ES2874537T3 - Sal oxalato de ruxolitinib - Google Patents
Sal oxalato de ruxolitinib Download PDFInfo
- Publication number
- ES2874537T3 ES2874537T3 ES15753374T ES15753374T ES2874537T3 ES 2874537 T3 ES2874537 T3 ES 2874537T3 ES 15753374 T ES15753374 T ES 15753374T ES 15753374 T ES15753374 T ES 15753374T ES 2874537 T3 ES2874537 T3 ES 2874537T3
- Authority
- ES
- Spain
- Prior art keywords
- ruxolitinib
- oxalate
- solid state
- salt
- pharmaceutical
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 title claims description 93
- 229960000215 ruxolitinib Drugs 0.000 title claims description 92
- -1 Ruxolitinib oxalate salt Chemical class 0.000 title description 5
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims abstract description 76
- 150000004935 Ruxolitinib derivatives Chemical class 0.000 claims abstract description 8
- 235000006408 oxalic acid Nutrition 0.000 claims abstract description 6
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 claims description 91
- 239000007787 solid Chemical class 0.000 claims description 53
- 239000008194 pharmaceutical composition Substances 0.000 claims description 35
- 150000003839 salts Chemical class 0.000 claims description 16
- 238000009472 formulation Methods 0.000 claims description 15
- 238000000034 method Methods 0.000 claims description 15
- 238000002360 preparation method Methods 0.000 claims description 13
- 239000000843 powder Substances 0.000 claims description 11
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 9
- 238000002441 X-ray diffraction Methods 0.000 claims description 8
- 206010028980 Neoplasm Diseases 0.000 claims description 7
- 201000011510 cancer Diseases 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- 208000032027 Essential Thrombocythemia Diseases 0.000 claims description 5
- 208000002250 Hematologic Neoplasms Diseases 0.000 claims description 5
- 239000003814 drug Substances 0.000 claims description 5
- 206010028537 myelofibrosis Diseases 0.000 claims description 5
- 230000008569 process Effects 0.000 claims description 5
- 201000004681 Psoriasis Diseases 0.000 claims description 4
- 230000001154 acute effect Effects 0.000 claims description 4
- 239000000203 mixture Substances 0.000 description 21
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 20
- 239000000047 product Substances 0.000 description 17
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- 238000010438 heat treatment Methods 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- 239000000725 suspension Substances 0.000 description 10
- JFMWPOCYMYGEDM-XFULWGLBSA-N ruxolitinib phosphate Chemical compound OP(O)(O)=O.C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 JFMWPOCYMYGEDM-XFULWGLBSA-N 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- 239000013078 crystal Substances 0.000 description 8
- 235000019439 ethyl acetate Nutrition 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 229960002539 ruxolitinib phosphate Drugs 0.000 description 8
- 239000008186 active pharmaceutical agent Substances 0.000 description 7
- 238000012545 processing Methods 0.000 description 7
- 238000003860 storage Methods 0.000 description 7
- LGJWVXWQCTZSGC-XFULWGLBSA-N (3r)-3-cyclopentyl-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile;sulfuric acid Chemical compound OS(O)(=O)=O.C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 LGJWVXWQCTZSGC-XFULWGLBSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 5
- 238000004090 dissolution Methods 0.000 description 5
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- 208000017733 acquired polycythemia vera Diseases 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 4
- 210000001035 gastrointestinal tract Anatomy 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 208000037244 polycythemia vera Diseases 0.000 description 4
- 238000001179 sorption measurement Methods 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 238000007906 compression Methods 0.000 description 3
- 230000006835 compression Effects 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 230000002349 favourable effect Effects 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 229910001496 lithium tetrafluoroborate Inorganic materials 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- 229940127557 pharmaceutical product Drugs 0.000 description 3
- 239000013557 residual solvent Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000011877 solvent mixture Substances 0.000 description 3
- 238000005550 wet granulation Methods 0.000 description 3
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 2
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 2
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 2
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 230000018044 dehydration Effects 0.000 description 2
- 238000006297 dehydration reaction Methods 0.000 description 2
- 238000000113 differential scanning calorimetry Methods 0.000 description 2
- 238000007907 direct compression Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 238000002329 infrared spectrum Methods 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- QHQSWYIJDFMBNE-OAQYLSRUSA-N (3r)-3-cyclopentyl-3-[4-[7-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl]propanenitrile Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C2=C3C=CN(C3=NC=N2)COCC[Si](C)(C)C)CCCC1 QHQSWYIJDFMBNE-OAQYLSRUSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- PTAPBGKYBVWNJY-UHFFFAOYSA-N 2,3-dibenzoyl-2,3-dihydroxybutanedioic acid;hydrate Chemical compound O.C=1C=CC=CC=1C(=O)C(O)(C(O)=O)C(O)(C(=O)O)C(=O)C1=CC=CC=C1 PTAPBGKYBVWNJY-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 229910002483 Cu Ka Inorganic materials 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 1
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- 108010024121 Janus Kinases Proteins 0.000 description 1
- 102000015617 Janus Kinases Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 102100031013 Transgelin Human genes 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 208000037979 autoimmune inflammatory disease Diseases 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 210000004913 chyme Anatomy 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000005056 compaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 230000011132 hemopoiesis Effects 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 229950005770 hyprolose Drugs 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 229940045773 jakafi Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 239000011812 mixed powder Substances 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 238000000643 oven drying Methods 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000000371 solid-state nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000002411 thermogravimetry Methods 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Sal de ruxolitinib con ácido oxálico.
Description
DESCRIPCIÓN
Sal oxalato de ruxolitinib
Campo de la invención
La presente invención se refiere a la sal oxalato de ruxolitinib, a formas de estado sólido de la misma, a procedimientos para la preparación de las mismas y a formulaciones de las mismas.
Antecedentes de la invención
El (R)-3-(4-(7H-pirrolo[2,3-d] pirimidín-4-il)-1H-pirazol-1-il)-3-ciclopentilpropanonitrilo presenta la fórmula química C17H18N6 y ha sido informado en el documento n° WO2007/070514 como inhibidor selectivo de las proteína-quinasas, tales como el enzima quinasa Janus en sus subtipos 1 y 2 (en lo sucesivo en la presente memoria también denominados "JAK1" y "JAK2") con valores de IC50 de 3,3 nM y 2,8 nM, respectivamente. El compuesto modula la señalización celular mediada por JAK1/JAK2, que regula varias citoquinas y factores de crecimiento que son importantes para la hematopoyesis y la función inmunitaria, y de esta manera se ha considerado que resulta útil en la terapia de diversas enfermedades, p.ej. el cáncer. El compuesto también se conoce con el nombre INN ruxolitinib y aparentemente presenta la estructura (I) a continuación:
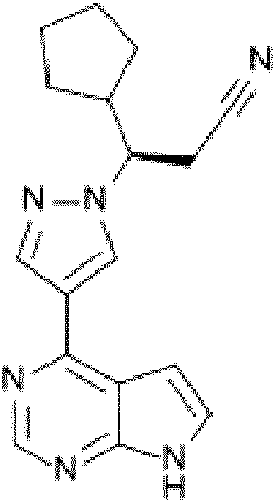
El ruxolitinib se comercializa bajo el nombre comercial Jakafi® en los EE.UU. y Jakavi® en Europa para el tratamiento de pacientes con mielofibrosis, policitemia vera, trombocitemia esencial y neoplasias malignas hematológicas. Recientemente se ha comentado que el ruxolitinib también puede resultar eficaz para el tratamiento de enfermedades inflamatorias de tipo autoinmune, tales como la soriasis.
El documento n° WO2007/070514 da a conocer la sal trifluoroacetato del ruxolitinib como un intermediario sintético. Se ha informado de sales farmacéuticamente aceptables del ruxolitinib en el documento n° WO2008/157208, es decir, fosfato de ruxolitinib, sulfato de ruxolitinib y maleato de ruxolitinib.
Una sal farmacéuticamente aceptable no sólo se define por su composición química, sino también por el denominado polimorfismo. El polimorfismo, la existencia de diferentes formas cristalinas, es una propiedad de algunas moléculas y complejos moleculares. Una única molécula, tal como el ruxolitinib o sales del mismo, puede dar lugar a una diversidad de polimorfos con diferentes estructuras cristalinas y propiedades físicas, tales como el punto de fusión, los comportamientos termales (p.ej., medido mediante análisis termogravimétrico, "TGA", o calorimetría de escaneo diferencial, "DSC", el patrón de difracción de rayos X de los polvos (PXRD), la huella digital de absorción de infrarrojos y el espectro de RMN de estado sólido. Puede utilizarse una o más de dichas técnicas para caracterizar un polimorfo particular y para distinguir las diferentes formas polimórficas de un compuesto.
Diferentes formas en estado sólido (incluyendo formas solvatadas) de un ingrediente farmacéutico activo pueden poseer diferentes propiedades. Dichas variaciones de las propiedades de diferentes formas de estado sólido y solvatos pueden proporcionar una base para la mejora de determinados aspectos del ingrediente farmacéutico activo (API, por sus siglas en inglés), tal como su formulación, por ejemplo mediante la facilitación de un mejor procesamiento o de las características de manipulación, la modificación del perfil de disolución en una dirección favorable, o la mejora de la estabilidad (polimorfos así como estabilidad química) y tiempo de almacenamiento. Dichas variaciones de las propiedades de diferentes formas de estado sólido también pueden ofrecer mejoras de la forma de dosificación final, por ejemplo, en el caso de que sirvan para mejorar la biodisponibilidad. Las diferentes formas de estado sólido y solvatos de un ingrediente farmacéutico activo también pueden dar lugar a una diversidad de polimorfos o formas cristalinas, que a su vez proporcionan oportunidades adicionales para evaluar las variaciones de las propiedades y características de un ingrediente farmacéutico activo sólido.
La identificación de nuevas formas polimórficas y solvatos de un producto farmacéutico puede proporcionar materiales que presentan, entre otros, propiedades de procesamiento deseables, tales como facilidad de manipulación, facilidad de procesamiento, estabilidad química y polimórfica con el almacenamiento y el procesamiento, y facilidad de
purificación, o resultan útiles como formas cristalinas intermediarias ventajosas que facilitan la conversión en otras formas de estado sólido (incluyendo otros solvatos) de dicho producto farmacéutico.
Las nuevas formas polimórficas y solvatos de un compuesto farmacéuticamente útil también pueden proporcionar una oportunidad para mejorar las características de rendimiento de un producto farmacéutico. Amplía el repertorio de materiales disponible para el científico de formulación para la optimización de las formulaciones, por ejemplo mediante la provisión de un producto con diferentes propiedades, p.ej., un mejor procesamiento o características de manipulabilidad, un perfil de disolución mejorado o un tiempo de almacenamiento mejorado. Finalmente, pueden prepararse nuevas formas polimórficas con fiabilidad y reproducibilidad mejorados en comparación con otras formas, por ejemplo, en términos de cristalinidad o pureza polimórfica.
El tracto intestinal presenta diferentes intervalos de pH, que varían entre pH 1 y 2 del estómago vacío, pH 3 a 5 para el estómago lleno de quimo, y pH 5 a 7,5 del colon. Puede diseñarse un régimen de ensayo de cribado de disolución para las formas de estado sólido de un ingrediente activo, por ejemplo a pH 1,2, a pH 4,5 y a pH 6,8, a fin de cubrir un intervalo amplio del tracto gastrointestinal.
Los estudios de solubilidad muestran una buena solubilidad del fosfato de ruxolitinib a pH 1,2, aunque una solubilidad considerablemente inferior en puntos de pH más alto. El maleato de ruxolitinib mostró menor solubilidad a pH 1,2 en comparación con el fosfato de ruxolitinib, y además una dependencia del pH diferente a pH 4,5 y a pH 6,8. El sulfato de ruxolitinib mostró una solubilidad moderada con menor dependencia del pH a 37°C.
Resulta favorable que las formas de estado sólido de un ingrediente activo muestren buena solubilidad a fin de encontrarse disponibles para la permeación hacia el interior del cuerpo.
Resulta adicionalmente favorable que las sales de los ingredientes activos muestren buena solubilidad en todos los intervalos de pH del tracto gastrointestinal.
El objetivo de la presente invención es encontrar una sal de ruxolitinib que proporcione una mejor solubilidad en todos los intervalos de pH en comparación con las sales de ruxolitinib del estado de la técnica, de manera que la sal se mantenga farmacéuticamente aceptable.
Descripción resumida de la invención
La presente invención proporciona una forma de estado sólido del oxalato de ruxolitinib, procedimientos para la preparación de la misma y composiciones y formulaciones farmacéuticas que comprenden la forma de estado sólido del oxalato de ruxolitinib, y procedimientos de preparación de las composiciones y formulaciones farmacéuticas. La presente invención proporciona además la utilización de dicha forma de estado sólido de oxalato de ruxolitinib para la preparación de composiciones y formulaciones farmacéuticas. De acuerdo con lo anteriormente expuesto, la presente invención proporciona además una composición farmacéutica que comprende dicha forma de estado sólido de oxalato de ruxolitinib de la presente invención. La composición farmacéutica puede comprender adicionalmente por lo menos un excipiente farmacéuticamente aceptable para formar una formulación farmacéutica que puede, por ejemplo, administrarse en pacientes que necesitan de dicho tratamiento.
La presente invención comprende un procedimiento de preparación de las formulaciones farmacéuticas anteriormente indicadas. El procedimiento comprende combinar la forma de estado sólido de oxalato de ruxolitinib con por lo menos un excipiente farmacéuticamente aceptable.
La forma de estado sólido tal como se define en la presente memoria, así como las composiciones y formulaciones farmacéuticas de oxalato de ruxolitinib pueden utilizarse como medicamentos, particularmente para el tratamiento de cáncer, mielofibrosis, policitemia vera, trombocitemia esencial y neoplasias malignas hematológicas. La presente invención proporciona además un método de tratamiento de cáncer, mielofibrosis, policitemia vera, trombocitemia esencial, neoplasias malignas hematológicas y soriasis, que comprende administrar una cantidad terapéuticamente eficaz de la forma de estado sólido de oxalato de ruxolitinib de la presente invención, o una cantidad terapéuticamente eficaz de por lo menos una de las composiciones o formulaciones farmacéuticas de la presente invención que comprenden dicha forma de estado sólido de oxalato de ruxolitinib de la presente invención en un paciente que necesita de la misma.
La presente invención proporciona además la utilización de dicha forma de estado sólido de ruxolitinib y/o sal de ruxolitinib, particularmente oxalato de ruxolitinib, o por lo menos una de las composiciones y/o formulaciones farmacéuticas anteriormente indicadas para la preparación de un medicamento destinado al tratamiento de cáncer, mielofibrosis, policitemia vera, trombocitemia esencial neoplasias malignas hematológicas y soriasis.
Breve descripción de las figuras
La figura 1 muestra una comparación de la solubilidad de oxalato de ruxolitinib, sulfato de ruxolitinib, maleato de ruxolitinib y fosfato de ruxolitinib a pH 1,2, pH 4,5 y pH 6,8.
La figura 2a muestra un patrón de difracción de rayos X de los polvos de oxalato de ruxolitinib.
La figura 2b muestra un espectro de RMN 13C del oxalato de ruxolitinib.
La figura 2c muestra un espectro de RMN 1H del oxalato de ruxolitinib.
La figura 2d muestra un termograma de DSC del oxalato de ruxolitinib.
La figura 2e muestra un espectro de IR del oxalato de ruxolitinib.
La figura 3a muestra un experimento de DVS con oxalato de ruxolitinib.
La figura 3b muestra una comparación entre los patrones de difracción de rayos X de los polvos de oxalato de ruxolitinib antes y después del experimento de DVS.
La figura 3c muestra una comparación entre los patrones de difracción de rayos X de los polvos de oxalato de ruxolitinib antes y después del experimento de estabilidad de 12 semanas a 40°C y 75% de humedad.
Descripción detallada de la invención
La presente invención proporciona una forma de estado sólido de oxalato de ruxolitinib, procedimientos para la preparación de la forma de estado sólido, así como composiciones y formulaciones farmacéuticas que comprenden dicha forma de estado sólido.
Según el documento n° WO 2008/157208, el sulfato, fosfato y maleato de ruxolitinib presentan una elevada cristalinidad y presentan buenas propiedades de manipulación. Sin embargo, el documento n° WO 2008/157208 es silencioso respecto a la solubilidad en todos los intervalos de pH del tracto gastrointestinal.
El sulfato de ruxolitinib no muestra la clara dependencia del pH de la sal fosfato (figura 1), sino que sólo presenta una solubilidad moderada.
El fosfato de ruxolitinib es la mejor sal soluble conocida hasta hoy, aunque la solubilidad se reduce a mayor pH. La proporción de solubilidad a pH 1,2 y a pH 4,5 es 7, y la proporción de pH 1,2 a pH 6,8 es 9. Puede observarse una tendencia similar para el maleato de ruxolitinib, aunque la solubilidad es más baja en todos los puntos de pH, medida con respecto al sulfato de ruxolitinib y al fosfato de ruxolitinib. La proporción de solubilidad a pH 1,2 y a pH 4,5 es 4, y la proporción de pH 1,2 a pH 6,8 también es 4.
La presente invención proporciona oxalato de ruxolitinib cristalino con una solubilidad mejorada (figura 1). La solubilidad a pH 1,2 es 1,3 veces mejor para el fosfato de ruxolitinib, y mejor en un factor de 5 a pH 4,5 y a pH 6,8. Sin embargo, la proporción de solubilidades a pH 1,2:4,5, así como a pH 1,2:6,8 es 2 para ambas proporciones para el oxalato de ruxolitinib, por lo tanto una proporción considerablemente mejor que para el fosfato de ruxolitinib con respecto al perfil de solubilidad dependiente del pH, así como las solubilidades globales.
Además, dependiendo de con que otra forma de estado sólido se compare, la forma oxalato de ruxolitinib de la presente invención puede presentar propiedades ventajosas seleccionadas de por lo menos una de entre: pureza química o polimórfica, cristalinidad incrementada, fluidez, solubilidad, tasa de disolución, biodisponibilidad, morfología o hábito cristalino, superficie específica y densidad picnométrica, densidad aparente/de compactación, estabilidad (tal como la estabilidad química, así como la estabilidad térmica y mecánica con respecto a la conversión polimórfica), la estabilidad frente a la deshidratación y/o la estabilidad de almacenamiento, un grado menor de higroscopicidad, un contenido bajo de solventes residuales y un procesamiento ventajoso y características de manipulación, tales como la compresibilidad y la densidad aparente.
Las formas de estado sólido del oxalato de ruxolitinib comprenden formas de cristal, o formas cristalinas, o formas amorfas de oxalato de ruxolitinib. Tal como se utiliza en la presente memoria, las formas de estado sólido, las formas de cristal, las formas cristalinas, los polimorfos y las formas polimórficas se utilizan intercambiablemente.
En la presente memoria puede hacerse referencia a una forma de cristal como caracterizada por datos gráficos "sustancialmente tal como se ilustra" en una figura. Dichos datos incluyen, por ejemplo, difractogramas de rayos X de polvos, espectros de infrarrojos y termogramas de DSC. Los datos gráficos proporcionan potencialmente información técnica adicional para definir adicionalmente la forma de estado sólido respectiva que no puede describirse necesariamente, o fácilmente, en referencia a valores numéricos de las posiciones de pico y/o a intensidades relativas. En cualquier caso, el experto en la materia entenderá que dichas representaciones gráficas de los datos pueden estar sujetas a pequeñas variaciones, p.ej., de las intensidades relativas de los picos y las posiciones de los picos, debido a factores tales como variaciones en la respuesta del instrumento y variaciones en la concentración y pureza de la muestra, las cuales son bien conocidas por el experto en la materia. Sin embargo, el experto en la materia será fácilmente capaz de comparar los datos gráficos en las figuras en la presente memoria con datos gráficos generados para una forma de estado sólida desconocida y confirmar si los dos conjuntos de datos gráficos están caracterizando la misma forma de estado sólido o dos formas de estado sólido diferentes.
En la presente memoria puede hacerse referencia a una forma de estado sólido (o polimorfo) como sustancialmente libre de cualesquiera otras formas cristalinas (o polimórficas). Tal como se utiliza en la presente memoria en el presente
contexto, la expresión "sustancialmente libre de cualesquiera otras formas" se entenderá que se refiere a que la forma de estado sólido contiene 20% o menos, 10% o menos, 5% o menos, 2% o menos, o 1% o menos de cualesquiera otras formas del compuesto de la invención según las mediciones mediante, por ejemplo, PXRD. De esta manera, las formas de estado sólido del oxalato de ruxolitinib indicadas en la presente memoria como sustancialmente libres de cualesquiera otras formas polimórficas se entenderá que contienen más de 80% (p/p), más de 90% (p/p), más de 95% (p/p), más de 98% (p/p), o más de 99% (p/p) de la forma de estado sólido de la invención de oxalato de ruxolitinib. De acuerdo con lo anteriormente expuesto, en algunas realizaciones de la invención, las formas de estado sólido indicadas de oxalato de ruxolitinib pueden contener de 1% a 20% (p/p), de 5% a 20% (p/p), o de 5% a 10% (p/p) de otra u otras formas de estado sólido de ruxolitinib o sales de las mismas.
Tal como se utiliza en la presente memoria, la expresión "temperatura ambiente" o "TA" se refiere a una temperatura de entre aproximadamente 20°C y aproximadamente 30°C. Habitualmente, la temperatura ambiente está comprendida entre aproximadamente 20°C y aproximadamente 25°C.
Tal como se utiliza en la presente memoria, la expresión "durante la noche" se refiere a un periodo de entre aproximadamente 15 y aproximadamente 20 horas, típicamente entre aproximadamente 16 y aproximadamente 20 horas.
Tal como se utiliza en la presente memoria, la expresión "presión reducida" se refiere a una presión de entre aproximadamente 10 mbar y aproximadamente 50 mbar.
Tal como se utiliza en la presente memoria, el término "aislado" corresponde a un producto o forma de estado sólido que está físicamente separado de la mezcla de reacción en la que se forma.
Tal como se utiliza en la presente memoria, a menos que se indique lo contrario, Los picos de XRPD informados en la presente memoria preferentemente se miden mediante la utilización de radiación de CuK, A=1,5418 A.
Tal como se utiliza en la presente memoria, la expresión "forma cristalina seca" se refiere a un polimorfo que se seca mediante la utilización de cualesquiera técnicas convencionales a fin de eliminar el solvente residual. Entre dichas técnicas convencionales se incluyen, aunque sin limitarse a ellas, la evaporación, el secado al vacío, el secado en un horno, el secado bajo flujo de nitrógeno, etc.
Tal como se utiliza en la presente memoria, y a menos que se indique lo contrario, el término "anhidro" en relación al oxalato de ruxolitinib cristalino se refiere a un oxalato de ruxolitinib cristalino que contiene no más de 1% (p/p) de agua o solventes orgánicos según mediciones mediante métodos convencionales, por ejemplo TGA, GC o KF. Una forma anhidra de los estados sólidos de oxalato de ruxolitinib de la presente invención se refiere a una forma que no contiene agua cristalina (u otros solventes) en una cantidad estequiométrica definida en el cristal.
Preferentemente, el oxalato de ruxolitinib es un mono-oxalato de ruxolitinib.
Tal como se utiliza en la presente memoria, y a menos que se indique lo contrario, la expresión "estabilidad polimórfica" en relación a las formas cristalinas de oxalato de ruxolitinib se refiere a que la conversión es inferior a 20%, 10%, 5%, 1%, 0,5% o 0,1% (p/p) del oxalato de ruxolitinib cristalino en cualquier otra forma de estado sólido de oxalato de ruxolitinib bajo las condiciones especificadas.
La presente invención comprende una forma cristalina de oxalato de ruxolitinib.
Preferentemente, el oxalato de ruxolitinib cristalino es mono-oxalato de ruxolitinib cristalino.
El oxalato de ruxolitinib puede caracterizarse por datos seleccionados de entre uno o más de los siguientes: un patrón de difracción de rayos X de los polvos que presenta picos en 5,2, 18,2, 21,0, 22,5 y 25,0 grados dos theta ± 0,2 grados dos theta; puede caracterizarse adicionalmente por el patrón de difracción de rayos X de los polvos con picos en 5,2, 18,2, 21,0, 22,5 y 25,0 grados dos theta ± 0,2 grados dos theta y que también presenta uno, dos, tres, cuatro, cinco o seis picos adicionales seleccionados de entre 9,0, 9,6, 13,8, 15,7, 17,7 y 22,9 grados dos theta ± 2,2 grados dos theta, y combinaciones de dichos datos; un patrón de difracción de rayos X de los polvos sustancialmente tal como se ilustra en la figura 2a.
El oxalato de ruxolitinib puede caracterizarse adicionalmente como uno o más de los siguientes: un termograma de DSC sustancialmente tal como se ilustra en la figura 2d: por dos endotermas agudas con temperaturas iniciales en 170,2 ± 2°C y 346,4 ± 2°C y una exoterma aguda con temperatura inicial de 317,6 ± 2°C, así como dos señales anchas en 186,5 ± 2°C y 265,0 ± 2°C, respectivamente.
El oxalato de ruxolitinib puede caracterizarse mediante cualesquiera combinaciones de los datos anteriormente proporcionados. Por ejemplo, por un patrón de difracción de rayos X de los polvos con picos en los grados 5,2, 18,2, 21,0, 22,5 y 25,0 dos theta ± 0,2 grados dos theta y también por un termograma de DSC sustancialmente tal como se ilustra en la figura 2d.
El oxalato de ruxolitinib anterior presenta propiedades ventajosas seleccionadas de por lo menos uno de entre: pureza química o polimórfica, fluidez, solubilidad, tasa de disolución, biodisponibilidad, morfología o hábito cristalino (tal como estabilidad química, así como estabilidad térmica y mecánica con respecto a la conversión polimórfica), estabilidad de almacenamiento, estabilidad frente a la deshidratación, baja higroscopicidad y bajo contenido de solventes residuales y procesamiento y características de manipulación ventajosas, tales como compresibilidad o densidad aparente. Particularmente, el oxalato de ruxolitinib cristalino presenta una elevada pureza química y excelentes propiedades de estabilidad. Específicamente, es estable tras el almacenamiento a 25°C y 60% de humedad relativa (HR) y a 40°C/75% de HR durante por lo menos 12 semanas. Además, el oxalato de ruxolitinib cristalino presenta una buena solubilidad y puede utilizarse para preparar una formulación oral, es decir, una tableta o una cápsula.
El oxalato de ruxolitinib en forma de estado sólido indicada puede utilizarse para preparar base de ruxolitinib u otras sales diferentes de ruxolitinib, así como formas de estado sólido de las mismas y/o formulaciones farmacéuticas que comprenden una o más de las sales y/o formas de estado sólido de las mismas.
La presente invención comprende además: 1) una composición farmacéutica que comprende dicha forma de estado sólido indicada en la presente memoria; 2) una formulación farmacéutica que comprende dichas formas de estado sólido o composiciones farmacéuticas indicadas en la presente memoria, y por lo menos un excipiente farmacéuticamente aceptable, 3) un procedimiento para preparar dichas formulaciones, que comprende combinar las formas de estado sólido anteriormente indicadas y por lo menos un excipiente farmacéuticamente aceptable, 4) la utilización de la forma de estado sólido anteriormente indicada en la preparación de una composición farmacéutica, y 5) un método de tratamiento de cáncer, que comprende administrar una cantidad terapéuticamente eficaz de las formas de estado sólido anteriormente indicadas, opcionalmente en forma de composiciones o formulaciones farmacéuticas. La presente invención proporciona además una forma cristalina de oxalato de ruxolitinib tal como se ha indicado anteriormente, para la utilización como medicamento, preferentemente para el tratamiento del cáncer. Las composiciones farmacéuticas también pueden utilizarse para preparar dicho medicamento.
Tras describir la invención en referencia a determinadas realizaciones preferentes, otras realizaciones resultarán evidentes para el experto en la materia a partir de la consideración de la especificación. La invención se define adicionalmente en referencia a los ejemplos, posteriormente, que describen en detalle la preparación de la composición y métodos de utilización de la invención. Resultará evidente para el experto en la materia que pueden ponerse en práctica muchas modificaciones, tanto de materiales como de los métodos, sin apartarse del alcance de la invención.
Métodos analíticos
Espectroscopía de RMN
Instrumento: espectrómetro de RMN Varian Mercury 400 Plus, Oxford AS, 400 MHz. Las muestras se midieron en DMSO-cfó a temperatura ambiente
Espectroscopía de IR
Aparato: FT-IR Thermo Nicolet, Avatar 330 con ATR.
Calorimetría diferencial de barrido (CDB)
Instrumento: Mettler Toledo DSC 822E acoplado con un controlador del flujo de gases Mettler Toledo TS0800GC1 (Mettler-Toledo GmbH, GielJen, Alemania)
Crisol de aluminio: 40 |jl
Tapa: perforada
Intervalo de 30°C a 350°C
temperaturas:
Tasa de calentamiento: 10°C/min
Enjuague de nitrógeno: 50 ml/min
Software: STARe, versión. 8.10
Interpretación: modo endotérmico
Difracción de rayos X de los polvos (PXRD)
La muestra se analizó en un difractómetro de rayos X de polvos D8 Advance (Bruker-AXS, Karlsruhe, Alemania). El soporte de muestras se hizo girar en un plano paralelo a su superficie a 20 rpm durante las mediciones. Se resumen condiciones adicionales para las mediciones en la tabla, posteriormente. Los datos en bruto se analizaron con el programa EVA (Bruker-AXS, Alemania). Las muestras se aplicaron en capas sobre un soporte de especímenes de silicio.
mediciones estándares
radiación de Cu Ka (A=1,5418 A)
fuente 38 kV / 40 mA
detector Vantec
rendija de detección variable
rendija de divergencia v6
rendija antidispersión v6
intervalo 20 / ° 2 < 20 < 55
Tamaño de paso / ° 0,017____________
Higroscopicidad
Se llevaron a cabo experimentos de sorción de vapor en el instrumento SPSx-1 p (Projekt Messtechnik, Ulm, Alemania) a una temperatura de 25°C y los cilos de humedad mostrados posteriormente.

Ejemplos
Ejemplo 1: preparación de base ruxolitinib
Se preparó base de ruxolitinib en cuatro etapas. La ruta de síntesis se proporciona en el esquema siguiente. Las tres etapas finales se describen en detalle.

Intermediario 2: (rac-3-ciclopentil-3-{4-[7-(2-trimetilsilanil-etoximetM)-7H-pirrolo[2,3-d]pirimidín-4-M]-pirazol-1-il}-propionitrilo)
Se añadieron 231,67 g (1,90 moles) de 3-ciclopentil-acrilonitrio y 23,70 ml (0,16 moles) de 1,8-diazabiciclo[5.4.0]undec-7-en (DBU) a una suspensión del material de partida (1) (500 g, 1,56 mole) en 4000 ml de acetonitrilo (MeCN) a temperatura ambiente bajo agitación y en una atmósfera de nitrógeno. La mezcla de reacción se sometió a calentamiento a 60°C durante 22 horas y después a 80°C durante 4 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente. La mezcla de reacción enfriada se evaporó bajo presión reducida a 42°C, rindiendo un aceite negro viscoso. El aceite se disolvió en 1300 ml de diclorometano (DCM) y se añadieron 650 g de sílice (63 a 200 pm). La suspensión se evaporó bajo presión reducida a 44°C. El producto en bruto obtenido en sílice se cargó en seco en una columna de sílice (1,25 kg (2,51) sílice 63 a 200 pm, diámetro de la columna: 7 cm; longitud: 92 cm). Toda la separación se llevó a cabo mediante un sistema flash ("combi flash" acompañante) con un flujo de solvente de 100 ml/min (presión de 5 a 15 bar). Se llevó a cabo la elución con EtOAc (acetato de etilo)/n-hexano 1/2. Se evaporó el solvente a 50°C, rindiendo el producto en forma de aceite amarillo altamente viscoso.
Intermediario 3 ((R)-3-ciclopentil-3-{4-[7-(2-trimetilsilanil-etoximetil)-7H-pirrolo[2,3-d]pirimidín-4-il]-pirazol-1-il}-propionitrilo)
El material de partida racémico (1159 g, 2, 7 moles) se disolvió en 2.000 ml de MeCN a 50°C. La solución obtenida se transfirió a un reactor de 25 l. Se añadieron 9560 ml de MeCN, 1960 ml de THF y 1960 ml de acetona, dando como resultado una solución amarilla. La camisa calefactora se ajustó a 55°C (Ti=51°C). Se añadieron 2137 g (2,14 eq.) de monohidrato de ácido (+)-dibenzoil-tartárico, dando como resultado una solución ligeramente naranja. Tras 30 minutos, la solución se volvió turbia y se ajustó la camisa de calentamiento a 80°C; tras 25 minutos resultó una solución naranja transparente y la camisa de calentamiento se ajustó a 55°C. A 56°C (Ti), se añadieron cristales puros enantioméricos para iniciar la cristalización y se ajustó la camisa de calentamiento a 50°C. Tras 1,5 h, se desactivó el calentamiento y se dejó que la mezcla se enfriase hasta la temperatura ambiente y se sometió a agitación durante la noche (Ti después de 1 h 45 min, a 37°C; temperatura final durante la noche: 22°C). Se separó el precipitado mediante filtración y se lavó con 2xd100 ml de una mezcla de MeCN:THF:acetona (5.8:1:1), rindiendo 1197 g del tartrato (85% del teórico, 52,4% ee).
Se agrupó lo anterior con otro lote con una pureza enantiomérica de 52,0%.
Se cargaron 2000 g de los lotes agrupados en un reactor y se añadieron14,48 l de una mezcla de MeCN, THF y acetona (10:1,69:1,69). El reactor se calentó a 80°C (Tj) (Ti=71°C); el producto se disolvió prácticamente por completo. Además, se añadieron 200 ml de la mezcla de solventes anteriormente indicada, resultando en una solución transparente. El reactor se enfrió a 52°C (tasa de enfriamiento: tras 1,5 h, 52°C). Se formó una suspensión incolora tras -1,5 h. Tras -3,5 h, se desactivó el calentamiento y la suspensión se sometió a agitación durante la noche (Ti final=20°C). Se separó el producto mediante filtración y se lavó con 2x600 ml de la mezcla de solventes anteriormente indicada. El producto obtenido mostró una pureza enantiomérica de 80,2% ee y un rendimiento de 1528 g (76% del teórico).
Se cargaron 1903 g del material cristalizado con una pureza óptica de 80% ee en un reactor y se añadieron 13,38 l de una mezcla de MeCN/THF y acetona (10:1,69:1,69). La mezcla se calentó a 80°C (Tj) (Ti=72,7°C) tras la adición de 200 ml adicionales de la mezcla de solventes anteriormente indicada; la suspensión se volvió una solución transparente. Se ajustó la temperatura a 55°C y tras 2,5 h a 52,8°C, se formó una suspensión incolora; se desactivó el calentamiento y se continuó con la agitación durante la noche. Se separó mediante filtración el producto precipitado (T=21°C) (1356 g, 92,0% ee).
Se cargaron 1356 g de dicho material en un reactor y se añadieron 11,99 l de una mezcla de MeCN/THF y acetona (10:1,69:1,69). La mezcla se calentó a 80°C (Tj) (Ti=73,0°C) (solución transparente). Se ajustó la temperatura a 55°C y tras 2,5 h a 52,8°C, se formó una suspensión incolora. Se ajustó la temperatura a 50°C. Tras 2 horas, se desactivó el calentamiento y se continuó con la agitación durante la noche. Se separó el producto mediante filtración (Ti=20,3°C) y se lavó con 2x600 ml de MeCN/THF/acetona (10:1,69:1,69), rindiendo 1063 g del producto con una pureza enantiomérica de 96,6% ee.
Se cargaron 1063 g de dicho material en un reactor y se añadieron 9390 ml de una mezcla de MeCN/THF/acetona (10:1,69:1,69). La mezcla se calentó a 80°C (Tj) (Ti=73,0°C) (solución transparente). Se ajustó la temperatura a 55°C y tras 1 h a 52,8°C, se formó una suspensión incolora. Se ajustó la temperatura a 50°C. Tras 2 horas, se desactivó el calentamiento y se continuó con la agitación durante la noche. Se separó el producto mediante filtración (Ti=20,3°C) y se lavó con 2x600 ml de MeCN/THF/acetona (10:1,69:1,69), rindiendo 813 g del producto con una pureza enantiomérica de 98,6% ee.
Procedimiento para liberar la base libre:
A 100 g de la sal de ácido tartárico se añadieron 2000 ml de acetato de etilo (suspensión blanca) y después 600 ml de agua. La mezcla se enfrió a 0°C y se añadió NaOH 2 N. Se extrajo la capa acuosa con 2x 400 ml de acetato de etilo. Se agruparon las capas orgánicas y se lavaron con NaOH 0,01 M (700 ml). Se evaporó la capa orgánica, rindiendo 54,5 g del producto en forma de aceite amarillo. Pureza química: 99,45%; HPLC/UV (A= 266 nm).
Intermediario 4: ((R)-3-ciclopentil-3-[4-(7H-pirrolo[2,3-d]pirimidín-4-M)-pirazol-1-il]-propionitrilo)
El material de partida (244 g, 0,559 moles) se disolvió en 2370 ml de MeCN, seguido de la adición de 210 ml de agua y tetrafluoroborato de litio (LiBF4, 520,48 g; 5,55 moles) a temperatura ambiente (ta). La temperatura de reacción se redujo mediante la adición de agua a 14°C (Ti=22°C) y se incrementó mediante la adición de LiBF4 a 27°C. La mezcla de reacción se calentó bajo reflujo (~80°C) durante 11 h. La reacción se monitorizó mediante HPLC. Tras consumir por completo el material de partida, la mezcla de reacción se enfrió a 5°C, seguido de la adición gota a gota de hidróxido amónico acuoso al 20% (274 ml) para ajustar el pH a 9. Se retiró el baño de hielo y la mezcla de reacción se calentó gradualmente hasta la t.a. Tras completar la desprotección, que se monitorizó mediante HPLC/CCF, la mezcla de reacción se filtró y se lavaron los sólidos con MeCN (530 ml). Se evaporaron los filtrados agrupados bajo presión reducida a 46°C y se añadieron al residuo 3160 ml de EtOAc y 1580 ml de solución hipersalina semisaturada. Se separaron las dos capas y se extrajo la capa acuosa con EtOAc (1050 ml). Las capas orgánicas agrupadas se lavaron con NaHCO3 semisaturado (1600 ml) y solución hipersalina (1600 ml); se secaron sobre Na2SO4 y se concentraron bajo presión reducida, rindiendo el producto en bruto en forma de aceite naranja. El producto en bruto se purificó mediante cromatografía flash en sílice (eluyente: EtOAc). Procedimiento de purificación: el producto en bruto se disolvió en 250 ml de acetato de etilo y se dividió en dos porciones prácticamente iguales; cada porción se purificó por separado mediante una columna de sílice (columna puriflash preempaquetada/puriflash/1600 g/50 pm) en un sistema flash Armen spot, flujo: 80 ml/min. El aceite incoloro obtenido se evaporó, resultando en una espuma, que se trituró con 200 ml de n-pentano (cada porción), resultando en un sólido incoloro que se separó mediante filtración y se secó al vacío a ta. El producto obtenido se almacenó bajo una atmósfera de argón en la nevera.
Ejemplo 2: preparación de oxalato de ruxolitinib.

A una solución de base libre de ruxolitinib (3 g, 9,8 mmoles) en isopropanol (42 ml) a 60°C se añadió ácido oxálico (0,97 g, 10,8 mmoles) en isopropanol (8,4 ml). Inmediatamente después de la adición, el producto empieza a precipitar en forma de sólido voluminoso blanco. Además, se añadieron 20 ml de isopropanol y la mezcla de reacción se sometió a agitación adicional (a 60°C) durante 1,5 horas. A continuación, la mezcla de reacción se enfrió hasta la temperatura ambiente y se sometió a agitación durante la noche. Se separó el producto precipitado mediante filtración y se secó en un horno de vacío a 50°C durante 3 días, rindiendo 3,38 g (87% del teórico) del producto en forma de sólido cristalino incoloro.
Alternativamente:
se disolvieron 100 g (327,2 mmoles) de base ruxolitinib (lote n° RS352) en 2 l de isopropanol a 56°C (temperatura dentro del matraz). Se disolvieron 20,62 g (229,1 mmoles, 0,7 equiv.9 de ácido oxálico por separado en 280 ml de isopropanol. Tras la disolución completa del ruxolitinib, se introdujo el ácido oxálico en dicha solución a 56°C en un periodo de 45 minutos. La solución se tornó ligeramente turbia y tras la adición de aprox. 100 ml (-30 minutos), se formó una suspensión espesa blanca. Tras completar la adición del ácido oxálico, la mezcla de reacción se sometió a agitación durante 60 minutos adicionales a dicha temperatura. Se desactivó la fuente de calentamiento y el matraz se enfrió hasta la ta (el matraz se dejó en el baño de aceite y se enfrió hasta la t.a. con el baño de aceite de enfriamiento). Se separó el producto mediante filtración, se lavó con 250 ml de isopropanol y se secó a 50°C bajo vacío. Rendimiento: 67,8 g (52,3%).
RMN 1H (400 MHz, DMSO-d6) 8 ppm: 1,12 - 1,37 (m, 3 H) 1,38 - 1,64 (m, 4 H) 1,69 - 1,89 (m, 1 H) 2,35 - 2,47 (m, 1 H) 3,12 - 3,29 (m, 2 H) 4,52 (m, 1 H) 6,88 - 7,07 (m, 1 H) 6,95 - 7,00 (m, 1 H) 7,56 - 7,61 (m, 1 H) 8,35 (s, 1 H) 8,67 (s, 1 H) 8,78 (s, 1 H) 12,10 (br. s., 1 H).
RMN 13C (400 MHz, DMSO-d6) 8 ppm: 23,0, 24,8, 25,4, 29,5, 44,8, 63,0, 100,3, 113,2, 118,6, 120,7, 127,3, 131,5, 139,7, 150,1, 151,1, 152,5, 161,5.
IR (ATR) [cm-1] ± 2 cm'1: 3115, 2953, 2870, 2816, 2253, 1718, 1618, 1591, 1556, 1450, 1439, 1427, 1387, 1342, 1259, 1192, 1113, 1022, 895, 814, 737, 590.
HPLC/UV a A= y 230 nm: > 99,9 %
Ejemplo 3: experimento de sorción dinámica de vapor con oxalato de ruxolitinib
Se llevó a cabo un experimento de sorción dinámica de vapor con el oxalato de ruxolitinib. A una temperatura constante de 25°C, la sustancia se expuso a dos ciclos de humedad relativa (hr) variable (40% ^ 0% ^-95% (75% de hr mantenida constante durante 48 h) ^ 0% ^40% ). La figura 3a presenta el cambio relativo de peso como función del tiempo y la humedad relativa, y en la figura 3a, se ilustran los cambios de peso como función de la humedad.
Para el oxalato de ruxolitinib, se observó un cambio relativo de peso de 1% (al final de la etapa de 75% de hr) y de 1,6% (al final de la etapa de 95% de hr). La adsorción y desorción del agua estaban directamente relacionadas con la humedad, lo que sugiere que el agua estaba reversiblemente unida a la superficie de la muestra. Dicha premisa se ve apoyada por el resultado del análisis de XRPD de la muestra, realizada después del experimento de DVS. La superposición de difractogramas proporcionada en la figura 3b demuestra que el estado sólido del oxalato de ruxolitinib era idéntico al de antes del experimento de DVS.
Tabla 1. Cambio de peso relativo como función del tiempo y la humedad relativa

Ejemplo 4: estabilidad.
Las muestras se almacenaron en recipientes cerrados y abiertos a 25°C/60% de hr y a 40°C/75% de rh y se analizaron tras 4, 8 y 12 semanas.
Se resumen los resultados de la determinación de la pureza química durante el almacenamiento a 40°C/75% de hr. Ambas sales se mantuvieron químicamente estables al almacenarlas bajo dichas condiciones. Por lo tanto, se omitió el análisis de las muestras, que se almacenaron a 25°C/60% de hr.

Se investigó la estabilidad en estado sólido mediante análisis de XRPD. Se resumen los resultados de la evaluación. El fosfato de ruxolitinib y el oxalato de ruxolitinib se mantuvieron sin cambios durante todo el periodo de observación.

Ejemplo 5: dependencia del pH de la solubilidad (mg/ml) a 37°C

(continuación)

Ejemplo 6. Formulaciones
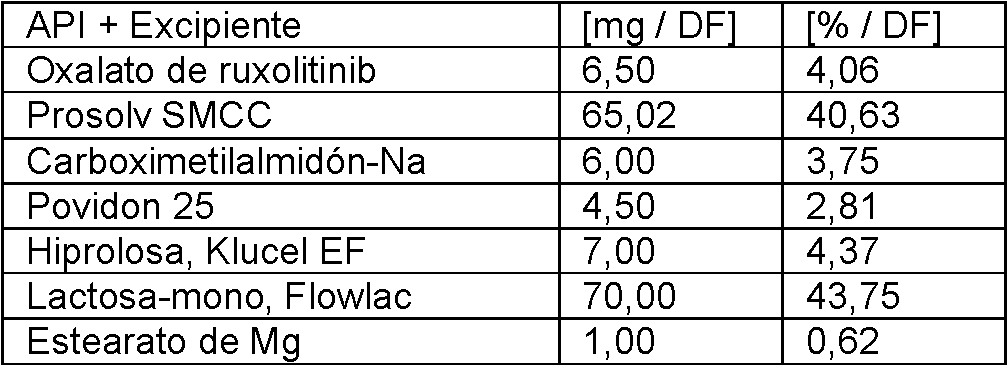
Compresión directa
Las tabletas se prensaron mediante una prensa de punzón único (Korsch EKO) con un diámetro de punzón de 8 mm, redonda.
Las tabletas siguientes se fabricaron mediante compresión directa.
Granulación húmeda
Las tabletas siguientes se fabricaron mediante granulación húmeda.

Procedimiento
• API, Prosolv, lactosa y hiprolosa se mezclaron en un mezclador de alta cizalla durante 15 minutos
• Se llevó a cabo granulación húmeda con una solución de Povidon en agua
• La mezcla se tamizó por un tamiz de 1 mm.
• Los polvos se secaron a 40°C durante 1,5 horas.
• Se añadió carboximetil-almidón y estearato de magnesio y se mezclaron durante 5 minutos adicionales.
• La compresión se llevó a cabo mediante una prensa excéntrica, Korsch EKO.
• Fuerza de compresión: 6 Kn
• Dureza de tableta: 57 N
• Diámetro de tableta: 8 mm
• Peso de la diana: 160,16 mg

Procedimiento
• El oxalato de ruxolitinib se molió en un molino de bolas durante 15 minutos a 250 rpm y se tamizó por un tamiz de 250 pm.
• El API molido, Prosolv SMCC, hiprolosa y Granulac se mezclaron en un mezclador de alta cizalla durante 15 minutos.
• S e disolvió povidona en agua a fin de obtener el líquido de granulación.
• Los polvos mezclados se granularon con líquido de granulación y se tamizaron por un tamiz de 1 mm. • Los gránulos se secaron en una cámara de secado a 40°C durante 1 hora.
• Se añadió carboximetilil-almidón y estearato de magnesio y se mezclaron durante 5 minutos adicionales.
• Las tabletas se comprimieron en una prensa de punzón único Korsch, EKO.
• Diámetro de tableta: 8 mm
• Fuerza de compresión: 7 kN
• Dureza de tableta: 47 N
• Peso de la diana: 160,0 mg.
Claims (12)
1. Sal de ruxolitinib con ácido oxálico.
2. Sal de ruxolitinib según la reivindicación 1, caracterizada por un patrón de difracción de rayos X de los polvos con picos en 5,2, 18,2, 21,0, 22,5 y 25,0 ± 0,2 grados dos theta.
3. Sal de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 2, que se caracteriza además por uno, dos, tres, cuatro, cinco o seis picos de PXRD adicionales, seleccionados de entre 9,0, 9,6, 13,8, 15,7, 17,7 y 22,9 grados dos theta ± 0,2 grados dos theta.
4. Sal de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 3, que se caracteriza además por uno o más de los siguientes: dos endotermas agudas con temperaturas iniciales en 170,2 ± 2°C y 346,4 ± 2°C, una exoterma aguda con una temperatura inicial de 317,6 ± 2°C, así como dos señales anchas en 186,5 ± 2°C y 265,0 ± 2°C, respectivamente.
5. Sal de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 4, en la que la forma salina es un monooxalato.
6. Composición farmacéutica que comprende el oxalato según cualquiera de las reivindicaciones anteriores 1 a 5.
7. Formulación farmacéutica que comprende oxalato de ruxolitinib según cualquiera de las reivindicaciones 1 a 5 y por lo menos un excipiente farmacéuticamente aceptable.
8. Utilización de oxalato de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 5 en la preparación de una composición o formulación farmacéutica.
9. Procedimiento para la preparación de la formulación farmacéutica según la reivindicación 7, que comprende oxalato de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 5 y por lo menos un excipiente farmacéuticamente aceptable.
10. Oxalato de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 5, la composición farmacéutica según la reivindicación 6 o la formulación farmacéutica según la reivindicación 7, para la utilización como medicamento.
11. Oxalato de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 5, la composición farmacéutica según la reivindicación 6 o la formulación farmacéutica según la reivindicación 7, para la utilización en el tratamiento de un sujeto que sufre de cáncer, mielofibrosis, polictemia vera, trombocitemia esencial, neoplasias malignas hematológicas y soriasis.
12. Utilización de oxalato de ruxolitinib según cualquiera de las reivindicaciones anteriores 1 a 5 en la preparación de: ruxolitinib, otras sales de ruxolitinib, formas de estado sólido de ruxolitinib u otras sales de ruxolitinib, o formulaciones farmacéuticas de: ruxolitinib, otras sales de ruxolitinib, formas de estado sólido de ruxolitinib u otras sales de ruxolitinib.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14181843 | 2014-08-21 | ||
| PCT/EP2015/069296 WO2016026974A1 (en) | 2014-08-21 | 2015-08-21 | Oxalate salt of ruxolitinib |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2874537T3 true ES2874537T3 (es) | 2021-11-05 |
Family
ID=51383641
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15753374T Active ES2874537T3 (es) | 2014-08-21 | 2015-08-21 | Sal oxalato de ruxolitinib |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP3183252B1 (es) |
| ES (1) | ES2874537T3 (es) |
| WO (1) | WO2016026974A1 (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PE20231743A1 (es) | 2020-08-18 | 2023-10-31 | Incyte Corp | Proceso e intermediarios para preparar un inhibidor de jak |
| TW202212341A (zh) | 2020-08-18 | 2022-04-01 | 美商英塞特公司 | 製備jak1抑制劑之方法及中間體 |
| WO2023288197A1 (en) | 2021-07-12 | 2023-01-19 | Incyte Corporation | Process and intermediates for preparing baricitinib |
| WO2023121574A1 (en) * | 2021-12-23 | 2023-06-29 | Deva Holding Anonim Sirketi | Novel polymorph of ruxolitinib hemifumarate and method of preparation |
| WO2024028193A1 (en) | 2022-08-03 | 2024-02-08 | Medichem, S.A. | Stable oral pharmaceutical formulation containing ruxolitinib hemifumarate |
| WO2024172778A1 (en) * | 2023-02-16 | 2024-08-22 | Deva Holding | Novel polymorph of ruxolitinib hemifumarate and method of preparation |
| WO2024187415A1 (en) | 2023-03-15 | 2024-09-19 | Zhejiang Qizheng Pharmaceutical Co., Ltd. | Pharmaceutical composition comprising ruxolitinib |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA98449C2 (en) * | 2005-12-13 | 2012-05-25 | Инсайт Корпорейшин | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| RS53245B2 (sr) * | 2007-06-13 | 2022-10-31 | Incyte Holdings Corp | Soli inhibitora janus kinaze (r)-3-(4-(7h-pirolo(2,3-d) pirimidin-4-il)-1h-pirazol-1-il)-3-ciklopentilpropan-nitrila |
| AU2012271814A1 (en) * | 2011-06-14 | 2013-12-12 | Novartis Ag | Combination of panobinostat and ruxolitinib in the treatment of cancer such as a myeloproliferative neoplasm |
-
2015
- 2015-08-21 ES ES15753374T patent/ES2874537T3/es active Active
- 2015-08-21 WO PCT/EP2015/069296 patent/WO2016026974A1/en active Application Filing
- 2015-08-21 EP EP15753374.6A patent/EP3183252B1/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3183252A1 (en) | 2017-06-28 |
| EP3183252B1 (en) | 2021-05-12 |
| WO2016026974A1 (en) | 2016-02-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2874537T3 (es) | Sal oxalato de ruxolitinib | |
| TWI833686B (zh) | Lsd1 抑制劑之調配物 | |
| EP3183253B1 (en) | Salt of (r)-3-(4-(7h-pyrrolo [2,3-d]pyrimidin-4-yl)-lh-pyrazol-l-yl)-3-cyclopentylpropanenitrile with benzenesulfonic acid | |
| ES2562462T3 (es) | Formas cristalinas de 3-(2,6-dicloro-3,5-dimetoxifenil)-1-{6-[4-(4-etilpiperazin-1-il)-fenilamino]-pirimidin-4-il}-1-metilurea y sus sales | |
| JP5462168B2 (ja) | N−(5−tert−ブチル−イソオキサゾール−3−イル)−N’−{4−[7−(2−モルホリン−4−イル−エトキシ)イミダゾ[2,1−b][1,3]ベンゾチアゾール−2−イル]フェニル}ウレアを含む固形、その組成物、及びその用途 | |
| ES2761903T3 (es) | Derivados de carbazol | |
| ES2586124T3 (es) | Polimorfo de rifaximina y proceso para la preparación del mismo | |
| ES2277301T3 (es) | Solvato de hemitartrato de zolpidem. | |
| EP3205653B1 (en) | Crystal form of bisulfate of jak inhibitor and preparation method therefor | |
| CA3173755A1 (en) | Integrin inhibitor and uses thereof | |
| WO2016079313A1 (en) | Crystalline forms of afatinib dimaleate | |
| ES2814499T3 (es) | Formas en estado sólido de sales de Nilotinib | |
| ES2956847T3 (es) | Método mejorado para la fabricación de 3-[(1s)-1-imidazo[1,2-a]piridin-6-iletil]-5-(1-metilpirazol-4-il)triazolo[4,5-b]pirazina y formas polimórficas de la misma | |
| KR20220066058A (ko) | 피루베이트 키나제 r(pkr) 활성화 조성물 | |
| EP2970245A1 (en) | Polymorphs and salts of a compound | |
| US20080200474A1 (en) | Novel flunarizine salt forms and methods of making and using the same | |
| ES2948919T3 (es) | Forma cristalina de derivado de aminopirano sustituido | |
| CN114075199A (zh) | Jak抑制剂化合物及其用途 | |
| CN114401720A (zh) | 多酪氨酸激酶抑制剂的晶型、制备方法及其用途 | |
| WO2018220252A1 (es) | Derivados de piridoquinazolina eficaces como inhibidores de proteína quinasa | |
| WO2012003413A1 (en) | Novel solid forms of tacedinaline | |
| AU2018436086B2 (en) | Process for manufacture of (S)-N-(3-((2-((4-((1-acetylpyrrolidin-3-yl)(methyl)amino)phenyl)amino)-5-methoxypyrimidin-4-yl)oxy) phenyl)acrylamide, and formulations thereof | |
| WO2023204303A1 (ja) | 7H-ピロロ[2,3-d]ピリミジン-4-アミン誘導体の結晶 | |
| WO2023030363A1 (en) | Solid forms of bcl-2 inhibitors, method of preparation, and use thereof | |
| EP3517529B1 (en) | Salt of quinazoline derivative, preparation method therefor and application thereof |