ES2850426T3 - Derivados de piperidinilo - Google Patents
Derivados de piperidinilo Download PDFInfo
- Publication number
- ES2850426T3 ES2850426T3 ES17722397T ES17722397T ES2850426T3 ES 2850426 T3 ES2850426 T3 ES 2850426T3 ES 17722397 T ES17722397 T ES 17722397T ES 17722397 T ES17722397 T ES 17722397T ES 2850426 T3 ES2850426 T3 ES 2850426T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- methyl
- indicates
- compounds
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(CN(CC1)C([C@](C)(C(F)(F)F)O)=O)C1N(C)C(C(C=*C)=CC=*)=O Chemical compound CC(CN(CC1)C([C@](C)(C(F)(F)F)O)=O)C1N(C)C(C(C=*C)=CC=*)=O 0.000 description 2
- VWSBNWIPICCWAM-UHFFFAOYSA-N CC(CN(CC1)C(OC(C)(C)C)=O)C1=O Chemical compound CC(CN(CC1)C(OC(C)(C)C)=O)C1=O VWSBNWIPICCWAM-UHFFFAOYSA-N 0.000 description 1
- MOSMMXCLLGCRRB-UHFFFAOYSA-N CC(CN(CC1)C(OC(C)(C)C)=O)C1N(C)C(c1ccccc1)=O Chemical compound CC(CN(CC1)C(OC(C)(C)C)=O)C1N(C)C(c1ccccc1)=O MOSMMXCLLGCRRB-UHFFFAOYSA-N 0.000 description 1
- NJBGCEIHJGURNN-UHFFFAOYSA-N CC(CN(CC1)C(OC(C)(C)C)=O)C1NC Chemical compound CC(CN(CC1)C(OC(C)(C)C)=O)C1NC NJBGCEIHJGURNN-UHFFFAOYSA-N 0.000 description 1
- DFODBEYNCMUIKJ-UTVJUBTNSA-N CC(CN(CC1)C([C@H](C(F)(F)F)O)=O)C1N(C)C(c(nc1)ccc1F)=O Chemical compound CC(CN(CC1)C([C@H](C(F)(F)F)O)=O)C1N(C)C(c(nc1)ccc1F)=O DFODBEYNCMUIKJ-UTVJUBTNSA-N 0.000 description 1
- JFKCOZFUPKSNIN-KDTYNDQISA-N CC(CN(CC1)C([C@](C)(C(F)(F)F)O)=O)C1C(N(C)c(cc1)ccc1Cl)=O Chemical compound CC(CN(CC1)C([C@](C)(C(F)(F)F)O)=O)C1C(N(C)c(cc1)ccc1Cl)=O JFKCOZFUPKSNIN-KDTYNDQISA-N 0.000 description 1
- JFKCOZFUPKSNIN-FABXCBLPSA-N C[C@@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@@H]1C(N(C)c(cc1)ccc1Cl)=O Chemical compound C[C@@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@@H]1C(N(C)c(cc1)ccc1Cl)=O JFKCOZFUPKSNIN-FABXCBLPSA-N 0.000 description 1
- MEVKEPOQYHRQEV-WBHUJUFNSA-N C[C@@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@@H]1N(C)C(c(cc1)ccc1C#N)=O Chemical compound C[C@@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@@H]1N(C)C(c(cc1)ccc1C#N)=O MEVKEPOQYHRQEV-WBHUJUFNSA-N 0.000 description 1
- IFXPJNYQDKWVAU-FABXCBLPSA-N C[C@@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@@H]1N(C)C(c1ccc(C(F)(F)F)cc1)=O Chemical compound C[C@@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@@H]1N(C)C(c1ccc(C(F)(F)F)cc1)=O IFXPJNYQDKWVAU-FABXCBLPSA-N 0.000 description 1
- MEVKEPOQYHRQEV-HNJNHCNJSA-N C[C@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@H]1N(C)C(c(cc1)ccc1C#N)=O Chemical compound C[C@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@H]1N(C)C(c(cc1)ccc1C#N)=O MEVKEPOQYHRQEV-HNJNHCNJSA-N 0.000 description 1
- OMQXOXRKWPHSAL-HYSWKAIVSA-N C[C@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@H]1N(C)C(c(cc1)ccc1F)=O Chemical compound C[C@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@H]1N(C)C(c(cc1)ccc1F)=O OMQXOXRKWPHSAL-HYSWKAIVSA-N 0.000 description 1
- TUKRVNCBIZEUNL-HACGYAERSA-N C[C@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@H]1N(C)C(c1ccccc1)=O Chemical compound C[C@H](CN(CC1)C([C@](C)(C(F)(F)F)O)=O)[C@H]1N(C)C(c1ccccc1)=O TUKRVNCBIZEUNL-HACGYAERSA-N 0.000 description 1
- SLPXDSRIGUTRDJ-MRXNPFEDSA-N C[C@](C(F)(F)F)(C(N(CC1)CCC1C(N(C)c(cc1)ccc1Cl)=O)=O)O Chemical compound C[C@](C(F)(F)F)(C(N(CC1)CCC1C(N(C)c(cc1)ccc1Cl)=O)=O)O SLPXDSRIGUTRDJ-MRXNPFEDSA-N 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N O=C(c1ccccc1)Cl Chemical compound O=C(c1ccccc1)Cl PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Abstract
Compuestos de fórmula I **(Ver fórmula)** donde X-Y indica CO- NA'' o NA''- CO, R1 indica Ar o Het, R2 indica H o CH3, R3 indica H o CH3, Ar indica fenilo, que no está sustituido o está mono, di o trisustituido por Hal, CN, A y/u OR3, Het indica pirimidilo, piridilo, piridazinilo, pirazinilo, piperidinilo, pirrolidinilo, pirazolilo, tiazolilo, imidazolilo, fura- nilo, tiofenilo, pirrolilo, oxazolilo, triazolilo, oxadiazolilo o tiadiazolilo, cada uno de los cuales no está sustituido o está mono o disustituido por Hal, A, CN y/u OR3, A indica alquilo ramificado o no ramificado con 1-6 átomos de C, donde 1-5 átomos de H pueden estar sustituidos por F, A'' indica alquilo sin ramificar o ramificado que tiene 1-4 átomos de C, Hal indica F, Cl, Br o I, y sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones.
Description
DESCRIPCIÓN
Derivados de piperidinilo
CAMPO TÉCNICO DE LA INVENCIÓN
La presente invención se refiere a nuevos derivados de piperidinilo que inhiben la piruvato deshidrogenasa quinasa (PDHK), a composiciones farmacéuticas que los comprenden, a procesos para su preparación y a su uso en terapia para el tratamiento de cánceres.
ANTECEDENTES DE LA INVENCIÓN
La piruvato deshidrogenasa quinasa (también denominada complejo piruvato deshidrogenasa quinasa, PDC quinasa o PDHK) es una enzima quinasa que actúa para inactivar la enzima piruvato deshidrogenasa mediante su fosforilación usando ATP.
La PDHK participa, por tanto, en la regulación del complejo piruvato deshidrogenasa del cual la piruvato deshidrogenasa es el primer componente. Tanto la PDHK como el complejo piruvato deshidrogenasa se localizan en la matriz mitocondrial de eucariotas. El complejo actúa convirtiendo el piruvato (un producto de la glucólisis en el citosol) en acetil-CoA, que posteriormente se oxida en la mitocondria para producir energía en el ciclo del ácido cítrico. Mediante la regulación por disminución de la actividad de este complejo, la PDHK disminuirá la oxidación del piruvato en las mitocondrias y aumentará la conversión de piruvato a lactato en el citosol.
La acción opuesta de la PDHK, concretamente la desfosforilación y activación de la piruvato deshidrogenasa, está catali zada por una fosfoproteína fosfatasa denominada piruvato deshidrogenasa fosfatasa.
(La piruvato deshidrogenasa quinasa no debe confundirse con la quinasa 1 dependiente de fosfoinosítido, que también se conoce a veces como «PDK1»).
Existen cuatro isoenzimas conocidas de la PDHK en humanos: PDHK1 a PDHK4.
Algunos estudios han demostrado que las células que carecen de insulina (o que son insensibles a la insulina) sobreex presan PDHK4. Como consecuencia, el piruvato formado a partir de la glucólisis no se puede oxidar, lo que conduce a una hiperglucemia debido al hecho de que la glucosa en sangre no se puede usar de forma efectiva. Por tanto, varios fármacos van dirigidos a PDHK4 con la esperanza de poder tratar la diabetes de tipo II.
La PDHK1 ha mostrado tener una mayor actividad en células cancerosas hipóxicas debido a la presencia de HIF-1. La PDHK1 desvía el piruvato del ciclo del ácido cítrico y mantiene vivas las células hipóxicas. Por tanto, la inhibición de la PDHK1 se ha sugerido como terapia antitumoral, ya que PDHK1 previene la apoptosis en estas células cancerosas. De forma similar, se ha demostrado que la PDHK3 se sobreexpresa en líneas celulares de cáncer de colon. Los tres inhibidores propuestos son AZD7545 y dicloroacetato, que se unen a PDHK1, y radicicol, que se une a PDHK3.
El aumento de PDC en la forma activa mediante la inhibición de la actividad PDHK es una diana farmacológica para la diabetes, las cardiopatías y el cáncer.
En el documento EP 2345629 A1 se describen inhibidores de PDHK que se consideran útiles para el tratamiento o la profilaxis de enfermedades relacionadas con trastornos de la utilización de la glucosa, por ejemplo, diabetes (p. ej., diabetes de tipo 1, diabetes de tipo 2, etc.), síndrome de resistencia a insulina, síndrome metabólico, hiperglucemia e hiperlactacidemia. Además, un inhibidor de PDHK se considera útil para el tratamiento o la profilaxis de las complicaciones diabéticas (p. ej., neuropatía, retinopatía, nefropatía, cataratas, etc.). Asimismo, un inhibidor de PDHK se considera útil para el trata miento o la profilaxis de enfermedades causadas por un aporte limitado a los tejidos de sustratos energéticos, por ejemplo, insuficiencia cardíaca, cardiomiopatía, isquemia miocárdica, dislipidemia y ateroesclerosis. Adicionalmente, un inhibidor de PDHK se considera útil para el tratamiento o la profilaxis de la isquemia cerebral o apoplejía cerebral. Además, un inhibidor de PDHK se considera útil para el tratamiento o la profilaxis de enfermedades mitocondriales, encefalopatía mitocondrial, cáncer y similares. También se considera útil para el tratamiento y la profilaxis de la hipertensión pulmonar.
Bibliografía:
Wikipedia, pyruvate dehydrogenase kinase;
T.E. Roche y cols., Cell. Mol. Life Sci. 64 (2007) 830-849;
A. Kumar y cols., Chemico-Biological Interactions 199 (2012) 29-37;
I. Papandreou y cols., Int. J. Cancer: 128, 1001-1008 (2011);
G. Sutendra y cols., Frontiers in Oncology, 2013, vol. 3, 1-11.
La invención tiene el objeto de encontrar compuestos nuevos que tengan propiedades valiosas, en particular aquellos que puedan usarse para la preparación de medicamentos.
Se ha encontrado que los compuestos según la invención y sus sales tienen propiedades farmacológicas muy valiosas a la vez que se toleran bien.
La presente invención se refiere específicamente a compuestos de fórmula I que inhiben la PDHK, preferiblemente la PDHK2, a composiciones que comprenden estos compuestos y a procesos para el uso de los mismos para el tratamiento de dolencias y enfermedades inducidas por PDHK.
Asimismo, los compuestos de fórmula I se pueden usar para el aislamiento e investigación de la actividad o expresión de PDHK. Además, son especialmente adecuados para su uso en métodos diagnósticos para enfermedades en conexión con la actividad incontrolada o alterada de PDHK.
El huésped o paciente puede pertenecer a cualquier especie de mamífero, por ejemplo, una especie de primate, especial mente humanos; roedores, como ratones, ratas y hámsteres; conejos; caballos, vacas, perros, gatos, etc. Los modelos de animales son interesantes para las investigaciones experimentales, proporcionando un modelo para el tratamiento de una enfermedad humana.
La susceptibilidad de una célula en particular al tratamiento con los compuestos según la invención puede determinarse mediante análisis in vitro. Normalmente, se combina un cultivo de la célula con un compuesto según la invención a diversas concentraciones durante un periodo de tiempo que es suficiente para permitir que los agentes activos, tal como anti-IgM, induzcan una respuesta celular como por ejemplo la expresión de un marcador de superficie, habitualmente entre aproxi madamente una hora y una semana. El análisis in vitro se puede realizar usando células en cultivo procedentes de la sangre o de una muestra de biopsia. La cantidad de marcador de superficie expresado se evalúa mediante citometría de flujo usando anticuerpos específicos que reconocen el marcador.
La dosis varía dependiendo del compuesto específico utilizado, la enfermedad específica, el estado del paciente, etc. Una dosis terapéutica normalmente es suficiente para reducir la población celular no deseada en el tejido diana, a la vez que se mantiene la viabilidad del paciente. El tratamiento se continúa generalmente hasta conseguir una reducción considera ble, por ejemplo, al menos aproximadamente una reducción del 50 % en la carga de células, y puede continuarse esen cialmente hasta que no se detecten más células no deseadas en el organismo.
TÉCNICA PREVIA
En el documento EP 2345629 A1 se describen derivados de fluoreno como inhibidores de PDHK para el tratamiento de enfermedades como la diabetes y el cáncer.
En el documento WO 2012/082947 se describen otros derivados de pirazol para su uso como agonistas de TGR5.
En el documento WO 2012/062783 se describe la preparación de derivados de pirazolilaminopirimidina para su uso como moduladores de LRRK2.
En el documento WO 2012/058127 se describe la preparación de derivados de fenilmetil-piperidinil-triazolil-piridinil-indazol. En el documento WO 2011/143057 se describe la preparación de pirazoles y triazoles sustituidos como nuevos inhibidores de la prolilcarboxipeptidasa.
En el documento WO 2008/083238 se describen derivados y análogos de piperidiniltiazol sustituidos para el tratamiento de la diabetes y trastornos metabólicos.
En el documento US 6979686 B1 se describen heteroarilpirazoles como inhibidores de la quinasa p38.
El compuesto
(R)-[4-(4-cianobenzoilamino)-1-(3,3,3-trifluoro-2-hidroxi-2-metilpropionil)piperidina] se describe en el ejemplo 20 del docu mento WO 2001/017942, como inhibidor de la piruvato deshidrogenasa.
Inhibición: IC50 (PDHK2): 9,70E-06.
RESUMEN DE LA INVENCIÓN
La invención se refiere a compuestos de fórmula I

donde
X-Y indica CO- NA'' o NA''- CO,
R1 indica Ar o Het,
R2 indica H o CH3 ,
R3 indica H o CH3 ,
Ar indica fenilo, que no está sustituido o está mono, di o trisustituido por Hal, CN, A y/u OR3,
Het indica pirimidilo, piridilo, piridazinilo, pirazinilo, piperidinilo, pirrolidinilo, pirazolilo, tiazolilo, imidazolilo, furanilo, tiofenilo, pirrolilo, oxazolilo, triazolilo, oxadiazolilo o tiadiazolilo, cada uno de los cuales no está sustituido o está mono o disustituido por Hal, A, CN y/u OR3,
A indica alquilo ramificado o no ramificado con 1-6 átomos de C, donde 1-5 átomos de H pueden estar sustituidos por F,
A'' indica alquilo sin ramificar o ramificado que tiene 1-4 átomos de C,
Hal indica F, Cl, Br o I,
y sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones.
La invención también se refiere a las formas óptimamente activas (estereoisómeros), los enantiómeros, los racematos, los diastereómeros y los hidratos y solvatos de estos compuestos.
También se describen derivados farmacéuticamente aceptables de los compuestos de fórmula I.
El término solvatos de los compuestos se refiere a aducciones de moléculas solventes inertes en los compuestos que se forman gracias a su fuerza de atracción mutua. Los solvatos son, por ejemplo, mono o dihidratos o alcóxidos.
Se entiende que la invención también se refiere a los solvatos de las sales.
El término derivados farmacéuticamente aceptables se considera que significa, por ejemplo, las sales de los compuestos según la invención y también los denominados compuestos profármacos.
Como se usa en este documento y a menos que se indique otra cosa, el término «profármaco» se refiere a un derivado de un compuesto de fórmula I que se puede hidrolizar, oxidar o hacer reaccionar de alguna otra forma en condiciones bioló gicas (in vitro o in vivo) para proporcionar un compuesto activo, especialmente un compuesto de fórmula I. Entre los ejem plos de profármacos se incluyen, pero sin limitaciones, derivados y metabolitos de un compuesto de fórmula I que incluyen restos biohidrolizables tales como amidas biohidrolizables, ésteres biohidrolizables, carbamatos biohidrolizables,
carbonatos biohidrolizables, ureidos biohidrolizables y análogos de fosfato biohidrolizables. En determinadas realizaciones, los profármacos de compuestos con grupos funcionales carboxilo son los ésteres de alquilo menores del ácido carboxílico. Los ésteres de carboxilato se forman convenientemente mediante esterificación de cualquiera de los restos de ácido car boxílico presentes en la molécula. Los profármacos se pueden preparar normalmente usando métodos bien conocidos, como los descritos en Burger's Medicinal Chemistry and Drug Discovery 6.a ed. (Donald J. Abraham ed., 2001, Wiley) y en Design and Application of Prodrugs (H. Bundgaard ed., 1985, Harwood Academic Publishers Gmfh).
La expresión «cantidad eficaz» indica la cantidad de un medicamento o de un principio activo farmacéutico que causa en un tejido, sistema, animal o ser humano la respuesta biológica o médica que busca o desea, por ejemplo, un investigador o un médico.
Además, la expresión «cantidad terapéuticamente eficaz» indica una cantidad que, en comparación con un sujeto corres pondiente que no ha recibido esta cantidad, tiene las siguientes consecuencias:
mejora del tratamiento, curación, prevención o eliminación de una enfermedad, síndrome, afección, dolencia, trastorno o efectos secundarios o también reducción de la progresión de una enfermedad, dolencia o trastorno.
La expresión «cantidad terapéuticamente eficaz» también abarca las cantidades que son eficaces para aumentar la función fisiológica normal.
La invención también se refiere al uso de mezclas de los compuestos de fórmula I, por ejemplo, mezclas de dos diastereoisómeros, por ejemplo, en la proporción 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 o 1:1000.
Estas son mezclas especialmente preferidas de compuestos estereoisoméricos.
El término «tautómeros» se refiere a formas isoméricas de un compuesto que están en equilibrio entre sí. Las concentra ciones de las formas isoméricas dependerán del entorno en el que se encuentre el compuesto y pueden ser diferente dependiendo de, por ejemplo, si el compuesto es un sólido o está en una solución orgánica o acuosa.
La invención se refiere a los compuestos de fórmula I y a sus sales, y a un proceso para la preparación de compuestos de fórmula I y sus sales, solvatos, tautómeros y estereoisómeros farmacéuticamente aceptables, caracterizados porque un compuesto de fórmula II

donde X, Y, R1 y R2 tienen los significados indicados en la reivindicación 1,
reacciona con el ácido 3,3,3-trifluoro-2-hidroxi-2-metil-propiónico
y/o
una base o ácido de fórmula I se transforma en una de sus sales.
Anteriormente y a continuación, los radicales X, Y, R1 y R2 tienen los significados indicados para la fórmula I, siempre que no se establezca expresamente otra cosa.
X-Y preferiblemente indica CO-NCH3 o NCH3-CO.
A preferiblemente indica metilo, además etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo o ferc-butilo, también además pentilo, 1-, 2- o 3-metilbutilo, 1,1-, 1,2- o 2,2-dimetilpropilo, 1 -etilpropilo, hexilo, 1-, 2-, 3- o 4-metilpentilo, 1,1-, 1,2-, 1,3-, 2,2-, 2,3- o 3,3-dimetilbutilo, 1- o 2-etilbutilo, 1 -etil-1-metilpropilo, 1 -etil-2-metil-propilo, 1,1,2- o 1,2,2-trimetilpropilo, además preferiblemente, por ejemplo, trifluorometilo.
A” preferiblemente indica metilo, además etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo o ferc-butilo.
R3 preferiblemente indica H.
Ar preferiblemente indica o-, m- o p-tolilo, o-, m- o p-etilfenilo, o-, m- o p-propilfenilo, o-, m- o p-isopropilfenilo, o-, m- o pferc-butilfenilo, o-, m- o p-hidroxifenilo, o-, m- o p-metoxifenilo, o-, m- o p-etoxifenilo, o-, m- o p-fluorofenilo, o-, m- o pbromofenilo, o-, m- o p-clorofenilo, o-, m- o p-cianofenilo, además preferiblemente 2,3-, 2,4-, 2,5-, 2,6-, 3,4- o 3,5-difluorofenilo, 2,3-, 2,4-, 2,5-, 2,6-, 3,4- o 3,5-diclorofenilo, 2,3-, 2,4-, 2,5-, 2,6-, 3,4- o 3,5-dibromofenilo, 2,5- o 3,4-dimetoxifenilo, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- o 3,4,5-triclorofenilo, 2,4,6-trimetoxifenilo, 2-hidroxi-3,5-diclorofenilo, p-yodofenilo, 4-fluoro-3-clorofenilo, 2-fluoro-4-bromofenilo, 2,5-difluoro-4-bromofenilo, 3-bromo-6-metoxifenilo, 3-cloro-6-metoxifenilo 3-fluoro-4-metoxifenilo o 2,5-dimetil-4-clorofenilo.
Independientemente de otras sustituciones, Het indica, por ejemplo, 2- o 3-furilo, 2- o 3-tienilo, 1-, 2- o 3-pirrolilo, 1-, 2-, 4- o 5-imidazolilo, 1-, 3-, 4- o 5-pirazolilo, 2-, 4- o 5-oxazolilo, 3-, 4- o 5-isoxazolilo, 2-, 4- o 5-tiazolilo, 3-, 4- o 5-isotiazolilo, 2-, 3- o 4-piridilo, 2-, 4-, 5- o 6-pirimidinilo, además preferiblemente 1,2,3-triazol-1-, -4- o -5-ilo, 1,2,4-triazol-1-, -3- o 5-ilo, 1- o 5-tetrazolilo, 1,2,3-oxadiazol-4- o -5-ilo, 1,2,4-oxadiazol-3- o -5-ilo, 1,3,4-tiadiazol-2- o -5-ilo, 1,2,4-tiadiazol-3- o -5-ilo, 1,2,3-tiadiazol-4- o -5-ilo, 3- o 4-piridazinilo o pirazinilo.
Adicionalmente, Het preferiblemente indica pirimidilo, pirazolilo, piridilo, piridazinilo o pirazinilo, cada uno de ellos no susti tuido o mono o disustituido por A.
Hal preferiblemente indica F, Cl o Br, pero también I, en particular preferiblemente F o Cl.
A lo largo de la invención, todos los radicales que aparecen más de una vez pueden ser idénticos o diferentes, es decir, son independientes entre sí.
Los compuestos de fórmula I pueden tener uno o más centros quirales y pueden, por tanto, darse en diversas formas estereoisoméricas. La fórmula I abarca todas estas formas.
La configuración preferida en el alcohol quiral es R.
Por consiguiente, la invención se refiere, en particular, a los compuestos de fórmula I en los que al menos uno de dichos radicales tiene uno de los significados preferidos indicados anteriormente.
Los compuestos de fórmula I y también los materiales de partida para su preparación se preparan, además, mediante métodos conocidos per se, como se describe en la bibliografía (por ejemplo, en trabajos convencionales, como Houben-Weyl, Methoden der Organischen Chemie [Métodos de química orgánica], Georg Thieme Verlag, Stuttgart), para ser pre cisos en las condiciones de reacción que son conocidas y adecuadas para dichas reacciones. También puede hacerse uso aquí de variantes conocidas per se, que no se mencionan en este documento con mayor detalle.
Los compuestos de partida para la preparación de compuestos de fórmula I son conocidos en general. Si son nuevos, sin embargo, se pueden preparar por métodos conocidos per se.
Los compuestos de fórmula I pueden obtenerse preferiblemente mediante la reacción de un compuesto de fórmula II con ácido 3,3,3-trifluoro-2-hidroxi-2-metil-propiónico.
La reacción se realiza generalmente en presencia de un agente de unión a ácido, preferiblemente una base orgánica, como N-etildiisopropilamina, trietilamina, dimetilanilina, piridina o quinolina.
Asimismo, la adición de un agente de activación de carboxilo, como [hexafluorofosfato de dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio («HATU») o 1-etil-3-(3-dimetilaminopropil)carbodiimida («EDCI») es favorable.
Dependiendo de las condiciones utilizadas, el tiempo de reacción está entre unos minutos y 14 días, la temperatura de reacción está entre aproximadamente -30° y 140°, normalmente entre 0° y 110°, en particular entre aproximadamente 20° y aproximadamente 100°.
Son ejemplos de solventes inertes adecuados hidrocarburos, como hexano, éter de petróleo, benceno, tolueno o xileno; hidrocarburos clorados, como tricloroetileno, 1,2-dicloroetano, tetracloruro de carbono, cloroformo o diclorometano; alcoho les, como metanol, etanol, isopropanol, n-propanol, n-butanol o ferc-butanol; éteres, como éter dietílico, éter diisopropílico, tetrahidrofurano (THF) o dioxano; éteres glicólicos, como éter monometílico o monoetílico de etilenglicol o éter dimetílico de etilenglicol (diglima); cetonas, como acetona o butanona; amidas, como acetamida, dimetilacetamida o dimetilformamida (DMF); nitrilos, como acetonitrilo; sulfóxidos, como dimetilsulfóxido (DMSO); disulfuro de carbono; ácidos carboxílicos, como ácido fórmico o ácido acético; compuestos nitrogenados, como nitrometano o nitrobenceno; ésteres, como acetato de etilo, o mezclas de dichos solventes.
Se da especial preferencia a etanol, acetonitrilo, diclorometano y/o DMF.
Sales farmacéuticas y otras formas
Los compuestos mencionados según la invención pueden usarse en sus formas finales no salinas. Por otro lado, la pre sente invención también abarca el uso de estos compuestos en la forma de sus sales farmacéuticamente aceptables, que se pueden obtener a partir de diversos ácidos y bases orgánicos e inorgánicos mediante procedimientos conocidos en la técnica. Las formas salinas farmacéuticamente aceptables de los compuestos de fórmula I se preparan en su mayor parte mediante métodos convencionales. Si el compuesto de fórmula I contiene un grupo carboxilo, una de sus sales adecuadas puede formarse mediante la reacción del compuesto con una base adecuada para obtener la sal de adición de base co rrespondiente. Estas bases son, por ejemplo, hidróxidos de metales alcalinos, incluyendo hidróxido de potasio, hidróxido sódico e hidróxido de litio; hidróxidos de metales alcalinotérreos, como hidróxido de bario e hidróxido de calcio; alcóxidos de metales alcalinos, por ejemplo etóxido de potasio y propóxido de sodio; y diversas bases orgánicas, como piperidina, dietanolamina y N-metilglutamina. Las sales de aluminio de los compuestos de fórmula I también están incluidas. En el caso de determinados compuestos de fórmula I, las sales de adición de ácido pueden formarse tratando estos compuestos con ácidos orgánicos e inorgánicos farmacéuticamente aceptables, por ejemplo, haluros de hidrógeno, como cloruro de hidrógeno, bromuro de hidrógeno o yoduro de hidrógeno, otros ácidos minerales y sus correspondientes sales, como sul fato, nitrato o fosfato y similares, y alquil y monoarilsulfonatos, como etanosulfonato, toluenosulfonato y bencenosulfonato, y otros ácidos orgánicos y sus correspondientes sales, como acetato, trifluoroacetato, tartrato, maleato, succinato, citrato, benzoato, salicilato, ascorbato y similares. En consecuencia, entre las sales de adición de ácido farmacéuticamente acep tables de los compuestos de fórmula I se incluyen las siguientes: acetato, adipato, alginato, arginato, aspartato, benzoato, bencenosulfonato (besilato), bisulfato, bisulfito, bromuro, butirato, alcanforato, alcanforsulfonato, caprilato, cloruro, clorobenzoato, citrato, ciclopentanopropionato, digluconato, dihidrogenofosfato, dinitrobenzoato, dodecilsulfato, etanosulfonato, fumarato, formato, galacterato (de ácido múcico), galacturonato, glucoheptanoato, gluconato, glutamato, glicerofosfato, hemisuccinato, hemisulfato, heptanoato, hexanoato, hipurato, clorhidrato, bromhidrato, yodhidrato, 2-hidroxietanosulfonato, yoduro, isetionato, isobutirato, lactato, lactobionato, malato, maleato, malonato, mandelato, metafosfato, metanosulfonato, metilbenzoato, monohidrogenofosfato, 2-naftalensulfonato, nicotinato, nitrato, oxalato, oleato, palmoato, pectinato, persulfato, fenilacetato, 3-fenilpropionato, fosfato, fosfonato, ftalato, sin embargo, esto no representa restricción alguna.
Adicionalmente, entre las sales de base de los compuestos según la invención se incluyen sales de aluminio, amonio, calcio, cobre, hierro(III), hierro(II), litio, magnesio, manganeso(III), manganeso(II), potasio, sodio y cinc, aunque no se pre tende que esto represente una restricción. De las sales mencionadas anteriormente, se da preferencia a las de amonio, a las sales de metales alcalinos sodio y potasio y a las sales de metales alcalinotérreos calcio y magnesio. Entre las sales de los compuestos de fórmula I que derivan de bases orgánicas no tóxicas farmacéuticamente aceptables se incluyen sales de aminas primarias, secundarias y terciarias, aminas sustituidas, incluyendo también aminas sustituidas naturales, aminas cíclicas y resinas de intercambio iónico básicas, por ejemplo, arginina, betaína, cafeína, cloroprocaína, colina, N,N'-dibenciletilendiamina (benzatina), diciclohexilamina, dietanolamina, dietilamina, 2-dietilaminoetanol, 2-dimetilaminoetanol, etanolamina, etilendiamina, N-etilmorfolina, N-etilpiperidina, glucamina, glucosamina, histidina, hidrabamina, isopropilamina, lidocaína, lisina, meglumina, N-metil-D-glucamina, morfolina, piperazina, piperidina, resinas de poliaminas, procaína, purinas, teobromina, trietanolamina, trietilamina, trimetilamina, tripropilamina y tris(hidroximetil)metilamina (trometamina), aunque no se pretende que esto represente una restricción.
Los compuestos de la presente invención que contienen grupos básicos con nitrógeno pueden cuaternizarse usando agen tes como haluros de alquilo (C1-C4), por ejemplo cloruro, bromuro y yoduro de metilo, etilo, isopropilo y ferc-butilo; sulfatos de dialquilo (C1-C4), por ejemplo, sulfato de dimetilo, dietilo y diamilo; haluros de alquilo (C10-C18), por ejemplo, cloruro, bromuro y yoduro de decilo, dodecilo, laurilo, miristilo y estearilo; y haluros de aril-alquilo (C1-C4), por ejemplo cloruro de bencilo y bromuro de fenetilo. Usando estas sales pueden prepararse tanto compuestos hidrosolubles como liposolubles según la invención.
Entre las sales farmacéuticas preferidas mencionadas anteriormente se incluyen acetato, trifluoroacetato, besilato, citrato, fumarato, gluconato, hemisuccinato, hipurato, clorhidrato, bromhidrato, isetionato, mandelato, meglumina, nitrato, oleato, fosfonato, pivalato, fosfato sódico, estearato, sulfato, sulfosalicilato, tartrato, tiomalato, tosilato y trometamina, aunque no se pretende que esto represente una restricción.
Se da particular preferencia a clorhidrato, diclorhidrato, bromhidrato, maleato, mesilato, fosfato, sulfato y succinato.
Las sales de adición de ácido de compuestos básicos de fórmula I se preparan poniendo la forma de base libre en contacto con una cantidad suficiente del ácido deseado, lo que provoca la formación de la sal de manera convencional. La base libre puede regenerarse poniendo la forma de sal en contacto con una base y aislando la base libre de manera convencio nal. Las formas de base libre difieren en un determinado aspecto de sus correspondientes formas salinas con respecto a determinadas propiedades físicas, como solubilidad en solventes polares; sin embargo, para los fines de la invención, las sales por lo demás se corresponden con las respectivas formas de base libre de las mismas.
Como se ha mencionado, las sales de adición de base farmacéuticamente aceptables de los compuestos de fórmula I se forman con metales o aminas, como metales alcalinos y metales alcalinotérreos o aminas orgánicas. Los metales preferidos son sodio, potasio, magnesio y calcio. Las aminas orgánicas preferidas son N,N'-dibenciletilendiamina, cloroprocaína, co lina, dietanolamina, etilendiamina, N-metil-D-glucamina y procaína.
Las sales de adición de base de compuestos ácidos según la invención se preparan poniendo en contacto la forma de ácido libre con una cantidad suficiente de la base deseada, provocando la formación de la sal de manera convencional. El ácido libre puede regenerarse poniendo la forma de sal en contacto con un ácido y aislando el ácido libre de manera convencional. Las formas de ácido libre difieren en algún aspecto de sus correspondientes formas salinas con respecto a determinadas propiedades físicas, como solubilidad en solventes polares; sin embargo, para los fines de la invención, las sales por los demás se corresponden con las respectivas formas de ácido libre de las mismas.
Si un compuesto según la invención contiene más de un grupo que es capaz de formar sales farmacéuticamente aceptables de este tipo, la invención también incluye sales múltiples. Entre las formas de sales múltiples típicas se incluyen, por ejem plo, bitartrato, diacetato, difumarato, dimeglumina, bifosfato, disodio y trihidrocloruro, aunque no se pretende que esto represente una restricción.
Con respecto a lo establecido anteriormente, puede observarse que la expresión «sal farmacéuticamente aceptable» en la conexión presente se refiere a un principio activo que comprende un compuesto de fórmula I en forma de una de sus sales, en especial si esta forma de sal confiere propiedades farmacocinéticas mejoradas al principio activo en comparación con la forma libre del principio activo o cualquier otra forma de sal del principio activo utilizada anteriormente. La forma de sal farmacéuticamente aceptable del principio activo también puede proporcionar este principio activo por primera vez con una propiedad farmacocinética deseada que no tenía antes y puede incluso tener una influencia positiva sobre la farmacodinámica de este principio activo con respecto a su eficacia terapéutica en el organismo.
Isótopos
Se pretende además que un compuesto de fórmula I incluya sus formas marcadas con isótopo. Una forma marcada con isótopo de un compuesto de fórmula I es idéntica a este compuesto excepto por el hecho de que uno o más átomos del compuesto se ha sustituido por un átomo o átomos con una masa atómica o un número másico que difiere de la masa atómica o el número másico del átomo natural. Entre los ejemplos de isótopos que se encuentran fácilmente en el mercado y que pueden incorporarse a un compuesto de fórmula I mediante métodos bien conocidos se incluyen isótopos de hidró geno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, por ejemplo, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36Cl, respectivamente. Se pretende que un compuesto de fórmula I o una sal farmacéuticamente aceptable que contenga uno o más de los isótopos mencionados anteriormente y/u otros isótopos de otros átomos sea parte de la presente invención. Se puede usar un compuesto de fórmula I marcado con isótopo de diversas formas beneficiosa. Por ejemplo, un compuesto de fórmula I marcado con isótopo dentro del cual se ha incorporado, por ejemplo, un radioisótopo, como 3H o 14C, es adecuado para ensayos de distribución en tejidos de medicamentos y/o sustratos. Estos radioisótopos, es decir, tritio (3H) y carbono 14 (14C), son especialmente preferidos debido a su sencilla preparación y a la excelente capacidad de detección. La incorporación de isótopos más pesados, por ejemplo deuterio (2H), a un compuesto de fórmula I tiene ventajas terapéu ticas debido a la mayor estabilidad metabólica de este compuesto marcado con el isótopo. Una mayor estabilidad metabólica se traduce directamente en dosis más bajas o un aumento de la semivida in vivo, lo que en la mayoría de las circuns tancias representaría una realización preferida de la presente invención. Un compuesto de fórmula I marcado con isótopo puede prepararse habitualmente realizando los procedimientos descritos en los esquemas de síntesis y la descripción relacionada, en la parte de ejemplos y en la parte de preparación del presente texto, sustituyendo un reactivo no marcado con isótopo por un reactivo marcado con isótopo fácilmente disponible.
También se puede incorporar deuterio (2H) en un compuesto de fórmula I con el fin de manipular el metabolismo oxidativo del compuesto por medio del efecto isotópico cinético primario. El efecto isotópico cinético primario es un cambio de la velocidad de una reacción química que es consecuencia del intercambio de núcleos isotópicos, lo que a su vez está cau sado por el cambio en las energías del estado fundamental para la formación de enlaces covalentes después de este intercambio isotópico. El intercambio de un isótopo más pesado normalmente tiene como consecuencia una reducción de la energía del estado fundamental para un enlace químico y causa, por tanto, una reducción de la velocidad de rotura de enlaces limitantes de la velocidad. Si la rotura de enlaces se produce en o en las proximidades de una región de punto de silla a lo largo de la coordenada de una reacción multiproducto, los cocientes de distribución de productos se pueden alterar de forma sustancial. Como explicación: si el deuterio se une a un átomo de carbono en una posición no intercambiable, las diferencias de velocidad de kM/kD = 2-7 son típicas. Si esta diferencia de velocidad se aplica con éxito a un compuesto de fórmula I que es susceptible a la oxidación, el perfil de este compuesto in vivo se puede modificar considerablemente y tiene como resultado una mejora de las propiedades farmacocinéticas.
Cuando se descubren y desarrollan fármacos, la persona experta en la materia intenta optimizar los parámetros farmacocinéticos, conservando a la vez las propiedades in vitro deseables. Es razonable asumir que muchos compuestos con malos perfiles farmacocinéticos son susceptibles del metabolismo oxidativo. Los ensayos de microsomas hepáticos in vitro actualmente disponibles proporcionan información valiosa sobre el curso del metabolismo oxidativo de este tipo, lo que a su vez permite el diseño racional de compuestos deuterados de fórmula I con mejor estabilidad a través de la resistencia a dicho metabolismo oxidativo. Se obtienen de este modo mejoras significativas en los perfiles farmacocinéticos de com puestos de fórmula I, y se pueden expresar cuantitativamente en términos de aumentos en la semivida (t1/2) in vivo, con centración en el efecto terapéutico máximo (Cmáx), área bajo la curva de dosis-respuesta (AUC) y F; y en términos de reducción del aclaramiento, dosis y costes de materiales.
La siguiente explicación está destinada a ilustrar lo anterior: un compuesto de fórmula I que tiene múltiples sitios potenciales de ataque para el metabolismo oxidativo, por ejemplo, átomos de hidrógeno bencílico y átomos de hidrógeno unidos a un átomo de nitrógeno, se prepara como una serie de análogos en los que diversas combinaciones de átomos de hidrógeno se sustituyen por átomos de deuterio, de modo que algunos, la mayoría o todos estos átomos de hidrógeno se han susti tuido por átomos de deuterio. Las determinaciones de la semivida permiten la determinación favorable y precisa del grado al cual ha mejorado la resistencia al metabolismo oxidativo. De este modo, se determina que la semivida del compuesto original se puede extender hasta el 100 % como consecuencia de un intercambio de hidrógeno deuterio de este tipo.
El intercambio de hidrógeno deuterio en un compuesto de fórmula I también se puede usar para conseguir una modificación favorable del espectro del metabolito del compuesto de partida con el fin de disminuir o eliminar los metabolitos tóxicos no deseados. Por ejemplo, si aparece un metabolito tóxico por escisión oxidativa del enlace carbono-hidrógeno (C-H), puede ser razonable asumir que el análogo deuterado disminuirá o eliminará en gran medida la producción del metabolito no deseado, incluso si la oxidación en particular no es un paso determinante de la velocidad. Se puede encontrar más infor mación sobre el estado de la técnica con respecto al intercambio de hidrógeno deuterio por ejemplo en Hanzlik y cols., J. Org. Chem. 55, 3992-3997, 1990, Reider y cols., J. Org. Chem. 52, 3326-3334, 1987, Foster, Adv. Drug Res. 14, 1-40, 1985, Gillette y cols., Biochemistry 33(10) 2927-2937, 1994, y Jarman y cols. Carcinogenesis 16(4), 683-688, 1993.
La invención además se refiere a medicamentos que comprenden al menos un compuesto de fórmula I y/o sales, solvatos y estereoisómeros farmacéuticamente aceptables del mismo, incluidas sus mezclas en todas las proporciones, y opcional mente excipientes y/o adyuvantes.
Las formulaciones farmacéuticas pueden administrarse en forma de unidades de dosis que comprenden una cantidad predeterminada de principio activo por unidad de dosis. Dicha unidad puede comprender, por ejemplo, de 0,5 mg a 1 g, preferiblemente de 1 mg a 700 mg, en especial preferiblemente de 5 mg a 100 mg, de un compuesto según la invención, dependiendo de la enfermedad tratada, el método de administración y la edad, peso y estado del paciente, o las formula ciones farmacéuticas se pueden administrar en forma de unidades de dosis que comprendan una cantidad predeterminada de principio activo por unidad de dosis. Las formulaciones de unidad de dosis preferidas son aquellas que comprenden una dosis, o parte de la dosis, diaria como se indica anteriormente, o una fracción correspondiente de la misma de un principio activo. Además, las formulaciones farmacéuticas de este tipo pueden prepararse usando un proceso que es ge neralmente conocido en la técnica farmacéutica.
Las formulaciones farmacéuticas pueden adaptarse para su administración mediante cualquier método adecuado deseado, por ejemplo, mediante métodos orales (incluyendo bucal o sublingual), rectales, nasales, tópicos (incluyendo bucal, sublin gual o transdérmico), vaginales o parenterales (incluyendo subcutáneo, intramuscular, intravenoso o intradérmico). Estas formulaciones pueden prepararse usando todos los procesos conocidos en la técnica farmacéutica mediante, por ejemplo, combinación del principio activo con el excipiente (o excipientes) o el adyuvante (o adyuvantes).
Las formulaciones farmacéuticas adaptadas para la administración oral pueden administrarse como unidades independien tes, como por ejemplo, cápsulas o comprimidos; polvos o gránulos; soluciones o suspensiones en líquidos acuosos o no acuosos; espumas comestibles o alimentos en forma de espuma; o emulsiones líquidas de aceite en agua o emulsiones líquidas de agua en aceite.
Por tanto, por ejemplo, en el caso de administración oral en forma de comprimido o cápsula, el principio activo puede combinarse con un excipiente inerte, oral, no tóxico y farmacéuticamente aceptable, como por ejemplo, etanol, glicerol, agua y similar. Los polvos se preparan triturando el compuesto hasta un tamaño fino adecuado y mezclándolo con un excipiente farmacéutico triturado de forma similar, como por ejemplo, un hidrato de carbono comestible, como por ejemplo, almidón o manitol. También pueden estar presentes un agente aromatizante, un conservante, un dispersante y un colo rante.
Las cápsulas se producen preparando una mezcla en polvo como se describe anteriormente y rellenando el envoltorio de gelatina conformado con la mezcla. Pueden añadirse a la mezcla en polvo agentes deslizantes y lubricantes, como por ejemplo, ácido silícico altamente disperso, talco, estearato de magnesio, estearato de calcio o polietilenglicol en forma sólida antes de la operación de llenado. Asimismo, puede añadirse un desintegrante o solubilizante, como por ejemplo, agar-agar, carbonato cálcico o carbonato sódico para mejorar la disponibilidad del medicamento después de que se haya tomado la cápsula.
Además, si se desea o es necesario, pueden incorporarse también a la mezcla agentes aglutinantes, lubricantes y desin tegrantes, así como colorantes adecuados. Entre los aglutinantes idóneos se incluyen almidón, gelatina, azúcares natura les, como por ejemplo, glucosa o beta-lactosa, edulcorantes a base de maíz, caucho natural y sintético, como por ejemplo, de acacia, tragacanto o alginato sódico, carboximetilcelulosa, polietilenglicol, ceras y similares. Entre los agentes lubrican tes utilizados en estas formas de dosis se incluyen oleato sódico, estearato sódico, estearato de magnesio, benzoato sódico, acetato sódico, cloruro sódico y similares. Entre los agentes desintegrantes se incluyen, sin restricciones, almidón, metilcelulosa, agar, bentonita, goma de xantano y similares. Los comprimidos se formulan, por ejemplo, preparando una
mezcla en polvo, granulando o prensando en seco la mezcla, añadiendo un lubricante y un desintegrante y prensando la mezcla completa para obtener los comprimidos. Se prepara una mezcla en polvo mezclando el compuesto triturado de forma adecuada con un diluyente o una base, como se describe anteriormente, y opcionalmente con un aglutinante, como por ejemplo, carboximetilcelulosa, un alginato, gelatina o polivinilpirrolidona, un retardante de disolución, como por ejemplo, parafina, un acelerador de la absorción, como por ejemplo, una sal cuaternaria, y/o un absorbente, como por ejemplo, bentonita, caolina o fosfato dicálcico. La mezcla en polvo puede granularse humedeciéndola con un aglutinante, como por ejemplo, jarabe, pasta de almidón, mucílago de acacia o soluciones de celulosa o materiales poliméricos y haciéndola pasar a través de un tamiz. Como alternativa a la granulación, la mezcla en polvo puede hacerse pasar a través de una máquina de comprimidos, dando lugar a trozos de forma no uniforme que se rompen para formar los gránulos. Los gránulos pueden lubricarse mediante la adición de ácido esteárico, una sal de estearato, talco o aceite mineral para evitar que se peguen a los moldes de vaciado de comprimidos. La mezcla con lubricante se prensa a continuación para obtener los comprimidos. Los compuestos según la invención también pueden combinarse con un excipiente inerte de flujo libre y, a continuación, prensarse directamente para obtener los comprimidos sin llevar a cabo los pasos de granulación o prensado en seco. Puede presentarse una capa protectora transparente u opaca compuesta por una capa de sellado de laca shellac, una capa de azúcar o material polimérico y una capa brillante de cera. Pueden añadirse colorantes a estos recubrimientos para diferenciar entre distintas unidades de dosis.
Pueden prepararse líquidos orales, como por ejemplo, soluciones, jarabes y elixires, en forma de unidades de dosis, de modo que una cantidad determinada comprenda una cantidad previamente especificada del compuesto. Los jarabes pue den prepararse disolviendo el compuesto en una solución acuosa con un aromatizante adecuado, mientras que los elixires se preparan usando un vehículo alcohólico no tóxico. Las suspensiones pueden formularse mediante dispersión del com puesto en un vehículo no tóxico. Pueden, asimismo, añadirse solubilizantes y emulsionantes como, por ejemplo, alcoholes de isostearilo etoxilados y éteres de polioxietilensorbitol, conservantes, aditivos aromatizantes como, por ejemplo, aceite de menta, edulcorantes naturales o sacarina, u otro aromatizante artificial y similares.
Las formulaciones de unidad de dosis para administración oral pueden, si se desea, estar encapsuladas en microcápsulas. La formulación también puede prepararse de manera que se prolongue o retrase la liberación, por ejemplo, recubriendo o incluyendo el material particulado en polímeros, cera y similares.
Los compuestos de fórmula I y las sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos pueden administrarse en forma de sistemas de administración de liposomas, como por ejemplo, vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas pueden formarse a partir de diversos fosfolípidos, como por ejemplo, colesterol, estearilamina o fosfatidilcolinas.
Los compuestos de fórmula I y las sales, tautómeros y derivados fisiológicamente funcionales farmacéuticamente acepta bles de los mismos también pueden administrarse usando anticuerpos monoclonales como vehículos individuales a los que están acopladas las moléculas del compuesto. Los compuestos también pueden acoplarse con polímeros solubles como vehículos que dirigen el medicamento. Estos polímeros pueden abarcar polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamidofenol, polihidroxietilaspartamidofenol o poli-(óxido de etileno)polilisina, sustituidos con radica les palmitoilo. Los compuestos pueden además acoplarse con una clase de polímeros biodegradables que son adecuados para conseguir la liberación controlada de un medicamento, por ejemplo, ácido poliláctico, poli-épsilon-caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidroxipiranos, policianoacrilatos y copolímeros de bloque de hidrogeles entrecruzados o anfipáticos.
Las formulaciones farmacéuticas adaptadas para administración transdérmica pueden administrarse como yesos indepen dientes para un contacto próximo y prolongado con la epidermis del receptor. Por tanto, por ejemplo, el principio activo puede administrarse a partir del yeso mediante iontoforesis, como se describe en términos generales en Pharmaceutical Research, 3(6), 318 (1986).
Los compuestos farmacéuticos adaptados para la administración tópica pueden formularse como pomadas, cremas, sus pensiones, lociones, polvos, soluciones, pastas, geles, pulverizadores, aerosoles o aceites.
Para el tratamiento de los ojos u otros tejidos externos, por ejemplo, la boca y la piel, las formulaciones se aplican preferi blemente como una pomada o crema tópica. En el caso de la formulación para obtener una pomada, el principio activo puede emplearse con una base de crema parafínica o miscible con agua. Alternativamente, el principio activo puede for mularse para obtener una crema con una base de crema de aceite en agua o una base de agua en aceite.
Las formulaciones farmacéuticas adaptadas para aplicación tópica en los ojos incluyen colirios, en los que el principio activo se disuelve o suspende en un vehículo adecuado, en particular, un solvente acuoso.
Las formulaciones farmacéuticas adaptadas para aplicación tópica en la boca abarcan pastillas para chupar, pastillas y colutorios.
Las formulaciones farmacéuticas adaptadas para la administración rectal pueden administrarse en forma de supositorios o enemas.
Las formulaciones farmacéuticas adaptadas para administración nasal en las que la sustancia vehículo es un sólido com prenden un polvo grueso con un tamaño de partícula, por ejemplo, en el intervalo de 20 a 500 micrómetros, que se admi nistra de manera que se aspira, es decir, mediante inhalación rápida a través de las fosas nasales a partir de un recipiente que contiene el polvo mantenido cerca de la nariz. Las formulaciones adecuadas para su administración como aerosol nasal o gotas nasales con un líquido como sustancia vehículo abarcan soluciones de principios activos en agua o aceite.
Las formulaciones farmacéuticas adaptadas para administración mediante inhalación abarcan vapores o polvos finamente particulados, que pueden generarse mediante diversos tipos de dispensadores presurizados con aerosoles, nebulizadores o insufladores.
Las formulaciones farmacéuticas adaptadas para la administración vaginal pueden administrarse como óvulos vaginales, tampones, cremas, geles, pastas, espumas o aerosol.
Entre las formulaciones farmacéuticas adaptadas para administración parenteral se incluyen soluciones acuosas y no acuosas estériles para inyección que comprenden antioxidantes, tampones, bacteriostáticos y solutos, mediante las cuales la formulación se hace isotónica con la sangre del receptor que se va a tratar, y suspensiones acuosas y no acuosas estériles, que pueden comprender medios de suspensión y espesantes. Las formulaciones pueden administrarse en reci pientes de dosis única o multidosis, por ejemplo, en ampollas y viales sellados, y conservarse liofilizadas, de modo que solo sea necesaria la adición del líquido vehículo estéril, por ejemplo agua para inyección, inmediatamente antes de que sea necesario su uso. Las soluciones y suspensiones para inyección preparadas según la receta pueden prepararse a partir de polvos, gránulos y comprimidos estériles.
Resulta evidente que, además de los constituyentes especialmente mencionados anteriormente, las formulaciones también pueden comprender otros agentes habituales en la técnica con respecto al tipo de formulación en particular; por tanto, por ejemplo, las formulaciones que son adecuadas para la administración oral pueden comprender aromatizantes.
Una cantidad terapéuticamente eficaz de un compuesto de fórmula I depende de varios factores, como por ejemplo, la edad y peso del animal, la enfermedad precisa que requiere tratamiento y su gravedad, la naturaleza de la formulación y el método de administración y es determinada finalmente por el doctor o veterinario responsable del tratamiento. Sin em bargo, una cantidad eficaz de un compuesto según la invención está generalmente en el intervalo de 0,1 a 100 mg/kg de peso corporal del receptor (mamífero) al día y, en particular, típicamente en el intervalo de 1 a 10 mg/kg de peso corporal al día. Por tanto, la cantidad real al día para un mamífero adulto que pesa 70 kg normalmente está entre 70 y 700 mg, donde esta cantidad puede administrarse como una dosis única al día o normalmente en una serie de dosis divididas (como por ejemplo, dos, tres, cuatro, cinco o seis) al día, de modo que la dosis total diaria sea la misma. Una cantidad eficaz de una sal o solvato, o de un derivado fisiológicamente funcional del mismo puede determinarse como la fracción de la canti dad eficaz del compuesto según la invención per se. Puede asumirse que son adecuadas dosis similares para el tratamiento de otras afecciones mencionadas anteriormente.
Un tratamiento combinado de este tipo se puede conseguir con la ayuda de una dispensación simultánea, consecutiva o independiente de los compuestos individuales del tratamiento. Los productos combinados de este tipo emplean los com puestos según la invención.
La invención además se refiere a medicamentos que comprenden al menos un compuesto de fórmula I y/o sales, tautómeros y estereoisómeros farmacéuticamente aceptables del mismo, incluidas sus mezclas en todas las proporciones, y al menos un principio activo adicional de un medicamento .
La invención también se refiere a un set (kit) compuesto de envases independientes de
(a) una cantidad eficaz de un compuesto de fórmula I y/o sales, tautómeros y estereoisómeros farmacéuticamente acep tables del mismo, incluidas sus mezclas en todas las proporciones,
y
(b) una cantidad eficaz de un principio activo adicional de un medicamento.
El set comprende recipientes adecuados, como cajas, botellas individuales, bolsas o ampollas. El set puede, por ejemplo, comprender ampollas independientes, cada una con una cantidad efectiva de un compuesto de fórmula I y/o sales, solvatos y estereoisómeros farmacéuticamente aceptables del mismo, incluidas sus mezclas en todas las proporciones, y una cantidad eficaz de un principio activo adicional de un medicamento en forma disuelta o liofilizada.
«Tratar» según se usa en este documento, significa el alivio, total o parcial, de los síntomas asociados con un trastorno o enfermedad, o la ralentización o cese de la progresión adicional o empeoramiento de estos síntomas, o la prevención o profilaxis de la enfermedad o trastorno en un sujeto en riesgo de desarrollar la enfermedad o trastorno.
El término «cantidad eficaz» en conexión con un compuesto de fórmula (I) puede significar una cantidad capaz de aliviar, totalmente o en parte, los síntomas asociados con un trastorno o enfermedad, o ralentizar o detener la progresión adicional o empeoramiento de estos síntomas, o prevenir o proporcionar profilaxis para la enfermedad o trastorno en un sujeto que tiene o está en riesgo de desarrollar una enfermedad descrita en este documento, como afecciones inflamatorias, afeccio nes inmunológicas, cáncer o afecciones metabólicas.
En una realización, una cantidad eficaz de un compuesto de fórmula (I) es una cantidad que inhibe la PDHK en una célula, como por ejemplo, in vitro o in vivo. En algunas realizaciones, la cantidad eficaz del compuesto de fórmula (I) inhibe la PDHK en una célula en un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 99 %, en comparación con la actividad de la PDHK en una célula no tratada. La cantidad eficaz del compuesto de fórmula (I), por ejemplo en una composición farmacéutica, puede estar a un nivel que ejercerá el efecto deseado; por ejemplo, aproximadamente de 0,005 mg/kg de peso corporal de un sujeto a aproximadamente 10 mg/kg de peso corporal de un sujeto en dosis unitaria tanto para admi nistración oral como parenteral.
USO
La presente invención se refiere específicamente a compuestos para el uso de la fórmula I y sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones, para el trata miento del cáncer, diabetes e isquemia cardíaca.
Asimismo, la presente invención se refiere a compuestos para el uso de la fórmula I y sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones, para el tratamiento del síndrome de resistencia a la insulina, síndrome metabólico, hiperglucemia, dislipidemia, ateroesclerosis, insuficiencia car díaca, cardiomiopatía, isquemia miocárdica, hiperlactacidemia, enfermedad mitocondrial, encefalomiopatía mitocondrial.
La presente invención se refiere específicamente a métodos para el tratamiento o prevención del cáncer, diabetes e isque mia cardíaca, que comprende la administración a un sujeto que lo necesita de una cantidad eficaz de un compuesto de fórmula I o una sal, tautómero, estereoisómero o solvato farmacéuticamente aceptable del mismo.
Además abarca el uso de los compuestos de fórmula I y/o las sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos para la preparación de un medicamento para el tratamiento o prevención de una enfermedad inducida por PDHK o una afección inducida por PDHK en un mamífero, en el cual en este método se administra una cantidad terapéuticamente eficaz de un compuesto según la invención a un mamífero enfermo que necesita dicho trata miento. La cantidad terapéutica varía según la enfermedad específica y puede ser determinada por la persona experta en la materia sin excesivo esfuerzo.
La expresión «enfermedades o afecciones inducidas por PDHK» se refiere a situaciones patológicas que dependen de la actividad de la PDHK. Entre las enfermedades asociadas con la actividad de PDHK se incluyen cáncer, diabetes e isquemia cardíaca.
La presente invención se refiere específicamente a compuestos para el uso de fórmula I y sales, tautómeros y estereoisó meros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones, para el tratamiento de enfermedades en las que la inhibición, regulación y/o inhibición de la modulación de PDHK tiene un papel.
La presente invención se refiere específicamente a compuestos de fórmula I y sales, tautómeros y estereoisómeros farma céuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones, para su uso para la inhibición de PDHK.
Los cánceres representativos para los que son útiles los compuestos de fórmula I para su tratamiento o prevención inclu yen, pero sin limitaciones, cáncer de cabeza, cuello, ojos, boca, garganta, esófago, bronquios, laringe, faringe, tórax, hueso, pulmón, colon, recto, estómago, próstata, vejiga urinaria, útero, cuello uterino, mama, ovarios, testículos u otros órganos reproductores, piel, tiroides, sangre, ganglios linfáticos, riñón, hígado, páncreas, cerebro, sistema nervioso central, tumores sólidos y tumores en la sangre.
Asimismo, los correspondientes cánceres para los que los compuestos de fórmula I son útiles para su tratamiento o pre vención incluyen cáncer de cerebro (gliomas), glioblastomas, leucemias, síndrome de Bannayan-Zonana, enfermedad de Cowden, enfermedad de Lhermitte-Duclos, cáncer de mama, cáncer de mama inflamatorio, tumor de Wilson, sarcoma de Ewing, rabdomiosarcoma, ependimoma, meduloblastoma, cáncer de colon, cabeza y cuello, riñón, pulmón, hígado, melanoma, de ovario, pancreático, de próstata, sarcoma, osteosarcoma, tumor de células gigantes de hueso y de tiroides.
Preferiblemente, la presente invención se refiere a un método en el que la enfermedad es un cáncer.
En especial preferiblemente la presente invención se refiere a un método en el que la enfermedad es un cáncer, en el que la administración es simultánea, secuencial o alternada con la administración de al menos un principio activo distinto.
Los compuestos descritos de fórmula I se pueden administrar en combinación con otros agentes terapéuticos conocidos, incluidos agentes antineoplásicos. Según se usa aquí, el término «agente antineoplásico» se refiere a cualquier agente que se administre a un paciente con cáncer para los fines de tratamiento del cáncer.
El tratamiento antineoplásico definido anteriormente se puede aplicar como monoterapia o puede implicar, además de los compuestos de fórmula I descritos en este documento, cirugía convencional, radioterapia o tratamiento farmacológico. Dicho tratamiento farmacológico, por ejemplo, una quimioterapia o una terapia dirigida, puede incluir uno o más, pero preferiblemente uno, de los siguientes agentes antitumorales:
Agentes alquilantes
como altretamina, bendamustina, busulfán, carmustina, clorambucilo, clormetina, ciclofosfamida, dacarbazina, ifosfamida, improsulfán, tosilato, lomustina, melfalán, mitobronitol, mitolactol, nimustina, ranimustina, temozolomida, tiotepa, treosulfán, mecloretamina, carbocuona; apazicuona, fotemustina, glufosfamida, palifosfamida, pipobromán, trofosfamida, uramustina, TH-3024, VAL-0834.
Compuestos de platino
como carboplatino, cisplatino, eptaplatino, hidrato de miriplatino, oxaliplatino, lobaplatino, nedaplatino, picoplatino, satraplatino; lobaplatino, nedaplatino, picoplatino, satraplatino.
Agentes que alteran el ADN
como amrubicina, bisantreno, decitabina, mitoxantrona, procarbazina, trabectedina, clofarabina;
amsacrina, brostalicina, pixantrona, laromustina13
Inhibidores de la topoisomerasa
como etopósido, irinotecán, razoxano, sobuzoxano, tenipósido, topotecán; amonafida, belotecán, acetato de eliptinio, voreloxina.
Modificadores de microtúbulos
como cabazitaxel, docetaxel, eribulina, ixabepilona, paclitaxel, vinblastina, vincristina, vinorelbina, vindesina, vinflunina; fosbretabulina, tesetaxel.
Antimetabolitos
como asparaginasa3, azacitidina, levofolinato de calcio, capecitabina, cladribina, citarabina, enocitabina, floxuridina, fludarabina, fluorouracilo, gemcitabina, mercaptopurina, metotrexato, nelarabina, pemetrexed, pralatrexato, azatioprina, tioguanina, carmofur;
doxifluridina, elacitarabina, raltitrexed, sapacitabina, tegafur23, trimetrexato.
Antibióticos antineoplásicos
como bleomicina, dactinomicina, doxorubicina, epirubicina, idarubicina, levamisol, miltefosina, mitomicina C, romidepsina, estreptozocina, valrubicina, zinostatina, zorubicina, daunorubicina, plicamicina; aclarubicina, peplomicina, pirarubicina.
Hormonas/antagonistas
como abarelix, abiraterona, bicalutamida, buserelina, calusterona, clorotrianiseno, degarelix, dexametasona, estradiol, fluocortolona fluoximesterona, flutamida, fulvestrant, goserelina, histrelina, leuprorelina, megestrol, mitotano, nafarelina, nandrolona, nilutamida, octreotida, prednisolona, raloxifeno, tamoxifeno, tirotropina alfa, toremifeno, trilostano, triptorelina, dietilestilbestrol;
acolbifeno, danazol, deslorelina, epitiostanol, orteronel, enzalutamida13
Inhibidores de la aromatasa
como aminoglutetimida, anastrozol, exemestano, fadrozol, letrozol, testolactona; formestano.
Moléculas pequeñas inhibidoras de quinasas
como crizotinib, dasatinib, erlotinib, imatinib, lapatinib, nilotinib, pazopanib, regorafenib, ruxolitinib, sorafenib, sunitinib, vandetanib, vemurafenib, bosutinib, gefitinib, axitinib;
afatinib, alisertib, dabrafenib, dacomitinib, dinaciclib, dovitinib, enzastaurina, nintedanib, lenvatinib, linifanib, linsitinib, masitinib, midostaurina, motesanib, neratinib, orantinib, perifosina, ponatinib, radotinib, rigosertib, tipifarnib, tivantinib, tivozanib, trametinib, pimasertib, alaninato de brivanib, cediranib, apatinib4, S-malato de cabozantinib13, ibrutinib13, icotinib4, buparlisib2, cipatinib4, cobimetinib13 idelalisib13 fedratinib1, XL-6474
Fotosensibilizadores
como metoxsaleno3;
porfímero de sodio, talaporfina, temoporfina.
Anticuerpos
como alemtuzumab, besilesomab, brentuximab vedotina, cetuximab, denosumab, ipilimumab, ofatumumab, panitumumab, rituximab, tositumomab; trastuzumab, bevacizumab, pertuzumab23;
catumaxomab, elotuzumab, epratuzumab, farletuzumab, mogamulizumab, necitumumab, nimotuzumab, obinutuzumab, ocaratuzumab, oregovomab, ramucirumab, rilotumumab, siltuximab, tocilizumab, zalutumumab, zanolimumab, matuzumab, dalotuzumab123 onartuzumab13, racotumomab1, tabalumab13, EMD-5257974, nivolumab13;
Citoquinas
como aldesleuquina, interferón alfa2, interferón alfa2a3, interferón alfa2b23; celmoleuquina, tasonermina, teceleuquina, oprelvequina1’3, interferón beta-1a recombinante4.
Fármacos conjugados
como denileuquina diftitox, ibritumomab tiuxetán, iobenguano 123I, prednimustina, trastuzumab emtansina, estramustina, gemtuzumab, ozogamicina, aflibercept;
cintredequina besudotox, edotreotida, inotuzumab ozogamicina, naptumomab estafenatox, oportuzumab monatox, tecnecio (99mTc) arcitumomab13, vintafolida13
Vacunas
como sipuleucel3; vitespen3, emepepimut-S3, oncoVAX4, rindopepimut3, troVax4, MGN-16014, MGN-17034.
Varios
alitretinoína, bexaroteno, bortezomib, everolimús, ácido ibandrónico, imiquimod, lenalidomida, lentinano, metirosina, mifamurtida, ácido pamidrónico, pegaspargasa, pentostatina, sipuleucel3, sizofirano, tamibaroteno, temsirolimús, talidomida, tretinoína, vismodegib, ácido zoledrónico, vorinostat; celecoxib, cilengitida, entinostat, etanidazol, ganetespib, idronoxilo, iniparib, ixazomib, lonidamina, nimorazol, panobinostat, peretinoína, plitidepsina, pomalidomida, procodazol, ridaforolimús, tasquinimod, telotristat, timalfasina, tirapazamina, tosedostat, trabedersén, ubenimex, valspodar, gendicina4, picibanilo4, reolisina4, clorhidrato de retaspimicina13, trebananib23, virulizina4, carfilzomib13, endostatina4, immucothel4, belinostat3, MGN-17034.
1 DCI Prop. (Denominación Común Internacional Propuesta)
2 DCI Rec. (Denominación Común Internacional Recomendada)
3 USAN (United States Adopted Name [Nombre adoptado en Estados Unidos])
4 no DCI.
Las siguientes abreviaturas se refieren respectivamente a las definiciones que aparecen a continuación:
ac (acuoso), h (hora), g (gramo), l (litro), mg (miligramo), MHz (megahercio), min (minuto), mm (milímetro), mmol (milimol), mM (milimolar), p.f. (punto de fusión), eq (equivalente), ml (mililitro), l (microlitro), ACN (acetonitrilo), AcOH (ácido acético), CDCl3 (cloroformo deuterado), CD3OD (metanol deuterado), CH3CN (acetonitrilo), c-hex (ciclohexano), DCC (diciclohexil carbodiimida), DCM (diclorometano), DIC (diisopropil carbodiimida), DIEA (diisopropiletil-amina), DMF (dimetilformamida), DMSO (dimetilsulfóxido), DMSO-d6 (dimetilsulfóxido deuterado), EDC (1-(3-dimetil-amino-propil)-3-etilcarbodiimida), ESI (ionización por electropulverización), EtOAc (etil acetato), Et2O (dietil éter), EtOH (etanol), HATU (hexafluorofosfato de dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio), HPLC (cromatografía líquida de alta resolución), i-PrOH (2-propanol), K2CO3 (carbonato de potasio), CL (cromatografía líquida), MeOH (metanol), MgSO4 (sulfato de mag nesio), EM (espectrometría de masas), MTBe (éter metil terc-butílico), NaHCO3 (bicarbonato de sodio), NaBH4 (borohidruro de sodio), NMM (N-metil morfolina), RMN (resonancia magnética nuclear), PyBOP (hexafluorofosfato de benzotriazol-1-iloxi-tris-pirrolidino-fosfonio), TA (temperatura ambiente), tR (tiempo de retención), SPE (extracción en fase sólida), TBTU (tetrafluoroborato de 2-(1-H-benzotriazol-1-il)-1,1,3,3-tetrametiluromio), TEA (trietilamina), TFA (ácido trifluoroacético), THF (tetrahidrofurano), TLC (cromatografía en capa fina), UV (ultravioleta).
Descripción de los ensayos in vitro
Abreviaturas:
GST = Glutatión-S-transferasa
FRET = Transferencia de energía por resonancia de fluorescencia
HTRF® = Fluorescencia homogénea resuelta en el tiempo
HEPES = Tampón ácido 4-(2-hidroxietil)-1-piperazin etanosulfónico
DTT = Ditiotreitol
BSA = Albúmina sérica bovina
CHAPS = 3-[(3-colamidopropil)dimetilamonio]-1-propanosulfonato
Prueba de actividad bioquímica de PDHK2: ensayo de inactivación de PDC
El ensayo de actividad bioquímica de PDHK2 se basa en la inactivación del PDC a través de la fosforilación de PDHK2. El ensayo se realiza en dos etapas: la reacción enzimática de PDHK2 en la que el PDC aislado es fosforilado por PDHK2 con ATP como cosustrato y el ensayo de actividad PDC en el que el piruvato y NAD se transforman en acetil-CoA y NADH. La actividad PDC se correlaciona con el aumento de NADH y por tanto es detectable directamente a través del aumento de la señal de fluorescencia (excitación: 340 nm, emisión: 450 nm). La inhibición de PDHK2 tiene como consecuencia un estado
de fosforilación más bajo y por tanto una menor disminución de la actividad de PDC y un mayor aumento en la señal de fluorescencia de NADH.
El ensayo de inactivación de PDC se realiza en placas de microtitulación Greiner de 384 pocillos y se usa para la detección de alto rendimiento. Se incuban 4 |jl de PDHK2 (humana, recombinante, Carna Bioscience, concentración final: 10 ng/|jl a 137 nM) y PDC (aislado de corazón de cerdo, Sigma-Aldrich, concentración final: 20 mU/ml) en ausencia o presencia del compuesto problema (10 concentraciones de dilución) durante 30 min a temperatura ambiente en tampón quinasa (tampón fosfato de potasio 15 mM, pH 7,0; KCl 60 mM; Dt T 1,5 mM; MgCh 2,5 mM; BSA al 0,0125 % (p/v); Pluronic F-68 al 0,125 %). La reacción quinasa se inicia mediante la adición de 4 j l de solución sustrato ATP (c.f. 5 jM en el tampón quinasa). Después de 30 min de incubación a 37 °C se añadieron 40 j l de solución de reacción PDC (Tris/HCl 100 mM, pH 7,8; EDTA 0,5 mM; MgCL 1 mM; NaF 50 mM; coenzima A 0,25 mM; piruvato 5 mM; NAD 1 mM; DTT 5 mM; pirofosfato de tiamina 1 mM). La primera medición de la fluorescencia se realiza en un lector Envision de Perkin Elmer (excitación: 340 nm, emisión: 450 nm). La reacción se incuba durante 45 min a temperatura ambiente. Después se realiza una segunda medición de la fluorescencia y se calcula la actividad PDC mediante la diferencia entre ambas mediciones. Como valor total para el ensayo de PDHK2 se usa la reacción de PDHK2 sin inhibidor. El valor cero farmacológico usado es DCA (Sigma-Aldrich) a una concentración final de 3 mM. Los valores de inhibición (IC50) se determinaron usando el programa Symyx Assay Explorer® o Condosseo® de GeneData.
Calorimetría de titulación isotérmica
Las mediciones de calorimetría de titulación isotérmica (ITC, por sus siglas en inglés) se realizaron con un microcalorímetro VP-ITC (Microcal, LLC/GE Healthcare Bio-Sciences AB, Uppsala, Suecia). Las titulaciones en general se realizaron titu lando la proteína (50 jM ) con respecto al compuesto problema (5 jM ) en inyecciones de 12 jl. Todos los experimentos de unión se realizaron a 30 °C. En general, los compuestos problema se diluyeron a partir de soluciones madre de DMSO en el tampón de medición con una concentración final máxima del 1 % de DMSO. El tampón de medición era HEPES 20 mM, KCl 135 mM, TCEP 1 mM, MgCl22 mM, NaH2PO415 mM, pH 7,5. La PDHK2 humana (12-407) se produjo en E. co licomo proteína con etiqueta His y se purificó mediante cromatografía de afinidad. La etiqueta se eliminó mediante proteólisis específica de lado. Antes de la titulación, el tampón de proteína se cambió por el tampón de medición que contenía la misma concentración de DMSO que la dilución del compuesto problema. El análisis de los datos de la ITC se realizó usando el software de calorimetría Origin 7 del mismo proveedor. Para la mayoría de las mediciones se asumió un modelo de unión de un único sitio de unión. Según el modelo matemático aplicado es posible calcular la constante de unión (Ka), la entalpía de unión observada (AH°bs) y la estequiometría (N) del complejo formado. Antes del análisis se corrigieron los datos sin procesar con respecto al calor de dilución mediante la extrapolación del valor de saturación desde el final de la titulación. Para permitir la comparación directa entre las correspondientes series experimentales y las preparaciones proteicas, se corrigió la concentración de proteína referenciando las titulaciones al inhibidor estándar con buen comportamiento. Los valores aparentes de estequiometría definieron la fracción de la proteína que compite por la unión y compensaron errores relativos en las mediciones de la concentración de proteína. Esta concentración corregida de proteína se usó para confi gurar la serie de experimentos de ITC con compuestos problema. Cualquier desviación de la estequiometría ideal 1:1 observada aquí se atribuyó a errores en la concentración de los compuestos. Esta concentración nominal de compuesto se corrigió también para obtener una estequiometría 1:1 en el ajuste.
Ensayo celular para la determinación de las actividades de los compuestos
Las actividades de los compuestos se determinaron en un ensayo de inmunofluorescencia celular. Las células HEK293T humanas se dispusieron en placas de 384 pocillos negras con fondo transparente y se dejaron crecer toda la noche.
Al día siguiente, los compuestos problema se añadieron a los pocillos y las placas se incubaron durante 5 horas. Después de esto, las células se fijaron con formaldehído, se permeabilizaron y se bloquearon. Se añadió el anticuerpo primario, anti-PDH-E1alfa (pSer300), AP1064 (Merck Millipore) y se incubó durante toda la noche en los pocillos de la placa. A continua ción, las células se lavaron y se añadió el anticuerpo secundario, Alexa Fluor 488, anticuerpo de cabra anticonejo (A-11008, Invitrogen) junto con Hoechst 33258 (H3569, Invitrogen) y se incubó durante 1 hora en los pocillos de la placa. Por último, las células se lavaron y las placas se midieron en el citómetro de escáner láser acumen hci (TTpLabtech).
Los datos sin procesar se normalizaron frente a un control inhibidor farmacológico y se generaron curvas de dosis-res puesta representando los valores porcentuales del efecto usando el paquete de software Genedata Screener (Genedata).
Anteriormente y a continuación, todas las temperaturas se indican en °C. En los siguientes ejemplos, «proceso convencio nal» significa que: se añade agua si es necesario, se ajusta el pH, si es necesario, a valores de entre 2 y 10, dependiendo de la constitución del producto final, la mezcla se extrae con acetato de etilo o diclorometano, las fases se separan, la fase orgánica se seca sobre sulfato sódico y se evapora, y el residuo se purifica mediante cromatografía en gel de sílice y/o mediante cristalización.
La RMN 1H se registró en un espectrómetro Bruker DPX-300, DRX-400, AVII-400 o BRUKER 500 MHz, usando la señal residual de solvente deuterado como referencia interna. Los desplazamientos químicos (5) se documentan en ppm con
respecto a la señal residual de solvente (5 = 2,49 ppm para RMN 1H en DMSO-d6). Los datos de RMN 1H se documentan como sigue: desplazamiento químico (multiplicidad, constantes de acoplamiento y número de hidrógenos). La multiplicidad se abrevia como sigue: s (singlete), d (doblete), t (triplete), c (cuadruplete), m (multiplete), a (ancho).
Las configuraciones absolutas se han determinado mediante análisis de estructura por rayos X.
Ejemplos
Condiciones de HPLC/EM:
Gradiente: 3,3 min; 0 min 4 % de B, 2,8 min 100 % de B, 3,3 min 100 % de B; caudal: 2,4 ml/min;
A: agua HCOOH (0,05 % vol.); B: acetonitrilo HCOOH (0,04 % vol.)
Columna: Chromolith SpeedROD RP-18e 50-4,6
Longitud de onda: 220 nm
Condiciones de HPLC/EM (B):
Gradiente: 0 min: 5 % de B, 8 min: 100% de B, 8,1 min: 100% de B, 8,5 min: 5 % de B, 10 min 5 % de B; caudal: 2,0 ml/min
A: agua TFA (0,1 % vol.); B: acetonitrilo TFA (0,1 % vol.)
Columna: XBridge C8, 3,5 |jm; 4,6 * 50 mm
Longitud de onda: 220 nm
Esquema de reacción:

Metil-p-tolil-amida del ácido 1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-carboxílico («A1»)

1.1 4-[(4-Metilfenil)carbamoil]piperidin-1-carboxilato de ferc-butilo
Se disolvieron ácido 1-[(ferc-butoxi)carbonil]piperidin-4-carboxílico (500,0 mg; 2,181 mmol) y p-tolilamina (303,8 mg; 2,835 mmol) en acetato de etilo (10,0 ml). La solución transparente se enfrió hasta 0-5 °C en un baño de agua con hielo. Se añadió trietilamina (1,06 ml; 7,633 mmol) en una única porción y se añadió gota a gota anhídrido propilfosfónico (solu ción al 50 % en acetato de etilo; 1,56 ml; 2,617 mmol) a 0-5 °C. La solución transparente se agitó durante otros 15 min a esta temperatura, se calentó posteriormente hasta temperatura ambiente y se agitó durante 14 h. La mezcla de reacción
se diluyó con acetato de etilo y se añadió a una solución saturada de carbonato sódico. Se separó la capa orgánica, se lavó con una solución saturada de carbonato sódico y con una solución de ácido cítrico al 10 %. La capa orgánica se secó con sulfato sódico, se filtró mediante succión y se evaporó hasta sequedad. Rendimiento: 690 mg (99 %) de un sólido de color beis; CL/EM, tR: 2,31 min, (M+H-t-Bu) 263,0.
1.2 4-[metil(4-metilfenil)carbamoil]piperidin-1-carboxilato de ferc-butilo
El compuesto 1.1 (100,0 mg; 0,314 mmol) se disolvió en DMF seco (3,0 ml) y se añadió carbonato de cesio (307,0 mg; 0,942 mmol). A la suspensión se añadió gota a gota yodometano (25 pl; 0,408 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante toda la noche. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con salmuera, se secaron con sulfato sódico, se filtraron mediante succión y se evaporaron hasta sequedad. Rendimiento: 65 mg (62 %) de un sólido de color beis; CL/EM, tR: 2,41 min, (M+H-t-Bu) 277,1.
1.3 N-Metil-N-(4-metilfenil)piperidin-4-carboxamida, clorhidrato
Se disolvió el compuesto 1.2 (65,0 mg; 0,196 mmol) en 1,4-dioxano seco (3,0 ml). Se añadió HCl en metanol (1,25 M; 1,5 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 14 h. El metanol se evaporó y el sólido precipitado se filtró mediante succión y se secó durante toda la noche a alto vacío. Rendimiento: 50 mg (95 %) de un sólido blanque cino; CL/EM, tR: 1,22 min; (M+H) 233,2.
1.4 Metil-p-tolil-amida del ácido 1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-carboxílico
Se disolvieron el compuesto 1.3 (50,0 mg; 0,186 mmol) y ácido (R)-3,3,3-trifluoro-2-hidroxi-2-metil-propiónico (60,0 mg; 0,372 mmol) en DMF (3,0 ml) y la solución se enfrió con un baño de agua con hielo. Se añadió N-etildiisopropilamina (0,22 ml; 1,302 mmol) seguido de la adición de hexafluorurofosfato de [dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio (155,6 mg; 0,409 mmol). La mezcla de reacción se agitó durante otros 15 min a esta temperatura, posteriormente se calentó hasta temperatura ambiente y se agitó durante 14 h. La solución marrón se diluyó con agua, se añadió una solución saturada de carbonato sódico y la fase acuosa se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron con sulfato sódico, se filtraron mediante succión y se evaporaron hasta sequedad. El residuo oleoso de color naranja se purificó mediante cromatografía. Rendimiento: 56 mg (81 %) de un sólido de color beis; CL/EM, tR: 2,06 min; (M+H) 373,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,30-7,22 (m, 2H), 7,22 7,13 (m, 2H), 6,69 (s, 1H), 4,50-4,35 (m, 2H), 3,14 (s, 3H), 2,74 - 2,60 (m, 2H), 2,58-2,45 (m, 1H), 2,35 (s, 3H), 1,66-1,53 (m, 4H), 1,51 (s, 3H).
Los siguientes compuestos se prepararon de forma análoga:
Ejemplo 2
(2-Fluoro-4-metil-fenil)-metil-amida del ácido 1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-carboxílico («A2»)
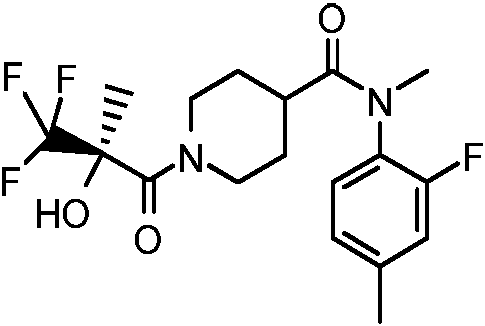
Rendimiento: 71 mg (61 %) de sólido incoloro; CL/EM, tR: 2,11 min; (M+H) 391,2; RMN 1H (400 MHz, DMSO-d6) 87,38 (t, J = 8,3 Hz, 1H), 7,28-7,20 (m, 1H), 7,16-7,10 (m, 1H), 7,02 (s, 1H), 4,76-4,15 (m, 2H), 3,07 (s, 3H), 2,94-2,58 (m, 1H), 2,44 2,25 (m, 5H), 1,67-1,35 (m, 7H).
Ejemplo 3
(2-cloro-4-trifluorometil-fenil)-metil-amida del ácido 1 -((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-pi peridin-4-carboxílico («A3»)

Rendimiento: 15 mg (11%) de un sólido de color beis; CL/EM, tR: 2,30 min; (M+H) 461,1/463,1; RMN 1H (400 MHz, DMSO-d6) 88,17-8,10 (m, 1H), 7,91-7,81 (m, 2H), 7,02 (s, 1H), 4,72-4,14 (m, 2H), 3,09 (s, 3H), 2,96-2,67 (m, 1H), 2,58-2,32 (m, 1H), 2,27-2,14 (m, 1H), 1,72-1,37 (m, 7H).
Ejemplo 4
(4-Ciano-fenil)-metil-amida del ácido 1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-carboxílico («A4»)

Rendimiento: 16 mg (16 %) de un sólido blanquecino; CL/EM, tR: 1,87 min; (M+H) 384,2; RMN 1H (400 MHz, DMSO-d6) 8 7,97-7,90 (m, 2H), 7,61-7,56 (m, 2H), 7,02 (s, 1H), 4,77-4,13 (m, 2H), 3,22 (s, 3H), 2,97-2,40 (m, 3H), 1,72-1,37 (m, 7H). Esquema de reacción:

Ejemplo 5
(4-Cloro-fenil)-metil-amida del ácido 1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-carboxílico («A5»)

5.1 4-[(4-Clorofenil)(metil)carbamoil]piperidin-1-carboxilato de ferc-butilo
Se disolvieron ácido 1-[(terc-butoxi)carbonil]piperidin-4-carboxílico (200,0 mg; 0,872 mmol) y 4-cloro-N-metilanilina (0,136 ml; 1,090 mmol) en acetato de etilo (10 ml). La solución transparente se enfrió hasta 0-5 °C en un baño de agua con hielo y se añadió trietilamina (0,42 ml; 3,053 mmol) en una única porción. Se añadió gota a gota anhídrido propilfosfónico (solución al 50 % en acetato de etilo; 0,62 ml; 1,047 mmol) a 0-5 °C y la solución transparente se agitó durante 15 minutos más a esta temperatura. La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 29 h. La solución transparente se diluyó con acetato de etilo y se extrajo con solución saturada de carbonato sódico. La capa orgánica se lavó con solución saturada de carbonato sódico y con solución de ácido cítrico al 10 %, se secó con sulfato sódico, se filtró mediante succión y se evaporó hasta sequedad. El residuo oleoso de color amarillo se purificó mediante cromatografía ultrarrápida (gel de sílice Si5080 g, CombiFlash Companion RF). Rendimiento: 258 mg (84 %) de sólido incoloro; CL/EM, tR: 2,39 min, (M+H-t-Bu) 297,0/299,0.
5.2 Clorhidrato de N-(4-clorofenil)-N-metilpiperidin-4-carboxamida
Se resuspendió el compuesto 1.1 (258,0 mg; 0,731 mmol) en solución de cloruro de hidrógeno (4 M en dioxano; 4,0 ml) y se agitó a temperatura ambiente durante 2 h. La solución transparente se evaporó hasta sequedad y el residuo se utilizó sin purificación adicional. Rendimiento: 211 mg de aceite incoloro.
5.3 (4-cloro-fenil)-metil-amida del ácido 1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-carboxílico
Se disolvieron el compuesto 1.2 (211,0 mg; 0,730 mmol) y ácido (R)-3,3,3-trifluoro-2-hidroxi-2-metil-propiónico (230,7 mg; 1,460 mmol) en DMF (5,0 ml) y se enfrió hasta 0 °C. Se añadieron N-etildiisopropilamina (0,86 ml; 5,085 mmol) y hexafluorofosfato de [dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio (607,7 mg; 1,598 mmol) y la mezcla de reacción se agitó durante 15 min a esta temperatura, se calentó hasta temperatura ambiente y se agitó durante 17 h. La solución de color marrón se diluyó con agua, se añadió solución saturada de carbonato sódico y se extrajo la capa acuosa con acetato de etilo. Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron con sulfato sódico, se filtraron mediante succión y se evaporaron hasta sequedad. El residuo oleoso se purificó mediante cromatografía en fase inversa. Rendimiento: 160 mg (56 %) de sólido incoloro; CL/EM, tR: 2,12 min; (M+H) 393,1/395,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,52-7,45 (m, 2H), 7,44-7,37 (m, 2H), 6,76 (s, 1H), 4,71-4,58 (m, 2H), 4,28-4,03 (m, 1H), 2,86-2,69 (m, 5H), 1,81-1,64 (m, 4H), 1,56 (s, 3H).
Ejemplo 6
N,3-Dimetil-N-(4-metilfenil)-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida; mezcla de diastereómeros («A6»)

Preparación según los procedimientos descritos para el ejemplo 1. Rendimiento: 75 mg (22 %) de un sólido blanquecino; CL/EM, tR: 2,15 min; (M+H) 387,2.
Ejemplos 7 y 8
Preparación de (3S,4S)-N,3-dimetil-N-(4-metilfenil)-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida («A7»)

y (3R,4R)-N,3-dimetil-N-(4-metilfenil)-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida («A8»)

La separación preparativa de los diastereoisómeros del ejemplo 6 se realizó mediante SFC (columna: ChiralPak IA; eluyente: CO2 :metanol - 90:10). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disolvie ron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A7»: 18 mg de un sólido incoloro; CL/EM, tR: 2,15 min; (M+H) 387,2; RMN 1H (400 MHz, DMSO-d6/TFA, 90 °C) 87,28 7,20 (m, 2H), 7,17-7,09 (m, 2H), 4,12-3,96 (m, 1H), 3,88 (dd, J = 12,8, 7,2 Hz, 1H), 3,52 (ddd, J = 12,7, 8,0, 3,8 Hz, 1H), 3,39-3,23 (m, 1H), 3,21-3,10 (m, 3H), 2,80-2,68 (m, 1H), 2,40-2,30 (m, 3H), 1,92-1,81 (m, 1H), 1,81-1,69 (m, 1H), 1,57-1,40 (m, 4H), 0,86 (d, J = 7,0 Hz, 3H).
«A8»: 24 mg de un sólido incoloro; CL/EM, tR: 2,15 min; (M+H) 387,2; RMN 1H (400 MHz, DMSO-d6, 120 °C) 87,28-7,18 (m, 2H), 7,18-7,08 (m, 2H), 6,45 (s, 1H), 4,03-3,91 (m, 1H), 3,80 (dd, J = 12,9, 7,3 Hz, 1H), 3,51 (ddd, J = 13,4, 8,0, 3,8 Hz, 1H), 3,37-3,25 (m, 1H), 3,14 (s, 3H), 2,76-2,68 (m, 1H), 2,33 (s, 3H), 1,89-1,77 (m, 1H), 1,77-1,66 (m, 1H), 1,57-1,38 (m, 4H), 0,84 (d, J = 7,0 Hz, 3H).
Los siguientes compuestos se prepararon de forma análoga:
Ejemplo 9
N-(4-Clorofenil)-N,3-dimetil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida; mezcla de diastereómeros («A9»)

Rendimiento: 110 mg (41 %) de un sólido blanquecino; CL/EM, tR: 2,19 min, (M+H) 407,1/409,2.
Ejemplos 10 y 11
Preparación de (3S,4S)-N-(4-clorofenil)-N,3-dimetil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida («A10»)

y (3R,4R)-N-(4-clorofenil)-N,3-dimetil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida («A11»)

La separación preparativa de los diastereoisómeros del ejemplo 9 se realizó mediante SFC (columna: ChiralPak IA; eluyente: CO2 :metanol - 80:20). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disolvie ron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A10»: 34 mg de un sólido incoloro; CL/EM, tR: 2,20 min; (M+H) 407,1/409,1; RMN 1H (400 MHz, DMSO-da, 90 °C) 87,51 7,44 (m, 2H), 7,35-7,28 (m, 2H), 6,62 (s, 1H), 4,11-3,95 (m, 1H), 3,87 (dd, J = 13,0, 6,7 Hz, 1H), 3,51-3,39 (m, 1H), 3,33 3,19 (m, 1H), 3,16 (s, 3H), 2,78-2,67 (m, 1H), 1,90-1,69 (m, 2H), 1,52-1,38 (m, 4H), 0,83 (d, J = 7,0 Hz, 3H).
«A11»: 30 mg de un sólido incoloro; CL/EM, tR: 2,21 min; (M+H) 407,1/409,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,50 7,44 (m, 2H), 7,34-7,28 (m, 2H), 6,63 (s, 1H), 4,11-3,95 (m, 1H), 3,83 (dd, J = 12,9, 7,0 Hz, 1H), 3,53-3,43 (m, 1H), 3,36 3,22 (m, 1H), 3,16 (s, 3H), 2,78-2,70 (m, 1H), 1,90-1,69 (m, 2H), 1,54-1,41 (m, 4H), 0,83 (d, J = 6,9 Hz, 3H).
Ejemplo 12
N-(2-Fluoro-4-metilfenil)-N,3-dimetil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida; mezcla de diastereómeros («A12»)

Rendimiento: 79,5 mg (37 %) de un sólido blanquecino; CL/EM, tR: 2,20 min; (M+H) 405,2.
Ejemplos 13 y 14
Preparación de (3S,4S)-N-(2-fluoro-4-metilfenil)-N,3-dimetil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida («A13»)

y (3R,4R)-N-(2-fluoro-4-metilfenil)-N,3-dimetil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida («A14»)

La separación preparativa de los diastereoisómeros del ejemplo 12 se realizó mediante SFC (columna: ChiralPak AD-H; eluyente: CO2 :metanol - 85:15). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se di solvieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A13»: 26 mg de un sólido incoloro; CL/EM, tR: 2,19 min; (M+H) 405,2; RMN 1H (400 MHz, DMSO-d6) 87,41-7,31 (m, 1H), 7,26-7,20 (m, 1H), 7,15-7,07 (m, 1H), 7,03-6,90 (m, 1H), 4,40-4,12 (m, 1H), 3,88-3,65 (m, 1H), 3,56-3,35 (m, 2H), 3,07 (d, J = 8,3 Hz, 3H), 2,64-2,51 (m, 1H), 2,35 (s, 3H), 1,92-1,57 (m, 2H), 1,47 (s, 3H), 1,44-1,34 (m, 1H), 0,88-0,70 (m, 3H).
«A14»: 22 mg de un sólido incoloro; CL/EM, tR: 2,20 min; (M+H) 405,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,28 (t, J = 8,2 Hz, 1H), 7,16 (d, J = 11,4 Hz, 1H), 7,09 (d, J = 8,0 Hz, 1H), 6,64 (s, 1H), 4,11-3,91 (m, 1H), 3,88-3,76 (m, 1H), 3,60 3,47 (m, 1H), 3,42-3,24 (m, 1H), 3,12 (s, 3H), 2,72-2,51 (m, 1H), 2,37 (s, 3H), 1,90-1,64 (m, 2H), 1,53 (s, 3H), 1,50-1,41 (m, 1H), 0,84 (d, J = 7,0 Hz, 3H).
Ejemplo 15
N-[2-Cloro-4-(trifluorometil)fenil]-N,3-dimetil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-carboxamida; mezcla de diastereómeros («A15»)

Rendimiento: 35 mg (25 %) de aceite incoloro; CL/EM, tR: 2,38 min, (M+H) 475,1/477,1.
Esquema de reacción:

Ejemplo 16
4-Cloro-N-metil-N-{1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida («A16»)

16.1 4-(N-Metil-4-clorobenzamido)piperidin-1-carboxilato de ferc-butilo
Se disolvió éster ferc-butílico del ácido 4-metilamino-piperidin-1-carboxílico (190,0 mg; 0,887 mmol) en diclorometano (10,0 ml), se trató con cloruro de 4-clorobenzoílo (170,7 mg; 0,975 mmol) y trietilamina (0,18 ml; 1,330 mmol) y se agitó durante 14 h a temperatura ambiente. La reacción se detuvo a continuación con salmuera y la capa acuosa se extrajo con diclorometano. Las capas orgánicas combinadas se secaron sobre sulfato de sodio, se filtraron y el solvente se evaporó a presión reducida. El residuo se purificó mediante cromatografía. Rendimiento: 295 mg (94 %) de un sólido blanquecino; CL/EM, tR: 2,34 min, (M+H-t-Bu) 297,1/299,1.
16.2 Clorhidrato de 4-cloro-N-metil-N-(piperidin-4-il)benzamida
La escisión del grupo Boc del compuesto 16.1 (295,0 mg; 0,836 mmol) se llevó a cabo como se describe para el ejemplo 1.3. Rendimiento: 233 mg (96 %) de un sólido blanquecino; CL/EM, tR: 1,17 min, (M+H) 253,1/255,1.
16.34-Cloro-N-metil-N-{1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida
Preparación con compuesto 16.2 (233,0 mg; 0,806 mmol) y ácido (R)-3,3,3-trifluoro-2-hidroxi-2-metil-propiónico (156,1 mg; 0,968 mmol; 120,00 mol%) como se describe para el compuesto 1.4. El producto sin procesar se purificó mediante HPLC preparativa (columna: sunfire). Rendimiento: 175 mg (55 %) de sólido incoloro; CL/EM, tR: 1,99 min; (M+H) 393,1/395,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,52-7,45 (m, 2H), 7,44-7,37 (m, 2H), 6,76 (s, 1H), 4,71-4,58 (m, 2H), 4,28-4,03 (m, 1H), 2,86-2,69 (m, 5H), 1,81-1,64 (m, 4H), 1,56 (s, 3H).
Los siguientes compuestos se prepararon de forma análoga:
Ejemplo 17
N,4-Dimetil-N-{1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida («A17»)

Rendimiento: 200,5 mg (57 %) de un sólido blanquecino; CL/EM, tR: 1,93 min; (M+H) 373,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,30-7,18 (m, 4H), 6,76 (s, 1H), 4,73-4,59 (m, 2H), 4,27-4,08 (m, 1H), 2,79 (s, 3H), 2,78-2,67 (m, 2H), 2,36 (s, 3H), 1,80-1,64 (m, 4H), 1,56 (s, 3H).
Ejemplo 18
2-Fluoro-4,N-dimetil-N-{1-[(R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil]-piperidin-4-il}-benzamida («A18»)

Rendimiento: 82 mg (21 %) de un sólido blanquecino; CL/EM, tR: 1,98 min; (M+H) 391,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,22 (t, J = 7,6 Hz, 1H), 7,12-7,04 (m, 2H), 6,76 (s, 1H), 4,76-4,55 (m, 2H), 3,95-3,12 (m, 1H), 2,95-2,58 (m, 5H), 2,36 (s, 3H), 1,83-1,61 (m, 4H), 1,55 (s, 3H).
Ejemplo 19
2-Cloro-N-metil-N-{1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}-4-(trifluorometil)benzamida («A19»)

Rendimiento: 43 mg (11 %) de un sólido blanquecino; CL/EM, tR: 2,24 min; (M+H) 461,1/463,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,89 (s, 1H), 7,81-7,73 (m, 1H), 7,65-7,56 (m, 1H), 6,83-6,70 (m, 1H), 4,79-4,49 (m, 3H), 2,99-2,84 (m, 3H), 2,72-2,61 (m, 2H), 1,86-1,63 (m, 4H), 1,60-1,48 (m, 3H).
Esquema de reacción:

Ejemplo 20
4-Cloro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diastereoisómeros («A20»)

Preparación según los procedimientos descritos para el ejemplo 16. Rendimiento: 135 mg (56 %) de un sólido blanquecino; CL/EM, tR: 2,08 min, (M+H) 407,1/409,1.
Ejemplos 21 y 22
Preparación de 4-cloro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A21»)

y 4-cloro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A22»)

La separación preparativa de los diastereoisómeros del ejemplo 20 se realizó mediante SFC (columna: ChiralCel OD-H; eluyente: CO2:2-propanol [con dietilamina al 0,5 %] - 90:10). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disolvieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A21»: 27 mg de un sólido incoloro; CL/EM, tR: 2,10 min; (M+H) 407,1/409,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,50 7,46 (m, 2H), 7,42-7,37 (m, 2H), 6,76 (s, 1H), 4,77-4,67 (m, 1H), 4,38-4,27 (m, 2H), 3,00-2,87 (m, 2H), 2,85 (s, 3H), 2,34 2,24 (m, 1H), 2,07 (cd, J = 12,4, 4,3 Hz, 1H), 1,72-1,64 (m, 1H), 1,58-1,54 (m, 3H), 0,96 (d, J = 7,1 Hz, 3H).
«A22»: 23,5 mg de un sólido incoloro; CL/EM, tR: 2,09 min; (M+H) 407,1/409,1; RMN 1H (400 MHz, DMSO-da) 87,62-7,47 (m, 2H), 7,47-7,35 (m, 2H), 7,22-6,81 (m, 1H), 5,01-4,69 (m, 1H), 4,68-4,10 (m, 2H), 3,18-2,94 (m, 1H), 2,94-2,67 (m, 4H), 2,40-2,21 (m, 1H), 2,18-1,88 (m, 1H), 1,80-1,62 (m, 1H), 1,56 (s, 3H), 1,03-0,79 (m, 3H).
Los siguientes compuestos se prepararon de forma análoga:
Ejemplo 23
N,4-Dimetil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diastereoisómeros («A23»)

Rendimiento: 150 mg (65 %) de un sólido de color amarillo pálido; CL/EM, tR: 2,03 min; (M+H) 387,2.
Ejemplos 24 y 25
Preparación de N,4-dimetil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A24»)

y N,4-dimetil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A25»)

La separación preparativa de los diastereoisómeros del ejemplo 23 se realizó mediante SFC (columna: ChiralPak AD-H; eluyente: CO2 :metanol - 90:10). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se di solvieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A24»: 42 mg de un sólido incoloro; CL/EM, tR: 2,03 min; (M+H) 387,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,28-7,21 (m, 4H), 6,76 (s, 1H), 4,72 (d, J = 13,3 Hz, 1H), 4,38-4,30 (m, 2H), 3,01-2,87 (m, 2H), 2,85 (s, 3H), 2,35 (s, 3H), 2,34-2,25 (m, 1H), 2,13-2,00 (m, 1H), 1,72-1,63 (m, 1H), 1,59-1,54 (m, 3H), 0,96 (d, J = 7,1 Hz, 3H).
«A25»: 49 mg de un sólido incoloro; CL/EM, tR: 2,03 min; (M+H) 387,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,28-7,21 (m, 4H), 6,75 (s, 1H), 4,72 (d, J = 13,7 Hz, 1H), 4,39-4,24 (m, 2H), 3,00-2,88 (m, 2H), 2,86 (s, 3H), 2,35 (s, 3H), 2,34-2,26 (m, 1H), 2,14-2,01 (m, 1H), 1,73-1,65 (m, 1H), 1,62-1,54 (m, 3H), 0,95 (d, J = 7,1 Hz, 3H).
Ejemplo 26
2-Fluoro-N,4-dimetil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diaste reoisómeros («A26»)

Rendimiento: 117 mg (67 %) de un sólido de color marrón pálido; CL/EM, tR: 2,08 min; (M+H) 405,1.
Ejemplo 27
2-Fluoro-N,4-dimetil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A27»)

La separación preparativa de «A27» a partir de la mezcla de diastereoisómeros (ejemplo 26) se realizó mediante SFC (columna: ChiralPak AD-H; eluyente: CÜ2 :metanol - 90:10). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disolvieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
Rendimiento: 33 mg de un sólido incoloro; CL/EM, tR: 2,08 min; (M+H) 405,2; RMN 1H (400 MHz, DMSO-d6, 120 °C) 8 7,22-7,14 (m, 1H), 7,09-6,99 (m, 2H), 6,61-6,53 (m, 1H), 4,67 (d, J = 13,7 Hz, 1H), 4,30 (d, J = 13,4 Hz, 2H), 3,07-2,88 (m, 2H), 2,81 (s, 3H), 2,40-2,21 (m, 4H), 2,07 (cd, J = 12,4, 4,5 Hz, 1H), 1,71-1,60 (m, 1H), 1,55 (s, 3H), 0,94 (d, J = 7,1 Hz, 3H).
Ejemplo 28
4-Fluoro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diaste reoisómeros («A28»)

Rendimiento: 280 mg (48 %) de aceite de color amarillo; CL/EM, tR: 1,96 min; (M+H) 391,2.
Ejemplos 29 y 30
Preparación de 4-fluoro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A29»)

y 4-fluoro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A30»)

La separación preparativa de los diastereoisómeros del ejemplo 28 se realizó mediante SFC (columna: ChiralPak AD-H; eluyente: CO2 :metanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se di solvieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A29»: 71,5 mg de un sólido incoloro; CL/EM, tR: 1,95 min; (M+H) 391,2; RMN 1H (400 MHz, DMSO-da) 87,48-7,41 (m, 2H), 7,30-7,22 (m, 2H), 7,10-7,01 (m, 1H), 5,01-4,14 (m, 3H), 3,18-2,87 (m, 1H), 2,83 (s, 3H), 2,80-2,59 (m, 1H), 2,35-1,96 (m, 2H), 1,70-1,57 (m, 1H), 1,53 (s, 3H), 1,01-0,86 (m, 3H).
«A30»: 69 mg de un sólido incoloro; CL/EM, tR: 1,96 min; (M+H) 391,1; RMN 1H (400 MHz, DMSO-d6) 87,48-7,42 (m, 2H), 7,30-7,22 (m, 2H), 7,16-7,05 (m, 1H), 4,96-4,16 (m, 3H), 3,17-2,89 (m, 1H), 2,84 (s, 3H), 2,81-2,61 (m, 1H), 2,40-1,98 (m, 2H), 1,72-1,64 (m, 1H), 1,61-1,52 (m, 3H), 0,97-0,85 (m, 3H).
Ejemplo 31
N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diastereoisómeros («A31»)

Rendimiento: 150 mg (59 %) de un sólido de color amarillo; CL/EM (B), tR: 3,62 min; (M+H) 373,3.
Ejemplos 32 y 33
Preparación de N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A32»)

y N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A33»)

La separación preparativa de los diastereoisómeros del ejemplo 31 se realizó mediante HPLC preparativa (columna: Chi ralPak IA; eluyente: hexano (con 0,1 % de dietilamina):etanol - 90:10).
«A32»: 30 mg de un sólido de color blanquecino; CL/EM (B), tR: 3,62 min; (M+H) 373,3; RMN 1H (400 MHz, DMSO-d6) 8 7,49-7,40 (m, 3H), 7,39-7,30 (m, 2H), 7,20-7,08 (m, 1H), 4,98-4,15 (m, 3H), 3,25-2,98 (m, 1H), 2,84 (s, 3H), 2,41-2,26 (m, 1H), 2,18-1,98 (m, 1H), 1,78-1,66 (m, 1H), 1,57 (s, 3H), 0,92 (d, J = 6,0 Hz, 3H).
«A33»: 39 mg de un sólido de color blanquecino; CL/EM (B), tR: 3,62 min; (M+H) 373,3; RMN 1H (400 MHz, DMSO-d6) 8 7,47-7,42 (m, 3H), 7,39-7,34 (m, 2H), 7,16-7,05 (m, 1H), 4,98-4,05 (m, 3H), 3,15-2,98 (m, 1H), 2,96-2,62 (m, 4H), 2,43-2,21 (m, 1H), 2,15-1,88 (m, 1H), 1,77-1,59 (m, 1H), 1,53 (s, 3H), 1,09-0,81 (m, 3H).
Ejemplo 34
4-Metoxi-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diastereoisómeros («A34»)

Rendimiento: 150 mg (39 %) de un sólido incoloro; CL/EM (B), tR: 3,69 min; (M+H) 403,3.
Ejemplos 35 y 36
Preparación de 4-metoxi-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A35»)

y 4-metoxi-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A36»)
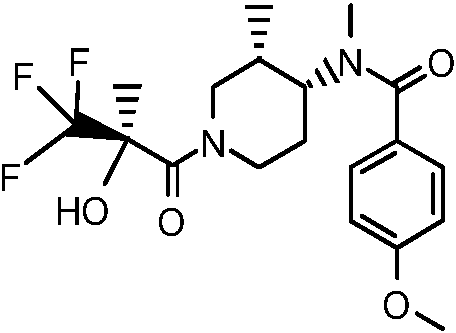
La separación preparativa de los diastereoisómeros del ejemplo 34 se realizó mediante HPLC preparativa (columna: ChiralPak IA; eluyente: hexano (con 0,1 % de dietilamina):etanol - 80:20).
«A35»: 35 mg de un sólido de color blanquecino; CL/EM (B), tR: 3,69 min; (M+H) 403,3; RMN 1H (400 MHz, DMSO-d6) 8 7.35 (d, J = 8,8 Hz, 2H), 7,19-7,08 (m, 1H), 6,98 (d, J = 8,8 Hz, 2H), 4,97-4,18 (m, 3H), 3,80 (s, 3H), 3,12-2,98 (m, 1H), 2,87 (s, 3H), 2,82-2,70 (m, 1H), 2,39-2,22 (m, 1H), 2,17-1,98 (m, 1H), 1,74-1,62 (m, 1H), 1,57 (s, 3H), 1,01-0,80 (m, 3H). «A36»: 35 mg de un sólido de color blanquecino; CL/EM (B), tR: 3,69 min; (M+H) 403,3; RMN 1H (400 MHz, DMSO-d6) 8 7.35 (d, J = 8,4 Hz, 2H), 7,17-7,05 (m, 1H), 6,97 (d, J = 8,8 Hz, 2H), 4,96-4,18 (m, 3H), 3,79 (s, 3H), 3,19-2,98 (m, 1H), 2,85 (s, 3H), 2,82-2,67 (m, 1H), 2,39-2,22 (m, 1H), 2,13-1,90 (m, 1H), 1,74-1,60 (m, 1H), 1,53 (s, 3H), 1,04-0,82 (m, 3H). Ejemplo 37
2,4-Difluoro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diaste reoisómeros («A37»)

Rendimiento: 108 mg (81 %) de aceite incoloro; CL/EM, tR: 2,00 min; (M+H) 409,2.
Ejemplos 38 y 39
Preparación de 2,4-difluoro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A38»)

y 2,4-difluoro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A39»)

La separación preparativa de los diastereoisómeros del ejemplo 37 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CO2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A38»: 38 mg de un sólido incoloro; CL/EM, tR: 2,00 min; (M+H) 409,2; RMN 1H (400 MHz, DMSO-da, 90 °C) 87,49-7,39 (m, 1H), 7,25 (td, J = 9,7, 2,4 Hz, 1H), 7,15 (td, J = 8,5, 2,4 Hz, 1H), 6,78 (s, 1H), 4,97-4,09 (m, 3H), 3,13-2,89 (m, 2H, solapado con agua), 2,83 (s, 3H), 2,44-2,18 (m, 1H), 2,09 (cd, J = 12,5, 4,4 Hz, 1H), 1,75-1,62 (m, 1H), 1,57 (s, 3H), 0,96 (d, J = 7,1 Hz, 3H).
«A39»: 36 mg de un sólido incoloro; CL/EM, tR: 2,00 min; (M+H) 409,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,48-7,39 (m, 1H), 7,25 (td, J = 9,7, 2,4 Hz, 1H), 7,15 (td, J = 8,5, 2,4 Hz, 1H), 6,76 (s, 1H), 4,94-4,18 (m, 3H), 3,09-2,89 (m, 2H, solapado con agua), 2,84 (s, 3H), 2,44-2,18 (m, 1H), 2,11 (cd, J = 12,5, 4,4 Hz, 1H), 1,75-1,64 (m, 1H), 1,59 (s, 3H), 0,95 (d, J = 7,1 Hz, 3H).
Ejemplo 40
3,5-Difluoro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}piridin-2-carboxamida, mezcla de diastereoisómeros («A40»)

Rendimiento: 106 mg (82 %) de aceite de color amarillo; CL/EM, tR: 1,78 min; (M+H) 410,1.
Ejemplos 41 y 42
Preparación de 3,5-difluoro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]piridin-2-carboxamida («A41»)

y 3,5-difluoro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]piridin-2-carboxamida («A42»)

La separación preparativa de los diastereoisómeros del ejemplo 40 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CO2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A41»: 35 mg de un sólido incoloro; CL/EM, tR: 1,78 min; (M+H) 410,1; RMN 1H (400 MHz, DMSO-da, 90 °C) 88,55-8,45 (m, 1H), 7,97 (td, J = 9,3, 2,3 Hz, 1H), 6,85-6,69 (m, 1H), 5,05-3,52 (m, 4H), 3,15-2,64 (m, 5H, solapado con agua), 2,11 (cd, J = 12,4, 4,4 Hz, 1H), 1,76-1,61 (m, 1H), 1,57 (s, 3H), 0,97 (d, J = 7,1 Hz, 3H).
«A42»: 35 mg de un sólido incoloro; CL/EM, tR: 1,78 min; (M+H) 410,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 88,52 (d, J = 2,2 Hz, 1H), 7,97 (td, J = 9,4, 2,3 Hz, 1H), 6,77 (s, 1H), 4,91-3,44 (m, 4H), 3,16-2,71 (m, 5H solapado con agua), 2,13 (cd, J = 12,4, 4,4 Hz, 1H), 1,77-1,62 (m, 1H), 1,59 (s, 3H), 0,96 (d, J = 7,1 Hz, 3H).
Ejemplo 43
4-Ciano-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diastereoisómeros («A43»)

Rendimiento: 117 mg (99 %) de aceite incoloro; CL/EM, tR: 1,85 min; (M+H) 398,1.
Ejemplos 44 y 45
Preparación de 4-ciano-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A44»)

y 4-ciano-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A45»)

La separación preparativa de los diastereoisómeros del ejemplo 43 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CÜ2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A44»: 34 mg de un sólido incoloro; CL/EM, tR: 1,86 min; (M+H) 398,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,88 (d, J = 8.4 Hz, 2H), 7,56 (d, J = 8,4 Hz, 2H), 6,77 (s, 1H), 4,73 (d, J = 12,4 Hz, 1H), 4,40-4,23 (m, 2H), 3,00-2,87 (m, 2H), 2,85 (s, 3H), 2,38-2,21 (m, 1H), 2,09 (cd, J = 12,4, 4,4 Hz, 1H), 1,70 (dc, J = 13,2, 3,4 Hz, 1H), 1,57 (s, 3H), 0,98 (d, J = 7,1 Hz, 3H).
«A45»: 34 mg de un sólido incoloro; CL/EM, tR: 1,86 min; (M+H) 398,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,88 (d, J = 8.4 Hz, 2H), 7,56 (d, J = 8,5 Hz, 2H), 6,76 (s, 1H), 4,73 (d, J = 12,3 Hz, 1H), 4,37-4,24 (m, 2H), 3,01-2,87 (m, 2H), 2,86 (s, 3H), 2,30 (s, 1H), 2,11 (cd, J = 13,4, 12,9, 4,8 Hz, 1H), 1,72 (dc, J = 12,6, 3,0 Hz, 1H), 1,61-1,57 (m, 3H), 0,97 (d, J = 7,1 Hz, 3H).
Ejemplo 46
2-Fluoro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}-4-(trifluorometil)benzamida, mezcla de diastereoisómeros («A46»)

Rendimiento: 156 mg (84 %) de aceite incoloro; CL/EM, tR: 2,23 min; (M+H) 459,1.
Ejemplos 47 y 48
Preparación de 2-fluoro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]-4-(trifluorometil)benzamida («A47»)

y 2-fluoro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]-4-(trifluorometil)benzamida («A48»)

La separación preparativa de los diastereoisómeros del ejemplo 46 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CO2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A47»: 49 mg de un sólido incoloro; CL/EM, tR: 2,23 min; (M+H) 459,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,71 (d, J = 9,7 Hz, 1H), 7,69-7,45 (m, 2H), 6,78 (s, 1H), 4,85-4,23 (m, 3H), 3,13-2,93 (m, 2H), 2,83 (sa, 3H), 2,53-2,26 (m, 1H), 2,11 (cd, J = 12,4, 4,3 Hz, 1H), 1,70 (dc, J = 10,0, 3,9 Hz, 1H), 1,57 (s, 3H), 0,97 (d, J = 7,1 Hz, 3H).
«A48»: 57 mg de un sólido incoloro; CL/EM, tR: 2,24 min; (M+H) 459,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,71 (d, J = 9,6 Hz, 1H), 7,65 (c, J = 8,6, 8,0 Hz, 2H), 6,77 (s, 1H), 4,86-4,20 (m, 3H), 3,16-2,96 (m, 2H), 2,84 (sa, 3H), 2,57-2,24 (m, 1H), 2,13 (cd, J = 12,4, 4,3 Hz, 1H), 1,72 (dc, J = 12,4, 3,3 Hz, 1H), 1,59 (s, 3H), 0,97 (d, J = 7,1 Hz, 3H).
Ejemplo 49
2-Cloro-4-fluoro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diastereoisómeros («A49»)

Rendimiento: 82 mg (60 %) de espuma incolora; CL/EM, tR: 2,06 min, (M+H) 425,1/427,0.
Ejemplos 50 y 51:
Preparación de 2-cloro-4-fluoro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A50»)

y 2-cloro-4-fluoro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A51»)

La separación preparativa de los diastereoisómeros del ejemplo 49 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CO2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A50»: 29,5 mg de un sólido incoloro; CL/EM, tR: 2,07 min; (M+H) 425,1/427,1; RMN 1H (400 MHz, DMSO-da, 90 °C) 8 7,47-7,34 (m, 2H), 7,25 (td, J = 8,5, 2,5 Hz, 1H), 6,74 (s, 1H), 4,90-4,18 (m, 3H), 3,21-2,61 (m, 5H), 2,57-2,34 (m, 1H), 2,16 1,99 (m, 1H), 1,72-1,60 (m, 1H), 1,54 (s, 3H), 0,95 (d, J = 7,1 Hz, 3H).
«A51»: 31 mg de un sólido incoloro; CL/EM, tR: 2,07 min; (M+H) 425,1/427,0; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,47 7,34 (m, 2H), 7,25 (td, J = 8,5, 2,5 Hz, 1H), 6,73 (s, 1H), 4,92-4,04 (m, 3H), 3,17-2,64 (m, 5H), 2,49-2,34 (m, 1H), 2,20-1,98 (m, 1H), 1,76-1,61 (m, 1H), 1,57 (s, 3H), 0,95 (d, J = 7,1 Hz, 3H).
Ejemplo 52
5-Fluoro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}piridin-2-carboxamida, mezcla de diastereoisómeros («A52»)

Rendimiento: 222 mg (99 %) de un sólido de color amarillo; CL/EM, tR: 1,72 min; (M+H) 392,1.
Ejemplos 53 y 54
Preparación de 5-fluoro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]piridin-2-carboxamida («A53»)

y 5-fluoro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]piridin-2-carboxamida («A54»)

La separación preparativa de los diastereoisómeros del ejemplo 52 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CO2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A53»: 63 mg de un sólido incoloro; CL/EM, tR: 1,72 min; (M+H) 392,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 88,52 (d, J = 2,9 Hz, 1H), 7,77 (td, J = 8,8, 2,9 Hz, 1H), 7,61 (dd, J = 8,6, 4,5 Hz, 1H), 6,74 (s, 1H), 4,71 (d, J = 11,6 Hz, 1H), 4,58-4,11 (m, 2H), 3,11-2,79 (m, 5H), 2,43-2,23 (m, 1H), 2,07 (cd, J = 12,1, 4,0 Hz, 1H), 1,70-1,59 (m, 1H), 1,54 (s, 3H), 0,95 (d, J = 7.1 Hz, 3H).
«A54»: 56 mg de un sólido incoloro; CL/EM, tR: 1,72 min; (M+H) 392,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 88,52 (d, J = 2,8 Hz, 1H), 7,77 (td, J = 8,8, 2,9 Hz, 1H), 7,61 (dd, J = 8,6, 4,6 Hz, 1H), 6,72 (s, 1H), 4,71 (d, J = 11,5 Hz, 1H), 4,53-4,18 (m, 2H), 3,15-2,76 (m, 5H), 2,41-2,25 (m, 1H), 2,09 (cd, J = 12,5, 4,4 Hz, 1H), 1,71-1,61 (m, 1H), 1,57 (s, 3H), 0,94 (d, J = 7.1 Hz, 3H).
Ejemplo 55
N,1-Dimetil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}-1H-pirazol-4-carboxamida, mezcla de diastereoisómeros («A55»)

Rendimiento: 140 mg (47 %) de espuma incolora; CL/EM, tR: 1,53 min; (M+H) 377,1.
Ejemplos 56 y 57
Preparación de N,1-dimetil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]-1H-pirazol-4-carboxamida («A56»)

y N,1-dimetil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]-1H-pirazol-4-carboxamida («A57»)

La separación preparativa de los diastereoisómeros del ejemplo 55 se realizó mediante SFC (columna: ChiralPak AD-H; eluyente: CO2 :etanol - 80:20). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A56»: 41 mg de un sólido incoloro; CL/EM, tR: 1,54 min; (M+H) 377,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 88,00 (s, 1H), 7,68 (s, 1H), 6,77 (s, 1H), 4,75 (d, J = 12,4 Hz, 1H), 4,47 (dt, J = 12,6, 4,4 Hz, 1H), 4,37 (d, J = 13,2 Hz, 1H), 3,87 (s, 3H), 3,12-2,93 (m, 5H), 2,37-2,25 (m, 1H), 2,09 (cd, J = 12,5, 4,4 Hz, 1H), 1,67-1,56 (m, 4H), 0,93 (d, J = 7,1 Hz, 3H).
«A57»: 46,5 mg de un sólido incoloro; CL/EM, tR: 1,54 min; (M+H) 377,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 88,00 (s, 1H), 7,68 (s, 1H), 6,76 (s, 1H), 4,75 (d, J = 12,5 Hz, 1H), 4,48 (dt, J = 12,5, 4,4 Hz, 1H), 4,38-4,28 (m, 1H), 3,87 (s, 3H), 3,03 (d, J = 10,6 Hz, 5H), 2,31 (dc, J = 7,0, 3,5 Hz, 1H), 2,10 (cd, J = 12,3, 4,2 Hz, 1H), 1,71-1,55 (m, 4H), 0,92 (d, J = 7,1 Hz, 3H).
Ejemplo 58
4-Cloro-2-fluoro-N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}benzamida, mezcla de diastereoisómeros («A58»)

Rendimiento: 121 mg (79 %) de sólido incoloro; CL/EM, tR: 2,13 min, (M+H) 425,1/427,1.
Ejemplos 59 y 60
Preparación de 4-cloro-2-fluoro-N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A59»)

y 4-cloro-2-fluoro-N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]benzamida («A60»)

La separación preparativa de los diastereoisómeros del ejemplo 58 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CO2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A59»: 44 mg de un sólido incoloro; CL/EM, tR: 2,14 min; (M+H) 425,1/427,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,46 (dd, J = 9,5, 1,8 Hz, 1H), 7,44-7,39 (m, 1H), 7,36 (dd, J = 8,2, 1,9 Hz, 1H), 6,77 (s, 1H), 4,74 (d, J = 11,0 Hz, 1H), 4,62-4,04 (m, 2H), 3,14-2,73 (m, 5H), 2,47-2,19 (m, 1H), 2,09 (cd, J = 12,4, 4,4 Hz, 1H), 1,76-1,61 (m, 1H), 1,57 (s, 3H), 0,96 (d, J = 7,1 Hz, 3H).
«A60»: 45 mg de un sólido incoloro; CL/EM, tR: 2,14 min; (M+H) 425,1/427,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,46 (dd, J = 9,5, 1,8 Hz, 1H), 7,44-7,39 (m, 1H), 7,36 (dd, J = 8,2, 1,9 Hz, 1H), 6,77 (s, 1H), 4,73 (d, J = 11,9 Hz, 1H), 4,63-4,15 (m, 2H), 3,19-2,73 (m, 5H), 2,47-2,21 (m, 1H), 2,11 (cd, J = 12,4, 4,4 Hz, 1H), 1,75-1,64 (m, 1H), 1,59 (s, 3H), 0,95 (d, J = 7,1 Hz, 3H).
Ejemplo 61
N-metil-N-{3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il}-4-(trifluorometil)benzamida, mezcla de diastereoisómeros («A61»)

Rendimiento: 118 mg (46 %) de aceite de color amarillo; CL/EM, tR: 2,18 min; (M+H) 441,1.
Ejemplos 62 y 63
Preparación de N-metil-N-[(3R,4S)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]-4-(trifluorometil)benzamida («A62»)

y N-metil-N-[(3S,4R)-3-metil-1-[(2R)-3,3,3-trifluoro-2-hidroxi-2-metilpropanoil]piperidin-4-il]-4-(trifluorometil)benzamida («A63»)

La separación preparativa de los diastereoisómeros del ejemplo 61 se realizó mediante SFC (columna: Lux Cellulose-2; eluyente: CO2 :etanol - 88:12). Las fracciones combinadas se evaporaron hasta sequedad. Los residuos oleosos se disol vieron en acetonitrilo, se diluyeron con agua y se liofilizaron.
«A62»: 39 mg de un sólido incoloro; CL/EM, tR: 2,18 min; (M+H) 441,1; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,76 (d, J = 8.0 Hz, 2H), 7,57 (d, J = 7,9 Hz, 2H), 6,74 (s, 1H), 4,71 (d, J = 12,1 Hz, 1H), 4,44-4,21 (m, 2H), 3,09-2,80 (m, 5H), 2,39 2,23 (m, 1H), 2,07 (cd, J = 12,4, 4,3 Hz, 1H), 1,74-1,63 (m, 1H), 1,62-1,45 (m, 3H), 0,96 (d, J = 7,1 Hz, 3H).
«A63»: 44 mg de un sólido incoloro; CL/EM, tR: 2,19 min; (M+H) 441,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 87,76 (d, J = 8.1 Hz, 2H), 7,57 (d, J = 7,9 Hz, 2H), 6,73 (s, 1H), 4,79-4,21 (m, 3H), 3,04-2,69 (m, 5H), 2,39-2,22 (m, 1H), 2,09 (cd, J = 12,5, 4,4 Hz, 1H), 1,76-1,65 (m, 1H), 1,59-1,55 (m, 3H), 0,95 (d, J = 7,1 Hz, 3H).
Los compuestos siguientes se obtuvieron de forma análoga a la descrita para el ejemplo 16:
Esquema de reacción:

Ejemplo 64
N-Etil-2-fluoro-4-metil-N-[1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-il]-benzamida («A64»)

Rendimiento: 26,5 mg (25 %) de sólido incoloro; CL/EM, tR: 2,09 min; (M+H) 405,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 8 7,23-7,16 (m, 1H), 7,11-7,03 (m, 2H), 6,80-6,72 (m, 1H), 4,72-4,55 (m, 2H), 3,38-3,15 (m, 2H), 2,84-2,31 (m, 6H), 1,88 1,63 (m, 4H), 1,59-1,49 (m, 3H), 1,16-0,96 (m, 3H).
Ejemplo 65
2-Fluoro-N-isopropil-4-metil-N-[1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-il]-benzamida («A65»)

Rendimiento: 113 mg (82 %) de sólido incoloro; CL/EM, tR: 2,26 min; (M+H) 419,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 8 7,19-7,13 (m, 1H), 7,08-7,02 (m, 2H), 6,81-6,71 (m, 1H), 4,69-4,55 (m, 2H), 3,70-3,60 (m, 1H), 3,51-3,40 (m, 1H), 3,12 2,66 (m, 4H), 2,35 (s, 3H), 1,66-1,57 (m, 2H), 1,55 (s, 3H), 1,33-1,14 (m, 6H).
Ejemplo 66
4-Ciano-N-etil-N-[1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-il]-benzamida («A66»)

Rendimiento: 10,5 mg (47 %) de un sólido incoloro; CL/EM, tR: 1,89 min; (M+H) 398,1; RMN 1H (400 MHz, DMSO-da, 90 °C) 87,90-7,83 (m, 2H), 7,57-7,50 (m, 2H), 6,77 (s, 1H), 4,69-4,57 (m, 2H), 3,99-3,75 (m, 1H), 3,31-3,23 (m, 2H), 2,77 2,62 (m, 2H), 1,85-1,71 (m, 4H), 1,56-1,52 (m, 3H), 1,12-1,05 (m, 3H).
Ejemplo 67
4-Ciano-N-isopropil-N-[1-((R)-3,3,3-trifluoro-2-hidroxi-2-metil-propionil)-piperidin-4-il]-benzamida («A67»)

Rendimiento: 53 mg (45 %) de sólido incoloro; CL/EM, tR: 2,04 min; (M+H) 412,2; RMN 1H (400 MHz, DMSO-d6, 90 °C) 8 7,89-7,83 (m, 2H), 7,51-7,45 (m, 2H), 6,76 (s, 1H), 4,69-4,55 (m, 2H), 3,68-3,57 (m, 1H), 3,50-3,40 (m, 1H), 2,84-2,69 (m, 2H), 2,39-2,17 (m, 2H), 1,71-1,63 (m, 2H), 1,55 (s, 3H), 1,26 (d, J = 6,5 Hz, 6H).
Datos farmacológicos
Tabla 1 Inhibición de PDHK de algunos compuestos representativos de fórmula I


IC50 [M] p. ej.: 5,90E-07 = 5,90 x 10 -7
Los compuestos mostrados en la tabla 1 son compuestos especialmente preferidos según la invención.
Los ejemplos siguientes hacen referencia a medicamentos:
Ejemplo A: viales para inyección
Una solución de 100 g de un principio activo de fórmula I y 5 g de hidrogenofosfato disódico en 3 litros de agua bidestilada se ajusta a pH 6,5 usando ácido clorhídrico 2 N, se esteriliza por filtración, se transfiere a viales para inyección, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada vial para inyección contiene 5 mg del principio activo.
Ejemplo B: supositorios
Se funde una mezcla de 20 g de un principio activo de fórmula I con 100 g de lecitina de soja y 1400 g de manteca de cacao, se vierte en los moldes y se deja enfriar. Cada supositorio contiene 20 mg del principio activo.
Ejemplo C: solución
Se prepara una solución de 1 g de un principio activo de fórmula I, 9,38 g de NaH2PO4-2 H2O, 28,48 g de Na2HPO4-12 H2O y 0,1 g de cloruro de benzalconio en 940 ml de agua bidestilada. El pH se ajusta a 6,8, la solución se lleva a 1 litro y se esteriliza mediante radiación. Esta solución puede usarse en forma de colirio.
Ejemplo D: pomada
Se mezclan 500 mg de un principio activo de fórmula I con 99,5 g de vaselina en condiciones asépticas.
Ejemplo E: comprimidos
Una mezcla de 1 kg de un principio activo de fórmula I, 4 kg de lactosa, 1,2 kg de almidón de patata, 0,2 kg de talco y 0,1 kg de estearato de magnesio se prensa de forma habitual para obtener comprimidos, de manera que cada comprimido contiene 10 mg del principio activo.
Ejemplo F: grageas
Los comprimidos se prensan de forma análoga al ejemplo E y, posteriormente, se recubren de forma habitual con un recubrimiento de sacarosa, almidón de patata, talco, goma de tragacanto y colorante.
Ejemplo G: cápsulas
Se introducen 2 kg de principio activo de fórmula I dentro de cápsulas duras de gelatina de forma habitual, de modo que cada cápsula contiene 20 mg de principio activo.
Ejemplo H: ampollas
Una solución de 1 kg de principio activo de fórmula I en 60 litros de agua bidestilada se esteriliza por filtración, se transfiere a ampollas, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada ampolla contiene 10 mg del prin cipio activo.
Claims (8)
1. Compuestos de fórmula I

donde
X-Y indica CO- NA'' o NA''- CO,
R1 indica Ar o Het,
R2 indica H o CH3 ,
R3 indica H o CH3 ,
Ar indica fenilo, que no está sustituido o está mono, di o trisustituido por Hal, CN, A y/u OR3,
Het indica pirimidilo, piridilo, piridazinilo, pirazinilo, piperidinilo, pirrolidinilo, pirazolilo, tiazolilo, imidazolilo, furanilo, tiofenilo, pirrolilo, oxazolilo, triazolilo, oxadiazolilo o tiadiazolilo, cada uno de los cuales no está sustituido o está mono o disustituido por Hal, A, CN y/u OR3,
A indica alquilo ramificado o no ramificado con 1-6 átomos de C, donde 1-5 átomos de H pueden estar sustituidos por F,
A'' indica alquilo sin ramificar o ramificado que tiene 1-4 átomos de C,
Hal indica F, Cl, Br o I,
y sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones.
2. Compuestos según la reivindicación 1, seleccionados entre el grupo compuesto por



y sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones.
3. Proceso para la preparación de compuestos de fórmula I según las reivindicaciones 1-2 y las sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos,
caracterizado porque un compuesto de fórmula II

donde X, Y, R1 y R2 tienen los significados indicados en la reivindicación 1,
reacciona con el ácido 3,3,3-trifluoro-2-hidroxi-2-metil-propiónico
y/o
una base o ácido de fórmula I se transforma en una de sus sales.
4. Medicamentos que comprenden al menos un compuesto de fórmula I según la reivindicación 1 y/o sales, tautómeros y estereoisómeros farmacéuticamente aceptables del mismo, incluidas sus mezclas en todas las proporciones, y opcionalmente un transportador, excipientes o vehículo farmacéuticamente aceptable.
5. Compuestos para el uso de la fórmula I según la reivindicación 1 y sales, tautómeros y estereoisómeros farmacéuti camente aceptables de los mismos, incluidas sus mezclas en todas las proporciones, para el tratamiento y/o preven ción del cáncer, diabetes, isquemia cardíaca, síndrome de resistencia a insulina, síndrome metabólico, hiperglucemia, dislipidemia, ateroesclerosis, insuficiencia cardíaca, cardiomiopatía, isquemia miocárdica, hiperlactacidemia, enfer medad mitocondrial, encefalopatía mitocondrial.
6. Compuestos para su uso según la reivindicación 5 para el tratamiento y/o prevención de enfermedades seleccionadas a partir del grupo de cáncer de cabeza, cuello, ojos, boca, garganta, esófago, bronquios, laringe, faringe, tórax, hueso, pulmón, colon, recto, estómago, próstata, vejiga urinaria, útero, cuello uterino, mama, ovarios, testículos u otros ór ganos reproductores, piel, tiroides, sangre, ganglios linfáticos, riñón, hígado, páncreas, cerebro, sistema nervioso central, tumores sólidos y tumores en la sangre.
7. Medicamentos que comprenden al menos un compuesto de fórmula I según la reivindicación 1 y/o sales, tautómeros y estereoisómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones y al menos un principio activo adicional.
8. Set (kit) compuesto por envases independientes de
(a) una cantidad eficaz de un compuesto de fórmula I según la reivindicación 1 y/o sales, tautómeros y estereoi sómeros farmacéuticamente aceptables de los mismos, incluidas sus mezclas en todas las proporciones, y
(b) una cantidad eficaz de un principio activo adicional de un medicamento.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16167455 | 2016-04-28 | ||
| PCT/EP2017/059689 WO2017186653A1 (en) | 2016-04-28 | 2017-04-25 | Piperidinyl derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2850426T3 true ES2850426T3 (es) | 2021-08-30 |
Family
ID=55854732
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17722397T Active ES2850426T3 (es) | 2016-04-28 | 2017-04-25 | Derivados de piperidinilo |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10428024B2 (es) |
| EP (1) | EP3448850B1 (es) |
| JP (1) | JP7028792B2 (es) |
| CN (1) | CN109071490B (es) |
| AU (1) | AU2017258697B2 (es) |
| CA (1) | CA3022231A1 (es) |
| ES (1) | ES2850426T3 (es) |
| IL (1) | IL262597B (es) |
| WO (1) | WO2017186653A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111655692B (zh) * | 2018-02-01 | 2023-10-10 | 日本烟草产业株式会社 | 含氮杂环酰胺化合物及其医药用途 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6979686B1 (en) | 2001-12-07 | 2005-12-27 | Pharmacia Corporation | Substituted pyrazoles as p38 kinase inhibitors |
| JP2003508509A (ja) * | 1999-09-04 | 2003-03-04 | アストラゼネカ アクチボラグ | ピルビン酸デヒドロゲナーゼの阻害剤としてのアミド |
| PE20070458A1 (es) * | 2005-09-14 | 2007-07-05 | Takeda Pharmaceutical | Composicion farmaceutica que comprende 2-[[6-(3r)-3-amino-1-piperidinil]-3,4-dihidro-3-metil-2,4-dioxo-1(2h)-pirimidinil]metil]-benzonitrilo como inhibidor de dipeptidil peptidasa |
| US7638541B2 (en) | 2006-12-28 | 2009-12-29 | Metabolex Inc. | 5-ethyl-2-{4-[4-(4-tetrazol-1-yl-phenoxymethyl)-thiazol-2-yl]-piperidin-1-yl}-pyrimidine |
| JP2010090860A (ja) * | 2008-10-10 | 2010-04-22 | Mitsubishi Heavy Ind Ltd | 低カロリーガス燃料を用いたガスエンジン |
| AR074797A1 (es) | 2008-10-10 | 2011-02-16 | Japan Tobacco Inc | Compuesto de fluoreno , composiciones farmaceuticas , inhibidores de pdhk y pdhk2 , metodos de tratamiento , usos de los mismos y kit comercial |
| WO2011143057A1 (en) | 2010-05-11 | 2011-11-17 | Merck Sharp & Dohme Corp. | Novel prolylcarboxypeptidase inhibitors |
| EP2632268A4 (en) | 2010-10-29 | 2014-09-10 | Merck Sharp & Dohme | NEW CONNECTIONS AS ERK HEMMER |
| HUE037844T2 (hu) | 2010-11-10 | 2018-09-28 | Genentech Inc | Pirazol-aminopirimidin-származékok mint LRRK2 modulátorok |
| WO2012082947A1 (en) | 2010-12-16 | 2012-06-21 | Irm Llc | Compounds and compositions as tgr5 agonists |
-
2017
- 2017-04-25 JP JP2018556282A patent/JP7028792B2/ja active Active
- 2017-04-25 WO PCT/EP2017/059689 patent/WO2017186653A1/en active Application Filing
- 2017-04-25 EP EP17722397.1A patent/EP3448850B1/en active Active
- 2017-04-25 ES ES17722397T patent/ES2850426T3/es active Active
- 2017-04-25 US US16/097,440 patent/US10428024B2/en active Active
- 2017-04-25 CN CN201780026304.7A patent/CN109071490B/zh active Active
- 2017-04-25 AU AU2017258697A patent/AU2017258697B2/en active Active
- 2017-04-25 CA CA3022231A patent/CA3022231A1/en active Pending
-
2018
- 2018-10-25 IL IL262597A patent/IL262597B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| AU2017258697B2 (en) | 2021-04-15 |
| EP3448850B1 (en) | 2020-11-04 |
| IL262597B (en) | 2021-08-31 |
| CN109071490B (zh) | 2021-04-06 |
| US10428024B2 (en) | 2019-10-01 |
| WO2017186653A1 (en) | 2017-11-02 |
| JP7028792B2 (ja) | 2022-03-02 |
| JP2019518005A (ja) | 2019-06-27 |
| CN109071490A (zh) | 2018-12-21 |
| CA3022231A1 (en) | 2017-11-02 |
| US20190135753A1 (en) | 2019-05-09 |
| EP3448850A1 (en) | 2019-03-06 |
| AU2017258697A1 (en) | 2018-12-13 |
| IL262597A (en) | 2018-12-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2797301T3 (es) | Derivados de piperidinil-propanona | |
| ES2633969T3 (es) | Derivados de ciclopentano 1,3-disustituidos | |
| ES2654403T3 (es) | Derivados de imidazopirazinona | |
| ES2671322T3 (es) | Derivados de piperidin urea | |
| WO2014093230A2 (en) | Compositions and methods for the production of pyrimidine and pyridine compounds with btk inhibitory activity | |
| ES2665148T3 (es) | Derivados de N1-(3,3,3-trifluoro-2-hidroxo-2-metilpropionil)-piperidina como inhibidores de la piruvato deshidrogenasa quinasa | |
| ES2934986T3 (es) | Derivados heterocíclicos | |
| ES2773511T3 (es) | Derivados de 1,4-dicarbonil-piperidilo | |
| ES2757052T3 (es) | Derivados heterocíclicos bicíclicos | |
| ES2850426T3 (es) | Derivados de piperidinilo | |
| CN107207482A (zh) | 用于治疗癌症的嘧啶衍生物 | |
| ES2671398T3 (es) | Derivados de ciclopentilamina 3-sustituida |