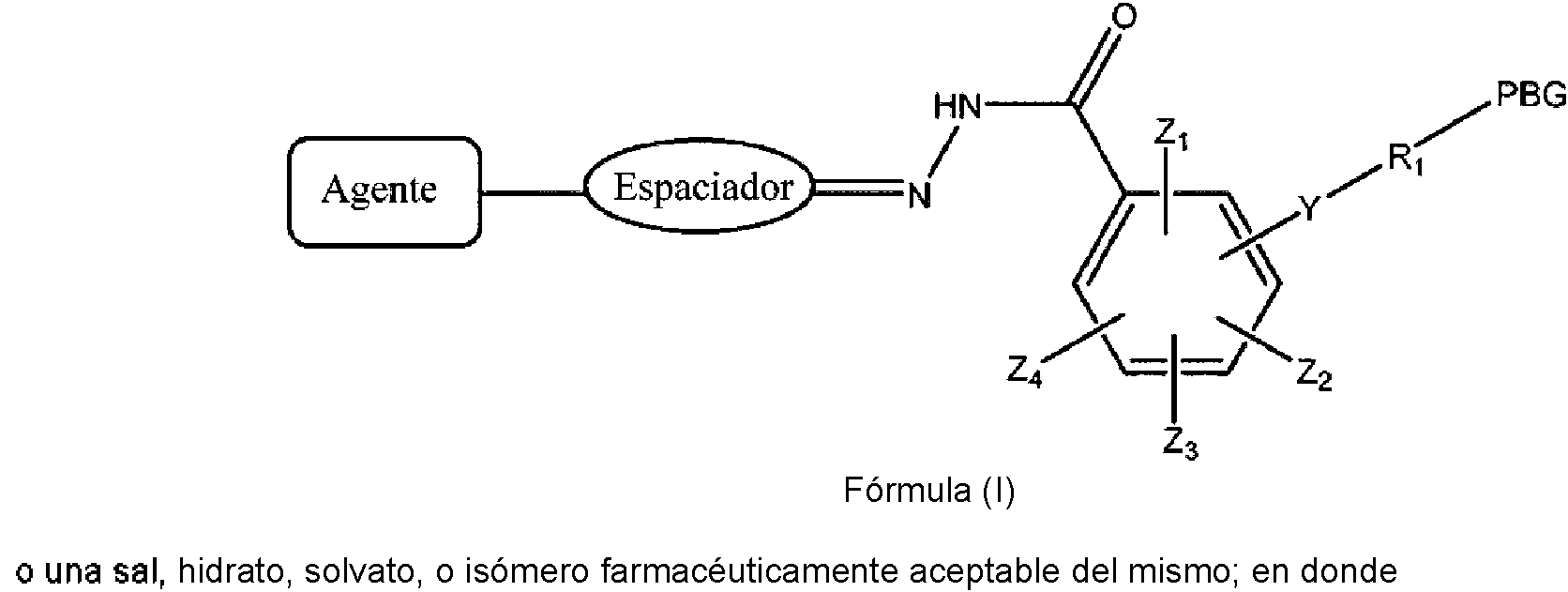
el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de proteasa, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;
el espaciador está ausente 
n es 0 o 1 ;
X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;
R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;
Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;
R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-,
R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;
Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, y un grupo aceptor de electrones;
PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido y un anticuerpo o un fragmento del mismo;
en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; y
en donde al menos uno de Zi, Z2, Z3 y Z4 es un grupo aceptor de electrones.
En algunas formas de realización de los compuestos descritos en el presente documento, PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, y un grupo éster de N-hidroxisuccinimida opcionalmente sustituido.
En algunas formas de realización, PBG está asociado con un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está covalentemente unido a un anticuerpo o fragmento del mismo. En algunas formas de realización, PBG está asociado con albúmina. En otras formas de realización, PBG está covalentemente unido a albúmina endógena o exógena. En otras formas de realización, PBG está covalentemente unido a la cisteína 34 de albúmina endógena o exógena.
En ciertas formas de realización, la invención proporciona un compuesto que tiene una estructura representada por la fórmula (II):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde agente, PBG, Y, Ri, Zi, Z2 , Z3 y Z4 son como se ha definido para un compuesto de fórmula (I).
En ciertas formas de realización, la invención proporciona un compuesto que tiene una estructura representada por la fórmula (III):

en donde agente, espaciador, PBG, Y, Ri, Z2, Z3 y Z4 son como se ha definido para un compuesto de fórmula (I); y en donde Zi es un grupo aceptor de electrones.
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (IV):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde agente, PBG, Y, R1, Z2, Z3 y Z4 son como se ha definido para un compuesto de fórmula (I); y
en donde Zi es un grupo aceptor de electrones.
En ciertas formas de realización, la invención proporciona un compuesto que tiene una estructura representada por la fórmula (V):

en donde agente, PBG, n, Y, Ri, R2, Zi, Z2, Z3 y Z4 son como se ha definido para un compuesto de fórmula (I). En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (VIa):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde:
M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno;
X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
R' es -R3 o -CH2R3 ;
en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y
en donde X, n, Z1, Z2, Z3 , Z4, Y, R1, R2, y PBG son como se ha definido para un compuesto de fórmula (V).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (VIb):

M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario.
X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3.
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3.
R3 se selecciona del grupo que consiste en -OH, -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y
en donde X, n, Z1, Z2, Z3 , Z4, Y, R1, R2, y PBG son como se ha definido para un compuesto de fórmula (V).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (VIIa):
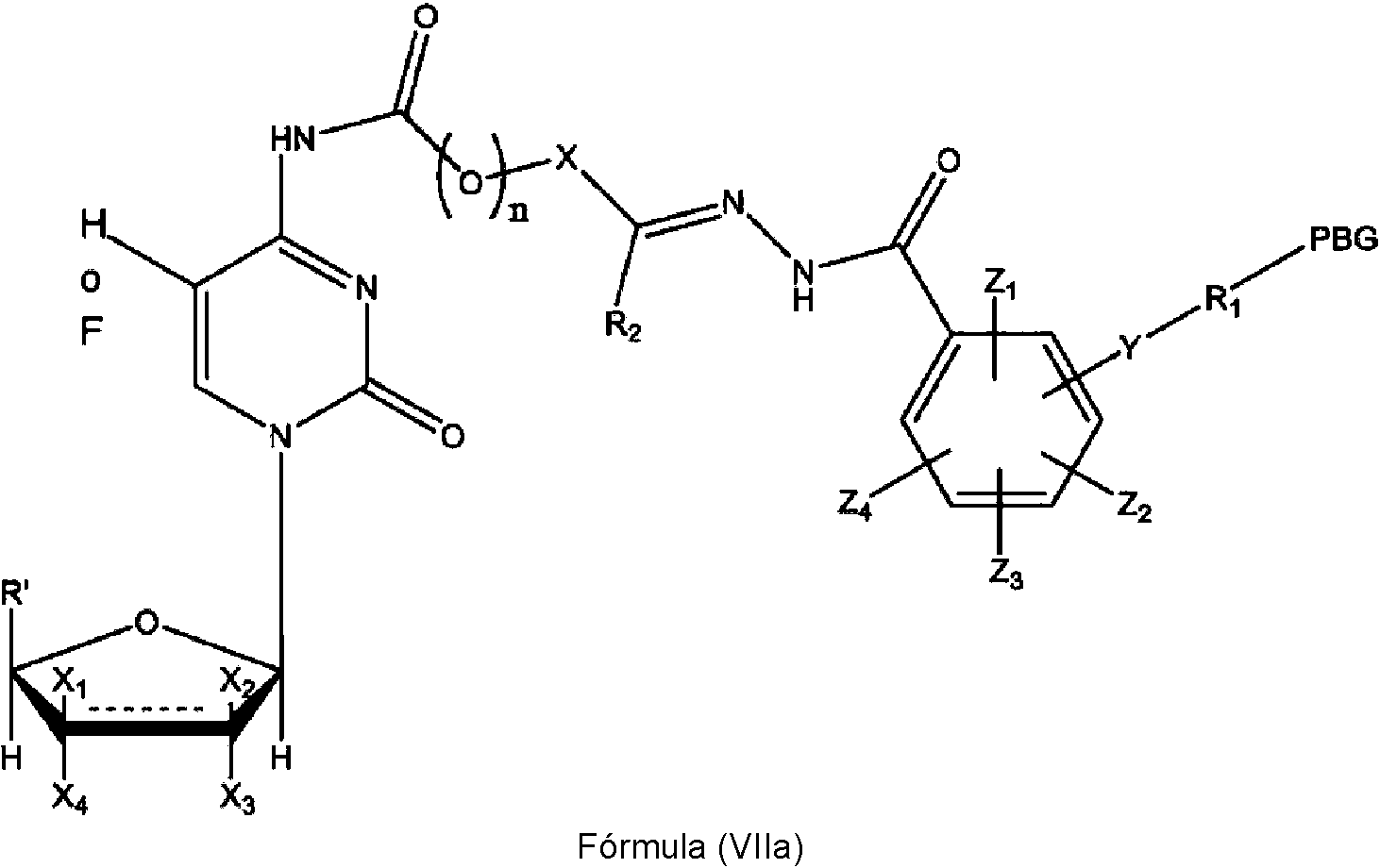
o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde R' es -R3 o 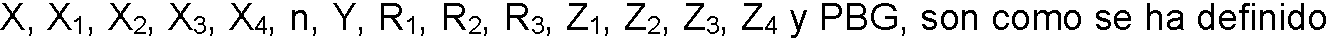 2 , Z3, Z4 un compuesto de fórmula (VI).
2 , Z3, Z4 un compuesto de fórmula (VI).
En ciertas formas de realización, la invención proporciona un compuesto que tiene una estructura representada por la fórmula (VIIb):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde X, X1, X2 , X3 , X4, 4 y PBG son como se ha definido p (V).
4 y PBG son como se ha definido p (V).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (VIII):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo; en donde
Zi es un grupo aceptor de electrones; y en donde agente, X, n, R2, PBG, Y, R1, Z2, Z3 y Z4 son como se ha definido para un compuesto de fórmula (V).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (IXa):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde:
M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno;
X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
R' es -R3 o -CH2R3 ;
en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y
en donde X, n, Y, Z1, Z2, Z3 , Z4, R1, R2, y PBG son como se ha definido para un compuesto de fórmula (VIII).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (IXb):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo; en donde
M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario;
X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;
R3 se selecciona de -OH, -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; y en donde X, n, Y, Z1, Z2, Z3 , Z4, R1, R2, y PBG, son como se ha definido para un compuesto de fórmula (VIII).
En ciertas formas de realización, X1, X2 , X3 , X4 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, -CH3, -F, -Cl, -Br, -I, y -N3.
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (Xa):
o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde R' es -R3 o -CH2R3;  2 , Z3 , Z4, compuesto de fórmula (IX).
2 , Z3 , Z4, compuesto de fórmula (IX).
En ciertas formas de realización, la invención proporciona un compuesto que tiene la estructura representada por la fórmula (Xb):

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo;
en donde X1, X2, X3 , X, n, Y, Z1, Z2, Z3 , Z4 , R1 y R2 son como se ha definido para un com
X, n, Y, Z1, Z2, Z3 , Z4 , R1 y R2 son como se ha definido para un com
En algunas formas de realización, el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 5'-desoxi-5-fluorocitidina, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 7-acetilcamptotecina, 9-formilcamptotecina, 9-acetilcamptotecina, 9-formil-10-hidroxicamptotecina, 10-formilcamptotecina, 10-acetilcamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansina, maitansinol; auristatina (incluyendo, pero no limitado a auristatina D, auristatina E, auristatina F, monometil auristatina D, monometil auristatina E, monometil auristatina F, éster metílico de monometil auristatina F, auristatina PYE auristatina PHE, el producto natural relacionado dolastatina 10, y derivados de las mismas); amatoxinas (incluyendo, pero no limitado a a-amanitina, p-amanitina, Y-amanitina, £-amanitin, amanina, amaninamida, arnanulina, y ácido amanulínico y derivados de los mismos); duocarmicina A, duocarmicina B1, duocarmicina B2, duocarmicina C, duocarmicina SA, CC1065, adozelesina, bizelesina, carzelesina; eribulina; trabectedina; pirrolobenzodiazepina, antramicina, tomaimicina, sibiromicina, DC-81, DSB-120; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.
En algunas formas de realización, el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansinoides; auristatinas; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.
En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6, y -Cn . En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl,
-Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y -CN. En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -F, -NO2 , y -CF3. En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo. En algunas formas de realización, al menos uno de Z1, Z2, Z3 y Z4 no es -H.
En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -Cn . En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y -CN. En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3. En algunas formas de realización, al menos uno de Z1, Z2, Z3 y Z4 no es -H.
En algunas formas de realización, Z1 se selecciona de halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6, y -CN; y Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6 , y -CN. En algunas formas de realización, Z1 se selecciona del grupo que consiste en -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3, y -CN; y Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3, y -CN. En algunas formas de realización, Z1 se selecciona del grupo que consiste en -Cl, -F, y -NO2; y Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2 , y -CF3.
En algunas formas de realización, R2 es metilo.
En algunas formas de realización, X1 es -H, X2 es -F, X3 es -F, y X4 es -OH.
La fracción hidrazona
El sistema de administración de fármacos contiene una fracción hidrazona cortable, lábil a ácido. El corte de la fracción hidrazona y la semivida de la liberación del fármaco varía según los sustituyentes aceptores de electrones y su posición en el anillo fenilo al que la hidrazona está unido. El anillo fenilo puede estar sustituido como sigue

En algunas formas de realización, el anillo fenilo comprende al menos un grupo aceptor de electrones. En algunas formas de realización preferidas, el anillo fenilo comprende un grupo aceptor de electrones unido a la posición 1. En algunas formas de realización, el anillo fenilo comprende un grupo aceptor de electrones unido a la posición 2. En algunas formas de realización, el anillo fenilo comprende un grupo aceptor de electrones unido a la posición 3. En algunas formas de realización preferidas, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 1 y 2. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 1 y 3. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 1 y 4. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 1 y 5. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 2 y 3. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 2 y 4. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 2 y 5. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 3 y 4. En algunas formas de realización, el anillo fenilo comprende dos grupos aceptores de electrones unidos a las posiciones 3 y 5. En algunas formas de realización, el anillo fenilo comprende tres grupos aceptores de electrones unidos a las posiciones 1, 3 y 5.
En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6, y -Cn . En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y -CN. En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -F, -NO2 , y -CF3. En algunas formas de realización, Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo. En algunas formas de realización, al menos uno de Z1, Z2, Z3 y Z4 no es -H.
En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3, y -CN, en donde al menos uno de Z1, Z2 , Z3 y Z4 no es -H. En algunas
formas de realización, Zi, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3, en donde al menos uno de Z1, Z2 , Z3 y Z4 no es -H. En algunas formas de realización, Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -F, -NO2, y -CF3, en donde al menos uno de Z1, Z2, Z3 y Z4 no es -H.
En algunas formas de realización, la semivida de la liberación del fármaco es aproximadamente 1,5 horas, aproximadamente 2,0 horas, aproximadamente 2,5 horas, aproximadamente 3,0 horas, aproximadamente 3,5 horas, aproximadamente 4,0 horas, aproximadamente 4,5 horas, aproximadamente 5,0 horas, aproximadamente 5,5 horas, aproximadamente 6,0 horas, aproximadamente 6,5 horas, aproximadamente 7,0 horas, aproximadamente 7,5 horas, aproximadamente 8,0 horas, aproximadamente 8,5 horas, aproximadamente 9,0 horas, aproximadamente 9,5 horas, aproximadamente 10,0 horas, aproximadamente 10,5 horas, aproximadamente 11,0 horas, aproximadamente 11,5 horas, aproximadamente 12,0 horas, aproximadamente 12,5 horas, aproximadamente 13,0 horas, aproximadamente 13,5 horas, aproximadamente 14,0 horas, aproximadamente 14,5 horas, aproximadamente 15,0 horas, aproximadamente 15,5 horas, aproximadamente 16,0 horas, aproximadamente 16,5 horas, aproximadamente 17,0 horas, aproximadamente 17,5 horas, aproximadamente 18,0 horas, aproximadamente 18,5 horas, aproximadamente 19,0 horas, aproximadamente 19,5 horas, aproximadamente 20,0 horas.
Sin querer estar vinculado por ninguna teoría, un anillo fenilo que comprende un grupo aceptor de electrones unido a la posición 1 estabiliza la fracción hidrazona, produciendo una liberación lenta y prolongada del agente en condiciones ácidas. En algunas formas de realización, la posición del grupo aceptor de electrones en el anillo fenilo con respecto a la posición de la fracción hidrazona en el anillo fenilo proporciona un método para controlar la liberación del fármaco.
En algunas formas de realización, la invención proporciona un método de hacer un compuesto para controlar la
liberación de un fármaco modificando el fármaco para añadir una fracción  o
o

Algunas formas de realización de la invención incluyen conjugados con los patrones de sustitución y las correspondientes semividas de la liberación del fármaco mostradas en la tabla A (HSA significa seroalbúmina
humana). La estructura de nemorrubicina  Los conjugados de nemorrubicina representados en la tabla A tienen la siguiente estructura
Los conjugados de nemorrubicina representados en la tabla A tienen la siguiente estructura

Tabla A
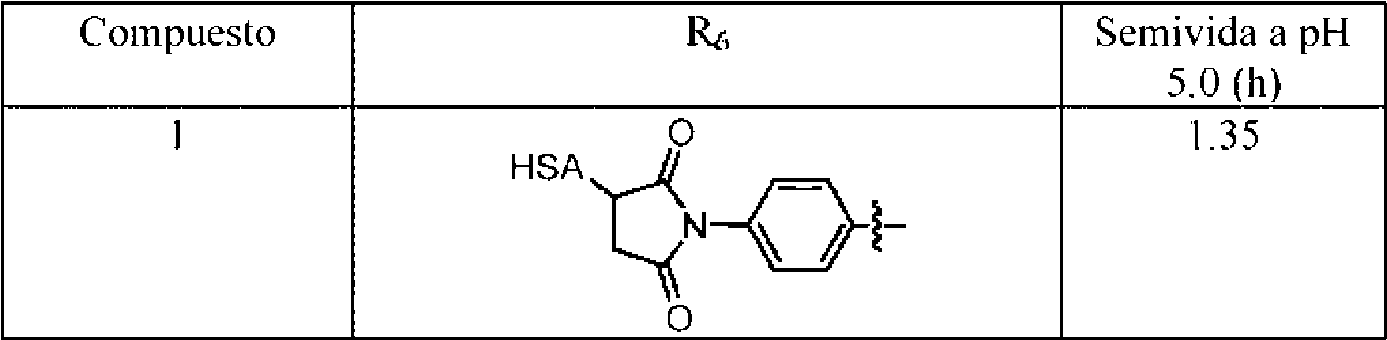


En algunas formas de realización, el compuesto de la invención se selecciona de: Ċ 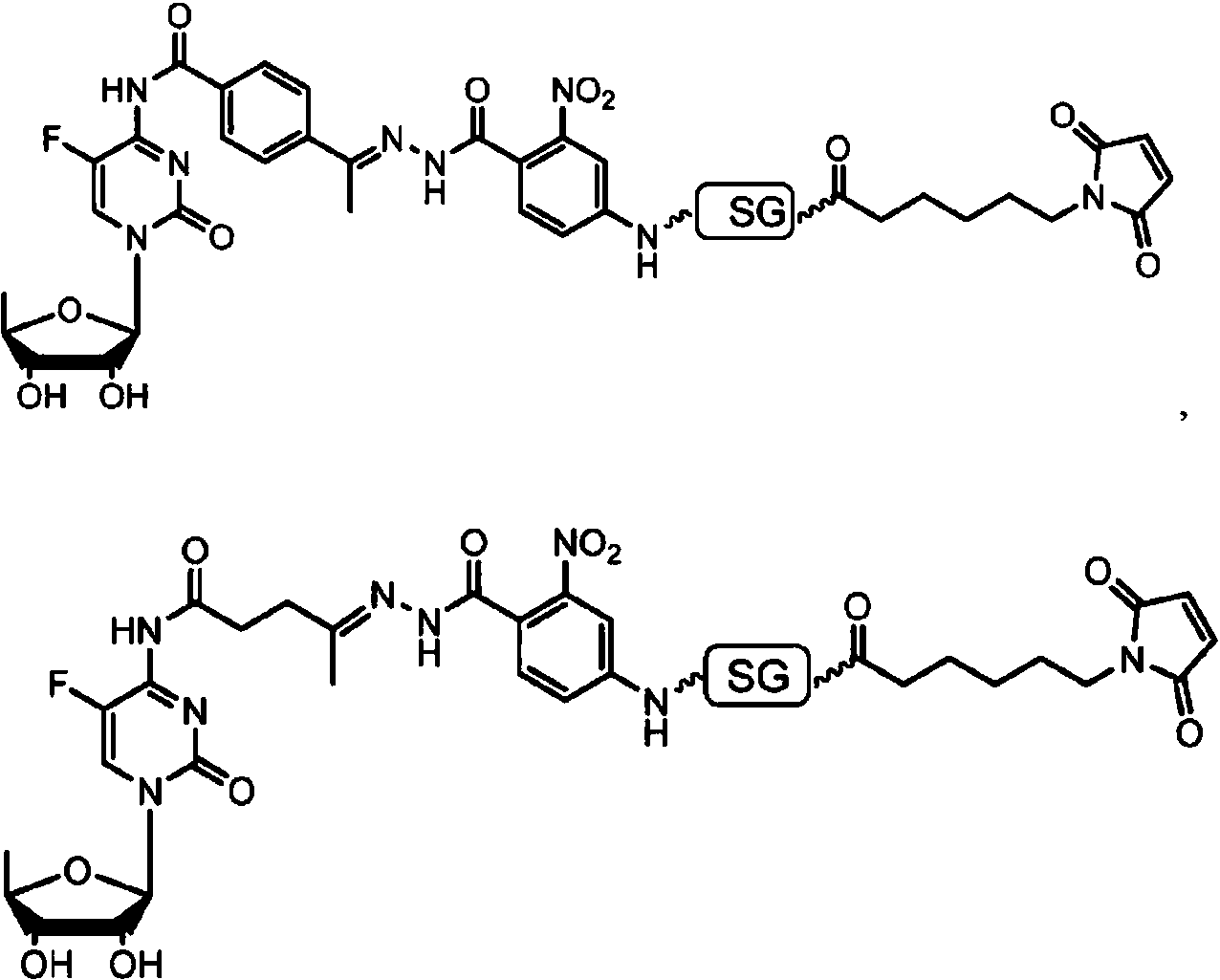

o una sal, hidrato, solvato, o isómero farmacéuticamente aceptable del mismo, en donde:
10 
c o está ausente o se selecciona del grupo que consiste en:

R es -OPO3M1 en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, 2NH4+, Na+, K+, NH4+ y/o H+ o ~ S 03M2 en donde M2 = Na+, K+, H+, y/o NH4+
En algunas formas de realización, el compuesto se selecciona del grupo que consiste en:




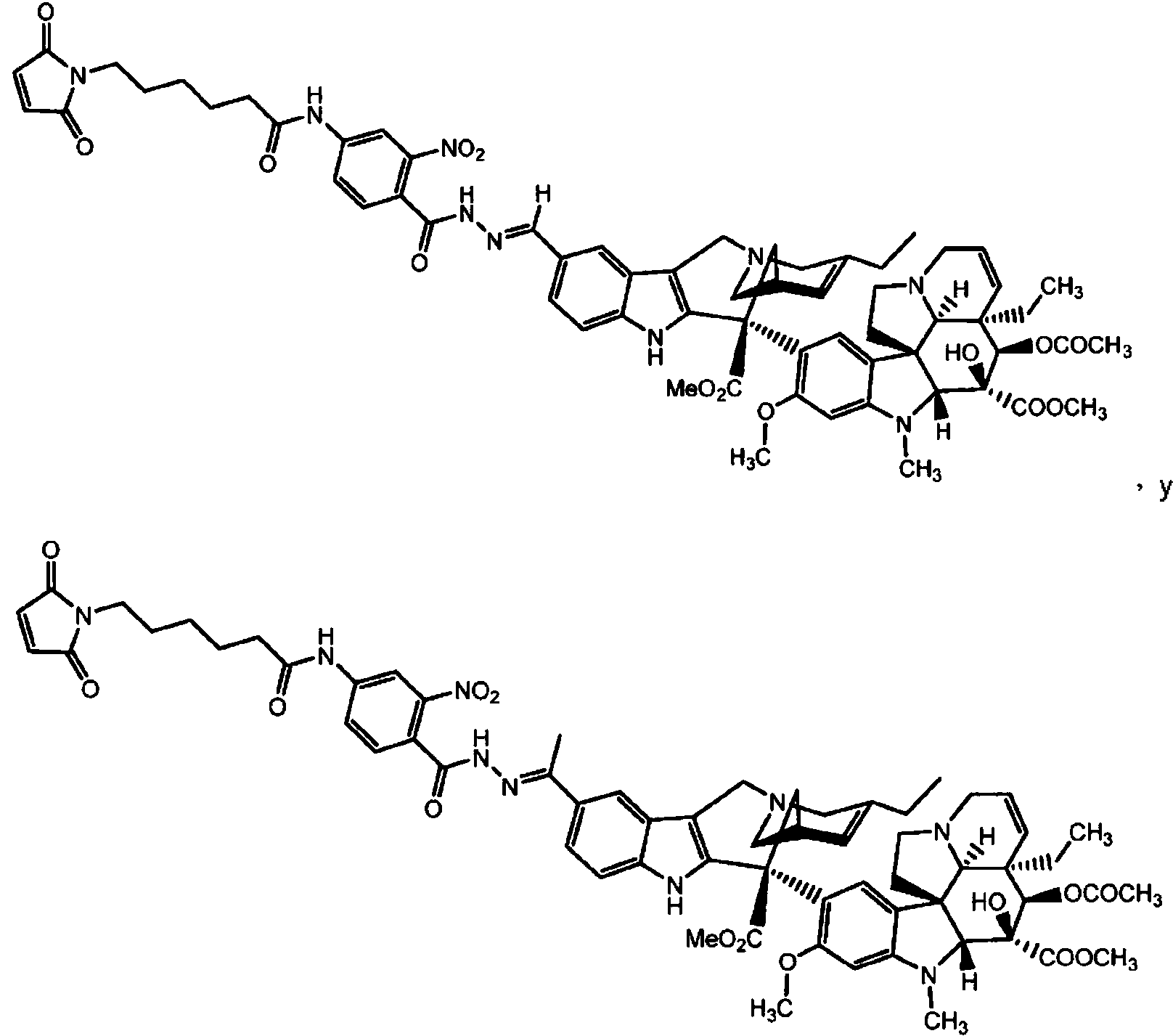
o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo.
En algunas formas de realización, el compuesto de la presente invención se selecciona del grupo que consiste en:

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo.
En algunas formas de realización, el compuesto de la presente invención se selecciona del grupo que consiste en:

o una sal, hidrato, solvato o isómero farmacéuticamente aceptable del mismo.
En algunas formas de realización, el compuesto es el compuesto 15, que tiene la estructura:

En algunas formas de realización, el compuesto 15 previene la desactivación de gemcitabina, lo que produce exposición del tumor sostenida.
El agente
En algunas formas de realización, el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente antiinflamatorio, un agente virostático, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o un fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, un derivado de cualquiera de los anteriores, una sal, hidrato, solvato, o isómero farmacéuticamente aceptable de cualquiera de los anteriores.
En algunas formas de realización, el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 5'-desoxi-5-fluorocitidina, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 7-acetilcamptotecina, 9-formilcamptotecina, 9-acetilcamptotecina, 9-formil-10-hidroxicamptotecina, 10-formilcamptotecina, 10-acetilcamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansina, maitansinol; auristatina (incluyendo, pero no limitado a auristatina D, auristatina E, auristatina F, monometil auristatina D, monometil auristatina E, monometil auristatina F, éster metílico de monometil auristatina F, auristatina PYE auristatina PHE, el producto natural relacionado dolastatina 10, y derivados de las mismas); amatoxinas (incluyendo, pero no limitado a a-amanitina, p-amanitina, Y-amanitina, £-amanitin, amanina, amaninamida, arnanulina, y ácido amanulínico y derivados de los mismos); duocarmicina A, duocarmicina B1, duocarmicina B2, duocarmicina C, duocarmicina SA,
CC1065, adozelesina, bizelesina, carzelesina; eribulina; trabectedina; pirrolobenzodiazepina, antramicina, tomaimicina, sibiromicina, DC-81, DSB-120; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.
En algunas formas de realización, el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 9-formilcamptotecina, 9-formil-10-hidroxicamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansinoides; auristatinas; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.
En algunas formas de realización, el agente es una N-nitrosourea.
En algunas formas de realización, el agente es una antraciclina, tal como, pero no limitada a, doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona o ametantrona.
En algunas formas de realización, el agente es un agente alquilante, tal como, pero no limitado a, clorambucilo, bendamustina, melfalán u oxazafosforinas.
En algunas formas de realización, el agente es un antimetabolito, tal como, pero no limitado a, 5-fluorouracilo, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona o tioguanina. En algunas formas de realización, el antimetabolito es un análogo de pirimidina o purina que contiene al menos un grupo amino primario o secundario.
En algunas formas de realización, el agente es un antagonista de ácido fólico, tal como, pero no limitado a, metotrexato, raltitrexed, pemetrexed o plevitrexed.
En algunas formas de realización, el agente es un taxano, tal como, pero no limitado a, paclitaxel o docetaxel.
En algunas formas de realización, el agente es una camptotecina, tal como, pero no limitado a, topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, -formilcamptotecina, 9-formilcamptotecina, 9-formil-10-hidroxicamptotecina, 7-butil-10-aminocamptotecina o 7-butil-9-amino-10,11,-metilenedioxocamptotecina.
En algunas formas de realización, el agente es un alcaloide de la vinca, tal como, pero no limitado a, vinblastina, vincristina, vindesina, vinorelbina o caliqueamicinas.
En algunas formas de realización, el agente es un maitansinoide, una auristatina, una epotilona, una bleomicina, dactinomicina, plicamicina, miromicina C, un complejo de platino(II) configurado en cis, o un derivado de cualquiera de los anteriores.
El espaciador
El enlace amida
En algunas formas de realización, el espaciador  donde n, X y R2 son como se ha definido en el presente documento.
donde n, X y R2 son como se ha definido en el presente documento.
En algunas formas de realización, el espaciador  donde n, X y R2 son como se ha definido en el presente documento.
donde n, X y R2 son como se ha definido en el presente documento.
En algunas formas de realización, n es 0 o 1.
En algunas formas de realización, X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido.
En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido, en donde un átomo de carbono en dicho alquilo de C1-C18 está sustituido con -OCH2CH2-. En algunas formas de realización, X1 es alquilo de C1-C18 opcionalmente sustituido, en donde dos átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido, en donde tres átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido, en donde cuatro átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido, en donde cinco átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido, en donde seis átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-.
En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-, en donde un átomo de carbono en dicho alquilo de C1-C18 está sustituido con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-, en donde dos átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-, en donde tres átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-, en donde cuatro átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-, en donde cinco átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-, en donde seis átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-.
En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido- C(O)-NH-R5-, en donde un átomo de carbono en dicho alquilo de C1-C18 está sustituido con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde dos átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde tres átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde cuatro átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde cinco átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, X es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde seis átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-.
En algunas formas de realización, X es arilo opcionalmente sustituido. En algunas formas de realización, X es

halógeno, -OH, -NO2 , -CN, -Oalquilo de C1-C6, alquilo de C1-C6 opcionalmente sustituido, haloalquilo de C1-C6 opcionalmente sustituido, y haloalcoxi de C1-C6 opcionalmente sustituido.
En algunas formas de realización, X es , en donde Z5 , Ze, Z7 y Zs se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -OH, -NO2 , -CN, -Oalquilo de C1-C6 opcionalmente sustituido, alquilo de C1-C6 opcionalmente sustituido, haloalquilo de C1-C6 opcionalmente sustituido, y haloalcoxi de C1-C6 opcionalmente sustituido. En algunas formas de realización, Z5 , Z6, Z7 y Z8 son cada uno -H.
, en donde Z5 , Ze, Z7 y Zs se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -OH, -NO2 , -CN, -Oalquilo de C1-C6 opcionalmente sustituido, alquilo de C1-C6 opcionalmente sustituido, haloalquilo de C1-C6 opcionalmente sustituido, y haloalcoxi de C1-C6 opcionalmente sustituido. En algunas formas de realización, Z5 , Z6, Z7 y Z8 son cada uno -H.
En algunas formas de realización, X es heteroarilo opcionalmente sustituido.
En algunas formas de realización, X es cicloalquilo opcionalmente sustituido.
En algunas formas de realización, R5 se selecciona del grupo que consiste en arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido. En algunas formas de realización, R5 es arilo opcionalmente sustituido.
En algunas formas de realización, R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido.
En algunas formas de realización, el espaciador está ausente.
En algunas formas de realización, el espaciador comprende una fracción o un enlace que es cortable por enzima. En algunas formas de realización, el enlace que se corta es un enlace peptídico, un enlace imida, o un enlace amida. En algunas formas de realización, el enlace amida se puede diseñar para ser específicamente cortable por una esterasa y/o amidasas, En algunas formas de realización, el espaciador comprende un enlace amida que se corta específicamente por carboxilesterasas. En formas de realización preferidas, el espaciador comprende un enlace amida que se corta específicamente por carboxilesterasa 2. En formas de realización preferidas, el espaciador comprende
un enlace amida que está unido a un anillo arilo como sigue: algunas formas de realización,
algunas formas de realización,
el espaciador comprende un enlace amida que está unido a un anillo arilo como sigue:
en donde Z5 , Za, Z7 y Z8 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -OH, -NO2, -CN, -Oalquilo de C1-Ca, alquilo de C1-Ca opcionalmente sustituido, haloalquilo de C1-Ca opcionalmente sustituido, y haloalcoxi de C1-C6 opcionalmente sustituido. En algunas formas de realización, el espaciador comprende
un enlace amida que está unido a un anillo arilo como sigue 
seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -OH, NO2 , -CN, -Oalquilo de C1-Ca, alquilo de C1-Ca opcionalmente sustituido, haloalquilo de C1-Ca opcionalmente sustituido y haloalcoxi de C1-Ca opcionalmente sustituido. En algunas formas de realización, Z5 , Za, Z7 y Z8 son cada uno -H.
En algunas formas de realización, el espaciador  donde R2 se selecciona del grupo que consiste en: -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido y heteroarilo opcionalmente sustituido; y W 1, W 2 , W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-Ca, -NO2, haloalquilo, -S(O)2-alquilo de C1-Ca, y -CN. En algunas formas de realización, W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN. En algunas formas de realización, W 1, W 2 , W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -F, -NO2 , y -CF3. En algunas formas de realización, W 1, W 2, W3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: un grupo fenoxi, un grupo amina primaria, secundaria o terciaria, un grupo éter, un grupo fenol, un grupo amida, un grupo éster, un grupo alquilo, un grupo alquilo sustituido, un grupo fenilo, y un grupo vinilo. En algunas formas de realización, W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo. En algunas formas de realización, al menos uno de W 1, W 2, W 3 y W 4 no es -H.
donde R2 se selecciona del grupo que consiste en: -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido y heteroarilo opcionalmente sustituido; y W 1, W 2 , W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-Ca, -NO2, haloalquilo, -S(O)2-alquilo de C1-Ca, y -CN. En algunas formas de realización, W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN. En algunas formas de realización, W 1, W 2 , W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -F, -NO2 , y -CF3. En algunas formas de realización, W 1, W 2, W3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: un grupo fenoxi, un grupo amina primaria, secundaria o terciaria, un grupo éter, un grupo fenol, un grupo amida, un grupo éster, un grupo alquilo, un grupo alquilo sustituido, un grupo fenilo, y un grupo vinilo. En algunas formas de realización, W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo. En algunas formas de realización, al menos uno de W 1, W 2, W 3 y W 4 no es -H.
En algunas formas de realización, Wi se selecciona del grupo que consiste en: halógeno, -C(O)OH, -C(O)O-alquilo de Ci-Ca, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -CN; y W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C i Ca, y -Cn . En algunas formas de realización, Wi se selecciona del grupo que consiste en: -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3, y -CN; y W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN. En algunas formas de realización, Wi se selecciona del grupo que consiste en: -Cl, -F, y -NO2; y Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en: -H, -Cl, -F, -NO2 , y -CF3.
En algunas formas de realización, el espaciador es en donde R2 es como se define en el
en donde R2 es como se define en el
presente documento. En algunas formas de realización, el espaciador es 
En algunas formas de realización, el espaciador es  y m es i, 2, 3, 4, 5, o 6.
y m es i, 2, 3, 4, 5, o 6.
En algunas formas de realización, el espaciador es 
En algunas formas de realización, R2 se selecciona del grupo que consiste en -H, alquilo de C i-C i2 opcionalmente sustituido, arilo opcionalmente sustituido y heteroarilo opcionalmente sustituido.
En un aspecto, el espaciador puede operar como un grupo protector, que previene la degradación metabólica del agente, permitiendo de esta manera un uso dirigido de una dosis mayor del agente. En algunas formas de realización, el agente es gemcitabina.
El enlazador
En algunas formas de realización, el enlazador comprende Y y Ri. En algunas formas de realización, Y está ausente. En algunas formas de realización, Ri está ausente. En algunas formas de realización, tanto Y como Ri están ausentes.
En algunas formas de realización Y se selecciona del grupo que consiste en metilo, etilo, -NH-C(O)-, -C(O)-NH, -C(O)-O-, y -O-C(O)-.
En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido, en donde un átomo de carbono en dicho alquilo de Ci-Ci8 está sustituido con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido, en donde dos átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido, en donde tres átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido, en donde cuatro átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido, en donde cinco átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido, en donde seis átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-.
En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido-NH-C(O)-R5-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido-NH-C(O)-R5-, en donde un átomo de carbono en dicho alquilo de Ci-Ci8 está sustituido con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido-NH-C(O)-R5-, en donde dos átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido-NH-C(O)-R5-, en donde tres átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido-NH-C(O)-R5-, en donde cuatro átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos con -OCH2CH2-. En algunas formas de realización, Ri es alquilo de Ci-Ci8 opcionalmente sustituido-NH-C(O)-R5-, en donde cinco átomos de carbono en dicho alquilo de Ci-Ci8 están sustituidos
con -OCH2CH2-. En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5-, en donde seis átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-.
En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-. En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido- C(O)-NH-R5-, en donde un átomo de carbono en dicho alquilo de C1-C18 está sustituido con -OCH2CH2-. En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde dos átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde tres átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde cuatro átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde cinco átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-. En algunas formas de realización, R1 es alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5-, en donde seis átomos de carbono en dicho alquilo de C1-C18 están sustituidos con -OCH2CH2-.
En algunas formas de realización, R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido.
En algunas formas de realización, la fracción compuesto de la invención se selecciona del grupo que consiste en:
compuesto de la invención se selecciona del grupo que consiste en:
M2 = Na+, K+, H+, NH4+,

Mi = Mg2+; 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+,

Mi = Mg2+, 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+

Mi = Mg2+; 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+,

Mi = Mg2+, 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+

Mi = Mg2+, 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+ 6i

El grupo de unión a proteínas
Los grupos de unión a proteínas (“PBG”) incluyen, pero no están limitados a, un grupo maleimida sustituido o sin sustituir, un grupo haloacetamida sustituido o sin sustituir, un grupo haloacetato sustituido o sin sustituir, un grupo piridiltio sustituido o sin sustituir, un grupo isotiocianato sustituido o sin sustituir, un grupo vinilcarbonilo sustituido o sin sustituir, un grupo aziridina sustituido o sin sustituir, un grupo disulfuro sustituido o sin sustituir, un grupo acetileno sustituido o sin sustituir, un grupo éster de N-hidroxisuccinimida sustituido o sin sustituir, un anticuerpo o fragmento del mismo. En algunas formas de realización, los grupos de unión a proteínas (“PBG”) incluyen, pero no están limitados a, un grupo maleimida sustituido o sin sustituir, un grupo haloacetamida sustituido o sin sustituir, un grupo haloacetato sustituido o sin sustituir, un grupo piridiltio sustituido o sin sustituir, un grupo isotiocianato sustituido o sin sustituir, un grupo vinilcarbonilo sustituido o sin sustituir, un grupo aziridina sustituido o sin sustituir, un grupo disulfuro sustituido o sin sustituir, un grupo acetileno sustituido o sin sustituir, un grupo éster de N-hidroxisuccinimida sustituido o sin sustituir. En algunas formas de realización, el grupo de unión a proteínas es un grupo maleimida sustituido o sin
sustituir. En algunas formas de realización, el grupo de unión a proteínas es
En algunas formas de realización, PBG está sustituido con alquilo de C1-C6 o halógeno. En algunas formas de realización, PBG está sustituido con metilo, -Cl o -Br. En algunas formas de realización, PBG incluye un anticuerpo o un fragmento del mismo. En algunas formas de realización, PBG incluye un ligando con especificidad para el receptor, por ejemplo, un compuesto de bajo o alto peso molecular tal como ácido fólico, vitaminas, péptidos, azúcares, proteínas nativas o modificadas.
Un grupo disulfuro se puede activar por un ácido tionitrobenzoico (por ejemplo, ácido 5'-tio-2-nitrobenzoico) como el grupo intercambiable. Un grupo maleimida, piridiltio, o éster de N-hidroxisuccinimida puede, donde sea apropiado, estar sustituido por un grupo alquilo o por los grupos solubles en agua anteriores. En general, un grupo de unión a proteínas posee propiedades de unión a proteínas, es decir, se une covalentemente (“un grupo de unión a proteínas covalente”) o no covalentemente (“un grupo de unión a proteínas no covalente”), en un entorno fisiológico, a aminoácidos particulares en la superficie de la proteína. El grupo maleimida, el grupo haloacetamida, el grupo haloacetato, el grupo piridiltio, el grupo disulfuro, el grupo vinilcarbonilo, el grupo aziridina, y/o el grupo acetileno preferiblemente reacciona con grupos tiol (-SH) de cisteínas, mientras el grupo éster de N-hidroxisuccinimida y/o el grupo isotiocianato preferiblemente reaccionan con el grupo amino (-NH) de lisinas, en la superficie de una proteína. Por ejemplo, el grupo de unión a proteínas, tal como un grupo maleimida, se puede unir a la cisteína 34 de albúmina. En algunas formas de realización, la albúmina no está modificada (por ejemplo, no está modificada para estar cargada,
sea positiva o negativamente). En algunas formas de realización, un PBG como se describe en el presente documento se puede unir a un anticuerpo o fragmento del mismo, tal como los descritos en el presente documento.
El compuesto usado en la invención incluye cualquiera y todas las combinaciones de uno o más agentes, espaciadores, enlazadores, grupos de unión a proteínas, fracciones cortables, es decir, hidrazona, enlace amida. Los compuestos pueden comprender una antraciclina, y una fracción cortable con ácido que se puede cortar de tal manera que se controle la liberación de la antraciclina. En ciertas formas de realización, la sustancia terapéuticamente eficaz comprende una antraciclina, una hidrazona como la fracción cortable con ácido cuyo corte y semivida de la liberación del fármaco varían según los sustituyentes aceptores de electrones y su posición en el anillo fenilo al que la hidrazona está unida, y un grupo maleimida como el grupo de unión a proteínas covalente. En algunas otras formas de realización, los compuestos pueden comprender gemcitabina, y una fracción cortable con ácido que se puede cortar de tal manera que se controle la liberación de gemcitabina. En algunas otras formas de realización, los compuestos pueden comprender un alcaloide de la vinca, y una fracción cortable con ácido que se puede cortar de tal manera que se controle la liberación del alcaloide de la vinca.
Anticuerpos como portadores
Las células cancerosas poseen marcadores específicos, conocidos como antígenos, que desempeñan un papel en crecimiento y evolución tumoral. Los antígenos son proteínas, con frecuencia localizados en la superficie del tumor. Una característica importante de los anticuerpos es su capacidad de unirse a antígenos diana con alta especificidad. Sin embargo, a pesar de la unión específica a antígenos, los anticuerpos con frecuencia carecen de actividad terapéutica (Panowski et al., mAbs, 6, 34-45 (2014); Chari et al., Angewandte Chem. Int. Ed., 53, 3796-3827 (2014)). Debido a su alta especificidad por antígenos, los anticuerpos se pueden usar como portadores para administración de fármacos. Se pueden conjugar a una sustancia terapéuticamente eficaz -formando un conjugado anticuerpo-fármaco (ADC)- para administrar la sustancia terapéuticamente eficaz a un sitio diana tal como un tumor. Tras la unión del ADC al antígeno, el complejo antígeno-ADC se internaliza mediante endocitosis. Una vez dentro del tumor, el ADC libera la sustancia terapéuticamente eficaz. Para que un ADC tenga éxito, la elección de la sustancia terapéuticamente eficaz, el enlazador y el anticuerpo es importante. La selección de antígeno, y por consiguiente un anticuerpo correspondiente, es importante ya que la eficacia de la internalización del complejo antígeno-anticuerpo influye cuanta sustancia terapéuticamente eficaz se administra. Además, para que un a Dc sea eficaz, el número de moléculas de fármaco requerido para destruir una célula tiene que estar por debajo de la cantidad de fármaco que se puede administrar. Por tanto, un fármaco potente, con potencia en el intervalo picomolar, es preferido. Por último, una vez el ADC se internaliza, el enlazador se debe cortar para liberar la sustancia terapéuticamente eficaz. Los enlazadores preferidos proporcionan liberación eficaz y controlada de la sustancia terapéuticamente eficaz al medio ácido del tumor, mientras previenen la liberación de la sustancia terapéuticamente eficaz en el plasma que podría producir toxicidad inespecífica y una ventana terapéutica más estrecha para el uso de la sustancia terapéuticamente eficaz. Los enlazadores divulgados en el presente documento se pueden conjugar con anticuerpos que se dirigen hacia cualquier antígeno o receptor expresado en una célula maligna. El anticuerpo puede ser quimérico, humanizado, humano o un anticuerpo genéticamente manipulado o químicamente modificado tal como un tioanticuerpo (THIOMAB), y permitir la unión del enlazador sin reducir significativamente la capacidad de unirse a la diana del anticuerpo.
En algunas formas de realización, los grupos de unión a proteínas (PBG) de los compuestos descritos en el presente documento se asocian con un anticuerpo o fragmento del mismo como se describe en el presente documento formando de esta manera un ADC. En algunas formas de realización, el PBG está covalentemente unido a un anticuerpo o fragmento del mismo. En otras formas de realización, el PBG está no covalentemente unido a un anticuerpo o fragmento del mismo.
En ciertos aspectos, el PBG de la presente solicitud se puede unir a un portador. En algunas formas de realización, el portador es un agente de unión a polipéptidos. La divulgación abarca agentes de unión a polipéptidos, tal como anticuerpos, porciones de unión a antígenos de anticuerpos, y andamiajes de unión a antígenos no inmunoglobulina en parte, que pueden dirigirse eficazmente a antígenos.
Se puede usar la unión de anticuerpos monoclonales a antígenos presentes en la superficie del tumor en los métodos y composiciones divulgados.
En algunas formas de realización, los anticuerpos pueden ser anticuerpos modificados, o fragmentos de unión a antígenos de los mismos que se unen al dominio extracelular de un antígeno.
En algunas formas de realización, el anticuerpo desinmunizado o fragmento de unión a antígeno del mismo que se une al dominio extracelular del antígeno, incluyendo una región variable de la cadena pesada y una región variable de la cadena ligera, en donde cada región variable tiene entre 2 a 20 sustituciones de aminoácidos en la región marco en comparación con un anticuerpo no humano o parental que se une al dominio extracelular del antígeno.
En algunas formas de realización, el anticuerpo desinmunizado o fragmento de unión a antígeno del mismo tiene una o más regiones determinantes de complementariedad (CDR) de un anticuerpo no humano o parental que se une al
dominio extracelular del antígeno. En una forma de realización, entre 1-5 sustituciones están presentes en las regiones determinantes de complementariedad (CDR).
En algunas formas de realización, anticuerpos monocatenarios, y anticuerpos quiméricos, humanizados o primatizados (injertados con CDR), así como anticuerpos monocatenarios quiméricos o injertados con CDR, que comprenden porciones derivadas de diferentes especies, se pueden usar como porciones de unión a antígeno de un anticuerpo. Las varias porciones de estos anticuerpos se pueden unir químicamente por técnicas convencionales, o se pueden preparar como una proteína contigua usando técnicas de ingeniería genética. Por ejemplo, se pueden expresar ácidos nucleicos que codifican una cadena quimérica o humanizada para producir una proteína contigua. Véase, por ejemplo, Cabilly et al., Patente en EE UU No. 4.816.567; Cabilly et al., Patente Europea No. 0.125.023 B1; Boss et al., Patente en EE UU No. 4.816.397; Boss et al., Patente Europea No. 0.120.694 B1; Neuberger, M. S. et al., documento WO 86/01533; Neuberger, M. S. et al., Patente Europea No. 0.194.276 B1; Winter, Patente en EE UU No.
5.225.539; y Winter, Patente Europea No. 0.239.400 B1. Véase también, Newman, R. et al., BioTechnology, 10: 1455 1460 (1992), respecto a anticuerpo primatizado. Véase, por ejemplo, Ladner et al., Patente en EE UU No. 4.946.778; y Bird, R. E. et al., Science, 242: 423-426 (1988)), respecto a anticuerpos monocatenarios.
Además, también se pueden usar fragmentos funcionales de anticuerpos, incluyendo fragmentos de anticuerpos quiméricos, humanizados, primatizados o monocatenarios. Los fragmentos funcionales de los anticuerpos objeto retienen al menos una función de unión y/o función de modulación del anticuerpo de longitud completa del que derivan. En ciertas formas de realización, los fragmentos funcionales retienen una función de unión a antígeno de un anticuerpo de longitud completa correspondiente (por ejemplo, especificidad para un tumor).
Por ejemplo, los fragmentos de anticuerpo capaces de unirse a un receptor antígeno o porción del mismo, incluyendo, pero no limitado a, fragmentos Fv, Fab, Fab' y F(ab')2 están abarcados por la invención. Tales fragmentos se pueden producir por corte enzimático o por técnicas recombinantes. Por ejemplo, el corte con papaína o pepsina puede generar fragmentos Fab o F(ab')2 , respectivamente. Los anticuerpos producidos en una variedad de formas truncadas usando genes de anticuerpos en los que se han introducido uno o más codones de terminación antes del sitio de terminación natural están abarcados por la invención. Por ejemplo, se puede diseñar un gen quimérico que codifica una porción de cadena pesada F(ab')2 para incluir secuencias de ADN que codifican el dominio CH1 y la región bisagra de la cadena pesada.
El término “ inmunoglobulina humanizada”, como se usa en el presente documento, se refiere a una inmunoglobulina que comprende porciones de inmunoglobulinas de diferente origen, en donde al menos una porción es de origen humano. Según esto, la presente invención abarca una inmunoglobulina humanizada que tiene especificidad de unión por un antígeno (por ejemplo, antígeno humano), dicha inmunoglobulina comprende una región de unión a antígeno de origen no humano (por ejemplo, roedor) y al menos una porción de una inmunoglobulina de origen humano (por ejemplo, una región marco humana, una región constante humana o una parte de la misma). Por ejemplo, el anticuerpo humanizado pueden comprender porciones derivadas de una inmunoglobulina de origen no humano con el requisito de especificidad, tal como un ratón, y de secuencias de inmunoglobulina de origen humano (por ejemplo, una inmunoglobulina quimérica), unidas químicamente por técnicas convencionales (por ejemplo, sintética) o preparada como un polipéptido contiguo usando técnicas de ingeniería genética (por ejemplo, ADN que codifica porciones de proteína del anticuerpo quimérico se puede expresar para producir una cadena polipeptídica contigua).
Otro ejemplo de una inmunoglobulina humanizada es una inmunoglobulina que contiene una o más cadenas de inmunoglobulina que comprende una CDR de origen no humano (por ejemplo, una o más CDR derivadas de un anticuerpo de origen no humano) y una región marco derivada de una cadena ligera y/o pesada de origen humano (por ejemplo, anticuerpos con CDR injertada con o sin cambios en el marco).
En algunas formas de realización, el anticuerpo es un anticuerpo antagonista. Como se describe en el presente documento, el término “anticuerpo antagonista” se refiere a un anticuerpo que puede inhibir una o más funciones de la diana, tal como una actividad de unión (por ejemplo, unión a ligando) y una actividad de señalización (por ejemplo, transporte de aminoácidos). Por ejemplo, un anticuerpo antagonista puede inhibir (reducir o prevenir) el transporte de glutamina por antígeno.
En algunas formas de realización, los anticuerpos anti-idiotípicos también son útiles. Los anticuerpos anti-idiotípicos reconocen determinantes antigénicos asociados con el sitio de unión a antígeno de otro anticuerpo. Los anticuerpos anti-idiotípicos se pueden preparar contra un segundo anticuerpo inmunizando un animal de la misma especie, y puede ser de la misma cepa, que el animal usado para producir el segundo anticuerpo. Véase, por ejemplo, la patente en EE UU No. 4.699.880. En una forma de realización, se generan anticuerpos contra un receptor o una porción del mismo, y estos anticuerpos se usan a su vez para producir un anticuerpo anti-idiotípico. Tal anticuerpo anti-idiotípico también puede ser un inhibidor de una función transportadora de antígeno, aunque no se una al antígeno mismo. Tal anticuerpo anti-idiotípico también se puede llamar un anticuerpo antagonista.
En algunas formas de realización, los anticuerpos adecuados se pueden fragmentar usando técnicas convencionales y los fragmentos cribar para utilidad de la misma manera que se ha descrito anteriormente para anticuerpos enteros.
Por ejemplo, se pueden generar fragmentos F(ab)2 tratando el anticuerpo con pepsina. El fragmento F(ab)2 resultante se puede tratar para reducir los puentes disulfuro para producir fragmentos Fab.
En algunas formas de realización, se pretende además que los anticuerpos adecuados incluyan moléculas biespecíficas, monocatenarias y quiméricas y humanizadas que tienen afinidad por un polipéptido antígeno conferida por al menos una región CDR del anticuerpo. Las técnicas para la producción de anticuerpos monocatenarios (patente en EE UU No. 4.946.778) también se pueden adaptar para producir anticuerpos monocatenarios. Además, se pueden usar ratones transgénicos u otros organismos incluyendo otros mamíferos, para expresar anticuerpos humanizados. Los métodos de generar estos anticuerpos se conocen en la técnica. Véase, por ejemplo, Cabilly et al., Patente en EE UU No. 4.816.567; Cabilly et al., Patente Europea No. 0.125.023 B1; Queen et al., Patente europea No. 0.451.216 B1; Boss et al., Patente en EE UU No. 4.816.397; Boss et al., Patente Europea No. 0.120.694 B1; Neuberger, M. S. et al., documento WO 86/01533; Neuberger, M. S. et al., Patente Europea No. 0.194.276 B1; Winter, Patente en EE UU No.
5.225.539; Winter, Patente Europea No. 0.239.400 B1, Padlan, E. A. et al., Solicitud de patente europea No. 0.519.596 A1. Véase también, Ladner et al., Patente en EE UU No. 4.946.778; y Bird, R. E. et al., Science, 242: 423-426 (1988)).
Tales inmunoglobulinas humanizadas se pueden producir usando ácidos nucleicos sintéticos y/o recombinantes para preparar genes (por ejemplo, ADNc) que codifican la cadena humanizada deseada. Por ejemplo, secuencias de ácido nucleico (por ejemplo, ADN) que codifican regiones variables humanizadas se pueden construir usando métodos de mutagénesis por PCR para alterar las secuencias de ADN que codifican una cadena humana o humanizada, tal como un molde de ADN de una región variable previamente humanizada (véase, por ejemplo, Kamman, M., et al., Nucl. Acids Res., 17: 5404 (1989)); Sato, K., et al., Cancer Research, 53: 851-856 (1993); Daugherty, B. L. et al., Nucleic Acids Res., 19(9): 2471-2476 (1991); y Lewis, A. P. y J. S. Crowe, Gene, 101: 297-302 (1991)). Usando estos y otros métodos adecuados, también se pueden producir variantes fácilmente. En una forma de realización, las regiones variables clonadas se pueden mutagenizar, y las secuencias que codifican variantes con la especificidad deseada se pueden seleccionar (por ejemplo, de una genoteca de fagos; véase, por ejemplo, Krebber et al., Patente en EE UU No. 5.514.548; Hoogenboom et al., documento WO 93/06213, publicado el 1 de abril, 1993)).
En algunas formas de realización, un anticuerpo es un anticuerpo monoclonal. Por ejemplo, un método para generar un anticuerpo monoclonal que se une específicamente a un polipéptido antígeno puede comprender administrar a un ratón una cantidad de una composición inmunógena que comprende el polipéptido antígeno eficaz para estimular un respuesta inmunitaria detectable, obtener células productoras de anticuerpo (por ejemplo, células del bazo) del ratón y fusionar las células productoras de anticuerpo con células de mieloma para obtener hibridomas productores de anticuerpos, y ensayar los hibridomas productores de anticuerpo para identificar un hibridoma que produce un anticuerpo monoclonal que se une específicamente al polipéptido antígeno. Una vez obtenido, un hibridoma se puede propagar en un cultivo celular, opcionalmente en condiciones de cultivo donde las células derivadas de hibridoma producen el anticuerpo monoclonal que se une específicamente a un polipéptido antígeno. El anticuerpo monoclonal se puede purificar del cultivo celular.
Además, las técnicas usadas para cribar anticuerpos con el fin de identificar un anticuerpo deseable pueden influir las propiedades del anticuerpo obtenido. Por ejemplo, un anticuerpo que se va a usar para ciertos fines terapéuticos puede ser capaz de dirigirse a un tipo celular particular. Según esto, para obtener anticuerpos de este tipo, puede ser deseable cribar para anticuerpos que se unen a células que expresan el antígeno de interés (por ejemplo, por separación celular activada por fluorescencia). Asimismo, si un anticuerpo se va a usar para unirse a un antígeno en solución, puede ser deseable ensayar unión en solución. Una variedad de diferentes técnicas están disponibles para ensayar interacciones anticuerpo:antígeno para identificar anticuerpos particularmente deseables. Tales técnicas incluyen ELISA, ensayos de unión de resonancia de plasmón de superficie (por ejemplo, el ensayo de unión Biacore, Biacore AB, Upsala, Suecia), ensayos sándwich (por ejemplo, el sistema de perlas paramagnéticas de IGEN International, Inc., Gaithersburg, Maryland), inmunotransferencias, ensayos de inmunoprecipitación e inmunohistoquímica.
Composiciones farmacéuticas
En algunas formas de realización, la invención proporciona una composición farmacéutica que comprende un compuesto descrito en el presente documento.
La cantidad total de un compuesto en una composición que se va a administrar a un paciente es una que es adecuada para el paciente. Un experto en la materia apreciaría que diferentes individuos pueden requerir diferentes cantidades totales de la sustancia terapéuticamente eficaz. En algunas formas de realización, la cantidad del compuesto es una cantidad farmacéuticamente eficaz. El experto en la materia sería capaz de determinar la cantidad del compuesto en una composición necesaria para tratar un paciente basado en factores tales como, por ejemplo, la edad, peso y estado físico del paciente. La concentración del compuesto depende de su solubilidad en la solución de administración intravenosa y el volumen de fluido que se puede administrar. Por ejemplo, la concentración del compuesto puede ser desde aproximadamente 0,1 mg/ml hasta aproximadamente 50 mg/ml en la composición inyectable. En algunas formas de realización, la concentración del compuesto puede ser desde aproximadamente 0,1 mg/ml hasta aproximadamente 25 mg/ml, desde aproximadamente 7 mg/ml hasta aproximadamente 17 mg/ml, desde aproximadamente 0,1 mg/ml hasta aproximadamente 5 mg/ml, o desde aproximadamente 0,25 mg/ml hasta
aproximadamente 4,5 mg/ml. En formas de realización particulares, la concentración del compuesto puede ser aproximadamente 0,1 mg/ml, aproximadamente 0,2 mg/ml, aproximadamente 0,3 mg/ml, aproximadamente 0,4 mg/ml, aproximadamente 0,5 mg/ml, aproximadamente 0,6 mg/ml, aproximadamente 0,7 mg/ml, aproximadamente 0,8 mg/ml, aproximadamente 0,9 mg/ml, aproximadamente 1,0 mg/ml, aproximadamente 1,1 mg/ml, aproximadamente 1,2 mg/ml, aproximadamente 1,3 mg/ml, aproximadamente 1,4 mg/ml, aproximadamente 1,5 mg/ml, aproximadamente 1,6 mg/ml, aproximadamente 1,7 mg/ml, aproximadamente 1,8 mg/ml, aproximadamente 1,9 mg/ml, aproximadamente 2,0 mg/ml, aproximadamente 2,1 mg/ml, aproximadamente 2,2 mg/ml, aproximadamente 2,3 mg/ml, aproximadamente 2,4 mg/ml, aproximadamente 2,5 mg/ml, aproximadamente 2,6 mg/ml, aproximadamente 2,7 mg/ml, aproximadamente 2,8 mg/ml, aproximadamente 2,9 mg/ml, aproximadamente 3,0 mg/ml, aproximadamente 3,1 mg/ml, aproximadamente 3,2 mg/ml, aproximadamente 3,3 mg/ml, aproximadamente 3,4 mg/ml, aproximadamente 3,5 mg/ml, aproximadamente 3,6 mg/ml, aproximadamente 3,7 mg/ml, aproximadamente 3,8 mg/ml, aproximadamente 3,9 mg/ml, aproximadamente 4,0 mg/ml, aproximadamente 4,1 mg/ml, aproximadamente 4,2 mg/ml, aproximadamente 4,3 mg/ml, aproximadamente 4,4 mg/ml, aproximadamente 4,5 mg/ml, aproximadamente 4,6 mg/ml, aproximadamente 4,7 mg/ml, aproximadamente 4,8 mg/ml, aproximadamente 4,9 mg/ml, aproximadamente 5,0 mg/ml, aproximadamente 5,1 mg/ml, aproximadamente 5,2 mg/ml, aproximadamente 5,3 mg/ml, aproximadamente 5,4 mg/ml, aproximadamente 5,5 mg/ml, aproximadamente 5,6 mg/ml, aproximadamente 5,7 mg/ml, aproximadamente 5,8 mg/ml, aproximadamente 5,9 mg/ml, o aproximadamente 6,0 mg/ml. En formas de realización particulares, la concentración del compuesto puede ser aproximadamente 7 mg/ml, aproximadamente 8 mg/ml, aproximadamente 9 mg/ml, aproximadamente 10 mg/ml, aproximadamente 11 mg/ml, aproximadamente 12 mg/ml, aproximadamente 13 mg/ml, aproximadamente 14 mg/ml, aproximadamente 15 mg/ml, aproximadamente 16 mg/ml, aproximadamente 17 mg/ml, aproximadamente 18 mg/ml, aproximadamente 19 mg/ml, aproximadamente 20 mg/ml, aproximadamente 21 mg/ml, aproximadamente 22 mg/ml, aproximadamente 23 mg/ml, aproximadamente 24 mg/ml, aproximadamente 25 mg/ml, aproximadamente 26 mg/ml, aproximadamente 27 mg/ml, aproximadamente 28 mg/ml, aproximadamente 29 mg/ml, o aproximadamente 30 mg/ml.
Las composiciones farmacéuticas y kits de la presente invención también pueden contener diluyentes, rellenos, sales, tampones, estabilizantes, solubilizantes, y otros materiales bien conocidos en la técnica. El término “farmacéuticamente aceptable” significa un material no tóxico que no interfiere con la eficacia de la actividad biológica del principio(s) activo(s). Las características del soporte dependerán de la vía de administración.
Las composiciones se pueden administrar en una variedad de maneras convencionales. Las rutas de administración ejemplares que se pueden usar incluyen oral, parenteral, intravenosa, intraarterial, cutánea, subcutánea, intramuscular, tópica, intracraneal, intraorbital, oftálmica, intravítrea, intraventricular, intracapsular, intrarraquídea, intracisternal, intraperitoneal, intranasal, aerosol, administración al sistema nervioso central (SNC), o administración por supositorio. En algunas formas de realización las composiciones son adecuadas para administración parenteral. Estas composiciones se pueden administrar, por ejemplo, por vía intraperitoneal, intravenosa, o intratecal. En algunas formas de realización, las composiciones se inyectan por vía intravenosa. En algunas formas de realización, una formulación reconstituida se puede preparar reconstituyendo una composición de compuesto de antraciclina liofilizado en un líquido de reconstitución que comprende etanol y agua. Tal reconstitución puede comprender añadir el líquido de reconstitución y mezclar, por ejemplo, girando o mezclando con el vórtex la mezcla. La formulación reconstituida se puede hacer después adecuada para inyección mezclando, por ejemplo, solución de Ringer con lactato con la formulación para crear una composición inyectable. Un experto en la materia apreciaría que un método de administrar una formulación o composición de una sustancia terapéuticamente eficaz dependería de factores tales como la edad, peso y estado físico del paciente que se trata, y la enfermedad o afección que se trata. El experto en la materia, por tanto, sería capaz de seleccionar un método de administración óptimo para un paciente en una base de caso a caso.
En algunas formas de realización, la invención proporciona compuestos y composiciones para uso como un medicamento. En algunas formas de realización, la invención proporciona compuestos y composiciones para uso en tratar un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, enfermedad inflamatoria aguda o crónica, y una enfermedad causada por bacterias, hongos, u otros microorganismos.
En algunas formas de realización, el compuesto divulgado en el presente documento se puede usar en la fabricación o preparación de un medicamento para tratar una enfermedad o afección seleccionada de un grupo que consiste en un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, enfermedad inflamatoria aguda o crónica, y una enfermedad causada por bacterias, hongos, u otros microorganismos.
En algunas formas de realización, el cáncer es un cáncer sanguíneo o un cáncer de tumor sólido. En algunas formas de realización, el cáncer se selecciona de carcinoma, sarcoma, leucemia, linfoma, mieloma múltiple, o melanoma.
En algunas formas de realización, el cáncer es adenocarcinoma, melanoma uveal, leucemia aguda, neuroma acústico, carcinoma ampular, carcinoma anal, astrocitomas, basalioma, cáncer pancreático, tumor de tejido conjuntivo, cáncer de vejiga, carcinoma bronquial, carcinoma bronquial no microcítico, cáncer de mama, linfoma de Burkitt, carcinoma de cuerpo, síndrome CUP, cáncer de colon, cáncer del intestino delgado, cáncer ovárico, carcinoma endometrial, cáncer de la vesícula biliar, carcinomas de la vesícula biliar, cáncer uterino, cáncer cervical, tumores de cuello, nariz y oídos, neoplasias hematológicas, leucemia de célula pilosas, cáncer uretral, cáncer de piel, gliomas, cáncer testicular, sarcoma de Kaposi, cáncer de laringe, cáncer de huesos, carcinoma colorrectal, tumores de cabeza/cuello, carcinoma de colon, craneofaringioma, cáncer de hígado, leucemia, cáncer de pulmón, cáncer de pulmón no microcítico, linfoma
de Hodgkin, linterna no hodgkiniano, cáncer de estómago, cáncer de colon, meduloblastoma, melanoma, meningioma, cáncer de riñón, carcinomas de células renales, oligodendroglioma, carcinoma esofágico, carcinomas osteolíticos y carcinomas osteoplásticos, osteosarcoma, carcinoma ovárico, carcinoma pancreático, cáncer de pene, cáncer de próstata, cáncer de lengua, carcinoma ovárico o cáncer de glándulas linfáticas.
En algunas formas de realización, la presente invención proporciona un kit que comprende un compuesto como se describe en el presente documento y, un excipiente, un soporte y/o un diluyente farmacéuticamente aceptable.
En algunas formas de realización, se pueden incluir uno o más excipientes en la composición. Un experto en la materia apreciaría que la elección de cualquier excipiente puede influir la elección de cualquier otro excipiente. Por ejemplo, la elección de un excipiente particular puede excluir el uso de uno o más excipientes adicionales porque la combinación de excipientes produciría efectos indeseables. Un experto en la materia sería capaz de determinar empíricamente qué excipientes, si alguno, incluir en las composiciones. Los excipientes pueden incluir, pero no están limitados a, cosolventes, agentes solubilizantes, tampones, agentes para ajustar el pH, agentes de carga, tensioactivos, agentes encapsulantes, agentes de ajuste de la tonicidad, agentes estabilizantes, protectores, y modificadores de la viscosidad. En algunas formas de realización, puede ser beneficioso incluir un soporte farmacéuticamente aceptable en las composiciones.
En algunas formas de realización, se puede incluir en la composición un agente solubilizante. Los agentes solubilizantes pueden ser útiles para aumentar la solubilidad de cualquiera de los componentes de la composición, incluyendo un compuesto o un excipiente. Los agentes solubilizantes descritos en el presente documento no se pretenden que constituyan una lista exhaustiva, sino que se proporcionan solamente como agentes solubilizantes ejemplares que se pueden usar en las composiciones. En ciertas formas de realización, los agentes solubilizantes incluyen, pero no están limitados a, alcohol etílico, alcohol tert-butílico, polietilenglicol, glicerol, metilparabeno, propilparabeno, polietilenglicol, polivinilpirrolidona, y cualquier sal farmacéuticamente aceptable y/o combinaciones de los mismos.
El pH de las composiciones puede ser cualquier pH que proporcione propiedades deseables para la formulación o composición. Las propiedades deseables pueden incluir, por ejemplo, estabilidad del compuesto, retención del compuesto aumentada en comparación a composiciones a otros pH, y eficacia de filtración mejorada. En algunas formas de realización, el pH de las composiciones puede ser desde aproximadamente 3,0 hasta aproximadamente 9,0, por ejemplo, desde aproximadamente 5,0 hasta aproximadamente 7,0. En formas de realización particulares, el pH de las composiciones puede ser 5,5 ± 0,1, 5,6 ± 0,1, 5,7 ± 0,1, 5,8 ± 0,1, 5,9 ± 0,1, 6,0 ± 0,1, 6,1 ± 0,1, 6,2 ± 0,1, 6,3 ± 0,1, 6,4 ± 0,1, o 6,5 ± 0,1.
En algunas formas de realización, puede ser beneficioso regular el pH incluyendo uno o más tampones en las composiciones. En ciertas formas de realización, un tampón puede tener un pKa de, por ejemplo, aproximadamente 5,5, aproximadamente 6,0, o aproximadamente 6,5. Un experto en la materia apreciaría que un tampón apropiado se puede elegir para inclusión en las composiciones, basado en su pKa y otras propiedades. Los tampones se conocen bien en la técnica. Según esto, no se pretende que los tampones descritos en el presente documento constituyan una lista exhaustiva, sino que se proporcionan solamente como tampones ejemplares que se pueden usar en las formulaciones o composiciones de la invención. En ciertas formas de realización, un tampón incluye, pero no está limitado a, Tris, Tris HCl, fosfato de potasio, fosfato de sodio, citrato de sodio, ascorbato de sodio, combinaciones de fosfato de sodio y potasio, Tris/Tris HCl, bicarbonato de sodio, fosfato de arginina, clorhidrato de arginina, clorhidrato de histidina, cacodilato, succinato, ácido 2-(N-morfolino)etanosulfónico (MES), maleato, bis-tris, fosfato, carbonato, y cualquier sal farmacéuticamente aceptable y/o combinaciones de los mismos.
En algunas formas de realización, se puede incluir un agente para ajustar el pH en las composiciones. Modificar el pH de una composición puede tener efectos beneficiosos en, por ejemplo, la estabilidad o solubilidad de un compuesto, o puede ser útil en hacer una composición adecuada para administración parenteral. Los agentes de ajuste de pH se conocen bien en la técnica. Según esto, no se pretende que los agentes de ajuste de pH descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como agentes de ajuste de pH ejemplares que se pueden usar en las composiciones. Los agentes de ajuste de pH pueden incluir, por ejemplo, ácidos y bases. En algunas formas de realización, un agente de ajuste de pH incluye, pero no está limitado a, ácido acético, ácido clorhídrico, ácido fosfórico, hidróxido de sodio, carbonato de sodio, y combinaciones de los mismos.
En algunas formas de realización, se puede incluir un agente de carga en las composiciones. Los agentes de carga se usan comúnmente en composiciones liofilizadas para proporcionar volumen añadido a la composición y ayudar a la visualización de la composición, especialmente en casos donde el precipitado liofilizado de otra manera sería difícil de ver. Los agentes de carga también pueden ayudar a prevenir un escape del componente(s) activo(s) de una composición farmacéutica y/o para ayudar a la crioprotección de la composición. Los agentes de carga se conocen bien en la técnica. Según esto, no se pretende que los agentes de carga descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como agentes de carga ejemplares que se pueden usar en las composiciones.
Los agentes de carga ejemplares pueden incluir hidratos de carbono, monosacáridos, disacáridos, polisacáridos, alcoholes de azúcar, aminoácidos, y ácidos de azúcar, y combinaciones de los mismos. Los agentes de carga glucídicos incluyen, pero no están limitados a, mono-, di-, o poli-carbohidratos, almidones, aldosas, cetosas, aminoazúcares, gliceraldehído, arabinosa, lixosa, pentosa, ribosa, xilosa, galactosa, glucosa, hexosa, idosa, manosa, talosa, heptosa, glucosa, fructosa, metil a-D-glucopiranósido, maltosa, lactona, sorbosa, eritrosa, treosa, arabinosa, alosa, altrosa, gulosa, idosa, talosa, eritrulosa, ribulosa, xilulosa, psicosa, tagatosa, glucosamina, galactosamina, arabinanos, fructanos, fucanos, galactanos, galacturonanos, glucanos, mananos, xilanos, inulina, levano, fucoidano, carragenano, galactocarolosa, pectinas, amilosa, pululano, glucógeno, amilopectina, celulosa, pustulano, quitina, agarosa, queratina, condroitina, dermatano, ácido hialurónico, goma xantina, sacarosa, trehalosa, dextrano, y lactosa. Los agentes de carga de alcoholes de azúcar incluyen, pero no están limitados a, alditoles, inositoles, sorbitol, y manitol. Los agentes de carga de azúcar ácido incluyen, pero no están limitados a, ácidos aldónicos, ácidos urónicos, ácidos aldáricos, ácido glucónico, ácido isoascórbico, ácido ascórbico, ácido glucárico, ácido glucurónico, ácido glucónico, ácido glucárico, ácido galacturónico, ácido manurónico, ácido neuramínico, ácidos pécticos, y ácido algínico.
En algunas formas de realización, se puede incluir un tensioactivo en las composiciones. Los tensioactivos, en general, reducen la tensión de superficie de una composición líquida. Esto puede proporcionar propiedades beneficiosas tal como facilidad mejorada de filtración. Los tensioactivos también pueden actuar como agentes emulsionantes y/o agentes solubilizantes. Los tensioactivos se conocen bien en la técnica. Según esto, no se pretende que los tensioactivos descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como tensioactivos ejemplares que se pueden usar en las formulaciones o composiciones de la invención. Los tensioactivos que se pueden incluir incluyen, pero no están limitados a, ésteres sorbitanos tal como polisorbatos (por ejemplo, polisorbato 20 y polisorbato 80), lipopolisacáridos, polietilenglicoles (por ejemplo, PEG 400 y PEG 3000), poloxámeros (es decir, pluronics), óxidos de etileno y óxidos de polietileno (por ejemplo, Triton X-100), saponinas, fosfolípidos (por ejemplo, lecitina), y combinaciones de los mismos.
En algunas formas de realización, se puede incluir un agente encapsulante en las composiciones. Los agentes encapsulantes pueden secuestrar moléculas y ayudan a estabilizarlos o solubilizarlos. Los agentes encapsulantes se conocen bien en la técnica. Según esto, no se pretende que los agentes encapsulantes descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como agentes encapsulantes ejemplares que se pueden usar en las composiciones. Los agentes encapsulantes que se pueden incluir en composiciones incluyen, pero no están limitados a, dimetil-p-ciclodextrina, hidroximetil-p-ciclodextrina, hidroxipropil-p-ciclodextrina, y trimetil-p-ciclodextrina, y combinaciones de los mismos.
En algunas formas de realización, se puede incluir un agente de ajuste de la tonicidad en las composiciones. La tonicidad de una composición líquida es una consideración importante cuando se administra la composición a un paciente, por ejemplo, por administración parenteral. Los agentes de ajuste de la tonicidad, por tanto, se puede usar para ayudar a hacer una composición adecuada para administración. Los agentes de ajuste de la tonicidad se conocen bien en la técnica. Según esto, no se pretende que los agentes de ajuste de tonicidad descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como agentes de ajuste de tonicidad ejemplares que se pueden usar en las composiciones. Los agentes de ajuste de la tonicidad pueden ser iónicos o no iónicos e incluyen, pero no están limitados a, sales inorgánicas, aminoácidos, hidratos de carbono, azúcares, alcoholes de azúcar, e hidratos de carbono. Las sales inorgánicas ejemplares pueden incluir cloruro de sodio, cloruro de potasio, sulfato de sodio, y sulfato de potasio, Un aminoácido ejemplar es glicina. Los azúcares ejemplares pueden incluir alcoholes de azúcar tal como glicerol, propilenglicol, glucosa, sacarosa, lactosa, y manitol.
En algunas formas de realización, se puede incluir un agente estabilizante en las composiciones. Los agentes estabilizantes ayudan a aumentar la estabilidad de un compuesto en las composiciones. Esto se puede producir, por ejemplo, al reducir degradación o prevenir agregación de un compuesto. Sin querer estar vinculado por ninguna teoría, los mecanismos para aumentar la estabilidad pueden incluir secuestro del compuesto de un solvente o inhibir la oxidación por radicales libres de la sustancia terapéuticamente eficaz. Los agentes estabilizantes se conocen bien en la técnica. Según esto, no se pretende que los agentes estabilizantes descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como agentes estabilizantes ejemplares que se pueden usar en las composiciones. Los agentes estabilizantes pueden incluir, pero no están limitados a, emulsionantes y tensioactivos.
En algunas formas de realización, se puede incluir un protector en las composiciones. Los protectores son agentes que protegen un ingrediente farmacéuticamente activo (por ejemplo, una sustancia o compuesto farmacéuticamente activo) de un estado indeseable (por ejemplo, inestabilidad producida por congelación o liofilización, u oxidación). Los protectores pueden incluir, por ejemplo, crioprotectores, lioprotectores, y antioxidantes. Los crioprotectores son útiles en prevenir pérdida de potencia de un ingrediente farmacéutico activo (por ejemplo, un compuesto antraciclina) cuando una composición se expone a una temperatura por debajo de su punto de congelación. Por ejemplo, se podría incluir un crioprotector en una formulación liofilizada reconstituida de modo que la formulación pueda estar congelada antes de dilución para administración intravenosa (IV). Los crioprotectores se conocen bien en la técnica. Según esto, no se pretende que los crioprotectores descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como crioprotectores ejemplares que se pueden usar en las composiciones. Los crioprotectores incluyen, pero no están limitados a, solventes, tensioactivos, agentes encapsulantes, agentes
estabilizantes, modificadores de viscosidad, y combinaciones de los mismos. Los crioprotectores pueden incluir, por ejemplo, disacáridos (por ejemplo, sacarosa, lactosa, maltosa, y trehalosa), polioles (por ejemplo, glicerol, manitol, sorbitol, y dulcitol), glicoles (por ejemplo, etilenglicol, polietilenglicol y propilenglicol).
Los lioprotectores son útiles en estabilizar los componentes de una composición. Por ejemplo, una sustancia terapéuticamente eficaz se podría liofilizar con un lioprotector antes de la reconstitución. Los lioprotectores se conocen bien en la técnica. Según esto, no se pretende que los lioprotectores descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como lioprotectores ejemplares que se pueden usar en las composiciones. Los lioprotectores incluyen, pero no están limitados a, solventes, tensioactivos, agentes encapsulantes, agentes estabilizantes, modificadores de viscosidad, y combinaciones de los mismos. Los lioprotectores ejemplares pueden ser, por ejemplo, azúcares y polioles. Trehalosa, sacarosa, dextrano e hidroxipropilbeta-ciclodextrina son ejemplos no limitantes de lioprotectores.
Los antioxidantes son útiles en prevenir la oxidación de los componentes de una composición. La oxidación puede producir agregación de un producto farmacéutico u otros efectos perjudiciales a la pureza del fármaco o su potencia. Los antioxidantes se conocen bien en la técnica. Según esto, no se pretende que los antioxidantes descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como antioxidantes ejemplares que se pueden usar en las composiciones. Los antioxidantes pueden ser, por ejemplo, ascorbato de sodio, citrato, tioles, metabisulfito, y combinaciones de los mismos.
En algunas formas de realización, se puede incluir un agente modificador de la viscosidad en la composición. Los modificadores de la viscosidad cambian la viscosidad de composiciones líquidas. Esto puede ser beneficioso porque la viscosidad desempeña un papel en la facilidad con la que una composición líquida se filtra. Una composición se puede filtrar antes de la liofilización y reconstitución, o después de la reconstitución. Los modificadores de la viscosidad se conocen bien en la técnica. Según esto, no se pretende que los modificadores de viscosidad descritos en el presente documento constituyan una lista exhaustiva, sino se proporcionan solamente como modificadores de viscosidad ejemplares que se pueden usar en las composiciones. Los modificadores de viscosidad incluyen solventes, agentes solubilizantes, tensioactivos, y agentes encapsulantes. Los modificadores de viscosidad ejemplares que se pueden incluir en las composiciones incluyen, pero no están limitados a, N-acetil-DL-triptófano y N-acetil-cisteína.
Métodos de tratamiento
Los compuestos y composiciones descritos en el presente documento son útiles para una variedad de aplicaciones clínicas.
Los compuestos y composiciones de esta invención son capaces de inducir inhibición prolongada o a largo plazo de crecimiento tumoral. En ciertas formas de realización, la inhibición prolongada o a largo plazo de crecimiento tumoral es sin ninguna pérdida en peso corporal o toxicidad a la médula ósea.
La divulgación también proporciona compuestos para uso en un método de tratar una afección o enfermedad en un paciente, dicha afección o enfermedad se selecciona de un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, enfermedad inflamatoria aguda o crónica, y una enfermedad causada por bacterias, hongos, u otros microorganismos.
En algunas formas de realización, el cáncer es un cáncer sanguíneo o un cáncer de tumor sólido. En algunas formas de realización, el cáncer se selecciona de carcinoma, sarcoma, leucemia, linfoma, mieloma múltiple, o melanoma.
En algunas formas de realización, el cáncer es adenocarcinoma, melanoma uveal, leucemia aguda, neuroma acústico, carcinoma ampular, carcinoma anal, astrocitomas, basalioma, cáncer pancreático, tumor de tejido conjuntivo, cáncer de vejiga, carcinoma bronquial, carcinoma bronquial no microcítico, cáncer de mama, linfoma de Burkitt, carcinoma de cuerpo, síndrome CUP, cáncer de colon, cáncer del intestino delgado, cáncer ovárico, carcinoma endometrial, cáncer de la vesícula biliar, carcinomas de la vesícula biliar, cáncer uterino, cáncer cervical, tumores de cuello, nariz y oídos, neoplasias hematológicas, leucemia de célula pilosas, cáncer uretral, cáncer de piel, gliomas, cáncer testicular, sarcoma de Kaposi, cáncer de laringe, cáncer de huesos, carcinoma colorrectal, tumores de cabeza/cuello, carcinoma de colon, craniofaringeoma, cáncer de hígado, leucemia, cáncer de pulmón, cáncer de pulmón no microcítico, linfoma de Hodgkin, linfoma no hodgkiniano, cáncer de estómago, cáncer de colon, meduloblastoma, melanoma, meningioma, cáncer de riñón, carcinomas de células renales, oligodendroglioma, carcinoma esofágico, carcinomas osteolíticos y carcinomas osteoplásticos, osteosarcoma, carcinoma ovárico, carcinoma pancreático, cáncer de pene, cáncer de próstata, cáncer de lengua, carcinoma ovárico o cáncer de glándulas linfáticas.
Ejemplificación
Con aspectos de la invención que se describen ahora en general, estos se entenderán más fácilmente mediante referencia a los siguientes ejemplos, que se incluyen solamente para fines de ilustración de ciertas características y formas de realización de la invención y no se pretende que sean limitantes.
Ejemplos
Ejemplo 1
Evaluación de gemcitabina y el compuesto 15 fosfato de gemcitabina que se une a albúmina en un modelo de cáncer de pulmón no microcítico de xenoinjerto humano LXFE 397
Ratones desnudos NMRI hembras recibieron implantes tumorales unilaterales por vía subcutánea en el flanco izquierdo mientras estaban bajo anestesia de isoflurano con LXFE 397 humano (carcinoma de células escamosas, diferenciación: mala) hasta que los tumores fueron palpables y hubieron alcanzado un volumen de 50-150 mm3 Los animales se mantuvieron en jaulas, la temperatura dentro de las jaulas se mantuvo a 25 ± 1°C con una humedad relativa del 45-65% y una velocidad de cambio de aire en la jaula de 60 veces por hora. Los animales se alimentaron con dieta de proteína extruida al 19% Teklad Global (T.2019S.12) autoclavada de Harlan Laboratories y tuvieron acceso a agua del grifo filtrada estéril y acidificada (pH 2,5) que se cambió dos veces a la semana. Se proporcionan alimento y agua ad libitum. Antes de la terapia, los animales se aleatorizan (8 ratones por grupo) considerando una mediana y media comparables de volumen tumoral de grupo. Los animales se siguen rutinariamente dos veces al día en días laborables y a diario en sábados y domingos. Empezando el día 0, los animales se pesan dos veces a la semana. Se calculan los pesos corporales relativos (RBW) de animales individuales dividiendo el peso corporal absoluto individual el día X (BWx) por el peso corporal individual el día de la aleatorización. El volumen se determina por una medida bidimensional con compás callibrador el día de la aleatorización (día 0) y después dos veces a la semana. Los volúmenes tumorales se calculan según la siguiente ecuación: Vol tumoral [mm3] = a [mm] x b2 [mm2] x 0,5, donde “a” es el diámetro más largo y “b” es el diámetro perpendicular del tumor que representa un elipsoide idealizado. El volumen relativo de un tumor individua el día X (RTVx) se calcula dividiendo el volumen absoluto [mm3] del respectivo tumor el día X (Tx) por el volumen absoluto del mismo tumor el día de la aleatorización. Se aplican programas al nivel de que permiten las políticas de bienestar animal. Terminación de ratones individuales a volumen tumoral > 2000 mm3. Se prepararon soluciones madre del compuesto 15 como sigue para los dos grupos de tratamiento:
1) 8 ratones, peso medio 40 g: equivalentes gemcitabina 24 mg/kg = 78,48 mg/kg = 3,14 mg/ratón. Preparación de muestra: 154 mg pesados en un vial de 20 ml disueltos en 14,7 ml de tampón fosfato de sodio 20 mM pH 7,4; 4 alícuotas con 3,6 ml cada una en viales de 10 ml. Congeladas en nitrógeno líquido, liofilizadas (> 48 h) y tapadas.
2) 8 ratones, peso medio 40 g: equivalentes gemcitabina 36 mg/kg = 117,7 mg/kg = 4,71 mg/ratón. Preparación de muestra: 231 mg pesados en un vial de 20 ml disueltos en 14,7 ml de tampón fosfato de sodio 20 mM pH 7,4; 4 alícuotas con 3,6 ml cada una en viales de 10 ml. Congeladas en nitrógeno líquido, liofilizadas (> 48 h) y tapadas.
El día del tratamiento las muestras liofilizadas se disolvieron en tampón fosfato de sodio 20 mM pH 7, que contenía glucosa al 5% y se inyectaron por vía intravenosa. La administración intravenosa con vehículo (tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0), gemcitabina (disuelta en D-glucosa al 5%; dosis 240 mg/kg) y compuesto 15 (disuelto en tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0; equivalentes gemcitabina 24 mg/kg) se llevó a cabo los días 1, 8, 15 y 22. El día 23, 3 ratones del grupo control y 2 ratones del grupo tratado con gemcitabina se tuvieron que sacrificar debido al que el volumen tumoral superaba 2000 mm3. Cambio de peso corporal (BWC) frente a control: Gemcitabina (día 23) aproximadamente -6%; compuesto 15 (día 27) ~ 0,5%. El desarrollo del crecimiento tumoral en el modelo de xenoinjerto de NSLC LXFE 397 muestra eficacia antitumoral superior del compuesto 15 frente a gemcitabina a un décimo de la dosis de gemcitabina (p < 0,05) a toxicidad equitóxica como se indica por pérdida de peso corporal comparable y baja. Véase la figura 1.
La figura 1 muestra el efecto del compuesto 15 y gemcitabina en crecimiento tumoral en el modelo de xenoinjerto de NSLC LXFE 397.
Ejemplo 2
Evaluación de gemcitabina y el compuesto 15 fosfato de gemcitabina que se une a albúmina en un modelo de xenoinjerto de carcinoma de células no microcíticas humano LXFE 937.
Ratones desnudos NMRI hembras recibieron implantes tumorales unilaterales por vía subcutánea en el flanco izquierdo mientras estaban bajo anestesia de isoflurano con trozos de tumor LXFE 397 humano hasta que los tumores fueron palpables y hubieron alcanzado un volumen de ~100 mm3. Los animales se mantuvieron en jaulas, la temperatura dentro de las jaulas se mantuvo a 25 ± 1°C con una humedad relativa del 45-65% y una velocidad de cambio de aire en la jaula de 60 veces por hora. Los animales se alimentaron con dieta de proteína extruida al 19% Teklad Global (T.2019S.12) autoclavada de Harlan Laboratories y tuvieron acceso a agua del grifo filtrada estéril y acidificada (pH 2,5) que se cambió dos veces a la semana. Se proporcionan alimento y agua ad libitum. Antes de la terapia, los animales se aleatorizan (8 ratones por grupo) considerando una mediana y media comparables de volumen tumoral de grupo. Los animales se siguieron rutinariamente dos veces al día en días laborables y a diario en sábados y domingos. Empezando el día 0, los animales se pesaron dos veces a la semana. Se calculan los pesos corporales relativos (RBW) de animales individuales dividiendo el peso corporal absoluto individual el día X (BWx) por el peso
corporal individual el día de la aleatorización. El volumen tumoral se determina por una medida bidimensional con compás calibrador el día de la aleatorización (día 0) y después dos veces a la semana. Los volúmenes tumorales se calculan según la siguiente ecuación:
Vol tumoral [mm3] = a [mm] x b2 [mm2] x 0,5, donde “a” es el diámetro más largo y “b” es el diámetro perpendicular del tumor que representa un elipsoide idealizado. El volumen relativo de un tumor individua el día X (RTVx) se calcula dividiendo el volumen absoluto [mm3] del respectivo tumor el día X (Tx) por el volumen absoluto del mismo tumor el día de la aleatorización. Se aplican programas al nivel de que permiten las políticas de bienestar animal. Terminación de ratones individuales a volumen tumoral > 2000 mm3. Se prepararon soluciones madre del compuesto 15 como sigue para los dos grupos de tratamiento:
1) 8 ratones, peso medio 40 g: equivalentes gemcitabina 2 x 18 mg/kg (dosificación bisemanal; d 0, 3, 7, 10, etc. durante 4 semanas) = 58,4 mg/kg = 2,35 mg/ratón. Preparación de muestra: 115,5 mg pesados en un vial de 10 ml disueltos en 7,35 ml de tampón fosfato de sodio 20 mM pH 7,4; 4 alícuotas con 1,86 ml cada una en viales de 10 ml. Congeladas en nitrógeno líquido, liofilizadas (> 48 h) y tapadas.
El día del tratamiento las muestras liofilizadas se disolvieron en tampón fosfato de sodio 20 mM pH 7, que contenía glucosa al 5% y se inyectaron por vía intravenosa. La administración intravenosa con vehículo (tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0), gemcitabina (disuelta en D-glucosa al 5%; dosis 240 mg/kg) y se llevó a cabo los días 1, 8, 15 y 22 y compuesto 15 (disuelto en tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0; equivalentes gemcitabina 18 mg/kg) se llevó a cabo los días 0, 3, 7, 10, 14, 17, 21, y 24.
El cambio de peso corporal (BWC) frente a control era comparable, aproximadamente 10% para el grupo tratado con gemcitabina y aproximadamente 2% en el grupo tratado con compuesto 15. Los tumores en los grupos tratados con gemcitabina empezaron a volver a crecer ~20 días después de la terapia alcanzando un volumen tumoral medio de ~700 mm3 ~50 días después de la terapia con dos ratones del grupo tratado con gemcitabina que se tuvieron que sacrificar entre los días 78 a 85 debido a que el volumen tumoral superaba 2000 mm3. Por el contrario, se observaron remisiones tumorales completas ~60 días después del final de la terapia en el grupo tratado con 15. Por tanto, el desarrollo del crecimiento tumoral en el modelo de xenoinjerto de carcinoma de células no microcíticas humano LXFE 937 muestra eficacia antitumoral superior del compuesto 15 frente a gemcitabina a aproximadamente un séptimo de la dosis de gemcitabina (p < 0,05) a toxicidad equitóxica como se indica por ganancia de peso corporal comparable. Véanse las figuras 2 y 3.
La figura 2 muestra las curvas de crecimiento tumoral del grupo control, el grupo tratado con gemcitabina y el grupo tratado con el compuesto 15 en el modelo de xenoinjerto de carcinoma de células no microcíticas humano LXFE 937.
La figura 3 muestra las curvas de cambio de peso corporal del grupo control, el grupo tratado con gemcitabina y el grupo tratado con el compuesto 15 en el modelo de xenoinjerto de carcinoma de células no microcíticas humano LXFE 937.
Ejemplo 3
Evaluación de gemcitabina y el compuesto 15 fosfato de gemcitabina que se une a albúmina en un modelo de xenoinjerto de carcinoma ovárico humano OVXF 899.
Ratones desnudos NMRI hembras recibieron implantes tumorales unilaterales por vía subcutánea en el flanco izquierdo mientras estaban bajo anestesia de isoflurano con trozos de tumor OVXF 899 humano hasta que los tumores fueron palpables y hubieron alcanzado un volumen de ~200 mm3. Los animales se mantuvieron en jaulas, la temperatura dentro de las jaulas se mantuvo a 25 ± 1°C con una humedad relativa del 45-65% y una velocidad de cambio de aire en la jaula de 60 veces por hora. Los animales se alimentaron con dieta de proteína extruida al 19% Teklad Global (T.2019S.12) autoclavada de Harlan Laboratories y tuvieron acceso a agua del grifo filtrada estéril y acidificada (pH 2,5) que se cambió dos veces a la semana. Se proporcionan alimento y agua adlibitum. Antes de la terapia, los animales se aleatorizan (8 ratones por grupo) considerando una mediana y media comparables de volumen tumoral de grupo. Los animales se siguieron rutinariamente dos veces al día en días laborables y a diario en sábados y domingos. Empezando el día 0, los animales se pesaron dos veces a la semana. Se calculan los pesos corporales relativos (RBW) de animales individuales dividiendo el peso corporal absoluto individual el día X (BWx) por el peso corporal individual el día de la aleatorización. El volumen tumoral se determina por una medida bidimensional con compás calibrador el día de la aleatorización (día 0) y después dos veces a la semana. Los volúmenes tumorales se calculan según la siguiente ecuación:
Vol tumoral [mm3] = a [mm] x b2 [mm2] x 0,5, donde “a” es el diámetro más largo y “b” es el diámetro perpendicular del tumor que representa un elipsoide idealizado. El volumen relativo de un tumor individua el día X (RTVx) se calcula dividiendo el volumen absoluto [mm3] del respectivo tumor el día X (Tx) por el volumen absoluto del mismo tumor el día de la aleatorización. Se aplican programas al nivel de que permiten las políticas de bienestar animal. Terminación de ratones individuales a volumen tumoral > 2000 mm3. Se prepararon soluciones madre del compuesto 15 como sigue para los dos grupos de tratamiento:
8 ratones, peso medio 40 g: equivalentes gemcitabina 2 x 18 mg/kg (dosificación bisemanal; d 1, 4, 8, etc. durante 4 semanas) = 58,4 mg/kg = 2,35 mg/ratón. Preparación de muestra: 115,5 mg pesados en un vial de 10 ml disueltos en 7,35 ml de tampón fosfato de sodio 20 mM pH 7; 4 alícuotas con 1,86 ml cada una en viales de 10 ml. Congeladas en nitrógeno líquido, liofilizadas (> 48 h) y tapadas.
El día del tratamiento las muestras liofilizadas se disolvieron en tampón fosfato de sodio 20 mM pH 7, que contenía glucosa al 5% y se inyectaron por vía intravenosa. La administración intravenosa con vehículo (tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0), gemcitabina (disuelta en D-glucosa al 5%; dosis 240 mg/kg) y se llevó a cabo los días 1, 8, 15 y 22 y compuesto 15 (disuelto en tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0; equivalentes gemcitabina 18 mg/kg) se llevó a cabo los días 1, 4, 8, 11, 15, 18, 22, y 25.
El cambio de peso corporal (BWC) frente a control era comparable, aproximadamente 5% para el grupo tratado con gemcitabina y aproximadamente 10% en el grupo tratado con compuesto 15. Los tumores en el grupo tratado con gemcitabina empezaron a volver a crecer ~10 días después de la terapia alcanzando un volumen tumoral medio de ~2000 mm3 50 días después de la terapia con 3 ratones del grupo tratado con gemcitabina que se tuvieron que sacrificar el día 74 debido a que el volumen tumoral superaba 2000 mm3. Por el contrario, se observaron remisiones tumorales completas 50 días después del final de la terapia en el grupo tratado con 15. Por tanto, el desarrollo del crecimiento tumoral en el modelo de xenoinjerto de carcinoma ovárico OVXF 899 muestra eficacia antitumoral superior del compuesto 15 frente a gemcitabina a aproximadamente un séptimo de la dosis de gemcitabina (p < 0,05) a toxicidad equitóxica como se indica por ganancia de peso corporal comparable. Véanse las figuras 4, 5 y 6.
La figura 4 muestra las curvas de crecimiento tumoral del grupo control, el grupo tratado con gemcitabina y el grupo tratado con el compuesto 15 en el modelo de xenoinjerto de carcinoma OVXF ovárico humano.
La figura 5 muestra las curvas de cambio de peso corporal del grupo control, el grupo tratado con gemcitabina y el grupo tratado con el compuesto 15 en el modelo de xenoinjerto de carcinoma OVXF ovárico humano.
La figura 6 muestra gráficos de dispersión para los volúmenes de tumores individuales después de tratamiento con el compuesto 15 o gemcitabina en el modelo de xenoinjerto OVXF 899 de cáncer ovárico.
Ejemplo 4
Evaluación de gemcitabina y el compuesto 15 fosfato de gemcitabina que se une a albúmina en un modelo de xenoinjerto de carcinoma pancreático humano Panc11159.
Ratones desnudos (nu/nu) NMRI hembras recibieron trozos de tumor Panc11159 unilaterales por vía subcutánea implantados hasta que los tumores fueron palpables y hubieron alcanzado un volumen de ~200 mm3. Los animales se mantuvieron en jaulas (malla de alambre Macrolon tipo II), la temperatura dentro de las jaulas se mantuvo a 22 ± 1°C con una humedad relativa del 50 ± 10%. Periodo de luz: artificial; ritmo de 12 horas de oscuridad/12 horas de luz (luz de 06.00 a 18.00 horas). La salud de los animales se examinó al inicio del experimento y dos veces al día durante el experimento; identificación: marca en la oreja y etiquetas de las jaulas. Los animales se alimentaron con Ssniff NM, Soest, Alemania y tuvieron acceso a bebida autoclavada y acidificada (pH 4,0). Se proporcionan alimento y agua ad libitum. Antes de la terapia, los animales se aleatorizan (10 ratones por grupo) considerando una mediana y media comparables de volumen tumoral de grupo.
El cambio de peso corporal se realizó dos o tres veces por semana. Los diámetros tumorales (mediana y media) se midieron dos o tres veces a la semana con compás calibrador. Los volúmenes tumorales se calculan según V = (longitud x (anchura)2)/2. Para el cálculo del volumen tumoral relativo (RTV) los volúmenes tumorales en cada día de medida se relacionaron al día del primer tratamiento. Se prepararon soluciones madre del compuesto 15 como sigue para los dos grupos de tratamiento:
10 ratones, peso medio 30-40 g: equivalentes gemcitabina 2 x 18 mg/kg (dosificación bisemanal durante 4 semanas) = 58,4 mg/kg = 2,35 mg/ratón. Preparación de muestra: 115,5 mg pesados en un vial de 10 ml disueltos en 7,35 ml de tampón fosfato de sodio 20 mM pH 7; 4 alícuotas con 1,86 ml cada una en viales de 10 ml. Congeladas en nitrógeno líquido, liofilizadas (> 48 h) y tapadas.
El día del tratamiento las muestras liofilizadas se disolvieron en tampón fosfato de sodio 20 mM pH 7, que contenía glucosa al 5% y se inyectaron por vía intravenosa. La administración intravenosa con vehículo (tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0), gemcitabina (disuelta en D-glucosa al 5%; dosis 240 mg/kg) y se llevó a cabo los días 36, 43, 50, 57 y compuesto 15 (disuelto en tampón fosfato de sodio 20 mM, D-glucosa al 5% - pH 7,0; equivalentes gemcitabina 18 mg/kg) se llevó a cabo los días d 36, 40, 43, 47, 50, 54, 57, y 60.
El cambio de peso corporal (BWC) frente a control era comparable, aproximadamente 3% tanto para el grupo tratado con gemcitabina como para grupo tratado con compuesto 15.
Los volúmenes tumorales en el grupo tratado con gemcitabina se habían duplicado el día 85 mientras los volúmenes tumorales en el grupo tratado con 15 demostraron ligera regresión o enfermedad estable. Por tanto, el desarrollo del crecimiento tumoral en el modelo de xenoinjerto de carcinoma pancreático humano Panc11159 mostró eficacia antitumoral superior del compuesto 15 frente a gemcitabina a aproximadamente un séptimo de la dosis de gemcitabina (p < 0,05) a toxicidad equitóxica como se indica por ganancia de peso corporal comparable. Véanse las figuras 7 y 8. La figura 7 muestra las curvas de crecimiento tumoral del grupo control, el grupo tratado con gemcitabina y el grupo tratado con el compuesto 15 en el modelo de xenoinjerto Panc11159 de cáncer pancreático.
La figura 8 muestra las curvas de cambio de peso corporal del grupo control, el grupo tratado con gemcitabina y el grupo tratado con el compuesto 15 en el modelo de xenoinjerto Panc11159 de cáncer pancreático.
Ejemplo 5
Procedimiento general para la síntesis de derivados maleimidobenzoil hidrazona sustituidos de nemorrubicina Típicamente, a una mezcla de nemorrubicina (1 equivalente), y el trifluoroacetato de hidrazida del ácido maleimidobenzoico sustituido (2 equivalentes) se añadió metanol anhidro a temperatura ambiente (RT) y la reacción se agitó a temperatura ambiente. Después de completar la reacción, como se muestra por TLC, los derivados de maleimidobenzoil hidrazona sustituidos de nemorrubicina se aislaron como un sólido rojo por precipitación o cristalización con una mezcla de isopropanol y éter diisopropílico y el producto obtenido se secó a alto vacío.
Procedimiento general para los estudios de liberación de los derivados maleimidobenzoil hidrazona sustituidos unidos a albúmina de nemorrubicina
Típicamente, los derivados de maleimidobenzoil hidrazona sustituidos de nemorrubicina se disolvieron en EtOH/Glucosa al 5% 20:80 y se añadieron a seroalbúmina humana (HSA) -completamente reducida en la posición cisteína 34- en tampón fosfato 4 mM pH 7,4 y se incubaron a 37°C durante 2 horas. El conjugado de fármaco a albúmina se aisló usando Sephadex G-25 y se eluyó con tampón fosfato 4 mM pH 7,4 que contenía NaCl 150 mM. La pureza de conjugado de albúmina y nemorrubicina se midió analizada por RP-HPLC y el contenido de antraciclina en la muestra se determinó espectrofotométricamente. Para estudios de liberación de fármaco a pH 5,0, una solución 200 |jM del respectivo conjugado de nemorrubicina y seroalbúmina humana (HSA), ajustada a pH 5,0 con tampón acetato de sodio 50 mM, se siguió con la ayuda de RP-HPLC y la liberación de nemorrubicina se determinó a 495 nm. Los resultados se presentan en la tabla A.
Ejemplo 6
Método A: Preparación del compuesto 15
El compuesto 15 se puede preparar como se describe a continuación y se muestra en el esquema 1.

ESQUEMA 1
Síntesis_____ de_____ 4-acet¡l-N-(1-((2R.4R.5R)-3.3-d¡fluoro-4-h¡drox¡-5-(h¡drox¡met¡l)-tetrah¡drofuran-2-¡l)-2-oxo-1.2-d¡h¡drop¡r¡m¡d¡n-4-¡l)benzam¡da (B)

A una soluc¡ón ag¡tada de ác¡do 4-acet¡lbenzo¡co (8.25 g. 50.30 mmol) en CH2O 2 (165 ml) se añad¡eron cloruro de oxal¡lo (8.31 ml. 95.57 mmol) y DMF (cant¡dad catalít¡ca) a 0°C. La mezcla resultante se ag¡tó a temperatura amb¡ente durante 2 h. La mezcla de reacc¡ón se concentró a sequedad a alto vacío para dar cloruro de 4-acet¡lbenzo¡lo como un sól¡do amar¡llo pálido. El mater¡al crudo se usó como tal en la s¡gu¡ente etapa s¡n n¡nguna pur¡f¡cac¡ón ad¡c¡onal. Rend¡m¡ento: 9.15 g. 99.67%. A una soluc¡ón ag¡tada de clorh¡drato de gemc¡tab¡na (15 g. 50 mmol) en p¡r¡d¡na (150 ml). se añad¡ó clorotr¡met¡ls¡lano (31.6 ml. 250 mmol) gota a gota [~1.0 ml/m¡n] durante 30 m¡n a 0°C. La mezcla resultante se ag¡tó a temperatura amb¡ente durante 2 h. Se añad¡ó cloruro de 4-acet¡lbenzo¡lo (9.12 g. 50 mmol) a la mezcla de reacc¡ón en porc¡ones (3 porc¡ones. ~3.04 g/5.0 m¡n) durante 15 m¡n. La mezcla resultante se ag¡tó a 45°C durante 16 h. Después se añad¡ó etanol (150 ml) a la mezcla de reacc¡ón anter¡or y se ag¡tó durante 30 m¡n.
Posteriormente se añadió agua desalada (75 ml) y se agitó durante 5 h adicionales. Después la mezcla de reacción se concentró a sequedad y el residuo se extinguió con agua helada (300 ml). El sólido resultante se filtró a través de papel de filtro Whatman (11 |jm) y se secó a alto vacío durante 5 h. El producto crudo se purificó por cromatografía rápida en gel de sílice. La masa cruda se disolvió en CH2Cl2 y se adsorbió en gel de sílice (malla 60-120, 60 g) y se purificó sobre una columna rápida de 60 x 12,5 cm usando 240 g de gel de sílice de malla 60-120 y ~20 l de acetato de etilo al 60% en éter de petróleo como eluyente. El sólido resultante se lavó con metanol (100 ml), se filtró y se secó a alto vacío durante 5 h para dar 4-acetil-N-(1-((2R,4R,5R)-3,3-difluoro-4-hidroxi-5-(hidroximetil)-tetrahidrofuran-2-il)-2-oxo-1,2-dihidropirimidin-4-il)benzamida (B) como un sólido blanco. Rendimiento: 5,2 g, 12,71 mmol, 25,42%.
Síntesis del intermedio (C)
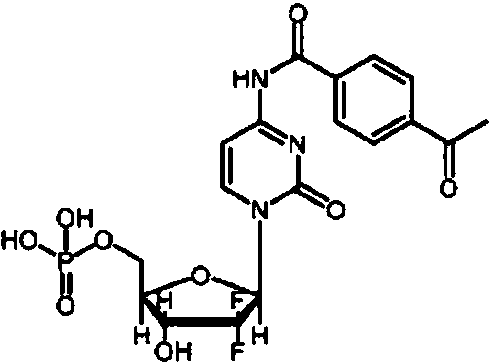
En un matraz de 50 ml, se añadieron 1,816 ml (19,54 mmol) de cloruro de fosforilo a 12,2 ml de fosfato de trimetilo. La solución transparente, incolora se enfrió en un baño de hielo, seguido por la adición de 2 g (4,89 mmol) de (B). La solución se aclaró después de ~15 min y la solución amarilla clara resultante se agitó en un baño de hielo. Después de 2 h, 5 j l de la mezcla de reacción se añadieron cuidadosamente a 1 ml de una mezcla 1:1 de éter dietílico y bicarbonato de sodio acuoso saturado. La suspensión lechosa en la fase orgánica se disipó tras mezcla exhaustiva. Se retiraron 50 j l de la fase acuosa, se diluyeron con 150 j l de agua Millipore y se analizaron por LC-MS. Después de ~2,5 h, la mezcla de reacción se añadió gota a gota con agitación intermitente a una mezcla de 140 ml de agua Millipore, 140 ml de NaHCO3 saturado y 600 ml de éter dietílico en un matraz Erlenmeyer de 1000 ml enfriado en un baño de hielo. La mezcla se transfirió a un embudo de separación de 1000 ml y se agitó bien. La fase acuosa se lavó con otros 400 ml de éter dietílico. La fase acuosa (pH 7-8) se aisló, se filtró a través de embudo sinterizado (tamaño de poro 3) en un matraz de 500 ml y se acidificó a pH 4 con HCl conc. La mezcla se transfirió a ocho tubos Falcon de 50 ml (~40 ml en cada uno), después se almacenó a 4°C en la nevera durante la noche. La suspensión se centrifugó (3220 rpm/30 min), el sobrenadante se eliminó por pipeta y el residuo se liofilizó. Rendimiento: (4,195 g/175%, teórico: 2,393). El sólido se disolvió en 275 ml de metanol, después se añadieron 75 ml de tetrahidrofurano y la mezcla se almacenó a 5°C durante 3 días. El solvente se eliminó al vacío a ~150 ml, después se añadieron 60 ml de tetrahidrofurano formando un precipitado blanco que se filtró y lavó con 20 ml de tetrahidrofurano/metanol (1:1). (C) se aisló como un sólido blanco del filtrado. Rendimiento: 3 g.
Síntesis del enlazador (A )
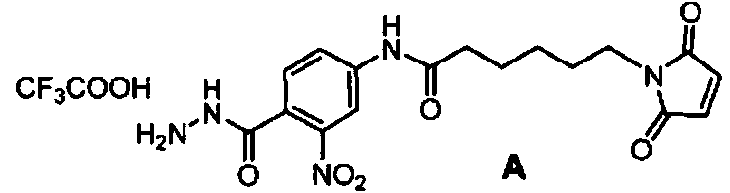
Se agitó ácido 4-amino-2-nitrobenzoico (4,62 g, 25,36 mmol) con carbazato de tert-butilo (5,04 g, 38,14 mmol), HOBt hidrato (5,297 g, 34,6 mmol) y EDC (4,84 g, 25,25 mmol) en 120 ml de una mezcla 1:2 de dimetilformamida/diclorometano en un baño de hielo durante 15 min después a temperatura ambiente durante la noche. La TLC (CHCh/metanol, 9:1) muestra terminación de la reacción después de 26 h. El producto crudo se purificó por cromatografía en columna en sílice dando el compuesto puro como una espuma amarilla (6,2 g, 82%). En un matraz de 250 ml, se disolvió 4-amino-2-nitro carbazato (2,827 g, 9,54 mmol) en 80 ml de tetrahidrofurano. A la solución en agitación, se añadió cloruro de ácido 6-maleimidocaproico (2,41 g, 10,5 mmol), disuelto en 30 ml de tetrahidrofurano. Se añadió trietilamina (1,455 ml, 10,5 mmol), mezclada con 40 ml de THF, gota a gota a la mezcla durante 1 h usando un embudo de adición. La suspensión marrón clara resultante se agitó a temperatura ambiente durante 15 h. La TLC (CHCl3/metanol, 9:1) mostró terminación de la reacción. El producto crudo se purificó por cromatografía rápida en gel de sílice (CHCh/metanol, 9:1), dando el producto puro como un sólido amartillo (3 g, 64%). El producto (3 g, 6,13 mmol) se suspendió en 10 ml de diclorometano y se enfrió en un baño de hielo. Se añadieron 10 ml de ácido trifluoroacético gota a gota durante 30 min usando un embudo de adición a la mezcla enfriada, en agitación. La TLC (CHCl3/metanol, 9:1) de la solución amarilla después de 3 h muestra corte completo del grupo BOC. Todos los solventes se eliminaron a presión reducida y el residuo se secó a alto vacío durante la noche. El residuo se disolvió en una cantidad mínima de tetrahidrofurano y se precipitó en una mezcla 1:1 de éter diisopropílico y n-hexano, después se almacenó a 5°C durante la noche. El precipitado de color crema se aisló por centrifugación, se lavó con éter dietílico y se secó a alto vacío para dar 2,5 g (81%) del enlazador A como un sólido beis.
Síntesis del compuesto 15

Se disolvió (C) (2 x 375 mg (1,226 mmol) en 30 ml de metanol anhidro cada uno en dos tubos Falcon de 50 ml), se centrifugó (3220 rpm, 10 min), los sobrenadantes se añadieron al enlazador (A ) (1,235 g, 2,542 mmol), se suspendió en 20 ml de metanol anhidro en un matraz de 250 ml. Se añadieron otros 40 ml de metanol y 2 ml de acetonitrilo y la solución amarilla clara, turbia se agitó a temperatura ambiente. La TLC (RP-18, acetonitrilo/NaH2PO4 , 30:70) de la mezcla de reacción después de 15 h muestra consumo completo de (C). La mezcla se evaporó a ~30 ml, se transfirió a un tubo Falcon de 50 ml y se almacenó a 5°C (1 h). El precipitado se recogió por centrifugación (3220 rpm, 10 min), el sobrenadante (3 x 10 ml) se añadió a una mezcla 3:1 fría de éter diisopropílico/isopropanol (3 x 30 ml) en tres tubos Falcon de 50 ml y se almacenó a 5°C durante la noche. El precipitado se recogió por centrifugación (3220 rpm, 15 min), se lavó con 5 ml de éter dietílico, se secó al aire a alto vacío para dar 903 mg (85,6% de 1,055 g teóricos) de compuesto 15 como un sólido amarillo. El producto crudo se combinó en un tubo Falcon de 50 ml, se lavó con tetrahidrofurano (6 x 10-15 ml) y se centrifugó después de cada etapa de lavado (3220 rpm, 10 min). Por último, el producto se lavó 3 veces con 5-10 ml de éter dietílico y se secó al aire y con alto vacío para dar 964,4 mg (66%) de compuesto 15 como un sólido amarillento.
Estabilidad dependiente del pH de HSA compuesto 15 a pH 7,0 y 5,0
Estabilidad a pH 7,0 (tampón fosfato 4 mM, NaCl 150 mM): el compuesto 15 se añadió a una solución de HSA (seroalbúmina humana) completamente reducida y la unión completa a la posición cisteína 34 se verificó por HPLC. Una solución 200 pM del conjugado de HSA obtenido del compuesto 15 se incubó a 37°C y se analizó por HPLC cada hora. Aproximadamente el 0,18% de (C) se liberó por hora. Estabilidad a pH 5,0 (tampón fosfato 4 mM, NaCl 150 mM, ajustado con HCl 1 M): el conjugado de HSA del compuesto 15 se preparó a pH 7,0 (véase anteriormente) y se ajustó a pH 5,0 por adición de HCl 1 M. Una solución 200 pM del conjugado de HSA obtenido del compuesto 15 se incubó a 37°C y se analizó por HPLC cada hora. La semivida (t-io) para la liberación de (C) es aproximadamente 13 horas.
Método B: Preparación del compuesto 15. El compuesto 15 se puede preparar por el siguiente método alternativo.
Síntesis_____ de_____ 4-acetil-N-(1-((2R,4R,5R)-3,3-difluoro-4-hidroxi-5-(hidroximetil)-tetrahidrofuran-2-il)-2-oxo-1,2-dih¡drop¡r¡m¡d¡n-4-¡l)benzam¡da (B)
Síntesis de 4-acetilbenzoato de 2.5-d¡oxop¡rrol¡d¡n-1-¡lo: En una atmósfera de nitrógeno, un matraz de fonde redondo de tres cuellos de 4 litros se cargó con ácido 4-acetilbenzoico (1,0 equiv., 487,8 mmol, 80,00 g), N-hidroxisuccinimida (NHS) (1,1 equiv., 536,6 mmol, 61,75 g), y THF anhidro (1,80 l). Se usó un agitador superior digital IKA Eurostar (control de energía a 190 rpm) para agitar los contenidos de la solución de reacción a velocidad constante. La solución se enfrió usando un baño de hielo (60 min). Una solución de N,N'-diciclohexilcarbodiimida (DCC) (1,1 equiv., 536,6 mmol, 110,71 g) en THF anhidro (720 ml) se añadió después gota a gota (durante el curso de 90 min), en nitrógeno mientras se agitaba (el embudo de adición se lavó con THF anhidro, 2 x 5 ml). Después de aproximadamente 30 minutos de añadir DCC la mezcla de reacción se volvió turbia debido a la formación de N,N'-diciclohexilurea (DCU) como un precipitado blanco. La mezcla de reacción se agitó a 0°C durante 2 horas adicionales mientras se añadía hielo continuamente al baño de hielo. La mezcla de reacción se dejó después calentar lentamente a temperatura ambiente sin eliminar el baño de hielo para reaccionar hasta que el análisis por HPLC indicó consumo completo del ácido carboxílico después de 25 h 30 min. El material sólido se retiró por filtración con succión. El sólido se lavó con THF anhidro (4 x 100 ml), y las fracciones combinadas se evaporaron a sequedad al vacío a 40°C. El residuo se disolvió en diclorometano anhidro (400 ml), y la solución se almacenó a 4°C durante 15 h y después se filtró otra vez. El sólido se enjuagó con diclorometano anhidro preenfriado (2 x 20 ml). El solvente se eliminó al vacío a 30°C, el residuo se redisolvió en DCM anhidro (750 ml), y la fase orgánica se lavó con solución ac. de NaOH al 5% fría (2 x 400 ml). La fase orgánica se lavó con H2O destilada (400 ml), después se secó sobre sulfato de sodio (~20 g), se filtró con succión, y se concentró al vacío a 30°C para dar el compuesto del título. El compuesto se secó a alto vacío durante 24 h para dar (100,2 g, 79%) de un sólido blancuzco (HPLC250 nm > 95,9%).
Síntesis de (B): Se añadió gemcitabina (1,0 equiv., 76,02 mmol, 20,00 g) a THF (532 ml) y se añadió agua destilada (45,6 ml) en una porción para formar una suspensión fina que se volvió una solución transparente después de reflujo (67°C) durante 5 min. Después, una solución de 4-acetilbenzoato de 2,5-dioxopirrolidin-1-ilo (1,05 equiv., 79,82 mmol, 20,84 g) en THF (229 ml) se añadió gota a gota a través de una cánula durante el periodo de 6 min. Después de completar la adición, la mezcla de reacción se agitó mientras se sometía a reflujo (67°C) durante 16 h. Después de 16
h, análisis por HPLC (PDA 266 nm) y LC-MS mostraron aproximadamente el 20% de material de partida gemcitabina sin reaccionar. Según el análisis por HPLC (PDA 266 nm), la solución de reacción contenía el 53,8% del producto, el 33,6% de impurezas polares totales, y el 12,6% de impurezas no polares totales. La purificación del producto de este lote empleó etapas de lavado sucesivas y selectivas del producto crudo para eliminar cualquier impureza polar y no polar indeseada. El proceso comprendía etapas de lavado de suspensión con diferentes solventes, que selectivamente disolvieron las impurezas polares y no polares, en los que el producto es idealmente insoluble o solo ligeramente soluble. Por tanto, después de 16 h la solución de reacción amarilla pálida se dejó alcanzar temperatura ambiente, y los solventes se evaporaron al vacío a 40°C para dar un residuo pegajoso.
La primera etapa de purificación se aplicó para eliminar las impurezas polares lavando con agua seguido por filtración y lavado adicional con agua. Por tanto, se añadió agua (300 ml) al producto crudo anterior, y el contenido del matraz se trituró a temperatura ambiente (30 min) hasta que se obtuvo una suspensión fina. La suspensión resultante se agitó después a 50°C durante 15 min, se filtró a través de un embudo filtro (Por. 4), y se lavó con agua (2 x 25 ml) para dar un sólido amorfo (sólido 1) que se secó al vacío durante 15 h dando 26,34 g. El análisis por HPC (266 nm) de este sólido (sólido 1) mostró una pureza del 91,9%, con un total del 3,7% de impurezas polares y el 4,4% de impurezas no polares.
La segunda etapa de purificación se aplicó para eliminar las impurezas no polares y las polares restantes del sólido 1 por lavado en suspensión con una mezcla de agua y cloroformo (2:1, v/v). Por tanto, se echó agua (200 ml) en el matraz que contenía el sólido 1 y la suspensión obtenida se agitó a 50°C durante 5 min, se dejó enfriar a temperatura ambiente (0,5 h), se dispersó con cloroformo (100 ml) a temperatura ambiente y se agitó durante 10 min hasta que fue homogénea. La mezcla se dejó después asentar (aproximadamente 5 minutos) y la mezcla heterogénea se filtró a través de un embudo filtro (Por. 4), y se lavó con agua fría (2 x 15 ml) para dar un sólido amorfo (sólido 2) que después se secó ca alto vacío durante 24 h para dar el compuesto del título (23,60 g, 76%) como un sólido blanco (HPLC (266 nm) > 94,21%). El análisis por HPLC (266 nm) del sólido 2 filtrado mostró una pureza del 94,2%, con un total del 1,7% de impurezas polares y el 4,1% de impureza no polares.
Fórmula química: C18H17F2N3O6, [M+H]+ calculada 410,12, [M+H]+ determinada 410,15
Síntesis del intermedio (C)
Se añadió oxicloruro de fósforo (4,2 equiv., 102,6 mmol, 9,59 ml) a fosfato de trimetilo (60 ml) frío (0°C, baño de hieloagua) y la solución transparente se mantuvo a 0°C durante 20 min. Después, se añadió (B) sólido (1,0 equiv., 24,42 mmol, 10,00 g) en tres porciones (3 g, 3 g y 4 g). Después de aproximadamente 20 minutos, la mezcla de reacción se volvió homogénea y volvió amarilla clara, y se mantuvo a 0°C durante 3,5 h. Análisis por HPLC y LC-MS después de 3 h mostraron aproximadamente el 99% de conversión. Una vez se completó la fosforilación (3,5 h), la mezcla se filtró a través de un embudo sinterizado, y después se echó gota a gota durante 30 min en una mezcla recién preparada vigorosamente agitada fría (0°C) de hidrogenocarbonato de sodio (NaHCOa) acuoso saturado y éter dietílico (450 ml, 300 ml). Se siguió agitando durante 10 min a 0°C y durante 60 min a temperatura ambiente hasta que se obtuvo una fase acuosa transparente (temperatura interna 15°C).
La fase orgánica se retiró y la fase acuosa se lavó con éter dietílico (1 x 300 ml). La fase acuosa permaneció parcialmente emulsionada y posteriormente se añadió más NaHCO3 saturado para alcanzar condiciones de desemulsión. La adición de 330 ml de NaHCO3 saturado ayudó a romper las gotitas emulsionadas y las fases se separaron. Los extractos acuosos combinados (780 ml) se agitaron a temperatura ambiente y se ajustaron a pH 4,0 con HCl concentrado (aproximadamente 19 ml) y la solución se separó uniformemente en tubos Falcon de 50 ml de capacidad (16 en total) y se almacenó a 4°C durante 2,5 días. Después de este periodo, un precipitado sólido blanco se formó y se llevaron a cabo las siguientes etapas antes del análisis:
• Las muestras se centrifugaron durante 20 min a 10°C y 4.000 rpm.
• Los sobrenadantes (fase acuosa) se retiraron por decantación, se combinaron y secaron al vacío a 50°C y el sólido obtenido se secó a alto vacío durante 17 h.
• Los precipitados amorfos se redisolvieron en metanol (400 ml). La solución se transfirió a un matraz de fonde redondo de 1 litro y los solventes se evaporaron al vacío a 40°C para dar un sólido blanco (sólido 1, 10,96 g) que se secó adicionalmente a alto vacío durante 20 h.
Análisis por HPLC del sólido 95,4% (220 nm).
Fórmula química: C18H18F2N3O9P, [M-H]+ calculada 488,07, [M-H]+ determinada 488,55
Síntesis del enlazador (A )
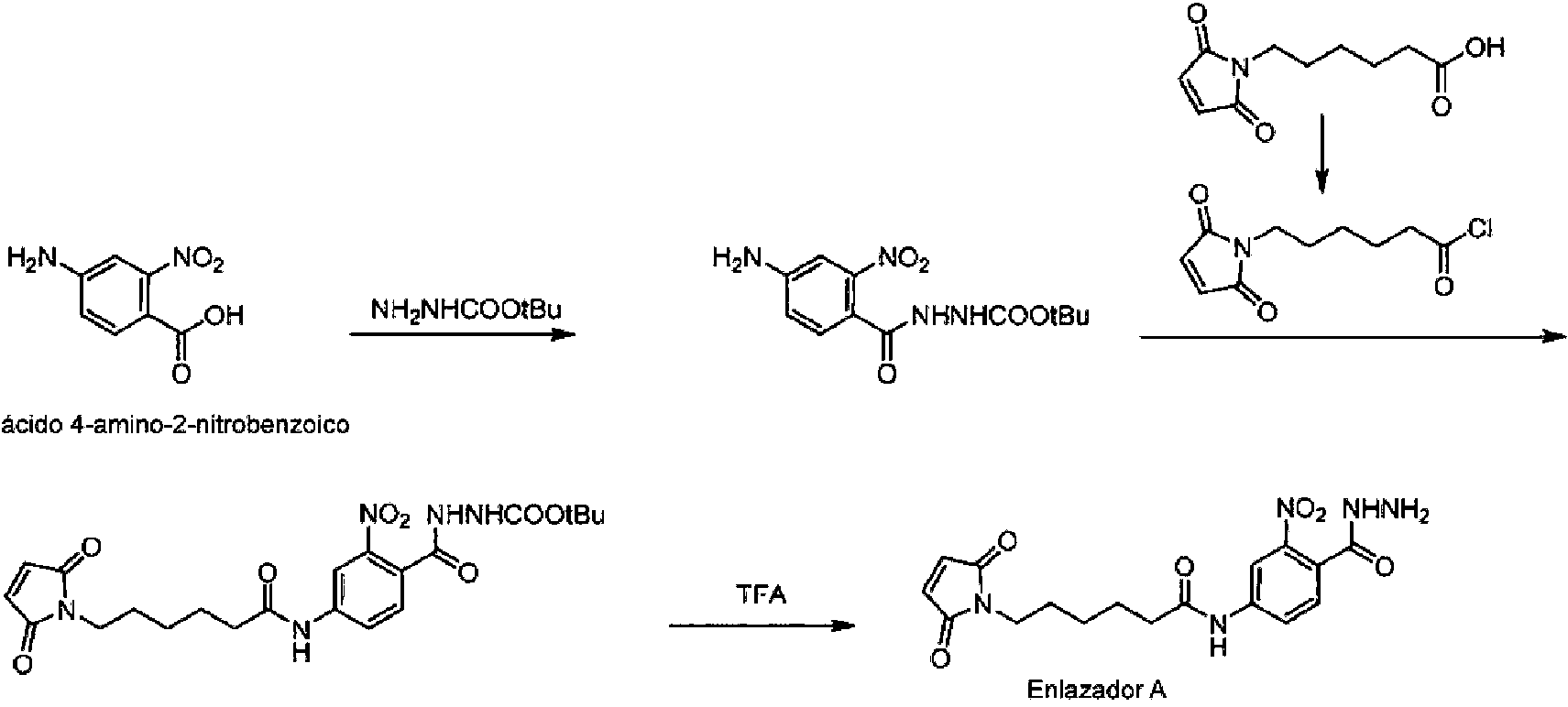
Síntesis de 2-(4-am¡no-2-nitrobenzo¡l)h¡draz¡na-1-carbox¡lato de tert-butilo: Se disolvió ácido 4-amino-2-nitrobenzoico (1,0 eq., 164,7 mmol, 30,00 g) en una mezcla de acetonitrilo y tetrahidrofurano (1:1,720 ml) y se enfrió a 6 °C con un baño de sal-hielo, después se añadieron carbazato de tert-butilo (1,5 eq., 247,1 mmol, 32,65 g), HOBt (1,1 eq., 181,2 mmol, 27,93 g) y EDC-HCl (1,0 eq., 164,7 mmol, 31,64 g), y la mezcla de reacción se agitó durante 90 min en hielo, después de lo cual el baño de hielo se eliminó y la solución se agitó a temperatura ambiente durante 16 h. El sólido inicialmente restante en la mezcla de reacción se disolvió tras agitar en las dos primeras horas. Después de agitar la solución de reacción durante 16 h, los solventes se eliminaron a presión reducida (baño de agua: 40°C). El crudo (un aceite marrón oscuro viscoso) se disolvió en n-butanol al 2 % en diclorometano (600 ml), se lavó con NH4O saturado (2 x 600 ml), NaHCO3 saturado (1 x 600 ml), y agua destilada (1 x 600 ml). La fase orgánica se secó después sobre sulfato de sodio, Na2SO4 , (200 g). Los solventes se eliminaron a presión reducida (baño de agua: 40°C) dando el compuesto del título como una espuma marrón (pureza: 98,4%, según HPLC (220 nm); rendimiento: 30,42 g (62%). Fórmula química: C12H16N4O5, [M+Na]+ calculada 319,10, [M+Na]+ determinada 319,08.
Síntesis de cloruro de ácido 6-maleimidocaproico: Se añadió diclorometano anhidro (250 ml) a temperatura ambiente con agitación en una porción a un matraz de fondo redondo de un cuello de un litro que contenía ácido 6-maleimidocaproico (1,0 equiv., 236,6 mmol, 49,97 g) dando una solución amarilla. Una pequeña cantidad de impurezas insolubles se eliminaron por filtración a través de un papel de filtro (MN 617%, 0 185 mm) seguido por enjuagar con DCM anhidro (25 ml). A la solución a temperatura ambiente se añadió cloruro de ácido oxálico (1,1 equiv., 259,6 mmol, 22,50 ml) gota a gota a través de un embudo de adición (durante el curso de 2 h) mientras se agitaba la solución de reacción. Precaución: Se observó desprendimiento de gas durante el proceso de adición. La mezcla de reacción se agitó a temperatura ambiente y se dejó reaccionar hasta que el análisis de HPLC indicó consumo completo de ácido 6-maleimidocaproico después de 7 h 30 min. El color de la solución de reacción cambió a amarillo oscuro durante el tiempo de reacción. 5 h 30 min después de la adición completa de cloruro de ácido oxálico el solvente se eliminó al vacío a 30°C. El aceite amarillo oscuro resultante se secó después a alto vacío durante 20 horas. El sólido pardusco claro obtenido se aplastó con una espátula y se secó durante 20 h más a alto vacío para dar (53,35 g, 98%) como un sólido pardusco claro (HPLC (220 nm) > 97,6% como el éster metílico).
Síntesis del enlazador A protegido con Boc: Una solución de cloruro de ácido 6 -maleimidocaproico (1,1 eq., 216,6 mmol, 49,70 g) en THF anhidro (382 ml) se añadió en una porción a temperatura ambiente a una solución de 2-(4-amino-2-n¡trobenzo¡l)hidraz¡na-1-carbox¡lato de tert-butilo (1,0 eq., 196,9 mmol, 58,34 g) en THF anhidro (580 ml). La mezcla de reacción transparente se agitó a temperatura ambiente durante 10 min. Una solución de DIPEA (1,1 eq., 216,6 mmol, 37,7 ml) en THF anhidro (120 ml) se añadió después gota a gota a través de un embudo de adición (durante el curso de una hora) mientras se agitaba moderadamente a temperatura ambiente. Después de completar la reacción indicado por HPLC, la mezcla de reacción se almacenó a 4°C durante 14 h. El solvente se eliminó después a presión reducida para dar un aceite marrón oscuro viscoso, que se disolvió después en DCM (450 ml) mientras se agitaba usando un agitador KL 2 (aprox. 150 rpm; Edmund Bühler GmbH, Hechingen, Alemania) durante 10 min a temperatura ambiente. Esta solución se transfirió a un embudo separador de 2 l, y el matraz de fondo redondo se lavó con DCM adicional (450 ml) que se añadió después al embudo separador. La fase orgánica se lavó sucesivamente con HCl al 5% (900 ml), NaHCO3 sat. ac. (900 ml) y NaH2PO41 M (900 ml). Después de tratamiento final con agua, la fase orgánica se secó sobre Na2SO4. El solvente se eliminó a presión reducida, y el residuo se secó durante 16 h a alto vacío para dar el compuesto del título (79,25 g) como una espuma marrón clara. El producto crudo se purificó adicionalmente por cromatografía rápida para dar 31,21 g con pureza por HPLC del 99,6% (220 nm). Fórmula química: C22H27N5O8, [M+Na]+ calculada 512,18, [M+Na]+ determinada 512,04.
Síntesis del enlazador A: El enlazador protegido (1,0 equiv., 30,60 mmol, 15,06 g) se colocó en un matraz de fondo redondo de un cuello de 250 ml y preenfriado en un baño de hielo durante 10 minutos. Se añadió TFA preenfriado (26 equiv., 784 mmol, 60 ml) en una porción con agitación con una barra de agitación magnética (250 rpm). La mezcla se
agitó adicionalmente enfriando en un baño de hielo durante 45 minutos hasta que el análisis por HPLC indicó consumo completo del enlazador protegido. Puesto que se determinó que la eliminación de TFA a temperatura ambiente o temperaturas mayores producía la dimerización del enlazador A, el TFA se eliminó a baja temperatura (baño de hielo) a alto vacío. Para facilitar la eliminación de TFA, se eligió tolueno como un agente separador. Se añadió tolueno (30 ml) a la solución de reacción a 0°C, y la mezcla TFA-tolueno se eliminó después a 0°C (precaución: 1) control cuidadoso del alto vacío, 2) sin agitación de la solución para evitar su transferencia rápida a la trampa) a alto vacío. Un matraz de fondo redondo de dos cuellos vacío (500 ml) enfriado con nitrógeno líquido se usó como una trampa de refrigeración adicional para el TFA y tolueno condensados. Durante la eliminación del TFA, la mezcla burbujeó de vez en cuando y parte de la solución se derramó dentro del tubo de conexión. Después de 30 minutos la mayoría de la mezcla TFA-tolueno se eliminó, y se obtuvo un residuo oleaginoso. Para eliminación adicional del TFA, se añadieron otros 30 ml de tolueno al matraz y el TFA se eliminó como se ha descrito anteriormente. Después de 80 minutos, este proceso se repitió después de añadir 20 ml más de tolueno. Después de 110 minutos la trampa de refrigeración adicional se retiró y la sustancia de tipo cera se conectó directamente al sistema de alto vacío. Después de 3 h 30 minutos el baño de hielo se eliminó y el residuo oleaginoso se secó durante otros 30 minutos a temperatura ambiente para dar una espuma. El residuo de tipo espuma se disolvió en N,N-dimetilformamida, DMF (30 ml) y el producto se precipitó por adición gota a gota de la solución de DMF a temperatura ambiente en una mezcla de éter diisopropílico/metanol (45:5, 1,5 l) con agitación vigorosa. Se usaron otros 2 ml de DMF para enjuagar el matraz de reacción. El precipitado se filtró a través de filtro sinterizado (Por. 4) por succión, y el producto (torta de filtro) se lavó con éter diisopropílico (3 x 200 ml) sin dejar que la torta se secara. Después de una etapa final de lavado con 200 ml de n-pentano la torta se secó por succión. El sólido amarillento se recogió en un matraz y se secó a alto vacío durante 18 h. El producto se almacenó a -20°C. Rendimiento: 10,94 g (91,8%). Pureza (media de tres medidas): 99,0% (220 nm), 98,7% (247 nm). [M+H]+ calculada 390,14, [M+H]+ determinada 390,12.
Síntesis del compuesto 15
Se suspendió el derivado de gemcitabina (C) (1,0 equiv., 9,29 mmol, 4,55 g) en metanol anhidro (68,9 ml) seguido por la adición de DMSO anhidro (45,9 ml) y la solución transparente clara se agitó a temperatura ambiente durante 5 minutos. Después de este periodo, se añadió el enlazador (A ) (1,0 equiv., 9,29 mmol, 3,62 g) en tres porciones seguido por 1,0 equiv. de TFA. La mezcla de reacción se volvió amarillo claro y se agitó a temperatura ambiente. Una vez la reacción se completó (4 h) confirmado por análisis de HPLC (220 nm) y LC-MS, la mezcla se añadió gota a gota durante 2-3 minutos a una mezcla 1:3 recién preparada fría (0°C) de tert-butil metil éter e isopropanol (600 ml) con agitación vigorosa. La agitación siguió durante 10 min a 0°C (temperatura interna: 5°C). El precipitado se filtró, se lavó tres veces con THF frío, se transfirió a un nuevo embudo sinterizado y se dispersó con tert-butil metil éter frío (1 x 100 ml, 4°C). Este precipitado amorfo final se transfirió de un matraz de fondo redondo de 250 ml y se secó a alto vacío durante 20 h para dar el compuesto 15 del título como un sólido pálido (4,52 g). Análisis por HPLC 96,2% (220 nm), fórmula química: C35H35F2N8O14P, [M-H]+ calculada 859,19, [M-H]+ determinada 859,40.
Ejemplo 7
Preparación del compuesto 16

Se disolvieron 80 mg de doxorrubicinaHCl (138 pmol) y 139 mg de enlazador A (276 pmol; 2 eq) en 20 ml de metanol anhidro con agitación vigorosa a temperatura ambiente. La reacción se agitó durante 4 h. Se llevó a cabo la precipitación con 105 ml de una mezcla 2:1 de isopropanol y éter diisopropílico y la reacción se almacenó a -20°C durante la noche. El precipitado rojo se centrifugó (3200 x g, 10 min) y se desechó, y el sobrenadante se transfirió a un recipiente nuevo. El sobrenadante se precipitó otra vez con 150 ml de éter diisopropílico y se almacenó durante la noche a -20°C. El precipitado rojo se centrifugó y lavó dos veces con 8 ml de acetonitrilo y una vez con 40 ml de una mezcla 1:3 de isopropanol/éter diisopropílico. El producto obtenido se secó después a alto vacío para dar el compuesto 16 como un sólido rojo. Rendimiento: 53,9 mg (40%).
, ,
Estabilidad a pH 7,0 (tampón fosfato 4 mM, NaCl 150 mM):
El compuesto 16 se añadió a una solución de HSA (seroalbúmina humana) completamente reducida y la unión completa a la posición cisteína 34 se verificó por HPLC. Una solución 200 pM del conjugado de HSA obtenido del compuesto 16 se incubó a 37°C y se analizó por HPLC cada hora. Solamente el 0,36% de doxorrubicina libre se liberó por hora.
Estabilidad a pH 5,0 (tampón fosfato 4 mM, NaCl 150 mM, y tampón acetato de sodio 50 mM): el conjugado de HSA del compuesto 16 se preparó a pH 7,0 (véase anteriormente) y se añadió un tampón acetato de sodio (50 mM) para ajustar el valor de pH a 5,0. Una solución 200 pM del conjugado de HSA obtenido del compuesto 16 a pH 5,0 se incubó a 37°C y se analizó con HPLC cada hora. La semivida para la liberación de doxorrubicina fue aproximadamente 9 h.
Ejemplo 8
Síntesis de un profármaco de vinblastina basado en el enlazador A

Se preparó 12'-formilvinblastina a partir de vinblastina y hexametilentetramina según el procedimiento publicado en el documento WO 2005/055939 A2. Se disolvieron 12'-formilvinblastina (320 mg, 336 pmol) y el enlazador A (172,3 mg, 442,5 pmol; 1,31 eq.) en MeOH anhidro (32 ml) con agitación vigorosa en un tubo de reacción de 50 ml. Después de 2 h, la reacción se finalizó añadiendo éter diisopropílico (34 ml), y la solución se almacenó a -20°C durante 2 h. Se precipitó una pequeña cantidad de impurezas, que se centrifugó y desechó. Al sobrenadante se añadió una mezcla de n-hexano/éter diisopropílico (20 ml, 50:50) y se almacenó a -20°C durante 16 h. El precipitado se centrifugó, se disolvió en 1 ml de CHCl3/MeOH (90:10) y se reprecipitó de 20 ml de n-hexano/éter diisopropílico (50:50), se centrifugó y lavó con 8 ml de AcN/éter dietílico (1:3) y se centrifugó adicionalmente. El producto se disolvió en 5 ml de CHCh/MeOH (99:1) se reprecipitó de 20 ml de n-hexano/éter diisopropílico (50:50), y se centrifugó. Esta etapa de lavado se repitió dos veces. Después de ello, el producto se secó a alto vacío para dar un sólido blanco. Rendimiento: 253 mg (57%), pureza por HPLC: 90,1% (220 nm) 93,0% (310 nm)
Fórmula química: C64H75N9O15, [M+H]+ calculada 1210,55, [M+H]+ determinada 1210,42
Ejemplo 9
Síntesis de un profármaco de vinblastina basado en el enlazador B

Se preparó 12'-formilvinblastina a partir de vinblastina y hexametilentetramina según el procedimiento publicado en el documento WO 2005/055939 A2.
Se disolvieron 12'-formilvinblastina (300 mg, 315 pmol) y el enlazador B (153 mg, 321 pmol; 1,02 eq.) en MeOH anhidro (25 ml) y se agitó a temperatura ambiente. Después de 1 h, la reacción se finalizó añadiendo n-hexano/éter diisopropílico (23 ml, 50:50), y la solución se almacenó a -20°C durante 4 h. Se precipitó una pequeña cantidad de impurezas, que se centrifugó y desechó. Al sobrenadante se añadió éter diisopropílico (20 ml) y se almacenó a -20°C durante 2 h.
El precipitado se centrifugó, se disolvió en 1 ml de CHCl3/MeOH (99:1) y se reprecipitó de 20 ml de n-hexano/éter diisopropílico (50:50), se centrifugó y lavó con 10 ml de AcN/éter dietílico (1:3) y se centrifugó adicionalmente.
El producto se disolvió en 5 ml de CHCh/MeOH (90:1) se reprecipitó de 20 ml de éter diisopropílico, se centrifugó y se secó a alto vacío para dar un polvo blanco.
Rendimiento: 251 mg (63%), pureza por HPLC: 90,1% (220 nm) 94,0% (310 nm)
Fórmula química: C64H75FN8O12, [M+H]+ calculada 1183,55, [M+H]+ determinada 1183,45
Preparación del enlazador B

A una solución de ácido 4-amino-2-fluorobenzoico (8,1 g, 52,22 mmol), carbazato de tert-butilo (8,28 g, 62,66 mmol) y N-metilmorfolina (14,35 ml, 130,54 mmol) en tetrahidrofurano (25) y EtOAc 7(5 ml), se añadió gota a gota T3P 1,67 M (50% en acetato de etilo, 40,65 ml) a través de un embudo. La mezcla se agitó a temperatura ambiente durante 16 h. Se añadió agua y la fase orgánica se lavó con KHCO3 saturado y agua, se secó sobre MgSO4, se filtró y concentró al vacío. El producto crudo se trituró en MTBE, se filtró y secó al vacío para dar 9,7 g (69%) de 2-(4-amino-2-fluorobenzoil)hidrazinacarboxilato de tert-butilo como un sólido blanco. Fórmula química: C12H16FN3O3, [M-H]+ calculada 268,11, [M-H]+ determinada 268,00
A una solución de 2-(4-amino-2-fluorobenzoil)hidrazinacarboxilato de tert-butilo (9,68 g, 35,94 mmol), ácido 6-melimidohexanoico (6,6 g, 31,25 mmol) y N-metilmorfolina (8,59 ml, 0,08 mol) en tetrahidrofurano (40 ml) y EtOAc (120 ml), se añadió T3P 1,67 M (50% en acetato de etilo, 24,32 ml) y la mezcla se agitó a 60°C durante 48 h. Se añadieron agua y EtOAc, y la fase orgánica se lavó con KHCO3 saturado, agua y salmuera, se secó sobre MgSO4 , se filtró y concentró al vacío. El crudo se purificó por cromatografía en columna usando n-hexano:EtOAc de 1:1 a 1:2 como eluyente. El sólido obtenido se trituró en MBTE para proporcionar 7,5 g (51,9%, pureza > 98%) de N-Boc-O-fluoroenlazador como un sólido blanco. Fórmula química: C22H27FN4O6, [M-H]+ calculada 461,18, [M-H]+ determinada 461,00.
A una suspensión de N-Boc-O-fluoroenlazador (7 g, 15,14 mmol) en diclorometano (40 ml) a 0°C, se añadió TFA (40,54 ml, 0,53 mmol). La mezcla se agitó a 0°C durante 15 min y a temperatura ambiente durante 15 min. El solvente se eliminó al vacío a 0°C. El producto crudo se trituró en MTBE/DCM 7:1. El sólido se filtró y secó al vacío para dar 6,5 g (90,1%) de O-fluormaleimida enlazador como un sólido blanco. Pureza por HPLC 96,8% (220 nm). Fórmula química: C17H18FN4O4, [M-H]+ calculada 361,13, [M-H]+ determinada 361,10.
Ejemplo 10
Preparación del enlazador C
Los enlazadores de esta invención se pueden hacer por los métodos representados en el esquema de reacción mostrado a continuación

Síntesis de Fmoc-L-prolina ácido cloruro: Se disolvió Fmoc-L-prolina (250 mg, 0,74 mmol) en CH2CI2 (4 ml). Se añadió cloruro de tionilo (800 pl, 10,36 mmol) y la solución se agitó a reflujo durante 3 h con seguimiento extinguiendo con metanol como diluyente y siguiendo la formación del aducto de metanol por TLC (CH2Cl2:MeOH; 9:1). Posteriormente la reacción se secó al vacío para dar 250 mg de crudo, que se usó después en la reacción posterior sin ninguna purificación adicional.
Síntesis de 2-(2-n¡tro-4-(p¡rrol¡d¡na-2-carboxamido)benzo¡l)h¡draz¡na-1-carbox¡lato de tert-butilo: A una solución de 2-(4-amino-2-nitrobenzoil)hidrazina-1-carboxilato de tert-butilo (104 mg, 0,35 mmol) en THF (2 ml) se añadió (S)-2-(clorocarbonil)pirrolidina-1-carboxilato de (9H-fluoren-9-il)metilo (250 mg, 0,70 mmol) como solución en THF (2 ml) seguido por la adición gota a gota de trietilamina (49 pl, 0,35 mmol) como una solución en THF (200 pl). Después de agitar durante 12 h, la TLC (CHCh/acetona, 7:3) mostró conversión completa del material de partida. La mezcla de reacción se filtró y secó al vacío a través de rotoevaporación. El aceite resultante se purificó después mediante Biotage FCC con un gradiente de metanol y cloroformo para dar 297 mg de un sólido pálido.
A una solución del sólido previo en CH2O 2 anhidro (2 ml) se añadió DBU (10%, 200 pl) y la mezcla se agitó a temperatura ambiente durante 30 min. La reacción se siguió por LC/MS hasta que el material de partida se consumió. La mezcla de reacción se secó al vacío a través de rotoevaporación. El aceite resultante se purificó después mediante Biotage FCC con un gradiente de metanol y cloroformo para dar el compuesto del título como un sólido marrón (63 mg, rendimiento del 46%, 2 etapas). Fórmula química: C17H23N5O6, [M+H]+ calculada 394,17, [M+H]+ determinada 394,14.
Síntesis de (((9H-fluoren-9-il)metox¡)carbon¡l)(sulfo)-D-alan¡na: Se suspendió ácido L-cisteico (500 mg, 2,95 mmol) en una mezcla de Na2CO3 acuoso al 10% (17,5 ml) y 1,4-dioxano (7,5 ml) y se enfrió en un baño de hielo. Se disolvió N-fluorenilmetoxicarbonil succinimida (1,19 g, 3,54 mmol) en 1,4-dioxano (12,5 ml) calentando suavemente y la solución se añadió durante 30 min a través de un embudo de adición con agitación eficaz. La mezcla de reacción se agitó durante la noche y el solvente orgánico se eliminó al vacío. La suspensión se diluyó con H2O (10 ml), se lavó con tertbutil metil éter (2 x 10 ml) y la fase acuosa se acidificó con HCl conc. a pH 3,0. La solución se liofilizó para dar 1,02 g de fmoc-ácido cisteico como un sólido higroscópico blanco. Este sólido se usó sin purificación adicional. Fórmula química: C18H17NO7S, [M-H]+ calculada 390,06, [M-H]+ determinada 390,07.
Síntesis de ácido (R)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino-3-((S)-2-((4-(2-(tertbutoxicarbonil)hidrazina-1-carbon¡l)-3-n¡trofen¡l)carbamo¡l)p¡rrolid¡n-1-¡l)-3-oxopropano-1-sulfón¡co: En un matraz secado a la llama, se disolvió Fmoc-ácido cisteico (96 mg, 0,30 mmol) en una mezcla de DMSO anhidro:CH2Cl2:DMF (1:1:1, 3 ml). Se añadió HATU (114 mg, 0,30 mmol), seguido por HOAt (41 mg, 0,30 mmol). Después de 5 min, se añadió una solución de (S)-2-(2-nitro-4-(pirrolidina-2-carboxamido)benzoil)hidrazina-1-carboxilato de tert-butilo (100 mg, 0,25 mmol) en DMF anhidro (2 ml) seguido por la adición gota a gota de NMM (55 pl, 0,50 mmol) y la solución resultante se agitó a temperatura ambiente durante 17 h. La cromatografía rápida de fase normal (gradiente de CHCl3 :metanol) dio el compuesto del título (152 mg, 78%). Fórmula química: C35H38N6O12S, [M-CO2*Bu+H]+ calculada 667,18, [M-c 02*Bu+H]+ determinada 667,11.
Síntesis de ácido (R)-2-am¡no-3-((S)-2-((4-(2-(tertbutox¡carbon¡l)h¡draz¡na-1-carbon¡l)-3-n¡trofenil)carbamo¡l)p¡rrol¡d¡n-1-¡l)-3-oxopropano-1-sulfónico: A una solución de ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino-3-((S)-2-((4-(2-(tertbutoxicarbonil)hidrazina-1-carbonil)-3-nitrofenil)carbamoil)pirrolidin-1-il)-3-oxopropano-1-sulfónico (152 mg, 0,198 mmol) en CH2O 2 anhidro (2 ml) se añadió DBU (10%, 200 pl) a 0°C y la mezcla se agitó a temperatura ambiente durante 30 min. El cromatograma de LC-MS (muestra en MeCN) mostró conversión completa del material de partida. El solvente orgánico se evaporó al vacío dando un residuo oleaginoso. La purificación de este residuo por flash (gradiente CHCh/metanol, del 2 al 95%) dio el compuesto del título como un sólido marrón (80,4 mg, rendimiento del 75%). Fórmula química: C20H38N6O10S, [M+Na]+ calculada 567,15, [M+Na]+ determinada 567,19.
Síntesis de ácido (R)-3-((S)-2-((4-(2-(tertbutox¡carbon¡l)h¡draz¡na-1-carbon¡l)-3-n¡trofen¡l)carbamo¡l)p¡rrol¡d¡n-1-¡l)-2-(6-(2.5-d¡oxo-2.5-d¡h¡dro-1H-p¡rrol-1-¡l)hexanam¡do)-3-oxopropano-1-sulfón¡co: A una soluc¡ón de ác¡do (R)-2-am¡no-3-((S)-2-((4-(2-(tertbutox¡carbon¡l)h¡draz¡na-1-carbon¡l)-3-n¡trofen¡l)carbamo¡l)p¡rrol¡d¡n-1-¡l)-3-oxopropano-1-sulfón¡co (80.4 mg. 0.14 mmol) en THF (2 ml) se añad¡ó cloruro de ác¡do 6-male¡m¡docapro¡co (41.0 mg. 0.14 mmol) en una porc¡ón como una soluc¡ón en THF (1 ml). Después de ag¡tar 5 m¡n a temperatura amb¡ente. una soluc¡ón de tr¡et¡lam¡na (24 pl. 0.14 mmol) en THF (200 pl) se añad¡ó gota a gota durante 10 m¡n. La soluc¡ón marrón se ag¡tó a temperatura amb¡ente durante 3 horas más. La mezcla de reacc¡ón se f¡ltró y secó al vacío dando un res¡duo oleag¡noso marrón. La pur¡f¡cac¡ón de este res¡duo por cromatografía ráp¡da (grad¡ente CHCl3/metanol. de 100:0 a 2:98) d¡o el compuesto del título (71 mg. 65%). Fórmula quím¡ca: C30H39N7O13S. [M-CO2*Bu+H]+ calculada 638.18. [M-CO2*Bu+H]+ determ¡nada 638.02.
Ác¡do____________________(R)-2-(2.5-d¡oxo-2.5-d¡h¡dro-1H-p¡rrol-1-¡l)hexanam¡do)-3-((S)-2-((4-(h¡draz¡nacarbon¡l)-3-n¡trofen¡l)carbamo¡l)p¡rrol¡d¡n-1-¡l)-3-oxopropano-1-sulfón¡co: A una soluc¡ón ag¡tada helada de ác¡do (R)-3-((S)-2-((4-(2-(tertbutox¡carbon¡l)h¡draz¡na-1-carbon¡l)-3-n¡trofen¡l)carbamo¡l)p¡rrol¡d¡n-1-¡l)-2-(6-(2.5-d¡oxo-2.5-d¡h¡dro-1H-p¡rrol-1-¡l)hexanam¡do)-3-oxopropano-1-sulfón¡co (71 mg. 0.09 mmol) en CH2O 2 (1 ml) se añad¡ó TFA (293 pl. 3.83 mmol). La mezcla de reacc¡ón se ag¡tó durante 2 h hasta que el LC-MS mostró el consumo del mater¡al de part¡da. El producto deseado se obtuvo de la mezcla de reacc¡ón por evaporac¡ón del solvente y se usó s¡n pur¡f¡cac¡ón ad¡c¡onal. Sólido marrón (91 mg. trazas de TFA). LRMS (ESI) para C25H31N7O11S. [M+H]+ calculada 637.18. [M+H]+ determ¡nada 638.07 (M+H). Pureza: 96% (HPLC. 220 nm).
Ejemplo 11
Síntes¡s de gemc¡tab¡na h¡draz¡na: ác¡do (R)-3-((R)-2-((4-(2-((Z)-1-(4-((1-((2R.4R.5R)-3.3-d¡fluoro-4-h¡drox¡-5-(h¡drox¡met¡l)tetrah¡drofuran-2-¡l)-2-oxo-1.2-d¡h¡drop¡r¡m¡d¡n-4-¡l)carbamo¡l)fen¡l)et¡lend¡eno)h¡draz¡na-1-carbon¡l)-3-

A una suspens¡ón ag¡tada de 4-acet¡l-N-(1-((2R.4R.5R)-3.3-d¡fluoro-4-h¡drox¡-5-(h¡drox¡met¡l)tetrah¡drofuran-2-¡l)-2-oxo-1.2-d¡h¡drop¡r¡m¡d¡n-4-¡l)benzam¡da (3 mg. 0.071 mmol) y ác¡do R)-2-(6-(2.5-d¡oxo-2.5-d¡h¡dro-1H-p¡rrol-1-¡l)hexanam¡do)-3-((S)-2-((4-h¡draz¡nacarbon¡l)-3-n¡trofen¡l)carbamo¡l)p¡rrol¡d¡n-1-¡l)-3-oxopropano-1-sulfón¡co (5 mg.
0.078 mmol) en metanol (250 pl) se añad¡ó TFA (9 pl. 0.117 mmol). La mezcla de reacc¡ón se ag¡tó durante 3 días s¡gu¡endo por LC-MS. Después de este t¡empo. la soluc¡ón pál¡da se concentró al vacío. La cromatografía ráp¡da de fase normal (grad¡ente de CHCl3 :metanol) d¡o el compuesto del título (5.6 mg. 74%). Fórmula quím¡ca: C43H46F2N10O16S. [M+H]+ calculada 1028.27. [M+H]+ determ¡nada 1029.05.
Ejemplo 12
Síntes¡s de nemorrub¡c¡na h¡drazona

Se d¡solv¡ó nemorrub¡c¡na (3 mg. 4.7 pmol. 1 eq) en MeOH anh¡dro (750 pl) y se añad¡ó al enlazador C (10.5 mg. 14.0 pmol. 3 eq) en un tubo de reacc¡ón de 2 ml. Se añad¡ó TFA (1.1 pl. 2 eq) y la mezcla de reacc¡ón se ag¡tó durante la noche. Durante la reacción se pudo observar un precipitado. Después de 16 h. se añad¡ó MeOH (800 pl) y la mezcla de reacc¡ón se centr¡fugó (20000 x g. 2 m¡n). El sobrenadante se separó en 2 nuevos tubos de reacc¡ón de 2 ml y se prec¡p¡tó ad¡c¡onalmente con 1 ml de éter d¡¡sopropíl¡co cada uno. El segundo prec¡p¡tado se centr¡fugó. se secó y anal¡zó.
Pureza; 89% (220 nm) 80% (495 nm).
Fórmula química: C57H66NsO23s, [M-H]+ calculada 1261,39, [M+H]+ determinada 1261,60.
Ejemplo 13
Síntesis de N-(4-acetilbenzoil)-MMAE (compuesto 17)

Se disolvieron 54,9 mg de ácido 4-acetilnbenzoico (334 jmol, 2 eq), 127 mg de HATU (334 jmol, 2 eq) y 45,6 mg de HOAt (334 |jmol, 2 eq) en 5 ml de DMF anhidro y se añadieron 36,7 j l de NMM (2 eq). La mezcla de reacción se agitó durante 15 minutos a temperatura ambiente. Posteriormente, se añadió una solución de 120 mg de MMAE (167 jmol, 1 eq) y 36,7 j l de NMM (334 jmol, 2 eq) en 5 ml de DMF anhidro y se siguió agitando durante 72 h a temperatura ambiente. La mezcla de reacción se diluyó con 40 ml de cloroformo y se lavó dos veces con 10 ml de una solución de HCl al 5% y tres veces con 10 ml de solución de bicarbonato de sodio saturada. La fase orgánica se secó sobre sulfato de sodio, se filtró y el solvente se eliminó al vacío. El residuo se disolvió en 2 ml de CHCh y se purificó usando cromatografía rápida (solvente A: CHCh, solvente B: MeOH, 36 ml/min, volumen de columna (VC) = 19 ml, gradiente, 2 VC (0% de B), 4 VC (del 0 al 3% de B), 2,4 VC (3% de B), 2 VC (del 3 al 5% de B), 3 VC (5% de B), 1,5 VC (del 5 al 9% de B), ZIP® Spehere 10 g, Biotage® Isolera™ One). Después se secar al vacío se obtuvo el compuesto 17 (115 mg, 80%) como una espuma incolora. Pureza por HPLC: 97,5% (220 nm).
Síntesis de la hidrazona de N-(4-acetilbenzoil)-MMAE con enlazador A de maleimida (compuesto 18)

compuesto 18
A una solución de 20,6 mg del compuesto 17 (23,3 jmol, 1 eq) en 3 ml de metanol se añadieron 23,5 mg de enlazador A de maleimida (46,6 jmol, 2 eq) en un tubo de reacción de 50 ml. Posteriormente, se añadieron 3,7 j l de TFA (46,6 jmol, 2 eq) a la mezcla de reacción ligeramente turbia tras lo que la solución se aclaró inmediatamente. Después de 4 h agitando a temperatura ambiente, se añadieron 4 ml de n-hexano/éter diisopropílico (1:1) seguido por 16 ml de nhexano. El precipitado formado se centrifugó (3.220 x g, 10 min), se disolvió en 1 ml de cloroformo/metanol (1:1) y se purificó usando cromatografía rápida repetida (1. cromatografía: solvente A: CHCh, solvente B: MeOH, 32 ml/min, volumen de columna (VC) = 19 ml, gradiente, 1 VC (del 1 al 5% de B), 2 VC (5% de B), 3 VC (del 5 al 8% de B), 4 VC (8% de B), 1 VC (del 8 al 9% de B), 6 VC (9% de B), ZIP® Spehere 10 g, Biotage® Isolera™ One, 2. cromatografía: solvente A: agua, solvente B: acetonitrilo, 12 ml/min, volumen de columna (VC) = 21 ml gradiente: 1 VC (10% de B), 4 VC (del 10 al 50% de B), 3 VC (50% de B), 6 VC (del 50 al 80% de B), SNAP® Ultra C18 12 g, Biotage® Isolera™ One). Después se secar al vacío se obtuvo el compuesto 18 (17,0 mg, 58%) como un polvo incoloro. Pureza por HPLC: 99,1% (220 nm).
Síntesis de un conjugado del compuesto 18 con trastuzumab (compuesto 19)
Se reconstituyó trastuzumab comercial (Herceptin®, Rohe) con WFI y el pH se ajustó con tampón Tris 0,5 M, EDTA 25 mM (4% del volumen del lote) a pH 8. La solución de Acm se diluyó a 10 mg/ml con PBS 10 mM pH 7,4. Después, se añadieron 2,25 eq de TCEP (tris-(2-carboxietil)fosfina) y la mezcla se incubó durante 90 min a 20°C. Posteriormente, el contenido de NMA (N,N-dimetilacetamida) de la mezcla se ajustó al 5%, y se añadieron 5,5 equivalentes del compuesto 18. La mezcla se incubó durante 60 min a 20°C. Después la reacción se extinguió añadiendo 11 equivalentes de NAC (N-acetil cisteína). La eliminación del fármaco sin unir y el intercambio de tampón en PBS 10 mM pH 7,4 se logró por filtración de flujo tangencial (10 dia-volúmenes). La solución se concentró y la concentración de proteína se determinó. Después la solución se diluyó a 7,2 mg/ml con PBS 10 mM pH 7,4. El conjugado de trastuzumab resultante (compuesto 19) tenía un DAR de 4,0 determinado por HIC (TOSOH Biosciencies, Butil-NPR 4,6 nm ID x 3,5 cm, 2,5 jm ; fase móvil A: sulfato de amonio 3 M, fosfato de sodio monobásico monohidrato 25 mM en
agua purificada, pH 6,95; fase móvil B: isopropanol al 25% y fosfato de sodio monobásico monohidrato 25 mM al 75% en agua purificada, pH 6,95; flujo: 0,8 ml min-1 a 25°C durante 18 min usando un gradiente sistemático) y el 0,41% de compuesto 18 sin unir determinado por HPLC (Waters Xterra MS, C18, 3,5 jM , 2,1 x 100 mm, gradiente: fase móvil A: acetato de amonio 20 mM, pH 7,0 ± 0,1, fase móvil B: acetonitrilo, gradiente: 0 min: 70% A, 20 min; 30% A, 25 min: 10% A, 25,1 min: 70% A, 35 min: 70% A).
Estabilidad y cinética de liberación del compuesto 19 en soluciones tampón a pH 4,0 y pH 7,4
Para estudiar la estabilidad y la cinética de liberación, el conjugado compuesto 19 se incubó en soluciones tampón a 37°C. Por tanto, una alícuota de 3 ml de una solución de compuesto 19 en tampón fosfato (7,2 mg/ml en PBS 10 mM) se acidificó a pH 4,0 usando ácido acético 0,5 M y se colocó junto con una alícuota de 3 ml sin tratar (pH 7,4) en un bloque calentador a 37°C. Después de intervalos apropiados, se sacaron muestras (alícuotas de 50 j l) a ambos pH y se almacenaron a -20°C hasta el análisis. Antes del análisis, las muestras se retiraron del congelador y se añadieron 7 j l de NaCl 5 M, así como 93 j l de metanol frío. Después las muestras se almacenaron durante 30 min a -20°C, se centrifugaron a 4°C y 200 rpm durante 60 min. Posteriormente, 75 j l del sobrenadante se diluyeron con 75 j l de agua purificada y la mezcla se agitó con el vórtex y analizó usando HPLC (Waters Xterra MS, C18, 3,5 jM , 2,1 x 100 mm, gradiente: fase móvil A: acetato de amonio 20 mM, pH 7,0 ± 0,1, fase móvil B: acetonitrilo, gradiente: 0 min: 70% A, 20 min; 30% A, 25 min: 10% A, 25,1 min: 70% A, 35 min: 70% A). Se preparó una curva estándar del compuesto 17 a 2,00, 1,00, 0,50, 0,30, 0,20, 0,10, 0,05 y 0,01 jM . Se usó un intervalo de 2,00 a 0,05 jM para la cuantificación UV a 214 nm. Se encontró que el compuesto 17 era el único producto liberado. Después de 24 h, el 2,7% del compuesto 17 se ha liberado del ADC a pH 7,4 mientras que a pH 4,0 se observó el 39,6% del compuesto 17 libre.
Claims (1)
- REIVINDICACIONESUn compuesto que tiene la estructura de la fórmula (I):
 Fórmula (I)o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde:a) el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;el espaciador está ausente, o se selecciona del grupo que consiste eny
Fórmula (I)o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde:a) el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;el espaciador está ausente, o se selecciona del grupo que consiste eny
 X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; y alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-, o Ri es un aminoácido natural o no natural,o R1 tiene la siguiente fórmula:
X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; y alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-, o Ri es un aminoácido natural o no natural,o R1 tiene la siguiente fórmula: en donde:ciona del grupo que consiste en:
en donde:ciona del grupo que consiste en:
 R es: ~ 0 P 03Mi en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, y/o 2NH4+ o- S O 3M2 en donde M2 = Na+, K+, H+, NH4+R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, un grupo aceptor de electrones, y un grupo soluble en agua;PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido, un anticuerpo o un fragmento del mismo, y un anticuerpo derivatizado o fragmento derivatizado del mismo;en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; yen donde al menos uno de Z1 , Z2, Z3 y Z4 es un grupo aceptor de electrones; ob) el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;el espaciador está ausente
R es: ~ 0 P 03Mi en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, y/o 2NH4+ o- S O 3M2 en donde M2 = Na+, K+, H+, NH4+R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, un grupo aceptor de electrones, y un grupo soluble en agua;PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido, un anticuerpo o un fragmento del mismo, y un anticuerpo derivatizado o fragmento derivatizado del mismo;en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; yen donde al menos uno de Z1 , Z2, Z3 y Z4 es un grupo aceptor de electrones; ob) el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;el espaciador está ausente n es 0 o 1 ;X se selecciona del grupo que consiste en: alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-, R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H y un grupo aceptor de electrones;PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido, y un anticuerpo o fragmento del mismo;en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; yen donde al menos uno de Z1, Z2, Z3 y Z4 es un grupo aceptor de electrones.El compuesto según la reivindicación 1, en donde dicho compuesto tiene una estructura de fórmula (II):
n es 0 o 1 ;X se selecciona del grupo que consiste en: alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;Y está ausente o se selecciona del grupo que consiste en alquilo de C1-C6 opcionalmente sustituido, -NH-C(O)-, -C(O)-NH-, -C(O)-O-, y -O-C(O)-;R1 está ausente o se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-, R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H y un grupo aceptor de electrones;PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, un grupo éster de N-hidroxisuccinimida opcionalmente sustituido, y un anticuerpo o fragmento del mismo;en donde cuando el espaciador está ausente, el agente está unido al nitrógeno adyacente al espaciador por un doble enlace; yen donde al menos uno de Z1, Z2, Z3 y Z4 es un grupo aceptor de electrones.El compuesto según la reivindicación 1, en donde dicho compuesto tiene una estructura de fórmula (II): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, PBG, Y, R1, Z1, Z2, Z3 y Z4 son como se han definido en la reivindicación 1.El compuesto según la reivindicación 1, dicho compuesto que tiene una estructura de fórmula (III):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, PBG, Y, R1, Z1, Z2, Z3 y Z4 son como se han definido en la reivindicación 1.El compuesto según la reivindicación 1, dicho compuesto que tiene una estructura de fórmula (III): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, espaciador, Z2, Z3, Z4, Y, R1 y PBG son como se han definido en la reivindicación 1; y en donde Z1 es un grupo aceptor de electrones.El compuesto según la reivindicación 1, dicho compuesto que tiene una estructura de fórmula (IV):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, espaciador, Z2, Z3, Z4, Y, R1 y PBG son como se han definido en la reivindicación 1; y en donde Z1 es un grupo aceptor de electrones.El compuesto según la reivindicación 1, dicho compuesto que tiene una estructura de fórmula (IV): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, PBG, Y, R1, Z2 , Z3 y Z4 son como se han definido en la reivindicación 1; yen donde Z1 es un grupo aceptor de electrones.El compuesto según la reivindicación 1, dicho compuesto que tiene una estructura de fórmula (V):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, PBG, Y, R1, Z2 , Z3 y Z4 son como se han definido en la reivindicación 1; yen donde Z1 es un grupo aceptor de electrones.El compuesto según la reivindicación 1, dicho compuesto que tiene una estructura de fórmula (V): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, n, X, R1, R2 , Z1, Z2, Z3 , Z4, Y, R1 y PBG son como se han definido en la reivindicación 1.El compuesto según la reivindicación 5, dicho compuesto que tiene una estructura dea) Fórmula (VIa):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde el agente, n, X, R1, R2 , Z1, Z2, Z3 , Z4, Y, R1 y PBG son como se han definido en la reivindicación 1.El compuesto según la reivindicación 5, dicho compuesto que tiene una estructura dea) Fórmula (VIa): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde:M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno;X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;R' es -R3 o -CH2R3 ;en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; yen donde X, n, Z1, Z2, Z3 , Z4, Y, R1, R2, y PBG son como se ha definido en la reivindicación 5; ob) Fórmula (VIb):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde:M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno;X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;R' es -R3 o -CH2R3 ;en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; yen donde X, n, Z1, Z2, Z3 , Z4, Y, R1, R2, y PBG son como se ha definido en la reivindicación 5; ob) Fórmula (VIb): o una sal, hidrato o solvato farmacéuticamente aceptable del mismo; en dondeM es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario;X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;R3 se selecciona del grupo que consiste en -H -OH, -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un amonio ion o amonio sustituido con alquilo; yen donde X, n, Z1, Z2, Z3 , Z4, Y, R1, R2, y PBG, son como se ha definido en la reivindicación 5.El compuesto según la reivindicación 6, dicho compuesto que tiene la estructura dea) fórmula (VIIa):
o una sal, hidrato o solvato farmacéuticamente aceptable del mismo; en dondeM es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario;X1 y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;R3 se selecciona del grupo que consiste en -H -OH, -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2 , -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un amonio ion o amonio sustituido con alquilo; yen donde X, n, Z1, Z2, Z3 , Z4, Y, R1, R2, y PBG, son como se ha definido en la reivindicación 5.El compuesto según la reivindicación 6, dicho compuesto que tiene la estructura dea) fórmula (VIIa): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde R' es -R3 , Z3, en la reivindicación
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde R' es -R3 , Z3, en la reivindicación b) Fórmula (VIIb):
b) Fórmula (VIIb): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde X, X1, X2 , X3 , X4,Z4 y PBG, son como se ha def El compuesto de la reivindicación 5, dicho compuesto tiene la estructura de fórmula (VIII):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde X, X1, X2 , X3 , X4,Z4 y PBG, son como se ha def El compuesto de la reivindicación 5, dicho compuesto tiene la estructura de fórmula (VIII):
 o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde agente, X, n, R2, PBG, Y, R1, Z2 , Z3 y Z4 son como se han definido en la reivindicación 5; yen donde Zi es un grupo aceptor de electrones.El compuesto según la reivindicación 8 , dicho compuesto tiene la estructura dea) Fórmula (IXa):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde agente, X, n, R2, PBG, Y, R1, Z2 , Z3 y Z4 son como se han definido en la reivindicación 5; yen donde Zi es un grupo aceptor de electrones.El compuesto según la reivindicación 8 , dicho compuesto tiene la estructura dea) Fórmula (IXa): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde:M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno;Xi y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupoque consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;R' es -R3 o -CH2R3 ;en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; yen donde X, n, Z1, Z2, Z3 , Z4, Ri, R2, y PBG son como se ha definido en la reivindicación 8; ob) Fórmula (IXb):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde:M es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario y opcionalmente contiene uno o más sustituyentes seleccionados de halógeno;Xi y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupoque consiste en -H, -OH, alquilo de C1-C6 , halógeno, y -N3;R' es -R3 o -CH2R3 ;en donde cada aparición de R3 se selecciona independientemente del grupo que consiste en -OH, -CH3 , -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; yen donde X, n, Z1, Z2, Z3 , Z4, Ri, R2, y PBG son como se ha definido en la reivindicación 8; ob) Fórmula (IXb): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en dondeM es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario;Xi y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de Ci-Ca, halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de Ci-Ca, halógeno, y -N3;R3 se selecciona del grupo que consiste en -H, -OH, -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; yen donde X, n, Zi, Z2, Z3 , Z4, Ri, R2, y PBG son como se ha definido en la reivindicación 8.10. El compuesto según la reivindicación 9, dicho compuesto que tiene una estructura dea) Fórmula (Xa):
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en dondeM es un grupo pirimidina o purina que contiene al menos un grupo amino primario o secundario;Xi y X2 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, alquilo de Ci-Ca, halógeno, y -N3;X3 y X4 están cada uno independientemente, según permita la valencia, ausente o se seleccionan del grupo que consiste en -H, -OH, alquilo de Ci-Ca, halógeno, y -N3;R3 se selecciona del grupo que consiste en -H, -OH, -OP(O)(OH)2 , -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), un aminoácido, un grupo acilo, y una sal farmacéuticamente aceptable del mismo, en donde la sal contiene un ion de metal alcalino, un ion de metal alcalino, un ion amonio o amonio sustituido con alquilo; yen donde X, n, Zi, Z2, Z3 , Z4, Ri, R2, y PBG son como se ha definido en la reivindicación 8.10. El compuesto según la reivindicación 9, dicho compuesto que tiene una estructura dea) Fórmula (Xa): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde R' e , R1 y reivindicación
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo;en donde R' e , R1 y reivindicación b) Fórmula (Xb):
b) Fórmula (Xb): o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo; yen donde X1, X2 , X3 , X4, R3, X, n, Y, Z1, Z2 , Z3, Z4, R1 y R2 son como se ha definido en la reivindicación 9.11. El compuesto según cualquiera de las reivindicaciones 1-10, en donde:a) el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 5'-desoxi-5-fluorocitidina, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 7-acetilcamptotecina, 9-formilcamptotecina, 9-acetilcamptotecina, 9-formil-10-hidroxicamptotecina, 10-formilcamptotecina, 10-acetilcamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansina, maitansinol; auristatina (incluyendo, pero no limitado a auristatina D, auristatina E, auristatina F, monometil auristatina D, monometil auristatina E, monometil auristatina F, éster metílico de monometil auristatina F, auristatina PYE auristatina PHE, el producto natural relacionado dolastatina 10, y derivados de las mismas); amatoxinas (incluyendo, pero no limitado a a-amanitina, p-amanitina, Y-amanitina, £-amanitin, amanina, amaninamida, arnanulina, y ácido amanulínico y derivados de los mismos); duocarmicina A, duocarmicina B1, duocarmicina B2, duocarmicina C, duocarmicina SA, CC1065, adozelesina, bizelesina, carzelesina; eribulina; trabectedina; pirrolobenzodiazepina, antramicina, tomaimicina, sibiromicina, DC-81, DSB-120; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores; ob) el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, g G211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 9-formilcamptotecina, 9-formil-10-hidroxicamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansinoides; auristatinas; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.12. El compuesto según cualquiera de las reivindicaciones 1-11, en donde:a) Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -CN, en donde al menos uno de Z1, Z2, Z3 y Z4 no es -H;b) Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN;c) Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3;d) Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), -P(O)(OH)2 , -SO3H, y una sal farmacéuticamente aceptable del mismo;e) Z1 se selecciona del grupo que consiste en halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6 , y -Cn ; yZ2 , Z3 y Z4 se seleccionan cada uno independientemente de -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6, o -CN;f) Z1 se selecciona del grupo que consiste en -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3, y -CN; yZ2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN; og) Z1 se selecciona del grupo que consiste en -Cl, -F y -NO2, yZ2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3.13. El compuesto según cualquiera de las reivindicaciones 1-12, en dondese
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo; yen donde X1, X2 , X3 , X4, R3, X, n, Y, Z1, Z2 , Z3, Z4, R1 y R2 son como se ha definido en la reivindicación 9.11. El compuesto según cualquiera de las reivindicaciones 1-10, en donde:a) el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 5'-desoxi-5-fluorocitidina, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, GG211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 7-acetilcamptotecina, 9-formilcamptotecina, 9-acetilcamptotecina, 9-formil-10-hidroxicamptotecina, 10-formilcamptotecina, 10-acetilcamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansina, maitansinol; auristatina (incluyendo, pero no limitado a auristatina D, auristatina E, auristatina F, monometil auristatina D, monometil auristatina E, monometil auristatina F, éster metílico de monometil auristatina F, auristatina PYE auristatina PHE, el producto natural relacionado dolastatina 10, y derivados de las mismas); amatoxinas (incluyendo, pero no limitado a a-amanitina, p-amanitina, Y-amanitina, £-amanitin, amanina, amaninamida, arnanulina, y ácido amanulínico y derivados de los mismos); duocarmicina A, duocarmicina B1, duocarmicina B2, duocarmicina C, duocarmicina SA, CC1065, adozelesina, bizelesina, carzelesina; eribulina; trabectedina; pirrolobenzodiazepina, antramicina, tomaimicina, sibiromicina, DC-81, DSB-120; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores; ob) el agente se selecciona del grupo que consiste en N-nitrosoureas; doxorrubicina, 2-pirrolpirrolinoantraciclina, morfolinoantraciclina, diacetatoxialquilantraciclina, daunorrubicina, epirrubicina, idarrubicina, nemorrubicina, PNU-159682, mitoxantrona; ametantrona; clorambucilo, bendamustina, melfalán, oxazafosforinas; 5-fluorouracilo, 2'-desoxi-5-fluoridina, citarabina, cladribina, fludarabina, pentostatina, gemcitabina, 4-amino-1-(((2S,3R,4S,5R)-3,4-dihidroxi-5-metiltetrahidrofuran-2-il)metil)-5-fluoropirimidin-2(1H)-ona, tioguanina; metotrexato, raltitrexed, pemetrexed, plevitrexed; paclitaxel, docetaxel; topotecano, irinotecano, SN-38, 10-hidroxicamptotecina, g G211, lurtotecano, 9-aminocamptotecina, camptotecina, 7-formilcamptotecina, 9-formilcamptotecina, 9-formil-10-hidroxicamptotecina, 7-butil-10-aminocamptotecina, 7-butil-9-amino-10,11,-metilenedioxocamptotecina; vinblastina, vincristina, vindesina, vinorelbina; caliqueamicinas; maitansinoides; auristatinas; epotilonas; bleomicina; dactinomicina; plicamicina, miromicina C, y complejos de platino(II) configurados en cis; o un derivado de cualquiera de los anteriores.12. El compuesto según cualquiera de las reivindicaciones 1-11, en donde:a) Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y -CN, en donde al menos uno de Z1, Z2, Z3 y Z4 no es -H;b) Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN;c) Z1, Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3;d) Z1, Z2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2, -OP(O)(OH)(NH2), -P(O)(OH)2 , -SO3H, y una sal farmacéuticamente aceptable del mismo;e) Z1 se selecciona del grupo que consiste en halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2, haloalquilo, -S(O)2-alquilo de C1-C6 , y -Cn ; yZ2 , Z3 y Z4 se seleccionan cada uno independientemente de -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6, o -CN;f) Z1 se selecciona del grupo que consiste en -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3, y -CN; yZ2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y -CN; og) Z1 se selecciona del grupo que consiste en -Cl, -F y -NO2, yZ2 , Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3.13. El compuesto según cualquiera de las reivindicaciones 1-12, en dondese a) se selecciona del grupo que consiste en:
a) se selecciona del grupo que consiste en: b) se selecciona del grupo que consiste en
b) se selecciona del grupo que consiste en
 14. El compuesto según cualquiera de las reivindicaciones 1-13, en donde Y es:a) -C(O)-NH-;b) -C(O)-O-; oc) ausente.15. El compuesto según cualquiera de las reivindicaciones 1-14, en donde R1 se:a) selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-;b)
14. El compuesto según cualquiera de las reivindicaciones 1-13, en donde Y es:a) -C(O)-NH-;b) -C(O)-O-; oc) ausente.15. El compuesto según cualquiera de las reivindicaciones 1-14, en donde R1 se:a) selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquilo de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-;b) c) ausente;d) un aminoácido natural o no natural; oe)
c) ausente;d) un aminoácido natural o no natural; oe) R es: -OPO3M1 en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, y/o 2NH4+ o~ S 03M2 en donde M2 = Na+, K+, H+, NH4+16. El compuesto según cualquiera de las reivindicaciones 1-15, en donde dicho PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, y un grupo éster de N-hidroxisuccinimida opcionalmente sustituido.17. Un conjugado que comprende el compuesto según la reivindicación 16 y un anticuerpo o fragmento del mismo, en donde el PBG está asociado con el anticuerpo o fragmento del mismo.18. Un conjugado que comprende el compuesto según la reivindicación 16 y un anticuerpo o fragmento del mismo, en donde el PBG está covalentemente unido al anticuerpo o fragmento del mismo.19. Un conjugado que comprende el compuesto según la reivindicación 16, y albúmina, en donde el PBG está asociado con la albúmina.20. Un conjugado que comprende el compuesto según la reivindicación 16, y albúmina endógena o exógena, en donde el PBG está covalentemente unido a la albúmina endógena o exógena.21. El conjugado según la reivindicación 20, en donde el PBG está covalentemente unido a la cisteína 34 de la albúmina endógena o exógena.22. El compuesto según cualquiera de las reivindicaciones 1-16, en donde dicho PBG es un grupo maleimida opcionalmente sustituido.23. El compuesto según cualquiera de las reivindicaciones 1-16, en donde PBG es
R es: -OPO3M1 en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, y/o 2NH4+ o~ S 03M2 en donde M2 = Na+, K+, H+, NH4+16. El compuesto según cualquiera de las reivindicaciones 1-15, en donde dicho PBG es un grupo de unión a proteínas seleccionado del grupo que consiste en un grupo maleimida opcionalmente sustituido, un grupo haloacetamida opcionalmente sustituido, un grupo haloacetato opcionalmente sustituido, un grupo piridiltio opcionalmente sustituido, un grupo isotiocianato opcionalmente sustituido, un grupo vinilcarbonilo opcionalmente sustituido, un grupo aziridina opcionalmente sustituido, un grupo disulfuro opcionalmente sustituido, un grupo acetileno opcionalmente sustituido, y un grupo éster de N-hidroxisuccinimida opcionalmente sustituido.17. Un conjugado que comprende el compuesto según la reivindicación 16 y un anticuerpo o fragmento del mismo, en donde el PBG está asociado con el anticuerpo o fragmento del mismo.18. Un conjugado que comprende el compuesto según la reivindicación 16 y un anticuerpo o fragmento del mismo, en donde el PBG está covalentemente unido al anticuerpo o fragmento del mismo.19. Un conjugado que comprende el compuesto según la reivindicación 16, y albúmina, en donde el PBG está asociado con la albúmina.20. Un conjugado que comprende el compuesto según la reivindicación 16, y albúmina endógena o exógena, en donde el PBG está covalentemente unido a la albúmina endógena o exógena.21. El conjugado según la reivindicación 20, en donde el PBG está covalentemente unido a la cisteína 34 de la albúmina endógena o exógena.22. El compuesto según cualquiera de las reivindicaciones 1-16, en donde dicho PBG es un grupo maleimida opcionalmente sustituido.23. El compuesto según cualquiera de las reivindicaciones 1-16, en donde PBG es 24. Un compuesto que tiene la estructura de fórmula (I):
24. Un compuesto que tiene la estructura de fórmula (I): en donde:el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;el espaciador está ausente, o se selecciona del grupo que consiste eny
en donde:el agente se selecciona del grupo que consiste en un agente citostático, un agente citotóxico, una citoquina, un agente inmunosupresor, un antirreumático, un antiflogístico, un antibiótico, un analgésico, un agente virostático, un agente antiinflamatorio, un agente antimicótico, un inhibidor de factor de transcripción, un modulador del ciclo celular, un modulador de MDR, un inhibidor de proteasoma o proteasa, un modulador de apoptosis, un inhibidor enzimático, un inhibidor de transducción de señales, un inhibidor de angiogénesis, una hormona o derivado de hormona, un anticuerpo o fragmento del mismo, un péptido terapéutica o diagnósticamente activo, una sustancia radioactiva, una sustancia que emite luz, una sustancia que absorbe luz, y un derivado de cualquiera de los anteriores;el espaciador está ausente, o se selecciona del grupo que consiste eny
 n es 0 o 1 ;X se selecciona del grupo que consiste en en donde opcionalmente hasta seis átomos de carbono en dicho alquileno de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquileno de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquileno de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquileno de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquileno de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;
n es 0 o 1 ;X se selecciona del grupo que consiste en en donde opcionalmente hasta seis átomos de carbono en dicho alquileno de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquileno de C1-C18 opcionalmente sustituido-NH-C(O)-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquileno de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; alquileno de C1-C18 opcionalmente sustituido-C(O)-NH-R5- en donde opcionalmente hasta seis átomos de carbono en dicho alquileno de C1-C18 están cada uno independientemente sustituido con -OCH2CH2-; arilo opcionalmente sustituido; heteroarilo opcionalmente sustituido; y cicloalquilo opcionalmente sustituido;R5 se selecciona del grupo que consiste en un arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido;R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido; M1 = Mg2+; 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+,
M1 = Mg2+; 2 Na+, 2K+, 2H+, 2 NH4+, Na+, K+, NH4+ y/o H+, 25. El compuesto según cualquiera de las reivindicaciones 1-24, en donde el espaciador es:
25. El compuesto según cualquiera de las reivindicaciones 1-24, en donde el espaciador es: X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente reemplazados con -OCH2CH2-; arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido; yR2 es como se define en la reivindicación 1;
X se selecciona del grupo que consiste en alquilo de C1-C18 opcionalmente sustituido en donde opcionalmente hasta seis átomos de carbono en dicho alquilo de C1-C18 están cada uno independientemente reemplazados con -OCH2CH2-; arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, y cicloalquilo opcionalmente sustituido; yR2 es como se define en la reivindicación 1; opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido; y W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6 , y CN;
opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido; y W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6 , y CN; opcionalmente sustituido, arilo opcionalmente sustituido, o heteroarilo opcionalmente sustituido; y W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente de un grupo fenoxi, un grupo amino primario, secundario o terciario, un grupo éter, un grupo fenol, un grupo amida, un grupo éster, un grupo alquilo, un grupo alquilo sustituido, un grupo fenilo, y un grupo vinilo;. en donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, o heteroarilo opcionalmente sustituido; y W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente de -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo;
opcionalmente sustituido, arilo opcionalmente sustituido, o heteroarilo opcionalmente sustituido; y W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente de un grupo fenoxi, un grupo amino primario, secundario o terciario, un grupo éter, un grupo fenol, un grupo amida, un grupo éster, un grupo alquilo, un grupo alquilo sustituido, un grupo fenilo, y un grupo vinilo;. en donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, o heteroarilo opcionalmente sustituido; y W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente de -OP(O)(OH)2, -P(O)(OH)OP(O)(OH)2, -OP(O)(OH)OP(O)(OH)OP(O)(OH)2 , -OP(O)(OH)(NH2), -P(O)(OH)2, -SO3H, y una sal farmacéuticamente aceptable del mismo; donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;
donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido; W 1 se selecciona del grupo que consiste en halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y CN; y,W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6 , y CN;en donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;
W 1 se selecciona del grupo que consiste en halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6 , -NO2, haloalquilo, -S(O)2-alquilo de C1-C6, y CN; y,W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, halógeno, -C(O)OH, -C(O)O-alquilo de C1-C6, -NO2 , haloalquilo, -S(O)2-alquilo de C1-C6 , y CN;en donde R2 se selecciona del grupo que consiste en -H, alquilo de C1-C12 opcionalmente sustituido, arilo opcionalmente sustituido, y heteroarilo opcionalmente sustituido;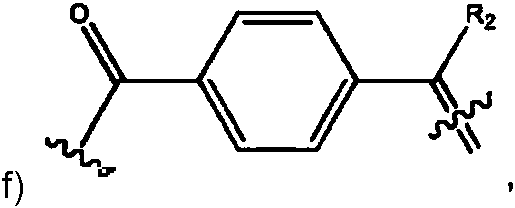 onde m es 1, 2, 3, 4, 5, o 6 y R2 se define como en la reivindicación 1; o
onde m es 1, 2, 3, 4, 5, o 6 y R2 se define como en la reivindicación 1; o 26. El compuesto según la reivindicación 25, en donde:a) W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y CN;b) W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3;c) W 1 se selecciona del grupo que consiste en -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y CN; y W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y CN; od) W 1 se selecciona del grupo que consiste en -Cl, -Br, y -NO2; y Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3.27. El compuesto según cualquiera de las reivindicaciones 6-26, en donde X1, X2 , X3 y X4 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, -CH3 , -F, -Cl, -Br, -I, y -N3.28. Un compuesto seleccionado del grupo que consiste en:
26. El compuesto según la reivindicación 25, en donde:a) W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y CN;b) W 1, W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3;c) W 1 se selecciona del grupo que consiste en -Cl, -Br, -I, -F, -C(O)OH, -NO2, -CF3 , y CN; y W 2, W 3 y W 4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -Br, -I, -F, -C(O)OH, -NO2 , -CF3 , y CN; od) W 1 se selecciona del grupo que consiste en -Cl, -Br, y -NO2; y Z2, Z3 y Z4 se seleccionan cada uno independientemente del grupo que consiste en -H, -Cl, -F, -NO2, y -CF3.27. El compuesto según cualquiera de las reivindicaciones 6-26, en donde X1, X2 , X3 y X4 se seleccionan cada uno independientemente del grupo que consiste en -H, -OH, -CH3 , -F, -Cl, -Br, -I, y -N3.28. Un compuesto seleccionado del grupo que consiste en: o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo,en donde:
o una sal, hidrato, o solvato farmacéuticamente aceptable del mismo,en donde: está ausente o se selecciona del grupo que consiste en:
está ausente o se selecciona del grupo que consiste en: R es -O P O 3M1 en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, 2NH4+, Na+, K+, NH4+, y/o H+ o - S O 3M2 en donde M2 = Na+, K+, H+, NH4+29. Un compuesto seleccionado del grupo que consiste en:
R es -O P O 3M1 en donde Mi = Mg2+, 2 Na+, 2K+, 2H+, 2NH4+, Na+, K+, NH4+, y/o H+ o - S O 3M2 en donde M2 = Na+, K+, H+, NH4+29. Un compuesto seleccionado del grupo que consiste en:


 30. Un compuesto seleccionado del grupo que consiste en:
30. Un compuesto seleccionado del grupo que consiste en: 31. Un compuesto seleccionado del grupo que consiste en:
31. Un compuesto seleccionado del grupo que consiste en: y una sal, hidrato, o solvato farmacéuticamente aceptable del mismo.32. Una composición farmacéutica que comprende un compuesto de cualquiera de las reivindicaciones 1-31, y un soporte farmacéuticamente aceptable.33. Un compuesto según cualquiera de las reivindicaciones 1-31 para uso en tratar una enfermedad o afección seleccionada del grupo que consiste en un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, una enfermedad inflamatoria aguda o crónica, y una enfermedad causada por bacterias, hongos, u otros microorganismos.
y una sal, hidrato, o solvato farmacéuticamente aceptable del mismo.32. Una composición farmacéutica que comprende un compuesto de cualquiera de las reivindicaciones 1-31, y un soporte farmacéuticamente aceptable.33. Un compuesto según cualquiera de las reivindicaciones 1-31 para uso en tratar una enfermedad o afección seleccionada del grupo que consiste en un cáncer, una enfermedad vírica, una enfermedad autoinmunitaria, una enfermedad inflamatoria aguda o crónica, y una enfermedad causada por bacterias, hongos, u otros microorganismos.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562182219P | 2015-06-19 | 2015-06-19 | |
| US201562261213P | 2015-11-30 | 2015-11-30 | |
| US201562261563P | 2015-12-01 | 2015-12-01 | |
| PCT/US2016/038223 WO2016205738A2 (en) | 2015-06-19 | 2016-06-17 | Delivery systems for controlled drug release |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2909486T3 true ES2909486T3 (es) | 2022-05-06 |
Family
ID=57546445
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16812576T Active ES2909486T3 (es) | 2015-06-19 | 2016-06-17 | Sistemas de administración para liberación controlada de fármaco |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US11384104B2 (es) |
| EP (2) | EP3310800B1 (es) |
| JP (3) | JP6948708B2 (es) |
| CN (2) | CN107922457B (es) |
| AU (1) | AU2016279051B2 (es) |
| CA (1) | CA2989963A1 (es) |
| DK (1) | DK3310800T3 (es) |
| ES (1) | ES2909486T3 (es) |
| IL (1) | IL256371B (es) |
| MA (1) | MA45472A (es) |
| MX (1) | MX386808B (es) |
| NZ (1) | NZ738229A (es) |
| PT (1) | PT3310800T (es) |
| RU (1) | RU2748992C2 (es) |
| WO (1) | WO2016205738A2 (es) |
| ZA (1) | ZA201708234B (es) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106279352B (zh) | 2015-05-29 | 2020-05-22 | 上海新理念生物医药科技有限公司 | 海兔毒素10的衍生物及其应用 |
| WO2017177316A1 (en) * | 2016-04-11 | 2017-10-19 | 3R Valo, S.E.C. | Aminobenzoic acid derivatives for use as anti-inflammatory agents, anti-metastatic agents and/or anticancer agents |
| KR20190038537A (ko) | 2016-06-17 | 2019-04-08 | 마젠타 테라퓨틱스 인코포레이티드 | Cd117+ 세포의 고갈을 위한 조성물 및 방법 |
| SG10202102897PA (en) | 2017-01-20 | 2021-04-29 | Magenta Therapeutics Inc | Compositions and methods for the depletion of cd137+ cells |
| EP3617221A4 (en) * | 2017-04-19 | 2021-04-14 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | CYTOTOXIN AND CONJUGATE, USES THEREOF AND MANUFACTURING PROCESSES THEREFORE |
| EP3668538A4 (en) | 2017-08-15 | 2021-06-16 | NantCell, Inc. | HANK-CETUXIMAB COMBINATIONS AND PROCEDURES |
| US11377473B2 (en) * | 2017-11-30 | 2022-07-05 | Centurion Biopharma Corporation | Albumin-binding prodrugs of auristatin E derivatives |
| IL305268A (en) * | 2017-11-30 | 2023-10-01 | Centurion Biopharma Corp | Maytansinoid-based drug delivery systems |
| EP4667495A2 (en) | 2017-12-15 | 2025-12-24 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Bioactive molecule conjugate, preparation method and use thereof |
| CN110090308B (zh) * | 2018-01-30 | 2023-03-24 | 四川科伦博泰生物医药股份有限公司 | 制备偶联物的方法 |
| CN110227164B (zh) | 2018-03-06 | 2021-11-23 | 江苏吉贝尔药业股份有限公司 | 含酮羰基的疏水性抗肿瘤药物及其缀合物、含有缀合物的纳米制剂及其制备方法及应用 |
| AU2023359114A1 (en) | 2022-10-09 | 2025-04-24 | LaNova Medicines Limited | Compounds, compositions and methods |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4125704A (en) * | 1977-09-14 | 1978-11-14 | Sri International | Phenyl-substituted rubidazone analogs |
| GB8308235D0 (en) | 1983-03-25 | 1983-05-05 | Celltech Ltd | Polypeptides |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| GB8422238D0 (en) | 1984-09-03 | 1984-10-10 | Neuberger M S | Chimeric proteins |
| US4699880A (en) | 1984-09-25 | 1987-10-13 | Immunomedics, Inc. | Method of producing monoclonal anti-idiotype antibody |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| JPH02500329A (ja) | 1987-05-21 | 1990-02-08 | クリエイテイブ・バイオマリキユールズ・インコーポレーテツド | ターゲット化多機能蛋白質 |
| US5094950A (en) | 1988-06-07 | 1992-03-10 | Nihon Medi-Physics Co., Ltd. | Diethylenetriamine pentaacetic acid derivatives |
| US4966999A (en) * | 1988-06-07 | 1990-10-30 | Cytogen Corporation | Radiohalogenated compounds for site specific labeling |
| IL162181A (en) | 1988-12-28 | 2006-04-10 | Pdl Biopharma Inc | A method of producing humanized immunoglubulin, and polynucleotides encoding the same |
| DE69233482T2 (de) | 1991-05-17 | 2006-01-12 | Merck & Co., Inc. | Verfahren zur Verminderung der Immunogenität der variablen Antikörperdomänen |
| EP0605522B1 (en) | 1991-09-23 | 1999-06-23 | Medical Research Council | Methods for the production of humanized antibodies |
| DE614989T1 (de) | 1993-02-17 | 1995-09-28 | Morphosys Proteinoptimierung | Verfahren für in vivo Selektion von Ligandenbindende Proteine. |
| DE19926154A1 (de) | 1999-06-09 | 2000-12-14 | Ktb Tumorforschungs Gmbh | Verfahren zur Herstellung einer injizierbaren Arzneimittelzubereitung |
| GB0004297D0 (en) | 2000-02-23 | 2000-04-12 | Ucb Sa | 2-oxo-1 pyrrolidine derivatives process for preparing them and their uses |
| US6884869B2 (en) | 2001-04-30 | 2005-04-26 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| WO2005055939A2 (en) | 2003-12-04 | 2005-06-23 | Amr Technology, Inc. | Vinca derivatives |
| CN101490087B (zh) | 2006-05-30 | 2013-11-06 | 健泰科生物技术公司 | 抗体和免疫偶联物及其用途 |
| KR20100029764A (ko) * | 2007-05-16 | 2010-03-17 | 케이티비 투머포슝스케쉘샤프트 엠비에이치 | 저점성 안트라사이클린 제제 |
| EP2289558A1 (en) | 2009-08-25 | 2011-03-02 | KTB Tumorforschungsgesellschaft mbH | Bisphosphonate-prodrugs |
| EP2780039B9 (en) | 2011-11-17 | 2018-04-18 | Pfizer Inc | Cytotoxic peptides and antibody drug conjugates thereof |
| EP2630971B8 (en) * | 2012-02-21 | 2017-12-13 | Vergell Medical S.A. | Combinations of albumin-based drug delivery systems |
| WO2014080251A1 (en) | 2012-11-24 | 2014-05-30 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and their uses for conjugation of drugs to cell binding molecules |
| CN104688740A (zh) | 2012-12-21 | 2015-06-10 | 百奥泰生物科技(广州)有限公司 | 类美登素衍生物及其制备方法和用途 |
| US20140221429A1 (en) * | 2013-02-04 | 2014-08-07 | Saud Ibrahim Al-Resayes | Heterocyclic schiff's bases as novel and new antiglycation agents |
| FR3005051A1 (fr) | 2013-04-25 | 2014-10-31 | Pf Medicament | Derives de la dolastatine 10 et d'auristatines |
| FR3017298B1 (fr) | 2014-02-07 | 2016-03-04 | Centre Nat Rech Scient | Conjugues et pro-drogues pour le traitement du cancer et de maladies inflammatoires |
| WO2016160615A1 (en) | 2015-03-27 | 2016-10-06 | Regeneron Pharmaceuticals, Inc. | Maytansinoid derivatives, conjugates thereof, and methods of use |
| WO2016205378A1 (en) | 2015-06-15 | 2016-12-22 | Abbott Diabetes Care Inc. | Stabilized lactate responsive enzymes, electrodes and sensors, and methods for making and using the same |
| IL305268A (en) | 2017-11-30 | 2023-10-01 | Centurion Biopharma Corp | Maytansinoid-based drug delivery systems |
| US11377473B2 (en) | 2017-11-30 | 2022-07-05 | Centurion Biopharma Corporation | Albumin-binding prodrugs of auristatin E derivatives |
-
2016
- 2016-06-17 PT PT168125763T patent/PT3310800T/pt unknown
- 2016-06-17 CN CN201680041045.0A patent/CN107922457B/zh active Active
- 2016-06-17 AU AU2016279051A patent/AU2016279051B2/en active Active
- 2016-06-17 WO PCT/US2016/038223 patent/WO2016205738A2/en not_active Ceased
- 2016-06-17 MX MX2017016524A patent/MX386808B/es unknown
- 2016-06-17 EP EP16812576.3A patent/EP3310800B1/en active Active
- 2016-06-17 ES ES16812576T patent/ES2909486T3/es active Active
- 2016-06-17 CA CA2989963A patent/CA2989963A1/en active Pending
- 2016-06-17 JP JP2017565192A patent/JP6948708B2/ja active Active
- 2016-06-17 NZ NZ738229A patent/NZ738229A/en unknown
- 2016-06-17 RU RU2018101862A patent/RU2748992C2/ru active
- 2016-06-17 CN CN202210430597.8A patent/CN115089722A/zh active Pending
- 2016-06-17 US US15/735,885 patent/US11384104B2/en active Active
- 2016-06-17 DK DK16812576.3T patent/DK3310800T3/da active
- 2016-06-17 EP EP21180000.8A patent/EP3954394A1/en active Pending
-
2017
- 2017-06-17 MA MA045472A patent/MA45472A/fr unknown
- 2017-12-04 ZA ZA2017/08234A patent/ZA201708234B/en unknown
- 2017-12-17 IL IL256371A patent/IL256371B/en active IP Right Grant
-
2020
- 2020-09-10 JP JP2020152004A patent/JP2021001185A/ja not_active Withdrawn
-
2022
- 2022-06-03 US US17/831,710 patent/US12497417B2/en active Active
-
2023
- 2023-01-16 JP JP2023004453A patent/JP7595098B2/ja active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2909486T3 (es) | Sistemas de administración para liberación controlada de fármaco | |
| JP2024038168A (ja) | 生物活性分子コンジュゲート、その調製法及び使用 | |
| ES2402254T3 (es) | Conjugados de amatoxinas con ligadores mejorados | |
| ES2821878T3 (es) | Conectores autoinmolativos no lineales y conjugados de los mismos | |
| ES2970618T3 (es) | Profármacos de derivados de auristatina E que se unen a la albúmina | |
| WO2022262516A1 (zh) | 连接子及其缀合物 | |
| KR20240100413A (ko) | 인(v) 및 약물 모이어티를 포함하는 접합체 | |
| TW202339803A (zh) | 包含磷(v)及喜樹鹼分子部分的共軛物 | |
| JP2023164604A (ja) | メイタンシノイド系薬物送達システム | |
| HK40069235A (en) | Delivery systems for controlled drug release | |
| KR102905519B1 (ko) | 제어된 약물 방출을 위한 전달 시스템 | |
| BR112017027445B1 (pt) | Compostos, composição farmacêutica e uso dos mesmos para tratar um câncer, uma doença viral, uma doença autoimune, uma doença inflamatória aguda ou crônica e uma doença causada por bactérias, fungos, ou outros micro-organismos | |
| HK1254624B (en) | Delivery systems for controlled drug release | |
| RU2795101C2 (ru) | Альбумин-связывающие пролекарства на основе производных ауристатина е | |
| AU2023408641A1 (en) | Conjugates comprising a phosphorus(v) moiety and a drug | |
| JP2025542295A (ja) | リン(v)部分および薬物を含むコンジュゲート |