ES2829198T3 - Análisis digital de secuencia de la metilación de ADN - Google Patents
Análisis digital de secuencia de la metilación de ADN Download PDFInfo
- Publication number
- ES2829198T3 ES2829198T3 ES18179746T ES18179746T ES2829198T3 ES 2829198 T3 ES2829198 T3 ES 2829198T3 ES 18179746 T ES18179746 T ES 18179746T ES 18179746 T ES18179746 T ES 18179746T ES 2829198 T3 ES2829198 T3 ES 2829198T3
- Authority
- ES
- Spain
- Prior art keywords
- methylation
- loci
- cpg
- nucleic acid
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Genetics & Genomics (AREA)
- Analytical Chemistry (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- General Chemical & Material Sciences (AREA)
Abstract
Un procedimiento de selección de un subconjunto definido de loci CpG en un ácido nucleico marcador para su uso en un ensayo de detección de ácido nucleico que detecta la metilación coordinada de un subconjunto definido de loci CpG, en el que la metilación coordinada del subconjunto definido de loci CpG es indicativo de adenoma o cáncer, comprendiendo el procedimiento: a) la determinación del estado de metilación de una pluralidad de loci CpG en cada una de una pluralidad de copias individuales de un ácido nucleico marcador de una pluralidad de muestras normales; b) la determinación del estado de metilación de dicha pluralidad de loci CpG en cada una de una pluralidad de copias individuales de dicho ácido nucleico marcador de una pluralidad de muestras no normales de sujetos que tienen adenoma o cáncer; en el que la pluralidad de copias individuales de dicho ácido nucleico marcador analizado en cada una de dichas muestras normales y dichas muestras de adenoma o dichas muestras de cáncer comprende al menos 1.000; c) la determinación de las relaciones de metilación de cada locus CpG en dicha pluralidad de dichos loci CpG en dicho ácido nucleico marcador; en el que la determinación de dichas relaciones de metilación comprende la determinación de la relación entre la media de metilación en cada uno de dicha pluralidad de loci CpG en dichas muestras normales respecto a la media de metilación de cada locus CpG correspondiente en dicha pluralidad de loci CpG en dichas muestras no normales; d) la selección de un conjunto definido de loci CpG en dicho ácido nucleico marcador, en el que dicho conjunto definido de loci CpG comprende al menos tres loci CpG que tienen relaciones de metilación ventajosas que se correlacionan con adenoma o cáncer; y e) la selección de un subconjunto definido de loci CpG de dicho conjunto definido, en el que dicho subconjunto definido de loci CpG comprende al menos tres loci CpG, y en el que dicho subconjunto definido se selecciona de manera que el porcentaje de copias individuales en dicho ácido nucleico marcador de dicha pluralidad de muestras normales que están metiladas en todos los dichos al menos tres loci CpG en dicho subconjunto definido es menor que el porcentaje de copias individuales de dicho ácido nucleico marcador de dicha pluralidad de muestras no normales de sujetos que tienen adenoma o cáncer que están metiladas en todos los dichos al menos tres loci CpG en dicho subconjunto definido.
Description
DESCRIPCIÓN
Análisis digital de secuencia de la mutilación de ADN
Referencia a solicitudes relacionadas
La presente solicitud reivindica el beneficio de prioridad de la Solicitud de Patente Provisional de EE. UU. con el Número de Serie 61/438.649, presentada el 2 de febrero de 2011.
Campo de la invención
La presente invención se refiere a procedimientos para la determinación y usos de patrones de metilación específicos indicativos de adenoma y carcinoma de acuerdo con las reivindicaciones. En particular, la invención se refiere a análisis de loci CpG definidos que están metilados coordinadamente en los ADN de muestras de cáncer y adenoma, y procedimientos de utilización de análisis de loci metilados coordinadamente o más regiones marcadoras en el diseño de ensayo para adenomas y cánceres que tienen una sensibilidad y especificidad mejoradas.
Antecedentes de la invención
En el orden superior de eucariotas, el ADN se puede metilar en las citosinas localizadas 5' a la guanosina en los dinucleótidos CpG. Esta modificación tiene importantes efectos reguladores sobre la expresión genética, especialmente cuando se implican áreas ricas en CpG, conocidas como islas CpG, que se encuentran a menudo en las regiones de promotor de los genes. Mientras que aproximadamente un 75 % de los sitios CpG a lo largo del genoma humano están metilados, los sitios CpG en las islas CpG normalmente o están metilados, y se ha asociado una metilación aberrante de las islas CpG con ciertas enfermedades, incluyendo los cánceres. Por ejemplo, se asocia una hipermetilación de islas CpG con la inactivación transcripcional de genes supresores tumorales definidos en cánceres humanos, por ejemplo, en cáncer colorrectal. Por lo tanto, la detección de un ácido nucleico hipermetilado podría indicar una susceptibilidad o aparición de distintas formas de cánceres.
A pesar de las indicaciones que sugieren una relación entre el fenotipo metilador de islas CpG (CIMP) y los cánceres (véase, por ejemplo, Baylin SB, y col., Adv Cancer Res 1998;72:141-196 y Jones PA, y col., Nat Rev Genet 2002;3:415-428), la idea de que el análisis del estado de metilación solo podría ser una herramienta diagnóstica o pronóstica útil ha sido controvertida. Como expone Issa, y col. en una editorial en Gastroenterology 179(3):2005, los investigadores habían mezclado resultados en la confirmación de la relación entre CIMP y cánceres. Aunque se ha demostrado supuestamente en otras múltiples enfermedades malignas (Shen, I., y col. J Natl Cancer Inst 2002;94:755-761; Garcia-Manero G, y col., Clin Cancer Res 2002;8:2217-2224; Toyota M, y col., Blood 2001;97:2823-2829; Ueki T, y col., Cancer Res 2000;60:1835-1839; Toyota M, y col., Cancer Res 1999;59:5438-5442; Strathdee G, y col., Am J Pathol 2001;158:1121-1127; Abe M, y col., Cancer Res 2005;65:828-834) y varios grupos confirmaron los hallazgos originales utilizando marcadores y tecnologías similares (Whitehall VL, y col., Cancer Res 2002;62:6011-6014; van Rijnsoever M, y col., Gut 2002;51:797-802) otros grupos no fueron capaces de establecer dichas relaciones (Eads c A , y col., Cancer Res 2001;61:3410-3418; Esteller M, y col., Cancer Res 2000;60:129-133). Últimamente en 2003, una publicación concluía que todos los casos de metilación en el cáncer colorrectal se relacionaban con la vejez más que con una neoplasia (Yamashita K, y col., Cancer Cell 2003;4:121-131).
Los resultados discrepantes se habían atribuido en parte al hecho de que se había demostrado que el 70 % a 80 % de los eventos de metilación aberrante de ADN en el cáncer colorrectal están relacionados con la edad (Toyota M, y col., Proc Natl Acad Sci U S A 1999;96:8681-8686) y que los fenotipos relacionados con el cáncer solo están claros cuando se filtran. También se ha señalado que los procedimientos demasiado sensibles, no cuantitativos pueden sobre-estimar la metilación y enmascarar las distinciones entre la metilación que se asocia con el cáncer y la que no. Issa establece que los “eventos de metilación (solos) pueden no proporcionar el marcador de cáncer universal ideal se pensó una vez que era debido a que los genes diana CIMP no serán útiles para explorar todos los cánceres colorrectales (se prevén muchos falsos negativos), y los genes diana no CIMP probablemente darán una alta tasa de falsos positivos debido a que también están metilados en la mucosa de apariencia normal de individuos ancianos sin tumores” (Issa, y col., supra).
Una estrategia para aumentar la especificidad clínica de los análisis de metilación en la detección del cáncer es mirar múltiples genes marcadores. Por ejemplo, Zou y col., examinaron el estado de metilación de BMP3, EYA2, ALX4, y vimentin en muestras de cáncer. Mientras que los niveles de metilación eran significativamente más altos tanto en cáncer como en adenoma que en el epitelio normal, para cada uno de los cuatro genes, la sensibilidad, según se determinó por las curvas operadoras de receptor, no mejoraba significativamente combinando cualquiera o todos los marcadores en comparación con el mejor marcador sencillo (Zou, y col., Cancer Epidemiol Biomarkers Prev 2007;16(12):2686).
Zou también miró las neoplasias que presentan metilación en más de uno de los genes marcadores y descubrió que era frecuente la co-metilación, presentando un 72 % de los cánceres y un 84 % de los adenomas ensayados una hipermetilación en dos o más genes. Zou informó que la metilación de uno o más de cuatro (al menos uno), dos o más de cuatro, tres o más de cuatro. o cuatro de cuatro de estos genes marcadores se apreciaba en un 88 %, 72 %,
53 %, y 41 % de 74 cánceres y un 98 %, 84 %, 60 % y 39 % de 62 adenomas, en comparación con el 24 %, 7 %, 3 % y 0 % de 70 epitelios normales, respectivamente, demostrando que, aunque el ensayo se hace progresivamente más específico cuantos más genes se incluyen en el conjunto de co-metilación, la sensibilidad declina vertiginosamente.
El documento WO 2010/118016 A2 se refiere al uso del análisis de secuencia digital para determinar la fracción de moléculas metiladas en una muestra.
El documento WO 03/064701 A2 desvela un procedimiento para la identificación sistemática de posiciones de dinucleótidos CpG metilados diferencialmente en secuencias de ADN genómico para su uso como marcadores diagnósticos, pronósticos y/o de estado fiables.
El documento WO 03/044232 A1 desvela procedimientos y kits útiles para detectar neoplasias midiendo el nivel de metilación de biomarcadores, especialmente la región promotora de GSTP1 para la detección de adenocarcinoma de próstata.
Ahlquist y col., Gastroenterology, 138(6), 2010, 2127-2139, está interesado en la detección molecular de neoplasia colorrectal e inter alia expone la detección de neoplasia colorrectal por ensayo del ADN fecal con respecto a varios marcadores diferentes.
Zou y col., Clinical Chemistry, 56(6), 2010, Supplement, abstract n° D-144, A199, desvela un procedimiento para detectar vimentin metilada y no metilada basándose en una estrategia que combina una PCR con química Invader.
El documento US 2010/273164 A1 desvela procedimientos y composiciones para la determinación de un perfil de citosina metilada de una secuencia de ácido nucleico diana.
Weisenberg y col., Nucleic Acid Research, 36(14), 2008, 4689-4698, se refiere a un análisis de metilación de ADN por secuenciación digital genómica con bisulfitos y MethylLight digital.
El documento US 2010/0124747 A1 se refiere a composiciones y procedimientos para el diagnóstico o pronóstico de cáncer testicular o derivado de células germinales masculinas.
Sumario de la invención
La presente invención se expone en las reivindicaciones adjuntas. Los casos y realizaciones de la descripción que no se encuentran en el ámbito de dichas reivindicaciones se proporcionan solo con fines ilustrativos, y no forman parte de la presente invención. La divulgación se refiere a procedimientos de identificación de regiones de genes específicos y regiones específicas de ácido nucleico genómico útil en la detección de la metilación asociada con el cáncer colorrectal. Los procedimientos comprenden, por ejemplo, la detección de secuencias metiladas, por ejemplo, en biopsias tisulares, extractos fecales, u otros fluidos corporales con una sensibilidad y especificidad mejoradas. La presente divulgación proporciona procedimientos de análisis de metilación que comprende la identificación de los loci de metilación que presentan relaciones de metilación ventajosas cuando la metilación en células no normales, por ejemplo, células cancerosas o de adenoma se comparan con la metilación de fondo en células normales. La divulgación se refiere a procedimientos de análisis de la metilación en cada uno de varios loci en un conjunto de sitios posibles de metilación en una secuencia marcadora, en la que la presencia de metilación en todos los loci del conjunto de sitios definidos se produce más frecuentemente en células cancerosas y de adenoma que en células normales, dicho hallazgo de metilación en todos los loci del subconjunto de loci definidos de una muestra es indicativo de adenoma o cáncer.
La divulgación proporciona un procedimiento de identificación de un conjunto de loci CpG metilados en un ácido nucleico marcador en el que la metilación es indicativa de adenoma. que comprende:
a) determinación del estado de metilación de un conjunto de loci CpG definido en cada uno de una pluralidad de copias individuales de un ácido nucleico marcador de una pluralidad de muestras normales;
b) determinación del estado de metilación de dicho conjunto de loci CpG definido en cada una de una pluralidad de copias individuales de dicho ácido nucleico marcador de una pluralidad de muestras no normales (por ejemplo, de adenoma o cáncer) para identificar un subconjunto de loci CpG definido de dicho conjunto definido,
en el que el porcentaje de copias individuales de dicho ácido nucleico marcador de dicha pluralidad de muestras normales que están metiladas en todos dichos loci CpG en dicho subconjunto definido es menor que el porcentaje de copias individuales de dicho ácido nucleico marcador de dicha pluralidad de muestras no normales que están metiladas en todos los dichos loci CpG en dicho subconjunto definido, y en el que la metilación en todos los dichos loci CpG de dicho subconjunto definido de dicho ácido nucleico marcador es indicativo de un estado no normal, por ejemplo, un adenoma y/o cáncer. El porcentaje medio de copias individuales del ácido nucleico marcador metilado en todos los loci en dicho conjunto de loci CpG definido en dicha pluralidad de muestras no normales puede ser mayor que la media de porcentaje de copias individuales del ácido nucleico marcador metilado en todos los loci de dicho conjunto de loci CpG definido en la pluralidad de muestras normales. El porcentaje medio de copias
individuales del ácido nucleico marcador metilado en todos los loci de dicho conjunto de loci CpG definido en la pluralidad de muestras no normales puede ser al menos una desviación típica, preferentemente dos desviaciones típicas, más preferentemente tres desviaciones típicas mayor que la media de porcentaje de copias individuales de dicho ácido nucleico marcador metilado en todos los loci de dicho conjunto definido de loci CpG en dicha pluralidad de muestras normales.
En algunas realizaciones, el subconjunto de loci CpG definido consiste en los mismos loci que el conjunto de loci CpG definido.
La determinación del estado de metilación del conjunto de loci CpG puede conseguirse por cualquier procedimiento conocido por los expertos en la técnica. En algunas realizaciones, el procedimiento comprende el tratamiento del ADN de las muestras con bisulfito. El tratamiento de modificación con bisulfito se describe, por ejemplo, en la Pat. de EE. UU. N.° 6.017.704, cuya divulgación completa se incorpora en el presente documento por referencia. En algunas realizaciones, la determinación del estado de metilación del conjunto de loci CpG definido comprende el análisis digital de cada uno de una pluralidad de loci CpG en una pluralidad de copias individuales de un ácido nucleico marcador. En algunas realizaciones preferidas, el análisis digital comprende la secuenciación digital, y/o la PCR digital.
En ciertas realizaciones preferidas, la muestra no normal comprende una muestra de adenoma, y en realizaciones preferidas particulares, comprende una muestra de adenoma colorrectal. En algunas realizaciones preferidas, una muestra no normal comprende una muestra de cáncer, y en ciertas realizaciones preferidas comprenden una muestra de cáncer colorrectal.
La presente invención proporciona procedimientos para la detección del cáncer o adenoma en una muestra, por ejemplo, de un sujeto. En algunas realizaciones, la presente invención proporciona procedimientos que comprende la determinación del estado de metilación de cada locus CpG en un subconjunto de loci CpG definido en al menos una molécula de ácido nucleico marcador de adenoma o cáncer, en el que la metilación de cada uno de los loci CpG del subconjunto de loci CpG definido en la molécula de ácido nucleico marcador de cáncer o adenoma es indicativa de cáncer o adenoma en la muestra. El subconjunto definido comprende al menos tres loci CpG mientras que en realizaciones preferidas, el subconjunto definido comprende al menos cuatro loci CpG o al menos cinco loci CpG.
En ciertas realizaciones, la determinación comprende el análisis de un ensayo de detección de los loci CpG en un ácido nucleico, configurado para determinar el estado de metilación de cada uno de los loci en un ensayo de detección en un único ácido nucleico. En algunas realizaciones preferidas, la determinación comprende el análisis de los loci CpG en un ensayo de detección de ácido nucleico configurado para determinar el estado metilación de cada uno de dichos loci en una única mezcla de reacción. En algunas realizaciones, el ensayo de detección de ácido nucleico comprende un ensayo de extensión por cebador. En ciertas realizaciones preferidas, el ensayo de detección de ácido nucleico puede comprender uno o más de entre un ensayo de amplificación de ácido nucleico, un ensayo de secuenciación de ácido nucleico, un ensayo de escisión específico de la estructura, un ensayo de escisión de 5' nucleasa, un ensayo de escisión invasiva y/o un ensayo de unión.
Los procedimientos de la presente invención no se limitan al análisis de un único ácido nucleico marcador de cáncer o adenoma. Por ejemplo, en algunas realizaciones, el estado de metilación de cada locus CpG en un subconjunto de loci CpG definido en al menos una molécula de ácido nucleico marcador de cáncer o adenoma que comprende el análisis de las moléculas de un ácido nucleico de una pluralidad de marcadores de cáncer o adenoma. En algunas realizaciones, la pluralidad de los marcadores de cáncer o adenoma comprende al menos tres marcadores de cáncer o adenoma, mientras que, en algunas realizaciones, la pluralidad comprende al menos cuatro marcadores de cáncer o adenoma. En algunas realizaciones preferidas, los marcadores de cáncer o adenoma y las moléculas de ácido nucleico se seleccionan de entre el grupo que comprende los marcadores Vimentin, BMP3, Septin 9, TFPI2, 2 regiones de LRAT, y EYA4 y moléculas de ácido nucleico. En algunas realizaciones, los procedimientos de ensayo de la invención se combinan con el análisis de uno o más marcadores de cáncer, tales como marcadores de sangre oculta en heces (por ejemplo, hemoglobina, alfa-defensina, calprotectina, a1-antitripsina, albúmina, MCM2, transferrina, lactoferrina, y lisozima.
En ciertas realizaciones preferidas del procedimiento descrito en el presente documento, una molécula de ácido nucleico marcador de cáncer o adenoma comprende una molécula de ácido nucleico de vimentin, y en algunas realizaciones particularmente preferidas, el subconjunto de loci CpG definido en la molécula de ácido nucleico de vimentin comprende los loci 37, 40 y 45.
En ciertas realizaciones preferidas del procedimiento descrito en el presente documento, una molécula de ácido nucleico marcador de cáncer o adenoma comprende una molécula de ácido nucleico de BMP3, y en algunas realizaciones particularmente preferidas, el subconjunto de loci CpG definido en la molécula de ácido nucleico de BMP3 comprende los loci 34, 53 y 61.
En ciertas realizaciones preferidas del procedimiento descrito en el presente documento, una molécula de ácido nucleico marcador de cáncer o adenoma comprende una molécula de ácido nucleico de Septin 9, y en algunas realizaciones particularmente preferidas, el subconjunto de loci CpG definido en la molécula de ácido nucleico de
Septin 9 comprende los loci 59, 61, 68 y 70.
En ciertas realizaciones preferidas del procedimiento descrito en el presente documento, una molécula de ácido nucleico marcador de cáncer o adenoma comprende una molécula de ácido nucleico de TFPI2, y en algunas realizaciones particularmente preferidas, el subconjunto de loci CpG definido en la molécula de ácido nucleico de TFPI2 comprende los loci 55, 59, 63 y 67.
En ciertas realizaciones preferidas del procedimiento descrito en el presente documento, una molécula de ácido nucleico marcador de cáncer o adenoma comprende una molécula de ácido nucleico de EYA4, y en algunas realizaciones particularmente preferidas, el subconjunto de loci CpG definido en la molécula de ácido nucleico de EYA4 comprende los loci 31, 34, 37, y 44.
En ciertas realizaciones preferidas del procedimiento descrito en el presente documento, el al menos un marcador de cáncer o adenoma o molécula de ácido nucleico comprende una pluralidad de marcadores o moléculas de ácido nucleico que comprende marcadores o moléculas de ácido nucleico de Vimentin, BMP3, Septin 9, y TFPI2.
La presente invención proporciona adicionalmente procedimientos de selección de un subconjunto de loci CpG definido en un ácido nucleico marcador para su uso en un ensayo de detección de ácido nucleico detectando la metilación coordinada del subconjunto de loci CpG definido en el que la metilación coordinada del subconjunto de loci CpG definido es indicativo de un estado no normal, por ejemplo, adenoma o cáncer, comprendiendo el procedimiento A) la determinación del estado de metilación de una pluralidad de loci CpG en cada una de una pluralidad de copias de un ácido nucleico marcador de una pluralidad de muestras normales; b) la determinación del estado de metilación de una pluralidad de loci CpG encada una de una pluralidad de copias individuales de dicho ácido nucleico marcador de una pluralidad de muestras no normales (por ejemplo, adenoma o cáncer), en el que la pluralidad de copias individuales de un ácido nucleico marcador analizado en cada una de dichas muestras de adenoma o cáncer comprende al menos 1.000; c) la determinación de las relaciones de metilación para cada locus de la pluralidad de dichos loci CpG en el ácido nucleico marcador; en el que la determinación de las relaciones de metilación comprende la determinación de la relación entre la media de metilación de cada uno de la pluralidad de loci CpG en las muestras normales respecto a la media de metilación de cada locus CpG correspondiente en dicha pluralidad de loci CpG en las muestras no normales; d) la selección de un conjunto de loci CpG definido en el ácido nucleico marcador, en el que el conjunto de loci CpG definido comprende una pluralidad de loci CpG que tiene relaciones de metilación ventajosas que se correlacionan con el estado no normal (por ejemplo, adenoma o cáncer) y seleccionar un subconjunto de loci CpG definida de dicho conjunto definido, en el que dicho subconjunto de loci CpG definido comprende al menos tres loci CpG, y en el que dicho subconjunto definido se selecciona de manera que el porcentaje de copias individuales de dicho ácido nucleico marcador de dicha pluralidad de muestras normales que están metiladas en todos los dichos menos tres loci CpG de dicho subconjunto definido es menor que el porcentaje de copias individuales de dicho ácido nucleico marcador de dicha pluralidad de muestras de adenoma o dichas muestras de cáncer que están metiladas en todos los dichos al menos tres loci CpG en dicho subconjunto definido.
En realizaciones preferidas, la pluralidad de copias individuales de un ácido nucleico marcador que se analiza en muestras normales y no normales (por ejemplo, adenoma o cáncer) comprende preferentemente al menos 10.000 y más preferentemente al menos 100.000 copias. El número de copias analizadas no se limita a estos números completos, sino que puede ser cualquier entero por encima de 10. El número de copias de diferentes tipos de muestra, por ejemplo, normales y no normales no tiene por qué ser igual.
En ciertas realizaciones preferidas de los procedimientos de selección de un conjunto de loci CpG definido en un ácido nucleico marcador que se describe en el presente documento, la pluralidad de muestras normales y no normales (por ejemplo, de adenoma o cáncer) que se comparan comprende al menos 10, preferentemente al menos 25, más preferentemente al menos 100 muestras. El número de muestras analizadas no se limita a estos números enteros, sino que puede ser cualquier entero por encima de aproximadamente 10. El número de muestras diferentes de los diferentes tipos de muestra, por ejemplo, normales y no normales, no tiene por qué ser igual.
En ciertas realizaciones el conjunto de loci CpG definido comprende al menos cuatro loci CpG, preferentemente al menos cinco loci CpG.
La determinación del estado de metilación de la pluralidad de loci CpG se puede conseguir por cualquier procedimiento conocidos por los expertos en la técnica, incluyendo los descritos como más detalle posteriormente. En algunas realizaciones, el procedimiento comprende el tratamiento del ADN de las muestras con bisulfito. En algunas realizaciones, la determinación del estado de metilación del conjunto de loci CpG definido comprende el análisis digital de cada uno de una pluralidad de loci CpG en una pluralidad de copias individuales de un ácido nucleico marcador. En algunas realizaciones preferidas, el análisis digital comprende la secuenciación digital, y/o la PCR digital. Los procedimientos para preparar las muestras, por ejemplo, muestras fecales, para el análisis también se conocen en la técnica. Véase, por ejemplo, los documentos US7005266; 6.303.304; 5.741.650; 5.952.178; y 6.268.136.
Definiciones
Para facilitar el entendimiento de la presente invención, se definen posteriormente varios términos y frases.
Como se utiliza en el presente documento, las expresiones “secuenciación digital” y “secuenciación de única molécula” se utilizan de manera intercambiable y se refieren a la determinación de la secuencia de nucleótidos de moléculas de ácido nucleico individuales. Los sistemas para la secuenciación de moléculas individuales incluyen, pero no se limitan a los instrumentos 454 FLX™ o 454 TITANIUM™ (Roche), el SOLEXA™/Analizador de Genoma Illumina, el Secuenciador de Molécula Única HELISCOPE™ (Helicos Biosciences), y el secuenciador de ADN SOLID™ (Life Techologies/Applied Biosystems), así como otras plataformas aún en desarrollo por compañías tales como Intelligent Biosystems y Pacific Biosystems.
Como se utiliza en el presente documento, el término “de fondo” como se utiliza en referencia a la metilación de un locus o una región se refiere a la metilación que se observa en una célula o muestra normal en un locus o región de un ácido nucleico que generalmente no está metilado en células normales. Por ejemplo, se considera generalmente que las islas CpG están sin metilar en las células humanas normales pero la metilación no está completamente ausente en las islas CpG de células normales.
Como se utiliza en el presente documento, “metilación” o “metilado”, como se utiliza en referencia al estado de metilación de una citosina, por ejemplo, en un locus CpG, se refiere en general a la presencia o ausencia de un grupo metilo en la posición 5 del resto de citosina (es decir, si una citosina particular es 5-metilcitosina). la metilación se puede determinar directamente, por ejemplo, como se prueba por procedimientos de rutina para el análisis del estado de metilación de citosinas, por ejemplo, determinando la sensibilidad (o falta de la misma) de un resto C en particular a la conversión a uracilo por tratamiento con bisulfito. Por ejemplo, un resto de citosina en una muestra que no se convierte en uracilo cuando la muestra se trata con bisulfito de la manera que se esperaría que se convirtiera ese resto si no estuviera metilado (por ejemplo, en condiciones en las que la mayoría o todas las citosinas no metiladas en la muestra se convierten en uracilos) se puede en general considerar como “metilado”.
Como se utiliza en el presente documento, las expresiones “PCR digital”, “PCR de molécula única” y “amplificación de molécula única” se refiere a una PCR y otros procedimientos de amplificación de ácido nucleico que están configurados para proporcionar un producto de amplificación o señal de una única molécula de partida. Normalmente, las muestras se dividen, por ejemplo, por dilución seriada o por partición en porciones suficientemente pequeñas (por ejemplo, en microcámaras o en emulsiones) de manera que cada porción o dilución tiene, de media, no más de una única copia del ácido nucleico diana. Los procedimientos de PCR de molécula única se describen en, por ejemplo, el documento US 6.143.496, que se refiere un conjunto para contener y particionar fluidos; y el documento US 7.459.315, que se refiere a un procedimiento de división de una muestra en un conjunto con cámaras de muestra en las que las muestras se particionan por afinidad de superficie a las cámaras, entonces se sellan las cámaras con un “fluido de desplazamiento” curable. Véase también los documentos US 6.440.706 y US 6.753.147, y Vogelstein, y col., Proc. Natl. Acad. Sci. USA Vol. 96, pp. 9236-9241, agosto de 1999. Véase también el documento US 20080254474, que describe una combinación de PCR digital combinada con detección de la metilación.
Como se utiliza en el presente documento, “sensibilidad” como se utiliza en referencia a un ensayo diagnóstico, por ejemplo, un ensayo de metilación se refiere a sensibilidad clínica - la proporción de muestras positivas quedan un resultado positivo utilizando un ensayo diagnóstico. La sensibilidad se calcula en general como el número de positivos verdaderos identificados por el ensayo, dividido por la suma del número de positivos verdaderos y el número de falsos negativos determinados por el ensayo de muestras conocidas positivas. De manera similar, el término “especificidad” se refiere a la proporción o número de negativos verdaderos determinados por el ensayo dividido por la suma del número de negativos verdaderos y el número de falsos positivos determinados por el ensayo de muestras negativas conocidas.
Como se utiliza en el presente documento en referencia a ensayos diagnósticos o análisis, el término “complementario” se refiere a diferentes ensayos que, cuando se utilizan en conjunto, proporciona un resultado más sensible y/o específico que el que se puede proporcionar por cualquiera de los diferentes ensayos utilizados solos.
Como se utiliza en el presente documento, el término “ informativo” o “informatividad” se refiere a una cualidad de un marcador o panel de marcadores, y específicamente a la probabilidad de encontrar un marcador (o panel de marcadores) en una muestra positiva.
El término “muestra” como se utiliza en el presente documento se utiliza en su sentido más amplio. Por ejemplo, una muestra sospechosa de contener un gen o cromosoma humano o secuencias asociadas con un cromosoma humano puede comprender una célula, cromosomas aislados de una célula (por ejemplo, un manojo de cromosomas en metafase), ADN genómico (en solución o unido a un soporte sólido tal como por análisis de transferencia de Southern), ADNc (en solución o unido a un soporte sólido) y similares.
Como se utiliza en el presente documento, la expresión “ isla CpG” se refiere a una región de un ADN genómico que contiene un alto porcentaje de sitios CpG respecto a la incidencia media de CpG genómico (por la misma especie, por el mismo individuo, o por subpoblación (por ejemplo, cepa, subpoblación óptica, o similares). Existen distintos parámetros y definiciones para las islas CpG; por ejemplo, en algunas realizaciones, las islas CpG se definen como que tienen un porcentaje de GC que es mayor del 50 % y con una relación de CpG observada/esperada que es
mayor del 60% (Gardiner-Garden y col. (1987) J Mol. Biol. 196:261-282; Baylin y col. (2006) Nat. Rev. Cáncer 6:107-116; Irizarry y col. (2009) Nat. Genetics 41:178-186; cada uno incorporado en el presente documento por referencia en su totalidad). En algunas realizaciones, las islas CpG pueden tener un contenido en GC >55 % y la CpG observada/ CpG esperada de 0,65 (Takai y col. (2007) PNAS 99:3740-3745; que se incorpora en el presente documento por referencia en su totalidad). También existen distintos parámetros con respecto a la longitud de las islas CpG. Como se utiliza en el presente documento, las islas CpG pueden tener menos de 100 pb; 100-200 pb, 200-300 pb, 300-500 pb, 500-750 pb; 750-100 pb; 1000 o más pb de longitud. En algunas realizaciones, las islas CpG muestran patrones de metilación alterados con respecto a los controles (por ejemplo, la metilación alterada en los sujetos cancerosos con respecto a los sujetos sin cáncer; los patrones de metilación alterados específicos de tejido; metilación alterada en las heces de los sujetos con neoplasia colorrectal (por ejemplo, cáncer colorrectal, adenoma colorrectal) con respecto a los sujetos sin neoplasia colorrectal). En algunas realizaciones, la metilación alterada implica hipermetilación. En algunas realizaciones, la metilación alterada implica hipometilación.
Como se utiliza en el presente documento, la expresión “costa CpG” o “costa de la isla CpG” se refiere a una región genómica externa a una isla CpG que es o que tiene el potencial de tener patrones de metilación alterados (véase, por ejemplo, Irizarry y col. (2009) Nat. Genetics 41:178-186; que se incorpora en el presente documento por referencia en su totalidad). Las costas de islas CpG puede mostrar patrones de metilación alterada con respecto a los controles (por ejemplo, la metilación alterada en los sujetos de cáncer con respecto a los sujetos sin cáncer; patrones de metilación alterados específicos de tejidos; metilación alterada en heces de sujetos con neoplasia colorrectal (por ejemplo, cáncer colorrectal, adenoma colorrectal) con respecto a sujetos sin neoplasia colorrectal). En algunas realizaciones, la metilación alterada implica hipermetilación. En algunas realizaciones, la metilación alterada implica hipometilación. Las costas de islas CpG puede localizarse en distintas regiones con respecto a las islas CpG (véase, por ejemplo, Irizarry y col. (2009) Nat. Genetics 41;178-186; que se incorpora en el presente documento por referencia en su totalidad). En consecuencia, en algunas realizaciones, las costas de islas CpG se localizan a menos de 100 pb; 100-250 pb; 250-500 pb; 500-1000 pb; 1000-1500 pb; 1500-2000 pb; 2000-3000 pb; 3000 pb o más, distancia de la isla CpG.
El término “diana”, cuando se utiliza en referencia a un procedimiento de detección o análisis de ácido nucleico, se refiere a un ácido nucleico que tiene una secuencia particular de nucleótido que se va a detectar o analizar, por ejemplo, en una muestra sospechosa de contener el ácido nucleico diana. En algunas realizaciones, una diana es un ácido nucleico que tiene una secuencia particular para la cual es deseable determinar un estado de metilación. Cuando se utiliza en referencia a una reacción en cadena de polimerasa, “diana” se refiere en general a la región de ácido nucleico unida por cebadores utilizados para la reacción en cadena de polimerasa. Por lo tanto, se busca que la “diana” se clasifique fuera de otras secuencias de ácido nucleico que puede estar presente en una muestra. Un “segmento” se define como una región de ácido nucleico en la secuencia diana. La expresión “matriz de muestra” se refiere al ácido nucleico que se origina de una muestra que se analiza en cuanto a la presencia de una diana.
Como se utiliza en el presente documento, el término “ locus” se refiere a una posición particular, por ejemplo, de una mutación, polimorfismo o un resto de C en un dinucleótido CpG, en una región definida o segmento de ácido nucleico, tal como un gen o cualquier otra secuencia caracterizada en un cromosoma o molécula de ARN. Un locus no se limita a cualquier tamaño o longitud particular, y se puede referir a una parte de un cromosoma, un gen, elemento genético funcional, o un único nucleótido o par de bases. Como se utiliza en el presente documento, en referencia a sitios CpG que se pueden metilar, un locus se refiere al resto C del dinucleótido CpG.
Como se utiliza en el presente documento, la expresión “relación de metilación” se refiere a la cantidad o grado de metilación observada para la metilación de una región o locus en particular (por ejemplo, un locus CpG en un gen o región marcadora) en una pluralidad de células no normales (por ejemplo, células en un estado de enfermedad particular, tal como células cancerosas o pre-cancerosas) en comparación con la cantidad o grado de metilación observada para la misma región o locus en una pluralidad de células normales (por ejemplo, células que no están en un estado de enfermedad particular de interés). Por ejemplo, para un locus CpG que presenta una metilación media del 8,39889 % en un muestreo de células normales y una metilación media de 74,0771 % en un muestreo de una pluralidad de células de adenoma, una relación de metilación se puede expresar como la relación de la media determinada para células normales: células de adenoma, o 0,11348. Una relación de metilación no necesita expresarse de una manera en particular o por cualquier cálculo en particular. A modo de ejemplo y sin limitación, la relación de metilación anterior puede expresarse de manera alternativa, por ejemplo, como 8,39889:74,0771; 8,39889/74,0771:8,39889; como un “veces de metilación sobre la de fondo” 8,81987 calculada, etc.
Como se utiliza en el presente documento, “relación de metilación ventajosa” se refiere a una relación de metilación para un locus en el que la metilación se correlaciona con un estado celular, por ejemplo, un estado de enfermedad particular (por ejemplo, normal, pre-cancerosa, cancerosa) que, cuando se compara con la metilación de otro loci que se correlaciona con el mismo estado de enfermedad, presenta un porcentaje de metilación más alto en una población de células no normales en comparación con los niveles de metilación de fondo en el mismo locus en una población de células normales. En algunos ejemplos, ciertos loci CpG, por ejemplo, en una secuencia marcadora de metilación, se presenta una señal-respecto a-ruido mayor, es decir, el grado de metilación en comparación con el de fondo que otro loci en la misma secuencia marcadora. En otros casos, ciertos genes marcadores o genes asociados con enfermedad presentan relaciones de metilación ventajosas en algunos o todos los loci en comparación con las relaciones de metilación observada en algunos o todos los loci en otra secuencia marcadora.
Como se utiliza en el presente documento, la expresión “metilado coordinadamente” se utiliza en referencia a la mutilación de loci, por ejemplo, loci CpG en una secuencia marcadora, que presentan un patrón de metilación particular que se correlaciona con un estado celular, por ejemplo, un estado de enfermedad en particular (por ejemplo, normal. pre-canceroso, canceroso). En realizaciones preferidas la metilación de loci que están todos metilados de manera correlacionada con un estado de enfermedad se puede considerar que están metilados coordinadamente en células que tienen ese estado de enfermedad. “metilación coordinada” no se limita a situaciones en las que todos los loci coordinados están metilados. Cualquier patrón de metilación entre un conjunto de loci particular que se correlaciona con un estado celular, incluyendo los patrones en los que todos los loci coordinados están metilados, patrones en los que los loci presentan un patrón de metilación y no metilación reproducible, y los patrones en los que ninguno de los loci del conjunto está metilado, se incluyen todos en el significado de “metilado coordinadamente”.
Como se utiliza en el presente documento, la expresión “análisis de metilación coordinada” se utiliza de manera intercambiable con “análisis de multimetilación” y se refiere a un ensayo en el que los estados de metilación de una pluralidad de metilación de loci individuales en una secuencia marcadora, por ejemplo, loci CpG, se determinan juntos. En realizaciones preferidas, el análisis de metilación coordinada se lleva a cabo utilizando un procedimiento de copia única/digital (por ejemplo, una secuenciación digital) o un procedimiento de ensayo configurado para interrogar todos los loci CpG seleccionados en cada molécula ensayada, de manera que se revele al patrón de metilación de cada molécula única.
Como se utiliza en el presente documento, la expresión “conjunto definido” de loci CpG (u otra metilación de loci) se refiere al conjunto de loci CpG en un gen o región marcadora para el análisis de metilación. Un conjunto definido de loci CpG en un gen o región marcadora puede comprender todos los loci CpG del gen o región, o puede comprender menos de todos los loci de ese gen o región.
Como se utiliza en el presente documento, la expresión “subconjunto definido” de loci CpG (u otra metilación de loci) se refiere a un subconjunto del conjunto definido de loci CpG en un gen o región marcadora cuya metilación se ha determinado que es indicativa de un estado o normal, por ejemplo, de un adenoma o cáncer. Por ejemplo, en el análisis de metilación coordinada para determinar la presencia de cáncer colorrectal, se determina el estado de metilación de un subconjunto definido de loci CpG en al menos una molécula de ácido nucleico marcador, siendo la metilación simultánea de todos los dicho loci CpG en el subconjunto definido indicativo de cáncer en la muestra. Un subconjunto definido de loci CpG en un gen o región marcadora puede comprender todos los loci CpG en el conjunto definido, o puede comprender menos de todos los loci en el conjunto definido de loci en ese gen o región.
Como se utiliza en el presente documento, la expresión “cáncer colorrectal” significa que incluye la definición médica bien aceptada que define el cáncer colorrectal como una afección médica caracterizada por el cáncer de células del tracto intestinal por debajo del intestino delgado (por ejemplo, en el intestino grueso (colon), incluyendo el ciego, colon ascendente, colon trasverso, colon descendente, y colon sigmoideo, y recto). Adicionalmente, como se utiliza en el presente documento, la expresión “cáncer colorrectal significa que incluye adicionalmente las afecciones médicas que se caracterizan por el cáncer de células del duodeno e intestino delgado (yeyuno e íleon).
Como se utiliza en el presente documento, el término “metástasis” significa que se refiere al proceso en el que las células cancerosas que se originan en un órgano o parte del cuerpo se relocalizan en otra parte del cuerpo y continúan replicándose. Las células metastatizadas posteriormente forman tumores que se pueden metastatizar adicionalmente. La metástasis por lo tanto se refiere a la diseminación del cáncer dese la parte del cuerpo donde existía originalmente a otras partes del cuerpo. Como se utiliza en el presente documento, la expresión “células de cáncer colorrectal metastatizadas” significa que se refiere a células del cáncer colorrectal que han metastatizado; las células de cáncer colorrectal localizadas en una parte del cuerpo distinta del duodeno, intestino delgado (yeyuno e íleon), intestino grueso (colon), incluyendo el ciego, colon ascendente, colon transverso, colon descendente, y colon sigmoideo, y recto.
Como se utiliza en el presente documento, “un individuo sospechoso de ser susceptible a un cáncer colorrectal metastatizado” significa que se refiere a un individuo que tiene un riesgo por encima de la media de desarrollar un cáncer colorrectal metastatizado. Ejemplos de individuos con un riesgo particular de desarrollar un cáncer colorrectal metastatizado son los que cuya historia médica de familia indica una incidencia por encima de la media de cáncer colorrectal entre los miembros de la familia y/o los que ya han desarrollado un cáncer colorrectal y se han tratado eficazmente que por lo tanto afronta un riesgo de recaída o recurrencia. Otros factores que pueden contribuir a un riesgo por encima de la media de desarrollar un cáncer colorrectal metastatizado que por lo tanto daría lugar a la clasificación de un individuo como que es sospechoso de ser susceptible a un cáncer colorrectal metastatizado se puede basar en la genética específica del individuo, los antecedentes médicos o de comportamiento y las características.
El término “neoplasia” como se utiliza en el presente documento se refiere a cualquier crecimiento nuevo y anormal de tejido. Por lo tanto, una neoplasia puede ser una neoplasia premaligna o una neoplasia maligna.
La expresión “marcador específico de neoplasia” se refiere a cualquier material biológico que se puede utilizar para indicar la presencia de una neoplasia. Ejemplos de materiales biológicos incluyen, sin limitación, ácidos nucleicos,
polipéptidos, carbohidratos, ácidos grasos, componentes celulares (por ejemplo, membranas celulares y mitocondrias), y células completas. En algunos casos, los marcadores son regiones de ácido nucleico particulares, por ejemplo, genes, regiones intragénicas, etc. Se hace referencia a las regiones de ácido nucleico que son marcadoras, por ejemplo, como “genes marcadores”, “regiones marcadoras”, “secuencias marcadoras”, etc.
La expresión “marcador específico de neoplasia colorrectal” se refiere a cualquiera material biológico que se puede utilizar para indicar la presencia de una neoplasia colorrectal (por ejemplo, una neoplasia colorrectal premaligna, una neoplasia colorrectal maligna). Ejemplos de marcadores específicos de neoplasia colorrectal incluyen, pero no se limitan a, marcadores epiteliales exfoliados (por ejemplo, bmp-3, bmp-4, SFRP2, vimentin, septin 9, a LX4, EYA4, TFPI2, NDRG4, FOXE1, ADN largo, BAT-26, K-ras, a Pc , antígeno genético de melanoma, p53, BRAF, y PIK3CA) y marcadores de sangre oculta fecal (por ejemplo, hemoglobina, alfa-defensina, calprotectina, a1-antitripsina, albúmina, MCM2, transferrina, lactoferrina y lisozima). Véase también los documentos US 7485420; US7432050; US5352775; US5648212; USRE36713; US5527676; US5955263; US6090566; US6245515; US6677312; US6800617; US7087583; y US7267955.
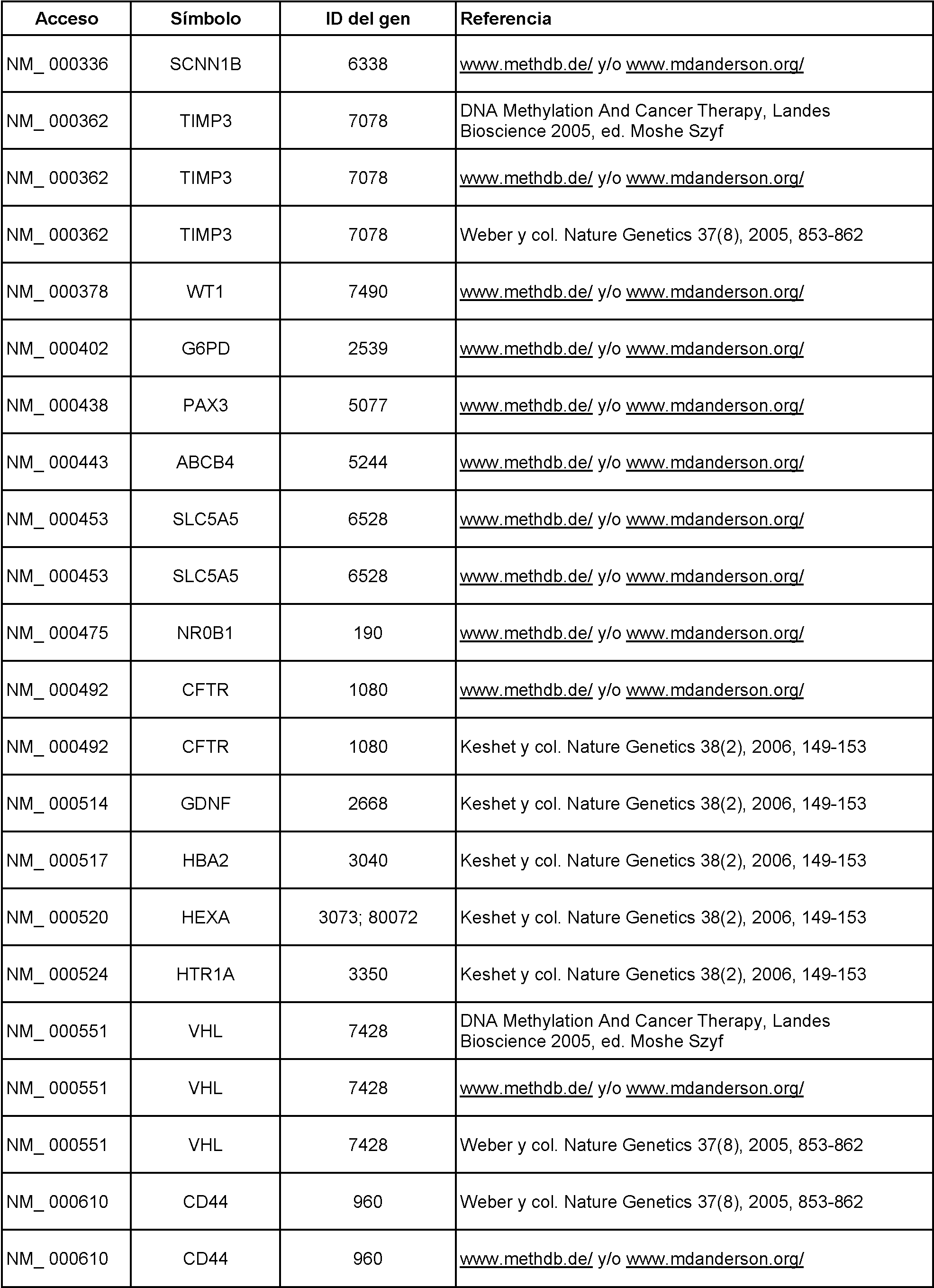
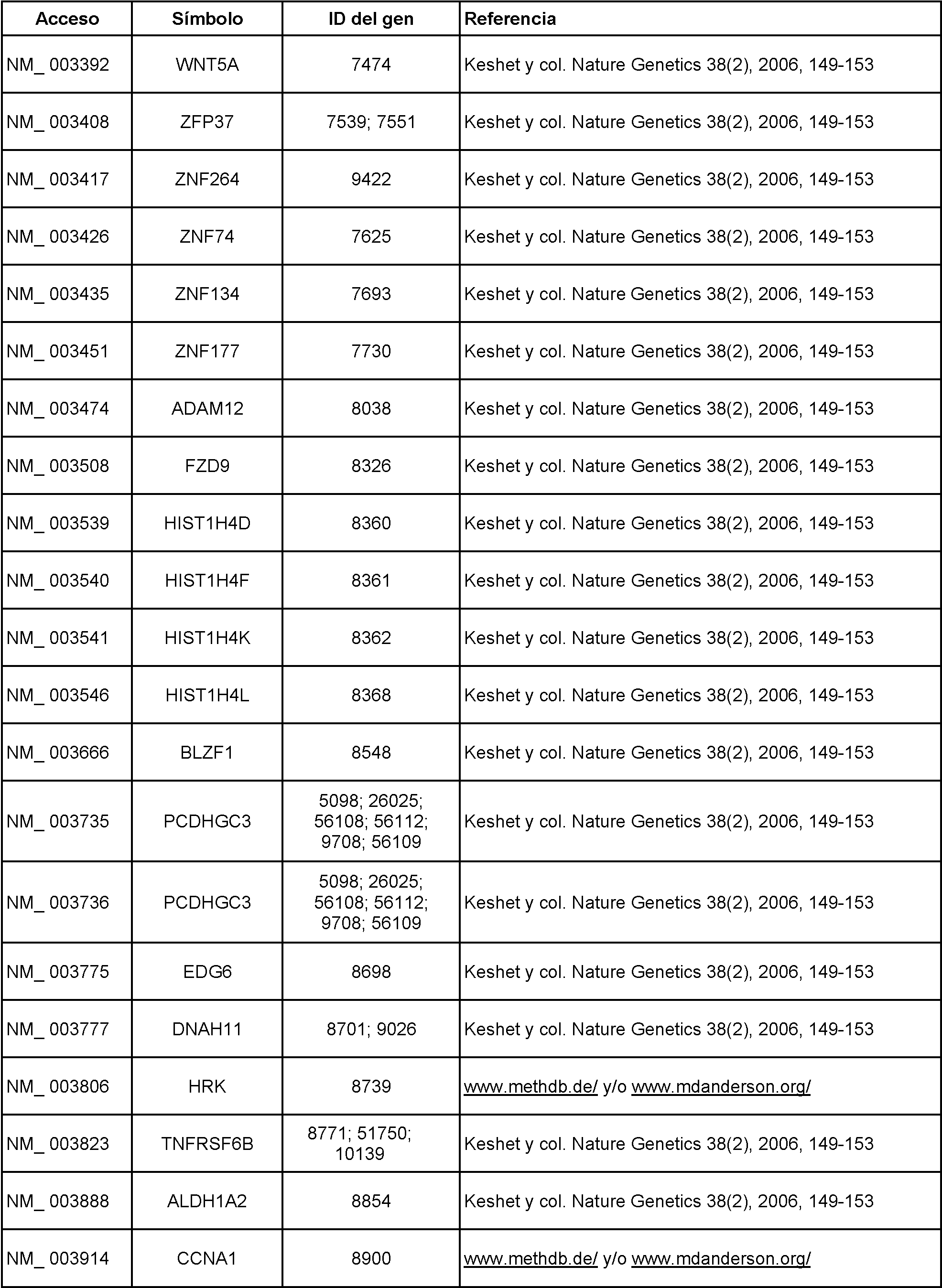
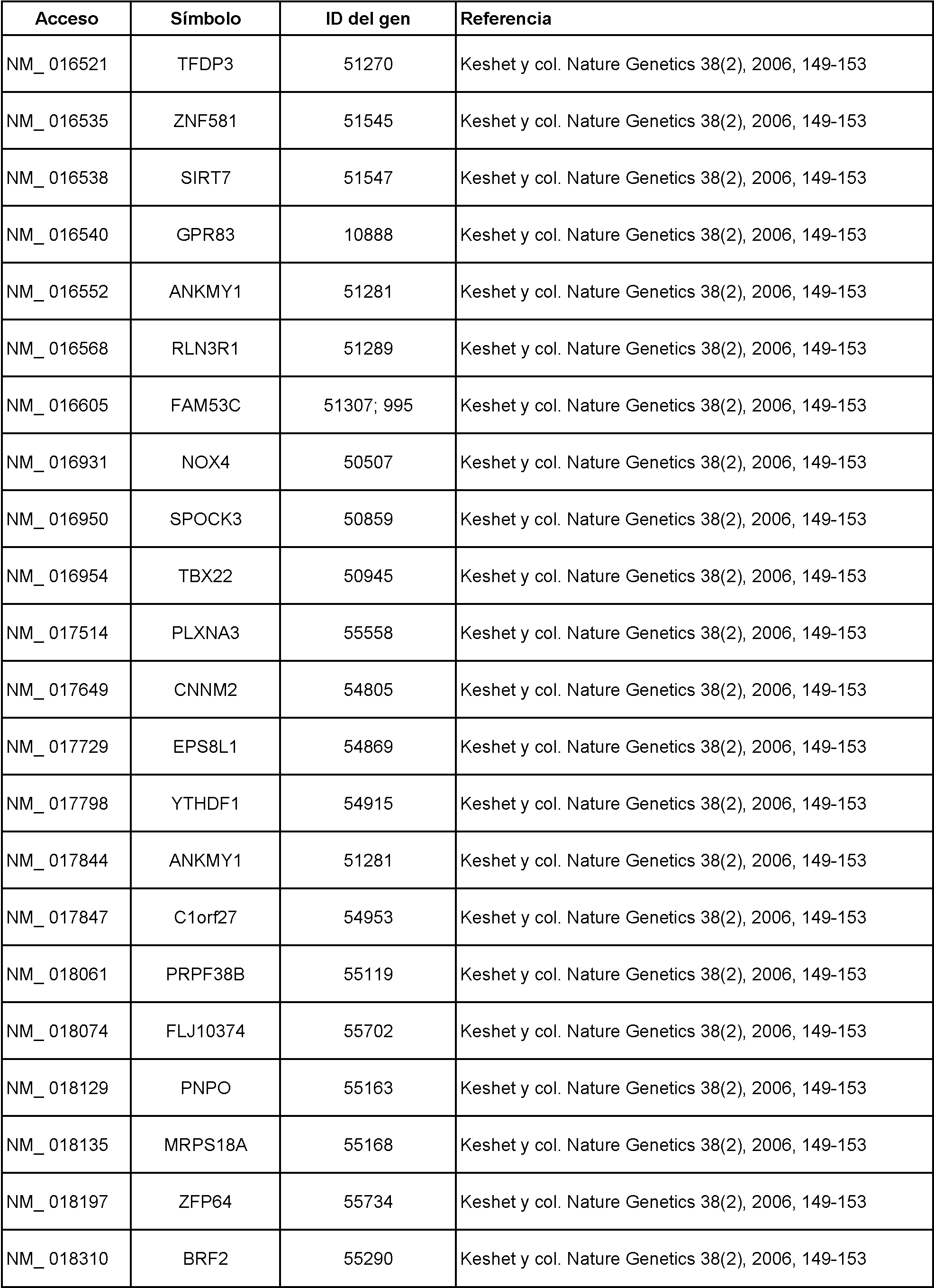
Marcadores adicionales incluyen, pero no se limitan a los de la Tabla 1, a continuación:
Tabla 1

(continuación)

(continuación)
(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)
(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)
(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

Véase también llana Keshet, y col., Nature Genetics 38, 149-153 (1 de febrero de 2006) y Gerd P Pfeifer, y col., Expert Opinion on Medical Diagnostics, septiembre de 2007, Vol. 1, N.° 1, páginas 99-108.
Como se utiliza en el presente documento, el término “adenoma” se refiere a un tumor benigno de origen glandular. Aunque estos crecimientos son benignos, con el tiempo pueden progresar y convertirse en malignos. Como se utiliza en el presente documento, la expresión “adenoma colorrectal” se refiere a un tumor colorrectal benigno en el que las células formas estructuras glandulares reconocibles o en el que las células claramente derivan de un epitelio glandular.
El término “amplificar” o “amplificación” en el contexto de los ácidos nucleicos se refiere a la producción de múltiples copias de un polinucleótido, o una parte del polinucleótido, normalmente comenzando a partir de una pequeña cantidad del polinucleótido (por ejemplo, una única molécula de polinucleótido), donde los productos de amplificación o amplicones son generalmente detectables. La amplificación de polinucleótidos engloba una variedad de procesos químicos y enzimáticos. La generación de múltiples copias de ADN de una o unas cuantas copias de una molécula de ADN diana o matriz durante la reacción en cadena de polimerasa (PCR) o una reacción en cadena de ligasa (LCR; véase, por ejemplo, la Patente de EE. UU. N.° 5.494.810;) son formas de amplificación. Tipos adicionales de amplificación incluyen, pero no se limitan a, PCR específica de alelo (véase, por ejemplo, la Patente de EE. UU. N.° 5.639.611); amplificación dependiente de helicasa (véase, por ejemplo, la Patente de EE. UU. N.° 7.662.594), PCR de inicio caliente (véase, por ejemplo, las Patentes de Ee . UU. N.° 5,773,258 y 5,338,671), PR específica de intersecuencia, PCR inversa (véase, por ejemplo, Triglia, y col. (1988) Nucleic Acids Res., 16:8186), PCR mediada por ligamiento (véase, por ejemplo, Guilfoyle, R. y col., Nucleic Acids Research, 25:1854-1858 (1997); Patente de EE. UU. N.° 5.508.169), PCR específica de metilación (,véase, por ejemplo, Herman, y col., (1996) PnAS 93(13) 9821-9826), PCR de minicebador, amplificación en sonda dependiente de ligamiento múltiple (véase, por ejemplo, Schouten, y col., (2002) Nucleic Acids Research 30(12): e57), PCR múltiple (véase, por ejemplo, Chamberlain, y col., (1988) Nucleic Acids Research 16(23) 11141-11156; Ballabio, y col., (1990) Human Genetics 84(6) 571-573; Hayden, y col., (2008) BMC Genetics 9:80), PCR anidada, PCR de extensión solapada (véase, por ejemplo, Higuchi, y col., (1988) Nucleic Acids Research 16(15) 7351-7367), PCR en tiempo real (véase, por ejemplo, Higuchi, y col., (1992) Biotechnology 10:413-417; Higuchi, y col., (1993) Biotechnology 11:1026-1030), Pc R de transcripción inversa (véase, por ejemplo, Bustin, S.A. (2000) J. Molecular Endocrinology 25:169-193; que se incorpora en el presente documento por referencia en su totalidad), PCR en fase sólida, PCR entrelazada asimétrica térmica y PCR de toma de contacto (véase, por ejemplo, Don, y col., Nucleic Acids Research (1991) 19(14) 4008; Roux, K. (1994) Biotechniques 16(5) 812-814; Hecker, y col., (1996) Biotechniques 20(3) 478-485). La amplificación de polinculeótidos también se puede conseguir utilizando PCR digital (véase, por ejemplo, Kalinina, y col., Nucleic Acids Research. 25; 1999-2004, (1997); Vogelstein y Kinzler, Proc Natl Acad Sci USA. 96; 9236-41, (1999); Publicación de Patente internacional N.° WO05023091A2; Publicación de Patente de EE. UU. N.° 20070202525).
La expresión “reacción en cadena de polimerasa” (“PCR”) se refiere al procedimiento de las patentes N.° 4,683,195, 4,683,202, y 4,965,188 de K.B. Mullis, que describe un procedimiento para aumentar la concentración de un segmento de una secuencia diana en una mezcla de ADN genómico sin clonación ni purificación. Este proceso de
amplificación de la secuencia diana consiste en la introducción de un gran exceso de dos oligonucleótidos cebadores a la mezcla de ADN que contiene la secuencia diana deseada, seguido por una secuencia precisa de ciclado térmico en presencia de una ADN polimerasa. Los dos cebadores son complementarios a sus respectivas cadenas de la secuencia diana de doble cadena. Para efectuar la amplificación, la mezcla se desnaturaliza y entonces los cebadores se hibridan con sus secuencias complementarias de la molécula diana. A continuación de la hibridación, los cebadores se extienden con una polimerasa de manera que forman un nuevo par de cadenas complementarias. Las etapas de desnaturalización, hibridado con cebadores, y extensión con polimerasa se pueden repetir muchas veces (es decir, la desnaturalización, hibridado y extensión constituye un “ciclo”; puede haber numerosos “ciclos”) para obtener una alta concentración de un segmento amplificado de la secuencia diana deseada. La longitud del segmento amplificado de la secuencia diana deseada se determina por las posiciones relativas de los cebadores con respecto uno con el otro, y, por lo tanto, esta longitud es un parámetro controlable. Mediante el aspecto repetitivo del procedimiento, se hace referencia al procedimiento como la “reacción en cadena de polimerasa” (“PCR”). Debido a que los segmentos amplificados deseados de la secuencia diana se convierten en las secuencias predominantes (en términos de concentración) en la mezcla, se dice que han sido “amplificados por PCR” y son “productos de la PCR” o “amplicones”.
Como se utiliza en el presente documento, la expresión “ensayo de detección de ácido nucleico” se refiere a cualquier procedimiento de determinación de la composición de nucleótidos de un ácido nucleico de interés. El ensayo de detección de ácido nucleico incluye, por no se limita a, procedimientos de secuenciación de ADN, procedimientos de hibridación con sondas, ensayos de escisión específica de la estructura (por ejemplo, el ensayo INVADER (Hologic, Inc.) y se describen, por ejemplo, en las Patentes de EE. UU. N.° 5.846.717, 5.985.557, 5.994.069, 6.001.567, 6.090.543, y 6.872.816; Lyamichev y col., Nat. Biotech., 17:292 (1999), Hall y col., PNAS, USA, 97:8272 (2000), y documento US 2009/0253142); procedimientos de escisión enzimático no coincidente (por ejemplo, Variagenics, Pat. de EE. UU. N.° 6.110.684, 5.958.692, 5.851.770); reacción en cadena de la polimerasa; procedimientos de hibridación ramificada (por ejemplo, Chiron, Pat. de EE. UU. N.° 5.849.481, 5.710.264, 5.124.246, y 5.624.802, replicación de círculo rotativo (por ejemplo, Pat. de EE. UU. N.° NASBA (por ejemplo, Pat. de EE. UU. N.° 5.409.818); tecnología de baliza molecular (por ejemplo, Pat. de EE. UU. N.° 6.150.097); tecnología de E-sensor (Motorola, Pat. de EE. UU. N.° 6.248.229, 6.221.583, 6.013.170, y 6.063.573); tecnología de ciclado de sondas (por ejemplo, Pat. de EE. UU. N.° 5.403.711, 5.011.769, y 5.660.988); procedimientos de amplificación de señal de Dade Behring (por ejemplo, Pat. de EE. UU. N.° 6.121.001, 6.110.677, 5.914.230, 5.882.867, y 5.792.614); reacción en cadena de la ligasa ( por ejemplo, Barnay Proc. Natl. Acad. Sci USA 88, 189-93 (1991)); y procedimientos de hibridación en sándwich (por ejemplo, Pat. de EE. UU. N.° 5,288,609).
Como se utilizan en el presente documento, los términos “complementario” o “complementariedad” utilizados en referencia a polinucleótidos (es decir, una secuencia de nucleótidos) se refiere a polinucleótidos relacionados por las reglas de emparejamiento de bases. Por ejemplo, la secuencia “5'-A-G-T-3'” es complementaria a la secuencia “3'-T-C-A-5'”. La complementariedad puede ser “parcial”, nen la que solo algunos de las bases de ácido nucleico se emparejan según las reglas de emparejamiento de bases. O, puede ser una complementariedad “completa” o “total” entre los ácidos nucleicos. El grado de complementariedad entre cadenas de ácidos nucleicos tiene efectos significativos sobre la eficacia y fuerza de la hibridación entre cadenas de ácido nucleico. Esto es de particular importancia en las reacciones de amplificación, así como procedimientos de detección que dependen de la unión entre ácidos nucleicos.
Como se utiliza en el presente documento, el término “cebador” se refiere a un oligonucleótido, sea de origen natural, como en una digestión de restricción purificada, o producido sintéticamente, que es capaz de actuar como un punto de inicio de síntesis cuando se coloca en condiciones en las que la se induce la complementariedad con una cadena de ácido nucleico (por ejemplo, en presencia de nucleótidos y un agente inductor tal como un biocatalizador (por ejemplo, una ADN polimerasa o similar). El cebador normalmente es de cadena sencilla para una máxima eficacia de la amplificación, pero puede ser alternativamente parcial o completamente de cadena doble. La parte del cebador que se hibrida con un ácido nucleico matriz es lo suficientemente larga para cebar la síntesis de productos de extensión en presencia del agente de inducción. Las longitudes exactas de los cebadores dependerán de muchos factores, incluyendo temperatura, fuente del cebador y uso del procedimiento. Los cebadores pueden comprender marcadores, marcas, restos de captura, etc.
Como se utiliza en el presente documento, la expresión “molécula de ácido nucleico” se refiere a cualquier molécula que contiene un ácido nucleico, incluyendo, pero no limitada a, ADN o ARN. La expresión engloba secuencias que incluyen cualquiera de los análogos de bases conocidas de ADN y ARN incluyendo, pero sin limitarse a, 4-acetilcitosina, 8-hidroxi-N6-metiladenosina, aziridinilcitosina, pseudoisocitosina, 5-(carboxihidroxil-metil) uracilo, 5-fluorouracilo, 5-bromouracilo, 5-carboximetilaminometil-2-tiouracilo, 5-carboximetil-aminometiluracilo, dihidrouracilo, inosina, N6-isopenteniladenina, 1-metiladenina, 1-metilpseudouracilo, 1-metilguanina, 1-metilinosina, 2,2-dimetilguanina, 2-metiladenina, 2-metilguanina, 3-metil-citosina, 5-metilcitosina, N6-metiladenina, 7-metilguanina, 5-metilaminometiluracilo, 5-metoxi-amino-metil-2-tiouracilo, beta-D-manosilqueosina, 5'-metoxicarbonilmetiluracilo, 5-metoxiuracilo, 2-metiltio-N- isopenteniladenina, uracilo-5- metiléster de ácido oxiacético, uracilo-5-ácido oxiacético, oxibutoxosina, pseudouracilo, queosina, 2-tiocitosina, 5-metil-2-tiouracilo, 2-tiouracilo, 4-tiouracilo, 5-metiluracilo, N-uracilo-5-metiléster del ácido oxiacético, uracilo-5-ácido oxiacético, pseudouracilo, queosina, 2-tiocitosina, y 2,6-diaminopurina.
Como se utiliza en el presente documento, el término “nucleobase” es sinónimo de otros términos que se utilizan en la técnica que incluyen “nucleótido”, “desoxinucleótido”, “resto de nucleótido”, “resto de desoxinucleótido”, “nucleótido trifosfato (NTP)”, o “desoxinucleótido trifosfato (dNTP)”.
Un “oligonucleótido” se refiere a un ácido nucleico que incluye al menos dos unidades monoméricas de ácido nucleico (por ejemplo, nucleótidos), normalmente más de tres unidades monoméricas, y más normalmente más de diez unidades monoméricas. El tamaño exacto de un oligonucleótido depende en general de distintos factores, incluyendo la última función o uso del nucleótido. Como ilustración adicional, los oligonucleótidos tienen normalmente menos de 200 restos de longitud (por ejemplo, entre 15 y 100), sin embargo, como se utiliza en el presente documento, el término también tiene la intención de englobar cadenas de polinucleótido más largas. A menudo se hace referencia a los oligonucleótidos por su longitud. Por ejemplo, se hace referencia a un oligonucleótido de 24 restos como un “24-mero”. Normalmente, los monómeros de nucleótidos se unen por enlaces fosfodiéster o análogos de los mismos, incluyendo, fosforotioato, fosforoditioato, fosforoselenoato, fosforodiselenoato, fosforoanilotioato, fosforoanilidato, fosforoamidato, y similares, incluyendo los contraiones asociados, por ejemplo, H+, NH4+, Na+, y similares, si dichos contraiones están presentes. Adicionalmente, los oligonucleótidos son normalmente de cadena sencilla. Los oligonucleótidos se preparan opcionalmente por cualquier procedimiento adecuado, incluyendo, pero sin limitarse a, aislamiento de una secuencia existente o natural, replicación o amplificación de ADN, transcripción inversa, clonación y digestión de restricción de secuencias apropiadas, o síntesis química directa por un procedimiento tal como el procedimiento de fosfotriéster de Narang y col. (1979) Meth Enzymol. 68: 90-99; el procedimiento fosfodiéster de Brown y col. (1979) Meth Enzymol. 68: 109 151; el procedimiento dietilfosforoamidita de Beaucage y col. (1981) Tetrahedron Lett. 22: 1859-1862; el procedimiento triéster de Matteucci y col. (1981) J Am Chem Soc. 103:3185-3191; procedimientos de síntesis automática; o el procedimiento de soporte sólido de la Pat. de EE. UU. N.° 4,458,066, titulada "PROCESS FOR PREPARING POLYNUCLEOTIDES," expedida el 3 de jul. de 1984 de Caruthers y col., u otros procedimientos conocidos por los expertos en la técnica.
Una “secuencia” de un biopolímero se refiere al orden e identificación de unidades monoméricas (por ejemplo, nucleótidos, aminoácidos, etc.) en el biopolímero. La secuencia (por ejemplo, una secuencia de base) de un ácido nucleico se lee normalmente en la dirección 5' a 3'.
La expresión “de tipo silvestre” se refiere a un gen o producto genético que tiene las características del gen o producto genético cuando se aísla de una fuente de origen natural. Un gen de tipo silvestre es el que se observa más frecuentemente en una población y por lo tanto se denomina arbitrariamente la forma “normal” o “de tipo silvestre” del gen. Por el contrario, los términos “modificado”, “mutante”, y “variante” se refiere a un gen o producto genético que presenta modificaciones en la secuencia y/o propiedades funcionales (es decir, características alteradas) cuando se comprara con el gen o producto genético de tipo silvestre. Se señala que se pueden aislar mutantes de origen natural; estos se identifican por el hecho de que tienen características alteradas cuando se comparan con el gen o producto genético de tipo silvestre.
Como se utiliza en el presente documento, el término “gen” se refiere a una secuencia de ácido nucleico (por ejemplo, ADN) que comprende las secuencias codificantes necesarias para la producción de un polipéptido, precursor. o ARN (por ejemplo, ARNr, ARNt). El polipéptido puede codificarse por una secuencia codificante de longitud completa o por cualquier parte de la secuencia codificante a condición de que se mantengan la actividad deseada o las propiedades funcionales (por ejemplo, actividad enzimática, unión a ligandos, transducción de señal, inmunogenicidad, etc.) del polipéptido de longitud completa o fragmento. El término también engloba la región codificante de un gen estructural y las secuencias localizadas adyacentes a la región codificante en los extremos 5' y 3' a una distancia de aproximadamente 1 kb o más en cualquier extremo de manera que el gen se corresponde con la longitud del ARNm de longitud completa. Se hace referencia a las secuencias localizadas 5' de la región codificante y presentes en el ARNm como secuencias 5' no traducidas. Se hace referencia a las secuencias localizadas 3' o corriente abajo de la región codificante y presentes en el ARNm como secuencias 3' no traducidas. El término “gen” engloba las formas de ADNc y genómica de un gen. Una forma genómica o clon de un gen contiene la región codificante interrumpida por secuencias no codificantes denominadas “ intrones o “regiones de intervención” o “secuencias de intervención”. Los intrones son segmentos de un gen que se transcriben en un ARN nuclear (por ejemplo, ARNhn); los intrones pueden contener elementos reguladores (por ejemplo, amplificadores). Los intrones se retiran o “recortan” de la transcripción nuclear o primaria; los intrones por lo tanto están ausentes de la transcripción en ARN mensajero (ARNm). El ARNm funciona durante la traducción para especificar la secuencia u orden de aminoácidos en un polipéptido que surge.
Además de contener intrones, las formas genómicas de un gen también pueden incluir secuencias localizadas en el extremo 5' y 3' de las secuencias que están presentes en la transcripción en ARN. Se hacer referencia a estas secuencias como secuencias o regiones “flanqueantes” (estas secuencias flanqueantes se localizan 5' o 3' respecto a las secuencias no traducidas presentes en la transcripción en ARNm). La región flanqueante 5' puede contener secuencias reguladoras tales como promotores y amplificadores que controlan o tienen influencia en la transcripción del gen. La región flanqueantes 30 puede contener secuencias que dirigen la terminación de la transcripción, la escisión post-transcripcional y la poliadenilación.
Como se utiliza en el presente documento, los términos “multimetilación”, “metilación en serie” y “metilación
específica” se utilizan de manera intercambiable para referirse a que las combinaciones definidas de sitios o loci CpG en una secuencia marcadora deben estar metiladas para decir que la secuencia está metilada en un ensayo coordinado o de multimetilación. Por ejemplo, un ensayo de metilación específica de los sitios CpG para BMP3 puede necesitar que las posiciones CpG en 23, 34, 53, 61, 70 y 74, numeradas en referencia a las Figuras 1A y 1B están todas metiladas con el fin de que una muestra se clasifique como metilada en el marcador BMP3. La metilación específica de BMP3 no se limita a este conjunto de loci particulares, sino que puede incluir más, menos o una colección diferente de loci CpG. Los loci CpG seleccionados para el co-análisis en un ensayo de multimetilación se seleccionan preferentemente, por ejemplo, por análisis de muestras normales (sin adenoma, sin cáncer) para identificar las combinaciones de metilación de CpG que se representan menos frecuentemente en las muestras normales. En realizaciones preferidas, las combinaciones de sitios de metilación se seleccionan de entre las que producen una buena señal-respecto a-ruido en muestras de cáncer y adenoma (es decir, cuando la media de multimetilación en una combinación de loci particular en muestras de cáncer dividida por la media de multimetilación en los loci de muestras normales sea alta).
Como se utiliza en el presente documento, los términos metilación “individual” y “media” se utilizan de manera intercambiable para referirse a análisis en los que cada locus CpG se analiza individualmente, de manera que todas las moléculas en las que la base está metilada se incluyen en un recuento, independientemente del estado de metilación de otros loci, por ejemplo, en el mismo marcador. En general los porcentajes de metilación de todos los loci en el marcador/región se promedian entonces, para producir una cifra de porcentaje de metilación para ese marcador.
Como se utiliza en el presente documento, el término “kit” se refiere a cualquier sistema de suministro para suministrar materiales. En el contexto de ensayos de reacción, dichos sistemas de suministro incluyen sistemas que permiten el almacenamiento, transporte, o suministro de reactivos de reacción (por ejemplo, oligonucleótidos, enzimas, etc. en los envases apropiados) y/o materiales de soporte (por ejemplo, tampones, construcciones escritas para llevar a cabo el ensayo, etc.) de una localización a otra. Por ejemplo, los kits incluyen uno o más recipientes (por ejemplo, cajas) que contienen los reactivos de reacción y/o materiales de soporte relevantes. Como se utiliza en el presente documento, la expresión “kit fragmentado” se refiere a un sistema de suministro que comprende dos o más envases separados que contiene cada uno una subparte del total de los componentes del kit. Los envases se pueden suministrar para el receptor que se pretende juntos o por separado. Por ejemplo, un primera envase puede contener una enzima para su uso en un ensayo, mientras que un segundo envase contiene oligonucleótidos. Se tiene la intención que la expresión “kit fragmentado” englobe kits que contengan reactivos específicos de analitos (ASR) regulados bajo la sección 520(e) del Acta Federal de Alimentos, Fármacos y Cosméticos, pero no se limita a estos. Además, cualquier sistema de suministro que comprende dos o más envases separados que contenga cada uno una subparte de los componentes totales del kit se incluyen en la expresión “kit fragmentado”. Por el contrasto, un “kit combinado” se refiere a un sistema de suministro que contiene todos los componentes de un ensayo de reacción en un único envase (por ejemplo, en un único recipiente que alberga cada uno de los componentes deseados). El término “kit” incluye tanto los kits fragmentados como los combinados.
Como se utiliza en el presente documento, el término “ información” se refiere a una colección de hechos o de datos. En referencia a la información almacenada o procesado utilizando un sistema de computadora, incluyendo, pero no limitada a, internet, el término se refiere a cualquier dato almacenado en cualquier formato (por ejemplo, analógico, digital, óptico, etc.). Como se utiliza en el presente documento, la expresión “información relativa a un sujeto” se refiere a los hechos o datos pertenecientes a un sujeto (por ejemplo, un ser humano, planta, o animal). La expresión “información genómica” se refiere a la información perteneciente a un genoma que incluye, pero no se limita a, secuencias de ácido nucleico, genes, frecuencias de alelo, niveles de expresión de ARN, expresión proteica, fenotipos que se correlacionan con Genotipos, etc. “ Información de frecuencia de alelo” se refiere a los hechos o datos que pertenecen a las frecuencias de alelos, incluyendo, pero sin limitarse a, identidades de alelos, correlaciones estadísticas entre la presencia de un alelo y una característica de un sujeto (por ejemplo, un sujeto humano), la presencia o ausencia de un alelo en un individuo o población, el porcentaje de probabilidad de que un alelo esté presente en un individuo que tenga una o más características particulares, etc.
Descripción de los dibujos
Las Figuras 1A y 1B proporciona información de secuencia y CpG para las regiones marcadoras ejemplares en el presente análisis. Para cada gen diana, se muestra la secuencia nativa de la región en la línea superior. Los restos C no metilados que se convertirían por bisulfito y amplificación en Ts se muestran como restos T. Las posiciones candidatas a la metilación se muestran encuadradas. El número de referencia por base y posiciones CpG se muestra encima de cada secuencia nativa. Las localizaciones de cebador para la amplificación se muestran como una fila de posiciones de bases subrayadas.
Las Figuras 2A-J proporciona tablas que muestran los análisis de muestras normales, de adenoma y cáncer en el que la media de metilación se determinó en cada una de las posiciones CpG indicadas, en las regiones marcadoras indicadas. Para cada marcador, las posiciones CpG numeradas son las que se indica en referencia a los números de las Figuras 1A y 1B. La media de metilación de cada locus específico se muestra en la parte de abajo de cada columna para las muestras normales, de adenoma y cáncer. La relación de metilación normal/mutante para cada locus (una relación de metilación en cada locus) se muestra en la parte inferior de
cada columna de los datos de muestras de Adenoma y Cáncer. La columna de Medias a la derecha de cada tabla indica la media de metilación a lo largo de todos los loci CpG indicados para cada una de las muestras. Los valores de la Media y SD a lo largo de todas las muestras normales en todos los loci se indican en la parte inferior de cada tabla de valores de las muestras normales.
Las Figuras 3A-I proporcionan tablas que muestras los análisis de las muestras normales, de adenoma y cáncer en los que se calculó la media de metilación a lo largo de todos los loci CpG indicados en las Figuras 2A-J para cada marcador en cada muestra. Para las muestras normales en la Fig. 3A, se indican la media, desviación típica y la media más 2 o 3 desviaciones típicas para cada marcador. Para las muestras de adenoma y cáncer, las celdas sombreadas en las Fig. 3B y 3C indican un resultado positivo, que se refleja como un valor medio de metilación para ese marcador que es mayor que la media de metilación 3 desviaciones típicas determinadas para ese marcador en las muestras normales.
Las Figuras 3D y 3E muestran el efecto calculado de una dilución de 20 veces de ADN de adenoma y cáncer en ADN normal, Las Figuras 3F y 3G muestran una dilución de 10 veces calculada, y las Fig. 3H y 3I muestran una dilución calculada de 5 veces. En cada una de las diluciones calculadas, la media de metilación para un marcador se divide por 20, 10 o 5, se añade a la media de metilación de ADN normal para ese marcador. Las celdas sombreadas de las Fig. 3D-3I indican un valor de la media de metilación para ese marcador que es mayor que la media de metilación 2 desviaciones típicas (especificidad del 97,5 %) determinada para ese marcador en las muestras normales.
Debajo de cada una de las Figuras 3B-3I, se indican el porcentaje de valores positivos para cada marcador en la muestra tipo y la dilución para es panel. El porcentaje de muestras que dan una señal positiva para al menos uno de los marcadores Vimentin, BMP3, Septin 9 y TFPI2 se indican en la parte inferior de cada panel.
Las Figuras 4A y 4B proporcionan la secuencia y la información de CpG para genes ejemplares utilizados en el presente análisis. Los loci CpG de cada gen marcador incluidos en los subconjuntos definidos de loci CpG para el análisis de metilación coordinada en muestras de adenoma y cáncer colorrectal se muestran con un fondo negro y letras en blanco.
Las Figuras 5A-I proporcionan tablas que muestran los análisis de muestras normales, de adenoma y de cáncer en los que se determinó la metilación de cada una de las posiciones CpG indicadas en las regiones marcadoras indicadas ( es decir, las muestras se ensayaron en cuanto al porcentaje de copias de ADN que presentan metilación en todos los loci CpG del subconjunto definido). Cada marcador se ensayó en cada uno de los loci CpG en los subconjuntos definidos en las Figuras 4A y 4B y los datos del porcentaje de metilación refleja el porcentaje de copias del marcador que tienen metilación en todos los loci CpG ensayados (análisis de metilación coordinada o de “multimetilación”). Para las muestras normales, en la Fig. 5A, se indican la media de multimetilación, la desviación típica y la media más 2 o 3 desviaciones típicas para cada marcador. Para las muestras de adenoma y cáncer, las celdas sombreadas de las Fig. 5B y 5C indican un resultado positivo, reflejado como el valor de multimetilación para ese marcador que es mayor que la media de multimetilación 3 desviaciones típicas determinadas para ese marcador en las muestras normales.
Las Figuras 5D y 5E muestran el efecto calculado de una dilución de 20 veces de ADN de adenoma y cáncer en ADN normal. Las Fig. 5F y 5G muestran una dilución calculada de 10 veces, y 5H y 5I muestran una dilución calculada de 5 veces. En cada una de las diluciones calculadas, la media de multimetilación para un marcador se divide por 30, 10, 9 5, se añade a la media de multimetilación del ADN normal para ese marcador. Las celdas sombreadas de las Fig. 5D-5I indican un valor de la media de multimetilación para ese marcador que es mayor que la media de multimetilación 2 desviaciones típicas (especificidad del 97,5 %) determinada para ese marcador en las muestras normales.
Debajo de cada una de las Fig. 5B-5I, se indica el porcentaje de valores positivos de cada marcador en el tipo de muestra y dilución para ese panel. Se indican el porcentaje de muestras que dan una señal positiva para al menos uno de los marcadores de vimentin, BMP3, Septin 9 y TFPI2 en la parte inferior de cada panel.
La Figura 6 muestra una tabla y un gráfico que comparan los valores del porcentaje de positivos de cada marcador en las muestras de adenoma y cáncer, como se indica, utilizando procedimientos de análisis de metilación individual/media o multimetilación para ensayar cada uno de los marcadores indicados, en cada una de las diluciones calculadas indicadas.
La Figura 7 muestra una tabla y un gráfico que compara los valores del porcentaje de positivos determinados en muestras de adenoma y cáncer, determinados utilizando cuatro marcadores con la media inferior de fondo en estas muestras (vimentin, BMP3, Septin 9, TFPI2), utilizando el procedimiento de análisis de metilación individual/media o multimetilación, en cada una de las diluciones calculadas indicadas en ADN normal.
Descripción detallada de la invención
Se describen las realizaciones de la invención en el presente sumario, y en el Sumario de la invención, anterior, que se incorpora en el presente documento por referencia. Aunque la invención se ha descrito en conexión con
realizaciones específicas, se debería entender que la invención reivindicada no debería limitarse indebidamente a dichas realizaciones específicas.
La presente invención se refiere a procedimientos de determinación, y usos de patrones de metilación específicos indicativos de adenoma y carcinoma de acuerdo con las reivindicaciones. En particular, la invención se refiere a análisis de subconjuntos definidos de loci CpG que están metilados coordinadamente en los ADN de muestras de cáncer y adenoma, y procedimientos para utilizar el análisis de loci metilados coordinadamente en uno o más marcadores o regiones en el diseño de ensayos para adenoma y cáncer que tienen una sensibilidad y especificidad mejoradas.
La presente invención se refiere a la observación de que, en los ácidos nucleicos marcadores para los que el estado de metilación es indicativo del estado celular, por ejemplo, canceroso, pre-canceroso, normal, etc., un subconjunto de la metilación individual de loci, por ejemplo, loci CpG, en células no normales presentan en general un grado de metilación mayor con respecto a los niveles de fondo de metilación observada en los loci correspondiente de las células normales, mientras que la metilación de otros loci en las células no normales pueden presentar niveles de metilación que están más cercanos a los niveles de fondo. En algunas realizaciones, el grado de metilación observada para un locus particular en una pluralidad de células cancerosas o pre-cancerosas con respecto a las células normales se expresa como una relación de metilación.
Algunas realizaciones de la presente invención se refieren a la exploración de genes marcadores conocidos o sospechosos para identificar la metilación específica de loci que presentan relaciones mayores de metilación asociada a enfermedad con respecto a la metilación de fondo, en comparación con otros genes marcadores u otros loci en el mismo gen marcador. En algunas realizaciones preferidas, la presente invención se refiere al análisis de metilación coordinada, para medir el grado hasta el cual una molécula marcadora o una muestra presenta metilación en todos los loci seleccionados de una pluralidad.
La presente invención se refiere al análisis de estados de metilación de un conjunto definido de loci CpG individuales en marcadores de metilación (o regiones diana con dichos marcadores) con un número significativamente suficiente de moléculas de ADN individuales en muestras de adenoma o muestras de cáncer para identificar subconjuntos definidos de loci CpG que tengan relaciones de metilación ventajosas en comparación con otros loci de las mismas muestras de adenoma o cáncer. Un subconjunto definido de loci CpG que tenga relaciones de metilación ventajosas en una muestra puede comprender la totalidad de un conjunto de loci CpG en un marcador particular o región diana de un marcador, o puede ser menor de todos los loci CpG en la región caracterizada del marcador.
Los procedimientos convencionales de análisis del estado de metilación de un marcador generalmente implican el análisis de una población mixta de moléculas. Por ejemplo, la amplificación de un ácido nucleico marcador de una muestra produce en general una mezcla de amplicones que vienen de muchas copias de una molécula diana. Si las condiciones de amplificación no son selectivas de una variante genética, el producto amplicón contiene una mezcla de la variante y el ADN normal o de tipo silvestre. Incluso si los cebadores son específicos de una mutación o para un sitio de metilación particular, cuando se amplifica el ADN a partir de muchas copias de ADN diana derivado de muchas células, puede haber una heterogeneidad en otras posiciones de bases del amplicón resultante. Si esta población mixta, y las secuencias particulares o mutaciones presentes en una pequeña parte de la población son esencialmente no detectables. Aunque algunos investigadores han secuenciados los clones individuales de dichas amplificaciones para examinar la información de secuencia de moléculas individuales de la mezcla, solo se analizaban unas pocas moléculas y los datos acumulados no sugerían que alguno de los loci específicos en los marcadores presentara de manera previsible relaciones de metilación ventajosas en comparación con otros loci metilados de la misma diana, o que el análisis coordinado de loci tuviera relaciones de metilación ventajosas que pudieran ser útiles en mejorar la especificidad y sensibilidad de los ensayos de detección de neoplasias. Un aspecto de la presente invención se basa en la observación de que la recolección de la información de la relación de metilación de una cantidad muy grande de moléculas individuales tanto de muestras normales y no normales revela que la metilación de algunos loci en las regiones marcadores o secuencias presenta un grado mayor de metilación en células no normales en comparación con la de fondo (metilación en los mismos loci de células normales) que otros loci individuales de la misma región marcadora o gen. Estos loci en las secuencias no normales que tienen un nivel de metilación mayor en comparación con la de fondo se puede ver como particularmente ventajoso ya que son más fáciles de identificar sobre el nivel de metilación de fondo observado en las células normales. Un aspecto de esta ventaja es que ese análisis de estos loci particulares permite la identificación de metilación asociada al cáncer con más sensibilidad, y en un nivel mayor al de fondo de las células normales.
La presente invención también se refiere a la observación de que el análisis coordinado de múltiples loci proporciona un nivel significativamente aumentado de sensibilidad de la identificación de células cancerosas o precancerosas, especialmente en muestras que pueden comprender también una cantidad significativa de células normales. Por ejemplo, la Figura 6 compara la sensibilidad de detección de ce de adenoma y cancerosas. Para cada uno de los genes marcadores indicados, la metilación se determinaba como una media a lo largo de la región marcadora (por ejemplo, la media de metilación en el marcador vimentina a lo largo de todos los loci de 26, 37, 40, 45, 52, 54, 59, 63, y 74; véase las Figuras 3A-I), indicada como media de metilación “ individual”, o como un porcentaje de moléculas que presentan metilación en todos un subconjunto de loci seleccionados (por ejemplo, metilación en el marcador vimentin en los tres loci 37, 40, y 45; véase las Figuras 5A-I), es decir, el análisis de metilación coordinada
de múltiples loci individuales, indicado como “multi”. Las sensibilidades de las mismas muestras también se muestran en las diluciones de 5, 10 o 20 veces en ADN normal. La Figura 6 muestra que, mientras que las sensibilidades del ensayo pueden ser similares en el ADN analizado directamente del tejido sin dilución, como la cantidad de fondo del ADN normal se aumenta en las diluciones mayores, el análisis de metilación coordinada muestra que es más sensible que el análisis de media de metilación. Por ejemplo, en el análisis de Septin 9, las muestras de adenoma y cáncer se pueden detectar por encima del fondo solamente en los perfiles sin diluir y de dilución de cinco veces cuando se analiza la media de metilación a lo largo del marcador, mientras que estas mismas muestras se pueden detectar con más de aproximadamente un 69-74 % de sensibilidad con una dilución de 20 veces, y un 90-93 % de sensibilidad con una dilución de 10 veces, cuando se utiliza un análisis de metilación coordinada de los loci 37, 40 y 45.
La divulgación proporciona un procedimiento para diseñar un ensayo de metilación para identificar un estado de enfermedad, que comprende I) la selección de al menos una secuencia de análisis; II)la determinación del estado de metilación de una pluralidad de loci en la secuencia en una población de células normales y una población de células no normales para determinar una tasa media de metilación para cada uno de la pluralidad de loci de cada células normal y no normal; y III) la identificación de al menos dos loci en dicha pluralidad de loci que tiene relaciones de metilación ventajosas.
I. Selección de secuencias. Los marcadores de metilación asociados con estados de enfermedad particulares se han identificado para varios estados de enfermedad. Por ejemplo, los marcadores específicos de neoplasias colorrectales incluyen, por ejemplo, bmp-3, bmp-4, SFRP2, vimentin, septin 9, ALX4, EYA4, TFPI2, NDRG4, FOXE1, ADN largo, BAT-26, K-ras, Ap C, antígeno genético de melanoma, p53, BRAF, y PIK3CA. Marcadores adicionales incluyen, pero no se limitan a los de la Tabla 1 anterior. El análisis de los loci candidatos a metilación para identificar los que tienen relaciones de metilación ventajosas puede comprender el análisis de cada locus en la secuencia diana (por ejemplo, cada CpG)o puede comprender el análisis de un subconjunto de metilación de loci. Los CpG se pueden seleccionar para el análisis por su localización en punto calientes de metilación particulares, mientras que otros loci para el análisis puede localizarse convenientemente con respecto a los sitios de unión al cebador u otras características de secuencia. La Figura 1A proporciona una selección ejemplar de marcadores asociados a una neoplasia con cada uno de los restos C en los loci CpG se indican por un cuadrado. Para cada gen diana, la secuencia nativa de la región se muestra en la línea superior y las secuencias de ADN no metilado y metilado según aparecen después de la conversión con bisulfito y amplificación se muestran debajo. Los restos C no metilados que se convertirían por bisulfito y amplificación en restos T se muestran como Ts.
En algunas realizaciones, la presente invención proporciona el uso de un ensayo de detección de ácidos nucleicos para analizar de manera coordinada una pluralidad de loci ventajosos en una muestra, determinando de esta manera el estado de enfermedad de las células de una muestra.
II. Determinación de las relaciones de metilación para los loci de la secuencia(s) seleccionada. Como se ha expuesto anteriormente, la determinación de la relación de metilación para un locus comprende la determinación de la tasa media de metilación para ese locus en una población de células normales y la determinación de la tasa media de metilación en el mismo locus en una población de células no normales. Como se ha señalado anteriormente, los procedimientos utilizados comúnmente de análisis de metilación de genes marcadores se llevan a cabo en ácidos nucleico mezclados, por ejemplo, amplicones producidos a partir de ADN no fraccionado de una población mixta de células l(tales como el ADN purificado de una muestra tisular multicelular). Mientras que algunos estudios han analizado clones individuales de amplicones producidos a partir de una muestra de ADN no fraccionado, la cantidad de clones analizados ha sido normalmente demasiado pequeña para revelar diferencias significativas o reproducibles en las relaciones de metilación en loci CpG individuales en las secuencias. Por ejemplo, en su comparación de genes altamente metilados en el cáncer colorrectal, Zou, y col. analizaban solamente seis clones de cada muestra (Zou, y col., Cancer Epidemiol Biomarkers Prev 2007;16(12):2686), mientras que Weisenberg, y col. utilizaba la presente invención que comprende el análisis a gran escala de moléculas de ADN individuales, por ejemplo, por secuenciación directa de moléculas de ADN individuales, o por secuenciación del ADN amplificado clónicamente.
Aunque no limitando la presente invención a ningún procedimiento en particular, los procedimientos de amplificación clónica de copias de ácidos nucleicos individuales (por ejemplo, utilizando la PCR) se pueden utilizar en el análisis rápido de grandes cantidades de marcadores individuales de muestras normales y no normales. Los procedimientos de amplificación de molécula única pueden comprender el uso de microcámaras, reacciones de mulsión, “PCR puente” en soportes sólidos, o cualquiera de un número de procedimientos establecidas de segregación de productos de amplificación que aparecen de las moléculas diana individuales. A continuación de la amplificación de molécula única, se pueden secuenciar los amplicones.
Los procedimientos mejorados de secuenciación de molécula individuales directamente obvian la necesidad de clonar moléculas en células o, en algunos procedimientos, la necesidad de amplificar clónicamente antes de la secuenciación. La eliminación de clonación en las células hace que el análisis de colecciones de moléculas mucho mayores sea más eficaz. Las plataformas para la secuenciación de moléculas individuales incluyen 454 FLX™ o 454 TITANIUM™ (Roche), el SOLEXA™/Analizador de Genoma Illumina, el Secuenciador de Molécula Única HELISCOPE™ (Helicos Biosciences), la máquina Genoma Personal Ion (Ion Torrent) y el secuenciador de ADN SOLID™ (Life Techologies/Applied Biosystems), así como otras plataformas aún en desarrollo por compañías tales como Intelligent Biosystems y Pacific Biosystems. Aunque la química por la que se genera la
información de secuencia varía en las plataformas de secuenciación de la próxima generación, todas ellas comparten la característica común de generar datos de secuencia a partir de un gran número de matrices de secuenciación individuales, en reacciones de secuenciación que se ejecutan simultáneamente. Lods datos de las reacciones se recopilan utilizando, por ejemplo, una celda de flujo, un sensor químico u óptico, y/o explorador, y las secuencias se ensamblan y analizan utilizando un software bioinformático. En ciertas realizaciones preferidas, la presente invención proporciona procedimientos de análisis de marcadores de metilación utilizando secuenciación digital para identificar la metilación de loci asociada a neoplasia que tienen relaciones de metilación que son significativamente ventajosos estadísticamente en comparación con otros loci en los mismos marcadores. En realizaciones preferidas, la secuenciación digital se hace de una manera alta o masivamente paralela, proporcionando una precisión mayor en la identificación de la metilación de sitio CpG que tienen relaciones de metilación ventajosas.
Para los procedimientos de secuenciación digital masivamente paralela mencionada anteriormente, cada molécula se analiza en cuanto a la metilación en cada locus CpG, de manera que el porcentaje de copias de ADN que tienen metilación en cualquier combinación de loci CpG se puede analizar después de la ejecución experimental. Además, cada secuencia marcadora particular, por ejemplo, cada molécula de ácido nucleico diana, o amplicón clónico se pueden interrogar muchas, muchas veces, por ejemplo, al menos 100 veces, a veces más de 1000 veces, y en algunos casos por encima de 100.000 veces, o hasta 500.000 veces. Por lo tanto, los patrones de metilación coordinada indicativa de cáncer o adenoma que sería detectable en el análisis de un grupo de moléculas diana individuales se puede revelar.
III. Selección de un subconjunto de loci de metilación por análisis coordinado
Como se ha señalado anteriormente, la determinación del estado de metilación de un grupo de loci CpG en un gran número de copias de ADN marcador de muestras normales y muestras no normales (por ejemplo, de muestras de adenoma o cáncer) revela que ciertos loci CpG de los genes o regiones marcadoras pueden tender a estar metilados de manera coordinada. Además, el diseño de ensayos de detección de ácidos nucleicos para interrogar una pluralidad de loci CpG para los que la metilación coordinada es indicativa de adenoma o cáncer en una muestra puede proporcionar un ensayo que tenga una señal-respecto al ruido mejorada en comparación con ensayos que supervisan el porcentaje medio de metilación a lo largo de genes marcadores completos.
Un aspecto de la selección de un subconjunto de loci CpG comprende la selección de loci que se ha determinado que están metilados coordinadamente por el uso, por ejemplo, de procedimientos de análisis digital. Otro aspecto comprende la selección de loci CpG de los que se ha determinado que tienen relaciones de metilación ventajosas cuando se compara el ADN normal con el ADN de adenoma o cáncer. Los diseños de ensayos pueden, pero no necesariamente, utilizar un locus CpG que tenga la relación de metilación más ventajosa en comparación con otros loci en el mismo marcador. En algunas realizaciones, la selección de una pluralidad de loci CpG como un subconjunto comprende la selección de la pluralidad de loci que tiene las relaciones de metilación más ventajosas. En otras realizaciones, la selección de una pluralidad de loci CpG como un subconjunto comprende la selección del locus que tiene la relación de metilación más ventajosa, seleccionando entonces al menos loci CpG adicionales que está situados convenientemente con respecto al locus seleccionado en primer lugar para la configuración de un ensayo de detección de ácido nucleico particular (por ejemplo, la selección de loci CpG que tiene una proximidad particular entre ellos para la configuración de un ensayo de escisión invasivo, un ensayo de ligadura, un ensayo de amplificación, etc.) con el fin de interrogar a todos los loci CpG seleccionados en copias del ADN diana en un ensayo único. En algunas realizaciones, un subconjunto de loci CpG candidatos se analiza adicionalmente para determinar el porcentaje de copias del ADN marcador de muestras no normales que están metiladas coordinadamente en los loci candidatos, y que tienen poca o ninguna metilación coordinada en las muestras normales.
Análisis de muestras para la detección de adenoma o cáncer
Los procedimientos convencionales de análisis de metilación (por ejemplo, PCR específica de metilación convencional, PCR específica de metilación entiempo real, véase, por ejemplo, los documentos US 5.786.146, 6.017.704, 6.200.756, 6.265.171), analizan normalmente de manera no digital, por ejemplo, analizando una mezcla de moléculas co-amplificadas derivadas de una mezcla de ácidos nucleicos ADN diana, de manera que el análisis de los productos amplificados proporciona la información de secuencia que refleja el estado de metilación agregado o medio en la población de amplicones, por no proporciona información del porcentaje de moléculas de partida que tenían metilación coordinada en todos de una pluralidad de loci CpG. En algunos casos los investigadores han analizado varios amplicones clonados, que pueden revelar la diversidad de metilación en los loci CpG en un gen marcador diana. Sin embargo, la secuenciación de clones individuales no proporciona suficientes datos para revelar una metilación coordinada estadísticamente significativa de subconjuntos de loci CpG específicos.
En contraste con los procedimientos convencionales, los inventores buscaron analizar los genes marcadores de metilación en de una manera de secuenciación digital masivamente paralela para identificar metilación coordinada estadísticamente significativa de loci CpG específicos asociados con neoplasias (adenoma y carcinoma). Este procedimiento de análisis permite:
1. Analizar muestras en cuanto a la metilación coordinada en un gen marcador como medio para detectar neoplasias sin necesidad de ensayar cada marcador (mutación) genético.
2. Analizar muestras en cuanto a metilación coordinada en una pluralidad de genes marcadores como medio de
detección neoplasias sin la necesidad de ensayar ningún marcador (mutación) genético.
Los inventores decidieron utilizar una secuenciación “digital” en un gran número de muestras tisulares obtenidas de biopsias de adenomas colorrectales, cánceres colorrectales, epitelios colorrectales normales y otros cánceres GI y secuenciaron varias regiones específicas con varios genes. Este tipo de secuenciación proporciona un patrón de metilación para cada gen metilado individual. Para la primera ejecución los inventores tenían 9 muestras de tejidos normales, 38 de adenomas y 36 de cáncer con los siguientes marcadores - Vimentin, BMP3, Septin 9, TFPI2, 2 regiones de LRAT, y EYA4.
Sorprendentemente, los inventores descubrieron que, en algunos de los genes, el fondo observado como metilación en muestras normales se distribuye aleatoriamente en las secuencias, mientras que la metilación asociada con el cáncer y adenoma no. Por lo tanto, si se aplican ciertas reglas, por ejemplo, si todos los restos C a, b y c tienen que estar metilados en un ensayo diagnóstico, entonces el número de copias que presentan metilación en las tres posiciones de esa secuencia se reduce en comparación con el número de copias de ADN que presentan metilación en un subconjunto de posiciones. En algunos de los genes o regiones marcadoras ensayadas, la reducción del número de copias de ADN en el ADN que presenta metilación en todos los sitios seleccionados arroja un grado mayor que en el ADN de la muestra de cáncer y/o adenoma, dando como resultado un aumento significativo de la relación de la señal específica respecto al ruido de fondo. Para ciertos genes, el fondo del ADN normal se reduce drásticamente utilizando un análisis de multimetilación (metilación coordinada), mientras que no se ve una reducción equivalente en la señal del ADN de cáncer y adenoma. Para otros genes el fondo en las muestras normales se reduce menos y/o la señal del ADN del cáncer también disminuye con el análisis de multimetilación, de manera que hay menos o ninguna mejoría neta en la señal respecto a ruido y las ventajas de utilizar el análisis de multimetilación son menores. Los genes que tienen una señal respecto a ruido favorable en los análisis de multimetilación se determinan fácilmente empíricamente.
A partir de los datos de multimetilación presentados en el presente documento (véase, por ejemplo, las Figuras 2A-J) es posible:
a. Identificar regiones en las secuencias genéticas que dan una mayor discriminación entre células normales y no normales, por ejemplo, de cáncer y adenoma);
b. Identificar genes particulares que tienen mayor señal respecto a ruido (señal de células no normales en comparación con el fondo de células normales);
c. Identificar la metilación de loci particulares que tiene mayor señal respecto a ruido;
d. Identificar la metilación en loci particulares que tienen metilación coordinadamente en muestras de adenoma y cáncer, pero no en muestras normales, de manera que la detección de metilación coordinada en estos loci es un indicador sensible de adenoma o cáncer;
e. Identificar los genes como metilación de fondo muy bajo que permita una mayor dilución de ADN metilado en ADN normal con menos disminución en la sensibilidad del ensayo;
f. Identificar los genes que son complementarios diagnósticamente entre ellos, es decir, que cuando se analizan en combinación producen una información diagnóstica de elevada sensibilidad y/o elevada especificidad en comparación con los genes analizados solos.
g. Identificar combinaciones de genes que dan una elevada sensibilidad a elevada especificidad, por ejemplo, un 100 % de sensibilidad para el cáncer y adenoma a un 100 % de especificidad.
Ejemplos experimentales
Ejemplo 1
Uso de PCR digital y secuenciación para la identificación de subconjuntos de loci CpG específicos metilados en muestras de cáncer y adenoma
El ADN extraído de las muestras de tejido congeladas se trató con un kit de conversión con bisulfito EPITECT (Qiagen) para convertir las citosinas no metiladas en uracilo. Las citosinas metiladas se mantenían sin convertir. Se diseñaron los cebadores de cada región genética de manera que la composición de los productos de amplificación se mantuviera igual que las secuencias diana originales y las secuencias metiladas y no metiladas se amplificaron con eficacias iguales. La amplificación del ADN convertido que contenía d/U producía amplicones que tienen restos T en lugar de los restos dU. Los amplicones se prepararon entonces por secuenciación en el instrumento Illumina. Para cada muestra de tejido, se preparó la reacción de amplificación de cada diana a partir de la misma muestra de ADN tratado con bisulfito.
Después de la secuenciación, se analizaron cuantitativamente los datos como la media de metilación similar a la secuenciación de Sanger, pero con una precisión y resolución más altas en la que se calcula la señal combinada en cada posición a partir de moléculas individuales. Para cada secuencia de amplicón, se evaluó un conjunto de loci CpG en cuanto al porcentaje de metilación en los diferentes tejidos, para identificar un subconjunto de loci que estaban co-metilados más frecuentemente en muestras de cáncer y/o adenoma que en tejidos normales.
Protocolo de secuenciación Illumina
La secuenciación se llevó a cabo de acuerdo con el procedimiento recomendado por el Analizador de Genoma Illumina IIx, GAIIx, software de colección de datos ver. 2.5, y softwares de análisis Pipeline ver. 1.5. En resumen, el procedimiento Illumina comprende a) preparación de una biblioteca a partir del ADN de la muestra por unión de marcadores de secuencia conocida que permitan la indexación, la unión a la celda de flujo, la amplificación, y la secuenciación; b) unión de la biblioteca a una superficie de celda de flujo; c) amplificación en puente para producir agrupamientos de fragmentos de ADN derivado de moléculas únicas, y d) secuenciación en la utilización de reacciones de extensión por cebador iterativo utilizando terminados reversibles marcados para determinar la secuencia de nucleótidos de cada agrupamiento de amplicones. Véase, por ejemplo, Bentley, y col, Nature 456, 53 59 (6 nov. 2008)/ ido:10.1038/nature07517con procedimientos y datos suplementarios, incorporados en el presente documento por referencia. El uso de secuencias marcadoras únicas para la indexación permite el análisis de muestras múltiples en una única celda de flujo. Véase, por ejemplo, Craig, y col., Nat. Methods Nat Methods. 2008 oct.; 5(10):887-93 (Epub 14 sept. 2008), incorporado en el presente documento por referencia.
Conjunto de muestra: N = 82, compuesto de ADN de tejido extraído de 42 cánceres colorrectales, 31 adenomas pre-cancerosos y 9 de mucosa colónica normal.
Configuración de celda de flujo
Las muestras se indexaron a 12 por calle para un total de 7 calles. Una celda de flujo está compuesta por 8 calles, una de las cuales se dedica a un control de calidad phiX.
Preparación de biblioteca:
El ADN extraído del tejido de pacientes se trató con bisulfito y se llevó a cabo una amplificación en 2 etapas utilizando aproximadamente 10.000 copias genómicas del material inicial. La primera ronda utilizaba cebadores con cola (T1) (Illumina) específicos de las secuencias marcadoras. Estas colas eran secuencias derivadas de Illumina necesarias para la ronda dos. La segunda ronda (T2) (Illumina) la PCR utiliza cebadores específicos para las colas Illumina añadidas en T1, e incorpora el índice, el cebador de secuenciación, y la unión de secuencias a la celda de flujo. Durante la preparación de la biblioteca, se ejecutaron comprobaciones de qPCR múltiples en las muestras para asegurar representaciones equimolares de todos los amplicones de las bibliotecas.
Diseño de cebadores:
Se diseñaron cebadores directo e inverso específicos para las regiones con citosinas no CpG convertidas (utilizando, por ejemplo, software MethPrimer) para amplificar cada uno de los sitios de biomarcador específico de una manera específica de no metilación. Cuando las citosinas CpG no se evitan en el diseño de cebador, se utilizan mezclas degeneradas (C/T; G/A) los sitios en los cebadores. Si es necesario indagar secuencias adicionales fuera del amplicón primario, se pueden diseñar cebadores adicionales. Si las CpG de la secuencia diana no se pueden evitar, los cebadores pueden incorporar bases degeneradas en los sitios CpG (software BiSearch).
Los cebadores para la segunda ronda de PCR comprenden secuencias para la unión a la celda de flujo Illumina (sitios de amplificación en puente), secuenciación de sitios de cebador (para la lectura de muestras), sitios de índice, y sitios de cebadores de secuenciación (par la lectura de indexación). Cada uno de los conjuntos de cebador (x) tiene 12 marcas de índice diferentes, para un total de 12x conjuntos. Los conjuntos de cebador independientes del índice (n = x) se optimizan en ADN no metilado convertido (por ejemplo, ADN humano) y ADN metilado convertido. Por ejemplo, los ADN de control se amplifican, purifican, (por ejemplo, utilizando el tratamiento AMPURE (Agencourt)), y se ejecuta en un Bioanalizador 2100 Agilent para evaluar el tamaño y cantidad de los ácidos nucleicos amplificados.
Etapas experimentales:
Aislamiento de ADN y conversión con bisulfito:
1) Se extrajo el ADN y purificó a partir de los tejidos utilizando los kits DNAZOL (Invitrogen), o QIAAMP (Qiagen) y se midió la concentración y pureza por absorbancia (A230/A260/A280) utilizando un espectrofotómetro Nanodrop ND-1000 (Thermo Scientific). Se utilizó la fluorescencia PIc Og REEN (Molecular Probes) en conjunción con el lector de placas TECAN F-200 (Tecan) para muestras altas que presentaban los valores A230 altos.
2) Se ajustaron las muestras a una concentración de al menos 200 ng/ul utilizando un concentrador por evaporación Speedvac (Thermo Scientific), según fuera necesario.
3) Para cada muestra, se trataron 2 ug de ADN con bisulfito utilizando las placas de 96 pocillos EPITECT (Qiagen).
4) Se evaluó la recuperación por absorbancia con fluorescencia OLIGOGREEN (Molecular Probes) y se evaluó la eficacia de conversión con una PCR cuantitativa utilizando cebadores no CpG que contenían citosina específicos para el ADN no convertido. Se determinó que la eficacia de conversión era mayor del 99 %.
Primera ronda de PCR:
5) Las 84 muestras se amplificaron en reacciones con 30 ng de ADN utilizando conjuntos de cebadores (T1)
Claims (9)
1. Un procedimiento de selección de un subconjunto definido de loci CpG en un ácido nucleico marcador para su uso en un ensayo de detección de ácido nucleico que detecta la metilación coordinada de un subconjunto definido de loci CpG, en el que la metilación coordinada del subconjunto definido de loci CpG es indicativo de adenoma o cáncer, comprendiendo el procedimiento:
a) la determinación del estado de metilación de una pluralidad de loci CpG en cada una de una pluralidad de copias individuales de un ácido nucleico marcador de una pluralidad de muestras normales;
b) la determinación del estado de metilación de dicha pluralidad de loci CpG en cada una de una pluralidad de copias individuales de dicho ácido nucleico marcador de una pluralidad de muestras no normales de sujetos que tienen adenoma o cáncer; en el que la pluralidad de copias individuales de dicho ácido nucleico marcador analizado en cada una de dichas muestras normales y dichas muestras de adenoma o dichas muestras de cáncer comprende al menos 1.000;
c) la determinación de las relaciones de metilación de cada locus CpG en dicha pluralidad de dichos loci CpG en dicho ácido nucleico marcador; en el que la determinación de dichas relaciones de metilación comprende la determinación de la relación entre la media de metilación en cada uno de dicha pluralidad de loci CpG en dichas muestras normales respecto a la media de metilación de cada locus CpG correspondiente en dicha pluralidad de loci CpG en dichas muestras no normales;
d) la selección de un conjunto definido de loci CpG en dicho ácido nucleico marcador, en el que dicho conjunto definido de loci CpG comprende al menos tres loci CpG que tienen relaciones de metilación ventajosas que se correlacionan con adenoma o cáncer; y
e) la selección de un subconjunto definido de loci CpG de dicho conjunto definido, en el que dicho subconjunto definido de loci CpG comprende al menos tres loci CpG, y en el que dicho subconjunto definido se selecciona de manera que el porcentaje de copias individuales en dicho ácido nucleico marcador de dicha pluralidad de muestras normales que están metiladas en todos los dichos al menos tres loci CpG en dicho subconjunto definido es menor que el porcentaje de copias individuales de dicho ácido nucleico marcador de dicha pluralidad de muestras no normales de sujetos que tienen adenoma o cáncer que están metiladas en todos los dichos al menos tres loci CpG en dicho subconjunto definido.
2. El procedimiento de la reivindicación 1, en el que la pluralidad de copias individuales de un ácido nucleico marcador analizado en dichas muestras normales y muestras no normales de sujetos que tienen adenoma o cáncer comprende al menos 10.000, más preferentemente al menos 100.000.
3. El procedimiento de una cualquiera de la reivindicación 1 o la reivindicación 2, en el que la pluralidad de muestras normales y de adenoma o de cáncer comprende al menos 10, preferentemente al menos 25, más preferentemente al menos 100.
4. El procedimiento de una cualquiera de las reivindicaciones 1 a 3, en el que dicho conjunto definido de loci CpG comprende al menos cuatro loci CpG, preferentemente al menos cinco loci CpG.
5. El procedimiento de una cualquiera de las reivindicaciones 1 a 4, en el que dicha determinación del estado de metilación de dicha pluralidad de loci CpG comprende tratar ADN de dichas muestras con bisulfito.
6. El procedimiento de una cualquiera de las reivindicaciones 1 a 5, en el que dicha determinación del estado de metilación de dicha pluralidad de loci CpG comprende el análisis digital de dicha pluralidad de loci CpG en dicho ácido nucleico marcador.
7. El procedimiento de la reivindicación 6, en el que dicho análisis digital comprende al menos una de secuenciación digital y PCR digital.
8. El procedimiento de una cualquiera de las reivindicaciones 1 a 7, en el que dichas muestras no normales de sujetos que tienen adenoma o cáncer comprenden muestras colorrectales.
9. El procedimiento de la reivindicación 1, en el que dichas muestras se seleccionan del grupo que consiste en biopsia tisular, extracto de heces u otros fluidos corporales con sensibilidad y especificidad mejoradas.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161438649P | 2011-02-02 | 2011-02-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2829198T3 true ES2829198T3 (es) | 2021-05-31 |
Family
ID=46577813
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18179746T Active ES2829198T3 (es) | 2011-02-02 | 2012-02-02 | Análisis digital de secuencia de la metilación de ADN |
| ES12742758.1T Active ES2686309T3 (es) | 2011-02-02 | 2012-02-02 | Análisis digital de secuencia de la metilación de ADN |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12742758.1T Active ES2686309T3 (es) | 2011-02-02 | 2012-02-02 | Análisis digital de secuencia de la metilación de ADN |
Country Status (8)
| Country | Link |
|---|---|
| US (5) | US9637792B2 (es) |
| EP (3) | EP3795699B1 (es) |
| JP (1) | JP2014519310A (es) |
| CN (2) | CN110129436A (es) |
| AU (1) | AU2012212127B2 (es) |
| CA (1) | CA2826696C (es) |
| ES (2) | ES2829198T3 (es) |
| WO (1) | WO2012106525A2 (es) |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8673555B2 (en) | 2008-02-15 | 2014-03-18 | Mayo Foundation For Medical Education And Research | Detecting neoplasm |
| US8361720B2 (en) | 2010-11-15 | 2013-01-29 | Exact Sciences Corporation | Real time cleavage assay |
| ES2829198T3 (es) | 2011-02-02 | 2021-05-31 | Exact Sciences Dev Co Llc | Análisis digital de secuencia de la metilación de ADN |
| RU2700088C2 (ru) * | 2012-08-31 | 2019-09-12 | Нэшнл Дифенс Медикл Сентэ | Способы скрининга рака |
| JP6269491B2 (ja) * | 2012-09-19 | 2018-01-31 | シスメックス株式会社 | 大腸癌に関する情報の取得方法、ならびに大腸癌に関する情報を取得するためのマーカーおよびキット |
| CA2935141C (en) * | 2013-10-21 | 2021-06-08 | Sung-Chun Kim | Method and apparatus for analyzing biomolecule by using oligonucleotide |
| US10253358B2 (en) | 2013-11-04 | 2019-04-09 | Exact Sciences Development Company, Llc | Multiple-control calibrators for DNA quantitation |
| US10138524B2 (en) | 2013-12-19 | 2018-11-27 | Exact Sciences Development Company, Llc | Synthetic nucleic acid control molecules |
| EP4234722A3 (en) | 2014-03-31 | 2023-09-20 | Mayo Foundation for Medical Education and Research | Detecting colorectal neoplasm |
| US10184154B2 (en) | 2014-09-26 | 2019-01-22 | Mayo Foundation For Medical Education And Research | Detecting cholangiocarcinoma |
| WO2016160454A1 (en) | 2015-03-27 | 2016-10-06 | Exact Sciences Corporation | Detecting esophageal disorders |
| KR20250073555A (ko) | 2015-10-30 | 2025-05-27 | 이그잭트 사이언시즈 코포레이션 | 복합 증폭 검출 분석 및 혈장으로부터 dna의 분리 및 검출 방법 |
| CN105603061A (zh) * | 2015-12-11 | 2016-05-25 | 宁云山 | 一种基于数字PCR技术检测人Septin 9基因甲基化的方法及所用探针与引物及其试剂盒 |
| DE102015226843B3 (de) * | 2015-12-30 | 2017-04-27 | Technische Universität Dresden | Verfahren und Mittel zur Diagnostik von Tumoren |
| WO2017201400A1 (en) * | 2016-05-19 | 2017-11-23 | The Regents Of The University Of California | Determination of cell types in mixtures using targeted bisulfite sequencing |
| KR20250171455A (ko) | 2017-01-27 | 2025-12-08 | 이그젝트 싸이언스 디블롭먼트 컴패니, 엘엘씨 | 메틸화된 dna 분석에 의한 결장 신조직형성의 검출 |
| US11965157B2 (en) | 2017-04-19 | 2024-04-23 | Singlera Genomics, Inc. | Compositions and methods for library construction and sequence analysis |
| US10975443B2 (en) | 2017-11-30 | 2021-04-13 | Mayo Foundation For Medical Education And Research | Detecting breast cancer |
| US10648025B2 (en) | 2017-12-13 | 2020-05-12 | Exact Sciences Development Company, Llc | Multiplex amplification detection assay II |
| TWI834642B (zh) | 2018-03-13 | 2024-03-11 | 美商格瑞爾有限責任公司 | 異常片段偵測及分類 |
| CN108676878B (zh) * | 2018-05-23 | 2019-12-17 | 杭州诺辉健康科技有限公司 | 检测ndrg4基因甲基化位点的产品在制备结直肠癌早期检测的产品的用途 |
| EP3899953A1 (en) | 2018-12-21 | 2021-10-27 | Grail, Inc. | Source of origin deconvolution based on methylation fragments in cell-free-dna samples |
| KR20220092561A (ko) | 2019-10-31 | 2022-07-01 | 메이오 파운데이션 포 메디칼 에쥬케이션 앤드 리써치 | 난소암 검출 |
| EP4111457A1 (en) * | 2020-02-28 | 2023-01-04 | Grail, LLC | Identifying methylation patterns that discriminate or indicate a cancer condition |
| CN112195243A (zh) * | 2020-09-22 | 2021-01-08 | 北京华大吉比爱生物技术有限公司 | 一种检测多基因甲基化的试剂盒及其应用 |
| CN114561464A (zh) * | 2021-12-20 | 2022-05-31 | 上海锐翌生物科技有限公司 | 用于进展期腺瘤诊断、检测或筛查的引物探针组及试剂盒 |
| CN114317775B (zh) * | 2022-01-12 | 2023-06-30 | 中国人民解放军军事科学院军事医学研究院 | 一种NCOA4的RNA m6A修饰作为γ射线辐射标志物的应用 |
Family Cites Families (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US563961A (en) | 1896-07-14 | Alfred john keys | ||
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US5288609A (en) | 1984-04-27 | 1994-02-22 | Enzo Diagnostics, Inc. | Capture sandwich hybridization method and composition |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4965188A (en) | 1986-08-22 | 1990-10-23 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences using a thermostable enzyme |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US5011769A (en) | 1985-12-05 | 1991-04-30 | Meiogenics U.S. Limited Partnership | Methods for detecting nucleic acid sequences |
| US5124246A (en) | 1987-10-15 | 1992-06-23 | Chiron Corporation | Nucleic acid multimers and amplified nucleic acid hybridization assays using same |
| US5359100A (en) | 1987-10-15 | 1994-10-25 | Chiron Corporation | Bifunctional blocked phosphoramidites useful in making nucleic acid mutimers |
| US5403711A (en) | 1987-11-30 | 1995-04-04 | University Of Iowa Research Foundation | Nucleic acid hybridization and amplification method for detection of specific sequences in which a complementary labeled nucleic acid probe is cleaved |
| CA1340807C (en) | 1988-02-24 | 1999-11-02 | Lawrence T. Malek | Nucleic acid amplification process |
| US5639611A (en) | 1988-12-12 | 1997-06-17 | City Of Hope | Allele specific polymerase chain reaction |
| US5527676A (en) | 1989-03-29 | 1996-06-18 | The Johns Hopkins University | Detection of loss of the wild-type P53 gene and kits therefor |
| US6800617B1 (en) | 1989-03-29 | 2004-10-05 | The Johns Hopkins University | Methods for restoring wild-type p53 gene function |
| US5362623A (en) | 1991-06-14 | 1994-11-08 | The John Hopkins University | Sequence specific DNA binding by p53 |
| US6677312B1 (en) | 1989-03-29 | 2004-01-13 | The Johns Hopkins University | Methods for restoring wild-type p53 gene function |
| CA2036946C (en) | 1990-04-06 | 2001-10-16 | Kenneth V. Deugau | Indexing linkers |
| US5494810A (en) | 1990-05-03 | 1996-02-27 | Cornell Research Foundation, Inc. | Thermostable ligase-mediated DNA amplifications system for the detection of genetic disease |
| US5849481A (en) | 1990-07-27 | 1998-12-15 | Chiron Corporation | Nucleic acid hybridization assays employing large comb-type branched polynucleotides |
| US5352775A (en) | 1991-01-16 | 1994-10-04 | The Johns Hopkins Univ. | APC gene and nucleic acid probes derived therefrom |
| US6872816B1 (en) | 1996-01-24 | 2005-03-29 | Third Wave Technologies, Inc. | Nucleic acid detection kits |
| US5846717A (en) | 1996-01-24 | 1998-12-08 | Third Wave Technologies, Inc. | Detection of nucleic acid sequences by invader-directed cleavage |
| US5994069A (en) | 1996-01-24 | 1999-11-30 | Third Wave Technologies, Inc. | Detection of nucleic acids by multiple sequential invasive cleavages |
| US5338671A (en) | 1992-10-07 | 1994-08-16 | Eastman Kodak Company | DNA amplification with thermostable DNA polymerase and polymerase inhibiting antibody |
| WO1995014106A2 (en) | 1993-11-17 | 1995-05-26 | Id Biomedical Corporation | Cycling probe cleavage detection of nucleic acid sequences |
| ATE248224T1 (de) | 1994-04-25 | 2003-09-15 | Avitech Diagnostics Inc | Die bestimmung von mutationen durch spaltung mit resolvase |
| US5851770A (en) | 1994-04-25 | 1998-12-22 | Variagenics, Inc. | Detection of mismatches by resolvase cleavage using a magnetic bead support |
| DE69528670T2 (de) | 1994-12-23 | 2004-03-11 | Dade Behring Inc., Deerfield | Nachweis von nukleinsäuren durch nuklease-katalysierte produktbildung |
| US5882867A (en) | 1995-06-07 | 1999-03-16 | Dade Behring Marburg Gmbh | Detection of nucleic acids by formation of template-dependent product |
| US5773258A (en) | 1995-08-25 | 1998-06-30 | Roche Molecular Systems, Inc. | Nucleic acid amplification using a reversibly inactivated thermostable enzyme |
| US5854033A (en) | 1995-11-21 | 1998-12-29 | Yale University | Rolling circle replication reporter systems |
| US5965408A (en) | 1996-07-09 | 1999-10-12 | Diversa Corporation | Method of DNA reassembly by interrupting synthesis |
| US5914230A (en) | 1995-12-22 | 1999-06-22 | Dade Behring Inc. | Homogeneous amplification and detection of nucleic acids |
| US5985557A (en) | 1996-01-24 | 1999-11-16 | Third Wave Technologies, Inc. | Invasive cleavage of nucleic acids |
| US5741650A (en) | 1996-01-30 | 1998-04-21 | Exact Laboratories, Inc. | Methods for detecting colon cancer from stool samples |
| EP0892808B1 (en) | 1996-04-12 | 2008-05-14 | PHRI Properties, Inc. | Detection probes, kits and assays |
| US5786146A (en) | 1996-06-03 | 1998-07-28 | The Johns Hopkins University School Of Medicine | Method of detection of methylated nucleic acid using agents which modify unmethylated cytosine and distinguishing modified methylated and non-methylated nucleic acids |
| US6017704A (en) | 1996-06-03 | 2000-01-25 | The Johns Hopkins University School Of Medicine | Method of detection of methylated nucleic acid using agents which modify unmethylated cytosine and distinguishing modified methylated and non-methylated nucleic acids |
| US5952178A (en) | 1996-08-14 | 1999-09-14 | Exact Laboratories | Methods for disease diagnosis from stool samples |
| US6096273A (en) | 1996-11-05 | 2000-08-01 | Clinical Micro Sensors | Electrodes linked via conductive oligomers to nucleic acids |
| US6143496A (en) | 1997-04-17 | 2000-11-07 | Cytonix Corporation | Method of sampling, amplifying and quantifying segment of nucleic acid, polymerase chain reaction assembly having nanoliter-sized sample chambers, and method of filling assembly |
| US6013459A (en) | 1997-06-12 | 2000-01-11 | Clinical Micro Sensors, Inc. | Detection of analytes using reorganization energy |
| US6268136B1 (en) | 1997-06-16 | 2001-07-31 | Exact Science Corporation | Methods for stool sample preparation |
| US6063573A (en) | 1998-01-27 | 2000-05-16 | Clinical Micro Sensors, Inc. | Cycling probe technology using electron transfer detection |
| ATE224458T1 (de) | 1998-02-04 | 2002-10-15 | Variagenics Inc | Techniken zu detektion von fehlpaarungen |
| US6235502B1 (en) | 1998-09-18 | 2001-05-22 | Molecular Staging Inc. | Methods for selectively isolating DNA using rolling circle amplification |
| US6440706B1 (en) | 1999-08-02 | 2002-08-27 | Johns Hopkins University | Digital amplification |
| US7005266B2 (en) | 2000-02-04 | 2006-02-28 | Qiagen Gmbh | Nucleic acid isolation from stool samples and other inhibitor-rich biological materials |
| US7611869B2 (en) * | 2000-02-07 | 2009-11-03 | Illumina, Inc. | Multiplexed methylation detection methods |
| US7432050B2 (en) | 2001-10-05 | 2008-10-07 | Case Western Reserve University | Methods and compositions for detecting colon cancers |
| ATE393240T1 (de) * | 2001-11-16 | 2008-05-15 | Univ Johns Hopkins Med | Verfahren zum nachweis von prostatakrebs |
| US20030215842A1 (en) * | 2002-01-30 | 2003-11-20 | Epigenomics Ag | Method for the analysis of cytosine methylation patterns |
| EP1340818A1 (en) * | 2002-02-27 | 2003-09-03 | Epigenomics AG | Method and nucleic acids for the analysis of a colon cell proliferative disorder |
| AU2003302146B2 (en) * | 2002-06-26 | 2008-06-26 | Cold Spring Harbor Laboratory | Methods and compositions for determining methylation profiles |
| US7662594B2 (en) | 2002-09-20 | 2010-02-16 | New England Biolabs, Inc. | Helicase-dependent amplification of RNA |
| US7485420B2 (en) | 2003-08-14 | 2009-02-03 | Case Western Reserve University | Methods and compositions for detecting colon cancers |
| WO2005023091A2 (en) | 2003-09-05 | 2005-03-17 | The Trustees Of Boston University | Method for non-invasive prenatal diagnosis |
| US20070059753A1 (en) * | 2005-09-15 | 2007-03-15 | Tatiana Vener | Detecting gene methylation |
| LT3002338T (lt) | 2006-02-02 | 2019-10-25 | Univ Leland Stanford Junior | Neinvazinė vaisiaus genetinė atranka, panaudojant skaitmeninę analizę |
| US20100092981A1 (en) * | 2006-04-03 | 2010-04-15 | Genzyme Corporation | Methods of detecting hypermethylation |
| EP2099931A4 (en) * | 2006-12-19 | 2010-11-24 | Cornell Res Foundation Inc | USE OF LECITHIN: RETINOL-ACYLTRANSFERASE GENEPROMOTERMETHYLATION IN THE EVALUATION OF A PERSON'S CANCER STATUS |
| GB0700374D0 (en) * | 2007-01-09 | 2007-02-14 | Oncomethylome Sciences S A | NDRG family methylation markers |
| US9290803B2 (en) * | 2007-04-12 | 2016-03-22 | University Of Southern California | DNA methylation analysis by digital bisulfite genomic sequencing and digital methylight |
| MX2010010161A (es) | 2008-03-15 | 2011-03-21 | Hologic Inc Star | Composiciones y metodos para el analisis de moleculas de acido nucleico durante reacciones de amplificacion. |
| US8609327B2 (en) * | 2008-07-10 | 2013-12-17 | International Business Machines Corporation | Forming sub-lithographic patterns using double exposure |
| US20100124747A1 (en) * | 2008-11-03 | 2010-05-20 | University Of Southern California | Compositions and methods for diagnosis or prognosis of testicular cancer |
| US20100273164A1 (en) * | 2009-03-24 | 2010-10-28 | President And Fellows Of Harvard College | Targeted and Whole-Genome Technologies to Profile DNA Cytosine Methylation |
| US20120164638A1 (en) * | 2009-04-06 | 2012-06-28 | Case Western Reserve University | Digital Quantification of DNA Methylation |
| US8916344B2 (en) * | 2010-11-15 | 2014-12-23 | Exact Sciences Corporation | Methylation assay |
| ES2829198T3 (es) | 2011-02-02 | 2021-05-31 | Exact Sciences Dev Co Llc | Análisis digital de secuencia de la metilación de ADN |
-
2012
- 2012-02-02 ES ES18179746T patent/ES2829198T3/es active Active
- 2012-02-02 US US13/364,978 patent/US9637792B2/en active Active
- 2012-02-02 AU AU2012212127A patent/AU2012212127B2/en active Active
- 2012-02-02 CN CN201910145167.XA patent/CN110129436A/zh active Pending
- 2012-02-02 EP EP20193533.5A patent/EP3795699B1/en active Active
- 2012-02-02 EP EP18179746.5A patent/EP3441479B1/en active Active
- 2012-02-02 WO PCT/US2012/023646 patent/WO2012106525A2/en not_active Ceased
- 2012-02-02 EP EP12742758.1A patent/EP2670893B1/en active Active
- 2012-02-02 ES ES12742758.1T patent/ES2686309T3/es active Active
- 2012-02-02 CA CA2826696A patent/CA2826696C/en active Active
- 2012-02-02 JP JP2013552632A patent/JP2014519310A/ja active Pending
- 2012-02-02 CN CN201280016745.6A patent/CN103703173A/zh active Pending
-
2016
- 2016-09-28 US US15/278,697 patent/US10519510B2/en active Active
-
2019
- 2019-10-28 US US16/665,738 patent/US10870893B2/en active Active
-
2020
- 2020-11-23 US US17/101,904 patent/US11952633B2/en active Active
-
2024
- 2024-04-05 US US18/628,011 patent/US20240368702A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| JP2014519310A (ja) | 2014-08-14 |
| EP2670893A2 (en) | 2013-12-11 |
| WO2012106525A2 (en) | 2012-08-09 |
| WO2012106525A3 (en) | 2014-02-20 |
| US10870893B2 (en) | 2020-12-22 |
| US10519510B2 (en) | 2019-12-31 |
| US11952633B2 (en) | 2024-04-09 |
| EP2670893A4 (en) | 2015-11-11 |
| EP3795699B1 (en) | 2023-12-20 |
| CN110129436A (zh) | 2019-08-16 |
| US20240368702A1 (en) | 2024-11-07 |
| AU2012212127A1 (en) | 2013-08-22 |
| US20120196756A1 (en) | 2012-08-02 |
| CA2826696C (en) | 2019-12-03 |
| US9637792B2 (en) | 2017-05-02 |
| EP3441479B1 (en) | 2020-09-02 |
| US20200048720A1 (en) | 2020-02-13 |
| EP3441479A1 (en) | 2019-02-13 |
| EP2670893B1 (en) | 2018-06-27 |
| CA2826696A1 (en) | 2012-08-09 |
| ES2686309T3 (es) | 2018-10-17 |
| EP3795699A1 (en) | 2021-03-24 |
| CN103703173A (zh) | 2014-04-02 |
| AU2012212127B2 (en) | 2016-06-02 |
| US20210095350A1 (en) | 2021-04-01 |
| US20170073771A1 (en) | 2017-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2829198T3 (es) | Análisis digital de secuencia de la metilación de ADN | |
| ES2812753T3 (es) | Detección de neoplasma colorectal | |
| ES2681035T3 (es) | Método para detectar pólipos gástricos y cáncer gástrico utilizando un gen marcador de pólipos gástricos y metilación específica del cáncer gástrico | |
| WO2016101258A1 (zh) | 一种检测与人体异常状态相关的差异甲基化CpG岛的方法 | |
| ES2778028T3 (es) | Un método de cribado para cáncer colorrectal | |
| BR122020021163B1 (pt) | métodos de triagem de início ou predisposição para início de neoplasma do intestino grosso ou monitoramento do progresso de neoplasma em indivíduo | |
| US12252747B2 (en) | Unbiased DNA methylation markers define an extensive field defect in histologically normal prostate tissues associated with prostate cancer: new biomarkers for men with prostate cancer | |
| CN105452485A (zh) | 用于检测癌前病损的方法 | |
| WO2022022386A1 (zh) | 早期结直肠癌和腺瘤的dna甲基化标志物、检测其的方法及其应用 | |
| US20130225420A1 (en) | Molecular subtyping of oral squamous cell carcinoma to distinguish a subtype that is unlikely to metastasize | |
| Tan et al. | Quantitative methylation analyses of resection margins predict local recurrences and disease-specific deaths in patients with head and neck squamous cell carcinomas | |
| CN113430272A (zh) | 一种食管癌或癌前病变的诊断或辅助诊断的试剂、试剂盒及其应用 | |
| EP2978861B1 (en) | Unbiased dna methylation markers define an extensive field defect in histologically normal prostate tissues associated with prostate cancer: new biomarkers for men with prostate cancer | |
| HK40050531A (en) | Digital sequence analysis of dna methylation | |
| CN117604095A (zh) | 用于食管癌诊断的甲基化检测试剂及试剂盒 | |
| KR20110084486A (ko) | 장암 진단을 위한 장암 특이적 메틸화 마커 유전자의 메틸화 검출방법 |